ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет предварительной заявки США №63/036, 522, поданной 9 июня 2020 г., и предварительной заявки США №63/116,204, поданной 20 ноября 2020 г., и предварительной заявки США №63/175,655, поданной 16 апреля 2021 г., содержание каждого из которых полностью включено в настоящий документ посредством ссылки.
ОБЛАСТЬ ТЕХНИКИ
Настоящее описание относится к новым соединениям хиназолинона или их фармацевтически приемлемым солям, к фармацевтическим композициям, содержащим такие соединения и соли, и к способам применения таких соединений, солей и композиций для лечения аномального роста клеток, включая рак, у субъекта.
УРОВЕНЬ ТЕХНИКИ
Настоящее изобретение относится к хиназолинонам для лечения BRAF-ассоциированных заболеваний и нарушений, включая BRAF-ассоциированные опухоли, включая злокачественные и доброкачественные BRAF-ассоциированные опухоли ЦНС и злокачественные экстракраниальные BRAF-ассоциированные опухоли.
Белок BRAF, член RAF семейства серии/треонинкиназ, участвует в каскаде пути Ras-Raf-MEK-киназа, регулируемая внеклеточными сигналами (ERK) или сигнального пути митоген-активируемой протеинкиназы (MAPK)/ERK, который влияет на деление и дифференциацию клеток. Мутации в гене BRAF могут привести к неконтролируемому росту и последующему образованию опухоли. При раке было идентифицировано более 100 уникальных мутаций в гене BRAF (Cerami, Е., et al., Cancer Discov. 2012, 2, 401-404). Эти мутации приводят к активации ERK через различные функциональные механизмы, и сгруппированы в три класса, два из которых называются мутациями I класса и II класса, на основании их зависимости от димеризации и активации RAS для активности; эти свойства определяют их чувствительность к ингибиторам RAF (Yao, A., et al., Cancer Cell 2015, 28, 370-383).
Активирующие мутации BRAF I класса, такие как V600E и/или V600K, были обнаружены при раке человека, таком как меланома, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников, почечно-клеточный рак и его метастатический рак, а также первичные опухоли головного мозга. Мутации I класса, такие как мутанты BRAF V600, подают сигналы как RAS-независимые активные мономеры.
Мутации BRAF II класса включают не-V600 мутации, которые активируют MEK через димеризацию, но без необходимости в RAS (Yao, A., et al., Cancer Cell 2015, 28, 370-383). Эти мутации II класса подвергаются конститутивной, RAS-независимой димеризации, что приводит к повышенной активации ERK с низкой активностью RAS из-за отрицательной обратной связи. Распространенные точечные мутации II класса включают G469A/V/R, K601E/N/T и L597Q/V. Не-V600 мутанты резистентны к ингибиторам BRAF I класса, таким как вемурафениб. He-V600 мутанты BRAF также были обнаружены при многих видах рака и более распространены, чем мутации V600, в определенных типах опухолей. He-V600 мутации BRAF обнаруживаются в 5-16% меланом, а также во множестве других типов опухолей (Siroy АЕ, et al., J Invest Dermatol. 2015; 135:508-515; Dahlman KB, et al. Cancer Discov. 2 012;2:791-797). Приблизительно 50-80% мутаций BRAF при немелкоклеточном раке легкого, и 22-30% при колоректальном раке кодируют не-V600 мутации. (Jones JC, et al. J Clin Oncol. 2017; 35:2624-2630; Paik PK, et al. J Clin Oncol. 2011;29:2046-2051). Мутации BRAF II класса, такие как G469A, G469R, G469V, K601E, K601N, K601T, L597Q и L597V, были идентифицированы в глиомах (Schreck, К.С.et al., Cancers (2019) 11:1262) и других опухолях, таких как рак молочной железы, мелкоклеточный рак легкого, рак поджелудочной железы, рак щитовидной железы, рак предстательной железы, аденокистозная карцинома, рак аппендикса, рак тонкой кишки, плоскоклеточный рак головы и шеи и ангиосаркома (Sullivan, R.J., Cancer Discov February 1 2018 (8) (2) 184-195). Мутации BRAF II класса также были выявлены при метастатическом раке (Dagogo-Jack, I., Clin Cancer Res. Sept. 2018; Schirripa, M., Clin Cancer Res., May 2019; Menzer, C., J. Clin Oncol 2019, 37 (33): 3142-3151).
Кроме того, делеции BRAF внутри рамки считывания могут функционировать как мутации II класса. Например, у пациентов, получавших ингибиторы BRAF V600, наблюдалась приобретенная резистентность. Механизмы приобретенной резистентности включают альтернативный сплайсинг. Сплайс-варианты BRAF кодируют активную киназу, но не имеют интактного домена связывания RAS. Было обнаружено, что клетки, резистентные к вемурафенибу, экспрессируют вариантные формы BRAF V600E, в которых отсутствуют экзоны, охватывающие RAS-связывающий домен, в частности, отсутствуют экзоны 4-10, экзоны 4-8, экзоны 2-8 или экзоны 2-10 (Poulikakos, P.I., et al., Nature, 480(7377):387-390.
В настоящее время не существует эффективного таргетного лечения для пациентов, имеющих не-V600 изменения BRAF или мутации резистентности к ингибитору BRAF.
Хотя некоторые ингибиторы мутаций BRAF V600 вызывают отличные экстракраниальные ответы, метастазы рака в головной мозг все же могут развиваться во время или после терапии ингибиторами BRAF (Oliva I.C.G, et al., Annals of Oncology, 29: 1509-1520 (2018)). По оценкам, у 20% всех субъектов с раком развиваются метастазы в головной мозг, при этом большинство метастазов в головной мозг возникает у лиц с меланомой, колоректальным раком, раком легких и почечно-клеточным раком (Achrol A.S., et al., Nature Reviews (2019), 5:5, pp. 1-26). Хотя это наиболее вероятные типы рака, любой тип рака может распространиться на мозг.Развитие метастазов в головной мозг остается существенным фактором общей смертности от рака у пациентов с раком на поздних стадиях, поскольку прогноз остается неблагоприятным, несмотря на мультимодальные методы лечения и достижения в области системной терапии, которая включает в себя комбинации хирургии, лучевой терапии, химиотерапии, иммунотерапии и/или таргетных терапий.
BRAF также был идентифицирован как потенциальная мишень для лечения первичных опухолей головного мозга. Преобладание мутации BRAF-V600E в первичных опухолях головного мозга было описано Schindler et al. (Acta Neuropathol 121(3):397-405, 2011) на основе анализа 1320 опухолей центральной нервной системы (ЦНС), и Behling et al. (Diagn Pathol 11(1):55, 2016), который проанализировал 969 опухолей ЦНС у детей и взрослых. Эти исследования, в комбинации с другими, сообщают о наличии мутаций BRAF-V600E при различных видах рака, включая папиллярные краниофарингиомы, плеоморфные ксантоастроцитомы (РХА), ганглиоглиомы, астробластомы и другие. (Behling et al., Diagn Pathol 11(1):55, 2016; Brastianos et al., Nat Genet 46(2):161-165, 2014; Dougherty et al., Neuro Oncol 12(7):621-630, 2010; Lehman et al., Neuro Oncol 19(1):31-42, 2017; Mordechai et al., Pediatr Hematol Oncol 32(3):207-211, 2015; Myung et al., Transl Oncol 5(6):430-436, 2012; Schindler et al., Acta Neuropathol 121(3):397-405, 2011).
Также были описаны раки, включая метастатические раки, имеющие BRAF-слитые белки (J.S. Ross, et al., Int. J. Cancer: 138, 881-890 (2016)).
Гематоэнцефалические интерфейсы включают эндотелий церебральных микрососудов, образующий гематоэнцефалический барьер (ВВВ), и эпителий сосудистых сплетений, образующий гемато-СМЖ барьер (BCSFB). Гематоэнцефалический барьер (ВВВ) представляет собой высокоселективный физический, транспортный и метаболический барьер, отделяющий ЦНС от крови. ВВВ может препятствовать проникновению некоторых лекарственных средств в ткань головного мозга и является ограничивающим фактором в доставке многих периферически вводимых агентов в ЦНС. Многие лекарства, обычно используемые для лечения рака, не способны проникать через ВВВ. Это означает, что лекарственные средства не способны проникать в мозг и, следовательно, не могут эффективно убивать раковые клетки в мозге. Современные методы лечения субъектов с опухолями головного мозга включают хирургическую резекцию, лучевую терапию и/или химиотерапию такими агентами, как темозоломид и/или бевацизумаб. Однако хирургическое лечение рака головного мозга не всегда возможно или желательно, например, опухоль может быть недоступна или субъект может быть неспособен выдержать нейрохирургическую травму. Кроме того, известно, что лучевая терапия и лечение цитотоксическими агентами имеют нежелательные побочные эффекты. Например, появляется все больше свидетельств того, что применение темозоломида само по себе может вызывать мутации и ухудшать прогноз у значительной части субъектов (В.Е. Johnson et al., Science 343: 189-193 (2014)), и маркировка бевацизумаба содержит предупреждения о перфорация желудочно-кишечного тракта, хирургических и раневых осложнениях, кровотечениях. Ингибиторы киназы применимы для лечения многих периферических раков. Однако из-за своих структурных характеристик, многие ингибиторы киназ, такие как ингибиторы BRAF (например, вемурафениб и дабрафениб), являются субстратами активных транспортеров, таких как Р-гликопротеины (Р-gp) или белок резистентности рака молочной железы (BCRP). Например, сообщалось, что дабрафениб имеет коэффициент эффлюкса MDR1 11,4, коэффициент эффлюкса BCRP 21,0 и общее отношение мозга к плазме 0,023; свободное отношение мозга к плазме не сообщалось (Mittapalli, RK, et al., J Pharmacol. Exp Ther 344:655-664, March 2013), и вемурафениб, как сообщается, имеет коэффициент эффлюкса MDR1, равный 83, и коэффициент эффлюкса BCRP 4 95 и общее отношение мозга к плазме 0,004; свободное соотношение мозга к плазме не сообщалось (Mittapalli, RK. et al., J Pharmacol. Exp Ther 342:33-40 (March 2012).
Учитывая, что и P-gp, и BCRP экспрессируются в эндотелиальных клетках, выстилающих капилляры головного мозга, активность и P-gp, и BCRP в ВВВ играет критическую роль в предотвращении распространения большинства ингибиторов киназ в паренхиму головного мозга. Следовательно, ингибиторы киназы обычно не подходят для лечения опухолей или рака в головном мозге, который защищен ВВВ.
Таким образом, остается потребность в лечении опухолей, несущих мутации BRAF, включая мутации I класса и II класса, включая мутации резистентности. Кроме того, потребность в лечении опухолей ЦНС, включая опухоли ЦНС, несущие мутации BRAF, в том числе мутации резистентности, остается неудовлетворенной.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Соответственно, в настоящем документе предложено соединение формулы I:
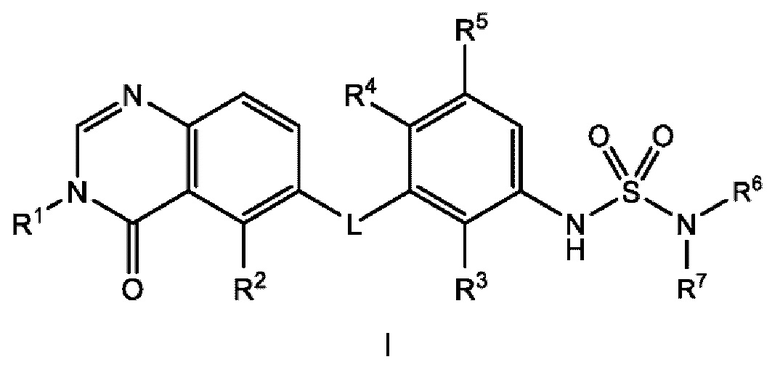
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) С1-С6 алкил-, AR1, AR1CH2-, hetAR1 или hetCyc1;
AR1 представляет собой фенил, который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота, и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -ОСН2СН3, и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
при условии, что соединение не представляет собой:
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид,
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид, или
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-этил-N-метиламино-1-сульфонамид.
Также в настоящем документе предложено соединение формулы I-A
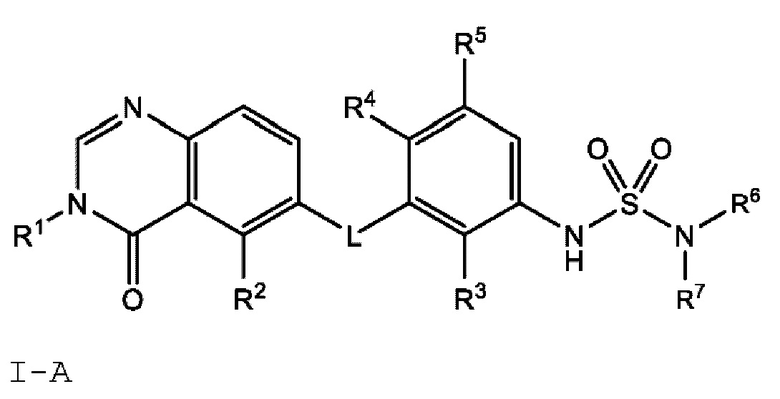
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) С1-С6 алкил-, AR1, AR1CH2-, hetAR1 или hetCyc1;
AR1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота, и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCF2H, -OCD3, -СН3 и -СН2СН3, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 7-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
при условии, что соединение не представляет собой:
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид,
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид, или
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-этил-N-метиламино-1-сульфонамид.
Также в настоящем документе предложено соединение формулы II:
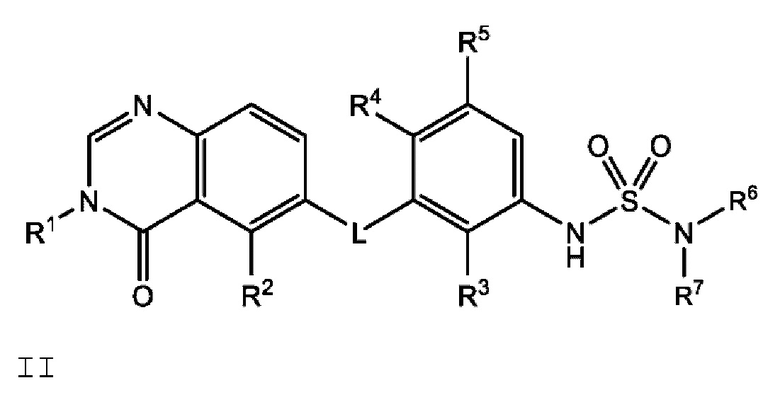
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил или С1-С6 фторалкил;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -ОСН2СН3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
при условии, что соединение не представляет собой:
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид,
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид, или
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-этил-N-метиламино-1-сульфонамид.
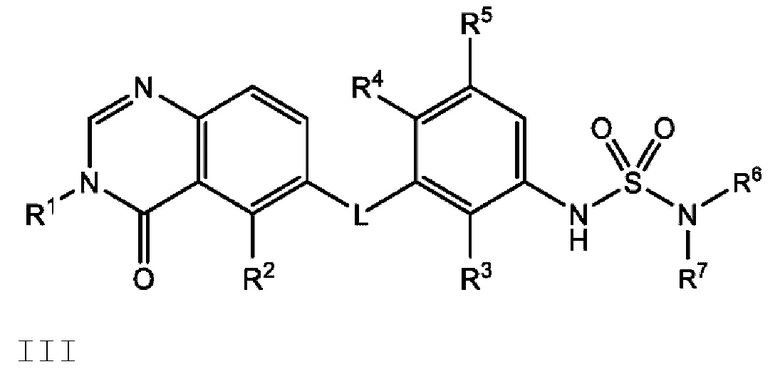
Также в настоящем документе предложено соединение формулы III
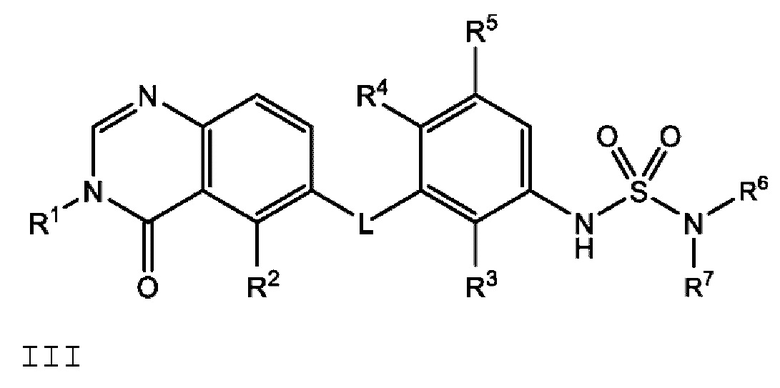
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) С1-С6 алкил-, AR1, AR1CH2-, hetAR1 или hetCyc1;
AR1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота, и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой C1-С6 алкил, и
R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О.
Также в настоящем документе предложено соединение формулы IV
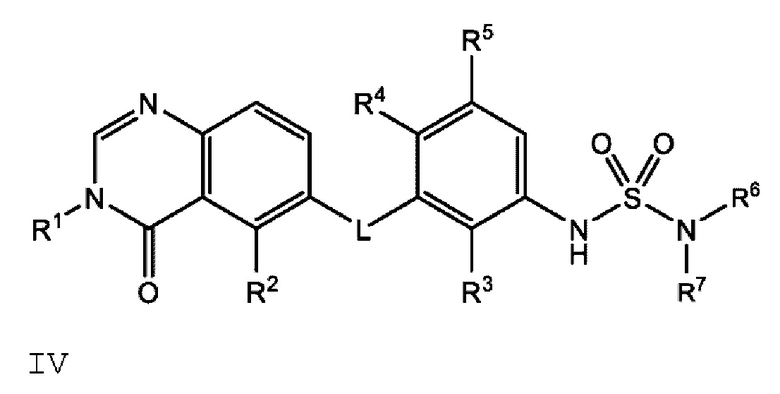
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой C1-С6 алкил, C1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) C1-С6 алкил-, AR1, AR1CH2-, hetAR1 или hetCyc1;
AR1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
где если R1 представляет собой метил, L представляет собой NH, R3 представляет собой Cl, R4 представляет собой F, R5 представляет собой Н, и R6 представляет собой метил и R7 представляет собой этил, или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют пирролидинил или 3-фторпирролидинил, то R2 представляет собой -СН2СН3, -СН=СН2, F, Cl, Br или CN.
Также в настоящем документе предложено соединение формулы V
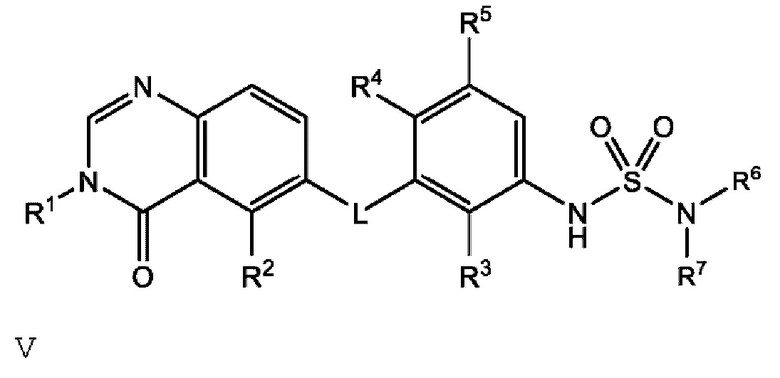
или его фармацевтически приемлемая соль, где:
L представляет собой NH;
R1 представляет собой С1-С6 алкил;
R2 представляет собой F или Cl;
R3 представляет собой Cl;
R4 представляет собой F;
R5 представляет собой Н;
R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, и (iii) 6-7-членного мостикового кольца.
Также в настоящем документе предложена фармацевтическая композиция, содержащая соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых носителей.
В настоящем документе также предложен способ лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в таком лечении, включающий введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемой соли. Соединения по изобретению можно вводить в виде отдельных агентов, или их можно вводить в комбинации с другими противораковыми терапиями, такими как одна или несколько дополнительных противораковых терапий, независимо выбранных из одного или нескольких противораковых агентов и/или хирургического вмешательства и/или лучевой терапии.
В настоящем документе также предложен способ ингибирования метастазов, связанных с BRAF-ассоциированной опухолью, у субъекта, нуждающегося в таком лечении, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемой соли.
В настоящем документе также предложен способ ингибирования активности киназы BRAF in vitro или in vivo, включающий контакт клетки с терапевтически эффективным количеством соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли.
В настоящем документе также предложен способ ингибирования клеточной пролиферации in vitro или in vivo, включающий контакт клетки с терапевтически эффективным количеством соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли.
Также в настоящем документе предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения в терапии.
Также в настоящем документе предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения при лечении опухолей.
Также в настоящем документе предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения для ингибирования метастазов, связанных с BRAF-ассоциированной опухолью.
Также в настоящем документе предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения при ингибировании активности киназы BRAF.
Также в настоящем документе предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения при лечении BRAF-ассоциированного заболевания или нарушения (например, BRAF-ассоциированной опухоли).
Также в настоящем документе предложено применение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, как определено в настоящем документе, в производстве лекарственного средства для лечения BRAF-ассоциированной опухоли (например, BRAF-ассоциированной злокачественной опухоли или BRAF-ассоциированной доброкачественной опухоли).
Также в настоящем документе предложено применение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, как определено в настоящем документе, в производстве лекарственного средства для ингибирования метастазов, связанных с BRAF-ассоциированной опухолью.
Также в настоящем документе предложено применение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, как определено в настоящем документе, в производстве лекарственного средства для ингибирования активности киназы BRAF.
Также в настоящем документе предложено применение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, как определено в настоящем документе, в производстве лекарственного средства для лечения BRAF-ассоциированного заболевания или нарушения.
В настоящем документе также предложен способ лечения BRAF-ассоциированной опухоли у нуждающегося в этом субъекта, включающий (а) определение того, что опухоль ассоциирована с мутацией BRAF; и (b) введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемой соли, или его фармацевтической композиции.
В настоящем документе также предложена фармацевтическая комбинация для лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом, которая содержит (а) соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль, и (b) дополнительный противораковый агент, где соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемая соль, и дополнительный противораковый агент составляют в виде отдельных композиций или доз для отдельного или последовательного применения для лечения BRAF-ассоциированной опухоли, где количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемой соли и дополнительного противоракового агента вместе эффективны при лечении BRAF-ассоциированной опухоли. Также в настоящем документе предложено применение такой комбинации для лечения BRAF-ассоциированной опухоли. Также в настоящем документе предложена коммерческая упаковка или продукт, содержащий такую комбинацию в виде комбинированного препарата для раздельного или последовательного применения при лечении BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом.
В настоящем документе также предложены способы лечения субъекта с BRAF-ассоциированной опухолью, которые включают введение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли до, во время или после введения другой противоопухолевой терапии (например, хирургического вмешательства, лучевой терапии и/или другого противоракового лекарственного средства).
Также в настоящем документе предложен способ получения соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли.
Также в настоящем документе предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль, полученная способом получения соединения, как определено в настоящем документе.
Если не указано иное, все технические и научные термины, используемые в настоящем документе, имеют то же значение, которое обычно понимается специалистом в области, к которой относится данное изобретение. Способы и материалы описаны в настоящем документе для использования в настоящем изобретении; также могут быть использованы другие подходящие способы и материалы, известные в данной области техники. Материалы, способы и примеры являются только иллюстративными и не предназначены для ограничения. Все публикации, патентные заявки, патенты, последовательности, записи в базе данных и другие ссылки, упомянутые в настоящем документе, полностью включены посредством ссылки. В случае конфликта, настоящая спецификация, включая определения, будет иметь преимущественную силу.
Другие признаки и преимущества изобретения будут очевидны из следующего подробного описания и чертежей, а также из формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 показана порошковая рентгенограмма (XRPD) кристаллической формы (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида, формы А.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем документе предложено соединение Формулы I:
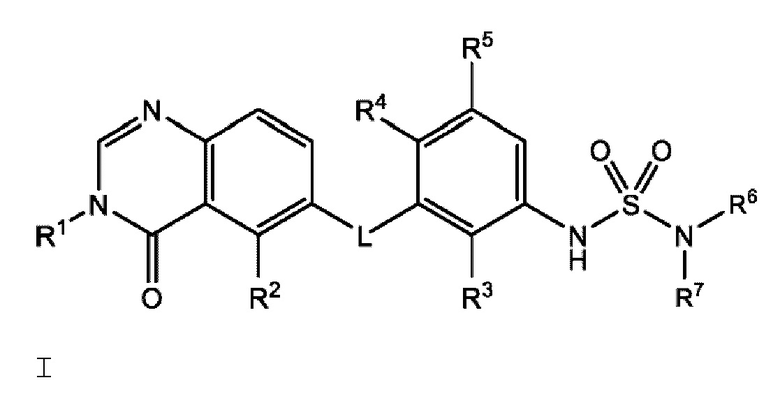
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) С1-С6 алкил-, AR1, AR1CH2-, hetAR1 или hetCyc1;
AR1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCF2H, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
при условии, что соединение не представляет собой:
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид,
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид, или
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-этил-N-метиламино-1-сульфонамид.
В одном варианте осуществления, в настоящем документе предложено соединение формулы I-A
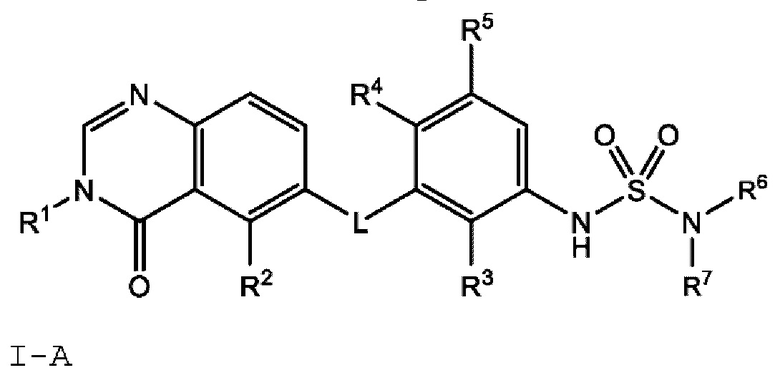
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) С1-С6 алкил-, AR1, AR1CH2-, hetAR1 или hetCyc1;
AR1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой C1-С6 алкил, и
R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCF2H, -OCD3, -СН3 и -СН2СН3, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 7-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
при условии, что соединение не представляет собой:
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид,
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид, или
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-этил-N-метиламино-1-сульфонамид.
Для сложных химических названий, используемых в настоящем документе, замещающая группа обычно указывается перед группой, к которой она присоединена. Например, метоксиэтил включает этильную цепь с метокси заместителем.
Термин «галоген» означает -F (иногда обозначаемый в настоящем документе как «фтор» или «фтор»), -Cl, -Br и -I.
Термины «C1-С3 алкил» и «C1-С6 алкил», используемые в настоящем документе, относятся к насыщенным одновалентным углеводородным радикалам с линейной или разветвленной цепью, содержащим от одного до трех или от одного до шести атомов углерода, соответственно. Примеры алкильных групп включают, но не ограничены ими, метил, этил, 1-пропил, изопропил, 1-бутил, изобутил, втор-бутил, трет-бутил, 2-метил-2-пропил, пентил, неопентил и гексил.
Термин «C1-С6 фторалкил», используемый в настоящем документе, относится к C1-С6 алкильному радикалу, как определено в настоящем документе, где от одного до трех атомов водорода заменены от одного до трех атомов фтора, соответственно. Примеры включают, но не ограничены ими, фторметил, дифторметил, трифторметил, 2-фторэтил, 2,2-дифторэтил, и 2,2,2-трифторэтил.
Термин «С1-С6 дейтероалкил», используемый в настоящем документе, относится к С1-С6 алкильному радикалу, как определено в настоящем документе, который замещен от одного до шести атомов дейтерия. Пример включает, но не ограничен этим, -CD3.
Термин «С3-С6 циклоалкил» означает насыщенное карбоциклическое кольцо, имеющее от 3 до 6 атомов углерода в кольце, например циклопропил, циклобутил, циклопентил или циклогексил.
Термин «C1-С6 алкокси», используемый в настоящем документе, относится к насыщенным одновалентным алкокси радикалам с линейной или разветвленной цепью, содержащим от одного до шести атомов углерода, где радикал находится на атоме кислорода. Примеры алкоксигрупп включают метокси, этокси, пропокси и изопропокси.
Термин «(С1-С6 алкокси) С1-С6 алкил», используемый в настоящем документе, относится к С1-С6 алкильному радикалу, как определено в настоящем документе, где один из атомов углерода замещен С1-С6 алкоксигруппой. Примеры (С1-С6 алкокси)С1-С6 алкильных групп включают метоксиметил (СН3ОСН2-) и метоксиэтил (СН3ОСН2СН2-).
Термин «гетероарил», используемый в настоящем документе, относится к ароматической молекуле, содержащей, по меньшей мере, один гетероатом в составе ароматического кольца.
Термин «гетероцикл», используемый в настоящем документе, относится к насыщенной циклоалкильной группе, в которой одна или несколько кольцевых метиленовых групп (-СН2-) замещены гетероатомом. Например, используемый в настоящем документе термин «hetCyc1» относится к насыщенному 4-6-членному моноциклическому циклоалкильному кольцу, в котором одна из метиленовых групп замещена -O-, и используемый в настоящем документе термин «hetCyc2» относится к 5-6-членному насыщенному моноциклическому циклоалкильному кольцу, в котором одна или две метиленовые группы замещены группой, независимо выбранной из -O- и -N-, при условии, что кольцо не содержит двух соседних кольцевых гетероатомов.
На протяжении всего описания следует понимать, что количество и природа необязательных замещающих групп будут ограничены в той степени, в какой такие замещения имеют химический смысл.
Термин «соединение», используемый в настоящем документе, подразумевает включение всех стереоизомеров, геометрических изомеров, таутомеров и изотопов изображенных структур. Предполагается, что соединения, идентифицированные в настоящем документе по названию или структуре как одна конкретная таутомерная форма, включают другие таутомерные формы, если не указано иное.
В одном варианте осуществления формулы I, L представляет собой NH.
В одном варианте осуществления формулы I, L представляет собой О.
В одном варианте осуществления формулы I, R1 представляет собой С1-С6 алкил. Неограничивающие примеры включают метил, этил и изопропил. В одном варианте осуществления формулы I, R1 представляет собой метил.
В одном варианте осуществления формулы I, R1 представляет собой С1-С6 дейтероалкил. Неограничивающий пример включает -CD3.
В одном варианте осуществления формулы I, R1 представляет собой С1-С6 фторалкил. В одном варианте осуществления формулы I, R1 представляет собой фторметил.
В одном варианте осуществления формулы I, R1 представляет собой С3-С6 циклоалкил. Неограничивающие примеры включают циклопропил, циклобутил и циклопентил.
В одном варианте осуществления формулы I, R1 представляет собой (С3-С6 циклоалкил)СН2-. Неограничивающий пример включает циклопропилметил.
В одном варианте осуществления формулы I, R1 представляет собой (С1-С6 алкокси)С1-С6 алкил-. Неограничивающий пример включает метоксиэтил.
В одном варианте осуществления формулы I, R1 представляет собой AR1. В одном варианте осуществления AR1 представляет собой фенил, который необязательно замещен 1, 2 или 3 заместителями, независимо выбранными из галогена и C1-С3 алкила. Неограничивающим примером AR1 является фенил.
В одном варианте осуществления формулы I, R1 представляет собой AR1CH2-. В одном варианте осуществления, часть AR1 необязательно замещена 1 или 2 заместителями, независимо выбранными из галогена и С1-С3 алкила. Неограничивающим примером AR1CH2- является бензил (-СН2С6Н5).
В одном варианте осуществления формулы I, R1 представляет собой hetAR1. В одном варианте осуществления, hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 атома азота в кольце и необязательно замещенное 1 или 2 заместителями, независимо выбранными из галогена и С1-С3 алкила. В одном варианте осуществления, hetAR1 является незамещенным. Неограничивающим примером является пиридил.
В одном варианте осуществления формулы I, R1 представляет собой hetCyc1. Неограничивающий пример включает тетрагидрофуранил.
В одном варианте осуществления формулы I, R2 представляет собой -СН3.
В одном варианте осуществления формулы I, R2 представляет собой -СН2СН3.
В одном варианте осуществления формулы I, R2 представляет собой -СН=СН2.
В одном варианте осуществления формулы I, R2 представляет собой F.
В одном варианте осуществления формулы I, R2 представляет собой Cl.
В одном варианте осуществления формулы I, R2 представляет собой Br.
В одном варианте осуществления формулы I, R2 представляет собой CN.
В одном варианте осуществления формулы I, R2 представляет собой -СН3, F или Cl.
В одном варианте осуществления формулы I, R2 представляет собой F или Cl.
В одном варианте осуществления формулы I, R3 представляет собой F.
В одном варианте осуществления формулы I, R3 представляет собой Cl.
В одном варианте осуществления формулы I, R4 представляет собой Н.
В одном варианте осуществления формулы I, R4 представляет собой F.
В одном варианте осуществления формулы I, R5 представляет собой Н.
В одном варианте осуществления формулы I, R5 представляет собой F.
В одном варианте осуществления формулы I, R5 представляет собой Cl.
В одном варианте осуществления формулы I, R6 представляет собой С1-С6 алкил, и R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил.
В одном варианте осуществления формулы I, R6 представляет собой метил или этил.
В одном варианте осуществления формулы I, R7 представляет собой C1-С6 алкил. В одном варианте осуществления, R7 представляет собой метил.
В одном варианте осуществления формулы I, R7 представляет собой hetCyc2. В одном варианте осуществления, R7 представляет собой тетрагидрофуранил.
