ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым соединениям, которые активируют калиевые каналы Kv7. Отдельные аспекты настоящего изобретения направлены на фармацевтические композиции, содержащие указанные соединения, и варианты применения соединений для лечения нарушений, реагирующих на активацию калиевых каналов Kv7.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Потенциал-зависимые калиевые (Kv) каналы пропускают ионы калия (K+) через клеточные мембраны в ответ на изменения мембранного потенциала, и за счет этого они могут контролировать клеточную возбудимость путем модулирования (увеличения или уменьшения) электрической активности клетки. Функциональные каналы Kv существуют в виде мультимерных структур, образованных за счет ассоциации четырех альфа- и четырех бета-субъединиц. Альфа-субъединицы содержат шесть трансмембранных доменов, порообразующую петлю и сенсор потенциала и расположены симметрично вокруг центральной поры. Бета- или вспомогательные субъединицы взаимодействуют с альфа-субъединицами и могут модифицировать свойства канального комплекса для обеспечения без ограничения изменений в электрофизиологических или биофизических свойствах каналов, уровнях экспрессии или профилях экспрессии.
Было идентифицированы девять семейств альфа-субъединиц каналов Kv, и они имеют название Kv1-Kv9. По этой причине существует громадное многообразие функции каналов Kv, которое возникает как следствие множественности подсемейств, образования как гомомерных, так и гетеромерных субъединиц в пределах подсемейств и дополнительных эффектов ассоциации с бета-субъединицами (Christie, 25 Clinical and Experimental Pharmacology and Physiology, 1995, 22, 944-951).
Семейство каналов Kv7 состоит из по меньшей мере пяти членов, которые включают один или несколько из следующих каналов млекопитающих: Kv7.1, Kv7.2, Kv7.3, Kv7.4, Kv7.5, а также их любые относящиеся к млекопитающим или не относящиеся к млекопитающим эквиваленты или варианты (в том числе сплайс-варианты). В качестве альтернативы члены данного семейства обозначают по названиям их генов, представляющих собой KCNQ1, KCNQ2, KCNQ3, KCNQ4 и KCNQ5 соответственно (Dalby-Brown, et al., Current Topics in Medicinal Chemistry, 2006, 6, 9991023).
Как упоминалось выше, калиевые каналы Kv7 нейронов играют определенные роли в осуществлении контроля возбуждения нейронов. Каналы Kv7, в частности гетеродимеры Kv7.2/Kv7.3, лежат в основе М-тока (Wang et al Science. 1998 Dec 4; 282(5395): 1890-3). М-ток имеет характерную зависимость от времени и напряжения, которая в результате обеспечивает стабилизацию мембранного потенциала в ответ на множественные возбуждающие стимулы.
Таким образом, М-ток вовлечен в осуществление контроля возбудимости нейронов (Delmas & Brown, Nature, 2005, 6, 850-862). М-ток представляет собой неактивирующий калиевый ток, обнаруженный во множестве типов нервных клеток. В каждом типе клеток он является преобладающей формой осуществления контроля возбудимости мембраны, являясь единственным устойчивым током, находящимся в пределах диапазона, обеспечивающего инициирование потенциала действия (Marrion, Annual Review Physiology 1997, 59, 483-504).
Ретигабин (сложный этиловый эфир N-(2-амино-4-(4-фторбензиламино)-фенил)карбаминовой кислоты) представляет собой соединение, которое связывается с калиевыми каналами Kv7 (Wuttke, et al., Molecular Pharmacology, 2005, 67, 1009-1017). Ретигабин активирует ток K+ в нервных клетках, и фармакологические характеристики данного индуцированного тока демонстрируют соответствие с опубликованными фармакологическими характеристиками М-канала, который был соотнесен с гетеромультимерным K+-каналом Kv7.2/3, что указывает на то, что активация каналов Kv7.2/3 обуславливает по меньшей мере некоторую противосудорожную активность данного средства (Wickenden, et al., Molecular Pharmacology 2000, 58, 591-600). Ретигабин является эффективным в отношении снижения частоты возникновения судорожных припадков у пациентов с эпилепсией (Bialer, et al., Epilepsy Research 2002, 51, 31-71). Ретигабин характеризуется широким спектром действия и сильными противосудорожными свойствами. Он проявляет активность после перорального и интраперитонеального введения у крыс и мышей в ряде тестов на противосудорожную активность (Rostock, et al., Epilepsy Research 1996, 23, 211-223).
Пять членов данного семейства ионных каналов различаются в отношении их паттернов экспрессии. Экспрессия Kv7. 1 ограничена сердцем, периферическим эпителием и гладкими мышцами, при этом экспрессия Kv7.2, Kv7.3, Kv7.4 и Kv7.5, по-видимому, преобладает в нервной системе, включая нейроны гиппокампа, коры, вентральной области покрышки и спинномозгового узла (обзор см. в Greene & Hoshi, Cellular and Molecular Life Sciences, 2017, 74(3), 495-508).
Гены KCNQ2 и KCNQ3, по-видимому, являются мутированными при наследственной форме эпилепсии, известной как доброкачественные семейные неонатальные судороги (Rogawski, Trends in Neurosciences 2000, 23, 393-398). Белки, кодируемые генами KCNQ2 и KCNQ3, локализуются в пирамидальных нейронах коры и гиппокампа головного мозга человека, областей головного мозга, ассоциированных с возникновением и распространением активности, обуславливающей развитие судорожного припадка (Cooper et al., Proceedings National Academy of Science USA 2000, 97, 4914-4919).
Кроме того, молекулы мРНК, соответствующие Kv7.3 и 5, в дополнение к таковым, соответствующим Kv7.2, экспрессируются в астроцитах и глиальных клетках. Таким образом, каналы Kv7.2, Kv7.3 и Kv7.5 могут способствовать модулированию синаптической активности в ЦНС и содействовать нейропротекторным эффектам соединений, индуцирующих открытие каналов KCNQ (Noda, et al., Society for Neuroscience Abstracts 2003, 53.9), что может являться значимым для лечения нейродегенеративных нарушений, таких как без ограничения болезнь Альцгеймера, болезнь Паркинсона и хорея Гентингтона.
мРНК, соответствующая субъединицам Kv7.2 и Kv7.3, обнаружена в областях головного мозга, ассоциированных с тревожностью и формами эмоционального поведения, такими как депрессия и биполярное расстройство, например в гиппокампе, вентральной области покрышки и миндалевидном теле (Saganich, et al. Journal of Neuroscience 2001, 21, 4609-4624; Friedman et al., Nat Commun. 2016; 7: 11671.), а ретигабин, как сообщается, проявляет активность в животных моделях поведения, подобного тревожному (Korsgaard et al J Pharmacol Exp Ther. 2005 Jul; 314(1): 282-92. Epub 2005 Apr 6.). Таким образом, каналы Kv7 являются значимыми для лечения нарушений, связанных с эмоциями, таких как без ограничения биполярная депрессия, большое депрессивное расстройство, тревожность, суицидальное поведение, панические атаки, социофобия.
Также сообщалось относительно наличия повышенной экспрессии каналов Kv7.2/3 в моделях нейропатической боли (Wickenden, et al., Society for Neuroscience Abstracts 2002, 454.7), и было предположено, что модуляторы калиевых каналов проявляют активность как в случае нейропатической боли, так и в случае эпилепсии (Schroder, et al., Neuropharmacology 2001, 40, 888-898).
В дополнение к роли в развитии нейропатической боли экспрессия мРНК, соответствующей Kv7.2-5, в тройничном и спинномозговом узлах, а также в каудальном ядре тройничного нерва означает, что соединения, индуцирующие открытие таких каналов, могут также затрагивать обработку сенсорной информации из болевых очагов при мигрени (Goldstein, et al. Society for Neuroscience Abstracts 2003, 53.8). В своей совокупности эти свидетельства указывают на значимость соединений, индуцирующих открытие канала KCNQ, для лечения хронической боли и нарушений, связанных с нейропатией.
WO 07/90409 относится к применению соединений, индуцирующих открытие канала Kv7, для лечения шизофрении. Соединения, индуцирующие открытие канала Kv7, модулируют функционирование дофаминергической системы (Friedman et al., Nat Commun. 2016; Scotty et al J Pharmacol Exp Ther. 2009 Mar; 328(3): 951-62. doi: 10.1124/jpet.108.146944. Epub 2008 Dec 19; Koyama et al., J Neurophysiol. 2006 Aug;96(2):535-43. Epub 2006 Jan 4; Li et al Br J Pharmacol. 2017 Dec; 174(23): 4277-4294. doi: 10.1111/bph.14026. Epub 2017 Oct 19.; Hansen et al J Pharmacol Exp Ther. 2006 Sep; 318(3): 1006-19. Epub 2006 Jun 14), что может являться значимым для лечения психических нарушений, таких как без ограничения психоз, мания, нарушения, связанные с воздействием стрессовых факторов, острые реакции на стресс, синдром дефицита внимания и гиперактивности, посттравматическое стрессовое расстройство, обсессивно-компульсивное расстройство, нарушения импульсного контроля, расстройства личности, шизотипическое нарушение, агрессивность, нарушения аутического спектра. В WO 01/96540 раскрыто применение модуляторов М-тока, формируемого за счет экспрессии генов KCNQ2 и KCNQ3, при бессоннице, тогда как в WO 01/092526 раскрыто, что модуляторы Kv7.5 могут быть использованы для лечения нарушений сна. В WO 09/015667 раскрыто применение соединений, индуцирующих открытие Kv7, в лечении нарушения половой функции.
Хотя пациенты, страдающие от вышеуказанных нарушений, могут иметь доступные варианты лечения, у многих таких вариантов отсутствует требуемая эффективность и они сопровождаются нежелательными побочными эффектами. Следовательно, существует неудовлетворенная потребность в новых видах терапии для лечения указанных нарушений.
В попытке идентифицировать новые виды терапии авторы настоящего изобретения идентифицировали серию новых соединений, представленных формулой I. Соответственно, в настоящем изобретении представлены новые соединения в качестве лекарственных препаратов для лечения нарушений, которые модулируются калиевыми каналами KCNQ.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы I,
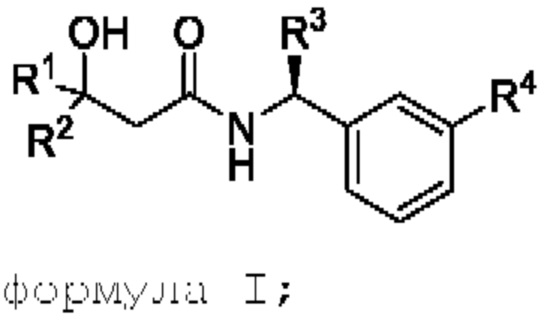
где
R1 выбрана из группы, состоящей из C1-С6алкила, CF3, CH2CF3, CF2CHF2, С3-С8циклоалкила, где указанный С3-С8циклоалкил может быть замещен 1 или 2 из C1-С3алкила, F, CHF2 или CF3, и
R2 представляет собой Н, С1-С6алкил или CF3; или
R1 и R2 объединены (вместе с атомами углерода, к которым они присоединены)с образованием С3-С5циклоалкила, необязательно замещенного 1 или 2 F, CHF2 или CF3; и
R3 представляет собой C1-С3алкил или СН2О-С1-3алкил, при этом указанные C1-С3алкил или СН2О-С1-3алкил замещены C≡N, 3F или С3-С5циклоалкилом;
R4 выбрана из группы, состоящей из OCF3 или OCHF2.
В соответствии с другим аспектом настоящего изобретения настоящее изобретение относится к новым соединениям согласно настоящему изобретению.
В настоящем изобретении также рассматривается фармацевтическая композиция, содержащая соединение согласно настоящему изобретению и фармацевтически приемлемые носитель или вспомогательное вещество.
Кроме того, в настоящем изобретении рассматривается способ лечения пациента, как описано в формуле изобретения и вариантах осуществления, а также предусматриваются варианты лечения пациентов, страдающих эпилепсией, биполярным расстройством, мигренью и шизофренией, предусматривающие введение субъекту терапевтически эффективного количества соединения в соответствии с настоящим изобретением.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно одному варианту осуществления соединение формулы I может содержать группу R4, которая представляет собой OCF3 или OCHF2.
В соответствии с другим вариантом осуществления соединение в соответствии с формулой I может содержать группу R3, выбранную из группы, предусматривающей СН2-O-CF3, СН2-O-циклопропил, CH2-C≡N.
В соответствии с дополнительным вариантом осуществления соединение в соответствии с формулой I может содержать группу R1, которая представляет собой С3-С4циклоалкил, необязательно замещенный 1 или 2 из C1-С3алкила, F, CHF2 или CF3.
В еще одном варианте осуществления соединение в соответствии с любой Формулой I может содержать группу R1 и группу R2, которые объединены с образованием циклобутила, необязательно замещенного 1 или 2 F, и при этом R4 представляет собой OCF3 или OCHF2.
В соответствии с конкретным вариантом осуществления настоящего изобретения соединение в соответствии с настоящим изобретением выбрано из группы, состоящей из
(S)-N-((R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((R)-1-(3-(дифторметокси)фенил)-2-(трифторметокси)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((R)-1-(3-(трифторметокси)фенил)-2-(трифторметокси)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-2-циано-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-3-циано-1-(3-(трифторметокси)фенил)пропил)-3-гидрокси-4,4-диметилпентанамида;
(R)-N-(2-циклопропокси-1-(3-(трифторметокси)фенил)этил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамида;
(R)-N-(2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамида;
(R)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил)-2-(трифторметокси)этил)ацетамида или
(S)-N-(2-циано-1-(3-(трифторметокси)фенил)этил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамида
или фармацевтически приемлемой соли любого из этих соединений.
Другой аспект настоящего изобретения относится к соединению, выбранному из группы, состоящей из
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-3-гидрокси-4,4-диметил-N-((S)-1-(3-(2,2,2-трифторэтокси)фенил)этил)пентанамида;
(R)-3-гидрокси-4,4-диметил-N-((S)-1-(3-(2,2,2-трифторэтокси)фенил)этил)пентанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида;
(R)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида;
(S)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида;
(R)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксипропанамида;
(S)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксипропанамида;
(S)-3-(3,3-дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамида;
(R)-3-(3,3-дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-гидроксипропанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-гидроксипропанамида;
(S)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-1-(3-(дифторметокси)фенил)-4,4-дифторбутил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-1-(3-(дифторметокси)фенил)-3,3-дифторпропил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(R)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)ацетамида;
(R)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)ацетамида;
(S)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил)бутил)ацетамида;
(S)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил)-4,4-дифторбутил)ацетамида;
(S)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(1-(3-(трифторметокси)фенил)пропил)ацетамида;
(S)-N-(3,3-дифтор-1-(3-(трифторметокси)фенил)пропил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида;
(R)-3-циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамида;
(S)-3-циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамида;
(S)-3-циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида;
(R)-3-циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида;
(R)-3-(1-фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамида
и (S)-3-(1-фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамида
или фармацевтически приемлемой соли любого из этих соединений.
Ссылка на соединения, охваченные настоящим изобретением, включает рацемический смеси, а также оптически чистые изомеры соединений, для которых это применимо, а также таутомерный формы соединений, для которых это применимо. Кроме того, соединения по настоящему изобретению потенциально могут существовать в полиморфной и аморфной формах или в виде несольватированной, а также сольватированной форм с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п.
В настоящем изобретении также предусмотрены меченные изотопами формы соединений по настоящему изобретению, как например с помощью дейтерия. Соединение также может содержать позитронно-активные изотопы для медицинской визуализации и исследований посредством позитронно-эмиссионной томографии (PET) с целью определения распределения рецепторов. Подходящими позитронно-активными изотопами, которые могут быть включены в соединения по настоящему изобретению, являются 11С, 13N, 15O и 18F. Меченные изотопами соединения по настоящему изобретению, как правило, могут быть получены посредством традиционных методик, известных специалистам в данной области техники, или посредством способов, аналогичных таковым, описанным в сопутствующих примерах, с применением подходящего меченного изотопами реагента вместо немеченного изотопами реагента.
Соединение согласно настоящему изобретению может находиться в фармацевтической композиции, содержащей соединение и фармацевтически приемлемые вспомогательное вещество или носитель.
В одном варианте осуществления настоящее изобретение относится к соединению согласно настоящему изобретению для применения в терапии.
В другом варианте осуществления настоящее изобретение относится к способу лечения нуждающегося в этом пациента, страдающего эпилепсией, биполярным расстройством, мигренью или шизофренией, предусматривающему введение субъекту терапевтически эффективного количества соединения в соответствии с настоящим изобретением.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения нуждающегося в этом пациента, страдающего психозом, манией, нарушениями, связанными с воздействием стрессовых факторов, острыми реакциями на стресс, биполярной депрессией, большим депрессивным расстройством, тревожностью, паническими атаками, социофобией, нарушениями сна, СДВГ, ПТСР, ОКР, нарушениями импульсного контроля, расстройствами личности, шизотипическим нарушением, агрессивностью, хронической болью, нейропатией, нарушениями аутического спектра, хореей Гентингтона, склерозом, рассеянным склерозом, болезнью Альцгеймера, предусматривающему введение субъекту терапевтически эффективного количества соединения в соответствии с настоящим изобретением.
Согласно одному варианту осуществления соединение по настоящему изобретению применяют в терапии. Применение соединения в соответствии с настоящим изобретением предусмотрено для лечения эпилепсии, биполярного расстройства, мигрени или шизофрении или, в другом варианте осуществления, для лечения психоза, мании, нарушений, связанных с воздействием стрессовых факторов, острых реакций на стресс, биполярной депрессии, большого депрессивного расстройства, тревожности, панических атак, социофобии, нарушений сна, СДВГ, ПТСР, ОКР, нарушений импульсного контроля, расстройств личности, шизотипического нарушения, агрессивности, хронической боли, нейропатии, нарушений аутического спектра, хореи Гентингтона, склероза, рассеянного склероза, болезни Альцгеймера.
В другом варианте осуществления соединение по настоящему изобретению предназначено для изготовления лекарственного препарата для лечения эпилепсии, синдрома ломкой Х-хромосомы, синдрома Ангельмана, биполярного расстройства, мигрени или шизофрении или, в другом варианте осуществления, для изготовления лекарственного препарата для лечения психоза, мании, нарушений, связанных с воздействием стрессовых факторов, острых реакций на стресс, биполярной депрессии, большого депрессивного расстройства, тревожности, панических атак, социофобии, нарушений сна, СДВГ, ПТСР, ОКР, нарушений импульсного контроля, расстройств личности, шизотипического нарушения, агрессивности, хронической боли, нейропатии, нарушений аутического спектра, хореи Гентингтона, склероза, рассеянного склероза, болезни Альцгеймера.
В контексте настоящего изобретения "необязательно замещенный" означает, что указанный фрагмент может быть замещен или может не быть замещен, и если он замещен, то содержит один или два заместителя. Подразумевается, что если для "необязательно замещенного" фрагмента заместители не указаны, то положение занято атомом водорода.
Заданный диапазон может взаимозаменяемо обозначаться с помощью "-" (дефиса) или "от … до", например, термин "C1-3алкил" эквивалентен "от C1 до С3алкил".
Термины "C1-С3алкил" и "C1-С6алкил" относятся к насыщенному углеводороду с неразветвленной или разветвленной цепью, содержащему от одного до шести атомов углерода включительно. Примеры таких групп включают без ограничения, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил и трет-бутил.
Термин "C1-С3алкокси" относится к фрагменту формулы -OR, где R означает C1-С3алкил, определенный выше.
Термины "С3-С4циклоалкил", "С3-С5циклоалкил", "С3-С8циклоалкил" или "циклопропил" относятся к насыщенному моноциклическому кольцу. Примеры таких групп включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и циклооктил.
Пути введения
Фармацевтические композиции, содержащие соединение по настоящему изобретению, определенное выше, могут быть специально составлены для введения любым подходящим путем, таким как пероральный, ректальный, назальный, трансбуккальный, сублингвальный, трансдермальный и парентеральный (например, подкожный, внутримышечный и внутривенный) путь, при этом пероральный путь является предпочтительным.
Следует принимать во внимание, что путь будет зависеть от общего состояния здоровья и возраста субъекта, подлежащего лечению, природы подлежащего лечению состояния и активного ингредиента.
Фармацевтические составы и вспомогательные вещества
В дальнейшем термин "вспомогательное вещество" или "фармацевтически приемлемое вспомогательное вещество" относится к фармацевтическим вспомогательным веществам, в том числе без ограничения наполнителям, антиадгезивным средствам, связующим средствам, покрытиям, красителям, разрыхлителям, ароматизаторам, веществам, улучшающим скольжение, смазывающим средствам, консервантам, сорбентам, подсластителям, растворителям, средам-носителям и вспомогательным средствам.
В настоящем изобретении также предусмотрена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению, такое как одно из соединений, раскрытых в экспериментальном разделе в данном документе. В настоящем изобретении также предусмотрен способ получения фармацевтической композиции, содержащей соединение согласно настоящему изобретению. Фармацевтические композиции согласно настоящему изобретению можно составлять с фармацевтически приемлемыми вспомогательными веществами в соответствии с общепринятыми методиками, такими как методики, раскрытые в Remington, "The Science and Practice of Pharmacy", 22-е изд. (2012), под редакцией Allen, Loyd V., Jr.
Фармацевтические композиции для перорального введения включают твердые пероральные лекарственные формы, такие как таблетки, капсулы, порошки и гранулы; и жидкие пероральные лекарственные формы, такие как растворы, эмульсии, суспензии и сиропы, а также порошки и гранулы, подлежащие растворению или суспендированию в подходящей жидкости.
Твердые лекарственные формы для перорального введения могут быть представлены в виде отдельных единиц (например, таблеток или твердых или мягких капсул), каждая из которых содержит предварительно определенное количество активного ингредиента и предпочтительно одно или несколько подходящих вспомогательных веществ. При необходимости твердые лекарственные формы могут быть покрыты оболочками, такими как кишечнорастворимые оболочки, или они могут быть составлены таким образом, чтобы обеспечивать модифицируемое высвобождение активного ингредиента, как например отсроченное или пролонгированное высвобождение, согласно способам, хорошо известным из уровня техники. При необходимости твердая лекарственная форма может представлять собой лекарственную форму, распадающуюся под действием слюны, такую как, например, таблетка, диспергируемая в полости рта.
Примеры вспомогательных веществ, пригодных для перорального твердого состава, включают без ограничения микрокристаллическую целлюлозу, кукурузный крахмал, лактозу, маннит, повидон, кроскармеллозу натрия, сахарозу, циклодекстрин, тальк, желатин, пектин, стеарат магния, стеариновую кислоту и низшие алкиловые эфиры целлюлозы. Аналогичным образом, твердый состав может включать вспомогательные вещества для составов с отсроченным или пролонгированным высвобождением, известные из уровня техники, такие как глицерилмоностеарат и гипромеллоза. Если для перорального введения используется твердый материал, то состав можно получать, например, с помощью смешивания активного ингредиента с твердыми вспомогательными веществами, а затем прессования смеси в стандартной таблетирующей машине; или состав, например, в виде порошка, гранулы или мини таблетки, может быть помещен, например, в твердую капсулу. Количество твердого вспомогательного вещества в стандартной дозе будет существенно варьировать, однако, как правило, будет находиться в пределах от приблизительно 25 мг до приблизительно 1 г.
Жидкие лекарственные формы для перорального введения могут быть представлены в виде, например, настоек, сиропов, капель для перорального применения или наполненных жидкостью капсул. Жидкие лекарственные формы для перорального введения также могут быть представлены в виде порошков для растворения или суспендирования в водной или неводной жидкости. Примеры вспомогательных веществ, пригодных для перорального жидкого состава, включают без ограничения этанол, пропиленгликоль, глицерин, полиэтиленгликоли, полоксамеры, сорбит, полисорбат, моно- и диглицериды, циклодекстрины, кокосовое масло, пальмовое масло и воду. Жидкие лекарственные формы для перорального введения можно получать, например, с помощью растворения или суспендирования активного ингредиента в водной или неводной жидкости, или с помощью включения активного ингредиента в жидкую эмульсию типа "масло-в-воде" или "вода-в-масле".
В твердых и жидких пероральных составах могут использоваться дополнительные вспомогательные вещества, такие как красители, ароматизирующие добавки и консерванты и т.п.
Фармацевтические композиции для парентерального введения включают стерильные водные и неводные растворы, дисперсии, суспензии или эмульсии для инъекции или инфузии, концентраты для инъекции или инфузии, а также стерильные порошки, перед применением подлежащие ресуспендированию в стерильных растворах или дисперсиях для инъекции или инфузии. Примеры вспомогательных веществ, пригодных для состава для парентерального введения, включают без ограничения воду, кокосовое масло, пальмовое масло и растворы циклодекстринов. Водные составы должны быть подходящим образом забуферены, если это необходимо, и приведены в состояние изотоничности с помощью достаточного количества физиологического раствора или глюкозы.
Другие типы фармацевтических композиций включают суппозитории, составы для ингаляционного введения, кремы, гели, кожные пластыри, имплантаты и составы для трансбуккального или сублингвального введения.
Необходимо, чтобы вспомогательные вещества, используемые для любого фармацевтического состава, соответствовали предполагаемому пути введения и были совместимы с активными ингредиентами.
Дозы
В одном варианте осуществления соединение по настоящему изобретению вводят в количестве от приблизительно 0,001 мг/кг массы тела до приблизительно 100 мг/кг массы тела в день. В частности, ежедневная дозировка может находиться в диапазоне от 0,01 мг/кг массы тела до приблизительно 50 мг/кг массы тела в день. Точные дозировки будут зависеть от частоты и способа введения, пола, возраста, веса и общего состояния здоровья подлежащего лечению субъекта, природы и тяжести подлежащего лечению состояния и каких-либо сопутствующих подлежащих лечению заболеваний, предполагаемого эффекта лечения, а также других факторов, известных специалистам в данной области техники.
Типичная дозировка соединения по настоящему изобретению для перорального введения взрослым будет находиться в диапазоне 0,1-1000 мг/день, как например 1-500 мг/день, как например 1-100 мг/день или 1-50 мг/день. Для удобства соединения по настоящему изобретению вводят в единичной лекарственной форме, содержащей указанные соединения в количестве, составляющем от приблизительно 0,1 до 500 мг, например, 10 мг, 50 мг, 100 мг, 150 мг, 200 мг или 250 мг, соединения по настоящему изобретению.
Изомерная и таутомерная формы
Если соединения по настоящему изобретению содержат один или несколько хиральных атомов, если не указано иное, ссылка на соединения будет охватывать энантиомерно или диастереомерно чистое соединение, а также смеси энантиомеров или диастереомеров в любом соотношении.
Усовершенствованное стереохимическое представление MDL применяется для описания неизвестной стереохимии соединений по настоящему изобретению. Таким образом, обозначение "or1" у хирального атома углерода используют для указания того, что абсолютная стереоконформация при данном атоме неизвестна; например стереоконформация при данном атоме углерода предусматривает либо (S), либо (R).
Кроме того, варианты представления хиральной связи, отходящей от обозначенного "or1" атома углерода, с применением направленной вверх клиновидной линии или направленной вниз клиновидной линии являются равноценными; например два изображения характеризуются одинаковым значением, при этом значение предусматривает, что абсолютная стереоконформация при обозначенном "or1" атоме углерода неизвестна и может представлять собой (S) или (R).
Таким образом, применение связей в виде направленной вверх клиновидной линии и связей в виде направленной вниз клиновидной линии, отходящих от обозначенных "or1" атомов, предназначено только для обеспечения визуального ориентира, указывающего на то, что на изображениях представлены разные стереоизомеры, в которых конформация при обозначенном "or1" атоме углерода неизвестна.
Кроме того, некоторые из соединений по настоящему изобретению могут существовать в разных таутомерных формах, и предполагается, что в объем настоящего изобретения включены любые таутомерные формы, которые могут образовывать соединения.
Терапевтически эффективное количество
В контексте настоящего изобретения термин "терапевтически эффективное количество" соединения означает количество, достаточное для облегчения, остановки, частичной остановки, устранения или задержки клинических проявлений данного заболевания и его осложнений при терапевтическом вмешательстве, предусматривающем введение указанного соединения. Количество, достаточное для осуществления этого, определяется как "терапевтически эффективное количество". Эффективные количества для каждой цели будут зависеть от тяжести заболевания или повреждения, а также от веса и общего состояния здоровья субъекта. Следует понимать, что определение соответствующей дозировки можно осуществлять с применением общепринятых экспериментов, путем построения матрицы значений и тестирования различных точек в матрице, что находится в пределах компетенции квалифицированного врача.
Лечение и осуществление лечения
В контексте настоящего изобретения "лечение" или "осуществление лечения" предназначено для обозначения ведения пациента и ухода за ним с целью облегчения, остановки, частичной остановки, устранения или задержки развития клинического проявления заболевания. Пациентом, подлежащим лечению, предпочтительно является млекопитающее, в частности человек.
Все ссылки, включая публикации, патентные заявки и патенты, цитируемые в данном документе, включены в данный документ посредством ссылки во всей своей полноте и в той же степени, как если бы было указано, что каждая ссылка отдельно и конкретно включена посредством ссылки и приведена во всей своей полноте (в максимальной степени, допускаемой законом).
Применение при эпилепсии, эпилептических синдромах, эпилептических, симптомах или судорожных припадках
В другом варианте осуществления настоящее изобретение относится к способу лечения нуждающегося в этом пациента, страдающего эпилепсией, эпилептическими синдромами, эпилептическими симптомами, устойчивой к лечению или рефрактерной эпилепсией или судорожными припадками, предусматривающему введение субъекту терапевтически эффективного количества соединения в соответствии с настоящим изобретением.
В еще одном варианте осуществления настоящее изобретение относится к способу лечения нуждающегося в этом пациента, страдающего фокальной (парциальной) эпилепсией с простыми парциальными припадками, фокальной (парциальной) эпилепсией с комплексными парциальными припадками, генерализованной идиопатической эпилепсией, большими судорожными припадками, эпилептическим статусом, неонатальными припадками, KCNQ-ассоциированной эпилептической энцефалопатией (KCNQ2EE) и доброкачественными семейными неонатальными судорогами, а также другими эпилептическими синдромами (такими как тяжелая миоклоническая эпилепсия раннего детского возраста, эпилепсия с непрерывными комплексами "спайк-волна" во время медленноволнового сна, синдром Веста, синдром Леннокса-Гасто, синдром Драве и ранняя миоклоническая энцефалопатия, синдром Отахара) или судорожными припадками, связанными с воздействием стрессовых факторов, гормональными изменениями, употреблением наркотических средств (таких как амфетамины или кокаин), употреблением алкоголя, инфекцией или метаболическими расстройствами (такими как гипонатриемия), или предназначенному для применения в лечении эпилептических симптомов, являющихся компонентом нейродегенеративных нарушений, таких как болезнь Альцгеймера, болезнь с тельцами Леви, ювенильная форма болезни Гентингтона, лобно-височная лобарная дегенерация, предусматривающему введение субъекту терапевтически эффективного количества соединения по настоящему изобретению.
Применение соединения в соответствии с настоящим изобретением предусмотрено для лечения эпилепсии, включая применение в лечении фокальной (парциальной) эпилепсии с простыми парциальными припадками, фокальной (парциальной) эпилепсии с комплексными парциальными припадками, генерализованной идиопатической эпилепсии, больших судорожных припадков, эпилептического статуса, неонатальных припадков, KCNQ-ассоциированной эпилептической энцефалопатии (KCNQ2EE) и доброкачественных семейных неонатальных судорог, а также других эпилептических синдромов (таких как тяжелая миоклоническая эпилепсия раннего детского возраста, эпилепсия с непрерывными комплексами "спайк-волна" во время медленноволнового сна, синдром Веста, синдром Леннокса-Гасто, синдром Драве и ранняя миоклоническая энцефалопатия, синдром Отахара) или судорожных припадков, связанных с воздействием стрессовых факторов, гормональными изменениями, употреблением наркотических средств, употреблением алкоголя, инфекцией, травматическим повреждением головного мозга, инсультом, формами рака головного мозга, нарушениями аутического спектра или метаболическими расстройствами (такими как гипонатриемия), или применение предусмотрено при эпилептических симптомах, являющихся компонентом нейродегенеративных нарушений, таких как болезнь Альцгеймера, болезнь с тельцами Леви, ювенильная форма болезни Гентингтона, лобно-височная лобарная дегенерация.
В другом варианте осуществления соединение по настоящему изобретению предназначено для изготовления лекарственного препарата для лечения эпилепсии, эпилептических синдромов, эпилептических симптомов, устойчивой к лечению или рефрактерной эпилепсии или судорожных припадков, включая фокальную (парциальную) эпилепсию с простыми парциальными припадками, фокальную (парциальную) эпилепсию с комплексными парциальными припадками, генерализованную идиопатическую эпилепсию, большие судорожные припадки, эпилептический статус, неонатальные припадки, KCNQ-ассоциированную эпилептическую энцефалопатию (KCNQ2EE) и доброкачественные семейные неонатальные судороги, а также другие эпилептические синдромы (такие как тяжелая миоклоническая эпилепсия раннего детского возраста, эпилепсия с непрерывными комплексами "спайк-волна" во время медленноволнового сна, синдром Веста, синдром Леннокса-Гасто, синдром Драве и ранняя миоклоническая энцефалопатия, синдром Отахара) или судорожные припадки, связанные с воздействием стрессовых факторов, гормональными изменениями, употреблением наркотических средств, употреблением алкоголя, инфекцией, травматическим повреждением головного мозга, инсультом, формами рака головного мозга, нарушениями аутического спектра или метаболическими расстройствами (такими как гипонатриемия), или предназначено для применения в лечении эпилептических симптомов, являющихся компонентом нейродегенеративных нарушений, таких как болезнь Альцгеймера, болезнь с тельцами Леви, ювенильная форма болезни Гентингтона, лобно-височная лобарная дегенерация.
Классификация форм эпилепсии может быть основана на ICD-10 (2016 г., опубликована WHO), и ее описание приводится в разделах G40 и G41, а также она учитывается при лечении эпилепсии в соответствии с настоящим изобретением.
G40.0. Локализованная (фокальная)(парциальная) идиопатическая эпилепсия и эпилептические синдромы с судорожными припадками с фокальным началом.
G40.1. Локализованная (фокальная)(парциальная)
симптоматическая эпилепсия и эпилептические синдромы с простыми парциальными припадками.
G40.2. Локализованная (фокальная) (парциальная) симптоматическая эпилепсия и эпилептические синдромы с комплексными парциальными судорожными припадками.
G40.3. Генерализованная идиопатическая эпилепсия и эпилептические синдромы.
G40.4. Другие виды генерализованной эпилепсии и эпилептических синдромов.
G40.5. Особые эпилептические синдромы.
G40.6. Большие судорожные припадки неуточненные (с малыми припадками или без них)
G40.7. Малые припадки неуточненные без больших судорожных припадков.
G40.8. Другие формы эпилепсия.
G40.9. Эпилепсия неуточненная.
G41. Эпилептический статус.
Лечение судорожных припадков
Судорожный припадок представляет собой внезапное неконтролируемое нарушение электрической активности в головном мозге. Он может вызвать изменения в отношении поведения, двигательной активности или ощущений, а также в отношении уровней сознания. Если у человека наблюдается два или более судорожных припадков или тенденция к периодическому возникновению судорожных припадков, то в этом случае ставят диагноз эпилепсия.
Существует множество типов судорожных припадков, которые варьируют по степени тяжести. Типы судорожных припадков варьируются в отношении того, где и каким образом они возникают в головном мозге. Продолжительность большинства судорожных припадков составляет от 30 секунд до двух минут.
Фокальные судорожные припадки
Фокальные судорожные припадки возникают в результате аномальной электрической активностью в одной области головного мозга. Фокальные судорожные припадки могут возникать с потерей сознания или без нее, предусматривая:
• Фокальные судорожные припадки с нарушением осознанности. В такие судорожные припадки вовлечены изменение или потеря сознания или осознанности. Человек может смотреть в пустоту и не реагировать на окружающую обстановку обычным образом или выполнять повторяющиеся движения, такие как потирание рук, жевание, глотание или хождение по кругу.
• Фокальные судорожные припадки без потери сознания.
Такие судорожные припадки могут изменять эмоции или изменять то, как предметы выглядят, пахнут, ощущаются, чувствуются на вкус или звучат, однако при этом человек не теряет сознания. Такие судорожные припадки также могут приводить к непроизвольным подергиваниям некоторой части тела, такой как рука или нога, и спонтанным симптомам, связанным с сенсорной системой, таким как покалывание, головокружение и световые вспышки.
Генерализованные судорожные припадки
Судорожные припадки, в развитие которых, по-видимому, вовлекаются все области головного мозга, называют генерализованными припадками. Разные типы генерализованных судорожных припадков включают:
• Абсансные судорожные припадки. Абсансные судорожные припадки, ранее известные как малые судорожные припадки, часто возникают у детей и характеризуются устремленным в пустоту взглядом или слабовыраженными движениями тела, такими как моргание глазами или чмокание губами. Такие судорожные припадки могу возникать в виде кластеров и обуславливать кратковременную утерю осознанности.
• Тонические судорожные припадки. Тонические судорожные припадки вызывают скованность мышц. Такие судорожные припадки обычно затрагивают мышцы спины, рук и ног.
• Атонические судорожные припадки. Атонические судорожные припадки, также известные как "дроп- припадки", вызывают потерю мышечного контроля, что может приводить к внезапному коллапсу или падению.
• Клонические судорожные припадки. Клонические судорожные припадки ассоциированы с повторяющимися или ритмическими движениями мышц рывками. Такие судорожные припадки обычно затрагивают шею, лицо и руки.
• Миоклонические судорожные припадки. Миоклонические судорожные припадки обычно проявляются в виде внезапных кратковременных подергиваний или судорог рук и ног.
• Тонико-клонические судорожные припадки. Тонико-клонические судорожные припадки, ранее известные как большие судорожные припадки, являются наиболее выраженным типом эпилептических припадков и могут вызывать внезапную потерю сознания, скованность и дрожь в теле, а также в некоторых случаях потерю контроля над мочевым пузырем или прикусывание языка.
Во многих случаях припадки могут быть ассоциированы со следующими факторами или обусловлены ими:
высокой температурой тела, которая может быть ассоциирована с инфекцией, как например менингит;
недостатком сна;
низкой концентрацией натрия в крови (гипонатриемией), что может произойти при диуретической терапии;
приемом лекарственных препаратов, таких как некоторые болеутоляющие средства, антидепрессанты или средства терапии никотиновой зависимости, которые снижают порог судорожной готовности;
травмой головы, которая приводит к кровоизлиянию в область головного мозга;
инсультом;
наличием опухоли головного мозга;
употреблением наркотических средств, таких как амфетамины или кокаин;
употреблением спирта.
Заголовки и подзаголовки применяются в данном документе исключительно для удобства, и их не следует рассматривать как ограничивающие каким-либо образом настоящее изобретение.
Использование всевозможных примеров или вводного слова перед примерами (в том числе "как например", "например", "к примеру" и "собственно") в данном описании предназначено исключительно для лучшего иллюстрирования настоящего изобретения и не предусматривает ограничение объема настоящего изобретения, если не указано иное.
Цитирование и включение патентных документов в данный документ служит исключительно для удобства и не отражает какую-либо оценку значимости, патентоспособности и/или юридической силы таких патентных документов.
Настоящее изобретение включает все модификации и эквиваленты объекта изобретения, изложенного в прилагаемой к данному документу формуле изобретения согласно действующему законодательству
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Биологическая оценка
Культура клеток
Синтезированный фрагмент кДНК, кодирующий человеческий Kv7.3 и человеческий Kv7.2, разделенные последовательностью Р2А, встраивали в вектор pcDNA5/FRT/TO с использованием сайтов рестрикции BamHI и XhoI. Затем конструкцию трансфицировали в клетки HEK Flp-In 293 с применением липофектамина 2000. Трансфицированные клетки выращивали в течение 48 часов в DMEM, содержащей 10% (об./об.) FBS и 1% PenStrep, а затем поддерживали при селекции в DMEM, содержащей 10% (об./об.) FBS, 1% PenStrep и 200 мкг/мг гигромицина В, при 37°С в увлажненной атмосфере 5% СО2. Полученную устойчивую клеточную линию hKv7.2/hKv7.3 (HEK-hKv7.2/hKv7.3) функционально тестировали с помощью автоматических патч-зажимов целой клетки и с помощью них показывали типичный ток Kv7, который был чувствительным к ХЕ991 и усилен ретигабином.
Анализ поступления таллия
Анализ поступления таллия в отношении активации калиевого канала проводили аналогично опубликованной процедуре (CD. Weaver, et al., J Biomol Screen 2004, 9, 671-677) с применением набора для анализа калия FLIPR (Molecular Devices). Клетки HEK-hKv7.2/hKv7.3 высевали в 96-луночные культуральные планшеты с черными стенками и прозрачным дном(Corning, Актон, Массачусетс, США) при плотности 80000 клеток/лунка (100 мкл/лунка) в случае, если клетки анализировали на следующий день, или 4 0000 клеток/лунка (100 мкл/лунка) в случае, если клетки анализировали через два дня после высева.
