В настоящем изобретении предложено соединение формулы (I), которое представляет собой ингибитор BRAF и имеет свойства «разрушения парадокса», его производство, фармацевтические композиции, содержащие его, и его применение в качестве терапевтически активного вещества.
В настоящем изобретении предложено новое соединение формулы (I)
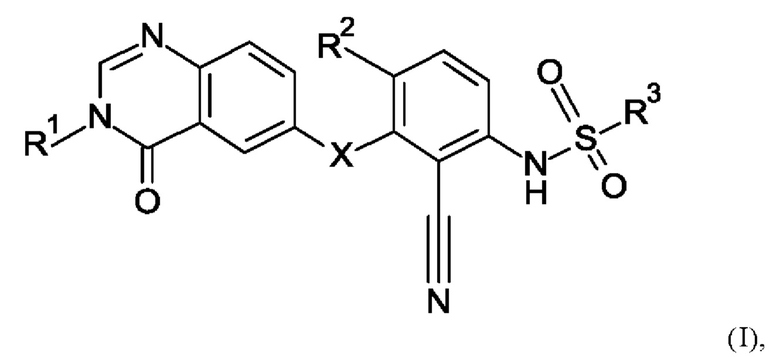
R1 представляет собой C1-6-алкил;
X выбран из
i) -NH- и
ii) -О-;
R2 выбран из
iii) Н,
iv) циано и
v) галогена;
R3 выбран из
vi) NR4R5 и
vii) CHR6R7;
R4 выбран из
viii) C1-6-алкила,
ix) С3-8-циклоалкила и
x) С3-8-циклоалкил-C1-6-алкила;
R5 выбран из
xi) C1-6-алкила,
xii) С3-8-циклоалкила и
xiii) С3-8-циклоалкил-C1-6-алкила;
или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил, необязательно замещенный R8, где гетероциклоалкил выбран из пирролидинила и пиперидинила;
R6 выбран из
xiv) C1-6-алкила,
xv) С3-8-циклоалкила и
xvi) С3-8-циклоалкил-C1-6-алкила;
R7 выбран из
xvii) C1-6-алкила,
xviii) С3-8-циклоалкила и
xix) С3-8-циклоалкил-C1-6-алкила;
или R6 и R7 вместе с атомом углерода, к которому они присоединены, образуют С3-8-циклоалкил, необязательно замещенный R8; и
R8 представляет собой галоген;
при условии, что N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамид исключен, или его фармацевтически приемлемая соль.
Класс серин-треониновых киназ быстро распространяющейся фибросаркомы (RAF) включает три представителя (ARAF, BRAF, RAF1), которые составляют первый узел сигнального пути МАР-киназ. Несмотря на очевидную избыточность трех изоформ RAF в распространении сигналов через фосфорилирование MEK1 и 2, частые онкогенные активирующие мутации обычно обнаруживают только для BRAF. В частности, замена V600 глутаминовой кислотой или лизином делает киназу высокоактивируемой с дальнейшей гиперстимуляцией пути MAPK, независимо от внешних стимуляций (Cell. 2015 Jun 18; 161(7): 1681-1696).
Мутантный BRAF является целевым онкогенным фактором, и три ингибитора BRAF (вемурафениб, дабрафениб и энкорафениб) вышли на рынок к настоящему времени, демонстрируя эффективность при BRAFV600E-положительной меланоме. Однако почти повсеместно наблюдается быстрое приобретение лекарственной устойчивости, а длительность терапевтических эффектов целевой терапии остается ограниченной.
Кроме того, разработанные ингибиторы BRAF выявили неожиданную и «парадоксальную» способность подавления сигнального пути MAPK в опухолях, обусловленных BRAFV600E, в то время, как те же ингибиторы демонстрировали стимулирующую активность MAPK в моделях BRAF дикого типа (ДТ) (N Engl J Med 2012; 366:271-273; и British Journal of Cancer, т. 111, c. 640 645(2014)).
Затем механистические исследования «парадокса RAF» выявили, что онкогенный BRAFV600E фосфорилирует MEK 1/2 в мономерной цитозольной форме, тогда как активация BRAF ДТ и RAF1 требует сложного этапа событий, включающих транслокацию клеточной мембраны и гомо- и/или гетеродимеризацию, стимулированную активированным RAS (KRAS, NRAS, HRAS) (Nature Reviews Cancer, т. 14, с. 455-467(2014)).
Связывание ингибиторов, подобных вемурафенибу, дабрафенибу или энкорафенибу, с протомером BRAF ДТ или RAF1 быстро индуцирует гомо- и/или гетеродимеризацию RAF и мембранную ассоциацию новообразованного димера RAF. В димерной конформации один протомер RAF аллостерически индуцирует конформационные изменения другого, что приводит в результате к активному состоянию киназы и, что важно, к конформации, неблагоприятной для связывания ингибитора. Димер, индуцированный медикаментозным лечением, как результат, способствует фосфорилированию МЕК за счет катализа, осуществляемого несвязанным протомером с гиперактивацией пути.
«Парадокс RAF» приводит в результате к двум клинически значимым последствиям: 1) ускоренному росту вторичных опухолей при монотерапии BRAFi (преимущественно кератоакантомы и плоскоклеточных карцином) (N Engl J Med 2012; 366:271-273) и 2) приобретение лекарственной устойчивости в условиях монотерапии BRAF, а также в комбинациях BRAFi+MEKi, представляет собой активацию димер-опосредованного сигнального пути RAF генетически обусловленными событиями, включая мутации RAS, амплификации BRAF, экспрессию сплайс-вариантов BRAF с димерным действием (Nature Reviews Cancer, т. 14, с. 455-467(2014)).
Настоящее изобретение относится к неожиданному открытию, что ингибитор BRAF формулы (I) демонстрирует значительно менее «парадоксальную» активацию сигнального пути MAPK, при этом сохраняя высокую активность. Это соединение также может называться «разрушителем парадокса» или «разрушителем парадокса RAF» в сравнении с соединениями, индуцирующими «парадокс RAF» (и которые могут называться «индукторами парадокса» или «индукторами парадокса RAF»).
Термин «фармацевтически приемлемая соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые не являются биологически или в других отношениях нежелательными. Эти соли образуются с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., в частности хлористоводородная кислота, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N-ацетилцистеин и т.п. Кроме того, эти соли могут быть получены добавлением неорганического основания или органического основания к свободной кислоте. Соли, полученные из неорганического основания, включают, но не ограничиваются этим, соли натрия, калия, лития, аммония, кальция, магния и т.п. Соли, полученные из органических оснований, включают, но не ограничиваются этим, соли первичных, вторичных и третичных аминов, замещенных аминов, в том числе встречающихся в природе замещенных аминов, циклических аминов и основных ионообменных смол, например изопропиламина, триметиламина, диэтиламина, триэтиламина, трипропиламина, этаноламина, лизина, аргинина, N-этилпиперидина, пиперидина, полииминовых смол и т.п.
Термин «защитная группа» (ЗГ) обозначает группу, которая селективно блокирует реакционноспособный сайт в мультифункциональном соединении, так что химическая реакция может быть осуществлена селективно по другому незащищенному реакционноспособному сайту в значении, традиционно связанном с ним в синтетической химии. Защитные группы могут быть удалены в подходящий момент. Иллюстративные защитные группы представляют собой амино-защитные группы, карбокси-защитные группы или гидрокси-защитные группы. Конкретные защитные группы представляют собой трет-бутоксикарбонильную (Boc), бензилоксикарбонильную (Cbz), флуоренилметоксикарбонильную (Fmoc) и бензильную (Bn) группы. Дополнительные конкретные защитные группы представляют собой трет-бутоксикарбонильную (Boc) и флуоренилметоксикарбонильную (Fmoc) группы.
Более конкретная защитная группа представляет собой трет-бутоксикарбонильную (Boc) группу.
Сокращение мкМ означает микромолярный и является эквивалентом символа μМ.
Сокращение мкл означает микролитр и является эквивалентом символа μл.
Сокращение мкг означает микрограмм и является эквивалентом символа μт.
Соединение формулы (I) может содержать несколько асимметрических центров и может находиться в форме оптически чистых энантиомеров, смесей энантиомеров, таких как, например, рацематы, оптически чистых диастереоизомеров, смесей диастереоизомеров, диастереоизомерных рацематов или смесей диастереоизомерных рацематов.
В соответствии с правилом Кана - Ингольда - Прелога асимметрический атом углерода может находиться в «R»- или «S»-конфигурации.
Также вариант осуществления настоящего изобретения представляет собой соединение в соответствии с формулой (I), описанной в данном документе, и фармацевтически приемлемые соли, в частности соединение в соответствии с формулой (I), описанной в данном документе.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где
R1 представляет собой C1-6-алкил;
X выбран из
i) -NH- и
i) -О-;
R2 выбран из
ii) Н и
iii) галогена;
R3 выбран из
iv) NR4R5 и
v) CHR6R7;
R4 выбран из
vi) C1-6-алкила,
vii) С3-8-циклоалкила и
viii) С3-8-циклоалкил-C1-6-алкила;
R5 выбран из
ix) C1-6-алкила,
x) С3-8-циклоалкила и
xi) С3-8-циклоалкил-C1-6-алкила;
или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил, необязательно замещенный R8, где гетероциклоалкил выбран из пирролидинила и пиперидинила;
R6 выбран из
xii) C1-6-алкила,
xiii) С3-8-циклоалкила и
xiv) С3-8-циклоалкил-C1-6-алкила;
R7 выбран из
xv) C1-6-алкила,
xvi) С3-8-циклоалкила и
xvii) С3-8-циклоалкил-C1-6-алкила;
или R6 и R7 вместе с атомом углерода, к которому они присоединены, образуют циклопентильное или циклогексильное кольцо, необязательно замещенное R8;
R8 представляет собой галоген;
при условии, что (3R)-N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамид исключен,
или его фармацевтически приемлемая соль.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где
R1 представляет собой метил;
X выбран из
i) -NH- и
ii) -О-;
R2 выбран из
iii) Н,
iv) хлора и
v) фтора;
R3 выбран из
vi) NR4R5 и
vii) CHR6R7;
R4 выбран из
viii) метила,
ix) этила,
x) пропила,
xi) циклопропила и
xii) циклопропилметила;
R5 выбран из
xiii) метила,
xiv) этила,
xv) пропила,
xvi) циклопропила и
xvii) циклопропилметила;
или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил, необязательно замещенный R8, где гетероциклоалкил выбран из пирролидинила и пиперидинила;
R6 выбран из
xviii) метила,
xix) этила,
xx) пропила,
xxi) циклопропила и
xxii) циклопропилметила;
R7 выбран из
xxiii) метила,
xxiv) этила,
xxv) пропила,
xxvi) циклопропила и
xxvii) циклопропилметила;
или R6 и R7 вместе с атомом углерода, к которому они присоединены, образуют циклопентильное или циклогексильное кольцо, необязательно замещенное R8;
R8 представляет собой фтор;
при условии, что (3R)-N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамид исключен,
или его фармацевтически приемлемая соль.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где
R1 представляет собой метил;
X выбран из
i) -NH- и
ii) -О-;
R2 выбран из
iii) Н,
iv) хлора и
v) фтора;
R3 выбран из
vi) NR4R5 и
vii) CHR6R7;
R4 представляет собой метил;
R5 представляет собой этил;
или R4 и R5 вместе с атомом азота, к которому они присоединены, образуют пирролидинильное кольцо, необязательно замещенное R8;
R6 представляет собой метил;
R7 представляет собой этил;
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют циклопентильное или циклогексильное кольцо;
R8 представляет собой фтор;
при условии, что (3R)-N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамид исключен,
или его фармацевтически приемлемая соль.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R1 представляет собой C1-6-алкил.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R1 представляет собой метил.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R2 выбран из
i) H,
ii) хлора и
iii) фтора.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R2 выбран из Н и хлора.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R3 представляет собой NR4R5.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R3 представляет собой CHR6R7.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R4 представляет собой метил.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R5 представляет собой этил.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют гетероциклоалкил, необязательно замещенный R8, где гетероциклоалкил выбран из пирролидинила и пиперидинила.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют пирролидинильное кольцо, необязательно замещенное R8.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R4 и R5 вместе с атомом азота, к которому они присоединены, образуют незамещенный гетероциклоалкил, где гетероциклоалкил представляет собой пирролидинил.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R6 и R7 независимо выбраны из
i) метила,
ii) этила,
iii) пропила,
iv) циклопропила и
v) циклопропилметила.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R6 представляет собой метил.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R7 представляет собой этил.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R6 и
R7 вместе с атомом азота, к которому они присоединены, образуют циклопентильное или циклогексильное кольцо, необязательно замещенное R8.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R6 и R7 вместе с атомом азота, к которому они присоединены, образуют циклопентильное или циклогексильное кольцо.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где R8 представляет собой фтор.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где соединение выбрано из
6-[2-циано-3-[[этил(метил)сульфамоил]амино]-6-фторфенокси]-3-метил-4-оксохиназолина;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
(3R)-N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамида;
N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
(3R)-N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклогексансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]бутан-2-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
(3R)-N-[2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]-3-фторпирролидин-1-сульфонамида;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
N-[4-хлор-2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]пирролидин-1-сульфонамида;
N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида и
6-[2-циано-3-(диметилсульфамоиламино)-6-фторфенокси]-3-метил-4-оксохиназолина;
или его фармацевтически приемлемая соль.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где соединение выбрано из
6-[2-циано-3-[[этил(метил)сульфамоил]амино]-6-фторфенокси]-3-метил-4-оксохиназолина;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклогексансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]бутан-2-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
N-[4-хлор-2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]пирролидин-1-сульфонамида;
N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида и
6-[2-циано-3-(диметилсульфамоиламино)-6-фторфенокси]-3-метил-4-оксохиназолина;
или его фармацевтически приемлемая соль.
В отдельном варианте осуществления настоящего изобретения представлено соединение в соответствии с формулой (I), описанной в данном документе, где соединение выбрано из
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклогексансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]бутан-2-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина и
N-[4-хлор-2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]пирролидин-1-сульфонамида;
или его фармацевтически приемлемая соль.
Термин «C1-6-алкил», отдельно или в комбинации, означает одновалентную линейную или разветвленную насыщенную углеводородную группу с 1-6 атомами углерода. Примеры C1-6-алкила включают метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил и пентил. Конкретными C1-6-алкильными группами являются метил, этил, пропил и н-бутил. Более конкретными C1-6-алкильными группами являются метил, этил и пропил.
Термин «С3-8-циклоалкил», отдельно или в комбинации, означает одновалентную насыщенную моноциклическую или бициклическую углеводородную группу с 3-8 кольцевыми атомами углерода. Бициклический означает кольцевую систему, состоящую из двух насыщенных карбоциклов, имеющих один или два совместных атомов углерода. Примерами моноциклического С3-8-циклоалкила является циклопропил, циклобутанил, циклопентил, циклогексил или циклогептил. Конкретными моноциклическими циклоалкильными группами являются циклопропил, циклопентил и циклогексил.
Термин «С3-8-циклоалкил-C1-6-алкил», отдельно или в комбинации, означает -C1-6-алкильную группу, где один из атомов водорода C1-6-алкильной группы заменен С3-8-циклоалкильной группой. Примеры С3-8-циклоалкил-C1-6-алкила включают циклопропилметил, циклопропилэтил, циклопропилбутил, циклобутилпропил, 2-циклопропилбутил, циклопентилбутил, циклогексилметил, циклогексилэтил, бицикло[4.1.0]гептанилметил, бицикло[4.1.0]гептанилэтил, бицикло[2.2.2]октанилметил и бицикло[2.2.2]октанилэтил. Конкретный пример С3-8-циклоалкил-C1-6-алкила представляет собой циклопропилметил.
Термины «галоген» и «галогено», отдельно или в комбинации, в данном документе используются взаимозаменяемо и означают фтор, хлор, бром или йод. Конкретные примеры галогена представляют собой хлор и фтор. Конкретный галоген представляет собой фтор.
