Область техники
Настоящее изобретение, принадлежащее к области исследования и разработки противораковых соединений, относится к ряду ДНК-алкилирующих агентов, активируемых под действием фермента AKR1C3, полученных в результате проведения дальнейшего исследования и разработки соединений, описанных в заявке на патент №PCT/US2016/021581 (публикации под №WO2016145092A1), которая соответствует заявке на патент Китая №2016800150788 (публикации под №CN107530556A).
Предшествующий уровень техники
Заявка Китая на выдачу патента на изобретение CN201910392606.7 под заголовком "Фторсодержащее соединение и его применение в медицине против рака", поданная заявителем 13 мая 2019 г.,
и
заявка Китая на выдачу патента на изобретение CN201911324466.6 под заголовком "Фторсодержащее соединение и его применение в медицине против рака", поданная заявителем 20 декабря 2019 г.,
включены в данное описание посредством ссылки.
Все соединения среди ДНК-алкилирующих противораковых лекарственных средств, направленно воздействующих на сверхэкспрессируемую альдокеторедуктазу 1С3 (AKR1C3) (ДНК-алкилирующие агенты из заявки на патент №PCT/US2016/021581 (публикация под №WO2016/145092), которая соответствует заявке на патент Китая №CN2016800150788 (публикация №CN107530556A)), разработанные в компании авторов настоящего изобретения, представляют собой масла желтого цвета. Поскольку эти соединения не являются твердыми формами, при последующих работах по исследованию и разработке композиции возникают приведенные далее затруднения.
Разделение и очистка представляют собой сложный и дорогостоящий процесс. Ввиду того, что эти соединения представляют собой масла, их невозможно очистить методами высокоэффективной/малозатратной перекристаллизации или суспензионной очистки, и они могут быть очищены только посредством колоночной хроматографии. Это приводит к усложнению процесса и, таким образом, к высоким затратам при получении фармацевтически активных ингредиентов.
Композиция в такой форме неудобна для использования и обладает плохой стабильностью. Процедуры перенесения/отмеривания масел не могут быть выполнены с легкостью. Важно отметить, что форма масла делает невозможным или неудобным разработку и приготовление разнообразных лекарственных форм композиции. Как правило, возможна разработка только в виде лиофилизированного порошка для инъекций или раствора для инъекций для введения, при этом способ введения будет менее разнообразным и дорогостоящим. Кроме того, некоторые пациенты не очень хорошо реагируют на введение лиофилизированного порошка или раствора для инъекций.
Краткое описание сущности изобретения
Для решения вышеупомянутых технических проблем в настоящем изобретении предложены разработка и синтез ряда фторсодержащих соединений путем структурной модификации соединений, описанных в патентной заявке №PCT/US2016/021581 (публикация №WO2016/145092), которая соответствует заявке на патент Китая №CN2016800150788 (публикация №CN107530556A) под заголовком "ДНК-алкилирующие агенты".
Соответственно, вышеупомянутая патентная заявка №PCT/US2016/021581 (публикация №WO2016/145092), которая соответствует заявке на патент Китая №CN2016800150788 (публикация №CN107530556A), включена в данное описание посредством ссылки. Если какое-либо определение или понятие, представленное в данном описании, отличается от определения или понятия, представленного в вышеупомянутых заявочных материалах, преимущественную силу имеет определение или понятие, представленное в данном описании. Если какое-либо понятие или определение, представленное в данном описании, четко не определено или ограничено, оно должно быть определено в соответствии с вышеупомянутыми заявочными материалами. Другие понятия или определения, которые четко не определены или не ограничены ни в данном описании, ни в вышеупомянутых заявочных материалах, следует интерпретировать в соответствии, среди прочего, с учебниками и справочниками по органической химии и медицинской химии.
Доказано, что соединения 3424, описанные в патентных заявках PCT/US2016/021581, PCT/US2016/025665 и PCT/US2016/062114 в качестве признанных во всем мире низкомолекулярных терапевтических лекарственных средств направленного действия с высокой селективностью в отношении опухолей, продемонстрировали превосходные противораковые эффекты в различных доклинических исследованиях на клетках и животных моделях. Эти соединения, как специфичные субстраты альдокеторедуктазы AKR1C3, могут быть быстро и эффективно восстановлены только в раковых клетках, которые сверхэкспрессируют AKR1C3, тем самым происходит высвобождение цитотоксинов, что приводит к высокоселективному действию по уничтожению раковых клеток.
Как продемонстрировано в документах (включая документ 1: Richard В. Lock, Kathryn Evans, Raymond Yung, Tara Pritchard, Beverly A. Teicher, JianXin Duan, Yuelong Guo, Stephen W. Erickson, Malcolm A. Smith. The AKR1C3-Activated Prodrug OBI-3424 Exerts Profound In Vivo Efficacy Against Preclinical Models of T-Cell Acute Lymphoblastic Leukemia (T-ALL); a Pediatric Preclinical Testing Consortium LCM Study (реферат). В: Proceedings of the AACR-NCl-EORTC International Conference: Molecular Targets and Cancer Therapeutics; 2017, Oct 26-30; Philadelphia, PA. Philadelphia (PA): AACR; Mol. Cancer Ther., 2018; 17 (1 Suppl): номер реферата LB-B16; документ 2: Evans К, Duan J, Pritchard T, Jones CD, McDermottL, Gu Z, Toscan CE, EI-ZeinN, MayohC, Erickson SW, Guo Y, Meng F, Jung D, Rathi KS, Roberts KG, Mullighan CG, Shia CS, Pearce T, Teicher BA, Smith MA, Lock RB. OBI-3424, a novel AKR1C3-activated prodrug, exhibits potent efficacy against preclinical models of T-ALL. Clin. Cancer Res., 2019, Apr 23. pii:clin canres.0551.2019.doi:10.1158/1078-0432.CCR-19-0551), соединение AST-3424 (OBI-3424) (т.е. S-изомер соединения 2870) показало удовлетворительное действие в отношении таких раковых заболеваний, как лейкоз и рак легкого, в I фазе клинических испытаний.
Поскольку все соединения этого ряда представляют собой масла, имеющие массу недостатков вследствие неудобств при их хранении, транспортировке или дозировании, существует необходимость в разработке аналогичных соединений, которые при комнатной температуре являются твердыми веществами.
Исследовательская группа полагала применить такое решение: превращение масел в твердые соли с использованием процесса образования солей. Тем не менее, на основании этих экспериментов было обнаружено, что реакция образования солей, протекающая между неорганическими кислотами, такими как серная кислота/соляная кислота, и азотсодержащим трехчленным кольцом, не может приводить к получению желаемой соли: эксперименты продемонстрировали, что в кислотных условиях азотсодержащее трехчленное кольцо в соединениях, описанных в вышеупомянутом документе CN107530556A, будет раскрываться с образованием побочных продуктов с открытым кольцом. Многочисленные экспериментальные испытания указывают на то, что вышеупомянутые традиционные методы нецелесообразны.
На основании опыта и экспериментальных результатов исследовательская группа нестандартным образом ввела фторсодержащие группы, такие как конкретные трифторметильные группы, фторзамещенные арильные группы или гетероарильные группы, в конкретные положения (как например, в положение между нитробензольным кольцом и амин-содержащей фосфатной группой) в структурах указанных выше соединений. После проведения этой модификации было обнаружено, что все полученные соединения представляли собой твердые вещества (включая твердые и воскообразные вещества).
Дальнейшие эксперименты in vitro показывают, что эти соединения обладают высокой активностью в отношении ингибирования пролиферации раковых клеток in vitro. Кроме того, обнаружено, что комбинация данных соединений и ингибитора AKR1C3 ТН3021 обладает более низкой ингибирующей активностью. Это демонстрирует, что введение специфичной фторсодержащей группы в конкретное положение может способствовать не только получению соединения в твердом состоянии, чтобы облегчить приготовление композиций и содействовать дозированию и хранению, но также может способствовать получению ДНК-алкилирующих агентов, активируемых ферментом AKR1C3.
Предложены соединение формулы I, II или III или его фармацевтически приемлемая соль или сольват,
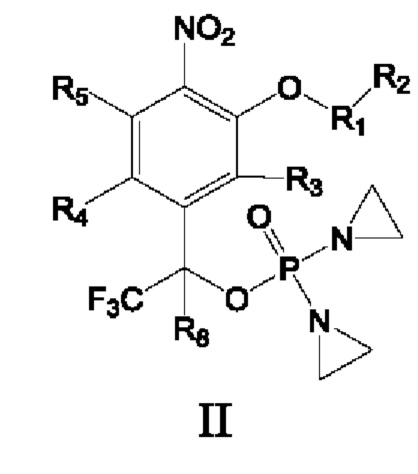
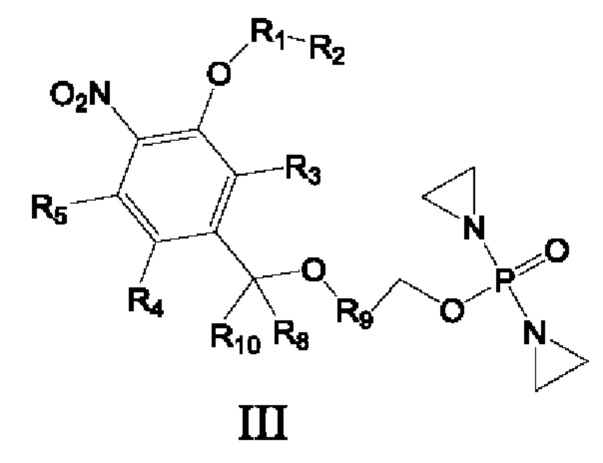
где
R1 представляет собой С6-С10арил или Z-замещенный арил, 4-15-членный гетероцикл или Z-замещенный гетероцикл, 5-15-членный гетероарил или Z-замещенный гетероарил либо 7-15-членное конденсированное кольцо или Z-замещенное конденсированное кольцо;
R2 представляет собой атом водорода, галогена, циано или изоциано, гидрокси, сульфгидрил, амино, OTs, OLCMS, С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил, С2-С6алкинил или Z-замещенный алкинил, С3-С8циклоалкил или Z-замещенный циклоалкил, С6-С10арил или Z-замещенный арил, 4-15-членный гетероцикл или Z-замещенный гетероцикл, 5-15-членный гетероарил или Z-замещенный гетероарил, простой эфир, имеющий от 1 до 6 атомов углерода, или Z-замещенный алкокси, имеющий от 1 до 6 атомов углерода, -CONR6R7, -SO2NR6R7, -SO2R6, -SO2R6, -OCOO-R6, -COOR6, -NR6COR7, -OCOR6, -NR6SO2R7 или -NR6SO2NR6R7, или R2 вместе с атомом в группе R1, к которому он присоединен, образуют 7-15-членное конденсированное кольцо или Z-замещенное конденсированное кольцо;
R3 представляет собой атом водорода, галогена, циано или изоциано, гидрокси, сульфгидрил, амино, OTs, OLCMS, С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил, С2-С6алкинил или Z-замещенный алкинил, С3-С8циклоалкил или Z-замещенный циклоалкил, С6-С10арил или Z-замещенный арил, 4-15-членный гетероцикл или Z-замещенный гетероцикл, 5-15-членный гетероарил или Z-замещенный гетероарил, С1-С6алкокси или Z-замещенный С1-С6алкокси, -CONR6R7, -SO2NR6R7, -SO2R6, -OCOR6, -OCOO-R6, -COOR6, -NR6COR7, -OCOR6 или -NR6SO2R7;
R4 и R5 каждый независимо представляет собой атом водорода, галогена, циано или изоциано, гидрокси, сульфгидрил, амино, OTs, OLCMS, С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил,
С2-С6алкинил или Z-замещенный алкинил, С3-С8циклоалкил или Z-замещенный циклоалкил, С6-С10арил или Z-замещенный арил, 4-15-членный гетероцикл или Z-замещенный гетероцикл, 5-15-членный гетероарил или Z-замещенный гетероарил, С1-С6алкокси или Z-замещенный С1-С6алкокси, -CONR6R7, -SO2NR6R7, -SO2R6, -OCOO-R6, -COOR6, -NR6COR6, -OCOR6 или -NR6SO2R7, или R4 и R5 вместе с атомом в бензольном кольце, к которому они присоединены, образуют 7-15-членное конденсированное кольцо или Z-замещенное конденсированное кольцо;
R6 и R7 каждый независимо представляет собой атом водорода, циано или изоциано, С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил, С2-С6алкинил или Z-замещенный алкинил, С3-С8циклоалкил или Z-замещенный циклоалкил, С6-С10арил или Z-замещенный арил, 4-15-членный гетероцикл или Z-замещенный гетероцикл, 5-15-членный гетероарил или Z-замещенный гетероарил либо С1-С6алкокси или Z-замещенный С1-С6алкокси, или R6 и R7 вместе с атомом, к которому они присоединены, образуют 5-7-членный гетероциклил или Z-замещенный 5-7-членный гетероциклил;
R8 и R10 каждый независимо представляет собой атом водорода, дейтерия, арил или Z-замещенный арил, С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил, С2-С6алкинил или Z-замещенный алкинил, С3-С8циклоалкил или Z-замещенный циклоалкил, и по меньшей мере один из R8 и R10 должен представлять собой атом водорода или дейтерия;
R9 представляет собой замещенный С6-С10арил, который замещен по меньшей мере одним атомом фтора или одной нитрогруппой, замещенный 4-15-членный гетероцикл, который замещен по меньшей мере одним атомом фтора или одной нитрогруппой, или замещенный 5-15-членный гетероарил, который замещен по меньшей мере одним атомом фтора или одной нитрогруппой;
заместитель Z представляет собой атом галогена, циано или изоциано, гидрокси, сульфгидрил, амино, OTs, OLCMS, С1-С3алкил или замещенный алкил, С1-С3алкокси или замещенный алкокси, С2-С3алкенил или замещенный алкенил, С2-С3алкинил или замещенный алкинил, С3-С8циклоалкил или замещенный циклоалкил, ароматическое кольцо, гетероцикл, гетероароматическое кольцо и конденсированное кольцо или замещенное ароматическое кольцо, гетероциклическое кольцо или гетероароматическое кольцо и конденсированное кольцо, при этом замещение по своему типу может представлять собой моно- или дизамещение;
замещение в замещенном С6-С10ариле, замещенном 4-15-членном гетероцикле или замещенном 5-15-членном гетероариле в группе R9 представляет собой замещение атомом галогена, нитро, циано или изоциано, гидрокси, амино, С1-С3алкилом или алкокси, алкенилом, алкинилом, циклоалкилом или бензольным кольцом, замещенным бензольным кольцом, С1-С3алкокси или алкокси, замещенным атомом галогена.
Термин "замещение" имеет широкое значение. Это может быть монозамещение (когда может быть замещен только один атом Н у атома С в бензольном кольце и тому подобное) или множественное замещение, которое включает множественные замещения у определенного атома С, т.е. дизамещение и тризамещение (как например, в гем-дифторметиле и гем-трифторметиле), или отдельные замещения у разных атомов С в кольце (как например, в перфторбензоле).
Гетероцикл и гетероарил включают трехчленные кольца, четырехчленные кольца, пятичленные кольца, шестичленные кольца и семичленные кольца. Примеры приведены ниже.
Трехчленные кольца включают этиленоксид, азиран и этиленсульфид;
четырехчленные кольца включают азетидин, оксаэтидин, тиаэтидин и этидин;
пятичленные кольца включают пирролидин, пирролин, 1-пирролин, 3-пирролин, 2-пирролин, пиррол, пиразолидин, 2-пиразолин, имидазол, пиразол, фуран, тетрагидрофуран, дигидрофуран, тетрагидротиофен, тиофен, сульфолан, фосфол, оксазол, 1,2,3-триазол, 1,2,4-триазол и 1,3,4-тиадиазол;
шестичленные кольца включают пиперидин, тетрагидропиран, тетрагидротиопиран, пиридин, пиран, тиопиран, дигидропиридин, морфолин, пиперазин, пиридазин, пиразин, 1,3,5-триазин и 1,3,5-тритиан;
семичленные кольца включают азепан (азациклогептан), оксагептан, тиагептан, азепин,оксепин и тиепин.
Конденсированное кольцо определяют как слитую конструкцию из указанных выше гетероцикла и гетероарила или конструкцию, полученную слиянием указанных выше гетероцикла и гетероарила с циклоалкановой структурой. Слитая конструкция может быть в форме, полученной путем связывания через простую связь или совместного использования одного, двух или даже трех атомов (т.е. в форме спироциклических, конденсированных или мостиковых колец). Ниже приведены некоторые наиболее распространенные конденсированные кольцевые структуры: нафталин, хинолин, индол, изоиндол, изохинолин, циннолин, хиноксалин, бифенил, кумарин, флуорен, дифенилкаран, карбазол, антрацен, акридин, тиофеназин, адамантан, азулен, фенантрен, антрахинон, флавоноиды и изофлавоны.
Очевидно, что указанные выше соединения также включают соединения, замещенные изотопом Z. Типичный характер замены на Z состоит в том, что атом Н в группе "атом водорода-галоген" заменяется на более тяжелый атом водорода дейтерий (D).
В частности, положение с заменой на дейтерий располагается в группе Ph-C*- в соединениях формулы II и III, как показано на приведенных ниже формулах:
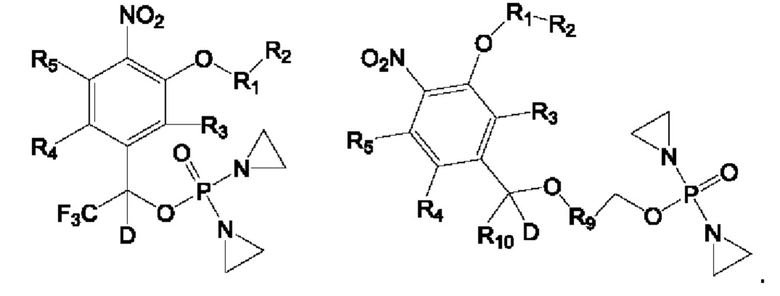
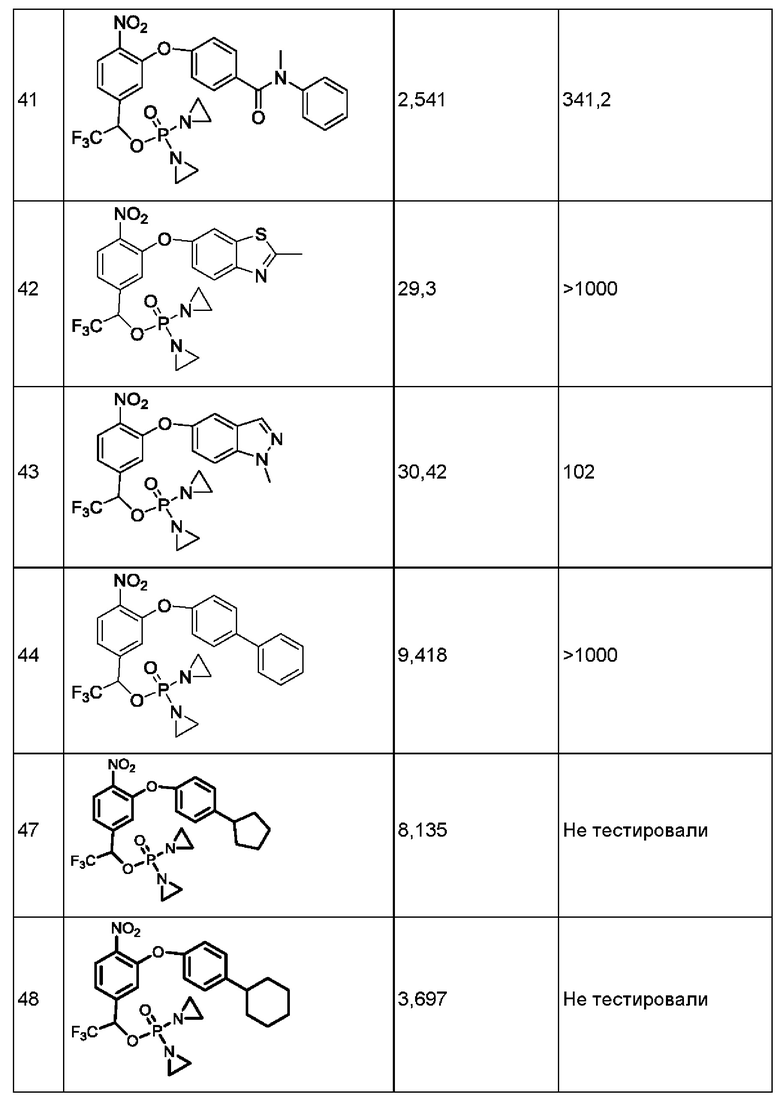
Кроме того, в приведенных выше соединениях
R1 представляет собой фенил или Z-замещенный фенил, шестичленный азотсодержащий гетероцикл или Z-замещенный гетероцикл, шестичленный азотсодержащий гетероарил или Z-замещенный гетероарил либо 9-14-членное конденсированное кольцо или Z-замещенное конденсированное кольцо;
R2 представляет собой атом водорода, галогена, циано или изоциано, гидрокси, С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил, С2-С6алкинил или Z-замещенный алкинил, С3-С8циклоалкил или Z-замещенный циклоалкил, С6-С10арил или Z-замещенный арил, 4-15-членный азотсодержащий гетероцикл или Z-замещенный азотсодержащий гетероцикл, 5-15-членный азотсодержащий гетероарил или замещенный азотсодержащий гетероарил, С1-С6алкокси или фторзамещенный С1-С6алкокси, -CONR6R7, -SO2NR6R7, -SO2R6, -OCOO-R6, -COOR6, -NR6COR7, -OCOR6, -NR6SO2R7 или -NR6SO2NR6R7;
R6 и R7 каждый независимо представляет собой С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил, С2-С6алкинил или Z-замещенный алкинил, С3-С8циклоалкил или Z-замещенный циклоалкил, С6-С10арил или Z-замещенный арил, 4-15-членный гетероцикл или Z-замещенный гетероцикл, 5-15-членный гетероарил или Z-замещенный гетероарил, С1-С6алкокси или фторзамещенный С1-С6алкокси, или R6 и R7 вместе с атомом азота, в которому они присоединены, образуют 5-7-членную гетероциклическую группу или Z-замещенную 5-7-членную гетероциклическую группу.
Кроме того, в приведенных выше соединениях
R1 представляет собой фенил или Z-замещенный фенил, шестичленный азотсодержащий гетероцикл или Z-замещенный гетероцикл, шестичленный азотсодержащий гетероарил или Z-замещенный гетероарил либо 9-14-членное конденсированное кольцо или Z-замещенное конденсированное кольцо;
R2 представляет собой атом водорода, галогена, циано или изоциано, гидрокси, С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил, С2-С6алкинил или Z-замещенный алкинил, С3-С8циклоалкил или Z-замещенный циклоалкил, С6-С10арил или Z-замещенный арил, 4-15-членный азотсодержащий гетероцикл или Z-замещенный азотсодержащий гетероцикл, 5-15-членный азотсодержащий гетероарил или замещенный азотсодержащий гетероарил, С1-С6алкокси или фторзамещенный С1-С6 алкокси, -CONR6R7, -SO2R6, -OCOO-R6, -COOR6, -NR6COR6, -OCOR6, -NR6SO2R6 или -NR6SO2NR6R7;
R6 и R7 каждый независимо представляет собой С1-С6алкил или Z-замещенный алкил, С2-С6алкенил или Z-замещенный алкенил, С2-С6алкинил или Z-замещенный алкинил, Сз-С8циклоалкил или Z-замещенный циклоалкил, С6-С10арил или Z-замещенный арил, 4-15-членный гетероцикл или Z-замещенный гетероцикл, 5-15-членный гетероарил или Z-замещенный гетероарил, С1-С6алкокси или фторзамещенный С1-С6алкокси, или R6 и R7 вместе с атомом азота, в которому они присоединены, образуют 5-7-членную гетероциклическую группу или Z-замещенную 5-7-членную гетероциклическую группу.
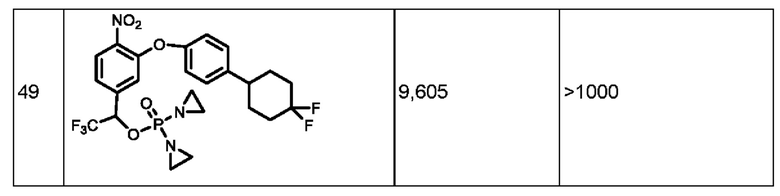
Кроме того, в приведенных выше соединениях представляет собой фенил, тетрагидропиран, тетрагидротиопиран, тетрагидрофуран, пиридин, фуран, пиран, тиопиран, тиазол, дигидропиридин, морфолин, пиперазин, пиридазин, пиразин, 1,3,5-триазин, нафталин, хинин, бензотиазол, бензотиопиран, бензофуран, бензимидазол, индол или имидазопиридин либо Z-замещенный фенил, пиперидин, тетрагидропиран, тетрагидротиопиран, тетрагидрофуран, пиридин, фуран, пиран, тиопиран, тиазол, дигидропиридин, морфолин, пиперазин, пиридазин, пиразин, 1,3,5-триазин, нафталин, хинин, бензотиазол, бензотиопиран, бензофуран, бензимидазол, индол или имидазопиридин;
R2 представляет собой -CON(CH3)2, -SO2CH3, -ОСОО-СН3, -СООСН3, -NHCOCH3, -NMeCOCH3, -NHCOCF3, -ОСОСН3, -NHSO2CH3, -NMeSO2CH3, -NHSO2CF3, -NMeSO2CF3, -CF3, F, Cl, Me, бензол, фторбензол, хлорбензол, -OCF3, пиридил, фторпиридил, хлорпиридил, фурил, тиопиран, тиазол, -CONMePh, С5-С6циклоалкил или F-замещенный С5-С6циклоалкил, 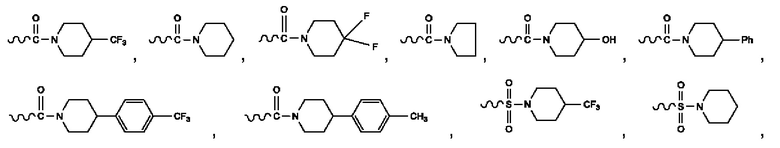
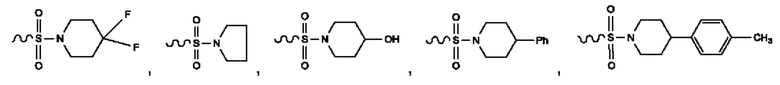 или
или 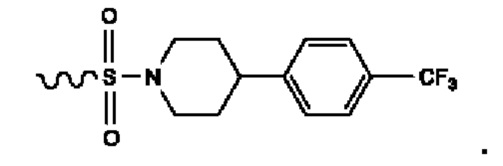 Указанная выше волнистая линия представляет собой химическую связь, которая слева соединена с другим атомом в положении, которое может относиться к любому атому в кольце, в котором этот присоединенный атом располагается.
Указанная выше волнистая линия представляет собой химическую связь, которая слева соединена с другим атомом в положении, которое может относиться к любому атому в кольце, в котором этот присоединенный атом располагается.
Кроме того, в формуле I R2 представляет собой Н.
Кроме того, R3, R4 и R5 каждый независимо представляет собой Н.
Кроме того, R8 и R10 каждый независимо представляет собой Н.
Кроме того, R9 представляет собой монофтор-, фторхлор-, дифтор- или тетрафторзамещенный фенил.
Кроме того, R9 представляет собой 

 или
или .
.
Сплошные линии с проведенными поверх волнистыми линиями представляют химические связи. Один конец такой химической связи соединен с другим атомом, представляющим собой любой атом в кольце, или соединен с каким-либо атомом для образования любой конфигурации (E/Z или R/S).
Кроме того, соединения выбраны из следующих соединений:
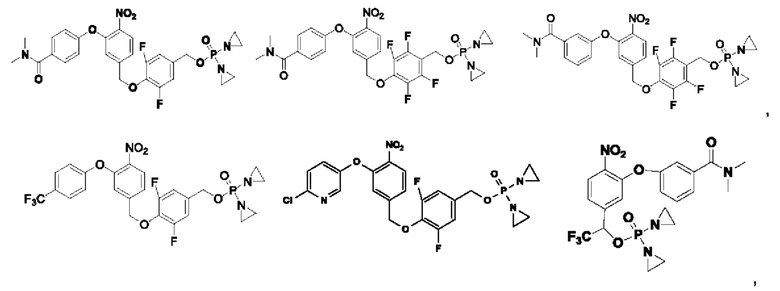
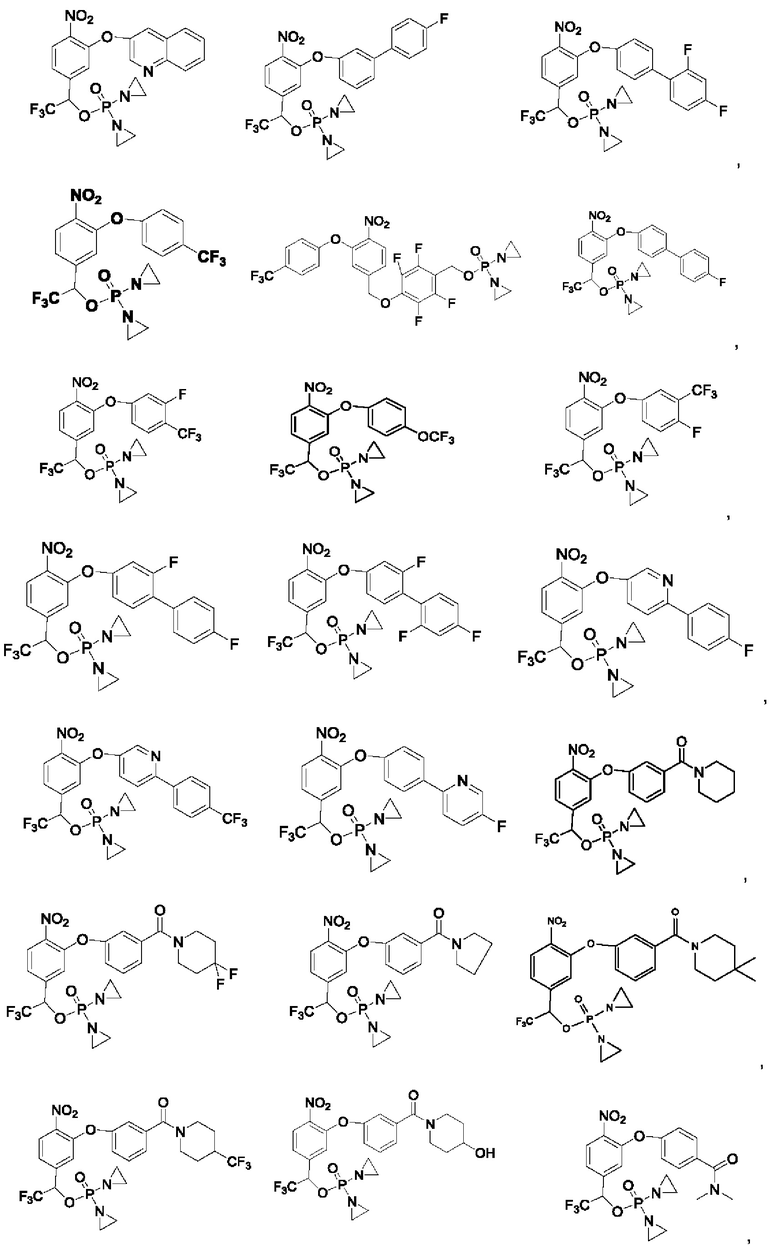
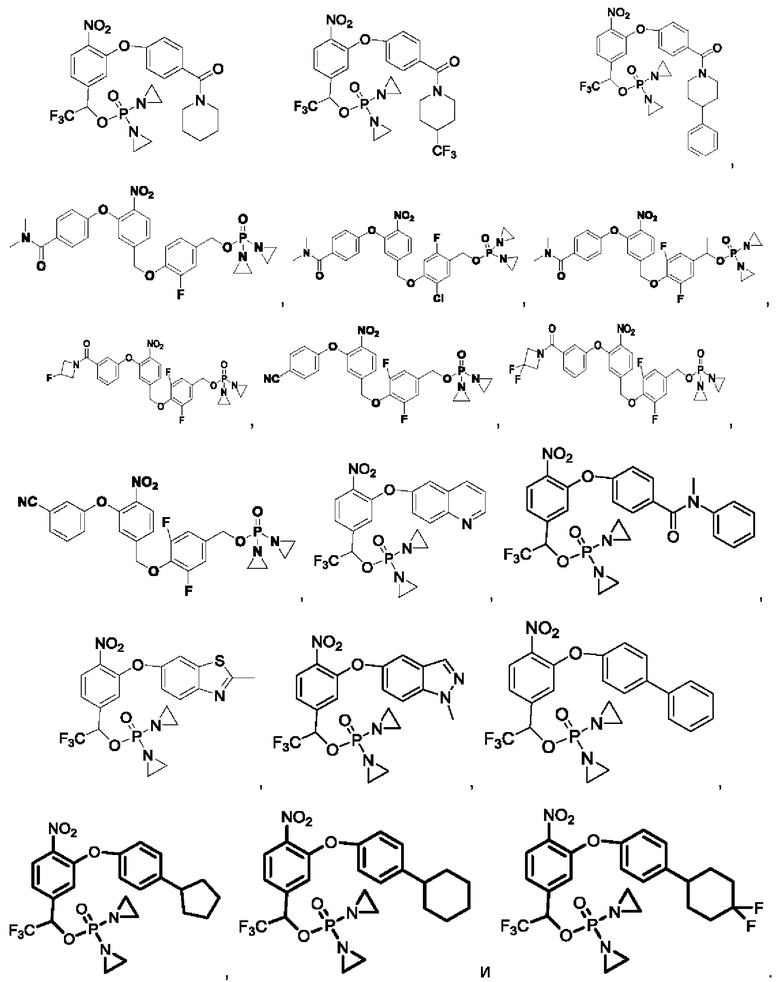
Кроме того, что касается представленных выше соединений, то соли могут представлять собой основные соли или представлять собой кислые соли.