В день анализа среду удаляли, после чего 50 мкл/лунка тестируемого соединения разбавляли до 2х конечной концентрации в HBSS, содержащем 20 мМ HEPES, и добавляли 50 мкл/лунка 2х загрузочного буфера красителя. Затем клетки инкубировали в течение 60 мин. при комнатной температуре в темноте. Стимулирующий буфер без хлорида, содержащий Т1+ и K+, при конечной концентрации 5х (концентрация 5х: 5 мМ в обоих случаях) и тестируемое соединение в конечной концентрации 1х, получали во время инкубации. Затем клетки анализировали на функциональной системе скрининга лекарственного средства FDSS7000EX (Hamamatsu). Через 60 сек. считывания базового сигнала флуоресценции при 0,1 Гц и 10 сек. при 1 Гц добавляли 25 мкл/лунка стимулирующего буфера и флуоресценцию непрерывно измеряли в течение 50 сек. при 1 Гц, затем в течение 4 мин. при 0,1 Гц. Эффект соединения количественно определяли с применением AUC в качестве регистрируемых показаний и нормализовали по эталонному соединению, которое включали в каждый планшет.
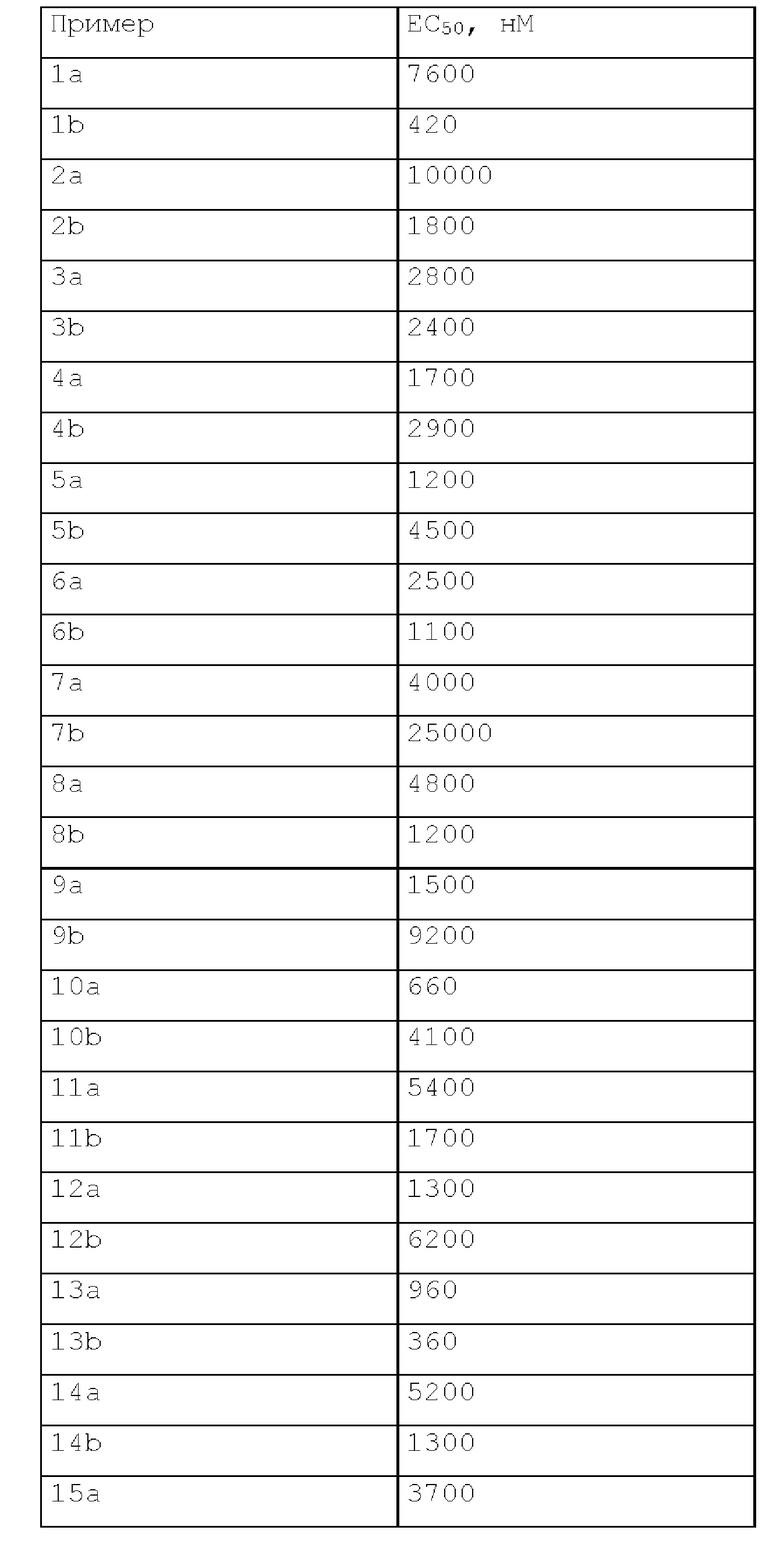
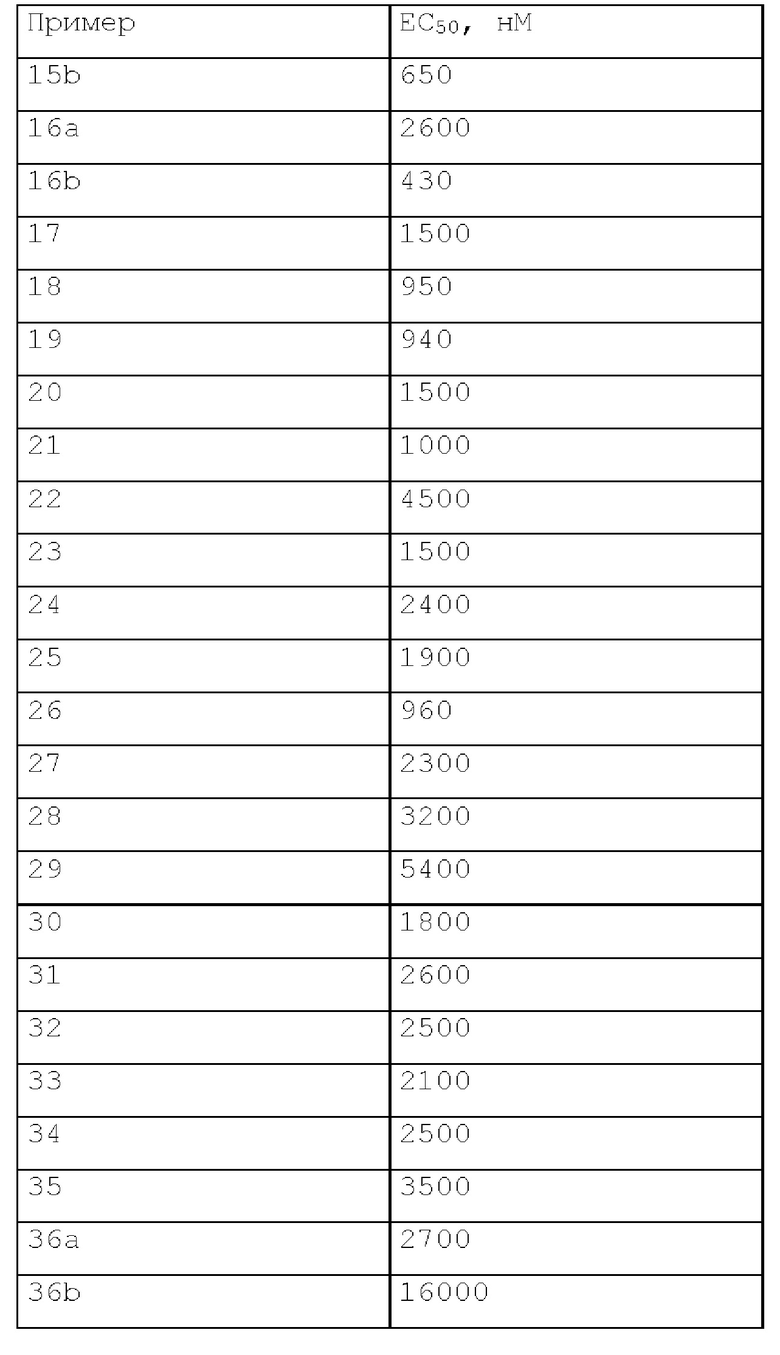
Эффекты соединения
В анализе, описанном выше, соединения по настоящему изобретению характеризовались следующей биологической активностью.
Синтез соединений по настоящему изобретению:
Общие способы
Общие процедуры синтеза промежуточных соединений и соединений общей формулы I описаны не схеме реакции 1 и конкретно проиллюстрированы в способах получения и примерах. В пределах объема настоящего изобретения существуют вариации описанных процедур, которые известны специалисту в данной области техники.
Соединения по настоящему изобретению получают, как описано на схеме 1. Несколько соединений общей формулы I содержат два хиральных атома углерода и образуются в виде смеси диастереомеров. В этом случае диастереомеры можно разделить с получением отдельных стереоизомеров Ia и Ib.
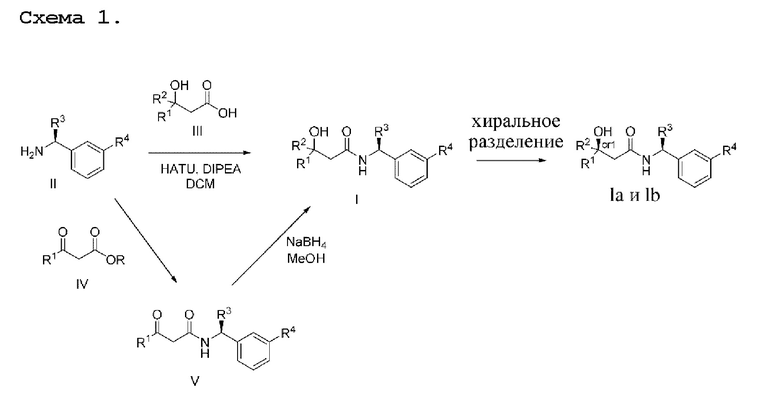
На схеме I показано получение соединения общей формулы I посредством двух общих путей получения. Первый путь получения представляет собой синтез соединений формулы I посредством реакции энантиомерно чистого амина общей формулы II и кислоты общей формулы III, посредством широко известной в уровне техники методики превращения кислоты и амина в амид. Данная методика включает образование реакционно-способных производных кислоты формулы III, включая без ограничения активированные сложные эфиры и реакционно-способные смешанные ангидриды, с последующей конденсацией с аминами общей формулы II. Одной такой методикой является осуществление конденсации в присутствии (1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний 3-оксида гексафторфосфата (HATU) и подходящего основания, такого как диизопропилэтиламин (DIPEA), в растворителе, таком как дихлорметан (DCM). В качестве альтернативы, если R2 представляет собой Н, соединения общей формулы I могут быть получены посредством второго общего пути, в котором промежуточные соединения общей формулы V обрабатывают с помощью подходящего восстановителя, такого как NaBH4, в подходящем растворителе, таком как метанол.
Промежуточные соединения формулы V могут быть получены из энантиомерно чистых аминов общей формулы II и карбоновой кислоты общей формулы IV (R=Н). Данное преобразование можно осуществить с применением условий реакции, аналогичных описанным выше для конденсации соединений II и III с образованием соединения I.
Вариацией данной процедуры является реакция прямого сочетания между хиральным амином общей формулы II и сложным эфиром карбоновой кислоты общей формулы IV (R=Me, Et). Данная реакция может быть осуществлена путем нагревания реагентов с обратным холодильником в подходящем растворителе, таком как толуол, в присутствии подходящего основания, такого как DIPEA, и в присутствии каталитического количества подходящего катализатора, такого как 4-диметиламинопиридин (DMAP).
Оптически активные амины общей формулы II могут быть получены как описано на схеме 2а.
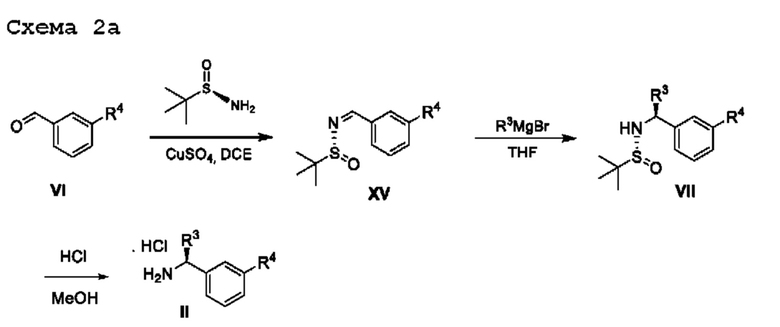
Альдегиды общей формулы VI можно вводить в реакцию конденсации с (R)-2-метилпропан-2-сульфинамидом в подходящем растворителе, таком как дихлорэтан, в присутствии осушителя, такого как изопропоксид титана(IV) или сульфат меди. Образовавшийся сульфинилимин XV обрабатывали R3MgBr в подходящем инертном растворителе, таком как THF, с получением соответствующим образом замещенных (R)-2-метил-N-((S)-1-арил-алкил)пропан-2-сульфинамидов VII, которые превращаются в соединения общей формулы II посредством обработки подходящей кислотой в подходящем растворителе, таком как HCl в МеОН.
В вариациях данной процедуры заместитель R3, несущий функциональную группу, может быть дополнительно модифицирован с помощью методики превращения стандартной функциональной группы, известной специалисту в области химии. Примеры таких манипуляций для получения хиральных аминов проиллюстрированы на схеме 2b. Промежуточное соединение общей формулы VII, в котором R3=аллил, можетбыть образовано из соединения XV и аллил-MgBr, и может подвергаться каталитическому восстановлению до R3=н- пропил с применением Н2(g) в присутствии подходящего катализатора, такого как Pd/C, в подходящем растворителе, таком как этилацетат. В качестве альтернативы, промежуточное соединение общей формулы VII, в котором R3=аллил, может подвергаться дигидроксилированию с последующим окислительным расщеплением с получением промежуточного соединения VII, в котором R3=ацетальдегид, с помощью обработки подходящими катализаторами, такими как K2OSO4 и NaIO4, в присутствии подходящего основания, такого как 2,6-лутидин, и в подходящем растворителе, таком как диоксан/Н2О. Полученный альдегид может подвергаться дополнительным преобразованиям; например, обмену кислорода на два атома фтора путем обработки подходящим реагентом, таким как (диэтиламино)сера трифторид (DAST), в подходящем растворителе, таком как DCM. На конечной стадии хиральные амины могут быть получены путем гидролиза, как описано выше.
В качестве другого примера промежуточное соединение общей формулы XVI, в котором R3 представляет собой сложный эфир уксусной кислоты, может образоваться из соединения XV во время реакции с этил-2-бромацетатом в присутствии CuCl и активированного Zn в подходящем растворителе, таком как THF. Сложноэфирная группа может быть дополнительно преобразована путем преобразований, известных специалисту в области химии. Например, сложный эфир может быть восстановлен до первичного спирта путем обработки подходящим восстановительным средством, таким как LiAlH4, в подходящем растворителе, таком как THF.
Полученный спирт также может быть дополнительно преобразован путем преобразований, известных квалифицированному практику. Например, спирт можно превращать в уходящую группу посредством активации с помощью реагента, такого как мезилхлорид, в присутствии подходящего основания, такого как триэтиламин (TEA), в подходящем растворителе, таком как DCM. Полученные мезилаты можно подвергать реакции с подходящим нуклеофилом, таким как цианид калия, в подходящем растворителе, таком как DMSO.
Схема 2b.

Альдегиды общей формулы VI также можно превращать в хиральные амины общей формулы II посредством альтернативной стратегии, проиллюстрированной на схеме 2 с.
Схема 2с.
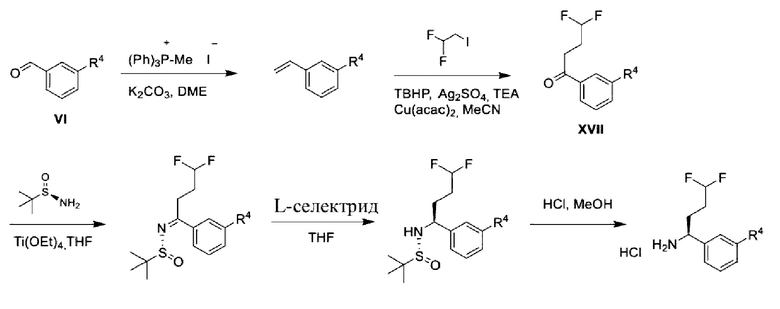
Следуя этой процедуре, соответствующим образом замещенные бензальдегиды VI можно превращать в соответствующие соответствующим образом замещенные стиролы с применением методики Виттига, известной специалисту в области химии. Такие промежуточные соединения могут подвергаться реакции алкилирования-окисления с образованием кето-промежуточных соединений XVII. Промежуточные соединения общей формулы XVII можно вводить в реакцию конденсации с (R)-2-метилпропан-2-сульфинамидом в подходящем растворителе, таком как дихлорэтан, в присутствии подходящего осушителя, такого как титан(IV)этоксид. Образовавшийся сульфинилимин может быть восстановлен с помощью подходящего восстановителя, такого как L-селектрид, в подходящем инертном растворителе, таком как THF, с получением соответствующего соответствующим образом замещенного (R)-2-метил-N-((S)-1-арил-алкил)пропан-2-сульфинамида, который можно превращать в хиральные амины общей формулы II посредством обработки подходящей кислотой в подходящем растворителе, таком как HCl в МеОН.
Специалисту в данной области техники будет понятно, что другие преобразования возможны из нескольких из промежуточных соединений; настоящее изобретение предназначено для включения таких альтернативных преобразований.
Альдегиды формулы VI, применяемые для получения соединений по настоящему изобретению, являются коммерчески доступными, или могут быть получены как описано в литературе, см. Journal of Medicinal Chemistry, 45(18), 3891-3904; 2002.
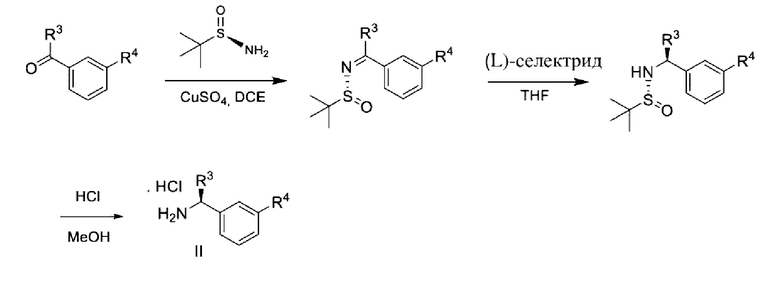
В другой процедуре хиральные амины формулы II могут быть получены из арилкетона посредством восстановления гидридом промежуточного соединения, представляющего собой сульфинилимин, полученный из реакции (R)-2-метилпропан-2-сульфинамида с реагентом, таким как L-селектрид; как показано на схеме 3а.
Схема 3а
Вариация данной процедуры проиллюстрирована на схеме 3b. В данной процедуре дополнительные заместители R3, которые представляют собой эфиры, могут быть установлены из обычного промежуточного соединение альфа- бромацетофенона XI, полученного путем бромирования соответствующим образом замещенного ацетофенона. Примеры включают без ограничения введение групп фторалкилметиленового эфира в качестве R3. Таким образом, с применением данной методики, соответствующим образом замещенный бромацетофенон можно подвергать реакции с донором фторалкокси, таким как трифторметил трифторметансульфонат и KF, в условиях Финкелынтейна в подходящем растворителе, таком как диметилацетамид (DMA), с получением промежуточного соединения XII, которое можно дополнительно превращать в хиральные амины, как описано выше. Другие примеры включают без ограничения введение алкилметиленовых эфиров в качестве R3. Таким образом, соответствующим образом замещенный бромацетофенон можно превращать в промежуточное соединение XIII, представляющее собой ацилдиазо, путем реакции с 4-метил-N'-(п-толилсульфонил)бензолсульфоногидразидом в присутствии подходящего основания, такого как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), и в подходящем растворителе, таком как THF. Промежуточное соединение XIII может подвергаться реакции образования карбена путем обработки подходящим катализатором, таким как трифлат индия, в подходящем растворителе, таком как толуол. Образованный карбен может быть захвачен in situ подходящим спиртом с получением промежуточных соединений XIV, представляющих собой алкоксикето. Из промежуточного соединения XIV могут быть получены хиральные амины формулы II посредством стандартных манипуляций, как описано выше.
Схема 3b.
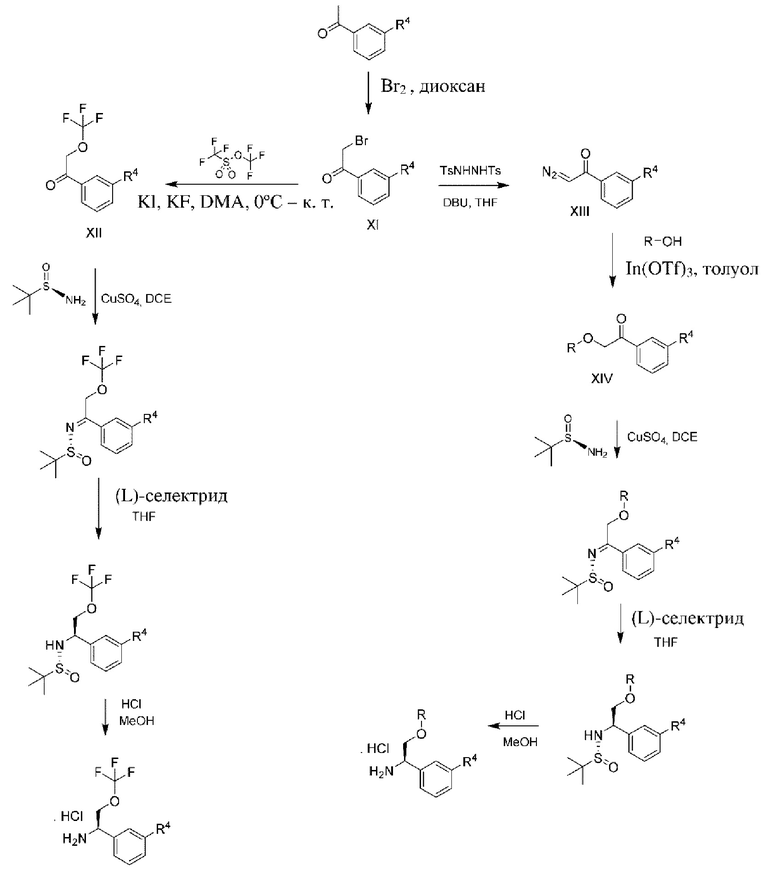
Соответствующим образом замещенные кетоны или ацетофеноны, применяемые для получения соединений по настоящему изобретению, являются коммерчески доступными, или могут быть получены посредством способов, известных специалисту в данной области техники.
Специалисту в данной области техники будет понятно, что другие преобразования возможны из промежуточных соединений общей формулы XI; настоящее изобретение предназначено для включения таких альтернативных преобразований.
Другая процедура, подходящая для получения хиральных аминов общей формулы II, в которой R3 представляет собой эфир, описанный на схеме 4.
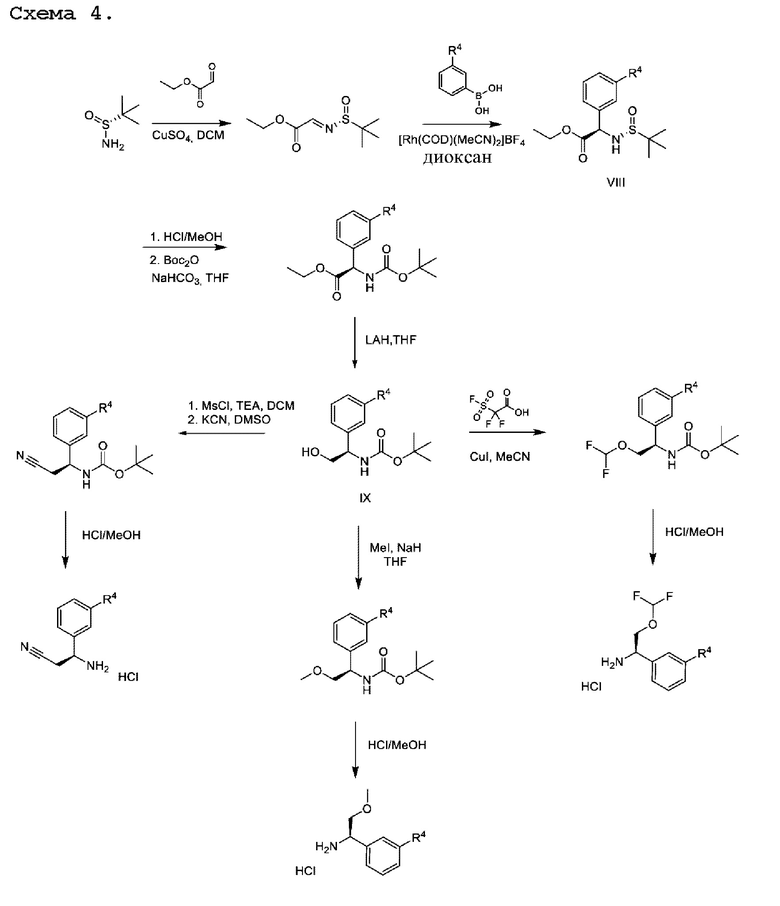
В данной процедуре глиоксилат сульфинилимина, образованный при реакции конденсация между глиоксиловым сложным эфиром и (R)-2-метилпропан-2-сульфинамидом, может подвергаться реакции с соответствующим образом замещенной бороновой кислотой с применением подходящего катализатора, такого как бис(ацетонитрил)(1,5-циклооктадиен)родий(I)тетрафторборат, в подходящем растворителе, таком как диоксан, как описано в JP 2017/095366 А. Полученные промежуточные соединения VIII могут быть гидролизованы, повторно защищены подходящей защитной группой, такой как Boc, в стандартных условиях и подвергнуты восстановлению сложным эфиром в подходящих условиях, таких как LiAlH4 в THF, с получением промежуточных соединений общей формулы IX, представляющих собой спирт.Промежуточные соединения формулы IX могут быть дополнительно модифицированы для получения требуемого заместителя R3. Например, спиртовая группа в промежуточных соединениях IX может быть дифторметилирована с применением подходящего реагента, такого как 2,2-дифтор-2-(фторсульфонил)уксусная кислота, в условиях, таких как активация CuI, в подходящем растворителе, таком как ацетонитрил, или спиртовая группа в соединении IX может быть алкилирована с применением простых алкилгалогенидов, таких как метилиодид, в присутствии подходящего основания, такого как NaH, в подходящем растворителе, таком как THF. В качестве альтернативы спирт промежуточных соединений IX можно превращать в уходящую группу посредством активации с помощью реагента, такого как мезилхлорид, в присутствии подходящего основания, такого как TEA, в подходящем растворителе, таком как DCM. Полученные мезилаты можно подвергать реакции с подходящим нуклеофилом, таким как цианид калия, в подходящем растворителе, таком как DMSO.
Специалисту в данной области техники будет понятно, что другие преобразования возможны из промежуточных соединений общей формулы IX; настоящее изобретение предназначено для включения таких альтернативных преобразований.
Карбоновые кислоты общей формулы III могут быть получены как описано на схеме 5.
Схема 5
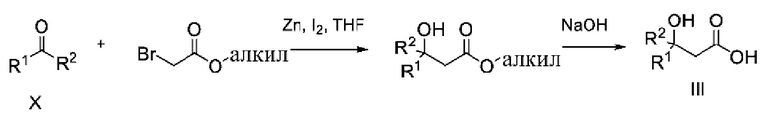
Кетоны общей формулы X подвергают реакции со сложным эфиром алкила бромуксусной кислоты, активированным с помощью, например, Zn и йода, с получением соответствующего альдольного аддукта. В альтернативной процедуре сложный эфир бромуксусной кислоты может быть активирован с применением Zn и TMSCl (триметилсилилхлорид). На конечной стадии гидролиз сложного эфира алкила осуществляют путем обработки подходящим основанием, таким как NaOH или LiOH, в подходящем растворителе, таком как вода или спирт в воде, и с последующим подкислением с помощью подходящей кислоты с получением соединений формулы III.
Сложный эфир карбоновой кислоты общей формулы IV (R=Me, Et) является коммерчески доступным или может быть получен как описано на схеме 6.
Схема 6
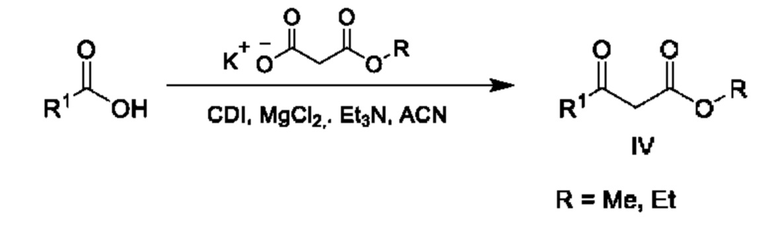
Соответствующим образом замещенные карбоновые кислоты могут быть активированы с применением подходящего реагента, такого как CDI, и их вводят в реакцию конденсации с 3-этокси-3-оксо-пропаноатом калия в присутствии MgCl2 с получением промежуточных соединений общей формулы IV.
1Н ЯМР спектры регистрировали при 400,13 МГц на устройстве Bruker Avance III 400 или при 300,13 МГц на устройстве Bruker Avance 300. В качестве растворителя использовали дейтерированный диметилсульфоксид или дейтерированный хлороформ. В качестве внутреннего стандартного образца использовали тетраметилсилан. Значения химического сдвига выражены в значениях ррт относительно тетраметилсилана. Для множественных сигналов ЯМР используются следующие сокращения: s = синглет, d = дублет, t = триплет, q = квартет, qui = квинтет, h = гептет, dd = двойной дублет, ddd = двойной дублет дублетов, dt = двойной триплет, dq = двойной квартет, tt = триплет триплетов, m = мультиплет и brs = широкий сигнал.
Хроматографические системы и способы для оценки химической чистоты (способы LCMS) и хиральной чистоты (способы SFC и HPLC) описаны ниже.
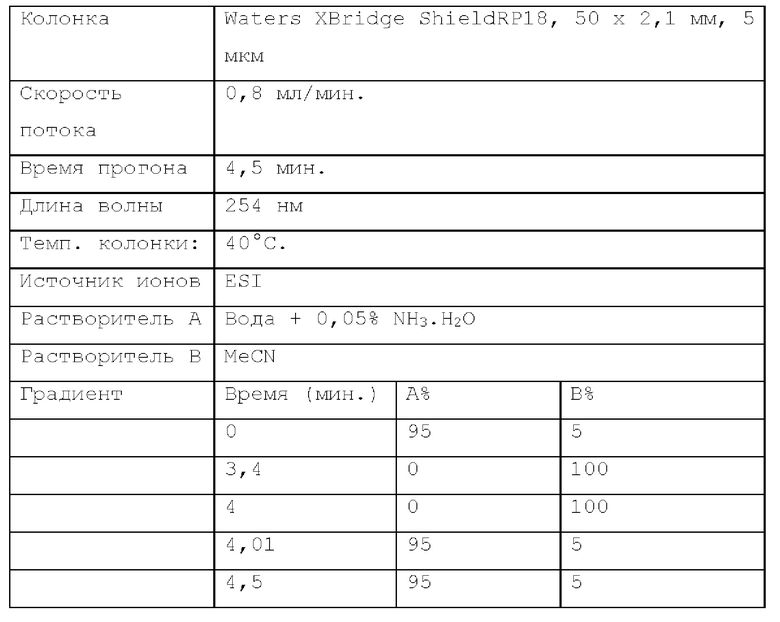
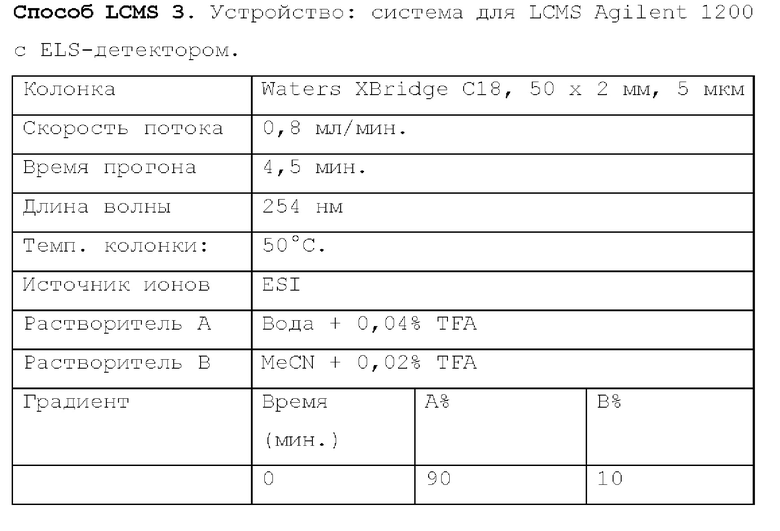
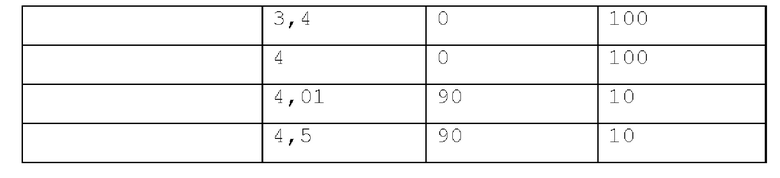
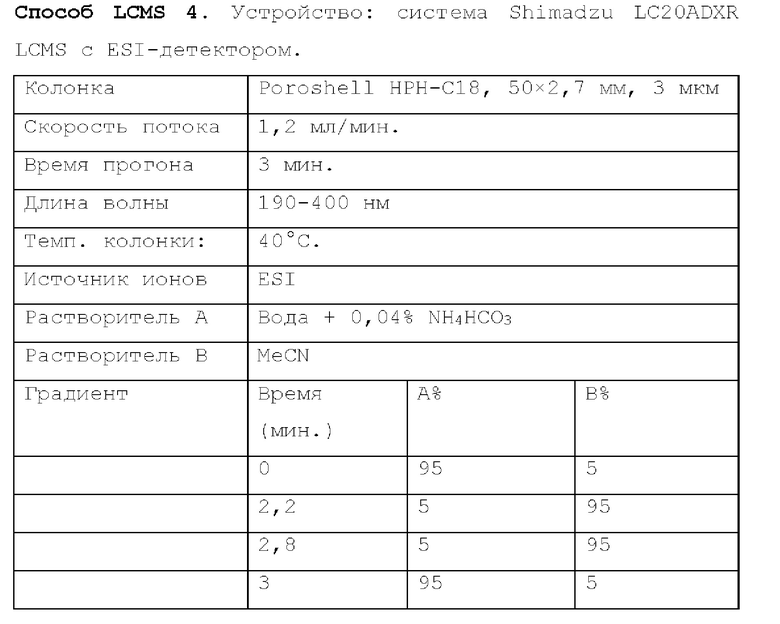
Способ LCMS 2. Устройство: Система для LCMS Agilent 1200 с ELS-детектором.
Хиральные методы анализа.