Термин «гетероциклоалкил» означает одновалентную насыщенную или частично ненасыщенную моно- или бициклическую кольцевую систему с 4-9 кольцевыми атомами, содержащую 1, 2 или 3 кольцевых гетероатома, выбранных из N, О и S, при этом остальные кольцевые атомы представляют собой углерод. Бициклический означает состоящий из двух циклов, имеющих один или два совместных кольцевых атома. Примеры моноциклического насыщенного гетероциклоалкила представляют собой 4,5-дигидрооксазолил, оксетанил, азетидинил, пирролидинил, 2-оксопирролидин-3-ил, тетрагидрофуранил, тетрагидротиофенил, пиразолидинил, имидазолидинил, оксазолидинил, изоксазолидинил, тиазолидинил, пиперидинил, тетрагидропиранил, тетрагидротиопиранил, пиперазинил, морфолинил, тиоморфолинил, 1,1-диоксотиоморфолин-4-ил, азепанил, диазепанил, гомопиперазинил или оксазепанил. Примеры бициклического насыщенного гетероциклоалкила представляют собой оксабицикло[2.2.1]гептанил, оксаспиро[3.3]гептанил, 8-азабицикло[3.2.1]октил, хинуклидинил, 8-окса-3-азабицикло[3.2.1]октил, 9-азабицикло[3.3.1]нонил, 3-окса-9-азабицикло[3.3.1]нонил или 3-тиа-9-азабицикло[3.3.1]нонил. Примеры частично ненасыщенного гетероциклоалкила представляют собой дигидрофурил, имидазолинил, дигидрооксазолил, тетрагидропиридинил или дигидропиранил. Конкретный гетероциклоалкил представляет собой пирролидинил и пиперидинил.
Способы изготовления описанного в данном документе соединения формулы (I) также являются объектом изобретения.
Получение соединения формулы (I) по данному изобретению можно осуществлять путями последовательного или конвергентного синтеза. Синтез по настоящему изобретению проиллюстрирован на следующих общих схемах. Навыки, необходимые для проведения реакций и очистки получаемых в результате продуктов, известны специалистам в данной области техники. Заместители и индексы, используемые в нижеприведенном описании способов, имеют значение, указанное в данном документе, если не указано иное.
Конкретнее, соединение формулы (I) можно получать способами, приведенными ниже, способами, приведенными в примерах, или аналогичными способами. Соответствующие условия реакций для отдельных шагов реакций известны специалисту в данной области техники. Последовательность реакций не ограничивается последовательностью, показанной на схеме 1, однако, в зависимости от исходных веществ и их соответствующей реакционной способности, последовательность шагов реакций может быть свободно изменена. Исходные материалы являются либо коммерчески доступными, либо их можно получить способами, аналогичными способам, приведенным ниже, способами, описанными в ссылках, приведенных в описании, или в примерах, или способами, известными в данной области техники.
Общий синтез соединений
Соединения формулы (I) могут быть получены путем реакции арилфторидов формулы А с сульфонамидами или сульфамидами Б в присутствии основания, такого как Cs2CO3 или NaH, в растворителе, таком как ДМФА или NMP (схема 1).
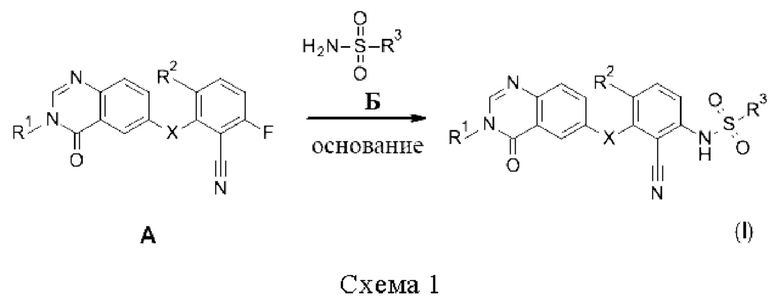
Соединения формулы (I) могут быть дополнительно получены путем реакции анилинов В с сульфонилхлоридами или сульфамоилхлоридами Г в присутствии основания, такого как пиридин, в растворителе, таком как ДХМ (схема 2).

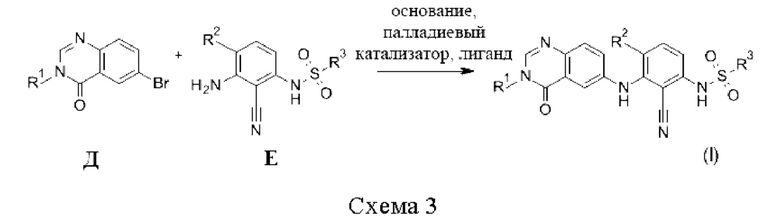
Соединения формулы (I), когда X=NH, могут быть дополнительно получены путем реакции бромидов Д с анилинами Е в присутствии основания, такого как Cs2CO3, палладиевого катализатора, такого как трис(дибензилиденацетон)дипалладий (0), и лиганда, такого как BippyPhos, в растворителе, таком как диоксан (схема 3).
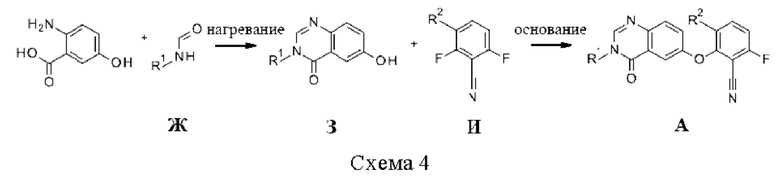
Промежуточные соединения А, где X=О, могут быть получены путем конденсации 2-амино-5-гидроксибензойной кислоты и N-алкилформамидов Ж, например, путем нагрева в отсутствии растворителя, для получения 6-гидроксихиназолин-4-онов 3, которые могут реагировать со фторбензонитрилами И в присутствии основания, такого как NaH или Cs2CO3, в растворителе, таком как ДМФА или NMP (схема 4).
Промежуточные соединения В могут быть получены путем реакции промежуточных соединений А с водным раствором аммиака в растворителе, таком как 2-пропанол (схема 5).
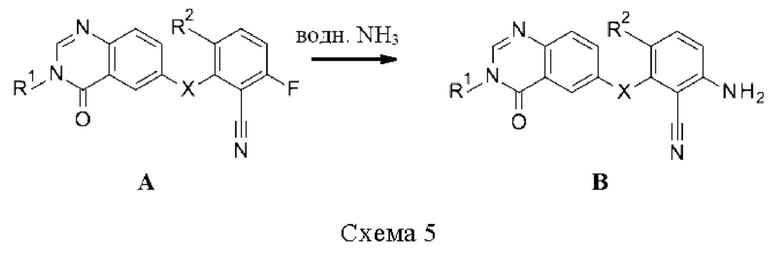
Промежуточные соединения В, где X=NH, могут быть получены путем восстановления 2,6-динитробензонитрилов К восстановителем, таким как железо, в водном растворе HCl в растворителе, таком как смесь метанола и диоксана, для получения 2,6-диаминобензонитрилов Л. При помощи последовательной реакции с 6-бромхиназолин-4-онами Д в присутствии катализатора, такого как трис(дибензилиденацетон)дипалладий (0), лиганда, такого как BippyPhos, и основания, такого как Cs2CO3, в растворителе, таком как диоксан, получают промежуточные соединения В (схема 6).
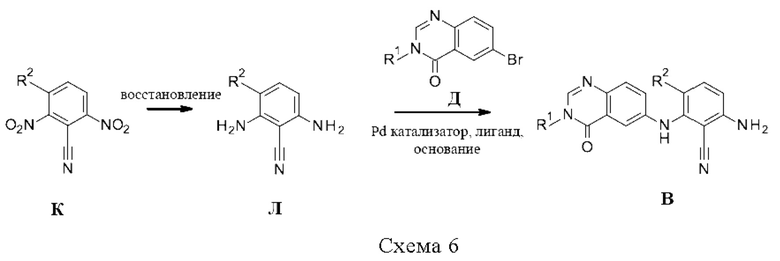
Промежуточные соединения В, где X=NH, также могут быть получены, как показано на схеме 7. 2-Амино-6-нитробензойную кислоту и N-алкилформамид Ж конденсируют с образованием 6-нитрохиназолин-4-онов М, например, путем нагрева в отсутствии растворителя, которые затем восстанавливают до 6-аминохиназолин-4-она Н, например, путем использования водорода и катализатора (такого как палладий на угле) в растворителе, таком как смесь метанола и уксусной кислоты. При помощи реакции со фторбензонитрилами И в присутствии основания, такого как трет-бутоксид калия, в растворителе, таком как ДМСО, получают промежуточные соединения О. При помощи реакции с азидной солью, такой как NaN3, в растворителе, таком как ДМФА, получают азиды П, которые могут быть восстановлены до амина В (например, с использованием процедуры, описанной в Pei and Wickham, Tetrahedron Lett. (1993), 34, 7509-7512).
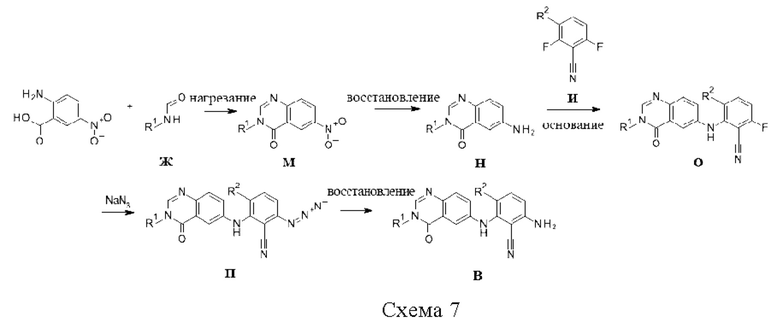
Промежуточные соединения E могут быть получены путем обработки 2,6-динитробензонитрила Р сульфонамидами или сульфамидами Б в присутствии основания, такого как Cs2CO3, в растворителе, таком как ДМФА. Полученные в результате нитросоединения С могут быть восстановлены, например, путем гидрогенизации с катализатором, таким как Pd(OH)2, в растворителе, таком как смесь метанола и ТГФ (схема 8).
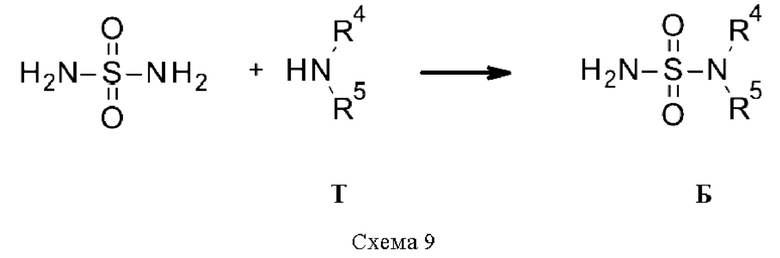
Промежуточные соединения Б, где R3 относится к типу NR4R5 (т.е. сульфамидам), если они не доступны коммерчески, могут быть получены в результате реакции диамида серной кислоты с амином Т в диоксане, в присутствии или отсутствии основания, такого как триэтиламин (схема 9).
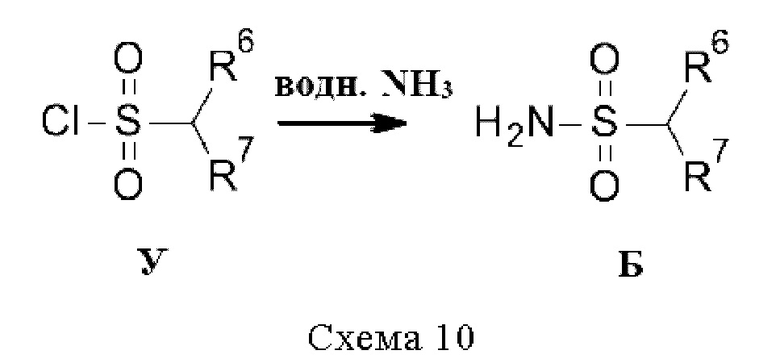
Промежуточные соединения Б, где R3 относится к типу CHR6R7 (т.е. сульфонамидам), если они не доступны коммерчески, могут быть получены из соответствующих сульфонилхлоридов У путем реакции с водным раствором аммиака (схема 10).
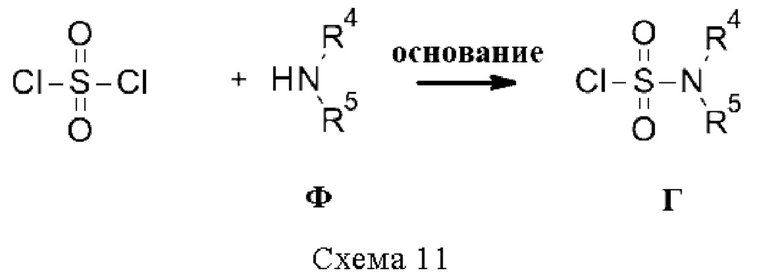
Промежуточные соединения Г, где R3 относится к типу NRdRe (т.е. сульфамоилхлоридам), если они не доступны коммерчески, могут быть получены из вторичного амина Ф и сульфурилдихлорида в присутствии основания, такого как ДИПЭА, в растворителе, таком как ДХМ (схема 11).
Следует понимать, что соединение формулы (I) в данном изобретении может быть дериватизировано в функциональных группах, чтобы получить производные, которые способны к обратному превращению в исходное соединение in vivo.
В частности, настоящее изобретение также относится к:
соединению формулы (I), описанному в данном документе, или его фармацевтически приемлемой соли для применения в качестве терапевтически активного вещества;
фармацевтической композиции, содержащей соединение формулы (I), описанной в настоящем документе, или его фармацевтически приемлемую соль и терапевтически инертный носитель;
соединению формулы (I), описанной в настоящем документе, или его фармацевтически приемлемой соли для применения в лечении или профилактике рака;
соединению формулы (I), описанной в настоящем документе, или его фармацевтически приемлемой соли для применения в лечении или профилактике рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или немелкоклеточного рака легкого (НМРЛ).
применению соединения формулы (I), описанной в настоящем документе, или его фармацевтически приемлемой соли для лечения или профилактики рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ;
применению соединения формулы (I), описанной в настоящем документе, или его фармацевтически приемлемой соли для получения лекарственного средства для лечения или профилактики рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ; и
способу лечения или профилактики рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ, при этом способ включает введение эффективного количества соединения формулы (I),
описанной в настоящем документе, или его фармацевтически приемлемой соли пациенту, который в этом нуждается.
Определенный вариант осуществления изобретения относится к соединению формулы (I), описанной в данном документе, или его фармацевтически приемлемой соли для применения в терапевтическом и/или профилактическом лечении рака, в частности, видов рака, обусловленных мутацией BRAF, более конкретно рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ.
Определенный вариант осуществления изобретения относится к соединению формулы (I), описанной в данном документе, или его фармацевтически приемлемой соли для производства лекарственного средства для терапевтического и/или профилактического лечения рака, в частности, видов рака, обусловленных мутацией BRAF, более конкретно рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ.
Определенный вариант осуществления изобретения относится к фармацевтической композиции, содержащей соединение формулы (I), описанной в данном документе, или его фармацевтически приемлемую соль и фармацевтически приемлемый эксципиент.
Определенный вариант осуществления изобретения относится к способу терапевтического и/или профилактического лечения рака, в частности, видов рака, обусловленных мутацией BRAF, более конкретно рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ, включающему введение эффективного количества соединения формулы (I), описанной в данном документе, или его фармацевтически приемлемой соли пациенту, который в этом нуждается.
Определенный вариант осуществления изобретения относится к соединению формулы (I), описанной в данном документе, или его фармацевтически приемлемой соли для применения в качестве лекарственного средства в терапевтическом и/или профилактическом лечении пациента с раком, обусловленным мутацией BRAF, в частности, более конкретно рака щитовидной железы, колоректального рака, рака головного мозга, меланомы или НМРЛ, включающем определение статуса мутации BRAF у указанного пациента, а затем введение указанному пациенту соединения формулы (I), описанной в данном документе, или его фармацевтически приемлемой соли.
Кроме того, изобретение включает все заместители соединения формулы (I) в их соответствующей дейтерированной форме, где применимо.
Определенный вариант осуществления изобретения относится к соединению формулы (I), описанной в данном документе, или его фармацевтически приемлемой соли, где по меньшей мере один заместитель содержит по меньшей мере один радиоизотоп. Конкретные примеры радиоизотопов представляют собой 2Н, 3Н, 13С, 14С и 18F.
