Изобретение относится к фармацевтической и пищевой промышленности и может быть использовано для получения смеси этиловых эфиров йодированных и нейодированных жирных кислот фармацевтического качества, имеющей широкий спектр применения в качестве диагностического и терапевтического агентов, а также в виде пищевых добавок.
В настоящее время проводятся интенсивные исследования в области разработки технологии синтеза смеси эфиров йодированных и нейодированных жирных кислот, обладающих высокой стабильностью и чистотой финального продукта. В Государственной фармакопее Российской Федерации отсутствует фармакопейная статья, касающаяся эфиров йодированных жирных кислот, а среди зарегистрированных лекарственных препаратов есть только сверхжидкий «Липиодол УльтраФлюид» французской компании Гербе (Регистрационное удостоверение П N008677).
Ключевыми показателями качества готового продукта, представляющего собой смесь этиловых эфиров йодированных и нейодированных жирных кислот, являются внешний вид, массовое содержание йода, вязкость и содержание свободного йода. При этом немаловажным фактом является разработка простой и недорогой технологии синтеза, реализуемой в относительно короткий временной интервал.
Известен способ получения йодированных растительных жирных кислот и их эфиров, в соответствии с которым на первом этапе осуществляют омыление триглицеридов жирных кислот растительного масла путем добавления воды и гидроксида натрия или гидроксида калия. Затем проводят йодирование полученных жирных кислот с помощью йодистого водорода, полученного, например, из смеси воды, йодида натрия и треххлористого фосфора с последующей промывкой водой и осушкой сульфатом натрия. Этерификацию полученной смеси йодированных жирных кислот проводят с помощью безводного спирта под действием кислотного катализатора (серной кислоты) [CN101676255A, 2010].
Данный метод, по мнению его авторов, характеризуется низкой стоимостью, простотой получения готового продукта, отличающегося хорошей стабильностью и приемлемой вязкостью. Вместе с тем, в данном методе отсутствуют какие-либо методы очистки конечного продукта, что может привести к наличию в нем нежелательных токсических примесей, образовавшихся при получении йодистоводородной кислоты из йода, воды и треххлористого фосфора, что исключает возможность использования готового продукта в качестве фармацевтического препарата. Кроме того, недостатком данного метода является разделение стадий гидролиза триглицеридов и йодирования, последствием чего является низкая скорость реакции йодирования (до 96 часов).
Известен способ получения этиловых эфиров йодированных жирных кислот растительного происхождения, в соответствии с которым переэтерификацию растительного масла проводят путем обработки макового масла этанолом в присутствии катализатора, например, серной кислоты. В результате получают неочищенный продукт - смесь этиловых эфиров жирных кислот макового масла. Их йодирование проводят путем смешивания с фосфорной кислотой и йодидом щелочного металла (натрия) с последующей промывкой водным раствором тиосульфата натрия. Очистку смеси этиловых эфиров йодированных жирных кислот макового масла проводят путем введения раствора тиосульфата натрия с последующим нагревом до 90°С и продувкой воздухом. В результате получают смесь эфиров йодированных жирных кислот растительного масла с массовым содержанием йода 37,8-38,6% и вязкостью 38-42 сП [CN107287029A, 2017 г.].
Недостатками данного способа являются очистка от свободного йода путем продувки воздухом при нагреве на конечной стадии очистки, которая может привести к термоокислению и получению нежелательных примесей, а также продолжительная процедуры завершающей стадии очистки (58 часов).
Известен способ получения эфира йодированного растительного масла, в котором переэтерификацию растительного масла проводят путем его обработки абсолютным этанолом в присутствии кислотного катализатора (метансульфокислоты или серной кислоты). На следующей стадии проводят йодирование смеси эфиров жирных кислот растительного масла, причем этот процесс проводят в безводной реакционной системе путем добавления безводного йодида натрия с сопутствующим добавлением безводной органической среды (5 мл/г в пересчете на йодид натрия) в среде азота с последующим добавлением безводной кислоты. Экстракцию проводят органическим растворителем. Затем осуществляют промывку тиосульфатом натрия и сушку с использованием безводного сульфата магния с последующим фильтрованием через силикагель. В качестве безводной органической среды может быть использован безводный апротонный органический растворитель, способный растворять йодистый водород, поскольку протонный растворитель может вызывать разложение йодистого водорода. В качестве безводной кислоты могут быть использованы любые кислоты, способные образовывать йодистый водород в соответствующих органических средах. В качестве органического растворителя могут быть использованы неполярные алкановые растворители, такие как петролейный эфир, н-гексан, циклогексан, н-гептан, н-октан и циклооктан [CN112251295A, 2021].
По мнению авторов данного патента, предложенный способ получения смеси эфиров йодированных жирных кислот не требует использования сложного оборудования и характеризуется безопасной работой, однако он имеет следующие недостатки: проведение процедуры йодирования в безводной реакционной системе требует использования более дорогих реактивов и оборудования.
Наиболее близким к заявляемому нами способу является известный способ получения йодированного этилового эфира жирной кислоты, который включает следующие стадии:
1) Переэтерификацию растительного масла под действием этанола в присутствии гидроксида натрия. Избыточный гидроксид натрия и глицерин удаляют добавлением н-гексана и промывкой водой.
2) Протонирование этилового эфира жирной кислоты фосфорной кислотой с образованием протонированного этилового эфира жирной кислоты.
3) Иодирование протонированного этилового эфира жирной кислоты проводят путем добавления йодида щелочного металла к раствору в фосфорной кислоте. По окончании реакции водный слой, содержащий остаточную фосфорную кислоту, экстрагируют н-гексаном и удаляют. Слой н-гексана промывают дистиллированной водой для удаления остаточной фосфорной кислоты и солей щелочного металла с последующим концентрированием при пониженном давлении для удаления н-гексана и получением сырого продукта. В качестве щелочного металла могут быть использованы йодиды калия или натрия.
4) Очистку полученного сырого продукта проводят путем добавления последовательно н-гексана, гидроксида натрия и этанола с дальнейшим добавлением дистиллированной воды до образования водного слоя. Затем добавляют водный раствор тиосульфата натрия для удаления свободного йода. Затем осуществляют промывку дистиллированной водой для удаления остаточных солей в слое н-гексана с последующим концентрированием при пониженном давлении для удаления н-гексана с получением смеси этиловых эфиров йодированных жирных кислот [ЕР3428256 В1, 2019 г.].