Соединения, описанные в данной заявке, также могут включать форму своих солей формулы II или формулы III. Другими словами, согласно настоящему изобретению предложены фармацевтически приемлемые соли соединений, показанных в данном описании. Такие соли могут представлять собой основные соли, включая соли соединений с неорганическим основанием (таким как гидроксид щелочного металла и гидроксид щелочноземельного металла) или с органическим основанием (таким как моноэтаноламин, диэтаноламин или триэтаноламин). Альтернативно, такие соли могут представлять собой кислые соли, включая соли соединений с неорганической кислотой (такой как соляная кислота, бромистоводо родная кислота, йодистоводородная кислота, азотная кислота, перхлорная кислота, серная кислота или фосфорная кислота) или с органической кислотой (такой как метансульфоновая кислота, трифторметансульфоновая кислота, этансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, фумаровая кислота, щавелевая кислота, малеиновая кислота и лимонная кислота). В данной области техники существует хорошо известная технология для выбора и получения приемлемых соли, сольвата и тому подобных форм какого-либо соединения.
Кроме того, что касается приведенных выше соединений, то сольват представляет собой гидрат, алкоголят и тому подобное.
Соединения, описанные в данной заявке, также можно использовать в форме сольватов. Другими словами, согласно настоящему изобретению предложены фармацевтически приемлемые сольваты соединений формулы II или III, показанных в данном описании. Сольват представляет собой гидрат, алкоголят и тому подобное, при этом алкоголят включает этанолат.
Кроме того, согласно настоящему изобретению также предложено применение вышеупомянутых соединений формулы II или формулы III для приготовления лекарственного средства для лечения опухолей и раковых заболеваний.
Кроме того, согласно настоящему изобретению также предложено лекарственное средство или композиция, содержащая вышеупомянутые соединения формулы II или формулы III, для применения в лечении опухолей и раковых заболеваний у пациентов.
Опухоли и раковые заболевания включают
рак легкого, немелкоклеточный рак легкого, рак печени, рак поджелудочной железы, рак желудка, рак кости, рак пищевода, рак молочной железы, рак предстательной железы, рак яичка, рак толстой кишки, рак яичников, рак мочевого пузыря, рак шейки матки, меланому, плоскоклеточную карциному, базальноклеточную карциному, аденокарциному, карциному потовых желез, карциному сальных желез, папиллярную карциному, папиллярную аденокарциному, кистозную аденокарциному, кистозную карциному, медуллярную карциному, бронхогенную карциному, остеокарциному, эпителиальную карциному, карциному желчного протока, хориокарциному, эмбриональную карциному, семиному, опухоль Вильмса, глиобластому, астроцитому, медуллобластому, краниофарингиому, эпендимому, опухоль шишковидной железы, гемоцитобластому, неврому голосовых складок, менингиому, нейробластому, нейробластому зрительных путей, ретинобластому, нейрофиброму, фибросаркому, фибробластому, фиброму, фиброаденому, фиброхондрому, фиброцистому, фибромиксому, фиброостеому, фибромиксосаркому, фибропапиллому, миксосаркому, миксоцистому, миксохондрому, миксохондросаркому, миксохондрофибросаркому, миксаденому, миксобластому, липосаркому, липому, липоаденому, липобластому, липохондрому, липофиброму, липоангиому, миксолипому, хондросаркому, хондрому, хондромиому, хордому, хориокарциному, хориоэпителиому, хориобластому, остеосаркому, остеобластому, остеохондрофиброму, остеохондросаркому, остеохондрому, остеоцистому, остеодентиному, остеофиброму, фибросаркому кости, ангиосаркому, гемангиому, ангиолипому, ангиохондрому, гемангиобластому, ангиокератому, ангиоглиому, ангиоэндотелиому, ангиофиброму, ангиомиому, ангиолипому, ангиолимфангиому, ангиолиполейомиому, ангиомиолипому, ангиомионеврому, ангиомиксому, ангиоретикулему, лимфангиосаркому, лимфогранулему, лимфангиому, лимфому, лимфомиксому, лимфосаркому, лимфангиофиброму, лимфоцитому, лимфоэпителиому, лимфобластому, эндотелиому, эндобластому, синовиому, синовиальную саркому, мезотелиому, опухоль соединительной ткани, опухоль Юинга, лейомиому, лейомиосаркому, лейомиобластому, лейомиофиброму, рабдомиому, рабдомиосаркому, рабдомиомиксому, острый лимфолейкоз, острый миелогенный лейкоз, анемию хронического заболевания, полицитемию, лимфому, рак эндометрия, глиому, колоректальный рак, рак щитовидной железы, уротелиальный рак или множественную миелому; предпочтительно, чтобы раковые заболевания или опухоли представляли собой раковые заболевания или опухоли центральной нервной системы.
Результаты экспериментов подтвердили, что некоторые из соединений по настоящему изобретению обладают относительно хорошей проницаемостью через клеточные мембраны и могут быть способны в относительно хорошей степени проникать через гематоэнцефалический барьер в центральную нервную систему. Поэтому данные соединения могут более легко воздействовать на опухоли и раковые образования в полости черепа головного мозга и в спинном мозге центральной нервной системы.
Согласно настоящему изобретению предложен способ лечения рака или опухоли, включающий стадию внесения вышеупомянутого(ой) лекарственного средства или композиции; и стадию определения содержания или уровня экспрессии редуктазы AKR1C3 в раковых клетках у пациента с использованием антитела к AKR1C3, и
введение вышеупомянутого(ой) лекарственного средства или композиции пациенту, если измеренное(ый) содержание или уровень экспрессии редуктазы AKR1C3 точно соответствует предварительно заданному значению или превышает его.
Содержание редуктазы AKR1C3 можно определить с использованием методов, включающих методы иммуноферментного твердофазного анализа (ELISA) и методы иммуногистохимии (ICH).
Детекцию в жидких образцах, таких как плазма и кровь, можно проводить непосредственно, используя имеющийся в продаже набор для анализа альдокеторедуктазы 1С3 (AKR1C3) человека методом ELISA. Детекцию в других образцах проводят после подвергания лечению.
Иммуногистохимический (ICH) метод подходит для детектирования образцов солидных опухолей.
Согласно настоящему изобретению предложен способ лечения рака или опухоли, включающий стадию внесения вышеупомянутого(ой) лекарственного средства или композиции; и стадию корректировки содержания редуктазы AKR1C3, и
введение вышеупомянутого(ой) лекарственного средства или композиции пациенту, когда содержание редуктазы AKR1C3 подведено до величины, точно равной предварительно заданному значению или превышающей его.
Исследования показывают, что после лучевой терапии содержание ферментов AKR1C3 в опухолевой ткани пациентов с раком головы и шеи повышается. Следовательно, можно повысить уровень экспрессии ферментов AKR1C3 путем облучения опухолевой ткани пациентов радиоактивными лучами, применяемыми в лучевой терапии. Радиоактивные лучи включают α-, β-, γ-лучи, генерируемые радиоактивными изотопами, и рентгеновские лучи, электронные лучи, протонные пучки и пучки других частиц, генерируемые различными установками или ускорителями для рентгенотерапии.
Этот способ предназначен главным образом для той ситуации, когда содержание редуктазы AKR1C3 у пациента является относительно низким, и выполняется путем подведения уровня содержания редуктазы AKR1C3 у пациента до соответствующего уровня посредством определенного корректирующего способа лечения/введения.
Согласно настоящему изобретению также предложены схемы для получения приведенных далее соединений.
Соединения V и VI подвергают реакции конденсации с образованием замкнутых колец для получения соединений приведенных выше формул II и III:
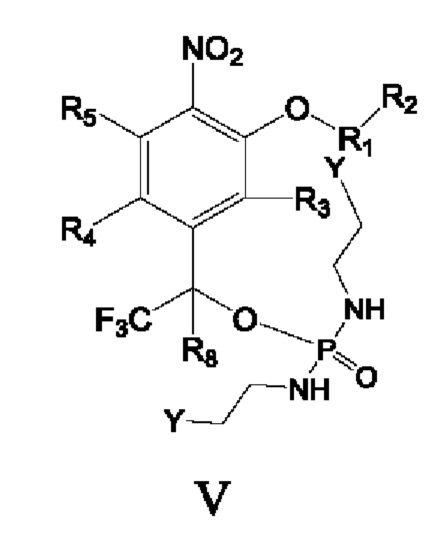

где Y представляет собой уходящую группу, а остальные переменные определены как в соединениях формул II и III.
Кроме того, в вышеупомянутом способе получения
Y представляет собой Cl, Br, I, -OTs, -ONO2, -OLCMS или -OTf,
и в реакции конденсации в качестве связывающих кислоту веществ используют органические амины, и очевидно, что в качестве связывающих кислоту веществ также можно использовать неорганические основания. Неорганические основания включают гидроксиды щелочных металлов, гидроксиды щелочноземельных металлов, карбонаты и бикарбонаты.
Кроме того, в вышеупомянутом способе получения Y представляет собой Br, и в реакции конденсации в качестве связывающего кислоту вещества используют N,N-диизопропилэтиламин (DIPEA), а в качестве катализатора используют оксид серебра (Ag2O).
Согласно настоящему изобретению также предложены приведенные далее схемы получения:
способ получения вышеупомянутого соединения, характеризующийся осуществлением взаимодействия
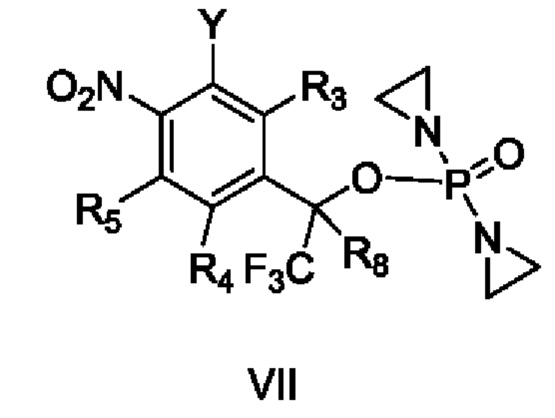

соединения VII с R2R1OH с получением соединения II и осуществлением взаимодействия соединения VIII с R2R1OH с получением соединения III, или осуществлением взаимодействия соединений IX и X
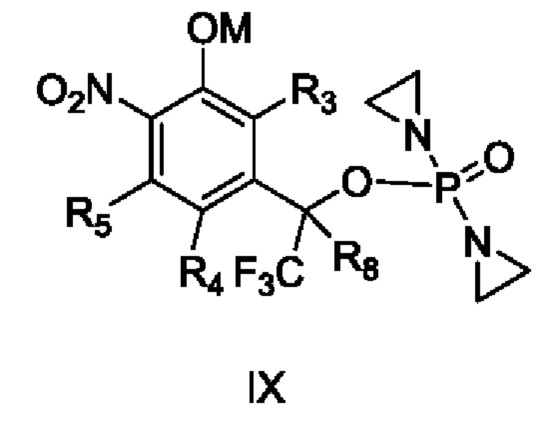

с YR1R2 с получением соединений II и III,
где Y представляет собой уходящую группу, М означает Н или щелочной металл, а остальные заместители являются такими, как определено в приведенных выше схемах.
Кроме того, в приведенном выше способе получения Y представляет собой F, Cl, Br, I, -OTs, -ONO2, -OLCMS или -OTf, и в ходе реакции добавляют основание.
Упомянутое в данном описании основание может представлять собой органическое основание (в том числе органический амин) или неорганическое основание (МОН, при этом М означает щелочной металл или щелочноземельный металл): карбонат, бикарбонат, сульфит и бисульфит щелочного металла или щелочноземельного металла, гидроксид и гидрид щелочного металла или щелочноземельного металла, либо другие дегидрирующие реагенты: алкилат щелочного металла (RM, при этом R означает алкильную группу, М означает щелочной металл) и алкоголят щелочного металла (MOR, при этом R означает гидрокарбильную группу, и М означает щелочной металл).
Что касается лекарственного средства или композиции, описанных в данной заявке, то приготовленное лекарственное средство характеризуется конкретным диапазоном дозировок показанных соединений либо их солей или сольватов, и/или приготовленное лекарственное средство находится в конкретной лекарственной форме, и его вводят, используя конкретный способ введения.
Что касается применения, описанного в данной заявке, то полученное лекарственное средство также может содержать фармацевтически приемлемые вспомогательные средства или эксципиенты. Лекарственное средство может находиться в любой лекарственной форме, пригодной для клинического введения, такой как таблетки, суппозитории, диспергируемые таблетки, таблетки с энтеросолюбильным покрытием, жевательные таблетки, перорально распадаемые таблетки, капсулы, покрытые сахарной оболочкой вещества, гранулы, сухие порошки, пероральные растворы, шприц с маленькой иглой для инъекций, лиофилизированный порошок для инъекций или инфузионные растворы. В соответствии с конкретной лекарственной формой и способом введения фармацевтически приемлемые вспомогательные средства или эксципиенты в составе лекарственного средства могут включать одно или более чем одно из приведенных ниже веществ: разбавитель, солюбилизатор, разрыхлитель, суспендирующий агент, смазывающее вещество, связующее вещество, наполнитель, корригент, подсластитель, антиоксидант, поверхностно-активное вещество, консервант, средство для оболочек (wrapping agent) и пигмент.
Предпочтительно, пациентом является млекопитающее, более предпочтительно человек.
Подробное описание изобретения
Ниже настоящее изобретение будет описано со ссылкой на конкретные примеры. Специалисты в данной области техники могут понять, что эти примеры используются только для описания изобретения, а не для ограничения каким-либо образом его объема.
Все экспериментальные способы в приведенных далее примерах представляют собой традиционные способы, если не указано иное. Все исходные вещества для лекарственных средств, реагенты и тому подобное, использованные в приведенных далее примерах представляют собой имеющиеся в продаже продукты, если не указано иное.
Термины "пациент" и "субъект" используются взаимозаменяемо для обозначения млекопитающего, нуждающегося в лечении рака. Как правило пациентом является человек. Как правило, пациентом является человек, у которого диагностирован рак. В некоторых воплощениях термин "пациент" или "субъект" может относиться к не являющемуся человеком млекопитающему, используемому при скрининге, характеристике и оценке лекарственных средств и методов терапии, как например, не являющийся человеком примат, собака, кошка, кролик, свинья, мышь или крыса.
Термин "пролекарство" относится к соединению, которое после введения метаболизируется или иным образом преобразуется в биологически активное или более активное соединение (или лекарственное средство) с точки зрения по меньшей мере одного свойства. Пролекарство, по сравнению с лекарственным средством, химически модифицировано таким образом, что оно, по сравнению с лекарственным средством, становится менее активным или неактивным, но такая химическая модификация заключается в том, что в результате осуществления метаболических или других биологических процессов после введения пролекарства образуется соответствующее лекарственное средство. Пролекарство может обладать, по сравнению с активным лекарственным средством, измененной метаболической стабильностью или измененными связанными с транспортом характеристиками, иметь меньше побочных эффектов или более низкую токсичность либо улучшенный вкус (например, см. ссылку Nogrady, 1985, Medicinal Chemistry A Biochemical Approach, Oxford University Press, New York, страницы 388-392, включенную в данное описание посредством ссылки). Пролекарство может быть синтезировано с использованием других реагентов, отличных от соответствующего лекарственного средства.
Термин "солидная опухоль" относится к солидным опухолям, включая, но не ограничиваясь этим, метастатические опухоли в кости, головном мозге, печени, легких, лимфатическом узле, поджелудочной железе, предстательной железе, коже и мягкой ткани (саркома).
Термин "терапевтически эффективное количество" лекарственного средства относится к количеству лекарственного средства, которое при введении пациенту с диагнозом рак будет иметь предполагаемый терапевтический эффект, например, облегчение, уменьшение интенсивности симптомов, временное облегчение или устранение одного или более проявлений рака у пациента. Терапевтический эффект не обязательно возникает при введении одной дозы и может проявляться только после введения ряда доз. Таким образом, терапевтически эффективное количество может быть введено за один или несколько приемов.
Термин "лечение" состояния или пациента относится к выполнению стадий с получением целебных или желаемых результатов, в том числе клинических результатов. Для целей данного изобретения целебные или желаемые клинические результаты включают, но не ограничиваются этим, облегчение или улучшение одного или более симптомов рака; снижение степени заболевания; задержку или замедление прогрессирования заболевания; облегчение, временное облегчение или стабилизацию болезненного состояния; или другие целебные результаты. В некоторых случаях лечение рака может приводить к частичному ответу или стабильному заболеванию.
Термин "опухолевые клетки" относится к опухолевых клеткам любого соответствующего вида, например, вида млекопитающих, таким как клетки мышей, собак, кошек, лошадей или человека.
Приведенное выше описание воплощений настоящего изобретения не ограничивает настоящее изобретение. Специалисты в данной области техники могут выполнить различные модификации и внести изменения в соответствии с настоящим изобретением, и любая модификация и любое изменение в пределах сущности настоящего изобретения должны быть включены в объем формулы изобретения, прилагаемой к настоящему изобретению.
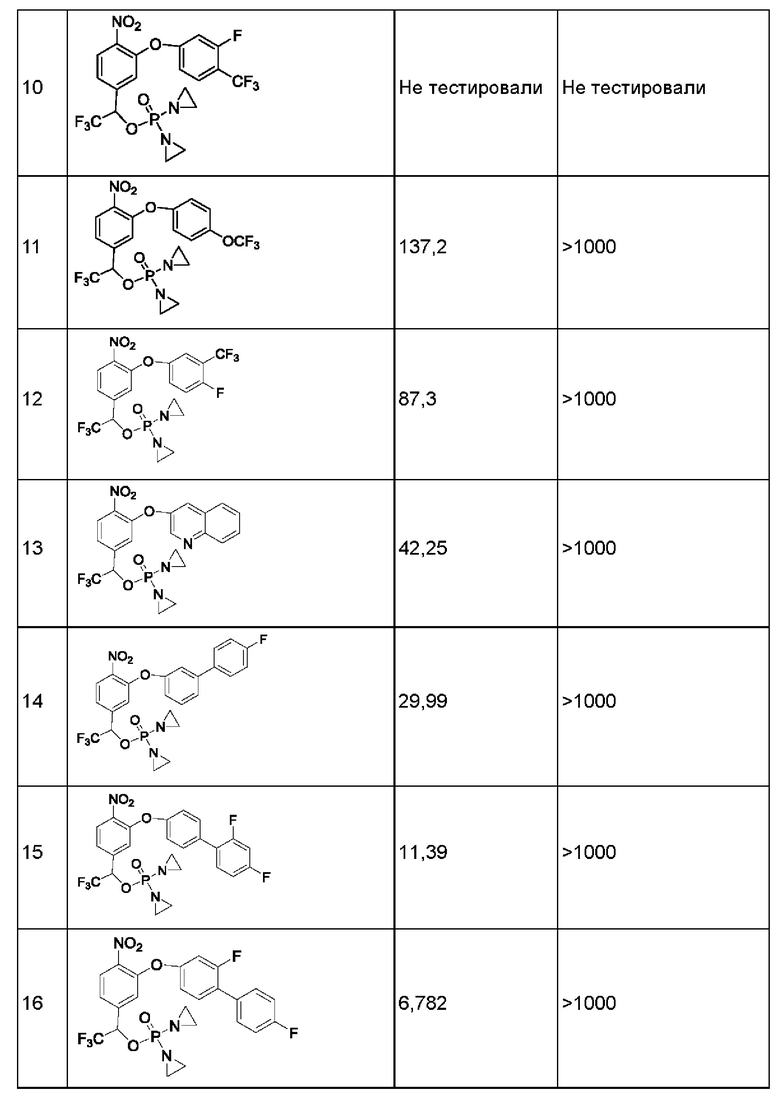
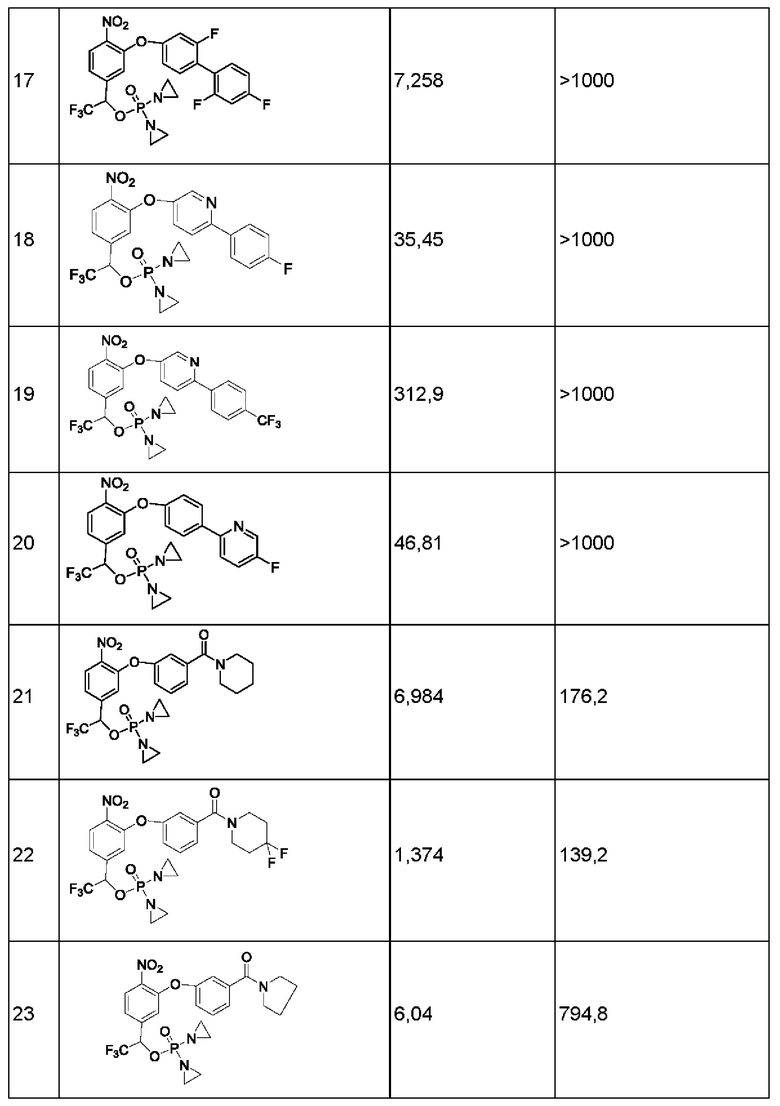
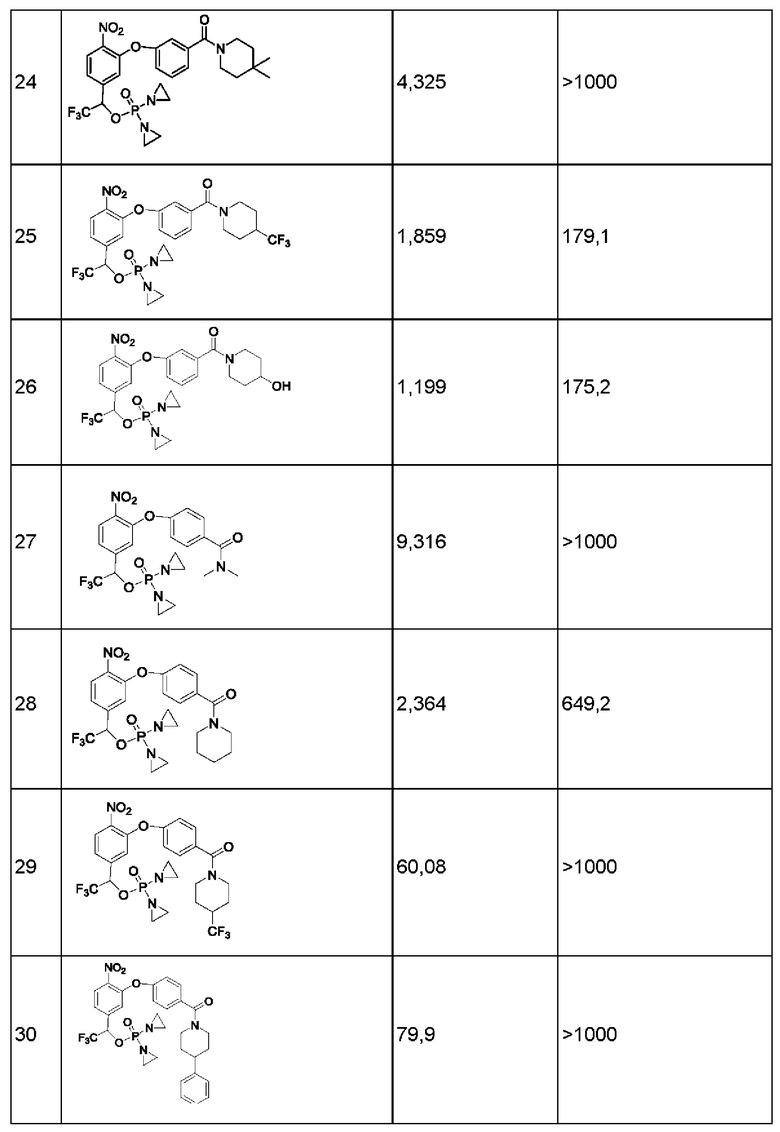
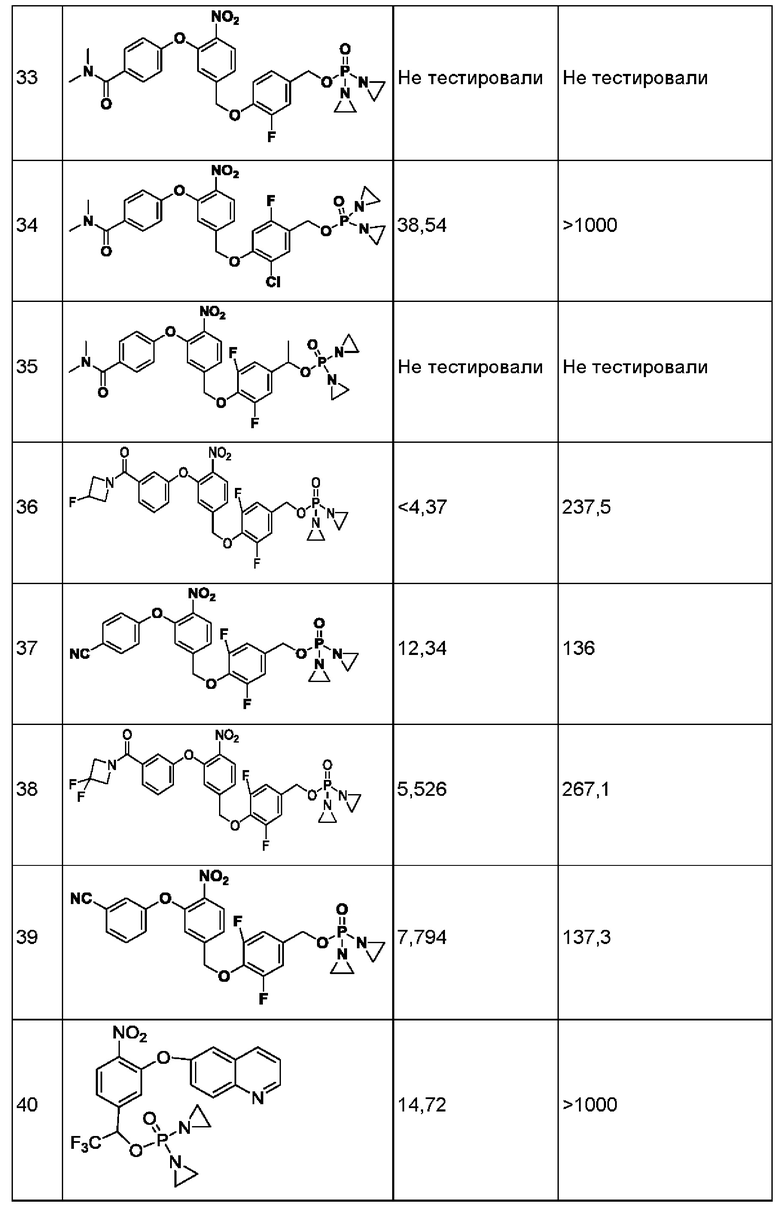
1. Данные тестирования по ингибированию раковых клеток линии Н460
Анализы in vitro на предмет цитотоксичности в отношении линии опухолевых клеток человека
Данные по пролиферации in vitro опухолевых клеток человека из линии Н460 немелкоклеточного рака легкого под действием соединений представлены ниже в таблице.
Значения IC50 приведены в наномолярной концентрации и описывают результат воздействия соединений в различных концентрациях в течение 2 ч с последующим выполнением стадии промывки и добавления свежей среды, затем ростом, окрашиванием клеток на предмет жизнеспособности и сравнением с контролем, обработанным только средой.
Конкретно, клетки в экспоненциальной фазе роста рассевали в 96-луночный планшет с плотностью 4 × 103 клеток на одну лунку и инкубировали при 37°С в условиях 5% CO2, 95% воздуха и относительной влажности 100% в течение 24 часов, после чего добавляли тестируемые соединения. Соединения растворяли в 100%-ном диметилсульфоксиде (DMSO) в концентрации, в 200 раз превышающей желаемую конечную тестируемую концентрацию. Перед добавлением соединения далее разбавляли полной средой до концентрации, в 4 раза превышающей желаемую конечную концентрацию. В лунки микротитрационного планшета, уже содержащие по 150 мкл среды, добавляли аликвсты по 50 мкл растворов соединения в определенной концентрации, в результате чего получали указанную конечную концентрацию лекарственного средства. После добавления лекарственного средства планшеты инкубировали в течение еще 2 часов при 37°С, 5% CO2, 95% воздуха и относительной влажности 100%, затем лекарственное средство отмывали, добавляли свежую порцию среды и планшеты инкубировали в течение еще 70 ч при 37°С, 5% CO2, 95% воздуха и относительной влажности 100%. По окончании такого инкубирования выполняли количественное определение жизнеспособных клеток, используя анализ с реагентом AlamarBlue. Концентрацию лекарственного средства, вызывающую ингибирование роста на 50% (IC50), рассчитывали, применяя компьютерное программное обеспечение, и результаты показаны в приведенной ниже таблице.
Аналогичным образом, чтобы также проверить, что соединения активируются под действием AKR1C3 (члена СЗ из семейства 1 альдостерон-редуктаз), влияние некоторых соединений на пролиферацию раковых клеток Н460 тестировали в присутствии специфического ингибитора фермента AKR1C3 (в 3 микромолярной концентрации). Раствор ингибирующего соединения добавляли к клеточной культуре за два часа до обработки соединением. В качестве ингибитора использовали соединение 36, т.е. соединение 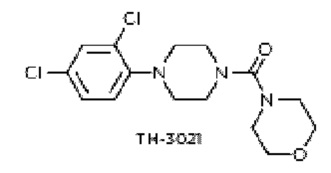 , описанное в работе Flanagan et al., Bioorganic and Medicinal Chemistry (2014), страницы 962-977.
, описанное в работе Flanagan et al., Bioorganic and Medicinal Chemistry (2014), страницы 962-977.
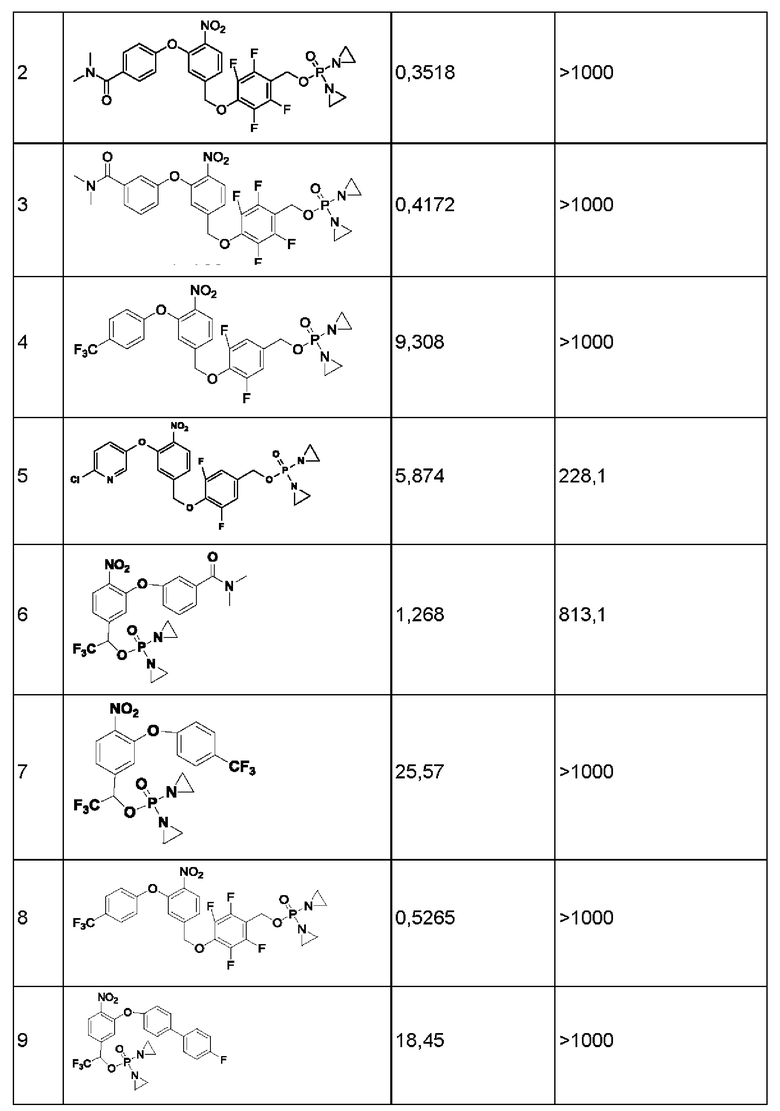
Результаты эксперимента демонстрируют, что AKR1C3 человека способствует активации вышеупомянутых соединений, и в то же время соединения этого ряда проявляют активность в отношении ингибирования пролиферации раковых клеток.
2. Примеры синтеза соединений
THF означает тетрагидрофуран; DCM означает дихлорметан; ЕА или EtOAC означает этилацетат; TEA означает триэтиламин; HPLC означает высокоэффективную жидкостную хроматографию; МТВЕ означает метил-трет-бутиловый эфир; DMAP означает 4-диметиламинопиридин; DBAD означает ди-трет-бутил-азодикарбоксилат; TFA означает трифторуксусную кислоту; LCMS означает жидкостную хроматографию в сочетании с масс-спектрометрией; EtOH означает этанол; t-BuOH означает трет-бутанол; DMF означает диметилформамид; РЕ означает петролейный эфир; экв. означает эквивалент, т.е. молярное соотношение; TBAF означает фторид тетрабутиламмония; и DIPEA означает N,N-диизопропилэтиламин.
В процессе синтеза все химические реагенты и лекарственные средства, источники которых не были указаны, характеризовались как "аналитически" или "химически чистые", и их приобретали у поставляющих реагенты компаний.
Другие английские сокращения, упомянутые в данном описании, являются предметом интерпретации сточки зрения области органической химии.