Кроме того, изобретение включает все оптические изомеры, т.е. диастереоизомеры, диастереомерные смеси, рацемические смеси, все их соответствующие энантиомеры и/или таутомеры, а также их сольваты, соединения формулы (I), где это применимо.
Соединение формулы (I) может содержать один или более асимметрических центров и, следовательно, может существовать в виде рацематов, рацемических смесей, отдельных энантиомеров, диастереомерных смесей и отдельных диастереомеров. Могут присутствовать дополнительные асимметрические центры в зависимости от природы различных заместителей в молекуле. Каждый такой асимметрический центр будет независимо давать два оптических изомера, и предполагается, что все возможные оптические изомеры и диастереомеры в смесях и в виде чистых или частично очищенных соединений включены в это изобретение. Предполагается, что данное изобретение охватывает все такие изомерные формы этих соединений. Независимые синтезы этих диастереомеров или их хроматографическое разделение можно осуществить, как известно в данной области техники, путем соответствующей модификации методологии, описанной в данном документе. Их абсолютную стереохимию можно определять с помощью рентгеноструктурной кристаллографии кристаллических продуктов или кристаллических промежуточных соединений, которые дериватизированы, если необходимо, реагентом, содержащим асимметрический центр с известной абсолютной конфигурацией. Если необходимо, рацемические смеси соединений можно разделять, чтобы выделить индивидуальные энантиомеры. Разделение можно осуществлять способами, хорошо известными в данной области техники, например сочетанием рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереомерной смеси с последующим разделением отдельных диастереомеров стандартными способами, такими как фракционная кристаллизация или хроматография.
В вариантах осуществления изобретения, когда представлены оптически чистые энантиомеры, оптически чистый энантиомер означает, что соединение содержит > 90% необходимого изомера по массе, в частности > 95% необходимого изомера по массе, или конкретнее > 99% необходимого изомера по массе, при этом указанное процентное содержание основано на общей массе изомера(-ов) соединения. Хирально чистые или хирально обогащенные соединения можно получить хирально-селективным синтезом или путем разделения энантиомеров. Разделение энатиомеров можно провести с конечным продуктом или альтернативно с пригодным промежуточным соединением.
Также в одном варианте осуществления настоящего изобретения предложено описанное в данном документе соединение формулы (I), изготовленное в соответствии с любым из описанных способов.
Аналитические процедуры
Материалы
Среду DMEM без фенолового красного, дополненную L-глутамином, приобретали у (Thermo Fisher Scientific). Фетальную бычью сыворотку (ФБС) приобретали у VWR. Расширенный набор ERK фосфо-Т202 /Y204 - 10000 тестов приобретали у Cisbio № по кат. 64AERPEH. Клетки А375 и НСТ116 сначала получали от АТСС и хранили в репозитории Roche. 384-луночные микропланшеты приобретали у Greiner Bio-One, 384-луночные (с крышкой, HiBase, малый объем, кат. 784-080).
HTRF-анализ для определения P-ERK в клетках А375 или НСТ116
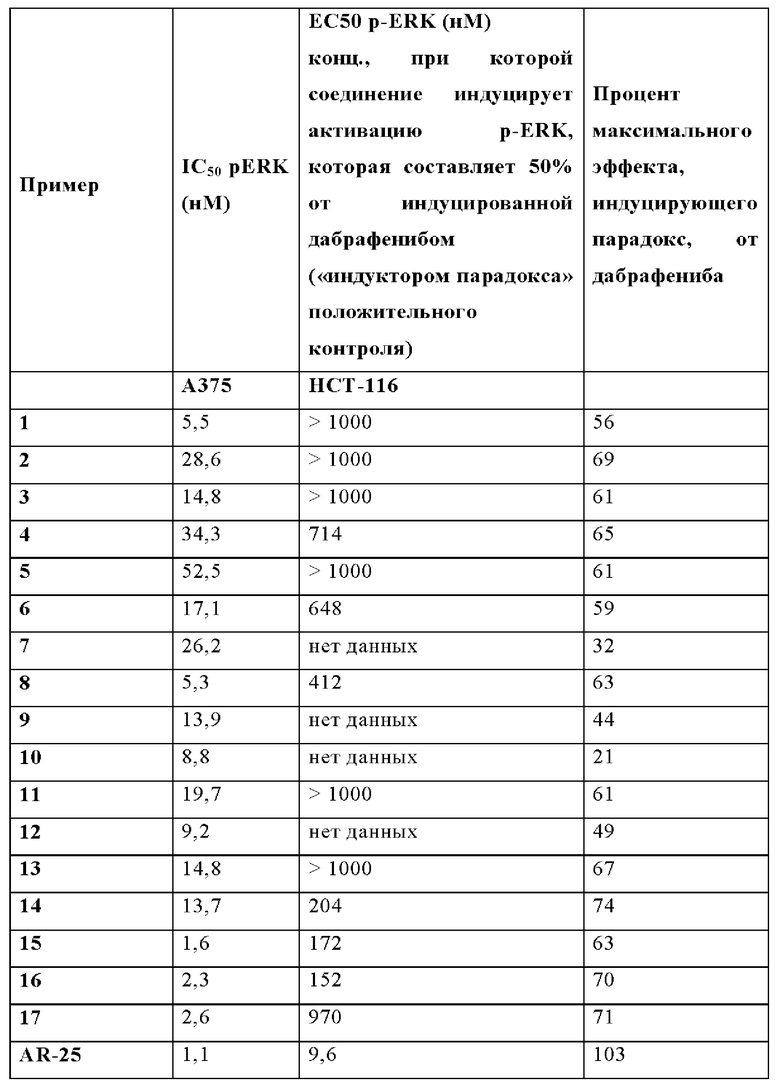
А375 представляет собой модель клеточного рака, экспрессирующую BRAF с мутацией V600E, а НСТ116 представляет собой модель клеточного рака, экспрессирующую BRAF ДТ. Ингибиторы BRAF первого поколения, такие как, например, дабрафениб, индуцируют «парадоксальный» эффект в отношении клеток опухолей, поскольку они ингибируют рост клеток BRAF с мутацией V600E (таких как, например, А375), при этом они активируют рост клеток BRAF ДТ (таких как, например, НСТ116). Фосфорилирование ERK 1,2 (конечное звено каскада фосфорилирования пути MAPK) далее сообщается как основной показатель состояния активации пути MAPK.
Перед анализом линии клеток А375 и НСТ116 выдерживают в среде DMEM без фенолового красного, дополненной 10% фетальной бычьей сывороткой (ФБС). После обработки соединениями уровни P-ERK определяют путем измерения сигнала флуоресценции FRET, индуцированного селективным связыванием 2 антител, предоставленных в указанном наборе (Cisbio, № по кат. 64AERPEH), с белком ERK при фосфорилировании по Thr202/Tyr204. Вкратце, 8000 клеток/лунка в 12 мкл среды/лунка высевают в 384-луночный планшет и оставляют на ночь в термостате (при 37°С с увлажненной средой с 5% СО2), на следующий день планшет обрабатывают в двух повторностях тестовыми соединениями, дабрафенибом и PLX8394 (два последние в качестве контролей) в таких конечных концентрациях лекарственного препарата: 10 мкМ-3 мкМ-1 мкМ-0,3 мкМ-0,1 мкМ-0,03 мкМ-0,01 мкМ-0,003 мкМ-0,001 мкМ, все лунки подвергают нормализации ДМСО и проводят инкубацию с лекарственным препаратом в течение 1 часа. Затем в лунки добавляют 4 мкл 4Х буфера для лизиса, поставляемого с набором, затем планшет центрифугируют в течение 30 секунд (300 rcf (относительное ускорение центрифуги)) и инкубируют на шейкере для планшетов в течение 1 ч при к.т.
В конце инкубации в тестовые лунки добавляют 4 мкл/лунка раствора улучшенного антитела к P-ERK (приготовленного в соответствии с инструкцией производителя), затем 4 мкл/лунка раствора меченного криптатом антитела P-ERK (приготовленного в соответствии с инструкцией производителя) (Cisbio, № по кат. 64AERPEH).
Для обеспечения надлежащей нормализации данных контрольные лунки, не обработанные лекарственным препаратом, указанные в таблице ниже, всегда включены в каждый планшет (в соответствии с инструкцией производителя):
Композиции лунок HTRF p-ERK (мкл):
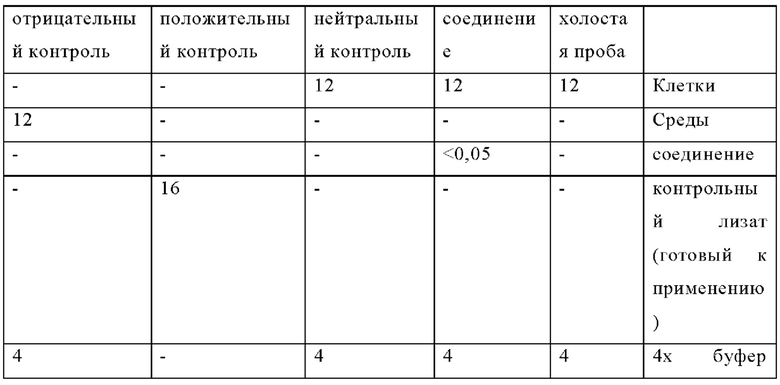
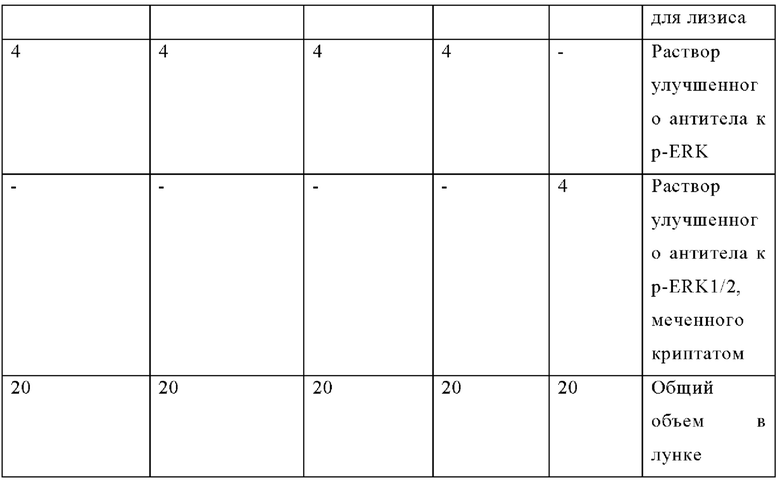
Затем планшет центрифугируют при 300 rcf в течение 30 секунд, герметизируют для предотвращения испарения и инкубируют в течение ночи в темноте при комнатной температуре.
Затем планшет анализируют и собирают значения излучаемой флуоресценции при помощи устройства Pherastast FSX (BMG Labtech) при 665 и 620 нМ.
Полученные значения флуоресценции обрабатывают в соответствии с формулой Соотношение=Сигнал(620 нм)/Сигнал(625 нм)*10000, затем среднее значение соотношения холостой пробы вычитают из всех значений.
Данные нормализуют в случае клеток А375 (ингибирование BRAF), учитывая среднее значение соотношения (вычтенное значение холостой пробы), полученное для клеток, обработанных только ДМСО, как 100%, и с учетом среднего значения соотношения (вычтенное значение холостой пробы), полученного для клеток, обработанных 10 мкМ дабрафенибом, как 0%. Среднее значение нормализованных точек аппроксимируют сигмоидальной кривой и определяют IC50.
Данные нормализуют в случае клеток НСТ116 (активация BRAF), учитывая среднее значение соотношения (вычтенное значение холостой пробы), полученное для клеток, обработанных только ДМСО, как 0%, и с учетом среднего значения соотношения (вычтенное значение холостой пробы), полученного для клеток, обработанных дабрафенибом, при концентрации, при которой обеспечивается наивысший сигнал, как 100%. Отдельные точки аппроксимируют сигмоидальными или колоколообразными кривыми, и определяют процент активации в сравнении с максимальной активацией, опосредованной дабрафенибом. ЕС50 представляет собой концентрацию, при которой достигается активация, равная 50% максимального значения, достигнутого при помощи дабрафениба.
В случае если активация не достигает 50% максимального значения, достигнутого при помощи дабрафениба, то расчет ЕС50 не применяется.
Процент максимального эффекта, индуцирующего парадокс, от дабрафениба определяют путем оценки процента, при котором тестируемое соединение индуцирует свой максимальный сигнал P-ERK, как процента наивысшего сигнала, производимого дабрафенибом в тестируемом диапазоне доз.
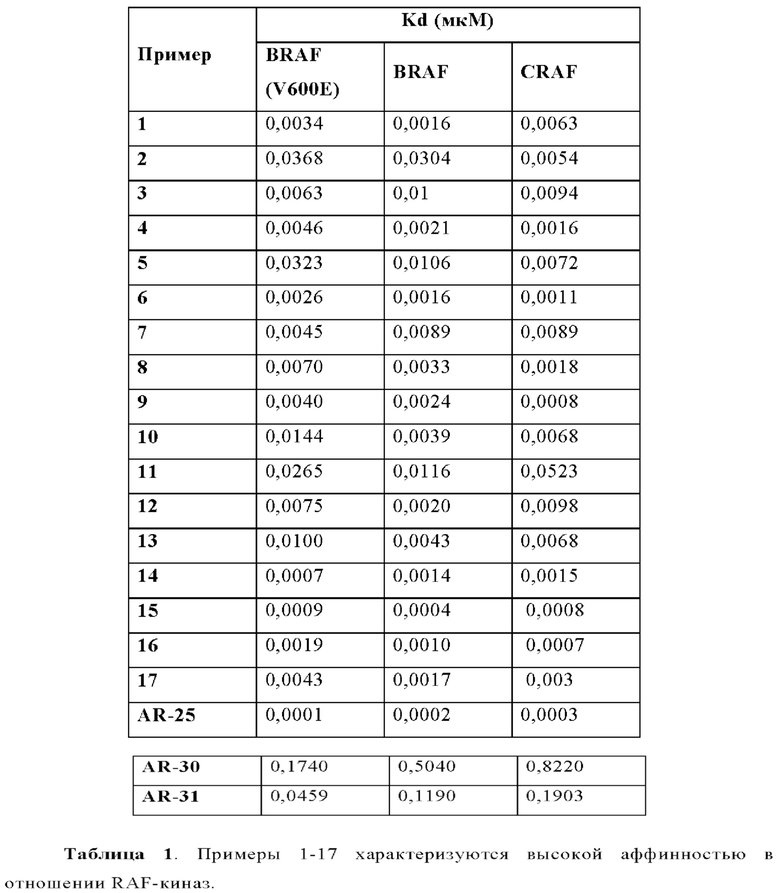

В WO 2012/118492 раскрыты эталонные соединения AR-25 как пример 25, AR-30 как пример 30 и AR-31 как пример 31.
Соединение формулы (I) или его фармацевтически приемлемую соль можно использовать в качестве лекарственного средства (например, в форме фармацевтического препарата). Фармацевтический препарат можно вводить внутрь, например, перорально (например, в форме таблеток, покрытых оболочкой таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий), назально (например, в форме назальных спреев), ректально (например, в форме суппозиториев) или местно через глаза (например, в форме растворов, мазей, гелей или водорастворимых полимерных вкладок). Однако введение также можно осуществлять парентерально, а именно, внутримышечно, внутривенно или внутриокулярно (например, в форме стерильных инъекционных растворов).
Соединение формулы (I) или его фармацевтически приемлемую соль можно обрабатывать фармацевтически инертными, неорганическими или органическими адъювантами для производства таблеток, покрытых оболочкой таблеток, драже, твердых желатиновых капсул, инъекционных растворов или составов для местного применения. В качестве таких адъювантов для таблеток, драже и твердых желатиновых капсул можно использовать, например, лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и т.д.
Подходящими адъювантами для мягких желатиновых капсул являются, например, растительные масла, воски, жиры, полутвердые вещества и жидкие полиолы и т.д.
Подходящими адъювантами для получения растворов и сиропов являются, например, вода, полиолы, сахароза, инвертированный сахар, глюкоза и т.д.
Подходящими адъювантами для инъекционных растворов являются, например, вода, спирты, полиолы, глицерин, растительные масла и т.д.