Известный способ также предлагает возможность выполнения стадии 1 (переэтерификации) уже после стадий 2 (протонирования) и 3 (йодирования). В данном случае процесс включает в себя следующие стадии:
1. Протонирование триглицеридов жирных кислот фосфорной кислотой с образованием протонированных триглицеридов жирных кислот.
2. Йодирование протонированных триглицеридов йодидом щелочного металла в фосфорной кислоте с получением смеси триглицеридов йодированных жирных кислот.
3. Переэтерификацию триглицеридов йодированных жирных кислот с образованием смеси этиловых эфиров йодированных жирных кислот.
4. Удаление остаточного йода.
К недостаткам данных способов можно отнести то, что йодирование в приведенных условиях неизбежно сопровождается гидролизом триглицеридов, что приводит к усложнению реакционной смеси и, как следствие, затрудняет контроль ее состава. Также эти способы не позволяют провести очистку получаемого продукта от парафинов и других нежелательных веществ, что не позволяет обеспечить гарантированное качество продукта.
Также в известном решении не оговорено использование инертных газов для защиты реакционных сред и продуктов, что также делает сомнительным получение данным способом продукта требуемого качества.
Задачей, на решение которой направлено заявленное изобретение, является получение смеси эфиров йодированных и нейодированных жирных кислот фармацевтического качества, обладающего высокой чистотой и соответствующего ключевым показателями качества (внешний вид, массовое содержание йода, рентгеноконтрастность, вязкость), при одновременном упрощении технологии и снижении стоимости синтеза этих эфиров.
Технический результат достигается тем, что в способе синтеза смеси этиловых эфиров йодированных и нейодированных жирных кислот из растительного масла, заключающийся в проведении нескольких стадий обработки растительного масла, полученного методом холодного отжима или методом сверхкритической CO2-экстракции, на первой стадии йодирование-гидролиз триглицеридов непредельных жирных кислот проводят в однореакторном режиме путем эмульгирования растительного масла в фосфорной кислоте, удаления из реакционной смеси и реакторного пространства кислорода, загрузки при температуре 0°С йодида щелочного металла и последующем повышении температуры до 60-100°С и перемешивании смеси в течение 20-24 часов, после чего производят стадию очистки полученной смеси от остаточного йода путем отделения органического слоя от воды и его обработки водным раствором тиосульфата натрия, далее проводят стадию удаления балластных веществ некислотного характера путем нейтрализации органического слоя водным раствором гидроксида щелочного металла с получением смеси солей щелочного металла йодированных и нейодированных жирных кислот с последующей экстракцией органическим растворителем балластных веществ, после чего соли йодированных и нейодированных жирных кислот возвращают в кислотную форму путем их обработки раствором минеральной кислоты, и далее проводят реакцию этерификации смеси очищенных солей йодированных и нейодированных жирных кислот под действием этилового спирта в присутствии минеральной кислоты, затем производят финишную очистку полученной смеси этиловых эфиров йодированных и нейодированных жирных кислот от примесей смолистых и других нежелательных веществ методом флэш-хроматографии на силикагеле с применением в качестве элюента смеси органических растворителей, удаляемых впоследствии вакуумной перегонкой при температуре не выше 40°С.
Кроме того, в изобретении может иметь место следующее:
- в качестве растительного масла используют следующие его виды: масло семян мака, масло виноградных косточек, масло грецкого ореха и другие растительные масла, содержащие смесь триглицеридов непредельных и предельных жирных кислот, таких как пальмитолеиновая (C16:1), стеариновая (C18:0), олеиновая (C18:1), линолевая (C18:2), линоленовая (С18:3), гондоиновая (C20:1), эйкозадиеновая (C20:2), эруковая (C22:1), докозадиеновая (С22:2), селахолевая (C24:1), бегеновая (С22:0), пальмитиновая (C16:0), арахиновая (C20:0) и лигноцериновая (С24:0);
- после этерификации и перед стадией финишной очистки проводят дополнительную очистку смеси от свободного остаточного йода обработкой водным раствором тиосульфата натрия, после чего отделяют водный слой и производят осушку органического слоя его пропусканием через безводный сульфат натрия;
- водные растворы тиосульфата натрия используют с концентрациями от 1 до 10% масс;
- в качестве минеральной кислоты, используемой при обработке солей йодированных и нейодированных жирных кислот с целью возвращения их в кислотную форму, используют серную, фосфорную или соляную кислоту с доведением рН раствора до 3,0-3,5;
- реакцию этерификации проводят в течение 2 часов при перемешивании средней интенсивности и температуре 60-90°С;
- для метода флэш-хроматографии используют силикагель в стерильных одноразовых картриджах с предпочтительным размером частиц 40-100 мкм, а в качестве органических растворителей используют смесь петролейного эфира фракции 40-70°С с хлористым метиленом, хлороформом или диэтиловым эфиром.
Проведение на первой стадии обработки масла в одном реакторе йодирования - гидролиза триглицеридов непредельных жирных кислот без разделения их на отдельные процессы позволяет сократить технологическую схему и время процесса синтеза смеси эфиров йодированных и нейодированных жирных кислот.
При продолжительности процесса меньше 20 часов гидролиз триглицеридов жирных кислот (как йодированных, так и нейодированных) будет неполным, что приводит к снижению выхода жирных кислот, а увеличение продолжительности свыше 24 часов не приводит к дальнейшему увеличению их выхода.
Все стадии синтеза проводятся на стандартном наборе оборудования: емкостного аппарата с мешалкой и рубашкой для нагрева/охлаждения; делительных воронок; нутч- или дрк-нутч-фильтров; ротационного вакуумного испарителя; картриджей, заполненных силикагелем для флэш-хроматографии не требует использования какого-либо специального оборудования, в том числе и вентиляционного, за счет отсутствия необходимости работы с легкоокисляющимися йодистым водородом и треххлористым фосфором, который к тому же еще и легко гидролизуется на воздухе.
