Настоящее изобретение относится к способу получения фракции, содержащей белки лактоферрин или лактопероксидазу, из источника, содержащего по меньшей мере один из указанных белков, а также белков высокой степени очистки лактоферрина или лактопероксидазы.
Введение
Лактоферрин (ЛФ) и лактопероксидаза (ЛПО) представляют собой функциональные минорные белки, содержащиеся в молоке, сыворотке и молозиве. ЛФ представляет собой гликозилированный белок с молекулярной массой 80 кДа, который способен реагировать на различные физиологические изменения и изменения окружающей среды, и поэтому считается ключевым компонентом первой линии защиты организма хозяина. Структурные характеристики ЛФ обеспечивают функциональность в дополнение к функции гомеостаза Fe3+, общей для всех трансферринов: высокая антимикробная активность против широкого спектра бактерий, грибов, дрожжей, вирусов и паразитов; противовоспалительное и антиканцерогенное действие; а также несколько ферментативных функций [1]. ЛПО играет жизненно важную роль в защите лактирующей молочной железы и кишечного тракта новорожденных от патогенных микроорганизмов, участвует в разрушении различных канцерогенов и защите клеток животных от пероксидирующих воздействий [2].
ЛФ способен связывать железо. Нативный ЛФ содержит от 15 до 20% хололактоферрина (holo LF), который содержит железо. Остальная часть представляет собой аполактоферрин (apo LF), не содержащий железо [32]. Теоретически аполактоферрин имеет довольно низкий уровень насыщения железом (уже связанное железо - значение A). Теоретическое значение A аполактоферрина составляет около <3%. Кроме того, аполактоферрин обладает высоким потенциалом связывания железа (железосвязывающая способность - значение C). Значение C аполактоферрина составляет около >50%. Высокочистый и неденатурированный аполактоферрин потенциально может иметь еще более высокие значения C, составляющие более 70%. С другой стороны, хололактоферрин имеет высокое значение A (>50%) и низкое значение C (<10%). Чем выше значение C выделенного ЛФ, тем выше его потенциал связывания железа. Более высокий потенциал связывания железа обусловливает более высокий уровень антимикробной активности, поскольку активный ЛФ удаляет железо, необходимое для роста микроорганизмов.
В настоящее время ЛФ и ЛПО выделяют из молока и побочных продуктов его переработки (например, сыворотки), используя множество различных способов, таких как: I) выделение с помощью парамагнитных частиц с поли(глицидилметакрилатом) и гепариновым лигандом [3], (II) при помощи катионного поверхностно-активного вещества (например, бромида цетилдиметиламмония) [4], (III) различные хроматографические методы (например, катионообменная или аффинная хроматография) [1, 2, 5-10], а также (IV) другие методы (например, при помощи гидрофобных ионных жидкостей [11]). В общих чертах хроматографические методы, в первую очередь ионообменная хроматография, представляют собой способ быстрого выделения ЛФ при сравнительно низких затратах [12]. Хроматографический подход также преобладает над другими благодаря его надежности и воспроизводимости. Наиболее распространенным методом хроматографической очистки ЛФ и ЛПО является использование частиц смолы с высокой катионообменной способностью, а также мембран или монолитных колонок [13-15].
Современная хроматография страдает от некоторых недостатков: (I) расширение пика с увеличением скорости потока вследствие диффузионного массопереноса в порах, поэтому при увеличении скорости потока перепад градиента должен уменьшаться, чтобы получить такое же разрешение, (II) повышение интенсивности потока сокращает время процесса хроматографии, но в то же время увеличивает объем элюирования, и (III) увеличение длины колонны приводит к сильному падению давления в колонне, что является ограничивающим фактором для скорости потока.
На этапе хроматографической очистки ЛФ и ЛПО, присутствующие в молоке или сыворотке, при определенных условиях связываются с поверхностью сильного катионита, а затем собираются во фракции элюирования при помощи буферов с более высоким pH или высокой концентрацией соли. Чтобы упростить процесс выделения ЛФ и ЛПО чаще всего элюируют с использованием нескольких буферных растворов с высокой ионной силой или pH>9 в ступенчатом режиме. Такой подход часто позволяет добиться сравнительно высокой чистоты (60-95%) фракции требуемого белка за один этап хроматографии. Для обессоливания, концентрирования и повышения чистоты часто применяется процесс ультрафильтрации с целью удалить небольшое количество низкомолекулярных примесей. Затем полученный белковый концентрат обычно сушат методом лиофилизации или распылительной сушки. Wang и др. [18] продемонстрировали, что лиофилизированный ЛФ содержит меньше воды (около 2-3%), чем высушенный распылением (≈5%). При этом, напротив, высушенный распылением ЛФ показал несколько меньшую степень денатурации и антиоксидантную активность на 6-7% выше, что близко к этому показателю у свежего жидкого ЛФ. Распылительная сушка [1] является проверенным способом, используемым для получения аморфных порошков ЛФ с неповрежденной молекулярной конфигурацией и сильными антиоксидантными свойствами.
В EP 0418704 A1 [19] описываются процессы выделения, очистки и восстановления молочных белков, способных связывать железо, с использованием ионообменных хроматографических колонок, которые содержат частицы смолы с поверхностными сульфогруппами. ЛФ выделяют с помощью элюирования в ступенчатом режиме на основе pH/электропроводности, и заявленная чистота конечного продукта составляет >90%. Кроме того, требуется процесс ультрафильтрации для удаления небольшого количества низкомолекулярных примесей, что в конечном итоге обеспечивает чистоту ЛПО и ЛФ 90% или выше.
Процесс, описанный в EP 1466923 A1 [20], включает в себя этап хроматографии с сильнокислой катионообменной смолой (ее частицами), где чистота выделенного ЛФ составляет от 79 до 91%.
В WO 2006/119644 A1 [21] описывается метод очистки ЛФ, стабилизации его в растворе и повышения его активности. Данный процесс предназначен для дополнительной очистки уже выделенного ЛФ с более низкой чистотой. Очистка производится при помощи гидрофобного адсорбента (его частиц) в присутствии водного кислого раствора, содержащего концентрацию заряженного удаляемого растворенного вещества. Достигнутая конечная чистота ЛФ составила >95%.
В патенте США 5861491 A [13] описываются способы выделения человеческого ЛФ, включая человеческий ЛФ, продуцируемый в результате экспрессии трансгена, кодирующего рекомбинантный человеческий ЛФ (rhLF), а также другие родственные виды ЛФ, из молока, обычно из коровьего молока. Как правило, молоко либо молочная фракция, содержащая человеческий ЛФ, контактирует с сильной катионообменной смолой в присутствии сравнительно высокой ионной силы для предотвращения связывания белков, не являющихся белками ЛФ, а также других веществ с сильной катионообменной смолой. Затем частицы смолы отделяются от молока центрифугированием, и ЛФ, связанный с катионообменной смолой, затем элюируется при помощи нескольких буферных растворов с различной концентрацией соли в ступенчатом режиме. Чистота верхних фракций человеческого и коровьего ЛФ превышает показатель примерно в 95%.
В EP 0348508 B1 [22] описано выделение ЛФ из сырого молока с помощью перекрестно-сшитой сульфированной полисахаридной смолы (ее частиц).
Адсорбированный на колонке ЛФ элюируется в ступенчатом режиме при помощи буфера с повышенной концентрацией NaCl. Измеренная чистота коровьего ЛФ составила 95%, а чистота ЛФ, полученного из обезжиренного человеческого молозива - 98%.
Способ, описанный в патенте США 6096870 A [23], относится к разделению сывороточных белков (иммуноглобулин, β-лактоглобулин, α-лактальбумин, альбумин бычьей сыворотки (BSA), ЛФ), в частности, к последовательному разделению сывороточных белков на отдельные фракции с помощью предварительно подготовленной хроматографической колонки с частицами сильной катионообменной смолы. Последовательное элюирование указанных белковых фракций достигается при помощи буферов с подходящим pH и ионной силой в ступенчатом режиме. Конечная чистота продуктов распылительной сушки составила: у иммуноглобулинов ≥80%, у BSA и ЛФ ≥75% и у β-лактоглобулина ≥85%.
В патенте США 8603560 B2 [24] описан процесс выделения молочных белков из молока или сыворотки с использованием катионообменной смолы, загруженной в колонку. Элюция стимулировалась ступенчатым изменением pH или ионной силы. Конечная чистота ЛФ составила 80%.
В CA 2128111 C [25] описывается процесс выделения ЛФ и ЛПО из молока и молочных продуктов в промышленных масштабах. Выделение достигается за счет адсорбции указанных белков на катионообменнике и элюированием указанных белков по отдельности либо одновременно путем ступенчатого элюирования одним или несколькими солевыми растворами. Данные о конечной чистоте выделенных белков отсутствуют.
В EP 0253395 B9 [26] раскрывается способ выделения коровьего ЛФ из молока высокой чистоты с помощью слабокислой катионообменной смолы. ЛФ извлекают из ионообменного вещества методом ступенчатого элюированя растворами хлорида натрия различной концентрации. Согласно старому методу Лаурелла, чистота получаемого ЛФ составляет 90-99%.
В патенте США 5596082 A [27] описан процесс выделения ЛФ и ферментной ЛПО из молока и молочных продуктов в промышленных масштабах. Данный процесс включает этапы адсорбции указанных белков на катионообменнике путем пропускания молока либо производных молока через катионообменник. Элюция указанных белков стимулировалась ступенчатой элюцией с помощью различных концентраций солей. Конечная чистота ЛПО и ЛФ составила 93% и 94%, соответственно.