Подходящими адъювантами для суппозиториев являются, например, натуральные или отвержденные масла, воски, жиры, полутвердые или жидкие полиолы и т.д.
Подходящими адъювантами для местных глазных составов являются, например, циклодекстрины, маннит или множество других носителей и эксципиентов, известных в данной области техники.
Кроме того, фармацевтические препараты могут содержать консерванты, солюбилизаторы, вещества, повышающие вязкость, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферы, маскирующие агенты или антиоксиданты. Также они могут содержать другие имеющие терапевтическую ценность вещества.
Дозировку можно варьировать в широком диапазоне и, конечно, ее следует подбирать в зависимости от индивидуальных требований в каждом конкретном случае. В целом, в случае перорального введения суточная доза составляет от около 0,1 мг до 20 мг на кг массы тела, предпочтительно от около 0,5 мг до 4 мг на кг массы тела (например, около 300 мг на человека), предпочтительно разделенная на 1-3 отдельные дозы, которые могут состоять, например, из одинаковых количеств, если это уместно. В случае местного применения состав может содержать от 0,001% до 15% по массе лекарственного средства, а необходимую дозу, которая может составлять от 0,1 до 25 мг, можно вводить один раз в день или в неделю, или в виде нескольких доз (от 2 до 4) в день, или в виде нескольких доз в неделю. Однако следует понимать, что в случае соответствующего показания верхняя или нижняя граница, приведенная в данном документе, может быть превышена.
Фармацевтические композиции
Соединение формулы (I) или его фармацевтически приемлемую соль можно использовать в качестве терапевтически активного вещества, например в форме фармацевтического препарата. Фармацевтический препарат можно вводить перорально, например в форме таблеток, таблеток с оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако введение также можно осуществлять ректально, например в форме суппозиториев, или парентерально, например в форме инъекционных растворов.
Соединение формулы (I) и его фармацевтически приемлемые соли можно подвергать обработке с фармацевтически инертным неорганическим или органическим носителем для получения фармацевтического препарата. Лактозу, кукурузный крахмал или его производные, тальк, стеариновую кислоту или ее соли и тому подобное можно использовать, например, в качестве таких носителей для таблеток, таблеток с оболочкой, драже и твердых желатиновых капсул. Пригодные носители для мягких желатиновых капсул представляют собой, например, растительные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное. Однако в зависимости от природы активного вещества обычно нет необходимости в носителях в случае мягких желатиновых капсул. Пригодные носители для получения растворов и сиропов представляют собой, например, воду, полиолы, глицерин, растительное масло и тому подобное. Пригодные носители для суппозиториев представляют собой, например, природные или гидрогенизированные масла, воски, жиры, полутвердые и жидкие полиолы и тому подобное.
Кроме того, фармацевтический препарат может содержать фармацевтически приемлемые вспомогательные вещества, такие как консерванты, вещества, увеличивающие растворимость, стабилизаторы, смачивающие агенты, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферные агенты, маскирующие агенты или антиоксиданты. Также они могут содержать другие имеющие терапевтическую ценность вещества.
Лекарственные средства, содержащие соединение формулы (I) или его фармацевтически приемлемую соль и терапевтически инертный носитель, также представлены в данном изобретении, как и способ их получения, который включает придание одному или более соединениям формулы (I) и/или их фармацевтически приемлемым солям и, если необходимо, одному или более другим терапевтически ценным веществам лекарственной формы для введения вместе с одним или более терапевтически инертными носителями.
Дозировка может варьироваться в широком диапазоне и, конечно, будет подбираться под индивидуальные требования в каждом конкретном случае. В случае перорального введения дозировка для взрослых может изменяться от около 0,01 мг до около 1000 мг в сутки соединения общей формулы (I) или соответствующего количества его фармацевтически приемлемой соли. Суточную дозировку можно вводить в виде однократной дозы или несколькими дозами, а кроме того, можно также превышать верхнюю границу, когда установлено, что существуют показания.
Следующие примеры иллюстрируют данное изобретение, не ограничивая его, а могут служить только в качестве иллюстрации. Фармацевтические препараты обычно содержат около 1-500 мг, в частности 1-100 мг, соединения формулы (I). Примерами композиций по настоящему изобретению являются:
Пример А
Таблетки со следующим составом изготавливают традиционным способом:
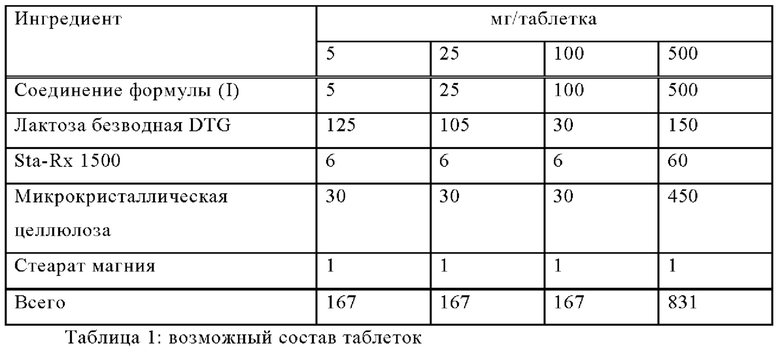
Методика приготовления
1. Смешать ингредиенты 1, 2, 3 и 4 и гранулировать с очищенной водой.
2. Высушить гранулы при 50°С.
3. Пропустить гранулы через соответствующее помольное оборудование.
4. Добавить ингредиент 5 и перемешивать в течение трех минут; спрессовать на соответствующем прессе.
Пример Б-1
Изготавливают капсулы со следующим составом:
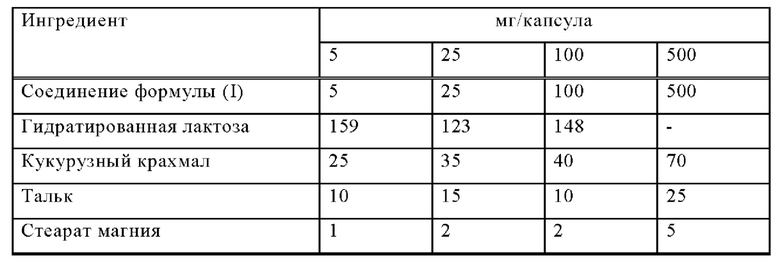

Методика приготовления
1. Смешать ингредиенты 1, 2 и 3 в соответствующем смесителе в течение 30 минут.
2. Добавить ингредиенты 4 и 5 и смешивать в течение 3 минут.
3. Наполнить соответствующие капсулы.
Соединение формулы (I), лактозу и кукурузный крахмал сначала смешивают в смесителе, а потом в измельчителе. Смесь возвращают в смеситель; к ней добавляют тальк и тщательно перемешивают. Смесью с помощью устройства заполняют подходящие капсулы, например твердые желатиновые капсулы.
Пример Б-2
Изготавливают мягкие желатиновые капсулы со следующим составом:
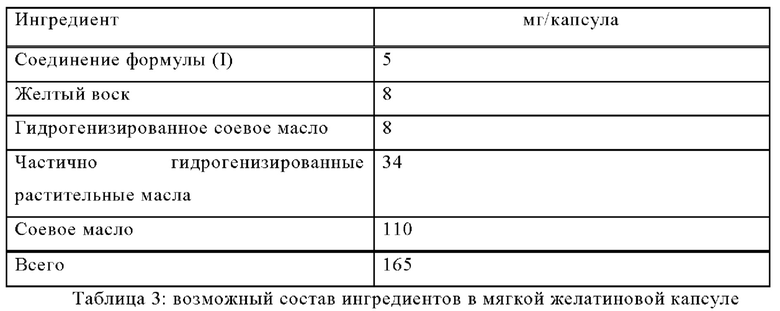
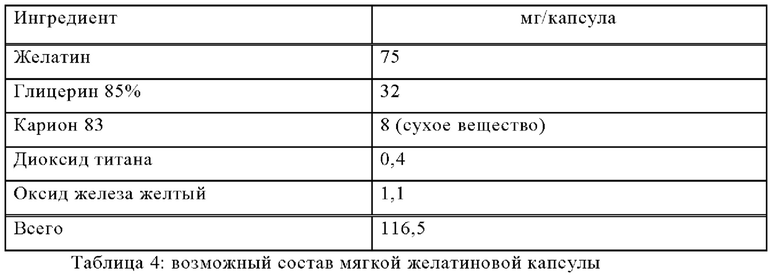
Методика приготовления
Соединение формулы (I) растворяют в теплом расплаве других ингредиентов, и смесью заполняют мягкие желатиновые капсулы подходящего размера.
Заполненные мягкие желатиновые капсулы обрабатывают в соответствии с обычными методиками.
Пример В
Изготавливают суппозитории со следующим составом:
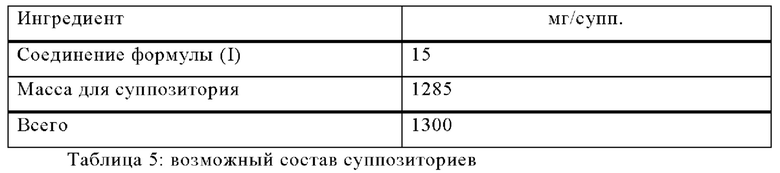
Методика приготовления
Массу для суппозитория плавят в стеклянном или стальном сосуде, тщательно перемешивают и охлаждают до 45°С. После этого в нее добавляют мелко измельченное соединение формулы (I) и перемешивают до полного диспергирования. Смесь выливают в формы для суппозиториев пригодного размера, оставляют охлаждаться; затем суппозитории вынимают из форм и отдельно упаковывают в вощеную бумагу или металлическую фольгу.
Пример Г
Изготавливают инъекционные растворы со следующим составом:
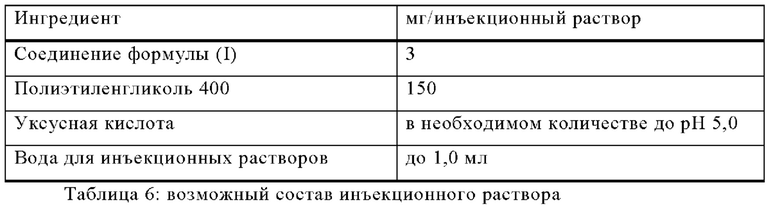
Методика приготовления
Соединение формулы (I) растворяют в смеси полиэтиленгликоля 400 и воды для инъекций (часть). рН доводят до 5,0 уксусной кислотой. Объем доводят до 1,0 мл путем добавления остаточного количества воды. Раствор фильтруют, наливают во флаконы с необходимым избытком и стерилизуют.
Пример Д
Изготавливают саше со следующим составом:
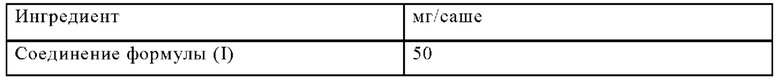

Методика приготовления
Соединение формулы (I) смешивают с лактозой, микрокристаллической целлюлозой и карбоксиметилцеллюлозой натрия и гранулируют со смесью поливинилпирролидона в воде. Гранулят смешивают со стеаратом магния и ароматизирующими добавками и помещают в саше.
Примеры
Следующие примеры представлены для иллюстрации изобретения. Их не следует рассматривать, как ограничивающие объем изобретения, а только как его иллюстрацию.
Сокращения
АсОН = уксусная кислота; ДХМ = дихлорметан; ДИПЭА = диизопропилэтиламин; ДМАП = диметиламинопиридин; ДМФА = диметилформамид; ДМСО = диметилсульфоксид; ЭРИ = электрораспылительная ионизация; EtOAc = этилацетат; EtOH = этанол; ГТФ = гуанозинтрифосфат; ГАТУ = гексафторфосфат азабензотриазолтетраметилурония; ВЭЖХ = высокоэффективная жидкостная хроматография; МеОН = метанол; МС = масс-спектрометрия; NMP = N-метил-2-пирролидон; ЯМР = ядерный магнитный резонанс; к.т. = комнатная температура; ТГФ = тетрагидрофуран; ТРИС = трис(гидроксиметил)аминометан.
Промежуточное соединение I1: 6-гидрокси-3-метилхиназолин-4-он
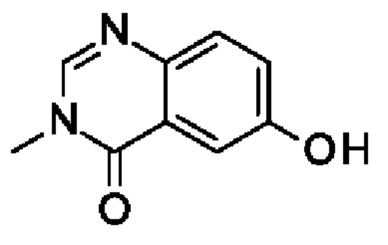
2-Амино-5-гидроксибензойную кислоту (10 г, 65,3 ммоль, экв.: 1,0) и N-метилформамид (30 г, 29,9 мл, 503 ммоль, экв.: 7,7) нагревали при 145°С в течение 21 ч 45 мин, затем охлаждали до к.т. Реакционную смесь разводили с помощью 50 мл Н2О и перемешивали при к.т. в течение 20 мин. Полученный в результате осадок собирали при помощи фильтрации. Светло-коричневое твердое вещество промывали 3×20 мл воды. Твердое вещество поглощали в толуоле и выпаривали до сухого состояния (3×). Твердое вещество сушили в вакууме при 40°С в течение ночи под высоким вакуумом с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (10,3 г, выход 89%). МС (ЭРИ) м/з: 177,1 [М+Н]+.
Промежуточное соединение I2: 3,6-дифтор-2-(3-метил-4-оксохиназолин-6-ил)оксибензонитрил

Карбонат цезия (3,22 г, 9,79 ммоль, экв.: 1,15) добавляли при к.т. к раствору I1 (1500 мг, 8,51 ммоль, экв.: 1) в N,N-диметилформамиде (35 мл). Смесь перемешивали в течение 30 мин при к.т., затем добавляли 2,3,6-трифторбензонитрил (1,47 г, 1,08 мл, 9,37 ммоль, экв.: 1,1). Через 1 ч реакционную смесь охлаждали на льду и разбавляли водой (120 мл). Полученное в результате твердое вещество собирали при помощи фильтрации, промывали ледяной водой (100 мл) и гептаном (100 мл) и высушивали с отсасыванием. Твердое вещество поглощали в толуоле и выпаривали до сухого состояния (3×), затем сушили в течение ночи в вакууме с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (2,58 г, выход 97%). МС (ЭРИ) м/з: 314,1 [М+Н]+.
Промежуточное соединение I3: 2-фтор-6-(3-метил-4-оксохиназолин-6-ил)оксибензонитрил
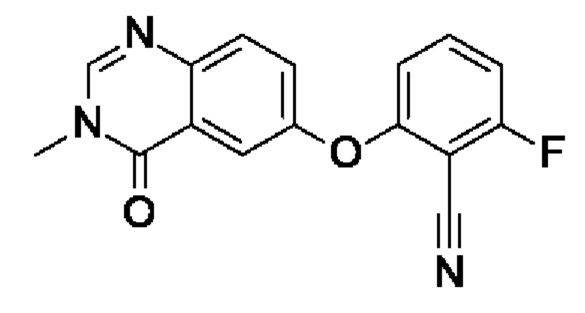
NaH (60% в минеральном масле, 285 мг, 6,53 ммоль, экв.: 1,15) добавляли при 0°С к раствору I1 (6-гидрокси-3-метилхиназолин-4-она) (1,00 г, 5,68 ммоль, экв.: 1,0) в ДМФА (15 мл). Охлаждающую баню удаляли и перемешивали реакционную смесь при к.т. в течение 15 мин. Реакционную смесь снова охлаждали до 0°С и добавляли 2,6-дифторбензонитрил (790 мг, 5,68 ммоль, экв.: 1,0) в ДМФА (2,5 мл). Реакционную смесь подогревали до к.т. и перемешивали в атмосфере аргона в течение 2 ч. Через 2 ч реакционную смесь охлаждали на льду и добавляли воду (100 мл). Полученный в результате осадок собирали при помощи фильтрации и промывали водой и гептанами с получением указанного в заголовке соединения в виде бежевого твердого вещества (1,62 г, чистота 93%, выход 90%). МС (ЭРИ) м/з: 296,1 [М+Н]+.