Повышение качества конечного продукта в предлагаемом способе достигается очисткой от балластных веществ некислотного характера после стадии йодирования-гидролиза растительного масла.
Проведение дополнительной очистки смеси от свободного остаточного йода после этерификации и перед стадией ее финишной очистки позволяет снизить содержание свободного йода в органическом продукте до концентрации не выше 0,05% масс, а содержание влаги - до 0,04-0,06% масс.
Проведение финишной очистки полученной смеси этиловых эфиров йодированных и нейодированных жирных кислот от примесей смолистых и других нежелательных веществ методом флэш-хроматографии на силикагеле с применением в качестве элюента смеси органических растворителей повышает качество получаемого продукта.
В предлагаемом способе получения стабильной смеси эфиров йодированных и нейодированных жирных кислот в качестве сырья может быть использовано масло семян мака, масло виноградных косточек, масло грецкого ореха и другие растительные масла, полученные методом холодного отжима и содержащие смесь триглицеридов непредельных и предельных жирных кислот, таких как пальмитолеиновая (C16:1), стеариновая (C18:0), олеиновая (C18:1), линолевая (C18:2), линоленовая (C18:3), гондоиновая (C20:1), эйкозадиеновая (C20:2), эруковая (C22:1), докозадиеновая (С22:2), селахолевая (C24:1), бегеновая (С22:0), пальмитиновая (C16:0), арахиновая (C20:0) и лигноцериновая (С24:0).
Изобретение поясняется графически, где:
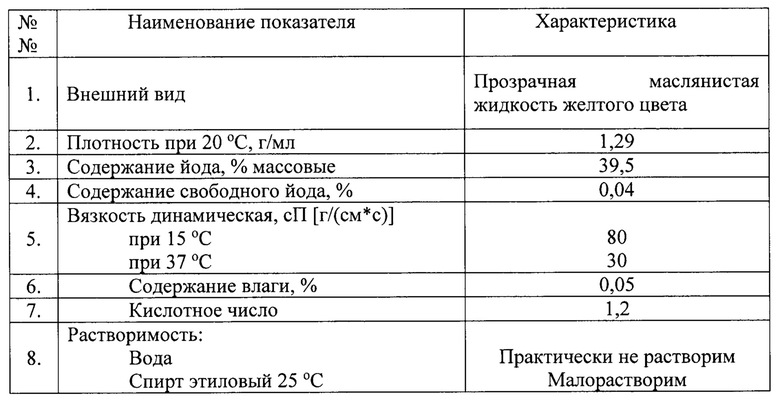
- на фиг. 1 изображен инфракрасный спектр смеси этиловых эфиров йодированных и нейодированных жирных кислот при использовании в качестве сырья масла семян мака, полученной в результате использования предлагаемого способа синтеза;
- на фиг. 2 изображен инфракрасный спектр смеси этиловых эфиров йодированных и нейодированных жирных кислот препарата «Липиодол Ультра-Флюид»;
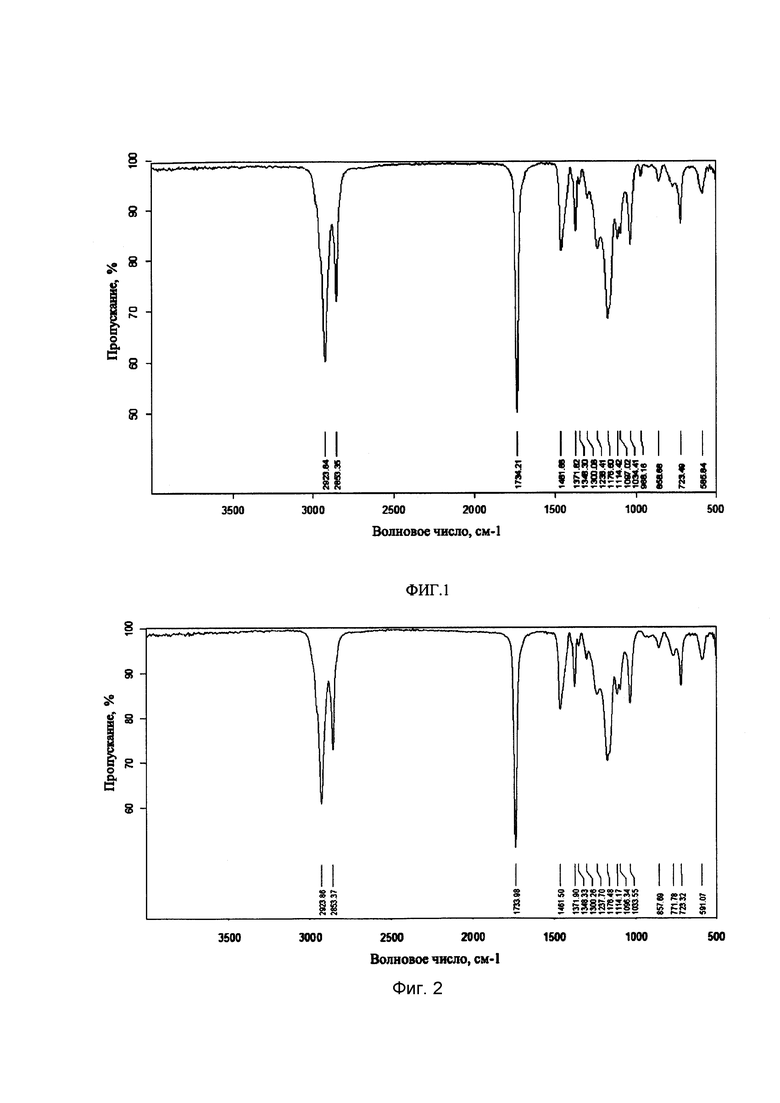
- на фиг. 3 изображен спектр ядерного магнитного резонанса на ядрах 1Н смеси этиловых эфиров йодированных и нейодированных жирных кислот при использовании в качестве сырья масла семян мака, полученной в результате использования предлагаемого способа;
- на фиг. 4 изображен спектр ядерного магнитного резонанса на ядрах 1Н смеси этиловых эфиров йодированных и нейодированных жирных кислот препарата «Липиодол Ультра-Флюид»;
Способ получения стабильной смеси этиловых эфиров йодированных и нейодированных жирных кислот заключается в обработке растительного масла, включающей в себя следующие технологические стадии:
- Стадия йодирования - гидролиза триглицеридов непредельных жирных кислот, которую осуществляют в реакторе путем эмульгирования растительного масла в фосфорной кислоте, удаления из реакционной смеси и реакторного пространства кислорода и продувки инертным газом, в качестве которого может быть использован, например, аргон или азот, в атмосфере которого и выполняется проведение всей этой стадии, после чего производят загрузку йодида щелочного металла (предпочтительно йодида натрия или йодида калия) в твердом виде, причем загрузку йодида щелочного металла осуществляют при температуре около 0°С при продувке инертным газом (предпочтительно азотом) при перемешивании, не нарушающем состояние эмульсии. После завершения загрузки йодида металла начинают повышать температуру до 60-100°С (предпочтительно 75-85°С), при этом осуществляют перемешивание, не нарушающее состояние эмульсии, и сохраняют инертную атмосферу. Процесс продолжают в течение 12-24 часов.