В одном варианте осуществления формулы I, R7 представляет собой С3-С6 циклоалкил. В одном варианте осуществления, R7 представляет собой циклопропил или циклобутил.
В одном варианте осуществления формулы I, R6 представляет собой метил или этил, и R7 представляет собой метил, тетрагидрофуранил, циклопропил или циклобутил.
В одном варианте осуществления формулы I, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, которым является О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3 и -СН2СН3, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца и (iv) 6-8-членного спироциклического кольца.
В одном варианте осуществления формулы I, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 4-6-членное моноциклическое кольцо, необязательно имеющее второй кольцевой гетероатом, которым является О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCF2H, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN. Неограничивающие примеры включают структуры:
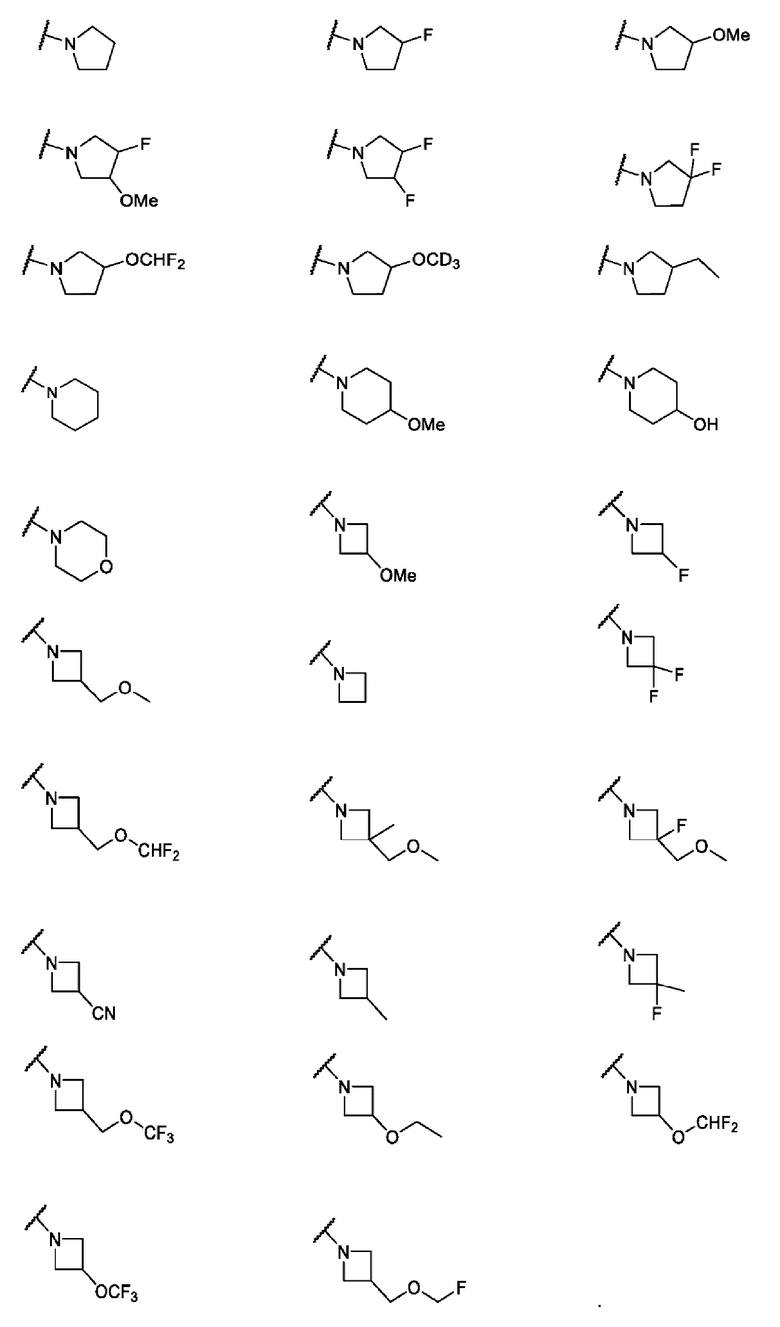
В одном варианте осуществления формулы I, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 4-6-членное моноциклическое кольцо, где указанное кольцо замещено заместителем, выбранным из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN.
В одном варианте осуществления формулы I, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, замещенное F. Примеры включают структуры:
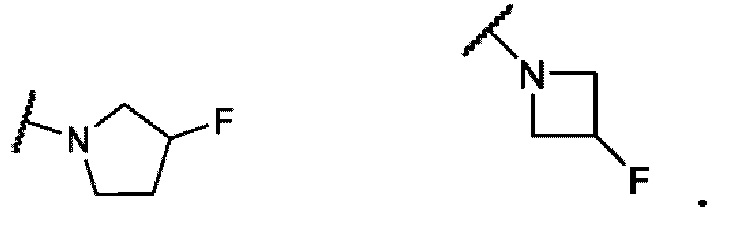
В одном варианте осуществления формулы I, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3. Не ограничивающие примеры включают структуры:
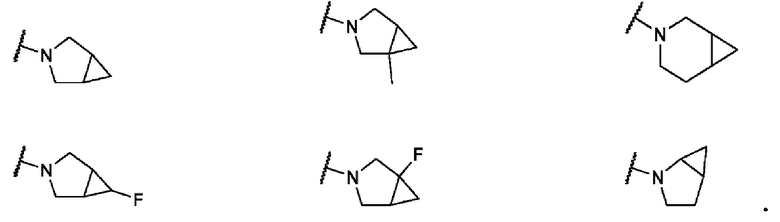
В одном варианте осуществления формулы I, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-7-членное мостиковое кольцо. Не ограничивающие примеры включают структуры:

В одном варианте осуществления формулы I, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-8-членное спироциклическое кольцо. Не ограничивающие примеры включают структуры:
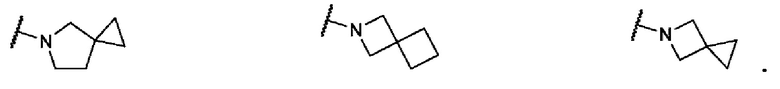
Любые из вышеуказанных вариантов осуществления формулы I могут быть скомбинированы друг с другом.
В одном варианте осуществления, в настоящем документе предложено соединение формулы II
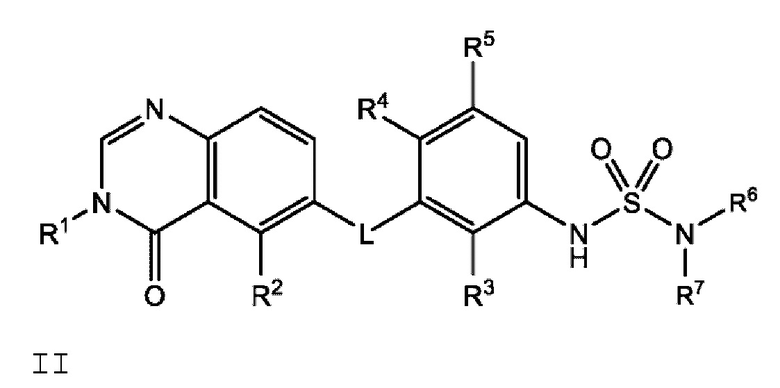
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил или С1-С6 фторалкил;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой C1-С6 алкил, и
R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
при условии, что соединение не представляет собой: N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид,
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид, или
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-этил-N-метиламино-1-сульфонамид.
В одном варианте осуществления формулы II, L представляет собой NH.
В одном варианте осуществления формулы II, L представляет собой О.
В одном варианте осуществления формулы II, R1 представляет собой С1-С6 алкил. Неограничивающие примеры включают метил, этил и изопропил. В одном варианте осуществления формулы II, R1 представляет собой метил.
В одном варианте осуществления формулы II, R1 представляет собой С1-С6 фторалкил. В одном варианте осуществления формулы II, R1 представляет собой фторметил.
В одном варианте осуществления формулы II, R2 представляет собой -СН3.
В одном варианте осуществления формулы II, R2 представляет собой -СН2СН3.
В одном варианте осуществления формулы II, R2 представляет собой -СН=СН2.
В одном варианте осуществления формулы II, R2 представляет собой F.
В одном варианте осуществления формулы II, R2 представляет собой Cl.
В одном варианте осуществления формулы II, R2 представляет собой Br.
В одном варианте осуществления формулы II, R2 представляет собой CN.
В одном варианте осуществления формулы II, R2 представляет собой -СН3, F или Cl.
В одном варианте осуществления формулы II, R2 представляет собой F или Cl.
В одном варианте осуществления формулы II, R3 представляет собой F.
В одном варианте осуществления формулы II, R3 представляет собой Cl.
В одном варианте осуществления формулы II, R4 представляет собой Н.
В одном варианте осуществления формулы II, R4 представляет собой F.
В одном варианте осуществления формулы II, R5 представляет собой Н.
В одном варианте осуществления формулы II, R5 представляет собой F.
В одном варианте осуществления формулы II, R5 представляет собой Cl.
В одном варианте осуществления формулы II, R6 представляет собой С1-С6 алкил и R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил.
В одном варианте осуществления формулы II, R6 представляет собой метил или этил.
В одном варианте осуществления формулы II, R7 представляет собой C1-С6 алкил. В одном варианте осуществления, R7 представляет собой метил.
В одном варианте осуществления формулы II, R7 представляет собой hetCyc2. В одном варианте осуществления, R7 представляет собой тетрагидрофуранил.
В одном варианте осуществления формулы II, R7 представляет собой С3-С6 циклоалкил. В одном варианте осуществления, R7 представляет собой циклопропил или циклобутил.
В одном варианте осуществления формулы II, R6 представляет собой метил или этил, и R7 представляет собой метил, тетрагидрофуранил, циклопропил или циклобутил.
В одном варианте осуществления формулы II, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3 и -СН2СН3, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца.
В одном варианте осуществления формулы II, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 4-6-членное моноциклическое кольцо, необязательно имеющее второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCF2H, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN. Не ограничивающие примеры включают структуры:
В одном варианте осуществления формулы II, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, необязательно замещенное F. Примеры включают структуры:
В одном варианте осуществления формулы II, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-7-членное конденсированное бициклическое кольцо, необязательно замещенное 1 или 2 заместителями, независимо выбранными из F и -СН3. Не ограничивающие примеры включают структуры:
В одном варианте осуществления формулы II, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-7-членное мостиковое кольцо. Не ограничивающие примеры включают структуры:

В одном варианте осуществления формулы II, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-8-членное спироциклическое кольцо. Неограничивающие примеры включают структуры:
Любые из вышеуказанных вариантов осуществления формулы II могут быть скомбинированы друг с другом.
В одном варианте осуществления, в настоящем документе предложено соединение формулы III:
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) C1-С6 алкил-, AR1, AR1CH2-, hetAR1 или hetCyc1;
AR1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -ОСН2СН3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О.
В одном варианте осуществления формулы III, L представляет собой NH.
В одном варианте осуществления формулы III, L представляет собой О.
В одном варианте осуществления формулы III, R1 представляет собой С1-С6 алкил или С1-С6 фторалкил.
В одном варианте осуществления формулы III, R1 представляет собой C1-С6 алкил. Неограничивающие примеры включают метил, этил и изопропил. В одном варианте осуществления, формулы III, R1 представляет собой метил.
В одном варианте осуществления формулы III, R1 представляет собой C1-С6 фторалкил. В одном варианте осуществления формулы III, R1 представляет собой фторметил.
В одном варианте осуществления формулы III, R2 представляет собой -СН=СН2.
В одном варианте осуществления формулы III, R2 представляет собой F.
В одном варианте осуществления формулы III, R2 представляет собой Cl.
В одном варианте осуществления формулы III, R2 представляет собой Br.
В одном варианте осуществления формулы III, R2 представляет собой CN.
В одном варианте осуществления формулы III, R2 представляет собой F или Cl
В одном варианте осуществления формулы III, R3 представляет собой F.
В одном варианте осуществления формулы III, R3 представляет собой Cl.
В одном варианте осуществления формулы III, R4 представляет собой Н.
В одном варианте осуществления формулы III, R4 представляет собой F.
В одном варианте осуществления формулы III, R5 представляет собой Н.
В одном варианте осуществления формулы III, R5 представляет собой F.
В одном варианте осуществления формулы III, R5 представляет собой Cl.
В одном варианте осуществления формулы III, R6 представляет собой С1-С6 алкил и R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил.
В одном варианте осуществления формулы III, R6 представляет собой метил или этил.
В одном варианте осуществления формулы III, R7 представляет собой C1-С6 алкил. В одном варианте осуществления, R7 представляет собой метил.
В одном варианте осуществления формулы III, R7 представляет собой hetCyc2. В одном варианте осуществления, R7 представляет собой тетрагидрофуранил.
В одном варианте осуществления формулы III, R7 представляет собой С3-С6 циклоалкил. В одном варианте осуществления, R7 представляет собой циклопропил или циклобутил.
В одном варианте осуществления формулы III, R6 представляет собой метил или этил и R7 представляет собой метил, тетрагидрофуранил, циклопропил или циклобутил.
В одном варианте осуществления формулы III, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3 и -СН2СН3, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца.
В одном варианте осуществления формулы III, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 4-6-членное моноциклическое кольцо, необязательно имеющее второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCF2H, -OCD3, -СН3, -СН2СН3, СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN. Не ограничивающие примеры включают структуры:
В одном варианте осуществления формулы III, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, необязательно замещенное F. Примеры включают структуры:
В одном варианте осуществления формулы III, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-7-членное конденсированное бициклическое кольцо, необязательно замещенное 1 или 2 заместителями, независимо выбранными из F и -СН3. Не ограничивающие примеры включают структуры:
В одном варианте осуществления формулы III, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-7-членное мостиковое кольцо. Не ограничивающие примеры включают структуры:

В одном варианте осуществления формулы III, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-8-членное спироциклическое кольцо. Неограничивающие примеры включают структуры:
Любые из вышеуказанных вариантов осуществления формулы III могут быть скомбинированы друг с другом.
В одном варианте осуществления, в настоящем документе предложено соединение формулы IV
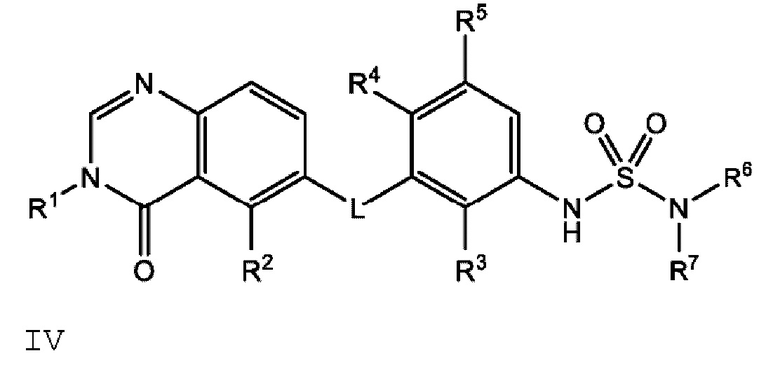
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) C1-С6 алкил-, AR1, AR1CH2-, hetAR1 или hetCyc1;
Ar1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetAR1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
где если R1 представляет собой метил, L представляет собой NH, R3 представляет собой Cl, R4 представляет собой F, R5 представляет собой Н, и R6 представляет собой метил и R7 представляет собой этил, или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют пирролидинил или 3-фторпирролидинил, то R2 представляет собой -СН2СН3, -СН=СН2, F, Cl, Br или CN.
В одном варианте осуществления формулы IV, L представляет собой NH.
В одном варианте осуществления формулы IV, L представляет собой О.
В одном варианте осуществления формулы IV, R1 представляет собой С1-С6 алкил или С1-С6 фторалкил.
В одном варианте осуществления формулы IV, R1 представляет собой C1-С6 алкил. Неограничивающие примеры включают метил, этил и изопропил, при условии что если R1 представляет собой метил, L представляет собой NH, R3 представляет собой Cl, R4 представляет собой F, R5 представляет собой Н, и R6 представляет собой метил и R7 представляет собой этил, или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют пирролидинил или 3-фторпирролидинил, то R2 представляет собой -СН2СН3, -СН=СН2, F, Cl, Br или CN. В одном варианте осуществления формулы IV, R1 представляет собой метил.
В одном варианте осуществления формулы IV, R1 представляет собой С1-С6 фторалкил. В одном варианте осуществления формулы IV, R1 представляет собой фторметил.
В одном варианте осуществления формулы IV, R2 представляет собой -СН3.
В одном варианте осуществления формулы IV, R2 представляет собой -СН2СН3.
В одном варианте осуществления формулы IV, R2 представляет собой -СН=СН2.
В одном варианте осуществления формулы IV, R2 представляет собой F.
В одном варианте осуществления формулы IV, R2 представляет собой Cl.
В одном варианте осуществления формулы IV, R2 представляет собой Br.
В одном варианте осуществления формулы IV, R2 представляет собой CN.
В одном варианте осуществления формулы IV, R2 представляет собой -СН3, F или Cl.
В одном варианте осуществления формулы IV, R2 представляет собой F или Cl
В одном варианте осуществления формулы IV, R3 представляет собой F.
В одном варианте осуществления формулы IV, R3 представляет собой Cl.
В одном варианте осуществления формулы IV, R4 представляет собой Н.
В одном варианте осуществления формулы IV, R4 представляет собой F.
В одном варианте осуществления формулы IV, R5 представляет собой Н.
В одном варианте осуществления формулы IV, R5 представляет собой F.
В одном варианте осуществления формулы IV, R5 представляет собой Cl.
В одном варианте осуществления формулы IV, R6 представляет собой С1-С6 алкил и R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил.
В одном варианте осуществления формулы IV, R6 представляет собой метил или этил.
В одном варианте осуществления формулы IV, R7 представляет собой С1-С6 алкил. В одном варианте осуществления, R7 представляет собой метил.
В одном варианте осуществления формулы IV, R7 представляет собой hetCyc2. В одном варианте осуществления, R7 представляет собой тетрагидрофуранил.
В одном варианте осуществления формулы IV, R7 представляет собой С3-С6 циклоалкил. В одном варианте осуществления, R7 представляет собой циклопропил или циклобутил.
В одном варианте осуществления формулы IV, R6 представляет собой метил или этил и R7 представляет собой метил, тетрагидрофуранил, циклопропил или циклобутил.
В одном варианте осуществления формулы IV, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца.
В одном варианте осуществления формулы IV, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCF2H, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN. Не ограничивающие примеры включают структуры:
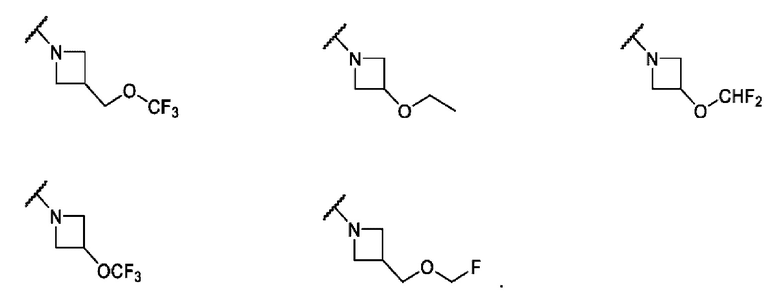
В одном варианте осуществления формулы IV, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, необязательно замещенное F. Примеры включают структуры:
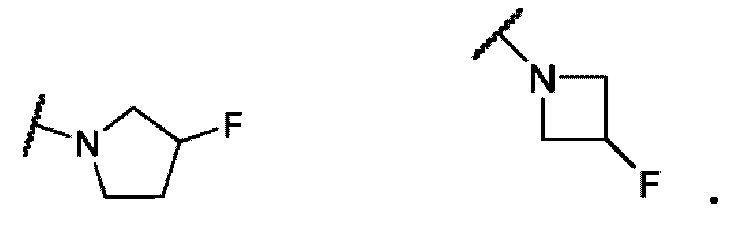
В одном варианте осуществления формулы IV, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-7-членное конденсированное бициклическое кольцо, необязательно замещенное 1 или 2 заместителями, независимо выбранными из F и -СН3. Не ограничивающие примеры включают структуры:
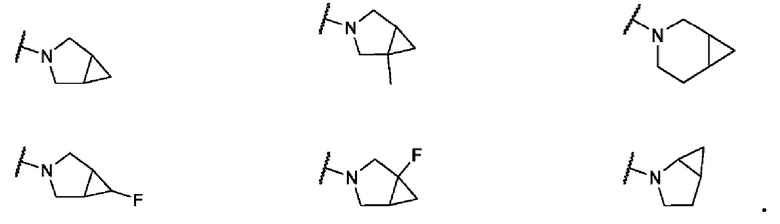
В одном варианте осуществления формулы IV, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-7-членное мостиковое кольцо. Не ограничивающие примеры включают структуры:

В одном варианте осуществления формулы IV, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 6-8-членное спироциклическое кольцо. Неограничивающие примеры включают структуры:

Любые из вышеуказанных вариантов осуществления формулы IV могут быть скомбинированы друг с другом.
В одном варианте осуществления, в настоящем документе предложено соединение формулы V
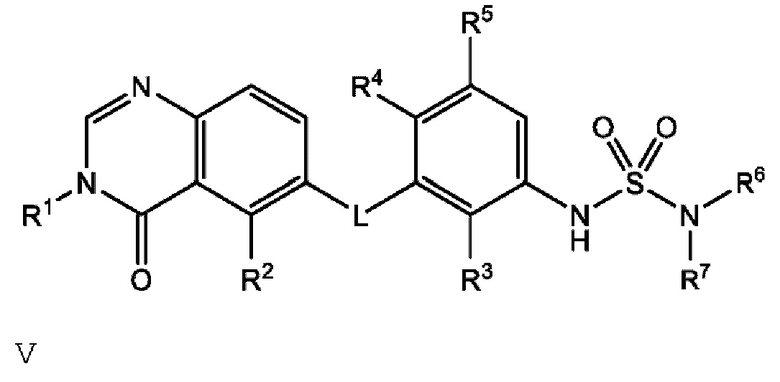
или его фармацевтически приемлемая соль, где:
L представляет собой NH;
R1 представляет собой С1-С6 алкил;
R2 представляет собой F или Cl;
R3 представляет собой Cl;
R4 представляет собой F;
R5 представляет собой Н;
R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, и (iii) 6-7-членного мостикового кольца.
В одном варианте осуществления формулы V, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, необязательно имеющее второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN.
В одном варианте осуществления формулы V, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F.
В одном варианте осуществления формулы V, R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 6-7-членное мостиковое кольцо.
В одном варианте осуществления формулы V, R1 представляет собой метил.
В одном варианте осуществления формулы V, R3 представляет собой F.
В одном варианте осуществления формулы V, R3 представляет собой Cl.
В одном варианте осуществления формулы V, R1 представляет собой метил, R3 представляет собой F, и R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F. В одном варианте осуществления, указанное кольцо замещено одним F.
В одном варианте осуществления формулы V, R1 представляет собой метил, R3 представляет собой хлор, и R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 6-7-членное мостиковое кольцо.
Соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV и формулы V, формулы IV и формулы V включают их фармацевтически приемлемые соли. Кроме того, соединения формулы I также включают другие соли таких соединений, которые не обязательно являются фармацевтически приемлемыми солями и которые могут быть использованы в качестве промежуточных продуктов для получения и/или очистки соединений формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V и/или для разделения энантиомеров соединений формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V.
Термин «фармацевтически приемлемая соль» относится к обычной кислотно-аддитивной или основно-аддитивной соли, которая сохраняет биологическую эффективность и свойства соединений формулы (I), и которая может быть получена с подходящими нетоксичными органическими или неорганическими кислотами или органическими или неорганическими основаниями. Примеры кислотно-аддитивных солей включают соли, полученные из неорганических кислот, таких как, но не ограничиваясь ими, хлористовдородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота, азотная кислота и хлорная кислота, и полученные из различных органических кислот, такие как, но не ограничиваясь ими, уксусная кислота, пропионовая кислота, бензойная кислота, гликолевая кислота, фенилуксусная кислота, салициловая кислота, малоновая кислота, малеиновая кислота, олеиновая кислота, памовая кислота, пальмитиновая кислота, бензолсульфоновая кислота, толуолсульфоновая кислота, метансульфоновая кислота, щавелевая кислота, винная кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, глутаминовая кислота, фумаровая кислота и подобные. Примерами основно-аддитивных солей являются соли, полученные из гидроксидов аммония, калия, натрия и четвертичного аммония, таких как гидроксид тетраметиламмония. Эти соли часто проявляют более благоприятные свойства растворимости, чем соединения, используемые для их получения, и поэтому они более подходят для использования в приготовлении различных фармацевтических составов.
Кроме того, следует понимать, что соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их соли могут быть выделены в форме сольватов и, соответственно, что любой такой сольват включен в объем настоящего изобретения. Например, соединения формулы I и их соли могут существовать как в не сольватированной, так и в сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и подобные.
Термин «сольват» относится к нековалентным стехиометрическим или не стехиометрическим комбинациям растворителя и растворенного вещества. Термин «гидрат» относится к нековалентным стехиометрическим или не стехиометрическим комбинациям воды и растворенного вещества. Например, соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемые соли или полиморфы могут существовать в не сольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как анизол, дихлорметан, толуол, 1,4-диоксан, вода и подобные.
Предлагаемые в настоящем документе соединения могут содержать один или несколько центров асимметрии и поэтому могут быть получены и выделены в смеси изомеров, такой как рацемическая смесь, или в энантиомерно чистой форме. Настоящее изобретение включает все индивидуальные стереоизомеры и геометрические изомеры соединений изобретения и их смесей. Индивидуальные энантиомеры могут быть получены хиральным разделением или с использованием соответствующего энантиомера в синтезе. Связи с атомом углерода в соединениях по изобретению могут быть изображены здесь сплошной линией 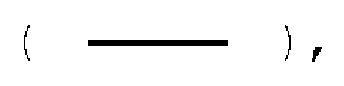 прямой жирной чертой
прямой жирной чертой  прямой пунктирной чертой
прямой пунктирной чертой  сплошным клином
сплошным клином 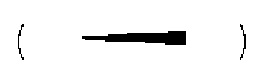 или пунктирным клином
или пунктирным клином  Использование сплошной линии для обозначения связей с асимметричными атомами углерода означает, что включены все возможные стереоизомеры (например, специфические энантиомеры, рацемические смеси и т.д.) на этом атоме углерода. Использование либо прямой толстой полосы, либо прямой пунктирной полосы предназначено для обозначения относительной стереохимии. Использование либо сплошного клина, либо пунктирного клина означает абсолютную стереохимию. Для соединений, описанных в примерах, содержащих один или несколько стереоцентров, если конкретная стереохимия не показана, подразумевается, что соединение включает смесь стереоизомеров. Используемый в настоящем документе термин «стереоцентр» относится к атому с тремя или несколькими различными соединениями, где замена двух из этих соединений приводит к другому стереоизомеру. Примеры включают, но не ограничены ими, sp3 (тетраэдрический) атом углерода, имеющий четыре различных соединения.
Использование сплошной линии для обозначения связей с асимметричными атомами углерода означает, что включены все возможные стереоизомеры (например, специфические энантиомеры, рацемические смеси и т.д.) на этом атоме углерода. Использование либо прямой толстой полосы, либо прямой пунктирной полосы предназначено для обозначения относительной стереохимии. Использование либо сплошного клина, либо пунктирного клина означает абсолютную стереохимию. Для соединений, описанных в примерах, содержащих один или несколько стереоцентров, если конкретная стереохимия не показана, подразумевается, что соединение включает смесь стереоизомеров. Используемый в настоящем документе термин «стереоцентр» относится к атому с тремя или несколькими различными соединениями, где замена двух из этих соединений приводит к другому стереоизомеру. Примеры включают, но не ограничены ими, sp3 (тетраэдрический) атом углерода, имеющий четыре различных соединения.
Соединения формулы I, формулы I-A, формулы II и формулы III могут существовать в различных геометрических изомерных формах. Кроме того, некоторые соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V могут содержать один или несколько центров асимметрии, таким образом, они существуют в стереоизомерных и диастереомерных формах. Термин «стереоизомер» обозначает соединение, которое обладает одинаковой молекулярной связностью и множественностью связей, но отличается расположением атомов в пространстве. Все эти соединения, такие как цис-изомеры, транс-изомеры, диастереомерные смеси, рацематы, нерацемические смеси энантиомеров, по существу чистые и чистые энантиомеры входят в объем изобретения. В одном варианте осуществления, по существу чистые энантиомеры содержат до 5% масс. соответствующего противоположного энантиомера. В одном варианте осуществления, по существу чистые энантиомеры содержат до 2% масс. соответствующего противоположного энантиомера. В одном варианте осуществления, по существу чистые энантиомеры содержат до 1% масс. соответствующего противоположного энантиомера.
Оптические изомеры могут быть получены путем разделения рацемических смесей известными способами, например, с использованием оптически активной кислоты или основания для образования диастереоизомерных солей или путем образования ковалентных диастереомеров. Подходящие кислоты включают, например, винную кислоту, диацетилвинную кислоту, дибензоилвинную кислоту, дитолуоилвинную кислоту и камфорсульфоновую кислоту. Смеси диастереоизомеров можно разделить на отдельные диастереомеры на основе их физических и/или химических различий способами, известными специалистам в данной области техники, такими как хроматография или фракционная кристаллизация. Затем из выделенных диастереоизомерных солей высвобождаются оптически активные основания или кислоты. Различные способы разделения оптических изомеров включают хиральную хроматографию (например, хиральные колонки для ВЭЖХ), необязательно используемую путем дериватизации с целью максимального разделения энантиомеров. Подходящими хиральными колонками для ВЭЖХ являются колонки Diacel, такие как колонки CHIRALPAK или CHIRALCEL, которые можно обычно выбирать по желанию. Там, где это применимо, можно также использовать ферментативное разделение, осуществляемое путем дериватизации. Оптически активные соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V также могут быть получены с использованием оптически активных исходных материалов с использованием хирального синтеза без условий реакции рацемизации.
Также включены кислотно-аддитивные соли или основно-аддитивные соли, в которых противоион является оптически активным, например, d-лактат или I-лизин, или рацемическим, например, dl-тартрат или dl-аргинин.
При кристаллизации любого рацемата возможны кристаллы двух различных типов. Первый тип представляет собой упомянутое выше рацемическое соединение (истинный рацемат), в котором образуется одна гомогенная форма кристалла, содержащая оба энантиомера в эквимолярных количествах. Второй тип представляет собой рацемическую смесь или конгломерат, где две формы кристаллов образуются в эквимолярных количествах, каждая из которых содержит один энантиомер.
Соединения по изобретению могут демонстрировать явления таутомерии и структурной изомерии. Например, соединения могут существовать в нескольких таутомерных формах, включая енольную и иминовую форму, а также кето- и енаминовую форму и геометрические изомеры и их смеси. Все такие таутомерные формы включены в объем соединений по изобретению. Таутомеры существуют в виде смесей таутомерного набора в растворе. В твердой форме обычно преобладает один таутомер. Хотя может быть описан один таутомер, настоящее изобретение включает все таутомеры соединений представленных формул.
Обычные методы получения/выделения индивидуальных энантиомеров включают хиральный синтез из подходящего оптически чистого предшественника или разделение рацемата (или рацемата соли или производного) с использованием, например, хиральной жидкостной хроматографии высокого давления (ВЭЖХ) или сверхкритической жидкостной хроматографии (СЖХ).
Альтернативно, рацемат (или рацемический предшественник) может быть подвергнут взаимодействию с подходящим оптически активным соединением, например, спиртом, или, в случае, когда соединение содержит кислотную или основную группу, кислотой или основанием, таким как винная кислота или 1-фенилэтиламин. Полученную диастереомерную смесь можно разделить хроматографией и/или фракционной кристаллизацией, и один или оба диастереоизомера превратить в соответствующий(ие) чистый(ые) энантиомер(ы) способами, хорошо известными специалисту в данной области техники.
Хиральные соединения по изобретению (и их хиральные предшественники) могут быть получены в энантиомерно обогащенной форме с использованием хроматографии, обычно ВЭЖХ, на асимметричной смоле с подвижной фазой, состоящей из углеводорода, обычно гептана или гексана, содержащего от 0 до 50% изопропанола, обычно, от 2 до 20%, и от 0 до 5% алкиламина, обычно 0,1% диэтиламина. Концентрация элюента дает обогащенную смесь.
Стереоизомерные конгломераты могут быть разделены обычными способами, известными специалистам в данной области; см., например, "Stereochemistry of Organic Compounds" E L Eliel (Wiley, New York, 1994), описание которой полностью включено в настоящий документ посредством ссылки.
Энантиомерная чистота соединений, описанных в настоящем документе, может быть описана с точки зрения энантиомерного избытка (эи), который указывает на степень, в которой образец содержит один энантиомер в большем количестве, чем другой. Рацемическая смесь имеет эи 0%, в то время как отдельный полностью чистый энантиомер имеет эи 100%. Точно так же диастереомерная чистота может быть описана в терминах диастереомерного избытка (ди).
Соединения по изобретению могут демонстрировать явления таутомерии и структурной изомерии. Например, соединения могут существовать в нескольких таутомерных формах, включая енольную и иминовую форму, а также кето- и енаминовую форму и геометрические изомеры и их смеси. Все такие таутомерные формы входят в объем соединений по изобретению. Таутомеры существуют в виде смесей таутомерного набора в растворе. В твердой форме обычно преобладает один таутомер. Хотя может быть описан один таутомер, настоящее изобретение включает все таутомеры соединений представленных формул.
Кроме того, некоторые из соединений по изобретению могут образовывать атропоизомеры (например, замещенные биарилы). Атропоизомеры представляют собой конформационные стереоизомеры, которые возникают, когда вращение вокруг простой связи в молекуле предотвращается или значительно замедляется в результате пространственных взаимодействий с другими частями молекулы, и заместители на обоих концах одинарной связи несимметричны. Взаимное превращение атропоизомеров происходит достаточно медленно, чтобы их можно было разделить и выделить в заданных условиях. Энергетический барьер для термической рацемизации может определяться пространственными затруднениями для свободного вращения одной или нескольких связей, образующих хиральную ось.