Промежуточное соединение I4: 6-амино-3-хлор-2-(3-метил-4-оксохиназолин-6-ил)оксибензонитрил
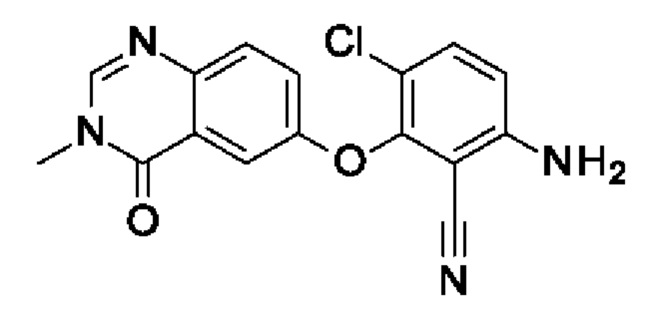
Шаг 1: 3-хлор-6-фтор-2-(3-метил-4-оксохиназолин-6-ил)оксибензонитрил

3-Хлор-2,6-дифторбензонитрил (200 мг, 1,15 ммоль, экв.: 1,0), I1 (6-гидрокси-3-метилхиназолин-4-он) (203 мг, 1,15 ммоль, экв.: 1,0) и K2CO3 (319 мг, 2,3 ммоль, экв.: 2,0) в NMP (1,15 мл) нагревали при 100°С в течение 14 ч, охлаждали до к.т. Реакционную смесь разбавляли водой (30 мл) и экстрагировали с помощью EtOAc (2×30 мл). Объединенные органические слои промывали солевым раствором (3×40 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Очисткой при помощи флеш-хроматографии (20 г диоксида кремния, 50-100% EtOAc в гептане) получали указанное в заголовке соединение в виде бесцветного твердого вещества (231 мг, чистота 100%, выход 61%). МС (ЭРИ) м/з: 330,1 [М+Н]+.
Шаг 2: 6-амино-3-хлор-2-(3-метил-4-оксохиназолин-6-ил)оксибензонитрил
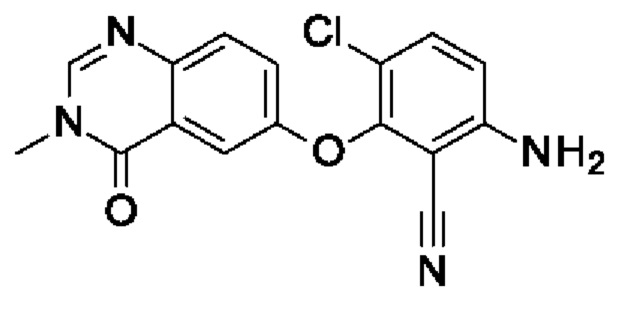
3-Хлор-6-фтор-2-(3-метил-4-оксохиназолин-6-ил)оксибензонитрил (550 мг, 1,67 ммоль, экв.: 1,0), гидроксид аммония (25% в воде, 2,7 г, 3 мл, 77 ммоль, экв.: 46,2) и 2-пропанол (3 мл) нагревали в герметично закрытом флаконе при помощи микроволнового излучения в течение 25 мин при 160°С. Реакционную смесь охлаждали до к.т., и полученный в результате осадок отфильтровывали, промывали водой, изопропанолом и диэтиловым эфиром с получением указанного в заголовке соединения в виде бесцветного твердого вещества (426 мг, чистота 96%, выход 78%). МС (ЭРИ) м/з: 327,1 [М+Н]+.
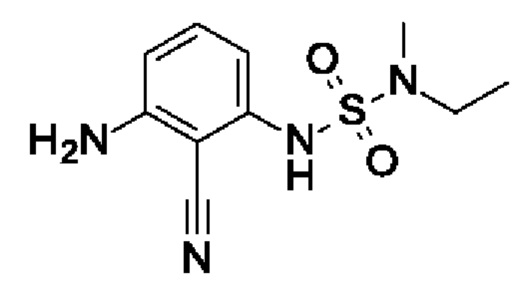
Промежуточное соединение I5: 1-амино-2-циано-3-[[этил(метил)сульфамоил]амино]бензол
Шаг 1: 2-циано-1-[[этил(метил)сульфамоил]амино]-3-нитробензол
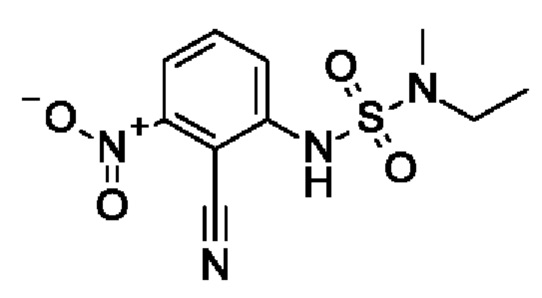
2,6-Динитробензонитрил (1,46 г, 7,56 ммоль, экв.: 1,1) растворяли в ДМФА (15 мл). Добавляли CS2CO3 (2,46 г, 7,56 ммоль, экв.: 1,1) и [метил(сульфамоил)амино]этан (1 г, 6,87 ммоль, экв.: 1,0). Реакционную смесь перемешивали в течение 2 ч при 65°С, затем концентрировали в вакууме. Остаток поглощали в 2-метил-ТГФ и промывали водно-солевым раствором, и водный слой экстрагировали 2×2-метил-ТГФ. Органические слои объединяли, сушили с помощью Na2SO4, фильтровали и концентрировали в вакууме. Остаток разбавляли ДХМ, выпаривали с силикагелем до сухого состояния и переносили в колонку. Очисткой при помощи флеш-хроматографии (40 г диоксида кремния, 0-100% EtOAc в ДХМ) получали указанное в заголовке соединение в виде светло-красного вязкого масла (720 мг, чистота 77%), которое использовали без дополнительной очистки. МС (ЭРИ) м/з: 285,1 [М+Н]+.
Шаг 2: 1-амино-2-циано-3-[[этил(метил)сульфамоил]амино]бензол
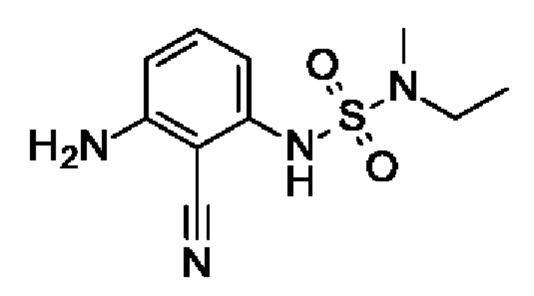
1-Амино-2-циано-3-[[этил(метил)сульфамоил]амино]бензол (703 мг, 1,9 ммоль, экв.: 1,0) растворяли в МеОН (17 мл) и ТГФ (7 мл), затем добавляли Pd(OH)2 (катализатор Перлмана, 26,7 мг, 190 мкмоль, экв.: 0,1) и реакционную смесь перемешивали в атмосфере водорода из баллона при к.т. Через 1 ч реакционную смесь фильтровали через фильтр Whatman Spartan 30/0,45RC, и фильтрат выпаривали. Остаток разбавляли EtOAc и переносили в колонку. Очисткой при помощи флеш-хроматографии (80 г диоксида кремния, 0-68% EtOAc в гептане) получали указанное в заголовке соединение в виде вязкого оранжевого масла (457 мг, чистота 100%, выход за два шага 27%). МС (ЭРИ) м/з: 255,1 [М+Н]+.
Промежуточное соединение I6: (3R)-N-(3-амино-2-цианофенил)-3-фторпирролидин-1-сульфонамид
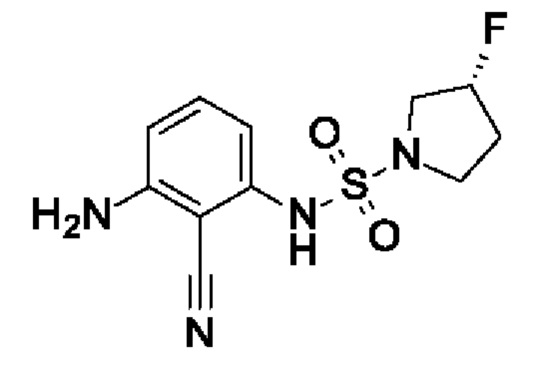
Шаг 1: (3R)-N-(2-циано-3-нитрофенил)-3-фторпирролидин-1-сульфонамид
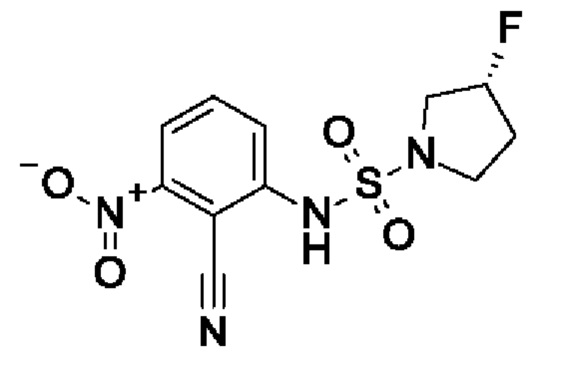
2,6-Динитробензонитрил (900 мг, 4,66 ммоль, экв.: 1,0) растворяли в ДМФА (10 мл). Cs2CO3 (2,28 г, 6,99 ммоль, экв.: 1,5) и I9 (1,18 г, 6,99 ммоль, экв.: 1,5). Реакционную смесь перемешивали в течение 1 ч при 60°С, затем концентрировали в вакууме. Остаток поглощали в 2-метил-ТГФ и промывали водным раствором NH4Cl, и водный слой экстрагировали 1×2-метил-ТГФ. Органические слои объединяли, сушили с помощью Na2SO4, фильтровали и концентрировали в вакууме. Остаток разбавляли ДХМ, выпаривали с силикагелем до сухого состояния и переносили в колонку. Очисткой при помощи флеш-хроматографии (40 г диоксида кремния, 0-100% EtOAc в ДХМ) получали указанное в заголовке соединение в виде светло-красного вязкого масла (795 мг). МС (ЭРИ) м/з: 315,1 [М+Н]+.
Шаг 2: (3R)-N-(3-амино-2-цианофенил)-3-фторпирролидин-1-сульфонамид
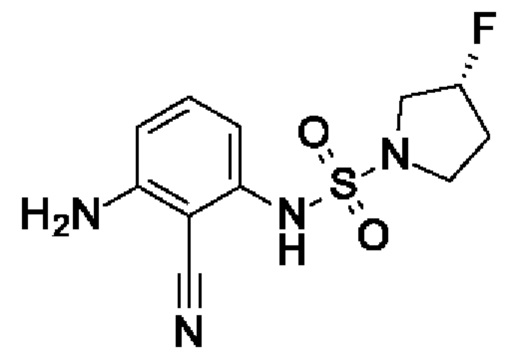
(3R)-N-(2-Циано-3-нитрофенил)-3-фторпирролидин-1-сульфонамид (751 мг, 2,39 ммоль, экв.: 1,0) растворяли в МеОН (13 мл) и ТГФ (6 мл), затем добавляли Pd(OH)2 (катализатор Перлмана, 33,6 мг, 239 мкмоль, экв.: 0,1) и реакционную смесь перемешивали в атмосфере водорода из баллона при к.т. Через 1 ч реакционную смесь фильтровали через фильтр Whatman Spartan 30/0,45RC, и фильтрат выпаривали. Остаток разбавляли EtOAc и переносили в колонку. Очисткой при помощи флеш-хроматографии (80 г диоксида кремния, 0-79% EtOAc в гептане) получали указанное в заголовке соединение в виде вязкого оранжевого масла (560 мг, чистота 100%, выход за два шага 42%). МС (ЭРИ) м/з: 285,1 [М+Н]+.
Промежуточное соединение I7: 2-амино-6-[(3-метил-4-оксохиназолин-6-ил)амино]бензонитрил
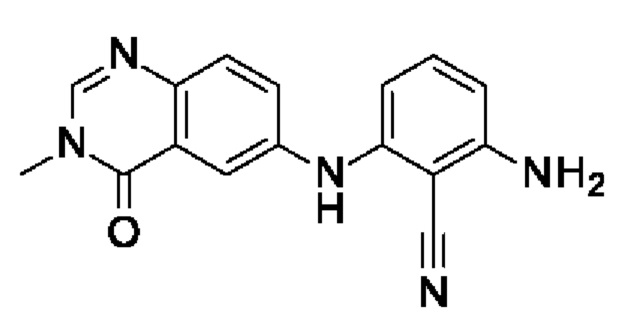
Шаг 1: 2,6-диаминобензонитрил
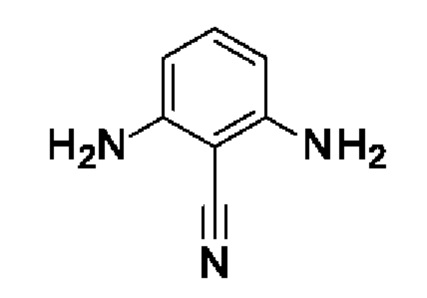
2,6-Динитробензонитрил (3 г, 15,5 ммоль, экв.: 1,0) растворяли в смеси метанола (60 мл) и диоксана (35 мл). Реакционную смесь нагревали до 75°С, затем добавляли по каплям HCl (37% водн., 11 г, 9,29 мл, 111 ммоль, экв.: 7,16), затем железо (2,78 г, 49,7 ммоль, экв.: 3,2) добавляли 4 порциями на протяжении 8 мин. Реакционную смесь перемешивали в течение 1 ч при 64°С, затем концентрировали в вакууме. Остаток поглощали в 2-метил-ТГФ и льду и промывали насыщ. водн. NaHCO3. Оба слоя фильтровали, и водный слой снова экстрагировали с помощью 2-метил-ТГФ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток разбавляли ДХМ, выпаривали с силикагелем до сухого состояния и переносили в колонку. Очисткой при помощи флеш-хроматографии (80 г диоксида кремния, ДХМ) получали указанное в заголовке соединение (268 мг, выход 13%) вместе с 2-амино-6-нитробензонитрилом (465 мг, выход 18%). 2,6-диаминобензонитрил: 1H-ЯМР (300 МГц, ДМСО-d6) δ ч./млн 5,55 (с, 4Н) 5,89 (д, J=8,1 Гц, 2Н) 6,89 (т, J=8,1 Гц, 1Н). 2-амино-6-нитробензонитрил: 1H-ЯМР (300 МГц, ДМСО-d6) δ ч./млн 6,74 (шир с, 2Н) 7,20 (дд, J=8,3, 1,0 Гц, 1Н) 7,40-7,55 (м, 2Н).
Шаг 2: 2-амино-6-[(3-метил-4-оксохиназолин-6-ил)амино]бензонитрил
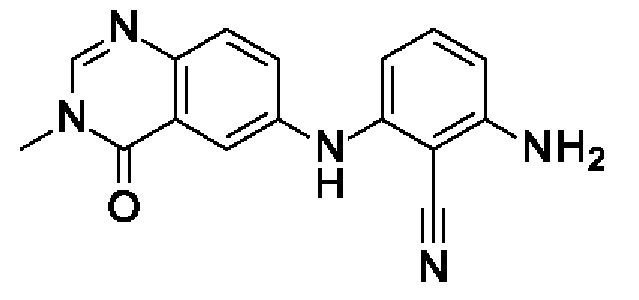
6-Бром-3-метилхиназолин-4(3Н)-он (200 мг, 820 мкмоль, экв.: 1,0) и 2,6-диаминобензонитрил (109 мг, 820 мкмоль, экв.: 1,0) растворяли в диоксане (10 мл), затем добавляли Cs2CO3 (809 мг, 2,46 ммоль, экв.: 3,0). Реакционную смесь продували аргоном, затем добавляли BippyPhos (25,7 мг, 49,2 мкмоль, экв.: 0,06) и хлороформовый аддукт трис(дибензилиденацетон)дипалладия (0) (26 мг, 24,6 мкмоль, экв.: 0,03). Реакционную смесь снова продували аргоном, и флакон закрывали. Реакционную смесь нагревали до 110°С и перемешивали в течение 9,5 ч. Реакционную смесь поглощали в 15 мл 2-метил-ТГФ и льда и промывали 4 мл 1% водн. лимонной кислоты. Водный слой снова экстрагировали 1×15 мл 2-метил-ТГФ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток разбавляли EtOAc и переносили в колонку. Очисткой при помощи флеш-хроматографии (40 г диоксида кремния, 0-100% EtOAc в гептане) получали указанное в заголовке соединение в виде светло-желтого твердого вещества (21 мг, чистота 97%, выход 8,6%). МС (ЭРИ) м/з: 292,1 [М+Н]+.