В изобретении, раскрытом в патенте США 9115211 B2 [28], описывается выделение ЛФ с помощью катионообменной смолы. Полученный описанным способом ЛФ имеет чистоту более 95%, практически не содержит LPS (липополисахарид), эндотоксины и ангиогенин, с уровнем насыщения железом от 9% до 15%.
В EP 2421894 A1 [29] описан метод получения ЛФ с низким содержанием железа с насыщением железом менее 10% или, более предпочтительно, с насыщением железом примерно от 9% до 3,89%. Полученный данным методом ЛФ с низким содержанием железа демонстрирует повышенную антимикробную активность по сравнению со стандартным ЛФ. В данном методе используются кислота и растворитель. После высвобождения Fe3+ добавленные вспомогательные средства удаляли методом ультрафильтрации и диафильтрации. Полученный продукт имеет светло-кремовый/бледно-бежевый цвет с насыщением железом от 3,89% до 5,1% (по данным ВЭЖХ/рентгеновской флуоресценции (XRF)).
В WO2014/207678 A1 [30] раскрывается способ очистки ЛФ от секреторной жидкости, включающий подщелачивание секреторной жидкости, контактирование подщелачиваемой секреторной жидкости с воздухом и осаждение ЛФ из данной подщелачиваемой секреторной жидкости с помощью органического растворителя (ацетона).
В WO1995/022258 A2 [31] раскрываются способы выделения человеческого ЛФ из молока, в особенности из молока нечеловеческих видов, и отделения человеческого ЛФ от нежелательных макромолекулярных видов, присутствующих в молоке, включая отделение от нечеловеческих видов ЛФ. Для выделения используется сильная катионообменная смола (например, S Sepharose™). Белки (ЛФ и другие) элюировали ступенчатым градиентом соли и pH.
В работе G. Majka и др. (2013) «Высокопроизводительный метод количественного определения насыщения железом в препаратах лактоферрина», Anal. Bioanal. Chem., 405, 5191-5200 раскрывается метод получения коровьего лактоферрина с низким содержанием железа. Для получения лактоферрина с низким содержанием железа одной только ионообменной хроматографии было недостаточно. Поэтому ионообменную хроматографию сочетали с расширенным диализом цитратным буфером 100 мМ в течение 24 часов.
Сущность изобретения
Одна из задач настоящего изобретения состоит в обеспечении композиции высокоактивного лактоферрина с высокой чистотой.
Другая задачей изобретения является обеспечение способа, подходящего для преодоления по меньшей мере некоторых недостатков уровня техники.
Высокоактивный лактоферрин характеризуется довольно низким уровнем насыщения железом (уже связанное железо - значение A) и высоким потенциалом связывания железа (железосвязывающая способность - значение C).
Еще одна задача настоящего изобретения состоит в обеспечении композиции, содержащей лактопероксидазу высокой чистоты.
И еще одна задача изобретения состоит в обеспечении способа получения лактоферрина или лактопероксидазы высокой чистоты из вышеуказанного источника, содержащего белки.
Эти цели достигаются при помощи способа получения фракции, содержащей белки лактоферрин или лактопероксидазу, из источника, содержащего по меньшей мере один из указанных белков, причем из источник выбирают из группы, состоящей из молока, молозива, кислой либо сладкой сыворотки, посредством процесса хроматографического разделения при помощи монолитной колонки, обладающей сильными катионообменными свойствами, где в процессе разделения применяют элюирование с градиентом pH или комбинированное элюирование с градиентом pH и соли после загрузки источника в колонку.
В одном из вариантов реализации настоящего изобретения градиент pH обычно начинается в диапазоне pH от 4,0 до <pH 8,0, предпочтительно в диапазоне pH от 4,0 до 7,5, более предпочтительно - в диапазоне pH от примерно 4,0 до примерно <pH 7, в частности в диапазоне pH от примерно 4,5 до примерно <pH 6,5.
В другом варианте реализации градиент pH обычно заканчивается в диапазоне от примерно pH 8 до pH <13, предпочтительно в диапазоне pH от 8 до pH 12, в частности, в диапазоне pH от 8 до pH <12.
Может быть целесообразным отфильтровать источник перед загрузкой его в колонку.
В еще одном варианте реализации градиент соли создают путем повышения концентрации соли, в частности, градиент соли соответствует электропроводности в диапазоне от примерно 5 мСм/см до примерно 55 мСм/см. Рекомендуется использовать нейтральные соли для регулирования концентрации соли, чтобы избежать влияния на pH буферного раствора. В частности, пригодными являются соли, используемые в технологических процессах пищевой промышленности, обычно хлорид натрия.
Градиент pH, используемый в сочетании с упомянутым ранее градиентом соли, начинается со значения pH обычно в диапазоне pH от 4,0 до <pH 8,0, предпочтительно в диапазоне pH от 4,0 до 7,5, более предпочтительно - в диапазоне pH от 4,0 до <pH. 7, в частности, в диапазоне pH от 4,5 до <pH 6,5. Градиент pH, используемый в сочетании с градиентом соли, обычно заканчивается в диапазоне pH от 8 до pH <13, предпочтительно в диапазоне pH от pH 8 до pH 12, в частности, в диапазоне pH от pH 8 до pH <12.
В еще одном варианте реализации изобретения может быть собрана фракция А, которая элюируется в диапазоне рН от примерно 8 до примерно рН <11, предпочтительно в диапазоне рН от 8,0 до рН 10,0, более предпочтительно - в диапазоне рН от 8,2 до pH 10,0, в частности от примерно 8,9 до примерно pH 10. Данная фракция обычно содержит лактопероксидазу.
В еще одном варианте реализации может быть собрана фракция B, которая элюируется в диапазоне pH от >10 до pH 12,0, предпочтительно в диапазоне pH от >10,4 до 12, более предпочтительно - от примерно >11 до примерно 12, точнее от примерно >11 до примерно 11,7. Данная фракция обычно содержит лактоферрин.
В особенно целесообразном варианте реализации способа по изобретению процесс хроматографического разделения включает следующие этапы:
Доведение значения pH источника до значения ниже pH 7, в частности, ниже pH 6,5;
Приведение в контакт источника по пункту (i) с монолитной колонкой, обладающей сильными катионообменными свойствами; за которым следует
Пропускание градиентного буфера через колонку, тем самым увеличивая значение pH; и
Сбор фракции А, которая элюируется в диапазоне pH от примерно 8 до примерно <11, предпочтительно в диапазоне pH от 8,0 до 10,0, более предпочтительно - в диапазоне pH от 8,2 до 10,0, в частности от примерно 8,9 до примерно 10; и/или
Сбор фракции B, которая элюируется в диапазоне pH от >10 до 12,0, предпочтительно - от примерно >10,4 до примерно 12, более предпочтительно - в диапазоне pH от >11,0 до 12,0, в частности - от примерно >11 до примерно 11,7;
Необязательно, дополнительная обработка фракций А и/или В, в частности обработка для нейтрализации, концентрирования, консервирования и т.п.
Может быть полезно отфильтровать источник, содержащий лактоферрин и/или лактопероксидазу, до этапа (ii).
В другом варианте реализации способа по изобретению перед этапом (ii) монолитная колонка может быть уравновешена уравновешивающим буфером с pH примерно <7, в частности - примерно <6.
Монолитная колонка, обладающая сильными катионообменными свойствами, в частности, выбирается из группы, состоящей из следующего: монолитная колонка, модифицированная SO3H, монолитная колонка, модифицированная -COOH, монолитная колонка, модифицированная -OSO3H или монолитная колонка, модифицированная -OPO3H. В контексте настоящего изобретения монолитная колонка, модифицированная SO3H, -COOH, -OSO3H либо -OPO3H также охватывает соответствующие соли кислотных фрагментов, в частности их щелочные соли, такие как соли натрия, калия, например SO3Na, -COONa, -OSO3Na, либо -OPO3Na или SO3K, -COOK, -OSO3K, либо -OPO3K.
Согласно способу по изобретению, фракция A, элюируемая при более низком значении pH, чем фракция B, обычно содержит лактопероксидазу, тогда как фракция B обычно содержит лактоферрин.
В другом варианте реализации перед этапом (iii) или (iv) колонка может быть промыта буфером для уравновешивания.
Обычно фракции, содержащие лактоферрин и лактопероксидазу, могут быть дополнительно обработаны, например высушены, в частности методом распылительной сушки.
Способ по изобретению позволяет получить лактоферрин или лактопероксидазу высокой чистоты. Чистота лактоферрина составляет 98%, а чистота лактопероксидазы - 78%. Кроме того, значение C лактоферрина составляет >50% или >60%, а значение A лактоферрина составляет >1%. В предпочтительном варианте реализации значение С лактоферрина составляет >70%, а значение А лактоферрина - >2%. Предпочтительно значение С лактоферрина составляет ≥70,0%, предпочтительно - от 70,0% до 80,0%, более предпочтительно - от 70,0% до 77,0%. Предпочтительно значение А лактоферрина составляет ≥2,0%, предпочтительно <3,9%, предпочтительно от 1,0% до 7,0%, предпочтительно от 2,0% до 7,0%, предпочтительно от 2,0% до 5,0%, более предпочтительно - от 2% до 4%.
В соответствии с другим вариантом реализации настоящего изобретения, монолитная колонка может быть продезинфицирована промывкой буфером с pH >12 обычно после этапа (iv) или (v).