Если не указано иное, все ссылки в настоящем документе на соединения по изобретению включают ссылки на их соли, сольваты, гидраты и комплексы, и на сольваты, гидраты и комплексы солей, включая их полиморфы, стереоизомеры и меченные изотопами варианты.
Соединения по изобретению могут существовать в форме фармацевтически приемлемых солей, таких как, например, кислотно-аддитивные соли и основно-аддитивные соли соединений одной из представленных в настоящем документе формул. Используемый в настоящем документе термин «фармацевтически приемлемая соль» относится к тем солям, которые сохраняют биологическую эффективность и свойства исходного соединения. Фраза «фармацевтически приемлемая(ые) соль(и)», используемая в настоящем документе, если не указано иное, включает соли кислотных или основных групп, которые могут присутствовать в соединениях формул, описанных в настоящем документе.
Например, соединения по изобретению, которые являются основными по своей природе, способны образовывать широкий спектр солей с различными неорганическими и органическими кислотами. Хотя такие соли должны быть фармацевтически приемлемыми для введения животным, на практике часто желательно сначала выделить соединение по настоящему изобретению из реакционной смеси в виде фармацевтически неприемлемой соли, и затем просто превратить последнюю обратно в соединение в виде свободного основания путем обработки щелочным реагентом и затем превратить последнее свободное основание в фармацевтически приемлемую кислотно-аддитивную соль. Кислотно-аддитивные соли основных соединений по изобретению могут быть получены обработкой основного соединения по существу эквивалентным количеством выбранной минеральной или органической кислоты в среде водного растворителя или в подходящем органическом растворителе, таком как метанол или этанол. После выпаривания растворителя получают желаемую твердую соль. Желаемую кислую соль также можно осадить из раствора свободного основания в органическом растворителе путем добавления к раствору подходящей минеральной или органической кислоты.
Кислоты, которые можно использовать для получения фармацевтически приемлемых кислотно-аддитивных солей таких основных соединений, которые образуют нетоксичные кислотно-аддитивные соли, т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидройодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, изоникотинат, ацетат, лактат, салицилат, цитрат, кислый цитрат, тартрат, пантотенат, битартрат, аскорбат, сукцинат, малеат, гентизинат, фумарат, глюконат, глюкуронат, сахарат, формиат, бензоат, глутамат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат.
Примеры солей включают, но не ограничены ими, ацетат, акрилат, бензолсульфонат, бензоат (например, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат и метоксибензоат), бикарбонат, бисульфат, бисульфит, битартрат, борат, бромид, бутин-1,4-диоат, эдетат кальция, камзилат, карбонат, хлорид, капроат, каприлат, клавуланат, цитрат, деканоат, дигидрохлорид, дигидрофосфат, эдетат, эдислат, эстолат, эзилат, этилсукцинат, формиат, фумарат, глюцептат, глюконат, глутамат, гликолят, гликоллиларсанилат, гептаноат, гексин-1,6-диоат, гексилрезорцинат, гидрабамин, гидробромид, гидрохлорид, g-гидроксибутират, йодид, изобутират, изотионат, лактат, лактобионат, лаурат, малат, малеат, малонат, манделат, мезилат, метафосфат, метансульфонат, метилсульфат, моногидрофосфат, мукат, напсилат, нафталин-1-сульфонат, нафталин-2-сульфонат, нитрат, олеат, оксалат, памоат (эмбонат), пальмитат, пантотенат, фенилацетаты, фенилбутират, фенилпропионат, фталат, фосфат/дифосфат, полигалактуронат, пропансульфонат, пропионат, пропиолат, пирофосфат, пиросульфат, салицилат, стеарат, субацетат, суберат, сукцинат, сульфат, сульфонат, сульфит, таннат, тартрат, теоклат, тозилат и валерат.
Иллюстративные примеры подходящих солей включают органические соли, полученные из аминокислот, таких как глицин и аргинин, аммиак, первичные, вторичные и третичные амины и циклические амины, такие как пиперидин, морфолин и пиперазин, и неорганические соли, полученные из натрия, кальция, калия, магния, марганца, железа, меди, цинка, алюминия и лития.
Соединения по изобретению, которые включают основную группу, такую как аминогруппа, могут образовывать фармацевтически приемлемые соли с различными аминокислотами в дополнение к кислотам, упомянутым выше.
Альтернативно, полезные соединения, которые являются кислыми по своей природе, могут быть способны образовывать основные соли с различными фармакологически приемлемыми катионами. Примеры таких солей включают соли щелочных или щелочноземельных металлов и, в частности, соли натрия и калия. Все эти соли получают обычными способами. Химические основания, которые используются в качестве реагентов для получения фармацевтически приемлемых солей оснований по настоящему изобретению, представляют собой такие, которые образуют нетоксичные основные соли с кислыми соединениями по настоящему изобретению. Эти соли могут быть получены любым подходящим способом, например обработкой свободной кислоты неорганическим или органическим основанием, таким как амин (первичный, вторичный или третичный), гидроксид щелочного металла или гидроксид щелочноземельного металла или подобный. Эти соли также могут быть получены обработкой соответствующих кислых соединений водным раствором, содержащим желаемые фармакологически приемлемые катионы, и последующим выпариванием полученного раствора досуха, предпочтительно при пониженном давлении. Альтернативно, их также можно приготовить путем смешивания низших алканольных растворов кислых соединений и желаемого алкоксида щелочного металла вместе с последующим выпариванием полученного раствора досуха тем же способом, что и ранее. В любом случае, предпочтительно использовать стехиометрические количества реагентов для обеспечения полноты реакции и максимального выхода желаемого конечного продукта.
Химические основания, которые могут быть использованы в качестве реагентов для получения фармацевтически приемлемых основных солей соединений по изобретению, которые являются кислыми по своей природе, представляют собой такие, которые образуют нетоксичные основные соли с такими соединениями. Такие нетоксичные основные соли включают, но не ограничены ими, соли, полученные из таких фармакологически приемлемых катионов, как катионы щелочных металлов (например, калия и натрия) и катионы щелочноземельных металлов (например, кальция и магния), аддитивные соли аммония или водорастворимого амина, такие как N-метилглюкамин (меглумин), и низший алканоламмоний и другие основные соли фармацевтически приемлемых органических аминов.
Могут также образовываться полусоли кислот и оснований, например, полусульфатные и полукальциевые соли.
Обзор подходящих солей см. в Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley VCH, 2002). Способы получения фармацевтически приемлемых солей соединений по изобретению и взаимопревращенных форм соли и свободного основания известны специалистам в данной области техники.
Соли по настоящему изобретению могут быть получены способами, известными специалистам в данной области техники. Фармацевтически приемлемая соль соединений по изобретению может быть легко получена путем смешивания вместе растворов соединения и желаемой кислоты или основания, в зависимости от ситуации.
Соль может осаждаться из раствора и собираться фильтрованием или может быть выделена выпариванием растворителя. Степень ионизации в соли может варьироваться от полностью ионизированной до почти неионизированной.
Специалистам в данной области техники должно быть понятно, что соединения по изобретению в форме свободного основания, обладающие основной функциональностью, могут быть превращены в кислотно-аддитивные соли путем обработки стехиометрическим избытком соответствующей кислоты. Кислотно-аддитивные соли соединений по изобретению могут быть повторно превращены в соответствующее свободное основание обработкой стехиометрическим избытком подходящего основания, такого как карбонат калия или гидроксид натрия, обычно в присутствии водного растворителя и при температуре между примерно 0°С и 100°С. Форма свободного основания может быть выделена обычными способами, такими как экстракция органическим растворителем. Кроме того, кислотно-аддитивные соли соединений по настоящему изобретению могут быть заменены за счет использования преимущества различной растворимости солей, свойств летучести или кислотности кислот, или путем обработки ионообменной смолой с соответствующей нагрузкой. Например, на взаимопревращение может влиять реакция соли соединений по изобретению с небольшим стехиометрическим избытком кислоты с более низким pK, чем кислотный компонент исходной соли. Это превращение обычно проводят при температуре примерно от 0°С до точки кипения растворителя, используемого в качестве среды для процедуры. Подобные обмены возможны с основно-аддитивными солями, как правило, через посредство формы свободного основания.
Соединения по изобретению могут существовать как в не сольватированной, так и в сольватированной формах. Когда растворитель или вода прочно связаны, комплекс будет иметь четко определенную стехиометрию, не зависящую от влажности. Однако, когда растворитель или вода слабо связаны, как в канальных сольватах и гигроскопичных соединениях, содержание воды/растворителя будет зависеть от влажности и условий сушки. В таких случаях, отсутствие стехиометрии будет нормой. Термин «сольват» используется в настоящем документе для описания молекулярного комплекса, включающего соединение по изобретению и одну или несколько молекул фармацевтически приемлемого растворителя, например, этанола. Термин «гидрат» используется, когда растворителем является вода. Фармацевтически приемлемые сольваты в соответствии с изобретением включают гидраты и сольваты, в которых растворитель кристаллизации может быть изотопно замещен, например D2O, d6-ацетон, d6-ДМСО.
Изобретение также относится к пролекарствам соединений предложенных в настоящем документе формул. Таким образом, некоторые производные соединений по изобретению, которые могут иметь небольшую фармакологическую активность или не иметь ее, при введении пациенту могут быть превращены в соединения по изобретению, например, путем гидролитического расщепления. Такие производные называются «пролекарствами». Дополнительную информацию об использовании пролекарств можно найти в 'Prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella); 'Bioreversible Carriers in Drug Design', Pergamon Press, 1987 (ed. E В Roche, American Pharmaceutical Association), and Guarino, V.R; Stella, V.J.: Biotech Pharm. Aspects 2007 5 (Pt2) 133-187, описания которых полностью включены в настоящий документ посредством ссылки.
В одном варианте осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль. В одном варианте осуществления, соединение любого из примеров 1-164 может быть в форме свободного основания. В одном варианте осуществления, соединение любого из примеров 1-164 может быть в форме кислой соли. В одном варианте осуществления, некоторые соединения из примеров 1-164 выделяют в виде трифторацетатов.
Предложенные в настоящем документе соединения могут также содержать не природные пропорции атомных изотопов на одном или нескольких атомах, составляющих такие соединения. То есть атом, в частности, когда он упоминается в отношении соединения согласно формулы I, формулы I-A, формулы II, формулы III, формулы IV и формулы V, включает все изотопы и изотопные смеси этого атома, встречающиеся в природе или синтетически производится, либо в естественном изобилии, либо в изотопно-обогащенной форме. Например, когда упоминается водород, подразумевается, что он относится к 1H, 2Н, 3H или их смесям; когда упоминается углерод, подразумевается, что он относится к 11С, 12C, 13С, 14С или их смесям; когда упоминается азот, подразумевается, что он относится к 13N, 14N, 15N или их смесям; когда упоминается кислород, подразумевается, что он относится к 14О, 15O, 16О, 17О, 18О или их смесям; и когда упоминается фтор, подразумевается, что он относится к 18F, 19F или их смесям. Как отмечалось выше, соединения, представленные в настоящем документе, таким образом, также включают соединения с одним или несколькими изотопами одного или нескольких атомов и их смеси, включая радиоактивные соединения, в которых один или несколько не радиоактивных атомов заменены одним из его радиоактивно обогащенных изотопов. Соединения с радиоактивной меткой можно использовать в качестве терапевтических агентов, например противораковых терапевтических агентов, исследовательских реагентов, например реагентов для анализа, и диагностических агентов, например, агентов для визуализации in vivo. Предполагается, что все изотопные варианты соединений, представленных в настоящем документе, независимо от того, являются ли они радиоактивными или нет, входят в объем настоящего изобретения. Некоторые изотопно-меченые соединения по изобретению, например такие, в которые включены радиоактивные изотопы, такие как 3H и 14С, можно использовать в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы трития, т.е. 3H, и углерода 14, т.е. 14C, особенно предпочтительны из-за простоты их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2Н, может обеспечить определенные терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличение периода полувыведения in vivo или снижение требований к дозировке, и, следовательно, в некоторых обстоятельствах может быть предпочтительным. Меченые изотопами соединения по изобретению, как правило, могут быть получены путем проведения процедур, описанных на схемах и/или в приведенных ниже примерах и получениях, путем замены реагента, не меченного изотопом, реагентом, меченным изотопом.
Изобретение также относится к пролекарствам соединений представленных в настоящем документе формул. Таким образом, некоторые производные соединений по изобретению, которые могут иметь или не иметь небольшую фармакологическую активность, при введении пациенту могут быть превращены в соединения по изобретению, например, путем гидролитического расщепления. Такие производные называются «пролекарствами». Дополнительную информацию об использовании пролекарств можно найти в Prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T Higuchi and W Stella) и «Bioreversible Carriers in Drug Design», Pergamon Press, 1987 (ed. EB Roche, American Pharmaceutical Association), описание которых полностью включено в настоящее описание посредством ссылки.
Пролекарства в соответствии с изобретением могут быть получены, например, путем замены соответствующих функциональных групп, присутствующих в соединениях по изобретению, на определенные группы, известные специалистам в данной области техники как «про-группы», как описано, например, в «Design of Prodrugs» by H Bundgaard (Elsevier, 1985), описание которой полностью включено в настоящий документ посредством ссылки.
Некоторые неограничивающие примеры пролекарств по изобретению включают:
(i) если соединение содержит функциональную группу карбоновой кислоты (-СООН), ее сложный эфир, например, замещение водорода на (С1-С8)алкил;
(ii) когда соединение содержит спиртовую функциональную группу (-ОН), ее эфир, например, замещение водорода на (С1-С6)алканоилоксиметил или группу фосфатного эфира; и
(iii) если соединение содержит первичную или вторичную аминогруппу (-NH2 или -NHR, где R1 H), его амид, например замену одного или обоих атомов водорода подходящей метаболической лабильной группой, такой как амид, карбамат, мочевина, фосфонат, сульфонат и т.д.
Дополнительные примеры замещающих групп в соответствии с приведенными выше примерами и примеры других типов пролекарств можно найти в вышеупомянутых ссылках.
Наконец, некоторые соединения по изобретению могут сами действовать как Пролекарства других соединений по изобретению.
Также в объем изобретения включены метаболиты соединений описанных в настоящем документе формул, т.е. соединения, образующиеся in vivo при введении лекарственного средства.
В иллюстративных целях, на схемах 1-10 показаны общие способы получения соединений, представленных в настоящем документе, а также ключевых промежуточных соединений. Более подробное описание отдельных стадий реакции см. в разделе «Примеры» ниже. Специалистам в данной области техники понятно, что для синтеза соединений изобретения могут быть использованы другие пути синтеза. Хотя конкретные исходные материалы и реагенты изображены на схемах и обсуждаются ниже, другие исходные материалы и реагенты могут быть легко заменены для получения различных производных и/или условий реакции. Кроме того, многие из соединений, полученных способами, описанными ниже, могут быть дополнительно модифицированы в свете этого описания с использованием обычной химии, хорошо известной специалистам в данной области техники.
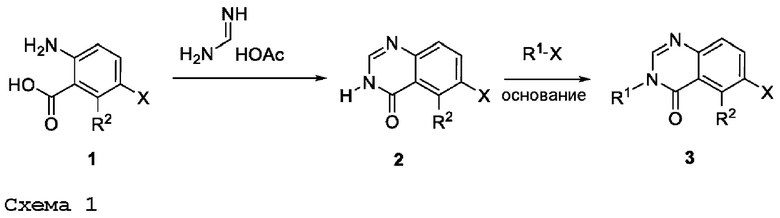
На схеме 1 описан синтез промежуточного соединения 3, где Х представляет собой галоген, который используется для получения соединений формулы I, где R1 и R2 имеют значения, определенные для формулы I. Соединение 1 можно циклизовать с ацетатом формамидина в органическом растворителе, таком как EtOH при повышенной температуре с получением соединения 2. Соединение 2 может быть алкилировано реагентом, имеющим формулу R1X, где R1 имеет значение, определенное для формулы I, и Х представляет собой галоген, в присутствии основания, такого как Cs2CO3, в растворителе, таком как ДМФ, с получением соединения 3.
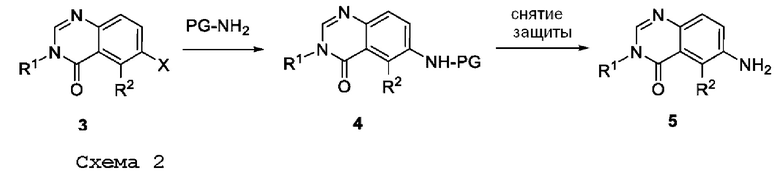
На схеме 2 описан синтез промежуточного соединения 5, которое можно использовать для получения соединений формулы I, где R1 и R2 имеют значения, определенные для формулы I, и L представляет собой NH. Соединение 3 (полученное, например, в соответствии со схемой 1) можно сочетать с реагентом, имеющим формулу (PG)NH2, где PG представляет собой защитную группу амина (такую как п-метоксибензил (РМВ) или трет-бутоксикарбонил (Boc)) в присутствии катализатора, такого как палладиевый катализатор (например, Pd2(dba)3) и лиганда (например, Xantphos), для получения соединения 4. С соединения 4 можно снять защиту в стандартных условиях, например, с использованием ТФК, чтобы получить соединение 5.
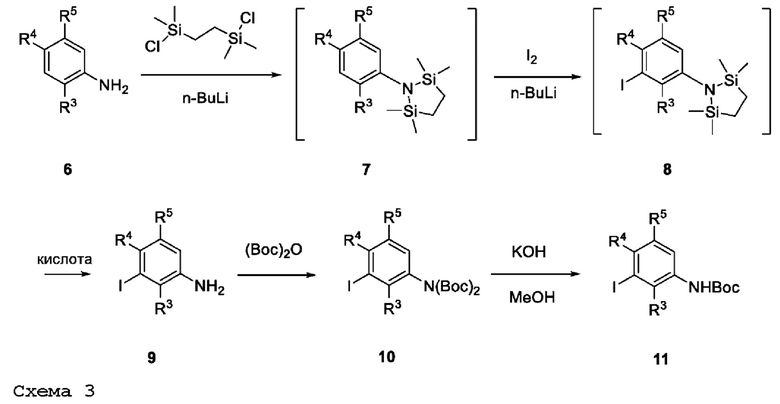
На схеме 3 описан синтез промежуточного соединения 11, которое можно использовать для получения соединений формулы I, где R3, R4 и R5 определены для формулы I. Соединение 6 (где R3, R4 и R5 определены для формулы I) может реагировать с 1,2-бис(хлордиметилсилил)этаном в присутствии сильного основания, такого как н-бутиллитий, в подходящем растворителе, таком как ТГФ, при низких температурах, например -78°С, с образованием 1-аза-2,5-дисилациклопентанового соединения 7. Соединение 7 можно подвергнуть взаимодействию с йодом в присутствии, например, н-бутиллития или аналогичного агента в подходящем растворителе, таком как ТГФ, с получением соединения 8. Соединение 8 может быть удалено путем взаимодействия с кислотой, такой как HCl, в подходящем растворителе с получением соединения 9. Соединение 9 может быть подвергнуто взаимодействию с ди-трет-бутилдикарбонатом ((Boc)2O) в присутствии катализатора, например, 4-диметиламинопиридина (ДМАП), в подходящем растворителе, таком как ТГФ, с получением соединения 10. С соединения 10 можно снять защиту в присутствии основания, такого как K2CO3, в подходящем растворителе, таком как МеОН, с получением соединения 11.
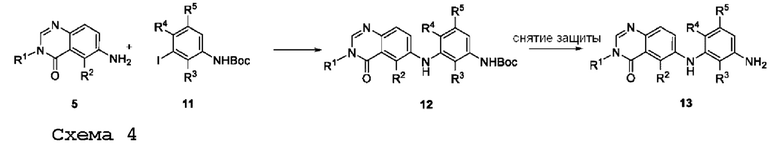
На схеме 4 описан синтез соединения 13, которое является промежуточным соединением, пригодным для получения соединений формулы I, где R1, R2, R3, R4 и R5 имеют значения, определенные для формулы I, и L представляет собой NH. Соединение 5 (полученное, например, в соответствии со схемой 2) можно сочетать с соединением 11 (полученным, например, в соответствии со схемой 3) в присутствии катализатора (например, палладиевого катализатора, например, Pd2(dba)3) и лиганда (например, Xantphos) с последующим удалением защиты в стандартных условиях (например, с помощью ТФК) с получением соединения 13.
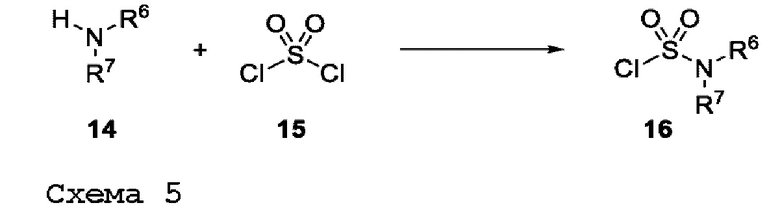
На схеме 5 описан синтез соединения 16, которое можно использовать для получения соединений формулы I, где R6 и R7 имеют значения, определенные для формулы I. Амин 14 можно сочетать с сульфурилдихлоридом 15 в присутствии основания, такого как ТЭА, в подходящем растворителе, таком как ДХМ, с получением соединения 16.
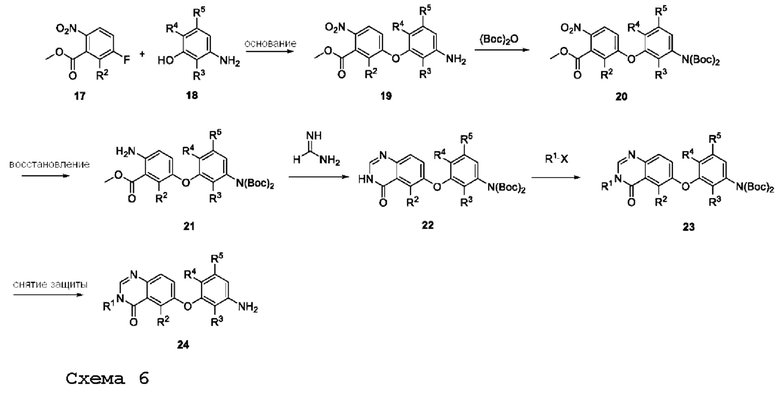
На схеме 6 описан синтез соединения формулы 24, которое можно использовать для получения соединений формулы I, где R1, R2, R3, R4 и R5 имеют значения, определенные для формулы I, и L представляет собой О. Соединение 17 (где R2 имеет значение, определенное для формулы I), может быть связано с соединением 18, где R3, R4 и R5 имеют значение, определенное для формулы I, в подходящем растворителе, таком как ДМСО, в присутствии основания, такого как Cs2CO3, при повышенной температуре с получением соединения 19. Соединение 19 может реагировать с (Boc)2O в присутствии катализатора, такого как ДМАП, в подходящем растворителе, таком как ТГФ, с получением соединения 20. Нитрогруппа соединения 20 может быть восстановлена в стандартных условиях восстановления нитрогруппы, таких как обработка Fe и NH4Cl, с получением соединения 21. Соединение 21 может быть подвергнуто циклизации с ацетатом формамидина в органическом растворителе, таком как EtOH, при повышенной температуре с получением соединения 22. Соединение 22 может быть алкилировано реагентом, имеющим формулу R1X, где R1 имеет значения, определенные для формул I, и Х представляет собой галоген, в присутствии основания, такого как Cs2CO3, в растворителе, таком как ДМФ, с получением соединения 23. С соединения 23 можно снять защиту в стандартных условиях (например, с помощью ТФК) с получением соединения 24.
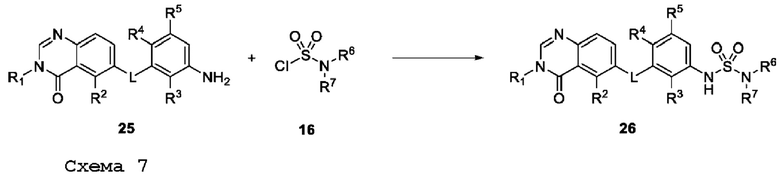
На схеме 7 описывает синтез соединения формулы 26, которое представляет собой соединение формулы I, где R1, R2, R3, R4, R5, R6 и R7 являются такими, как определено для формулы I, и L представляет собой NH (например, полученный в соответствии со схемой 4) или О (например, полученный в соответствии со схемой 6). Соединение 25 можно сочетать с соединением 16 в присутствии подходящего основания, такого как пиридин, или в присутствии трифлимида кальция в органическом растворителе, таком как толуол, при повышенных температурах с получением соединения 26.
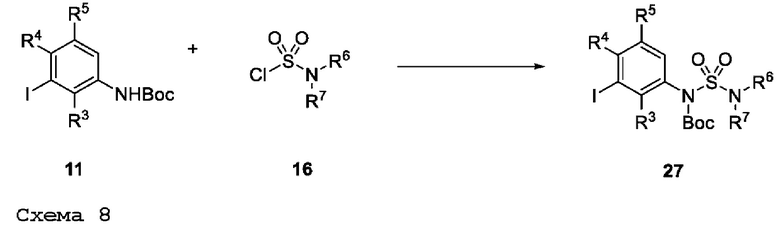
На схеме 8 описан синтез соединения 27, которое можно использовать для получения соединений формулы I, где R3, R4, R5, R6 и R7 имеют значения, определенные для формулы I. Амин 11, где R3, R4, R5, R6 и R7 имеют значения, определенные для формулы I, могут быть связаны с хлоридом сульфамида 16, где R6 и R7 имеют значения, определенные для формулы I, в присутствии основания, такого как NaH, в подходящем растворителе, таком как ТГФ, с получением соединения 27.
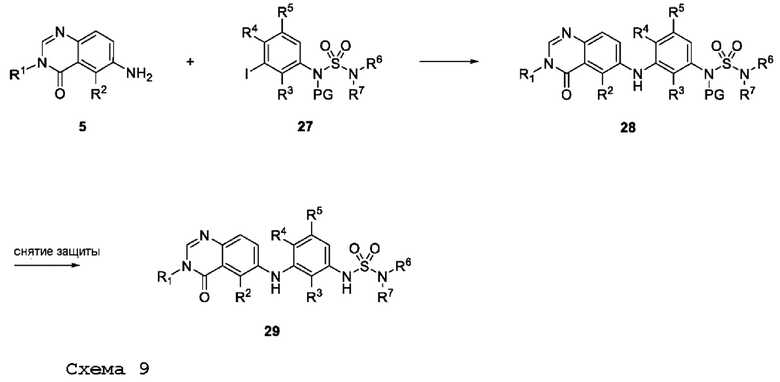
На схеме 9 описан синтез соединения формулы 29, которое представляет собой соединение формулы I, где R1, R2, R3, R4, R5, R6 и R7 имеют значения, определенные для формулы I. Соединение 5 (полученное, например, в соответствии со схемой 2), где R1 и R2 имеют значения, определенные для формулы I, можно сочетать с соединением 27 (полученным, например, в соответствии со схемой 8), где R3, R4, R5, R6 и R7 имеют значения, определенные для формулы I, и PG представляет собой защитную группу амина (такую как п-метоксибензил (РМВ) или трет-бутоксикарбонил (Boc)), в присутствии катализатора (например, палладиевого катализатора, например, Pd2(dba)3) и лиганда (например, Xantphos) с последующим снятием защиты в стандартных условиях (например, с помощью ТФК) с получением соединения 29.
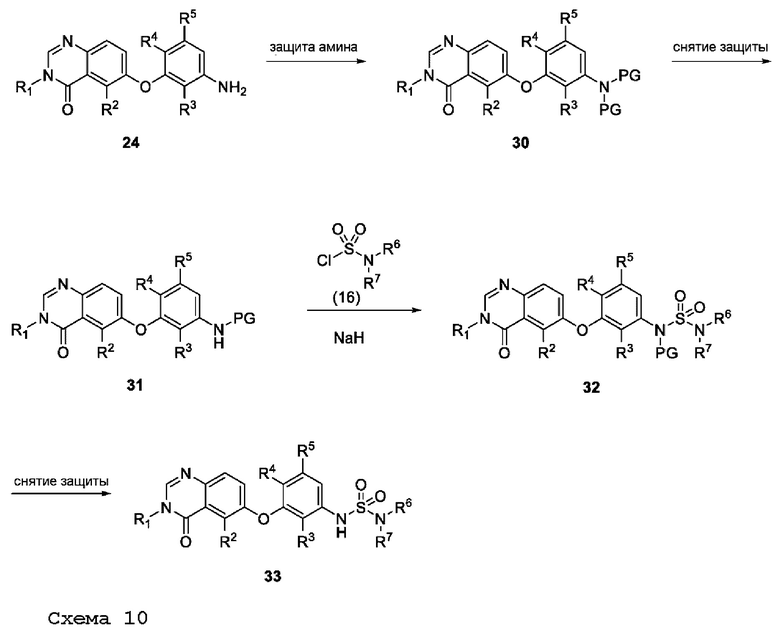
На схеме 10 описан синтез соединения 33, которое представляет собой соединение формулы I, где R1, R2, R3, R4 и R5 имеют значения, определенные для формулы I, и L представляет собой О. Аминогруппа соединения 24 (где R1, R2, R3, R4 и R5 имеют значения, определенные для формулы I), могут быть ди-защищены подходящей защитной группой амина (например, п-метоксибензилом (РМВ) или трет-бутоксикарбонилом (Boc)) путем взаимодействия с подходящего реагента (например, путем взаимодействия с (Вос)2О в присутствии катализатора, такого как ДМАП, в подходящем растворителе, таком как ТГФ), с получением соединения 30, где PG представляет собой защитную группу амина (например, p-метоксибензил (РМВ) или трет-бутоксикарбонил (Boc). Защита соединения 30 может быть удалена в подходящих условиях (например, в присутствии K2CO3 в органическом растворителе, таком как МеОН, при повышенной температуре) с получением монозащищенного соединения 31. Соединение 31 можно сочетать с сульфамоилхлоридом 16 в присутствии основания, такого как NaH, в подходящем растворителе, таком как ТГФ, с получением соединения 32. С соединения 32 можно снять защиту в стандартных условиях (например, с помощью ТФК), чтобы получить соединение 33.
Способы, показанные на схемах 1-10, применимы для получения соединений формул II, III и IV, а также для получения промежуточных соединений, пригодных для получения соединений формул II, III и IV.
В одном варианте осуществления, в настоящем документе предложен способ получения соединения формулы I или его фармацевтически приемлемой соли, который включает:
(а) для соединения формулы I, где L, R1, R2, R3, R4, R5, R6 и R7 имеют значения, определенные для формулы I, связывание соединения формулы (25)
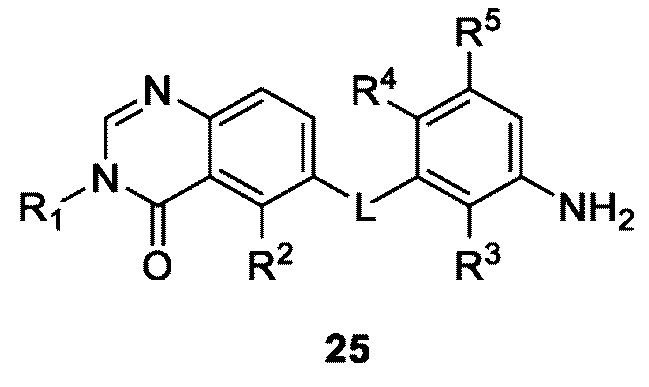
где L, R1, R2, R3, R4 и R5 такие, как определены для формулы I, с соединением, имеющим формулу (16)
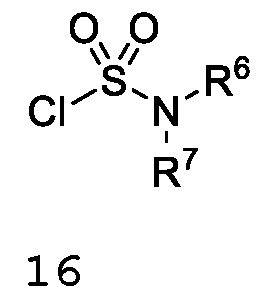
где R6 и R7 такие, как определены для формулы I, в присутствии подходящего основания; или
(b) для соединения формулы I где R1, R2, R3, R4 и R5 такие, как определены для формулы I и L представляет собой NH, взаимодействие соединения формулы (5)
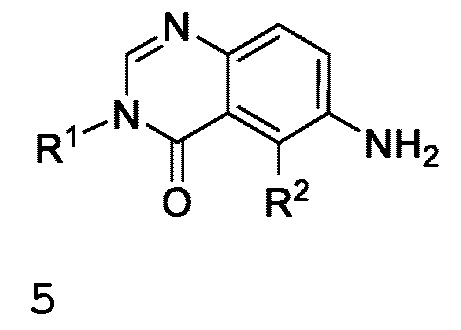
где R1 и R2 такие, как определены для формулы I, с соединением, имеющим формулу (27)
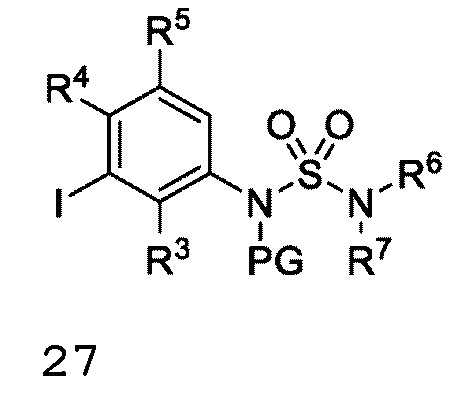
где R3, R4, R5, R6, и R7 такие, как определены для формулы I и PG представляет собой группу, защищающую амин, в присутствии палладиевого катализатора и лиганда с последующим удалением группы, защищающей амин; или
(с) для соединения формулы I где R1, R2, R3, R4 и R5 такие, как определены для формулы I и L представляет собой О, взаимодействие соединения, имеющего формулу (31)
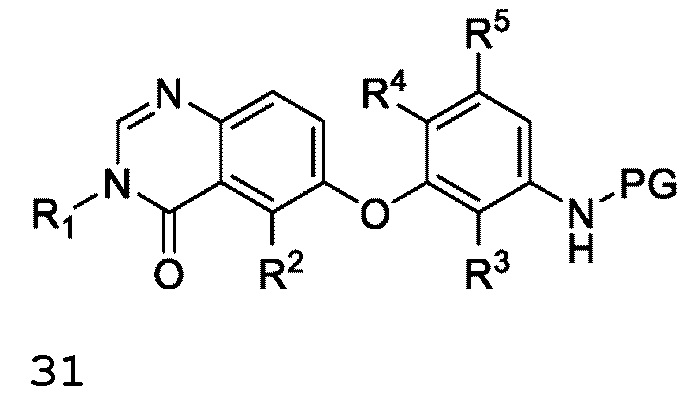
где R1, R2, R3, R4 и R5 такие, как определены для формулы I и PG представляет собой защитную группу амина с реагентом, имеющим формулу
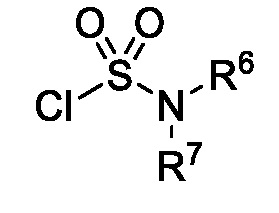
в присутствии основания с последующим удалением аминозащитной группы; и
необязательно, с образованием его фармацевтически приемлемой соли.
