Изобретение относится к химии гетероциклических органических соединений и их фармацевтическим композициям. Эти соединения и их фармацевтические композиции селективно ингибируют активность ataxia teleangiectasia mutated («АТМ» - мутантный при атаксии-телеангиэктазии белок) и DNA-dependent protein kinase («DNA-PK» - ДНК-зависимая протеинкиназа) серин/треониновых протеинкиназ. В связи с этим изобретение также относится к методу использования таких соединений и их композиций для лечения и/или профилактики онкологических заболеваний в качестве монотерапии и/или в комбинации с радиотерапией, химиотерапией и/или иммунотерапией.
Представители семейства киназ, родственных по отношению к фосфатидилинозитол-3-киназам (phosphatidylinositol 3-kinase related kinases, PIKK), - ATM и DNA-PK киназы, являются важными клеточными модуляторами, активность которых в частности связана с оксидативным стрессом и повреждением молекул ДНК, в особенности двухцепочечными разрывами. В условиях клинических испытаний было показано, что ингибирование активности указанных киназ (отдельно каждой или двух одновременно) с использованием малых лекарственных молекул значительно повышает чувствительность опухолевых клеток к ионизирующему облучению в рамках радиотерапии и действию некоторых классов противоопухолевых препаратов. При этом молекулы этого класса сами по себе не оказывают влияния на нормальные клетки. Таким образом, двойные ингибиторы ATM и DNA-PK в комбинации с радиотерапией, химиотерапией и/или иммунотерапией могут быть использованы для эффективного лечения онкологических заболеваний, значительно потенцируя эффект указанных терапевтических подходов. Применение таких молекул позволяет, в частности, снизить дозы радиации или ДНК-нацеленной противоопухолевой лекарственной молекулы, снижая тем самым нежелательные побочные эффекты. Терапия пациента двойными ингибиторами ATM/DNA-PK приводит к замедлению или прекращению репарации ДНК после радиотерапии. Таким образом, удается значительно усилить апоптоз выживших после терапевтического воздействия опухолевых клеток. Например, у мышей с мутациями в генах ATM и PRKDC, которые приводили к потере функции кодируемых ими белков ATM и DNA-PK соответственно, наблюдалась гиперчувствительность к ионизирующей радиации. Ожидается, что одновременное ингибирование ATM и DNA-PK будет более эффективно сенситизировать опухолевые клетки к радиации или другим ДНК-повреждающим агентам по сравнению с ингибированием какой-то одной из этих киназ. Минимизация нецелевого ингибирования родственных киназ, таких как mTOR или PI3Ks, может понизить токсичность ингибиторов этого класса. В рамках настоящего изобретения были разработаны двойные ингибиторы ATM и DNA-PK киназ, которые не оказывают фармацевтически значимого ингибирующего действия в отношении родственных киназ (PI3Kα, PI3Kβ, PI3Kδ, PI3Kγ и mTOR). Такие молекулы способны значительно повышать чувствительность опухолевых клеток к радиации и/или к определенным химиотерапевтическим агентам.
В настоящее время несколько малых лекарственных молекул находятся на ранних стадиях клинических испытаний в качестве монотерапии (МТ), хемо- (ХС) и радио-сенсибилизаторов (PC), включая: XRD-0394 (ATM/DNA-PK ингибитор, фаза-I, XRad Therapeutics, PC), лартесертиб (ATM ингибитор, фаза-I, Merck, МТ), М-3541 (ATM ингибитор, фаза-I, Merck, PC), AZD-1390 (ATM ингибитор, фаза-I, AstraZeneca, PC+XC), CC-115 (DNA-PK/mTOR ингибитор, фаза-П, Celgene, MT+PC+XC), BR-101801 (DN A-PK/PI3Kγ ингибитор, фаза-I, Boryung, МТ), M-9831 (DNA-PK ингибитор, фаза-I, Merck и Vertex, MT+XC), панулисиб (DNA-PK/mTOR/PI3Kα/ALK1 ингибитор, фаза-I, Piramal Enterprises и Piramal Life Sciences, МТ), пепосертиб (DNA-PK ингибитор, фаза-I, Merck, PC+MT+XC). На фармацевтическом рынке отсутствуют молекулы этого класса. Указанные соединения испытываются против онкологических заболеваний, включая: липосаркому, рак головного мозга, глиобластому, немелкоклеточный рак легких, рак голова и шеи, рак кожи, рак простаты, хронический лимфоцитарный лейкоз, лимфома (В- и Т-клеточная), гематологический рак крови, нейроэндокринный рак, аденокарцинома, рак мочевого пузыря, рак молочной железы и рак кишечника. Одной из особенностей некоторых молекул этого класса является способность проникать через гематоэнцефалический барьер (ГЭБ), например, AZD-1390, что может привести к разработке ЦНС-нацеленных хемо- и радиосенсибилизаторов.
В опубликованной заявке Китая CN 103936762 А, МПК C07D 498/04, A61K 31/5383, А61Р 35/00, А61Р 35/02, 23.07.2014, обсуждается синтез и результаты биологического тестирования для производных N-(5-(3,4-дигидро-2H-[1,4]оксазино[3,2-с]хинолин-9-ил)пиридин-3-ил)сульфонамидов в качестве ингибиторов mTOR и PI3K киназ, включая их 3-оксо-производные. В качестве возможных заместителей в положении 2 пиридинового кольца указаны в том числе различные эфиры. Однако активности по отношению к киназам ATM, DNA-PK и ATR не приводится. Кроме того, в примерах представлены исключительно 2-метокси-пиридил и 2-хлор-пиридил производные. С учетом того, что в рамках представленной заявки нами были получены селективные ингибиторы ATM (IC50<10 нМ) и DNA-PK (IC50<10 нМ), не показавшие активности против mTOR (IC50>1 мкМ, ИСмакс=2,000) и изоформ PI3K (IC50>1 мкМ, ИСмакс>10,000), не очевидно, что структуры, описанные в заявке CN 103936762 А, будут ингибировать ферментативную активность ATM, DNA-PK и ATR и наоборот. В нашем случае было показано, что 4-N-Me (R3=Me) и 5-N (Z1=N - циннолин) производные оказывают влияние на профиль селективности молекул этого класса. Кроме того, в CN 103936762 А циннолиновые производные не заявлены. Влияние метальной группы на селективность было ранее отмечено в серии 1H-имидазо[4,5-с]хинолин-2(3H)-онов (Pike K.G. et al. J Med Chem 2018; 61: 3823). Для представленной в качестве примера молекулы (8-(6-(3-(диметиламино)пропокси)пиридин-3-ил)-3-метил-1-(тетрагидро-2H-пиран-4-ил)-1H-имидазо[4,5-с]хинолин-2(3H)-он) были получены следующие активности (IC50 мкМ): 0,00004 (ATM), 0,14 (DNA-PK), 0,20 (mTOR), 0,32 (PI3Kα), 1,8 (PI3Kβ), 1,1 (PI3Kγ) и 0,27 (PI3Kδ). Несмотря на высокую активность против ATM киназы, молекула сравнительно сильно ингибировала активность mTOR, PI3Kα и PI3Kδ.
В опубликованной международной заявке WO 2020052688 А1, МПК C07D 487/04, A61K 31/5025, А61Р 35/00, 19.03.2020, представлены производные 8-(пиридин-3-ил)-1H-имидазо[4,5-с]циннолин-2(3H)-онов в качестве селективных ингибиторов ATM киназы. В частности, было показано, что циннолиновые аналоги, замещенные фтором по положению 7 в подструктуре 8-(пиридин-3-ил)-1H-имидазо[4,5-с]циннолин-2(3H)-она селективно ингибируют активность ATM киназы (IC50=1,9 нМ), при этом активность в отношении ATR, PI3Kα, PI3Kβ, PI3Kδ, PI3Kγ и mTOR была больше 10 мкМ. Однако, в WO 2020052688 А1 обсуждаются исключительно фторзамещенные в положении 7 1H-имидазо[4,5-с]хинолин-2(3H)-оны или 1H-имидазо[4,5-с]циннолин-2(3H)-оны. При этом авторы отмечают, что циннолиновая модификация позволяет снизить аффинность молекул по отношению к альдегидоксидазе, что препятствует их метаболизму, но не связывают ее с селективностью.
В опубликованной международной заявке WO 2021022078 А1, МПК A61K 31/444, A61K 31/4545, A61K 31/4745, C07D 471/04, C07D 471/10, C07D 491/20, 04.02.2021, представлены ингибиторы ATM и DNA-PK класса спиро-производных N-(5-(2-оксо-2,3-дигидро-1H-пирроло[2,3-с]хинолин-8-ил)пиридин-3-ил)сульфонамида, где введение атома фтора в положение 7 1Н-пирроло[2,3-с]хинолин-2(3H)-она приводило к существенной потере активности против mTOR. Однако для приведенных в примерах молекул ингибирующая способность в отношении PI3Kα/δ сохранялась (преимущественно значения параметра IC50<100 нМ), что указывает на сравнительно низкую селективность этого класса молекул. Примеры молекул, содержащих в своей структуре циннолин, не описаны. Мы показали, что сравнимую селективность можно получить и без атома фтора в аналогичном положении в структуре 9-(пиридин-3-ил)-2H-[1,4]оксазино[3,2-с]циннолин-3(4H)-онов.
В опубликованной международной заявке WO 2019201283 А1, МПК C07D 471/10, A61K 31/444, A61K 31/4545, A61K 31/4745, A61K 31/502, А61Р 35/00, 24.10.2019, описаны спиро-производные 1H-пирроло[2,3-с]хинолин-2(3H)-онов, 1,2-дигидробензо[ƒ][1,7]нафтиридин-3(4H)-оны и 1,2-дигидропиразино[2,3-с]хинолин-3(4H)-оны, а также их циннолиновые аналоги, в качестве ингибиторов ATM и DNA-PK киназ. Несмотря на то, что многие симропроизводные 1H-пирроло[2,3-с]хинолин-2(3H)-онов показали активность против ATM и DNA-PK киназ (IC50<0,5 нМ), их шестичленные аналоги: 1H-спиро[бензо[ƒ][1,7]нафтиридин-2,1'-циклобутан]-3(4H)-оны (один пример в заявке) и 1'H-спиро[циклопропан-1,2'-пиразино[2,3-с]хинолин]-3'(4'H)-оны (два примера в заявке) не продемонстрировали активности против ATM и DNA-PK киназ (IC50>100 нМ). Кроме того, в WO 2019201283 А1 не заявлены 2H-[1,4]оксазино[3,2-с]хинолин-3(4H)-оны и 2Н-[1,4]тиазино[3,2-с]хинолин-3(4H)-оны, а также их циннолиновые аналоги, и селективность в отношении ATR, PI3Kα, PI3Kβ, PI3Kδ, PI3Kγ и mTOR для примеров молекул в WO 2019201283 А1 не представлена. В опубликованной заявке Китая CN 109705139 А, МПК C07D 498/04, 03.05.2019, галогензамещенные по положению 9 производные 2H-[1,4]оксазино[3,2-с]хинолин-3(4H)-онов описаны в качестве ингибиторов активности c-Met киназы, без упоминания ATM и/или DNA-PK киназ.
В опубликованной международной заявке WO 2009155527, МПК C07D 487/04, C07D 487/02, C07D 403/02, A61K 31/437, А61Р 35/00, 23.12.2009, представлены замещенные хинолины в качестве мультикиназных ингибиторов. В частности, под структуру Маркуша подпадают 1,2-дигидробензо[ƒ][1,7]нафтиридин-3(4H)-оны и 1,2-дигидропиразино[2,3-с]хинолин-3(4H)-оны. При этом активности в отношении ATM, ATR и DNA-PK в заявке не указано. Кроме того, 2H-[1,4]оксазино[3,2-с]хинолин-3(4H)-оны и 2H-[1,4]тиазино[3,2-с]хинолин-3(4H)-оны не подпадают под приведенную в заявке структуру Маркуша. Учитывая сказанное выше, можно утверждать, что структуры, заявленные в настоящем изобретении, являются новыми, их активность против целевых киназ ATM и DNA-PK, а также нецелевых киназ ATR, PI3Kα, PI3Kβ, PI3Kδ, PI3Kγ и mTOR не очевидна и однозначно не следует из опубликованных ранее патентных заявок и научных статей.
Целью настоящего изобретения является разработка и создание новых гетероциклических селективных двойных ингибиторов ATM и DNA-PK киназ, перспективных в клинической практике для лечения и/или профилактики онкологических заболеваний в качестве монотерапии и/или в комбинации с радиотерапией, химиотерапией и/или иммунотерапией.