Объектом настоящего изобретения также является композиция, содержащая лактоферрин или лактопероксидазу, которая может быть получена способом по изобретению. Значение C лактоферрина составляет > 60%, а значение A > 1%. Предпочтительно значение C лактоферрина составляет >70%, а значение A >2%. Предпочтительно значение C лактоферрина составляет ≥70,0%, предпочтительно от 70,0% до 80,0%, более предпочтительно - от 70,0% до 77,0%.
Предпочтительно значение А лактоферрина составляет от 1,0% до 7,0%, предпочтительно - от 2,0% до 7,0%, предпочтительно от 2,0% до 5,0%, более предпочтительно - от 2% до 4%, предпочтительно составляет ≥2,0% и/или предпочтительно <3,9%. Предпочтительно значение А+С продукта по настоящему изобретению составляет по меньшей мере 61%, либо по меньшей мере 72%, либо по меньшей мере 73%.
Способ по настоящему изобретению обеспечивает:
- быстрый и простой метод выделения ЛФ и ЛПО из молока, молозива, а также кислой или сладкой сыворотки,
- одноэтапный хроматографический процесс выделения при помощи линейного градиента pH для выделения ЛФ и ЛПО, который обеспечивает получение высокочистых целевых белков (например, чистота ЛФ составляет >98%),
- продукт (белок ЛФ), полученный описанным способом, который согласно хорошо зарекомендовавшему себя методу измерения Способности связывать ненасыщенное железо (UIBC), является высокоактивным (значение C составляет >70%, набор для определения значения C для ЛФ, NRL Pharma) и содержит малое количество уже связанного железа (значение A составляет <3,9%, набор для определения значения A для ЛФ, NRL Pharma), которые являются главными характеристиками ЛФ, доступного на рынке. Данный продукт производится экономичным и технологически нетребовательным способом без использования особых химикатов и производится непосредственно на месте (на монолитном катионите), .
- способ, позволяющий получать ЛФ, который содержит малое количество железа (значение A <3,9%) и в то же время не содержит ангиогенин,
- способ, позволяющий получать ЛФ с высокой общей биологической активностью (значение C+A составляет >72%),
- способ, который легко может быть расширен до промышленных масштабов получения и позволяющий рентабельно производить высокочистые и биоактивные ЛФ и ЛПО,
- весь процесс выделения ЛФ/ЛПО, включающий хроматографию, концентрацию/обессоливание и процесс сушки, который обеспечивает извлечение ЛФ с итоговым значением >85%,
- хроматографический процесс, который допускает высокую скорость потока подвижной фазы (700 л/м2/ч), не препятствуя высокому разрешению белков, что обеспечивает высокую производительность процесса,
- процесс, при котором в среду, содержащую белки, предназначенные для выделения (например, в сыворотку), не вводятся химические вещества, и не изменяются ее характеристики каким-либо иным образом; благодаря этому возможно дальнейшее использование указанных сред в других биохимических промышленных процессах,
- способ, позволяющий получать ЛФ с заранее определенной долей уже связанного Fe.
Краткое описание иллюстраций
Фиг. 1. Хроматограмма пика элюции ЛФ, состоящая из субпиков элюции ЛФ. Данное явление является следствием небольших различий в значениях изоэлектрической точки (ИЭТ) ЛФ из-за содержания в нем железа. Voswinkel и др. [32] продемонстрировали, что снижение содержания железа вызывает небольшое снижение IEP (изоэлектрической точки) ЛФ.
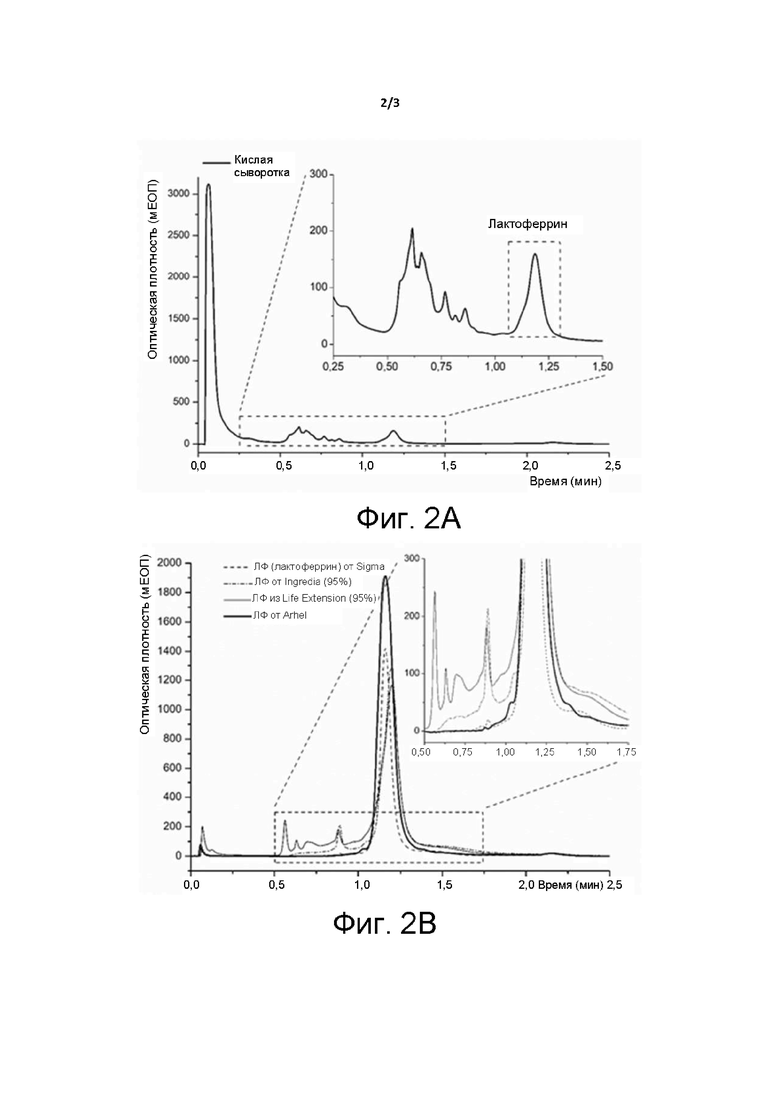
Фиг. 2. Хроматограммы (A) кислой сыворотки и (B) коммерческих продуктов ЛФ и собственно ЛФ.
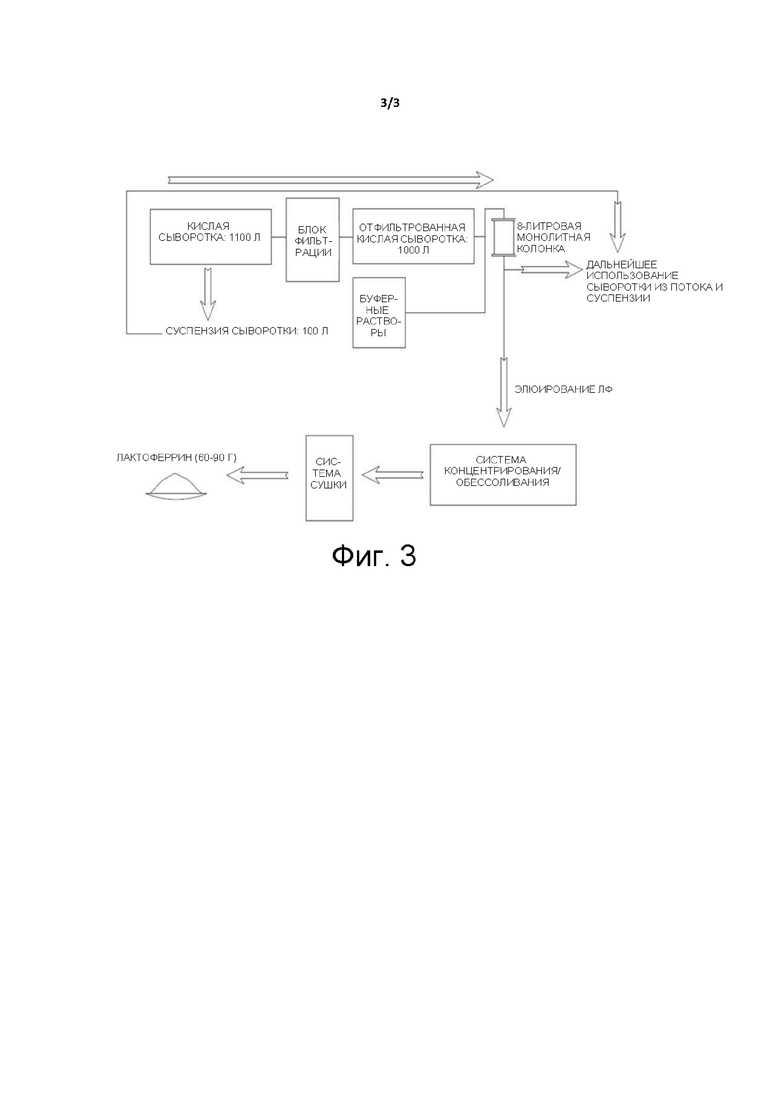
Фиг. 3. Схема процесса выделения ЛФ из кислой сыворотки с помощью одной монолитной колонки CIMmultus™ SO3 объемом 8 л (BIA Separations) и основная информация о массовом балансе процесса.
Подробное описание изобретения
Монолитные колонки с сильными катионообменными группами CIMmultus™ SO3 (сильный катионит от BIA Separations) были испытаны в различных режимах элюирования для выделения ЛФ и ЛПО из отфильтрованной сыворотки или молока по отдельности. Загрузка ЛФ/ЛПО производилась при естественном pH сыворотки, а два целевых белка элюировались отдельно посредством (I) ступенчатого увеличения pH, (II) этапов электропроводности, (III) линейного градиента электропроводности и (IV) линейного градиента pH.