Соединения формул 3, 5, 12, 13, 19, 20, 21, 22, 23, 24, 25, 28, 21 и 32 представляют собой синтетические промежуточные соединения, пригодные для получения соединений формулы I, и являются еще одним аспектом настоящего изобретения.
Термин «защитная группа амина», используемый в настоящем описании, относится к производным групп, обычно используемых для блокирования или защиты аминогруппы, когда проводят реакции с другими функциональными группами соединения. Примеры подходящих защитных групп для использования в любом из описанных здесь способов включают карбаматы, амиды, алкильные и арильные группы, имины, а также многие производные N-гетероатома, которые можно удалить для регенерации желаемой аминогруппы. Неограничивающими примерами защитных групп амина являются трет-бутилоксикарбонил («Boc»), 2-триметилсилилэтоксиметил (SEM) и п-метоксибензил (РМВ). Дополнительные примеры этих групп и других защитных групп можно найти в T.W. Greene, et al., Greene's Protective Groups in Organic Synthesis. New York: Wiley Interscience, 2014.
Соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV и формулы V или их фармацевтически приемлемые соли применимы для лечения заболеваний и нарушений, которые можно лечить с помощью ингибитора киназы BRAF, таких как BRAF-ассоциированные заболевания и нарушения, например, пролиферативные нарушения, такие как раки, включая солидные опухоли. Способность тестируемых соединений действовать как ингибиторы BRAF может быть продемонстрирована с помощью ферментного анализа, описанного в примере А1, клеточного анализа, описанного в примере А2, клеточного анализа, описанного в примере A3, и анализа пролиферации, описанного в примере А4. Значения IC50 показаны в таблицах А1 и А2.
В некоторых вариантах осуществления, некоторые соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV и формулы V или их фармацевтически приемлемые соли проявляют неожиданную пенетрантность в головной мозг и/или ЦНС. Такие соединения способны пересекать ВВВ и ингибировать киназу BRAF в головном мозге и/или других структурах ЦНС. В некоторых вариантах осуществления, соединения, предложенные в настоящем документе, способны пересекать ВВВ в терапевтически эффективном количестве. Например, лечение субъекта с раком (например, BRAF-ассоциированным раком, таким как BRAF-ассоциированный рак ЦНС) может включать введение (например, пероральное введение) соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемой соли субъекту. Соответственно, в некоторых вариантах осуществления, соединения, предложенные в настоящем документе, применимы для лечения рака ЦНС.
Используемые в настоящем документе термины «лечить» или «лечение» относятся к терапевтическим или паллиативным мерам. Благоприятные или желаемые клинические результаты включают, но не ограничены этим, полное или частичное облегчение симптомов, связанных с заболеванием или нарушением или состоянием, уменьшение степени заболевания, стабилизированное (т.е. не ухудшающееся) состояние заболевания, задержка или замедление прогрессирования заболевания, улучшение или временное облегчение болезненного состояния (например, одного или нескольких симптомов заболевания) и ремиссию (частичную или полную), определяемое или не определяемое. Однако «лечить» или «лечение» также может включать терапевтические меры (например, ингибирование киназы BRAF в BRAF-ассоциированной опухоли), которые временно ухудшают внешний вид и/или симптомы субъекта. Используемые в настоящем документе термины «лечение» и «лечение», когда они относятся, например, к лечению рака, не предназначены для использования в качестве абсолютных терминов. Например, «лечение рака» и «лечение рака», используемые в клинических условиях, предназначены для получения полезных или желаемых клинических результатов и могут включать улучшение состояния субъекта, страдающего раком. Благоприятные или желаемые клинические результаты включают, но не ограничены ими, одно или несколько из следующего: снижение пролиферации (или уничтожение) опухолевых или раковых клеток, ингибирование метастазирования опухолевых клеток, уменьшение метастазирования у субъекта, сокращение или уменьшение размера опухоли, изменение скорости роста одной или нескольких опухолей у субъекта, увеличение периода ремиссии у субъекта (например, по сравнению с одним или несколькими показателями у субъекта, имеющего аналогичный рак, не получающего лечения или получающего другое лечение, или по сравнению с одним или несколькими показателями у того же субъекта до лечения), уменьшение симптомов, возникающих в результате заболевания, повышение качества жизни тех, кто страдает от заболевания (например, оцениваемое с использованием FACT-G или EORTC-QLQC30), снижение дозы других лекарственных средств, необходимых для лечения заболевания, замедление прогрессирования заболевания и/или увеличение продолжительности жизни субъектов, страдающих заболеванием. «Лечение» также может означать увеличение продолжительности жизни по сравнению с ожидаемой выживаемостью при отсутствии лечения, например, увеличение общей выживаемости (OS) по сравнению с субъектом, не получающим лечения, как описано в настоящем документе, и/или увеличение выживаемости без прогрессирования заболевания. (PFS) по сравнению с субъектом, не получавшим лечения, как описано в настоящем документе.
Используемый в настоящем документе термин «субъект» относится к любому животному, включая млекопитающих, таких как мыши, крысы, другие грызуны, кролики, собаки, кошки, свиньи, крупный рогатый скот, овцы, лошади, приматы и люди. В некоторых вариантах осуществления, субъектом является человек. В некоторых вариантах осуществления, субъект испытывал и/или демонстрировал, по меньшей мере, один симптом заболевания или нарушения, подлежащего лечению и/или профилактике. В некоторых вариантах осуществления, у субъекта была идентифицирована или диагностирована опухоль с мутацией BRAF (BRAF-ассоциированная опухоль) (например, как определено с использованием анализа или набора, одобренного регулирующим органом, например, одобренного FDA). В некоторых вариантах осуществления, субъект имеет опухоль, положительную в отношении мутации BRAF (например, как определено с использованием анализа или набора, одобренного регулирующим органом). Субъектом может быть субъект, опухоли которого имеют мутацию BRAF (например, когда опухоль идентифицирована как таковая с использованием набора или анализа, одобренного регулирующим органом, например, одобренного FDA). В некоторых вариантах осуществления, у субъекта подозревают BRAF-ассоциированную опухоль. В некоторых вариантах осуществления, у субъекта есть история болезни, указывающая на то, что у субъекта есть опухоль с мутацией BRAF (и, необязательно, история болезни указывает, что субъекта следует лечить любой из предложенных в настоящем документе композиций). В некоторых вариантах осуществления, субъектом является человек. В некоторых вариантах осуществления, субъект-человек представляет собой педиатрического субъекта.
Термин «педиатрический субъект», используемый в настоящем документе, относится к субъекту в возрасте до 21 года на момент постановки диагноза или лечения. Термин «педиатрический» можно дополнительно разделить на различные субпопуляции, включающие: новорожденных (от рождения до первого месяца жизни); младенцев (от 1 месяца до двух лет); детей (от двух лет до 12 лет); и подростков (от 12 лет до 21 года (до, но не включая, двадцать второго дня рождения)). Berhman RE, Kliegman R, Arvin AM, Nelson WE, Nelson Textbook of Pediatrics, 15th Ed. Philadelphia: W.B. Sannders Company, 1996; Rudolph AM, et al. Rudolph's Pediatrics, 21st Ed. New York: McGraw-Hill, 2002; и Avery MD, First LR. Pediatric Medicine, 2nd Ed. Baltimore: Williams & Wilkins; 1994. В некоторых вариантах осуществления, педиатрический субъект находится в возрасте от рождения до первых 28 дней жизни, от 29 дней до возраста менее двух лет, от двух лет до возраста менее 12 лет или от 12 лет до возраста 21 года (до, но не включая, двадцать второго дня рождения). В некоторых вариантах осуществления, педиатрический субъект находится в возрасте от рождения до первых 28 дней жизни, от 29 дней до менее 1 года, от одного месяца до возраста четырех месяцев, от трех месяцев до семи месяцев, от шести месяцев до 1 года, от 1 года до 2 лет, от 2 лет до 3 лет, от 2 лет до семи лет, от 3 лет до 5 лет, от 5 лет до 10 лет, от 6 лет до 13 лет, от 10 лет до 15 лет или от 15 лет до 22 лет.
В некоторых вариантах осуществления, соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемая соль являются полезными для профилактики заболеваний и нарушений, как определено в настоящем документе. Термин «профилактика», используемый в настоящем документе, означает профилактику возникновения, рецидива или распространения, полностью или частично, заболевания или состояния, описанного в настоящем документе.
Термин «BRAF-ассоциированный» по отношению к заболеванию или нарушению, используемый в настоящем документе, относится к заболеваниям или нарушениям, связанным с или имеющим одну или несколько мутаций BRAF и/или слияний BRAF. Неограничивающие примеры BRAF-ассоциированного заболевания или нарушения включают, например, BRAF-ассоциированные опухоли.
Фраза «мутация BRAF» относится к генетической мутации (например, хромосомной транслокации, которая приводит к одной или нескольким мутациям в гене BRAF, что приводит к экспрессии белка BRAF с одной или несколькими точечными мутациями по сравнению с белком BRAF дикого типа), или альтернативной сплайсированной версии мРНК BRAF, которая дает белок BRAF, имеющий делецию, по меньшей мере, одной аминокислоты в белке BRAF по сравнению с белком BRAF дикого типа (т.е. вариант сплайсинга). Неограничивающие примеры мутаций BRAF включают мутации BRAF I класса (например, мутации BRAF V600, например, BRAF V600E и BRAF V600K), мутации BRAF II класса (например, не-V600 мутации BRAF и варианты сплайсинга BRAF) и мутации BRAF III класса.
Термин «мутации BRAF Т класса» относится к мутациям BRAF V600, которые сигнализируют как Ras-независимые активные мономеры. Примеры включают мутации BRAF V600E и BRAF V600K.
Термин «мутации BRAF TT класса» включает (i) не-V600 мутации BRAF, которые функционируют как RAS-независимые активированные димеры BRAF и/или CRAF, и (ii) сплайс-варианты BRAF, активность которых зависит от димеризации в RAS-независимой моде.
Примеры не-V600 мутаций BRAF (II класса), включают G469A, G469R, G469V, K601E, K601N, K601T, L597Q и L597V. В одном варианте осуществления, не-V600 мутация BRAF, представляет собой G469A.
Термин «сплайс-вариант BRAF» относится к аберрантно сплайсированным V600E изоформам BRAF. Варианты сплайсинга BRAF представляют собой мутации резистентности BRAF V600E, в которых отсутствуют экзоны, кодирующие часть RAS-связывающего домена, и которые проявляют повышенную димеризацию в клетках с низким уровнем активации RAS (Poulikakos et al., Nature, 480(7377):387-390. Примеры вариантов сплайсинга BRAF V600E включают отсутствие экзонов 4-8 (также известных как p61BRAF(V600E)), экзонов 4-10, экзонов 2-8 или экзонов 2-10. В одном варианте осуществления, мутация резистентности представляет собой p61BRAF(V600E).
Термин «мутация резистентности» относится к мутации в мутации BRAF V600E, которая возникает после воздействия на мутант BRAF V600E ингибитора BRAF, либо отдельно, либо в комбинации с другим противораковым агентом, таким как ингибитор МЕК. Опухоли с мутациями резистентности становятся менее чувствительными (например, резистентными к лечению) к ингибитору BRAF. В одном варианте осуществления, мутация резистентности возникает после воздействия вемурафениба.
Термин «мутации BRAF III класса» относится к не-V600 мутациям BRAF, которые функционируют как RAS-зависимые активированные димеры BRAF и/или CRAF. Неограничивающие примеры мутаций BRAF III класса включают G466A, G466E, G466R, G466V, D594A, D594E, D594G, D594H, G594N, D287H, V549L, S467A, S467E, S467L, G469E, N581S, N5811, F595L, G596A, G596C, G596D, G596R и K483M.
Термин «слияние BRAF» относится к транслокации гена BRAF, которая приводит к экспрессии слитого белка. В одном варианте осуществления, BRAF-ассоциированная опухоль или BRAF-ассоциированный рак имеют одно или несколько слияний BRAF, которые приводят к конститутивной активации и трансформации киназы, включая, но не ограничиваясь ими, KIAA11549-BRAF, MKRN1-BRAF, TRIM24-BRAF, AGAP3-BRAF, ZC3HAV1-BRAF, AKAP9-BRAF, CCDC6-BRAF, AGK-BRAF, EPS15-BRAF, NUP214-BRAF, ARMC10-BRAF, BTF3L4-BRAF, GHR-BRAF, ZC3HAV1-BRAF, ZNF767-BRAF, CCDC91-BRAF, DYNC112-BRAF, ZKSCAN1-BRAF, GTF2I-BRAF, MZT1-BRAF, RAD18-BRAF, CUX1-BRAF, SLC12A7-BRAF, MYRIP-BRAF, SND1-BRAF, NUB1-BRAF, KLHL7-BRAF, TANK-BRAF, RBMS3-BRAF, STRN3-BRAF, STK35-BRAF, ETFA-BRAF, SVOPL-BRAF, JHDM1D-BRAF или BCAP29-BRAF.
Термин «BRAF-ассоциированная опухоль» или «BRAF-ассоциированный рак», используемый в настоящем документе, относится к опухолям или ракам, ассоциированным с или имеющим мутацию BRAF, и включает опухоли, имеющие мутацию BRAF V600 I класса, например, мутацию BRAF V600E или V600K, и опухоли, имеющие мутацию BRAF II класса. BRAF-ассоциированные опухоли включают как доброкачественные BRAF-ассоциированные опухоли, так и злокачественные BRAF-ассоциированные опухоли (т.е. BRAF-ассоциированные раки).
Термин «опухоль», используемый в настоящем документе, относится к аномальному росту ткани, возникающему в результате неконтролируемой, обычно быстрой клеточной пролиферации. Опухоль может быть доброкачественной опухолью (не раковой) или злокачественной опухолью (т.е. раком). Опухоль может быть солидной опухолью или гемобластозом (т.е. гематологической опухолью, также известной как рак крови).
Термин «дикий тип» описывает нуклеиновую кислоту (например, ген BRAF или мРНК BRAF), которая обычно обнаруживается у субъекта, у которого нет заболевания или нарушения, относительно эталонной нуклеиновой кислоты или белка.
Термин «BRAF дикого типа» описывает нуклеиновую кислоту BRAF (например, ген BRAF или мРНК BRAF) или белок BRAF, обнаруженный у субъекта, у которого нет BRAF-ассоциированного заболевания, например, BRAF-ассоциированного рака (и, необязательно, также не имеет повышенного риска развития BRAF-ассоциированного заболевания и/или у него нет подозрения на BRAF-ассоциированное заболевание), или обнаруженный в клетке или ткани субъекта, у которого нет BRAF-ассоциированного заболевания, например BRAF-ассоциированного рака (и, необязательно, также не имеет повышенного риска развития BRAF-ассоциированного заболевания и/или не подозревается на наличие BRAF-ассоциированного заболевания).
Термин «регулирующее агентство» относится к агентству страны, утверждающему медицинское использование фармацевтических агентов в стране. Например, неограничивающим примером регулирующего органа является Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA).
В настоящем документе предложен способ лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в таком лечении, включающий введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или их фармацевтически приемлемой соли, или их фармацевтической композиции. Например, в настоящем документе предложены способы лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в таком лечении, где способ включает а) обнаружение мутации BRAF в образце, взятом у субъекта; и b) введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, мутация BRAF представляет собой мутацию I класса. В некоторых вариантах осуществления, мутация BRAF I класса представляет собой BRAFV600E. В некоторых вариантах осуществления, мутация BRAF представляет собой мутацию II класса. В некоторых вариантах осуществления, мутация II класса представляет собой не-V600 мутацию. В некоторых вариантах осуществления, не-V600 мутация представляет собой G469A. В некоторых вариантах осуществления, мутация II класса представляет собой вариант сплайсинга BRAF V600E. В некоторых вариантах осуществления, вариант сплайсинга BRAF V600E представляет собой p61BRAF(V600E).
В некоторых вариантах осуществления любого из описанных в настоящем документе способов применения, BRAF-ассоциированная опухоль представляет собой солидную опухоль. В некоторых вариантах осуществления, опухоль является внутричерепной. В некоторых вариантах осуществления, опухоль является экстракраниальной. В некоторых вариантах осуществления любого из способов применения, описанных в настоящем документе, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления любого из способов применения, описанных в настоящем документе, рак представляет собой меланому, рак толстой кишки, колоректальный рак, рак легкого (например, мелкоклеточный рак легкого или немелкоклеточный рак легкого), рак щитовидной железы (например, папиллярный рак щитовидной железы), медуллярный рак щитовидной железы, дифференцированный рак щитовидной железы, рецидивирующий рак щитовидной железы или трудно поддающийся лечению дифференцированный рак щитовидной железы), рак молочной железы, рак мочевого пузыря, рак яичников (карциному яичника), рак ЦНС (включая глиомы и LMD), рак кости, рак ануса, анального канала или аноректума, ангиосаркому, аденокистозную карциному, рак аппендикса, рак глаза, рак желчных протоков (холангиокарциному), рак шейки матки, протоковую карциному in situ, рак эндометрия, желчный пузырь, гепатобилиарный рак, гепато-панкреато-билиарную карциному, плоскоклеточную карциному головы и шеи, рак рта, рак полости рта, лейкоз, рак губы, рак ротоглотки, рак носа, полости носа или среднего уха, рак вульвы, рак пищевода, пищеводно-желудочный рак, рак шейки матки, карциноидную опухоль желудочно-кишечного тракта, нейроэндокринный рак желудочно-кишечного тракта, рак гортаноглотки, рак почки, рак гортани, рак печени, рак носоглотки, неходжкинскую лимфому, рак периферической нервной системы (например, нейробластому), нейроэндокринный рак, рак поджелудочной железы, брюшины, новообразование клеток плазмы, рак сальника и брыжейки, рак глотки, рак предстательной железы, рак почки (например, почечно-клеточный рак (RCC)), рак толстой кишки, рак тонкой кишки, саркому мягких тканей, рак желудка, рак яичка, рак матки рак, рак мочеточника или рак мочевого пузыря.
В одном варианте осуществления, BRAF-ассоциированный рак представляет собой рак ЦНС, меланому, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников, почечно-клеточную карциному или первичную опухоль головного мозга.
В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой экстракраниальный рак, выбранный из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого, рака яичников и нейробластомы. В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой меланому. В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой нейробластому.
В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой внутричерепной рак (рак головного мозга). В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой рак ЦНС.
В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой рак, имеющий мутацию BRAF I класса. В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой рак, имеющий мутацию BRAF V600E или BRAF V600K. В некоторых вариантах осуществления, BRAF-ассоциированный рак, имеющий мутацию BRAF V600E или BRAF V600K, выбран из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого, рака яичников, почечно-клеточной карциномы и их метастатических раков, а также первичных опухолей головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированный рак, имеющий мутацию BRAF V600E или BRAF V600K, представляет собой опухоль ЦНС. В некоторых вариантах осуществления, опухоль ЦНС представляет собой злокачественную опухоль (рак ЦНС). В некоторых вариантах осуществления, злокачественная опухоль представляет собой метастатический рак ЦНС. В некоторых вариантах осуществления, метастатический рак ЦНС выбран из метастатической меланомы, метастатического колоректального рака, метастатического немелкоклеточного рака легкого, метастатического рака щитовидной железы и метастатического рака яичников. В некоторых вариантах осуществления, опухоль ЦНС представляет собой внутричерепную LMD или внечерепную LMD.
В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой рак, имеющий мутацию BRAF II класса. В одном варианте осуществления, рак, имеющий мутацию BRAF II класса, выбран из рака легкого (например, немелкоклеточного рака легкого), меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденокистозной карциномы, рака аппендикса, рака тонкой кишки, нейроэндокринного рака желудочно-кишечного тракта, плоскоклеточного рака головы и шеи, ангиосаркомы, рака мочевого пузыря, новообразования клеток плазмы, гепатобилиарного рака, гепато-панкреато-билиарной карциномы, рака яичников, рака эндометрия, нейроэндокринного рака, холангиокарциномы, рака пищевода, саркомы мягких тканей, лейкоза, неходжкинской лимфомы и рака ЦНС (например, глиомы). В одном варианте осуществления, рак имеет мутацию BRAF G469A.
В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой рак, имеющий мутацию BRAF III класса. В одном варианте осуществления рак, имеющий мутацию BRAF III класса, выбран из меланомы, рака тонкой кишки, колоректального рака, немелкоклеточного рака легкого, рак эндометрия, рака шейки матки, лейкоза, рака мочевого пузыря, неходжкинской лимфомы, глиомы, рака яичников, рака предстательной железы, гепатобилиарного рака, рака пищевода и желудка, саркомы мягких тканей и рака молочной железы. В одном варианте осуществления, рак имеет мутацию BRAF G466V или BRAF D594G. В одном варианте осуществления, рак имеет мутацию BRAF G466V. В одном варианте осуществления, рак имеет мутацию BRAF D594G.
В одном варианте осуществления, BRAF-ассоциированная опухоль содержит BRAF-слитый белок, где опухоль представляет собой карциному молочной железы (например, инвазивную протоковую карциному молочной железы), колоректальную карциному (например, аденокарциному толстой кишки), карциному пищевода (например, аденокарциному пищевода), глиому (например, десмопластическую ганглиому головного мозга детского возраста, пилоцитарную астроцитому головного мозга, плеоморфную ксантоастроцитому головного мозга, глиому спинного мозга низкой степени злокачественности (NOS), анапластическую олигодендроглиому, анапластическую ганглиоглиому), карциному головы и шеи (например, нейроэндокринную карциному головы и шеи), карциному легких (например, аденокарциному легкого, немелкоклеточный рак легкого (NOS)), меланому (например, спитцоидную меланому кожи, неспитцоидную меланому слизистой оболочки, спитцоидную меланому кожи, неизвестную первичную меланому, неспитцоидную меланому кожи), карциному поджелудочной железы (например, аденокарциному, ацинарно-клеточную карциному поджелудочной железы), карциному предстательной железы (например, ацинарную аденокарциному предстательной железы), саркому (злокачественную твердую фиброзную опухоль), карциному щитовидной железы (папиллярную карциному щитовидной железы), неизвестную первичную карциному (например, неизвестную первичную аденокарциному), мезотелиому плевры, аденокарциному прямой кишки, карциному эндометрия матки (например, аденокарциному эндометрия матки (NOS)) или серозную карциному яичника.
В одном варианте осуществления, BRAF-ассоциированный рак выбран из видов рака, имеющих BRAF-слитые белки, описанные в таблице 1 (J.S. Ross, et al., Int. J. Cancer: 138, 881-890 (2016)).
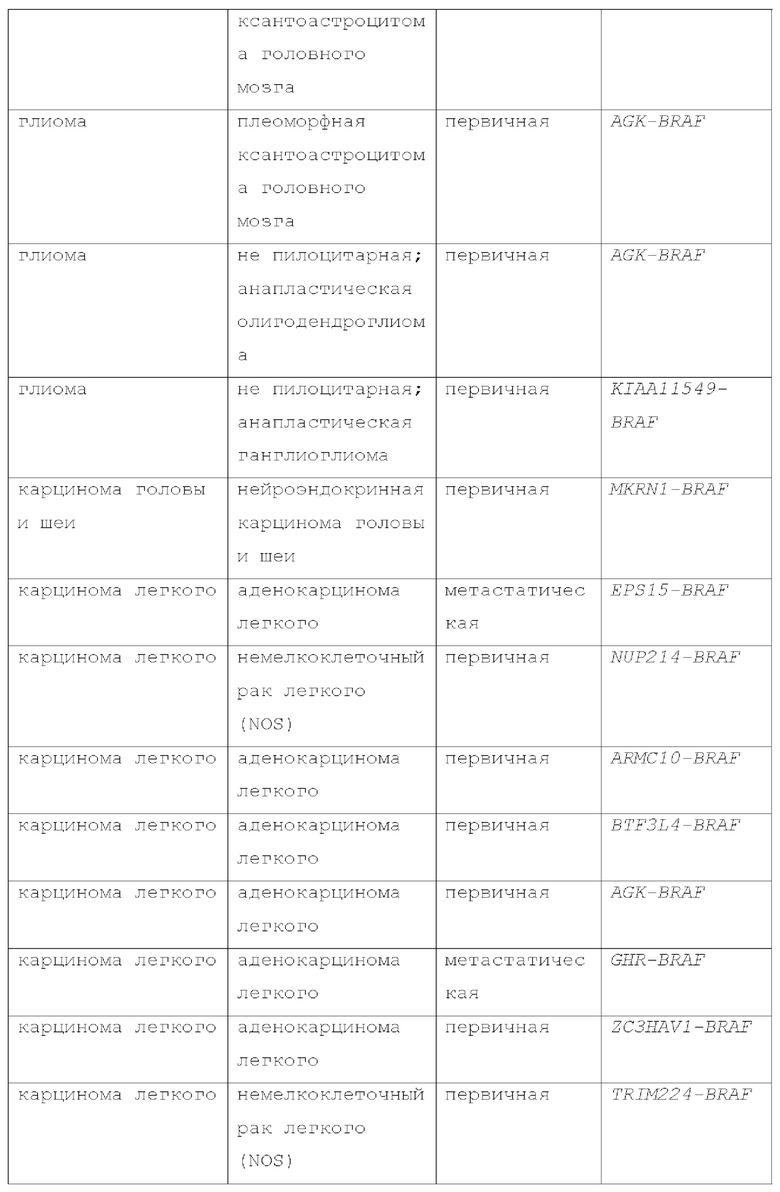

Термин «метастазирование» является известным в данной области техники термином, который относится к распространению раковых клеток из места, где они впервые образовались (первичный участок), в один или несколько других участков у субъекта (один или несколько вторичных участков). При метастазировании, раковые клетки отделяются от исходной (первичной) опухоли, проходят через кровь или лимфатическую систему и образуют новую опухоль (метастатическую опухоль) в других органах или тканях организма. Новая метастатическая опухоль включает те же или подобные раковые клетки, что и первичная опухоль. Во вторичном участке, опухолевая клетка может пролиферировать и начать рост или колонизацию вторичной опухоли в этом удаленном участке.
Термин «метастатический рак» (также известный как «вторичный рак»), используемый в настоящем документе, относится к типу рака, который возникает в одном типе ткани, но затем распространяется на одну или несколько тканей за пределами (первичного) происхождения рака. Метастатический рак головного мозга относится к раку головного мозга, т.е. раку, который возник в ткани, отличной от головного мозга, и дал метастазы в головной мозг.
В одном варианте осуществления, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль ЦНС (т.е. BRAF-ассоциированный рак ЦНС). Термин «рак ЦНС» или «рак ЦНС» или используемый в настоящем документе взаимозаменяемо относится к раку (т.е. злокачественной опухоли) ЦНС, включая раки головного мозга (также известный как внутричерепные опухоли), раки спинного мозга и раки мозговых оболочек, окружающих головной и спинной мозг. Термин «BRAF-ассоциированный рак ЦНС» относится к раку ЦНС, связанному с или имеющему мутацию BRAF. Рак ЦНС включает метастатический рак головного мозга и злокачественные первичные опухоли головного мозга.
В одном варианте осуществления, BRAF-ассоциированный рак ЦНС представляет собой BRAF-ассоциированный метастатический рак головного мозга. BRAF-ассоциированный метастатический рак головного мозга может быть результатом любого рака, описанного в настоящем документе, при котором у субъекта развился, по меньшей мере, один метастаз в головном мозге. В одном варианте осуществления, BRAF-ассоциированный метастатический рак головного мозга представляет собой метастатическую меланому, метастатический колоректальный рак или метастатический немелкоклеточный рак легкого. В одном варианте осуществления, BRAF-ассоциированный метастатический рак головного мозга представляет собой метастатическую меланому. В одном варианте осуществления, BRAF-ассоциированный метастатический рак головного мозга представляет собой метастатический колоректальный рак. В одном варианте осуществления, BRAF-ассоциированный метастатический рак головного мозга представляет собой метастатический немелкоклеточный рак легкого. В одном варианте осуществления, BRAF-ассоциированный метастатический рак головного мозга представляет собой метастатический рак яичников. В одном варианте осуществления, метастатический рак головного мозга представляет собой метастатический рак щитовидной железы. В одном варианте осуществления, BRAF-ассоциированный метастатический рак головного мозга представляет собой рак почки. В одном варианте осуществления, рак представляет собой BRAF-ассоциированный метастатический рак, по меньшей мере, с одним метастазом в головной мозг (т.е. метастатический рак головного мозга). В одном варианте осуществления, рак представляет собой BRAF-ассоциированную метастатическую меланому с, по меньшей мере, одним метастазом в головной мозг. В одном варианте осуществления, рак представляет собой BRAF-ассоциированный метастатический колоректальный рак, по меньшей мере, с одним метастазом в головной мозг. В одном варианте осуществления, рак представляет собой BRAF-ассоциированный метастатический немелкоклеточный рак легкого с, по меньшей мере, одним метастазом в головной мозг. В одном варианте осуществления, рак представляет собой BRAF-ассоциированный метастатический рак яичников, по меньшей мере, с одним метастазом в головной мозг. В одном варианте осуществления, рак представляет собой BRAF-ассоциированный метастатический рак щитовидной железы с, по меньшей мере, одним метастазом в головной мозг. В одном варианте осуществления, рак представляет собой BRAF-ассоциированную нейробластому с, по меньшей мере, одним метастазом в головной мозг.
Лептоменингиальные метастазы (лептоменингеальная болезнь (LMD)) представляют собой подмножество метастазов ЦНС, которые растут в оболочке головного или спинного мозга и/или в спинномозговой жидкости (CSF), или лептоменингеальный карциноматоз. У млекопитающих, мозговые оболочки представлены твердой мозговой оболочкой, паутинной мозговой оболочкой и мягкой мозговой оболочкой. CSF располагается в субарахноидальном пространстве между паутинной и мягкой мозговыми оболочками. Паутинную и мягкую мозговую оболочку вместе иногда называют лептоменинкс.Когда LMD возникает в лептоменинксе и/или CSF, окружающих спинной мозг, ее можно назвать «экстракраниальной LMD». Когда LMD возникает в лептоменинксе и/или CSF головного мозга, ее можно назвать «внутричерепной LMD». Поскольку раковые клетки LMD могут находиться в CSF, они могут быстро распространяться по ЦНС. В результате, LMD имеет плохой прогноз, выживаемость обычно измеряется месяцами. В одном варианте осуществления, метастатический рак представляет собой BRAF-ассоциированную LMD. В одном варианте осуществления, метастатический рак представляет собой внутричерепную BRAF-ассоциированную LMD. В одном варианте осуществления, метастатический рак представляет собой экстракраниальную BRAF-ассоциированную LMD. BRAF-ассоциированные раки с самой высокой частотой лептоменингеальных метастазов представляют собой рак легкого и меланому. В одном варианте осуществления, BRAF-ассоциированная LMD представляет собой LMD, происходящую из метастазов меланомы (т.е. LMD представляет собой метастатическую меланому). В одном варианте осуществления, BRAF-ассоциированная LMD представляет собой LMD, происходящую из метастазов колоректального рака (т.е. LMD представляет собой метастатический колоректальный рак). В одном варианте осуществления, BRAF-ассоциированная LMD представляет собой LMD, происходящую из метастазов немелко клеточного рака легкого (т.е. LMD представляет собой метастатический немелкоклеточный рак легкого).
В одном варианте осуществления, рак представляет собой BRAF-ассоциированный рак с высоким риском метастазирования. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой рак, имеющий мутацию BRAF V600E или BRAF V600K. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой меланому, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников или нейробластому. В одном варианте осуществления, BRAF-ассоциированный рак с высоким риском метастазирования представляет собой меланому, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников или нейробластому, каждый из которых имеет мутацию BRAF V600E или BRAF V600K. В одном варианте осуществления, BRAF-ассоциированный рак с высоким риском метастазирования представляет собой меланому. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой меланому, имеющую мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой колоректальный рак. В одном варианте осуществления, BRAF-ассоциированный рак с высоким риском метастазирования представляет собой колоректальный рак с мутацией BRAF V600E или мутацией BRAF V600K. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой рак щитовидной железы. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой рак щитовидной железы, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированный рак с высоким риском метастазирования представляет собой немелкоклеточный рак легкого. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой немелкоклеточный рак легкого, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой рак яичников. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой рак яичников, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой нейробластому. В одном варианте осуществления, BRAF-ассоциированный рак, имеющий высокий риск метастазирования, представляет собой нейробластому, имеющую мутацию BRAF V600E или мутацию BRAF V600K.