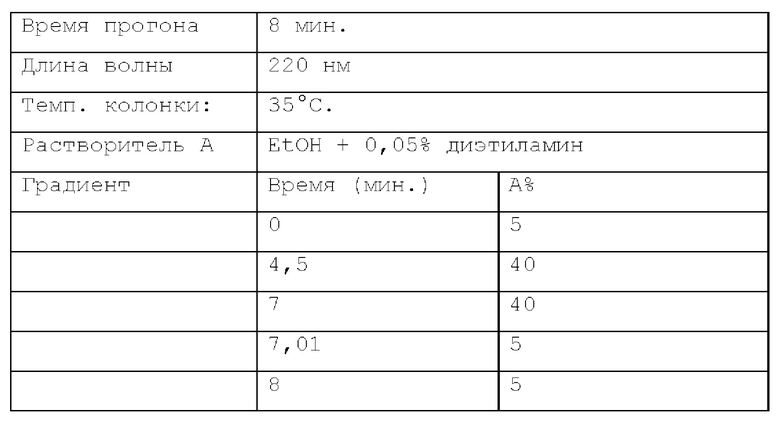
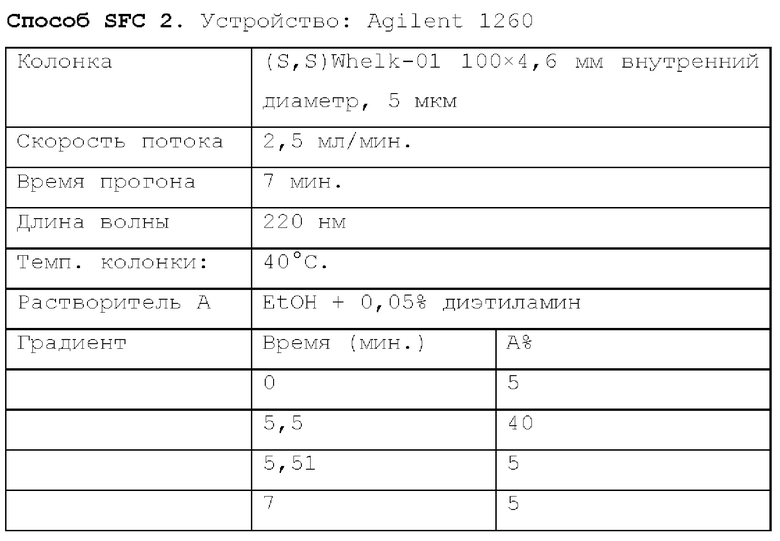

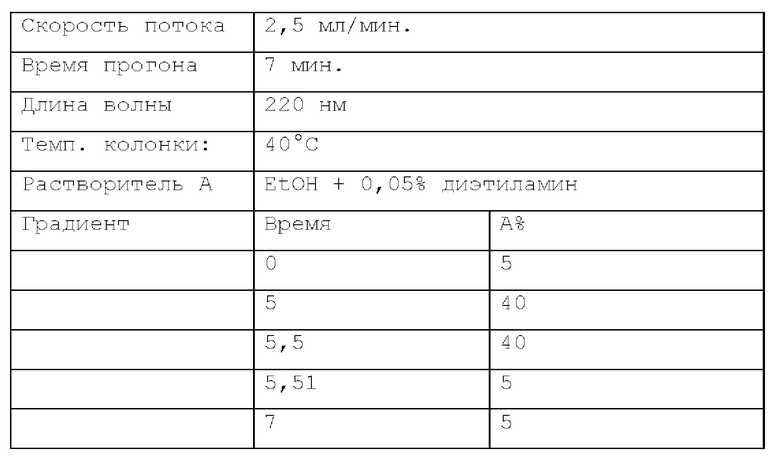
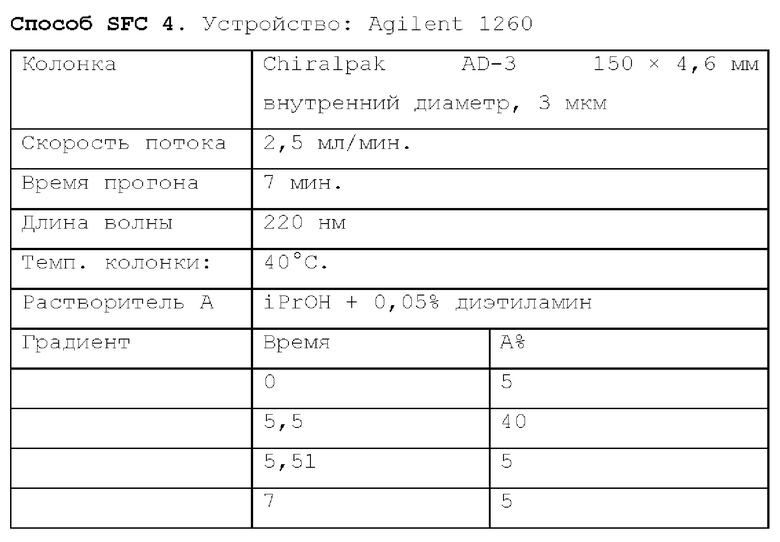
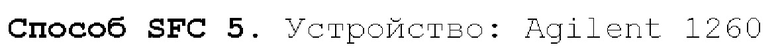
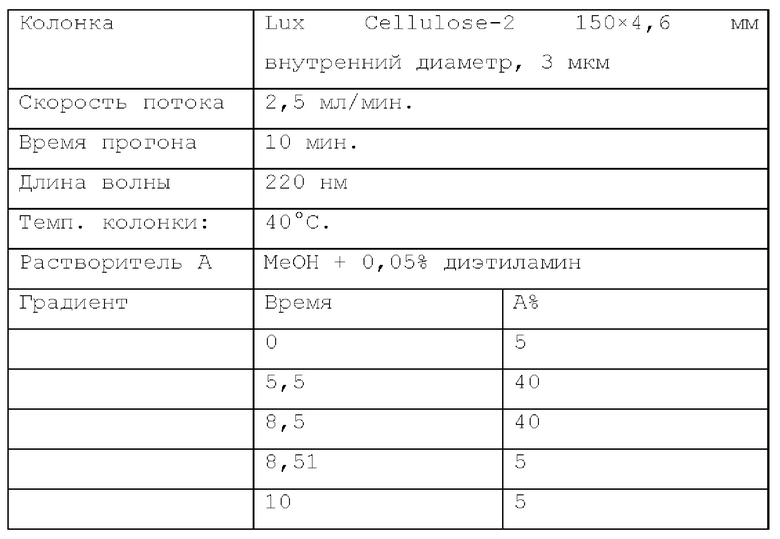
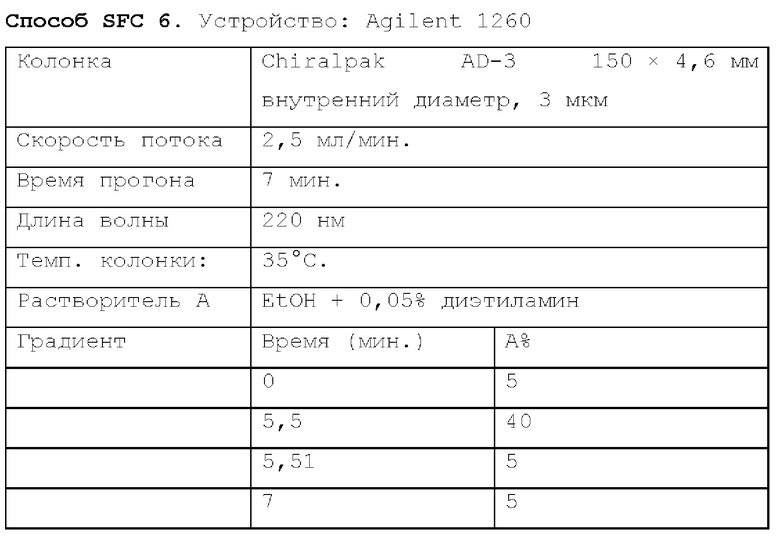
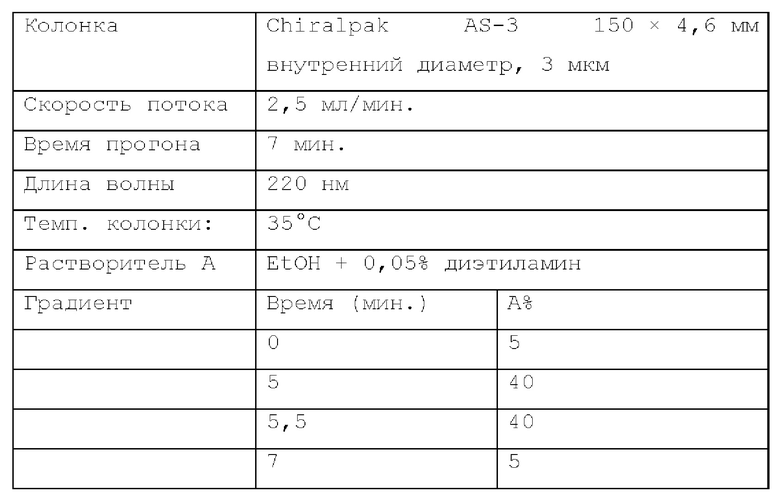
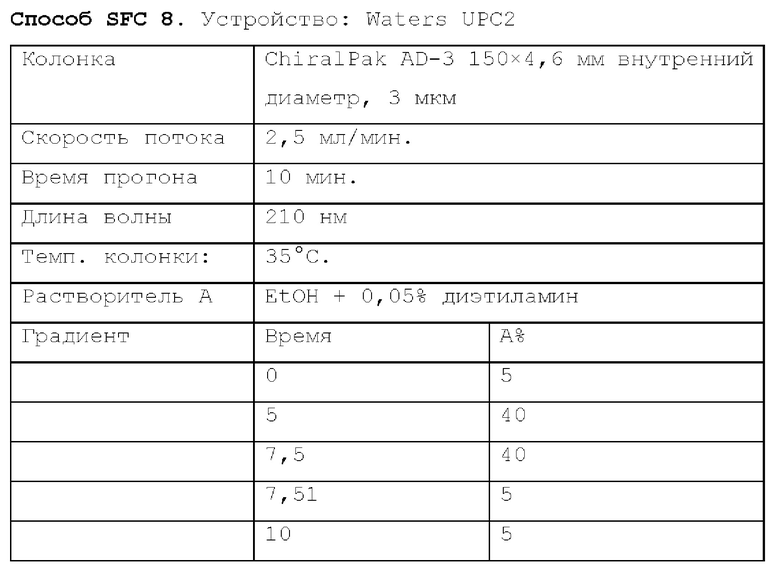
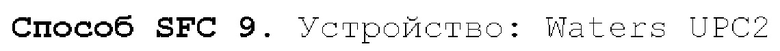

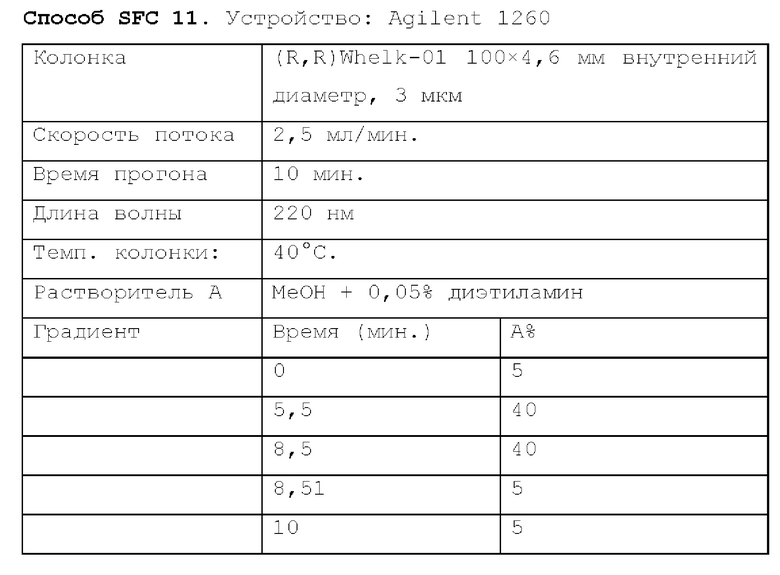
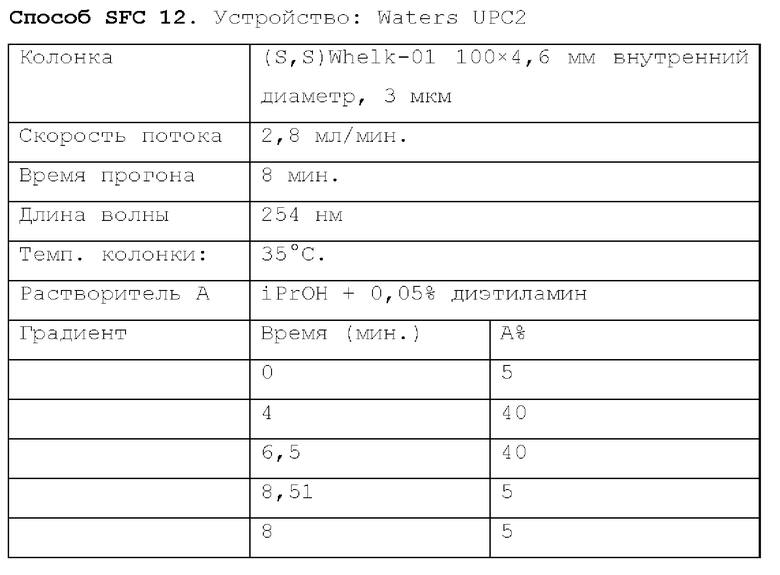
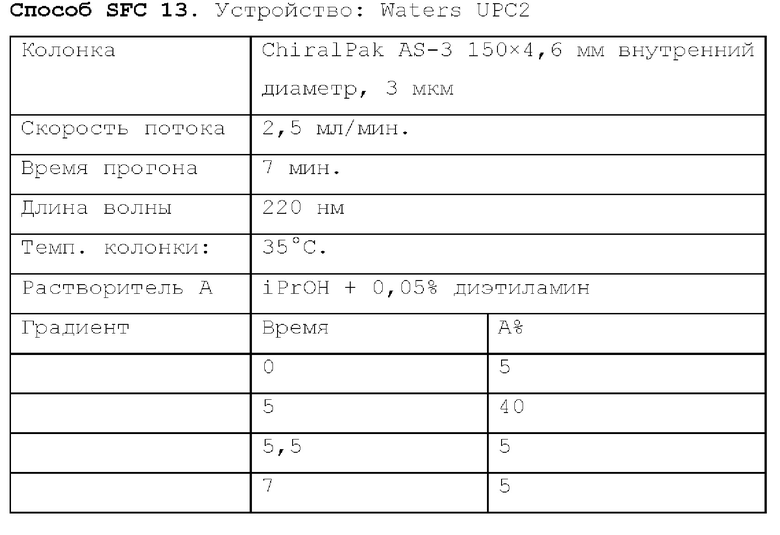
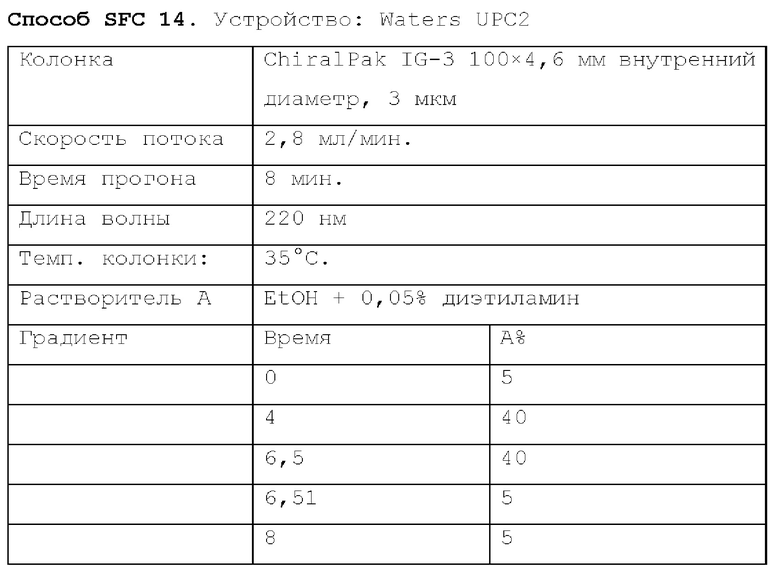
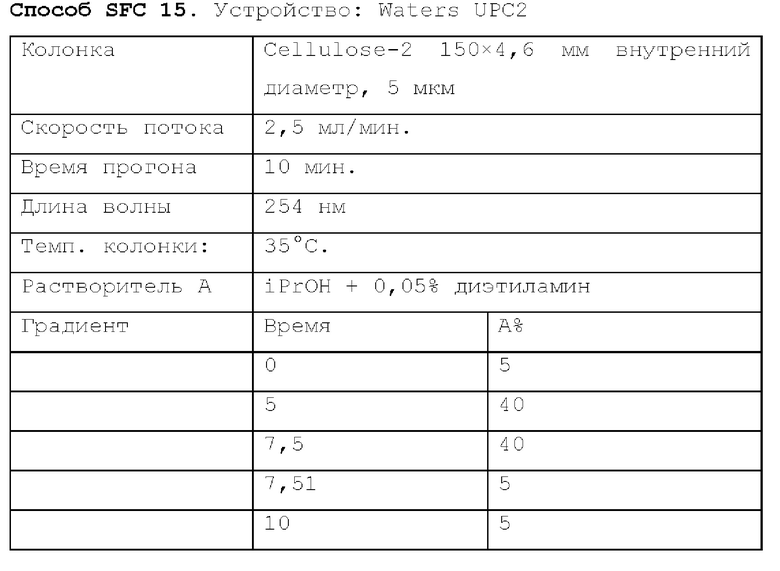
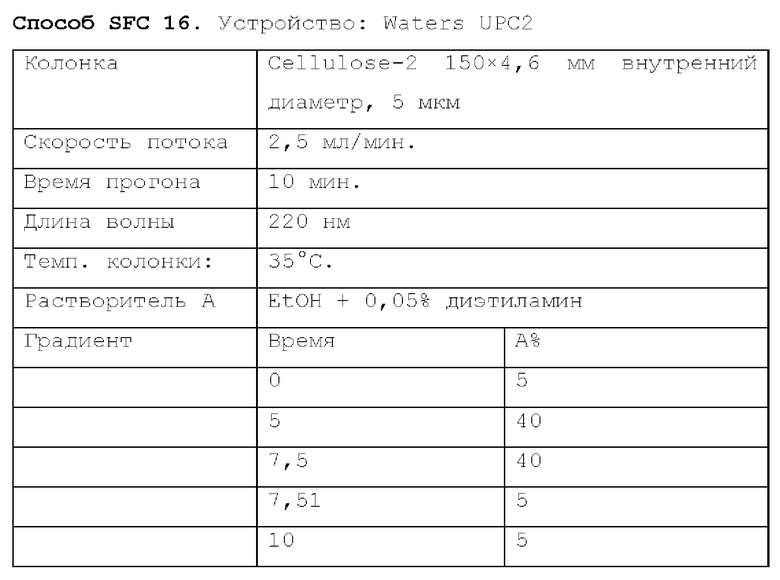
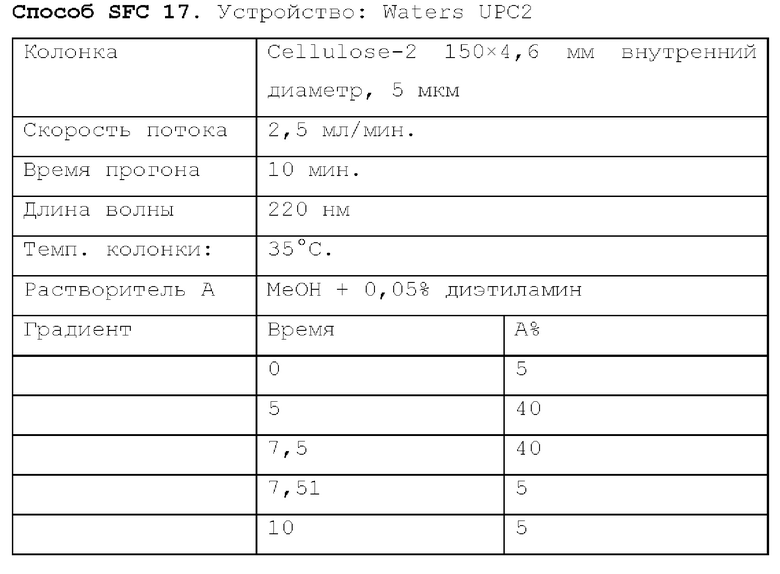
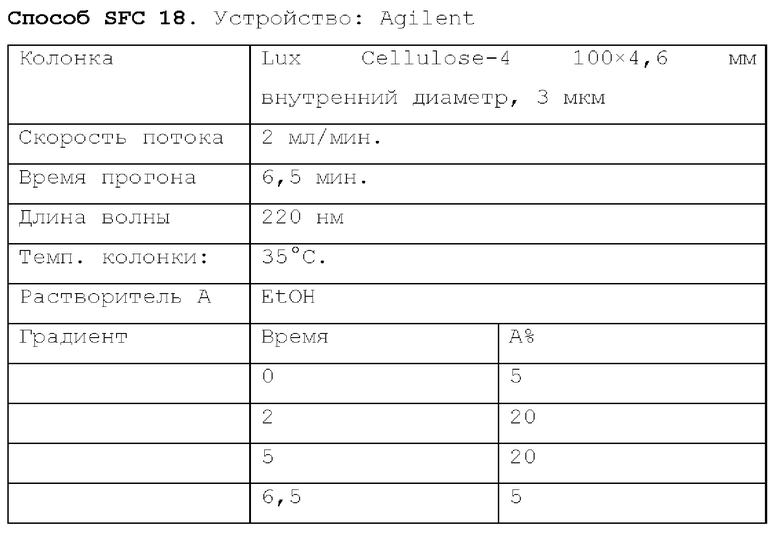

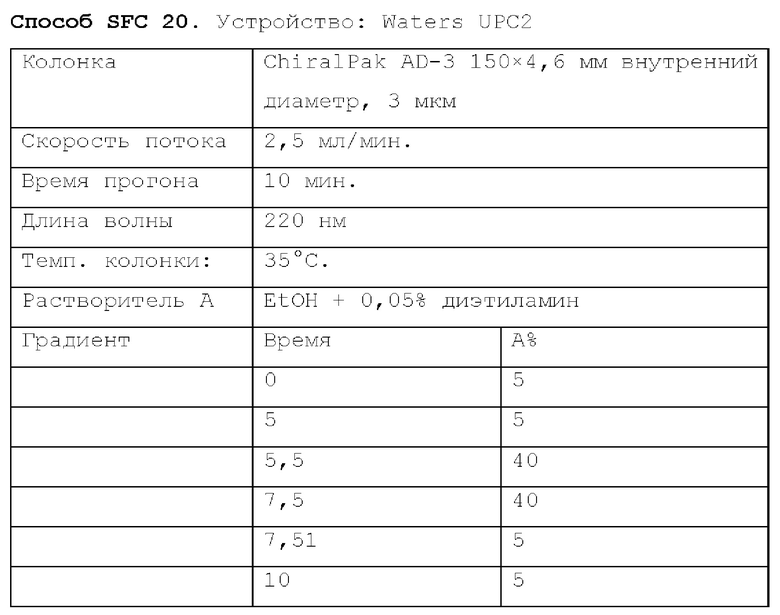
Способ SFC 21. Устройство: Waters UPC2
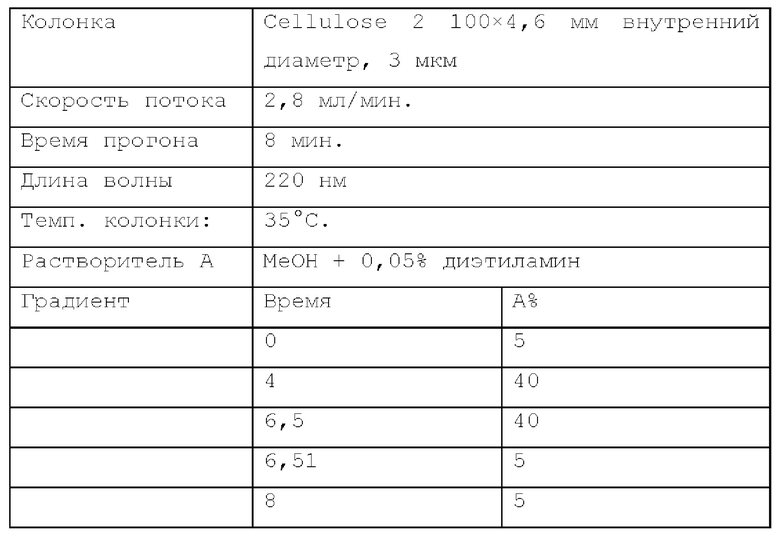
Способ SFC 22. Устройство: Agilent 1260
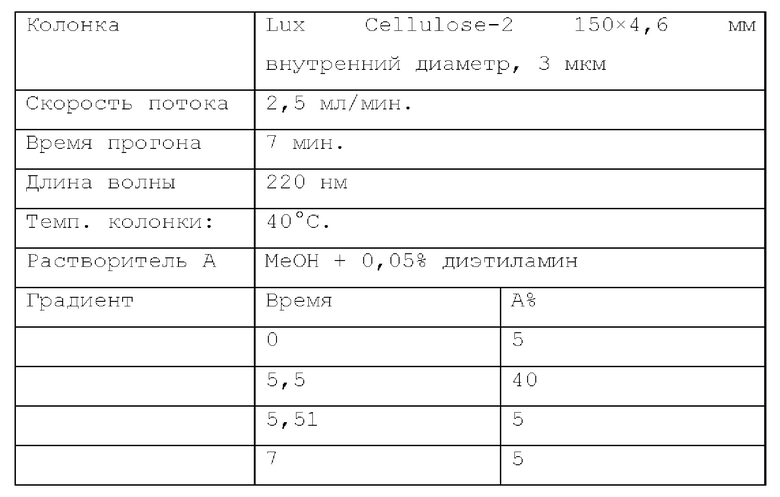
Способ хиральной HPLC 1. Устройство: SHIMADZU LC-20AB
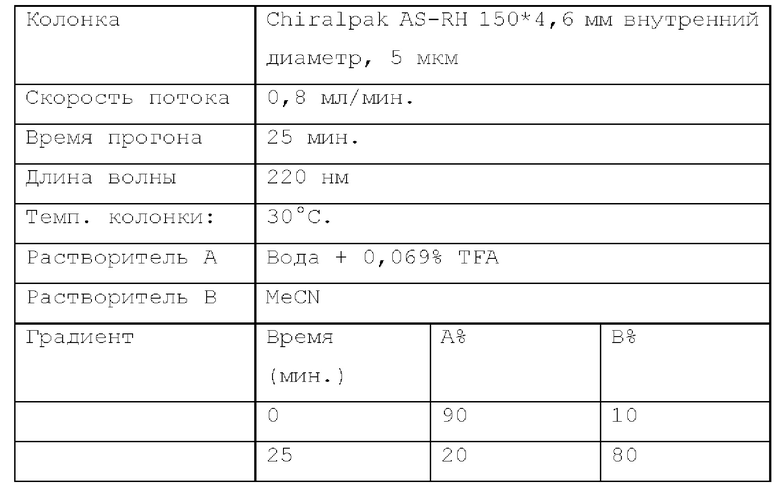
Способ хиральной HPLC 2. Устройство: SHIMADZU LC-20AB
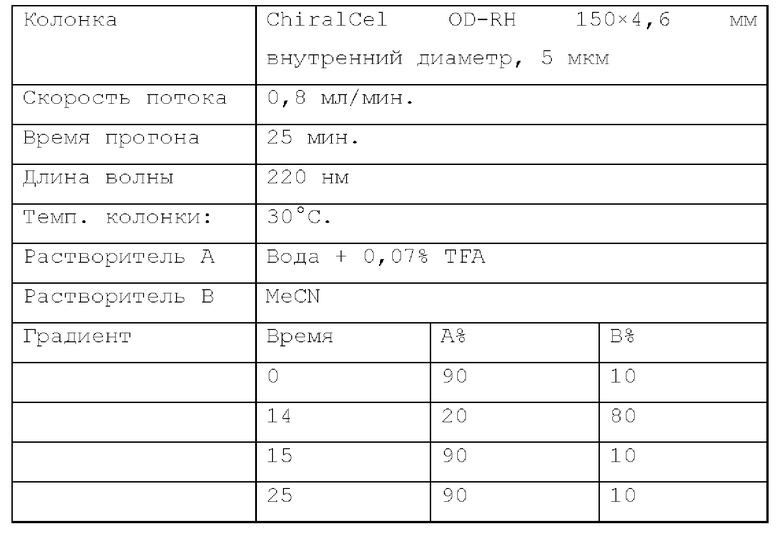
Способ хиральной HPLC 3. Устройство: SHIMADZU LC-20AB
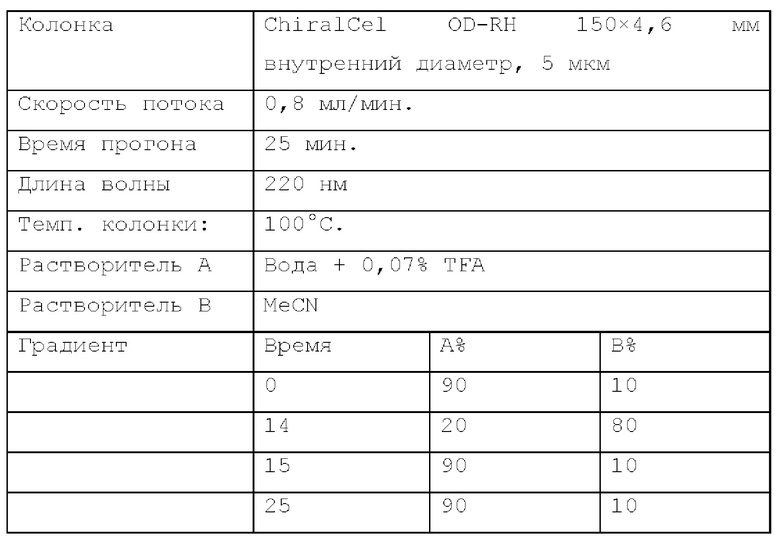
Способ хиральной HPLC 4. Устройство: SHIMADZU LC-20AB
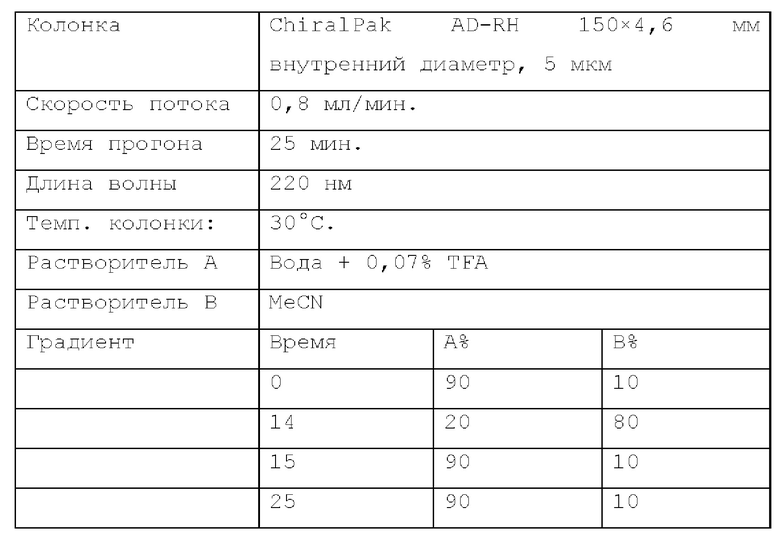
Получение промежуточных соединений
IIb: ((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этан-1-амин
Стадия 1. Получение этил-2-[(R)-трет-бутилсульфинил]иминоацетата
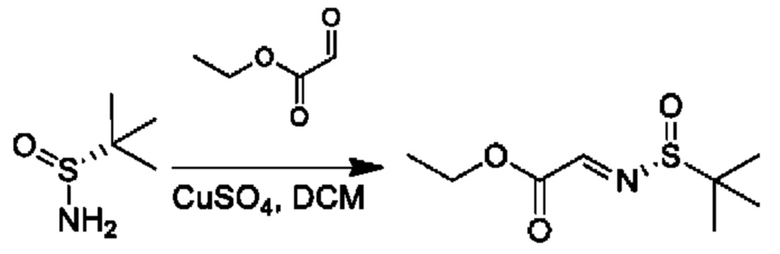
К раствору этил-2-оксоацетата (7,5 г, 36,7 ммоль) и (R)-2-метилпропан-2-сульфинамида (4,9 г, 40,4 ммоль) в DCM (150 мл) добавляли CuSO4 (12,9 г, 80,8 ммоль), и реакционную смесь перемешивали при 25°С в течение 24 часов. Твердое вещество отфильтровывали, промывали с помощью этилацетата (50 мл), и органический фазы объединяли и концентрировали. Полученный осадок очищали посредством колоночной хроматографии (силикагель, петролейный эфир/этилацетат, 5/1) с получением требуемого продукта (5,1 г, выход 67,6%).
Стадия 2. Получение этил-(2R)-2-[[(R)-трет-бутилсульфинил]амино]-2-[3-(трифторметокси)фенил]ацетата
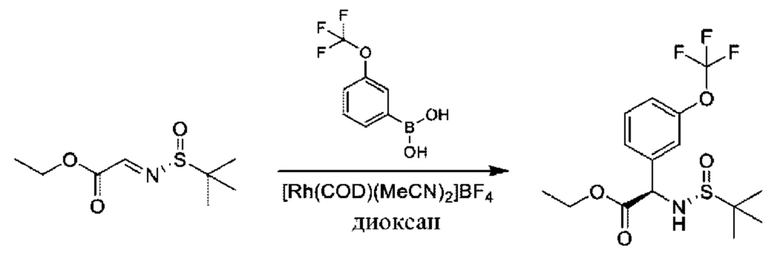
К раствору этил-2-[(R)-трет-бутилсульфинил]иминоацетата (7 г, 34,1 ммоль) и [3-(трифторметокси)фенил]бороновой кислоты (8,4 г, 40,9 ммоль) в диоксане (100 мл) добавляли [Rh(COD)(MeCN)2]BF4 (1,3 г, 3,4 ммоль), и данную смесь перемешивали при 80°С в течение 16 часов. Продукт очищали посредством хроматографии на силикагеле (петролейный эфир:этилацетат=5:1) с получением 9,8 г (78%).
Стадия 3. Получение гидрохлорида этил-(2R)-2-амино-2-[3-(трифторметокси)фенил]ацетата
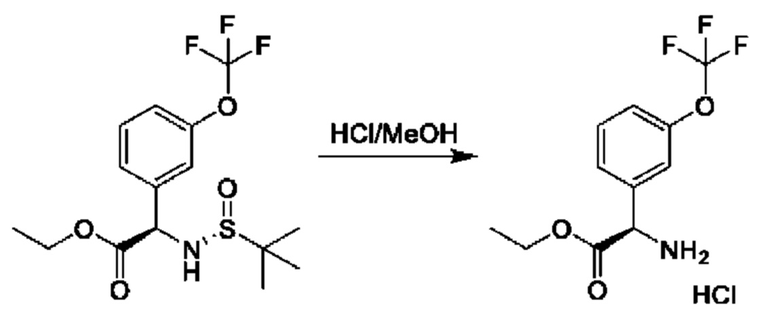
К раствору этил-(2R)-2-[[(R)-трет-бутилсульфинил]амино]-2-[3-
(трифторметокси)фенил]ацетата (9,8 г, 26,7 ммоль) в МеОН (100 мл) добавляли HCl/МеОН (4 М, 100 мл), и данную смесь перемешивали при 25°С в течение 2 часов, а затем концентрировали с получением этил-(2R)-2-амино-2-[3-(трифторметокси)фенил]ацетата (7,8 г, неочищенный).
Стадия 4. Получение этил-(R)-2-((трет-бутоксикарбонил)амино)-2-(3-(дифторметокси)фенил)ацетата
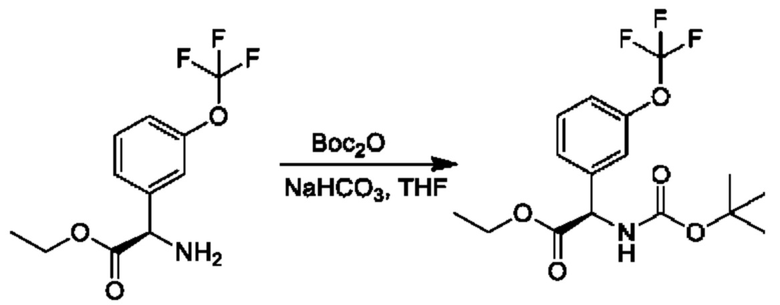
К смеси гидрохлорида этил-(2R)-2-амино-2-[3-(трифторметокси)фенил]ацетата (6 г) в THF (150 мл) и Вос2О (8,7 г) добавляли NaHCO3 (1,7 г), и осуществляли перемешивание при 25°С в течение 16 часов. Смесь концентрировали и очищали посредством хроматографии на диоксиде кремния (петролейный эфир:этилацетат=10:1) с получением продукта (7,2 г).
Стадия 5. Получение трет-бутил-(R)-(1-(3-(дифторметокси)фенил)-2-гидроксиэтил)карбамата
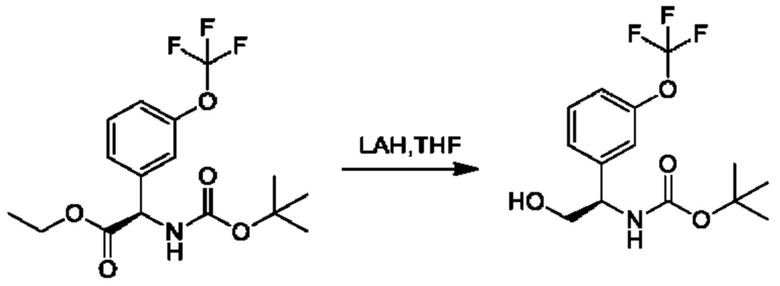
К суспензии LiAlH4 (1,7 г) в THF (200 мл) добавляли этил-(R)-2-((трет-бутоксикарбонил)амино)-2-(3-(дифторметокси)фенил)ацетат (4 г) в THF (25 мл) при охлаждении с помощью льда. Обеспечивали нагревание реакционной смеси до 25°С, и осуществляли ее перемешивание в течение 2 часов. Добавляли безводный сульфат магния, а затем последовательно добавляли по одной капле воды и этилацетата. Нерастворимые вещества отфильтровывали через слой целита. Фильтрат концентрировали и очищали посредством хроматографии на диоксиде кремния (петролейный эфир:этилацетат=5:1) (2,1 г).
Стадия 6. Получение трет-бутил-(R)-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)карбамата
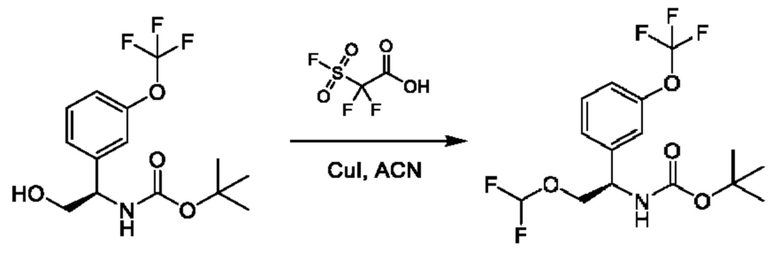
К раствору трет-бутил-(R)-(1-(3-(дифторметокси)фенил)-2-гидроксиэтил)карбамата (1,5 г) в MeCN (20 мл) добавляли CuI (360 мг), и осуществляли перемешивание при 25°С в атмосфере N2 в течение 30 минут. Раствор 2,2-дифтор-2-фторсульфонилуксусной кислоты (1,7 г) в MeCN (5 мл) добавляли при 45°С на протяжении 30 минут, и реакционную смесь перемешивали при 45°С в течение 1 часа. Смесь концентрировали, а затем разбавляли с помощью этилацетата (100 мл), фильтровали и концентрировали с получением требуемого продукта (1,5 г, неочищенный).
Стадия 7. Получение (R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этан-1-амина
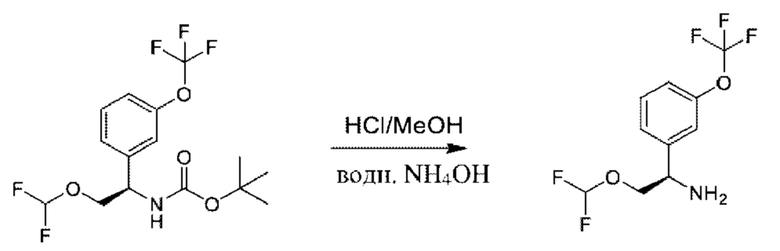
К раствору трет-бутил-(R)-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)карбамата (1,5 г) в МеОН (15 мл) добавляли HCl/МеОН (4 M в МеОН, 30 мл) при 25°С, и реакционную смесь перемешивали при 25°С в течение 30 минут. Гидроксид аммония (30%) добавляли до достижения рН=9, раствор концентрировали и очищали посредством хроматографии на диоксиде кремния (петролейный эфир:этилацетат=2:1) с получением (R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этан-1-амина (700 мг).
IIa: гидрохлорид (R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этан-1-амина
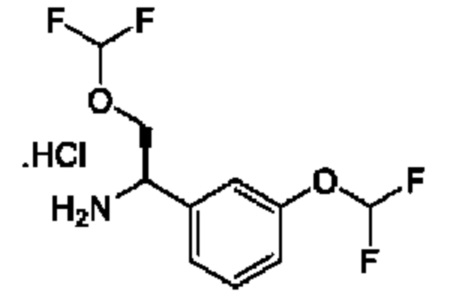
Получен, как описано для IIb, с применением подходящих реагентов.
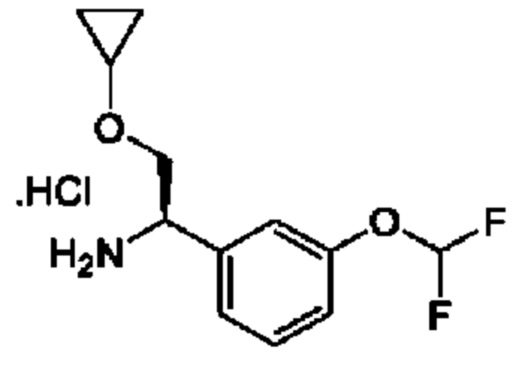
IIc: гидрохлорид (R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этан-1-амина
Стадия 1. Получение 4-метил-N'-(п-толилсульфонил)бензолсульфоногидразида
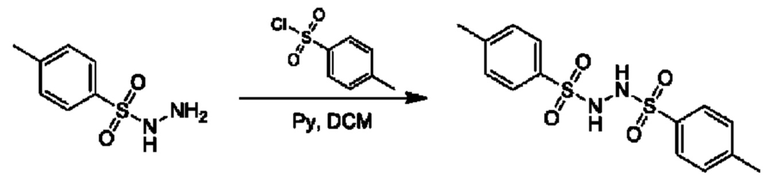
К смеси 4-метилбензолсульфоногидразида (70 г) и 4-метилбензолсульфонилхлорида (93 г) добавляли DCM (400 мл). Смесь охлаждали до 0°С, и по каплям добавляли пиридин (38,65 г), и реакционную смесь перемешивали при 0°С в течение 1 часа и при 20°С в течение 7 часов. К смеси добавляли воду (200 мл) и метил-трет-бутиловый эфир (МЕТВ) (200 мл) и ее фильтровали. Осадок на фильтре промывали с помощью МЕТВ (200 мл) и высушивали с получением 4-метил-N'-(п-толилсульфонил)бензолсульфоногидразида (125 г). 1Н ЯМР (DMSO-d6 400МГц): δ 9, 55 (s, 2Н), 7,61 (d, 4Н), 7,35 (d, 4Н), 2,36 (s, 6Н).
Стадия 2. Получение 2-бром-1-(3-(дифторметокси)фенил)этан-1-она
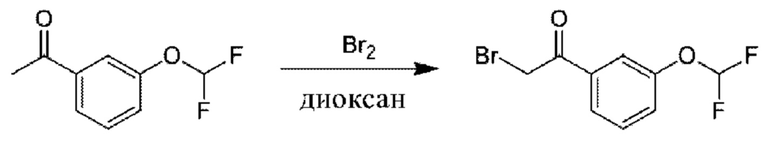
К раствору 1-[3-(дифторметокси)фенил]этанона (10 г) в диоксане (100 мл) добавляли раствор Br2 (8,58 г) в диоксане (100 мл). Полученную смесь перемешивали при 20°С в течение 2 часов. Добавляли насыщ. водн. раствор NaHCO3 (50 мл) и Н2О (100 мл), и водную фазу экстрагировали с помощью EtOAc (200 мл × 2). Органическую фазу промывали солевым раствором (200 мл), высушивали над Na2SO4 и концентрировали с получением 2-бром-1-[3-(дифторметокси)фенил]этанона (17 г). 1Н ЯМР (CDCl3 400 МГц): δ 7,81 (d, 1Н), 7,72 (s, 1Н), 7,49 (t, 1Н), 7,37 (d, 1Н), 6,56 (t, 1Н), 4,41 (s, 2Н).
Стадия 3. Получение 1-(3-(дифторметокси)фенил)-2-иминоэтан-1-она
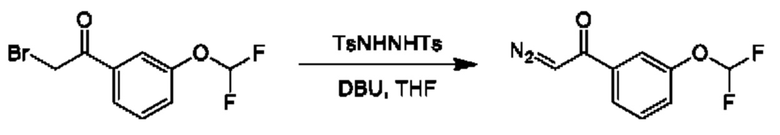
К раствору 2-бром-1-[3-(дифторметокси)фенил]этанона (5,6 г) и 4-метил-N'-(п-толилсульфонил)бензолсульфоногидразида (11,51 г) в THF (100 мл) по каплям добавляли DBU (12,87 г) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 часа и при 20°С в течение 4 часов. Смесь гасили с помощью нас.водн. NaHCO3 (200 мл) и разбавляли водой (200 мл), затем экстрагировали с помощью EtOAc (200 мл × 2). Органический слой промывали солевым раствором (100 мл), высушивали над Na2SO4 и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (20% EtOAc в петролейном эфире) с получением 1-(3-(дифторметокси)фенил)-2-иминоэтан-1-она (6,4 г).