Техническим результатом изобретения является активность химических соединений в отношении ATM и DNA-PK киназ, повышение селективности в отношении родственных киназ.
Указанный технический результат достигается посредством разработки и создания соединений общей формулы (I):
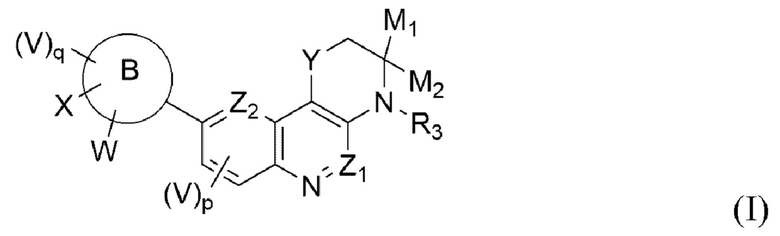
или соответствующая фармацевтическая композиция или соль, где:
- р и q выбирается независимо и равны 0, 1, 2 или 3;
- В выбирается независимо и представляет собой конденсированный ароматический или неароматический карбоцикл или гетероцикл;
- M1 и М2 выбирается независимо и представляет собой водород или M1 и М2 формируют вместе =O (кетогруппа);
- X, W и V выбирается независимо и представляет собой водород, галоген, опционально замещенный алкил, опционально замещенный алкенил, опционально замещенный алкинил, опционально замещенный галоалкил, опционально замещенный алкоксиалкил, опционально замещенный циклоалкил, опционально замещенный арил, опционально замещенный гетероциклил, опционально замещенный гетероарил, -R6-CN, -R6-NO2, -R6-OR5, -R6-N(R4)R5, -O-R6-N(R4)R5, -N=C(R4)R5, -S(O)rR4, -OS(O)2CF3, -R6-C(O)R4, -C(S)R4, -R6-C(O)OR4, -C(S)OR4, -R6-C(O)N(R4)R5, -C(S)N(R4)R5, -N(R5)C(O)R4, -N(R5)C(S)R4, -N(R5)C(O)OR4, -N(R5)C(S)OR4, -N(R5)C(O)N(R4)R5, -N(R5)C(S)N(R4)R5, -N(R5)S(O)tR4, -N(R5)S(O)tN(R4)R5, -R6-S(O)tN(R4)R5, -O-P(O)(R4)R5, -O-P(O)R40(R4), -O-P(O)R4N(R4)R5, -N(R5)-P(O)(R4)R5, -N(R5)-P(O)R4O(R4), -N(R5)-P(O)R4N(R4)R5, -N(R5)-P(O)O(R4)N(R4)R5, -N(R5)-P(O)N(R4)R5N(R4)R5, -N(R5)C(=NR5)R4, -N(R5)C(=NR5)N(R4)R5, и -N(R5)C(=N-CN)N(R4)R5, где r выбирается независимо и равны 0, 1, или 2, t выбирается независимо и равен 1 или 2; или два смежных заместителя V, или W, или X вместе с атомами углерода кольца, с которым они связаны напрямую, формируют конденсированный ароматический или неароматический карбоцикл или гетероцикл;
- Y выбирается независимо и представляет собой -О-, -S- или -SO2-;
- Z1 и Z2 выбирается независимо и представляет собой -C(R1a)- или N;
- R1a выбирается независимо и представляет собой водород, алкил, галоген, -CN, -NR2R4 или OR2;
- R2, R4 и R5 выбирается независимо и представляет собой водород, опционально замещенный алкил, опционально замещенный алкенил, опционально замещенный алкинил, опционально замещенный галоалкил, опционально замещенный алкоксиалкил, опционально замещенный циклоалкил, опционально замещенный арил, опционально замещенный гетероциклил, опционально замещенный гетероарил;
- R3 выбирается независимо и представляет собой водород или алкил;
- R6 выбирается независимо и представляет собой прямую связь или линейную или разветвленную опционально замещенную алкиленовую цепь, линейную или разветвленную опционально замещенную алкениленовую цепь, линейную или разветвленную опционально замещенную алкиниленовую цепь, или опционально замещенный гетероциклилен.
Согласно настоящему изобретению, к фармацевтически приемлемым носителям, растворителям или наполнителям относятся, помимо прочего, любой адъювант, носитель, наполнитель, скользящий агент, подсластитель, разбавитель, консервант, краситель, усилитель вкуса, поверхностно-активное вещество, смачивающий агент, диспергирующий агент, суспендирующий агент, стабилизатор, изотонический агент, растворитель или эмульгатор, одобренный для использования у людей или домашних животных.
К фармацевтически приемлемым солям относятся, помимо прочего, кислотно-аддитивные соли или основно-аддитивные соли.
К фармацевтически приемлемым кислотно-аддитивным солям относятся соли, которые сохраняют биологическую эффективность и свойства свободных оснований, которые не являются биологически или иным образом нежелательными и которые образуются с неорганическими кислотами, такими как, помимо прочего, соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и органические кислоты, такие как, помимо прочего, уксусная кислота, 2,2-дихлоруксусная кислота, адипиновая кислота, альгиновая кислота, аскорбиновая кислота, аспарагиновая кислота, бензолсульфоновая кислота, бензойная кислота, 4-ацетамидобензойная кислота, камфорная кислота, камфор-10-сульфоновая кислота, каприновая кислота, капроновая кислота, каприловая кислота, угольная кислота, коричная кислота, лимонная кислота, цикламовая кислота, додецилсерная кислота, этан-1,2-дисульфоновая кислота, этансульфоновая кислота, 2-гидроксиэтансульфоновая кислота, муравьиная кислота, фумаровая кислота, галактаровая кислота, гентизиновая кислота, глюкогептоновая кислота, глюконовая кислота, глюкуроновая кислота, глутаминовая кислота, глутаровая кислота, 2-оксо-глутаровая кислота, глицерофосфорная кислота, гликолевая кислота, гиппуровая кислота, изомасляная кислота, молочная кислота, лактобионовая кислота, лауриновая кислота, малеиновая кислота, яблочная кислота, малоновая кислота, миндальная кислота, метансульфоновая кислота, муциновая кислота, нафталин-1,5-дисульфоновая кислота, нафталин-2-сульфоновая кислота, 1-гидрокси-2-нафтойная кислота, никотиновая кислота, олеиновая кислота, оротовая кислота, щавелевая кислота, пальмитиновая кислота, памовая кислота, пропионовая кислота, пироглутаминовая кислота, пировиноградная кислота, салициловая кислота, 4-аминосалициловая кислота, себациновая кислота, стеариновая кислота, янтарная кислота, кислота, винная кислота, тиоциановая кислота, п-толуолсульфоновая кислота, трифторуксусная кислота, ундециленовая кислота и т.п.
К фармацевтически приемлемым основно-аддитивным солям относятся соли, которые сохраняют биологическую эффективность и свойства свободных кислот, которые сохраняют биологическую эффективность и свойства свободных кислот, которые не являются биологически или иным образом нежелательными. Эти соли получают добавлением неорганического или органического основания к свободной кислоте. Соли, полученные из неорганических оснований, включают, без ограничения, соли натрия, калия, лития, аммония, кальция, магния, железа, цинка, меди, марганца, алюминия и т.п. Предпочтительными неорганическими солями являются соли аммония, натрия, калия, кальция и магния. Соли, полученные из органических оснований, включают, помимо прочего, соли первичных, вторичных и третичных аминов, замещенные амины, включая встречающиеся в природе замещенные амины, циклические амины и основные ионообменные смолы, такие как аммиак, изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, диэтаноламин, этаноламин, деанол, N,N-диметиламиноэтанол, А^-диэтиламиноэтанол, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, бенетамин, бензатин, этилендиамин, глюкозамин, метилглюкамин, теобромин, триэтаноламин, трометамин, пурины, пиперазин, пиперидин, N-этилпиперидин, полиаминные смолы и т.п. Особенно предпочтительными органическими основаниями являются изопропиламин, диэтиламин, этаноламин, триметиламин, дициклогексиламин, холин и кофеин.
Часто в результате кристаллизации образуется сольват соединения, являющегося предметом изобретения. Используемый здесь термин «сольват» относится к агрегату, который содержит одну или несколько молекул соединения, являющееся предметом изобретения, с одной или несколькими молекулами растворителя. Растворителем может быть вода, и в этом случае сольват может представлять собой гидрат. В некоторых случаях растворитель может представлять собой органический растворитель. Таким образом, соединения настоящего изобретения могут существовать в виде гидратов, включая моногидрат, дигидрат, полугидрат, полуторный гидрат, тригидрат, тетрагидрат и т.п., а также соответствующих сольватированных форм. Соединение, являющееся предметом изобретения, может представлять собой настоящие сольваты, тогда как в других случаях соединение, являющееся предметом изобретения, может просто удерживать постороннюю воду или представлять собой смесь воды и некоторого постороннего растворителя.
К фармацевтической композиции относятся к препарату соединения, являющееся предметом изобретения, и среде, общепринятой в данной области техники для доставки биологически активного соединения млекопитающим, например, людям. Такая среда включает все фармацевтически приемлемые носители, разбавители или наполнители.
Среди соединений формулы (I) следующие соединения являются предпочтительными:
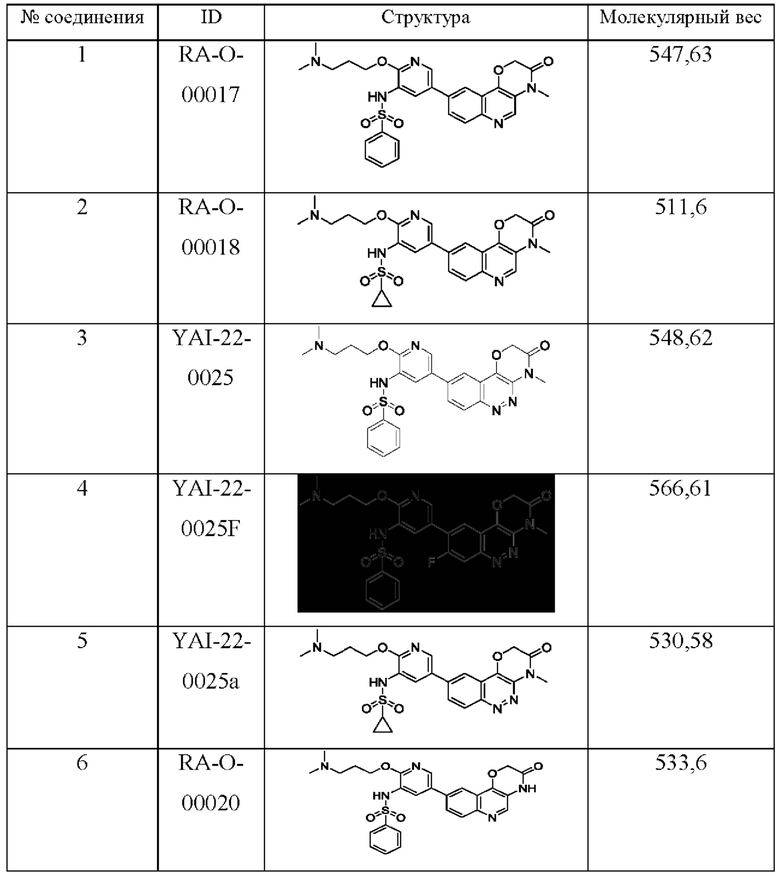
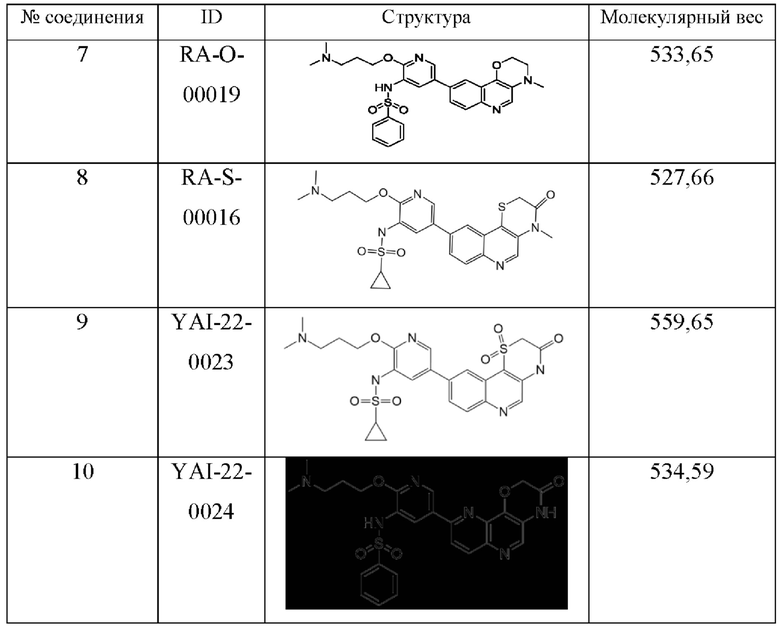
Соединения формулы (I) являются ингибиторами семейства ATM и DNA-PK киназ.