Количество необходимого для проведения реакции йодирования йодида щелочного металла определяют по результатам определения йодного числа исходного растительного масла. Избыток щелочного металла, используемого для йодирования, не должен превышать более чем на 10% количество йода по йодному числу, представляющего собой условную величину, характеризующую содержание в 100 г растительного масла непредельных соединений, выраженную в граммах йода, эквивалентного состоящему из галогенов реагенту, присоединившемуся к маслу;
- Стадия очистки полученной на предыдущей стадии реакционной смеси от остаточного йода, которую проводят последовательно путем разделения слоев эмульсии, отделения водного слоя от органического (содержащего целевой продукт) и обработкой последнего водным раствором тиосульфата натрия. При этом используют водные растворы тиосульфата натрия с концентрациями от 1 до 10% масс. (предпочтительно 5% масс).
- Стадия удаления балластных веществ некислотного характера, присутствующих в исходном масле, путем нейтрализации водным раствором гидроксида щелочного металла, например, гидроксида натрия или гидроксида калия, органического слоя с получением смеси солей щелочного металла йодированных и нейодированных жирных кислот, после чего выполняют экстракцию органическим растворителем балластных веществ. Далее соли йодированных и нейодированных жирных кислот возвращают в кислотную форму путем их обработки раствором минеральной кислоты, например, серной, фосфорной или соляной (предпочтительно, серной кислотой), и доведения рН раствора до 3-3,5. Все процессы выполняют при комнатной температуре. Суммарное время этой стадии составляет 1-2 часа.
- Стадия этерификации смеси очищенных солей йодированных и нейодированных жирных кислот, полученной на предыдущей стадии, которую выполняют под действием абсолютированного этилового спирта в присутствии минеральной кислоты (например, серной, фосфорной или соляной; предпочтительно серной). Реакцию проводят в течение 2 часов при перемешивании средней интенсивности и температуре 60-90°С (предпочтительно при 80°С).
- Стадия финишной очистки от примесей смолистых и других нежелательных веществ, присутствующих в количествах, не превышающих 0,01-0,5% масс., но имеющих склонность к нарушению стабильности полученного продукта при хранении, при которой используют метод очистки - флэш-хроматография на силикагеле с применением в качестве элюента смеси органических растворителей удаляемых впоследствии вакуумной перегонкой при температуре не выше 40°С с получением готового продукта, поскольку более высокая температура отгонки приводит к снижению качества продукта за счет образования окрашенных продуктов разрушения. Предпочтительно использование силикагеля в стерильных одноразовых картриджах с размером частиц 40-100 мкм. В качестве органических растворителей могут быть использованы петролейный эфир, хлористый метилен, хлороформ, этиловый эфир; предпочтительна для использования смесь петролейного эфира фракции 40-70°С и хлористого метилена.
Между стадией этерификации и стадией финишной очистки возможна стадия дополнительной очистки от свободного остаточного йода обработкой водным раствором тиосульфата натрия, с концентрациями от 1 до 10% масс, (предпочтительно 5% масс, добавление водного раствора тиосульфата натрия продолжают до прекращения обесцвечивания органического слоя), отделение водного слоя; осушку полученного масла пропусканием через безводный сульфат натрия, что позволяет снизить в органическом продукте содержание свободного йода до концентрации не выше 0,05% масс, а содержание влаги - до 0,04 - 0,06% масс.
В результате проведения всех вышеперечисленных стадий обработки растительного масла конечный продукт имеет следующие физико-химические характеристики:
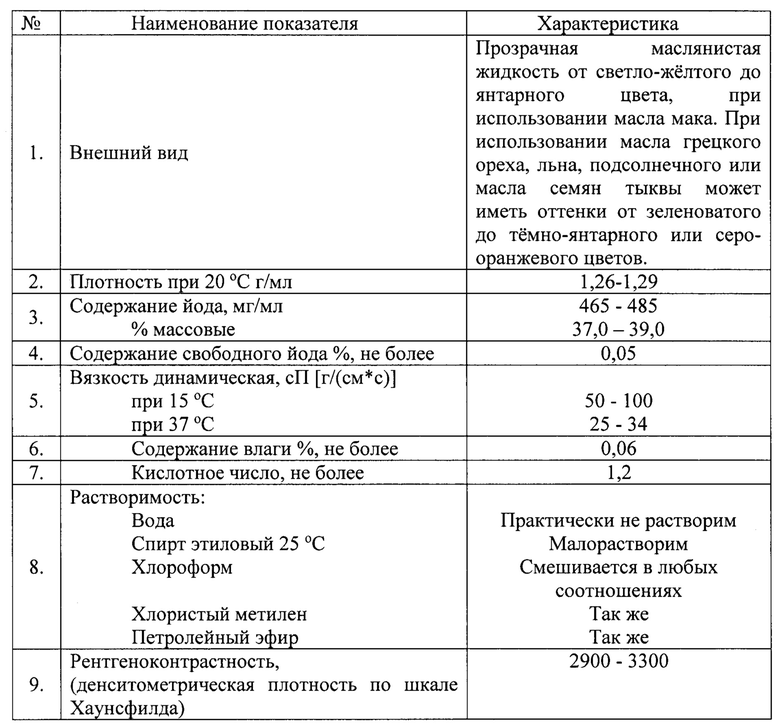
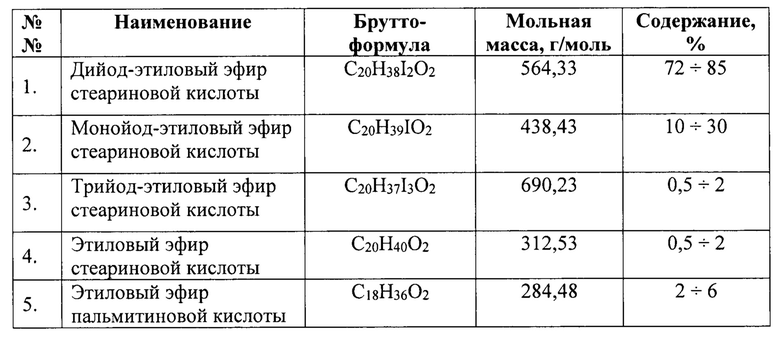
Конечный продукт, получаемый предлагаемым способом из растительного масла, имеет следующий химический состав (по основным жирно-кислотным компонентам, массовое содержание которых превышает 1%).