1Н ЯМР (CDCl3, 400 МГц): δ 7, 57 (dt, 1Н), 7,55 (s, 1H), 7,46 (t, 1H), 7,31 (dd, 1H), 6,56 (t, 1H), 5,90 (s, 1H).
Стадия 4. Получение 2-циклопропокси-1-(3-(дифторметокси)фенил)этан-1-она
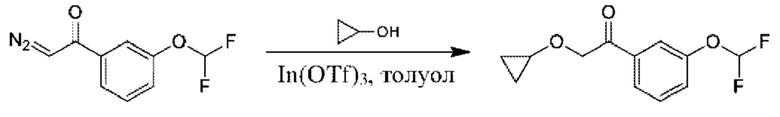
К раствору циклопропанола (1,75 г) и 2-диазо-1-[3-(дифторметокси)фенил]этанона (3,2 г) в толуоле (50 мл) добавляли трифлат индия(III) (1,7 г) в атмосфере N2. Полученную смесь перемешивали при 20°С в течение 16 часов. Смесь гасили с помощью насыщ. водн. раствора NaHCO3 (100 мл) и разбавляли с помощью Н2О (50 мл), а затем экстрагировали с помощью EtOAc (100 мл × 2). Органический слой промывали солевым раствором (200 мл × 2), высушивали над Na2SO4 и концентрировали. Неочищенный продукт очищали посредством колоночной хроматографии (5% EtOAc в петролейном эфире) с получением 2-циклопропокси-1-(3-(дифторметокси)фенил)этан-1-она (6,1 г).
1Н ЯМР (CDCl3, 400 МГц): δ 7,76 (d, 1Н), 7,68 (s, 1Н), 7,47 (t, 1Н), 7,34 (d, 1Н), 6,55 (t, 1Н), 4,74 (s, 1Н), 3,55-3,50 (m, 1Н), 0,70-0,66 (m, 2Н), 0,53-0,49 (m, 2Н).
Стадия 5. Получение (R, Z)-N-(2-циклопропокси-1-(3-(дифторметокси)фенил)этилиден)-2-метилпропан-2-сульфинамида
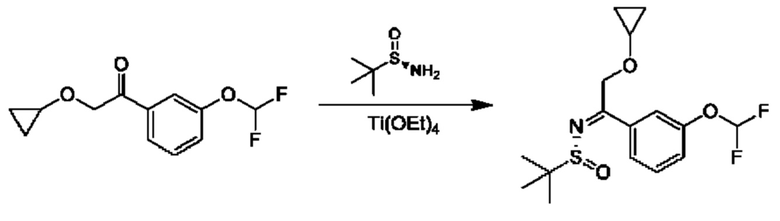
К раствору 2-(циклопропокси)-1-[3-(дифторметокси)фенил]этанона (6,1 г) и (R)-2-метилпропан-2-сульфинамида (4,6 г) в THF (100 мл) добавляли Ti(OEt)4 (11,5 г). Полученную смесь перемешивали при 60°С в течение 8 часов, а затем использовали непосредственно на следующей стадии.
Стадия 6. Получение (R)-N-((R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-2-метилпропан-2-сульфинамида

К раствору (R,Z)-N-(2-циклопропокси-1-(3-(дифторметокси)фенил)этилиден)-2-метилпропан-2-сульфинамида в THF (150 мл) из предыдущей стадии по каплям добавляли раствор L-селектрида (1 М в THF, 50,36 мл) при -45°С. Смесь перемешивали при -45°С в течение 1 часа, и гасили с помощью МеОН (100 мл) и Н2О (100 мл), и фильтровали на целите. Фильтрат экстрагировали с помощью EtOAc (200 мл × 2), органический слой промывали солевым раствором (200 мл × 2), высушивали над Na2SO4 и концентрировали.
Неочищенный продукт очищали посредством колоночной хроматографии (30~50% EtOAc в петролейном эфире) с получением (R)-N-((R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-2-метилпропан-2-сульфинамида (2,4 г).
Стадия 7. Получение гидрохлорида (R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этан-1-амина
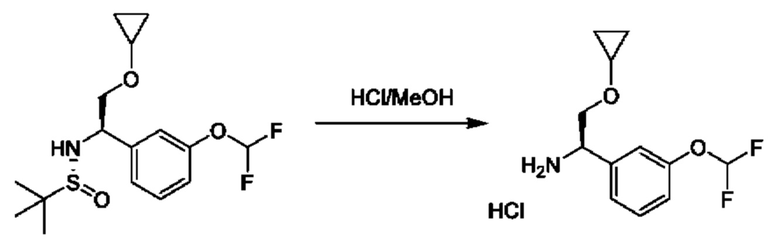
К раствору (R)-N-((R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-2-метилпропан-2-сульфинамида (0,46 г) в МеОН (10 мл) добавляли HCl/МеОН (10 мл). Полученную смесь перемешивали при 20°С в течение 1 часа и концентрировали с получением гидрохлорида (1R)-2-(циклопропокси)-1-[3-(дифторметокси)фенил]этанамина (0,46 г, неочищенный).
IId: гидрохлорид (R)-2-циклопропокси-1-(3-(трифторметокси)фенил)этан-1-амина
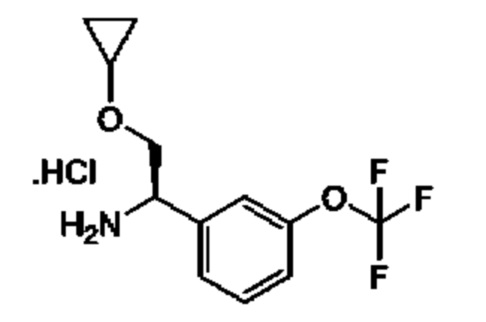
Получен, как описано для IIc, с применением 1-[3-(трифторметокси)фенил]этанона в качестве исходного материала.
IIe: гидрохлорид (R)-1-(3-(дифторметокси)фенил)-2-(трифторметокси)этан-1-амина
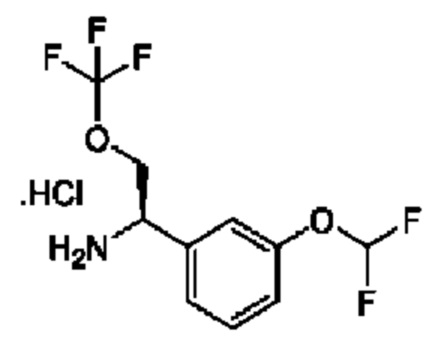
Стадия 1. Получение 2-бром-1-[3-(дифторметокси)фенил]этанона
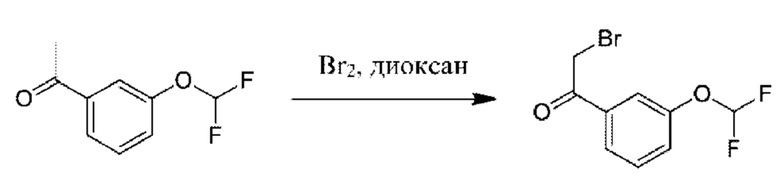
К раствору 1-[3-(дифторметокси)фенил]этанона (5 г) в диоксане (50 мл) добавляли раствор Br2 (4,29 г) в диоксане (50 мл) при 20°С. Данную смесь перемешивали при 20°С в течение 1 часа, затем разбавляли с помощью EtOAc (200 мл) и промывали водой (100 мл × 3). Органическую фазу высушивали над безводным Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир: EtOAc=10:1) с получением 2-бром-1-[3-(дифторметокси)фенил]этанона (5 г).
Стадия 2. Получение 1-(3-(дифторметокси)фенил)-2-(трифторметокси)этан-1-она
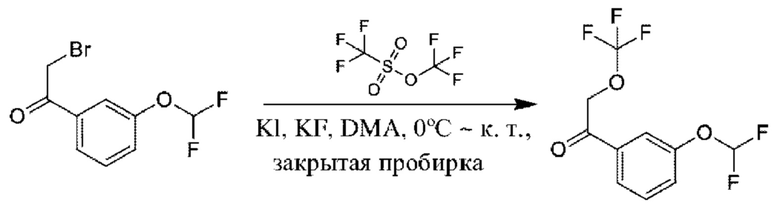
К раствору KF (712 мг) в DMA (20 мл) добавляли трифторметилтрифторметансульфонат (4,11 г) при 0°С. Реакционную смесь перемешивали в герметично закрытой пробирке в течение 1 часа, затем к данному раствору добавляли 2-бром-1-[3-(дифторметокси)фенил]этанон (2,5 г, 9,43 ммоль) и KI (157 мг) при 0°С, и температуру доводили до 20°С, и осуществляли перемешивание в течение 16 часов. Смесь разбавляли с помощью EtAOc (100 мл) и промывали водой (50 мл × 3). Органическую фазу концентрировали с получением продукта, который очищали посредством кислотной препаративной HPLC (1,4 г). 1Н ЯМР (CDCl3 400 МГц): δ 7,72 (d, 1Н), 7,66 (s, 1Н), 7,51 (t, 1Н), 7,40 (d, 1Н), 6,56 (t1H), 5,13 (s, 2Н). 19F ЯМР (CDCl3 400 МГц): δ -61,11, -81,52, -81,71.
Стадия 3. Получение (R)-N-(1-(3-(дифторметокси)фенил)-2-(трифторметокси)этилиден)-2-метилпропан-2-сульфинамида
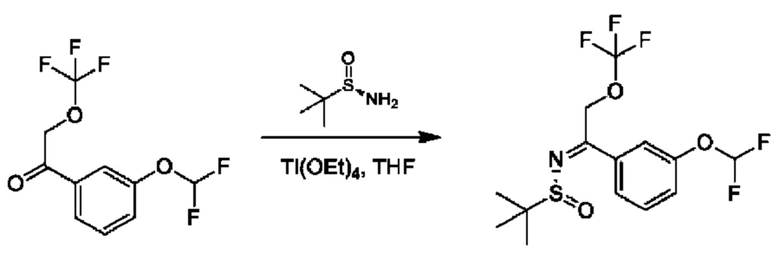
Смесь 1-(3-(дифторметокси)фенил)-2-(трифторметокси)этан-1-она (900 мг), (R)-2-метилпропан-2-сульфинамида (606 мг)и Ti(OEt)4 (2,28 г) в THF (50 мл) перемешивали при 60°С в атмосфере N2 в течение 16 часов.
Продукт (1,2 г, неочищенный) в THF (70 мл) получали и использовали непосредственно на следующей стадии.
Стадия 4. Получение (R)-N-((R)-1-(3-(дифторметокси)фенил)-2-(трифторметокси)этил)-2-метилпропан-2-сульфинамида
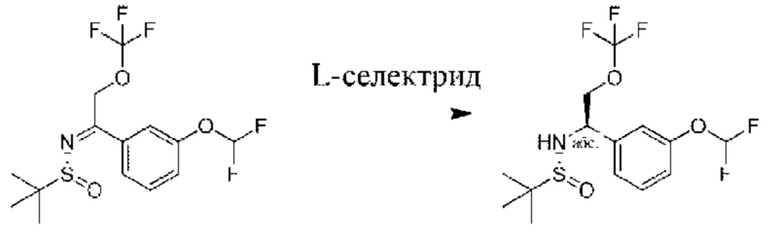
К раствору (R)-N-(1-(3-(дифторметокси)фенил)-2-(трифторметокси)этилиден)-2-метилпропан-2-сульфинамида (1,2 г) в THF (70 мл) добавляли L-селектрид (1 M в THF, 3,86 мл) при -60°С. Смесь перемешивали при -60°С в течение 0,5 часа, а затем разбавляли с помощью EtOAc (100 мл) и промывали водой (50 мл × 3). Органическую фазу высушивали, фильтровали и концентрировали. Неочищенный продукт очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат - 5:1) с получением (R)-N-((R)-1-(3-(дифторметокси)фенил)-2-(трифторметокси)этил)-2-метилпропан-2-сульфинамида (600 мг).
1Н ЯМР (CDCl3 400 МГц): δ 7,37 (t, 1Н), 7,20 (d, 1Н), 7,12-7,09 (m, 2Н), 6,50 (t, 1Н), 4,75-4,72 (m, 1Н), 4,20-4,16 (m, 1Н), 4,14-4,05 (m, 1Н), 3,87 (s, 1Н), 1,21 (s, 9Н).
Стадия 5. Получение гидрохлорида (R)-1-(3-(дифторметокси)фенил)-2-(трифторметокси)этан-1-амина
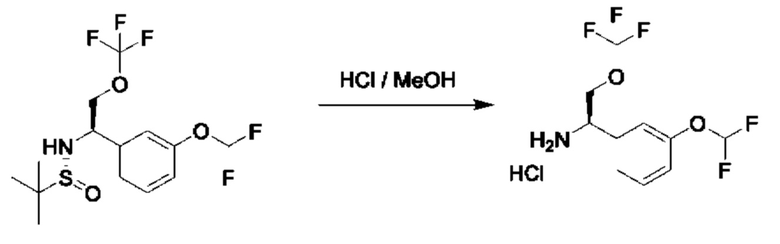
К раствору (R)-N-((R)-1-(3-(дифторметокси)фенил)-2-(трифторметокси)этил)-2-метилпропан-2-сульфинамида (600 мг) в МеОН (10 мл) добавляли HCl/МеОН (4 M в МеОН, 8,0 мл). Данную смесь перемешивали при 15°С в течение 1 часа, а затем концентрировали с получением гидрохлорида ((R)-1-(3-(дифторметокси)фенил)-2-(трифторметокси)этан-камина (500 мг, неочищенный).
1Н ЯМР (CDCl3 400 МГц): δ 9,17 (s, 3Н), 7,40-7,31 (m, 4Н), 6,55 (t, 1Н), 4,58 (s, 1Н), 4,46-4,42 (m, 1Н), 4,33-4,29 (m, 1Н).
IIf: гидрохлорид (R)-2-(трифторметокси)-1-(3-(трифторметокси)фенил)этан-1-амина
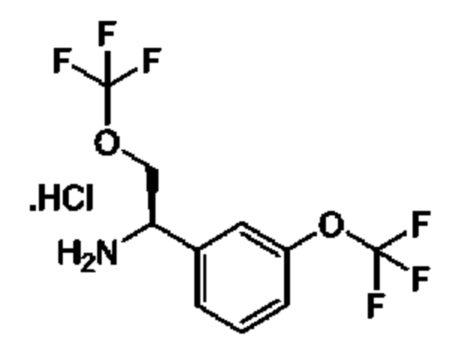
Получен, как описано для IIe, с применением 1-[3-(трифторметокси)фенил]этанона в качестве исходного материала.
1Н ЯМР (CDCl3 400 МГц): δ 9,18 (s, 3Н), 7,19-7,47 (m, 1Н), 7,19-7,38 (m, 2Н), 7,27 (s, 1Н), 4,65 (s, 1Н), 4,47-4,42 (m, 1Н), 4,32-4,30 (m, 1Н).
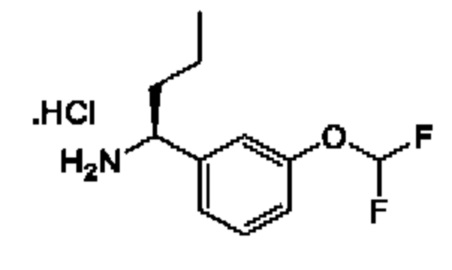
IIg: гидрохлорид (S)-1-(3-(дифторметокси)фенил)бутан-1-амина
Стадия 1. Получение (R)-N-(3-(дифторметокси)бензилиден)-2-метилпропан-2-сульфинамида
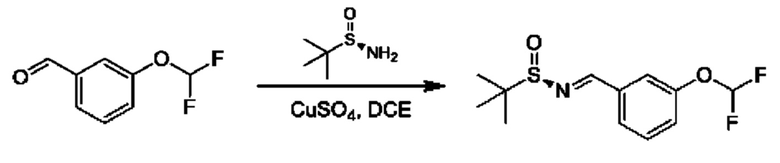
К раствору 3-(дифторметокси)бензальдегида (3 г) и (R)-2-метилпропан-2-сульфинамида (2,54 г) в DCE (120 мл) добавляли CuSO4 (13,91 г). Смесь перемешивали при 55°С в течение 16 часов. Реакционную смесь фильтровали и фильтрат концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат=от 100/1 до 10:1) с получением продукта(3 г).
Стадия 2. Получение (R)-N-((S)-1-(3-(дифторметокси)фенил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамида
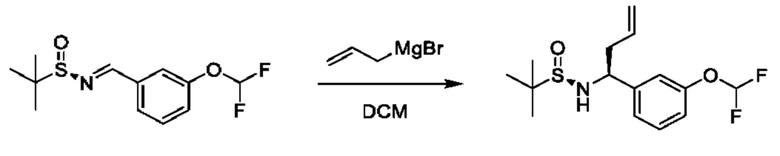
К раствору (R)-N-(3-(дифторметокси) бензилиден)-2-метилпропан-2-сульфинамида (1 г) в DCM (40 мл) медленно добавляли бромид аллилмагния (1 М раствор в THF, 10,9 мл) в THF при 0°С. Полученную смесь перемешивали при 0°С в течение 1 часа и при 25°С в течение 2 часов. Реакционную смесь гасили путем добавления насыщенного раствора NH4Cl (10 мл) при 0°С, затем разбавляли с помощью Н2О (50 мл) и экстрагировали с помощью DCM (40 мл × 3). Объединенные органические фазы промывали с помощью Н2О (40 мл), высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат=от 100/1 до 1:2) с получением (R)-N-((S)-1-(3-(дифторметокси)фенил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамида (580 мг).
1Н ЯМР (CDCl3 400 МГц): δ 7,31 (t, 1Н), 7,15 (d, 1Н), 7,08 (s, 1Н), 7,01 (d, 1Н), 6,48 (t, 1Н), 5,73-5,66 (m, 1Н), 5,19-5,15 (m, 2Н), 4,46 (t, 1Н), 3,65 (s, 1Н), 2,59-2,54 (m, 1Н), 2,46-2,38 (m, 1Н), 1,19 (s, 9Н).
Стадия 3. Получение (R)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-2-метилпропан-2-сульфинамида
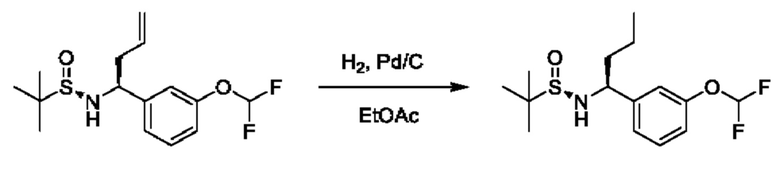
К раствору (R)-N-((S)-1-(3-(дифторметокси)фенил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамида (580 мг) в EtOAc (20 мл) добавляли Pd/C (0,4 г, чистота 10%) в атмосфере N2. Суспензию дегазировали под вакуумом и продували с помощью Н2 несколько раз. Смесь перемешивали в атмосфере Н2 (18 фунтов/кв. дюйм) при 25°С в течение 0,5 часа. Реакционную смесь фильтровали и концентрировали с получением (R)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-2-метилпропан-2-сульфинамида (560 мг).
Стадия 4. Получение гидрохлорида (S)-1-(3-(дифторметокси)фенил)бутан-1-амина
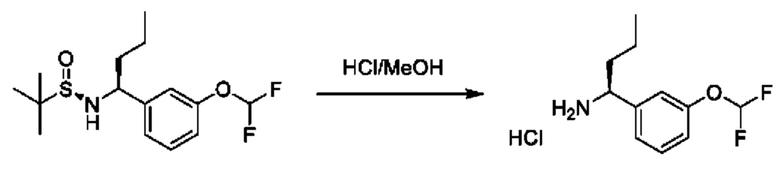
К раствору (R)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-2-метилпропан-2-сульфинамида (580 мг) в МеОН (8 мл) добавляли HCl/МеОН (4 М, 3,1 мл). Смесь перемешивали при 25°С в течение 3 часов, а затем концентрировали с получением гидрохлорида (1S)-1-[3-(дифторметокси)фенил]бутан-1-амина (250 мг).
IIh: гидрохлорид (S)-1-(3-(дифторметокси)фенил)-4,4-дифторбутан-1-амина
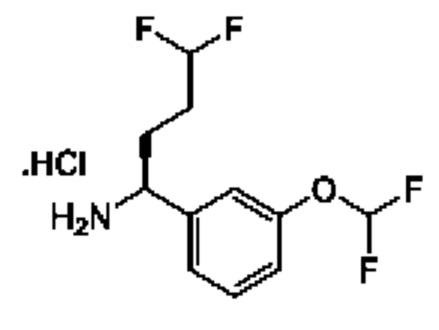
Стадия 1. Получение 1-(дифторметокси)-3-винилбензола
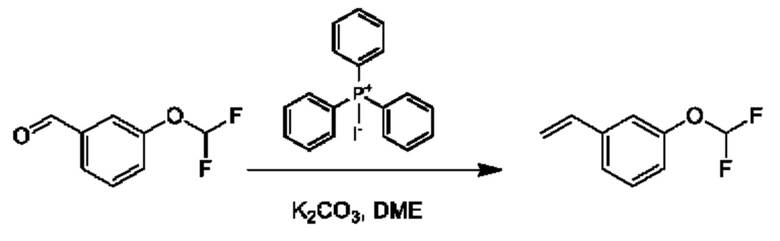
К раствору йодида метилтрифенилфосфония (7,05 г) в DME (50 мл) добавляли K2CO3 (2,41 г). Полученную смесь перемешивали при 20°С в течение 1 часа, затем добавляли 3-(дифторметокси)бензальдегид (1,5 г) и перемешивание продолжали при 80°С в течение 15 часов. Смесь фильтровали, и осадок на фильтре промывали с помощью петролейного эфира (100 мл). Фильтрат концентрировали и очищали с помощью колоночной хроматографии (SiO2, петролейный эфир/этилацетат=от 1/0 до 10/1) с получением 1-(дифторметокси)-3-винилбензола (1,4 г). 1Н ЯМР (CDCl3 400МГц): δ 7,32 (t, 1Н), 7,25 (d, 1Н), 7,16 (s, 1Н), 7,01 (d, 1Н), 6,70 (t, 1Н), 6,52 (t, 1Н), 5,77 (d, 1Н), 5,32 (d, 1Н).
Стадия 2. Получение 1-(3-(дифторметокси)фенил)-4,4-дифторбутан-1-она
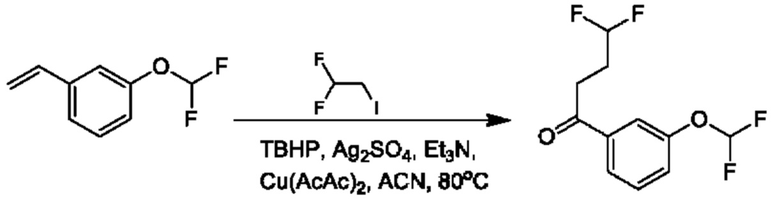
К раствору 1,1-дифтор-2-йодэтана (1 г), 1-(дифторметокси)-3-винилбензола (1,33 г), бис[(Z)-1-метил-3-оксобут-1-енокси]меди (273 мг) и Ag2SO4 (325 мг) в ACN (20 мл) добавляли Et3N (527 мг) и трет-бутилгидропероксид (ТВНР) (2,01 г, 70% в воде). Полученную смесь перемешивали при 80°С в течение 24 часов. Реакционную смесь гасили с помощью насыщенного водн. раствора Na2S2O3 и экстрагировали с помощью DCM (10 мл × 3). Органический фазы объединяли, промывали с помощью солевого раствора (5 мл × 2), высушивали над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат=от 1/0 до 5/1) с получением 1-(3-(дифторметокси)фенил)-4,4-дифторбутан-1-она (300 мг). 1Н ЯМР (CDCl3 400 МГц): δ 7,82 (d, 1Н), 7,73 (s, 1Н), 7,50 (t, 1Н), 7,36 (d, 1Н), 6,57 (t, 1Н), 6,02 (tt, 1Н), 3,18 (t, 2Н), 2,35-2,27 (m, 2Н).
Стадия 3. Получение (S,Е)-N-(1-(3-(дифторметокси)фенил)-4,4-дифторбутилиден)-2-метилпропан-2-сульфинамида
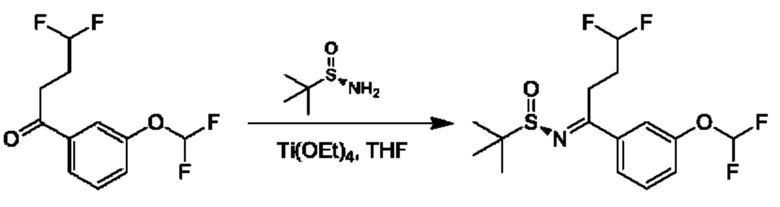
К раствору 1-(3-(дифторметокси)фенил)-4,4-дифторбутан-1-она (300 мг) и (R)-2-метилпропан-2-сульфинамида (218 мг) в THF (10 мл) добавляли Ti(OEt)4 (547 мг). Смесь перемешивали при 60°С в течение 6 часов. Реакционную смесь использовали непосредственно на следующей стадии.
Стадия 4. Получение N-[(1S)-1-[3-(дифторметокси)фенил]-4,4-дифторбутил]-2-метилпропан-2-сульфинамида
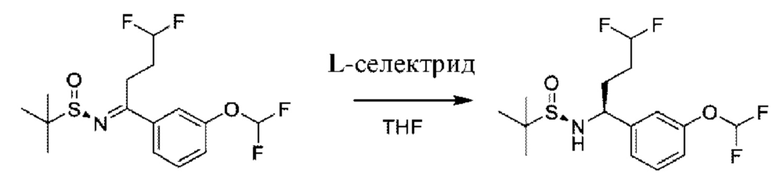
L-селектрид (1 M в THF, 3,59 мл) добавляли к раствору (S)-N-(1-(3-(дифторметокси)фенил)-4,4-дифторбутилиден)-2-метилпропан-2-сульфинамида (4 23 мг) в THF при -48°С. Реакционную смесь перемешивали в течение 0,5 часа, затем ее температуру доводили до 0°С и добавляли Н2О (~10 мл). Полученную смесь экстрагировали с помощью EtOAc (35 мл × 2). Органические экстракты промывали солевым раствором (10 мл), высушивали над Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат=от 1/0 до 1/1) с получением N-[(1S)-1-[3-(дифторметокси)фенил]-4,4-дифторбутил]-2-метилпропан-2-сульфинамида (140 мг).
Стадия 5. гидрохлорид (S)-1-(3-(дифторметокси)фенил)-4,4-дифторбутан-1-амина
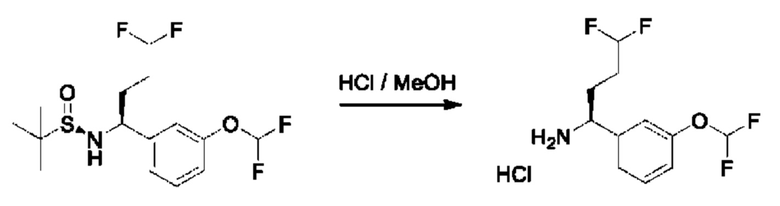
N-[(1S)-1-[3-(дифторметокси)фенил]-4,4-дифторбутил]-2-метилпропан-2-сульфинамид (140 мг) в МеОН (10 мл) и HCl/МеОН (5 мл, 4 М) перемешивали при 20°С в течение 1 часа. Смесь концентрировали с получением гидрохлорида (S)-1-(3-(дифторметокси)фенил)-4,4-дифторбутан-1-амина (110 мг).
IIi: гидрохлорид (S)-1-(3-(дифторметокси)фенил)-3,3-дифторпропан-1-амина
Стадия 1. Получение (S,Е)-N-(3-(дифторметокси)бензилиден)-2-метилпропан-2-сульфинамида
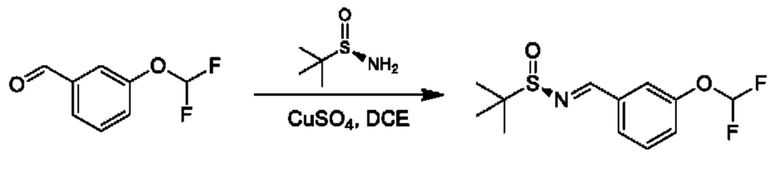
К раствору 3-(дифторметокси)бензальдегида (5 г) и (R)-2-метилпропан-2-сульфинамида (4,22 г) в DCE (150 мл) добавляли CuSO2 (23 г). Реакционную смесь перемешивали при 55°С в течение 20 часов, а затем фильтровали и концентрировали. Неочищенный продукт очищали посредством хроматографии на диоксиде кремния (SiO2, петролейный эфир/этилацетат=от 10/1 до 5/1) с получением (S,E)-N-(3-(дифторметокси)бензилиден)-2-метилпропан-2-сульфинамида (6,9 г).
1Н ЯМР (CDCl3 400 МГц): δ 8,55 (s, 1Н), 7,65 (d, 1Н), 7,62 (s, 1Н), 7,47 (t, 1Н), 7,25 (d, 1Н), 6,55 (t, 1Н), 1,25 (s, 9Н).
Стадия 2. Получение (S)-N-((S)-1-(3-(дифторметокси)фенил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамида
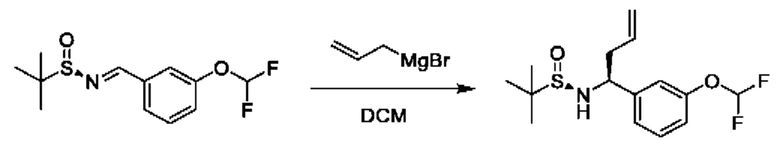
К раствору (S,Е)-N-(3-(дифторметокси)бензилиден)-2-метилпропан-2-сульфинамида (2 г) в DCM (60 мл) медленно добавляли бромид аллилмагния (1 М раствор в THF, 21,79 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 часа и при 25°С в течение 2 часов. Реакционную смесь гасили путем добавления насыщ. водн. раствора NH4Cl (10 мл) при 0°С, а затем разбавляли с помощью Н2О (50 мл) и экстрагировали с помощью DCM (40 мл × 3). Объединенные органические фазы промывали с помощью Н2О (40 мл), высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии (SiO2, петролейный эфир/этилацетат=от 100/1 до 1/2) с получением (S)-N-((S)-1-(3-(дифторметокси)фенил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамида (2,3 г).
1Н ЯМР (CDCl3 400 МГц): δ 7,31 (t, 1Н), 7,15 (d, 1Н), 7,08 (s, 1Н), 7,01 (d, 1Н), 6,49 (t, 1Н), 5,71-5,65 (m, 1Н), 5,19-5,15 (m, 2Н), 4,48-4,44 (m, 1Н), 3,66 (s, 1Н), 2,59-2,54 (m, 1Н), 2,46-2,40 (m, 1Н), 1,18 (s, 9Н).
Стадия 3. Получение (S)-N-((S)-1-(3-(дифторметокси)фенил)-3-оксопропил)-2-метилпропан-2-сульфинамида
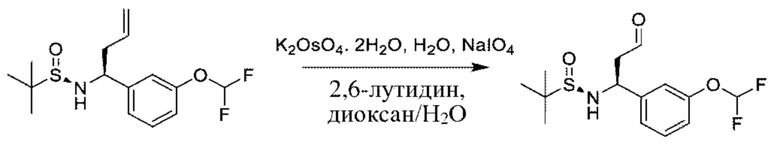
Гидрат осмата(VI) калия (27 6 мг) добавляли одной порцией к перемешиваемому раствору (S)-N-((S)-1-(3-(дифторметокси)фенил)бут-3-ен-1-ил)-2-метилпропан-2-сульфинамида (2,5 г), 2,6-лутидина (1,69 г) и периодата натрия (5,05 г) в смеси диоксана (10 мл) и Н2О (3 мл). Реакционную смесь перемешивали при 20°С в течение 1 часа, а затем разбавляли с помощью DCM (100 мл) и воды (20 мл). Водный слой экстрагировали с помощью DCM (25 мл × 2). Объединенные органические экстракты высушивали над Na2SO4. Неочищенную реакционную смесь использовали непосредственно на следующей стадии.
Стадия 4. Получение (S)-N-((S)-1-(3-(дифторметокси)фенил)-3,3-дифторпропил)-2-метилпропан-2-сульфинамида
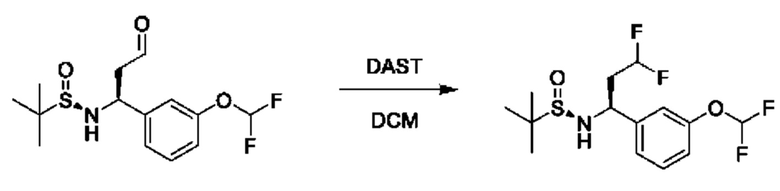
К раствору (S)-N-((S)-1-(3-(дифторметокси)фенил)-3-оксопропил)-2-метилпропан-2-сульфинамида (2 г) в DCM (200 мл) по каплям добавляли трифторид диэтиламиносеры (DAST) (3,03 г) при -78°С. Обеспечивали понижение температуры реакционной смеси до 20°С, и осуществляли ее перемешивание в течение 2 часов. Раствор выливали в насыщенный водный раствор NaHCO3 (50 мл), и органическую фазу отделяли. Данный раствор высушивали над безводным Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат=от 5/1 до 1/1) с получением (S)-N-((S)-1-(3-(дифторметокси)фенил)-3,3-дифторпропил)-2-метилпропан-2-сульфинамида (500 мг).
Стадия 5. Получение гидрохлорида (1S)-1-[3-(дифторметокси)фенил]-3,3-дифторпропан-1-амина
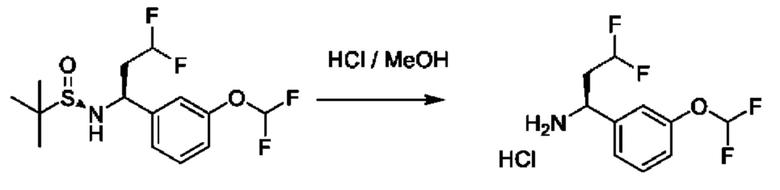
К раствору (S)-N-((S)-1-(3-(дифторметокси)фенил)-3,3-дифторпропил)-2-метилпропан-2-сульфинамида (380 мг) в МеОН (15 мл) добавляли HCl/МеОН (25 мл, 4 М) при 0°С, и реакционную смесь перемешивали в течение 0,5 часа, и обеспечивали понижение ее температуры до 25°С. Реакционную смесь концентрировали с получением гидрохлорида (1S)-1-[3-(дифторметокси)фенил]-3,3-дифторпропан-1-амина (300 мг, неочищенный, соль HCl). При этом его использовали непосредственно без дополнительной очистки.
IIj: гидрохлорид (S)-1-(3-(дифторметокси) фенил) этан-1-амина
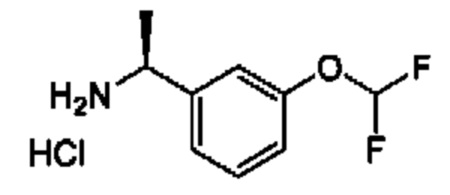
Стадия 1. Получение ((S,E)-N-(3-(дифторметокси)бензилиден)-2-метилпропан-2-сульфинамида
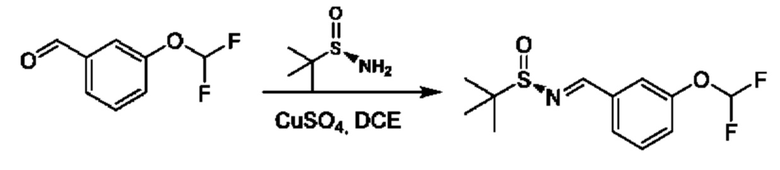
К смеси 3-(дифторметокси)бензальдегида (2 г) и (R)-2-метилпропан-2-сульфинамида (1,7 г) в DCE (60 мл) добавляли CuSO4 (9,3 г) при 55°С в атмосфере N2. Реакционную смесь перемешивали при 55°С в течение 12 часов, фильтровали, и фильтрат концентрировали. Неочищенный продукт очищали посредством хроматографии на колонке с силикагелем (петролейный эфир/этилацетат=20:1 - 10:1) с получением ((S,Е)-N-(3-(дифторметокси)бензилиден)-2-метилпропан-2-сульфинамида (4,5 г).
Стадия 2. Получение (S)-N-((S)-1-(3-(дифторметокси)фенил)этил)-2-метилпропан-2-сульфинамида
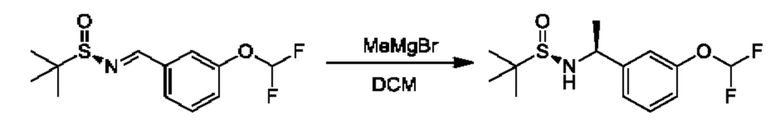
К раствору ((S,Е)-N-(3-(дифторметокси)бензилиден)-2-метилпропан-2-сульфинамида (2 г) в DCM (30 мл) по каплям добавляли бромид метилмагния (3 М в Et2O, 4,8 мл) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 часа и при 20°С в течение 16 часов. Реакционную смесь гасили с помощью водн. насыщ. раствора NH4Cl (10 мл), и водную фазу экстрагировали этилацетатом (30 мл × 3). Объединенные органические экстракты промывали солевым раствором (40 мл × 2), высушивали с помощью безводного Na2SO4, фильтровали, концентрировали и очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат = 5:1-1:1) с получением (S)-N-((S)-1-(3-(дифторметокси)фенил)этил)-2-метилпропан-2-сульфинамида (960 мг).
Стадия 3. Получение гидрохлорида (5)-1-(3-(дифторметокси)фенил)этан-1-амина
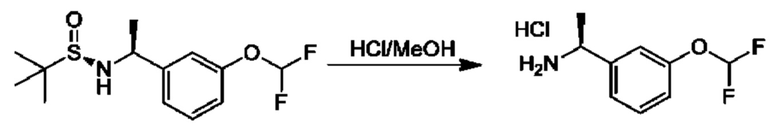
К раствору (S)-N-((S)-1-(3-(дифторметокси)фенил)этил)-2-метилпропан-2-сульфинамида (0,8 г) в МеОН (4 мл) добавляли HCl/МеОН (4 М, 2 мл). Полученную смесь перемешивали при 25°С в течение 3 часов и концентрировали с получением гидрохлорида (S)-1-(3-(дифторметокси)фенил)этан-1-амина (1,6 г, неочищенный).
IIk: гидрохлорид (R)-2-этокси-1-(3-(трифторметокси)фенил)этан-1-амина
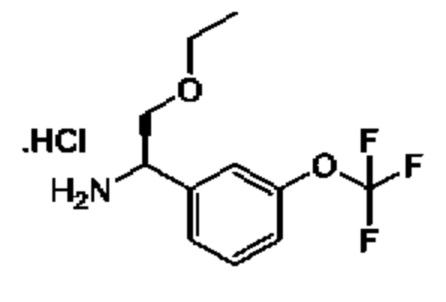
Стадия 1. Получение этил-(R,Е)-2-((трет-бутилсульфинил)имино)ацетата
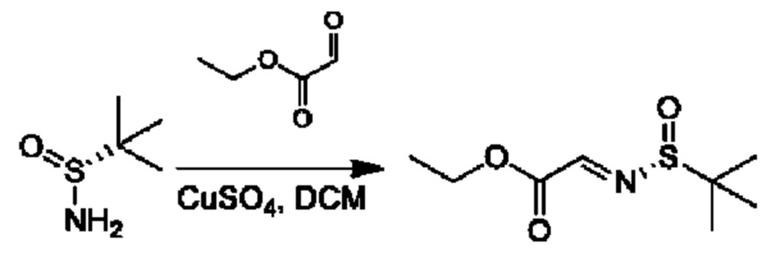
К раствору этил-2-оксоацетата (7,5 г) и (R)-2-метилпропан-2-сульфинамида (4,90 г) в DCM (150 мл) в атмосфере N2 добавляли CuSO4 (12,9 г), и реакционную смесь перемешивали при 25°С в течение 24 часов. Твердое вещество отфильтровывали, промывали с помощью этилацетата (50 мл), и органический слой концентрировали. Остаток очищали посредством хроматографии (SiO2, гексан/этилацетат, 5/1) с получением этил-(R,Е)-2-((трет-бутилсульфинил)имино)ацетата (5 г).