Изобретение включает применение соединения настоящего изобретения для ингибирования активности ATM и DNA-PK киназ в клетках или у субъекта, или при лечении или профилактике состояний, связанных с активностью или экспрессией ATM и DNA-PK киназ.
Соединения настоящего изобретения могут быть использованы для лечения или профилактики рака у млекопитающего, преимущественно человека, при этом метод лечения рака согласно настоящему изобретению включает в себя введение терапевтически эффективного количества соединения, являющегося предметом изобретения, в организм млекопитающего, нуждающегося в остановке, замедлении или обращении роста, развития или распространения рака, включая солидные опухоли или другие формы рака. В некоторых вариантах соединение вводят млекопитающему, получающему радиотерапию.
В другом аспекте изобретение предлагает способы лечения или профилактики рака у млекопитающих, причем способы включают введение нуждающемуся в этом млекопитающему терапевтически эффективного количества соединения, являющегося предметом изобретения. В некоторых вариантах осуществления соединение вводят млекопитающему в сочетании с агентом, повреждающим ДНК. Примеры агентов, повреждающих ДНК, включают цисплатин, оксалиплатин, карбоплатин, валрубицин, идарубицин, калихеамицин, ингибиторы PARP и т.д.
В другом аспекте изобретение предлагает фармацевтические композиции, содержащие соединения изобретения и фармацевтически приемлемые вспомогательные вещества. В одном из вариантов реализации фармацевтическая композиция содержит соединение, являющегося предметом изобретения, в фармацевтически приемлемом носителе и в количестве, эффективном для лечения онкологического заболевания у млекопитающего.
Соединение, являющееся предметом изобретения, при использовании в комбинированной терапии может повысить эффективность другого лекарственного лечения или может снизить частоту и/или степень тяжести нежелательных явлений, связанных с другим лекарственным лечением. Например, побочные эффекты радиотерапии (например, мукозит полости рта или желудочно-кишечного тракта, дерматит, пневмонит или усталость) могут быть уменьшены у пациентов, получающих комбинированную терапию, включающую соединение, являющееся предметом изобретения, и радиотерапию (например, частота побочных эффектов может быть уменьшена по меньшей мере на 1%, 5%, 10% или 20%) относительно пациентов, получающих радиотерапию без соединения, являющегося предметом изобретения. Кроме того, другие нежелательные явления, которые можно уменьшить у пациентов, получающих комбинированную терапию, включающую соединение по изобретению и радиотерапию (например, частота нежелательных явлений может быть снижена по меньшей мере на 1%, 5%, 10% или 20%) относительно у пациентов, получающих радиотерапию без соединения, являющегося предметом изобретения, могут возникнуть поздние эффекты радиотерапии, например, радиационно-индуцированный фиброз легких, повреждение сердца, непроходимость кишечника, повреждение нервов, сосудистое повреждение, лимфедема, некроз головного мозга или рак, вызванный радиотерапией. Аналогично, когда соединение вводят в комбинированной терапии с другим противоопухолевым препаратом (например, описанным в настоящем документе), комбинированная терапия может вызвать такую же или даже повышенную гибель опухолевых клеток, даже если доза другого противоопухолевого препарата понижен. Таким образом, снижение дозировок других противоопухолевых препаратов может снизить степень тяжести нежелательных явлений, вызванных другими противоопухолевыми препаратами.
В другом аспекте настоящее изобретение направлено на использование соединений изобретения, как изложено выше, в виде их стереоизомера, энантиомера, таутомера или их смесей, или их фармацевтически приемлемой соли или сольвата, или использование фармацевтического препарата композиция, содержащая фармацевтически приемлемый наполнитель и соединение по изобретению, как указано выше, в виде его стереоизомера, энантиомера, таутомера или их смесей, или его фармацевтически приемлемой соли или сольвата, в препарате лекарственного средства для использования в лечении заболевания. В некоторых вариантах реализации соединение, являющегося предметом изобретения, вводят в сочетании с радиотерапией. В других вариантах реализации соединение, являющегося предметом изобретения, вводят в сочетании с агентом, повреждающим ДНК. В некоторых вариантах осуществления заболеванием является рак.
Примеры разновидностей рака, подлежащего лечению с использованием способов и применений, раскрытых в настоящем документе, включают, помимо прочего, лейкозы и лимфомы - острый миелоидный лейкоз, острый лимфобластный лейкоз, острый мегакариоцитарный лейкоз, промиелоцитарный лейкоз, эритролейкоз, лимфобластный Т-клеточный лейкоз, хронический миелоидный лейкоз, хронический лимфоцитарный лейкоз, волосатоклеточный лейкоз, хронический нейтрофильный лейкоз, плазмоцитома, иммунобластный крупноклеточный лейкоз, мантийноклеточный лейкоз, множественная миелома, злокачественная лимфома, диффузная крупноклеточная В-клеточная лимфома, лимфома Ходжкина, неходжкинская лимфома, лимфобластная Т-клеточная лимфома, лимфома Беркитта и фолликулярная лимфома.
Примеры разновидностей рака, подлежащего лечению с использованием способов и применений, раскрытых в настоящем документе, включают, помимо прочего, рак головного мозга (например, астроцитому, глиому, глиобластому, медуллобластому, эпендимому), рак мочевого пузыря, рак молочной железы, опухоли центральной нервной системы, рак шейки матки, рак прямой кишки, рак толстой кишки, рак эндометрия, рак пищевода, стромальная опухоль желудочно-кишечного тракта, рак желудка, рак головы и шеи, рак полости рта, гепатоцеллюлярный рак, холангиокарцинома, метастатическое поражение печени, карцинома Меркеля, рак легких, меланома, мезотелиома, рак носоглотки, нейробластома, остеосаркома, рак яичников, рак поджелудочной железы, рак предстательной железы, рак почки, рак слюнной железы, саркомы, рак яичка, уротелиальный рак, рак вульвы и опухоль Вильмса.
В дополнительных вариантах реализации примеры рака, подлежащего лечению с использованием способов и применений, раскрытых в настоящем документе, не ограничиваются метастазами и метастатическим раком. Например, раскрытые здесь способы и применение для лечения рака могут включать лечение как первичных опухолей, так и метастазов.
В некоторых вариантах реализации способы и применения, раскрытые в настоящем документе, включают предварительное введение субъекту ингибитора ATM и DNA-PK перед применением радиотерапии или агента, повреждающего ДНК. Предварительное введение двойного ингибитора ATM и DNA-PK может задержать или устранить восстановление повреждений ДНК после радиотерапии.
Радиотерапия включает, помимо прочего, внешнюю радиотерапию с использованием рентгеновских лучей (фотонов), гамма-лучей кобальта-60 или других радиоактивных изотопов, нейтронов, электронов, протонов, ионов углерода, ионов гелия и других заряженных частиц. Радиотерапия также включает брахитерапию и радио фармацевтические препараты, которые испускают гамма-лучи, альфа-частицы, бета-частицы, оже-электроны или другие типы радиоактивных частиц из изотопов, включая иридий-192, йод-125, цезий-137, палладий-103, фосфорт-32, иттрий-90, галлий-67, астат-211, радий-223 и другие радиоактивные изотопы. Радиотерапия также включает в себя радиоиммунотерапию (РИТ) с антителами или небольшими молекулами, которые конъюгированы с радиоактивными изотопами, включая йод-131, иттрий-90, актиний-225, астат-211, галлий-67 и другие радиоактивные изотопы.
В некоторых вариантах реализации комбинированная терапия включает введение субъекту ингибитора ATM и DNA-PK и противоопухолевого агента, например, цисплатина, оксалиплатина, карбоплатина, ингибиторов топоизомеразы I, ингибиторов топоизомеразы II, антрациклинов, вальрубицин, идаруцин, калихеамицин, ингибиторы PARP (например, олапариб, рукапариб, нирапариб, велипариб, талазопариб), а также другие противоопухолевые агенты, известные специалистам в данной области.
В некоторых вариантах реализации комбинированная терапия включает введение субъекту ингибитора ATM и DNA-PK и противоопухолевых иммунотерапевтических средств, включая, помимо прочего, ипилимумаб, офатумумаб, ниволумаб, пембролизумаб, атезолизумаб, авелумаб, дурвалумаб и т.д.
В рамках комбинированной терапии, описанной в настоящем документе, ингибитор ATM и DNA-PK можно вводить субъекту одновременно или последовательно (например, до или после) другого лекарственного средства или радиотерапии.
Органический синтез
Соединения, являющиеся предметом настоящего изобретения, могут быть получены с использованием описанных ниже синтетических методов. Перечисленные методы не являются исчерпывающими и допускают введение разумных модификаций. Указанные реакции должны проводиться с использованием подходящих растворителей и материалов. При реализации данных общих методик для синтеза конкретных веществ необходимо учитывать присутствующие в веществах функциональные группы и их влияние на протекание реакции. Для получения некоторых веществ необходимо изменить порядок стадий либо отдать предпочтение одной из нескольких альтернативных схем синтеза. Следует понимать, что эти и все приведенные в материалах заявки примеры не являются ограничивающими и приведены только для иллюстрации настоящего изобретения.
Методика получения N-(2-(3-(диметиламино)пропокси)-5-(4-метил-3-оксо-3,4-дигидро-2H-[1,4]оксазино[3,2-с]хинолин-9-ил)пиридин-3-ил)бензолсульфонамида (RA-O-00017):
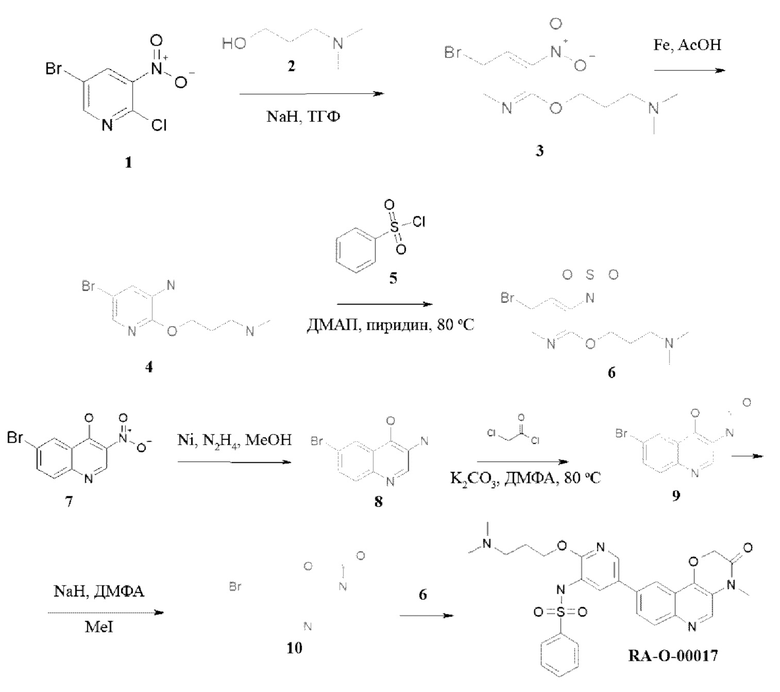
Соединение 3. К раствору спирта (2) 2,17 г (0,021 моль, 1,25 экв.) в 50 мл «сухого» ТГФ при 0°С прибавляли порциями 0,95 г 80% NaH (0,032 моль, 1,9 экв.), перемешивали 15 минут и прикапывали раствор 4 г (0,017 моль, 1 экв.) соединения (1) в 25 мл ТГФ. Перемешивали 12 часов, реакционную массу выливали в 200 мл воды и экстрагировали ЭА (3 раза по 100 мл). Объединенные органические фазы сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, элит ХМ - ХМ-МеОН 5%. MS m/z 305,2 (М+Н+). Выход 3 составил: 1 г, 16%.
Соединение 4. Соединение (3) 1 г (0,0033 моль, 1 экв.) растворяли в 50 мл смеси уксусная кислота - вода 4 к 1 и вносили порциями 0,74 г (0,0132 моль, 4 экв.) порошка железа. Реакционную смесь перемешивали 12 часов. В реакционную массу добавляли 50 мл ЭА, фильтровали через слой цеолита, упаривали, растворяли в ЭА и еще раз фильтровали через слой цеолита. Фильтрат промывали раствором K2CO3, сушили над безводным Na2SO4 и упаривали. Полученный остаток использовали в следующей стадии без дополнительной очистки. Выход 4 составил: 0,8 г, 88%.