В случае режимов ступенчатого элюирования, использованных для сравнения, достигнутая чистота ЛФ составила примерно 95%, а в случае градиента электропроводности достигнутая чистота была несколько выше 95%. Неожиданно было обнаружено, что при элюировании с повышающимся градиентом pH по изобретению, в частности с линейно повышающимся градиентом pH, достигнутая чистота белка составила более 98%. Данная чистота была подтверждена с помощью ВЭЖХ, анализа SDS-PAGE (ДНС-ПААГ - анализ методом электрофореза в полиакриламидном геле с додецилсульфатом натрия), биоанализатора и эксклюзионной хроматографии. Способ по настоящему изобретению обеспечивает получение указанных результатов за один этап хроматографии, к тому же в промышленном масштабе. Между прочим, согласно методу Лаурелла [26], используемому для расчета чистоты в ЕР 0253395, расчетная чистота продукта по изобретению, который может быть получен способом по изобретению, составляет >118%. Похоже, что установленный метод устарел и более не подходит для правильного определения чистоты ЛФ.
В случае элюирования с линейным градиентом pH было отмечено, что элюция ЛФ состоит из нескольких субпиков меньшей величины (Фиг. 1). Последнее предполагает, что ЛФ также разделялся по уровню содержания железа в белке - от более низкого к более высокому. Причина этого уже обсуждалась и доказывалась другими авторами [32].
Концентрированную водную дисперсию ЛФ сушили методом распылительной сушки или лиофилизатором и затем определяли ненасыщенную железосвязывающую способность (UIBC) ЛФ. Согласно данным набора для колориметрического анализа железа, предоставленного NRL Pharma Inc., Япония (подробный принцип работы опубликован Ito и др. [33]), уровень насыщения железом (уже связанное железо - значение A) ЛФ, полученного способом по настоящему изобретению, составлял от 2% до 4,9%. Его потенциал связывания железа (железосвязывающая способность - значение C), также называемый ненасыщенной железосвязывающей способностью (UIBC), был выше 70%. Данный результат превышает показатели представленных на рынке продуктов, у которых значения A и C находятся в диапазоне 4,6-11,7% и 34,4-52,1% соответственно (см. Таблицу 1). В то же время их общая биологическая активность (значение C + значение A) обычно ниже по сравнению с продуктом по изобретению.
Таблица 1: Биологическая активность образцов ЛФ, доступных на рынке и ЛФ по изобретению (Arhel d.o.o.)
* Общая биоактивность (A+C) выражается в виде суммы значений C и A, что дает процентную долю активного белка в образце.
** Значение пересчитано в соответствии с количеством чистого ЛФ в образце, которое составляет 64%.
В способе по настоящему изобретению предложен способ получения фракции, содержащей белки лактоферрин или лактопероксидазу, из источника, содержащего по меньшей мере один из указанных белков, в котором источник выбран из группы, состоящей из молока, молозиво, кислой или сладкой сыворотки, посредством процесса хроматографического разделения при помощи монолитной колонки, обладающей сильными катионообменными свойствами, в частности, монолитной колонки, модифицированной -SO3H, где в процессе разделения применяют элюирование с градиентом pH после загрузки источника в колонку.
Монолитная колонка обычно представляет собой устройство для хроматографического разделения, имеющее полый корпус, содержащий твердый пористый материал, представляющий собой продукт полимеризации мономеров. Поры данного материала образуются, например, в ходе процесса полимеризации (Патенты США №4923610, 4952344, 4889623).
Подходящие устройства описаны в предшествующем уровне техники, например в EP 1058844, EP 777725, и доступны на рынке. Используемый здесь монолитный материал для хроматографии модифицирован фрагментами -SO3H, которые подвергаются воздействию, помимо прочего, на поверхностях пористого материала. Модификация поверхности группами -SO3H придает материалу так называемые сильные катионообменные свойства (CAX). Специалистам в данной области известно, что другие материалы, например, классифицируются как слабый катионит. В отличие от вышеизложенного, для других целей может быть использован анионообменник (AEX).
Хроматография с помощью градиента pH осуществляется путем повышения pH от начального значения до конечной точки. Данное повышение может быть практически линейным, но может производиться и иным образом по мере получения результата изобретения, т.е. продуктов - лактоферрина и/или лактопероксидазы по изобретению. Специалистам в данной области известно, как производится градиентная хроматография как таковая. Оптимизация условий катионно-градиентной хроматографии, основанная на прямом и косвенном раскрытии настоящего изобретения, входит в обычную практику квалифицированного специалиста и не связана с чрезмерными усилиями и затратами на проведение эксперементов.
Чтобы создать условия для воспроизводимой обработки, может быть целесообразно уравновесить монолитную колонку перед фактическим разделением с помощью градиента pH. В этом случае колонка промывается уравновешивающим буфером. Обычно колонка промывается уравновешивающим буфером в объеме, равном 8-12 неиспользуемым объемам колонки. Выбор начального значения pH для хроматографии в некоторой степени зависит от значения pH источника, содержащего ЛФ и/или ЛПО. Например, если источником для получения ЛФ и/или ЛПО является кислая сыворотка, значение pH для уравновешивания может составлять 4,6, тогда как при использовании сладкой сыворотки значение pH может быть выше, а именно от 5,0 до 6,5.
Может быть целесообразным отфильтровать источник перед его загрузкой в колонку. Как правило, могут быть использованы средства для фильтрования, используемые в молочной промышленности. Особенно подходят керамические фильтры TFF (тангенциальной поточной фильтрации), спирально-витые мембраны или другие технологии непрерывной фильтрации.
После загрузки источника в монолитную колонку, в принципе, можно начинать хроматографию с помощью градиента pH. Однако может быть целесообразно, чтобы перед началом хроматографии для удаления примесей можно было использовать дополнительную промывку колонки буфером со значением pH, близким к условиям уравновешивания. Это способствует разделению получаемых белков, поскольку белки либо иные загрязнители, которые элюируются при данном значении pH, не препятствуют разделению ЛФ и/или ЛПО.
Градиент pH обычно начинается со значения pH в диапазоне от 4,0 до <8,0, предпочтительно в диапазоне от 4,0 до 7,5, более предпочтительно - в диапазоне от примерно 4,0 до примерно <7, в частности, в диапазоне от примеро 4,5 до примерно <6,5. Градиент pH обычно заканчивается в диапазоне pH от примерно 8 до <13, предпочтительно в диапазоне от 8 до 12, в частности от 8 до <12. На Фиг. 1 изображена типичная динамика процесса хроматографии с помощью градиента pH.
Следует отметить, что ионная сила буфера для элюирования также может оказывать некоторое влияние на разделение. В процессе хроматографического разделения ионная сила может также возрастать вслед за повышающимся градиентом, перекрывая величину необходимого градиента pH. Необходимый градиент pH для использования в сочетании с упомянутым ранее градиентом соли обычно начинается со значения pH в диапазоне от 4,0 до <8,0, предпочтительно в диапазоне от 4,0 до 7,5, более предпочтительно - в диапазоне от 4,0 до <7, в частности, в диапазоне от 4,5 до <6,5. Градиент pH, используемый в сочетании с градиентом соли, обычно заканчивается в диапазоне pH от 8 до <13, предпочтительно в диапазоне от 8 до 12, в частности, в диапазоне от 8 до <12.
К примеру, ЛПО элюируется в диапазоне pH от примерно 8 до примерно <11, если ионная сила эквивалентна электропроводности примерно 15 мСм/см, и при более низком pH в диапазоне от примерно 6,6 до примерно 7,5, если электропроводность возрастает с 4 до 55 мСм/см, предпочтительно с 5 до 55 мСм/см. Элюирование ЛФ проходит с аналогичной закономерностью. В случае если величина электропроводности средняя, но остается постоянной, ЛФ элюируется в диапазоне pH от примерно 10,7 до примерно 11,7, при возрастании электропроводности с 4 до 55 мСм/см, предпочтительно с 5 до 55 мСм/см, ЛФ элюируется при более низком pH в диапазоне pH от примерно 9,6 до примерно 10,7. Ионная сила, т. е. электропроводность можно регулировать добавлением подходящих солей. Используя подходящие соли, можно также отрегулировать значение pH буфера для элюирования.
Сбор фракции А, элюирующей в диапазоне pH от примерно 8 до примерно <11, предпочтительно в диапазоне от 8,0 до 10,0, более предпочтительно - в диапазоне от 8,2 до 10,0, в частности, от примерно 8,9 до примерно 10, или от примерно 6,6 до примерно 7,5 происходит при более высокой электропроводности от примерно 5 до 55 мСм/см. Данная фракция обычно содержит лактопероксидазу. Сбор фракции B, элюирующей в диапазоне pH от >10 до 12,0, предпочтительно в диапазоне от >10,4 до 12, более предпочтительно от примерно >11 до примерно 12, в частности, от примерно >11 до примерно 11,7, или от примерно 9,6 до примерно 10,7 происходит при более высокой электропроводности примерно от 5 до 55 мСм/см. Данная фракция обычно содержит лактоферрин.
Далее приводится описание стандартной реализации способа по настоящему изобретению. Диапазоны pH, в которых элюируются ЛФ и ЛПО, соответствуют средней электропроводности примерно 15 мСм/см.