Стадия 2. Получение этил-(R)-2-(((S)-трет-бутилсульфинил)амино)-2-(3-(трифторметокси)фенил)ацетата
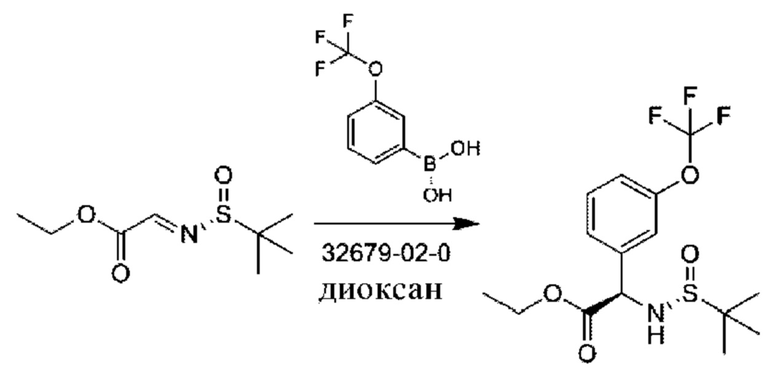
К раствору этил-(R,Е)-2-((трет-бутилсульфинил)имино)ацетата (5 г) и [3-(трифторметокси)фенил]бороновой кислоты (6,02 г) в диоксане (80 мл) добавляли бис(ацетонитрил)(1,5-циклооктадиен)родия(I) тетрафторборат (CAS:32679-02-0) (1,85 г), и данную смесь перемешивали при 80°С в течение 16 часов. Раствор фильтровали, и органическую фазу концентрировали. Остаток очищали посредством хроматографии (SiO2, петролейный эфир:EtOAc=6:1) с получением этил-(R)-2-(((S)-трет-бутилсульфинил)амино)-2-(3-(трифторметокси)фенил)ацетата (5,1 г).
Стадия 3. Получение этил-(R)-2-амино-2-(3-(трифторметокси)фенил)ацетата
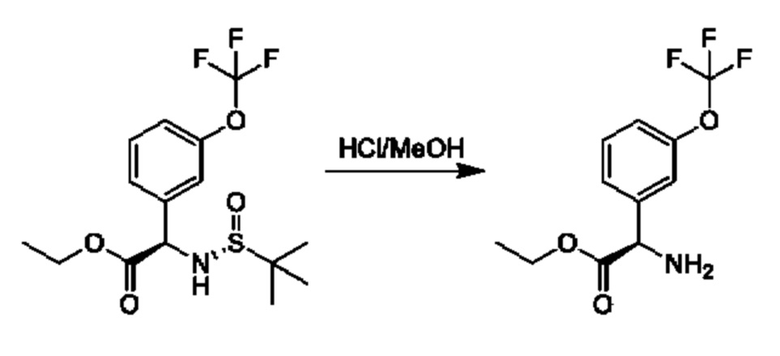
К раствору этил-(R)-2-(((S)-трет-бутилсульфинил)амино)-2-(3-(трифторметокси)фенил)ацетата (4,6 г) в МеОН (30 мл) добавляли HCl/МеОН (4 М, 25,04 мл) при 0°С. Реакционную смесь перемешивали при 25°С в течение 1 часа и концентрировали с получением этил-(R)-2-амино-2-(3-(трифторметокси)фенил)ацетата в виде хлористоводородной соли (3,3 г).
Стадия 4. Получение этил-(R)-2-((трет-бутоксикарбонил)амино)-2-(3-(трифторметокси)фенил)ацетата
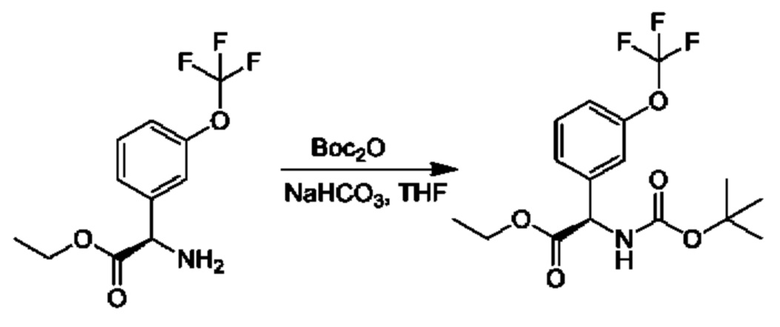
К смеси гидрохлорида этил-(R)-2-амино-2-(3-(трифторметокси)фенил)ацетата (3,3 г) в THF (80 мл) добавляли Вос2О (4,81 г) и NaHCO3 (925 мг), и реакционную смесь перемешивали при 25°С в течение 16 часов. Реакционную смесь концентрировали и разбавляли с помощью EtOAc (20 мл), промывали водой (20 мл), а затем концентрировали. Остаток очищали посредством хроматографии (SiO2; петролейный эфир:EtOAc=10:1) с получением этил-(R)-2-((трет-бутоксикарбонил)амино)-2-(3-(трифторметокси)фенил)ацетата (3,8 г).
Стадия 5. Получение трет-бутил-(R)-(2-гидрокси-1-(3-(трифторметокси)фенил)этил)карбамата
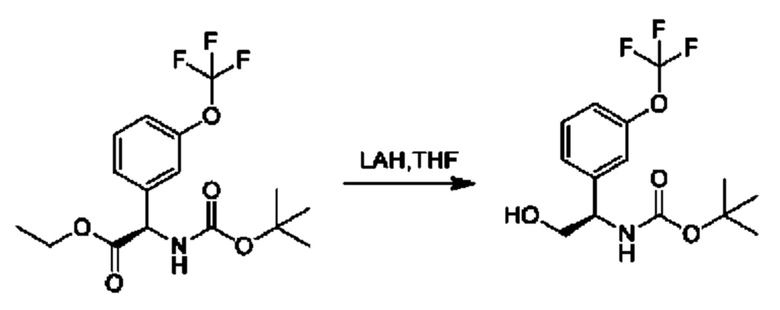
К суспензии LiAlH4 (2,1 г) в THF (200 мл) добавляли этил-(R)-2-((трет-бутоксикарбонил)амино)-2-(3-(трифторметокси) фенил) ацетат (5 г) в THF (20 мл) при охлаждении с помощью льда, и смесь перемешивали при температуре от 0°С до 25°С в течение 2 часов. Добавляли безводный сульфат магния, а затем последовательно добавляли воду (5 мл) и этилацетат (100 мл), и нерастворимые вещества отфильтровывали с использованием целита. Фильтрат концентрировали. Неочищенный продукт очищали посредством хроматографии (SiO2, петролейный эфир:EtOAc=5:1) с получением трет-бутил-(R)-(2-гидрокси-1-(3-(трифторметокси)фенил)этил)карбамата (3,37 г).
Стадия 6. Получение трет-бутил-(R)-(2-этокси-1-(3-(трифторметокси)фенил)этил)карбамата
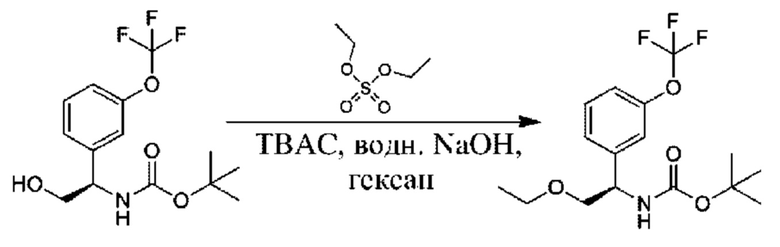
К раствору трет-бутил-(R)-(2-гидрокси-1-(3-(трифторметокси)фенил)этил)карбамата (1 г) в гексане (15 мл) добавляли диэтилсульфат (960 мг), хлорид тетрабутиламмония (ТВАС) (87 мг) и раствор NaOH (324 мг) в Н2О (1,5 мл). Полученную смесь перемешивали при 25°С в течение 20 часов. Реакционную смесь разбавляли с помощью EtOAc (200 мл), промывали водой (100 мл) и солевым раствором (100 мл), высушивали над Na2SO4 и концентрировали. Неочищенный продукт очищали посредством хроматографии (SiO2, 10% этилацетат в петролейном эфире) с получением трет-бутил-(R)-(2-этокси-1-(3-(трифторметокси)фенил)этил)карбамата (5,9 г).
Стадия 7. Получение гидрохлорида (R)-2-этокси-1-(3-(трифторметокси)фенил)этан-1-амина
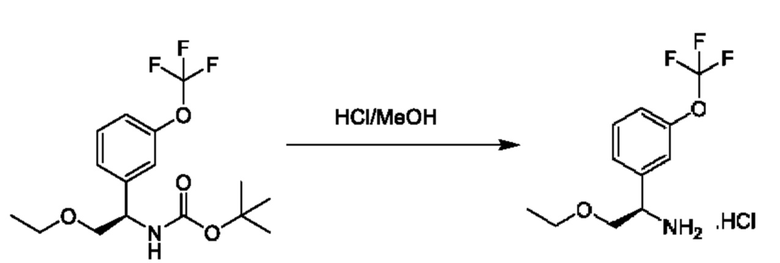
К раствору трет-бутил-(R)-(2-этокси-1-(3-(трифторметокси)фенил)этил)карбамата (5,9 г) в МеОН (100 мл) добавляли HCl/МеОН (4 М, 63,33 мл) при 25°С, и данную смесь перемешивали при 25°С в течение 16 часов. Раствор концентрировали с получением гидрохлорида (R)-2-этокси-1-(3-(трифторметокси)фенил)этан-1-амина (4,5 г).
II1: гидрохлорид (S)-1-(3-(трифторметокси)фенил)пропан-1-амина
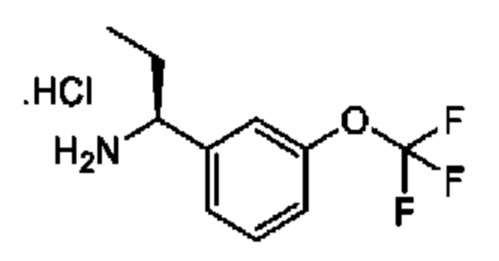
Стадия 1. Получение (S, Ε)-2-метил-N-(3-(трифторметокси)бензилиден)пропан-2-сульфинамида
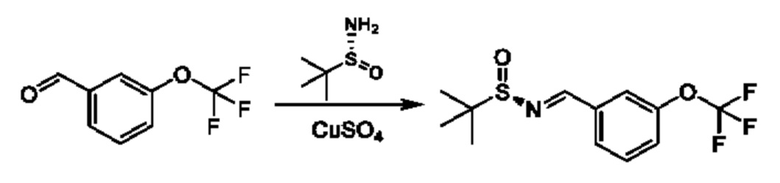
Смесь 3-(трифторметокси) бензальдегида (10,0 г), (R)-2-метилпропан-2-сульфинамида (7,7 г) и CuSO4 (12,6 г) в DCE (200 мл) перемешивали при 55°С в течение 16 часов.
Смесь фильтровали, и осадок на фильтре промывали с помощью DCM (200 мл). Фильтрат концентрировали. Остаток очищали посредством хроматографии (SiO2, градиент предусматривал 0-10% этилацетат/петролейный эфир) с получением (S, Ε)-2-метил-N-(3-(трифторметокси)бензилиден)пропан-2-сульфинамида (12,6 г).
Стадия 2. Получение (S)-2-метил-N-((S)-1-(3-(трифторметокси)фенил)пропил)пропан-2-сульфинамида
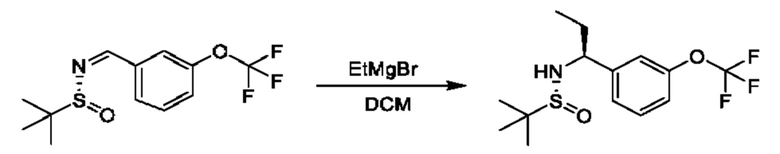
К раствору (S, Ε)-2-метил-N-(3-(трифторметокси)бензилиден)пропан-2-сульфинамида (2,0 г) в DCM (40 мл) при 0°С по каплям добавляли EtMgBr (3 Μ в Et2O, 9,1 мл). Полученную смесь перемешивали при 0°С в течение 1 часа и при 20°С в течение 3 часов. Смесь охлаждали до 0°С, и добавляли насыщ. водн. раствор NH4Cl (100 мл). Смесь экстрагировали с помощью DCM (100 мл × 2), фазы разделяли, и органический слой промывали солевым раствором (200 мл), высушивали над Na2SO4 и концентрировали. Остаток очищали посредством хроматографии (SiO2, градиент предусматривал 0-50% этилацетат/петролейный эфир) с получением продукта (1,4 г).
Стадия 3. Получение гидрохлорида (S)-1-(3-(трифторметокси)фенил)пропан-1-амина
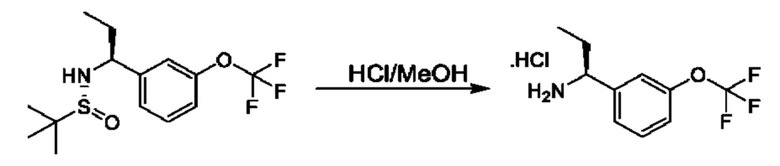
К раствору (S)-2-метил-N-((S)-1-(3-(трифторметокси)фенил)пропил)пропан-2-сульфинамида (1,4 г) в МеОН (40 мл) добавляли HCl/МеОН (4 М, 20 мл). Полученную смесь перемешивали при 30°С в течение 12 часов, а затем концентрировали с получением неочищенного гидрохлорида (S)-1-(3-(трифторметокси)фенил)пропан-1-амина, который применяли без дополнительной очистки (1 г).
IIm: гидрохлорид (S)-3-амино-3-(3-(трифторметокси)фенил)пропаннитрила
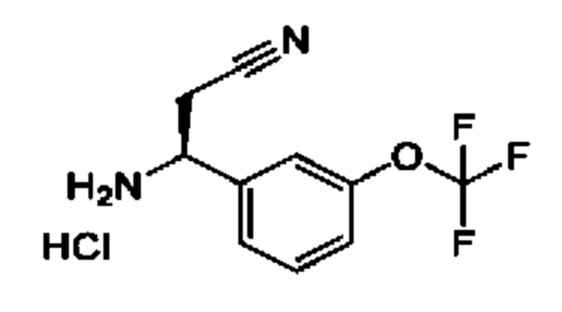
Стадия 1. Получение трет-бутил-(R)-(2-гидрокси-1-(3-(трифторметокси)фенил)этил)карбамата
Данное промежуточное соединение получали, как описано для промежуточного соединения IIk, стадии 1-5.
Стадия 2. Получение (R)-2-((трет-бутоксикарбонил)амино)-2-(3-(трифторметокси)фенил)этилметансульфоната
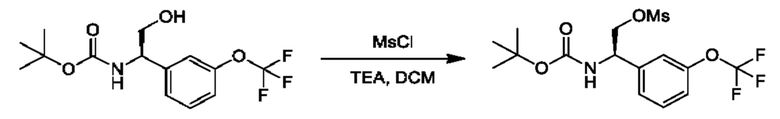
К раствору трет-бутил-(R)-(2-гидрокси-1-(3-(трифторметокси)фенил)этил)карбамата (2 г) в DCM (20 мл) добавляли Et3N (756 мг), затем добавляли метансульфонилхлорид (1,75 г) при 0°С. Смесь перемешивали при 20°С в течение 16 часов. Реакционную смесь промывали с помощью насыщенного водного раствора NH4Cl (15 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали с получением (R)-2-((трет-бутоксикарбонил)амино)-2-(3-(трифторметокси)фенил)этилметансульфоната (2,50 г, неочищенный), который использовали на следующей стадии без дополнительной очистки.
Стадия 3. Получение гидрохлорида (S)-3-амино-3-(3-(трифторметокси)фенил)пропаннитрила
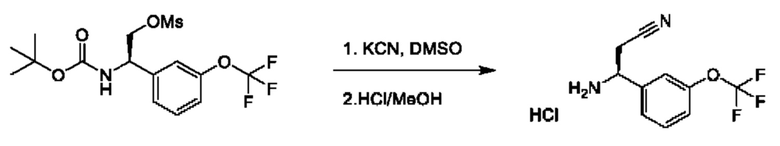
К раствору (R)-2-((трет-бутоксикарбонил)амино)-2-(3-(трифторметокси)фенил)этилметансульфоната (420 мг) в DMSO (5 мл) добавляли KCN (225 мг) при 20°С. Смесь перемешивали при 50°С в течение 16 часов. Реакционную смесь разбавляли с помощью 10% раствора Na2CO3 (40 мл) и экстрагировали с помощью EtOAc (30 мл × 3). Объединенные органические экстракты промывали водой (50 мл) и солевым раствором (50 мл), затем высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 4/1) с получением трет-бутил-(S)-(2-циано-1-(3-(трифторметокси)фенил)этил)карбамата (665 мг).
1Н ЯМР (400 МГц, CDCl3): δ 7,46 (t, 1Н), 7,33 (d, 1H), 7,24 (m, 2H), 7,04 (d, 1H), 5,31-5,38 (m, 1H), 3,73-3,71 (m, 1H), 3,11-3,05 (m, 1H), 2,84-2,92 (m, 2H), 2,49-2,43 (m, 1H), 2,28-2,37 (m, 1H), 0,93 (s, 9H).
К раствору трет-бутил-(S)-(2-циано-1-(3-(трифторметокси)фенил)этил)карбамата (400 мг) в МеОН (8 мл) добавляли HCl/МеОН (4 М, 4,00 мл) при 0°С. Смесь перемешивали при 25°С в течение 16 часов. Затем реакционную смесь концентрировали с получением гидрохлорида (S)-3-амино-3-(3-(трифторметокси)фенил)пропаннитрила (280 мг, неочищенный).
ΙΙn: гидрохлорид (S)-4-амино-4-(3-(трифторметокси)фенил)бутаннитрила
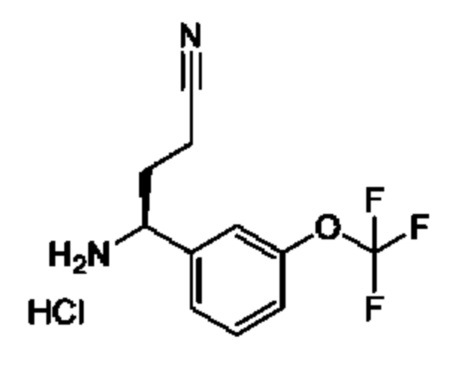
Стадия 1. Получение (R, Ε)-2-метил-N-(3-(трифторметокси)бензилиден)пропан-2-сульфинамида
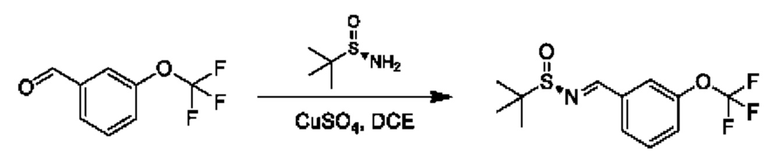
К раствору 3-(трифторметокси)бензальдегида (30 г) и (R)-2-метилпропан-2-сульфинамида (23,0 г) в DCE (600 мл) добавляли CuSO4 (37,8 г). Смесь перемешивали при 55°С в течение 24 часов и фильтровали. Осадок на фильтре промывали с помощью DCM (300 мл). Фильтраты объединяли и концентрировали, и остаток очищали посредством хроматографии (Si02, петролейный эфир/этилацетат = от 0/1 до 5:1) с получением (R, Ε)-2-метил-N-(3-(трифторметокси)бензилиден)пропан-2-сульфинамида (41,8 г).
Стадия 2. Получение этил-(S)-3-(((R)-трет-бутилсульфинил)амино)-3-(3-(трифторметокси)фенил)пропаноата
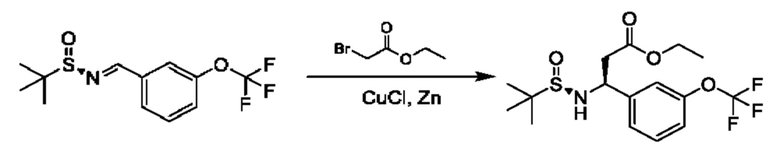
Раствор (R, Ε)-2-метил-N-(3-(трифторметокси)бензилиден)пропан-2-сульфинамида (5 г) в THF (60 мл) добавляли к суспензии активированного Ζn (11,15 г), CuCl (2,5 г) и этил-2-бромацетата (7,1 г) в THF (60 мл) при 0°С. Реакционную смесь перемешивали при 50°С в течение 2 часов и фильтровали. Осадок на фильтре промывали с помощью DCM (400 мл), объединенные органические фильтраты концентрировали. Остаток очищали посредством хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 5/1) с получением этил-(S)-3-(((R)-трет-бутилсульфинил)амино)-3-(3-(трифторметокси)фенил)пропаноата (7 г, неочищенный).
1Н ЯМР (CDCl3 400 МГц): δ 7,36 (t, 2Н), 7,20 (s, 1Н), 7,14 (d, 1H), 5,78 (d, 1H), 5,14-5,10 (m, 1H), 4,14-4,10 (m, 2H), 3,05-2,89 (m, 2H), 1,31 (s, 9H), 1,18 (t, 3H).
Стадия 3. Получение (R)-Ν-((S)-3-гидрокси-1-(3-(трифторметокси)фенил)пропил)-2-метилпропан-2-сульфинамида
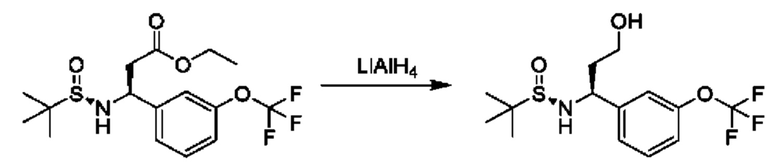
К раствору этил-(S)-3-(((R)-трет-бутилсульфинил)амино)-3-(3-(трифторметокси)фенил)пропаноата (7 г) в THF (70 мл) добавляли LiAlH4 (6 96 мг) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 часа, а затем гасили путем последовательного добавления Н2О (0,7 мл), 10% раствора NaOH (0,7 мл) и Н2О (2,1 мл) при 0°С. Смесь фильтровали. Остаток концентрировали с получением неочищенного (R)-N-((S)-3-гидрокси-1-(3-(трифторметокси)фенил)пропил)-2-метилпропан-2-сульфинамида(4,2 г).
Стадия 4. Получение гидрохлорида (S)-3-амино-3-(3-(трифторметокси)фенил)пропан-1-ола
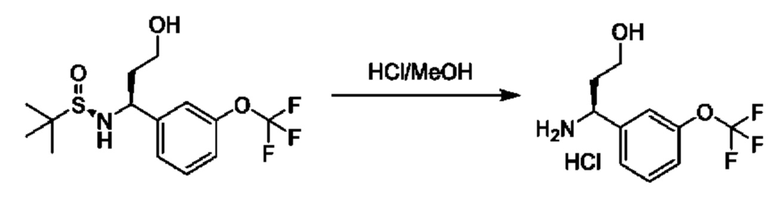
Неочищенный (R)-N-((S)-3-гидрокси-1-(3-(трифторметокси)фенил)пропил)-2-метилпропан-2-сульфинамид (4 г) из предыдущей стадии реакции растворяли в МеОН (40 мл), и добавляли HCl/МеОН (4 М, 23,6 мл). Реакционную смесь перемешивали при 20°С в течение 16 часов и концентрировали с получением гидрохлорида (S)-3-амино-3-(3-(трифторметокси)фенил)пропан-1-ола (3,2 г, неочищенный).
Стадия 5. Получение трет-бутил-(S)-(3-гидрокси-1-(3-(трифторметокси)фенил)пропил)карбамата
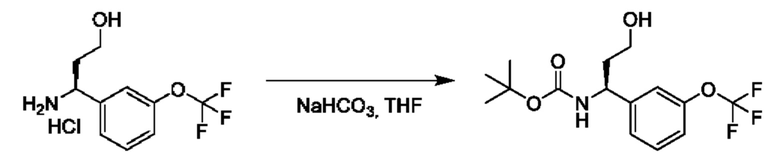
Неочищенный этил-(S)-3-(((R)-трет-бутилсульфинил)амино)-3-(3-(трифторметокси)фенил)пропаноат из предыдущей стадии реакции (3,2 г) растворяли в THF (35 мл), и добавляли Вос2О (10,28 г) и NaHCO3 (2 г). Реакционную смесь перемешивали при 20°С в течение 16 часов. Смесь концентрировали, и остаток разбавляли водой (70 мл), экстрагировали с помощью DCM (100 мл × 3), и объединенные органические экстракты высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 2/1) с получением трет-бутил-(S)-(3-гидрокси-1-(3-(трифторметокси)фенил)пропил)карбамата (3,2 г).
Стадия 6. Получение (S)-3-((трет-бутоксикарбонил)амино)-3-(3-(трифторметокси)фенил)пропилметансульфоната
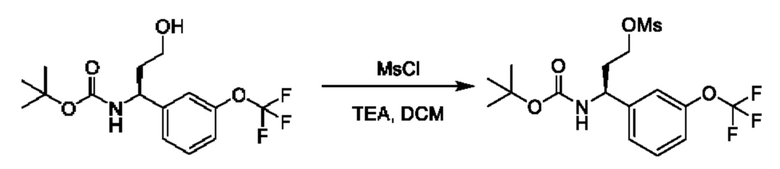
К раствору трет-бутил-(S)-(3-гидрокси-1-(3-(трифторметокси)фенил)пропил)карбамата (1 г) в DCM (30 мл) добавляли Et3N (905 мг) и метансульфонилхлорид (683 мг) при 0°С. Реакционную смесь перемешивали при 20°С в течение 16 часов, а затем промывали ледяной водой (15 мл), высушивали над безводным Na2SO4, фильтровали и концентрировали с получением ((S)-3-((трет-бутоксикарбонил)амино)-3-(3-(трифторметокси)фенил)пропилметансульфоната (1,2 г).
Стадия 7. Получение трет-бутил-(S)-(3-циано-1-(3-(трифторметокси)фенил)пропил)карбамата
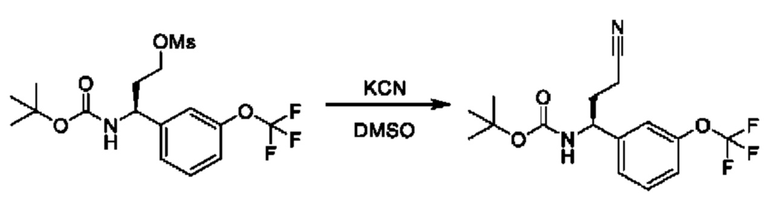
Полученный на предыдущей стадии ((S)-3-((трет-бутоксикарбонил)амино)-3-(3-(трифторметокси)фенил)пропилметансульфонат растворяли в DMSO (35 мл), и добавляли KCN (661 мг) при 20°С. Реакционную смесь перемешивали при 50°С в течение 16 часов, а затем разбавляли с помощью 10% раствора Na2CO3 (40 мл) и экстрагировали с помощью EtOAc (70 мл × 3). Объединенные органические экстракты промывали водой (50 мл) и солевым раствором (50 мл), затем высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 3/1) с получением трет-бутил-(S)-(3-циано-1-(3-(трифторметокси)фенил)пропил)карбамата (990 мг).
Стадия 8. Получение гидрохлорида (S)-4-амино-4-(3-(трифторметокси)фенил)бутаннитрила
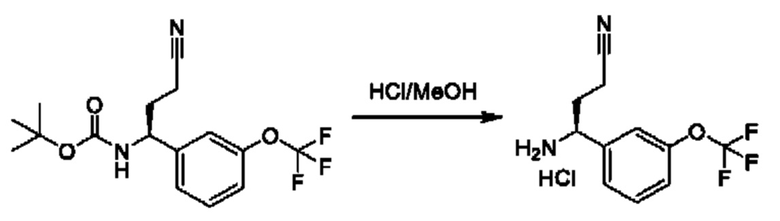
К раствору трет-бутил-(S)-(3-циано-1-(3-(трифторметокси)фенил)пропил)карбамата (900 мг) в МеОН (14 мл) добавляли HCl/МеОН (4 М, 6,53 мл). Смесь перемешивали при 20°С в течение 16 часов и концентрировали с получением гидрохлорида (S)-4-амино-4-(3-(трифторметокси)фенил)бутаннитрила (850 мг).
IIo: гидрохлорид (S)-3,3-дифтор-1-(3-(трифторметокси)фенил)пропан-1-амина
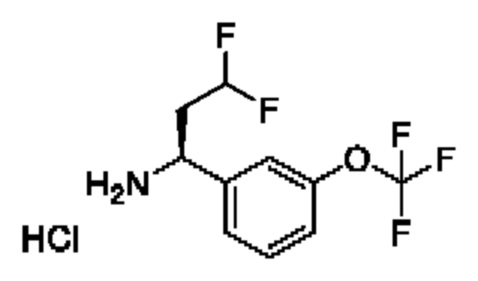
Получен, как описано для IIi, с применением 3-(трифторметокси)бензальдегида в качестве исходного материала.
IIp: гидрохлорид (R)-2-метокси-1-(3-(трифторметокси)фенил)зтан-1-амина
Стадия 1. Получение этил-(R)-2-((трет-бутоксикарбонил)амино)-2-(3-(трифторметокси)фенил)ацетата
Данное промежуточное соединение получали, как описано для IIk, стадии 1-4.
Стадия 2. Получение трет-бутил-(R)-(2-гидрокси-1-(3-(трифторметокси)фенил)этил)карбамата
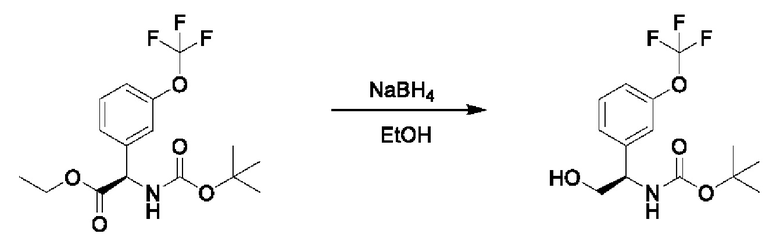
К раствору этил-(R)-2-((трет-бутоксикарбонил)амино)-2-(3-(трифторметокси)фенил)ацетата (10 г) в EtOH (90 мл) добавляли NaBH4 (4,17 г) при 0°С. Смесь извлекали из холодной бани и перемешивали в течение 2 часов. Реакцию гасили водой (20 мл) и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 1/0 до 3/1) с получением продукта (13,6 г).
1Н ЯМР (CDCl3 400 МГц): δ 7,39 (t, 1Н), 7,26 (d, 1H), 7,17-7,15 (m, 2H), 5,34 (s, 1H), 4,80 (s, 1H), 3,93-3,84 (m, 2H), 2,06 (s, 1H), 1,45 (s, 9H).
Стадия 3. Получение трет-бутил-(R)-(2-метокси-1-(3-(трифторметокси)фенил)этил)карбамата
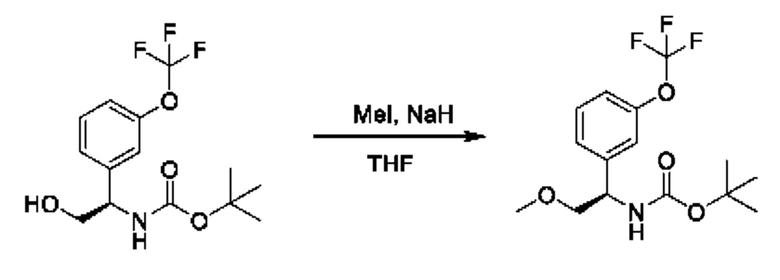
К раствору трет-бутил-(R)-(2-гидрокси-1-(3-(трифторметокси)фенил)этил)карбамата (1 г) и MeI (4 г) в THF (70 мл) добавляли NaH (149 мг, 60% в минеральном масле) при 0°С. Смесь перемешивали при 0°С в течение 1 часа и перемешивали при 25°С в течение 16 часов. Для гашения реакции добавляли воду (1 мл). THF удаляли, и к остатку добавляли EtOAc (200 мл). Раствор промывали водой (50 мл × 3) и концентрировали. Остаток очищали посредством колоночной хроматографии (SiO2, петролейный эфир/этилацетат = от 0/1 до 5/1) с получением трет-бутил-(R)-(2-метокси-1-(3-(трифторметокси)фенил)этил)карбамата (3,2 г).
1Н ЯМР (CDCl3 400 МГц): δ 7,36 (t, 1Н), 7,25 (m, 1H), 7,19 (s, 1H), 7,12 (d, 1H), 5,34 (s, 1H), 4,83 (s, 1H), 3,63-3,56 (m, 2H), 3,35 (s, 3H), 1,43 (s, 9H).
Стадия 4. Получение гидрохлорида (R)-2-метокси-1-(3-(трифторметокси)фенил)этан-1-амина
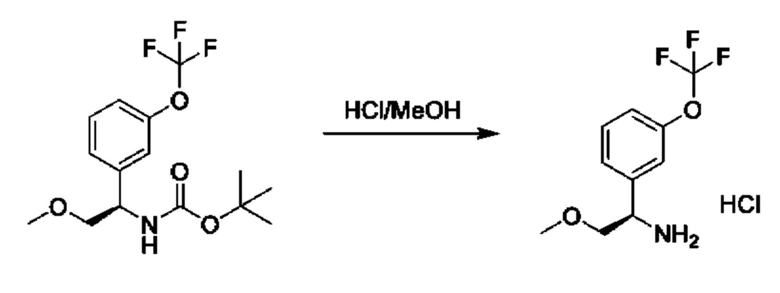
К раствору трет-бутил-(R)-(2-метокси-1-(3-(трифторметокси)фенил)этил)карбамата (2,7 г) в МеОН (40 мл) добавляли HCl/МеОН (4 М, 4 0 мл) при 25°С. Смесь перемешивали при 25°С в течение 16 часов. Смесь концентрировали с получением требуемого продукта (1,9 г, неочищенный).
IIIa: (S)-3-гидрокси-4,4-диметилпентановая кислота
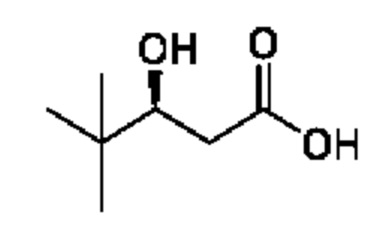
Получена в соответствии с имеющимися в литературе сведениями, как описано в Wang Ζ. et al: Tetrahedron: Asymmetry 10 (1999) 225-228.
IIIb: 2-(3,3-дифтор-1-гидроксициклобутил)уксусная кислота
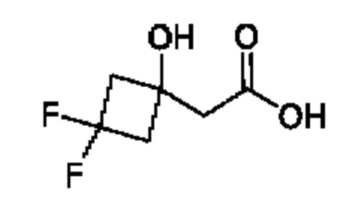
Стадия 1. Получение этил-2-(3,3-дифтор-1-гидроксициклобутил)ацетата
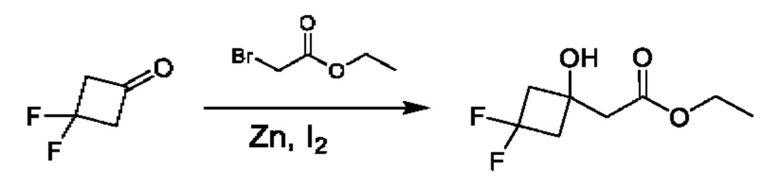
К раствору 3,3-дифторциклобутанона (0,2 г), Zn (198 мг) и 1 г (10 мг) в THF (13 мл) в атмосфере N2 по каплям добавляли этил-2-бромацетат (378 мг). Смесь перемешивали при 55°С в течение 6 часов. К реакционной смеси осторожно добавляли H2SO4 (10%, 10 мл) при 0°С, и смесь экстрагировали этилацетатом (20 мл × 3).
Органический экстракт промывали с помощью NaHCO3 (насыщ. водн. раствор, 10 мл), высушивали над Na2SO4 и концентрировали. Неочищенный продукт (0,26 г) использовали непосредственно без дополнительной очистки.
Стадия 2. Получение 2-(3,3-дифтор-1-гидроксициклобутил)уксусной кислоты
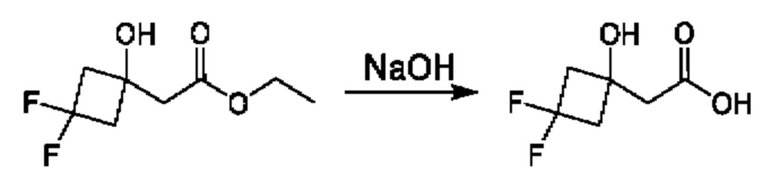
К раствору этил-2-(3,3-дифтор-1-гидроксициклобутил)ацетата (0,26 г) в МеОН (10 мл) и Н2О (2 мл) добавляли NaOH (107 мг) при 0°С. Смесь перемешивали при 20°С в течение 8 часов. Реакционный раствор охлаждали до 0°С, и в раствор добавляли 1 н. HCl до достижения рН 1-2. Остаток разбавляли солевым раствором (10 мл) и экстрагировали с помощью метил-трет-бутилового эфира (30 мл × 5). Объединенные органические экстракты высушивали над Na2SO4, фильтровали и концентрировали. Неочищенный продукт (0,24 г) применяли без дополнительной очистки.
IIIc: 3-(1-фторциклопропил)-3-гидроксибутановая кислота
Стадия 1. Получение этил-3-(1-фторциклопропил)-3-гидроксибутаноата
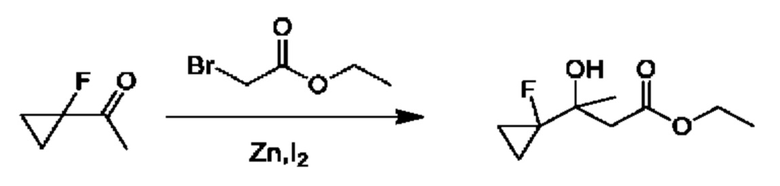
Раствор 1-(1-фторциклопропил)этанона (0,5 г), Zn (512 мг) и T2 (62 мг) в THF (30 мл) перемешивали при 20°С до тех пор, пока раствор не становился бесцветным, и по каплям добавляли этил-2-бромацетат (981 мг). Полученную смесь перемешивали при 20°С в течение 0,5 часа и при 65°С в течение 4,5 часа. Реакционную смесь промывали с помощью 10% водн. H2SO4 (20 мл) и экстрагировали с помощью EtOAc (50 мл × 2). Органические экстракты промывали солевым раствором (100 мл), высушивали над Na2SO4 и концентрировали с получением этил-3-(1-фторциклопропил)-3-гидроксибутаноата (0,83 г, неочищенный).
Стадия 2. Получение 3-(1-фторциклопропил)-3-гидроксибутановой кислоты
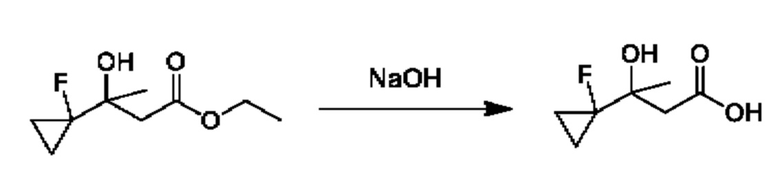
К раствору этил-3-(1-фторциклопропил)-3-гидроксибутаноата (0,83 г) в EtOH (10 мл) добавляли раствор NaOH (350 мг) в Н2О (3 мл). Реакционную смесь перемешивали при 20°С в течение 2 часов, а затем экстрагировали с помощью EtOAC (50 мл × 2). Водный слой подкисляли с помощью 10% HCl до рН=3 и экстрагировали с помощью EtOAC (50 мл × 2). Объединенные органические экстракты промывали солевым раствором (100 мл), высушивали над Na2SO4 и концентрировали с получением 3-(1-фторциклопропил)-3-гидроксибутановой кислоты (0,57 г, неочищенный).
IIId: 3-циклопропил-3-гидроксибутановая кислота
Стадия 1. Получение метил-3-циклопропил-3-гидроксибутаноата
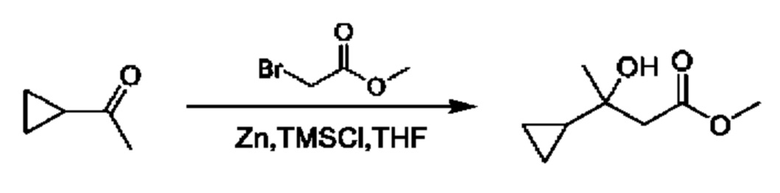
Zn (12,4 г) в THF (150 мл) добавляли к TMSCl (1,3 г), и полученную смесь перемешивали при 20°С в течение 15 минут, а затем нагревали до 70°С. Нагревание останавливали, и по каплям добавляли метил-2-бромацетат (21,8 г) с такой скоростью, что растворитель подвергался слабому кипению. Полученную смесь перемешивали при 70°С в течение 1 часа и при 20°С в течение 1 часа, затем добавляли раствор 1-циклопропилэтанона (10 г) в THF (50 мл). Реакционную смесь перемешивали при 20°С в течение 16 часов. Смесь выливали в NH3⋅H2O на льду (100 мл, 28%) и экстрагировали с помощью этилацетата (150 мл × 2). Органический экстракт промывали водой (150 мл) и солевым раствором (150 мл), высушивали над Na2SO4 и концентрировали с получением требуемого продукта (8,9 г, неочищенный).