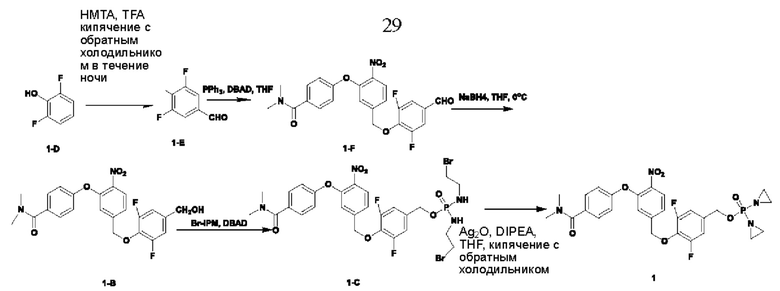
Соединение 1-D (5 г; 38,4 ммоль) растворяли в TFA (40 мл), добавляли уротропин (5,6 г; 38,4 ммоль; 1 экв.; имеется в продаже) и кипятили с обратным холодильником в течение ночи. После завершения реакции температуру понижали до комнатной температуры (КТ). Растворитель удаляли путем концентрирования. Остаток растворяли в DCM (70 мл), промывали раствором NaHCO3 и затем значение рН подводили до 1 концентрированной соляной кислотой. Водную фазу экстрагировали DCM (50 мл × 2), органические фазы объединяли, сушили с использованием Na2SO4 и концентрировали, получая соединение 1-Е (2,5 г; с выходом 41,3%), которое представляет собой белое твердое вещество. 1Н-ЯМР (ядерный магнитный резонанс) (300 МГц, CDCl3): δ млн-1 9.82 (s, 1Н), 7.49 (d, J=6,6 Гц, 2Н). LCMS: рассчитано 158,1; обнаружено 157,0 ([М-Н]-).
В атмосфере азота проводили растворение соединения 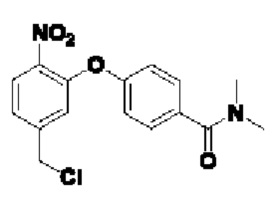 (1 г; 3,16 ммоль) и соединения 1-Е (1 г; 6,32 ммоль) в сверхсухом THF (15 мл). Добавляли трифенилфосфин (1,66 г; 6,32 ммоль; 2 экв.). По каплям при 0°С добавляли раствор DBAD (1,46 г; 6,32 ммоль; 2 экв.) в THF (6 мл). Реакцию проводили при комнатной температуре в течение ночи. По каплям при 0°С добавляли 8 мл воды. Проводили экстракцию с использованием DCM (20 мл×3), затем сушку и концентрирование. Образец перемешивали и пропускали через колонку (силикагель, 200-300 меш; петролейный эфир:ЕА=3:1), получая соединение 1-F (680 мг; с выходом 47% и содержанием 80%) в виде светло-желтого твердого вещества.
(1 г; 3,16 ммоль) и соединения 1-Е (1 г; 6,32 ммоль) в сверхсухом THF (15 мл). Добавляли трифенилфосфин (1,66 г; 6,32 ммоль; 2 экв.). По каплям при 0°С добавляли раствор DBAD (1,46 г; 6,32 ммоль; 2 экв.) в THF (6 мл). Реакцию проводили при комнатной температуре в течение ночи. По каплям при 0°С добавляли 8 мл воды. Проводили экстракцию с использованием DCM (20 мл×3), затем сушку и концентрирование. Образец перемешивали и пропускали через колонку (силикагель, 200-300 меш; петролейный эфир:ЕА=3:1), получая соединение 1-F (680 мг; с выходом 47% и содержанием 80%) в виде светло-желтого твердого вещества.
В атмосфере азота проводили растворение соединения 1-F (510 мг; 1,11 ммоль) в THF (5 мл). Температуру понижали до 0°С. Порциями добавляли боргидрид натрия (84 мг; 2,22 ммоль; 2 экв.) и температуру поддерживали при 0°С. Реакцию проводили в течение 1 часа. После завершения реакции по каплям добавляли насыщенный водный раствор хлорида аммония (3 мл). Проводили экстракцию с использованием ЕА (10 мл×3), затем промывку рассолом, сушку, концентрирование и разделение на колонке (силикагель, 100-200 меш; DCM:метанол=50:1), получая продукт 1-В (300 мг; выход=58,9%) в виде светло-желтого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ млн-1 8.01 (d, J=8,4 Гц, 1Н), 7.43 (d, J=8,7 Гц, 2Н), 7.34-7.37 (m, 1Н), 6.86 (d, J=8,4 Гц, 1Н), 5.14 (s, 2Н), 4.60 (s, 2Н), 3.09 (brs, 6Н). LCMS: рассчитано 458,4; обнаружено 459,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 1-В (300 мг; 0,65 ммоль) в THF (5 мл). Добавляли трифенилфосфин (375 мг; 1,43 ммоль; 2,2 экв.) и промежуточное соединение азотистого иприта бромизофосфорамид (Br-IPM; 442 мг; 1,43 ммоль; 2,2 экв.; имеется в продаже). Температуру понижали до 0°С. По каплям добавляли раствор DBAD (329 мг; 1,43 ммоль; 2,2 экв.) в THF (3 мл) и реакцию проводили при комнатной температуре в течение 3 ч. По каплям добавляли 3 мл воды. Проводили экстракцию с использованием DCM (10 мл×3), затем сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; DCM:метанол=30:1), получая продукт (400 мг; с выходом 82%) в виде красновато-коричневого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ млн-1 7.99 (d, J=8,4 Гц, 1Н), 7.48-7.46 (m, 2Н), 7.33-7.31 (m, 1Н), 7.12 (s, 1Н), 7.00 (d, J=8,1 Гц, 2Н), 6.93 (d, J=8,4 Гц, 2Н), 5.16 (s, 2Н), 4.93 (d, J=8,4 Гц, 2Н), 3.45-3.43 (m, 4Н), 3.37-3.32 (m, 4Н), 3.07 (brs, 6Н). LCMS: рассчитано 750,3; обнаружено 750,8 ([М+1]+).
В атмосфере азота проводили растворение соединения 1-С (400 мг; 0,533 ммоль) в сверхсухом THF (4 мл). Добавляли оксид серебра (1,46 г; 6,29 ммоль; 11,8 экв.) и по каплям добавляли диизопропилэтиламин (345 мг; 2,67 ммоль; 5 экв.). Температуру повышали до температуры дефлегмации, для мониторинга прохождения реакции использовали LCMS, и через 2,5 ч наблюдали завершение реакции. Выполняли фильтрование через целит с отсасыванием, затем несколько раз промывку THF и проводили концентрирование при низкой температуре, получая соединение №1 (19 мг; с выходом 6,07%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): α млн-1 7.99 (d, J=7,2 Гц, 1Н), 7.45 (d, J=7,8 Гц, 2Н), 7.32 (d, J=8,4 Гц, 1Н), 7.16 (d, J=1,6 Гц, 1Н), 7.04-7.06 (m, 2Н), 6.95 (d, J=8,4 Гц, 2Н), 5.15 (s, 2Н), 5.05 (d, J=8,0 Гц, 2Н), 3.11 (s, 3H), 3.03 (s, 3H), 2.23-2.13 (m, 8Н). LCMS: рассчитано 588,0; обнаружено 589,0 ([М+Н]+).
Синтез соединения №2
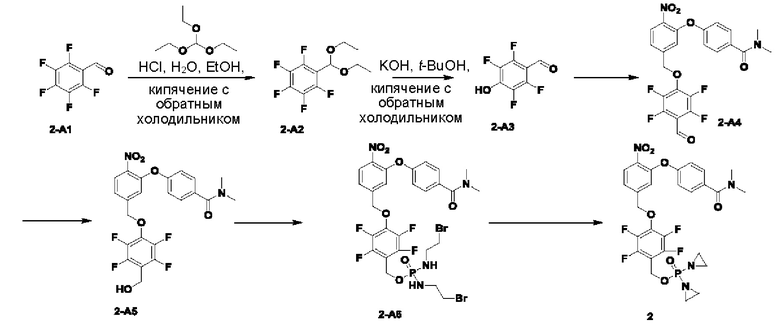
Соединение 2-А1 (10,0 г; 50,99 ммоль), триэтилортоформиат (9,8 г; 66,07 ммоль; 1,32 экв.), 12 н. раствор HCl (0,15 мл) добавляли к EtOH (30 мл) и кипятили с обратным холодильником в течение ночи. По завершении реакции растворитель выпаривали на роторном испарителе, получая соединение 2-А2 (11,2 г; неочищенный продукт).
Соединение 2-А2 (11,2 г; неочищенный продукт; 51,0 ммоль) и КОН (2,3 г; 204,0 ммоль; 4 экв.) кипятили с обратным холодильником в t-BuOH (150 мл) в течение 4 ч до завершения реакции. Температуру понижали до комнатной температуры, добавляли воду (100 мл) и проводили экстракцию с использованием EtOAc (100 мл×3). Значение рН водной фазы подводили до 3-4, используя 12 н. раствор HCl, и перемешивали в течение ночи. После этого ее экстрагировали EtOAc (100 мл×3), сушили с использованием Na2SO4, упаривали на роторном испарителе и затем суспендировали в петролейном эфире (РЕ) (20 мл), получая соединение 2-А3 (4,4 г; с выходом 44,4%) в виде беловатого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 10.21 (s, 1Н), 6.46 (s, 1Н). LCMS: рассчитано 194,0; обнаружено 192,8 ([М-Н]-).
В атмосфере азота добавляли соединение 2-В1 (500 мг; 1,5 ммоль), т.е. соединение 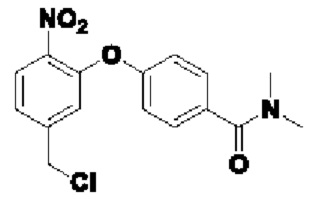 , соединение 2-А3 (1,45 г; 7,5 ммоль; 5 экв.) и диизопропилэтиламин (DIEA) (965 мг; 7,5 ммоль; 5 экв.) к DMF (5 мл), нагревали до 50°С и выдерживали в течение ночи. Выход в реакции больше не увеличивался после достижения примерно 50%. После охлаждения до комнатной температуры добавляли H2O (20 мл) и проводили экстракцию, используя EtOAc (20 мл×3). Органическую фазу промывали рассолом (15 мл×3) и промывали водой (15 мл×5). Органическую фазу сушили с использованием Na2SO4, упаривали на роторном испарителе для удаления растворителя и пропускали через колонку с силикагелем 200-300 меш (РЕ:EtOAc=3:1), получая соединение 2-А4 (290 мг; выход 33,9%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 10.15 (s, 1Н), 7.94 (d, J=8,4 Гц, 1Н), 7.41 (d, J=8,4 Гц, 2Н), 7.23 (d, J=8,4 Гц, 1 Н), 7.04-6.99 (m, 3H), 5.31 (s, 2Н), 3.06 (s, 3H), 2.96 (s, 3H). LCMS: рассчитано 492,1; обнаружено 493,1 ([М+Н]+).
, соединение 2-А3 (1,45 г; 7,5 ммоль; 5 экв.) и диизопропилэтиламин (DIEA) (965 мг; 7,5 ммоль; 5 экв.) к DMF (5 мл), нагревали до 50°С и выдерживали в течение ночи. Выход в реакции больше не увеличивался после достижения примерно 50%. После охлаждения до комнатной температуры добавляли H2O (20 мл) и проводили экстракцию, используя EtOAc (20 мл×3). Органическую фазу промывали рассолом (15 мл×3) и промывали водой (15 мл×5). Органическую фазу сушили с использованием Na2SO4, упаривали на роторном испарителе для удаления растворителя и пропускали через колонку с силикагелем 200-300 меш (РЕ:EtOAc=3:1), получая соединение 2-А4 (290 мг; выход 33,9%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 10.15 (s, 1Н), 7.94 (d, J=8,4 Гц, 1Н), 7.41 (d, J=8,4 Гц, 2Н), 7.23 (d, J=8,4 Гц, 1 Н), 7.04-6.99 (m, 3H), 5.31 (s, 2Н), 3.06 (s, 3H), 2.96 (s, 3H). LCMS: рассчитано 492,1; обнаружено 493,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 2-А4 (270 мг; 0,548 ммоль) в THF (3 мл), после чего температуру понижали до 0°С. Затем в эту систему порциями добавляли NaBH4 (42 мг; 1,1 ммоль; 2 экв.) и через 30 мин наблюдали завершение реакции. По каплям добавляли H2O (5 мл) и проводили экстракцию с использованием DCM (10 мл×3). Органическую фазу промывали водой (10 мл×3), органическую фазу сушили с использованием безводного Na2SO4, упаривали на роторном испарителе и пропускали через колонку (РЕ:EtOAc=1:1), получая соединение 2-А5 (95 мг; выход 35,0%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.01 (d, J=8,4 Гц, 1Н), 7.45 (d, J=8,4 Гц, 2Н), 7.33-7.30 (m, 1Н), 7.00 (d, J=8,4 Гц, 2Н), 6.94 (s, 1Н), 5.20 (s, 2Н), 4.74 (s, 2Н), 3.10 (s, 3H), 3.05 (s, 3H). LCMS: рассчитано 495,1; обнаружено 495,1 ([М+Н]+).
В атмосфере азота проводили растворение POCl3 (100 мг; 0,405 ммоль; 2 экв.) в DCM (2 мл) и затем температуру понижали до -40°С. Далее в эту систему добавляли соединение 2-А5 (100 мг; 0,202 ммоль; 1 экв.) и TEA (51 мг; 0,506 ммоль; 2,5 экв.) и выдерживали при -40°С в течение 6 ч до завершения реакции. В систему добавляли 2-бромэтиламина гидробромид (338 мг; 1,618 ммоль; 8 экв.) и затем в систему по каплям добавляли TEA (164 мг; 1,618 ммоль; 8 экв.). По окончании добавления температуру поддерживали при -40°С и через 30 мин наблюдали завершение реакции. После повышения температуры до комнатной температуры по каплям добавляли раствор NH4Cl (15 мл) и выполняли экстракцию с использованием DCM (10 мл×3). Органическую фазу промывали водой (10 мл×3) и органическую фазу сушили с использованием безводного Na2SO4. После упаривания на роторном испарителе ее пропускали через колонку с силикагелем 200-300 меш (EtOAc), получая соединение AST-2-A6 (50 мг; с содержанием 70% и выходом 31,4%) в виде желтого твердого вещества.
В атмосфере азота добавляли соединение 2-А6 (30 мг; 0,038 ммоль; 1 экв.), Ag2O (44 мг; 0,191 ммоль; 5 экв.) и DIEA (26 мг; 0,191 ммоль; 5 экв.) к THF (1 мл), нагревали до 65°С и кипятили с обратным холодильником, через 2 ч наблюдали завершение реакции. После охлаждения до комнатной температуры систему фильтровали через силикагель с отсасыванием и промывали THF. После упаривания маточной жидкости на роторном испарителе ее разделяли с применением препаративной жидкостной хроматографии на нейтральной фазе, экстрагировали DCM, упаривали на роторном испарителе и лиофилизировали, получая соединение №2 (6 мг; выход 25,2%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ 8.34 (d, J=8,4 Гц, 1Н), 7.50 (d, J=8,8 Гц, 2Н), 7.45-7.43 (m, 1Н), 7.30 (s, 1Н), 7.08 (d, J=8,8 Гц, 2Н), 5.38 (s, 2Н), 5.25 (d, J=7,6 Гц, 2Н), 3.10 (s, 3H), 3.05 (s, 3H), 2.20-2.15 (m, 8Н). LCMS: рассчитано 624,1; обнаружено 625,2 ([М+Н]+).
Синтез соединения №3
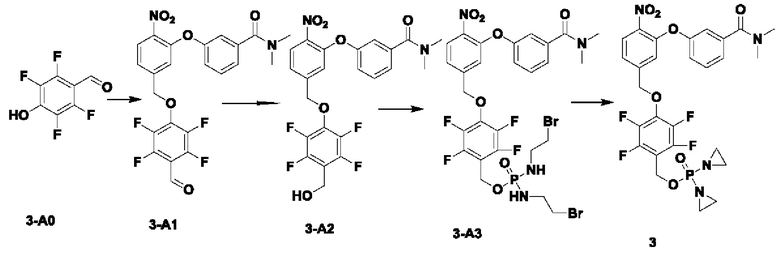
В атмосфере азота добавляли соединение 3-В1 (500 мг; 1,49 ммоль; 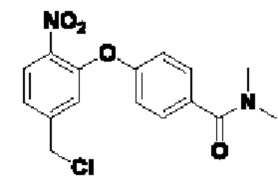 ), соединение 3-A0 (1,45 г; 7,470 ммоль; 5 экв.) и DIEA(965 мг; 7,470 ммоль; 5 экв.) к DMF (10 мл) и нагревали до 50°С и выдерживали в течение ночи. Выход в реакции больше не увеличивался после достижения примерно 50%. После охлаждения до комнатной температуры добавляли H2O (20 мл) и проводили экстракцию, используя EtOAc (20 мл×3). Органическую фазу промывали водой (15 мл×5) и промывали насыщенным рассолом (15 мл×3). Органическую фазу сушили, упаривали на роторном испарителе и пропускали через колонку с силикагелем 200-300 меш (РЕ:EtOAc=3:1), получая соединение 3-А1 (295 мг; выход 40,1%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 10.22 (s, 1Н), 8.00 (d, J=8,4 Гц, 1Н), 7.46-7.42 (m, 1Н), 7.30 (d, J=8,4 Гц, 1Н), 7.26-7.24 (m, 1Н), 7.10-7.08 (m, 3H), 5.37 (s, 2Н), 3.11 (s, 3H), 2.99 (s, 3H). LCMS: рассчитано 492,1; обнаружено 493,1 ([М+Н]+).
), соединение 3-A0 (1,45 г; 7,470 ммоль; 5 экв.) и DIEA(965 мг; 7,470 ммоль; 5 экв.) к DMF (10 мл) и нагревали до 50°С и выдерживали в течение ночи. Выход в реакции больше не увеличивался после достижения примерно 50%. После охлаждения до комнатной температуры добавляли H2O (20 мл) и проводили экстракцию, используя EtOAc (20 мл×3). Органическую фазу промывали водой (15 мл×5) и промывали насыщенным рассолом (15 мл×3). Органическую фазу сушили, упаривали на роторном испарителе и пропускали через колонку с силикагелем 200-300 меш (РЕ:EtOAc=3:1), получая соединение 3-А1 (295 мг; выход 40,1%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 10.22 (s, 1Н), 8.00 (d, J=8,4 Гц, 1Н), 7.46-7.42 (m, 1Н), 7.30 (d, J=8,4 Гц, 1Н), 7.26-7.24 (m, 1Н), 7.10-7.08 (m, 3H), 5.37 (s, 2Н), 3.11 (s, 3H), 2.99 (s, 3H). LCMS: рассчитано 492,1; обнаружено 493,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 3-А1 (290 мг; 0,589 ммоль) в THF (3 мл), после чего температуру понижали до 0°С. Затем в эту систему порциями добавляли NaBH4 (45 мг; 1,178 ммоль; 2 экв.) и через 30 мин наблюдали завершение реакции. В эту систему по каплям добавляли H2O (5 мл) и выполняли экстракцию с использованием DCM (10 мл×3). Органическую фазу промывали водой (10 мл×3), органическую фазу сушили, упаривали на роторном испарителе и пропускали через колонку (РЕ:EtOAc=1:1), получая соединение 3-А2 (m (масса)=100 мг; выход 35,0%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 7.99 (d, J=8,4 Гц, 1 Н), 7.44 (t, J=8,4 Гц, 1Н), 7.31 (d, J=7,6 Гц, 1Н), 7.22 (d, J=7,6 Гц, 1Н), 7.13 (dd, d, J=8,0; 1,6 Гц, 1Н), 7.05 (s, 1Н), 6.91 (s, 1Н), 5.24 (s, 2Н), 4.73 (s, 2Н), 3.02 (m, 6Н). LCMS: рассчитано 494,1; обнаружено 495,1 ([М+Н]+).
В атмосфере азота проводили растворение POCl3 (61 мг; 0,202 ммоль) в DCM (2 мл) и затем охлаждали до -40°С. Далее в эту систему добавляли соединение 3-А2 (50 мг; 0,101 ммоль) и TEA (26 мг; 0,253 ммоль; 2,5 экв.) и выдерживали при -40°С в течение 6 ч до завершения реакции. В систему добавляли 1-бромэтиламина гидробромид (169 мг; 0,81 ммоль) и затем в систему по каплям добавляли TEA (82 мг; 0,809 ммоль; 8 экв.). По окончании добавления температуру поддерживали при -40°С в течение 30 мин до завершения реакции. После повышения температуры до комнатной температуры по каплям добавляли раствор NH4Cl (10 мл) и выполняли экстракцию с использованием DCM (8 мл×3). Органическую фазу промывали водой (5 мл×3), органическую фазу сушили, упаривали на роторном испарителе и пропускали через колонку с силикагелем 200-300 меш (EtOAc), получая соединение 3-А3 (62 мг; 80,0%, с содержанием 70%) в виде желтого твердого вещества, которое использовали на следующей стадии.
В атмосфере азота добавляли соединение 3-А3 (60 мг; 0,08 ммоль; 1 экв.), Ag2O (88 мг; 0,382 ммоль; имеется в продаже) и DIEA (49 мг; 0,38 ммоль) к THF (2 мл), нагревали до 65°С и кипятили с обратным холодильником. Через 2 ч наблюдали завершение реакции. После охлаждения до комнатной температуры систему фильтровали через силикагель с отсасыванием и промывали THF. Остаточную жидкость упаривали на роторном испарителе, подготавливали, экстрагировали DCM, упаривали на роторном испарителе и лиофилизировали, получая соединение №3 (10 мг; выход 12%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ 8.03 (d, J=8,4 Гц, 1Н), 7.51 (t, J=8,0 Гц, 1Н), 7.43 (d, J=8,0 Гц, 1Н), 7.28-7.25 (m, 2Н), 7.13-7.10 (m, 2Н), 5.37 (s, 2Н), 5.25 (d, J=7,6 Гц, 2Н), 3.09 (s, 3H), 3.00 (s, 3H), 2.21-2.16 (m, 8Н). LCMS: рассчитано 624,1; обнаружено 625,1 ([М+Н]+).
Синтез соединения №4
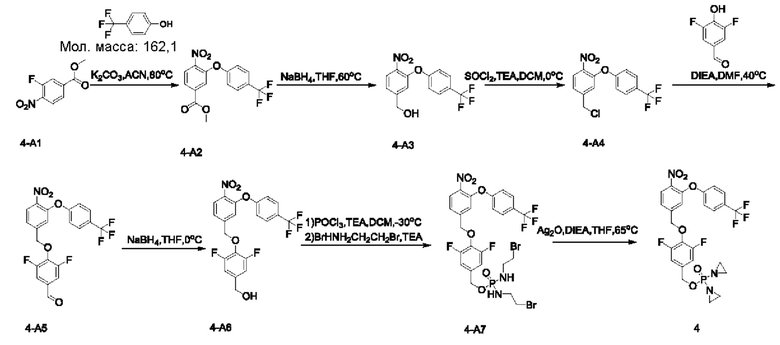
В атмосфере азота проводили растворение соединения 4-А1 (3,7 г; 18,45 ммоль) и патрифторметилфенола (2 г; 12,3 ммоль; имеется в продаже) в ацетонитриле (ACN; 30 мл). Добавляли К2СО3 (3,4 г; 24,6 ммоль), нагревали до 80°С и перемешивали в течение ночи. По завершении реакции систему подвергали фильтрованию через целит с отсасыванием и концентрировали, получая неочищенный продукт 4-А2 (5,6 г; 97,2%) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.03-7.95 (m, 2Н), 7.76 (d, J=1,6 Гц, 1Н), 7.66 (d, J=8,8 Гц, 2Н), 7.11 (d, J=8,8 Гц, 2Н), 3.94 (s, 3H). LCMS: рассчитано 341,1; обнаружено 342,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 4-А2 (1,6 г; 4,7 ммоль) в THF (30 мл). Порциями добавляли NaBH4 (1,4 г; 37,6 ммоль), нагревали до 60°С и перемешивали в течение ночи. По завершении реакции температуру понижали до 5°С и по каплям добавляли насыщенный водный раствор NH4Cl (15 мл). Осуществляли экстракцию с использованием DCM (20 мл), затем промывку водой (4×5 мл), сушку и концентрирование, получая неочищенный продукт 4-А3 (1,6 г) в виде желтой маслянистой жидкости, которую использовали непосредственно на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.02 (d, J=8,4 Гц, 1Н), 7.62 (d, J=8,8 Гц, 2Н), 7.31-7.28 (m, 1Н), 7.15 (s, 1Н), 7.08 (d, J=8,4 Гц, 2Н), 4.77 (s, 2Н), 1.86 (s, 1Н). LCMS: рассчитано 313,1; обнаружено 314,0342.1 ([М+Н]+).
В атмосфере азота проводили растворение неочищенного продукта 4-А3 (600 мг; 1,92 ммоль) с приведенной выше стадии в сверхсухом DCM (10 мл). Температуру понижали до 0°С и медленно по каплям добавляли SOCl2 (457 мг; 3,84 ммоль). Через 1 ч дополнительно добавляли 1 экв. TEA (194 мг; 1,92 ммоль), и через 30 мин наблюдали завершение реакции. Температуру понижали и по каплям добавляли насыщенный раствор NaHCO3 (10 мл). Осуществляли экстракцию с использованием DCM (15 мл×2), затем промывку водой (5 мл×3), сушку и концентрирование, получая неочищенный продукт 4-А4 (580 мг) в виде светло-желтой жидкости, которую использовали непосредственно на следующей стадии.
В атмосфере азота проводили растворение соединения 4-А4 (580 мг; 1,75 ммоль) и соединения 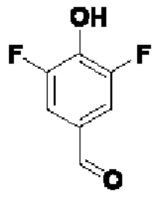 (692 мг; 4,375 ммоль) в DMF (10 мл), затем добавляли DIEA (1,4 г; 10,5 ммоль) и проводили реакцию в течение ночи при 40°С. По завершении реакции осуществляли экстракцию с использованием ЕА (15 мл×2), затем промывку водой (5 мл×6), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 10:1 до 5:1), получая продукт 4-А5 (300 мг; 55,0%) в виде желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 9.84-9.85 (m, 1Н), 8.04 (d, J=8,4 Гц, 1Н), 7.64 (d, J=8,8 Гц, 2Н), 7.46 (d, J=8,2 Гц, 2Н), 7.38 (d, J=8,4 Гц, 1Н), 7.21 (s, 1Н), 7.09 (d, J=8,4 Гц, 2Н), 5.32 (s, 2Н).
(692 мг; 4,375 ммоль) в DMF (10 мл), затем добавляли DIEA (1,4 г; 10,5 ммоль) и проводили реакцию в течение ночи при 40°С. По завершении реакции осуществляли экстракцию с использованием ЕА (15 мл×2), затем промывку водой (5 мл×6), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 10:1 до 5:1), получая продукт 4-А5 (300 мг; 55,0%) в виде желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 9.84-9.85 (m, 1Н), 8.04 (d, J=8,4 Гц, 1Н), 7.64 (d, J=8,8 Гц, 2Н), 7.46 (d, J=8,2 Гц, 2Н), 7.38 (d, J=8,4 Гц, 1Н), 7.21 (s, 1Н), 7.09 (d, J=8,4 Гц, 2Н), 5.32 (s, 2Н).
В атмосфере азота проводили растворение соединения 4-А5 (300 мг; 0,66 ммоль) в THF (6 мл). Температуру понижали до 0°С. Порциями добавляли NaBH4 (50 мг; 1,32 ммоль; имеется в продаже). Через 30 мин наблюдали завершение реакции. По каплям добавляли насыщенный раствор NH4Cl (5 мл) при 0°С. Осуществляли экстракцию с использованием DCM (15 мл×2), затем промывку водой (5 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; гептан:ЕА = от 10:1 до 5:1), получая продукт 4-А6 (190 мг; 90,6%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.02 (d, J=8,4 Гц, 1 Н), 7.63 (d, J=8,4 Гц, 2Н), 7.36-7.38 (m, 1 Н), 7.23 (s, 1 Н), 7.07 (d, J=8,4 Гц, 2Н), 6.90 (d, J=8,8 Гц, 2Н), 5.17 (s, 2Н), 4.63 (s, 2Н).
В атмосфере азота проводили растворение POCl3 (130 мг; 0,84 ммоль; имеется в продаже) в сверхсухом DCM (5 мл). Температуру понижали до -30°С. По каплям добавляли раствор соединения 4-А6 (190 мг; 0,42 ммоль) в DCM (5 мл), затем по каплям добавляли TEA (106 мг; 1,05 ммоль) и температуру поддерживали при -30°С в течение 6 ч до полного расходования исходных веществ. Добавляли 2-бромэтиламина гидробромид (688 мг; 3,36 ммоль) при -30°С и затем по каплям добавляли TEA (340 мг; 3,36 ммоль). По завершении реакции температуру понижали до 0°С и добавляли насыщенный раствор NH4Cl (10 мл). Осуществляли экстракцию с использованием DCM (15 мл×2), затем промывку водой (5 мл×4), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; гептан:ЕА = от 1:2 до ЕА), получая продукт (120 мг; выход 57,6%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.03 (d, J=8,4 Гц, 1Н), 7.64 (d, J=8,8 Гц, 2Н), 7.37 (d, J=8,0 Гц, 1Н), 7.24 (s, 1Н), 7.08 (d, J=8,4 Гц, 2Н), 6.95 (d, J=8,4 Гц, 2Н), 5.18 (s, 2Н), 4.94 (d, J=8,4 Гц, 2Н), 3.14-3.46 (m, 4Н), 3.33 (m, 4Н), 3.14 (m, 2Н). LCMS: рассчитано 747,0; обнаружено 748,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 4-А7 (120 мг; 0,16 ммоль) в THF (10 мл) и добавляли Ag2O (222 мг; 0,96 ммоль; имеется в продаже) и N,N-диизопропилэтиламин (124 мг; 0,96 ммоль; имеется в продаже). Температуру повышали до 65°С для осуществления реакции. Через 2 ч наблюдали завершение реакции. После этого проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM (20 мл), остаточную жидкость концентрировали и после проведения высокоэффективной жидкостной хроматографии получали чистый продукт (14,3 мг; 28,4%) в виде желтого воскообразного твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.02 (d, J=8,4 Гц, 1Н), 7.64 (d, J=8,8 Гц, 2Н), 7.37 (d, J=8,4 Гц, 1Н), 7.24 (s, 1Н), 7.08 (d, J=8,8 Гц, 2Н), 6.96 (d, J=8,4 Гц, 2Н), 5.18 (s, 2Н), 5.06 (d, J=8,4 Гц, 2Н), 2.13-2.23 (m, 8Н). LCMS: рассчитано 585,1; обнаружено 586,1 ([М+Н]+).
Синтез соединения №5
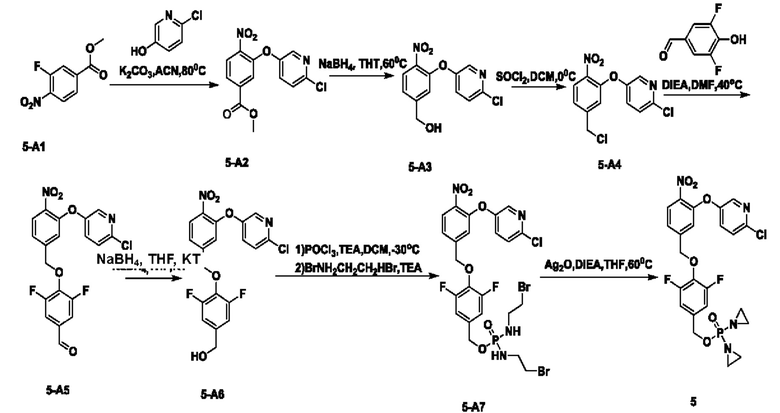
В атмосфере азота проводили растворение соединения 5-А1 (5,8 г; 28,9 ммоль) и 2-хлор-5-гидроксипиридина (2,5 г; 19,3 ммоль) в аиетонитриле (50 мл). После добавления карбоната калия (5,3 г; 38,6 ммоль) температуру повышали до 80°С и перемешивание осуществляли в течение ночи. По завершении реакции проводили фильтрование через целит с отсасыванием. Остаточную жидкость концентрировали, суспендировали в н-гептане и фильтровали с отсасыванием, получая чистый продукт 5-А2 (5,7 г; 95,7%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.20 (s, 1Н), 7.94-8.03 (m, 2Н), 7.69 (s, 1Н), 7.37 (s, 2Н), 3.93 (s, 3H). LCMS: рассчитано 308,0; обнаружено 309,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 5-А2 (2 г; 6,5 ммоль) в THF (30 мл), затем порциями добавляли NaBH4 (1,97 г; 52 ммоль). Температуру повышали до 60°С и перемешивание осуществляли в течение ночи. По завершении реакции температуру понижали и по каплям добавляли насыщенный водный раствор NH4Cl (15 мл). Осуществляли экстракцию с использованием DCM (20 мл), затем промывку водой (4×5 мл), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 5:1 до 1:1), получая продукт 5-А3 (m=800 мг; выход 44,4%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.16 (s, 1Н), 8.02 (d, J=8,4 Гц, 1Н), 7.34 (s, 2Н), 7.29 (d, J=8,4 Гц, 1Н), 7.12 (s, 1Н), 4.77 (s, 2Н), 1.84 (s, 1Н). LCMS: рассчитано 280,0; обнаружено 281,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 5-А3 (800 мг; 2,85 ммоль) в сверхсухом DCM (10 мл). Температуру понижали до 0°С и медленно по каплям добавляли SOCl2 (1,19 г; 9,98 ммоль). Через 4 ч наблюдали завершение реакции. Температуру понижали и по каплям добавляли насыщенный раствор NaHCO3 (10 мл). Осуществляли экстракцию с использованием DCM (15 мл×3). Органическую фазу промывали раствором NaHCO3 (5 мл×2), сушили и концентрировали, получая продукт 5-А4 (440 мг; светло-желтая жидкость; 51,6%), который использовали непосредственно на следующей стадии.