Процесс хроматографического разделения включает следующие этапы:
Доведение значения pH источника до значения ниже pH 7, в частности - ниже pH 6,5;
Приведение в контакт источника по пункту (i) с монолитной колонкой, обладающей сильными катионообменными свойствами, в частности с монолитной колонкой, модифицированной -SO3; за которым следует
Пропускание градиентного буфера через колонку, тем самым увеличивая значение pH; и
Сбор фракции А, которая элюируется в диапазоне pH от примерно 8 до примерно <11, предпочтительно в диапазоне pH от 8,0 до 10,0, более предпочтительно - в диапазоне pH от 8,2 до 10,0, в частности от примерно 8,9 до примерно 10; и/или
Сбор фракции B, которая элюируется в диапазоне pH от >10 до 12,0, предпочтительно - от примерно >10,4 до примерно 12, более предпочтительно - в диапазоне pH от >11,0 до 12,0, в частности - от примерно >11 до примерно 11,7;
Необязатель, дополнительная обработка фракций А и/или В, в частности обработка для нейтрализации, концентрирования, консервирования и т.п.
Для удаления нежелательного материала источника, содержащее лактоферрин и/или лактопероксидазу, фильтруется перед этапом (ii) через керамический фильтр.
Перед этапом (ii) монолитная колонка уравновешивается уравновешивающим буфером с pH примерно <7, в частности, примерно <6. Перед этапом (iii) или (iv) колонка промывается уравновешивающим буфером.
После сбора фракций, содержащих лактоферрин и лактопероксидазу, их дополнительно обрабатывают распылительной сушкой.
Полученный лактоферрин или лактопероксидаза имеет высокую чистоту. Чистота лактоферрина составляет > 98%, а чистота лактопероксидазы > 78%. Кроме того, значение C лактоферрина составляет ≥60%, а значение A лактоферрина составляет ≥1%. В предпочтительном варианте реализации значение C лактоферрина составляет >70%, а значение лактоферрина A составляет >2%. Предпочтительно значение C лактоферрина составляет ≥70,0%, предпочтительно от 70,0% до 80,0%, более предпочтительно от 70,0% до 77,0%. Предпочтительно значение A лактоферрина составляет от 1,0% до 7,0%, предпочтительно от 2,0% до 7,0%, предпочтительно от 2,0% до 5,0%, более предпочтительно - от 2% до 4%, предпочтительно ≥2,0% и/или предпочтительно <3,9%.
В соответствии с другим вариантом реализации монолитную колонку можно дезинфицировать промыванием буфером с pH >13, обычно после этапа (iv) или (v).
Этап дезинфекции осуществляется путем промывки колонки деионизированной водой (10-15 ПО (постоянных объемов)), 1М NaOH (4-10 ПО) со временем контакта от 1 до 3 часов и повторной промывкой водой (>30 ПО). Данный этап можно выполнять через каждые 8-10 рабочих циклов хроматографии.
Уникальность нового подхода согласно способу по настоящему изобретению также заключается в том, что он (I) не требует какого-либо химического изменения источника (например, сыворотки), из которого выделяется белок, а всего лишь предварительную фильтрацию с помощью стандартных методов фильтрации, (II) в отличие от других методов (например, описанных в EP2421894 A1 [29]) в нем используются недорогие химические вещества (буферы) в небольших количествах, (III) обеспечивает получение отдельных высокочистых фракций ЛПО и ЛФ, (IV) позволяет получить ЛФ с заранее определенным содержанием уже связанного Fe и (V) обеспечивает высокую степень извлечения белка, превышающую 85%, включая такие этапы получения, как обессоливание/концентрирование дисперсии элюирования белка и сушку.
Все литературные источники, приведенные в настоящем документе, включены в качестве ссылок в полном объеме, в котором их включение не противоречит изложенной здесь явно выраженной информации.
Далее настоящее изобретение поясняется и иллюстрируется с помощью нижеприведенных неограничивающих примеров.
Примеры:
Аналитическая обработка данных:
Аналитическая обработка данных ВЭЖХ:
Для определения ЛФ и ЛПО в образцах использовали хроматографическую систему, оснащенную многоволновым детектором MWD и устройство для контроля электропроводности с набором для измерения pH (PATfix™, BIA Separations d.o.o., Айдовщина, Словения). Хроматографические разделения производились на аналитической колонке CIMac™ SO3-0.1 (размер пор 1,3 мкм, BIA Separations d.o.o., Айдовщина, Словения) при помощи двух одноосновных буферных растворов фосфата натрия (25 мМ, pH 7,5) в режиме градиента электропроводности, которая повышалась с 3,5 до 152 мСм/см. Элюция анализируемых белков контролироваласть детектором MWD при 226 нм. Подробная информация о данном методе хроматографии представлена в Таблице 2, а хроматограммы некоторых проанализированных образцов показаны на Фиг. 1 и 2. Перед анализом все образцы были профильтрованы через CHROMAFIL® A-45/25, 0,45 мкм, фильтры из смешанных эфиров целлюлозы.
Таблица 2. Хроматографическая система и метод, использованные для анализа образцов.
Хроматографическая система: система ВЭЖХ PATfix™
Хроматографическая колонка: CIMac™ SO3, 100 мкл (BIA Separations d.o.o., Айдовщина, Словения)
Объем впрыска: 15 мкл
Детектирование: 226 нм
Определение значений C и A:
Для определения значений C и A сухих образцов ЛФ мы использовали Набор для колориметрического анализа железа, предоставленный NRL Pharma Inc., Япония (подробный принцип работы опубликован Ito и др. [33]), а для определения значения A сухих и жидких образцов мы использовали методику, описанную в литературе [33, 34] в сочетании с методом ВЭЖХ.
Определение значений C:
Для определения значения C ЛФ ЛФ насыщали известным избыточным количеством железа. Оставшееся железо окрашивали хелатирующим реагентом (максимальное поглощение 760 нм). Оставшееся железо определялось методом спектрофотометрии при 760 нм. Производилось серийное разведение комплекса железа, и методом спектрофотометрии при 760 нм строилась калибровочная кривая. Связанное железо рассчитывалось путем вычитания оставшегося железа из добавленного железа. Значение C указано в относительных единицах, причем расчетная теоретическая железосвязывающая способность ЛФ (каждая молекула ЛФ способна связывать 2 атома железа) принята за 100%.
Расчет значения C:
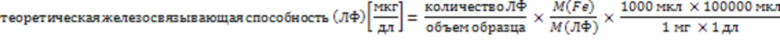 (1)
(1)
M(Fe) - молекулярная масса железа; M(ЛФ) - молекулярная масса ЛФ
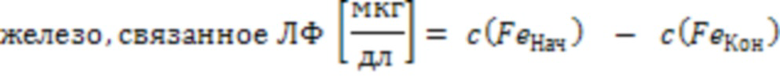 (2)
(2)
c(FeНач) - начальная концентрация железа; c(FeКон) - концентрация оставшегося железа
 (3)
(3)
Определение значений A:
Для определения значения A ЛФ ЛФ денатурировали денатурирующим реагентом и высвобожденное железо окрашивали хелатирующим агентом (максимальное поглощение 760 нм). Высвобожденное железо количественно определяли методом спектрофотометрии при 760 нм. Производилось серийное разведение комплекса железа, и методом спектрофотометрии при 760 нм строилась калибровочная кривая. Уже связанное железо (значение A) указано в относительных единицах, причем 2 атома железа, связанные с одной молекулой ЛФ приняты за 100%.
Расчет значения A:
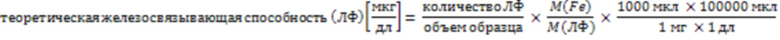 (1)
(1)
M(Fe) - молекулярная масса железа; M(ЛФ) - молекулярная масса ЛФ
 (4)
(4)
Схема экспериментальной версии системы
На Фиг. 3 представлена блок-схема принципа работы технологии выделения ЛФ/ЛПО и приблизительные значения массового баланса процесса в масштабе одного хроматографического цикла. Перед выделением ЛФ/ЛПО кислая сыворотка фильтруется при помощи керамической системы фильтров TFF (тангенциальная поточная фильтрация) с диаметром пор менее 0,8 мкм. Затем отфильтрованная сыворотка (1000 л) прокачивается через уравновешенную хроматографическую колонку. После этого связанные колонкой ЛФ и ЛПО элюируются комбинацией из различных буферных растворов. Затем элюированные фракции концентрируют и при необходимости обессоливают. После сушки концентрированного белка получается от 60 до 90 г продукции в виде сухого ЛФ. Незначительно измененная сыворотка, слитая из колонки, и суспензия сыворотки могут быть в дальнейшем использованы либо переработаны по отдельности или смешанными.
Пример 1
Перед загрузкой 80 мл монолитная колонка CIMmultus™ SO3 от BIA Separations была уравновешена при помощи 800 мл буферного раствора A (фосфат натрия либо цитратный буфер: 5-50 мМ, pH 4,6). После уравновешивания отфильтрованная кислая сыворотка прокачивалась через колонку до достижения предела емкости колонки в плане вмещения ЛФ и ЛПО. Насыщение емкости колонки было проверено путем анализа проб из потока на выходе из колонки методом ВЭЖХ, описанным в разделе «Аналитическая обработка данных». Объем сыворотки, прокачиваемой через колонку со скоростью 0,24 л/мин, обычно составлял от 10 до 20 л, что зависело в основном от концентрации ЛФ/ЛПО в обрабатываемой сыворотке. Затем колонку промывали буферным раствором A. Процесс выделения ЛФ и ЛПО запускали посредством промывки колонки двумя буферными растворами (смесь цитрата натрия, фосфатного, трис- и карбонатного буферов, от 4 до 50 мМ, pH=4,6 и 12,0) в режиме линейного градиента pH. Значение pH постепенно изменялось линейно в диапазоне от 4,6 до 12,0. Диапазоны pH для элюирования ЛФ/ЛПО были в пределах 8,9-10 и 11-11,7 соответственно. В результате выделения были получены две очень хорошо хроматографически разделенные элюционные фракции ЛФ и ЛПО, которые в дальнейшем легко обрабатывались по отдельности. На следующих этапах фракции выделенных белков нейтрализовали до pH=6 при помощи небольшого количества подходящего раствора кислоты, концентрировали с помощью мембраны TFF с размером пор от 1 до 50 кДа и сушили методом распылительной сушки. Конечная чистота ЛФ и ЛПО составила >98% и >70% соответственно. Значения C и A ЛФ составили 71% и 3,4% соответственно.