Соединение 6. К раствору 0,3 г (0,0011 моль, 1 экв.) соединения (4) в 25 мл пиридина добавляли 10 мг ДМАП и 0,58 г (0,0033 моль, 3 экв.) сульфохлорида (5), перемешивали при 80°С в течение суток. Пиридин упаривали, остаток выливали в воду и экстрагировали ЭА (3 раза по 100 мл). Объединенные органические фазы сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, элит ХМ-МеОН 25%. MS m/z 415,4 (М+Н+). Выход 6 составил: 0,4 г, 88%.
Соединение 8. К раствору 0,5 г (0,00185 моль) соединения (7) в 50 мл МеОН добавляли 0,5 г никеля Ренея и прикапывали 0,8 мл гидразингидрата. Перемешивали 12 часов, затем в реакционную массу добавляли 50 мл МеОН, перемешивали в течение 15 минут, затем фильтровали через слой цеолита, фильтрат упаривали. Остаток переупаривали 2 раза с толуолом. Получали 380 мг (выход 86%) соединения (8), которое сразу использовали в следующей стадии.
Соединение 9. К раствору 0,38 г (0,00158 моль, 1 экв.) соединения (8) в 50 мл ДМФА вносили 0,83 г (0,00347, 2 экв.) K2CO3 и затем прикапывали 0,24 г (0,0019 моль, 1,2 экв.) хлорацетилхлорида. Перемешивали при 80°С в течение 12 часов, затем реакционную массу выливали в 200 мл воды, выпавший осадок отфильтровывали и перекристаллизовывали из этанола. MS m/z 280,2 (М+Н+). 1H ЯМР (400 МГц, ДМСО-d6) δ 11,12 (с, 1H), 8,52 (с, 1H), 8,09 (д, J=1,7 Гц, 1Н), 7,86 (д, J=9,0 Гц, 1Н), 7,74 (дд, J=9,0, 1,8 Гц, 1Н), 4,92 (с, 2Н). Получали соединение 9 (0,28 г, выход 63%).
Соединение 10. К раствору 0,2 г (0,00071 моль, 1 экв.) соединения (9) в 25 мл ДМФА вносили 0,043 г (0,0142 моль, 2 экв.) NaH, затем прикапывали 0,126 г (0,00088 моль, 1,25 экв.) CH3I. Перемешивали 1 час, реакционную массу выливали в 100 мл воды, выпавший осадок отфильтровывали и перекристаллизовывали из этанола. MS m/z 294,2 (M+H+). Получили соединение 10 (0,16 г, выход 76%).
Соединение RA-O-00017. К раствору 0,15 г (0,51 ммоль, 1 экв.) соединения (10) в 15 мл «сухого» диоксана прибавляли 0,136 г (0,54 ммоль, 1.05 экв.) B2Pin2 и 0,16 г (1,63 ммоль, 3,2 экв.) KOAc. Раствор дегазировали, пропуская ток Ar 10 минут, и вносили 30 мг (0,041 ммоль, 0,08 экв.) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 12 часов, затем охлаждали до комнатной температуры и добавляли раствор 0,35 г (1 ммоль, 2 экв.) Cs2CO3 в 1,5 мл воды. Затем вносили 0,185 г (0,44 ммоль, 0,88 экв.) соединения (6). Раствор дегазировали пропуская ток Ar 10 минут и вносили 30 мг (0,041 ммоль, 0,08 экв.) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 8 часов. Реакционную массу разбавляли 15 мл EtOAc и фильтровали через слой цеолита. В фильтрат добавляли 30 мл воды. Органический слой отделяли, а водный экстрагировали EtOAc 2 раза по 30 мл. Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт ХМ-МеОН 10%. MS m/z 548,6 (М+Н+), 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,94 (с, 1Н), 9,50 (с, 1H), 8,89 (с, 1H), 8,42 (с, 1H), 8,08 (с, 2Н), 7,98-7,93 (м, 2Н), 7,77-7,60 (м, 5Н), 5,10 (с, 2Н), 4,14 (с, 2Н), 3,48 (с, 3Н), 3,19 (с, 2Н), 2,82 (с, 6Н), 1,93 (с, 2Н). Получали соединение RA-O-00017 (65 мг, выход 27%).
Методика получения N-(2-(3-(диметиламино)пропокси)-5-(4-метил-3-оксо-3,4-дигидро-2H-[1,4]оксазино[3,2-с]хинолин-9-ил) пиридин-3-ил)циклопропансульфонамида (RA-O-00018):
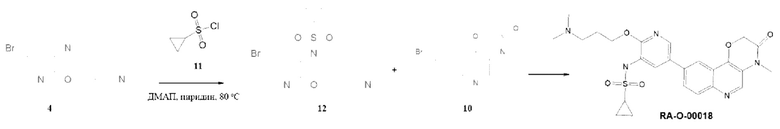
Соединения 4 и 10 были получены по методике, приведенной для RA-O-00017.
Соединение 12. К раствору 0,35 г (0,00127 моль, 1 экв.) соединения (4) в 25 мл пиридина добавляли 10 мг ДМАП и 0,53 г (0,0038 моль, 3 экв.) сульфохлорида (11), затем перемешивали при 80°С в течение 24 часов. Пиридин упаривали, остаток заливали 5% раствором K2CO3 и экстрагировали ЭА (3 раза по 100 мл). Объединенные органические фазы сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт ХМ-МеОН 25%. MS m/z 379,4 (М+H+). Выход 12 составил: 0,4 г (83%).
Соединение RA-O-00018. К раствору 0,15 г (0,51 ммоль, 1 экв.) соединения (10) в 15 мл «сухого» диоксана прибавляли 0,136 г (0,54 ммоль, 1,05 экв.) B2Pin2 и 0,16 г (1,63 ммоль, 3,2 экв.) KOAc. Раствор дегазировали пропуская ток Ar 10 минут и вносили 30 мг (0,041 ммоль, 0,08 экв.) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 12 часов, затем охлаждали до комнатной температуры и добавляли раствор 0,35 г (1 ммоль, 2 экв.) Cs2CO3 в 1,5 мл воды. Затем вносили 0,168 г (0,44 ммоль, 0,88 экв.) соединения (12). Раствор дегазировали пропуская ток Ar 10 минут и вносили 30 мг (0,041 ммоль, 0,08 экв.) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 8 часов. Реакционную массу разбавляли 15 мл EtOAc и фильтровали через слой цеолита. В фильтрат добавляли 30 мл воды. Органический слой отделяли, а водный экстрагировали EtOAc 2 раза по 30 мл. Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт ХМ-МеОН 10%. MS m/z 512,4 (М+Н+), 1H ЯМР (400 МГц, ДМСО-d6) δ 9,54 (с, 1Н), 9,35 (с, 1H), 8,89 (с, 1Н), 8,48 (д, J=1,7 Гц, 1H), 8,20 (с, 1Н), 8,14-8,00 (м, 3Н), 5,07 (с, 2Н), 4,44 (т, J=5,6 Гц, 2Н), 3,47 (с, 3Н), 3,35 (с, 2Н), 2,78 (м, 7Н), 2,16 (с, 2Н), 1,06-0,87 (м, 4Н). Получали 55 мг соединения RA-O-00018 (выход 24%).
Методика получения N-(2-(3-(диметиламино)ггоопокси)-5-(4-метил-3,4-дигидро-2H-[1,4] оксазино[3,2-с]хинолин-9-ил)пиридин-3-ил)бензолсульфонамида (RA-O-00019):
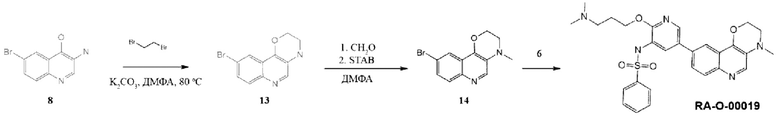
Соединения 8 и 6 были получены по методике, приведенной для RA-O-00017.
Соединение 13. К раствору 0,65 г (0,0027 моль, 1 экв.) соединения (8) в 50 мл ДМФА вносили 2,44 г (0,017 моль, 6 экв.) K2CO3 и затем прикапывали 1,5 г (0,008 моль, 3 экв.) дибромэтана. Перемешивали при 80°С в течение 12 часов, затем реакционную массу выливали в 200 мл воды, выпавший осадок отфильтровывали и перекристаллизовывали из этанола. MS m/z 266,2 (М+H+). Выход 13 составил: 0,63 г (87%).
Соединение 14. К раствору 0,35 г (0,0013 моль, 1 экв.) соединения (13) в 30 мл ДМФА вносили 0,5 мл 37% раствора формальдегида, затем 2,4 г (0,011 моль, 9 экв.) триацетоксиборгидрида натрия (STAB). Реакционную смесь перемешивали 3 часа, затем промывали насыщенным раствором NaHCO3. Органическую фазу сушили над безводным Na2SO4 и упаривали. Полученный остаток использовали в следующей стадии без дополнительной очистки. MS m/z 280,2 (М+Н+). Выход 14 составил: 0,15 г (42%).
Соединение RA-O-00019. К раствору 0,07 г (0,25 ммоль, 1 экв.) соединения (14) в 15 мл «сухого» диоксана прибавляли 0,068 г (0,27 ммоль, 1,05 экв.) B2Pin2 и 0,08 г (0,8 ммоль, 3,2 экв.) KOAc. Раствор дегазировали, пропуская ток Ar 10 минут, и вносили 30 мг (0,02 ммоль, 0,08 экв.) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 12 часов, затем охлаждали до комнатной температуры и добавляли раствор 0,17 г (1 ммоль, 2 экв.) Cs2CO3 в 1,5 мл воды. Затем вносили 0,093 г (0,44 ммоль, 0,88 экв.) соединения (6). Раствор дегазировали пропуская ток Ar 10 минут и вносили 15 мг (0,02 ммоль, 0,08 экв.) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном в течение 8 часов. Реакционную массу разбавляли 15 мл EtOAc и фильтровали через слой цеолита. В фильтрат добавляли 30 мл воды. Органический слой отделяли, а водный экстрагировали EtOAc 2 раза по 30 мл. Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт ХМ-МеОН 5%. MS m/z 534,6 (М+Н+), 1H ЯМР (400 МГц, ДМСО-d6) δ 8,59 (с, 1H), 8,18 (с, 1H), 7,90-7,85 (м, 2Н), 7,81-7,73 (м, 3Н), 7,67-7,52 (м, 5Н), 4,57 (с, 2Н), 4,17 (с, 2Н), 3,32 (с, 2Н), 3,15 (т, J=7,0 Гц, 2Н), 3,02 (с, 3Н), 2,78 (с, 6Н), 1,98 (с, 2Н). Получали 15 мг соединения RA-O-00019 (выход 6%).
Методика получения N(2-(3-(диметиламино)пропокси)-5-(3-оксо-3,4-дигидро-2H-[1,4]оксазино[3,2-с]хинолин-9-ил)пиридин-3-ил)бензолсульфонамида (RA-O-00020):
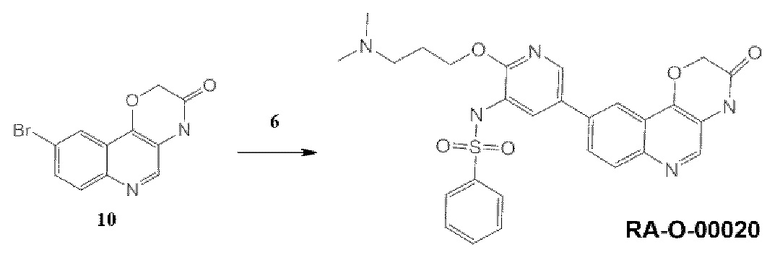
Соединения 10 и 6 были получены по методике, приведенной для RA-O-00017.