В атмосфере азота проводили растворение соединения 5-А4 (440 мг; 1,47 ммоль) и соединения 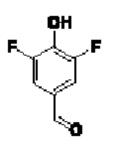 (581 мг; 3,675 ммоль) в DMF (10 мл) и затем добавляли DIEA (1,14 г; 8,82 ммоль). Реакцию проводили в течение ночи. По завершении реакции осуществляли экстракцию с использованием ЕА (15 мл×2), затем промывку водой (5 мл×6), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 10:1 до 5:1), получая продукт 5-А5 (m=340 мг; выход 55,0%) в виде желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3 δ млн-1 9.85 (t, J=1,6 Гц, 1Н), 8.16 (d, J=1,2 Гц, 1Н), 8.03 (d, J=8,4 Гц, 1Н), 7.46 (d, J=8,0 Гц, 2Н), 7.34-7.38 (m, 3H), 7.18 (s, 1Н), 5.32 (s, 2Н). LCMS: рассчитано 420,0; обнаружено 421,0 ([М+Н]+).
(581 мг; 3,675 ммоль) в DMF (10 мл) и затем добавляли DIEA (1,14 г; 8,82 ммоль). Реакцию проводили в течение ночи. По завершении реакции осуществляли экстракцию с использованием ЕА (15 мл×2), затем промывку водой (5 мл×6), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 10:1 до 5:1), получая продукт 5-А5 (m=340 мг; выход 55,0%) в виде желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3 δ млн-1 9.85 (t, J=1,6 Гц, 1Н), 8.16 (d, J=1,2 Гц, 1Н), 8.03 (d, J=8,4 Гц, 1Н), 7.46 (d, J=8,0 Гц, 2Н), 7.34-7.38 (m, 3H), 7.18 (s, 1Н), 5.32 (s, 2Н). LCMS: рассчитано 420,0; обнаружено 421,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 5-А5 (340 мг; 0,81 ммоль) в THF (6 мл). Температуру понижали до 0°С и порциями добавляли боргидрид натрия (61 мг; 1,62 ммоль). Через 30 мин наблюдали завершение реакции. По каплям при 0°С добавляли насыщенный раствор NH4Cl (5 мл). Осуществляли экстракцию с использованием DCM (10 млх2), затем промывку водой (5 мл×3), сушку и концентрирование, получая продукт 5-А6 (310 мг; светло-желтое твердое вещество; 90,6%), которое использовали непосредственно на следующей стадии. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 8.03 (d, J=8,4 Гц, 1Н), 7.55 (d, J=2,8 Гц, 1Н), 7.46-7.34 (m, 2Н), 7.01 (d, J=1,2 Гц, 1Н), 6.90-6.87 (m, 2Н), 5.17 (s, 2Н), 4.61 (d, J=6,4 Гц, 2Н). LCMS: рассчитано 422,0; обнаружено 423,0 ([М+Н]+).
В атмосфере азота к оксихлориду фосфора (224 мг; 1,46 ммоль) по каплям добавляли сверхсухой DCM (5 мл). Температуру понижали до -30°С. По каплям добавляли раствор соединения 5-А6 (310 мг; 0,73 ммоль) в DCM (5 мл), затем по каплям добавляли триэтиламин (185 мг; 1,825 ммоль) и температуру поддерживали при -30°С в течение 5 ч до полного расходования исходных веществ. Добавляли 2-бромэтиламина гидробромид (1,2 г; 5,84 ммоль) при -30°С и затем по каплям добавляли триэтиламин (591 мг; 5,84 ммоль). По завершении реакции осуществляли экстракцию с использованием DCM (15 мл×2), затем промывку водой (5 мл×4), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш), получая продукт 5-А4 (300 мг; выход 57,6%) в виде светло-желтого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.10 (s, 1Н), 8.00 (d, J=8,4 Гц, 1Н), 7.34-7.35 (m, 3H), 7.18 (s, 1Н), 6.93 (d, J=8,4 Гц, 2Н), 5.29 (s, 2Н), 5.17 (s, 2Н), 4.92 (d, J=8,4 Гц, 2Н), 3.42-3.48 (m, 4Н), 3.29-3.36 (m, 4Н), 3.20-3.22 (m, 2Н). LCMS: рассчитано 713,9; обнаружено 714,9 ([М+Н]+).
В атмосфере азота проводили растворение соединения 5-А7 (300 мг; 0,42 ммоль) в THF (10 мл) и добавляли оксид серебра (584 мг; 2,52 ммоль; имеется в продаже) и N,N-диизопропилэтиламин (326 мг; 2,52 ммоль). Температуру повышали до 60°С для осуществления реакции. Через 2,5 ч наблюдали завершение реакции. После этого проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM (20 мл), остаточную жидкость концентрировали и после проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №5 (66 мг; 28,4%) в виде светло-желтого твердого вещества 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.16 (d, J=2,4 Гц, 1Н), 8.02 (d, J=8,4 Гц, 1Н), 7.31-7.37 (m, 3H), 7.20 (s, 1Н), 6.96 (d, J=8,4 Гц, 2Н), 5.17 (s, 2Н), 5.06 (d, J=8,0 Гц, 2Н), 2.24-2.14 (m, 8Н). LCMS: рассчитано 552,1; обнаружено 553,1 ([М+Н]+).
Синтез соединения №6
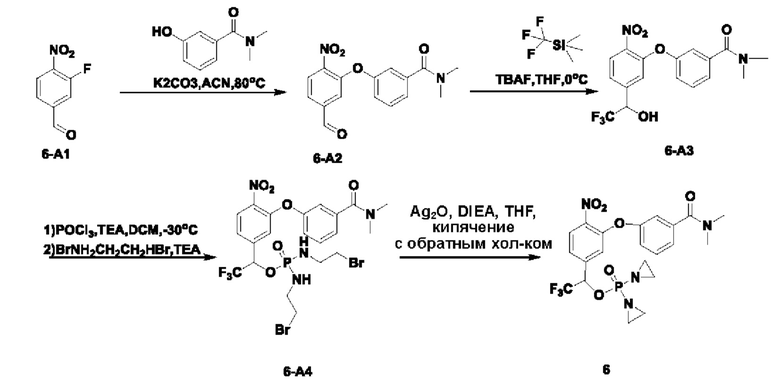
В атмосфере азота проводили растворение соединения 6-А1 (1,5 г; 9,15 ммоль; имеется в продаже) и соединения  (1 г; 6,10 ммоль) в ацетонитриле (20 мл). Температуру повышали до 80°С и через 5 ч наблюдали завершение реакции. Температуру понижали до комнатной температуры, проводили фильтрование через целит с отсасыванием и осадок на фильтре промывали ЕА. Остаточную жидкость концентрировали и проводили выделение на колонке с силикагелем (силикагель, 200-300 меш; гептан:ЕА = от 2:1 до 1:1), получая продукт 6-А2 (1,2 г; выход 63,2%) в виде светло-желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 9.96 (s, 1Н), 8.04 (d, J=8,2 Гц, 1Н), 7.73 (dd, J=8,2; 1,5 Гц, 1Н), 7.51 (d, J=1,4 Гц, 1 Н), 7.49-7.43 (m, 1 Н), 7.35-7.27 (m, 1Н), 7.14 (dd, J=7,9; 1,3 Гц, 2Н), 3.09 (s, 3H), 2.99 (s, 3H). LCMS: рассчитано 314,1; обнаружено 315,1 ([М+Н]+).
(1 г; 6,10 ммоль) в ацетонитриле (20 мл). Температуру повышали до 80°С и через 5 ч наблюдали завершение реакции. Температуру понижали до комнатной температуры, проводили фильтрование через целит с отсасыванием и осадок на фильтре промывали ЕА. Остаточную жидкость концентрировали и проводили выделение на колонке с силикагелем (силикагель, 200-300 меш; гептан:ЕА = от 2:1 до 1:1), получая продукт 6-А2 (1,2 г; выход 63,2%) в виде светло-желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 9.96 (s, 1Н), 8.04 (d, J=8,2 Гц, 1Н), 7.73 (dd, J=8,2; 1,5 Гц, 1Н), 7.51 (d, J=1,4 Гц, 1 Н), 7.49-7.43 (m, 1 Н), 7.35-7.27 (m, 1Н), 7.14 (dd, J=7,9; 1,3 Гц, 2Н), 3.09 (s, 3H), 2.99 (s, 3H). LCMS: рассчитано 314,1; обнаружено 315,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 6-А2 (550 мг; 1,75 ммоль) в THF (10 мл) и добавляли (трифторметил)триметилсилан (373 мг; 2,62 ммоль). Температуру понижали до 0°С и по каплям добавляли тетрабутиламмония фторид (0,04 мл; 0,04 ммоль; 1 М раствор в THF). Через 2 часа по каплям добавляли 3 н. соляную кислоту (0,5 мл). Осуществляли экстракцию с использованием DCM (10 мл×2), затем промывку раствором NaHCO3 (5 мл×3), промывку водой, промывку рассолом, сушку и концентрирование, получая 600 мг неочищенного продукта 6-А3 в виде светло-желтой маслянистой жидкости, которую использовали непосредственно на следующей стадии.
В атмосфере азота оксихлорид фосфора (376 мг; 2,45 ммоль) добавляли к сверхсухому DCM (15 мл). Температуру понижали до -30°С. По каплям добавляли раствор соединения 6-А3 (470 мг; 1,22 ммоль; неочищенного) в DCM (5 мл), затем по каплям добавляли триэтиламин (312 мг; 3,03 ммоль) и температуру поддерживали при -30°С в течение 4 ч до тех пор, пока исходные вещества не были израсходованы полностью. Добавляли 2-бромэтиламина гидробромид (2,0 г; 9,78 ммоль) при -30°С и затем по каплям добавляли триэтиламин (940 мг; 9,78 ммоль). Температуру поддерживали при -30°С. Через 1 ч наблюдали завершение реакции. При 0°С по каплям добавляли насыщенный водный раствор хлорида аммония (8 мл). Осуществляли экстракцию дихлорметаном (15 мл×2), затем промывку водой, сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; от смеси 1:2 РЕ:ЕА до ЕА), получая продукт 6-А4 (440 мг; выход 53,4%) в виде желтой маслянистой жидкости. 1Н-ЯМР: δ млн-1 7.98 (d, J=8,3 Гц, 1Н), 7.49-7.46 (m, 1Н), 7.29-7.21 (m, 4Н), 7.07 (s, 1Н), 5.71-5.67 (m, 1Н), 3.46-3.12 (m, 10H), 3.07 (s, 3H), 2.99 (s, 3H). MS: рассчитано: 676,0; обнаружено: 677,0 ([М+1]+).
В атмосфере азота проводили растворение соединения 6-А4 (440 мг; 0,65 ммоль) в THF (25 мл), добавляли оксид серебра (910 мг; 3,92 ммоль) и затем DIEA (510 мг; 3,92 ммоль). Температуру повышали до 65°С. Через 2 ч наблюдали завершение реакции. Температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM, остаточную жидкость концентрировали и после проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №6 (90,0 мг; 27%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.00 (d, J=8,4 Гц, 1Н), 7.52-7.32 (m, 2Н), 7.26-7.23 (m, 2Н), 7.08-7.06 (m, 2Н), 5.72 (dq, J=12,2; 6,1 Гц, 1Н), 3.09 (s, 3H), 2.97 (s, 3H), 2.32-1.89 (m, 8Н). LCMS: рассчитано 514,1; обнаружено 515,1 [(М+Н)+].
Синтез соединения №7
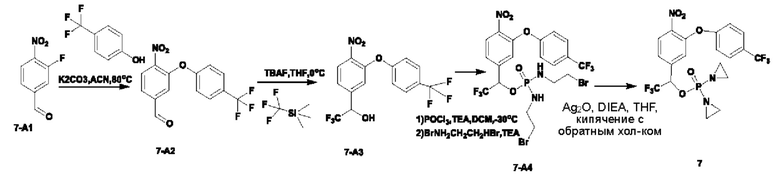
В атмосфере азота проводили растворение соединения 7-А1 (1,5 г; 9,15 ммоль; имеется в продаже) и п-трифторметилфенола (990 мг; 6,10 ммоль) в ацетонитриле (20 мл). Добавляли карбонат калия (1,7 г; 12,2 ммоль; имеется в продаже). Температуру повышали до 80°С и через 2,5 ч наблюдали завершение реакции. Температуру понижали до комнатной температуры, проводили фильтрование через целит с отсасыванием и осадок на фильтре промывали ЕА. Остаточную жидкость концентрировали и осуществляли разделение на колонке (силикагель, 200-300 меш; гептан:ЕА = от 5:1 до 1:1), получая продукт 7-А2 (1,1 г; выход 38,6%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 10.01 (s, 1Н), 8.09 (d, J=8,2 Гц, 1Н), 7.79 (dd, J=8,2; 1,6 Гц, 1Н), 7.68 (d, J=8,6 Гц, 2Н), 7.57 (d, J=1,5 Гц, 1 Н), 7.15 (d, J=8,6 Гц, 2Н).
В атмосфере азота проводили растворение соединения 7-А2 (400 мг; 1,29 ммоль) в THF (10 мл) и добавляли (трифторметил)триметилсилан (274 мг; 1,93 ммоль). Температуру понижали до 0°С и по каплям добавляли тетрабутиламмония фторид (0,04 мл; 0,04 ммоль; раствор в THF, 1 моль/л). Через 2 часа по каплям добавляли 3 н. соляную кислоту (0,5 мл). Осуществляли экстракцию с использованием DCM (10 мл×2), затем промывку раствором NaHCO3 (5 мл×3), промывку водой, промывку рассолом, сушку и концентрирование, получая 450 мг неочищенного продукта 7-А3 в виде светло-желтой маслянистой жидкости, которую использовали непосредственно на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.04 (d, J=7,8 Гц, 1Н), 7.64 (d, J=7,9 Гц, 2Н), 7.45 (d, J=7,7 Гц, 1 Н), 7.30 (s, 1 Н), 7.09 (d, J=8,0 Гц, 2Н), 5.09 (s, 1Н), 3.26 (s, 1Н). LCMS: рассчитано 381,2; обнаружено 382,0 ([М+1]+).
В атмосфере азота добавляли оксихлорид фосфора (362 мг; 2,36 ммоль) к сверхсухому DCM (15 мл). Температуру понижали до -30°С. По каплям добавляли раствор соединения 7-А3 (450 мг; 1,18 ммоль; неочищенного) в DCM (5 мл), затем по каплям добавляли триэтиламин (300 мг; 2,95 ммоль) и температуру поддерживали при -30°С в течение 4 ч до тех пор, пока исходные вещества не были израсходованы полностью. Добавляли 2-бромэтиламина гидробромид (1,9 г; 9,44 ммоль) при -30°С и затем по каплям добавляли триэтиламин (960 мг; 9,44 ммоль). Температуру поддерживали при -30°С. Через 1 ч наблюдали завершение реакции. По каплям при 0°С добавляли насыщенный водный раствор хлорида аммония (6 мл). Осуществляли экстракцию с использованием DCM (15 мл×2), затем промывку водой, сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш, гептан:ЕА = от 1:1 до 0:1), получая продукт 7-А4 (440 мг; 55,4%) в виде желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.05 (d, J=8,4 Гц, 1Н), 7.65 (d, J=8,6 Гц, 2Н), 7.44 (d, J=8,5 Гц, 1Н), 7.23 (s, 1Н), 7.10 (d, J=8,5 Гц, 2Н), 5.72-5.64 (m, 1Н), 3.40-3.10 (m, 10Н). LCMS: рассчитано 672,9; обнаружено 673,9 ([М+1]+).
В атмосфере азота проводили растворение соединения 7-А4 (400 мг; 0,59 ммоль) в THF (25 мл), добавляли оксид серебра (826 мг; 3,57 ммоль) и затем добавляли DIEA (461 мг; 3,57 ммоль). Температуру повышали до 65°С. Через 2 ч наблюдали завершение реакции. Температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM, остаточную жидкость концентрировали и после проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №7 (68 мг; выход 22,5%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.05 (d, J=8,5 Гц, 1Н), 7.65 (d, J=8,6 Гц, 2Н), 7.45 (d, J=8,5 Гц, 1Н), 7.27 (s, 1 Н), 7.09 (d, J=8,5 Гц, 2Н), 5.75 (d, J=4,4 Гц, 1Н), 2.33-1.98 (m, 8Н). LCMS: рассчитано LCMS: 511,1; обнаружено 512,0 ([М+Н]+).
Синтез соединения №8

Соединение 8-А1 (5,5 г; 27,6 ммоль) и п-трифторметилфенол (3,0 г; 18,5 ммоль; имеется в продаже) растворяли в ацетонитриле (30 мл). Добавляли K2CO3 (5,1 г; 37,0 ммоль; имеется в продаже). Температуру повышали до 80°С и осуществляли перемешивание в течение ночи до завершения реакции. Температура естественным образом снижалась до комнатной температуры, проводили фильтрование через целит с отсасыванием и осадок на фильтре промывали ЕА (10 мл×3). Остаточную жидкость концентрировали и после кристаллизации из метил-трет-бутилового эфира получали неочищенный продукт 8-А2 (4,6 г; 72,9%) в виде желтого твердого вещества. Этот неочищенный продукт использовали непосредственно в последующей реакции.
Соединение 8-А2 (2,5 г; 7,33 ммоль) растворяли в THF, порциями добавляли боргидрид натрия (2,2 г; 58,7 ммоль; имеется в продаже) и перемешивание проводили при комнатной температуре в течение 30 мин. Температуру повышали до 65°С, проводили перемешивание и мониторинг прохождения реакции. Через 2 ч наблюдали завершение реакции. Систему охлаждали до 0°С, по каплям добавляли H2O (20 мл) и перемешивание проводили в течение 20 мин. Осуществляли экстракцию с использованием DCM (50 мл×3), затем промывку водой, сушку с использованием безводного Na2SO4 и концентрирование, получая неочищенный продукт 8-А3 (1,5 г; 65,4%) в виде светло-желтого твердого вещества.
В атмосфере азота проводили растворение соединения 8-А3 (1,5 г; 4,79 ммоль) в DCM (20 мл). Температуру понижали до 0°С, по каплям добавляли SOCl2 (1,1 г; 9,58 ммоль) и TEA (485 мг; 4,79 ммоль) и перемешивали. Проводили мониторинг прохождения реакции. Через 1 ч наблюдали завершение реакции. При 0°С по каплям добавляли насыщенный водный раствор NaHCO3 (5 мл). Осуществляли экстракцию с использованием DCM (20 мл×3), затем промывку водным раствором NaHCO3, сушку с использованием безводного Na2SO4 и концентрирование, получая неочищенный продукт 8-А5 (1,3 г; 81,8%) в виде красновато-коричневой жидкости, которую использовали непосредственно на следующей стадии.
Соединение 8-А4 (220 мг; 0,66 ммоль) и 2,3,5,6-тетрафтор-4-гидроксибензальдегид (513 мг; 2,65 ммоль) растворяли в DMF (5 мл). По каплям добавляли DIEA(430 мг; 3,32 ммоль) и перемешивали. Температуру повышали до 45°С. Проводили мониторинг прохождения реакции. Через 3 ч наблюдали завершение реакции. Добавляли H2O (10 мл), и температура естественным образом снижалась до комнатной температуры. Осуществляли экстракцию с использованием ЕА (8 мл×3), затем сушку с использованием безводного Na2SO4 и концентрирование, получая неочищенный продукт 8-А5 (70 мг) в виде светло-желтой жидкости, которую использовали непосредственно на следующей стадии.
Соединение 8-А5 (70 мг; 0,14 ммоль) растворяли в THF (5 мл). Температуру понижали до 0°С. Порциями добавляли NaBH4 (11 мг; 0,37 ммоль). Проводили мониторинг прохождения реакции. Через 0,5 ч наблюдали завершение реакции. При 0°С по каплям добавляли H2O (3 мл) и перемешивали в течение 20 мин. Осуществляли экстракцию с использованием DCM (10 мл×3), затем промывку водой, сушку с использованием безводного Na2SO4 и концентрирование, получая 50 мг неочищенного продукта 8-А6 в виде светло-желтой жидкости, которую использовали непосредственно на следующей стадии.
POCl3 (31 мг; 0,20 ммоль; имеется в продаже) растворяли в DCM (3 мл). Температуру понижали до -30°С. По каплям добавляли раствор соединения 8-А6 (50 мг; 0,10 ммоль) в DCM (1 мл) и затем по каплям добавляли раствор триэтиламина (26 мг; 0,26 ммоль) в DCM (1 мл). Для проведения реакции температуру поддерживали при -30°С. Проводили мониторинг прохождения реакции. Через 2 ч отмечали расходование исходных веществ. Температуру понижали до -40°С. Добавляли 2-бромэтиламина гидробромид (167 мг; 0,82 ммоль) и TEA (83 мг; 0,82 ммоль). Температуру поддерживали при -40°С. Через 30 мин наблюдали завершение реакции. После повышения температуры до 5°С добавляли H2O (5 мл) и осуществляли экстракцию с использованием DCM (5 мл×3), затем промывку водой (3 мл×2), сушку с использованием безводного Na2SO4, концентрирование и разделение посредством колоночной хроматографии (силикагель, 200-300 меш; н-гептан:ЕА=1:1), получая продукт 8-А7 (50 мг; 62,7%) в виде светло-желтой жидкости.
Соединение 8-А7 (50 мг; 0,064 ммоль) растворяли в THF (5 мл) и добавляли Ag2O (74 мг; 0,32 ммоль; имеется в продаже) и DIEA (41 мг; 0,32 ммоль). Температуру повышали до 65°С. Проводили мониторинг прохождения реакции. Через 1 ч наблюдали завершение реакции. Температуру понижали до комнатной температуры, проводили фильтрование через целит с отсасыванием и твердое вещество промывали THF (2 мл×3). Остаточную жидкость концентрировали и после разделения высокоэффективной жидкостной хроматографией получали продукт (2,2 мг; 5,5%) в виде белого воскообразного вещества. 1Н-ЯМР (400 МГц, CDCl3) δ млн-1 8.04 (d, J=8,4 Гц, 1Н), 7.65 (d, J=8,6 Гц, 2Н), 7.36 (dd, J=8,4; 1,6 Гц, 1 Н), 7.20 (d, J=1,5 Гц, 1 Н), 7.09 (d, J=8,5 Гц, 2Н), 5.28 (s, 2Н), 5.23 (d, J=6,1 Гц, 2Н), 2.26-2.15 (m, 8Н). LCMS: рассчитано 621,1; обнаружено 622,1 ([М+Н]+).
Синтез соединения №9
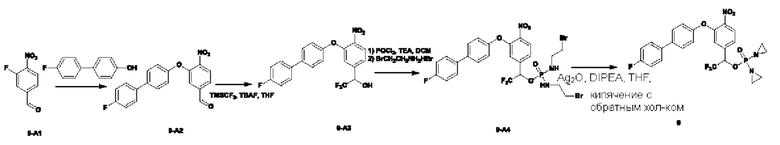
В атмосфере азота проводили растворение соединения 9-А1 (500 мг; 2,97 ммоль) и 4-фтор-4'-гидроксибифенила (723 мг; 3,84 ммоль; имеется в продаже для фармацевтики) в ацетонитриле (10 мл). Добавляли карбонат калия (820 мг; 5,94 ммоль; имеется в продаже). Температуру повышали до 85°С и перемешивание проводили в течение 2 ч до завершения реакции. Температуру понижали до комнатной температуры и проводили фильтрование с отсасыванием. Остаточную жидкость концентрировали и осуществляли выделение посредством колоночной хроматографии (силикагель, 200-300 меш; гептан:ЕА = 20:1), получая продукт (420 мг; выход 42,0%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 9.98 (s, 1Н), 8.05 (d, J=8,0 Гц, 1Н), 7.70 (dd, J1=8,0 Гц; J2=1,6 Гц, 1Н), 7.56-7.60 (m, 2Н), 7.52-7.56 (m, 3H), 7.12-7.17 (m, 4Н).
В атмосфере азота проводили растворение соединения 9-А2 (400 мг; 1,19 ммоль) в безводном THF (8 мл) и затем по каплям добавляли (трифторметил)триметилсилан (254 мг; 1,79 ммоль). Температуру понижали до 0°С и по каплям добавляли TBAF (0,03 мл; 1 М раствор в THF). Температуру поддерживали при 0°С в течение 1,5 ч до завершения реакции. По каплям добавляли 2 мл 3 н. соляной кислоты. Температура естественным образом повышалась до комнатной температуры. Проводили перемешивание в течение 1 ч. Добавляли 5 мл воды. Осуществляли экстракцию с использованием DCM (10мл×3), затем промывку водой (5 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = 10:1), получая продукт 9-А3 (380 мг; выход 78,4%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.00 (d, J=8,4 Гц, 1 Н), 7.51-7.57 (m, 4Н), 7.35 (d, J=8,4 Гц), 7.24 (s, 1Н), 7.10-7.16 (m, 4Н), 5.04-5.06 (m, 1Н). LCMS: рассчитано 407,0; обнаружено 430,0 ([M+Na]+).
В атмосфере азота к оксихлориду фосфора (285 мг; 1,866 ммоль) по каплям добавляли безводный DCM (10 мл). Температуру понижали до -40°С. По каплям добавляли раствор соединения 9-А3 (380 мг; 0,933 ммоль) в DCM (4 мл), затем по каплям добавляли триэтиламин (236 мг; 2,333 ммоль) и температуру поддерживали в диапазоне от -40°С до -35°С в течение 2 ч. По данным LC-LCMS соединение 9-А3 было израсходовано и превратилось в промежуточное соединение. При -40°С добавляли 2-бромэтиламина гидробромид (1,53 г; 7,464 ммоль) и затем по каплям добавляли раствор триэтиламина (755 мг; 7,464 ммоль) в DCM (2 мл). Температуру поддерживали при -40°С в течение 1 ч, и промежуточное соединение полностью расходовалось. Температура естественным образом повышалась до 0°С, и по каплям добавляли насыщенный водный раствор хлорида аммония (5 мл). Осуществляли экстракцию с использованием DCM (10 мл×3), затем промывку чистой водой (3 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА=1:1), получая продукт (m=350 мг; выход 53,7%) в виде беловатого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.01 (d, J=8,4 Гц, 1Н), 7.52-7.58 (m, 4Н), 7.34 (d, J=8,4 Гц, 1Н), 7.11-7.18 (m, 5Н), 5.61-5.68 (m, 1Н), 3.13-3.81 (m, 10H). LCMS рассчитано: 699,0; обнаружено 700,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 9-А4 (350 мг; 0,5 ммоль) в THF (10 мл) и затем добавляли оксид серебра (700 мг; 3,0 ммоль) и DIPEA (390 мг; 3,0 ммоль). Температуру повышали до 65°С и перемешивание проводили в течение 3 ч. После завершения реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM, остаточную жидкость концентрировали и после проведения препаративной хроматографии получали чистый продукт (m=31,4 мг; белое твердое вещество; выход 11,7%). 1Н-ЯМР (400 МГц, CD3OD): δ млн-1 8.08 (d, J=8,4 Гц, 1Н), 7.63-7.69 (m, 4Н), 7.50 (d, J=8,4 Гц, 1Н), 7.31 (s, 1Н), 7.16-7.20 (m, 4H), 5.99-6.04 (m, 1H), 2.03-2.24 (m, 8H). LCMS: рассчитано 537,0; обнаружено 538,0 ([М+Н]+).
Синтез соединения №11
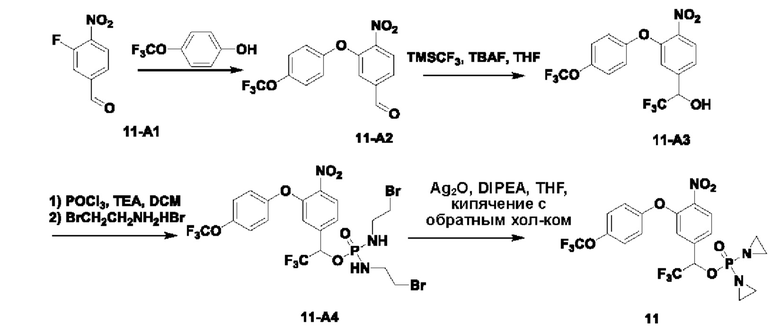
В атмосфере азота проводили растворение соединения 11-А1 (500 мг; 2,97 ммоль) и п-трифторметоксифенола (684 мг; 3,84 ммоль; имеется в продаже для фармацевтики) в ацетонитриле (10 мл). Добавляли карбонат калия (820 мг; 5,94 ммоль; имеется в продаже). Температуру повышали до 85°С и проводили перемешивание в течение 2 ч до завершения реакции. Температуру понижали до комнатной температуры и осуществляли фильтрование с отсасыванием. Остаточную жидкость концентрировали и проводили выделение колоночной хроматографией (силикагель, 200-300 меш; н-гептан:ЕА = 30:1), получая продукт 11-А2 (в целом 520 мг; светло-желтое твердое вещество; выход 53,5%). 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 9.99 (s, 1 Н), 8.06 (d, J=8,4 Гц, 1 Н), 7.73 (dd, J,=8,4 Гц, J2=1,6 Гц, 1Н), 7.50 (d, J2=1,2 Гц, 1Н), 7.29 (d, J=8,4 Гц, 1Н), 7.10-7.14 (m, 2Н).
В атмосфере азота проводили растворение соединения 11-А2 (500 мг; 1,528 ммоль) в безводном THF (8 мл) и затем по каплям добавляли (трифторметил)триметилсилан (370 мг; 2,598 ммоль). Температуру понижали до 0°С и по каплям добавляли TBAF (0,03 мл; 1 М раствор в THF). Температуру поддерживали при 0°С в течение 1,5 ч до завершения реакции. По каплям добавляли 2 мл 3 н. соляной кислоты. Температура естественным образом повышалась до комнатной температуры. В течение 1 ч осуществляли перемешивание. Добавляли 5 мл воды. Осуществляли экстракцию с использованием DCM (10 мл×3), затем промывку водой (5 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 20:1 до 10:1), получая продукт (400 мг; выход 65,9%) в виде желтого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.00 (d, J=8,4 Гц, 1Н), 7.38 (d, J =8,4 Гц, 1Н), 7.25 (d, J=8,8 Гц, 2Н), 7.21 (s, 1Н), 7.06 (d, J=8,8 Гц, 2Н), 5.04-5.09 (m, 1Н), 3.05 (brs, 1Н).
В атмосфере азота к оксихлориду фосфора (310 мг; 2,014 ммоль) по каплям добавляли безводный DCM (10 мл). Температуру понижали до -40°С. По каплям добавляли раствор соединения 11-А3 (400 мг; 1,007 ммоль) в DCM (4 мл), затем по каплям добавляли триэтиламин (255 мг; 2,518 ммоль) и температуру поддерживали в диапазоне от -40°С до -35°С в течение 2 ч. По данным LC-LCMS соединение 11-А3 было израсходовано и превратилось в промежуточное соединение. Добавляли 2-бромэтиламина гидробромид (1,65 г; 8,056 ммоль) при -40°С и затем по каплям добавляли раствор триэтиламина (815 мг; 8,056 ммоль) в DCM (2 мл). Температуру поддерживали при -40°С в течение 1 ч, и промежуточное соединение полностью расходовалось. Температура естественным образом повышалась до 0°С, и по каплям добавляли насыщенный водный раствор хлорида аммония (5 мл). Осуществляли экстракцию с использованием DCM (10мл×3), затем промывку чистой водой (3 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; от смеси 1:1 н-гептан:ЕА до 100%-ного ЕА), получая продукт 11-А4 (m=160 мг; выход 23,1%) в виде желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.02 (d, J=8,4 Гц, 1Н), 7.38 (d, J=8,4 Гц, 1Н), 7.27 (d, J=8,8 Гц, 2Н), 7.14 (s, 1Н), 7.08 (d, J=8,8 Гц, 2Н), 5.63-5.68 (m, 1Н), 3.46-3.49 (m, 2Н), 3.34-3.42 (m, 2Н), 3.26-3.32 (m, 2Н), 3.02-3.25 (m, 4Н). LCMS: рассчитано: 688,9; обнаружено 689,8 ([М+Н]+).
В атмосфере азота проводили растворение соединения 11-А4 (160 мг; 0,230 ммоль) в THF (10 мл) и затем добавляли оксид серебра (323 мг; 1,393 ммоль) и DIPEA (180 мг; 1,393 ммоль). Температуру повышали до 65°С и проводили перемешивание в течение 3 ч. После завершения реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM, остаточную жидкость концентрировали и получали чистое соединение №11 (m=26,3 мг; выход 21,7%) в виде желтого воскообразного вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.01 (d, J=8,4 Гц, 1Н), 7.39 (d, J=8,4 Гц, 1Н), 7.26 (d, J=8,8 Гц, 2Н), 7.18 (s, 1Н), 7.05-7.08 (m, 2Н), 5.60-5.74 (m, 1Н), 2.00-2.26 (m, 8Н). LCMS: рассчитано 527,1; обнаружено 528,0 ([М+Н]+).
Синтез соединения №12
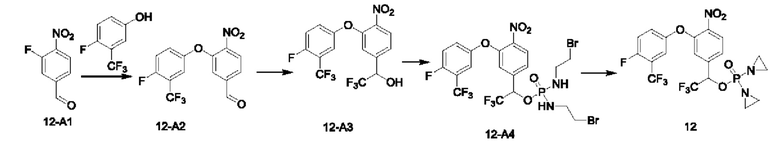
Соединение 12-А1 (1,0 г; 5,9 ммоль) и 4-фтор-3 (трифторметил)фенол (2,1 г; 11,8 ммоль; 2 экв.; имеется в продаже) растворяли в ацетонитриле (20 мл). В атмосфере азота к этой системе добавляли К2СО3 (1,6 г; 11,8 ммоль; 2 экв.). Температуру повышали до 80°С. Результаты мониторинга в течение 1 ч показали расходование исходных веществ и завершение реакции. Последующую обработку проводили так, как приведено ниже. После охлаждения до комнатной температуры систему подвергали фильтрованию через целит с отсасыванием и промывали DCM. После упаривания остаточной жидкости на роторном испарителе образец смешивали с силикагелем 300-400 меш (в 10-кратном количестве) и пропускали через колонку для флэш-хроматографии (н-гептан:ЕА=94%:6%), получая соединение 12-А2 (1,15 г; 59,2%) в виде светло-желтой маслянистой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 10.00 (s, 1Н), 8.08 (d, J=8,0 Гц, 1Н), 7.76 (dd, J,=8,4 Гц; J2=1,6 Гц, 1Н), 7.46 (d, J=1,6 Гц, 1Н), 7.33-7.34 (m, 1 Н), 7.27-7.2 8 (m, 1Н), 7.26-7.53 (m, 1Н).