Полученный продукт имеет фармацевтическое качество и соответствует требованиям, предъявляемым к ренттгеноконтрастным средствам, препаратам, применяемым в терапевтических целях при лечении болезней, связанных с дефицитом йода, и носителям жирорастворимых лекарственных средств.
При выполнении всех перечисленных выше условий проведения синтеза может быть получен продукт, представляющий собой смесь эфиров йодированных и нейодированных жирных кислот обозначенного выше качества с выходом не менее 55%.
Предлагаемый способ получения смеси эфиров йодированных жирных кислот с использованием в качестве сырья масла семян мака позволит удовлетворить потребности практической медицины Российской Федерации и осуществить замену единственного аналогичного зарегистрированного лекарственного препарата, сверхжидкого «Липиодола Ультра-Флюид» французской компании Гербе (Регистрационное удостоверение П N008677) отечественным, более дешевым и доступным полным аналогом.
На фиг. 1 и 2 приведены для сравнения инфракрасные спектры смеси этиловых эфиров йодированных и нейодированных жирных кислот, полученной в результате использования предлагаемого способа при использовании в качестве сырья масла семян мака, и аналогичный инфракрасный спектр препарата «Липиодол Ультра-Флюид», соответственно. Как видно из сравнения этих спектров, характерные полосы поглощения практически полностью совпадают, что свидетельствует об идентичности состава продуктов (на уровне «отпечатков пальцев»).
Представленный на фиг. 3 спектр ядерного магнитного резонанса на ядрах 1Н смеси этиловых эфиров йодированных и нейодированных жирных кислот, полученной в результате использования предлагаемого способа, при использовании в качестве сырья масла семян мака, в сравнении с представленными на фиг. 4 аналогичным спектром ядерного магнитного резонанса на ядрах 1Н смеси этиловых эфиров йодированных и нейодированных жирных кислот препарата «Липиодол Ультра-Флюид» показывают их подобие по всей ширине спектров, что также подтверждает идентичность продуктов.
Ниже приведены практические примеры реализации способа синтеза смеси эфиров йодированных и нейодированных жирных кислот при использовании в качестве сырья масла, содержащего смесь предельных и непредельных жирных кислот С16-С24.
Пример 1
В качестве исходного сырья использовано масло мака производства Турции, полученное по технологии холодного отжима.
В реактор объемом 5000 мл при комнатной температуре загружают фосфорную кислоту (1700 мл), включают перемешивание и через капельную воронку загружают товарное масло семян мака производства Турции (220 г). Смесь нагревают при перемешивании до 80°С и продолжают перемешивание в течение 30 мин. Реакционную смесь охлаждают до комнатной температуры, реактор продувают азотом и в токе азота разом загружают йодистый калий (222 г). Перемешивание продолжают 10-30 минут до получения однородной реакционной смеси и включают нагрев. По достижении температуры 80°С перемешивание продолжают еще 23-24 часа.
Затем нагрев и перемешивание отключают, дают реакционной массе охладиться до комнатной температуры и отстояться и сливают нижний кислотный слой. Кислотный слой экстрагируют петролейным эфиром фракции 40-70°С (3*1600 мл). Экстракт объединяют с органическим слоем и осушают сульфатом натрия (100-150 г) при периодическом перемешивании.
Осушенный фильтрат отделяют и обрабатывают 10% водным раствором тиосульфата натрия (200-300 мл) для удаления свободного йода. Органический слой отделяют и сушат безводным сульфатом натрия (100-150 г). Очищенный и осушенный экстракт упаривают при пониженном давлении досуха.
К остатку (294 г) при перемешивании разом добавляют раствор гидроксида натрия (36 г) в дистиллированной воде (520 мл). Осуществляют медленное перемешивание в течение 0,5 часа. К смеси добавляют петролейный эфир фракции 40-70°С (1400 мл) и продолжают медленное перемешивание органического слоя смеси, не допуская образования эмульсии, в течение 5 часов. Органический аморфный слой, содержащий примесные компоненты, отделяют. Процесс очистки повторяют трижды. Органические слои после экстракций объединяют, растворитель отгоняют при пониженном давлении, при этом получают кубовый остаток аморфного вещества светло-желтого цвета (12 г).
К водному слою при перемешивании добавляют водный 30% раствор серной кислоты до рН меньше 7, при этом происходит разделение на две фазы - темную легкую органическую и светлую тяжелую водную. Водную фазу сливают и подвергают экстракции петролейным эфиром фракции 40-70 (2*300 - 400 мл). Органическую фазу и экстракты объединяют, сушат безводным сульфатом натрия, отгоняют петролейный эфир досуха и получают очищенную смесь йодированных и нейодированных кислот в виде темного маслянистого продукта (282,3 г).
Очищенную смесь йодированных и нейодированных кислот (282,3 г) смешивают с абсолютированным этанолом (320 мл), затем добавляют серную кислоту (31,8 г). Реакционную смесь при перемешивании нагревают до 80°С и выдерживают 2 часа. После охлаждения летучие компоненты удаляют при пониженном давлении, маслянистый остаток подвергают экстракции петролейным эфиром фракции 40-70°С (последовательно 800, 500 и 400 мл). Затем все фракции объединяют, упаривают при пониженном давлении досуха и получают техническую смесь этиловых эфиров йодированных и нейодированных жирных кислот (348 г).