В одном варианте осуществления, рак представляет собой BRAF-ассоциированный рак, имеющий мутацию II класса. В одном варианте осуществления, мутация II класса представляет собой не-V600 мутацию. В одном варианте осуществления, He-V600 мутация представляет собой G469A, G469R, G469V, K601E, K601N, K601T, L597Q или L597V. В одном варианте осуществления, не-V600 мутация представляет собой G469A. В одном варианте осуществления, мутация II класса представляет собой вариант сплайсинга BRAF. В одном варианте осуществления, в варианте сплайсинга BRAF отсутствуют экзоны 4-8 (также известные как p61BRAF(V600E)), экзоны 4-10, экзоны 2-8 или экзоны 2-10. В одном варианте осуществления, вариант сплайсинга BRAF представляет собой p61BRAF(V600E). Неограничивающие примеры BRAF-ассоциированного рака, имеющего мутации II класса, включают рак легкого (например, немелкоклеточный рак легкого и мелкоклеточный рак легкого), меланому, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак щитовидной железы, рак предстательной железы, аденокистозную карциному, рак аппендикса, рак тонкой кишки, плоскоклеточную карциному головы и шеи, ангиосаркому, карциному мочевого пузыря, новообразование клеток плазмы, гепато-панкреато-билиарную карциному, рак яичников, нейроэндокринный рак, холангиокарциному и опухоли ЦНС.
В одном варианте осуществления, BRAF-ассоциированный рак представляет собой BRAF-ассоциированную опухоль ЦНС. В одном варианте осуществления, BRAF-ассоциированная опухоль ЦНС представляет собой BRAF-ассоциированную первичную опухоль головного мозга. В одном варианте осуществления, первичная опухоль головного мозга представляет собой злокачественную первичную опухоль головного мозга. В одном варианте осуществления, первичная опухоль головного мозга представляет собой доброкачественную первичную опухоль головного мозга. В одном варианте осуществления, первичная опухоль головного мозга имеет мутацию I класса. В одном варианте осуществления, первичная опухоль головного мозга имеет мутацию BRAF V600. В одном варианте осуществления, первичная опухоль головного мозга имеет мутацию BRAF V600E или BRAF V600K. В одном варианте осуществления, первичная опухоль головного мозга имеет мутацию II класса. В одном варианте осуществления, первичная опухоль головного мозга имеет мутацию II класса, выбранную из G469A, G469R, G469V, K601E, K601N, K601T, L597Q и L597V. В одном варианте осуществления, первичная опухоль головного мозга имеет мутацию G469A. Первичные опухоли головного мозга представляют собой опухоли, которые начинаются в головном мозге или позвоночнике и известны под общим названием глиомы. Термин «глиома» используется для описания опухолей, происходящих из глиальных клеток, присутствующих в ЦНС. Согласно классификации ВОЗ опухолей головного мозга, глиомы классифицируются по активности клеток и агрессивности по шкале, включающей I степень (доброкачественные опухоли ЦНС) и II-IV степени (злокачественные опухоли ЦНС):
Глиома I степени (пилоцитарная астроцитома): обычно возникает у детей в мозжечке или стволе мозга, и иногда и в больших полушариях, медленно растет. I степень может возникать у взрослых. Несмотря на то, что они доброкачественные (класс I по ВОЗ), сложность лечения этого заболевания делает их рост злокачественным в поведении с высокими показателями заболеваемости (Rostami, Acta Neurochir (Wien). 2017; 159(11): 2217-2221).
Глиома II степени (глиомы низкой степени злокачественности): включает астроцитому, олигодендроглиому и смешанную олигоастроцитму. Глиомы II степени обычно встречаются у молодых людей (20-50 лет) и чаще всего обнаруживаются в полушариях головного мозга. Из-за инфильтративной природы этих опухолей возможны рецидивы. Некоторые глиомы II степени рецидивируют и превращаются в более агрессивные опухоли (III или IV степени).
Глиома III степени (злокачественная глиома): включает анапластическую астроцитому, анапластическую олигодендроглиому и анапластическую смешанную олигоастроцитому. Опухоли III степени являются агрессивными раками высокой степени злокачественности и проникают в близлежащие ткани головного мозга с выступами в виде щупалец, что затрудняет полное хирургическое удаление.
Глиомы IV степени: включают мультиформную глиобластому (GBM) и глиосаркому; (GBM) представляет собой злокачественную глиому. GBM является наиболее агрессивной и наиболее распространенной первичной опухолью головного мозга. Мультиформная глиобластома обычно быстро распространяется и проникает в другие части мозга, образуя отростки в виде щупалец, что затрудняет полное хирургическое удаление. Глиосаркома представляет собой злокачественный рак, и определяется как глиобластома, состоящая из глиоматозных и саркоматозных компонентов.
В одном варианте осуществления, BRAF-ассоциированная первичная опухоль головного мозга представляет собой глиому. В некоторых вариантах осуществления, BRAF-ассоциированная первичная опухоль головного мозга представляет собой глиому, имеющую мутацию I класса. В некоторых вариантах осуществления, BRAF-ассоциированная первичная опухоль головного мозга представляет собой глиому, имеющую мутацию II класса.
Доброкачественные первичные опухоли головного мозга могут вызывать сильную боль, необратимое повреждение головного мозга и смерть, и в некоторых случаях, могут стать злокачественными. Неограничивающие примеры доброкачественных первичных опухолей головного мозга включают глиомы I степени, папиллярные краниофарингиомы, менингиому (включая рабдоидную менингиому), атипичные тератоидные/рабдоидные опухоли и дизэмбриопластическую нейроэпителиальную опухоль (DNT), пилоцитарную астроцитому, олигодендроглиому, смешанную олигоастроцитму, анапластическую астроцитому, анапластическую олигодендроглиому, анапластическую смешанную олигоастроцитому, диффузную астроцитому, эпендимому, плеоморфную ксантоастроцитому (РХА), ганглиоглиому, глиосаркому или анапластическую ганглиоглиому. В одном варианте осуществления, BRAF-ассоциированная опухоль представляет собой доброкачественную первичную опухоль головного мозга.
В одном варианте осуществления, BRAF-ассоциированный рак представляет собой рак периферической нервной системы. В одном варианте осуществления, рак периферической нервной системы представляет собой нейробластому. В одном варианте осуществления, рак представляет собой BRAF-ассоциированный рак.
Было обнаружено, что некоторые соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемые соли обладают хорошей проникающей способностью в головной мозг и/или ЦНС и/или проявляют низкий отток. Такие соединения способны пересекать ВВВ и могут быть полезны для ингибирования киназы BRAF в головном мозге и/или других структурах ЦНС.
Соответственно, некоторые соединения формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемые соли, описанные в настоящем документе, также можно использовать для лечения BRAF-ассоциированных опухолей ЦНС.Например, лечение субъекта с BRAF-ассоциированной опухолью ЦНС может включать введение (например, пероральное введение) соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли субъекту. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС имеет мутацию BRAF V600. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС имеет мутацию BRAF V600E и/или V600K. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС имеет мутацию BRAF V600E. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС имеет мутацию BRAF V600K. В некоторых вариантах осуществления, субъекта ранее лечили одной или несколькими другими противоопухолевыми терапиями, например, противораковым агентом, хирургическим вмешательством и/или лучевой терапией, например, как описано ниже. В некоторых вариантах осуществления, субъект получает лечение соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в комбинации с одной или несколькими другими противоопухолевыми терапиями, например, противораковым агентом, хирургическим вмешательством и/или лучевой терапией, например, как описано ниже. В некоторых вариантах осуществления, субъект подвергается лечению одной или несколькими противоопухолевыми терапиями, например противораковым агентом, хирургическим вмешательством и/или лучевой терапией, после введения соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, например, как описано ниже.
В некоторых вариантах осуществления любого из способов, описанных в настоящем документе, опухоль представляет собой BRAF-ассоциированную опухоль ЦНС, и способ включает введение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой опухоль ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль ЦНС представляет собой злокачественную опухоль ЦНС (рак ЦНС). В некоторых вариантах осуществления, злокачественная опухоль ЦНС представляет собой метастатический рак ЦНС. В некоторых вариантах осуществления, метастатический рак ЦНС представляет собой метастатическую меланому. В некоторых вариантах осуществления, метастатический рак ЦНС представляет собой колоректальный рак. В некоторых вариантах осуществления, метастатический рак ЦНС представляет собой метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, метастатический рак ЦНС представляет собой метастатический рак щитовидной железы. В некоторых вариантах осуществления, метастатический рак ЦНС представляет собой метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль ЦНС представляет собой LMD. В некоторых вариантах осуществления, LMD является внутричерепной. В некоторых вариантах осуществления, LMD является экстракраниальной. В некоторых вариантах осуществления, LMD представляет собой метастатическую меланому. В некоторых вариантах осуществления, LMD представляет собой метастатический колоректальный рак. В некоторых вариантах осуществления, LMD представляет собой метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой первичную опухоль головного мозга. В некоторых вариантах осуществления, первичная опухоль головного мозга представляет собой глиому 2 степени. В некоторых вариантах осуществления, первичная опухоль головного мозга представляет собой глиому 3 степени. В некоторых вариантах осуществления, первичная опухоль головного мозга представляет собой глиому 4 степени. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль ЦНС представляет собой доброкачественную опухоль. В некоторых вариантах осуществления, доброкачественная опухоль ЦНС представляет собой папиллярную краниофарингиому, менингиому (включая рабдоидную менингиому), атипичную тератоидную/рабдоидную опухоль или дизэмбриопластическую нейроэпителиальную опухоль (DNT). В некоторых вариантах осуществления, соединение выбрано из соединения примеров 1-164 или его фармацевтически приемлемой соли.
Способность определить, может ли соединение быть подходящим для лечения рака ЦНС, может быть определена, например, путем идентификации того, является ли соединение субстратом транспортера оттока, и/или измерением проницаемости клеток и/или измерением отношения свободного мозга к свободной плазме, как описано в настоящем документе.
В некоторых вариантах осуществления, соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемые соли обладают высокой клеточной проницаемостью. Способы определения проницаемости соединения можно определить в соответствии с анализом, описанным в примере В, и коэффициенты проницаемости для соединений формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V представлены в таблице B1.
Некоторые соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемые соли демонстрируют низкий отток. Способы оценки in vitro того, являются ли соединения субстратами транспортеров оттока Р-гликопротеина (P-gp или белка 1 с множественной лекарственной резистентностью (MDR1)) и белка резистентности к раку молочной железы (BCRP), описаны в примере В, и коэффициенты оттока соединений формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V представлены в таблице В2. В одном варианте осуществления, соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемые соли имеют коэффициент оттока ≤3,5 при тестировании в клетках, которые экспрессируют P-gp. В одном варианте осуществления соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемые соли имеют коэффициент оттока ≤3,5 при тестировании в клетках, которые экспрессируют P-gp, и коэффициент оттока ≤5,5 при тестировании клеток, экспрессирующих BCRP.
В некоторых вариантах осуществления, некоторые соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемые соли демонстрируют соотношения мозг (несвязанный)/плазма (несвязанный) от среднего до высокого (т.е. соотношение свободного мозга/плазмы от среднего до высокого). Способность соединения проникать через ВВВ субъекта (например, человека) можно определить на подходящей животной модели (например, на грызуне, таком как мышь). Например, способность определенных соединений формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V проникать через ВВВ у мышей определяли путем оценки соотношения концентрации несвязанного мозга к несвязанной плазме (свободного В/Р) у мышей, например, как описано в примере С, и соотношения свободного мозга к свободной плазме приведены в таблице С. Отношение свободного мозга к свободной плазме, равное или превышающее 0,3, свидетельствует о значительной степени свободного проникновения в ЦНС.
Соответственно, в некоторых вариантах осуществления, способы по настоящему изобретению включают способы лечения BRAF-ассоциированного рака ЦНС у субъекта, нуждающегося в этом. В одном варианте осуществления, способ включает введение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, так что, по меньшей мере, часть соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V проникает через ВВВ, как показано на подходящей животной модели. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,3 после введения (например, перорального или внутривенного введения) субъекту. Следует отметить, что долю соединения, проникающего через ВВВ, рассчитывают на основе площади под кривой зависимости концентрации от времени для данного периода времени (AUC0-t) в головном мозге по сравнению с плазмой. Соответственно, доли представляют собой соотношение концентраций. То есть, если (AUC0-24ч) для соединения составляет 30 нг/мл в головном мозге и 70 нг/мл в плазме, то доля соединения, проникающего через ВВВ, составляет 30% (30 нг/мл в головном мозге, деленные на общую концентрацию (30 нг/мл + 70 нг/мл)) (т.е. отношение мозга к плазме 0,30). В некоторых вариантах осуществления, доли рассчитывают на основании площади под кривой зависимости концентрации от времени для периода времени от t=0 (время введения дозы) до последней точки концентрации, поддающейся количественному определению, т.е. (AUC0-last).
Мутации в гене BRAF были идентифицированы при злокачественных меланомах, папиллярных карциномах щитовидной железы, колоректальных карциномах, немелкоклеточной карциноме легкого (NSCLC), карциномах яичников и их метастатических опухолях, а также в первичных опухолях головного мозга (Davies et al., 2002). Например, мутации BRAF наблюдались в многочисленных метастатических опухолях ЦНС, включая метастазы меланомы в головной мозг (Flaherty KT, et al., Nat Rev Cancer (2012) 12(5):349-61), метастазы колоректального рака в головной мозг и метастазы немелкоклеточного рака легкого в головной мозг (Berghoff, AS, Preusser М., Curr Opin Nenrol (2014) 27(6):689-696), папиллярный рак щитовидной железы (Kim, WW et al., J Otolaryngol Head Neck Surg. 2018 47: 4) и рак яичников (Grisham RN., et al., Cancer. 2013; 119:548-554).
Мутации BRAF также наблюдались в злокачественных первичных опухолях головного мозга, включая глиомы IV степени, например, глиобластомы и глиосаркомы, анапластические астроцитомы (опухоли высокой степени злокачественности) и анапластические ганглиоглиомы III степени по ВОЗ (Berghoff, AS, Preusser М., Curr Opin Neurol (2014) 27(6):689-696); Schindler et al. (Acta Neuropathol 121(3):397-405, 2011); Behling et al. (Diagn Pathol 11(1):55, 2016)), у детей и взрослых.
Мутации BRAF также наблюдались в доброкачественных первичных опухолях головного мозга, например в астроцитомах II степени по ВОЗ, плеоморфных ксантоастроцитомах (РХА) II степени по ВОЗ, плеоморфных ксантоастроцитомах с анаплазией, пилоцитарной астроцитоме (РА), папиллярных краниофарингиомах, ганглиоглиомах, астробластомах, пилоцитарных астроцитомах, атипичных тератоидных/рабдоидных опухолях, рабдоидных менингиомах (Berghoff, AS, Preusser М., Curr Opin Neurol (2014) 27(6): 689-696; Schindler et al. (Acta Neuropathol 121(3): 397-405, 2011); Behling et al. (Diagn Pathol 11(1):55, 2016); (Behling et al., Diagn Pathol 11(1): 55, 2016; Brastianos et al., Nat Genet 46(2): 161-165, 2014; Dougherty et al., Neuro Oncol 12(7): 621-630, 2010; Lehman et al., Neuro Oncol 19(1): 31-42, 2017; Mordechai et al., Pediatr Hematol Oncol 32(3): 207-211, 2015; Myung et al., Transl Oncol 5 (6): 430-436, 2012; Schindler et al., Acta Neuropathol 121(3): 397-405, 2011)), у детей и взрослых.
Мутации BRAF также были обнаружены в рецидивирующих нейробластомах (Eleveld, TF, et al., Nat Genet 47(8): 864-871, 2015). Нейробластома представляет собой детскую опухоль периферической нервной системы. У большинства субъектов с нейробластомой опухоли изначально отвечают на химиотерапию, но у значительной части субъектов возникают резистентные к терапии рецидивы.
Соответственно, в настоящем документе также предложен способ лечения субъекта, у которого диагностирована или идентифицирована BRAF-ассоциированная опухоль, например, любая из типовых BRAF-ассоциированных опухолей, описанных в настоящем документе, включающий введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или их фармацевтически приемлемой соли, или их фармацевтической композиции, как определено в настоящем документе. В некоторых вариантах осуществления, субъект, у которого была идентифицирована или диагностирована BRAF-ассоциированная опухоль посредством использования одобренного регулирующим органом, например, одобренного FDA, теста или анализа для выявления мутации BRAF у субъекта или образца биопсии у субъекта, или путем выполнения любого из неограничивающих примеров анализов, описанных в настоящем документе. В некоторых вариантах осуществления, тест или анализ предлагается в виде набора. В одном варианте осуществления, BRAF-ассоциированная опухоль может представлять собой рак, который имеет одну или несколько мутаций BRAF I класса (например, V600E и/или V600K). В одном варианте осуществления, BRAF-ассоциированная опухоль может представлять собой рак, имеющий одну или несколько мутаций II класса (например, G4 69A). В некоторых вариантах осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой BRAF-ассоциированный рак ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой BRAF-ассоциированный метастатический рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатическую меланому. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой внутричерепную или экстракраниальную LMD. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой первичную опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой доброкачественную опухоль ЦНС. В некоторых вариантах осуществления, рак выбран из рака легкого (например, немелкоклеточного рака легкого и мелкоклеточного рака легкого), меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденокистозной карциномы, рака аппендикса, рака тонкой кишки, плоскоклеточной карциномы головы и шеи, ангиосаркомы и опухоли ЦНС. В некоторых вариантах осуществления, соединение выбрано из соединения примеров 1-164 или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является педиатрический субъект.
Также предложены способы лечения опухоли у субъекта, нуждающегося в этом, включающие: (а) обнаружение BRAF-ассоциированной опухоли у субъекта; и (b) введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемой соли, или его фармацевтической композиции. В некоторых вариантах осуществления этих способов, опухоль представляет собой доброкачественную BRAF-ассоциированную опухоль. В некоторых вариантах осуществления этих способов, опухоль представляет собой злокачественную BRAF-ассоциированную опухоль. В некоторых вариантах осуществления этих способов, опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (например, любую из злокачественных BRAF-ассоциированных опухолей, описанных в настоящем документе), и способ дополнительно включает введение субъекту одной или нескольких дополнительных противоопухолевых терапий, например, хирургического вмешательства (например, по меньшей мере, частичной резекции опухоли) и/или лучевой терапии и/или противоракового агента. В некоторых вариантах осуществления этих способов, опухоль представляет собой доброкачественную BRAF-ассоциированную опухоль, например, доброкачественную BRAF-ассоциированную опухоль ЦНС, и способ дополнительно включает введение субъекту одной или нескольких дополнительных противоопухолевых терапий, например, хирургического вмешательства (например, по меньшей мере, частичной резекции опухоли) и/или лучевой терапии и/или противоракового агента. В некоторых вариантах осуществления, у субъекта определяют наличие BRAF-ассоциированной опухоли с помощью одобренного регулирующим органом, например, одобренного FDA, теста или анализа для выявления мутации BRAF у субъекта или в образце биопсии от субъекта (например, биоптате ткани или жидкости) или путем выполнения любого из неограничивающих примеров анализов, описанных в настоящем документе. В некоторых вариантах осуществления, тест или анализ предлагается в виде набора. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой BRAF-ассоциированный рак ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой BRAF-ассоциированный метастатический рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатическую меланому. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой внутричерепную или экстракраниальную LMD. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой первичную опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой доброкачественную опухоль ЦНС. В некоторых вариантах осуществления, рак выбран из рака легкого (например, немелкоклеточного рака легкого и мелкоклеточного рака легкого), меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденокистозной карциномы, рака аппендикса, рака тонкой кишки, плоскоклеточного рака головы и шеи, ангиосаркомы и опухоли ЦНС. В некоторых вариантах осуществления, соединение выбрано из соединения примеров 1-164 или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, субъект является взрослым субъектом. В некоторых вариантах осуществления, субъектом является педиатрический субъект.
Также предложены способы лечения субъекта, имеющего BRAF-ассоциированную опухоль, которые включают проведение анализа образца, полученного от субъекта, для определения наличия у субъекта опухоли, имеющей мутацию BRAF, и введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или их фармацевтически приемлемой соли, или их фармацевтической композиции субъекту, у которого определена мутация BRAF. В некоторых вариантах осуществления этих способов, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (т.е. BRAF-ассоциированный рак), и способ дополнительно включает введение субъекту одной или нескольких других противоопухолевых терапий, например, хирургического вмешательства (например, по меньшей мере, частичной резекции опухоли) и/или лучевой терапии и/или лечения противораковым агентом. В некоторых вариантах осуществления этих способов, субъект ранее подвергался другой противораковой терапии, например, хирургическому вмешательству (например, по меньшей мере, частичной резекции опухоли) и/или лучевой терапии, и/или лечению противораковым агентом. В некоторых вариантах осуществления, субъект представляет собой субъект с подозрением на наличие BRAF-ассоциированной опухоли, субъект, у которого проявляются один или несколько симптомов BRAF-ассоциированной опухоли, или субъект, имеющий повышенный риск развития BRAF-ассоциированной опухоли. В некоторых вариантах осуществления, в анализе используется секвенирование следующего поколения, пиросеквенирование, иммуногистохимия или FISH-анализ разделения. В некоторых вариантах осуществления, анализ представляет собой анализ, одобренный регулирующим органом, например набор, одобренный FDA. В некоторых вариантах осуществления, анализ представляет собой жидкую биопсию. В некоторых вариантах осуществления, биопсия представляет собой биопсию ткани. В некоторых вариантах осуществления, рак представляет собой рак ЦНС, и биопсия представляет собой жидкую биопсию (например, CSF). В некоторых вариантах осуществления, рак представляет собой рак ЦНС, и биопсия представляет собой биопсию ткани (например, образец опухоли, полученный во время традиционной хирургической операции, или стереотаксическую игольчатую биопсию, например стереотаксическую необходимую биопсию под контролем КТ или МРТ). В настоящем документе описаны дополнительные неограничивающие анализы, которые могут быть использованы в этих способах. Дополнительные анализы также известны в данной области техники. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой BRAF-ассоциированный рак ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой BRAF-ассоциированный метастатический рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатическую меланому. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой внутричерепную или экстракраниальную LMD. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой первичную опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой доброкачественную опухоль ЦНС. В некоторых вариантах осуществления, рак выбран из рака легкого (например, немелкоклеточного рака легкого и мелкоклеточного рака легкого), меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденокистозной карциномы, рака аппендикса, рака тонкой кишки, плоскоклеточного рака головы и шеи, ангиосаркомы и опухоли ЦНС. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является педиатрический субъект.
Также предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения при лечении BRAF-ассоциированной опухоли у субъекта, у которого выявлено или диагностировано наличие BRAF-ассоциированной опухоли посредством стадии проведения анализа (например, анализа in vitro) на образце, полученном от субъекта, для определения того, что у субъекта есть мутация BRAF, где наличие мутации BRAF указывает на то, что субъект имеет BRAF-ассоциированную опухоль. Также предложено применение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения BRAF-ассоциированной опухоли у субъекта, у которого идентифицировано или диагностировано наличие BRAF-ассоциированной опухоли на стадии проведения анализа образца, полученного от субъекта, для определения наличия у субъекта мутации BRAF, идентифицирующей наличие у субъекта BRAF-ассоциированной опухоли. Некоторые варианты осуществления любого из способов или применений, описанных в настоящем документе, дополнительно включают запись в истории болезни субъекта (например, на машиночитаемом носителе) о том, что субъекту, у которого определено наличие мутации BRAF при проведении анализа, следует ввести соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или пего фармацевтически приемлемую соль, или его фармацевтическую композицию. В некоторых вариантах осуществления, в анализе используется секвенирование следующего поколения, пиросеквенирование, иммуногистохимия или FISH-анализ разделения. В некоторых вариантах осуществления, анализ представляет собой анализ, одобренный регулирующим органом, например набор, одобренный FDA. В некоторых вариантах осуществления, анализ представляет собой жидкую биопсию. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой BRAF-ассоциированный рак ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой BRAF-ассоциированный метастатический рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатическую меланому. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой внутричерепную или экстракраниальную LMD. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой первичную опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой доброкачественную опухоль ЦНС. В некоторых вариантах осуществления, рак выбран из рака легкого (например, немелкоклеточного рака легкого и мелкоклеточного рака легкого), меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденокистозной карциномы, рака аппендикса, рака тонкой кишки, плоскоклеточного рака головы и шеи, ангиосаркомы и опухоли ЦНС. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является педиатрический субъект.
Также предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения при лечении BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом, или субъект, у которого выявлена или диагностирована BRAF-ассоциированная опухоль. Также предложено применение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли для изготовления лекарственного средства для лечения BRAF-ассоциированной опухоли, у субъекта, идентифицированного или диагностированного как имеющего BRAF-ассоциированную опухоль. В некоторых вариантах осуществления, у субъекта идентифицируют или диагностируют BRAF-ассоциированную опухоль с использованием одобренного регулирующим органом, например одобренного FDA, набора для выявления мутации BRAF у субъекта или образца биопсии субъекта. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой BRAF-ассоциированный рак ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой BRAF-ассоциированный метастатический рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатическую меланому. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой внутричерепную или экстракраниальную LMD. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой первичную опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой доброкачественную опухоль ЦНС. В некоторых вариантах осуществления, рак выбран из рака легкого (например, немелкоклеточного рака легкого и мелкоклеточного рака легкого), меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденокистозной карциномы, рака аппендикса, рака тонкой кишки, плоскоклеточного рака головы и шеи, ангиосаркомы и опухоли ЦНС. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является педиатрический субъект.
В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, анализ, используемый для определения наличия у субъекта мутации BRAF с использованием образца от субъекта, может включать, например, секвенирование нового поколения, иммуногистохимию, флуоресцентную микроскопию, FISH-анализ разделения Саузерн-блоттинг, Вестерн-блоттинг, FACS анализ, Нозерн-блоттинг и амплификацию на основе ПЦР (например, ОТ-ПЦР и количественной ОТ-ПЦР в реальном времени). Как хорошо известно в данной области техники, анализы обычно проводят, например, с использованием, по меньшей мере, одного меченого зонда нуклеиновой кислоты или, по меньшей мере, одного меченого антитела или его антигенсвязывающего фрагмента. В анализах могут использоваться другие способы обнаружения, известные в данной области техники для обнаружения мутации BRAF. В некоторых вариантах осуществления, образец представляет собой биологический образец или образец биопсии (например, образец биопсии, залитый парафином) субъекта. В некоторых вариантах осуществления, субъект представляет собой субъекта с подозрением на BRAF-ассоциированную опухоль, субъекта, имеющего один или несколько симптомов BRAF-ассоциированной опухоли, и/или субъекта, имеющего повышенный риск развития BRAF-ассоциированной опухоли).
В некоторых вариантах осуществления, биопсия представляет собой биопсию опухоли (например, образец опухоли, полученный во время традиционной хирургической операции или стереотаксической игольной биопсии, например, стереотаксической необходимой биопсии под контролем КТ или МРТ). Способы биопсии ткани можно использовать для выявления общей массы опухоли и/или мутации BRAF.
В некоторых вариантах осуществления, мутация BRAF может быть идентифицирована с помощью жидкой биопсии (по-разному называемой жидкостной биопсией или жидкофазной биопсией). См., например, Karachialiou et al., "Real-time liquid biopsies become a reality in cancer treatment", Ann. Transl. Med., 3(3):36, 2016. Способы жидкой биопсии можно использовать для определения общей массы опухоли и/или мутации BRAF. Жидкие биопсии могут быть выполнены на биологических образцах, относительно легко полученных от субъекта (например, посредством простого забора крови), и, как правило, менее инвазивны, чем традиционные способы, используемые для обнаружения опухолевой массы и/или мутации BRAF. В некоторых вариантах осуществления, жидкой биопсии можно использовать для выявления наличия мутации BRAF на более ранней стадии, чем традиционные способы. В некоторых вариантах осуществления, биологический образец, используемый для жидкой биопсии, может включать CSF, кровь, плазму, мочу, слюну, мокроту, бронхоальвеолярный лаваж, желчь, лимфатическую жидкость, кистозную жидкость, кал, асцит и их комбинации. В некоторых вариантах осуществления, для обнаружения циркулирующих опухолевых клеток (СТС) можно использовать жидкую биопсию. В некоторых вариантах осуществления, для обнаружения бесклеточной ДНК можно использовать жидкую биопсию. В некоторых вариантах осуществления, бесклеточная ДНК, обнаруженная с помощью жидкой биопсии, представляет собой циркулирующую опухолевую ДНК (цоДНК), полученную из опухолевых клеток. Анализ цоДНК (например, с использованием чувствительных методов обнаружения, таких как, помимо прочего, секвенирование нового поколения (NGS), традиционная ПЦР, цифровая ПЦР или анализ микрочипов) может быть использован для идентификации мутации BRAF.
В некоторых вариантах осуществления, мутация BRAF, идентифицированная с помощью жидкой биопсии, также присутствует в раковой клетке, которая присутствует у субъекта (например, в опухоли). В некоторых вариантах осуществления, любой из типов мутаций BRAF можно обнаружить с помощью жидкой биопсии. В некоторых вариантах осуществления, генетическая мутация, идентифицированная с помощью жидкой биопсии, может быть использована для идентификации субъекта как кандидата на конкретное лечение. Например, обнаружение мутации BRAF у субъекта может указывать на то, что субъект будет отвечать на лечение, которое включает введение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли.
«Опухолевое бремя», также называемое «опухолевой нагрузкой», относится к общему количеству опухолевого материала, распределенного по всему телу. Опухолевая нагрузка относится к общему количеству раковых клеток или общему размеру опухоли(ей) по всему телу, включая лимфатические узлы и костный мозг (bone narrow в оригинале, опечатка?). Опухолевая нагрузка может быть определена различными способами, известными в данной области техники, такими как, например, путем измерения размеров опухоли(ей) после удаления у субъекта, например, с помощью штангенциркуля, или при нахождении в теле с использованием методов визуализации, например, магнитно-резонансной томографии (МРТ), компьютерной томографии (КТ), мультидетекторной КТ (МДКТ), позитронно-эмиссионной томографии (ПЭТ), рентгена, ультразвука или сканирования костей.
Термин «размер опухоли» или «размер опухоли» относится к общему размеру опухоли, который можно измерить как длину и ширину опухоли. Размер опухоли можно определить различными способами, известными в данной области техники, такими как, например, измерение размеров опухоли(ей) при удалении из субъекта, например, с помощью штангенциркуля, или во время нахождения в теле с использованием методов визуализации, например, МРТ, сканирования костей, УЗИ или КТ.
Жидкие биопсии можно выполнять несколько раз в течение курса диагностики, курса мониторинга и/или курса лечения для определения одного или нескольких клинически значимых параметров, включая, без ограничения, прогрессирование заболевания или эффективность лечения, после назначение лечения субъекту. Например, первая жидкая биопсия может быть выполнена в первый момент времени, и вторая жидкая биопсия может быть выполнена во второй момент времени в ходе диагностики, курса наблюдения и/или курса лечения. В некоторых вариантах осуществления, первый момент времени может представлять собой момент времени до диагностики заболевания у субъекта (например, когда субъект здоров), и второй момент времени может представлять собой момент времени после того, как у субъекта развилось заболевание (например, второй момент времени можно использовать для диагностики заболевания у субъекта). В некоторых вариантах осуществления, первый момент времени может быть моментом времени до диагностики заболевания у субъекта (например, когда субъект здоров), после чего субъект находится под наблюдением, и второй момент времени может быть моментом времени после наблюдения субъекта. В некоторых вариантах осуществления, первый момент времени может представлять собой момент времени после диагностирования заболевания у субъекта, после чего субъекту проводят лечение, и второй момент времени может представлять собой момент времени после проведения лечения; в таких случаях второй момент времени можно использовать для оценки эффективности лечения (например, если генетическая(ие) мутация(и), обнаруженная(ые) в первый момент времени, снижена(ы) или не обнаруживается(ются)). В некоторых вариантах осуществления, лечение, которое должно быть назначено субъекту, может включать соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль.
В одном варианте осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль можно использовать отдельно или в комбинации с одной или несколькими различными формами лечения для лечения субъекта со злокачественной опухолью. Например, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль также может быть использовано в комбинации с одной или несколькими дополнительными противораковыми терапиями, например, хирургическим вмешательством, лучевой терапией, и/или противораковым агентом, который работает по такому же или другому механизму действия. В одном варианте осуществления, лечение субъекта, имеющего злокачественную BRAF-ассоциированную опухоль, соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в комбинации с одной или несколькими дополнительными терапиями, например хирургическим вмешательством, лучевой терапией и/или противораковым агентом, могут иметь повышенную терапевтическую эффективность по сравнению с лечением того же субъекта или аналогичного субъекта соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в качестве монотерапии.
Соответственно, в одном варианте осуществления, в настоящем документе предложены способы лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), которые включают: введение субъекту (i) терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в качестве монотерапии, или (ii) терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в комбинации с одной или несколькими дополнительными противораковыми терапиями. В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение периода времени, где субъекту в течение указанного периода времени вводят вторую противораковую терапию. В одном варианте осуществления, вторая противораковая терапия представляет собой второй противораковый агент.
Также в настоящем документе предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения в комбинации с дополнительной противораковой терапией. Также в настоящем документе предложена дополнительная противораковая терапия для применения в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью.
В настоящем документе также предложено соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль для применения при лечении BRAF-ассоциированной опухоли путем совместного введения с дополнительной противораковой терапией. В настоящем документе также предложена дополнительная противораковая терапия для применения при лечении BRAF-ассоциированной опухоли путем совместного введения с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью.