В атмосфере азота проводили растворение соединения 12-А2 (620 мг; 1,9 ммоль) и трифторметилтриметилсилана (TMSCF3; 539 мг; 3,8 ммоль; 2 экв.) в THF (6 мл). Температуру понижали до 0°С. К этой системе по каплям добавляли TBAF (0,04 мл; 0,04 ммоль; 1 М раствор в THF; имеется в продаже). После поддержания температуры при 0°С в течение 30 мин соединение 12-А2 полностью расходовалось. К системе по каплям добавляли 3 н. раствор HCl (3 мл), и система становилась прозрачной. После перемешивания при 0°С в течение 1 ч все исходные вещества превращались в продукты. Последующую обработку проводили так, как приведено ниже. Осуществляли экстракцию с использованием DCM (5 мл×3). Органическую фазу промывали водой (5 мл×3), сушили и упаривали на роторном испарителе. Образец смешивали с силикагелем 300-400 меш (в 7-кратном количестве) (н-гептан:ЕА=95:5), получая соединение 12-А3 (600 мг; 79,1%) в виде светло-желтого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.02 (d, J=8,4 Гц, 1Н), 7.41 (d, J=8,4 Гц, 1Н), 7.29 (dd, J,=6,4 Гц; J2=2,8 Гц, 1Н), 7.24-7.18 (m, 3H), 5.08 (dd, J,=12,4 Гц; J2=6,0 Гц, 1Н).
В атмосфере азота проводили растворение POCl3 (384 мг; 2,5 ммоль; 2 экв.) в DCM (5 мл). После этого температуру понижали до -40°С. Затем соединение 12-А3 (500 мг; 1,3 ммоль) растворяли в DCM (2 мл), далее по каплям добавляли к системе вместе с добавлением TEA (317 мг; 3,1 ммоль; 2,5 экв.). После поддержания температуры при -40°С в течение 2 ч соединение 12-А3 полностью превращалось в промежуточное соединение. Затем к этой системе добавляли 2-бромэтиламина гидробромид (2,1 г; 10,0 ммоль; 8 экв.) и TEA (1,0 г; 10,0 ммоль). Проводили мониторинг. Через 30 мин наблюдали завершение реакции. Последующую обработку проводили так, как приведено ниже. При 0°С добавляли насыщенный водный раствор хлорида аммония (10 мл). Осуществляли экстракцию с использованием DCM (20 мл×3). Органическую фазу промывали водой и рассолом и упаривали на роторном испарителе. После этого образец смешивали с силикагелем 200-300 меш (в 8-кратном количестве) (н-гептан:ЕА=1:1), получая соединение 12-А4 (350 мг; 39,0%) в виде светло-желтого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.06 (d, J=8,4 Гц, 1Н), 7.44 (d, J=8,4 Гц, 1Н), 7.33-7.35 (m, 1Н), 7.24-7.29 (m, 2), 7.17 (s, 1Н), 5.70 (dd, J=11,5; 6,0 Гц, 1Н), 3.86-2.96 (m, 10Н). LCMS: рассчитано 690,9; обнаружено 691,8 ([М+1]+).
В атмосфере азота проводили растворение соединения 12-А4 (350 мг; 0,5 ммоль) в THF (10 мл). Затем к этой системе добавляли Ag2O (587 мг; 2,5 ммоль) и DIEA(327 мг; 2,5 ммоль; 5 экв.). Температуру повышали до температуры дефлегмации, проводили мониторинг прохождения реакции, и через 2 ч наблюдали завершение реакции. Температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Проводили промывку DCM. Остаточную жидкость упаривали на роторном испарителе. Соединение №12 (74 мг; 26,4%) получали в виде светло-желтого твердого вещества, используя препаративную хроматографию на нейтральной фазе. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.04 (d, J=8,4 Гц, 1Н), 7.43 (dd, J=8,4 Гц, 1Н), 7.28-7.26 (m, 1Н), 7.23-7.22 (m, 2Н), 7.18 (s, 1Н), 5.76-5.71 (m, 1Н), 2.27-2.02 (m, 8Н). LCMS: рассчитано 529,1; обнаружено 530,0 ([М+1]+).
Синтез соединения №13

В атмосфере азота проводили растворение соединения 13-А1 (500 мг; 2,95 ммоль) и 3-гидроксихинолина (470 мг; 3,24 ммоль; имеется в продаже) в ацетонитриле (ACN; 5 мл). Добавляли карбонат калия (830 мг; 6 ммоль). Температуру повышали до 80°С и в течение 4 ч проводили перемешивание. По завершении реакции проводили фильтрование через целит с отсасыванием, затем концентрирование. Образец перемешивали и подвергали разделению посредством колоночной хроматографии (силикагель, 100-200 меш; н-гептан:ЕА=10:1), получая продукт (560 мг; 64,5%) в виде белого твердого вещества, которое использовали непосредственно на следующей стадии. 1Н-ЯМР (300 МГц, DMSO): δ млн-1 10.02 (s, 1Н), 8.92 (d, J=2,6 Гц, 1Н), 8.34 (d, J=8,2 Гц, 1Н), 8.09 (d, J=8,6 Гц, 2Н), 7.94 (t, J=8,2 Гц, 2Н), 7.82 (s, 1Н), 7.76 (t, J=7,1 Гц, 1Н), 7.65 (t, J=7,2 Гц, 1Н). LCMS: рассчитано 294,0; обнаружено 295,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 13-А2 (460 мг; 1,56 ммоль) и трифторметилтриметилсилана (430 мг; 3 ммоль) в THF (5 мл). При 0°С по каплям добавляли раствор фторида тетрабутиламмония в THF (0,1 мл; 0,1 ммоль; 1 М). Такую температуру поддерживали в течение 6 ч. Добавляли 1 н. соляную кислоту (2 мл) и перемешивали в течение 10 мин. THF удаляли путем концентрирования. К неочищенному продукту добавляли воду (10 мл) и осуществляли экстракцию с использованием DCM (20 мл). Органическую фазу отделяли и концентрировали и образец перемешивали. Неочищенный продукт подвергали разделению посредством колоночной хроматографии (силикагель, 100-200 меш; н-гептан:ЕА=10:1), получая продукт (400 мг) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO): δ млн-1 8.87 (d, J=2,8 Гц, 1 Н), 8.23 (d, J=8,5 Гц, 1Н), 8.08 (d, J=8,4 Гц, 1Н), 8.00 (d, J=2,7 Гц, 1Н), 7.96 (d, J=7,4 Гц, 1Н), 7.76 (ddd, J=8,4; 6,9; 1,4 Гц, 1Н), 7.69-7.62 (m, 1Н), 7.58 (d, J=8,5 Гц, 1Н), 7.43 (s, 1Н), 7.19 (d, J=5,9 Гц, 1Н), 5.43-5.31 (m, 1Н). LCMS: рассчитано 364,0; обнаружено 365,0 ([М+Н]+).
В атмосфере азота к POCl3 (168 мг; 1,1 ммоль) по каплям добавляли сверхсухой DCM (5 мл). Температуру понижали до -30°С. По каплям добавляли раствор соединения 13-А3 (200 мг; 0,55 ммоль) в DCM (5 мл), затем по каплям добавляли TEA (170 мг; 1,65 ммоль) и температуру поддерживали при -30°С в течение 6 ч до полного расходования исходных веществ. Добавляли 2-бромэтиламина гидробромид (897 мг; 4,4 ммоль) при -30°С и затем по каплям добавляли TEA (440 мг; 4,4 ммоль). По завершении реакции температуру понижали до 0°С. Добавляли насыщенный раствор NH4Cl (10 мл). Осуществляли экстракцию с использованием DCM (15 мл×2), затем промывку водой (5 млх4), сушку и концентрирование, получая 150 мг неочищенного продукта в виде желтого твердого вещества, которое использовали непосредственно в реакции на следующей стадии.
В атмосфере азота проводили растворение соединения 13-А4 (150 мг; 0,23 ммоль) в THF (10 мл) и добавляли Ag2O (170 мг; 1,38 ммоль) и N,N-диизопропилэтиламин (163 мг; 1,38 ммоль). Температуру повышали до 65°С. Через 2 ч, после завершения реакции, проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM (20 мл), остаточную жидкость концентрировали и после проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №13 (41 мг; 36%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 8.82 (d, J=2,8 Гц, 1Н), 8.22 (d, J=8,5 Гц, 1Н), 8.10 (d, J=8,6 Гц, 1Н), 7.94-7.87 (m, 2Н), 7.77 (ddd, J=8,4; 6,9; 1,4 Гц, 1Н), 7.69-7.62 (m, 2Н), 7.52 (s, 1Н), 6.13-6.02 (m, 1Н), 2.29-1.98 (m, 8Н). LCMS: рассчитано 494,1; обнаружено 495,0 ([М+Н]+).
Синтез соединения №14
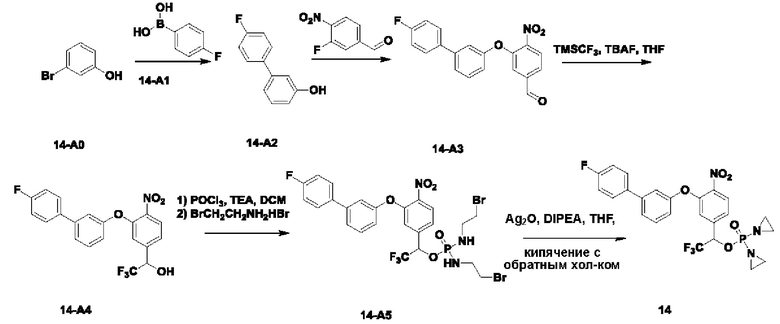
В атмосфере азота добавляли м-бромфенол (14-A0; 1,5 г; 8,57 ммоль) и п-фторфенилбороновую кислоту (14-А1; 1,0 г; 7,15 ммоль; имеется в продаже) к смеси жидкостей, состоящей издиоксана (30 мл) и H2O (5 мл). Трижды проводили вакуумирование и заполнение азотом. Затем добавляли Pd(AcO)2 (80 мг; 0,36 ммоль), PPh3 (94 мг; 0,36 ммоль) и К2СО3 (3,0 г; 21,44 ммоль), вновь 3 раза проводили вакуумирование и заполнение азотом и температуру повышали до 100°С. Мониторинг прохождения реакции проводили в течение 1 ч, после чего наблюдали ее завершение. Последующую обработку проводили так, как приведено ниже. Температуру понижали до комнатной температуры. После фильтрования с отсасыванием осуществляли экстракцию с использованием EtOAc (50 мл×3). Органическую фазу промывали водой, промывали рассолом и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 12:1 до 9:1), получая продукт 14-А2 (1,1 г; выход 62,7%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, MeOD): δ млн-1 7.59-7.54 (m, 2Н), 7.23-7.10 (m, 3H), 7.04-6.99 (m, 2Н), 6.76 (dd, J=8,1; 1,5 Гц, 1Н). LCMS: рассчитано 188,1; обнаружено 189,1 ([М+Н]+).
Соединение 14-А2 (580 мг; 3,08 ммоль) и 3-фтор-4-нитробензальдегид (434 мг; 2,57 ммоль; имеется в продаже) растворяли в ацетонитриле (10 мл). В атмосфере азота к этой системе добавляли К2СО3 (887 мг; 6,42 ммоль). Температуру повышали до 80°С. После проведения мониторинга в течение 2 ч наблюдали завершение реакции. После снижения температуры до комнатной температуры проводили фильтрование через целит с отсасыванием, затем промывку с использованием DCM. Остаточную жидкость концентрировали и подвергали разделению на колонке (колонка для обращенно-фазовой флэш-хроматографии; ацетонитрил:H2O =50%:50%), получая продукт 14-А3 (320 мг; 33,7%) в виде желтого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 9.98 (s, 1Н), 8.05 (d, J=8,4 Гц, 1Н), 7.71 (d, J=1,6 Гц, 1Н), 7.69-7.43 (m, 5Н), 7.28-7.27 (m, 1Н), 7.15-7.11 (m, 2Н), 7.08-7.05 (m, 1Н).
В атмосфере азота проводили растворение соединения 14-А3 (350 мг; 1,04 ммоль) и TMSCF3 (296 мг; 2,08 ммоль) в THF (4 мл). Температуру понижали до 0°С. К этой системе по каплям добавляли TBAF (0,01 мл; 0,01 ммоль; 1 М раствор в THF). Температуру поддерживали при 0°С в течение 30 мин, после чего реагент 14-А3 полностью расходовался. К этой системе по каплям добавляли 3 н. раствор HCl (2 мл), и система становилась прозрачной. После перемешивания при 0°С в течение 1 ч все исходные вещества превращались в продукты. Осуществляли экстракцию с использованием DCM (5 мл×3). Органическую фазу промывали водой (5 мл×3), сушили, концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 12:1 до 10:1), получая продукт (350 мг; выход 82,8%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.00 (d, J=8,4 Гц, 1Н), 7.53-7.34 (m, 5Н), 7.24-7.23 (m, 2Н), 7.15-7.11 (m, 2Н), 7.03-7.00 (m, 1Н), 5.03-5.02 (m, 1Н). LCMS: рассчитано 407,1; обнаружено 408,0 ([М+Н]4).
В атмосфере азота проводили растворение POCl3 (188 мг; 1,23 ммоль) в DCM (5 мл). После этого температуру понижали до -40°С. Далее, соединение 14-А4 (250 мг; 0,61 ммоль) растворяли в DCM (2 мл), после чего по каплям добавляли к системе вместе с добавлением TEA (155 мг; 1,53 ммоль). После выдерживания при температуре -40°С в течение 3 ч соединение 15-А3 полностью превращалось в промежуточное соединение. Затем к этой системе добавляли бромэтиламина гидробромид (1,0 г; 4,91 ммоль) и TEA (497 мг; 4,91 ммоль). Проводили мониторинг. Через 30 мин наблюдали завершение реакции. При 0°С добавляли насыщенный раствор NH4Cl (5 мл). Осуществляли экстракцию с использованием DCM (20 мл×3). Органическую фазу промывали водой, промывали рассолом, сушили, концентрировали и проводили разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 2:1 до 1:1), получая продукт (200 мг; 46,6%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.01 (d, J=8,4 Гц, 1Н), 7.54-7.33 (m, 5Н), 7.25 (d, J=2,0 Гц, 1Н), 7.18 (s, 1 Н), 7.15-7.10 (m, 2Н), 7.03 (d, J=1,2 Гц, 1Н), 5.70-5.60 (m, 1Н), 3.38-3.08 (m, 8Н). LCMS: рассчитано 699,0; обнаружено 699,9 ([М+Н]+).
В атмосфере азота проводили растворение соединения 15-А5 (150 мг; 0,22 ммоль) в THF (15 мл) и затем добавляли оксид серебра (497 мг; 2,1 ммоль) и DIPEA (277 мг; 2,1 ммоль). Температуру повышали до 65°С и проводили перемешивание в течение 1,5 ч. После завершения реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM, остаточную жидкость концентрировали и после проведения препаративной высокоэффективной жидкостной хроматографии получали чистый продукт (56 мг; 48,6%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD) δ млн-1 8.08 (d, J=8,4 Гц, 1Н), 7.67-7.59 (m, 2Н), 7.56-7.46 (m, 3H), 7.31 (d, J=2,2 Гц, 2Н), 7.22-7.13 (m, 2Н), 7.10-7.05 (m, 1Н), 6.05-5.95 (m, 1Н), 2.29-1.89 (m, 8Н). LCMS: рассчитано 537,1; обнаружено 538,1 ([М+Н]+).
Синтез соединения №15

Соединение 15-А1 (500 мг; 2,96 ммоль; имеется в продаже) и соединение 15-А0 (554 мг; 2,69 ммоль) растворяли в ацетонитриле (10 мл). К этой системе в атмосфере азота добавляли К2СО3 (743 мг; 5,38 ммоль). Температуру повышали до 80°С. Мониторинг прохождения реакции проводили в течение 1,5 ч, после чего наблюдали ее завершение. После снижения температуры до комнатной температуры проводили фильтрование через целит с отсасыванием и промывку с использованием DCM. Остаточную жидкость концентрировали и проводили разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 15:1 до 10:1), получая продукт (535 мг; выход 56,0%) в виде коричневого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 9.99 (s, 1Н), 8.05 (d, J=8,4 Гц, 1Н), 7.71 (d, J=8,4 Гц, 1Н), 7.56-7.55 (m, 3H), 7.41 (dd, J1=8,44,8 Гц, 1Н), 7.16 (d, J=8,4 Гц, 2Н), 7.00-6.91 (m, 2Н).
В атмосфере азота проводили растворение соединения 15-А2 (400 мг; 1,13 ммоль) и TMSCF3 (320 мг; 2,25 ммоль) в THF (4 мл). Температуру понижали до 0°С. К системе по каплям добавляли TBAF (0,02 мл; 0,02 ммоль; 1 М раствор в THF). После поддержания температуры при 0°С в течение 30 мин соединение 15-А2 полностью расходовалось. К этой системе по каплям добавляли 3 н. раствор HCl (2 мл), и система становилась прозрачной. После перемешивания при 0°С в течение 1 ч осуществляли экстракцию с использованием DCM (5 мл×3). Органическую фазу промывали водой (5 мл×3), сушили и концентрировали, получая неочищенный продукт (405 мг; выход 56,4%) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии. LCMS: рассчитано 425,3; обнаружено 426,0 ([М+1]+).
В атмосфере азота проводили растворение POCl3 (209 мг; 1,36 ммоль; имеется в продаже) в DCM (5 мл). После этого температуру понижали до -40°С. Затем соединение 15-А3 (290 мг; 0,68 ммоль) растворяли в DCM (2 мл), далее по каплям добавляли к системе вместе с добавлением TEA (173 мг; 1,70 ммоль). После поддержания температуры при -40°С в течение 2 ч соединение 15-А3 полностью превращалось в промежуточное соединение. Затем к этой системе добавляли бромэтиламина гидробромид (1,1 г; 5,46 ммоль) и TEA (552 мг; 5,46 ммоль). Проводили мониторинг. Через 30 мин наблюдали завершение реакции. При 0°С добавляли насыщенный раствор NH4Cl (5 мл). Осуществляли экстракцию с использованием DCM (20 мл×3). Органическую фазу промывали водой и промывали рассолом. Органическую фазу сушили, концентрировали и проводили разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 5:1 до 3:1), получая продукт (250 мг; выход 51,1%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.00 (d, J=8,4 Гц, 1Н), 7.54 (d, J=7,6 Гц, 2Н), 7.44-7.38 (m, 1Н), 7.35 (d, J=8,4 Гц, 1Н), 7.21 (s, 1Н), 7.13 (d, J=8,8 Гц, 1Н), 7.00-6.95 (m, 2Н), 5.68-5.62 (m, 1Н), 3.42-3.11 (m, 8Н). LCMS: рассчитано 717,2; обнаружено 717,9 ([М+Н]+).
В атмосфере азота проводили растворение соединения 15-А4 (230 мг; 0,32 ммоль) в THF (15 мл) и затем добавляли оксид серебра (372 мг; 1,60 ммоль) и DIPEA (207 мг; 1,60 ммоль). Температуру повышали до 65°С и проводили перемешивание в течение 1,5 ч. После завершения реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM, остаточную жидкость концентрировали и после проведения препаративной высокоэффективной жидкостной хроматографии получали чистый продукт (102 мг; 57,5%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.01 (d, J=8,4 Гц, 1Н), 7.54 (d, J=1,6 Гц, 2Н), 7.51-7.36(m, 2Н), 7.24 (s, 1Н), 7.12-7.11 (m, 2Н), 6.97-6.93 (m, 2Н), 5.72-5.69 (m, 1Н), 2.26-2.01 (m, 8Н). LCMS: рассчитано 555,1; обнаружено 556,1 ([М+Н]+).
Синтез соединения №16
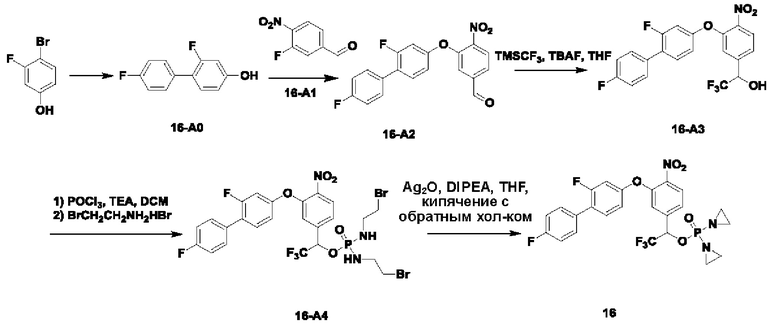
В атмосфере азота проводили растворение 3-фтор-4-бромфенола (5,0 г; 26,2 ммоль; имеется в продаже; 97%-ного) и п-фторфенилбороновой кислоты (4,0 г; 28,8 ммоль; имеется в продаже; 97%-ной) в смеси растворителей диоксана и воды (100 мл; диоксан:вода=9:1). После этого добавляли карбонат калия (10,8 г; 78,6 ммоль). Трижды проводили вакуумирование и заполнение газом. После добавления ацетата палладия (295 мг; 1,31 ммоль; имеется в продаже; 95%-ный) и трифенилфосфина (345 мг; 1,31 ммоль; имеется в продаже; 97%-ный) вновь трижды проводили вакуумирование и заполнение газом. Температуру повышали до 100°С и перемешивание осуществляли в течение ночи. По завершении реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием и промывку с использованием ЕА. Остаточную жидкость концентрировали и значение рН подводили до 3, добавляя 1 н. соляную кислоту. Осуществляли экстракцию с использованием ЕА (50 мл×3), затем промывку водой (10 мл×3) и промывку рассолом, сушку, концентрирование и разделение на колонке (силикагель, 100-200 меш; н-гептан:ЕА=20:1), получая продукт 16-A0 (3,5 г; 64,8%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, DMSO-d6): δ млн-1 10.02 (s, 1Н), 7.52-7.47 (m, 2Н), 7.34-7.23 (m, 3H), 6.71-6.64 (m, 2Н). LCMS: рассчитано 206,1; обнаружено 204,8 ([М-Н]-).
В атмосфере азота проводили растворение соединения 16-А1 (930 мг; 5,50 ммоль; имеется в продаже; 97%-ного) и соединения 16-A0 (1,36 г; 6,60 ммоль) в ацетонитриле (20 мл). Добавляли карбонат калия (1,52 г; 11,0 ммоль; имеется в продаже; 99%-ный). Температуру повышали до 85°С и перемешивание проводили в течение 2 ч до завершения реакции. После того, как температура понижалась до комнатной температуры, проводили фильтрование с отсасыванием. Остаточную жидкость концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА=15:1), получая соединение 16-А2 (1,10 г; 56,4%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 10.05 (s, 1Н), 8.30 (d, J=8,3 Гц, 1Н), 7.92 (dd, J=8,3; 1,5 Гц, 1Н), 7.78 (d, J=1,5 Гц, 1Н), 7.69-7.55 (m, 3H), 7.36-7.28 (m, 2Н), 7.27-7.21 (m, 1Н), 7.09 (dd, J=8,5; 2,3 Гц, 1Н).
В атмосфере азота проводили растворение соединения 16-А2 (700 мг; 1,970 ммоль) в безводном THF (10 мл) и затем по каплям добавляли (трифторметил)триметилсилан (476 мг; 3,35 ммоль; имеется в продаже; 98%-ный). Температуру понижали до 0°С и по каплям добавляли TBAF (0,04 мл; 1 М раствор в THF). Температуру поддерживали при 0°С в течение 1,5 ч до завершения реакции. По каплям добавляли 2 мл 3 н. соляной кислоты. Температура естественным образом повышалась до комнатной температуры. В течение 1 ч проводили перемешивание. Добавляли 5 мл воды. Осуществляли экстракцию с использованием DCM (10 мл×3), затем промывку водой (5 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 15:1 до 10:1), получая продукт 16-А3 (810 мг; 96,7%) в виде желтого масла. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 8.19 (d, J=8,5 Гц, 1Н), 7.63-7.52 (m, 4Н), 7.44 (s, 1Н), 7.32 (t, J=8,9 Гц, 2Н), 7.21 (d, J=5,8 Гц, 1Н), 7.14 (dd, J=11,7; 2,4 Гц, 1Н), 6.97 (dd, J=8,5; 2,1 Гц, 1Н), 5.44-5.37 (m, 1Н).
В атмосфере азота к оксихлориду фосфора (580 мг; 3,76 ммоль; имеется в продаже; 97%-ный) по каплям добавляли безводный DCM (10 мл). Температуру понижали до -40°С. По каплям добавляли раствор соединения 16-А3 (800 мг; 1,88 ммоль) в DCM (4 мл), затем по каплям добавляли триэтиламин (476 мг; 4,70 ммоль) и температуру поддерживали в диапазоне от -40°С до -35°С в течение 2 ч. По данным LC-LCMS соединение 16-А3 расходовалось и превращалось в промежуточное соединение. Добавляли 2-бромэтиламина гидробромид (3,08 г; 15,04 ммоль) при -40°С и затем по каплям добавляли раствор триэтиламина (1,52 г; 15,04 ммоль) в DCM (2 мл). Температуру поддерживали при -40°С в течение 1 ч, и промежуточное соединение расходовалось полностью. Температура естественным образом повышалась до 0°С, и по каплям добавляли насыщенный водный раствор хлорида аммония (5 мл). Осуществляли экстракцию с использованием DCM (10 мл×3), затем промывку чистой водой (3 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; от смеси 1:1 н-гептан:ЕА до 100%), получая продукт 16-А4 (в целом 600 мг; выход 44,4%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.04 (d, J=8,4 Гц, 1Н), 7.55-7.47 (m, 2Н), 7.46-7.38 (m, 2Н), 7.27 (s, 1Н), 7.19-7.10 (m, 2Н), 6.95-6.83 (m, 2Н), 5.69 (dd, J=11,4; 6,1 Гц, 1Н), 3.50-3.18 (m, 8Н). LCMS: рассчитано 716,9; обнаружено 717,8 ([М+Н]+).
В атмосфере азота проводили растворение соединения 16-А4 (600 мг; 0,837 ммоль) в THF (15 мл) и затем добавляли оксид серебра (1,16 мг; 5,02 ммоль) и DIPEA (649 мг; 5,02 ммоль). Температуру повышали до 65°С и проводили перемешивание в течение 3 ч. После завершения реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM и остаточную жидкость концентрировали. Проводили кристаллизацию, добавляя 1,5 мл безводного эфира, и осуществляли фильтрование с отсасыванием, получая чистое соединение №16 (159 мг; выход 34,2%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD) δ млн-1 8.12 (t, J=8,1 Гц, 1Н), 7.55 (m, 4Н), 7.45 (s, 1Н), 7.19 (t, J=8,8 Гц, 2Н), 6.98 (dd, J=7,8; 5,3 Гц, 2Н), 6.15-5.98 (m, 1Н), 2.39-1.93 (m, 8Н). LCMS: рассчитано 555,1; обнаружено 556,1 ([М+Н]+).
Синтез соединения №17
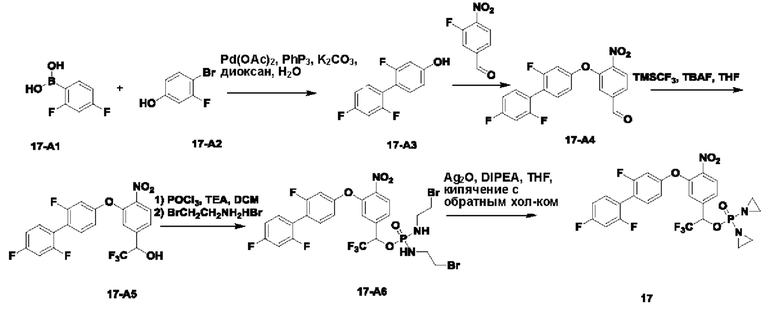
В атмосфере азота проводили растворение соединения 17-А1 (1,7 г; 11 ммоль; имеется в продаже), соединения 17-А2 (2 г; 10 ммоль), ацетата палладия (21,5 мг; 0,1 ммоль), трифенилфосфина (39 мг; 0,15 ммоль) и карбоната калия (2,8 г; 20 ммоль) в диоксане (20 мл) и воде (2 мл). Температуру повышали до 80°С и перемешивание осуществляли в течение ночи. По завершении реакции проводили фильтрование через целит с отсасыванием, затем концентрирование. Образец перемешивали и подвергали разделению посредством колоночной хроматографии (силикагель, 100-200 меш; н-гептан:ЕА=5:1), получая продукт 17-А3 (900 мг; 40%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, DMSO) δ млн-1 10.17 (s, 1Н), 7.55-7.08 (m, 3H), 6.79-6.50 (m, 3H). LCMS: рассчитано 224,0; обнаружено 222,6 [(М-Н)-].
В атмосфере азота проводили растворение соединения 17-А3 (500 мг; 2,2 ммоль) и 3-фтор-4-нитробензальдегиад (370 мг; 2,2 ммоль; имеется в продаже) в ACN (5 мл). Добавляли карбонат калия (830 мг; 6 ммоль). Температуру повышали до 80°С и в течение 4 ч проводили перемешивание. По завершении реакции проводили фильтрование через целит с отсасыванием, затем концентрирование. Образец перемешивали и подвергали разделению посредством колоночной хроматографии (силикагель, 100-200 меш; н-гептан:ЕА=10:1), получая 700 мг неочищенного продукта 17-А4 (белого твердого вещества с чистотой 80%), которое использовали непосредственно на следующей стадии. 1Н-ЯМР (300 МГц, DMSO) δ млн-1 10.03 (s, 1Н), 8.31 (d, J=8,2 Гц, 1Н), 7.94 (d, J=8,4 Гц, 1Н), 7.83 (s, 1Н), 7.59-7.50 (m, 2Н), 7.40 (t, J=8,8 Гц, 1Н), 7.32-7.16 (m, 2Н), 7.10 (d, J=8,6 Гц, 1Н).
В атмосфере азота проводили растворение соединения 17-А4 (680 мг; 1,82 ммоль) и трифторметилтриметилсилана (510 мг; 3,6 ммоль; имеется в продаже) в THF (8 мл). При 0°С по каплям добавляли раствор фторида тетрабутиламмония в THF (0,1 мл; 0,1 ммоль; 1 М; имеется в продаже). Такую температуру поддерживали в течение 6 ч. Добавляли 1 н. соляную кислоту (2 мл) и перемешивали в течение 10 мин. THF удаляли путем концентрирования. К неочищенному продукту добавляли воду (10 мл) и осуществляли экстракцию с использованием DCM (20 мл). Органическую фазу отделяли и концентрировали и образец перемешивали. Неочищенный продукт подвергали разделению посредством колоночной хроматографии (силикагель, 100-200 меш; н-гептан:ЕА=10:1), получая продукт 17-А5 (400 мг; выход 49,6%; чистота 90%) в виде светло-желтого твердого вещества. 1Н-ЯМР (300 МГц, DMSO) δ млн-1 8.19 (d, J=7,8 Гц, 1Н), 7.76 (t, J=8,5 Гц, 1H), 7.57-7.51 (m, 2Н), 7.38 (s, 1Н), 7.28-7.19 (m, 3H), 6.89 (dd, J=8,8; 1,7 Гц, 1 H), 5.40-5.36 (m, 1Н).
В атмосфере азота к POCl3 (168 мг; 1,1 ммоль; имеется в продаже) по каплям добавляли сверхсухой DCM (5 мл). Температуру понижали до -30°С. По каплям добавляли раствор соединения 17-А5 (220 мг; 0,5 ммоль) в DCM (5 мл), затем по каплям добавляли TEA (170 мг; 1,65 ммоль) и температуру поддерживали при -30°С в течение 6 ч до полного расходования исходных веществ. Добавляли 2-бромэтиламина гидробромид (897 мг; 4,4 ммоль) при -30°С и затем по каплям добавляли TEA (440 мг; 4,4 ммоль). По завершении реакции температуру понижали до 0°С. Добавляли насыщенный раствор NH4Cl (10 мл). Осуществляли экстракцию с использованием DCM (15 мл×2), затем промывку водой (5 мл×4), сушку и концентрирование, получая 250 мг неочищенного продукта 17-А6 в виде желтого твердого вещества, которое использовали непосредственно в реакции на следующей стадии. LCMS: рассчитано 732,9; обнаружено 733,9 ([М+Н]+).
В атмосфере азота проводили растворение соединения 17-А6 (250 мг; 0,34 ммоль) в THF (10 мл) и добавляли Ag2O (210 мг; 1,7 ммоль; имеется в продаже) и N,N-диизопропилэтиламин (220 мг; 1,7 ммоль). Температуру повышали до 65°С. Через 2 ч, после завершения реакции, проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM (20 мл), остаточную жидкость концентрировали и после проведения препаративной высокоэффективной жидкостной хроматографии получали 22 мг чистого соединения №17 (белого твердого вещества; 11%). 1Н-ЯМР (400 МГц, DMSO) δ 8.26 (d, J=8,5 Гц, 1Н), 7.67 (d, J=8,5 Гц, 1Н), 7.59-7.48 (m, 3H), 7.41 (dt, J=10,3; 2,5 Гц, 1Н), 7.24-7.19 (m, 2Н), 7.03 (dd, J=8,5; 2,3 Гц, 1Н), 6.36-6.32 (m, 1Н), 2.20-1.91 (m, 8Н). LCMS: рассчитано 573,1; обнаружено 574,1 ([М+Н]+).
Синтез соединения №18
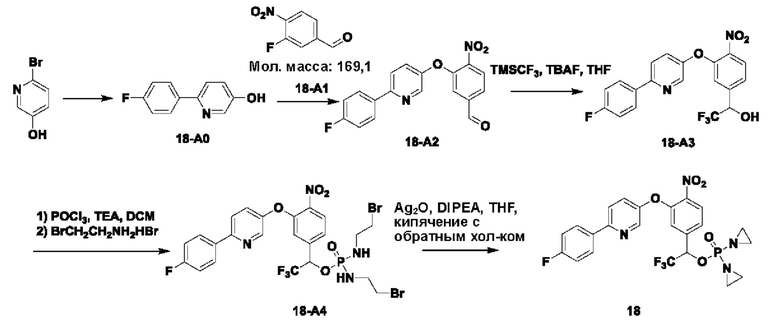
В атмосфере азота проводили растворение 2-бром-5-гидроксипиридина (3,0 г; 17,2 ммоль; от Xingtai All Fine) и п-фторфенилбороновой кислоты (18-A0; 2,7 г; 19,0 ммоль; имеется в продаже) в смеси растворителей диоксана и воды (60 мл; диоксан:вода=9:1). После этого добавляли карбонат калия (4,7 г; 34,4 ммоль). Трижды проводили вакуумирование и заполнение газом. Затем добавляли ацетат палладия (193 мг; 0,86 ммоль; имеется в продаже) и трифенилфосфин (225 мг; 0,86 ммоль; имеется в продаже), вновь трижды проводили вакуумирование и заполнение газом. Температуру повышали до 100°С и в течение ночи осуществляли перемешивание. По завершении реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием и промывку с использованием ЕА. Остаточную жидкость концентрировали. Добавляли 15 мл воды. Осуществляли экстракцию с использованием ЕА (50 мл×3), затем промывку водой (10 мл×3) и промывку рассолом, сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА=15:1), получая продукт (1,2 г; 36,9%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 10.03 (s, 1Н), 8.20 (d, J=2,7 Гц, 1Н), 8.00 (dd, J=8,8; 5,6 Гц, 2Н), 7.78 (d, J=8,6 Гц, 1Н), 7.32-7.18 (m, 3H). LCMS: рассчитано 189,1; обнаружено 190,2 ([М+Н]+).