Пример 2
Перед загрузкой 80 мл монолитная колонка CIMmultus™ SO3 от BIA Separations была уравновешена при помощи буферного раствора B (фосфат натрия либо цитратный буфер: 5-50 мМ, pH = 5,0-6,5 как pH сладкой сыворотки). После этого сладкую сыворотку пропускали через колонку до достижения предела емкости колонки в плане вмещения ЛФ и ЛПО. Насыщение емкости колонки было проверено путем анализа проб из потока на выходе из колонки методом ВЭЖХ, описанным в разделе «Аналитическая обработка данных». Объем сыворотки, прокачиваемой через колонку со скоростью 0,24 л/мин, обычно составлял от 10 до 40 л, что в основном зависело от концентрации ЛФ/ЛПО в обрабатываемой сыворотке. Затем колонку промывали буферным раствором B. Процесс выделения ЛФ и ЛПО запускали посредством промывки колонки двумя буферными растворами (смесь цитрата натрия, фосфатного, трис- и карбонатного буферов, от 4 до 50 мМ, pH=5,0 и 12,0) в режиме линейного градиента pH. Значение pH постепенно изменялось линейно в диапазоне от 5,0 до 12,0. Диапазоны pH для элюирования ЛФ/ЛПО были в пределах 8,9-10 и 11-11,7 соответственно. В результате выделения были получены две очень хорошо хроматографически разделенные элюционные фракции ЛФ и ЛПО, которые в дальнейшем легко обрабатывались по отдельности. На следующих этапах фракции выделенных белков нейтрализовали до pH=6 при помощи небольшого количества подходящего раствора кислоты, концентрировали с помощью мембраны TFF с размером пор от 1 до 50 кДа и сушили методом распылительной сушки. Конечная чистота ЛФ и ЛПО составила >98% и >70% соответственно. Значения C и A ЛФ составили 70,2% и 3,9% соответственно.
Пример 3
Перед загрузкой 8 л монолитная колонка CIMmultus™ SO3 - Strong CEX от BIA Separations была уравновешена 40-80 л буферного раствора C (натрий-фосфатный или цитратный буфер: 5-50 мМ с добавлением NaCl, pH=4,6, электропроводность 15 мСм/см). После этого кислую сыворотку пропускали через колонку до достижения предела емкости колонки в плане вмещения ЛФ и ЛПО. Насыщение емкости колонки было проверено путем анализа проб из потока на выходе из колонки методом ВЭЖХ, описанным в разделе «Аналитическая обработка данных». Объем сыворотки, прокачиваемой через колонку со скоростью потока 8 л/мин, обычно составлял от 1000 до 2000 л, что зависело в основном от концентрации ЛФ/ЛПО в обрабатываемой сыворотке. Затем колонку промывали буферным раствором C. Выделение ЛФ и ЛПО производилось посредством промывки колонки двумя буферными растворами (смесь цитрата натрия, фосфатного, трис- и карбонатного буферов, от 4 до 50 мМ с добавлением NaCl, pH=4,6 и 12.0) в режиме линейного градиента pH. Значение pH постепенно изменялось линейно в диапазоне от 4,6 до 12,0, в то время как электропроводность (15 мСм/см) оставалась постоянной на протяжении всего линейного градиента pH. Диапазоны pH для элюирования ЛПО/ЛФ были в пределах 8,2-9,3 и 10,7-11,2 соответственно. В результате вышеупомянутой процедуры были получены две очень хорошо хроматографически разделенные элюционные фракции ЛПО и ЛФ, которые в дальнейшем легко обрабатывались по отдельности. На следующих этапах фракции выделенных белков нейтрализовали до pH=6 при помощи небольшого количества подходящего раствора кислоты, концентрировали с помощью мембраны TFF с размером пор от 1 до 50 кДа и сушили методом распылительной сушки. Конечная чистота ЛФ/ЛПО составила >98% либо >75%. Значения C и A ЛФ составили 74,2% и 2,5% соответственно.
Пример 4
Перед загрузкой 8 л монолитная колонка CIMmultus™ SO3 - Strong CEX от BIA Separations была уравновешена с помощью буферного раствора C (натрий-фосфатный или цитратный буфер: 5-50 мМ, pH = 5,0-6,5, как pH сладкой сыворотки). После этого кислую сыворотку пропускали через колонку до достижения предела емкости колонки в плане вмещения ЛФ и ЛПО. Насыщение емкости колонки было проверено путем анализа проб из потока на выходе из колонки методом ВЭЖХ, описанным в разделе «Аналитическая обработка данных». Объем сыворотки, прокачиваемой через колонку со скоростью потока 8 л/мин, обычно составлял от 1000 до 2000 л, что зависело в основном от концентрации ЛФ/ЛПО в обрабатываемой сыворотке. Белки с IEP (изоэлектрической точкой) <7,5 сначала элюировались в режиме ступенчатой элюции pH при помощи буферного раствора C с pH = 7,5, а затем производилось выделение ЛФ и ЛПО путем промывки колонки в режиме линейного градиента pH с использованием тех же буферов, что и в Примере 2. Значение pH постепенно изменялось линейно в диапазоне от 7,5 до 12,0. Диапазоны pH для элюирования ЛПО/ЛФ были в пределах 8,9-10 и 11-11,7 соответственно. Затем отдельно собранные элюционные фракции ЛПО и ЛФ нейтрализовали до pH=6 небольшим количеством подходящего раствора кислоты, концентрировали с помощью мембраны TFF с размером пор от 1 до 50 кДа и сушили методом распылительной сушки. Конечная чистота ЛФ и ЛПО составила >98% и >75% соответственно. Значения C и A ЛФ составили 72,6% и 2,8% соответственно.
Пример 5
Перед загрузкой 8 л монолитная колонка CIMmultus™ SO3 - Strong CEX от BIA Separations была уравновешена с помощью буферного раствора C (натрий-фосфатный или цитратный буфер: 5-50 мМ с добавлением NaCl, pH=4,6, электропроводность 15 мСм/см). После этого кислую сыворотку пропускали через колонку до достижения предела емкости колонки в плане вмещения ЛФ и ЛПО. Насыщение емкости колонки было проверено путем анализа проб из потока на выходе из колонки методом ВЭЖХ, описанным в разделе «Аналитическая обработка данных». Объем сыворотки, прокачиваемой через колонку со скоростью потока 8 л/мин, обычно составлял от 1000 до 2000 л, что зависело в основном от концентрации ЛФ/ЛПО в обрабатываемой сыворотке. Затем колонку промывали буферным раствором C. Белки с IEP <7,5 сначала элюировали в режиме ступенчатой элюции pH с помощью буферного раствора C с pH=7,5, а затем производили выделение ЛФ и ЛПО путем промывки колонки в режиме линейного градиента pH с использованием тех же буферов, что и в Примере 3. Значение pH постепенно изменялось линейно в диапазоне от 7,5 до 12,0, в то время как электропроводность (15 мСм/см) оставалась постоянной на протяжении всего градиента pH. Диапазоны pH для элюирования ЛПО/ЛФ были в пределах 8,2-9,3 и 10,7-11,2 соответственно. Затем отдельно собранные элюционные фракции ЛПО и ЛФ нейтрализовали до pH=6 небольшим количеством подходящего раствора кислоты, концентрировали с помощью мембраны TFF с размером пор от 1 до 50 кДа и сушили методом распылительной сушки. Конечная чистота ЛФ и ЛПО составила >98% и >80% соответственно. Значения C и A ЛФ составили 71,6% и 3,2% соответственно.
Пример 6
Перед загрузкой 8 л монолитная колонка CIMmultus™ SO3 - Strong CEX от BIA Separations была уравновешена с помощью буферного раствора C (натрий-фосфатный или цитратный буфер: 5-50 мМ, pH = 4,6). После этого кислую сыворотку пропускали через колонку до достижения предела емкости колонки в плане вмещения ЛФ и ЛПО. Насыщение емкости колонки было проверено путем анализа проб из потока на выходе из колонки методом ВЭЖХ, описанным в разделе «Аналитическая обработка данных». Объем сыворотки, прокачиваемой через колонку со скоростью потока 8 л/мин, обычно составлял от 1000 до 2000 л, что зависело в основном от концентрации ЛФ/ЛПО в обрабатываемой сыворотке. Затем колонку промывали буферным раствором C. Белки с IEP <7,5 сначала элюировали в режиме ступенчатой элюции pH с помощью буферного раствора C с pH = 7,5, а затем производили выделение ЛФ и ЛПО путем промывки колонки двумя буферными растворами (смесь цитрата натрия, фосфатного, трис- и карбонатного буферов, от 4 до 50 мМ с добавлением NaCl для достижения требуемой электропроводности, pH = 4,6 и 12,0). Значение pH постепенно изменяли в диапазоне от 7,0 до 12,0, а градиент электропроводности увеличивался с 4 до 55 мСм/см. Диапазоны pH для элюирования ЛПО/ЛФ были в пределах 6,6-7,5 и 9,6-10,7 соответственно. Затем отдельно собранные элюционные фракции ЛПО и ЛФ нейтрализовали до pH=6 небольшим количеством подходящего раствора кислоты, обессоливали и концентрировали с помощью мембраны TFF с размером пор от 1 до 50 кДа и сушили методом распылительной сушки. Конечная чистота ЛФ и ЛПО составила >98% либо >85%. Значения C и A ЛФ составили 76,1% и 2,0% соответственно.