Очистку полученного технического продукта проводят методом флэш-хроматографии, используя колонку, заполненную силикагелем. Сырой продукт наносят на силикагель в виде раствора в элюенте (140 мл, состава: петролейный эфир фракции 40-70°С + хлористый метилен = 1+2) и проводят элюирование. Порции элюата, содержащие продукт (3360 мл), объединяют и упаривают досуха с получением товарного продукта, смеси этиловых эфиров йодированных и нейодированных жирных кислот (261 г, условный теоретический выход 75%). Полученный продукт имеет следующие характеристики.

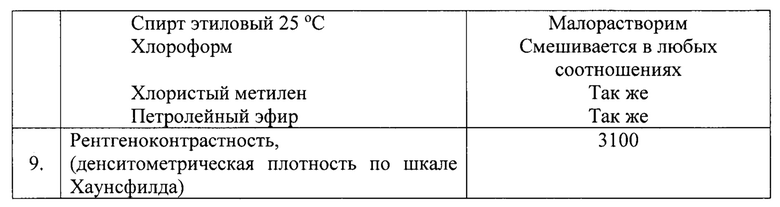
Пример 2
В качестве исходного сырья использовано масло семян мака производства Болгарии, полученное по технологии холодного отжима.
В реактор объемом 5000 мл при комнатной температуре загружают фосфорную кислоту (1700 мл), включают перемешивание и через капельную воронку загружают товарное масло семян мака производства Болгарии (220 г). Смесь нагревают при перемешивании до 80°С и продолжают перемешивание в течение 30 мин. Реакционную смесь охлаждают до комнатной температуры, реактор продувают азотом и в токе азота разом загружают йодистый калий (222 г). Перемешивание продолжают 10-30 минут до получения однородной реакционной смеси и включают нагрев. По достижении температуры 80°С перемешивание продолжают еще 23-24 часа.
Затем нагрев и перемешивание отключают, дают реакционной массе охладиться до комнатной температуры и отстояться и сливают нижний кислотный слой. Кислотный слой экстрагируют петролейным эфиром фракции 40-70°С (3*1600 мл). Экстракт объединяют с органическим слоем и осушают сульфатом натрия (100-150 г) при периодическом перемешивании.
Осушенный фильтрат отделяют и обрабатывают 10% водным раствором тиосульфата натрия (200-300 мл) для удаления свободного йода. Органический слой отделяют и сушат безводным сульфатом натрия (100-150 г). Очищенный и осушенный экстракт упаривают при пониженном давлении досуха.
К остатку (330 г) при перемешивании разом добавляют раствор гидроксида натрия (36 г) в дистиллированной воде (520 мл). Осуществляют медленное перемешивание в течение 0,5 часа. К смеси добавляют петролейный эфир фракции 40-70°С (1500 мл) и продолжают медленное перемешивание органического слоя смеси, не допуская образования эмульсии, в течение 5 часов. Органический аморфный слой, содержащий примесные компоненты, отделяют. Процесс очистки повторяют трижды. Органические слои после экстракций объединяют, растворитель отгоняют при пониженном давлении, при этом получают кубовый остаток аморфного вещества светло-желтого цвета (101 г).
К водному слою при перемешивании добавляют водный 30% раствор серной кислоты до рН меньше 7, при этом происходит разделение на две фазы - темную легкую органическую и светлую тяжелую водную. Водную фазу сливают и подвергают экстракции петролейным эфиром фракции 40-70°С (2*300 - 400 мл). Органическую фазу и экстракты объединяют, сушат безводным сульфатом натрия, отгоняют петролейный эфир досуха и получают очищенную смесь йодированных и нейодированных кислот в виде темного маслянистого продукта (192 г).
Очищенную смесь йодированных и нейодированных кислот (192 г) смешивают с абсолютированным этанолом (217 мл), затем добавляют серную кислоту (21,6 г). Реакционную смесь при перемешивании нагревают до 80°С и выдерживают 2 часа. После охлаждения летучие компоненты удаляют при пониженном давлении, маслянистый остаток подвергают экстракции петролейным эфиром фракции 40-70°С (последовательно 550, 350 и 250 мл). Затем все фракции объединяют, упаривают при пониженном давлении досуха и получают техническую смесь этиловых эфиров йодированных и нейодированных жирных кислот (246 г).
Очистку полученного технического продукта проводят методом флэш-хроматографии, используя колонку, заполненную силикагелем. Сырой продукт наносят на силикагель в виде раствора в элюенте (100 мл, состава: петролейный эфир фракции 40-70°С + хлористый метилен, 1+2) и проводят элюирование. Порции элюата, содержащие продукт (2155 мл), объединяют и упаривают досуха с получением товарного продукта, смеси этиловых эфиров йодированных и нейодированных жирных кислот (233 г, условный теоретический выход 55%). Полученный продукт имеет следующие характеристики:
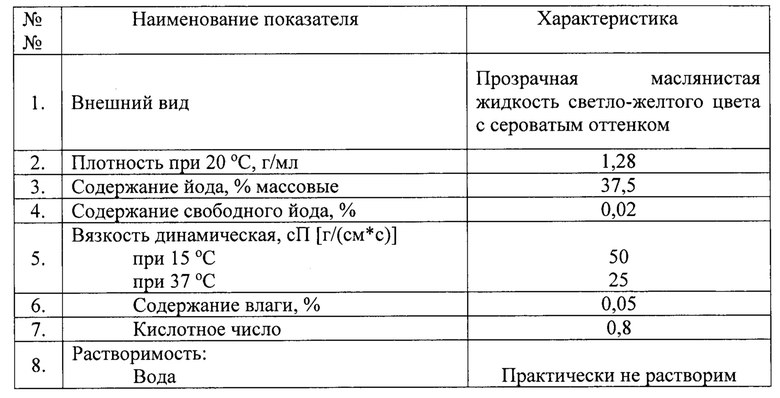

Пример 3
В качестве исходного сырья использовали масло семян виноградных косточек производства республики Дагестан, полученное методом сверхкритической СО2-экстракции.