Соединение RA-O-00020. К раствору 0,15 г (0,53 ммоль, 1 экв.) соединения (10) в 15 мл «сухого» диоксана прибавляли 0,136 г (0,53 ммоль, 1 экв.) B2Pin2 и 0,16 г (1,63 ммоль, 3 экв.) KOAc. Раствор дегазировали, пропуская ток Ar 10 минут, и вносили 30 мг (0,041 ммоль, 0,08 eq) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 12 часов, затем охлаждали до комнатной температуры и добавляли раствор 0,35 г (1 ммоль, 2 экв.) Cs2CO3 в 1,5 мл воды. Затем вносили 0,185 г (0,44 ммоль, 0,88 экв.) соединения (6). Раствор дегазировали, пропуская ток Ar 10 минут, и вносили 30 мг (0,041 ммоль, 0,08 экв.) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 8 часов. Реакционную массу разбавляли 15 мл EtOAc и фильтровали через слой цеолита. В фильтрат добавляли 30 мл воды. Органический слой отделяли, а водный экстрагировали EtOAc 2 раза по 30 мл. Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт ХМ-МеОН 10%. Получали 70 мг продукта, который дополнительно перекристаллизовывали из EtOH, и промывали полученный осадок диэтиловым эфиром. MS m/z 534,6 (М+Н+). 1Н ЯМР (400 МГц, ДМСО-d6) δ 11,08 (с, 1H), 8,49 (с, 1H), 7,98 (с, 1H), 7,96 (д, J=8,9 Гц, 1H), 7,87 (с, 1H), 7,84-7,72 (м, 3Н), 7,63-7,48 (м, 4Н), 5,00 (с, 2Н), 4,25 (т, J=5,6 Гц, 2Н), 3,13 (т, J=5,8 Гц, 2Н), 2,80 (с, 6Н), 2,05 (м, 2Н). Выход соединения RA-O-00020 составил 20 мг (9%).
Методика получения N-(2-(3-(диметиламино)пропокси)-5-(4-метил-3-оксо-3,4-дигидро-2H-[1,4]тиазино[3,2-c]хинолин-9-ил)пиридин-3-ил)циклопропансульфонамида (RA-S-00016):
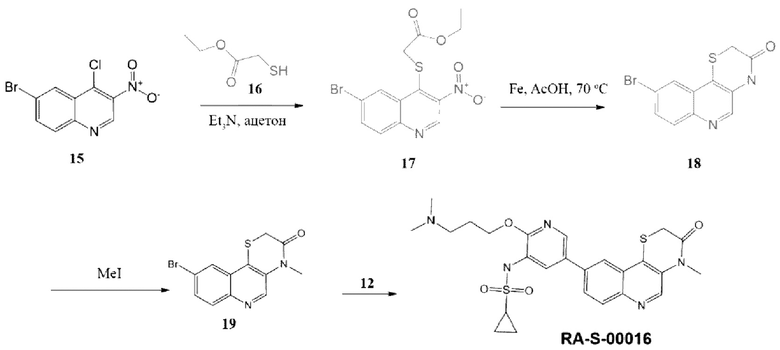
Соединение 12 было получено по методике, описанной для RA-O-00017.
Соединение 17. К раствору 0,225 г (0,00187 моль, 1,2 экв.) соединения (16) в 30 мл ацетона добавляли 0,26 мл Et3N (0,00187 моль, 1,2 экв.) и затем порциями вносили 0,45 г (0,00156 моль, 1 экв.) соединения (15). Реакционную смесь перемешивали в течение 12 часов, образовавшийся осадок отфильтровывали, фильтрат упаривали, заливали водой и экстрагировали ЭА (3 раза по 50 мл). Объединенные органические фазы сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт толуол. MS m/z 372,4 (М+Н+). Выход 17 составил 0,3 г (52%).
Соединение 18. Соединение 17 0,3 г (0,00081 моль, 1 экв.) растворяли в 20 мл уксусной кислоты и вносили порциями 0,226 г (0,00404 моль, 5 экв.) порошка железа и перемешивали 1 час, затем 12 часов при 70°С. Образовавшийся осадок отфильтровывали, осадок заливали диоксаном, кипятили, затем фильтровали. Фильтрат без предварительного упаривания очищали хроматографией на силикагеле, эл-нт диоксан, MS m/z 297,2 (М+Н+). Выход 18 составил 0,2 г (84%).
Соединение 19. К раствору 0,2 г (0,00068 моль, 1 экв.) соединения 18 в 25 мл ДМФА вносили 0,024 г (0,00102 моль, 1,5 экв.) 60% NaH и затем прикапывали 0,120 г (0,00085 моль, 1,25 экв.) CH3I. Реакционную смесь перемешивали 1 час, затем выливали в 100 мл воды, выпавший осадок отфильтровывали. MS m/z 309,2 (М+Н+). Получили 19 (0,14 г, 67%).
Соединение RA-S-00016. К раствору 0,1 г (0,32 ммоль, 1 экв.) соединения 19 в 15 мл «сухого» диоксана прибавляли 0,86 г (0,34 ммоль, 1,05 экв.) B2Pin2 и 0,095 г (0,97 ммоль, 3,2 экв.) KOAc. Раствор дегазировали, пропуская ток Ar 10 минут, и вносили 19 мг (0,025 ммоль, 0,08 eq) Pd(dppf)Cl2. Далее смесь перемешивали при 100°С под аргоном в течение 12 часов, затем охлаждали до комнатной температуры и добавляли раствор 0,2 г (0,64 ммоль, 2 экв.) Cs2CO3 в 1,5 мл воды. Затем вносили 0,108 г (0,28 ммоль, 0,88 экв.) соединения (12). Раствор дегазировали пропуская ток Ar 10 минут и вносили 19 мг (0,025 ммоль, 0,08 экв.) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 8 часов. Реакционную массу разбавляли 15 мл EtOAc и фильтровали через слой цеолита. В фильтрат добавляли 30 мл воды. Органический слой отделяли, а водный экстрагировали EtOAc 2 раза по 30 мл. Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт ХМ-МеОН 10%. MS m/z 528,4 (М+H+), 1H ЯМР (400 МГц, ДМСО-d6) δ 9,49 (с, 1H), 9,37 (с, 1H), 8,91 (с, 1H), 8,49 (д, J=2,3 Гц, 1H), 8,11 (м, 3Н), 8,03 (дд, J=8,7, 2,0 Гц, 1H), 4,45 (т, J=5,9 Гц, 2Н), 3,76 (с, 2Н), 3,55 (с, 3Н), 3,35 (м, 2Н), 2,84 (м, 6Н), 2,79 (м, 1H), 2,16 (т, J=7,8 Гц, 2Н), 0,97 (м, 4Н). Получали соединение RA-S-00016 (15 мг, 4%).
Методика получения N-(2-(3-(диметиламино)пропокси)-5-(4-метил-1,1-диоксидо-3 -оксо-3,4-дигидро-2H-[1,4]тиазино[3,2-с]хинолин-9-ил)пиридин-3-ил)циклопропансульфонамида (YAI-22-0023):
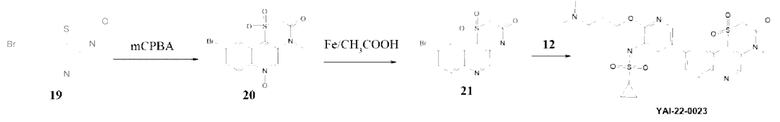
Соединение 19 было получено по методике, описанной для RA-O-00018. Соединение 12 было получено по методике, представленной для соединения RA-O-00017.
Соединение 20. К раствору 0,766 г (0,00247 моль) соединения (19) в 25 мл CH2Cl2 при 0°С прибавляли порциями 1,71 г (0,01 моль) мета-хлорнадбензойную кислоту (тСРВА), смесь перемешивали 4 часа. Реакционную массу промывали 5% раствором K2CO3, органическую фазу сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, ХМ-МеОН 10%. MS m/z 360,4 (М+Н+). Выход 20 составил 0,275 г (31%).
Соединение 21. Соединение 20 0,27 г (0,00075 моль, 1 экв.) растворяли в 20 мл уксусной кислоты и вносили порциями 0,167 г (0,003 моль, 5 экв.) порошка железа и перемешивали 1 час, затем 12 часов при 70°С. В реакционную массу добавляли 50 мл ЭА, осадок отфильтровывали. Фильтрат упаривали, остаток растворяли в 100 мл промывали 5% раствором K2CO3, органическую фазу сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, ХМ-МеОН 10%. MS m/z 342,4 (M+H+). Выход 21 составил 0,097 г (37%).
Соединение YAI-22-0023. К раствору 0,09 г (0,26 ммоль, 1 экв.) соединения (21) в 15 мл «сухого» диоксана прибавляли 0,070 г (0,27 ммоль, 1,05 экв.) B2Pin2 и 0,083 г (0,84 ммоль, 3,2 экв.) KOAc.Раствор дегазировали пропуская ток Ar 10 минут и вносили 19 мг (0,025 ммоль) Pd(dppf)Cl2. Далее смесь перемешивали при 100°С под аргоном 12 часов, затем охлаждали до комнатной температуры и добавляли раствор 0,17 г (0,52 ммоль, 2 экв.) Cs2CO3 в 1,5 мл воды. Затем вносили 0,10 г (0,26 ммоль, 1 экв.) соединения (12). Раствор дегазировали пропуская ток Ar 10 минут и вносили 19 мг (0,025 ммоль) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном 8 часов. Реакционную массу разбавляли 15 мл EtOAc и фильтровали через слой цеолита. В фильтрат добавляли 30 мл воды. Органический слой отделяли, а водный экстрагировали EtOAc 2 раза по 30 мл. Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, элюент ХМ-МеОН 10% (MS m/z 560,4 (М+Н+)), затем на препаративном хроматографе как в кислой, так и в нейтральной среде. Получили соединение YAI-22-0023 (12 мг, 14%). Спектр 1Н ЯМР полученного соединения соответствовал 1Н ЯМР спектру для молекулы RA-S-00016. Соединение YAI-22-0023 обладает сравнительно низкой стабильностью.
Методика получения N-(2-(3-(диметиламино)пропокси)-5-(4-метил-3-оксо-3,4-дигидро-2H-[1,4]оксазино[3,2-с]циннолин-9-ил)пиридин-3-ил)бензолсульфонамида (YAI-22-0025):
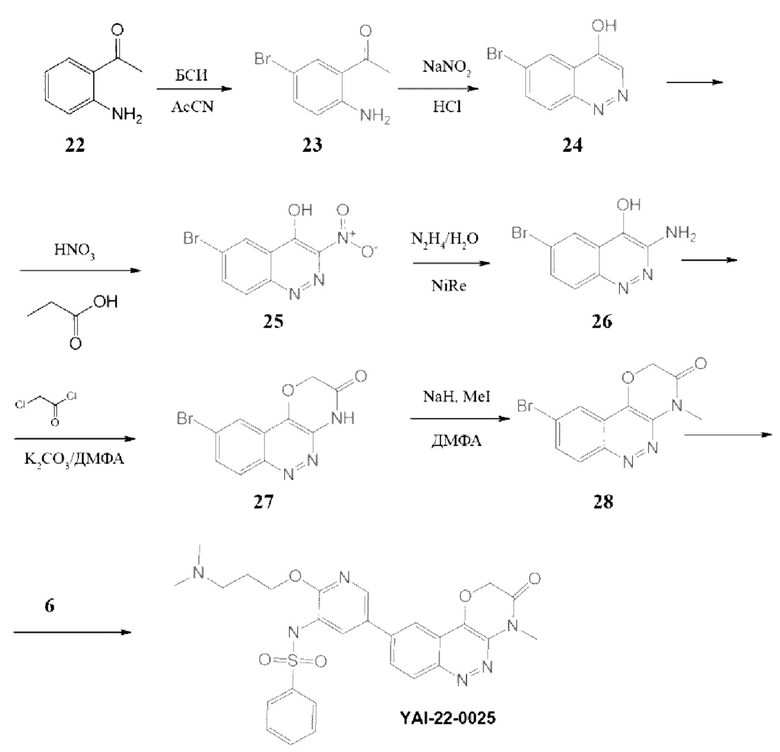
Соединение 23. К раствору 3,6 г (0,0266 моль) соединения (22) в 50 мл сухого ацетонитрила при 0°С прикапывали раствор 6,16 г (0,034 моль) N-бромсукцинимид (БСИ, NBS) в 10 мл сухого ацетонитрила. Смесь перемешивали при комнатной температуре 12 часов, реакционную массу упаривали, остаток очищали колоночной хроматографией на силикагеле, элюент СН2С12. MS m/z 215,06 (М+H+). Выход 23 составил 5,3 г (93%).
Соединение 24. К раствору 5,3 г (0,0247 моль) соединения (23) в 30 мл водной концентрированной HCl при 0°С прикапывали раствор 1,7 г (0,0247 моль) NaNO2 в 3 мл воды. Перемешивали при комнатной температуре 12 часов, затем нагревали при кипении 6 часов. Реакционную массу охлаждали, выпавший осадок отфильтровывали, сушили и затем промывали диэтиловым эфиром. MS m/z 226,06 (М+Н+). Выход 24 составил 4 г (72%).