В некоторых вариантах осуществления, субъекту вводят одну или несколько противораковых терапий, отличных от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, до введения соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, одна или несколько противораковых терапий выбраны из хирургии и/или лучевой терапии и/или противоракового агента, который работает по такому же или другому механизму действия. Например, в некоторых вариантах осуществления, субъекту, нуждающемуся в этом, может быть проведена, по меньшей мере, частичная резекция опухоли перед введением соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, лечение, по меньшей мере, частичной резекцией опухоли уменьшает размер опухоли (например, опухолевой массы) перед введением одной или нескольких доз соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, субъект, нуждающийся в этом, может пройти лучевую терапию перед введением соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, субъект, нуждающийся в этом, может пройти лечение одним или несколькими противоопухолевыми агентами, отличными от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, перед введением соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, у субъекта имеется рак, резистентный или демонстрирующий непереносимость к предыдущей терапии.
Соответственно, в некоторых вариантах осуществления, в настоящем документе представлены способы лечения субъекта, имеющего BRAF-ассоциированную опухоль, включающие (i) введение одной или нескольких противораковых терапий указанному субъекту в течение определенного периода времени, и (ii) после (i), введение (а) соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в виде монотерапии, или (b) соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, в комбинации с одной или несколькими дополнительными противораковыми терапиями.
В некоторых вариантах осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль можно вводить до введения одной или нескольких противоопухолевых терапий (например, хирургического вмешательства, лучевой терапии и/или противоракового агента, которое работает по такому же или другому механизму действия) для лечения субъекта с опухолью. Например, в некоторых вариантах осуществления, субъект, нуждающийся в этом, может подвергнуться, по меньшей мере, частичной резекции опухоли после введения соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, субъект, нуждающийся в этом, может пройти лучевую терапию после введения соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, субъект, нуждающийся в этом, может пройти курс лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью перед введением одного или нескольких противораковых агентов, отличных от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
Соответственно, в некоторых вариантах осуществления, в настоящем документе предложены способы лечения субъекта, имеющего BRAF-ассоциированную опухоль, включающие (i) введение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение определенного периода времени, и (ii) после указанного периода времени, введение одной или нескольких противораковых терапий. Например, нуждающемуся в этом субъекту можно вводить одну или несколько доз соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение определенного периода времени, и затем провести хотя бы частичную резекцию опухоли. В некоторых вариантах осуществления, лечение одной или несколькими дозами соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли уменьшает размер опухоли (например, бремя опухоли) до, по меньшей мере, частичной резекции опухоли. В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления любого из вышеописанных способов, дополнительная противораковая терапия представляет собой хирургическое вмешательство, лучевую терапию и/или противораковый агент, который работает по тому же или другому механизму действия.
Неограничивающие примеры дополнительных противораковых агентов, которые можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов, включают, но не ограничены ими, ингибиторы MEK, ингибиторы BRAF (например, ингибиторы BRAF, отличные от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V), ингибиторы EGFR, ингибиторы HER2 и/или HER3, ингибиторы Ax1, ингибиторы PI3K и ингибиторы S0S1), ингибиторы пути сигнальной трансдукции, ингибиторы контрольных точек, модуляторы пути апоптоза, цитотоксические химиотерапевтические средства, ангиогенез-таргетные терапии и иммунно-таргетные агенты, включая иммунотерапию.
В одном варианте осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов, является таргетным терапевтическим агентом. Используемый в настоящем документе термин «таргетный терапевтический агент» относится к молекуле, которая блокирует рост раковых клеток, вмешиваясь в конкретные молекулы-мишени, необходимые для канцерогенеза и роста опухоли, а не просто вмешиваясь во все быстро делящиеся клетки (например, традиционная цитотоксическая химиотерапия) и включает, но не ограничен ими, терапевтические агенты, таргетирующие на рецепторную тирозинкиназу, ингибиторы пути передачи сигнала (например, ингибиторы пути Ras-Raf-MEK-ERK, ингибиторы пути PI3K-Akt-mTOR-S6K («ингибиторы P13K»)), и модуляторы пути апоптоза.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов, является ингибитором MEK. В одном варианте осуществления, ингибитор MEK представляет собой биниметиниб, траметиниб, кобиниметиниб, селуметиниб, пимасертиб, рефаметиниб, мирдаметиниб, 2-(2-хлор-4-иодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамид (СТ-1040), 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-дион (TAK-733) или его фармацевтически приемлемую соль. Дополнительные примеры ингибиторов MEK включают соединения, описанные в WO 03/077 914, WO 2005/023759, WO 2005/051301, US 7,517,994, US 7,732,616, WO 2005/051906, WO 2005/051302, WO 2005/051300 и WO 2007/044084. В некоторых вариантах осуществления, ингибитор MEK представляет собой биниметиниб или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов, является другим ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V. Неограничивающие примеры других ингибиторов BRAF включают энкорафениб, дабрафениб, вемурафениб, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамид (PLX4720), (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамид (PLX8394) и их фармацевтически приемлемые соли, и соединения, описанные в международной заявке № PCT/IB2020/055992, опубликованной 30 декабря 2020 г. в виде публикации РСТ №WO 2020/261156 А1, включая, например, соединение, выбранное из:
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пропан-1-сульфонамид;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид;
N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-фенил)-3-фторпропан-1-сульфонамид;
N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-4-фторфенил}пропан-1-сульфонамид;
N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторпиридин-2-ил)пропан-1-сульфонамид; и
N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-4-фторфенил}-3-фторпропан-1-сульфонамид;
или его фармацевтически приемлемой соли. В одном варианте осуществления, ингибитор BRAF представляет собой энкорафениб или его фармацевтически приемлемую соль. В одном варианте осуществления, ингибитор BRAF представляет собой N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид или его фармацевтически приемлемую соль. Дополнительные примеры ингибиторов BRAF известны в данной области техники.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов является ингибитором EGFR. Неограничивающие примеры ингибиторов EGFR включают цетуксимаб (Erbitux®), панитумумаб (Vectibix®), осимертиниб (мерелектиниб, Tagrisso®), эрлотиниб (Tarceva®), гефитиниб (Iressa®), нецитумумаб (Portrazza™), нератиниб (Nerlynx®), лапатиниб (Tykerb®), вандетаниб (Caprelsa®), бригатиниб (Alunbrig®) и ингибиторы EGFR, описанные в публикациях РСТ №WO 2019/071351 и WO 2017/117 680, которые полностью включены в настоящий документ посредством ссылки. Дополнительные примеры ингибиторов EGFR известны в данной области техники. В одном варианте осуществления, ингибитор EGFR представляет собой цетуксимаб.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов, является ингибитором HER2 и/или HER3. Неограничивающие примеры ингибиторов HER2 и/или HER3 включают лапатиниб, канертиниб, (Е)-2-метокси-N-(3-(4-(3-метил-4-(6-метилпиридин-3-илокси)фениламино)хиназолин-6-ил)аллил)ацетамид (СР-724714), сапитиниб, 7-[[4-[(3-этинилфенил)амино]-7-метокси-6-хиназолинил]окси]-N-гидроксигептанамид (CUDC-101), мубритиниб, 6-[4-[(4-этилпиперазин-1-ил)метил]фенил]-N-[(1R)-1-фенилэтил]-7Н-пирроло[2,3-d]пиримидин-4-амин (АЕЕ788), ирбинитиниб (тукатиниб), позиотиниб, N-[4-[1-[4-(4-ацетил-1-пиперазинил)циклогексил]-4-амино-3-пиразоло[3,4-d]пиримидинил]-2-метоксифенил]-1-метил-2-индолкарбоксамид (KIN001-111), 7-циклопентил-5-(4-феноксифенил)-7Н-пирроло[2,3-d]пиримидин-4-иламин (KIN001-051), 6,7-диметокси-N-(4-феноксифенил)хиназолин-4-амин (KTN001-30), дазатиниб и бозутиниб.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов, является ингибитором Axl. Неограничивающие примеры ингибиторов Axl включают бемцентиниб, YW327,6S2 (моноклональное антитело), GL2I.T (рецептор-ловушку), 2-(5-хлор-2-(4-((4-метилпиперазин-1-ил)метил)фениламино)пиримидин-4-иламино)-N,N-диметилбензолсульфонамид (ТР-0903), 3-[2-[[3-фтор-4-(4-метил-1-пиперазинил) фенил]амино]-5-метил-7Н-пирроло[2,3-d]пиримидин-4-ил]-бензолацетонитрил (SGI-7079), гилтеритиниб, бозутиниб, кабозантиниб, сунитиниб, форетиниб, амуватиниб, глесатиниб, N-(4-((2-амино-3-хлорпиридин-4-ил)окси)-3-фторфенил)-4-этокси-1-(4-фторфенил)-2-оксо-1,2-дигидропиридин-3-карбоксамид (BMS777607), мерестиниб, (Z)-3-((3-((4-(морфолинометил)-1Н-пиррол-2-ил)метилен)-2-оксоиндолин-5-ил)метил)тиазолидин-2,4-дион (S49076) и (R)-N-(3-фтор-4-((3-((1-гидроксипропан-2-ил)амино)-1Н-пиразоло[3,4-b]пиридин-4-ил)окси)фенил)-3-(4-фторфенил)-1-изопропил-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-карбоксамид.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью для любого из вышеописанных способов, является ингибитором SOS1.
Неограничивающие примеры ингибиторов SOS1 включают ингибиторы, описанные в публикации РСТ №WO 2018/115380, которая полностью включена в настоящий документ посредством ссылки.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью для любого из вышеописанных способов, является ингибитором PI3K.
Неограничивающие примеры включают бупарлисиб (ВКМ120), алпелисиб (BYL719), самотолисиб (LY3023414), 8-[(1R)-1-[(3,5-дифторфенил)амино]этил]-N, N-диметил-2-(морфолин-4-ил)-4-оксо-4Н-хромен-6-карбоксамид (AZD8186), теналисиб (RP6530), вокталисиба гидрохлорид (SAR-245409), гедатолисиб (PF-05212384), панулисиб (Р-7170), тазелисиб (GDC-0032), транс-2-амино-8-[4-(2-гидроксиэтокси)циклогексил]-6-(6-метоксипиридин-3-ил)-4-метилпиридо[2,3-d]пиримидин-7(8Н)-он (PF-04691502), дувелисиб (ABBV-954), N2-[4-оксо-4-[4-(4-оксо-8-фенил-4Н-1-бензопиран-2-ил)морфолин-4-иум-4-илметокси]бутирил]-L-аргинил-глицил-L-аспартил-L-серина ацетат (SF-1126), пиктилисиб (GDC-0941), 2-метил-1-[2-метил-3-(трифторметил)бензил]-6-(морфолин-4-ил)-1Н-бензимидазол-4-карбоновую кислоту (GSK2636771), иделалисиб (GS-1101), умбралисиб тозилат (TGR-1202), пиктилисиб (GDC-0941), копанлисиба гидрохлорид (BAY 84-1236), дактолисиб (BEZ-235), 1-(4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2)ил)пиразин-2-ил]-1-этил-1Н-1,2,4-триазол-3-ил]пиперидин-1-ил)-3-гидроксипропан-1-он (AZD-8835), 5-[6,6-диметил-4-(морфолин-4-ил)-8,9-дигидро-6Н-[1,4]оксазино[4,3-е]пурин-2-ил]пиримидин-2-амин (GDC-0084), эверолимус, рапамицин, перифозин, сиролимус и темсиролимус.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью для любого из вышеописанных способов, является иммунотерапией. Термин «иммунотерапия» относится к агенту, который модулирует иммунную систему. В некоторых вариантах осуществления, иммунотерапия может повышать экспрессию и/или активность регулятора иммунной системы. В некоторых вариантах осуществления, иммунотерапия может снижать экспрессию и/или активность регулятора иммунной системы. В некоторых вариантах осуществления, иммунотерапия может рекрутировать и/или усиливать активность иммунной клетки.
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой терапию антителом (например, моноклональным антителом, конъюгированным антителом). В некоторых вариантах осуществления, терапия антителом представляет собой бевацизумаб (Mvasti™, Avastin®), трастузумаб (Herceptin®), авелумаб (Bavencio®), ритуксимаб (MabThera™, Rituxan®), эдреколомаб (Panorex), даратумумаб (Darzalex®), оларатумаб (Lartruvo™), офатумумаб (Arzerra®), алемтузумаб (Campath®), цетуксимаб (Erbitux®), ореговомаб, пембролизумаб (Keytruda®), динутиксимаб (Unituxin®), обинутузумаб (Gazyva®), тремелимумаб (CP-675,20б), рамикуримаб (Cyramza®), ублитуксимаб (TG-1101), панитумумаб (Vectibix®), элотузумаб (Empliciti™), нецитумумаб (Portrazza™), цирмтузумаб (UC-961), ибритумомаб (Zevalin®), изатуксимаб (SAR650984), нимотузумаб, фрезолимумаб (GC1008), лирилумаб (INN), могамулизумаб (Poteligeo®), фиклатузумаб (AV-299), деносумаб (Xgeva®), ганитумаб, урелумаб, пидилизумаб, аматуксимаб, блинатумомаб (AMG103; Blincyto®) или мидостаурин (Rydapt).
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой конъюгат антитело-лекарственное средство. В некоторых вариантах осуществления, конъюгат антитело-лекарственное средство представляет собой гемтузумаб озогамицин (Mylotarg™), инотузумаб озогамицин (Besponsa®), брентуксимаб ведотин (Adcetris®), адо-трастузумаб эмтансин (TDM-1; Kadcyla®), мирветуксимаб соравтансин (IMGN853) или анетумаб равтанзин.
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, включает токсин. В некоторых вариантах осуществления, иммунотерапия представляет собой денилейкин дифтитокс (Ontak®).
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой цитокиновую терапию. В некоторых вариантах осуществления, цитокиновая терапия представляет собой терапию интерлейкином 2 (IL-2), терапию интерфероном альфа (IFNα), терапию гранулоцитарным колониестимулирующим фактором (G-CSF), терапию интерлейкином 12 (IL-12), терапию интерлейкином 15 (IL-15), терапию интерлейкином 7 (IL-7) или терапию эритропоэтином-альфа (ЕРО). В некоторых вариантах осуществления, терапия IL-2 представляет собой альдеслейкин (Proleukin®). В некоторых вариантах осуществления, терапия IFNα представляет собой IntronA® (Roferon-A®). В некоторых вариантах осуществления, терапия G-CSF представляет собой филграстим (Neupogen®).
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой ингибитор иммунных контрольных точек. В некоторых вариантах осуществления, иммунотерапия включает один или несколько ингибиторов иммунных контрольных точек. В некоторых вариантах осуществления, ингибитор иммунной контрольной точки представляет собой ингибитор CTLA-4, ингибитор PD-1 или ингибитор PD-L1. В некоторых вариантах осуществления, ингибитор CTLA-4 представляет собой ипилимумаб (Yervoy®) или тремелимумаб (CP-675,206). В некоторых вариантах осуществления, ингибитор PD-1 представляет собой пембролизумаб (Keytruda®) или ниволумаб (Opdivo®). В некоторых вариантах осуществления, ингибитор PD-L1 представляет собой атезолизумаб (Tecentriq®), авелумаб (Bavencio®) или дурвалумаб (Imfinzi™). В некоторых вариантах осуществления, ингибитор PD-1 представляет собой RN888 (сасанлимаб).
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой иммунотерапию на основе мРНК. В некоторых вариантах осуществления, иммунотерапия на основе мРНК представляет собой CV9104 (см., например, Rausch et al. (2014) Human Vaccine Immunother 10(11): 3146-52; и Kubler et al. (2015) J. Immunother Cancer 3:26).
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой онколитическую вирусную терапию. В некоторых вариантах осуществления, онколитическая вирусная терапия представляет собой талимоген альгерпарепвек (T-VEC; Imlygic®).
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой противораковую вакцину. В некоторых вариантах осуществления, противораковая вакцина представляет собой вакцину против вируса папилломы человека (HPV). В некоторых вариантах осуществления, вакцина против HPV представляет собой Gardasil®, Gardasil9® или Cervarix®. В некоторых вариантах осуществления, противораковая вакцина представляет собой вакцину против вируса гепатита В (HBV). В некоторых вариантах осуществления, вакцина против HBV представляет собой Engerix-B®, Recombivax НВ® или GI-13020 (Tarmogen®). В некоторых вариантах осуществления, противораковая вакцина представляет собой Twinrix® или Pediarix®. В некоторых вариантах осуществления, противораковая вакцина представляет собой BiovaxID®, Oncophage®, GVAX, ADXS11-001, ALVAC-CEA, PROSTVAC®, Rindopepimut®, CimaVax-EGF, лапулейцел-Т (APC8024; Neuvenge™), GRNVAC1, GRNVAC2, GRN-1201, гепкортеспенлисимут-L (Hepko-V5), DCVAX®, SCIB1, BMT CTN 1401, PrCa VBIR, PANVAC, ProstAtak®, DPX-Survivac или виагенпуматуцел-L (HS-110).
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой пептидную вакцину. В некоторых вариантах осуществления, пептидная вакцина представляет собой нелипепимут-S (Е75) (NeuVax™), IMA901 или SurVaxM (SVN53-67). В некоторых вариантах осуществления, противораковая вакцина представляет собой иммуногенную персональную неоантигенную вакцину (см., например, Ott et al. (2017) Nature 547: 217-221; Sahin et al. (2017) Nature 547: 222-226). В некоторых вариантах осуществления, противораковая вакцина представляет собой RGSH4K или NEO-PV-01. В некоторых вариантах осуществления, противораковая вакцина представляет собой вакцину на основе ДНК. В некоторых вариантах осуществления, вакцина на основе ДНК представляет собой ДНК-вакцину маммаглобин-А (см., например, Kim et al. (2016) OncoImmunology 5(2): e1069940).
В некоторых вариантах осуществления, иммунотерапия, которую можно применять в комбинации с соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой клеточную иммунотерапию (например, адоптивную Т-клеточную терапию, терапию дендритными клетками, терапию естественными киллерами). В некоторых вариантах осуществления, клеточная иммунотерапия представляет собой сипулейцел-Т (АРС8015; Provenge™; Plosker (2011) Drugs 71(1): 101-108). В некоторых вариантах осуществления, клеточная иммунотерапия включает клетки, экспрессирующие химерный антигенный рецептор (CAR). В некоторых вариантах осуществления, клеточная иммунотерапия представляет собой терапию CAR-T-клетками. В некоторых вариантах осуществления, терапия CAR-T-клетками представляет собой тисагенлеклейцел (Kymriah™).
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, I-A, II, III, IV или V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой цитотоксическое химиотерапевтическое средство. Неограничивающие примеры цитотоксических химиотерапевтических средств включают триоксид мышьяка, блеомицин, кабазитаксел, капецитабин, карбоплатин, цисплатин, циклофосфамид, цитарабин, дакарбазин, даунорубицин, доцетаксел, доксорубицин, этопозид, 5-фторурацил, фолиновую кислоту, гемцитабин, иринотекан, ломустин, метотрексат, митомицин С, оксалиплатин, паклитаксел, пеметрексед, темозоломид и винкристин и их комбинации, например, Nordic FLOX (фторурацил, фолиновую кислоту и оксалиплатин), FOLFOXIRI (оксалиплатин, иринотекан и фторурацил), FOLFIRI (фолиновую кислоту, фторурацил и иринотекан) или САРЕОХ (капецитабин и оксалиплатин).
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов, представляет собой ангиогенез-таргетную терапию. Неограничивающие примеры ангиогенез-таргетной терапии включают афлиберцепт и бевацизумаб.
В некоторых вариантах осуществления, противораковый агент, который можно использовать в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемой солью в соответствии с любым из вышеописанных способов, включают модуляторы пути апоптоза (например, обатаклакс).
В некоторых вариантах осуществления, противораковая терапия, которую можно применять в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой лучевую терапию. Неограничивающие примеры лучевой терапии включают внешнюю лучевую терапию (например, внешнюю лучевую терапию с использованием киловольтных рентгеновских лучей или мегавольтных рентгеновских лучей) или внутреннюю лучевую терапию. Внутренняя лучевая терапия (также называемая брахитерапией) может включать использование, например, внутренней лучевой терапии с низкими дозами или внутренней лучевой терапии с высокими дозами. Внутренняя лучевая терапия с низкими дозами включает, например, введение небольших радиоактивных гранул (также называемых семенами) в или проксимально раковой ткани субъекта. Внутренняя лучевая терапия с высокими дозами включает, например, введение тонкой трубки (например, катетера) или имплантата в или проксимально раковой ткани субъекта, а также доставку высокой дозы излучения к тонкой трубке или имплантату с помощью радиационного аппарата. Способы проведения лучевой терапии у субъекта, имеющего рак, известны в данной области техники. В вариантах осуществления, в которых опухоль представляет собой опухоль ЦНС, лучевая терапия может включать лучевую терапию всего головного мозга (WBRT) или стереотаксическую радиохирургию (SRS), такую как Cyberknife®, XKnife®, Gamma knife® или ExacTrac®.
В некоторых вариантах осуществления, противораковая терапия, которую можно применять в комбинации с соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в любом из вышеописанных способов, представляет собой хирургию.
Неограничивающие примеры хирургии включают, например, открытую хирургию или минимально инвазивную хирургию. Хирургия может включать, например, по меньшей мере, частичную резекцию опухоли, удаление всей опухоли, уменьшение объема опухоли или удаление опухоли, вызывающей боль или давление у субъекта. В данной области техники известны способы проведения открытой хирургии и минимально инвазивной хирургии у субъекта, имеющего рак.
В некоторых вариантах осуществления, дополнительная терапия включает любую из перечисленных выше терапий или противораковых агентов, которые являются стандартами лечения рака, где рак имеет мутацию BRAF.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение периода времени, где субъекту вводят ингибитор MEK (например, любой из ингибиторов MEK, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, ингибитор MEK представляет собой биниметиниб или его фармацевтически приемлемую соль. В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение периода времени, где субъекту вводят ингибитор BRAF (например, любой из ингибиторов BRAF, описанных в настоящем документе, включая второе соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль) в течение указанного периода времени. В одном варианте осуществления, соединение представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение периода времени, где субъекту вводят ингибитор EGFR (например, любой из ингибиторов EGFR, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль. В одном варианте осуществления, опухоль представляет собой рак легкого.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемой соли в течение периода времени, где субъекту вводят ингибитор HER2 и/или HER3 в течение указанного периода времени. В одном варианте осуществления, соединение представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемой соли в течение периода времени, где субъекту вводят ингибитор Axl (например, любой из ингибиторов Axl, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение периода времени, где субъекту вводят ингибитор SOS1 (например, любой из ингибиторов SOS1, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение I представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемой соли в течение периода времени, где субъекту вводят ингибитор передачи сигнала (например, любой из ингибиторов передачи сигнала, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемой соли в течение периода времени, где субъекту вводят ингибитор контрольной точки (например, любой из ингибиторов контрольной точки, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемой соли в течение периода времени, где субъекту вводят модулятор пути апоптоза (например, любой из модуляторов пути апоптоза, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемой соли в течение периода времени, где субъекту вводят цитотоксическое химиотерапевтическое средство (например, любое из цитотоксических химиотерапевтических средств, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение периода времени, где субъекту в течение указанного периода времени вводят ангиогенез-таргетную терапию (например, любую из ангиогенез-таргетных терапий, описанных в настоящем документе). В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли в течение периода времени, где субъекту вводят иммуно-таргетный агент (например, любой из иммуно-таргетных агентов, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение формулы I представляет собой соединение, выбранное из примеров 1-164, или его фармацевтически приемлемую соль.
В настоящем документе также предложена фармацевтическая комбинация для лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом, которая включает (а) соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль и (b) по меньшей мере, один дополнительный противораковый агент (например, любой из типовых дополнительных противораковых агентов, описанных в настоящем документе или известных в данной области техники), где соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль, и, по меньшей мере, один дополнительный противораковый агент составлены отдельно для одновременного, раздельного или последовательного применения для лечения опухоли, где количества соединения формулы I, формулы I-A, формула II, формула III, формула IV или формула V или его фармацевтически приемлемой соли и дополнительного противоракового агента вместе эффективны при лечении опухоли; (ii) применение такой комбинации для приготовления лекарственного средства для лечения опухоли; и (iii) коммерческую упаковку или продукт, содержащий такую комбинацию в качестве комбинированного препарата для одновременного, раздельного или последовательного применения; и способ лечения опухоли у нуждающегося в этом субъекта.
Используемый в настоящем документе термин «фармацевтическая комбинация» относится к не фиксированной комбинации активных ингредиентов. Термин «не фиксированная комбинация» означает, что соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль и, по меньшей мере, один дополнительный противораковый агент составляют в виде отдельных композиций или дозировок, таких, которые можно вводить субъекту, нуждающемуся в этом, одновременно, одновременно или последовательно, с различными промежуточными временными рамками, где такое введение обеспечивает эффективные уровни двух или нескольких соединений в организме субъекта. Они также применимы к коктейльной терапии, например, введению трех или нескольких активных ингредиентов.
Соответственно, в настоящем документе также предложен способ лечения BRAF-ассоциированной опухоли, включающий введение субъекту, нуждающемуся в этом, фармацевтической комбинации для лечения указанной опухоли, которая включает (а) соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль, и (b) дополнительный противораковый агент для одновременного, раздельного или последовательного применения для лечения опухоли, где количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли и дополнительного противоракового агента вместе эффективны при лечении опухоли. В одном варианте осуществления, BRAF-ассоциированная опухоль представляет собой злокачественную опухоль, и дополнительный противораковый агент представляет собой противораковый агент, например, любой из противораковых агентов, описанных в настоящем документе. В некоторых вариантах осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль и дополнительный противораковый агент вводят одновременно в виде отдельных доз. В некоторых вариантах осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль и дополнительный противораковый агент вводят в виде отдельных доз последовательно, в любом порядке, например, ежедневно или периодическими дозами, в совместно терапевтически эффективных количествах. Дополнительные противораковые агенты можно вводить с одной или несколькими дозами соединения формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, или его фармацевтической композиции, как часть одной и той же или отдельных дозированных форм, одним и тем же или разными путями введения и/или по одной и той же или разным схемам введения в соответствии со стандартной фармацевтической практикой, известной специалисту в данной области техники. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой злокачественную BRAF-ассоциированную опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой BRAF-ассоциированный рак ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой BRAF-ассоциированный метастатический рак. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатическую меланому. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF ассоциированный метастатический рак представляет собой метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированный метастатический рак представляет собой внутричерепную или экстракраниальную LMD. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС представляет собой первичную опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированная опухоль представляет собой доброкачественную опухоль ЦНС. В некоторых вариантах осуществления, рак выбран из рака легкого (например, немелкоклеточного рака легкого и мелкоклеточного рака легкого), меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденокистозной карциномы, рака аппендикса, рака тонкой кишки, плоскоклеточного рака головы и шеи, ангиосаркомы и опухоли ЦНС.
В некоторых вариантах осуществления любого из способов, описанных в настоящем документе, у субъекта имеется BRAF-ассоциированная опухоль (например, доброкачественная, злокачественная или метастатическая опухоль), где субъект ранее проходил лечение или стандартную терапию (например, лечение одним или несколькими противораковыми агентами, отличными от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, и/или лучевой терапией, и/или хирургией), где указанная BRAF-ассоциированная опухоль стала резистентной или не переносит указанную предшествующую терапию. В некоторых вариантах осуществления, у субъекта имеется BRAF-ассоциированная опухоль (например, местно-распространенная или метастатическая опухоль), которая не поддается стандартной терапии. В одном варианте осуществления, способ включает введение соединения формулы I, выбранного из примеров 1-164, или его фармацевтически приемлемой соли.
Соответственно, в одном варианте осуществления, в настоящем документе предложен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль, где субъекта ранее лечили одной или несколькими противораковыми терапиями (например, противораковым агентом, лучевой терапией и/или хирургическим вмешательством), где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В одном варианте осуществления, BRAF-ассоциированная опухоль, стала резистентной к указанной предшествующей терапии. В одном варианте осуществления, рак представляет собой BRAF-ассоциированный рак, имеющий мутацию II класса. В одном варианте осуществления, мутация II класса представляет собой He-V600 мутацию. В одном варианте осуществления, не-VGOO мутация представляет собой G469A, G469R, G469V, K601E, K601N, K601T, L597Q или L597V. В одном варианте осуществления, He-V600 мутация, представляет собой G469A. В одном варианте осуществления, мутация II класса представляет собой вариант сплайсинга BRAF. В одном варианте осуществления, в варианте сплайсинга BRAF отсутствуют экзоны 4-8 (также известный как p61BRAF(V600E)), экзоны 4-10, экзоны 2-8 или экзоны 2-10. В одном варианте осуществления, вариант сплайсинга BRAF представляет собой p61BRAF(V600E). Неограничивающие примеры BRAF-ассоциированного рака, имеющего мутации II класса, включают рак легкого (например, немелкоклеточный рак легкого и мелкоклеточный рак легкого), меланому, колоректальный рак, рак молочной железы, рак поджелудочной железы, рак щитовидной железы, рак предстательной железы, аденокистозную карциному, рак аппендикса, рак тонкой кишки, плоскоклеточный рак головы и шеи, ангиосаркому и опухоли ЦНС.