В реактор объемом 5000 мл при комнатной температуре загружают фосфорную кислоту (1700 мл), включают перемешивание и через капельную воронку загружают товарное масло из виноградных косточек (220 г). Смесь нагревают при перемешивании до 80°С и продолжают перемешивание в течение 30 мин. Реакционную смесь охлаждают до комнатной температуры, реактор продувают азотом и в токе азота разом загружают йодистый калий (222 г). Перемешивание продолжают 10-30 минут до получения однородной реакционной смеси и включают нагрев. По достижении температуры 80°С перемешивание продолжают еще 23-24 часа.
Затем нагрев и перемешивание отключают, дают реакционной массе охладиться до комнатной температуры и отстояться и сливают нижний кислотный слой. Кислотный слой экстрагируют петролейным эфиром фракции 40-70°С (3*1600 мл). Экстракт объединяют с органическим слоем и осушают сульфатом натрия (100-150 г) при периодическом перемешивании.
Осушенный фильтрат отделяют и обрабатывают 10% водным раствором тиосульфата натрия (200-300 мл) для удаления свободного йода. Органический слой отделяют и сушат безводным сульфатом натрия (100-150 г). Очищенный и осушенный экстракт упаривают при пониженном давлении досуха.
К остатку (290 г) при перемешивании разом добавляют раствор гидроксида натрия (32 г) в дистиллированной воде (460 мл). Осуществляют медленное перемешивание в течение 0,5 часа. К смеси добавляют петролейный эфир фракции 40-70 (1300 мл) и продолжают медленное перемешивание органического слоя смеси, не допуская образования эмульсии, в течение 5 часов. Органический аморфный слой, содержащий примесные компоненты, отделяют. Процесс очистки повторяют трижды. Органические слои после экстракций объединяют, растворитель отгоняют при пониженном давлении, при этом получают кубовый остаток аморфного вещества светло-желтого цвета (62 г).
К водному слою при перемешивании добавляют водный 30% раствор серной кислоты до рН меньше 7, при этом происходит разделение на две фазы - темную легкую органическую и светлую тяжелую водную. Водную фазу сливают и подвергают экстракции петролейным эфиром фракции 40-70°С (2*300 - 400 мл). Органическую фазу и экстракты объединяют, сушат безводным сульфатом натрия, отгоняют петролейный эфир досуха и получают очищенную смесь йодированных и нейодированных кислот в виде темного маслянистого продукта (220 г).
Очищенную смесь йодированных и нейодированных кислот (220 г) смешивают с абсолютированным этанолом (250 мл), затем добавляют серную кислоту (24,7 г). Реакционную смесь при перемешивании нагревают до 80°С и выдерживают 2 часа. После охлаждения летучие компоненты удаляют при пониженном давлении, маслянистый остаток подвергают экстракции петролейным эфиром фракции 40-70°С (последовательно 630, 400 и 290 мл). Затем все фракции объединяют, упаривают при пониженном давлении досуха и получают техническую смесь этиловых эфиров йодированных и нейодированных жирных кислот (281 г).
Очистку полученного технического продукта проводят методом флэш-хроматографии, используя колонку, заполненную силикагелем. Сырой продукт наносят на силикагель в виде раствора в элюенте (115 мл, состава: петролейный эфир фракции 40-70°С + хлористый метилен = 1+2) и проводят элюирование. Порции элюата, содержащие продукт (2500 мл), объединяют и упаривают досуха с получением товарного продукта, смеси этиловых эфиров йодированных и нейодированных жирных кислот (267 г, условный теоретический выход 63%). Полученный продукт имеет следующие характеристики.

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ЛИНОЙОДОЛА | 2014 |
|
RU2558037C1 |
| СПОСОБ ПРОФИЛАКТИКИ ПОРАЖЕНИЯ РАДИОНУКЛИДАМИ ЙОДА И ОПТИМИЗАЦИИ ЙОДНОГО ОБМЕНА В ПОСТПРОФИЛАКТИЧЕСКИЙ ПЕРИОД | 2002 |
|
RU2323733C2 |
| Способ получения арахидоновой кислоты | 2016 |
|
RU2627273C1 |
| СПОСОБ ИЗВЛЕЧЕНИЯ ЙОДА ИЗ ВОДНЫХ РАСТВОРОВ | 2018 |
|
RU2771817C2 |
| Способ получения докозагексаеновой, эйкозапентаеновой и арахидоновой кислот или их смеси | 1988 |
|
SU1631067A1 |
| НАНОСТРУКТУРИРОВАННАЯ ЙОДОСОДЕРЖАЩАЯ БИОЛОГИЧЕСКИ АКТИВНАЯ ДОБАВКА К ПИЩЕ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2013 |
|
RU2536699C1 |
| БИОЛОГИЧЕСКИ АКТИВНАЯ КОРМОВАЯ ДОБАВКА ДЛЯ ПРОФИЛАКТИКИ ЙОДНОЙ НЕДОСТАТОЧНОСТИ И ОПТИМИЗАЦИИ ЙОДНОГО ОБМЕНА И КОРМОВОЙ ПРОДУКТ, ЕЕ СОДЕРЖАЩИЙ | 2002 |
|
RU2212818C1 |
| СПОСОБ ПОЛУЧЕНИЯ 1-ФЕНИЛ-2,3-ДИМЕТИЛ-4-ИОДПИРАЗОЛОНА-5 (ЙОДАНТИПИРИНА) | 2009 |
|
RU2401830C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДОКОЗАГЕКСАЕНОВОЙ КИСЛОТЫ | 2013 |
|
RU2537252C1 |
| БИОЛОГИЧЕСКИ АКТИВНАЯ ДОБАВКА К ПИЩЕ ДЛЯ ПРОФИЛАКТИКИ ЙОДНОЙ НЕДОСТАТОЧНОСТИ И ОПТИМИЗАЦИИ ЙОДНОГО ОБМЕНА И ПИЩЕВОЙ ПРОДУКТ, ЕЕ СОДЕРЖАЩИЙ | 2001 |
|
RU2192150C1 |
Изобретение относится к фармацевтической и пищевой промышленности и может быть использовано для получения смеси этиловых эфиров йодированных и нейодированных жирных кислот фармацевтического качества, имеющей широкий спектр применения в качестве диагностического и терапевтического агентов, а также в виде пищевых добавок. Предложен способ синтеза смеси этиловых эфиров йодированных и нейодированных жирных кислот из растительного масла, заключающийся в проведении нескольких стадий обработки растительного масла, полученного методом холодного отжима или методом сверхкритической СО2-экстракции. На первой стадии йодирование-гидролиз триглицеридов непредельных жирных кислот проводят в однореакторном режиме путем эмульгирования растительного масла в фосфорной кислоте, удаления из реакционной смеси и реакторного пространства кислорода, загрузки при температуре 0°С йодида щелочного металла и последующем