В атмосфере азота проводили растворение соединения 18-А1 (600 мг; 3,55 ммоль) и соединения 18-A0 (806 мг; 4,26 ммоль) в ацетонитриле (15 мл). После этого добавляли карбонат калия (980 мг; 7,1 ммоль; имеется в продаже). Температуру повышали до 85°С и перемешивание проводили в течение 2 ч до завершения реакции. Температуру понижали до комнатной температуры. Проводили фильтрование с отсасыванием. Остаточную жидкость концентрировали и проводили разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА=10:1), получая продукт 18-А2 (760 мг; 63,3%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 10.03 (s, 1Н), 8.59 (d, J=2,8 Гц, 1Н), 8.31 (d, J=8,3 Гц, 1Н), 8.19-8.11 (m, 2Н), 8.07 (d, J=8,7 Гц, 1Н), 7.90 (dd, J=8,3; 1,6 Гц, 1Н), 7.78-7.70 (m, 2Н), 7.37-7.26 (m, 2Н). LCMS: рассчитано 338,1; обнаружено 339,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 18-А2 (700 мг; 2,07 ммоль) в безводном THF (10 мл) и затем по каплям добавляли (трифторметил)триметилсилан (500 мг; 3,52 ммоль). Температуру понижали до 0°С и по каплям добавляли TBAF (0,03 мл; 1 М раствор в THF). Температуру поддерживали при 0°С в течение 1,5 ч до завершения реакции. По каплям добавляли 3 н. соляную кислоту (2 мл). Температура естественным образом повышалась до комнатной температуры. В течение 1 ч осуществляли перемешивание. Добавляли 5 мл воды. Осуществляли экстракцию с использованием DCM (10 мл×3), затем промывку водой (5 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 15:1 до 10:1), получая продукт (550 мг; 65,1%) в виде желтого масла. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 8.52 (d, J=2,9 Гц, 1Н), 8.19 (d, J=8,5 Гц, 1Н), 8.16-8.10 (m, 2Н), 8.06 (d, J=8,8 Гц, 1Н), 7.64 (dd, J=8,8; 2,9 Гц, 1Н), 7.55 (dd, J=8,7; 3,3 Гц, 1Н), 7.38 (s, 1Н), 7.36-7.28 (m, 2Н), 7.18 (dd, J=5,8; 3,4 Гц, 1Н), 5.43-5.34 (m, 1Н). LCMS: рассчитано 408,1; обнаружено 409,2 [М+Н]+).
В атмосфере азота к оксихлориду фосфора (414 мг; 2,70 ммоль; имеется в продаже) по каплям добавляли безводный DCM (10 мл). Температуру понижали до -40°С. По каплям добавляли раствор соединения 18-А3 (550 мг; 1,35 ммоль) в DCM (4 мл), затем триэтиламин (342 мг; 3,36 ммоль) и температуру поддерживали в диапазоне температур от -40°С до -35°С в течение 2 ч. По данным LC-LCMS соединение 18-А3 расходовалось и превращалось в промежуточное соединение. Добавляли 2-бромэтиламина гидробромид (2,2 г; 10,8 ммоль) при -40°С и затем по каплям добавляли раствор триэтиламина (1,1 г; 10,8 ммоль) в DCM (2 мл). Температуру поддерживали при -40°С в течение 1 ч, и промежуточное соединение полностью расходовалось. Температура естественным образом повышалась до 0°С, и по каплям добавляли насыщенный водный раствор хлорида аммония (5 мл). Осуществляли экстракцию с использованием DCM (10мл×3), затем промывку чистой водой (3 мл×3), сушку, концентрирование и разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 1:1 до 100% ЕА), получая продукт (520 мг; 55,0%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.49 (d, J=2,7 Гц, 1Н), 8.06 (dd, J=8,4; 2,7 Гц, 1H), 8.01-7.94 (m, 2H), 7.77 (d, J=8,7 Гц, 1H), 7.54-7.48 (m, 1 H), 7.43 (d, J=8,3 Гц, 1H), 7.26-7.24 (m, 1H), 7.18 (t, J=8,7 Гц, 2H), 5.69 (dq, J=12,3; 6,2 Гц, 1H), 3.49-3.16 (m, 8H). LCMS: рассчитано 700,0; обнаружено 700,9 ([М+Н]+).
В атмосфере азота проводили растворение соединения 18-А4 (520 мг; 0,743 ммоль) в THF (15 мл) и затем добавляли оксид серебра (1,03 г; 4,46 ммоль; имеется в продаже) и DIPEA(580 мг; 4,46 ммоль). Температуру повышали до 65°С и в течение 3 ч проводили перемешивание. После завершения реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №18 (106,7 мг; 26,7%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 8.43 (d, J=2,9 Гц, 1Н), 8.16 (d, J=8,5 Гц, 1Н), 8.07-7.97 (m, 2Н), 7.92 (d, J=8,8 Гц, 1Н), 7.65-7.54 (m, 2Н), 7.46 (s, 1Н), 7.24-7.19 (m, 2Н), 6.13-6.03 (m, 1Н), 2.26-2.08 (m, 8Н). LCMS: рассчитано 538,1; обнаружено 539,1 ([М+Н]+).
Синтез соединения №19
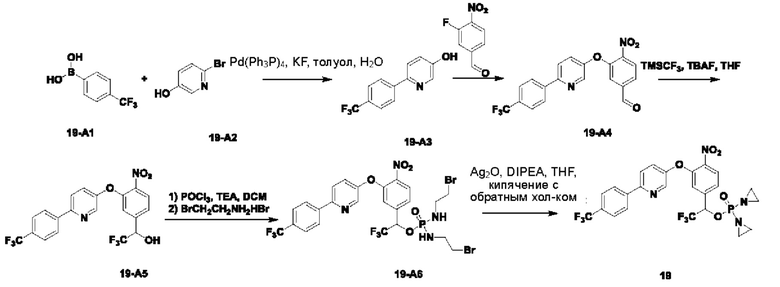
В атмосфере азота проводили растворение соединения 19-А1 (500 мг; 2,65 ммоль; имеется в продаже), соединения 19-А2 (460 мг; 2,65 ммоль; имеется в продаже), тетракистрифенилфосфинпалладия (373 мг; 0,3 ммоль) и фторида калия (300 мг; 5,2 ммоль) в толуоле (10 мл) и воде (1 мл). Температуру повышали до 80°С и в течение ночи осуществляли перемешивание. По завершении реакции проводили фильтрование через целит с отсасыванием, затем концентрирование. Образец перемешивали и подвергали разделению посредством колоночной хроматографии (силикагель, 100-200 меш; н-гептан:ЕА=10:1), получая продукт 19-А3 (600 мг; 94,7%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, DMSO) δ млн-1 10.24 (s, 1Н), 8.26 (d, J=2,4 Гц, 1Н), 8.19 (d, J=8,1 Гц, 2Н), 7.91 (d, J=8,7 Гц, 1Н), 7.78 (d, J=8,1 Гц, 2H), 7.29 (dd, J=8,6; 2,7 Гц, 1H). LCMS: рассчитано 239,0; обнаружено 240,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 19-А3 (870 мг; 2,9 ммоль) и 3-фтор-4-нитробензальдегидт (490 мг; 2,9 ммоль; имеется в продаже) в ACN (8 мл). Добавляли карбонат калия (830 мг; 6 ммоль). Температуру повышали до 80°С и в течение 4 ч осуществляли перемешивание. По завершении реакции проводили фильтрование через целит с отсасыванием, затем концентрирование. Образец перемешивали и подвергали разделению посредством колоночной хроматографии (силикагель, 100-200 меш; н-гептан:ЕА=10:1), получая продукт 19-А4 (640 мг) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3) δ млн-1 10.03 (s, 1Н), 8.57 (d, J=2,7 Гц, 1Н), 8.15-8.12 (m, 3H), 7.87-7.75 (m, 4Н), 7.60-7.51 (m, 2Н). LCMS: рассчитано 388,0; обнаружено 389,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 19-А4 (640 мг; 1,64 ммоль) и трифторметилтриметилсилана (430 мг; 3 ммоль; имеется в продаже) в THF (5 мл). При 0°С по каплям добавляли раствор фторида тетрабутиламмония в THF (0,1 мл; 0,1 ммоль; 1 М; имеется в продаже). Такую температуру поддерживали в течение 6 ч. Добавляли 1 н. соляную кислоту (2 мл) и перемешивали в течение 10 мин. THF удаляли путем концентрирования. К неочищенному продукту добавляли воду (10 мл) и осуществляли экстракцию с использованием DCM (20 мл). Органическую фазу отделяли и концентрировали и образец перемешивали. Неочищенный продукт выделяли колоночной хроматографией (силикагель, 100-200 меш; н-гептан:ЕА=10:1), получая продукт (640 мг; 85,2%) в виде светло-желтого твердого вещества. 1Н-ЯМР (300 МГц, DMSO) δ млн-1 8.59 (d, J=2,7 Гц, 1Н), 8.30 (d, J=7,5 Гц, 2Н), 8.19 (t, J=9,2 Гц, 2Н), 7.86 (d, J=8,5 Гц, 2Н), 7.72-7.65 (m, 1Н), 7.59-7.56 (m, 1Н), 7.43 (s, 1Н), 7.20 (d, J=5,8 Гц, 1Н), 5.39-5.37 (m, 1Н). LCMS: рассчитано 458,0; обнаружено 459,0 ([М+Н]+).
В атмосфере азота к POCl3 (200 мг; 1,3 ммоль) по каплям добавляли сверхсухой DCM (5 мл). Температуру понижали до -30°С. По каплям добавляли раствор соединения 19-А5 (300 мг; 0,65 ммоль) в DCM (5 мл), затем по каплям добавляли TEA (270 мг; 2,6 ммоль) и температуру поддерживали при -30°С в течение 6 ч до полного расходования исходных веществ. Добавляли 2-бромэтиламина гидробромид (1,1 г; 5,2 ммоль) при -30°С и затем по каплям добавляли TEA (530 мг; 5,2 ммоль). По завершении реакции температуру понижали до 0°С. Добавляли насыщенный раствор NH4Cl (10 мл). Осуществляли экстракцию с использованием DCM (15 мл×2), затем промывку водой (5 млх4), сушку и концентрирование, получая 200 мг неочищенного продукта, который в виде желтого твердого вещества использовали непосредственно в реакции на следующей стадии. 1Н-ЯМР (400 МГц, CDCl3) δ 8.46 (d, J=2,6 Гц, 1Н), 8.07-7.99 (m, 3H), 7.78-7.75 (m, 1Н), 7.68 (d, J=8,2 Гц, 2Н), 7.48-7.33 (m, 3H), 5.65-5.61 (m, 1Н), 3.51-3.04 (m, 10Н). LCMS: рассчитано 747,9; обнаружено 748,9 ([М+Н]+).
В атмосфере азота проводили растворение соединения 19-А6 (150 мг; 0,23 ммоль) в THF (10 мл) и добавляли Ag2O (170 мг; 1,38 ммоль; имеется в продаже) и N,N-диизопропилэтиламин (163 мг; 1,38 ммоль). Температуру повышали до 65°С. Через 2 ч, после завершения реакции, проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM (20 мл), остаточную жидкость концентрировали и после проведения препаративной высокоэффективной жидкостной хроматографии получали чистый продукт (41 мг; 36%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO) δ 8.61 (d, J=2,8 Гц, 1Н), 8.31 (d, J=8,2 Гц, 2Н), 8.27 (d, J=8,5 Гц, 1Н), 8.21 (d, J=8,8 Гц, 1Н), 7.87 (d, J=8,4 Гц, 2Н), 7.72 (dd, J=8,8; 2,9 Гц, 1Н), 7.65 (d, J=8,5 Гц, 1Н), 7.56 (s, 1Н), 6.32-6.28 (m, 1Н), 2.17-1.90 (m, 8Н). LCMS: рассчитано 588,1; обнаружено 589,1 ([М+Н]+).
Синтез соединения №20
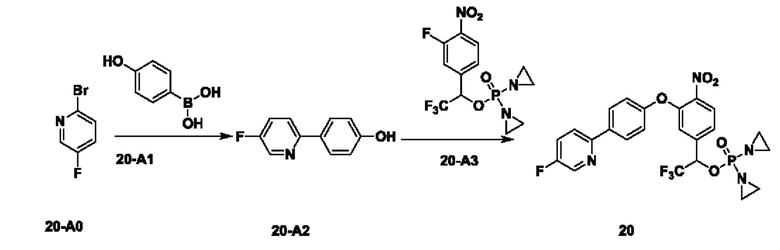
В атмосфере азота добавляли 2-бром-5-гидроксипиридин (1,5 г; 8,52 ммоль; имеется в продаже) и п-гидроксифенилбороновую кислоту (1,4 г; 10,23 ммоль; имеется в продаже) к смеси жидкостей, состоящей из DME (33 мл) и H2O (7 мл). Трижды проводили вакуумирование и заполнение азотом. После добавления Pd(PPh3)4 (300 мг; 0,26 ммоль; имеется в продаже) и Na2CO3 (1,8 г; 17,05 ммоль) вновь трижды проводили вакуумирование и заполнение азотом. Для осуществления реакции температуру повышали до 80°С. Мониторинг реакции проводили в течение 2,5 ч до ее завершения. Температуру понижали до комнатной температуры. Осуществляли экстракцию с использованием EtOAc (50 мл×3). Органическую фазу промывали водой, промывали рассолом и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 12:1 до 9:1), получая продукт (1,4 г; выход 86,8%) в виде белого твердого вещества. 1Н-ЯМР (300 МГц, MeOD): δ млн-1 8.43 (d, J=2,7 Гц, 1Н), 7.81-7.77 (m, 3H), 7.64-7.58 (m, 1Н), 6.87 (d, J=8,7 Гц, 2Н). LCMS: рассчитано 189,1; обнаружено 190,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 20-А3 (т.е. соединения №46; 100 мг; 0,27 ммоль) в ацетоне (15 мл) и затем добавляли соединение 20-А2 (102 мг; 0,54 ммоль) и Cs2CO3 (309 мг; 0,95 ммоль) и перемешивали в течение 2 ч при комнатной температуре. После завершения реакции проводили фильтрование через целит с отсасыванием. Твердое вещество промывали ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №20 (17 мг; 11,7%) в виде коричневого твердого вещества. 1Н-ЯМР (400 МГц, MeOD) δ млн-1 8.52 (d, J=2,9 Гц, 1Н), 8.10 (d, J=8,5 Гц, 1Н), 8.06 (d, J=8,8 Гц, 2Н), 7.92 (dd, J=8,8; 4,3 Гц, 1Н), 7.68 (dt, J=8,6; 2,9 Гц, 1Н), 7.54 (d, J=8,6 Гц, 1Н), 7.37 (s, 1Н), 7.19 (d, J=8,8 Гц, 2Н), 6.06-5.99 (m, 1Н), 2.27-1.97 (m, 8Н). LCMS: рассчитано 538,1; обнаружено 539,1 ([М+Н]+).
Синтез соединения №21

В атмосфере азота перемешивали соединение 21-А1 (2,0 г; 14,5 ммоль) и тионилхлорид (8 мл) при комнатной температуре в течение 1 ч и тионилхлорид удаляли на роторном испарителе. Добавляли 10 мл THF, систему охлаждали до 0°С и добавляли смесь пиперидина (1,85 г; 21,8 ммоль) и TEA (2,2 г; 21,8 ммоль). По завершении добавления температура системы естественным образом повышалась до комнатной температуры. Проводили мониторинг в течение 1 ч до завершения реакции. Систему разбавляли EtOAc (50 мл) и промывали H2O (30 мл×5). Органическую фазу сушили, концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 1:1 до 0:1), получая продукт (720 мг; выход 24,3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 9.64 (s, 1Н) 7.23 (t, J=8,0 Гц, 1H), 6.79-6.82 (m, 1H), 6.73 (d, J=7,6 Гц, 1H), 6.70 (d, J=1,2 Гц, 1H), 3.54 (m, 2H), 3.26 (m, 2H), 1.61-1.50 (m, 6H). LCMS: рассчитано 205,1; обнаружено 206,2 ([М+Н]+).
В атмосфере азота проводили растворение соединения 21-А2 (60 мг; 0,25 ммоль) в ацетоне (10 мл), затем добавляли соединение 21-А3 (т.е. соединение №46; 55,6 мг; 0,25 ммоль) и Cs2CO3 (245 мг; 0,75 ммоль) и перемешивали в течение 2,5 ч при комнатной температуре. После завершения реакции проводили фильтрование через целит с отсасыванием. Твердое вещество промывали ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №21 (25,0 мг; 16,9%) в виде беловатого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): δ млн-1 8.21 (d, J=8,0 Гц, 1Н), 7.60 (d, J=8,0 Гц, 1 Н), 7.53 (t, J=8,0 Гц, 1Н), 7.41 (s, 1Н), 7.24 (d, J=8,0 Гц, 1 Н) 7.20 (dd, J=8,0; 2,4 Гц, 1Н), 7.05 (s, 1Н), 6.33-6.25 (m, 1Н), 3.55 (m, 2Н), 3.25 (m, 2Н), 2.15-1.93 (m, 8Н), 1.59-1.52 (m, 6Н). LCMS: рассчитано 554,2; обнаружено 555,2 ([М+Н]+).
Синтез соединения №22
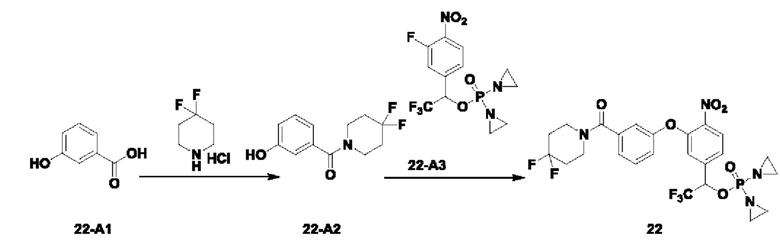
В атмосфере азота проводили растворение соединения 22-А1 (0,2 г; 1,45 ммоль) и раствора Т3Р (реагента сочетания - пропилфосфонового ангидрида; номер в Химической реферативной службе (CAS): 68957-94-8; 50%-ного раствора в ЕА) в 5 мл DCM. Добавляли 4,4-дифторпиперидин (0,25 г; 1,6 ммоль) и перемешивали при комнатной температуре. Проводили мониторинг в течение 2 ч до завершения реакции. Систему растворяли в DCM (20 мл) и промывали водой (10 мл×5), органическую фазу сушили, концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА=1:1), получая продукт (190 мг; выход 54,4%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 6.94-6.89 (m, 2Н), 6.87 (d, J=8,0 Гц, 1 Н), 6.36 (s, 1 Н), 3.87 (m, 2Н), 3.56 (m, 2Н), 2.04-1.97 (m, 4Н). LCMS: рассчитано 241,1; обнаружено 242,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 22-А2 (60 мг; 0,25 ммоль) в ацетоне (10 мл) и затем добавляли соединение 22-А3 (т.е. соединение №46; 55,6 мг; 0,25 ммоль) и Cs2CO3 (245 мг; 0,75 ммоль) и перемешивали в течение 2,5 ч при комнатной температуре. После завершения реакции проводили фильтрование через целит с отсасыванием. Твердое вещество промывали ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №22 (25,0 мг; 16,9%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 8.21 (d, J=8,4 Гц, 1Н), 7.60-7.54 (m, 2Н), 7.40 (s, 1Н), 7.33 (d, J=8,0 Гц, 1Н), 7.25-7.21 (m, 2Н), 6.33-6.26 (m, 1Н), 3.68 (m, 2Н), 3.40 (m, 2Н), 2.15-1.93 (m, 12Н). LCMS: рассчитано 590,1; обнаружено 591,1 ([М+Н]+).
Синтез соединения №23
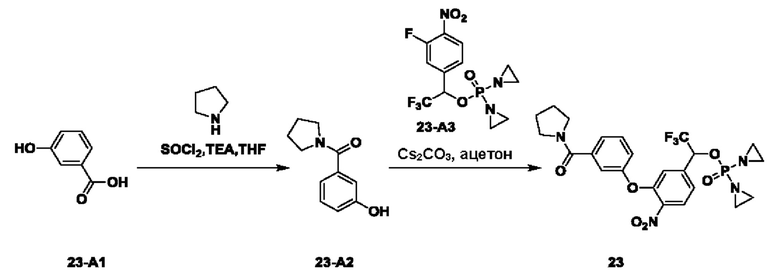
В атмосфере азота соединение 23-А1 (2,0 г; 14,5 ммоль) и тионилхлорид (8 мл) перемешивали при комнатной температуре в течение 1 ч и тионилхлорид удаляли на роторном испарителе. Добавляли 10 мл THF, систему охлаждали до 0°С и добавляли смесь тетрагидропиррола (1,5 г; 21,8 ммоль) и TEA (2,2 г; 21,8 ммоль). По завершении добавления температура в системе естественным образом повышалась до комнатной температуры. Проводили мониторинг в течение 1 ч до завершения реакции. Систему растворяли в EtOAc (40 мл) и промывали H2O (40 мл×5). Органическую фазу сушили, концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 1:1 до ЕА), получая неочищенный продукт (850 мг; 30,7%; при этом содержание неочищенного продукта составляло 80%) в виде бесцветного масла.
В атмосфере азота проводили растворение соединения 23-А2 (103 мг; 0,54 ммоль) в ацетоне (10 мл) и затем добавляли соединение 23-А3 (т.е. соединение №46; 120 мг; 0,54 ммоль) и Cs2CO3 (265 мг; 0,81 ммоль) и перемешивали в течение 2,5 ч при комнатной температуре. После завершения реакции проводили фильтрование через целит с отсасыванием. Твердое вещество промывали ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №23 (35,0 мг; 18,8%) в виде беловатого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 8.20 (d, J=8,0 Гц, 1Н), 7.59 (d, J=8,0 Гц, 1Н), 7.53 (t, J=8,0 Гц, 1Н), 7.39-7.38 (m, 2Н), 7.24-7.21 (m, 2Н), 6.32-6.25 (m, 1Н), 3.43 (t, J=6,8 Гц, 2Н), 3.36-3.34 (m, 2Н), 1.92-2.14 (m, 8Н), 1.84-1.77 (m, 4Н). LCMS: рассчитано 540,1; обнаружено 541,1 ([М+Н]+).
Синтез соединения №24
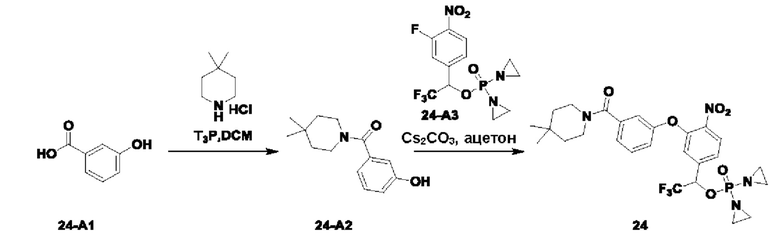
В атмосфере азота проводили растворение соединения 24-А1 (500 мг; 3,6 ммоль; имеется в продаже) и Т3Р (4,6 г; 14,4 ммоль; 50%-ного раствора в ЕА) в 10 мл DCM, добавляли 4,4-диметилпиперидина гидрохлорид (540 мг; 3,6 ммоль; от Shanghai Anmike) и перемешивали при комнатной температуре. Проводили мониторинг в течение 2 ч до завершения реакции. Систему разбавляли DCM (20 мл) и промывали H2O (10 мл×5). Органическую фазу сушили, концентрировали подвергали разделению на колонке (силикагель, 200-300 меш; от смеси 1:1 н-гептан:ЕА до ЕА), получая продукт (600 мг; выход 71,4%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 17.20 (t, J=8,0 Гц, 1Н), 6.82-6.81 (m, 1Н), 6.80-6.75 (m, 1Н), 6.73-6.72 (m, 1Н), 3.66 (m, 2Н), 3.34 (s, 2Н), 1.41 (m, 2Н), 1.30-1.29 (m, 2Н), 0.97 (s, 6Н). LCMS: рассчитано 233,1; обнаружено 234,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 24-А2 (103 мг; 0,54 ммоль) в ацетоне (10 мл), затем добавляли соединение 24-А3 (т.е. соединение №46; 120 мг; 0,54 ммоль) и Cs2CO3 (265 мг; 0,81 ммоль) и перемешивали в течение 2,5 ч при комнатной температуре. После завершения реакции проводили фильтрование через целит с отсасыванием. Твердое вещество промывали ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №24 (35,0 мг; 18,8%) в виде беловатого твердого вещества. 1Н-ЯМР (400 МГц, DMSO): δ млн-1 8.21 (d, J=8,4 Гц, 1Н), 7.59 (d, J=8,4 Гц, 1Н), 7.53 (t, J=8,0 Гц, 1Н), 7.40 (s, 1Н), 7.24 (d, J=7,6 Гц, 1Н), 7.19 (dd, J=7,6; 2,4 Гц, 1Н), 7.06-7.07 (m, 1Н), 6.32-6.27 (m, 1Н), 3.57 (m, 2Н), 3.26 (m, 2Н), 2.13-1.94 (m, 8Н), 1.23 (m, 2Н), 1.14 (m, 2Н), 0.94 (s, 6Н). LCMS: рассчитано 582,2; обнаружено 583,2 ([М+Н]+).
Синтез соединения №25
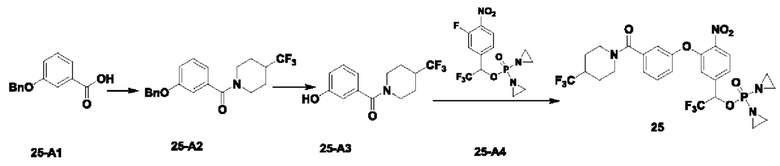
В атмосфере азота добавляли соединение 25-А1 (500 мг; 2,19 ммоль) и 4-(трифторметил)пиперидин (1,0 г; 6,57 ммоль; имеется в продаже) к DCM (10 мл) и затем к этой системе по каплям добавляли Т3Р (5,6 г; 8,77 ммоль; 50%-ный раствор в EtOAc; имеется в продаже). Далее систему охлаждали до 5°С и к этой системе по каплям добавляли DIEA(1,2 г; 8,77 ммоль; имеется в продаже). По завершении добавления температура естественным образом повышалась до комнатной температуры. Проводили мониторинг в течение 2 ч до завершения реакции. Растворитель удаляли путем концентрирования, после чего остаток растворяли в EtOAc (20 мл) и промывали H2O (20 мл×5). Органическую фазу сушили и концентрировали, получая продукт (790 мг; выход 98,9%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.42 (d, J=7,1 Гц, 2Н), 7.40-7.33 (m, 3H), 7.32-7.26 (m, 1Н), 7.13-7.10 (m, 1Н), 6.98-6.95 (m, 2Н), 5.14 (s, 2Н), 4.70 (m, 1Н), 3.75-7.72 (m, 1Н), 3.08-3.08 (m, 1Н), 2.84 (m, 1Н), 2.54-2.46 (m, 1 Н), 1.98 (m, 1Н), 1.81-1.76 (m, 1Н), 1.55-1.53 (m, 1Н), 1.44-1.35 (m, 1Н). LCMS: рассчитано 363,1; обнаружено 364,1 ([М+Н]+).
В атмосфере азота добавляли соединение 25-А2 (780 мг; 2,15 ммоль) и Pd(OH)2 (90 мг; имеется в продаже) к EtOH (10 мл). Трижды проводили вакуумирование и заполнение азотом. После трехкратного вакуумирования и заполнения водородом температуру повышали до 35°С и выдерживали в течение ночи. На следующий день проводили мониторинг до завершения реакции. Проводили фильтрование через целит с отсасыванием, выполняли промывку с использованием DCM. Остаточную жидкость концентрировали, получая соединение 25-А3 (390 мг; выход 66,6%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.26 (t, J=7,9 Гц, 1Н), 6.87 (dd, J=8,1; 2,3 Гц, 1 Н), 6.83 (d, J=7,5 Гц, 1Н), 6.80-6.76 (m, 1Н), 4.70 (m, 1Н), 3.84 (m, 1Н), 3.13 (m, 1Н), 2.85 (m, 1Н), 2.57-2.47 (m, 1Н), 2.06-1.79 (m, 2Н), 1.50-1.46 (m, 2Н). LCMS: рассчитано 273,1; обнаружено 274,2 ([М+Н]+).
В атмосфере азота проводили растворение соединения 25-А4 (т.е. соединения №46; 100 мг; 0,27 ммоль) в ацетоне (10 мл), затем добавляли соединение 25-А3 (148 мг; 0,54 ммоль) и Cs2CO3(309 мг; 0,95 ммоль; имеется в продаже) и перемешивали в течение 2 ч при комнатной температуре до завершения реакции. Последующую обработку осуществляли так, как приведено ниже. Проводили фильтрование через целит с отсасыванием, выполняли промывку ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №25 (40,8 мг; 24,2%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO) δ 8.20 (d, J=8,5 Гц, 1Н), 7.59 (d, J=8,6 Гц, 1 Н), 7.54 (t, J=7,9 Гц, 1Н), 7.39 (s, 1Н), 7.28 (d, J=7,6 Гц, 1Н), 7.22 (dd, J=8,2; 2,0 Гц, 1Н), 7.13 (s, 1Н), 6.28 (dq, J=12,9; 6,4 Гц, 1Н), 4.53 (m, 1Н), 3.60 (m, 1Н), 3.08 (m, 1Н), 2.78 (m, 1Н), 2.71-2.57 (m, 1Н), 2.13-1.92 (m, 8Н), 1.87-1.76 (m, 2Н), 1.39-1.37 (m, 2Н). LCMS: рассчитано 622,1; обнаружено 623,1 ([М+Н]+).
Синтез соединения №26
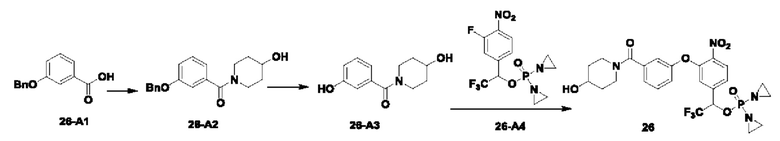
В атмосфере азота добавляли соединение 26-А1 (1,0 г; 4,38 ммоль) и п-гидроксипиперидин (1,3 г; 13,12 ммоль) к DCM (20 мл) и затем к этой системе по каплям добавляли Т3Р (11,2 г; 8,77 ммоль; 50%-ный раствор в EtOAc). Систему охлаждали до 5°С и затем к этой системе по каплям добавляли DIEA (2,4 г; 17,54 ммоль; имеется в продаже). По завершении добавления температура естественным образом повышалась до комнатной температуры. Проводили мониторинг в течение 5 ч до завершения реакции. Последующую обработку осуществляли так, как приведено ниже. Систему концентрировали, разбавляли EtOAc (40 мл) и промывали H2O (40 мл×5). Органическую фазу сушили, концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА=1:1-0:1), получая соединение 26-А2 (920 мг; выход 67,4%) в виде бесцветного масла. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.43 (d, J=8,0 Гц, 2Н), 7.38 (t, J=7,6 Гц, 3H), 7.32-7.28 (m, 1H), 7.10 (d, J=8,4 Гц, 1H), 6.98 (m, 1H), 5.95 (d, J=8,0 Гц, 1H), 5.13 (s, 2H), 4.14 (s, 1H), 3.89-3.85 (m, 1H), 3.56 (s, 1H), 3.17 (s, 1H), 1.92 (s, 1H), 1.74 (s, 1H), 1.54 (s, 1H), 1.40 (s, 1H), 1.03 (s, 1H). LCMS: рассчитано 311,2; обнаружено 312,2 ([M+H]+).
В атмосфере азота добавляли соединение 26-А2 (900 мг; 2,89 ммоль) и катализатор Pd(OH)2 (121 мг) к EtOH (10 мл). Трижды проводили вакуумирование и заполнение азотом. После трехкратного вакуумирования и заполнения водородом температуру повышали до 35°С и выдерживали в течение ночи. На следующий день проводили мониторинг до завершения реакции. Последующую обработку осуществляли так, как приведено ниже. В атмосфере азота проводили фильтрование через целит с отсасыванием и промывку с использованием DCM. Остаточную жидкость концентрировали, получая продукт 26-А3 (460 мг; выход 71,9%) в виде бесцветной маслянистой жидкости. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.26 (d, J=8,0 Гц, 1Н), 6.87 (dd, J=8,4; 2,0 Гц, 1Н), 6.82 (d, J=7,2 Гц, 1Н), 6.79 (d, J=2,0 Гц, 1Н), 4.16 (m, 1Н), 3.91-3.85 (m, 1Н), 3.62 (m, 2Н), 3.31-3.22 (s, 1Н), 1.80-1.64 (m, 1Н), 1.54-1.45 (m, 2Н). LCMS: рассчитано 221,1; обнаружено 222,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 26-А4 (т.е. соединения №46; 100 мг; 0,27 ммоль) и соединения 26-А3 (120 мг; 0,54 ммоль) в ацетоне (10 мл), затем добавляли Cs2CO3 (265 мг; 0,81 ммоль) и перемешивали в течение 2,5 ч при комнатной температуре до завершения реакции. Последующую обработку осуществляли так, как приведено ниже. Проводили фильтрование через целит с отсасыванием, выполняли промывку ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №26 (29,0 мг; 18,8%) в виде беловатого твердого вещества. 1Н-ЯМР (400 МГц, DMSO) δ млн-1 8.20 (d, J=8,5 Гц, 1Н), 7.59 (d, J=8,5 Гц, 1Н), 7.53 (t, J=7,9 Гц, 1Н), 7.40 (s, 1Н), 7.25 (d, J=7,7 Гц, 1Н), 7.20 (dd, J=7,9; 2,2 Гц, 1Н), 7.07 (d, J=1,4 Гц, 1Н), 6.29 (dq, J=13,0; 6,4 Гц, 1 Н), 4.77 (d, J=3,7 Гц, 1Н), 3.95 (s, 1 Н), 3.72-3.71 (m, 1Н), 3.44 (m, 1Н), 3.20-3.11 (m, 2Н), 2.23-1.90 (m, 8Н), 1.76-1.66 (m, 2Н), 1.52-1.18 (m, 2Н). LCMS: рассчитано 570,1; обнаружено 571,2 ([М+Н]+).