В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный рак, ранее лечился ингибитором BRAF (т.е. ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли), отдельно или в комбинации с другим противораковым агентом, перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида, (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба или его фармацевтически приемлемой соли. В одном варианте осуществления, BRAF-ассоциированный рак, который лечили предшествующим ингибитором BRAF, представлял собой рак с мутацией BRAF V600 (например, рак с мутацией BRAF V600E или BRAF V600K). В одном варианте осуществления, BRAF-ассоциированный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, BRAF-ассоциированный рак экспрессировал мутацию резистентности к BRAF V600 во время или после указанного предшествующего лечения. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированной метастатической меланомой получил лечение ингибитором BRAF (т.е. ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, или его фармацевтически приемлемой соли) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил)-2,4-дифторфенил]пропан-1-сульфонамида, (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба или его фармацевтически приемлемой соли. В одном варианте осуществления, меланома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, меланома экспрессировала мутацию резистентности BRAF V600E во время или после указанного предшествующего лечения. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированной метастатической меланомой получил лечение ингибитором BRAF (например, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли) и ингибитором MEK перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли, и ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-иодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (СТ-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба или их фармацевтически приемлемой соли, и ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиниметиниба или их фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее лечили энкорафенибом или его фармацевтически приемлемой солью и биниметинибом или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили дабрафенибом или его фармацевтически приемлемой солью и траметинибом или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили вемурафенибом или его фармацевтически приемлемой солью и кобиниметинибом или его фармацевтически приемлемой солью. В одном варианте осуществления, меланома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, меланома экспрессировала мутацию резистентности BRAF V600E во время или после указанного предшествующего лечения. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированной метастатической меланомой получил лечение одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, меланома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированной метастатической меланомой получил лечение одним или несколькими ингибиторами PI3K перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами PI3K, выбранными из бупарлисиба (ВКМ120), алпелисиба (BYL719), самотолисиба (LY3023414), 8-[(1R)-1-[(3,5-дифторфенил)амино]этил]-N, N-диметил-2-(морфолин-4-ил)-4-оксо-4Н-хромен-6-карбоксамида (AZD8186), теналисиба (RP6530), вокталисиба гидрохлорида (SAR-245409), гедатолисиба (PF-05212384), панулисиба (Р-7170), таселисиба (GDC-0032), транс-2-амино-8-[4-(2-гидроксиэтокси)циклогексил]-6-(6-метоксипиридин-3-ил)-4-метилпиридо[2,3-d]пиримидин-7(8Н)-она (PF-04691502), дувелисиба (ABBV-954), N2-[4-оксо-4-[4-(4-оксо-8-фенил-4Н-1-бензопиран-2-ил)морфолин-4-иум-4-илметокси]бутирил]-L-аргинил-глицил-L-аспартил-L-серина ацетата (SF-1126), пиктилисиба (GDC-0941), 2-метил-1-[2-метил-3-(трифторметил)бензил]-6-(морфолин-4-ил)-1Н-бензимидазол-4-карбоновой кислоты (GSK2636771), иделалисиба (GS-1101), умбралисиба тозилата (TGR-1202), пиктилисиба (GDC-0941), копанлисиба гидрохлорида (BAY 84-1236), дактолисиба (BEZ-235), 1-(4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиеразин-2-ил]-1-этил-1Н-1,2,4-триазол-3-ил]пиперидин-1-ил)-3-гидроксипропан-1-она (AZD-8835), 5-[6,6-диметил-4-(морфолин-4-ил)-8,9-дигидро-6Н-[1,4]оксазино[4,3-е]пурин-2-ил]пиримидин-2-амина (GDC-0084), эверолимуса, рапамицина, перифозина, сиролимуса и темсиролимуса. В одном варианте осуществления, субъекта ранее лечили бупарлисибом или алпелисибом по отдельности или в комбинации. В одном варианте осуществления, меланома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированной метастатической меланомой получил лечение ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) перед лечением соединением формулы I, формула I-A, формула II, формула III, формула IV или формула V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли и одним или несколькими ингибиторами контрольных точек, независимо выбранных из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, меланома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированной метастатической меланомой получил лечение ингибитором BRAF (например, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью), ингибитором MEK и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) до лечения соединением формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой солью, ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) или его фармацевтически приемлемой соли, и одного или нескольких ингибиторов контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба или его фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиниметиниба, и одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, меланома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированной метастатической меланомой подвергался лечению одним или несколькими алкилирующими агентами перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили одним или несколькими алкилирующими агентами, выбранными из темозоломида, фотемустина, ломустина и кармустина. В одном варианте осуществления, субъекта ранее лечили темозоломидом. В одном варианте осуществления, меланома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак, получил лечение ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью, ингибитором MEK и ингибитором EGFR перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) или его фармацевтически приемлемой соли, и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба или их фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиниметиниба или их фармацевтически приемлемой соли, и ингибитором EGFR, выбранным из цетуксимаба и панитумумаба. В одном варианте осуществления, субъекта ранее лечили энкорафенибом или его фармацевтически приемлемой солью, биниметинибом или его фармацевтически приемлемой солью и цетуксимабом. В одном варианте осуществления, субъекта ранее лечили дабрафенибом или его фармацевтически приемлемой солью, траметинибом или его фармацевтически приемлемой солью и панитумумабом. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком получил лечение ингибитором EGFR перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтическим приемлемой солью. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком получал лечение цетуксимабом или панитумумабом до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком получил лечение ингибитором EGFR и одним или несколькими цитотоксическими химиотерапевтическими агентами до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV, или формулы V или его фармацевтически приемлемой солью. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком получил лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и одним или несколькими цитотоксическими химиотерапевтическими агентами. В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получил лечение ингибитором EGFR, выбранным из цетуксимаба или панитумумаба, и одним или несколькими цитотоксическими химиотерапевтическими агентами, такими как Nordic FLOX (фторурацил, фолиновая кислота и оксалиплатин) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак получил лечение ингибитором EGFR и ингибитором BRAF до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальный раком получил лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее получал лечение ингибитором EGFR, выбранным из цетуксимаба и панитумумаба, и ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, или их фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее лечили энкорафенибом или его фармацевтически приемлемой солью и цетуксимабом. В одном варианте осуществления, субъекта ранее лечили вемурафенибом или его фармацевтически приемлемой солью и панитумумабом. В одном варианте осуществления, субъекта ранее лечили дабрафенибом или его фармацевтически приемлемой солью и панитумумабом. В одном варианте осуществления колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с метастатическим колоректальным раком получил лечение ингибитором MEK и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или или ингибитором PD-L1) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-иодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1). В одном варианте осуществления, субъекта ранее лечили ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиниметиниба, и одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK, которым является биниметиниб, и ингибиторами контрольных точек ниволумабом и ипилимумабом. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK биниметинибом и ингибитором контрольной точки пембролизумабом. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK биниметинибом и ингибитором контрольной точки авелумабом. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK траметинибом и ингибиторами контрольных точек ниволумабом и ипилимумабом. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком получил лечение одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или или ингибитором PD-L1) до лечения формулой I, формулой I-A, формулой II, формулой III, формулой IV или формулой V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, субъекта ранее лечили ниволумабом. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком подвергался лечению одним или несколькими цитотоксическими химиотерапевтическими агентами до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение оксалиплатином, иринотеканом, FOLFOXIRI (оксалиплатин, иринотекан и фторурацил), FOLFIRI (фолиновая кислота, фторурацил и иринотекан) или САРЕОХ (капецитабин и оксалиплатин) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, изобретения субъект с BRAF-ассоциированным метастатическим колоректальным раком получил лечение с помощью терапии антителами и одним или несколькими цитотоксическими химиотерапевтическими агентами до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV, или формулы V или его фармацевтически приемлемой солью. В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получил лечение терапией антителами, которая представляет собой бевацизумаб и одним или несколькими цитотоксическими химиотерапевтическими агентами. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF) получал лечение бевацизумабом и иринотеканом, бевацизумабом и FOLFOXIRI (оксалиплатин, иринотекан и фторурацил) или бевацизумабом и FOLFIRI (фолиновая кислота, фторурацил и иринотекан) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком получал лечение ингибитором EGFR, ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V, и одним или несколькими цитотоксическими химиотерапевтическими агентами перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком получил лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, ингибитором BRAF, выбранным из ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли, и одним или несколькими цитотоксическими химиотерапевтическими агентами, выбранными из цетуксимаба и панитумумаба, ингибитором BRAF, который представляет собой вемурафениб или его фармацевтически приемлемую соль, и цитотоксическим химиотерапевтическим агентом, которым является иринотекан. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получил лечение ингибитором EGFR и одним или несколькими цитотоксическими химиотерапевтическими агентами до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальный раком (например, метастатическим колоректальный раком с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и одним или несколькими цитотоксическими химиотерапевтическими агентами. В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальный раком (например, метастатическим колоректальный раком с мутацией BRAF) получил лечение ингибитором EGFR, выбранным из цетуксимаба и панитумумаба, и цитотоксического химиотерапевтического агента, который представляет собой иринотекан или FOLFIRI (фолиевая кислота, фторурацил и иринотекан). В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальный раком подвергался хирургическому лечению до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект стал невосприимчив к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком подвергался хирургическому лечению с последующим лечением ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, ингибитором MEK и ингибитором EGFR перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили хирургическим путем и ранее лечился ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниб, мирдаметиниб, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) или его фармацевтически приемлемой соли, и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили хирургическим путем и ранее лечился ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиниметиниба или его фармацевтически приемлемой соли, и ингибитором EGFR, выбранным из цетуксимаба и панитумумаба. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком подвергался лучевой терапии (например, лучевой терапии всего головного мозга или стереотаксической радиохирургии) до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект с BRAF-ассоциированным метастатическим колоректальным раком подвергался лучевой терапии (например, лучевой терапии всего головного мозга или стереотаксической радиохирургии) с последующим лечением ингибитором BRAF, ингибитором MEK и ингибитором EGFR перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили лучевой терапией и ранее лечился ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) или его фармацевтически приемлемой соли, и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъект ранее подвергался хирургическому лечению и ранее лечился ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, или его фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиниметиниба, или его фармацевтически приемлемой соли, и ингибитора EGFR, выбранного из цетуксимаба и панитумумаба. В одном варианте осуществления, колоректальный рак стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический немелкоклеточный рак легкого (например, метастатический немелкоклеточный рак легкого с мутацией BRAF), получил лечение одним или несколькими ингибиторами EGFR перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами EGFR, независимо выбранными из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили эрлотинибом. В одном варианте осуществления, субъекта ранее лечили гефитинибом. В одном варианте осуществления, субъекта ранее лечили эрлотинибом и гефитинибом. В одном варианте осуществления, немелкоклеточный рак легкого стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, изобретения субъект с BRAF-ассоциированным метастатическим немелкоклеточным раком легкого получил лечение ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой солью, ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) или его фармацевтически приемлемой соли, и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из вемурафениба, дабрафениба и энкорафениба или его фармацевтически приемлемой соли, и ингибитором EGFR, выбранным из цетуксимаба и панитумумаба, до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, немелкоклеточный рак легкого стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический рак щитовидной железы (например, метастатический рак щитовидной железы с мутацией BRAF), получил лечение ингибитором BRAF (т.е. ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) или его фармацевтически приемлемой соли, и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из вемурафениба, дабрафениба и энкорафениба, до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, рак щитовидной железы стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, у субъекта развились метастазы в головной мозг во время указанного предшествующего лечения.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную LMD и ранее лечился ингибитором BRAF (т.е. ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли) и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли, и одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, LMD стала резистентной к указанному предшествующему лечению.
В одном варианте осуществления, субъект имеет связанное с BRAF LMD и ранее лечился ингибитором BRAF (т.е. ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли), ингибитором MEK и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733) или его фармацевтически приемлемой соли, и ингибитором контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба или его фармацевтически приемлемой соли, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиниметиниба или его фармацевтически приемлемой соли, и одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, LMD стала резистентной к указанному предшествующему лечению.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную LMD и ранее лечился одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба, авелумаба и RN888. В одном варианте осуществления, LMD стала резистентной к указанному предшествующему лечению.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее подвергался хирургическому лечению перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF ассоциированную глиому и ранее подвергался лучевой терапии (например, лучевой терапии всего мозга или стереотаксической радиохирургии) до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или их фармацевтически приемлемой солью. В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее лечился одним или несколькими цитотоксическими химиотерапевтическими агентами до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили одним или несколькими цитотоксическими химиотерапевтическими агентами, независимо выбранными из цисплатина, пеметрекседа, винорелбина и паклитаксела. В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее лечился ингибитором орнитиндекарбоксилазы перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект ранее получал лечение ингибитором орнитиндекарбоксилазы, который представляет собой эфлорнитин (в виде рацемата или D- или L-энантиомера). В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее лечился алкилирующим агентом перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект ранее подвергался лечению алкилирующим агентом, выбранным из темозоломида, ломустина и кармустина. В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее лечился алкилирующим агентом и ингибитором орнитиндекарбоксилазы перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект ранее получал лечение алкилирующим агентом, выбранным из темозоломида, ломустина и кармустина, и ингибитором орнитиндекарбоксилазы, который представляет собой эфлорнитин (в виде рацемата или D- или L-энантиомера). В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее лечился лучевой терапией (например, лучевой терапией всего мозга или стереотаксической радиохирургией) и алкилирующим агентом до лечения соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект ранее получал лучевую терапию (например, лучевую терапию всего головного мозга или стереотаксическую радиохирургию) и алкилирующий агент, выбранный из темозоломида, ломустина и кармустина. В одном варианте осуществления, субъект стал резистентным к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее подвергался терапии антителами перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект ранее получал лечение терапией антителами, которая представляет собой бевацизумаб. В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее подвергался хирургическому лечению и лучевой терапии перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее подвергался хирургическому вмешательству, лучевой терапии и алкилирующему агенту перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект ранее подвергался хирургическому вмешательству, лучевой терапии (например, лучевой терапии всего мозга или стереотаксической радиохирургии) и алкилирующему агенту, выбранному из темозоломида, ломустина и кармустина. В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее лечился ингибитором BRAF (т.е. ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект ранее получал лечение ингибитором BRAF, выбранным из N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720), вемурафениба, дабрафениба, энкорафениба и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394). В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому и ранее лечился ингибитором BRAF (т.е. ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли) и ингибитором MEK перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъект ранее получал лечение ингибитором BRAF, выбранным из N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720), вемурафениба, дабрафениба, энкорафениба и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), и ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиниметиниба, селуметиниба, пимасертиба, рефаметиниба, мирдаметиниба, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-диона (TAK-733). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, или их фармацевтически приемлемой соли, и ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиниметиниба, или их фармацевтически приемлемой соли. В одном варианте осуществления, глиома стала резистентной к указанному предшествующему лечению. В одном варианте осуществления, глиома представляет собой глиому 2 степени, 3 степени или 4 степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную ганглиоглиому ствола головного мозга и ранее лечился ингибитором BRAF (т.е. ингибитором BRAF, отличным от соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли) перед лечением соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) или его фармацевтически приемлемой соли. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба или его фармацевтически приемлемой соли. В одном варианте осуществления, ганглиоглиома стала резистентной к указанному предшествующему лечению.
Хотя генетическая основа онкогенеза может различаться у разных типов рака, клеточные и молекулярные механизмы, необходимые для метастазирования, по-видимому, одинаковы для всех типов солидных опухолей. Во время метастатического каскада, раковые клетки теряют ответы ингибирования роста, претерпевают изменения в адгезивности и вырабатывают ферменты, которые могут разрушать компоненты внеклеточного матрикса. Это приводит к отделению опухолевых клеток от исходной опухоли, проникновению в кровоток через вновь образованную сосудистую сеть, миграции и экстравазации опухолевых клеток в благоприятные отдаленные места, где они могут образовывать колонии. Ряд генов был идентифицирован как промоторы или супрессоры метастазирования.
Соответственно, в настоящем документе также предложены способы лечения, ингибирования, профилактики, помощи в профилактике или уменьшения симптомов метастазирования BRAF-ассоциированного рака, у субъекта, нуждающегося в этом, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, или его фармацевтической композиции. В некоторых вариантах осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль используется в комбинации с другим противораковым лечением, например хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или лучевой терапией и/или лечением противораковым агентом. В одном варианте осуществления, рак представляет собой метастатический рак с метастазами в головной мозг, и способ включает введение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В одном варианте осуществления, рак представляет собой метастатическую меланому с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический колоректальный рак с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический немелкоклеточный рак легкого с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический рак яичников с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический рак щитовидной железы с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой нейробластому с метастазами в головной мозг, и способ включает введение соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В одном варианте осуществления, субъект ранее подвергался другому противораковому лечению, например, хирургическому вмешательству (например, по меньшей мере, частичной резекции опухоли) и/или лучевой терапии, и/или лечению противораковым агентом. В одном варианте осуществления, субъект стал резистентным к указанному предыдущему лечению. В одном варианте осуществления, субъект получает лечение соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в комбинации с другим противораковым лечением, например хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или лучевой терапией и/или лечением противораковым агентом.
В настоящем документе также предложены способы ингибирования метастазирования у субъекта, нуждающегося в этом, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли или его фармацевтической композиции. В некоторых вариантах осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль используется в комбинации с другим противораковым лечением, например хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или лучевой терапией и/или лечением противораковым агентом. В одном варианте осуществления, рак представляет собой метастатический рак с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатическую меланому с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический колоректальный рак с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический немелкоклеточный рак легкого с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический рак яичников с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический рак щитовидной железы с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой нейробластому с метастазами в головной мозг. В одном варианте осуществления, субъект ранее подвергался другому противораковому лечению, например, хирургическому вмешательству (например, по меньшей мере, частичной резекции опухоли) и/или лучевой терапии, и/или лечению противораковым агентом. В одном варианте осуществления, субъект стал резистентным к указанному предыдущему лечению. В одном варианте осуществления, субъект получает лечение соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в комбинации с другим противораковым лечением, например хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или лучевой терапией и/или лечением дополнительным противораковым агентом. В одном варианте осуществления, дополнительная противораковая терапия представляет собой противораковый агент. В одном варианте осуществления, дополнительный противораковый агент выбран из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов S0S1, ингибиторов пути сигнальной трансдукции, ингибиторов контрольных точек, модуляторов пути апоптоза, цитотоксических химиотерапевтических средств, ангиогенез-таргетной терапии, и иммуно-таргетных агентов. В одном варианте осуществления, дополнительный противораковый агент представляет собой ингибитор MEK. В одном варианте осуществления, ингибитор MEK представляет собой биниметиниб, траметиниб, кобиниметиниб или их фармацевтически приемлемую соль. В одном варианте осуществления, ингибитор MEK представляет собой биниметиниб или его фармацевтически приемлемую соль.
Используемый в настоящем документе термин «лечение метастазов» означает уменьшение размера, прогрессирования и/или дальнейшего распространения одного или нескольких метастазов.
Также в настоящем документе предложены способы ингибирования метастазирования у субъекта, нуждающегося в этом, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли или его фармацевтической композиции. В некоторых вариантах осуществления, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль используется в комбинации с другим противораковым лечением, например хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или лучевой терапией и/или лечением противораковым агентом. В одном варианте осуществления, рак представляет собой метастатический рак с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатическую меланому с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический колоректальный рак с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический немелкоклеточный рак легкого с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический рак яичников с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой метастатический рак щитовидной железы с метастазами в головной мозг. В одном варианте осуществления, рак представляет собой нейробластому с метастазами в головной мозг. В одном варианте осуществления, субъект ранее подвергался другому противораковому лечению, например, хирургическому вмешательству (например, по меньшей мере, частичной резекции опухоли) и/или лучевой терапии и/или лечению противораковым агентом. В одном варианте осуществления, субъект стал резистентным к указанному предыдущему лечению. В одном варианте осуществления, субъект получает лечение соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в комбинации с другим противораковым лечением, например хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или лучевой терапией и/или лечением противораковым средством. В одном варианте осуществления, противораковая терапия представляет собой противораковый агент. В одном варианте осуществления, противораковый агент выбран из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов S0S1, ингибиторов пути сигнальной трансдукции, ингибиторов контрольных точек, модуляторов пути апоптоза, цитотоксических химиотерапевтических средств, ангиогенез-таргетной терапии и иммуно-таргетных агентов. В одном варианте осуществления, противораковый агент представляет собой ингибитор MEK. В одном варианте осуществления, ингибитор MEK представляет собой биниметиниб, траметиниб, кобиниметиниб или их фармацевтически приемлемую соль. В одном варианте осуществления, ингибитор MEK представляет собой биниметиниб или его фармацевтически приемлемую соль.
Используемый в настоящем документе термин «ингибирование метастазирования» означает уменьшение возникновения (или повторного возникновения) одного или нескольких метастазов, профилактику возникновения (или повторного возникновения) одного или нескольких метастазов или уменьшение распространения одного или нескольких метастазов.
Также предложены способы снижения риска развития одного или нескольких метастазов или одного или нескольких дополнительных метастазов у субъекта, имеющего BRAF-ассоциированный рак, которые включают: отбор, идентификацию или диагностику субъекта как имеющего BRAF-ассоциированный рак, и введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли субъекту, выбранному, идентифицированному или диагностированному как имеющий BRAF-ассоциированный рак. Также предложены способы снижения риска развития одного или нескольких метастазов или одного или нескольких дополнительных метастазов у субъекта, имеющего BRAF-ассоциированный рак, которые включают введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, субъекту, имеющему BRAF-ассоциированный рак. Снижение риска развития одного или нескольких метастазов или одного или нескольких дополнительных метастазов у субъекта, имеющего BRAF-ассоциированный рак, можно сравнить с риском развития одного или нескольких метастазов или одного или нескольких дополнительных метастазов у субъекта до лечения, или по сравнению с субъектом или популяцией субъектов, имеющих аналогичный или такой же BRAF-ассоциированный рак, которые не получали лечения или получали другое лечение.
Фраза «риск развития одного или нескольких метастазов» означает риск того, что у субъекта или субъекта, имеющего первичную опухоль, разовьется дополнительная опухоль (например, солидная опухоль) в месте, удаленном от первичной опухоли, у субъекта или субъекта в течение установленного периода времени, где дополнительная опухоль включает те же или подобные раковые клетки, что и первичная опухоль. В настоящем документе описаны способы снижения риска развития одного или нескольких метастазов у субъекта или субъекта, имеющего рак.
Фраза «риск развития дополнительных метастазов» означает риск того, что субъект или субъект, имеющий первичную опухоль и одну или несколько дополнительных опухолей в местах, удаленных от первичной опухоли (где одна или несколько дополнительных опухолей включают те же или подобные раковые клетки, что и первичная опухоль) разовьется одна или несколько дополнительных опухолей, удаленных от первичной опухоли, где дополнительные опухоли включают те же или подобные раковые клетки, что и первичная опухоль. В настоящем документе описаны способы снижения риска развития дополнительных метастазов.
В настоящем документе также предложен способ лечения BRAF-ассоциированной опухоли, метастазов BRAF-ассоциированной опухоли или их комбинации у нуждающегося в этом субъекта, включающий введение соединения формулы I, формулы I-A, формулы II, субъекту формулы III, формулы IV или формулы V или их фармацевтически приемлемой соли. В одном варианте осуществления, у субъекта имеется, по меньшей мере, один метастаз или имеется риск развития, по меньшей мере, одного метастаза. В одном варианте осуществления, у субъекта имеется, по меньшей мере, один метастаз. В одном варианте осуществления, у субъекта имеется риск развития, по меньшей мере, одного метастаза. В одном варианте осуществления, субъект подвержен риску развития, по меньшей мере, одного метастаза, где указанный субъект имеет рак, выбранный из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого или рака яичников. В одном варианте осуществления, рак представляет собой рак, имеющий мутацию BRAF I класса (например, рак с мутацией BRAF V600, например, рак с мутацией BRAF V600E и/или BRAF V600K). В одном варианте осуществления, рак представляет собой рак, имеющий мутацию BRAF II класса (например, мутацию G469A или вариант сплайсинга BRAF V600E). В одном варианте осуществления, субъект ранее подвергался другому противораковому лечению, например, хирургическому вмешательству (например, по меньшей мере, частичной резекции опухоли) и/или лучевой терапии, и/или лечению противораковым агентом. В одном варианте осуществления, субъект стал резистентным к указанному предыдущему лечению. В одном варианте осуществления, субъект получает лечение соединением формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой солью в комбинации с другим противораковым лечением, например хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или лучевой терапией и/или лечением противораковым агентом. В одном варианте осуществления, противораковая терапия представляет собой противораковый агент, выбранный из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов S0S1, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов пути сигнальной трансдукции, ингибиторов контрольных точек, модуляторов пути апоптоза, цитотоксических химиотерапевтических средств, ангиогенез-таргетной терапии и иммуно-таргетных агентов. В одном варианте осуществления, противораковый агент представляет собой ингибитор MEK. В одном варианте осуществления, ингибитор MEK представляет собой биниметиниб, траметиниб, кобиниметиниб или их фармацевтически приемлемую соль. В одном варианте осуществления, ингибитор MEK представляет собой биниметиниб или его фармацевтически приемлемую соль.
В некоторых вариантах осуществления, субъекту вводят один или несколько агентов для ослабления побочных эффектов лечения (например, один или несколько из кортикостероидов, антагонистов серотонина, антагонистов дофамина, ингибиторов NK-1, каннабиноидов, противотревожных лекарственных средств (например, лоразепама или диазепама), антибиотиков, противогрибковых агентов, колониестимулирующего фактора, добавок железа, прокрита, эпоэтина альфа, дарбэпоэтина альфа, противорвотных агентов, диуретиков, NSAID, анальгетиков, метотрексата, антидиуретиков, пробиотиков, лекарств для снижения кровяного давления, противорвотных агентов, слабительных и т.д.).
В одном варианте осуществления, BRAF-ассоциированная опухоль представляет собой доброкачественную опухоль, и соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемая соль может быть использовано отдельно или в комбинации, с одной или несколькими различными формами лечения для лечения субъекта с доброкачественной опухолью.
В некоторых вариантах осуществления, субъект имеет опухоль ЦНС, и ему вводят один или несколько агентов для облегчения одного или нескольких симптомов, ассоциированных с опухолью ЦНС, включая, но не ограничиваясь ими, судороги, тошноту, головные боли, нечеткость зрения, потерю зрения, потерю ориентации в пространстве, изменения мелкой моторики и сонливость. Примеры таких агентов для облегчения одного или нескольких симптомов, ассоциированных с опухолью ЦНС, включают кортикостероиды, противосудорожные препараты (например, каннабидиол, габапентин или прегабалин), обезболивающие препараты (например, NSAID, ацетаминофен) и средства против тошноты.
Также предложен способ ингибирования активности киназы BRAF в клетке млекопитающего, включающий контакт клетки с эффективным количеством соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, контакт происходит in vitro. В некоторых вариантах осуществления, контакт происходит in vivo. В некоторых вариантах осуществления, контакт происходит in vivo, где способ включает введение терапевтически эффективного количества соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли субъекту, имеющему клетку, обладающую активностью киназы BRAF. В некоторых вариантах осуществления, клетка представляет собой раковую клетку. В некоторых вариантах осуществления, раковая клетка представляет собой любой рак, как описано в настоящем документе. В некоторых вариантах осуществления, раковая клетка представляет собой BRAF-ассоциированную раковую клетку. В некоторых вариантах осуществления, клетка представляет собой клетку головного мозга (например, нервную клетку или глиальную клетку).
Используемый в настоящем документе термин «контакт» относится к соединению указанных групп в системе in vitro или системе in vivo. Например, «контакт» киназы BRAF с соединением, представленным в настоящем документе, включает контакт клетки, содержащей киназу BRAF, с соединением, представленным в настоящем документе, а также, например, введение соединения, представленного в настоящем документе, в образец, содержащий клеточный или очищенный препарат, содержащий киназу BRAF.
В настоящем документе также предложен способ ингибирования клеточной пролиферации in vitro или in vivo, включающий контакт клетки с терапевтически эффективным количеством соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли, или его фармацевтической композиции, как определено в настоящем документе.
Используемый в настоящем документе термин «терапевтически эффективное количество» соединения, его фармацевтической композиции или его фармацевтической комбинации представляет собой количество, достаточное для достижения любого одного или нескольких полезных или желаемых результатов. При профилактическом применении, благоприятные или желаемые результаты включают устранение или снижение риска, уменьшение тяжести или отсрочку начала заболевания, включая биохимические, гистологические и/или поведенческие симптомы заболевания, его осложнения и промежуточные патологические фенотипы, проявляющиеся при развитии заболевания. Для терапевтического применения, полезные или желаемые результаты включают обеспечение терапевтического эффекта, который может включать уменьшение размера опухоли, ингибирование (например, замедление до некоторой степени, предпочтительно, остановку) прогрессирования опухоли, ингибирование (например, замедление до некоторой степени, предпочтительно, остановку) роста опухоли, ингибирование (например, замедление до некоторой степени, предпочтительно, остановку) инвазивности опухоли и/или ингибирование (например, замедление до некоторой степени, предпочтительно, остановку) метастазирования опухоли. Специалисту в данной области техники понятно, что прогрессирование опухоли у людей может быть определено различными способами. Например, размер опухоли, близкой к коже, можно измерить, установив ширину и глубину опухоли штангенциркулем, и затем рассчитав объем опухоли. Менее доступные опухоли, такие как раки легких и ЦНС, можно измерить путем наблюдения за изображениями, полученными с помощью магнитно-резонансной томографии (МРТ). Опухоли ЦНС, такие как опухоли головного мозга, можно измерить с помощью МРТ-сканирования и мониторинга неврологических характеристик. Рост опухоли головного мозга обычно связан со снижением неврологических характеристик. Обеспечение терапевтического эффекта также включает увеличение продолжительности жизни субъекта или субъектов сверх ожидаемого при отсутствии лечения, и/или облегчение до некоторой степени (или, предпочтительно, устранение) одного или нескольких признаков или симптомов, связанных с раком. В одном варианте осуществления, лечение субъекта или субъекта соединением или комбинацией по изобретению продлевает выживаемость по сравнению с ожидаемой при отсутствии лечения на 1 или более месяцев, например, на 3 или более месяцев, например, на 6 или более месяцев, например, на 1 или более лет, например, на 2 или более лет, например, на 3 или более лет, например, на 5 или более лет, например, на 10 или более лет. Оказание терапевтического эффекта также включает уменьшение количества раковых клеток. Оказание терапевтического эффекта также включает устранение раковых клеток. Оказание терапевтического эффекта также включает уменьшение опухолевой массы. Оказание терапевтического эффекта также включает в себя достижение стадии ремиссии рака. Терапевтически эффективное количество можно вводить за один или несколько введений. Для целей настоящего изобретения, дозировка терапевтически эффективного количества соединения или его фармацевтической композиции представляет собой количество, достаточное для проведения профилактического или терапевтического лечения прямо или косвенно. Как понятно в клиническом контексте, дозировка терапевтически эффективного количества соединения или его фармацевтической композиции может быть достигнута в сочетании с другой терапией. Таким образом, «терапевтически эффективное количество» можно рассматривать в контексте применения одной или нескольких терапий (например, одного или нескольких противоопухолевых агентов), и один агент можно рассматривать как вводимый в терапевтически эффективном количестве, если в сочетании с одним или несколькими другими агентами, желаемый результат может быть достигнут или достигается. Что касается лечения рака, терапевтически эффективное количество может также относиться к такому количеству, которое имеет эффект (1) уменьшения размера опухоли, (2) ингибирования (то есть, до некоторой степени замедления, предпочтительно, остановки) появления метастазов опухоли, (3) ингибирования до некоторой степени (то есть, замедление до некоторой степени, предпочтительно, прекращение) роста опухоли или инвазивности опухоли, и/или (4) облегчения до некоторой степени (или, предпочтительно, устранения) одного или нескольких признаков или симптомов, ассоциированных с раком. Терапевтическая или фармакологическая эффективность доз и схем введения также может быть охарактеризована как способность индуцировать, усиливать, поддерживать или продлевать контроль над заболеванием и/или общую выживаемость у субъектов с этими специфическими опухолями, что может быть измерено как увеличение времени до прогрессирования заболевания.
В одном варианте осуществления, субъект, получающий лечение в соответствии с любым из описанных в настоящем документе способов, может быть оценен в соответствии с одним или несколькими стандартными критериями оценки ответа, известными в данной области техники, включая RECIST (критерии оценки ответа при солидных опухолях, например, RECIST версии 1,0, RECIST версии 1,1 и модифицированный RECIST 1,1 (mRECIST 1,1)), RANO-BM (оценка ответа при нейроонкологических метастазах в головной мозг), Macdonald, RANO-LMD и NANO (неврологическая оценка в нейроонкологии). В одном варианте осуществления любого из указанных критериев, опухоль оценивают с помощью визуализирующего исследования (например, MPT, КТ, МДКТ или ПЭТ). В одном варианте осуществления, ответ на лечение оценивают в соответствии с RECIST версии 1,1, где: полный ответ (CR) определяется как полное исчезновение всех опухолевых поражений; частичный ответ (PR) определяется как уменьшение суммы измерений опухоли на, по меньшей мере, 30%; прогрессирующее заболевание (PD) определяется как увеличение, по меньшей мере, на 20% суммы измерений опухоли (где развитие новых поражений или существенное прогрессирование поражений вне мишени также определяется как PD), где увеличение, по меньшей мере, на 5 мм от исходного уровня оценивается как PD; и стабильное заболевание (SD) определяется ни как уменьшение, достаточное чтобы квалифицировать PR, ни как увеличение, достаточное чтобы квалифицировать PD, принимая в качестве эталона наименьшую сумму диаметров во время лечения. В одном варианте осуществления, оценки включают внутричерепной ответ (оцененный в соответствии с модифицированным RECIST с использованием МРТ с усилением гадолинием), внечерепной ответ, частоту общего ответа, частоту контроля заболевания (DCR), продолжительность ответа (DOR), выживаемость без прогрессирования (PFS) и общую выживаемость (OS).
В одном варианте осуществления, субъект имеет опухоль ЦНС и имеет, по меньшей мере, одну поддающуюся измерению внутричерепную опухоль. В одном варианте осуществления, по меньшей мере, одну поддающуюся измерению внутричерепную опухоль измеряют с помощью сканирования МРТ-КТ.
«Измеряемая» опухоль (опухолевое поражение) означает опухоль, которую можно точно измерить, по меньшей мере, в одном измерении (самый длинный диаметр в плоскости измерения не регистрируется) с минимальным размером: 10 мм при КТ (толщина среза КТ не более 5 мм); измерение 10 мм штангенциркулем при клиническом осмотре; 20 мм на рентгенограмме грудной клетки.
При использовании в качестве фармацевтических препаратов, соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль можно вводить в виде фармацевтических композиций. Эти композиции могут быть приготовлены способами, хорошо известными в области фармацевтики, и могут вводиться различными путями, в зависимости от того, требуется ли местное или системное лечение, и от обрабатываемой области. Введение может быть местным (в том числе чрескожным, эпидермальным, офтальмологическим и на слизистые оболочки, включая интраназальное, вагинальное и ректальное введение), легочным (например, путем ингаляции или вдувания порошков или аэрозолей, в том числе с помощью небулайзера; интратрахеально или интраназально), пероральным или парентеральным. Пероральное введение может включать дозированную форму, составленную для введения один раз в день или два раза в день (BID). Парентеральное введение включает внутривенное, внутриартериальное, подкожное, внутрибрюшинное, внутримышечное введение или инъекцию или инфузию; или внутричерепное, например интратекальное или интравентрикулярное введение. Парентеральное введение может осуществляться в форме однократной болюсной дозы, или может осуществляться, например, с помощью перфузионного насоса непрерывного действия. Фармацевтические композиции и составы для местного применения могут включать трансдермальные пластыри, мази, лосьоны, кремы, гели, капли, суппозитории, спреи, жидкости и порошки. Могут быть необходимы или желательны обычные фармацевтические носители, водные, порошковые или масляные основы, загустители и подобные.
В настоящем документе также предложены фармацевтические композиции, которые содержат в качестве активного ингредиента соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль в комбинации с одним или несколькими фармацевтически приемлемыми носителями (эксципиентами). Например, фармацевтическая композиция, приготовленная с использованием соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли. В некоторых вариантах осуществления, композиция подходит для местного применения. При изготовлении представленных в настоящем документе композиций, активный ингредиент обычно смешивают с эксципиентом, разбавляют эксципиентом или заключают в такой носитель в форме, например, капсулы, саше, бумаги или другого контейнера. Когда эксципиент служит разбавителем, он может быть твердым, полутвердым или жидким материалом, который действует как носитель, носитель или среда для активного ингредиента. Таким образом, композиции могут быть в виде таблеток, пилюль, порошков, пастилок, саше, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (в виде твердой или жидкой среды), мазей, содержащих, например, до 10% массовых действующего соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций, стерильных расфасованных порошков. В некоторых вариантах осуществления, композиция составлена для перорального введения. В некоторых вариантах осуществления, композиция представляет собой твердый пероральный состав. В некоторых вариантах осуществления, композиция составлена в виде таблетки или капсулы.
Кроме того, в настоящем документе предложены фармацевтические композиции, содержащие соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль с фармацевтически приемлемым носителем. Фармацевтические композиции, содержащие в качестве активного ингредиента соединение формулы I, формулы I-А, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль, можно приготовить путем тщательного смешивания соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли с фармацевтическим носителем в соответствии с обычными методами приготовления фармацевтических составов. Носитель может принимать самые разные формы в зависимости от желаемого пути введения (например, перорального, парентерального). В некоторых вариантах осуществления, композиция представляет собой твердую пероральную композицию.
Подходящие фармацевтически приемлемые носители хорошо известны в данной области техники. Описания некоторых из этих фармацевтически приемлемых носителей можно найти в The Handbook of Pharmaceutical Excipients, опубликованном Американской фармацевтической ассоциацией и Фармацевтическим обществом Великобритании.