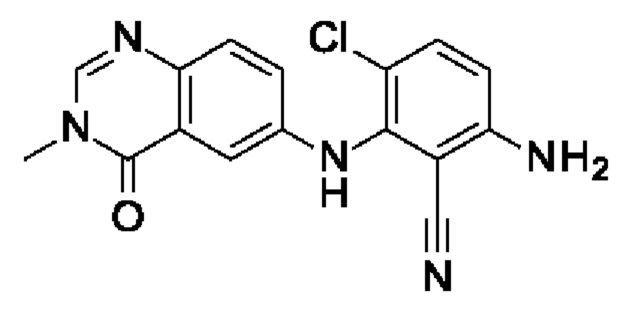
Промежуточное соединение I8: 6-амино-3-хлор-2-[(3-метил-4-оксохиназолин-6-ил)амино]бензонитрил
Шаг 1: 6-нитро-3-метилхиназолин-4-он
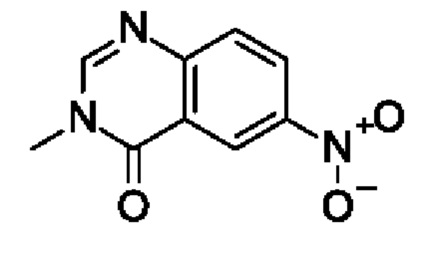
2-Амино-5-нитробензойную кислоту (5 г, 27,5 ммоль, экв.: 1,0) и N-метилформамид (15,2 г, 15 мл, 257 ммоль, экв.: 9,35) нагревали в течение 4 ч при 180°С в герметично закрытой пробирке. Затем реакционную смесь охлаждали до к.т. и вливали в ледяную воду (150 мл). Полученный в результате осадок собирали при помощи фильтрации и промывали дополнительным количеством ледяной воды. Твердое вещество концентрировали два раза до сухого состояния из толуола, затем сушили дополнительно под высоким вакуумом с получением указанного в заголовке соединения в виде желто-коричневого твердого вещества (3,17 г, выход 56%). МС (ЭРИ) м/з: 206,1 [М+Н]+.
Шаг 2: 6-амино-3-метилхиназолин-4-он

3-Метил-6-нитрохиназолин-4-он (1,00 г, 4,87 ммоль, экв.: 1,0) суспендировали в метаноле (25 мл) и АсОН (1 мл). Добавляли палладий на угле (10 мас. % Pd, 100 мг, 940 мкмоль, экв.: 0,193), и реакционную смесь перемешивали при к.т. в атмосфере водорода из баллона. Через 14 ч реакционную смесь фильтровали через целит (элюент МеОН) и концентрировали в вакууме. Остаток концентрировали два раза из толуола, затем сушили дополнительно под высоким вакуумом для получения указанного в заголовке соединения в виде темно-коричневого твердого вещества (821 мг, выход 96%). Материал применяли без дополнительной очистки, но его можно было дополнительно очищать перекристаллизацией из кипящей воды с промыванием полученных в результате темно-коричневых игл холодной водой, изопропанолом и диэтиловым эфиром. МС (ЭРИ) м/з: 176,1 [М+Н]+.
Шаг 3: 3-хлор-6-фтор-2-[(3-метил-4-оксохиназолин-6-ил)амино]бензонитрил
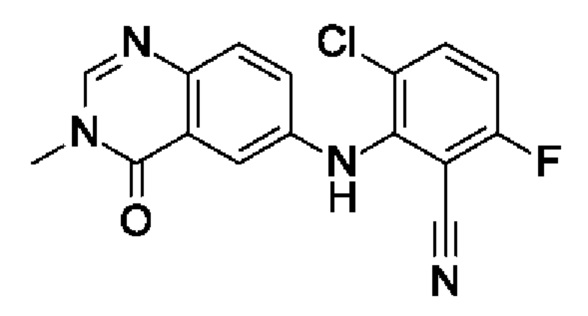
6-Амино-3-метилхиназолин-4-он (200 мг, 1,14 ммоль, экв.: 1,0) и 3-хлор-2,6-дифторбензонитрил (198 мг, 1,14 ммоль, экв.: 1,0) растворяли в ДМСО (3 мл). Добавляли трет-бутоксид калия (141 мг, 1,26 ммоль, экв.: 1,1) и реакционную смесь перемешивали при к.т. в течение 2 ч. Реакционную смесь разбавляли водой и экстрагировали 2×EtOAc. Объединенные органические слои промывали солевым раствором, сушили (MgSO4), фильтровали и концентрировали в вакууме. Очисткой при помощи флеш-хроматографии (24 г, 0-5% МеОН в ДХМ) получали указанное в заголовке соединение в виде желтого твердого вещества (108 мг, выход 29%). МС (ЭРИ) м/з: 329,2 [М+Н]+.
Шаг 4: 6-азидо-3-хлор-2-[(3-метил-4-оксохиназолин-6-ил)амино]бензонитрил
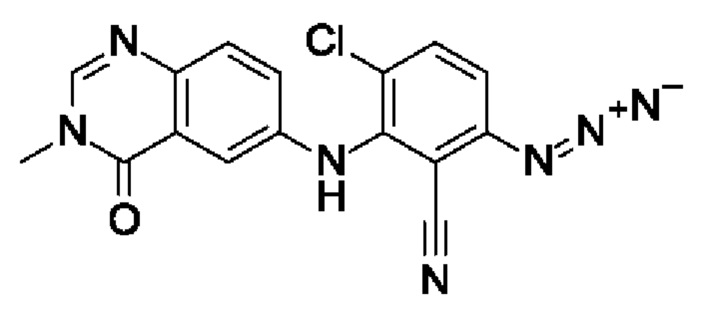
3-Хлор-6-фтор-2-[(3-метил-4-оксохиназолин-6-ил)амино]бензонитрил (320 мг, 973 мкмоль, экв.: 1,0), азид натрия (75,9 мг, 1,17 ммоль, экв.: 1,2) и сухой ДМФА (4 мл) перемешивали при 120°С в течение 1,5 ч в атмосфере азота. Реакционную смесь охлаждали до к.т., а затем разбавляли водой и экстрагировали 2×EtOAc. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения в виде коричневого твердого вещества (372 мг, чистота 92%, колич.). МС (ЭРИ) м/з: 352,2 [М+Н]+.
Шаг 5: 6-амино-3-хлор-2-[(3-метил-4-оксохиназолин-6-ил)амино]бензонитрил

К раствору 6-азидо-3-хлор-2-[(3-метил-4-оксохиназолин-6-ил)амино]бензонитрила (372 мг, 1,06 ммоль, экв.: 1) в 2-пропаноле (10 мл) добавляли триэтиламин (214 мг, 295 мкл, 2,12 ммоль, экв.: 2), 1,3-пропандитиол (80,1 мг, 74,9 мкл, 740 мкмоль, экв.: 0,7) и боргидрид натрия (40 мг, 1,06 ммоль, экв.: 1). Реакционную смесь перемешивали при к.т. в течение ночи, концентрировали в вакууме. EtOAc и лимонную кислоту (10% водн.) добавляли к остатку, фазы отделяли и водную фазу экстрагировали 2×EtOAc. Объединенные органические слои промывали солевым раствором, сушили над Na2SO4, фильтровали и выпаривали до сухого состояния с получением указанного в заголовке соединения в виде желтого твердого вещества (345 мг, колич.). МС (ЭРИ) м/з: 326,1 [М+Н]+.
Промежуточное соединение I9: (3R)-3-фторпирролидин-1-сульфонамид
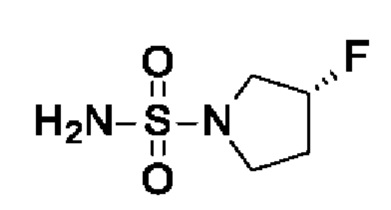
(R)-3-Фторпирролидина гидрохлорид (1,8 г, 14,3 ммоль, экв.: 1,2) добавляли к раствору диамида серной кислоты (1,148 г, 11,9 ммоль, экв.: 1,0) и триэтиламина (2,42 г, 3,33 мл, 23,9 ммоль, экв.: 2,0) в диоксане (10 мл). Реакционную смесь перемешивали в герметично закрытой пробирке при 115°С в течение 15,5 ч, затем охлаждали до к.т. и концентрировали в вакууме. Остаток разбавляли ДХМ, выпаривали с силикагелем до сухого состояния и переносили в колонку. Очисткой при помощи флеш-хроматографии (40 г диоксида кремния, 80% EtOAc) получали указанное в заголовке соединение в виде белого кристаллического твердого вещества (1,82 г, выход 91%). МС (ЭРИ) м/з: 169,1 [М+Н]+.
Промежуточное соединение I10: пирролидин-1-сульфонамид
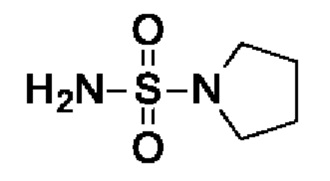
Пирролидин (1,78 г, 2,07 мл, 25 ммоль, экв.: 1,2) добавляли к раствору диамида серной кислоты (2 г, 20,8 ммоль, экв.: 1,0) в диоксане (20 мл). Реакционную смесь перемешивали в герметично закрытой пробирке при 115°С в течение 15,5 ч, затем охлаждали до к.т. и концентрировали в вакууме. Остаток разбавляли МеОН, выпаривали с силикагелем до сухого состояния и переносили в колонку. Очисткой при помощи флеш-хроматографии (40 г диоксида кремния, 0-100% EtOAc в гептане) получали указанное в заголовке соединение в виде белого твердого вещества (2,5 г, выход 80%). МС (ЭРИ) м/з: 151,1 [М+Н]+.
Промежуточное соединение I11: циклопентансульфонамид
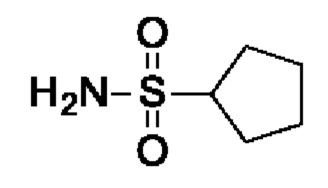
Циклопентансульфонилхлорид (675 мг, 619 мкл, 4 ммоль, экв.: 1,0) добавляли по каплям при к.т. к раствору гидроксида аммония (30-33% в воде, 10,8 г, 12 мл, 92,5 ммоль, экв.: 23,1). Реакционную смесь перемешивали в течение ночи при к.т. Через 20,5 ч добавляли по каплям HCl (25% водн.), пока значение рН раствора не составляло 7. Реакционную смесь экстрагировали 3×EtOAc, объединенные органические слои промывали 1× солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток сушили под высоким вакуумом с получением указанного в заголовке соединения в виде светло-коричневого твердого вещества (658 мг, чистота 91%, выход 100%). 1H-ЯМР (300 МГц, ДМСО-d6) δ ч./млн 1,42-1,74 (м, 4Н) 1,76-1,95 (м, 4Н) 3,33-3,45 (м, 1Н) 6,69 (с, 2Н).
Промежуточное соединение I12: (RS)-бутан-2-сульфонамид
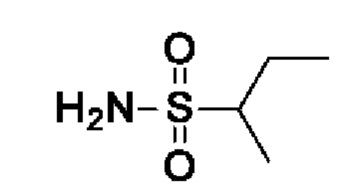
После процедуры, описанной для I11, указанное в заголовке соединение получали из бутан-2-сульфонилхлорида (626 мг, 4 ммоль) в виде светло-желтого масла (397 мг, выход 73%). 1Н ЯМР (300 МГц, ХЛОРОФОРМ-d) δ ч./млн 1,06 (т, J=7,5 Гц, 3Н) 1,41 (д, J=6,9 Гц, 3Н) 1,49-1,68 (м, 1Н) 1,98-2,27 (м, 1Н) 2,85-3,12 (м, 1Н) 4,44 (шир с, 2Н).
Промежуточное соединение I13: циклогексансульфонамид
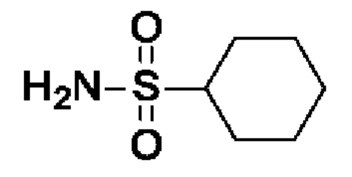
После процедуры, описанной для I11, указанное в заголовке соединение получали из циклогексансульфонилхлорида (568 мг, 2,8 ммоль) в виде белого твердого вещества (371 мг, выход 81%). 1Н-ЯМР (300 МГц, ДМСО-d6) δ ч./млн 1,14-1,41 (м, 5Н) 1,55-1,69 (м, 1Н) 1,72-1,86 (м, 2Н) 2,06 (шир д, J=10,7 Гц, 2Н) 2,63-2,89 (м, 1Н) 6,61 (с, 2Н).
Промежуточное соединение I14: (R)-3-фторпирролидин-1-сульфонилхлорид
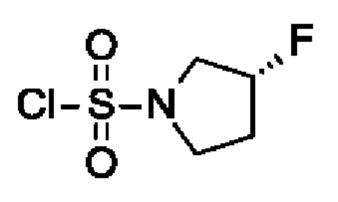
В 500-мл 4-горлой колбе (продутой аргоном; оснащенной термометром и капельной воронкой) (R)-3-фторпирролидина гидрохлорид (7 г, 55,7 ммоль, экв.: 1,0) объединяли с ДХМ (200 мл) в постоянном потоке аргона. ДИПЭА (21,6 г, 29,2 мл, 167 ммоль, экв.: 3,0) добавляли для получения светло-желтого раствора и реакционную смесь охлаждали до -70°С. Сульфурилдихлорид (15 г, 9,01 мл, 111 ммоль, экв.: 2,0) в ДХМ (20 мл) медленно добавляли через капельную воронку, при этом поддерживали температуру при -70°С. Реакционную смесь перемешивали при -70°С в течение 1 ч, а затем давали достичь к.т. на протяжении 1 ч. Реакционную смесь вливали в ледяную воду в колбе Эрленмейера. Смесь переносили в делительную воронку и фазы разделяли. Органический слой промывали 1 н. водн. HCl (100 мл). Водные слои экстрагировали еще двумя порциями ДХМ (каждая 80 мл). Органические слои объединяли, сушили над Na2SO4, фильтровали и выпаривали до сухого состояния, затем сушили под высоким вакуумом в течение 1 ч с получением указанного в заголовке соединения в виде коричневого твердого вещества (11,0 г, колич. выход), которое хранили при 4°С до применения. 1H-ЯМР (300 МГц, ХЛОРОФОРМ-d) δ ч./млн 1,97-2,60 (м, 2Н) 3,53-3,92 (м, 4Н) 5,04-5,64 (м, 1Н)
Промежуточное соединение I15: пирролидин-1-сульфонилхлорид
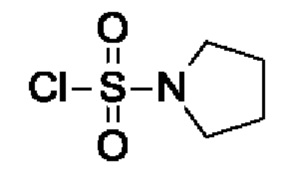
После процедуры, описанной для промежуточного соединения I14, указанное в заголовке соединение получали из пирролидина (2,17 г, 2,5 мл, 30,4 ммоль, экв.: 1,0) и сульфурилдихлорида (8,22 г, 4,92 мл, 60,9 ммоль, экв.: 2,0) в виде коричневой жидкости (4,4 г, выход 85%). 1Н-ЯМР (300 МГц, ХЛОРОФОРМ-d) δ ч./млн 1,90-2,14 (м, 4Н) 3,36-3,63 (м, 4Н).