Синтез соединения №27
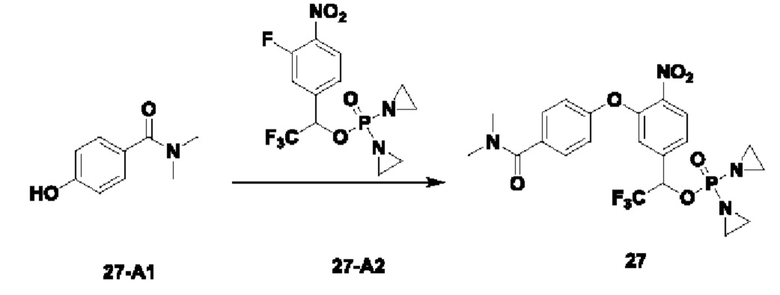
В атмосфере азота проводили растворение соединения 27-А2 (т.е. соединения №46; 100 мг; 0,27 ммоль) и соединения 27-А1 (89 мг; 0,54 ммоль) в ацетоне (10 мл), затем добавляли Cs2CO3 (265 мг; 0,81 ммоль) и перемешивали в течение 2 ч при комнатной температуре до завершения реакции. Последующую обработку осуществляли так, как приведено ниже. Проводили фильтрование через целит с отсасыванием, выполняли промывку ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №27 (21,0 мг; 15,9%) в виде желтого воскообразного вещества. 1Н-ЯМР (400 МГц, DMSO) δ млн-1 8.22 (d, J=8,5 Гц, 1Н), 7.61 (d, J=8,6 Гц, 1 Н), 7.50 (d, J=8,6 Гц, 2Н), 7.42 (s, 1Н), 7.14 (d, J=8,6 Гц, 2Н), 6.31 (dq, J=13,0; 6,4 Гц, 1Н), 2.97 (s, 3H), 2.96 (s, 3H), 2.22-1.90 (m, 8Н). LCMS: рассчитано 514,1; обнаружено 515,1 ([М+Н]+).
Соединение №28
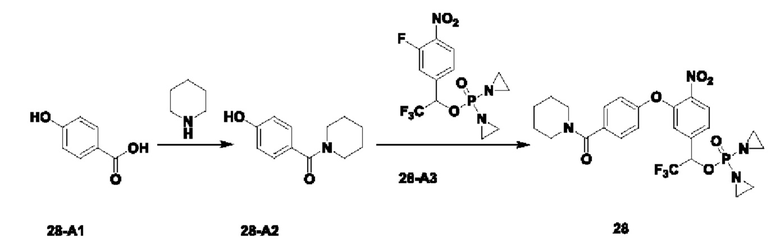
В атмосфере азота добавляли п-гидроксибензойную кислоту (28-А1; 2,0 г; 14,48 ммоль; имеется в продаже) к DCM (40 мл), затем добавляли оксалилхлорид (5,5 г; 43,44 ммоль) и DMF (53 мг; 0,72 ммоль) и перемешивали при комнатной температуре. По результатам мониторинга, проводимого в течение 2,5 ч, исходные вещества были израсходованы. Оксалилхлорид удаляли путем концентрирования. После того, как эту систему разбавляли DCM (50 мл), к системе по каплям в атмосфере азота добавляли пиперидин (3,7 г; 43,44 ммоль) и TEA (5,9 г; 57,92 ммоль). Проводили мониторинг в течение 30 мин до завершения реакции. Последующую обработку осуществляли так, как приведено ниже. После концентрирования реакционной смеси ее разбавляли 1 н. раствором NaOH (50 мл) и затем перемешивали в течение 5 мин. Осуществляли экстракцию с использованием DCM (50 мл×3). Водную фазу подкисляли 6 н. раствором HCl, вызывая осаждение белого твердого вещества. После того, как твердое вещество отфильтровывали с отсасыванием и сушили, получали продукт 28-А2 (2,0 г; выход 67,3%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.26 (dd, d1, J=6,8; 2,0 Гц, 2Н), 6.82 (dd, J=6,4; 2,0 Гц, 2Н), 3.64-3.48 (m, 4Н), 1.72-1.60 (m, 6Н). LCMS: рассчитано 205,1; обнаружено 206,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 28-А3 (т.е. соединения №46; 150 мг; 0,41 ммоль) и соединения 28-А2 (167 мг; 0,81 ммоль) в ацетоне (10 мл), затем к этой системе добавляли Cs2CO3 (463 мг; 1,42 ммоль) и перемешивали в течение 2 ч при комнатной температуре до завершения реакции. Последующую обработку осуществляли так, как приведено ниже. Проводили фильтрование через целит с отсасыванием, выполняли промывку ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №28 (57 мг; 25,3%) в виде желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO) δ 8.22 (d, J=8,5 Гц, 1Н), 7.61 (d, J=8,5 Гц, 1Н), 7.47-7.43 (m, 3H), 7.13 (t, J=5,6 Гц, 2Н), 6.30 (dt, J=12,9; 6,5 Гц, 1 Н), 3.84-3.34 (m, 4Н), 2.25-1.86 (m, 8Н), 1.70-1.38 (m, 6Н). LCMS: рассчитано 554,2; обнаружено 555,1 ([М+Н]+).
Синтез соединения №29
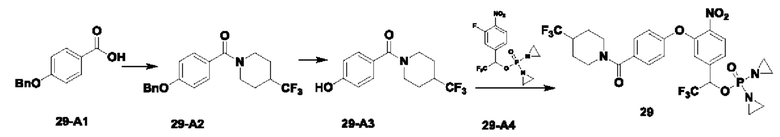
В атмосфере азота добавляли соединение 29-А1 (500 мг; 2,19 ммоль) и 4-(трифторметил)пиперидин (1,0 г; 6,57 ммоль; имеется в продаже) к DCM (10 мл) и затем к этой системе по каплям добавляли Т3Р (5,6 г; 4,38 ммоль; 50%-ный раствор в EtOAc; имеется в продаже). После охлаждения системы до 5°С к системе по каплям добавляли DIEA (1,2 г; 8,77 ммоль). По завершении добавления температура естественным образом повышалась до комнатной температуры. Реакцию проводили в течение ночи до ее завершения. Последующую обработку осуществляли так, как приведено ниже. Систему концентрировали, разбавляли EtOAc (20 мл) и промывали H2O (20 мл×5). Органическую фазу сушили и концентрировали, получая соединение 29-А2 (790 мг; выход 98,9%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.44-7.31 (m, 7Н), 7.09-7.06 (m, 2Н), 5.14 (s, 2Н), 4.66 (m, 1Н), 3.97 (m, 1 Н), 3.04 (m, 2Н), 2.55-2.47 (m, 1Н), 1.92 (m, 2Н), 1.54-1.51 (m, 2Н). LCMS: рассчитано 363,1; обнаружено 364,2 ([М+Н]+).
В атмосфере азота добавляли соединение 29-А2 (790 мг; 2,20 ммоль) и Pd(OH)2 (100 мг) к EtOH (10 мл). Трижды проводили вакуумирование и заполнение азотом. После трехкратного вакуумирования и заполнения водородом температуру повышали до 35°С и выдерживали в течение ночи. На следующий день выполняли мониторинг до завершения реакции. Проводили фильтрование через целит с отсасыванием, выполняли промывку с использованием DCM и остаточную жидкость концентрировали, получая соединение 29-А3 (450 мг; выход 75,2%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.29 (d, J=8,4 Гц, 2Н), 6.84 (d, J=8,4 Гц, 2Н), 4.59 (s, 1Н), 4.06 (s, 1Н), 3.00 (m, 2Н), 2.55-2.47 (m, 1Н), 1.92 (sm, 2Н), 1.54-1.50 (m, 2Н). LCMS: рассчитано 273,1; обнаружено 274,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 29-А4 (т.е. соединения №46; 100 мг; 0,27 ммоль) и соединения 29-А3 (118 мг; 0,54 ммоль) в ацетоне (10 мл), затем добавляли Cs2CO3 (265 мг; 0,81 ммоль) и перемешивали в течение 3 ч при комнатной температуре до завершения реакции. Последующую обработку осуществляли так, как приведено ниже. Проводили фильтрование через целит с отсасыванием, выполняли промывку ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №29 (55,0 мг; 32,6%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO) δ 8.22 (d, J=8,5 Гц, 1Н), 7.62 (d, J=8,5 Гц, 1Н), 7.51 (d, J=8,6 Гц, 2Н), 7.44 (s, 1Н), 7.14 (d, J=8,6 Гц, 2Н), 6.31 (dq, J=12,8; 6,2 Гц, 1Н), 4.54 (s, 1Н), 3.71 (s, 1Н), 2.86 (s, 2Н), 2.67-2.63 (m, 1Н), 2.20-1.93 (m, 8Н), 1.84 (m, 2Н), 1.43 (qd, J=12,6; 4,2 Гц, 2Н). LCMS: рассчитано 622,1; обнаружено 623,2 ([М+Н]+).
Синтез соединения №30
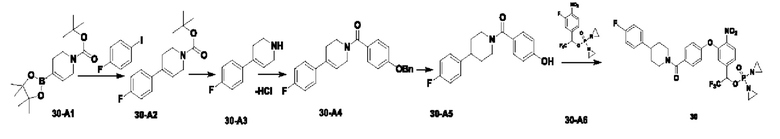
В атмосфере азота добавляли соединение 30-А1 (1,5 г; 4,95 ммоль; имеется в продаже) и п-фториодбензол (1,0 г; 4,50 ммоль; имеется в продаже) к DMF (90 мл). После трехкратного вакуумирования и заполнения азотом к этой системе добавляли dppfPdCl2, т.е. 1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, (330 мг; 0,45 ммоль; имеется в продаже) и К2СО3 (1,9 г; 13,51 ммоль). По завершении добавления вновь 3 раза проводили вакуумирование и заполнение азотом. Температуру системы повышали до 110°С и выдерживали в течение ночи. На следующий день проводили мониторинг до завершения реакции. После охлаждения системы до комнатной температуры ее выливали в воду (100 мл) и экстрагировали EtOAc (100 мл×3). Органическую фазу промывали водой, промывали рассолом, сушили, концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА = от 15:1 до 10:1), получая продукт 30-А2 (920 мг; выход 73,6%) в виде светло-зеленой жидкости. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 7.34-7.31 (m, 2Н), 7.03-6.99 (t, J=8,8 Гц, 2Н), 5.97 (m, 1Н), 4.06 (d, J=2,4 Гц, 2Н), 3.64-3.61 (m, 2Н), 2.49 (m, 2Н), 1.49 (s, 9Н). LCMS: рассчитано 277,1; обнаружено 222,1 ([М-56+Н]+).
Соединение 30-А2 (800 г; 2,88 ммоль) растворяли в МТВЕ (метил-трет-бутиловом эфире; 2 мл) и затем к этой системе по каплям добавляли раствор хлористого водорода в диоксане (4 М; 2 мл; 7,21 ммоль). Температуру поддерживали равной комнатной температуре в течение ночи. На следующий день проводили мониторинг до завершения реакции. Проводили фильтрование с отсасыванием и твердое вещество промывали МТВЕ, получая продукт 30-А3 (460 мг; 74,70%) в виде беловатого твердого вещества. 1Н-ЯМР (400 МГц, DMSO): δ млн-1 9.18 (s, 2Н), 7.52 (d, J=8,8; 5,6 Гц, 2Н), 7.23-7.19 (m, 2Н), 6.16 (s, 1 Н), 3.72 (s, 2Н), 3.29 (m, 2Н), 2.66 (m, 2Н). LCMS: рассчитано 213,1; обнаружено 178,1 ([М-HCl+H]+).
В атмосфере азота добавляли соединение  (350 мг; 1,53 ммоль) и соединение 30-А3 (407 мг; 2,30 ммоль) к DCM (10 мл) и затем к этой системе по каплям добавляли Т3Р (3,90 г; 6,12 ммоль; 50%-ный раствор в EtOAc). После охлаждения системы до 5°С к этой системе по каплям добавляли DIEA (791 мг; 6,12 ммоль). По завершении добавления температура естественным образом повышалась до комнатной температуры. Проводили мониторинг в течение 2 ч до завершения реакции. Систему концентрировали, разбавляли EtOAc (10 мл) и промывали H2O (10 мл×5). Органическую фазу сушили, концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА=1:1-0:1), получая продукт 30-А4 (460 мг; выход 77,9%) в виде светло-зеленого масла. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.45-7.43 (m, 6Н), 7.39-7.36 (m, 2Н), 7.32 (d, J=7,2 Гц, 1Н), 7.10-7.03 (m, 4Н), 6.13-5.97 (m, 1Н), 5.15 (s, 2Н), 4.29-4.22 (m, 2Н), 3.93-3.69 (m, 2Н), 2.60 (m, 2Н).
(350 мг; 1,53 ммоль) и соединение 30-А3 (407 мг; 2,30 ммоль) к DCM (10 мл) и затем к этой системе по каплям добавляли Т3Р (3,90 г; 6,12 ммоль; 50%-ный раствор в EtOAc). После охлаждения системы до 5°С к этой системе по каплям добавляли DIEA (791 мг; 6,12 ммоль). По завершении добавления температура естественным образом повышалась до комнатной температуры. Проводили мониторинг в течение 2 ч до завершения реакции. Систему концентрировали, разбавляли EtOAc (10 мл) и промывали H2O (10 мл×5). Органическую фазу сушили, концентрировали и подвергали разделению на колонке (силикагель, 200-300 меш; н-гептан:ЕА=1:1-0:1), получая продукт 30-А4 (460 мг; выход 77,9%) в виде светло-зеленого масла. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.45-7.43 (m, 6Н), 7.39-7.36 (m, 2Н), 7.32 (d, J=7,2 Гц, 1Н), 7.10-7.03 (m, 4Н), 6.13-5.97 (m, 1Н), 5.15 (s, 2Н), 4.29-4.22 (m, 2Н), 3.93-3.69 (m, 2Н), 2.60 (m, 2Н).
В атмосфере азота добавляли соединение 30-А4 (450 мг; 1,16 ммоль) и Pd(OH)2 (81 мг; имеется в продаже) к EtOH (10 мл). Трижды проводили вакуумирование и заполнение азотом. После трехкратного вакуумирования и заполнения водородом температуру повышали до 35°С и выдерживали в течение ночи. На следующий день проводили мониторинг до завершения реакции. Проводили фильтрование через целит с отсасыванием и выполняли промывку с использованием DCM. Остаточную жидкость концентрировали, получая продукт 30-А5 (310 мг; выход 89,2%) в виде бесцветного масла. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 7.33-7.31 (m, 2Н), 7.29-7.25 (m, 2Н), 7.04-6.99 (m, 2Н), 6.87-6.84 (m, 2Н), 4.72 (m, 2Н), 4.00 (m, 1Н), 3.19-2.96 (m, 2Н), 1.86 (m, 2Н), 1.68-1.66 (m, 2Н). LCMS: рассчитано 299,1; обнаружено 300,1 ([М+Н]+).
В атмосфере азота проводили растворение соединения 30-А6 (т.е. соединения №46; 100 мг; 0,27 ммоль) в ацетоне (10 мл), затем добавляли соединение 30-А5 (97 мг; 0,33 ммоль) и Cs2CO3 (265 мг; 0,81 ммоль) и перемешивали в течение 3 ч при комнатной температуре. По завершении реакции проводили фильтрование через целит с отсасыванием, твердое вещество промывали ацетоном и остаточную жидкость концентрировали. После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №30 (29,0 мг; 16,5%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 8.22 (d, J=8,5 Гц, 1Н), 7.61 (d, J=8,5 Гц, 1Н), 7.53 (d, J=8,6 Гц, 2Н), 7.44 (s, 1Н), 7.34-7.31 (m, 2Н), 7.16-7.11 (m, 4Н), 6.38-6.26 (m, 1Н), 4.62 (m, 1Н), 3.72 (m, 1Н), 3.24-3.10 (m, 2Н), 2.83 (m, 1Н), 2.17-1.91 (m, 8Н), 1.86-1.59 (m, 4Н). LCMS: рассчитано 648,2; обнаружено 649,2 ([М+Н]+).
Синтез соединения №33
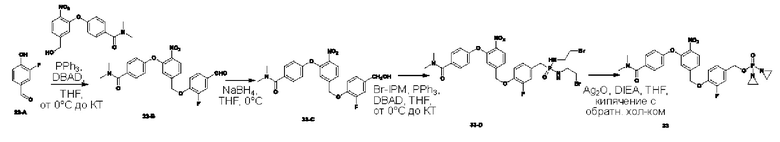
В атмосфере азота проводили растворение соединения  (1 г; 3,16 ммоль) и соединения 33-А (886 мг; 6,33 ммоль; 1,2 экв.; имеется в продаже) в THF (15 мл). Добавляли трифенилфосфин (1,66 г; 6,33 ммоль; 2 экв.; имеется в продаже). Температуру понижали до 0°С. По каплям добавляли раствор ди-трет-бутил-азодикарбоксилата (1,46 г; 6,33 ммоль; 2 экв.; имеется в продаже) в THF (8 мл). Реакцию проводили при комнатной температуре, и через 4 ч наблюдали ее завершение. Добавляли 10 мл воды. Осуществляли экстракцию с использованием DCM (25 мл×3), затем сушку и концентрирование. Образец перемешивали и пропускали через колонку (силикагель, 200-300 меш; ЕА:н-гептан=2:1), получая продукт 33-В (680 мг; выход 49,3%) в виде светло-желтого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ млн-1 9.87 (s, 1Н), 8.04 (d, J=7,8 Гц, 1Н), 7.63 (d, J=9,6 Гц, 2Н), 7.47 (d, J=8,1 Гц, 2Н), 7.34 (d, J=7,5 Гц, 1 Н), 7.02-7.11 (m, 4Н), 5.19 (s, 2Н), 3.08 (brs, 6Н). LCMS: рассчитано 438,4; обнаружено 439,0 ([М+Н]+).
(1 г; 3,16 ммоль) и соединения 33-А (886 мг; 6,33 ммоль; 1,2 экв.; имеется в продаже) в THF (15 мл). Добавляли трифенилфосфин (1,66 г; 6,33 ммоль; 2 экв.; имеется в продаже). Температуру понижали до 0°С. По каплям добавляли раствор ди-трет-бутил-азодикарбоксилата (1,46 г; 6,33 ммоль; 2 экв.; имеется в продаже) в THF (8 мл). Реакцию проводили при комнатной температуре, и через 4 ч наблюдали ее завершение. Добавляли 10 мл воды. Осуществляли экстракцию с использованием DCM (25 мл×3), затем сушку и концентрирование. Образец перемешивали и пропускали через колонку (силикагель, 200-300 меш; ЕА:н-гептан=2:1), получая продукт 33-В (680 мг; выход 49,3%) в виде светло-желтого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ млн-1 9.87 (s, 1Н), 8.04 (d, J=7,8 Гц, 1Н), 7.63 (d, J=9,6 Гц, 2Н), 7.47 (d, J=8,1 Гц, 2Н), 7.34 (d, J=7,5 Гц, 1 Н), 7.02-7.11 (m, 4Н), 5.19 (s, 2Н), 3.08 (brs, 6Н). LCMS: рассчитано 438,4; обнаружено 439,0 ([М+Н]+).
Соединение 33-В (680 мг; 1,55 ммоль) растворяли в THF (10 мл), температуру понижали до 0°С, порциями добавляли боргидрид натрия (117 мг; 3,10 ммоль; 2 экв.), температуру поддерживали при 0°С, и через 1 ч наблюдали завершение реакции. По каплям добавляли насыщенный водный раствор хлорида аммония (5 мл). Осуществляли экстракцию с использованием DCM (10 мл×3), затем сушку, концентрирование и разделение посредством колоночной хроматографии (силикагель, 100-200 меш; ЕА), получая продукт 33-С (410 мг; 60,3%) в виде светло-желтого твердого вещества. 1Н-ЯМР (300 МГц, CDCl3): δ млн-1 8.02 (d, J=8,1 Гц, 1Н), 7.43 (d, J=7,8 Гц, 2Н), 7.31 (d, J=6,0 Гц, 1Н), 7.12 (d, J=12,3 Гц, 1Н), 6.99-7.03 (m, 4Н), 6.84 (t, J=8,4 Гц, 1Н), 5.16 (s, 2Н), 4.62 (s, 2Н), 3.08 (brs, 6Н). LCMS: рассчитано 440,4; обнаружено 441,0 ([М+Н]+).
В атмосфере азота проводили растворение соединения 33-С (400 мг; 0,91 ммоль) в THF (5 мл). Добавляли трифенилфосфин (480 мг; 1,82 ммоль) и Br-IPM (564 мг; 1,82 ммоль; имеется в продаже). Температуру понижали до 0°С. По каплям при 0°С добавляли раствор ди-трет-бутил-азодикарбоксилата (420 мг; 1,82 ммоль) в THF (5 мл). Реакцию проводили при комнатной температуре в течение 3 ч. Добавляли воду (5 мл) при 0°С. Осуществляли экстракцию с использованием DCM (10 мл×3), затем сушку, концентрирование и разделение посредством колоночной хроматографии (силикагель, 200-300 меш; DCM:метанол=50:1), получая соединение 33-D (300 мг; выход 45,0%) в виде светло-желтого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.01 (d, J=8,4 Гц, 1Н), 7.45 (d, J=8,8 Гц, 2H), 7.32 (d, J=8,0 Гц, 1H), 7.26 (m, 1H), 7.16 (dd, J=11,5; 1,6 Гц, 1H), 7.10 (s, 1H), 7.02 (d, J=8,4 Гц, 2H), 6.89 (t, J=8,4 Гц, 1H), 5.13 (s, 2H), 4.95 (d, J=8,4 Гц, 2H), 3.45-3.41 (m, 4H), 3.34-3.30 (m, 4H), 3.08 (s, 6H). LCMS: рассчитано 732,0; обнаружено 732,8 ([М+Н]+).
В атмосфере азота проводили растворение соединения 33-D (200 мг; 0,27 ммоль) в THF (5 мл). Добавляли оксид серебра (742 мг; 3,2 ммоль; 11,8 экв.) и по каплям добавляли диизопропилэтиламин (176 мг; 1,365 ммоль; 5 экв.). Температуру повышали до температуры дефлегмации. Реакцию проводили в течение 2 ч, после чего температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием, затем несколько раз промывку THF и концентрирование при низкой температуре, получая соединение №33 (5 мг; 3,2%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 7.99 (d, J=8,4 Гц, 1Н), 7.44 (d, J=8,8 Гц, 2Н), 7.31 (d, J=8,0 Гц, 1Н), 7.15 (dd, J=11,6; 2,0 Гц, 1Н), 7.11 (s, 1Н), 7.03-7.07 (m, 3H), 6.91 (t, J=8,4 Гц, 1Н), 5.09 (s, 2Н), 5.05 (d, J=8,0 Гц, 2Н), 3.09 (s, 3H), 3.00 (s, 3H), 2.18-2.08 (m, 8Н). (Время удерживания: 7,359 мин). LCMS: рассчитано 570,0; обнаружено 571,0 ([М+Н]+).
Синтез соединения №34
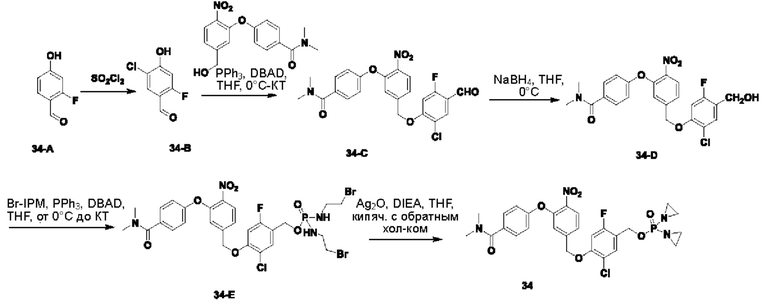
Соединение 34-А (10,0 г; 71,4 ммоль; имеется в продаже) добавляли к уксусной кислоте (30 мл), затем добавляли сульфонилхлорид (14,4 г; 107 ммоль; 1,5 экв.) и проводили реакцию при комнатной температуре в течение 17 ч. Добавляли 300 мл ледяной воды. Осуществляли экстракцию с использованием ЕА (300 мл×2). Органическую фазу промывали рассолом (20 мл×2), сушили с использованием безводного Na2SO4 и растворитель упаривали на роторном испарителе, получая неочищенный продукт. Неочищенный продукт суспендировали с метил-трет-бутиловым эфиром и фильтровали с отсасыванием, получая соединение 34-В (3,0 г; 25%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 9.98 (s, 1Н), 7.80 (d, J=4,4 Гц, 1Н), 6.89 (d, J=11,6 Гц, 1Н). LCMS: рассчитано 174,0; обнаружено 175,0 ([М+Н]+).
В атмосфере азота добавляли соединение 34-В (375 мг; 2,15 ммоль; 2 экв.) к THF (10 мл). Добавляли соединение  (340 мг; 1,08 ммоль) и трифенилфосфин (563 мг; 2,15 ммоль; 2 экв.; имеется в продаже). Добавляли DBAD (494,5 мг; 2,15 ммоль; 2 экв.; имеется в продаже) при 0°С. Реакцию проводили в течение 1 ч при комнатной температуре. С целью разбавления добавляли DCM (50 мл). Органическую фазу промывали водой (10 мл×2) и насыщенным рассолом (10 мл), сушили и упаривали на роторном испарителе. Образец перемешивали и пропускали через колонку для флэш-хроматографии (силикагель, 300-400 меш; ЕА и н-гептан, от 33% до 100%), получая соединение 34-С (400 мг; 78,7%) в виде белого продукта. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 10.04 (s, 1Н), 8.18 (d, J=8,4 Гц, 1Н), 7.87 (d, J=7,2 Гц, 1Н), 7.49 (d, J=8,4 Гц, 2Н), 7.44 (d, J=8,4 Гц, 1Н), 7.36 (d, J=8,4 Гц, 1Н), 7.31 (s, 1Н), 7.15 (d, J=8,4 Гц, 2Н), 5.44 (s, 2Н), 2.97 (s, 6Н). LCMS: рассчитано 472,1; обнаружено 472,9 ([М+Н]+).
(340 мг; 1,08 ммоль) и трифенилфосфин (563 мг; 2,15 ммоль; 2 экв.; имеется в продаже). Добавляли DBAD (494,5 мг; 2,15 ммоль; 2 экв.; имеется в продаже) при 0°С. Реакцию проводили в течение 1 ч при комнатной температуре. С целью разбавления добавляли DCM (50 мл). Органическую фазу промывали водой (10 мл×2) и насыщенным рассолом (10 мл), сушили и упаривали на роторном испарителе. Образец перемешивали и пропускали через колонку для флэш-хроматографии (силикагель, 300-400 меш; ЕА и н-гептан, от 33% до 100%), получая соединение 34-С (400 мг; 78,7%) в виде белого продукта. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 10.04 (s, 1Н), 8.18 (d, J=8,4 Гц, 1Н), 7.87 (d, J=7,2 Гц, 1Н), 7.49 (d, J=8,4 Гц, 2Н), 7.44 (d, J=8,4 Гц, 1Н), 7.36 (d, J=8,4 Гц, 1Н), 7.31 (s, 1Н), 7.15 (d, J=8,4 Гц, 2Н), 5.44 (s, 2Н), 2.97 (s, 6Н). LCMS: рассчитано 472,1; обнаружено 472,9 ([М+Н]+).
В атмосфере азота добавляли соединение 34-С (200 мг; 0,42 ммоль) к THF (10 мл), добавляли боргидрид натрия (32 мг; 0,84 ммоль; 2 экв.; имеется в продаже) при 0°С, температура естественным образом повышалась, и реакцию проводили в течение 0,5 ч. По завершении реакции по каплям добавляли насыщенный водный раствор хлорида аммония (10 мл). Осуществляли экстракцию с использованием DCM (20 мл×3), затем промывку водой (10 мл), промывку насыщенным рассолом (10 мл), сушку и упаривание на роторном испарителе, суспендирование с изопропанолом и фильтрование с отсасыванием, получая соединение 34-D (100 мг; 50%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 8.16 (d, J=8,4 Гц, 1Н), 7.49 (d, J=8,4 Гц, 2Н), 7.46 (d, J=8,0 Гц, 1Н), 7.43 (d, J=9,6 Гц, 1Н), 7.30 (s, 1Н), 7.15-7.12 (m, 3H), 5.32 (s, 2Н), 5.27 (t, J=5,6 Гц, 1Н), 4.45 (d, J=5,6 Гц, 2Н), 2.97 (s, 6Н). LCMS: рассчитано 474,1; обнаружено 475,0 ([М+Н]+).
В атмосфере азота добавляли соединение 34-D (100 мг; 0,21 ммоль) к THF (10 мл). Добавляли трифенилфосфин (110 мг; 0,42 ммоль; 2 экв.) и Br-IPM (129 мг; 0,42 ммоль; 2 экв.). Добавляли ди-трет-бутил-азодикарбоксилат (97 мг; 0,42 ммоль; 2 экв.) при 0°С. Реакцию проводили при комнатной температуре в течение 4 ч. По завершении реакции добавляли DCM (50 мл). Органическую фазу промывали насыщенным рассолом (10 мл), сушили, упаривали на роторном испарителе и пропускали через колонку для флэш-хроматографии (силикагель, 300-400 меш; ЕА и н-гептан 0-100% и затем DCM:MeOH (20:1)), получая соединение 34-Е (70 мг; 43,7%) в виде желтого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 7.96 (d, J=8,4 Гц, 1Н), 7.40-7.38 (m, 3H), 7.25 (d, J=7,6 Гц, 1 Н), 7.07 (s, 1 Н), 6.99 (d, J=8,8 Гц, 2Н), 6.57 (d, J=10,8 Гц, 1Н), 5.04 (s, 2Н), 4.92 (d, J=8,0 Гц, 2Н), 3.36-3.39 (m, 4Н), 3.24-3.29 (m, 4Н), 3.01 (s, 6Н). LCMS: рассчитано 766,0; обнаружено 766,8 ([М+Н]+).
Соединение 34-Е (70 мг; 0,091 ммоль) добавляли к THF (5 мл), добавляли оксид серебра (250 мг; 1,08 ммоль; 11,8 экв.) и DIEA(55 мг; 0,428 ммоль; 4,7 экв.), нагревали до 65°С и реакцию проводили в течение 6 ч. По завершении реакции проводили фильтрование с отсасыванием, осадок на фильтре промывали THF (10 млх2), фильтрат еще раз фильтровали с отсасыванием с использованием фильтрующей мембраны и упаривали на роторном испарителе, получая неочищенный продукт. После проведения препаративной HPLC получали соединение 34 (5 мг; 9,1%) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, CD3OD): δ млн-1 8.08 (d, J=8,4 Гц, 1Н), 7.54-7.47 (m, 4Н), 7.37 (s, 1Н), 7.12 (d, J=8,8 Гц, 2Н), 7.06 (d, J=11,2 Гц, 1Н), 5.29 (s, 2Н), 5.14 (d, J=8,0 Гц, 1Н), 3.13 (s, 3H), 3.07 (s, 3H), 2.15-2.14 (m, 8Н). LCMS: рассчитано 604,0; обнаружено 605,0 ([М+Н]+).
Для синтеза соединений №№35-44/47-49 следует обратиться к приведенным выше способам. Данные относительно свойств, ЯМР- и масс-спектров приведены ниже.
Соединение №35
Твердое вещество, 1Н-ЯМР (400 МГц, CDCl3) δ 7.99 (d, J=8,4 Гц, 1Н), 7.47 (d, J=8,6 Гц, 2Н), 7.32 (d, J=8,3 Гц, 1Н), 7.17 (s, 1Н), 7.05 (d, J=8,6 Гц, 2Н), 6.95 (t, J=6,9 Гц, 2Н), 5.54-5.48 (m, 1Н), 5.14 (s, 2Н), 3.11 (s, 3H), 3.03 (s, 3H), 2.21-2.00 (m, 8Н), 1.57 (d, J=6,5 Гц, 3H). LCMS: рассчитано 602,2; обнаружено 603,2 ([М+Н]+).
Соединение №36
Твердое вещество, 1Н-ЯМР (400 МГц, CDCl3) δ 8.01 (d, J=8,4 Гц, 1Н), 7.71-7.62 (m, 2Н), 7.34 (d, J=8,4 Гц, 1 Н), 7.22 (s, 1 Н), 7.04 (d, J=8,8 Гц, 2Н), 6.95 (d, J=8,5 Гц, 2Н), 5.47-5.28 (m, 1Н), 5.17 (s, 2Н), 5.06 (d, J=8,1 Гц, 2Н), 4.62-4.48 (m, 2Н), 4.38 (m, 2Н), 2.32-2.04 (m, 8Н). LCMS: рассчитано 518,1; обнаружено 619,2 ([М+Н]+).
Соединение №37
Воскообразное вещество, 1Н-ЯМР (400 МГц, CDCl3) δ 8.05 (d, J=8,4 Гц, 1Н), 7.71-7.63 (m, 2Н), 7.46-7.38 (m, 1Н), 7.31 (s, 1Н), 7.09-7.01 (m, 2Н), 6.97 (d, J=8,6 Гц, 2Н), 5.20 (s, 2Н), 5.07 (d, J=8,0 Гц, 2Н), 2.31-2.06 (m, 8Н). LCMS: рассчитано 542,1; обнаружено 543,1 ([М+Н]+).
Соединение №38
Беловатое твердое вещество, 1Н-ЯМР (400 МГц, CDCl3) δ 8.02 (d, J=8,4 Гц, 1 Н), 7.68 (d, J=8,7 Гц, 2Н), 7.36 (d, J=8,5 Гц, 1Н), 7.24 (s, 1 Н), 7.05 (d, J=8,7 Гц, 2Н), 6.96 (d, J=8,5 Гц, 2Н), 5.18 (s, 2Н), 5.06 (d, J=8,1 Гц, 2Н), 4.59-4.53 (m, 4Н), 2.38-2.02 (m, 8Н). LCMS: рассчитано 536,1; обнаружено 637,1 ([М+Н]+).