Использованная литература:
[1] Susana A. Gonzalez-Chavez, Sigifredo Arevalo-Gallegos, and QuintinRascon-Cruz. Лактоферрин: структура, функции и применение. Международный журнал противомикробных агентов, 33(4): 301.e1 - 301.e8, 2009.
[2] Nicolas Urtasun, Maria Fernanda Baieli, Daniela BelenHirsch, Maria Camila Martinez-Ceron, Osvaldo Cascone, and Federico Javier Wolman. Очистка лактопероксидазы из сыворотки с помощью аффинной хроматографии с красителем. Обработка пищевых продуктов и биопродуктов, 103:58 - 65, 2017.
[3] Lin Chen, Chen Guo, Yueping Guan, and Huizhou Liu. Выделение лактоферрина из кислой сыворотки методом магнитной аффинной сепарации. Технология разделения и очистки, 56 (2): 168 - 174, 2007. 56(2):168 - 174, 2007.
[4] Rhee-M.S. Imm J.Y. Noh, K.H. Отделение лактоферрина от модельной смеси сывороточного белка обратными мицеллами, образованными катионным поверхностно-активным веществом. Food Sci. Biotechnol., 14:131-136., 2005.
[5] Shalan Alwan Al-Mashikhi, Eunice Li-Chan, and ShuryoNakai. Разделение иммуноглобулинов и лактоферрина из сырной сыворотки с помощью хелатной хроматографии. Journal of Dairy Science, 71(7):1747 - 1755, 1988.
[6] S. Yoshida, Z. Wei, Y. Shinmura, and N. Fukunaga. Выделение лактоферринов а и b из коровьего молозива. Journal of Dairy Science, 83(10):2211 - 2215, 2000.
[7] Jonatan Andersson and Bo Mattiasson. Технология псевдодвижущегося слоя с упрощенным подходом к очистке протеина: Отделение лактопероксидазы и лактоферрина от концентрата сывороточного протеина. Journal of Chromatography A, 1107(1):88 - 95, 2006.
[8] Rong Rong Lu, Shi Ying Xu, Zhang Wang, and Rui Jin Yang. Выделение лактоферрина из коровьего молозива с помощью ультрафильтрации в сочетании с сильной катионообменной хроматографией в промышленных масштабах. Journal of Membrane Science, 297(1):152 - 161, 2007.
[9] T. Uchida, S. Dosako, K. Sato, and H. Kawakami. Последовательное разделение лактоферрина, лактопероксидазы и секреторного компонента методом сульфатно-связанной ионообменной хроматографии. Milchwissenschaft, 9(58):482-486, January 2003.
[10] O. Cascone Mariano Grasselli. Выделение лактоферрина из бычьей сыворотки с помощью аффинной хроматографии с красителем. Netherlands Milk and Dairy Journal, 4(50):551-561, January 1996.
[11] Enrique Alvarez-Guerra and Angel Irabien. Экстракция лактоферрина с помощью гидрофобных ионных жидкостей. Технология разделения и очистки, 98:432 - 440, 2012.
[12] Vojtech Adam, OndrejZitka, Petr Dolezal, Ladislav Zeman, Ales Horna, Jaromir Hubalek, Jan Sileny, Sona Krizkova, Libuse Trnkova, and Rene Kizek. Выделение лактоферрина с помощью монолитной колонки, соединенной со спектрометрическим или микроамперометрическим детектором. Sensors (Basel), 1(8):464-487, Январь 2008.
[13] J.H. Nuijens and H.H. Van Veen. Выделение лактоферрина из молока, 19 января 1999 г. Патент США 5861491.
[14] Conan J. Fee and Amita Chand. Улавливание лактоферрина и лактопероксидазы из сырого цельного молока с помощью катионообменной хроматографии. Технология разделения и очистки, 48(2):143-149, 2006. Разделение и очистка в пищевой промышленности.
[15] Chalore Teepakorn, Koffi Fiaty, and Catherine Charcosset. Сравнение мембранной хроматографии и монолитной хроматографии для разделения лактоферрина и альбумина бычьей сыворотки. Processes, 4(3), 2016.
[16] P. Milavec Zmak, H. Podgornik, J. Jancar, A. Podgornik, and A. Strancar. Перенос градиентных хроматографических методов разделения белков на монолитные колонки со средами конвективного взаимодействия. Journal of Chromatography A, 1006(1):195 - 205, 2003. Международный симпозиум по препаративной и промышленной хроматографии и смежным технологиям.
[17] Shuichi Yamamoto and Ayako Kita. Теоретические основы коротких хроматографических слоев: Оптимизация градиентного элюирования в коротких колонках. Journal of Chromatography A, 1065(1):45-50, 2005. 1st Monolith Summer School.
[18] Bo Wang, Yakindra Prasad Timilsena, Ewan Blanch, and Benu Adhikari. Характеристики порошков коровьего лактоферрина, полученных с помощью процессов распылительной и сублимационной сушки. Международный журнал биологических макромолекул, 95:985-994, 2017.
[19] K. Sato and S.I. Dousako. Процесс разделения, очистки и извлечения белков молока, способных связывать железо, 27 марта 1991. Заявка на европейский патент EP19900117442.
[20] K. Sato, M. Shiba, A. Shigematsu, N. Teduka, and A. Tomizawa. Способ получения лактоферрина, 13 октября 2004 г. Заявка на европейский патент EP 20040008008.
[21] G. Rowe, H. Aomari, and D. Petitclerc. Новый метод очистки лактоферрина, 16 ноября 2006. WO Patent App. PCT/CA2006/000,780.
[22] H. Kawakami, M. Tanimoto, and S. Dousako. Процесс выделения и очистки лактоферрина из молока с использованием сульфатного соединения, 9 сентября 1992 г. EP Patent 0,348,508.
[23] Z. Mozaffar, S.H. Ahmed, V. Saxena, and Q.R. Miranda. Последовательное сепарирование сыворотки, 1 августа 2000 г. Патент США 6096870.
[24] J. Souppe. Изолят молочного белка и способ его получения, 10 декабря 2013 г. Патент США 8603560.
[25] Kussendrager K.D., Kivits M.G.C., and Verver A.B. Процесс выделения лактоферрина и лактопероксидазы из молока и молочных продуктов, а также продуктов, полученных с помощью этого процесса, 16 июня 1998 г. Патент США 2128111.
[26] S. Okonogi, M. Tomita, T. Tomimura, Y. Tamura, and T. Mizota. Процесс получения коровьего лактоферрина высокой чистоты, 5 июля 2006 г. EP Patent 0,253,395.
[27] K.D. Kussendrager, M.G.C. Kivits, and A.B. Verver. Процесс выделения лактоферрина и лактопероксидазы из молока и молочных продуктов, а также продуктов, полученных таким способом, 21 января 1997 г. Патент США 5596082.
[28] Jean-Paul Perraudin. Метод получения лактоферрина, август 2015 г. Патент США 9,115.211 B2.
[29] Kevin Neil Pearce Shaojiang Chen. Метод получения лактоферрина с низким содержанием железа, февраль 2012. EP2421894A1.
[30] Hassan Mohamed Hassan Abdalla. Метод очистки лактоферрина, декабрь 2014. Патент США 9458225B2.
[31] Jan H. Nuijens Veen Harry H. Van. Выделение лактоферрина из молока, август 1995 г. WO1995022258A2.
[32] Linda Voswinkel, Thomas Vogel, and Ulrich Kulozik. Влияние насыщения железом коровьего лактоферрина на адсорбцию на мембране сильного й катионообменника. International Dairy Journal, 56:134-140, 2016.
[33] Satoshi Ito, Katsuya Ikuta, Daisuke Kato, Kotoe Shibusa, Noriyasu Niizeki, Hiroki Tanaka, Lynda Addo, Yasumichi Toki, Mayumi Hatayama, Junki Inamura, MotohiroShindo, Katsunori Sasaki, Naomi Iizuka, Mikihiro Fujiya, Yoshihiro Torimoto, and Yutaka Kohgo. Система анализа несвязанного трансферрином железа с помощью стандартного автоматического анализатора. ClinicaChimica Acta, 437:129 - 135, 2014.