Пример 1: 6-[2-циано-3-[[этил(метил)сульфамоил]амино]-6-фторфенокси]-3-метил-4-оксохиназолин
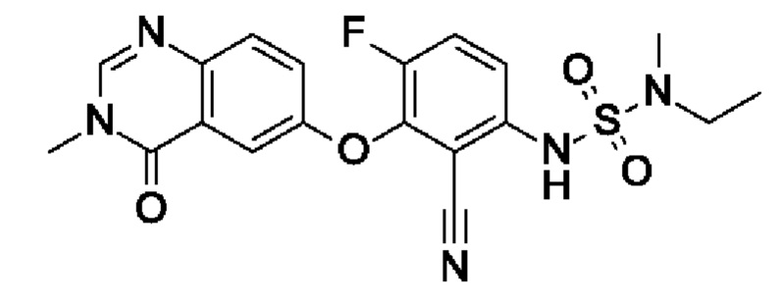
[Метил(сульфамоил)амино]этан (185 мг, 1,34 ммоль, экв.: 2,1) растворяли в NMP (8 мл). При 0°С добавляли NaH (60% в минеральном масле, 61,3 мг, 1,4 ммоль, экв.: 2,2), охлаждающую баню удаляли и реакционную смесь перемешивали при 50°С в течение 30 мин. Реакционную смесь охлаждали до 0°С, затем добавляли раствор I2 (200 мг, 638 мкмоль, экв.: 1,0) в NMP (2 мл). Реакционную смесь перемешивали при 125°С в течение 1 ч. Реакционную смесь охлаждали до к.т., и реакционную смесь поглощали в 10 мл 0,1 М водн. NaOH, льда и EtOAc. Водный слой отделяли и снова экстрагировали с помощью EtOAc. Водный слой подкисляли 2 М водн. HCl до рН 4 и экстрагировали 2×EtOAc, объединенные органические слои промывали 3×водой, 1×солевым раствором, затем сушили над Na2SO4, фильтровали и выпаривали. Остаток разбавляли ДХМ, выпаривали с силикагелем до сухого состояния и переносили в колонку. Очисткой при помощи флеш-хроматографии (40 г диоксида кремния, 0-100% EtOAc в гептане) получали указанное в заголовке соединение в виде белого твердого вещества (161 мг, чистота 97%, выход 57%). МС (ЭРИ) м/з: 432,2 [М+Н]+.
Пример 2: 6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолин
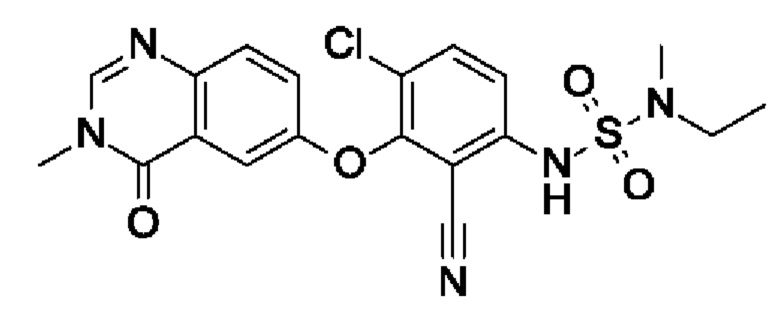
I4 (45 мг, 138 мкмоль, экв.: 1,0) растворяли в ДХМ (275 мкл) в герметично закрытой пробирке. Пиридин (490 мг, 501 мкл, 6,2 ммоль, экв.: 45), ДМАП (1,68 мг, 13,8 мкмоль, экв.: 0,1) и N-этил-N-метилсульфамоилхлорид (65 мг, 413 мкмоль, экв.: 3,0) добавляли при к.т. Пробирку герметично закрывали и реакционную смесь нагревали при 80°С. Через 24 ч реакционную смесь охлаждали до к.т., разбавляли ДХМ (20 мл), промывали 10% водн. лимонной кислотой (2×20 мл), водой (20 мл) и солевым раствором (20 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Неочищенную смесь загружали в сухом виде на Isolute и очищали при помощи флеш-хроматографии (50-100% EtOAc в гептане).
Полученный в результате материал дополнительно очищали при помощи обращенно-фазовой ВЭЖХ с получением указанного в заголовке соединения в виде бесцветного лиофилизированного твердого вещества (20 мг, чистота 100%, выход 32%). МС (ЭРИ) м/з: 448,2, 450,1 [М+Н]+.
Пример 3: (3R)-N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамид
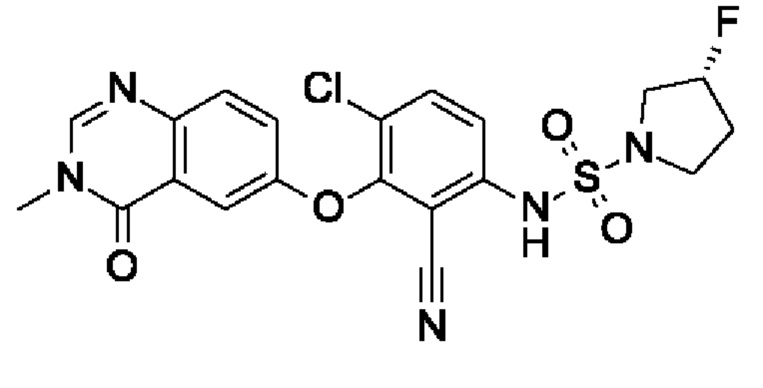
После процедуры, описанной для примера 2, указанное в заголовке соединение получали из I4 (55 мг, 168 мкмоль, экв.: 1,0) и I14 (129 мг, 688 мкмоль, экв.: 4,1) в виде грязно-белого лиофилизированного твердого вещества с последующей очисткой при помощи обращенно-фазовой ВЭЖХ (7 мг, чистота 98%, выход 8,5%). МС (ЭРИ) м/з: 478,0759 [М+Н]+.
Пример 4: N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамид
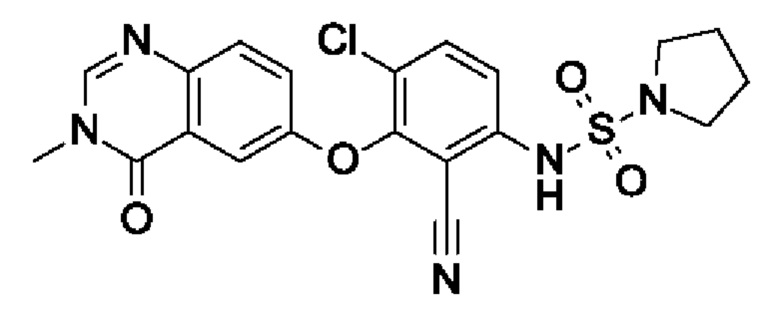
После процедуры, описанной для примера 2, указанное в заголовке соединение получали из I4 (100 мг, 306 мкмоль, экв.: 1,0) и I15 (156 мг, 918 мкмоль, экв.: 3,0) в виде грязно-белого твердого вещества с последующей флеш-хроматографией (1-6% МеОН в ДХМ) (25 мг, чистота 98%, выход 17%). МС (ЭРИ) м/з: 460,2, 462,2 [М+Н]+.
Пример 5: 6-[2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолин
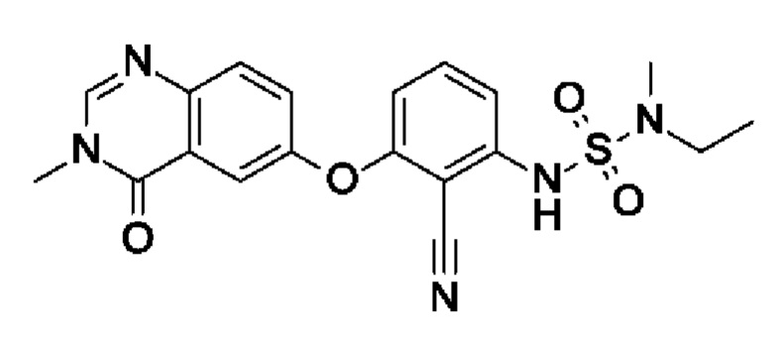
[Метил(сульфамоил)амино]этан (98,3 мг, 711 мкмоль, экв.: 2,1) растворяли в ДМФА (4 мл). При 0°С добавляли NaH (60% в минеральном масле, 32,5 мг, 745 мкмоль, экв.: 2,2), охлаждающую баню удаляли и реакционную смесь перемешивали при 50°С в течение 20 мин. Реакционную смесь охлаждали до 0°С, затем добавляли I3 (100 мг, 339 мкмоль, экв.: 1,0). Реакционную смесь перемешивали при 100°С в течение 21 ч, затем концентрировали в вакууме. Остаток суспендировали в насыщ. водн. NH4Cl (40 мл) и экстрагировали с помощью ДХМ (3 × 40 мл). Объединенные органические слои промывали солевым раствором (100 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Очисткой при помощи СФХ получали указанное в заголовке соединение в виде бесцветного твердого вещества (60 мг, чистота 98%, выход 42%). МС (ЭРИ) м/з: 414,2 [М+Н]+.
Пример 6: N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамид
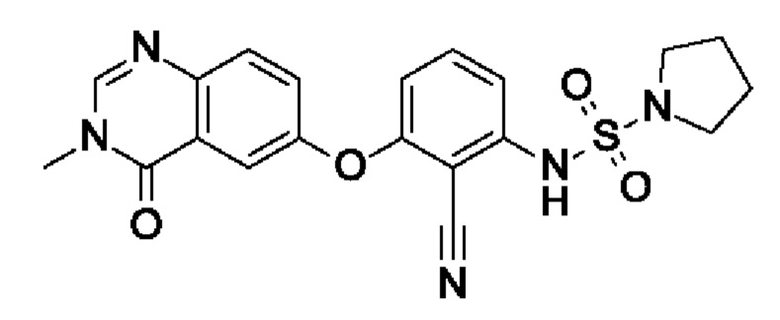
После процедуры, описанной для примера 5, указанное в заголовке соединение получали из I3 (100 мг, 339 мкмоль, экв.: 1,0) и I10 (107 мг, 711 мкмоль, экв.: 2,1) в виде бесцветного твердого вещества с последующей очисткой при помощи СФХ (48 мг, чистота 100%, выход 33%). МС (ЭРИ) м/з: 426,3 [М+Н]+.
Пример 7: (3R)-N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамид
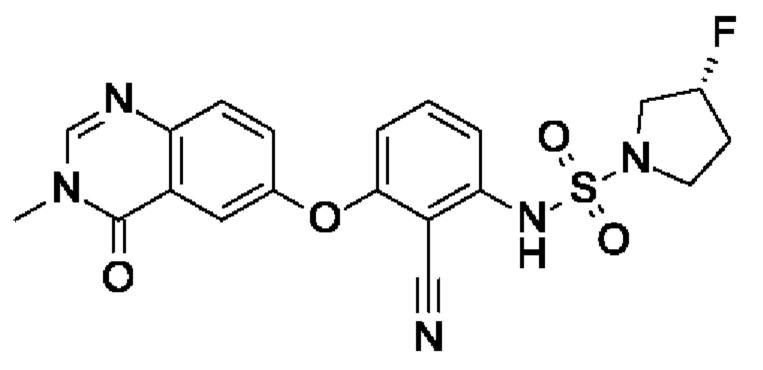
После процедуры, описанной для примера 5, указанное в заголовке соединение получали из I3 (100 мг, 339 мкмоль, экв.: 1,0) и I9 (120 мг, 711 мкмоль, экв.: 2,1) в виде бесцветного твердого вещества с последующей очисткой при помощи СФХ (80 мг, чистота 93%, выход 50%). МС (ЭРИ) м/з: 444,2 [М+Н]+.
Пример 8: N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамид
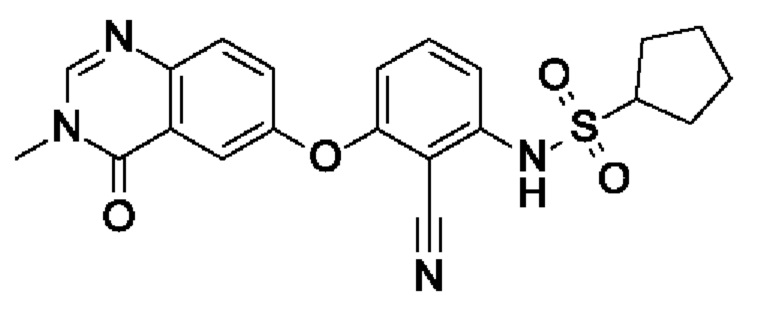
После процедуры, описанной для примера 5, указанное в заголовке соединение получали из I3 (75 мг, 254 мкмоль, экв.: 1,0) и I11 (79,6 мг, 533 мкмоль, экв.: 2,1) в виде бесцветного твердого вещества с последующей очисткой при помощи обращенно-фазовой ВЭЖХ (63 мг, чистота 100%, выход 58%). МС (ЭРИ) м/з: 425,3 [М+Н]+.
Пример 9: N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклогексансульфонамид
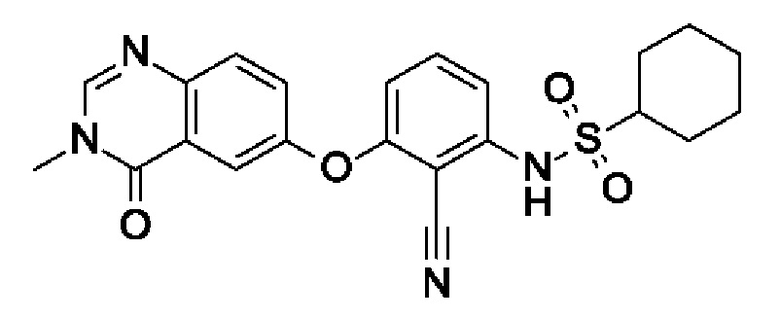
После процедуры, описанной для примера 5, указанное в заголовке соединение получали из I3 (75 мг, 254 мкмоль, экв.: 1,0) и I13 (87,1 мг, 533 мкмоль, экв.: 2,1) в виде бесцветного твердого вещества с последующей очисткой при помощи обращенно-фазовой ВЭЖХ (71 мг, чистота 98%, выход 63%). МС (ЭРИ) м/з: 439,3 [М+Н]+.
Пример 10: (RS)-N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]бутан-2-сульфонамид
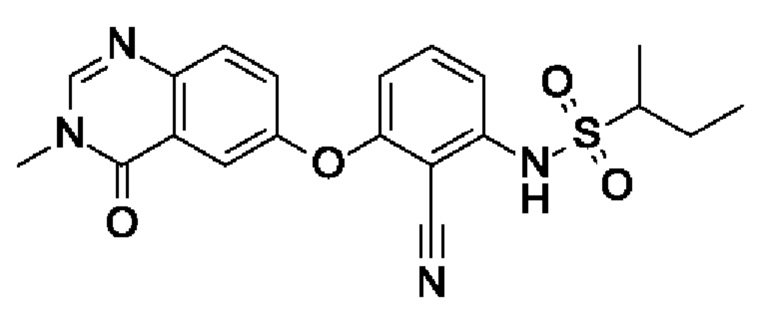
После процедуры, описанной для примера 5, указанное в заголовке соединение получали из I3 (75 мг, 254 мкмоль, экв.: 1,0) и I12 (73,2 мг, 533 мкмоль, экв.: 2,1) в виде бесцветного твердого вещества с последующей очисткой при помощи обращенно-фазовой ВЭЖХ (51 мг, чистота 100%, выход 49%). МС (ЭРИ) м/з: 413,3 [М+Н]+.
Пример 11: 6-[2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолин
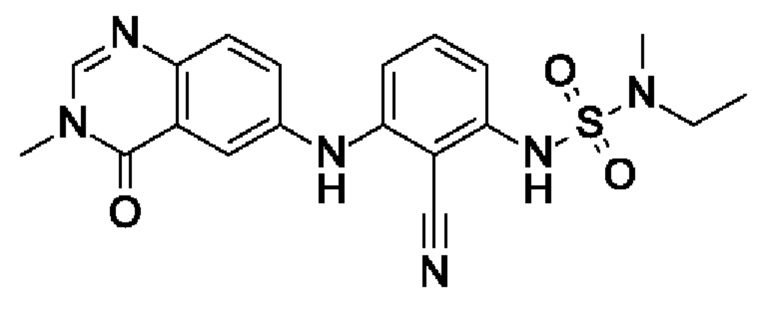
6-Бром-3-метилхиназолин-4(3Н)-он (21 мг, 86,1 мкмоль, экв.: 1,0) и 15 (21,9 мг, 86,1 мкмоль, экв.: 1,0) растворяли в диоксане (1,6 мл), затем добавляли Cs2CO3 (85 мг, 258 мкмоль, экв.: 3,0). Реакционную смесь продували аргоном, затем BippyPhos (2,7 мг, 5,16 мкмоль, экв.: 0,06) и хлороформовым аддуктом трис(дибензилиденацетон)дипалладия (0) (2,73 мг, 2,58 мкмоль, экв.: 0,03). Реакционную смесь снова продували аргоном, и флакон закрывали. Реакционную смесь нагревали до 110°С и перемешивали в течение 1 ч. Реакционную смесь поглощали в 15 мл 2-метил-ТГФ и льда и промывали 4 мл 1% водн. лимонной кислоты. Водный слой снова экстрагировали 1×15 мл 2-метил-ТГФ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток разбавляли EtOAc и переносили в колонку. Очисткой при помощи флеш-хроматографии (40 г диоксида кремния, 100% EtOAc) с последующей препаративной обращенно-фазовой ВЭЖХ получали указанное в заголовке соединение в виде белого твердого вещества (13 мг, чистота 94%, выход 53%). МС (ЭРИ) м/з: 413,2 [М+Н]+.