Соединение №39
Воскообразное вещество, 1Н-ЯМР (400 МГц, CDCl3) δ 8.03 (d, J=8,4 Гц, 1Н), 7.55-7.44 (m, 2Н), 7.37 (d, J=8,4 Гц, 1Н), 7.30-7.26 (m, 1Н), 7.25-7.20 (m, 2Н), 7.02-6.90 (m, 2Н), 5.20 (s, 2Н), 5.06 (d, J=8,0 Гц, 2Н), 2.28-2.10 (m, 8Н). LCMS: рассчитано 542,1; обнаружено 543,1 ([М+Н]+).
Соединение №40
Беловатое твердое вещество, 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 8.90-8.89 (s, 1Н), 8.33 (d, J=1,2 Гц, 1Н), 8.31 (d, J=1,2 Гц, 1Н), 8.25 (d, J=8,4 Гц, 1Н), 8.13 (d, J=10,0 Гц), 7.63-7.55 (m, 4Н), 7.46 (d, J=1,2 Гц, 1Н), 6.30-6.26 (m, 1Н), 2.11-1.92 (m, 8Н). LCMS: рассчитано 494,1; обнаружено 495,1 ([М+Н]+).
Соединение №41
Беловатое твердое вещество, 1Н-ЯМР (400 МГц, MeOD): δ млн-1 8.43 (d, J=2,9 Гц, 1Н), 7.24-7.19 (m, 2Н), 6.13-6.03 (m, 1Н), 2.26-2.08 (m, 8Н). LCMS: рассчитано 576,1; обнаружено 577,2 ([М+Н]+).
Соединение №42
Беловатое твердое вещество, 1Н-ЯМР (400 МГц, DMSO-d6): δ млн-1 8.20 (d, J=8,2 Гц, 1Н), 7.99-7.90 (m, 1Н), 7.89 (s, 1 Н) 7.55 (d, J=8,4 Гц, 1Н), 7.31-7.26 (m, 1Н), 6.28-6.24 (m, 1Н), 2.80 (s, 3H), 2.10-1.91 (m, 8Н). LCMS: рассчитано 514,1; обнаружено 515,0 ([М+Н]+).
Соединение №43
Беловатое твердое вещество, 1Н-ЯМР (400 МГц, MeOD): δ млн-1 8.05 (d, J=8,0 Гц, 1 Н), 8.01 (s, 1Н), 7.68 (d, J=8,0 Гц, 1Н), 7.49 (d, J=2,0 Гц, 1 Н), 7.43 (d, J=8,0 Гц, 1Н), 7.27 (dd, J=8,8; 2,0 Гц, 1Н), 7.17 (s, 1Н), 5.99-5.94 (m, 1Н), 2.20-2.02 (m, 8Н). LCMS: рассчитано 497,1; обнаружено 498,1 ([М+Н]+).
Соединение №44
Беловатое твердое вещество, 1Н-ЯМР (400 МГц, MeOD) δ 8.07 (d, J=8,4 Гц, 1Н), 7.75-7.67 (m, 2Н), 7.65-7.59 (m, 2Н), 7.49 (d, J=8,5 Гц, 1Н), 7.44 (t, J=7,6 Гц, 2Н), 7.35 (d, J=7,3 Гц, 1Н), 7.31 (s, 1Н), 7.20-7.14 (m, 2Н), 6.07-5.97 (m, 1Н), 2.26-2.03 (m, 8Н). LCMS: рассчитано 519,1; обнаружено 520,1 ([М+Н]+).
Соединение №47
Беловатое твердое вещество, 1Н-ЯМР (400 МГц, MeOD) δ 8.03 (d, J=8,4 Гц, 1Н), 7.43 (d, J=8,4 Гц, 1Н), 7.34 (d, J=8,5 Гц, 2Н), 7.19 (s, 1Н), 7.03 (t, J=5,7 Гц, 2Н), 6.07-5.94 (m, 1Н), 3.09-2.99 (m, 1Н), 2.24-2.02 (m, 10H), 1.87-1.83 (m, 2Н), 1.77-1.68 (m, 2Н), 1.65-1.56 (m, 2Н). LCMS: рассчитано 511,1; обнаружено 512,2 ([М+Н]+).
Соединение №48
Беловатое твердое вещество, 1Н-ЯМР (400 МГц, MeOD) δ 8.03 (d, J=8,4 Гц, 1Н), 7.43 (d, J=8,6 Гц, 1Н), 7.31 (d, J=8,5 Гц, 2Н), 7.19 (s, 1Н), 7.02 (d, J=8,5 Гц, 2Н), 6.00-5.96 (m, 1Н), 2.56 (m, 1Н), 2.23-2.03 (m, 8Н), 1.87 (m, 4Н), 1.78-1.75 (m, 2Н), 1.49-1.43 (m, 5Н). LCMS: рассчитано 525,2; обнаружено 526,2 ([М+Н]+).
Соединение №49
Желтое воскообразное вещество: 1Н-ЯМР (400 МГц, MeOD) δ 8.04 (d, J=8,4 Гц, 1 Н), 7.45 (d, J=8,2 Гц, 1Н), 7.35 (d, J=8,6 Гц, 2Н), 7.21 (s, 1 Н), 7.04 (d, J=8,6 Гц, 2Н), 5.99 (dd, J=9,9; 6,2 Гц, 1 Н), 2.74 (t, J=12,5 Гц, 1Н), 2.23-2.04 (m, 10Н), 2.02-1.73 (m, 6Н). LCMS: рассчитано 561,1; обнаружено 562,2 ([М+Н]+).
Синтез соединения №46
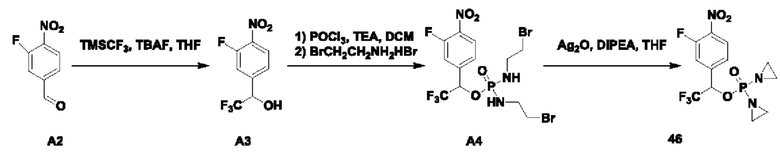
В атмосфере азота проводили растворение соединения А2 (2,0 г; 11,8 ммоль; имеется в продаже) и TMSCF3 (2,5 г; 17,7 ммоль; имеется в продаже) в THF (20 мл). Температуру понижали до 0°С. К этой системе по каплям добавляли TBAF (2,6 мл; 0,26 ммоль; раствор в THF; 1 моль/л; имеется в продаже). Температуру поддерживали при 0°С. Через 30 мин соединение А2 полностью расходовалось. К системе по каплям добавляли 3 н. раствор HCl (2 мл), и система становилась прозрачной. После перемешивания при 0°С в течение 1 ч все исходные вещества превращались в продукты. Осуществляли экстракцию с использованием DCM (10 мл×3). Органическую фазу промывали водой (10 мл×3), сушили, концентрировали и проводили разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА=12:1-10:1), получая продукт A3 (1,5 г; выход 53,2%) в виде желтого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.13-8.09 (m, 1Н), 7.60 (d, J=11,6 Гц, 1Н), 7.43 (d, J=8,4 Гц, 1Н), 5.12-5.18 (m, 1Н), 3.06 (d, J=4,4 Гц, 1Н).
В атмосфере азота проводили растворение POCl3 (963 мг; 4,61 ммоль; имеется в продаже) в DCM (10 мл). После этого температуру понижали до -40°С. Затем соединение A3 (1,5 г; 6,27 ммоль) растворяли в DCM (20 мл), после чего по каплям его добавляли к системе вместе с добавлением TEA (1,6 г; 15,70 ммоль). После поддержания температуры при -40°С в течение 2 ч соединение 46-А3 полностью превращалось в промежуточное соединение. Затем к системе добавляли бромэтиламина гидробромид (11,99 г; 50,16 ммоль) и TEA (10,1 г; 0,1 моль). Проводили мониторинг. Через 30 мин наблюдали завершение реакции. При 0°С добавляли насыщенный раствор NH4Cl (20 мл). Осуществляли экстракцию с использованием DCM (50 мл×3). Органическую фазу промывали водой и промывали рассолом, сушили, концентрировали и проводили разделение на колонке (силикагель, 200-300 меш; н-гептан:ЕА=5:1-1:1), получая продукт А4 (1,6 г; выход 65,3%) в виде желтого масла. 1Н-ЯМР (400 МГц, CDCl3): δ млн-1 8.15-8.11 (m, 1Н), 7.47 (d, J=11,6 Гц, 1Н), 7.44 (d, J=8,8 Гц, 1Н), 5.78-5.30 (m, 1Н), 3.53-3.05 (m, 10Н). LCMS: рассчитано 529,9; обнаружено 531,9 ([М+Н]+).
В атмосфере азота проводили растворение соединения А4 (1,6 г; 3,0 ммоль) в THF (20 мл) и затем добавляли оксид серебра (4,2 г; 18,0 ммоль) и DIPEA (2,3 г; 18,0 ммоль). Температуру повышали до 65°С и проводили перемешивание в течение 1,5 ч. После завершения реакции температуру понижали до комнатной температуры. Проводили фильтрование через целит с отсасыванием. Твердое вещество промывали DCM, остаточную жидкость концентрировали и проводили разделение на колонке, получая неочищенный продукт (520 мг; выход 46,8%) (силикагель, 200-300 меш; н-гептан:ЕА=5:1-1:1). После проведения препаративной высокоэффективной жидкостной хроматографии получали чистое соединение №46 (30 мг) в виде белого твердого вещества. 1Н-ЯМР (400 МГц, MeOD): δ млн-1 8.24-8.20 (m, 1Н), 7.70 (d, J=11,6 Гц, 1Н), 7.63 (d, J=8,8 Гц, 1Н), 6.14-6.10 (m, 1Н), 2.28-2.14 (m, 8Н). LCMS: рассчитано 369,1; обнаружено 370,1 ([М+Н]+).
Экспериментальные факты в приведенных выше примерах для конкретных соединений подтверждают, что все предложенные согласно настоящему изобретению соединения представляют собой либо твердые вещества, либо воскообразные вещества (полутвердые вещества), что устраняет недостаток, заключающийся в том, что предшествующие ДНК-алкилирующие агенты представляют собой масла (описанные в заявке РСТ №PCT/US2016/021581 (публикация №WO2016145092), которая соответствует заявке на патент Китая №2016800150788 (публикация №CN107530556A)), и это облегчает приготовление композиций.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ПРОИЗВОДНОЕ НА ОСНОВЕ ДИГИДРОПИРИМИДО-КОЛЬЦА В КАЧЕСТВЕ ИНГИБИТОРА HBV | 2015 |
|
RU2693897C2 |
| ХИНОЛИНОВЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ SMO | 2015 |
|
RU2695815C2 |
| АНАЛОГИ СОЕДИНЕНИЙ 4Н-ПИРАЗОЛО[1,5-А]БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ PARP | 2015 |
|
RU2672722C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| КОНДЕНСИРОВАННЫЕ ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ TNF | 2014 |
|
RU2679914C9 |
| МАКРОЦИКЛИЧЕСКИЕ АНТИБИОТИКИ ШИРОКОГО СПЕКТРА | 2018 |
|
RU2779477C2 |
| СОЕДИНЕНИЯ, МОДУЛИРУЮЩИЕ ЭСТРОГЕНОВЫЕ РЕЦЕПТОРЫ | 2019 |
|
RU2826635C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛА В ЛЕЧЕНИИ РАССТРОЙСТВ, АССОЦИИРОВАННЫХ С ВАНИЛОИДНЫМ РЕЦЕПТОРОМ TRPV1 | 2007 |
|
RU2458055C2 |
Настоящее изобретение относится к области медицинской химии, а именно к соединению формулы II или III или его фармацевтически приемлемой соли, где R1 представляет собой С6-С10арил или Z-замещенный С6-С10арил, шестичленный азотсодержащий гетероарил, хинолин, изохинолин, Z-замещенный хинолин или Z-замещенный изохинолин; R2 представляет собой атом водорода, атом галогена, циано, С1-С6алкил или галогензамещенный С1-С6алкил, С3-С8циклоалкил или Z-замещенный С3-С8циклоалкил, С6-С10арил или Z-замещенный С6-С10арил, Z-замещенный азотсодержащий гетероарил, галогензамещенный алкокси, имеющий от 1 до 6 атомов углерода, -CONR6R7; R3 представляет собой атом водорода; R4 и R5 каждый независимо представляет собой атом водорода; R6 и R7 каждый независимо представляет собой С1-С6алкил, С6-С10арил или R6 и R7 вместе с атомом, к которому они присоединены, образуют 4-6-членный гетероциклил или Z-замещенный 4-6-членный гетероциклил; R8 и R10 каждый независимо представляет собой атом водорода; R9 представляет собой замещенный С6-С10арил, который замещен по меньшей мере одним атомом фтора; заместитель Z представляет собой атом галогена, циано, гидрокси, С1-С3алкил, фенил или галогензамещенный С1-С3алкил, галогензамещенный С1-С3алкокси, С3-С8циклоалкил или замещенный С3-С8циклоалкил, ароматическое кольцо или замещенное ароматическое кольцо, при этом замещение по своему типу представляет собой моно- или дизамещение по одному и тому же атому; заместитель в замещенном С6-С10ариле в R9 представляет собой атом галогена. Также изобретение относится к индивидуальному соединению формулы  , к применению соединения формулы II или III для изготовления лекарственного средства, к лекарственному средству, содержащему эффективное количество соединения формулы II или III, к способу получения этого соединения. Технический результат изобретения заключается в разработке ДНК-алкилирующих агентов, активируемых под действием фермента AKR1C3, которые могут быть использованы для лечения рака. 6 н. и 10 з.п. ф-лы, 1 табл.
, к применению соединения формулы II или III для изготовления лекарственного средства, к лекарственному средству, содержащему эффективное количество соединения формулы II или III, к способу получения этого соединения. Технический результат изобретения заключается в разработке ДНК-алкилирующих агентов, активируемых под действием фермента AKR1C3, которые могут быть использованы для лечения рака. 6 н. и 10 з.п. ф-лы, 1 табл.
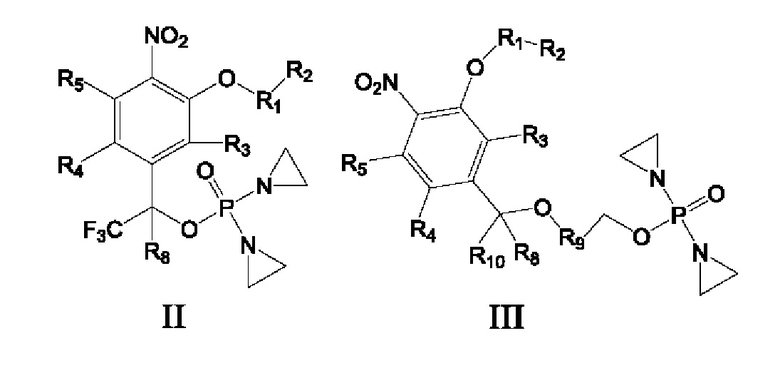
1. Соединение формулы II или III или его фармацевтически приемлемая соль,
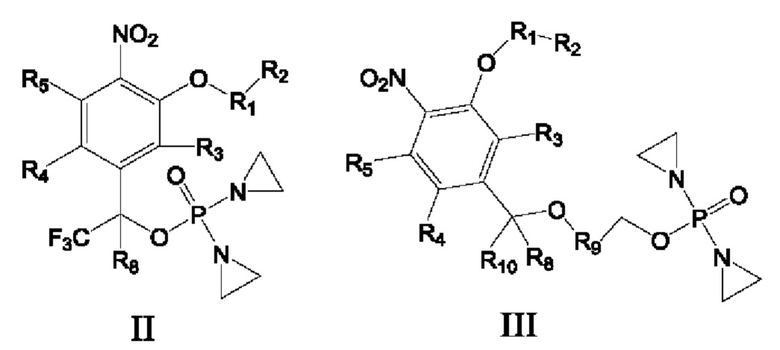
где
R1 представляет собой С6-С10арил или Z-замещенный С6-С10арил, шестичленный азотсодержащий гетероарил, хинолин, изохинолин, Z-замещенный хинолин или Z-замещенный изохинолин;
R2 представляет собой атом водорода, атом галогена, циано, С1-С6алкил или галогензамещенный С1-С6алкил, С3-С8циклоалкил или Z-замещенный С3-С8циклоалкил, С6-С10арил или Z-замещенный С6-С10арил, Z-замещенный азотсодержащий гетероарил, галогензамещенный алкокси, имеющий от 1 до 6 атомов углерода, -CONR6R7;
R3 представляет собой атом водорода;
R4 и R5 каждый независимо представляет собой атом водорода;
R6 и R7 каждый независимо представляет собой С1-С6алкил, С6-С10арил или R6 и R7 вместе с атомом, к которому они присоединены, образуют 4-6-членный гетероциклил или Z-замещенный 4-6-членный гетероциклил;
R8 и R10 каждый независимо представляет собой атом водорода;
R9 представляет собой замещенный С6-С10арил, который замещен по меньшей мере одним атомом фтора;
заместитель Z представляет собой атом галогена, циано, гидрокси, С1-С3алкил, фенил или галогензамещенный С1-С3алкил, галогензамещенный C1-С3алкокси, С3-С8циклоалкил или замещенный С3-С8циклоалкил, ароматическое кольцо или замещенное ароматическое кольцо, при этом замещение по своему типу представляет собой моно- или дизамещение по одному и тому же атому;
заместитель в замещенном С6-С10ариле в R9 представляет собой атом галогена.
2. Соединение по п. 1, отличающееся тем, что
R1 представляет собой фенил или Z-замещенный фенил, шестичленный азотсодержащий гетероарил, хинолин, изохинолин, Z-замещенный хинолин или Z-замещенный изохинолин;
R2 представляет собой атом водорода, атом галогена, циано, С1-С6алкил или галогензамещенный С1-С6алкил, С3-С8циклоалкил или Z-замещенный С3-С8циклоалкил, С6-С10арил или Z-замещенный С6-С10арил, Z-замещенный азотсодержащий гетероарил, фторзамещенный С1-С6алкокси, -CONR6R7;
R6 и R7 каждый независимо представляет собой С1-С6алкил, С6-С10арил, или R6 и R7 вместе с атомом азота, в которому они присоединены, образуют 5-6-членную гетероциклическую группу или Z-замещенную 4-6-членную гетероциклическую группу.
3. Соединение по п. 1, отличающееся тем, что в формуле II и формуле III
R1 представляет собой фенил, пиридин, хинолон, изохинолин, бензотиазол, индазол, Z-замещенный фенил;
R2 представляет собой -CON(CH3)2, -CF3, F, Cl, CN, Me, бензол, фторбензол, -OCF3, С5-С6циклоалкил или F-замещенный С5-С6циклоалкил, фторпиридил, -CONMePh,
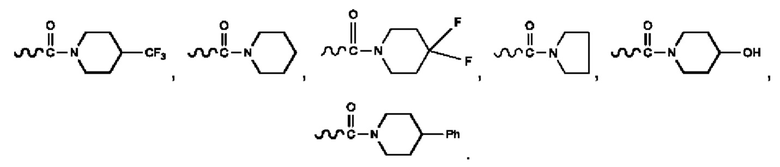
4. Соединение по п. 1, отличающееся тем, что
R3, R4 и R5 каждый независимо представляет собой Н.
5. Соединение по п. 1, отличающееся тем, что
R8 и R10 каждый независимо представляет собой Н.
6. Соединение по п. 1, отличающееся тем, что
R9 представляет собой монофтор-, монофтор-монохлор-, дифтор- или тетрафторзамещенный фенил.
7. Соединение по п. 1, отличающееся тем, что
R9 представляет собой
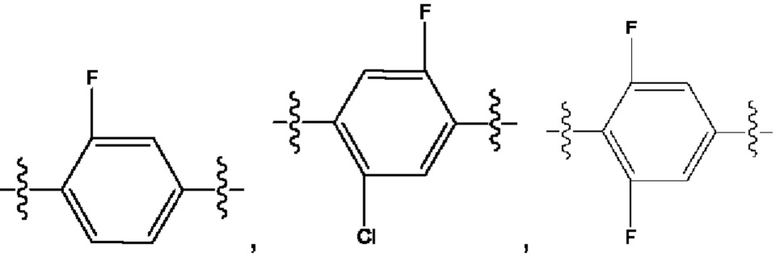 или
или 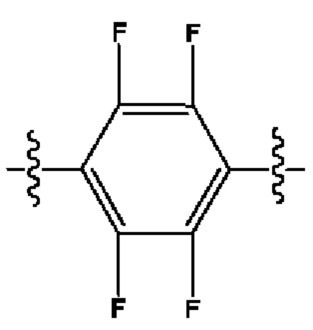 .
.
8. Соединение по п. 1, отличающееся тем, что соединение, замещенное изотопом дейтерием, имеет следующую структуру:
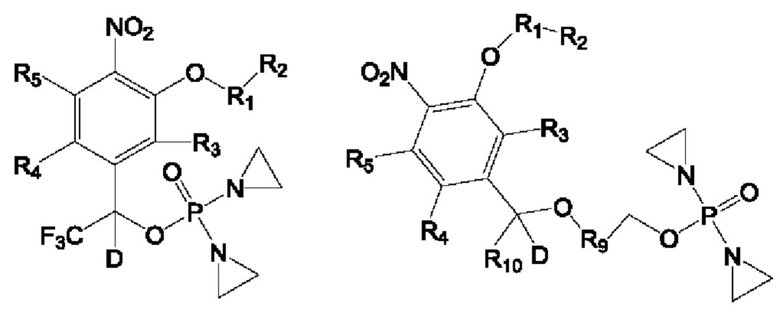 ,
,
где заместители являются такими, как определено в любом из пп. 1-7.
9. Соединение по п. 1, отличающееся тем, что соединение выбрано из следующих соединений: 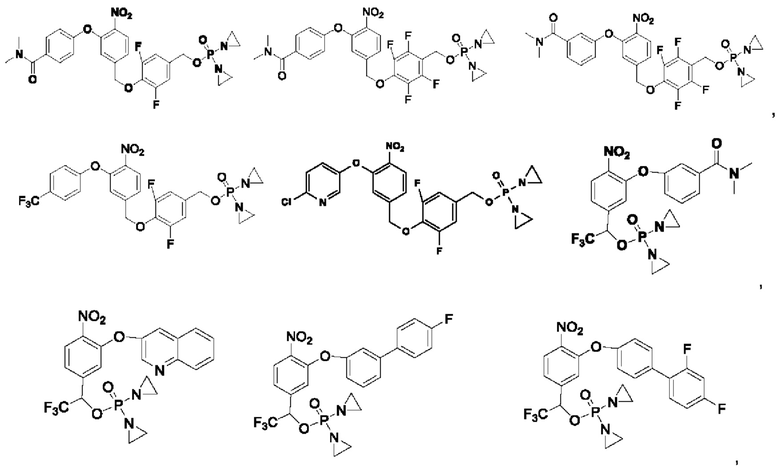
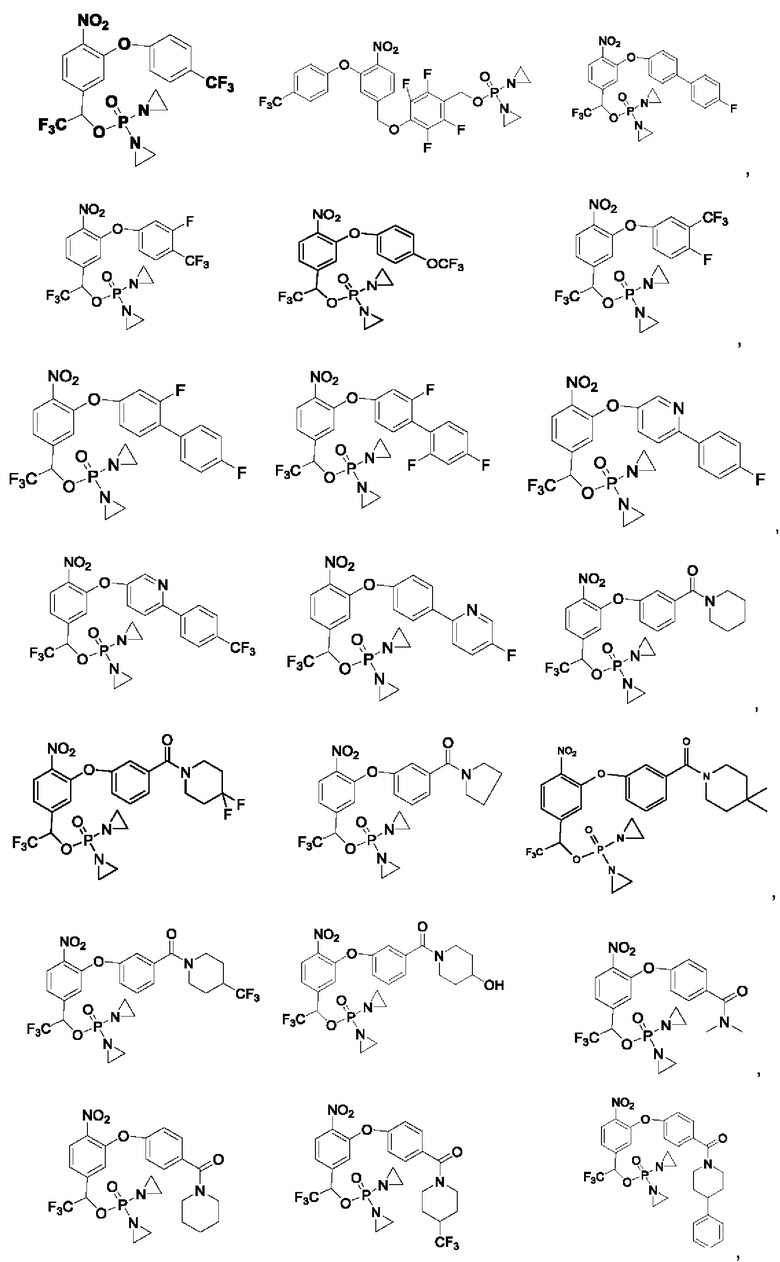
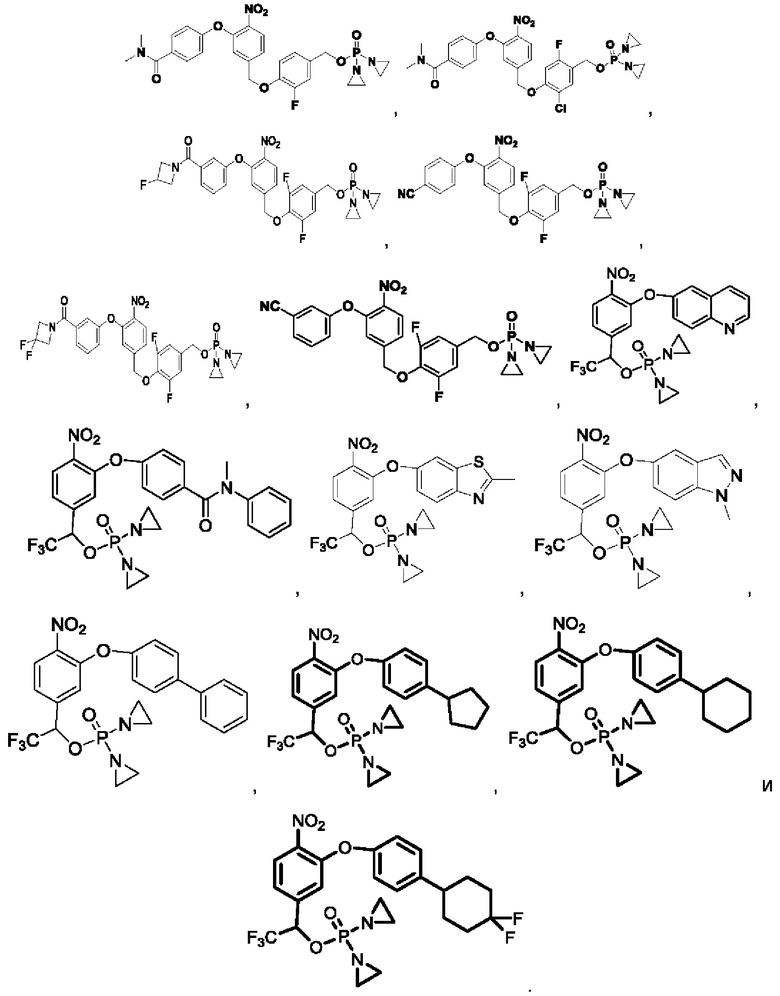
10. Соединение, имеющее следующую формулу, или его фармацевтически приемлемая соль 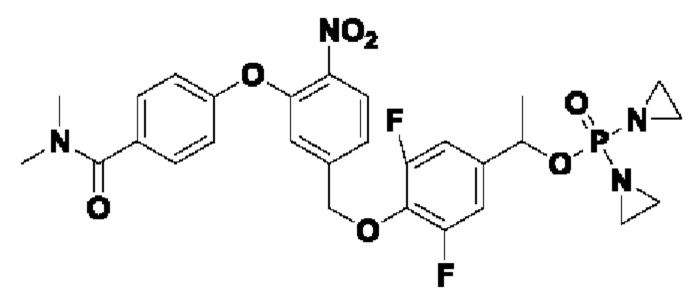 .
.
11. Соединение по любому из пп. 1-10, отличающееся тем, что соль представляет собой основную соль или кислую соль.
12. Применение соединения по любому из пп. 1-11 для изготовления лекарственного средства, являющегося ДНК-алкилирующим агентом, активируемым под действием фермента AKR1C3.
13. Лекарственное средство, являющееся ДНК-алкилирующим агентом, активируемым под действием фермента AKR1C3, содержащее эффективное количество соединения по любому из пп. 1-11.
14. Лекарственное средство по п. 13 для применения в лечении опухолей и раковых заболеваний у пациентов, при этом опухоли и раковые заболевания включают
рак легкого, немелкоклеточный рак легкого, рак печени, рак поджелудочной железы, рак желудка, рак кости, рак пищевода, рак молочной железы, рак предстательной железы, рак яичка, рак толстой кишки, рак яичников, рак мочевого пузыря, рак шейки матки, меланому, плоско клеточную карциному, базальноклеточную карциному, аденокарциному, карциному потовых желез, карциному сальных желез, папиллярную карциному, папиллярную аденокарциному, кистозную аденокарциному, кистозную карциному, медуллярную карциному, бронхогенную карциному, остеокарциному, эпителиальную карциному, карциному желчного протока, хориокарциному, эмбриональную карциному, семиному, опухоль Вильмса, глиобластому, астроцитому, медуллобластому, краниофарингиому, эпендимому, опухоль шишковидной железы, гемоцитобластому, неврому голосовых складок, менингиому, нейробластому, нейробластому зрительных путей, ретинобластому, нейрофиброму, фибросаркому, фибробластому, фиброму, фиброаденому, фиброхондрому, фиброцистому, фибромиксому, фиброостеому, фибромиксосаркому, фибропапиллому, миксосаркому, миксоцистому, миксохондрому, миксохондросаркому, миксохондрофибросаркому, миксаденому, миксобластому, липосаркому, липому, липоаденому, липобластому, липохондрому, липофиброму, липоангиому, миксолипому, хондросаркому, хондрому, хондромиому, хордому, хориокарциному, хориоэпителиому, хориобластому, остеосаркому, остеобластому, остеохондрофиброму, остеохондросаркому, остеохондрому, остеоцистому, остеодентиному, остеофиброму, фибросаркому кости, ангиосаркому, гемангиому, ангиолипому, ангиохондрому, гемангиобластому, ангиокератому, ангиоглиому, ангиоэндотелиому, ангиофиброму, ангиомиому, ангиолипому, ангиолимфангиому, ангиолиполейомиому, ангиомиолипому, ангиомионеврому, ангиомиксому, ангиоретикулему, лимфангиосаркому, лимфогранулему, лимфангиому, лимфому, лимфомиксому, лимфосаркому, лимфангиофиброму, лимфоцитому, лимфоэпителиому, лимфобластому, эндотелиому, эндобластому, синовиому, синовиальную саркому, мезотелиому, опухоль соединительной ткани, опухоль Юинга, лейомиому, лейомиосаркому, лейомиобластому, лейомиофиброму, рабдомиому, рабдомиосаркому, рабдомиомиксому, острый лимфолейкоз, острый миелогенный лейкоз, анемию хронического заболевания, полицитемию, лимфому, рак эндометрия, глиому, колоректальный рак, рак щитовидной железы, уротелиальный рак или множественную миелому.
15. Способ получения соединения по любому из пп. 1-9, отличающийся тем, что
соединения V и VI подвергают реакции конденсации с образованием замкнутых колец для получения соединений приведенных выше формул II и III:

где Y представляет собой уходящую группу, а остальные переменные являются такими, как определено в п. 1;
предпочтительно, где Y представляет собой Cl, Br, I, -OTs, -ONO2, -OLCMS или -OTf,
и в реакции конденсации в качестве связывающих кислоту веществ используют органические амины;
более предпочтительно, где Y представляет собой Br, и в реакции конденсации в качестве связывающего кислоту вещества используют N,N-диизопропилэтиламин (DIPEA), а в качестве катализатора используют оксид серебра (Ag2O).
16. Способ получения соединения по любому из пп. 1-9, отличающийся тем, что проводят взаимодействие
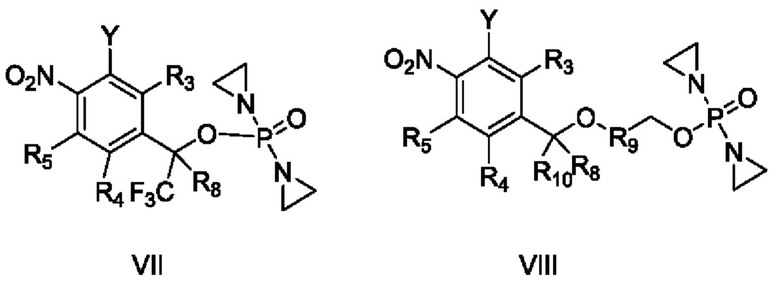
соединения VII с R2R1OH с получением соединения II и проводят взаимодействие соединения VIII с R2R1OH с получением соединения III,
где Y представляет собой уходящую группу.
| WO 2016145092 A1, 15.09.2016 | |||
| WO 2017087428 A1, 26.05.2017 | |||
| ПРОЛЕКАРСТВА ФОСФОРАМИДАТНЫХ АЛКИЛАТОРОВ | 2006 |
|
RU2414475C2 |
| Механизм для сообщения валу качательных движений около его оси | 1927 |
|
SU6852A1 |