Стадия 2. Получение 3-циклопропил-3-гидроксибутановой кислоты
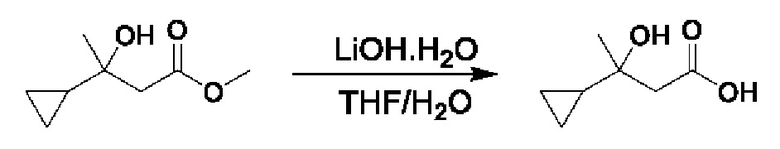
Смесь неочищенного метил-3-циклопропил-3-гидроксибутаноата (8,9 г) и LiOH⋅Н2О (11,8 г) в THF (100 мл) и Н2О (50 мл) перемешивали при 20°С в течение 16 часов. Добавляли Н2О (50 мл), и экстрагировали с помощью этилацетата (100 мл × 2). Органические экстракты удаляли. рН водного слоя доводили до ~5 с помощью 2 н. HCl,, осуществляли его экстрагирование с помощью этилацетата (100 мл × 3), и объединенные органический фракции промывали солевым раствором (100 мл × 10), высушивали над Na2SO4, фильтровали и концентрировали с получением требуемого продукта с общим выходом 30% (5,1 г).
1Н ЯМР (400 МГц, CDCl3) δ 2,67-2,51 (m, 2Н), 1,25 (s, 3Н), 0,90-1,00 (m, 1Н), 0,33-0,50 (m, 4H).
IIIe: 5,5,5-трифтор-3-гидрокси-3-метилпентановая кислота
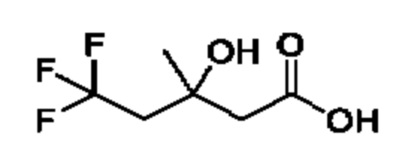
Стадия 1. Получение этил-5,5,5-трифтор-3-гидрокси-3-метилпентаноата
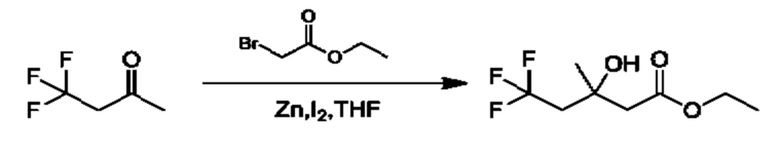
К смеси Zn (6,9 г) и 1 г (89 мг) в THF (80 мл) добавляли 4,4,4-трифторбутан-2-он (4,4 г) и этил-2-бромацетат (6,4 г) при 15°С. Смесь перемешивали при 60°С в течение 6 часов. Реакционную смесь охлаждали до 0°С и гасили с помощью H2SO4 (100 мл, 10% водн. раствор). Смесь экстрагировали этилацетатом (15 мл × 3). Объединенный органический экстракт промывали солевым раствором (15 мл) и высушивали над Na2SO4, фильтровали и концентрировали. Получали продукт (11,00 г, неочищенный), и использовали его непосредственно без дополнительной очистки.
Стадия 2. Получение 5,5,5-трифтор-3-гидрокси-3-метилпентановой кислоты
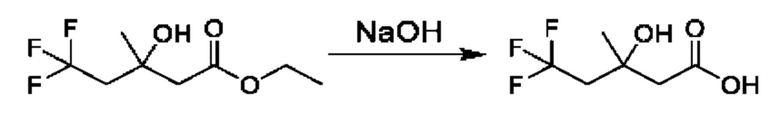
Смесь этил-5,5,5-трифтор-3-гидрокси-3-метилпентаноата (11 г, неочищенный) и NaOH (4,1 г) в Н2О (150 мл) перемешивали при 15°С в течение 16 часов. рН доводили до ~2 с помощью насыщ. KHSO4 при 0°С, и смесь экстрагировали с помощью этилацетата (200 мл × 3). Объединенный органический экстракт промывали солевым раствором (300 мл), высушивали над Na2SO4, фильтровали и концентрировали с получением продукта (10 г, неочищенный).
Следующие соединения получали посредством такой же методики, как описанная для IIIe, с применением соответствующих исходных материалов.
IIIf: 3-гидрокси-3,4-диметилпентановая кислота
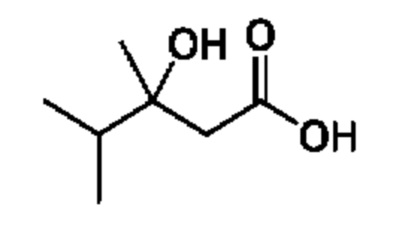
1Н ЯМР (CDCl3 400 МГц): δ 2,65-2,46 (m, 2Н), 2,09 (s, 1Н), 1,85-1,76 (m, 1H), 1,20 (s, 3H), 0,93 (dd, 6H).
IIIg: 3-гидрокси-З,5-диметилгексановая кислота
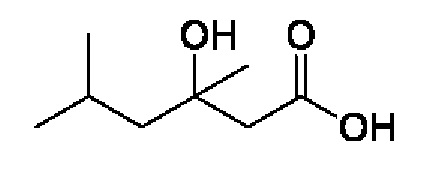
1H ЯМР (CDCl3 400 МГц): δ 2, 64-2, 50 (m, 2Н), 1,85-1,79 (m, 1Н), 1,49 (d, 2H), 1,32 (s, 3Н), 1,03-0,97 (m, 6H).
IIIh: 3-(3,3-диметилциклобутил)-3-гидроксипропановая кислота
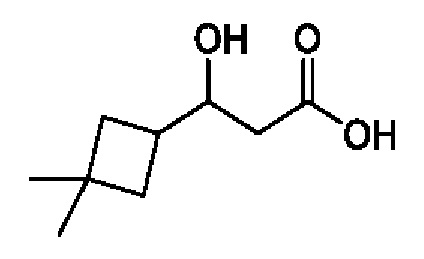
Стадия 1. Получение этил-3-(3,3-диметилциклобутил)-3-гидроксипропаноата
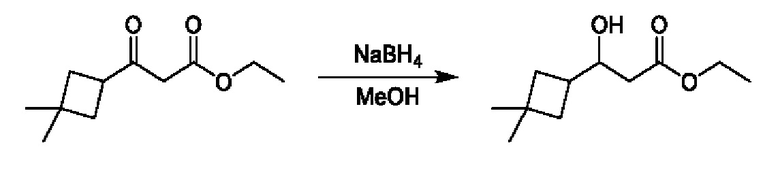
К раствору этил-3-(3,3-диметилциклобутил)-3-оксопропаноата (IVd) (1 г) в МеОН (8 мл) добавляли NaBH4 (95 мг). Смесь перемешивали при 0°С в течение 10 мин и гасили с помощью добавления Н2О (1 мл), концентрировали, а затем разбавляли с помощью EtOAc (30 мл), высушивали над Na2SO4, фильтровали и выпаривали. Остаток очищали посредством хроматографии (SiO2, петролейный эфир/этилацетат = от 5/1 до 4/1) с получением этил-3-(3,3-диметилциклобутил)-3-гидроксипропаноата (907 мг).
1Н ЯМР (DMSO-d6 400 МГц): δ 4,70 (d, 1Н), 4,06-4,00 (m, 2H), 3,71-3,68 (m, 1H), 2,26-2,23 (m, 1H), 2,15-2,12 (m, 2H), 1,63-1,59 (m, 3H), 1,52-1,49 (m, 1H), 1,17 (t, 3H), 1,09 (s, 3H), 0,99 (s, 3H).
Стадия 2. Получение 3-(3,3-диметилциклобутил)-3-гидроксипропановой кислоты
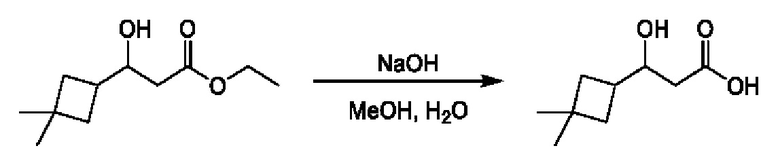
К раствору этил-3-(3,3-диметилциклобутил)-3-гидроксипропаноата (900 мг) в МеОН (10 мл) добавляли раствор NaOH (37 7 мг) в Н2О (5 мл). Смесь перемешивали при 25°С в течение 4 часов. В реакционную смесь добавляли 10% раствор HCl с достижением рН=3-4, затем ее разбавляли с помощью Н2О (30 мл) и экстрагировали с помощью EtOAc (30 мл × 2), объединенные органические слои высушивали над Na2SO4, фильтровали и концентрировали с получением 3-(3,3-диметилциклобутил)-3-гидроксипропановой кислоты (760 мг).
1Н ЯМР (DMSO-d6 400 МГц): δ 11,96-11,95 (m, 1Н), 4,65-4,61 (m, 1H), 3,71-3,66 (m, 1H), 2,19-2,05 (m, 3H), 1,63-1,51 (m, 4H), 1,09 (s, 3H), 1,00 (s, 3H).
Следующее соединение получали посредством такой же методики, как описанная для IIIh, с применением соответствующих исходных материалов.
IIIi: 3-циклопентил-3-гидроксипропановая кислота
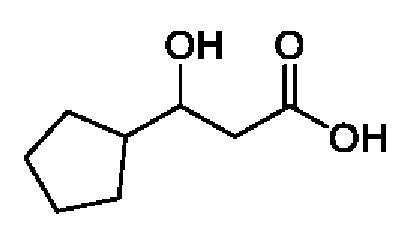
1Н ЯМР (DMSO-d6 400 МГц): δ 11,96 (s, 1Н), 4,63 (s, 1H), 3,66 (s, 1H), 2,36-2,32 (m, 2H), 1,83-1,75 (m, 1H), 1,62-1,35 (m, 8H).
IVa: этил-3-[1-(дифторметил)циклопропил]-3-оксопропаноат
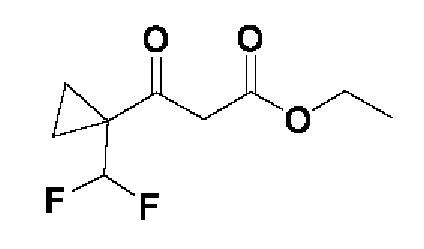
Стадия 1. Получение этил-3-[1-(дифторметил)циклопропил]-3-оксопропаноата
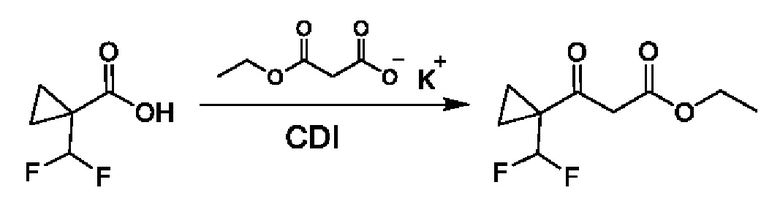
Et3N (2,34 г) и MgCl2 г (1,8 г) добавляли к суспензии калиевой соли (3-этокси-3-оксопропаноил)окси (2,6 г) в MeCN (30 мл), и осуществляли перемешивание при 20°С в течение 2 часов. Подвергнутую предварительному перемешиванию смесь карбонилдиимидазола (CDI) (1,4 г) и 1-(дифторметил)циклопропанкарбоновой кислоты (1 г) в MeCN (20 мл) добавляли при 0°С, и осуществляли перемешивание при 20°С в течение 14 часов. Реакционную смесь разбавляли с помощью Н2О (30 мл) и экстрагировали с помощью этилацетата (80 мл × 2). Объединенные органические экстракты промывали солевым раствором (30 мл), высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством флэш-хроматографии (SiO2, элюент характеризовался градиентом, предусматривающим 0-10% этилацетат/петролейный эфир) с получением продукта (0,98 г).
Следующие соединения получали посредством такой же методики, как описанная для IVa, с применением соответствующих исходных материалов.
IVb: этил-3-оксо-3-[1-(трифторметил)циклопропил]пропаноат
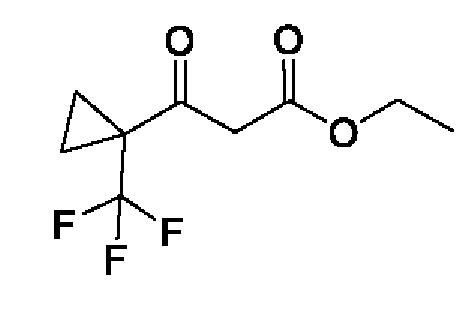
IVc: 3-(3,3-дифторциклобутил)-3-оксопропаноат
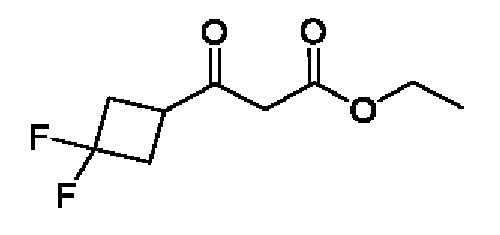
IVd: этил-3-(3,3-диметилциклобутил)-3-оксопропаноат
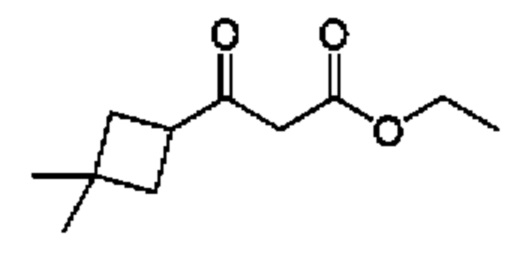
1Н ЯМР (CDCl3 400 МГц): δ 4,22-4,16 (m, 1Н), 3,39 (s, 2H), 3,34-3,25 (m, 1H), 2,08-1,90 (m, 4H), 1,29 (t, 3H), 1,27 (s, 3H), 1,06 (s, 3H).
IVe: этил-3-циклопешил-3-оксопропаноат
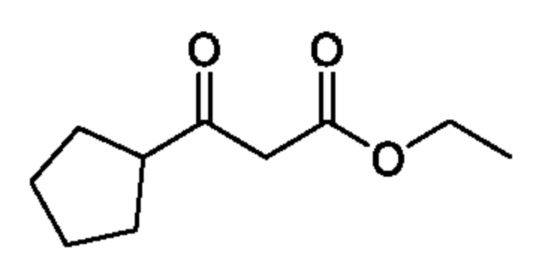
1H ЯМР (CDCl3 400 МГц): δ 4,24-4,18 (m, 2H), 3,49 (s, 2H), 3,03-2,95 (m, 1H), 1,84-1,60 (m, 8H), 1,28 (t, 3H).
IVf: этил-3-(1-этилциклопропил)-3-оксопропаноат
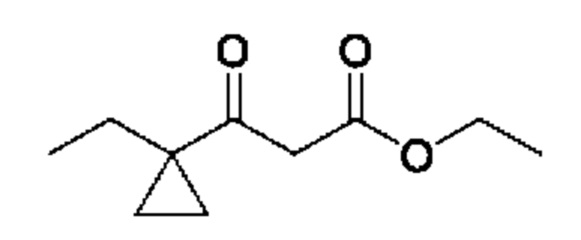
1H ЯМР (CDCl3 400 МГц): δ 4,22-4,16 (m, 2Н), 3,33 (s, 2Н), 1,64-1,60 (m, 2Н), 1,26-1,20 (m, 6Н), 0,94 (t, 3Н).
Va: (R)-Ν-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-4,4-диметил-3-оксопентанамид
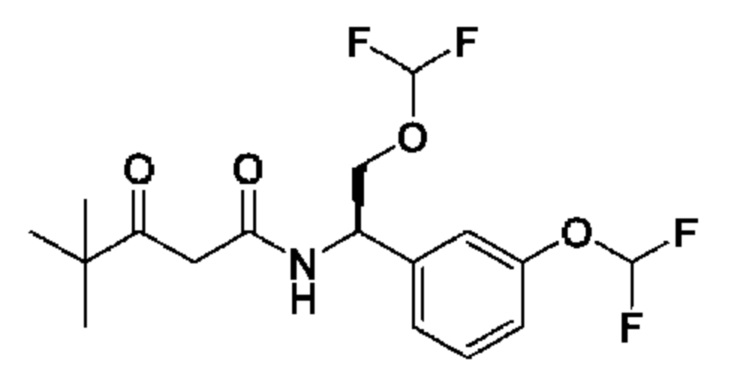
Стадия 1. Получение (R)-Ν-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-4,4-диметил-3-оксопентанамида
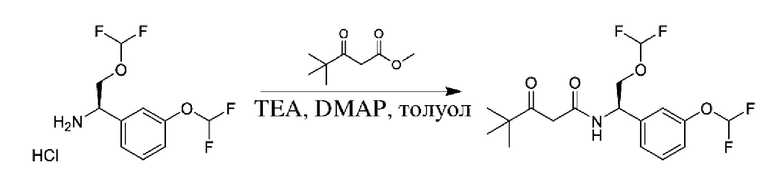
Раствор гидрохлорида (R)-2-(дифторметокси)-1-(3-(дифторметокси) фенил) этан-1-амина (IIa) (0,6 г), метил-4,4-диметил-3-оксопентаноата (750 мг), TEA (2,40 г) и DMAP (58 мг) в толуоле (10 мл) перемешивали при 90°С в течение 16 часов. Смесь разбавляли с помощью EtOAc (50 мл), промывали водой (30 мл) и солевым раствором (50 мл), высушивали над Na2SO4 и концентрировали. Неочищенный продукт очищали посредством хроматографии (SiO2, 30% ЕА в петролейном эфире) с получением (R)-N-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-4,4-диметил-3-оксопентанамида (0,28 г).
Следующее промежуточное соединение получали аналогично методике для Va с применением соответствующих промежуточных соединений.
Vb: (R)-Ν-(2-зтокси-1-(3-(трифторметокси)фенил)этил)-4,4-диметил-3-оксопентанамид
Получен из IIk и 4,4-диметил-3-оксопентановой кислоты.
Vc: (R)-Ν-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-оксо-3-(1-(трифторметил)циклопропил)пропанамид
Получен из IIa и IVb.
Vd: (R)-Ν-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-оксо-3-(1-трифторметил)циклопропил)пропанамид
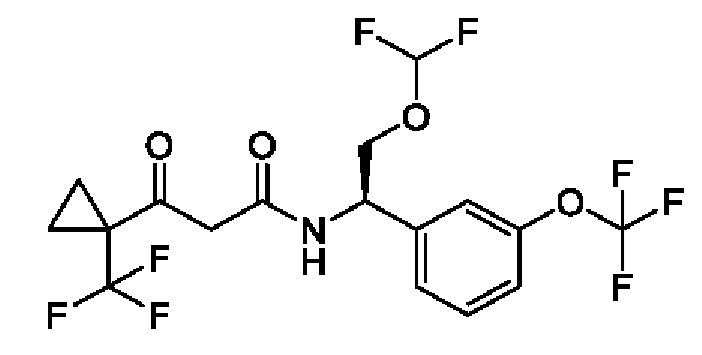
Получен из IVb и IIb.
Ve: (R)-3-(3,3-дифторциклобутил)-Ν-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)зтил)-3-оксопропанамид
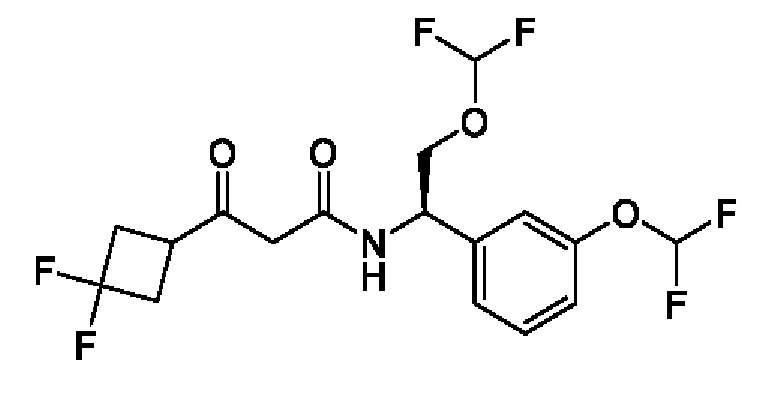
Получен из IVc и IIa.
Vf: (R)-3-(3,3-дифторциклобутил)-N-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-оксопропанамид
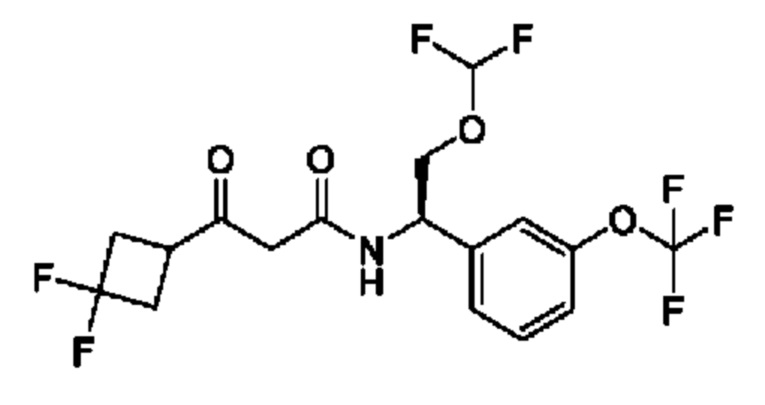
Получен из IVc и IIb.
Vg: (S)-3-(3,3-дифторциклобутил)-Ν-(1-(3-(дифторметокси)фенил)бутил)-3-оксопропанамид
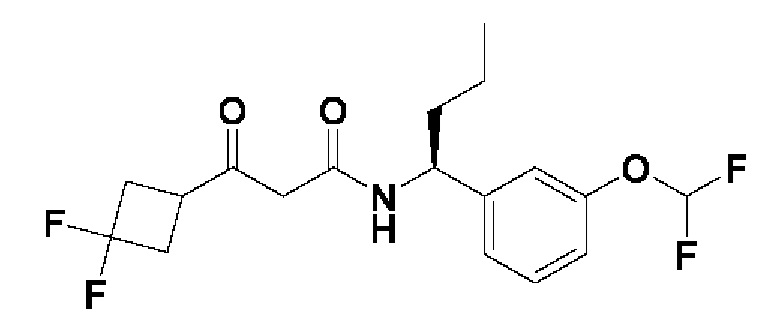
Получен из IVc и IIg.
Vh: (R)-Ν-(2-(дифτорметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-оксопропанамид
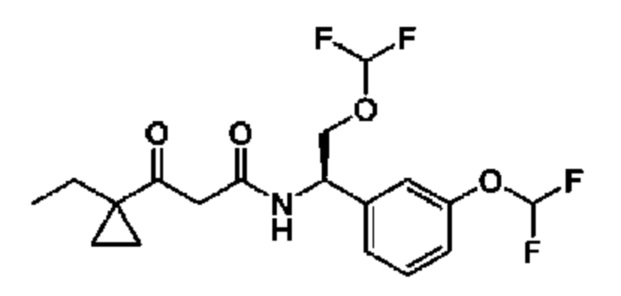
Получен из IVf и IIa.
Примеры
Пример 1a. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид
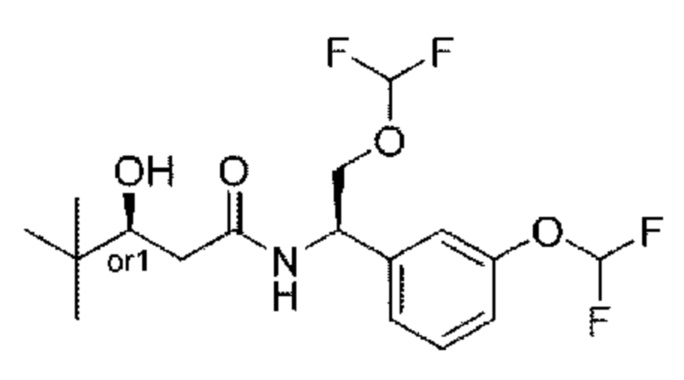
и Пример 1b. Ν-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид
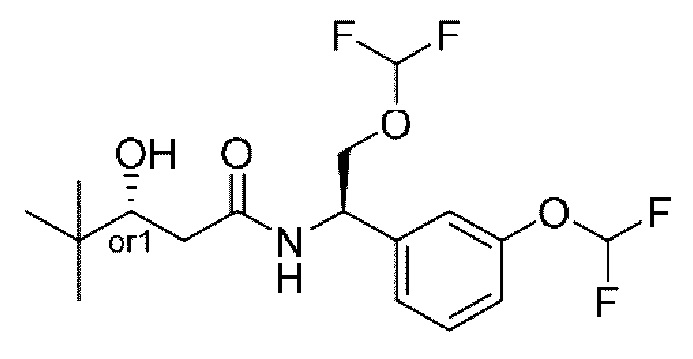
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида
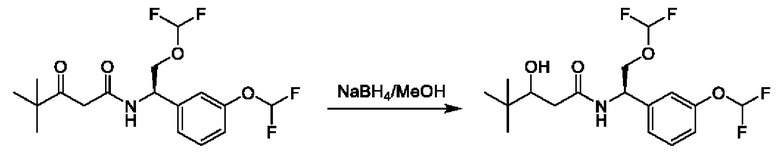
К раствору (R)-N-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-4,4-диметил-3-оксопентанамида (Va) (0,28 г) в МеОН (10 мл) добавляли NaBH4 (56 мг) при 0°С. Полученную смесь перемешивали при 0°С в течение 1 часа. Смесь концентрировали и остаток повторно растворяли в EtOAc (50 мл) и промывали с помощью воды (50 мл) и солевого раствора (50 мл), высушивали над Na2SO4 и концентрировали.
Стадия 2. Разделение (R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида и (S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида
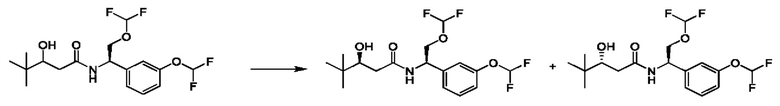
N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид отделяли посредством хроматографии.
Пример 1а.
1Н ЯМР (CDCl3 400 МГц): δ 7,34 (t, 1Н), 7,16 (d, 1H), 7,09 (s, 1H), 7,04 (d, 1H), 6,59 (d, 1H), 6,50 (t, 1H), 6,20 (t, 1H), 5,30-5,26 (m, 1H), 4,15-4,08 (m, 2H), 3,69-3,66 (m, 1H), 2,90 (d, 1H), 2,43 (dd, 1H), 2,28 (dd, 1H), 0, 91 (s, 9H).
LC-MS: tR=2,49 минуты (LC-MS Способ 1), масса/заряд = 382,2 [Μ+H]+.
SFC: tR=1,94 минуты (SFC Способ 1), ee% = 95,26%.
Пример 1b.
1H ЯМР (CDCl3 400 МГц): δ 7,35 (t, 1H), 7,16 (d, 1H), 7,06-7,04 (m, 2H), 6,60 (d, 1H), 6,50 (t, 1H), 6,20 (t, 1H), 5,28 (m, 1H), 4,10 (m, 2H), 3,67 (m, 1H), 3,04 (d, 1H), 2,45 (dd, 1H), 2,28 (dd, 1H), 0,91 (s, 9H).
LC-MS: tR=2,50 минуты (LC-MS Способ 1), масса/заряд = 382,2 [Μ+H]+.
SFC: tR=2,03 минуты (SFC Способ 1), ee% = 95,26%.
Следующие примеры получали аналогично методике, описанной для 1а и 1b, с применением соответствующих промежуточных соединений.
Пример 2а. N-((R)-2-Этокси-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид
и Пример 2b. N-((R)-2-Этокси-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид
Стадия 1. Получение Ν-((R)-2-этокси-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида
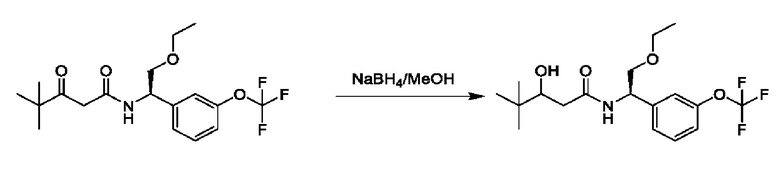
Получен из Vb.
Стадия 2. Разделение (S)-3-гидрокси-4,4-диметил-N-((S)-1-(3-(2,2,2-трифторэтокси)фенил)этил)пентанамида и (R)-3-гидрокси-4,4-диметил-N-((S)-1-(3-(2,2,2-трифторэтокси)фенил)этил)пентанамида
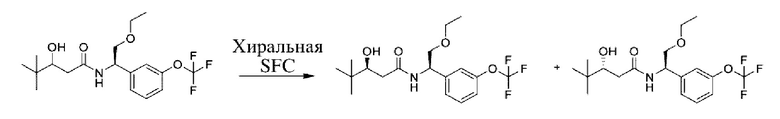
N-((R)-2-Этокси-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид отделяли посредством хроматографии.
Пример 2а.
1Н ЯМР (DMSO-d6 300 МГц): δ 8,34 (d, 1Н), 7,48-7,35 (m, 3Н), 7,23 (d, 1H), 5,07 (d, 1H), 4,61 (d, 1H), 3,34-3,54 (m, 5H), 2,31-2,20 (m, 1Η), 2,17-2,08 (m, 1H), 1,08 (t, 2H), 0,83 (s, 9H).
LC-MS: tR=1,87 минуты (LC-MS Способ 4), масса/заряд = 378,2 [Μ+H]+.
SFC: tR=1,71 минуты (SFC Способ 18), ee% = 96,0%.
Пример 2b.
1Н ЯМР (DMSO-d6 300 МГц): δ 8,39 (d, 1H), 7,46 (t, J=7,8 Гц, 5H), 7,35 (t, 2H), 7,23 (d, 3H), 5,06 (d, 1H), 4,63 (d, 1H), 3,56-3,52 (m, 3H), 3,49-3,43 (m, 2H), 2,29-2,25 (m, 1H), 2,19-2,08 (m, 1H), 1,07 (t, , 2H), 0,82 (s, 9H).
LC-MS: tR=1,87 минуты (LC-MS Способ 4), масса/заряд = 378,2 [Μ+H]+.
SFC: tR=1,82 минуты (SFC Способ 18), ee% = 99,1%.
Пример 3а. Ν-((R) -2-(Дифторметокси)-1-(3-(дифторметокси)фенил)зтил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамид
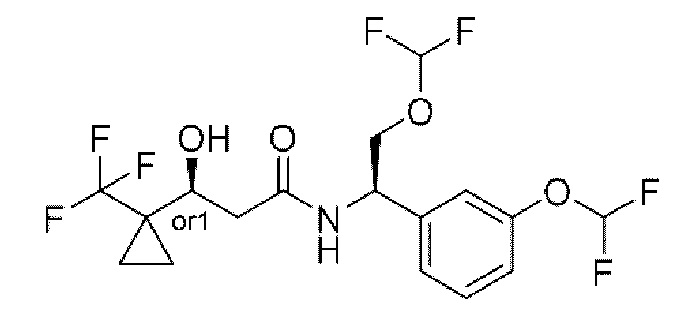
и
Пример 3b. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамид
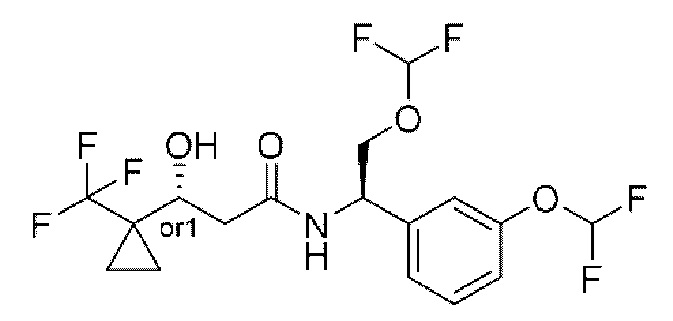
Стадия 1. Получение Ν-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида
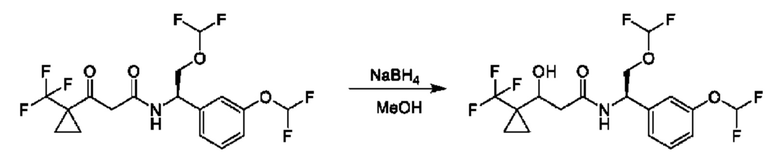
Получен из Vc.
Стадия 2. Разделение (R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида и (S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида
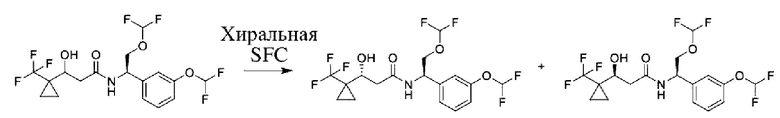
N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамид отделяли посредством хиральной SFC.
Пример 3а.
1Н ЯМР (CDCl3 400 МГц): δ 7,35 (t, 1Н), 7,16 (d, 1Η), 7,07-7,05 (m, 2H), 6,45 (t, 1Η), 6,33 (s, 1H), 6,22 (t, 1H), 5,28-5,24 (m, 1H), 4,17-4,04 (m, 3H), 3,68 (s, 1H), 2,67 (dd, 1H), 2,55 (dd, 1H), 0,92-0,88 (m, 3H), 0,85-0, 82 (m, 1H).
LC-MS: tR=2,51 минуты (LCMS Способ 1), масса/заряд = 434,1 [Μ+Η]+.
SFC: tR=1,95 минуты (SFC Способ 3), ee% = 100%.
Пример 3b.
1Н ЯМР (CDCl3 400 МГц): δ 7,36 (t, 1H), 7,16 (d, 1H), 7,06 (m, 2H), 6,50 (t, 1H), 6,38 (d, 1H), 6,21 (t, 1H), 5,27-5,23 (m, 1H), 4,13-4,04 (m, 3H), 3,76 (d, 1H), 2,67-2,56 (m, 2H), 0,98-0,87 (m, 4H).
LC-MS: tR=2,51 минуты (LCMS Способ 1), масса/заряд = 434,1 [Μ+Η]+.
SFC: tR=1,58 минуты (SFC Способ 3), ee% = 90,0%.
Пример 4a. Ν-((R)-2-(Дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамид
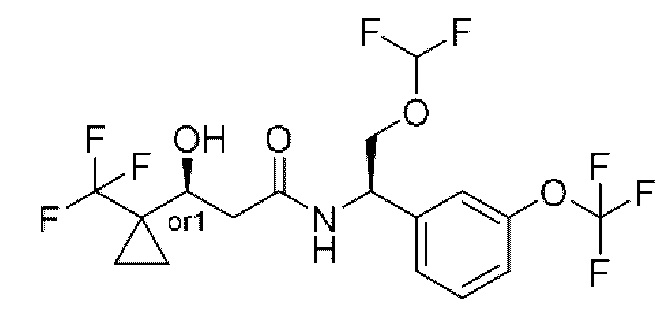
и
Пример 4b. N- ((R)-2-(Дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамид
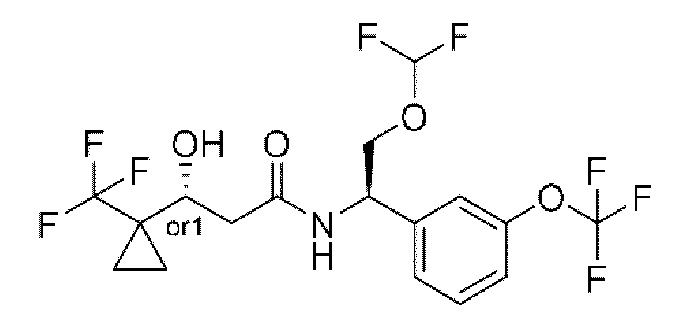
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида
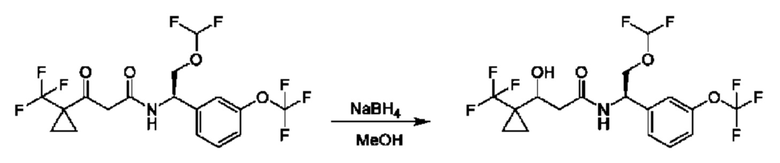
Получен из Vd.
Стадия 2. Разделение (R)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида и (S)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида
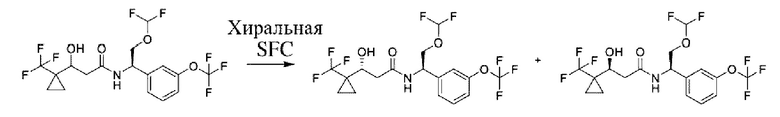
N-((R)-2-(Дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамид отделяли посредством хиральной SFC.
Пример 4а.
1Н ЯМР (CDCl3 400 МГц): δ 7,41 (t, 1Н), 7,27 (s, 1Η), 7,18 (m, 2H), 6,41 (s, 1Η), 6,23 (t, 1H), 5,30 (s, 1H), 4,16-4,06 (m, 3H), 3,74 (s, 1H), 2,69-2,56 (m, 2H), 1,00-0,90 (m, 4H).
LC-MS: tR=2,53 минуты (LCMS Способ 2), масса/заряд = 452,1 [Μ+H]+.
SFC: tR=1,21 минуты (SFC Способ 7), ee% = 100%.
Пример 4b.
1H ЯМР (CDCl3 400 МГц): δ 7,40 (t, 1Н), 7,27 (s, 1H), 7,18 (m, 2H), 6,42 (s, 1Η), 6,24 (t, 1H), 5,30 (s, 1H), 4,19-4,05 (m, 3H), 3,69 (s, 1H), 2,71-2,55 (m, 2H), 0,93-0,83 (m, 4H).
LC-MS: tR=2,63 минуты (LCMS Способ 1), масса/заряд = 452,1 [Μ+H]+.
SFC: tR=1,57 минуты (SFC Способ 7), ee% = 99,8%.
Пример 5a. 3-(3,3-Дифторциклобутил)-Ν-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамид
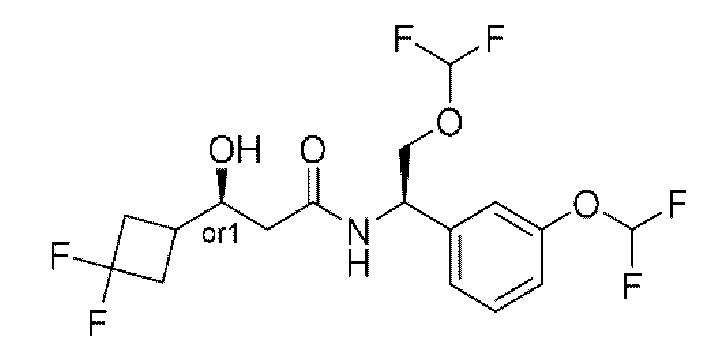
и
Пример 5b. 3-(3,3-Дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамид
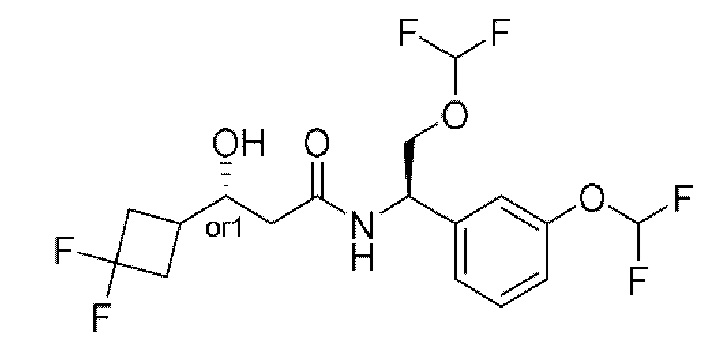
Стадия 1. Получение (R)-3-(3,3-дифторциклобутил)-N-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-оксопропанамида
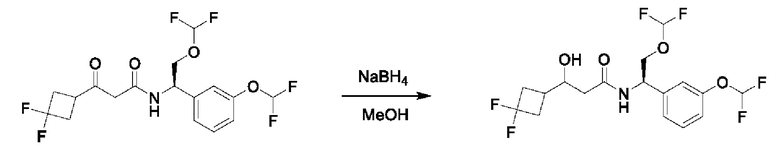
Получен из Ve.
Стадия 2. Разделение (R)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида и (S)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида
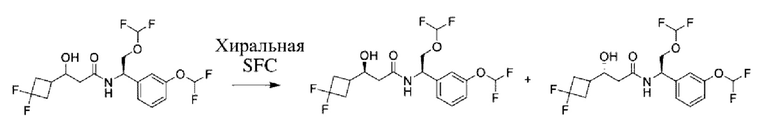
(R)-3-(3,3-Дифторциклобутил)-N-(2-(трифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-оксопропанамид отделяли посредством хиральной SFC.
Пример 5а.
1H ЯМР (CDCl3 400 МГц): δ 7, 37 (t, 1Н), 7,17 (d, 1Н), 7,09-7,07 (m, 2Н), 6,51 (t, 1Н), 6,32 (m, 1H), 6,13 (t, 1H), 5,30-5,25 (m, 1H), 4,18-4,14 (m, 1H), 4,10-4,06 (m, 1H), 4,00-3,96 (m, 1H), 3,64 (d, 1H), 2,59-2,55 (m, 3H), 2,45-2,35 (m, 2H), 2, 34-2, 26 (m, 1H), 2,21-2,08 (m, 1H).