[34] Robert J. Hilton, Matthew C. Seare, N. David Andros, Zachary Kenealey, Catalina Matias Orozco, Michael Webb, and Richard K. Watt. Фосфат ингибирует загрузку Fe3+ в трансферрин in vitro путем образования растворимого фосфатного комплекса Fe(iii): Потенциальные частицы железа, не связанные трансферрином. Journal of Inorganic Biochemistry, 110:1 - 7, 2012.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПРОИЗВОДСТВА ЛАКТОФЕРРИНА, ФРАКЦИЯ, СОДЕРЖАЩАЯ ЛАКТОФЕРРИН, И ЕЕ ПРИМЕНЕНИЯ | 2010 |
|
RU2579661C2 |
| СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ ЛАКТОФЕРРИНА ИЗ МОЛОЧНОГО СЫРЬЯ | 2016 |
|
RU2634859C1 |
| СПОСОБ ПЕРЕРАБОТКИ МОЛОКА И ПРОДУКТ, ПОЛУЧЕННЫЙ ЭТИМ СПОСОБОМ | 2012 |
|
RU2535876C2 |
| УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ОЧИСТКИ ЛАКТОФЕРРИНА МОЛОКА И ЕГО ПРОДУКТЫ | 2013 |
|
RU2683228C2 |
| Способ получения лактоферрина | 2018 |
|
RU2717340C1 |
| Применение проточной фильтрующей центрифуги для извлечения лактоферрина из молочного сырья | 2021 |
|
RU2780347C1 |
| СПОСОБ ПОЛУЧЕНИЯ БАД "Л-ПФИ" ИЗ ВТОРИЧНОГО МОЛОЧНОГО СЫРЬЯ И ПОЛУЧЕННАЯ ЭТИМ СПОСОБОМ БАД "Л-ПФИ" | 2009 |
|
RU2400106C1 |
| ОБОГАЩЕННЫЕ АНГИОГЕНИНОМ ФРАКЦИИ МОЛОКА | 2009 |
|
RU2538654C2 |
| СПОСОБ ОЧИСТКИ БЕЛКА СЛИЯНИЯ | 2015 |
|
RU2698654C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИММУНОГЛОБУЛИНОВ ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ И ДРУГИЕ ИММУНОГЛОБУЛИНОВЫЕ ПРОДУКТЫ | 1999 |
|
RU2197500C2 |
Группа изобретений относится к области биотехнологии, в частности к способам выделения белков из молочного сырья. Описан способ получения фракции, содержащей белки лактоферрин и/или лактопероксидазу, из источника, выбранного из группы: молоко, молозиво, кислая либо сладкая сыворотка, посредством процесса хроматографического разделения с помощью монолитной колонки, обладающей катионобменными свойствами, в котором в процессе разделения применяется комбинированное элюирование с градиентом pH и соли после загрузки источника в колонку, где градиент pH начинается в диапазоне pH от примерно 4,0 до примерно менее 8,0 и заканчивается в диапазоне от примерно 8 до менее 13. Кроме того, раскрывается композиция, содержащая лактоферрин, где лактоферрин имеет железосвязывающую способность более 60% и уровень насыщения железом более 1%, а также композиция, содержащая лактопероксидазу, где чистота лактопероксидазы составляет более 70%. Изобретения позволяют получить лактоферрин и лактопероксидазу с высокой степенью чистоты. 5 н. и 14 з.п. ф-лы, 3 ил., 2 табл., 6 пр.
1. Способ получения фракции, содержащей белок лактоферрин, из источника, содержащего указанный белок, где источник выбран из группы, состоящей из молока, молозива, кислой или сладкой сыворотки, посредством процесса хроматографического разделения с помощью монолитной колонки, обладающей сильными катионообменными свойствами, в котором применяют комбинированное элюирование с градиентом pH и соли после загрузки источника в колонку,
где градиент pH начинается в диапазоне pH от примерно 4,0 до примерно менее 8,0,
где градиент pH заканчивается в диапазоне от примерно 8 до менее 13.
2. Способ получения фракции, содержащей белок лактопероксидазу, из источника, содержащего указанный белок, где источник выбран из группы, состоящей из молока, молозива, кислой или сладкой сыворотки, посредством процесса хроматографического разделения с помощью монолитной колонки, обладающей сильными катионообменными свойствами, в котором применяют комбинированное элюирование с градиентом pH и соли после загрузки источника в колонку,
где градиент pH начинается в диапазоне pH от примерно 4,0 до примерно менее 8,0,
где градиент pH заканчивается в диапазоне от примерно 8 до менее 13.
3. Способ получения фракции, содержащей белки лактоферрин и лактопероксидазу, из источника, содержащего указанные белки, где источник выбран из группы, состоящей из молока, молозива, кислой или сладкой сыворотки, посредством процесса хроматографического разделения с помощью монолитной колонки, обладающей сильными катионообменными свойствами, в котором применяют комбинированное элюирование с градиентом pH и соли после загрузки источника в колонку,
где градиент pH начинается в диапазоне pH от примерно 4,0 до примерно менее 8,0,
где градиент pH заканчивается в диапазоне от примерно 8 до менее 13.
4. Способ по любому из пп. 1-3, отличающийся тем, что источник фильтруют перед загрузкой источника в колонку.
5. Способ по любому из пп. 2-4, отличающийся тем, что собирают фракцию А, которая элюируется в диапазоне pH от примерно 8 до примерно менее 11, в частности от примерно 8,9 до примерно 10 или от примерно 6,6 до примерно 7,5 при более высокой электропроводности в диапазоне примерно от 5 до 55 мСм/см.
6. Способ по любому из пп. 1, 3 или 4, отличающийся тем, что собирают фракцию В, которая элюируется в диапазоне pH от примерно более 10,4 до примерно 12, в частности от примерно более 11 до примерно 11,7 или от примерно 9,6 до примерно 10,7 при более высокой электропроводности в диапазоне примерно от 5 до 55 мСм/см.
7. Способ по любому из пп. 1-6, отличающийся тем, что процесс хроматографического разделения включает этапы:
(i) доведение значения pH источника до значения ниже 7, в частности ниже 6,5;
(ii) приведение в контакт источника с этапа (i) с монолитной колонкой, обладающей сильными катионообменными свойствами; за которым следует
(iii) пропускание буфера с градиентом pH через колонку, тем самым увеличивая значение pH; и
(iv) сбор фракции A, которая элюируется в диапазоне pH от примерно 8 до примерно менее 11, в частности от примерно 8,0 до примерно 10, предпочтительно в диапазоне pH от примерно 8,2 до примерно 10, более предпочтительно от примерно 8,9 до примерно 10 или от примерно 6,6 до примерно 7,5 при более высокой электропроводности в диапазоне примерно от 5 до 55 мСм/см и обычно содержит лактопероксидазу; и/или
(v) сбор фракции B, которая элюируется в диапазоне pH от примерно более 10 до примерно 12,0, предпочтительно от примерно более 10,4 до примерно 12, более предпочтительно от примерно более 11,0 до примерно 12,0, в частности от примерно более 11 до примерно 11,7 либо от примерно 9,6 до примерно 10,7 при более высокой электропроводности примерно от 5 до 55 мСм/см и обычно содержит лактоферрин;
(vi) необязательно, дополнительную обработку фракций А и/или В, в частности, путем обработки для нейтрализации, концентрирования, консервирования и тому подобного.
8. Способ по п. 7, отличающийся тем, что источник фильтруют перед этапом (ii) или монолитную колонку перед этапом (ii) уравновешивают уравновешивающим буфером с pH примерно менее 7, в частности примерно менее 6.5.
9. Способ по любому из пп. 1-8, отличающийся тем, что монолитную колонку, обладающую сильными катионообменными свойствами, выбирают из группы, состоящей из монолитной колонки, модифицированной -SO3H, монолитной колонки, модифицированной -COOH, монолитной колонки, модифицированной -OSO3H, или монолитной колонки, модифицированной -OPO3H.
10. Способ по любому из пп. 1-9, отличающийся тем, что градиент соли достигают посредством концентрации солей, в частности градиент соли соответствует электропроводности в диапазоне от примерно 5 мСм/см до примерно 55 мСм/см.
11. Способ по любому из пп. 7-10, отличающийся тем, что перед этапом (iii) или (iv) колонку промывают уравновешивающим буфером по п. 8.
12. Способ по любому из пп. 1-11, отличающийся тем, что фракцию, содержащую белок, сушат, в частности, методом распылительной сушки.
13. Способ по любому из пп. 3-12, отличающийся тем, что чистота лактоферрина составляет более 90% и чистота лактопероксидазы составляет более 50%, в частности железосвязывающая способность лактоферрина составляет более 50 и уровень насыщения железом лактоферрина составляет более 1.
14. Способ по п. 1, отличающийся тем, что чистота лактоферрина составляет более 90%, в частности железосвязывающая способность лактоферрина составляет более 50 и уровень насыщения железом лактоферрина составляет более 1.
15. Способ по п. 2, отличающийся тем, что чистота лактопероксидазы составляет более 70%.
16. Способ по любому из пп. 7-15, отличающийся тем, что после этапа (iv) или (v) колонку дезинфицируют путем промывания колонки буфером с pH более 12.
17. Композиция, содержащая лактоферрин, получаемая способом по пп. 1 или 3-16, где лактоферрин имеет железосвязывающую способность более 60% и уровень насыщения железом более 1%.
18. Композиция по п. 17, отличающаяся тем, что железосвязывающая способность составляет 70% или более и уровень насыщения железом составляет 5% или менее.
19. Композиция, содержащая лактопероксидазу, получаемая способом по пп. 2 или 3-16, где чистота лактопероксидазы составляет более 70%.
| СПОСОБ ПРОИЗВОДСТВА ЛАКТОФЕРРИНА, ФРАКЦИЯ, СОДЕРЖАЩАЯ ЛАКТОФЕРРИН, И ЕЕ ПРИМЕНЕНИЯ | 2010 |
|
RU2579661C2 |
| US 5516675 А1, 14.05.1996 | |||
| US 5149647 A1, 22.09.1992 | |||
| US 6096870 A1, 01.08.2000 | |||
| СПОСОБ ВЫДЕЛЕНИЯ И ОЧИСТКИ ЛАКТОФЕРРИНА ИЗ МОЛОЧНОГО СЫРЬЯ | 2016 |
|
RU2634859C1 |
| WO 2007103525 А2, 13.09.2007. | |||