Соединение 25. К 40 мл пропионовой кислоты добавляли 4 г (0,0177 моль) соединения (24) и перемешивали при 110°С 30 минут, далее прикапывали 5 мл концентрированной HNO3, реакционную смесь перемешивали при 110°С в течение 2 часов. Далее нагрев завершали и смесь перемешивали в течение 12 часов. Выпавший осадок отфильтровывали и сушили над твердым NaOH в эксикаторе. MS m/z 271,2 (М+Н+). Получали соединение 25 (2,7 г, 56%).
Соединение 26. К раствору 2,7 г (0,001 моль) соединения (25) в 50 мл МеОН добавляли 0,5 г никеля Ренея и прикапывали 0,8 мл гидразингидрата. Реакционную смесь перемешивали в течение 12 часов, далее добавляли 50 мл МеОН и смесь перемешивали в течение 15 минут, затем фильтровали через слой цеолита, фильтрат упаривали. Остаток переупаривали 2 раза с толуолом. Получали соединение 26 (0,73 г, 30%), которое сразу использовали в следующей стадии.
Соединение 27. К раствору 0,73 г (0,003 моль, 1 экв.) соединения (26) в 50 мл ДМФА вносили 1,57 г (0,0114, 3,8 экв.) K2CO3 и затем прикапывали 0,4 г (0,0036 моль, 1,2 экв.) хлорацетилхлорида. Смесь перемешивали при 80°С в течение 12 часов, реакционную массу выливали в 200 мл воды, выпавший осадок отфильтровывали и перекристаллизовывали из этанола. MS m/z 281,2 (M+H+). Получали соединение 27 (0,6 г, 70%).
Соединение 28. К раствору 0,6 г (0,0021 моль, 1 экв.) соединения (27) в 25 мл ДМФА вносили 0,15 г (0,0042 моль, 2 экв.) NaH и затем прикапывали 0,377 г (0,00267 моль, 1,25 экв.) СН31. Реакционную смесь перемешивали 1 час, далее выливали в 100 мл воды, выпавший осадок отфильтровывали и перекристаллизовывали из этанола. MS m/z 295,2 (M+H+). Получили соединение 28 (0,14 г, 22%).
Соединение YAI-22-0025. К раствору 0,14 г (0,00047 ммоль, 1 экв.) соединения (28) в 15 мл «сухого» диоксана прибавляли 0,126 г (0,0005 ммоль, 1,05 экв.) B2Pin2 и 0,15 г (0,0015 ммоль, 3,2 экв.) KOAc. Раствор дегазировали пропуская ток Ar 10 минут и вносили 30 мг (0,.041 ммоль) Pd(dppf)Cl2. Далее смесь перемешивали при 100°С под аргоном 12 часов, затем охлаждали до комнатной температуры и добавляли раствор 0,35 г (1 ммоль) Cs2CO3 в 1,5 мл воды. Затем вносили 0,23 г (0,0057 ммоль, 1,2 экв.) соединения (6). Раствор дегазировали пропуская ток Ar 10 минут и вносили 30 мг (0,041 ммоль) Pd(dppf)Cl2. Далее смесь перемешивали при 100°С под аргоном в течение 8 часов. Реакционную массу разбавляли 15 мл EtOAc и фильтровали через слой цеолита. В фильтрат добавляли 30 мл воды. Органический слой отделяли, а водный экстрагировали EtOAc 2 раза по 30 мл. Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт ХМ-МеОН 10% и препаративной хроматографией в присутствии трифторуксусной кислоты. MS m/z 549,4 (М+Н+), 1H ЯМР (400 МГц, ДМСО-d6) δ 9,96 (с, 1H), 9,45 (с, 1H), 8,49 (д, J=2,3 Гц, 1Н), 8,37 (д, J=8,9 Гц, 1H), 8,12-8,02 (м, 2Н), 7,99 (д, J=2,3 Гц, 1H), 7,82-7,73 (м, 2Н), 7,69 (т, J=7,4 Гц, 1H), 7,60 (т, J=7,6 Гц, 2Н), 5,17 (с, 2Н), 4,14 (т, J=5,9 Гц, 2Н), 3,62 (с, 3Н), 3,28-3,10 (м, 2Н), 2,82 (с, 6Н), 2,01-1,81 (м, 2Н). Получили 12 мг соединения YAI-22-0025 (CF3COO-). Выход составил 7%.
Методика получения N-(2-(3-(диметиламино)пропокси)-5-(3-оксо-3,4-дигидро-2H-[1,4]оксазино[3,2-с][1,5]нафтиридин-9 -ил)пиридин-3-ил)бензолсульфонамида (YAI-22-0024):
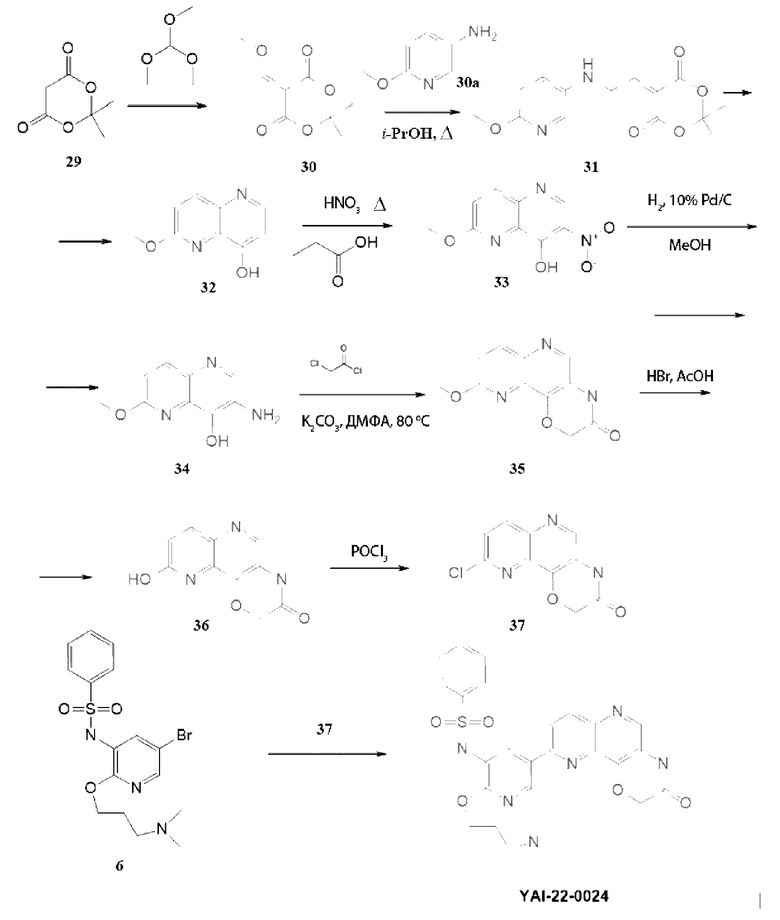
Соединение 30. Нагревали при кипении 2 часа 7 г (0,0485 моль) кислоты Мельдрума (29) в 25 мл триметилортоформиата. Реакционную массу упаривали, остаток переупаривали 2 раза с толуолом. Получали 9,22 г (выход 99%) соединения (30), которое сразу использовали в следующей стадии.
Соединение 31. К раствору 9 г (0,048 моль, 1 экв.) соединения (30) в 50 мл изопропанола вносили 6 г (0,048, 1 экв.) 6-метоксипиридин-3-амина (30а). Смесь перемешивали при кипении 2 часа, реакционную массу охлаждали, выпавший осадок отфильтровьшали. MS m/z 293,2 (М+H+). Получали соединение 31 (9,4 г, 67%).
Соединение 32. Нагревали до 220°С 30 мл дифенилового эфира и вносили 9 г (0,03 моль) соединения (31). Смесь перемешивали при данной температуре 15 минут, далее охлаждали и добавляли 30 мл гексана. Выпавший осадок отфильтровьшали, промывали диэтиловым эфиром и сушили. MS m/z 177,2 (М+H+). Получали соединение 32 (4,5 г, 83%).
Соединение 33. К 50 мл пропионовой кислоты добавляли 4,5 г (0,0255 моль) соединения (32) и перемешивали при 110°С 30 минут, далее прикапывали 5 мл концентрированной HNO3. Реакционную смесь перемешивали при данной температуре 2 часа, нагрев завершали и смесь перемешивали в течение 12 часов. Выпавший осадок отфильтровывали и сушили над твердым NaOH в эксикаторе. MS m/z 222,2 (М+Н+). Получали соединение 33 (1,1 г, 19,5%).
Соединение 34. К раствору 0,7 г (0,0031 моль) соединения (33) в 25 мл метанола вносили 0,1 г 10%) Pd/C и гидрировали при атмосферном давлении в течение 3 часов. Реакционную массу фильтровали через слой цеолита, остаток промывали метанолом, фильтрат упаривали и получали 0,56 г соединения (34), которое немедленно запускали в следующую стадию.
Соединение 35. К раствору 0,56 г (0,0029 моль, 1 экв.) соединения (34) в 50 мл ДМФА вносили 1,61 г (0,012, 4 экв.) K2CO3, затем прикапывали 0,4 г (0,0035 моль, 1,2 экв.) хлорацетилхлорида. Смесь перемешивали при 80°С в течение 12 часов, далее выливали в 200 мл воды, выпавший осадок отфильтровывали и перекристаллизовывали из этанола. MS m/z 232,2 (М+Н+). Получали соединение 35 (0,26 г, 38%).
Соединение 36. К раствору 0,26 г (0,00112 моль) соединения (35) в 6 мл уксусной кислоты прикапывали 2 мл конц. HBr, реакционную смесь перемешивали при 50°С в течение 24 часов, упаривали, остаток выливали в 25 мл 10%) раствора Na2CO3, выпавший осадок отфильтровьшали, сушили над NaOH в эксикаторе. MS m/z 218,2 (M+H+). Получали соединение 36 (0,162 г, 66%).
Соединение 37. Нагревали при кипении 0,16 г (0,00073 моль) соединения (36) в 20 мл POCl3 в течение 4 часов, далее реакционную массу упаривали, остаток выливали в 25 мл 10% раствора Na2CO3, вьтавший осадок отфильтровывали, сушили над NaOH в эксикаторе. MS m/z 236,4 (М+Н+). Получали соединение 37 (0,093 г, 53%).
Соединение YAI-22-0024. К раствору 0,136 г (0,000328 ммоль, 1 экв.) соединения (37) в 15 мл «сухого» диоксана прибавляли 0,087 г (0,000344 моль, 1,05 экв.) B2Pin2 и 0,1 г (0,001 моль, 3,2 экв.) KOAc. Раствор дегазировали, пропуская ток Ar 10 минут, и вносили 30 мг (0,041 ммоль) Pd(dppf)Cl2. Далее перемешивали при 100°С под аргоном в течение 12 часов, затем охлаждали до комнатной температуры и добавляли раствор 0,35 г (1 ммоль, 2 экв.) Cs2CO3 в 1.5 мл воды. Затем вносили 0,067 г (0,00028 ммоль, 0,88 экв.) соединения (6). Раствор дегазировали, пропуская ток Ar 10 минут, и вносили 30 мг (0,041 ммоль) Pd(dppf)Cl2. Далее смесь перемешивали при 100°С под аргоном в течение 8 часов. Реакционную массу разбавляли 15 мл EtOAc и фильтровали через слой цеолита. В фильтрат добавляли 30 мл воды. Органический слой отделяли, а водный экстрагировали EtOAc 2 раза по 30 мл. Органические фазы объединяли, промывали насыщенным раствором NaCl, сушили над безводным Na2SO4 и упаривали. Полученный остаток очищали хроматографией на силикагеле, эл-нт ХМ-МеОН 10%. MS m/z 548,6 (М+Н+), 1H ЯМР (400 МГц, ДМСО-d6) δ 11,20 (с, 1H), 8,68-8,51 (м, 2Н), 8,42-8,27 (м, 2Н), 8,15 (д, J=9,0 Гц, 1H), 7,83 (д, J=7,7 Гц, 2Н), 7,61-7,48 (м, 3Н), 5,03 (с, 2Н), 4,21 (т, J=5,8 Гц, 2Н), 3,09 (т, J=6,9 Гц, 3Н), 2,74 (с, 6Н), 2,04 - 1,87 (м, 2Н). Получали 15 мг соединения YAI-22-0024. Выход составил 10%.