LC-MS:tR=2,74 минуты (LCMS Способ 1), масса/заряд = 416,1 [M+H]+.
SFC:tR=2,49 минуты (SFC Способ 4), ee%=97,7%.
Пример 5b.
1H ЯМР (CDCl3 400 МГц): δ 7, 37 (t, 1Н), 7,17 (d, 1H), 7,08-7,07 (m, 2H), 6,51 (t, 1H), 6,32 (m, 1H), 6,23 (t, 1H), 5,30-5,26 (m, 1H), 4,18-4,15 (m, 1H), 4,11-4,07 (m, 1H), 4,02-3,98 (m, 1H), 2,64-2,51 (m, 3H), 2,45-2,36 (m, 2Н), 2,33-2,25 (m, 1Н), 2,21-2,16 (m, 1H).
LC-MS:tR=2,73 минуты (LCMS Способ 1), масса/заряд = 416,1 [M+H]+.
SFC:tR=2,59 минуты (SFC Способ 4), ee%=96,1%.
Пример 6a. 3-(3,3-Дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксипропанамид
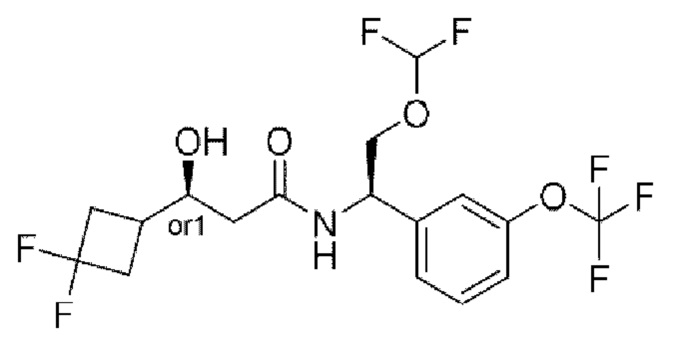
и
Пример 6b. 3-(3,3-Дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксипропанамид
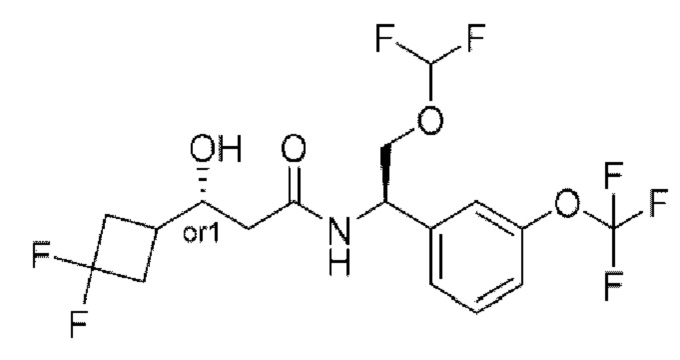
Стадия 1. Получение (R)-3-(3,3-дифторциклобутил)-N-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-оксопропанамида
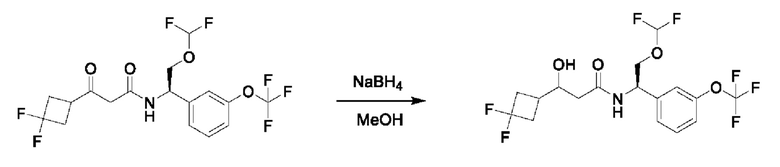
Получен из Vf.
Стадия 2. Разделение (R)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксипропанамида и (S)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксипропанамида
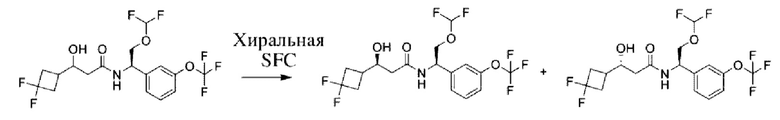
(R)-3-(3,3-Дифторциклобутил)-N-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-оксопропанамид отделяли посредством хиральной SFC.
Пример 6а.
1Н ЯМР (CDCl3 400 МГц): δ 7,41 (t, 1Н), 7,26 (d, 1Н), 7,19 (d, 2Н), 6,38 (d, 1Н), 6,24 (t, 1Н), 5,33-5,28 (m, 1Н), 4,20-4,16 (m, 1Н), 4,13-4,10 (m, 1Н), 4,01-4,00 (s, 1Н), 3,56 (d, 2Н), 2,61-2,54 (m, 3Н), 2, 44-2, 39 (m, 2Н), 2,39-2, 32 (m, 1Н), 2, 18 (m, 1Н).
LC-MS:tR=2,53 минуты (LCMS Способ 1), масса/заряд = 434,0 [М+Н]+.
SFC:tR=1,62 минуты (SFC Способ 5), ее%=92,9%.
Пример 6b.
1Н ЯМР (CDCl3 400 МГц): δ 7,42 (t, 1Н), 7,26 (d, 1Н), 7,20-7,18 (m, 2H), 6,34 (d, 1H), 6,24 (t, 1H), 5,33-5,28 (m, 1H), 4,20-4,16 (m, 1H), 4,13-4,00 (m, 1H), 4,00 (m, 1H), 3,61 (s, 1H), 2,61-2,57 (m, 3H), 2,43-2,39 (m, 2H), 2,39-2,32 (m, 1H), 2,20 (m, 1H).
LC-MS: tR=2,54 минуты (LCMS Способ 1), масса/заряд = 434,0 [М+Н]+.
SFC:tR=1,71 минуты (SFC Способ 5), ее%=97,9%.
Пример 7а. 3-(3,3-Дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамид
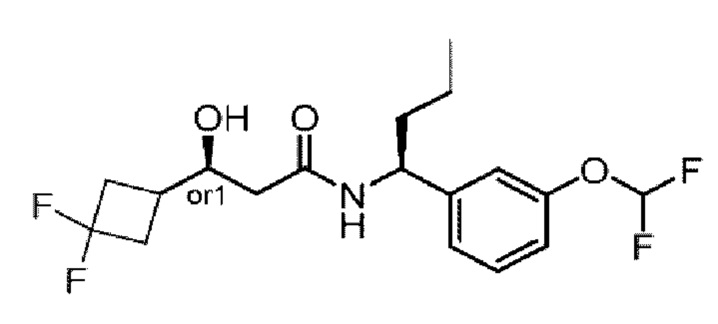
и
Пример 7b. 3-(3,3-Дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамид
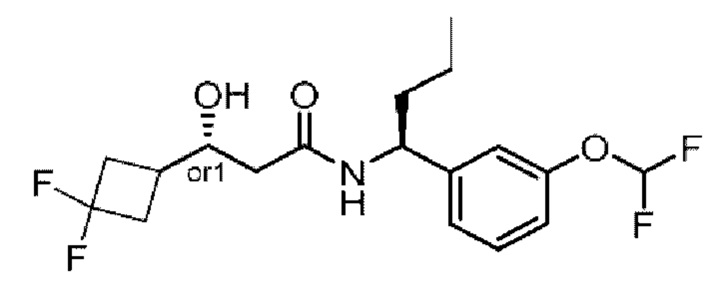
Стадия 1. Получение 3-(3,3-дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамида
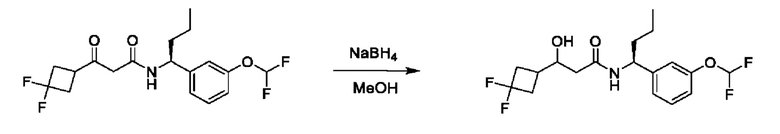
Получен из Vg.
Стадия 2. Разделение (S)-3-(3,3-дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамида и (R)-3-(3,3-дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамида
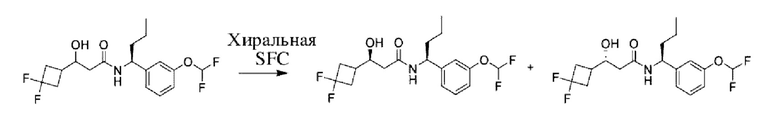
3-(3,3-Дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамид отделяли посредством хиральной SFC.
Пример 7а.
1Н ЯМР (CDCl3 400 МГц): δ 7,32 (t, 1Н), 7,11 (d, 1Н), 7,01-7,00 (m, 2H), 6,49 (t, 1H), 5,85 (d, 1H), 4,93 (q, 1H), 3,94-3,91 (m, 1H), 3,81 (d, 1H), 2,56-2,21 (m, 7H), 1,74-1,69 (m, 2H), 1,33-1,28 (m, 2H), 0,91 (t, 3H).
LC-MS:tR=2,41 минуты (LC-MS Способ 1), масса/заряд = 378,0 [M+Н]+.
SFC:tR=2,37 минуты (SFC способ 6), ее%=92,4%
Пример 7b.
1H ЯМР (CDCl3 400 МГц): δ 7,31 (t, 1Н), 7,10 (d, 1Н), 7,01-7,00 (m, 2H), 6,49 (t, 1H), 5,86 (d, 1H), 4,93 (q, 1H), 3,96-3,92 (m, 1H), 3,77 (d, 1H), 2,54-2,14 (m, 7H), 1,74-1,70 (m, 2H), 1,33-1,28 (m, 2H), 0,91(t, 3H).
LC-MS:tR=2,45 минуты (LC-MS Способ 1), масса/заряд = 378,0 [M+H]+.
SFC:tR=2,49 минуты (SFC Способ 6), ee%=99,5%
Пример 8a. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-зтилциклопропил)-3-гидроксипропанамид
и
Пример 8b. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)зтил)-3-(1-зтилциклопропил)-3-гидроксипропанамид
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-гидроксипропанамида
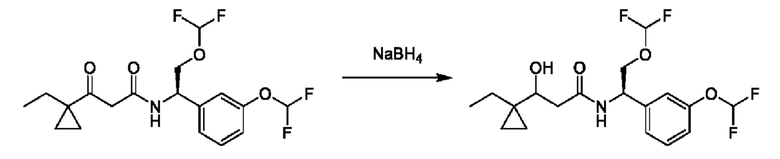
Получен из Vh.
Стадия 2. Разделение (S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-гидроксипропанамида и (R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-гидроксипропанамида
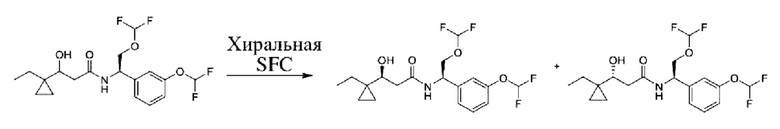
N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-гидроксипропанамид отделяли посредством хиральной SFC
Пример 8а.
1H ЯМР (CDCl3 400 МГц): δ 7,28 (t, 1Н), 7,11 (d, 1Н), 7,03 (s, 1Н), 6,98 (d, 1Н), 6,66 (d, 1Н), 6,44 (t, 1Н), 6,14 (t, 1Н), 5,24-5,19 (m, 1Н), 4,10-4,02 (m, 2Н), 3,47-3,44 (m, 1Н), 2,57 (d, 1Н), 2,50-2,41 (m, 2Н), 1, 53-1, 48 (m, 2Н), 1,34-1,31 (m, 1Н), 0,83-0,80 (m, 3Н), 0,38-0,29 (m, 4Н).
LC-MS:tR=2,38 минуты (LCMS Способ 1), масса/заряд = 394,0 [М+Н]+.
SFC:tR=2,38 минуты (SFC Способ 1), ее%=99,4%
Пример 8b.
1Н ЯМР (CDCl3 400 МГц): δ 7, 29 (t, 1Н), 7,11 (d, 1Н), 7,01-6,99 (m, 2Н), 6,67 (d, 1Н), 6,44 (t, 1Н), 6,15 (t, 1Н), 5,24-5,19 (m, 1Н), 4,10-4,00 (m, 2Н), 3, 49-3, 47 (d, 1Н), 2,64 (d, 1Н), 2,49-2,41 (m, 2Н), 1, 53-1, 48 (m, 2Н), 1,34-1,31 (m, 1Н), 0,83-0,80 (m, 3Н), 0,39-0,29 (m, 4Н).
LC-MS:tR=2,38 минуты (LCMS Способ 1), масса/заряд = 394, 0 [М+Н]+.
SFC: t=2,60 минуты (SFC Способ 1), ее%=98,7%
Пример 9а. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)зтил)-3-(1-фторциклопропил)-3-гидроксибутанамид
и
Пример 9b. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)зтил)-3-(1-фторциклопропил)-3-гидроксибутанамид
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида
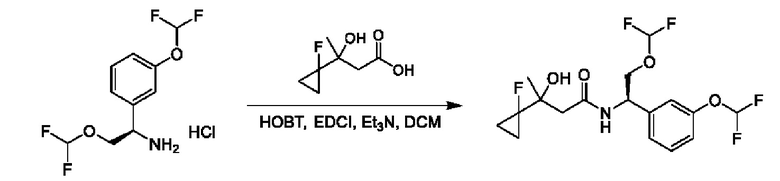
К раствору гидрохлорида (R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этан-1-амина (IIa) (400 мг) и 3-(1-фторциклопропил)-3-гидрокси-бутановой кислоты (IIIc) (307 мг) в DCM (20 мл) добавляли N-гидроксибензотриазол (HOBt) (213 мг), 1-этил-3-(3-диметиламинопропил)карбодиимид (EDCI) (363 мг) и Et3N (320 мг). Смесь перемешивали при 25°С в течение 16 часов и затем разбавляли с помощью воды (10 мл), и экстрагировали с помощью EtOAc (20 мл × 3). Объединенные органические экстракты высушивали над Na2SO4, фильтровали и концентрировали. Остаток очищали посредством основной препаративной HPLC с получением N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида (250 мг).
Стадия 2. Разделение (R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида и (S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида
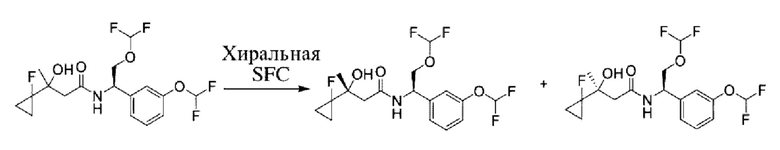
N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамид отделяли посредством хиральной SFC.
Пример 9а.
1Н ЯМР (CDCl3 400 МГц): δ 7,38 (t, 1Н), 7,20 (d, 1Н), 7,10 (m, 2Н), 6,52 (t, 1Н), 6,39 (d, 1Н), 6,25 (t, 1Н), 5,31 (m, 1Н), 4,80 (s, 1Н), 4,15 (m, 2Н), 2,71 (dd, 1Н), 2,54 (dd, 1Н), 1,36 (s, 3Н), 0,85-0,55 (m, 4Н).
LC-MS: tR=2,48 минуты (LCMS Способ 1), масса/заряд = 398,2 [М+Н]+.
SFC:tR=2,46 минуты (SFC Способ 12), ее%=100%.
Пример 9b.
1H ЯМР (CDCl3 400 МГц): δ 7,39 (t, 1Н), 7,19 (d, 1Н), 7,10-7,08 (m, 2Н), 6,52 (t, 1Н), 6,42 (m, 1Н), 6,24 (t, 1Н), 5,33-5,28 (m, 1Н), 4,71 (s, 1Н), 4,19-4,10 (m, 2Н), 2,72 (d, 1Н), 2,52 (d, 1Н), 1,36 (s, 3Н), 0,99-0,85 (m, 4Н).
LC-MS:tR=2,47 минуты (LCMS Способ 1), масса/заряд = 398,1 [M+Н]+.
SFC:tR=2,65 минуты (SFC Способ 12), ее%=98,8%.
Следующие примеры получали аналогично методике, описанной для примера 9а и примера 9b, с применением соответствующих промежуточных соединений.
Пример 10а. N-((R)-2-(Дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамид
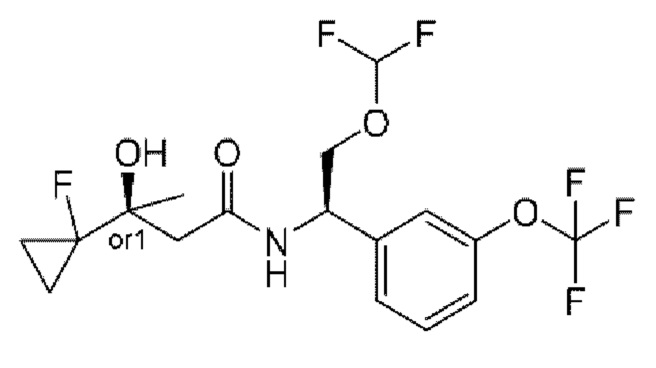
и
Пример 10b. N-((R)-2-(Дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамид
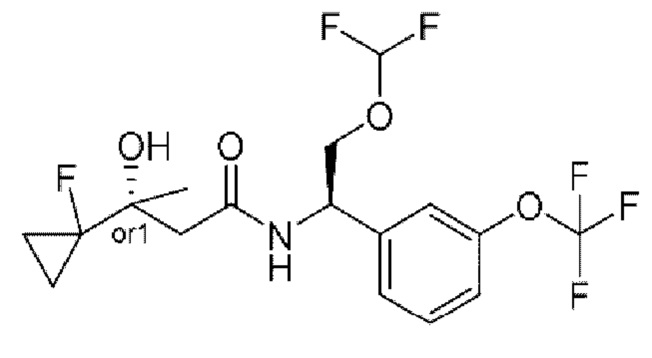
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида
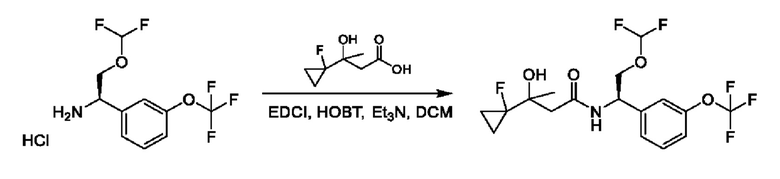
Получен из IIb и IIIc.
Стадия 2. Разделение (S)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида и (R)-N-((R) -2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида
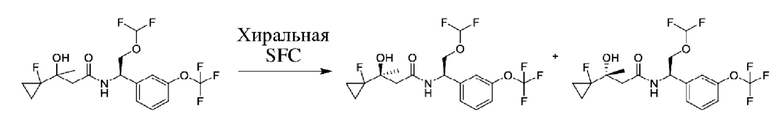
N-((R)-2-(Дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамид отделяли с применением хиральной SFC.
Пример 10а.
1Н ЯМР (CDCl3 400 МГц): δ 7,48 (t, 1Н), 7,35 (d, 1Н), 7,27-7,25 (m, 2Н), 6,64 (d, 1Н), 6,31 (t, 1Н), 5,40-5,38 (m, 1Н), 4,86 (s, 1Н), 4,26-4,18 (m, 2Н), 2,78 (d, 1Н), 2,60 (d, 1Н), 1,42 (s, 3Н), 0, 86-0, 57 (m, 4Н).
LC-MS:tR=2,64 минуты (LCMS Способ 1), масса/заряд = 416,2 [М+Н]+.
SFC:tR=2,38 минуты (SFC Способ 2), ее%=100%.
Пример 10b.
1Н ЯМР (CDCl3 400 МГц): δ 7,42 (t, 1Н), 7,29 (m, 1H), 7,20-7,19 (m, 2H), 6,44-6,06 (m, 2H), 5,35-5,31 (m, 1H), 4,67 (s, 1H), 4,20-4,12 (m, 2H), 2,73 (dd, 1H), 2,53 (dd, 1H), 1,36 (s, 3H), 0, 99-0, 86 (m, 4H).
LC-MS:tR=2,659 минуты (LCMS Способ 1), масса/заряд = 416,2 [M+H]+.
SFC:tR=2,561 минуты (SFC Способ 6), ee%=95,9%.
Пример 11a. 3-Циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)зтил)-3-гидроксибутанамид
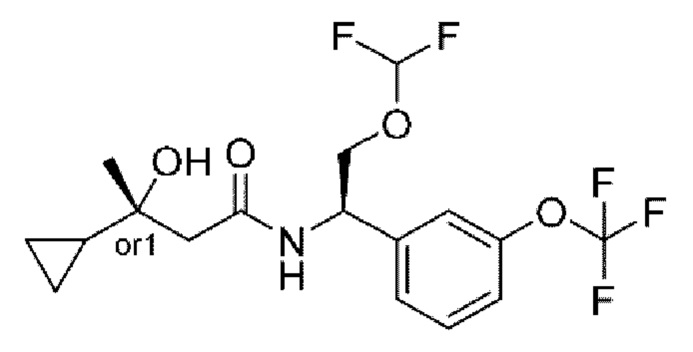
и
Пример 11b. 3-Циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамид
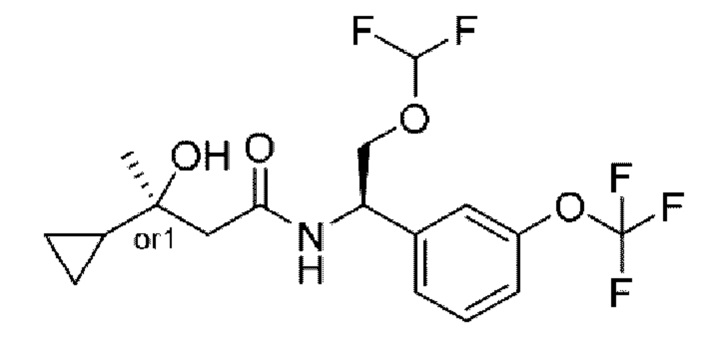
Стадия 1. Получение 3-циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамида
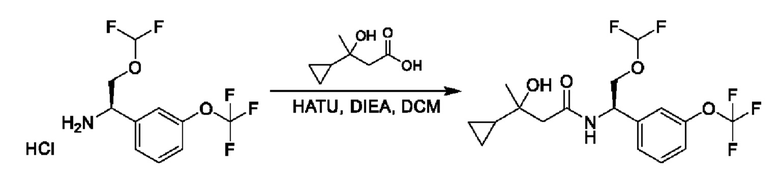
Получен из IIb и IIId.
Стадия 2. Разделение (R)-3-циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамида и (S)-3-циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамида
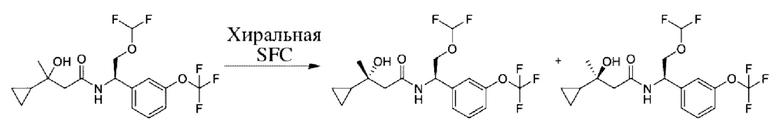
3-Циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамид отделяли посредством хиральной SFC.
Пример 11а.
1Н ЯМР (CDCl3 400 МГц): δ 7,38 (t, 1Н), 7,26 (d, 1Н), 7,17-7,15 (m, 2Н), 6,72 (d, 1Н), 6,21 (t, 1Н), 5,35-5,31 (m, 1Н), 4,17-4,09 (m, 2Н), 3,36 (s, 1Н), 2,52-2,42 (m, 2Н), 1,18 (s, 3Н), 0,90-0,88 (m, 1Н), 0,43-0,34 (m, 4Н).
LC-MS:tR=2,42 минуты (LCMS Способ 1), масса/заряд = 420, 1 [М+Na]+.
SFC:tR=2,17 минуты (SFC Способ 13), ее%=100%.
Пример 11b.
1Н ЯМР (CDCl3 400 МГц): δ 7,39 (t, 1Н), 7,29-7,20 (m, 1Н), 7,21-7,16 (m, 2Н), 6,74 (d, 1Н), 6,23 (t, 1Н), 5,36-5,32 (m, 1Н), 4,19-4,10 (m, 2Н), 3,40 (s, 1Н), 2,49 (s, 2Н), 1,18 (s, 3Н), 0,90-0,87 (m, 1Н), 0,41-0,27 (m, 4Н).
LC-MS:tR=2,95 минуты (LCMS Способ 1), масса/заряд = 420, 1 [М+Na]+.
SFC:tR=2,48 минуты (SFC Способ 13), ее%=100%.
Пример 12а. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамид
и
Пример 12b. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамид
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамида
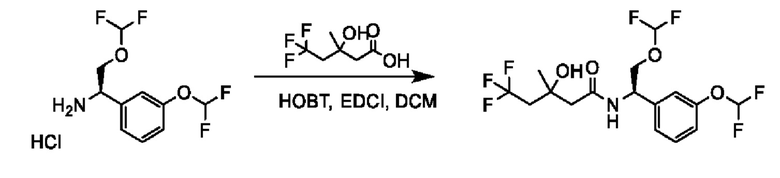
Получен из IIa и IIIe.
Стадия 2. Разделение (R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамида и (S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамида
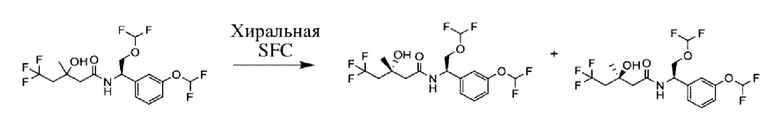
N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамид отделяли посредством хиральной SFC
Пример 12а.
1Н ЯМР (CDCl3 400 МГц): δ 7,38 (t, 1Н), 7,17 (d, 1Н), 7,10-7,08 (m, 2Н), 6,51 (t, 1Н), 6,37 (d, 1Н), 6,25 (t, 1Н), 5,32-5,27 (m, 1Н), 4,68 (s, 1Н), 4,18 (dd, 1Н), 2,52 (dd, 1H), 2,61-2,52 (m, 2H), 2,47-2,41 (m, 2H), 1, 40 (s, 3H).
LC-MS: tR=2,52 минуты (LCMS Способ 1), масса/заряд = 422,1 [M+H]+.
SFC:tR=1,10 минуты (SFC Способ 14), ee%=100%.
Пример 12b.
1H ЯМР (CDCl3 400 МГц): δ 7,39 (t, 1Н), 7,18 (d, 1H), 7,10-7,08 (m, 2H), 6,52 (t, 1H), 6,36 (d, 1H), 6,24 (t, 1H), 5,33-5,29 (m, 1H), 4,68 (s, 1H), 4,18 (dd, 1H), 2,52 (dd, 1H), 2,61-2,42 (m, 4H), 1,39 (s, 3H).
LC-MS:tR=2,52 минуты (LCMS Способ 1), масса/заряд = 422, 1 [M+H]+.
SFC:tR=1,24 минуты (SFC Способ 14), ee%=95,8%.
Пример 13a. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамид
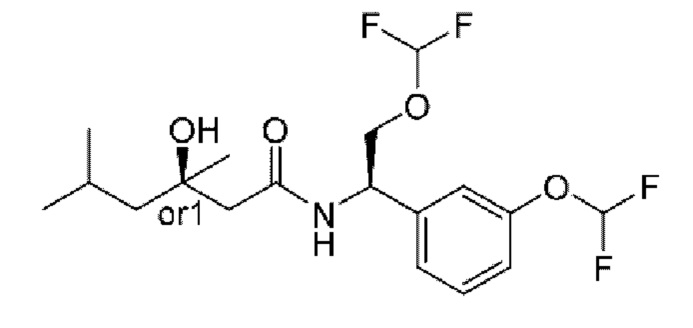
и
Пример 13b. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамид
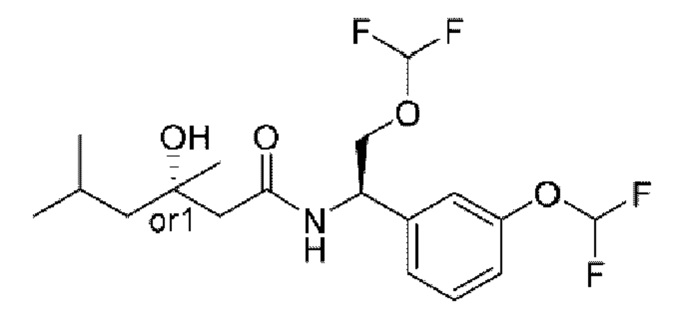
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамида
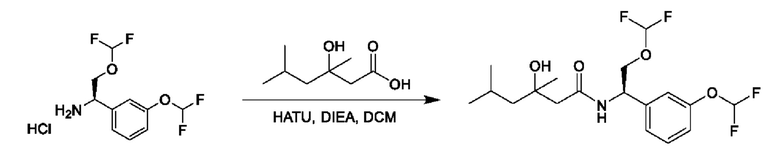
Получен из IIa и IIIg
Стадия 2. Разделение (R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамида и (S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамида
N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамид отделяли посредством хиральной SFC
Пример 13а.
1Н ЯМР (CDCl3 400 МГц): δ 7,37 (t, 1Н), 7,19 (d, 1Н), 7,10-7,07 (m, 2Н), 6,76 (d, 1Н), 6,51 (t, 1Н), 6,23 (t, 1Н), 5,35-5,30 (m, 1Н), 4,19-4,08 (m, 2Н), 3,39 (s, 1Н), 2,51-2,34 (m, 2Н), 1,84-1,76 (m, 1Н), 1,43 (d, 2Н), 1,27 (s, 3Н), 0, 98-0, 95 (m, 6Н).
LC-MS:tR=2,54 минуты (LCMS Способ 1), масса/заряд = 396,1 [М+Н]+.
SFC:tR=2,40 минуты (SFC Способ 15), ее%=99,3%.
Пример 13b.
1H ЯМР (CDCl3 400 МГц): δ 7, 37 (t, 1Н), 7,19 (d, 1Н), 7,10 (s, 1Н), 7,07 (d, 1Н), 6,73 (d, 1Н), 6,51 (t, 1Н), 6,23 (t, 1Н), 5,35-5,31 (m, 1Н), 4,19-4,09 (m, 2Н), 3,37 (s, 1Н), 2,50-2,34 (m, 2Н), 1,85-1,76 (m, 1Н), 1,45 (d, 2Н), 1,26 (s, 3Н), 1,00-0,94 (m, 6Н).
LC-MS:tR=2,54 минуты (LC-MS Способ 1), масса/заряд = 396,1 [М+Н]+.
SFC:tR=2,66 минуты (SFC Способ 15), ее%=98,8%.
Пример 14а. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)зтил)-3-гидрокси-3,4-диметилпентанамид
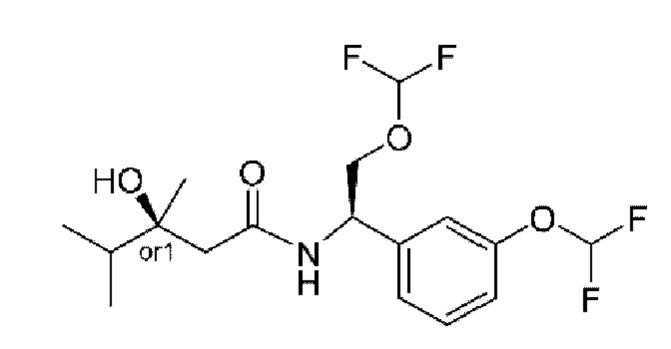
и
Пример 14b. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамид
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамида
Получен из IIa и IIIf
Стадия 2. Разделение (S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамида и (R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамида
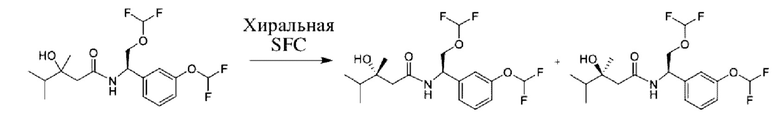
N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамид отделяли посредством хиральной SFC.
Пример 14а.
1Н ЯМР (CDCl3 400 МГц): δ 7,34 (t, 1Н), 7,16 (d, 1Н), 7.07 (s, 1Н), 7,04 (d, 1Н), 6,76 (d, 1Н), 6,48 (t, 1Н), 6,20 (t, 1Н), 5,32-5,27 (m, 1Н), 4,16-4,12 (m, 1Н), 4,09-4,04 (m, 1Н), 3,39 (s, 1Н), 2, 49-2, 28 (m, 2Н), 1,77-1,70 (m, 1Н), 1,15 (s, 3Н), 0,93-0,89 (m, 6Н).
LC-MS:tR=2,41 минуты (LCMS Способ 1), масса/заряд = 382,0 [М+Н]+.
SFC:tR=2,44 минуты (SFC Способ 15), ее%=100%.
Пример 14b.
1H ЯМР (CDCl3 400 МГц): δ 7,35 (t, 1Н), 7,16 (d, 1Н), 7.08 (s, 1H), 7,04 (d, 1H), 6,79 (d, 1H), 6,48 (t, 1H), 6,19 (t, 1H), 5,31-5,26 (m, 1H), 4,14-4,10 (m, 1H), 4,08-4,04 (m, 1H), 3,43 (s, 1H), 2,48-2,28 (m, 2H), 1,75-1,69 (m, 1H), 1,12 (s, 3H), 0,92-0,88 (m, 6H).
LC-MS:tR=2,41 минуты (LCMS Способ 1), масса/заряд = 382,0 [M+H]+.
SFC:tR=2,68 минуты (SFC Способ 15), ее%=97,4%.
Пример 15а. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамид
и
Пример 15b. N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамид
Стадия 1. Получение N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамида
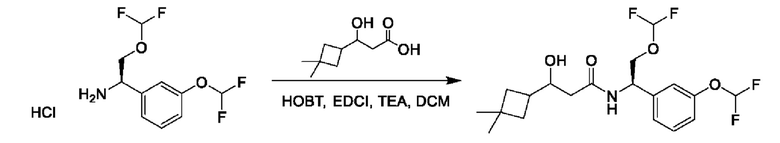
Получен из IIa и IIIh
Стадия 2. Разделение (S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамида и (R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамида
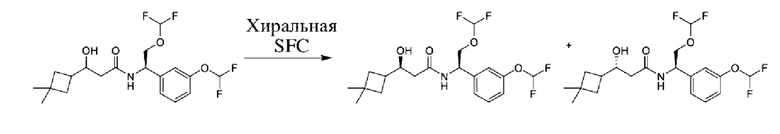
N-((R)-2-(Дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамид отделяли посредством хиральной SFC.
Пример 15а.
1Н ЯМР (DMSO-d6 400 МГц): δ 8,41 (d, 1Н), 7,40 (t, 1Н), 7,26 (d, 1Н), 7,21 (t, 1H), 7,20 (s, 1H), 7,09 (d, 1H), 6,67 (t, 1H), 5,17-5,12 (m, 1H), 4,61 (d, 1H), 3,98-3,97 (m, 2H), 3,70-3,67 (m, 1H), 2,16-2,11 (m, 3H), 1,64-1,52 (m, 4H), 1,09 (s, 3H), 0,99 (s, 3H).
LC-MS:tR=2,35 минуты (LC-MS Способ 3), масса/заряд = 408,1[M+H]+.
SFC:tR=2,32 минуты (SFC Способ 16), ee%=99,7
Пример 15b.
1H ЯМР (DMSO-d6 400 МГц): δ 8,43 (d, 1Н), 7,40 (t, 1H), 7,24 (d, 1H), 7,22 (t, 1H), 7,18 (s, 1H), 7,08 (d, 1H), 6,67 (t, 1H), 5,14-5,10 (m, 1H), 4,63 (d, 1H), 3,99-3,96 (m, 2H), 3,70-3,67 (m, 1H), 2,13-2,08 (m, 3H), 1,64-1,49 (m, 4H), 1,06 (s, 3H), 0,97 (s, 3H).
LC-MS:tR=2,34 минуты (LCMS Способ 3), масса/заряд = 408,1[M+H]+.
SFC:tR=2,64 минуты (SFC Способ 16), ee%=98,7%.
Пример 16a. 3-Циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамид
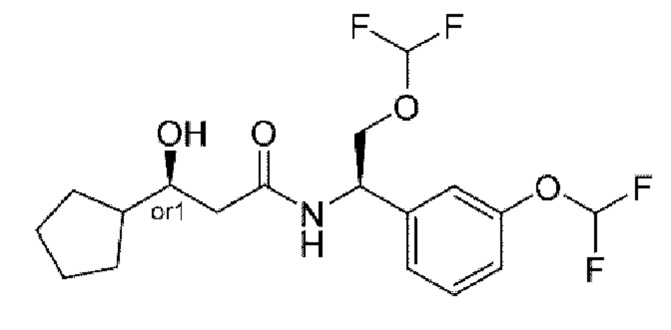
и
Пример 16b. 3-Циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамид
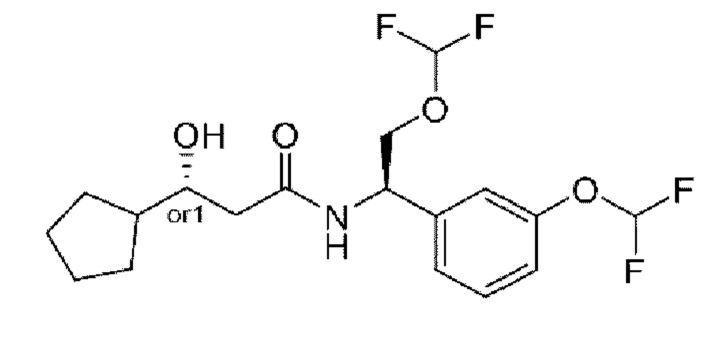
Стадия 1. Получение 3-циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида
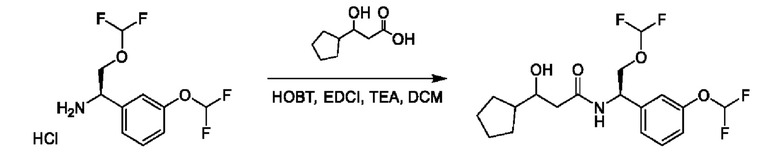
Получен из IIa и IIIi.
Стадия 2. Разделение (S)-3-циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида и (R)-3-циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида
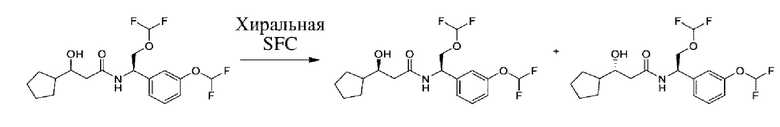
3-Циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамид отделяли посредством хиральной SFC.
Пример 16а.
1Н ЯМР (DMSO-d6 400 МГц) δ 8, 43 (d, 1Н), 7,39 (t, 1H), 7,27-7,25 (m, 1H), 7,20 (s, 1H), 7,20 (t, 1H), 7,08 (d, 1H), 6,66 (t, 1H), 5,16-5,14 (m, 1H), 4,61 (d, 1H), 3,99-3,97 (m, 2H), 3,69-3,65 (m, 1H), 2,26-2,25 (m, 2H), 1,80-1,76 (m, 1H), 1,64-1,22 (m, 8H).
LC-MS:tR=2,48 минуты (LC-MS Способ 1), масса/заряд = 394, 1 [M+H]+.
SFC:tR=2,64 минуты (SFC Способ 17), ee%=98,8%.
Пример 16b.
1H ЯМР (DMSO-d6 400 МГц): δ 8,41 (d, 1H), 7,36 (d, 1H), 7,20 (d, 1H), 7,18 (t, 1H), 7,15 (s, 1H), 7,04 (m, 1H), 6,63 (t, 1H), 5,13-5,07 (m, 1H), 4,59 (d, 1H), 3,98-3,91 (m, 2H), 3,66-3,62 (m, 1H), 2,23-2,19 (m, 2H), 1,73-1,69 (m, 1H), 1,60-1,33 (m, 8H).
LC-MS: tR=2,27 минуты (LC-MS Способ 2), масса/заряд = 394, 2 [M+H]+.
SFC:tR=3,08 минуты (SFC Способ 17), ee%=100%
Пример 36a. 3-(1-Фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамид
и
Пример 36b. 3-(1-Фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамид
Стадия 1. Получение 3-(1-фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамида 
Получен из IIp и IIIc.
Стадия 2. Разделение (R)-3-(1-фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамида и (S)-3-(1-фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамида
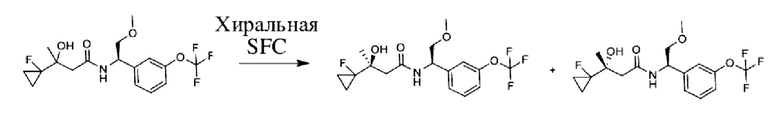
3-(1-Фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамид отделяли посредством хиральной SFC.
Пример 36а.
1Н ЯМР (CDCl3 400 МГц): δ 7,38 (t, 1Н), 7,29 (d, 1Н), 7,22 (s, 1Н), 7,16 (d, 1Н), 6,58 (d, 1Н), 5,18-5,13 (m, 1Н), 5,04(s, 1Н), 3, 70-3, 62 (m, 2Н), 3,38 (s, 3Н), 2,72-2,68 (m, 1H), 2,54-2,50 (m, 1H), 1,36 (s, 3H), 0,81-0,53 (m, 4H).
LC-MS:tR=2,43 минуты (LCMS Способ 1), масса/заряд = 380,0 [M+H]+.
SFC:tR=1,29 минуты (SFC Способ 21), ее%=99,6%.
Пример 36b.