Пример 12: (3R)-N-[2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]-3-фторпирролидин-1-сульфонамид
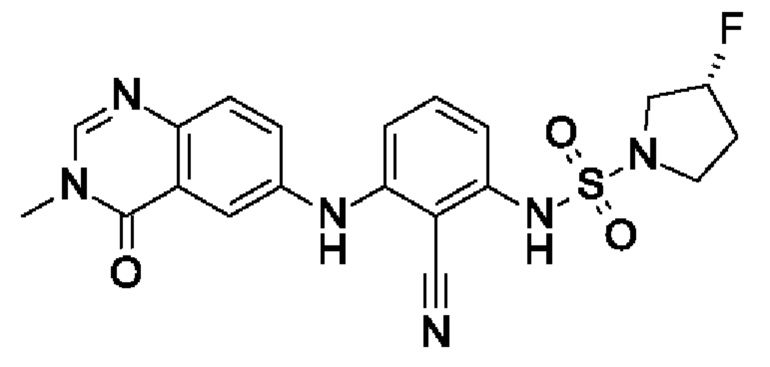
I7 (20,5 мг, 70,4 мкмоль, экв.: 1,0) растворяли в ДХМ (150 мкл). При к.т. добавляли пиридин (256 мг, 260 мкл, 3,24 ммоль, экв.: 46), ДМАП (877 мкг, 7,04 мкмоль, экв.: 0,1) и I14 (39,6 мг, 211 мкмоль, экв.: 3,0). Реакционную смесь перемешивали при 75°С в течение 21,5 ч. Реакционную смесь поглощали в 2-метил-ТГФ, льду и 1% водн. лимонной кислоте. Водный слой подвергали обратной экстракции 2×2-метил-ТГФ. Органические слои объединяли, промывали солевым раствором, сушили над Na2SO4 и концентрировали в вакууме. Остаток разбавляли ДХМ, выпаривали с силикагелем до сухого состояния и переносили в колонку. Очисткой при помощи флеш-хроматографии (25 г диоксида кремния, 100% EtOAc) получали указанное в заголовке соединение в виде светло-желтого твердого вещества (5,1 мг, чистота 95%, выход 16%). МС (ЭРИ) м/з: 443,2 [М+Н]+.
Пример 13: 6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолин
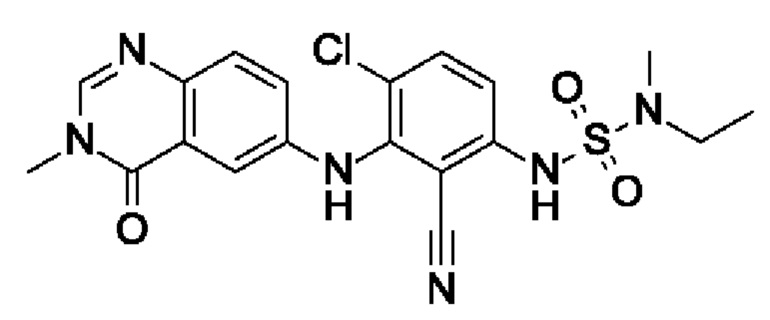
I8 (75 мг, 230 мкмоль, экв.: 1) растворяли в пиридине (1 мл) и ДХМ (1 мл) во флаконе. Этил(метил)сульфамоилхлорид (90,7 мг, 576 мкмоль, экв.: 2,5) и ДМАП (1,41 мг, 11,5 мкмоль, экв.: 0,05) в ДХМ (1 мл) добавляли к реакционной смеси, и флакон герметично закрывали. Реакционную смесь перемешивали в течение двух дней при 60°С. Реакционную смесь гасили водой, разбавляли ДХМ и промывали 2 × лимонной кислотой (10% водн.). Водные слои экстрагировали 2×ДХМ. Органические слои объединяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Очисткой при помощи флеш-хроматографии (12 г диоксида кремния, 0-5% МеОН в ДХМ) получали указанное в заголовке соединение в виде оранжевого твердого вещества (25 мг, чистота 95%, выход 24%). МС (ЭРИ) м/з: 447,1 [М+Н]+.
Пример 14: N-[4-хлор-2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]пирролидин-1-сульфонамид
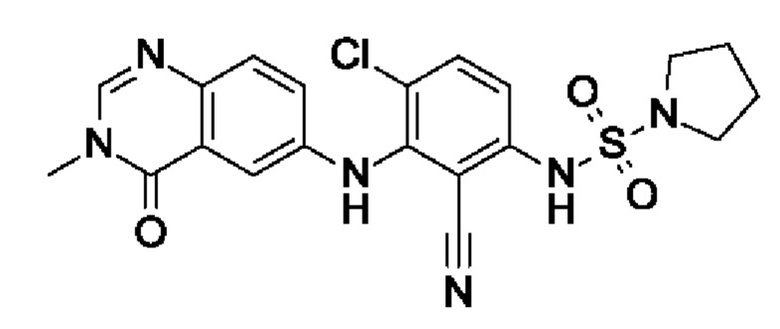
После процедуры, описанной для примера 13, указанное в заголовке соединение получали из I8 (75 мг, 230 мкмоль, экв.: 1,0) и I15 (122 мг, 576 мкмоль, экв.: 2,5) в виде белого твердого вещества (10 мг, чистота 100%, выход 9%) после очистки при помощи препаративной обращенно-фазовой ВЭЖХ. МС (ЭРИ) м/з: 459,2 [М+Н]+.
Пример 15: N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамид
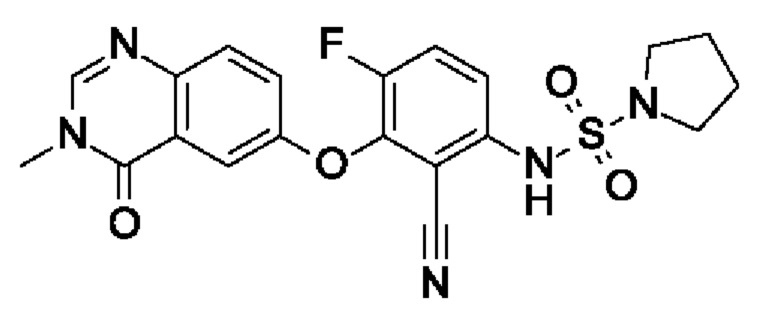
I10 (50,3 мг, 335 мкмоль, экв.: 2,1) и карбонат цезия (114 мг, 351 мкмоль, экв.: 2,2) объединяли в высушенной путем нагревания реакционной пробирке. Пробирку располагали в атмосфере аргона и добавляли ДМФА (456 мкл). Смесь перемешивали в течение 30 минут при 50°С. Затем обеспечивали охлаждение реакционной смеси до комнатной температуры. Добавляли I2 (50 мг, 160 мкмоль, экв.: 1,0) в ДМФА (1,14 мл). Пробирку герметично закрывали и перемешивали реакционную смесь при 100°С в течение 16 ч. Смесь поглощали в насыщ. водн. NH4Cl (10 мл) и EtOAc (10 мл). Фазы разделяли, и водный слой дополнительно экстрагировали 3×10 мл EtOAc. Объединенные органические слои промывали водой (30 мл) и солевым раствором (30 мл), сушили (Na2SO4), фильтровали и концентрировали в вакууме. Остаток разбавляли ДХМ и переносили в колонку. Очисткой при помощи флеш-хроматографии (12 г диоксида кремния, 0-3% МеОН/ДХМ) получали указанное в заголовке соединение (40 мг, чистота 100%, выход 56,5%) в виде бесцветного твердого вещества. МС (ЭРИ) м/з: 444,2 [М+Н]+.
Пример 16: N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамид
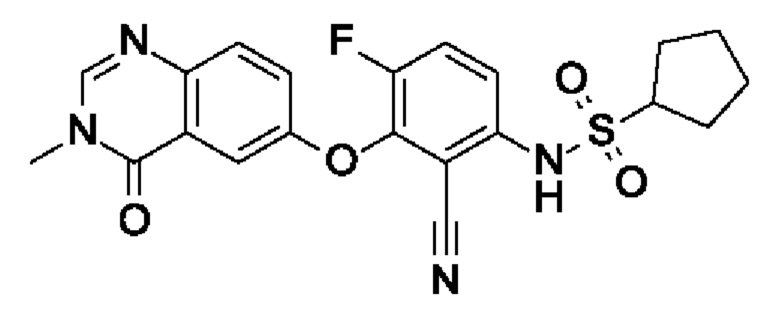
После процедуры, описанной для примера 15, указанное в заголовке соединение получали из I11 (50 мг, 335 мкмоль, экв.: 2,1) и I2 (50 мг, 160 мкмоль, экв.: 1,0) в виде бесцветного твердого вещества (43 мг, чистота 100%, выход 61%) после очистки при помощи флеш-хроматографии и СФХ. МС (ЭРИ) м/з: 443,2 [М+Н]+.
Пример 17: 6-[2-циано-3-(диметилсульфамоиламино)-6-фторфенокси]-3-метил-4-оксохиназолин
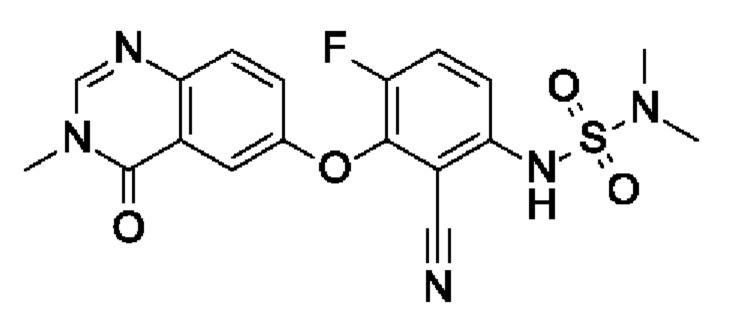
После процедуры, описанной для примера 15, указанное в заголовке соединение получали из N,N-диметилсульфамида (41,6 мг, 335 мкмоль, экв.: 2,1) и I2 (50 мг, 160 мкмоль, экв.: 1,0) в виде белой пены (34 мг, чистота 100%, выход 51%) после флеш-хроматографии. МС (ЭРИ) м/з: 418,2 [М+Н]+.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВЫЕ ПРОИЗВОДНЫЕ МЕТИЛХИНАЗОЛИНОНА | 2020 |
|
RU2802968C1 |
| СОЕДИНЕНИЯ 4-ОКСО-3,4-ДИГИДРОХИНАЗОЛИНОНА ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2021 |
|
RU2814662C1 |
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4-ОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2020 |
|
RU2797606C1 |
| НОВЫЕ СУЛЬФОНАМИДКАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ | 2018 |
|
RU2808572C2 |
| МАКРОЦИКЛИЧЕСКИЕ АМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕАЗЫ | 2013 |
|
RU2625796C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ПИРРОЛИДИНА | 2017 |
|
RU2736498C1 |
| СОЕДИНЕНИЯ | 2019 |
|
RU2800063C2 |
| Производные хинолона как ингибиторы рецептора фактора роста фибробластов | 2015 |
|
RU2721723C2 |
| Соединения, активные по отношению к бромодоменам | 2015 |
|
RU2743074C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АЗЕТИДИНА | 2013 |
|
RU2629114C2 |
Изобретение относится к соединению, выбранному из 6-[2-циано-3-[[этил(метил)сульфамоил]амино]-6-фторфенокси]-3-метил-4-оксохиназолина; 6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина; (3R)-N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамида; N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида; 6-[2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина; N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида; (3R)-N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамида; N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида; N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклогексансульфонамида; N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]бутан-2-сульфонамида; 6-[2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина; (3R)-N-[2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]-3-фторпирролидин-1-сульфонамида; 6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина; N-[4-хлор-2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]пирролидин-1-сульфонамида; N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида; N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида и 6-[2-циано-3-(диметилсульфамоиламино)-6-фторфенокси]-3-метил-4-оксохиназолина; или его фармацевтически приемлемой соли, которые являются ингибиторами BRAF. Изобретение относится также к фармацевтической композиции, содержащей указанное соединение, его применению для лечения или профилактики обусловленного мутацией BRAF рака, выбранного из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы и немелкоклеточного рака легкого (НМРЛ), и для получения лекарственного средства для лечения или профилактики указанных видов рака, а также к способу их лечения. 6 н. и 2 з.п. ф-лы, 9 табл., 17 пр.
1. Соединение, выбранное из
6-[2-циано-3-[[этил(метил)сульфамоил]амино]-6-фторфенокси]-3-метил-4-оксохиназолина;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
(3R)-N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамида;
N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
(3R)-N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]-3-фторпирролидин-1-сульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклогексансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]бутан-2-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
(3R)-N-[2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]-3-фторпирролидин-1-сульфонамида;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
N-[4-хлор-2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]пирролидин-1-сульфонамида;
N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида и
6-[2-циано-3-(диметилсульфамоиламино)-6-фторфенокси]-3-метил-4-оксохиназолина;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, выбранное из
6-[2-циано-3-[[этил(метил)сульфамоил]амино]-6-фторфенокси]-3-метил-4-оксохиназолина;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
N-[4-хлор-2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]фенокси]-3-метил-4-оксохиназолина;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклогексансульфонамида;
N-[2-циано-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]бутан-2-сульфонамида;
6-[2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
6-[6-хлор-2-циано-3-[[этил(метил)сульфамоил]амино]анилино]-3-метил-4-оксохиназолина;
N-[4-хлор-2-циано-3-[(3-метил-4-оксохиназолин-6-ил)амино]фенил]пирролидин-1-сульфонамида;
N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]пирролидин-1-сульфонамида;
N-[2-циано-4-фтор-3-(3-метил-4-оксохиназолин-6-ил)оксифенил]циклопентансульфонамида и
6-[2-циано-3-(диметилсульфамоиламино)-6-фторфенокси]-3-метил-4-оксохиназолина;
или его фармацевтически приемлемая соль.
3. Соединение по любому из пп. 1 и 2 для применения в качестве ингибитора BRAF.
4. Фармацевтическая композиция, ингибирующая BRAF, содержащая эффективное количество соединения по любому из пп. 1 и 2 и терапевтически инертный носитель.
5. Соединение по любому из пп. 1 и 2 для применения в лечении или профилактике обусловленного мутацией BRAF рака, выбранного из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы и немелкоклеточного рака легкого (НМРЛ).
6. Применение соединения по любому из пп. 1 и 2 для лечения или профилактики обусловленного мутацией BRAF рака, выбранного из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы и НМРЛ.
7. Применение соединения по любому из пп. 1 и 2 для получения лекарственного средства для лечения или профилактики обусловленного мутацией BRAF рака, выбранного из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы и НМРЛ.
8. Способ лечения или профилактики обусловленного мутацией BRAF рака, выбранного из рака щитовидной железы, колоректального рака, рака головного мозга, меланомы и НМРЛ, причем указанный способ включает введение эффективного количества соединения, определенного в любом из пп. 1 и 2, пациенту, который в этом нуждается.
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| S | |||
| WENGLOWSKY et al | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Пишущая машина для тюркско-арабского шрифта | 1922 |
|
SU24A1 |
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Раздвижной золотник-байпас | 1925 |
|
SU1923A1 |
| ИНГИБИТОР BRAF КИНАЗЫ N-(3-(5-(4-ХЛОРОФЕНИЛ)-1H-ПИРАЗОЛО[3,4-B]ПИРИДИН-3-КАРБОНИЛ)-2,4-ДИФТОРОФЕНИЛ) ПРОПАН-1-СУЛЬФОНАМИД | 2018 |
|
RU2687107C1 |