Методика получения N-(2-(3-(диметиламино)пропокси)-5-(4-метил-3-оксо-3,4-дигидро-2H-[1,4]оксазино[3,2-с]циннолин-9-ил)пиридин-3-ил)циклопропансульфонамида (YAI-22-0025а):
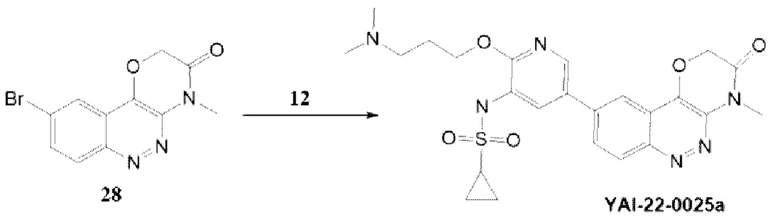
Соединение 28 было получено по методике, описанной для YAI-22-0025, соединение 12 было получено по методике, приведенной для RA-O-00017. Соединение YAI-22-0025a было получено по реакции кросс-каплинга по аналогии с YAI-22-0025. Для соединения YAI-22-0025a были получены следующие аналитические характеристики: MS m/z 513.16 (М+Н+), 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,81 (с, 1H), 9,33 (уш с, 1H), 8,3 (с, 1H), 8,35 (д, 7,2 Гц, 1H), 8,22 (д, J=7,2 Гц, 1Н), 8,17 (с, 1H), 7,75 (с, 1H), 5,21 (с, 2Н), 4,22 (т, J=5,8 Гц), 4,17-3,61 (м, 4Н), 3,66 (с, 3Н), 3,23 (уш с, 2Н), 2,33 (с, 6Н), 1,92 (уш с, 2Н). Получили 12 мг соединения YAI-22-0025a (CF3COO-). Выход соединения YAI-22-0025F составил 10 мг (10%).
Методика получения N-(2-(3-(диметиламино)пропокси)-5-(8-фтор-4-метил-3-оксо-3,4-дигидро-2H-[1,4]оксазино[3,2-с]циннолин-9-ил)пиридин-3-ил)бензолсульфонамида (YAI-22-0025F):
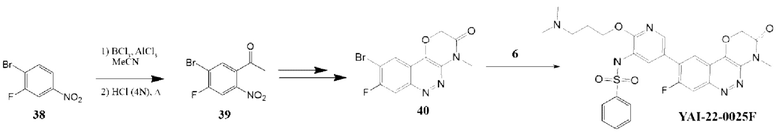
Соединение 39. К раствору BCl3 (5,9 мл, 1 М) в ДХМ (5,9 ммоль) по каплям добавляли 1-бром-2-фтор-4-нитробензол (38) (1 г, 5,26 ммоль) в CHCI2CHCI2 (10 мл) при перемешивании при температуре 0°С. Затем к смеси порциями добавляли MeCN (1 мл) и AlCl3 (0,8 г, 5,9 ммоль). Полученную смесь перемешивали при 120°Св течение 16 часов. После охлаждения смеси, по каплям при 0°С добавляли 2N водный раствор HCl (13 мл). Полученную смесь перемешивали при 100°С в течение 2 часов, затем выливали в ледяную воду и экстрагировали ДХМ (15 мл × 2). Объединенные органические фазы промывали соляным раствором, сушили над безводным Na2SO4, фильтровали и концентрировали. Остаток очищали колоночной хроматографией на силикагеле. Получили соединение 39 (0,27 г, 22%), MS m/z 232 (М+Н+).
Соединение 40 было получено по аналогии с методикой, описанной для соединения 28, с использованием в качестве стартового реагента 39. Соединение YAI-22-0025F было получено в условиях реакции Сузуки-Мияуры по аналогии с соединением YAI-22-0025. Для соединения YAI-22-0025F были получены следующие аналитические характеристики: MS m/z 567,2 (М+Н+), 1H ЯМР (400 МГц, ДМСО-d6) δ 9,89 (с, 1H), 9,47 (уш с, 1H), 8,5 (с, 1H), 8,48 (с, 1Н), 8,10 (с, 1H), 7,82 (с, 1H), 7,82-7,5 (м, 5Н), 5,21 (с, 2Н), 4,22 (т, J=5,8 Гц), 3,66 (с, 3Н), 3,23 (уш с, 2Н), 2,33 (с, 6Н), 1,92 (уш с, 2Н). Получили 10 мг (10%) соединения YAI-22-0025F.
Соединения настоящего изобретения могут быть получены приведенными выше способами или аналогичными способами с использованием соответствующих исходных веществ согласно выбранным заместителям и их положению.
Биологические тесты
Применение соединений настоящего изобретения в качестве ингибитора ATM и DNA-PK киназ может быть определено по методикам следующих примеров, и уровни активности соединений могут быть определены значениями IC50 (концентрация соединения, приводящая к ингибированию активности фермента на 50%).
Фармакологические тесты и результаты кратко изложены следующим образом:
(1) Анализ на ингибирующую активность в отношении ATM киназы
Ингибирующую активность синтезированных соединений в отношении ATM определяли методом резонансного переноса энергии флюоресценции (FRET) и сравнивали с положительным контролем, KU-55933.
ATM(h) инкубировали в буфере, содержащем 30 нМ GST-cMyc-p53 и Mg/АТФ (концентрация по необходимости). Реакцию инициировали добавлением смеси Mg/АТФ. После инкубации в течение 30 минут при комнатной температуре реакцию останавливали добавлением стоп-раствора, содержащего ЭДТА. Далее добавляли буферный раствор для детекции, который содержал меченное 6.2 моноклональное антитело против GST и меченное европием антитело антифосфо-8ег15 против фосфорилированного р53. Планшет считывали в режиме флуоресценции с временным разрешением и определяли гомогенный сигнал флуоресценции с временным разрешением (HTRF) по формуле HTRF = 10000 × (Em665nm/Em620nm). Значения параметра IC50 рассчитывались с использованием уравнения Хилла и стандартной кривой доза-ответ.
(2) Анализ на ингибирующую активность в отношении DNA-PK киназы
Ингибирующую активность синтезированных соединений в отношении DNA-PK определяли методом резонансного переноса энергии флюоресценции (FRET) и сравнивали с положительным контролем, PI-103.
DNA-PK(h) инкубировали в буфере, содержащем 50 нМ GST-cMyc-p53 и Mg/АТФ (концентрация по необходимости). Реакцию инициировали добавлением смеси Mg/АТФ. После инкубации в течение 30 минут при комнатной температуре реакцию останавливали добавлением стоп-раствора, содержащего ЭДТА. Далее добавляли буферный раствор для детекции, который содержал меченное 62 моноклональное антитело против GST и меченное европием антитело антифосфо-Ser15 против фосфорилированного р53. Планшет считывали в режиме флуоресценции с временным разрешением и определяли гомогенный сигнал флуоресценции с временным разрешением (HTRF) по формуле HTRF = 10000 × (Em665nm/Em620nm). Значения параметра IC50 рассчитывались с использованием уравнения Хилла и стандартной кривой доза-ответ.
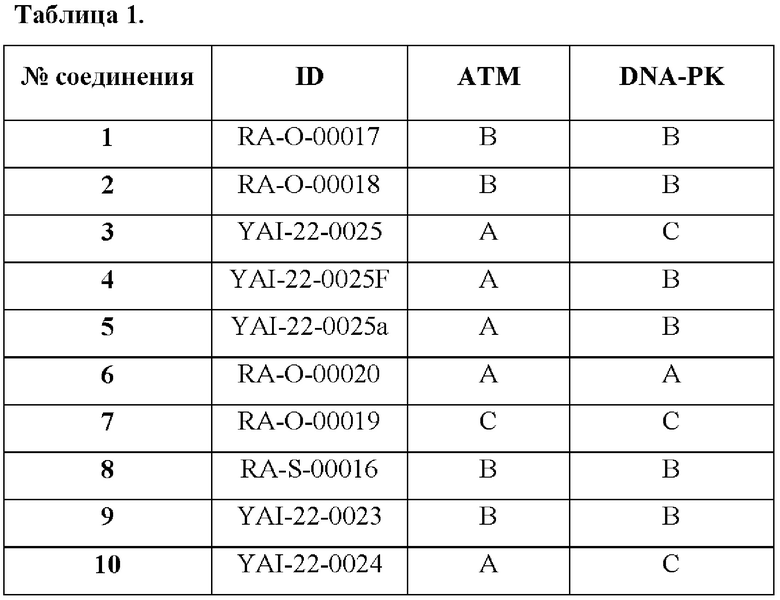
Иллюстративные примеры соединений, обладающих высокой ингибирующей активностью против киназ ATM и DNA-PK, приведены в Таблице 1.
Диапазоны активностей
А IC50<10нМ,
В 10нМ<IC50<100 нМ,
С 100 нМ<IC50<1,000 нМ.
(3) Анализ селективности соединений против родственных киназ семейств PIKK и PI3K
Соединения изобретения тестировали на ингибирующую активность (IC50) против родственных киназ PIKK и PI3K семейств.
Анализ на ингибирующую активность в отношении ATR киназы
Ингибирующую активность синтезированных соединений в отношении ATR определяли методом резонансного переноса энергии флюоресценции (FRET) и сравнивали с положительным контролем, VE-822.
ATR/ATRIP(h) инкубировали в буфере, содержащем 50 нМ GST-cMyc-р53 и Mg/АТФ (концентрация по необходимости). Реакцию инициировали добавлением смеси Mg/АТФ. После инкубации в течение 30 минут при комнатной температуре реакцию останавливали добавлением стоп-раствора, содержащего ЭДТА. Далее добавляли буфер для детекции, который содержал меченное 62 моноклональное антитело против GST и меченное европием антитело антифосфо-Ser15 против фосфорилированного р53. Затем планшет считывали в режиме флуоресценции с временным разрешением и определяли гомогенный сигнал флуоресценции с временным разрешением (HTRF) по формуле HTRF = 10000 × (Em665nm/Em620nm). Значения параметра IC50 рассчитывались с использованием уравнения Хилла и стандартной кривой доза-ответ.
Анализ на ингибирующую активность в отношении mTOR киназы
Ингибирующую активность синтезированных соединений в отношении mTOR определяли радиометрическим методом и сравнивали с положительным контролем, PI-103.
mTOR/FKBP12 (ч) инкубировали с 50 мМ 2-(4-(2-гидроксиэтил)-1-пиперазинил)этансульфоновой кислоты (HEPES), рН=7,5, 1 мМ ЭДТА, 0,01% Твин 20, субстратом 2 мг/мл, 10 мкМ FKBP12, 3 мМ MnCl2 и [гамма-33Р]-АТФ (специфическая активность и концентрация по необходимости). Реакцию инициировали добавлением смеси Mn/АТФ. После инкубации в течение 40 минут при комнатной температуре реакцию останавливали добавлением фосфорной кислоты до концентрации 0,5%. Затем аликвоту реакционной смеси наносили на фильтр и промывали четыре раза по 4 минуты в 0,425%) фосфорной кислоте и один раз в метаноле перед сушкой и сцинтилляционным подсчетом. Значения параметра IC50 рассчитывались с использованием уравнения Хилла и стандартной кривой доза-ответ.
Анализ на ингибирующую активность в отношении PI3K киназ
Ингибирующую активность синтезированных соединений в отношении PI3K киназ определяли методом резонансного переноса энергии флюоресценции (FRET) и сравнивали с положительным контролем, PI-103.
PI3K (p110α/p85α)(h), PI3K (p110α(E542K)/p85α)(h), PI3K (p110β/p85α)(h), PI3K (p110δ/p85α)(h) и PI3K (p120γ)(h) инкубировали в буфере, содержащем 10 мкМ фосфатидилинозитол-4,5-бисфосфата и Mg/АТФ (концентрация по необходимости). Реакцию инициировали добавлением раствора АТФ или смеси Mg/АТФ. После инкубации в течение 30 минут при комнатной температуре реакцию останавливали добавлением стоп-раствора, содержащего ЭДТА и биотинилированный фосфатидилинозитол-3,4,5-трифосфат. Затем, добавляли буфер для обнаружения, который содержит меченные европием моноклональные антитела против GST, меченный GST домен GRP1 РН и стрептавидин-аллофикоцианин. Затем планшет считывали в режиме флуоресценции с временным разрешением и сигнал гомогенной флуоресценции с временным разрешением (HTRF) определяют по формуле HTRF = 10000 × (Em665nm/Em620nm). Значения параметра IC50 рассчитывались с использованием уравнения Хилла и стандартной кривой доза-ответ.