1Н ЯМР (CDCl3 400 МГц): δ 7,38 (t, 1Н), 7, 28-7, 27 (m, 1Н), 7,20 (s, 1Н), 7,15 (d, 1Н), 6,58 (d, 1Н), 5,18-5,14 (m, 1Н), 4,93 (s, 1Н), 3,70-3,62 (m, 2Н), 3,38 (s, 3Н), 2,71 (dd, 1Н), 2,52 (dd, 1Н), 1,35 (s, 3Н), 1, 00-0, 86 (m, 4Н).
LC-MS:tR=2,53 минуты (LCMS Способ 1), масса/заряд = 380,0 [М+Н]+.
SFC:tR=1,76 минуты (SFC Способ 22), ее%=81,4%.
Пример 17. (S)-N-((R)-2-Циклопропокси-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид
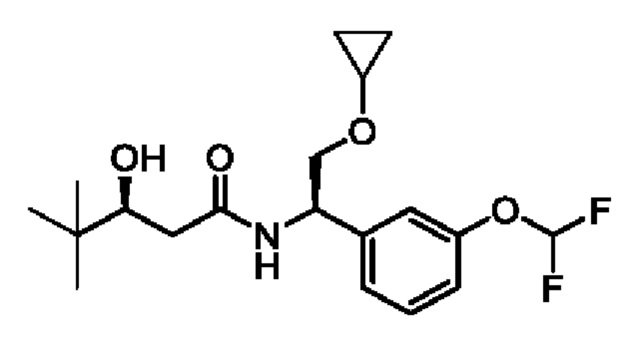
Стадия 1. Получение (S)-N-((R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида
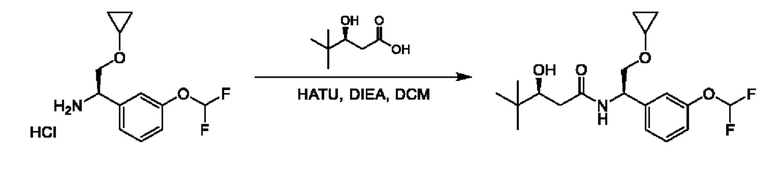
К раствору гидрохлорида (R)-2-циклопропокси-1-(3-(дифторметокси) фенил) этан-1-амина (IIc) (0,2 г), (3S)-3-гидрокси-4,4-диметил-пентановой кислоты (IIIa) (144 мг) и HATU (37 5 мг) в DCM (10 мл) добавляли DIEA (319 мг). Смесь перемешивали при 20°С в течение 16 часов и концентрировали. Неочищенный продукт очищали с получением (S)-N-((R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4 -диметилпентанамида.
1Н ЯМР (CDCl3 400 МГц): δ 7,33 (t, 1Н), 7,17 (d, 1Н), 7,08 (s, 1Н), 7,03 (d, 1Н), 6,49 (m, 1Н), 6,51 (t, 1Н), 5,18-5,14 (m, 1Н), 3, 80-3, 78 (m, 1Н), 3, 72-3, 66 (m, 2Н), 3,42 (d, 1Н), 3,35-3,25 (m, 1Н), 2, 44-2, 26 (m, 2Н), 0,93 (s, 9Н), 0,57-0,45 (m, 4Н).
LC-MS:tR=2,40 минуты (LCMS Способ 1), масса/заряд = 372,1 [М+Н]+.
SFC:tR=1,988 минуты (SFC Способ 7), ее%=97,5%.
Следующие примеры получали аналогично методике, описанной для примера 17, с применением соответствующих промежуточных соединений.
Пример 18. (S)-N-((R)-1-(3-(Дифторметокси)фенил)-2-(трифторметокси)этил)-3-гидрокси-4,4-диметилпентанамид
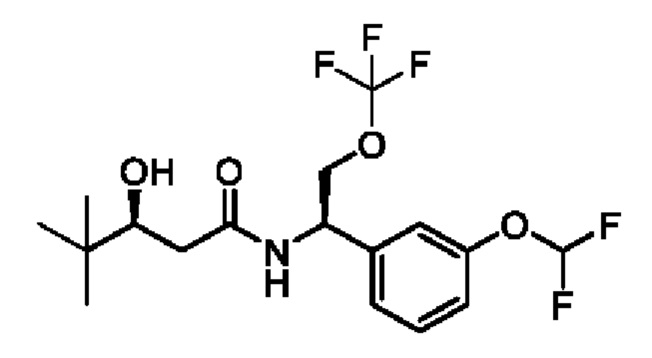
Получен из IIe и IIIa.
1H ЯМР (CDCl3 400 МГц): δ 7,41-7,36 (m, 1Н), 7,18 (d, 1Н), 7,10-7,08 (m, 2H), 6,67 (d, 1H), 6,52 (t, 1H), 5,37-5,32 (m, 1H), 4,28-4,20 (m, 1H), 3,69 (d, 1H), 2,93 (s, 1H), 2, 47-2, 43 (m, 1H), 2,36-2, 29 (m, 1H), 0,93 (s, 9H).
LC-MS:tR=2,38 минуты (LCMS Способ 3), масса/заряд = 400,0 [M+H]+.
SFC:tR=2,11 минуты, (SFC Способ 4), ee%=96,4%.
Пример 19. (S)-N-((R)-1-(3-(Трифторметокси)фенил)-2-(трифторметокси)зтил)-3-гидрокси-4,4-диметилпентанамид
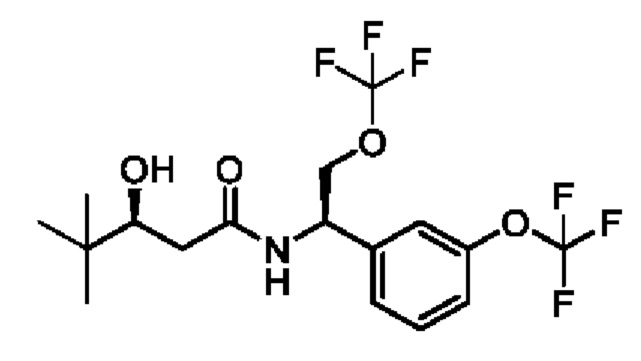
1Н ЯМР (CDCl3 400 МГц): δ 7,35 (t, 1Н), 7,21-7,19 (m, 1Н), 7,13-7,11 (m, 2Н), 6,65 (d, 1Н), 5,32-5,27 (m, 1Н), 4,21-4,13 (m, 1Н), 3,64-3,61 (m, 1Н), 2,83 (d, 1Н), 2,41-2,36 (m, 1Н), 2,29-2,25 (m, 1Н), 0,86 (s, 9Н).
LC-MS:tR=2,56 минуты (LCMS Способ 3), масса/заряд = 418,0 [М+Н]+.
HPLC:tR=13,54 минуты (HPLC Способ 2), ее%=65,9%
Пример 20. (S)-N-(S)-1-(3-(Дифторметокси)фенил)бутил)-3-гидрокси-4,4-диметилпентанамид
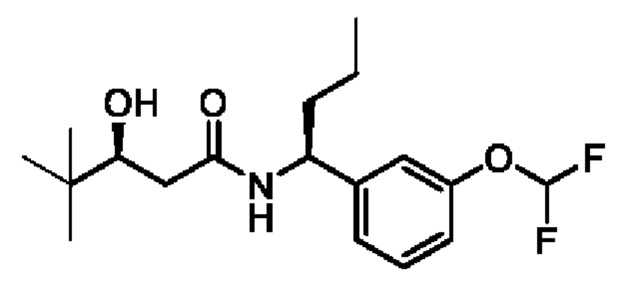
Получен из IIg и IIIa.
1Н ЯМР (CDCl3 400 МГц): δ 7, 33 (t, 1Н), 7,14 (d, 1Н), 7,03-7,00 (m, 2Н), 6,52 (t, 1Н), 6,22-6,20 (m, 1Н), 4,97 (q, 1Н), 3,68-3,64 (m, 1Н), 3,31 (d, 1Н), 2,39-2,24 (m, 2Н), 1,75-1,72 (m, 2Н), 1,36-1,29 (m, 2Н), 0,95-0,91 (m, 12Н).
LC-MS:tR=2,30 минуты (LCMS Способ 3), масса/заряд = 344,1 [М+Н]+.
SFC:tR=2,13 минуты (SFC Способ 1), ее%=98,7%.
Пример 21. (S) -N-((S)-1-(3-(Дифторметокси)фенил)-4,4-дифторбутил)-3-гидрокси-4,4-диметилпентанамид
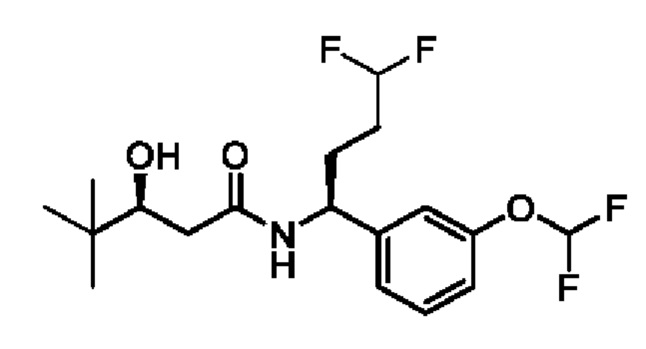
Получен из IIh и IIIa.
1Н ЯМР (CDCl3 400 МГц): δ 7, 36 (t, 1Н), 7,14 (d, 1Н), 7,05 (m, 2Н), 6,52 (t, 1Н), 6,25 (d, 1Н), 5,84 (tt, 1Н), 5,03 (q, 1Н), 3,68 (m, 1Н), 2,97 (d, 1Н), 2,40-2,21 (2Н), 1, 98-1, 77 (4Н), 0,91 (s, 9Н).
LC-MS:tR=2,43 минуты (LCMS Способ 1), масса/заряд = 380,0 [М+Н]+.
HPLC:tR=14,01 минуты (HPLC Способ 3), ее%=95,7%.
Пример 22. (S)-N-((S)-1-(3-(Дифторметокси)фенил)-3,3-дифторпропил)-3-гидрокси-4,4-диметилпентанамид
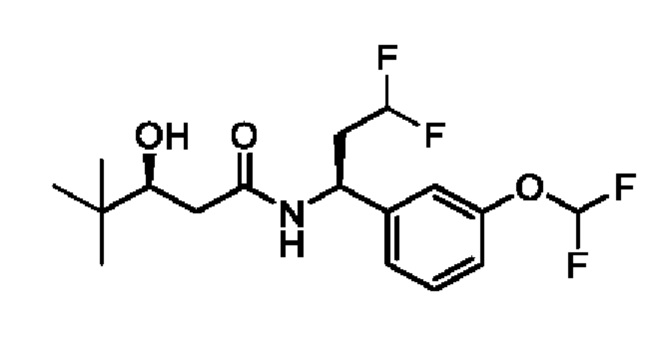
Получен из IIi и IIIa.
1Н ЯМР (CDCl3 400 МГц): δ 7, 35 (t, 1Н), 7,14 (d, 1Н), 7,05-7,03 (m, 2Н), 6,50 (t, 1Н), 6,53-6,50 (m, 1Н), 5,80 (tt, 1Н), 5, 30-5,24 (m, 1Н), 3,66 (dd, 1Н), 2,89 (s, 1Н), 2,40-2,22 (m, 4Н), 0,89 (s, 9Н).
LC-MS:tR=2,63 минуты (LCMS Способ 1), масса/заряд = 366,2 [М+Н]+.
HPLC:tR=13,43 минуты (HPLC Способ 1), ее%=96,7%.
Пример 23. (S)-N-((S)-1-(3-(Дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид
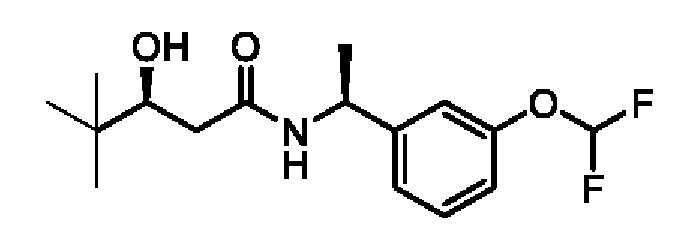
Получен из IIj и IIIa.
1Н ЯМР (CDCl3 400 МГц): δ 7, 34-7, 30 (m, 1Н), 7,16-7,12 (m, 1Н), 7,04-6,97(т, 2 Н), 6,31 (t, 1 Н), 6,14 (brs, 1 Н), 5,12-5,06 (m, 1 Н), 3,67-3,62 (m, 1 Н), 3,29 (s, 1 Н), 2,37-2,31 (m, 1 Н), 2,26-2,19 (m, 1 Н), 1,47-1,43 (m, 3 Н), 0,98 (s, 9 Н).
LC-MS:tR=2,155 минуты (LCMS Способ 2), масса/заряд = 316,1 [М+Н]+.
SFC:tR=2,416 минуты (SFC Способ 8), ее%=100%.
Пример 24. (S)-N-((S)-2-Циано-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамид
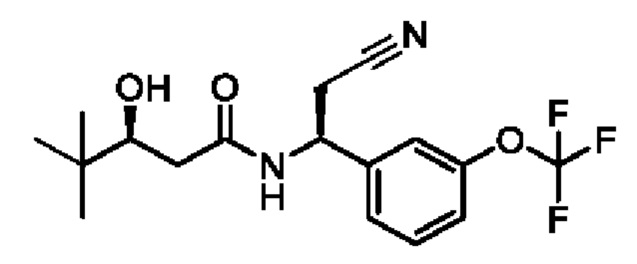
Получен из IIm и IIIa.
1Н ЯМР (400 МГц, DMSO-d6): δ 8,66 (d, 1Н), 7,51 (t, 1Н), 7,45-7,43 (m, 2Н), 7,30 (d, 1Н), 5,25-5,23 (m, 1Н), 4,62 (d, 1Н), 3,57-3,51 (m, 1Н), 3,00 (dd, 2Н), 2,31-2,11 (m, 2Н), 0,81 (s, 9Н).
LC-MS:tR=2,42 минуты (LSMS Способ 1), масса/заряд = 359,2 [М+Н]+.
HPLC:tR=12,56 минуты (HPLC Способ 4), ее%=100%.
Пример 25. (S)-N-((S)-3-Циано-1-(3-(трифторметокси)фенил)пропил)-3-гидрокси-4,4-диметилпентанамид
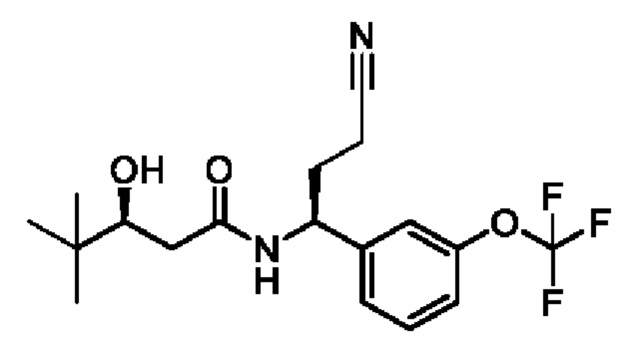
Получен из IIn и IIIa.
1Н ЯМР (CDCl3 400 МГц): δ 7, 42 (t, 1Н), 7,24 (m, 1Н), 7,18 (d, 1Н), 7,13 (s, 1Н), 6,49 (d, 1Н), 5,19-5,13 (m, 1Н), 3,75-3,71 (m, 1Н), 2,82 (d, 1Н), 2,45-2,40 (m, 3Н), 2,30-2,27 (m, 1Н), 2,23-2,16 (m, 2Н), 0,92 (s, 9Н).
Пример 26. (R)-2-(3,3-Дифтор-1-гидроксициклобутил)-N-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)зтил)ацетамид
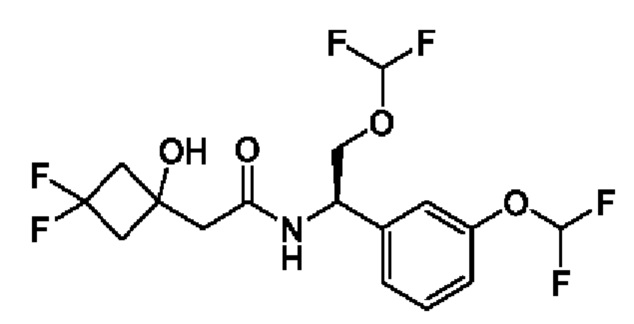
Получен из IIa и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7, 37 (t, 1Н), 7,15 (d, 1Н), 7,09-7,06 (m, 2Н), 6,50 (t, 1Н), 6,32 (t, 1Н), 6,23 (m, 1Н), 5, 29-5, 24 (m, 1Н), 4,74 (s, 1Н), 4,17 (dd, 1Н), 4,08 (dd, 1Н), 2,75-2,72 (m, 2Н), 2,68 (s, 2Н), 2,62-2, 56 (m, 2Н).
LC-MS:tR=2,39 минуты (LCMS Способ 1), масса/заряд = 402,1 [М+Н]+.
SFC:tR=1,87 минуты (SFC Способ 1), ее%=100%
Пример 27. (R)-2-(3,3-Дифтор-1-гидроксициклобутил)-N-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)ацетамид
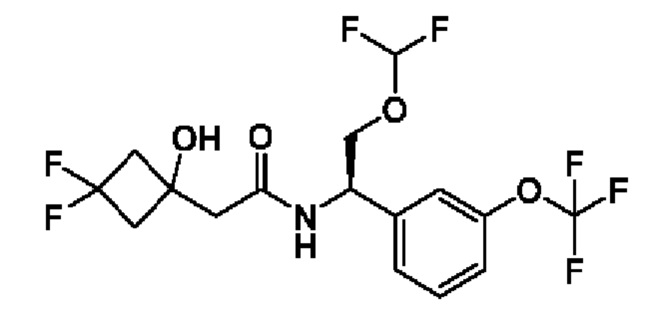
Получен из IIb и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7,35 (t, 1Н), 7,14-7,09 (m, 3Н), 6,18 (t, 1Н), 6,21 (d, 1Н), 5,23 (m, 1Н), 4,66 (br s, 1Н), 4,13 (dd, 1Н), 4,04(dd, 1Н), 2, 72-2, 66 (m, 2Н), 2,64 (s, 2Н), 2,56-2,50 (m, 2Н).
LC-MS:tR=2,53 минуты (LC-MS Способ 1), масса/заряд = 420,2 [М+Н]+.
HPLC:tR=12,77 минуты (HPLC Способ 2), ее%=86,7%.
Пример 28. (R)-N-(2-Циклопропокси-1-(3-(трифторметокси)фенил)этил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамид
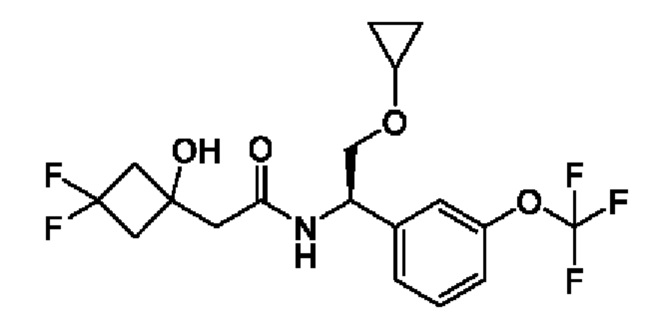
Получен из IId и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7,37 (t, 1Н), 7,22 (d, 1Н), 7,15 (m, 2Н), 6,40 (d, 1Н), 5,13 (m, 1Н), 4,92 (s, 1Н), 3,81 (m, 1Н), 3,69 (m, 1Н), 3,29 (m, 1Н), 2, 78-2, 52 (6Н), 0,57-0,44 (4Н).
LC-MS:tR=2,53 минуты (LC-MS Способ 1), масса/заряд = 410,0 [М+Н]+.
SFC:tR=1,50 минуты (SFC Способ 7), ее%=99,7%
Пример 29. (R)-N-(2-Циклопропокси-1-(3-(дифторметокси)фенил)этил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамид
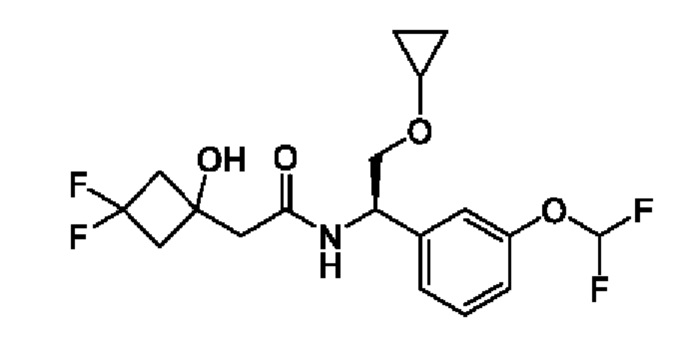
Получен из IIe и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7,35 (d, 1Н), 7,15 (d, 1Н), 7,06 (s, 1Н), 7,05 (d, 1Н), 6,51 (t, 1Н), 6,39 (d, 1Н), 5,15-5,10 (m, 1Н), 4,98 (m, 1Н), 3,83-3,67 (m, 2Н), 3,31-3,30 (m, 1Н), 2,79-2,75 (m, 2Н), 2,68 (d, 2Н), 2,64-2,52 (m, 2Н), 0,60-0,46 (m, 4Н).
LC-MS:tR=2,40 минуты (LC-MS Способ 1), масса/заряд = 392,1[М+Н]+.
SFC:tR=2,32 минуты (SFC Способ б), ее%=100,00%
Пример 30. (R)-2-(3,3-Дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил) -2-(трифторметокси)зтил)ацетамид
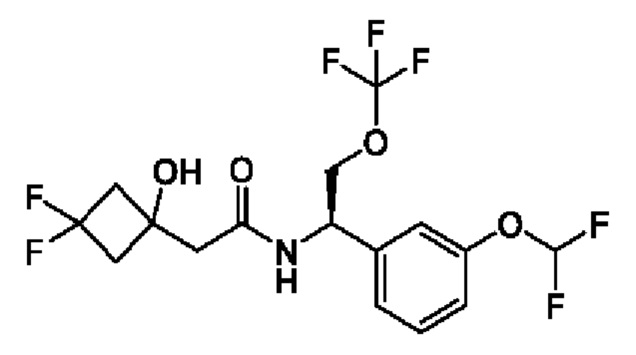
Получен из IIe и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7,39 (t, 1Н), 7,15 (d, 1Н), 7,08 (m, 2Н), 6,51 (t, 1Н), 6,30 (m, 1Н), 5,32 (m, 1Н), 4,63 (s, 1Н), 4,23 (m, 2Н), 2,76-2,57 (6Н).
LC-MS:tR=2,48 минуты (LC-MS Способ 1), масса/заряд = 420,0 [М+Н]+.
SFC:tR=12,96 минуты (HPLC Способ 2), ее%=75,5%
Пример 31. (S)-2-(3,3-Дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил)бутил)ацетамид
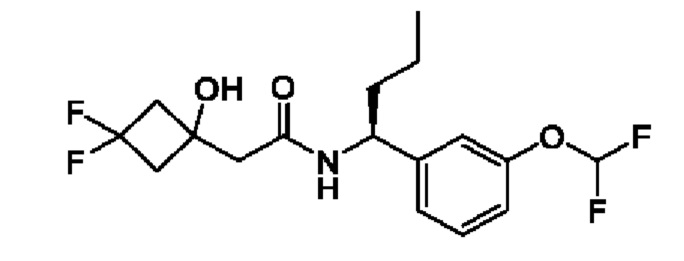
Получен из IIg и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7, 35 (t, 1Н), 7,13 (d, 1Н), 7,04 (m, 2Н), 6,52 (t, 1Н), 5,91 (m, 1Н), 5,04 (s, 1Н), 4,96 (q, 1Н), 2, 78-2, 56 (6Н), 1,76 (m, 2Н), 1,34 (m, 2Н), 0, 95 (t, 3Н).
LC-MS:tR=2,44 минуты (LC-MS Способ 1), масса/заряд = 364,0 [М+Н]+.
SFC:tR=1,71 минуты (SFC Способ 10), ее%=94,8%.
Пример 32. (S)-2-(3,3-Дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил)-4,4-дифторбутил)ацетамид
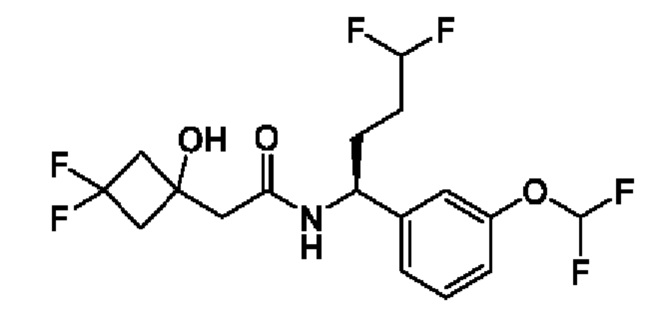
Получен из IIh и IIIb.
1H ЯМР (CDCl3 400 МГц): δ 7, 38 (t, 1Н), 7,13 (d, 1Н), 7,08 (d, 1Н), 7,04 (s, 1Н), 6,52 (t, 1Н), 5,90 (d, 1Н), 5,85 (tt, 1Н), 5,00 (q, 1Н), 4,82 (s, 1Н), 2,78-2,54 (6Н), 2,00 (m, 2Н), 1,86 (m, 2Н).
LC-MS:tR=2,50 минуты (LCMS Способ 1), масса/заряд = 400,1 [М+Н]+.
HPLC:tR=12,48 минуты (HPLC Способ 2), ее%=98,3%.
Пример 33. (S)-2-(3,3-Дифтор-1-гидроксициклобутил)-N-(1-(3-(трифторметокси)фенил)пропил)ацетамид
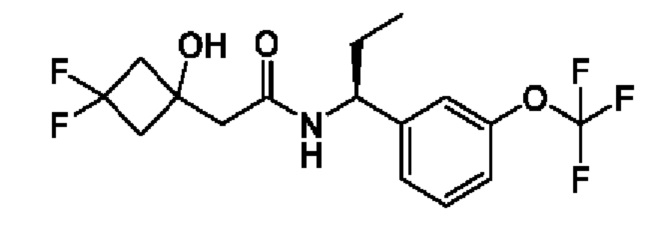
Получен из III и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7, 38 (t, 1Н), 7,20 (d, 1Н), 7,14 (d, 1Н), 7,10 (s, 1Н), 5,89 (d, 1Н), 4,99 (brs, 1Н), 4,89 (q, 1Н), 2,77-2,70 (m, 2Н), 2,63 (d, 2Н), 2,60-2,50 (m,2Н), 1,87-1,80 (m, 2Н), 0,92 (t, 3Н).
LC-MS:tR=2,57 минуты (LCMS Способ 1), масса/заряд = 368,1 [М+Н]+.
SFC:tR=13,09 минуты (SFC Способ 1), ее%=100%
Пример 34. (S)-N-(2-Циано-1-(3-(трифторметокси)фенил)зтил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамид
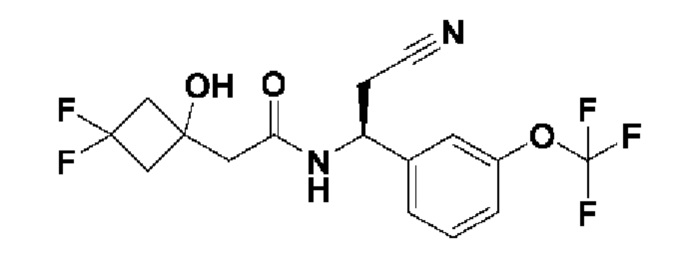
Получен из IIm и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7,46 (t, 1Н), 7,30 (d, 1Н),7,24 (m, 1Н), 7,19 (s, 1Н), 6,51 (s, 1Н), 5,34-5,29 (m, 1Н), 4,44 (s, 1Н), 3, 09-3, 03 (m, 1Н), 2,91-2,89 (m, 1Н), 2,77-2,73 (m, 2Н), 2,70-2,53 (m, 4Н).
LC-MS:tR=2,257 минуты (LC-MS Способ 1), масса/заряд = 379, 0 [М+Н]+.
SFC:tR=2,60 минуты (SFC Способ 11), ee%=100%.
Пример 35. (S)-N-(3,3-Дифтор-1-(3-(трифторметокси)фенил)пропил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамид
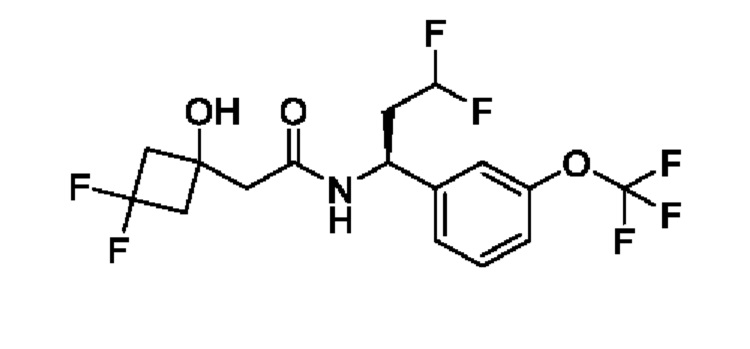
Получен из IIo и IIIb.
1Н ЯМР (CDCl3 400 МГц): δ 7, 44 (t, 1Н), 7,23 (m, 2Н), 7,15 (s, 1Н), 6,15 (br d, 1Н), 5,83 (tt, 1Н), 5,32 (m, 1Н), 4,69 (s, 1Н), 2,73 (m, 2Н), 2,66 (s, 2Н), 2,62-2,34 (4Н).
LC-MS:tR=2,53 минуты (LC-MS Способ 1), масса/заряд = 404,1 [М+Н]+.
SFC:tR=1,66 минуты (SFC Способ 19), ее%=98,5%
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ АРИЛАМИДОВ В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2011 |
|
RU2535671C1 |
| ЗАМЕЩЕННЫЕ ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2827641C1 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ БЛОКАТОРОВ TTX-S | 2013 |
|
RU2652117C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛОВ В КАЧЕСТВЕ МОДУЛЯТОРОВ СВЯЗАННОГО С РЕТИНОИДАМИ ОРФАННОГО РЕЦЕПТОРА ГАММА (RORγ) И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2019 |
|
RU2789059C2 |
| АРИЛ-ЗАМЕЩЕННЫЕ КАРБОКСАМИДНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ БЛОКАТОРОВ КАЛЬЦИЕВЫХ ИЛИ НАТРИЕВЫХ КАНАЛОВ | 2010 |
|
RU2575168C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИН КИНАЗЫ ДЛЯ ЛЕЧЕНИЯ ГИПЕРПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2013 |
|
RU2644947C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ ПИРИМИДИН-4(3H)-ОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2021 |
|
RU2839367C1 |
| ТЕТРАГИДРОИМИДАЗО[1,5-d][1,4]ОКСАЗЕПИНОВОЕ ПРОИЗВОДНОЕ | 2014 |
|
RU2659219C2 |
| ИНГИБИТОР EGFR И ЕГО ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2702631C2 |
Группа изобретений относится к органической химии и включает соединение формулы I или его фармацевтически приемлемые соли, индивидуальные соединения, указанные в формуле изобретения, или их фармацевтически приемлемые соли, фармацевтическую композицию на их основе и применение. В формуле I R1 выбран из группы, состоящей из C1-C6 алкила, CF3, CH2CF3, CF2CHF2, C3-C8 циклоалкила, где указанный C3-C8 циклоалкил может быть замещен 1 или 2 заместителями, выбранными из группы, состоящей из C1-C3 алкила, F, CHF2 и CF3; R2 представляет собой H, C1-C6 алкил или CF3; или R1 и R2 объединены с образованием C3-C5 циклоалкила, необязательно замещенного 1 или 2 F, CHF2 или CF3; R3 представляет собой CH2-O-циклопропил, C1-C3 алкил или CH2O-C1-3 алкил, при этом указанные C1-C3 алкил или CH2O-C1-C3 алкил замещены C≡N, 3 F или C3-C5 циклоалкилом, R4 выбран из группы, состоящей из OCF3 или OCHF2. Технический результат - соединения, определенные в формуле изобретения, для лечения нарушений, реагирующих на активацию калиевых каналов Kv7. 4 н. и 8 з.п. ф-лы, 35 пр. 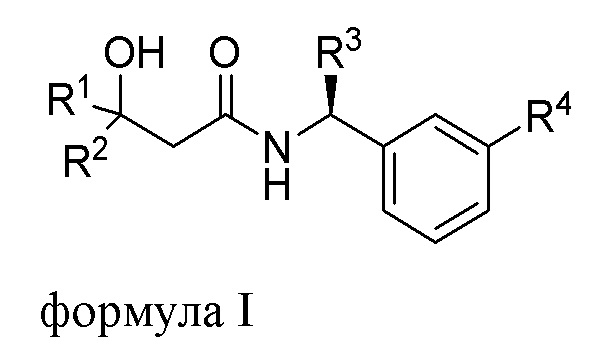
1. Соединение формулы I
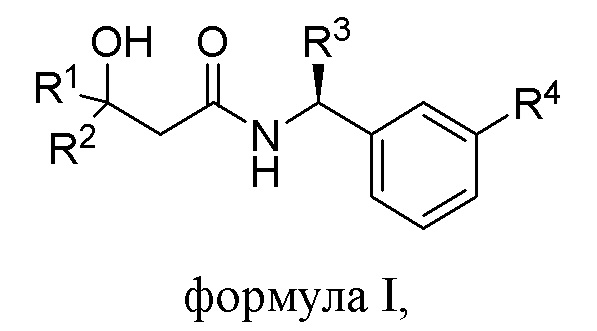
где
R1 выбран из группы, состоящей из C1-C6 алкила, CF3, CH2CF3, CF2CHF2, C3-C8 циклоалкила, где указанный C3-C8 циклоалкил может быть замещен 1 или 2 заместителями, выбранными из группы, состоящей из C1-C3 алкила, F, CHF2 и CF3; и
R2 представляет собой H, C1-C6 алкил или CF3; или
R1 и R2 объединены с образованием C3-C5 циклоалкила, необязательно замещенного 1 или 2 F, CHF2 или CF3; и
R3 представляет собой CH2-O-циклопропил, C1-C3 алкил или CH2O-C1-3 алкил, при этом указанные C1-C3 алкил или CH2O-C1-C3 алкил замещены C≡N, 3 F или C3-C5 циклоалкилом;
R4 выбран из группы, состоящей из OCF3 или OCHF2,
или его фармацевтически приемлемая соль.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R4 представляет собой OCF3 или OCHF2.
3. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, где R3 выбрана из группы, состоящей из CH2-O-CF3, CH2-O-циклопропила, CH2-C≡N.
4. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, где R1 представляет собой C3-C4циклоалкил, необязательно замещенный 1 или 2 из C1-C3 алкила, F, CHF2 или CF3.
5. Соединение по любому из предыдущих пунктов или его фармацевтически приемлемая соль, где R1 и R2 объединены с образованием циклобутила, необязательно замещенного 1 или 2 F, и R4 представляет собой OCF3 или OCHF2.
6. Соединение по п. 1, где соединение выбрано из группы, состоящей из:
(S)-N-((R)-2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((R)-1-(3-(дифторметокси)фенил)-2-(трифторметокси)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((R)-1-(3-(трифторметокси)фенил)-2-(трифторметокси)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-2-циано-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-3-циано-1-(3-(трифторметокси)фенил)пропил)-3-гидрокси-4,4-диметилпентанамида;
(R)-N-(2-циклопропокси-1-(3-(трифторметокси)фенил)этил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамида;
(R)-N-(2-циклопропокси-1-(3-(дифторметокси)фенил)этил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамида;
(R)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил)-2-(трифторметокси)этил)ацетамида или
(S)-N-(2-циано-1-(3-(трифторметокси)фенил)этил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамида
или фармацевтически приемлемой соли любого из этих соединений.
7. Соединение, выбранное из группы, состоящей из:
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
N-((R)-2-этокси-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
N-((R)-2-этокси-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидрокси-3-(1-(трифторметил)циклопропил)пропанамида;
(R)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида;
(S)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида;
(R)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксипропанамида;
(S)-3-(3,3-дифторциклобутил)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксипропанамида;
(S)-3-(3,3-дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамида;
(R)-3-(3,3-дифторциклобутил)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидроксипропанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-гидроксипропанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-этилциклопропил)-3-гидроксипропанамида;
(S)-N-((S)-1-(3-(дифторметокси)фенил)бутил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-1-(3-(дифторметокси)фенил)-4,4-дифторбутил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-1-(3-(дифторметокси)фенил)-3,3-дифторпропил)-3-гидрокси-4,4-диметилпентанамида;
(S)-N-((S)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-4,4-диметилпентанамида;
(R)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)ацетамида;
(R)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)ацетамида;
(S)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил)бутил)ацетамида;
(S)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(1-(3-(дифторметокси)фенил)-4,4-дифторбутил)ацетамида;
(S)-2-(3,3-дифтор-1-гидроксициклобутил)-N-(1-(3-(трифторметокси)фенил)пропил)ацетамида;
(S)-N-(3,3-дифтор-1-(3-(трифторметокси)фенил)пропил)-2-(3,3-дифтор-1-гидроксициклобутил)ацетамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-(1-фторциклопропил)-3-гидроксибутанамида;
(R)-3-циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамида;
(S)-3-циклопропил-N-((R)-2-(дифторметокси)-1-(3-(трифторметокси)фенил)этил)-3-гидроксибутанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-5,5,5-трифтор-3-гидрокси-3-метилпентанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,5-диметилгексанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидрокси-3,4-диметилпентанамида;
(S)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамида;
(R)-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-(3,3-диметилциклобутил)-3-гидроксипропанамида;
(S)-3-циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида;
(R)-3-циклопентил-N-((R)-2-(дифторметокси)-1-(3-(дифторметокси)фенил)этил)-3-гидроксипропанамида;
(R)-3-(1-фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамида
и (S)-3-(1-фторциклопропил)-3-гидрокси-N-((R)-2-метокси-1-(3-(трифторметокси)фенил)этил)бутанамида
или фармацевтически приемлемой соли любого из этих соединений.
8. Фармацевтическая композиция для лечения нарушения, реагирующего на активацию калиевых каналов Kv7, содержащая эффективное количество соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли и одно или несколько фармацевтически приемлемых вспомогательных веществ.
9. Применение соединения по пп. 1-7 или его фармацевтически приемлемой соли для лечения нарушения, реагирующего на активацию калиевых каналов Kv7.
10. Применение по п. 9, где нарушение выбрано из группы, состоящей из эпилепсии, эпилептических синдромов, эпилептических симптомов, устойчивой к лечению или рефрактерной эпилепсии, судорожных припадков, биполярного расстройства, мигрени или шизофрении.
11. Применение по п. 9, где нарушение выбрано из группы, состоящей из психоза, мании, нарушений, связанных с воздействием стрессовых факторов, острых реакций на стресс, биполярной депрессии, большого депрессивного расстройства, тревожности, панических атак, социофобии, нарушений сна, СДВГ, ПТСР, ОКР, нарушений импульсного контроля, расстройств личности, шизотипического нарушения, агрессивности, хронической боли, нейропатии, нарушений аутического спектра, хореи Гентингтона, склероза, рассеянного склероза, болезни Альцгеймера.
12. Применение по п. 9, где нарушение выбрано из группы, состоящей из фокальной (парциальной) эпилепсии с простыми парциальными припадками, фокальной (парциальной) эпилепсии с комплексными парциальными припадками, генерализованной идиопатической эпилепсии, больших судорожных припадков, эпилептического статуса, неонатальных припадков, KCNQ-ассоциированной эпилептической энцефалопатии (KCNQ2EE), доброкачественных семейных неонатальных судорог, а также других эпилептических синдромов, таких как тяжелая миоклоническая эпилепсия раннего детского возраста, эпилепсия с непрерывными комплексами "спайк-волна" во время медленноволнового сна, синдром Веста, синдром Леннокса-Гасто, синдром Драве и ранняя миоклоническая энцефалопатия, синдром Отахара, или судорожных припадков, связанных с воздействием стрессовых факторов, гормональными изменениями, употреблением наркотических средств, употреблением алкоголя, инфекцией или метаболическими расстройствами, такими как гипонатриемия, или эпилептических симптомов, являющихся компонентом нейродегенеративных нарушений, таких как болезнь Альцгеймера, болезнь с тельцами Леви, ювенильная форма болезни Гентингтона, лобно-височная лобарная дегенерация.
| WO 2006033844 A2, 30.03.2006 | |||
| Sigrid Marie Blom et al | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| PLoS One, 2014, vol.9, no.6, p.e100209 | |||
| КЛЮЧ ДЛЯ КРУГЛЫХ ГАЕК | 1930 |
|
SU22582A1 |
| УБЕЖИЩЕ ВРЕМЕННОГО ТИПА | 1991 |
|
RU2069728C1 |
| СПОСОБ ВЫДЕЛЕНИЯ ЭРИТРОМИЦИНА | 2001 |
|
RU2203952C2 |