повышении температуры до 60-100°С и перемешивании смеси в течение 20-24 часов. После этого производят стадию очистки полученной смеси от остаточного йода путем отделения органического слоя от воды и его обработки водным раствором тиосульфата натрия. Далее проводят стадию удаления балластных веществ некислотного характера путем нейтрализации органического слоя водным раствором гидроксида щелочного металла с получением смеси солей щелочного металла йодированных и нейодированных жирных кислот с последующей экстракцией органическим растворителем балластных веществ. Затем соли йодированных и нейодированных жирных кислот возвращают в кислотную форму путем их обработки раствором минеральной кислоты и далее проводят реакцию этерификации смеси очищенных солей йодированных и нейодированных жирных кислот под действием этилового спирта в присутствии минеральной кислоты. После чего производят финишную очистку полученной смеси этиловых эфиров йодированных и нейодированных жирных кислот от примесей смолистых и других нежелательных веществ методом флэш-хроматографии на силикагеле с применением в качестве элюента смеси органических растворителей, удаляемых впоследствии вакуумной перегонкой при температуре не выше 40°С. Технический результат - получение смеси эфиров йодированных и нейодированных жирных кислот фармацевтического качества, обладающей высокой чистотой и соответствующей ключевым показателям качества, а именно: внешний вид, массовое содержание йода, рентгеноконтрастность, вязкость, при одновременном упрощении технологии и снижении стоимости синтеза этих эфиров. 7 з.п. ф-лы, 4 ил., 3 пр.
1. Способ синтеза смеси этиловых эфиров йодированных и нейодированных жирных кислот из растительного масла, заключающийся в проведении нескольких стадий обработки растительного масла, полученного методом холодного отжима или методом сверхкритической СО2-экстракции, отличающийся тем, что на первой стадии йодирование-гидролиз триглицеридов непредельных жирных кислот проводят в однореакторном режиме путем эмульгирования растительного масла в фосфорной кислоте, удаления из реакционной смеси и реакторного пространства кислорода, загрузки при температуре 0°С йодида щелочного металла и последующем повышении температуры до 60-100°С и перемешивании смеси в течение 20-24 часов, после чего производят стадию очистки полученной смеси от остаточного йода путем отделения органического слоя от воды и его обработки водным раствором тиосульфата натрия, далее проводят стадию удаления балластных веществ некислотного характера путем нейтрализации органического слоя водным раствором гидроксида щелочного металла с получением смеси солей щелочного металла йодированных и нейодированных жирных кислот с последующей экстракцией органическим растворителем балластных веществ, после чего соли йодированных и нейодированных жирных кислот возвращают в кислотную форму путем их обработки раствором минеральной кислоты, и далее проводят реакцию этерификации смеси очищенных солей йодированных и нейодированных жирных кислот под действием этилового спирта в присутствии минеральной кислоты, после чего производят финишную очистку полученной смеси этиловых эфиров йодированных и нейодированных жирных кислот от примесей смолистых и других нежелательных веществ методом флэш-хроматографии на силикагеле с применением в качестве элюента смеси органических растворителей, удаляемых впоследствии вакуумной перегонкой при температуре не выше 40°С.
2. Способ по п. 1, отличающийся тем, что в качестве растительного масла используют следующие его виды: масло семян мака, масло виноградных косточек, масло грецкого ореха и другие растительные масла, содержащие смесь триглицеридов непредельных и предельных жирных кислот, таких как пальмитолеиновая (C16:1), стеариновая (C18:0), олеиновая (C18:1), линолевая (С18:2), линоленовая (С18:3), гондоиновая (С20:1), эйкозадиеновая (С20:2), эруковая (C22:1), докозадиеновая (С22:2), селахолевая (C24:1), бегеновая (С22:0), пальмитиновая (C16:0), арахиновая (С20:0) и лигноцериновая (С24:0).
3. Способ по п. 1, отличающийся тем, что после этерификации и перед стадией финишной очистки проводят дополнительную очистку от свободного остаточного йода обработкой водным раствором тиосульфата натрия, после чего отделяют водный слой и производят осушку органического слоя его пропусканием через безводный сульфат натрия
4. Способ по п. 1 или 3, отличающийся тем, что используют водные растворы тиосульфата натрия с концентрациями от 1 до 10% масс.
5. Способ по п. 1, отличающийся тем, что в качестве минеральной кислоты, используемой при обработке солей йодированных и нейодированных жирных кислот с целью возвращения их в кислотную форму, используют серную, фосфорную или соляную кислоту с доведением рН раствора до 3-3,5.
6. Способ по п. 1, отличающийся тем, что реакцию этерификации проводят в течение 2 часов при перемешивании средней интенсивности и температуре 60-90°С.
7. Способ по п. 1, отличающийся тем, что для метода флэш-хроматографии используют силикагель в стерильных одноразовых картриджах с предпочтительным размером частиц 40-100 мкм, а в качестве органических растворителей используют смесь петролейного эфира с хлористым метиленом, хлороформом или диэтиловым эфиром.
8. Способ по п. 7, отличающийся тем, что используют петролейный эфир фракции 40-70°С.
| EP 3428256 B1, 01.09.2021 | |||
| CN 0111187162 A, 22.05.2020 | |||
| CN 103045373 A, 17.04.2013 | |||
| GB 191019350 A, 22.12.1910 | |||
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ «-ЙОДПЕРФТОРМОНОКАРБОНОВЫХ КИСЛОТ | 0 |
|
SU322983A1 |