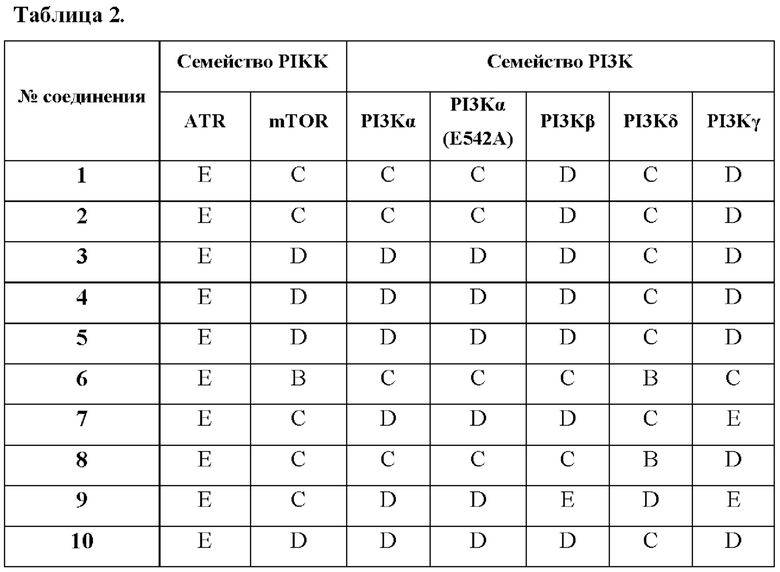
Активности иллюстративных примеров молекул в отношении ATR, mTOR и изоформ PI3K киназ приведены в Таблице 2.
Диапазоны активностей
А IC50<10нМ,
В 10нМ<IC50<100 нМ,
С 100 нМ<IC50<1,000 нМ,
D 1,000 нМ<IC50<10,000 нМ,
Е IC50>10,000 нМ.
Результаты указывают на то, что синтезированные соединения показывают фармакологически значимую активность и селективность в отношении ATM и DNA-PK, при этом не демонстрируя существенной активности в отношении ATR и mTOR, а также ряда киназ PI3K, таких как PI3Kα, PI3Kβ, PI3Kδ и PI3Kγ. Соединения показывают ингибирующую активность против ATM и DNA-PK, одновременно с высокой достоверностью, что установлено в прямом in vitro тесте, согласно общепринятой и апробированной методике.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДВОЙНЫЕ ИНГИБИТОРЫ ATM И DNA-PK ДЛЯ ПРИМЕНЕНИЯ В ПРОТИВООПУХОЛЕВОЙ ТЕРАПИИ | 2020 |
|
RU2800756C1 |
| Соединение-ингибитор мультикиназ и его кристаллическая форма и применение | 2017 |
|
RU2723985C1 |
| Ингибитор ДНК-зависимой протеинкиназы | 2020 |
|
RU2835434C2 |
| Гетероциклические ингибиторы МСТ4 | 2017 |
|
RU2771875C2 |
| НОВЫЕ ГИДРОКСИСЛОЖНОЭФИРНЫЕ ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2734418C2 |
| ДЕЙТЕРИРОВАННЫЕ СОЕДИНЕНИЯ ХИНАЗОЛИНОНА И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2015 |
|
RU2656485C2 |
| МОДУЛЯТОРЫ КИНАЗ AURORA И FLT3 | 2013 |
|
RU2643809C2 |
| СОКРИСТАЛЛЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2014 |
|
RU2675270C2 |
| СО-КРИСТАЛЛЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2014 |
|
RU2823603C2 |
| СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2010 |
|
RU2581367C2 |
Изобретение относится к соединению, определенному формулой (I): 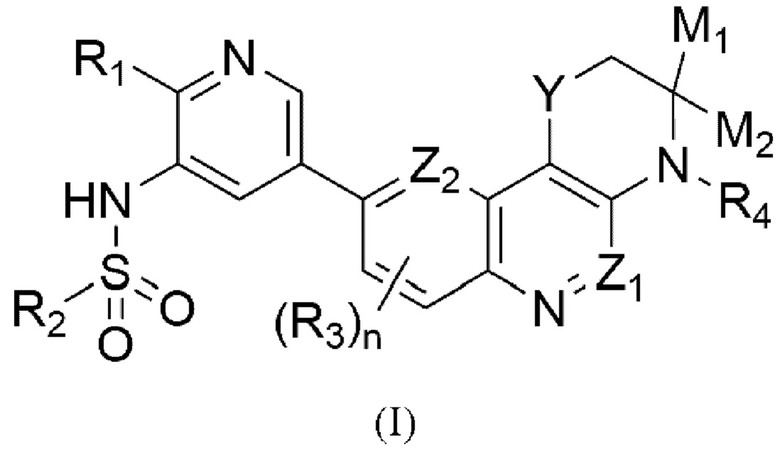 или его фармацевтически приемлемой соли, где Y может представлять собой -S- или -SO2-, тогда Z1 и Z2 выбирается независимо и представляет собой -СН- или N; или Y может представлять собой -О-, тогда какой-либо из Z1 и Z2 представляет собой N, а другой представляет собой -СН-; M1 и М2 представляют собой водород или M1 и М2 формируют вместе =O (кетогруппа); n равно 0 или 1; R1 представляет собой -O-L-N(Ra,Rb); R2 представляет собой С1-3 алкил, С3 циклоалкил или фенил; R3 представляет собой галоген; R4 представляет собой водород или С1-2 алкил; каждый Ra и Rb независимо представляет собой водород или С1-3 алкил; L представляет собой этилен или н-пропилен, а также фармацевтической композиции и применению соединения для производства лекарственного средства для лечения онкологического заболевания, поддающегося лечению двойным ингибитором ATM и DNA-PK. Технический результат: получены новые соединения формулы (I), которые являются двойным ингибитором ATM и DNA-PK и могут быть использованы для производства лекарственного средства для лечения онкологического заболевания, поддающегося лечению двойным ингибитором ATM и DNA-PK. 5 н. и 7 з.п. ф-лы, 2 табл.
или его фармацевтически приемлемой соли, где Y может представлять собой -S- или -SO2-, тогда Z1 и Z2 выбирается независимо и представляет собой -СН- или N; или Y может представлять собой -О-, тогда какой-либо из Z1 и Z2 представляет собой N, а другой представляет собой -СН-; M1 и М2 представляют собой водород или M1 и М2 формируют вместе =O (кетогруппа); n равно 0 или 1; R1 представляет собой -O-L-N(Ra,Rb); R2 представляет собой С1-3 алкил, С3 циклоалкил или фенил; R3 представляет собой галоген; R4 представляет собой водород или С1-2 алкил; каждый Ra и Rb независимо представляет собой водород или С1-3 алкил; L представляет собой этилен или н-пропилен, а также фармацевтической композиции и применению соединения для производства лекарственного средства для лечения онкологического заболевания, поддающегося лечению двойным ингибитором ATM и DNA-PK. Технический результат: получены новые соединения формулы (I), которые являются двойным ингибитором ATM и DNA-PK и могут быть использованы для производства лекарственного средства для лечения онкологического заболевания, поддающегося лечению двойным ингибитором ATM и DNA-PK. 5 н. и 7 з.п. ф-лы, 2 табл.
1. Соединение, определенное формулой (I):
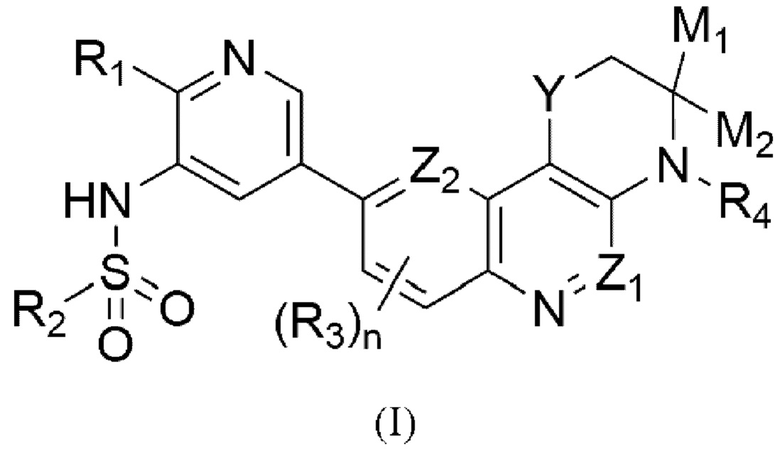
или его фармацевтически приемлемая соль, где
- Y может представлять собой -S- или -SO2-, тогда Z1 и Z2 выбирается независимо и представляет собой -СН- или N;
или
Y может представлять собой -О-, тогда какой-либо из Z1 и Z2 представляет собой N, а другой представляет собой -СН-;
- M1 и М2 представляют собой водород или M1 и М2 формируют вместе =O (кетогруппа);
- n равно 0 или 1;
- R1 представляет собой -O-L-N(Ra,Rb);
- R2 представляет собой С1-3 алкил, С3 циклоалкил или фенил;
- R3 представляет собой галоген;
- R4 представляет собой водород или С1-2 алкил;
- каждый Ra и Rb независимо представляет собой водород или С1-3 алкил;
- L представляет собой этилен или н-пропилен.
2. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении ATM и DNA-PK, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по п. 1 и фармацевтически приемлемое вспомогательное вещество.
3. Применение соединения, указанного в п. 1, для производства лекарственного средства для лечения онкологического заболевания, поддающегося лечению двойным ингибитором ATM и DNA-PK.
4. Применение по п. 3, где онкологическим заболеванием является острый миелоидный лейкоз, острый лимфобластный лейкоз, острый мегакариоцитарный лейкоз, промиелоцитарный лейкоз, эритролейкоз, лимфобластный Т-клеточный лейкоз, хронический миелоидный лейкоз, хронический лимфоцитарный лейкоз, волосатоклеточный лейкоз, хронический нейтрофильный лейкоз, плазмоцитома, иммунобластный крупно клеточный лейкоз, мантийноклеточный лейкоз, множественная миелома, злокачественная лимфома, диффузная крупноклеточная В-клеточная лимфома, лимфома Ходжкина, неходжкинская лимфома, лимфобластная Т-клеточная лимфома, лимфома Беркитта, фолликулярная лимфома, рак головного мозга (например, астроцитома, глиома, глиобластома, медуллобластома, эпендимома), рак мочевого пузыря, рак молочной железы, опухоли центральной нервной системы, рак шейки матки, рак прямой кишки, рак толстой кишки, рак эндометрия, рак пищевода, стромальная опухоль желудочно-кишечного тракта, рак желудка, рак головы и шеи, рак полости рта, гепатоцеллюлярный рак, холангиокарцинома, метастатическое поражение печени, карцинома Меркеля, рак легких, меланома, мезотелиома, рак носоглотки, нейробластома, остеосаркома, рак яичников, рак поджелудочной железы, рак предстательной железы, рак почки, рак слюнной железы, саркомы, рак яичка, уротелиальный рак, рак вульвы и опухоль Вильмса.
5. Применение соединения, указанного в п. 1, для производства лекарственного средства для лечения онкологического заболевания, поддающегося лечению двойным ингибитором ATM и DNA-PK, у пациента, который проходит радиотерапию.
6. Применение по п. 5, в котором соединение вводят пациенту одновременно с радиотерапией.
7. Применение по п. 5, в котором соединение вводят пациенту перед радиотерапией.
8. Применение по п. 5, в котором соединение вводят пациенту после радиотерапии.
9. Применение соединения, указанного в п. 1, для производства лекарственного средства для лечения онкологического заболевания, поддающегося лечению двойным ингибитором ATM и DNA-PK, у пациента, который получает противоопухолевый агент, выбранный из следующего перечня: цисплатин, оксалиплатин, карбоплатин, вальрубицин, идаруцин, калихеамицин, олапариб, рукапариб, нирапариб, велипариб, талазопариб, ипилимумаб, офатумумаб, ниволумаб, пембролизумаб, атезолизумаб, авелумаб, дурвалумаб.
10. Применение по п. 9, в котором соединение вводят пациенту одновременно с противоопухолевым агентом.
11. Применение по п. 9, в котором соединение вводят пациенту перед противоопухолевым агентом.
12. Применение по п. 9, в котором соединение вводят пациенту после противоопухолевого агента.
| CN 103936762 A, 23.07.2014 | |||
| WO 2015146929 A1, 13.04.2017 | |||
| ПРОИЗВОДНЫЕ 2-АМИНОПИРИДИНА ИЛИ 2-АМИНОПИРИМИДИНА В КАЧЕСТВЕ ЦИКЛИНЗАВИСИМЫХ ИНГИБИТОРОВ КИНАЗЫ | 2019 |
|
RU2762557C1 |
| ПРИСПОСОБЛЕНИЕ ДЛЯ УПЛОТНЕНИЯ И ПРОЧЕСЫВАНИЯ ВОЛОКНИСТЫХ ПОЛУФАБРИКАТОВ | 1931 |
|
SU29771A1 |