Способы составления фармацевтических композиций описаны в многочисленных публикациях, таких как Pharmaceutical Dosage Forms: Tablets, Second Edition, Revised and Expanded, тома 1-3, под редакцией Lieberman et al.; Pharmaceutical Dosage Forms: Parenteral Medications, тома 1-2, под редакцией Avis et al; и Pharmaceutical Dosage Forms: Disperse Systems, тома 1-2, под редакцией Lieberman et al.; опубликовано Marcel Dekker, Inc.
При приготовлении композиций в пероральной дозированной форме можно использовать любую из обычных фармацевтических сред. Таким образом, для жидких пероральных препаратов, таких как суспензии, эликсиры и растворы, подходящие носители и добавки включают воду, гликоли, масла, спирты, ароматизаторы, консерванты, стабилизаторы, красители и подобные; для твердых пероральных препаратов, таких как порошки, капсулы и таблетки, подходящие носители и добавки включают крахмалы, сахара, разбавители, гранулирующие агенты, смазывающие агенты, связующие агенты, разрыхлители и подобные. Подходящие связующие агенты включают, без ограничений, крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, натуральные и синтетические камеди, такие как аравийская камедь, трагакант или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и подобные. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и подобные. Твердые пероральные препараты также могут быть покрыты такими веществами, как сахара, или иметь энтеросолюбильное покрытие, чтобы модулировать основное место всасывания. Для парентерального введения, носитель обычно будет состоять из стерильной воды, и могут быть добавлены другие ингредиенты для увеличения растворимости или сохранения. Суспензии или растворы для инъекций также можно приготовить с использованием водных носителей вместе с соответствующими добавками. Фармацевтические композиции по настоящему документу будут содержать, на дозированную единицу, например, таблетку, капсулу, порошок, инъекцию, чайную ложку и подобные, количество активного ингредиента, необходимое для доставки терапевтически эффективного количества, как описано в настоящем документе.
Композиции, содержащие соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль, могут быть составлены в виде стандартной дозированной формы, где каждая доза содержит от примерно 5 до примерно 1000 мг (1 г), чаще от примерно 100 до примерно 500 мг активного ингредиента. Термин «стандартная дозированная форма» относится к физически дискретным единицам, подходящим в качестве разовых доз для субъектов-людей и других субъектов, где каждая единица содержит заданное количество активного материала (т.е. соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли), рассчитанная на получение желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим эксципиентом.
В некоторых вариантах осуществления, предлагаемые в настоящем документе композиции содержат от примерно 5 мг до примерно 50 мг активного ингредиента. Специалисту в данной области техники понятно, что речь идет о соединениях или композициях, содержащих от примерно 5 мг до примерно 10 мг, от примерно 10 мг до примерно 15 мг, от примерно 15 мг до примерно 2 0 мг, от примерно 2 0 мг до примерно 25 мг, от примерно 25 мг до примерно 3 0 мг, от примерно 3 0 мг до примерно 35 мг, от примерно 35 мг до примерно 4 0 мг, от примерно 4 0 мг до примерно 4 5 мг или от примерно 45 мг до примерно 50 мг активного ингредиента.
В некоторых вариантах осуществления, предлагаемые в настоящем документе композиции содержат от примерно 50 мг до примерно 500 мг активного ингредиента. Специалисту в данной области техники понятно, что речь идет о соединениях или композициях, содержащих от примерно 50 мг до примерно 100 мг, от примерно 100 мг до примерно 150 мг, от примерно 150 мг до примерно 200 мг, от примерно 200 мг до примерно 250 мг, от примерно 250 мг до примерно 300 мг, от примерно 350 мг до примерно 4 00 мг или от примерно 4 50 мг до примерно 500 мг активного ингредиента. В некоторых вариантах осуществления, композиции, предложенные в настоящем документе, содержат примерно 10 мг, примерно 2 0 мг, примерно 8 0 мг или примерно 160 мг активного ингредиента.
Суточная доза соединения формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемой соли может варьироваться в широком диапазоне от 1,0 до 10000 мг на взрослого человека в день или выше, или любой диапазон в этих пределах. Для перорального введения, композиции предпочтительно предложены в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100, 150, 160, 200, 250 и 500 мг активного ингредиента для симптоматической корректировки дозы для субъекта, подлежащего лечению. Терапевтически эффективное количество лекарственного средства обычно вводят в дозировке от примерно 0,1 мг/кг до примерно 1000 мг/кг массы тела в день или любой другой диапазон в этих пределах. Предпочтительно, диапазон составляет от примерно 0,5 до примерно 500 мг/кг массы тела в день или любой другой диапазон в этих пределах. Более предпочтительно, от примерно 1,0 до примерно 250 мг/кг массы тела в день или любой другой диапазон в этих пределах. Более предпочтительно, от примерно 0,1 до примерно 100 мг/кг массы тела в день или любой другой диапазон в этих пределах. Например, диапазон может составлять от примерно 0,1 до примерно 50,0 мг/кг массы тела в день или любой другой диапазон в этих пределах. В другом примере, диапазон может составлять от примерно 0,1 до примерно 15,0 мг/кг массы тела в день или любой другой диапазон в этих пределах. В еще одном примере, диапазон может составлять от примерно 0,5 до примерно 7,5 мг/кг массы тела в день или любое количество в этом диапазоне. Фармацевтические композиции, содержащие соединение формулы I, формулы I-A, формулы II, формулы III, формулы IV или формулы V или его фармацевтически приемлемую соль, можно вводить по схеме от 1 до 4 раз в день или в виде однократной суточной дозы.
Активное соединение может быть эффективным в широком диапазоне доз и обычно вводится в терапевтически эффективном количестве. Оптимальные дозы для введения могут быть легко определены специалистами в данной области техники. Таким образом, следует понимать, что количество фактически вводимого соединения обычно определяется врачом и будет варьироваться в зависимости от соответствующих обстоятельств, включая способ введения, фактическое вводимое соединение, активность препарата, состояние, подлежащее лечению, и прогрессирование болезненного состояния. Кроме того, факторы, ассоциированные с конкретным субъектом, которого лечат, включая ответ субъекта, возраст, вес, диету, время введения и тяжесть симптомов субъекта, приведут к необходимости корректировки доз.
В некоторых вариантах осуществления, соединения, предложенные в настоящем документе, можно вводить в количестве от примерно 1 мг/кг до примерно 100 мг/кг. В некоторых вариантах осуществления, соединение, предложенное в настоящем документе, можно вводить в количестве от примерно 1 мг/кг до примерно 20 мг/кг, от примерно 5 мг/кг до примерно 5 0 мг/кг, от примерно 10 мг/кг до примерно 40 мг/кг, от примерно 15 мг/кг до примерно 45 мг/кг, от примерно 20 мг/кг до примерно 60 мг/кг или от примерно 4 0 мг/кг до примерно 7 0 мг/кг. Например, примерно 5 мг/кг, примерно 10 мг/кг, примерно 15 мг/кг, примерно 2 0 мг/кг, примерно 25 мг/кг, примерно 30 мг/кг, примерно 35 мг/кг, примерно 4 0 мг/кг, примерно 4 5 мг/кг, примерно 5 0 мг/кг, примерно 55 мг/кг, примерно 60 мг/кг, примерно 65 мг/кг, примерно 7 0 мг/кг, примерно 7 5 мг/кг, примерно 8 0 мг/кг, примерно 8 5 мг/кг, примерно 9 0 мг/кг, примерно 95 мг/кг или примерно 100 мг/кг. В некоторых вариантах осуществления, такое введение может осуществляться один раз в день (QD) или два раза в день (BID). В некоторых вариантах осуществления, такое введение может быть прерывистой схемой введения.
Специалисту в данной области техники будет понятно, что испытания, как in vivo, так и in vitro, с использованием подходящих, известных и общепринятых клеточных и/или животных моделей позволяют прогнозировать способность тестируемого соединения лечить или предотвращать данное нарушение.
Специалисту в данной области техники также будет понятно, что клинические испытания на людях, включая первые испытания на людях, испытания в диапазоне доз и эффективности на здоровых субъектах и/или на субъектах, страдающих данным нарушением, могут быть проведены в соответствии со способами, хорошо известными в клинической и медицинской области техники.
В настоящем документе предложены фармацевтические наборы, применимые, например, для лечения BRAF-ассоциированных заболеваний или нарушений, таких как рак, которые включают один или несколько контейнеров, содержащих фармацевтическую композицию, содержащую терапевтически эффективное количество соединения, предложенного в настоящем документе. Такие наборы могут дополнительно включать, при желании, один или несколько различных компонентов обычных фармацевтических наборов, таких как, например, контейнеры с одним или несколькими фармацевтически приемлемыми носителями, дополнительные контейнеры и т.д., как будет очевидно специалистам в данной области техники. Инструкции в виде вкладышей или этикеток с указанием количества вводимых компонентов, рекомендации по применению и/или рекомендации по смешиванию компонентов также могут быть включены в набор.
Также в настоящем документе предложены следующие варианты осуществления:
Вариант осуществления 1. Соединение формулы I
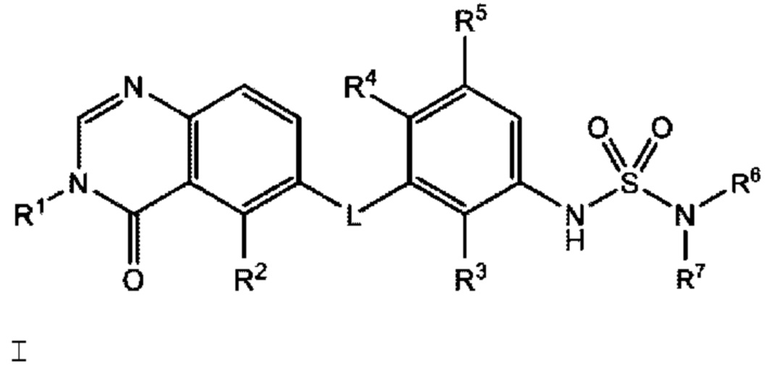
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) С1-С6 алкил-, Ar1, Ar1CH2-, hetAr1 или hetCyc1;
Ar1 представляет собой фенил, который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetAr1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
при условии, что соединение не представляет собой:
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид,
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид, или
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-этил-N-метиламино-1-сульфонамид.
Вариант осуществления 2. Соединение формулы II
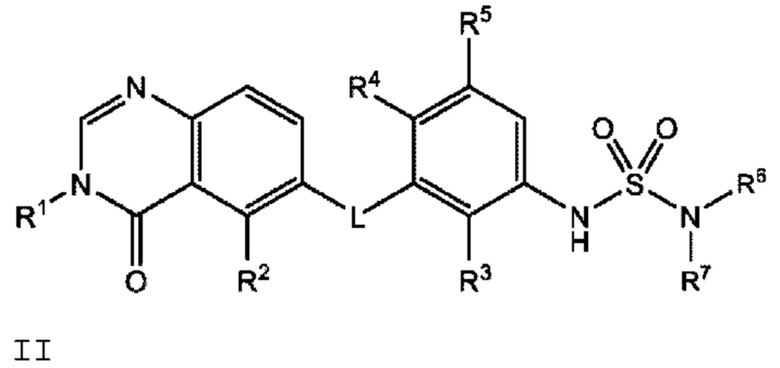
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой С1-С6 алкил или С1-С6 фторалкил;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой C1-С6 алкил, и
R7 представляет собой C1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой O, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
при условии, что соединение не представляет собой:
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид,
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид, или
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-этил-N-метиламино-1-сульфонамид.
Вариант осуществления 3. Соединение формулы III
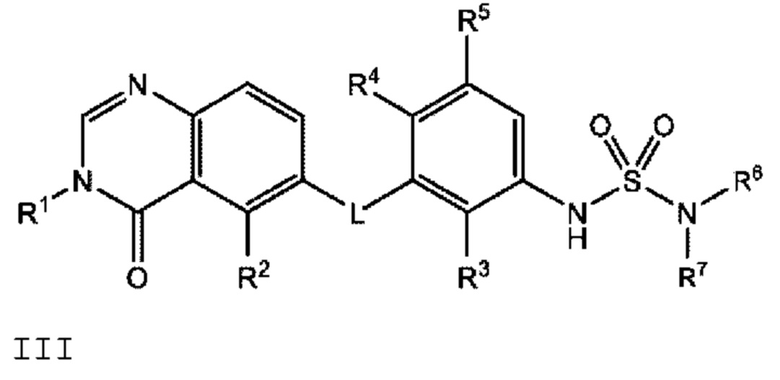
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой C1-С6 алкил, С1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) C1-С6 алкил-, Ar1, Ar1CH2-, hetAr1 или hetCyc1;
Ar1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и C1-С3 алкилом;
hetAr1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и C1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой O, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О.
Вариант осуществления 4. Соединение формулы IV
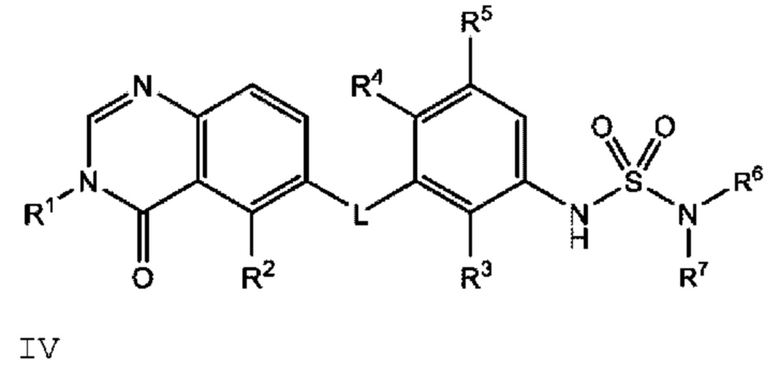
или его фармацевтически приемлемая соль, где:
L представляет собой NH или О;
R1 представляет собой C1-С6 алкил, C1-С6 дейтероалкил, С1-С6 фторалкил, С3-С6 циклоалкил, (С3-С6 циклоалкил)СН2-, (С1-С6 алкокси) C1-С6 алкил-, Ar1, Ar1CH2-, hetAr1 или hetCyc1;
Ar1 представляет собой фенил который необязательно замещен 1, 2, 3, 4 или 5 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetAr1 представляет собой 5-6-членное гетероарильное кольцо, имеющее 1 или 2 кольцевых атома азота и которое необязательно замещено 1, 2 или 3 заместителями, независимо выбранными из галогена и С1-С3 алкила;
hetCyc1 представляет собой 4-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее кольцевой атом кислорода;
R2 представляет собой -СН3, -СН2СН3, -СН=СН2, F, Cl, Br или CN;
R3 представляет собой F или Cl;
R4 представляет собой Н или F;
R5 представляет собой Н, F или Cl;
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил,
или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -ОСН2СН3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца; и
hetCyc2 представляет собой 5-6-членное насыщенное моноциклическое гетероциклическое кольцо, имеющее 1 или 2 кольцевых гетероатомов, независимо выбранных из N и О;
где если R1 представляет собой метил, L представляет собой NH, R3 представляет собой Cl, R4 представляет собой F, R5 представляет собой Н, и R6 представляет собой метил и R7 представляет собой этил, или R6 и R7 вместе с атомом азота, к которому они присоединены, образуют пирролидинил или 3-фторпирролидинил, то R2 представляет собой -СН2СН3, -СН=СН2, F, Cl, Bt или CN.
Вариант осуществления 5. Соединение по любому из вариантов осуществления 1-4 или его фармацевтически приемлемая соль, где R1 представляет собой С1-С6 алкил.
Вариант осуществления 6. Соединение по варианту осуществления 5 или его фармацевтически приемлемая соль, где R1 представляет собой метил.
Вариант осуществления 7. Соединение по любому из вариантов осуществления 1-4 или его фармацевтически приемлемая соль, где R1 представляет собой С1-С6 фторалкил.
Вариант осуществления 8. Соединение по варианту осуществления 7 или его фармацевтически приемлемая соль, где R1 представляет собой фторметил.
Вариант осуществления 9. Соединение по любому из вариантов осуществления 1-8 или его фармацевтически приемлемая соль, где R2 представляет собой F или С1.
Вариант осуществления 10. Соединение по любому из вариантов осуществления 1-9 или его фармацевтически приемлемая соль, где:
R6 представляет собой С1-С6 алкил, и
R7 представляет собой С1-С6 алкил, hetCyc2 или С3-С6 циклоалкил.
Вариант осуществления 11. Соединение по любому из вариантов осуществления 1-9 или его фармацевтически приемлемая соль, где R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, (ii) 6-7-членного конденсированного бициклического кольца, необязательно замещенного 1 или 2 заместителями, независимо выбранными из F и -СН3, (iii) 6-7-членного мостикового кольца, и (iv) 6-8-членного спироциклического кольца.
Вариант осуществления 12. Соединение по варианту осуществления 11 или его фармацевтически приемлемая соль, где R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, необязательно имеющее второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN.
Вариант осуществления 13. Соединение по варианту осуществления 11 или его фармацевтически приемлемая соль, где R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, необязательно замещенное F.
Вариант осуществления 14. Соединение по любому из вариантов осуществления 1-13 или его фармацевтически приемлемая соль, где L представляет собой О.
Вариант осуществления 15. Соединение по любому из вариантов осуществления 1-13 или его фармацевтически приемлемая соль, где L представляет собой NH.
Вариант осуществления 16. Соединение по варианту осуществления 1, выбранное из:
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксипирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-этил-N-метил)-сульфамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N,N-диметил)-сульфамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-(N-этил-N-метил)-сульфамида;
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксипирролидин-1-сульфонамида;
(R)-N-(2-хлор-4-фтор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксипирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида;
(R)-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-метоксипирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида;
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксипирролидин-1-сульфонамида;
N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)пирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида;
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида;
цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-4-метоксипирролидин-1-сульфонамида;
цис-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-фтор-4-метоксипирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида;
цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-4-метоксипирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторпирролидин-1-сульфонамида;
цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,4-дифторпирролидин-1-сульфонамида;
цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,4-дифторпирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,3-дифторпирролидин-1-сульфонамида;
(R)-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-фторпирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(дифторметокси)пирролидин-1-сульфонамида;
(S)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида;
(S)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-метоксипирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фторпирролидин-1-сульфонамида;
(R)-N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-3-метоксипирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фторпирролидин-1-сульфонамида;
(R)-N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-3-фторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксипирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пирролидин-1-сульфонамида;
N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида;
N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида;
N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)пирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пирролидин-1-сульфонамида;
N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)фенил)-(N-этил-N-метил)-сульфамида;
(R)-N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксипирролидин-1-сульфонамида;
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-метоксипирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида;
N-(2-хлор-3-((5-циано-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((3-метил-4-оксо-5-винил-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида;
N-(2-хлор-3-((5-этил-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метокси-d3)пирролидин-1-сульфонамида;
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(метокси-d3)пирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метокси-d3)пирролидин-1-сульфонамида;
(R)-N-(5-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-3-фторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамида;
(R)-N-(5-хлор-2-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида;
цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3,4-дифторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3,3-дифторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-этилпирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3,3-дифторпирролидин-1-сульфонамида;
цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3,4-дифторпирролидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида;
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-(дифторметокси)пирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида;
6-(2-хлор-3-{[этил(метил)сульфамоил]амино}-6-фторфенокси)-3,5-диметил-З,4 дигидрохиназолин-4-она;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N,N-диметил)-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-метил-N-(тетрагидрофуран-3-ил))-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-циклопропил-N-метил)-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-циклобутил-N-метил)-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-4-метоксипиперидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(трифторметил)пирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-4-гидроксипиперидин-1-сульфонамида;
(транс)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-4 -метоксипирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)морфолин-4-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-изопропил-N-метил)-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-азабицикло[2,2,1]heptane-2-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-5-азаспиро[2,4]гептан-5-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-(N,N-диметил)-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пиперидин-1-сульфонамида;
(транс)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,4-дифторпирролидин-1-сульфонамида;
(S)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-изопропил-N-метил)-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-изопропил-N-метил)-сульфамида;
(цис)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,4-дифторпирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-изопропил-N-метил)-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторпирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-этилпирролидин-1-сульфонамида;
цис-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3,4-дифторпирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-(N-этил-N-метил)-сульфамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-(N,N-диметил)-сульфамида;
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-метоксипирролидин-1-сульфонамида;
цис-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фтор-4-метоксипирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида;
(R)-N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-3-фторпирролидин-1-сульфонамида;
N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)пирролидин-1-сульфонамида;
(R)-N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-3-метоксипирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-этилпирролидин-1-сульфонамида;
(S)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-(N-этил-N-метил)-сульфамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-(N,N-диметил)-сульфамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-(N,N-диметил)-сульфамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-(N-этил-N-метил)-сульфамида;
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)пирролидин-1-сульфонамида;
(R)-N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксиазетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксиазетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторазетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-(метоксиметил)азетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторазетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-метоксиазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксиазетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксиазетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксиазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(метоксиметил)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторазетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторазетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,3-дифторазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((дифторметокси)метил)азетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((дифторметокси)метил)азетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-((дифторметокси)метил)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)-3-метилазетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-этоксиазетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-этоксиазетидин-1-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(дифторметокси)азетидин-1-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(дифторметокси)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-3-(метоксиметил)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-цианоазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метилазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-6-фтор-3-азабицикло[3,1,0]гексан-3-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-1-фтор-3-азабицикло[3,1,0]гексан-3-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторфенил)пирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторфенил)-3-фторпирролидин-1-сульфонамида;
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторфенил)-3-метоксипирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-азабицикло[3,1,0]гексан-2-сульфонамида;
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)пирролидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-3-метилазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((трифторметокси)метил)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-азаспиро[3,3]гептан-2-сульфонамида;
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-азаспиро[3,3]гептан-2-сульфонамида;
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азаспиро[3,3]гептан-2-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-этоксиазетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(дифторметокси)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(трифторметокси)азетидин-1-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-5-азаспиро[2,3]гексан-5-сульфонамида;
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((фторметокси)метил)азетидин-1-сульфонамида;
или его фармацевтически приемлемой соли.
Вариант осуществления 17. Соединение формулы V
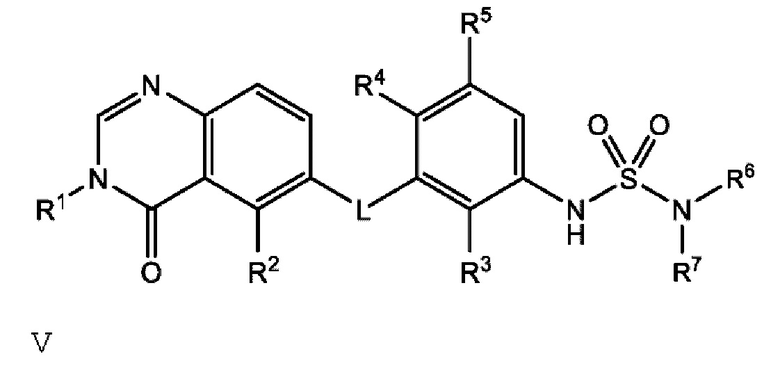
или его фармацевтически приемлемая соль, где:
L представляет собой NH;
R1 представляет собой С1-С6 алкил;
R2 представляет собой F или С1;
R3 представляет собой С1;
R4 представляет собой F;
R5 представляет собой Н;
R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенную кольцевую систему, выбранную из (i) 4-6-членного моноциклического кольца, необязательно имеющего второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -OCH2CH3 и CN, и (iii) 6-7-членного мостикового кольца.
Вариант осуществления 18. Соединение по варианту осуществления 17, где R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 4-6-членное моноциклическое кольцо, необязательно имеющее второй кольцевой гетероатом, который представляет собой О, где указанное кольцо необязательно замещено 1 или 2 заместителями, независимо выбранными из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -ОСН2СН3 и CN.
Вариант осуществления 19. Соединение по варианту осуществления 18, где R6 и R7 вместе с атомом азота, к которому они присоединены, образуют 4-6-членное моноциклическое кольцо, где указанное кольцо замещено заместителем, выбранным из F, -ОН, -ОСН3, -OCHF2, -OCD3, -СН3, -СН2СН3, -СН2ОСН3, -CH2OCH2F, -CH2OCHF2, -CH2OCF3, -OCF3, -ОСН2СН3 и CN.
Вариант осуществления 20. Соединение по варианту осуществления 19, где R6 и R7 вместе с атомом азота, к которому они присоединены, образуют насыщенное 4-6-членное моноциклическое кольцо, замещенное F.
Вариант осуществления 21. Соединение, которое выбрано из:
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида, имеющего структуру:
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида и кристаллической формы (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида, имеющего структуру
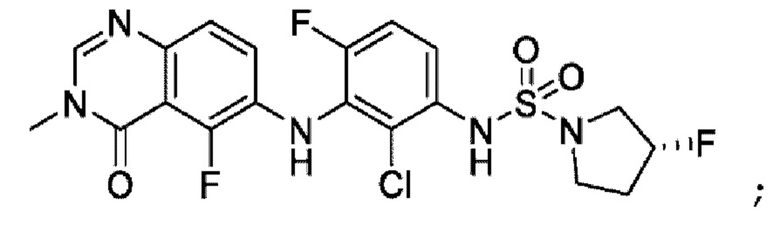 и
и
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамида, имеющего структуру:
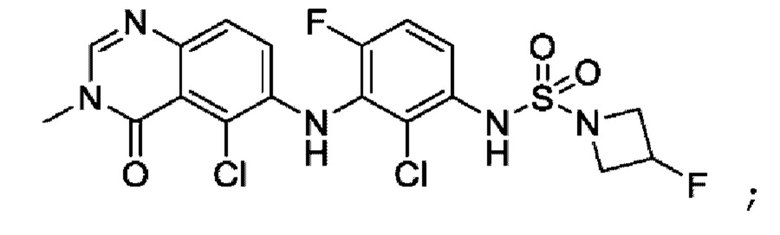
или его фармацевтически приемлемой соли.
Вариант осуществления 22. Соединение, которое представляет собой N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид, имеющий структуру:
или его фармацевтически приемлемую соль.
Вариант осуществления 23. Соединение, которое представляет собой N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид, имеющий структуру:
Вариант осуществления 24. Фармацевтическая композиция, содержащая соединение по любому из вариантов осуществления 1-23 или его фармацевтически приемлемую соль и один или несколько фармацевтически приемлемых носителей.
Вариант осуществления 25. Способ получения соединения по варианту осуществления 1 или его фармацевтически приемлемой соли, включающий:
(a) для соединения по варианту осуществления 1, где L, R1, R2, R3, R4, R5, R6 и R7 такие, как определены в варианте осуществления 1, сочетание соединения, имеющего формулу (25)
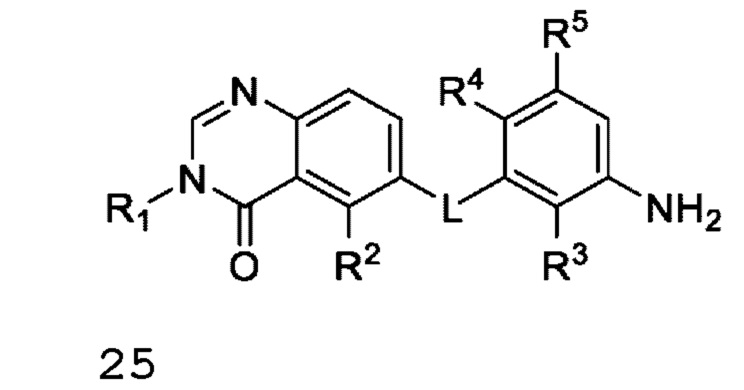
где L, R1, R2, R3, R4 и R5 такие, как определены в варианте осуществления 1, с соединением, имеющим формулу (16)

где R6 и R7 такие, как определены в варианте осуществления 1, в присутствии подходящего основания; или
(b) для соединения по варианту осуществления 1, где R1, R2, R3, R4 и R5 такие, как определены в варианте осуществления 1 и L представляет собой NH, взаимодействие соединения формулы (5)
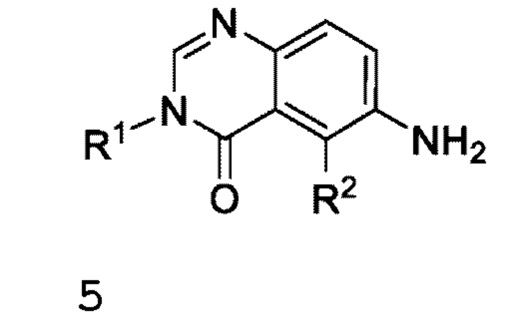
где R1 и R2 такие, как определены в варианте осуществления 1, с соединением, имеющим формулу (27)
где R3, R4, R5, R6, и R7 такие, как определены в варианте осуществления 1, и PG представляет собой защитную группу для амина в присутствии палладиевого катализатора и лиганда с последующим удалением защитной группы для амина; или
(с) для соединения формулы I, где R1, R2, R3, R4 и R5 такие, как определены в варианте осуществления 1, и L представляет собой О, взаимодействие соединения, имеющего формулу (31)
где R1, R2, R3, R4 и R5 такие, как определены в варианте осуществления 1, и PG представляет собой защитную группу амина с реагентом, имеющим формулу
в присутствии основания с последующим удалением аминозащитной группы; и
необязательно образуя его фармацевтически приемлемую соль.
Вариант осуществления 26. Способ лечения BRAF-ассоциированной опухоли у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения по любому из вариантов осуществления 1-23 или его фармацевтически приемлемой соли.
Вариант осуществления 27. Способ по варианту осуществления 26, где указанная BRAF-ассоциированная опухоль имеет мутацию BRAF II класса.
Вариант осуществления 28. Способ по варианту осуществления 27, где указанная мутация BRAF II класса представляет собой не-V600 мутацию BRAF.
Вариант осуществления 29. Способ по варианту осуществления 28, где указанная не-V600 мутация BRAF представляет собой BRAF G469A или G469R.
Вариант осуществления 30. Способ по варианту осуществления 29, где указанная мутация BRAF II класса представляет собой вариант сплайсинга BRAF V600E.
Вариант осуществления 31. Способ по варианту осуществления 30, где указанный вариант сплайсинга BRAF V600E представляет собой p61BRAF(V600E).
Вариант осуществления 32. Способ по любому из вариантов осуществления 26-31, где указанная BRAF-ассоциированная опухоль представляет собой рак, выбранный из рака легкого, меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденоидно-кистозной карциномы, рака аппендикса, рака тонкой кишки, нейроэндокринного рака желудочно-кишечного тракта, плоскоклеточной карциномы головы и шеи, ангиосаркомы, рака мочевого пузыря, новообразования плазматических клеток, гепатобилиарного рака, гепато-панкреато-билиарной карциномы, рака яичников, нейроэндокринного рака, холангиокарциномы, рака пищевода, саркомы мягких тканей, лейкоза, неходжкинской лимфомы и рака ЦНС.
Вариант осуществления 33. Способ по любому из вариантов осуществления 26-31, где указанный рак представляет собой метастатический рак.
Вариант осуществления 34. Способ по варианту осуществления 33, где указанный рак представляет собой метастатический рак ЦНС.
Вариант осуществления 35. Способ по любому из вариантов осуществления 26-31, где указанная BRAF-ассоциированная опухоль представляет собой первичную опухоль.
Вариант осуществления 36. Способ по варианту осуществления 35, где указанная первичная опухоль головного мозга представляет собой злокачественную опухоль.
Вариант осуществления 37. Способ по варианту осуществления 35, где указанная первичная опухоль головного мозга представляет собой глиому 2 степени, глиому 3 степени или глиому 4 степени.
Вариант осуществления 38. Способ по варианту осуществления 26, где указанная BRAF-ассоциированная опухоль имеет мутацию BRAF I класса.
Вариант осуществления 38. Способ по варианту осуществления 38, где указанная мутация BRAF I класса представляет собой BRAF V600E или BRAF V600K.
Вариант осуществления 40. Способ по варианту осуществления 38 или 39, где указанная BRAF-ассоциированная опухоль выбрана из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого, рака яичников, почечно-клеточного рака и их метастатического рака, а также первичных опухолей головного мозга.
Вариант осуществления 41. Способ по варианту осуществления 38 или 39, где указанная BRAF-ассоциированная опухоль представляет собой опухоль ЦНС.
Вариант осуществления 42. Способ по варианту осуществления 41, где указанная опухоль ЦНС представляет собой злокачественную опухоль.
Вариант осуществления 43. Способ по варианту осуществления 42, где указанная злокачественная опухоль представляет собой метастатический рак ЦНС.
Вариант осуществления 44. Способ по варианту осуществления 43, где указанный метастатический рак ЦНС выбран из метастатической меланомы, метастатического колоректального рака, метастатического немелкоклеточного рака легкого, метастатического рака щитовидной железы и метастатического рака яичников.
Вариант осуществления 45. Способ по варианту осуществления 41, где указанная опухоль ЦНС представляет собой внутричерепную или экстракраниальную LMD.
Вариант осуществления 46. Способ по варианту осуществления 41, где указанная опухоль ЦНС представляет собой первичную опухоль головного мозга.
Вариант осуществления 47. Способ по варианту осуществления 46, где указанная первичная опухоль головного мозга представляет собой злокачественную опухоль.
Вариант осуществления 48. Способ по варианту осуществления 47, где указанная первичная опухоль головного мозга представляет собой глиому 2 степени, глиому 3 степени или глиому 4 степени.
Вариант осуществления 49. Способ по варианту осуществления 26, где указанная BRAF-ассоциированная опухоль имеет мутацию BRAF III класса.
Вариант осуществления 50. Способ по варианту осуществления 29, где указанная BRAF-ассоциированная опухоль представляет собой рак, выбранный из меланомы, рака тонкой кишки, колоректального рака, немелкоклеточного рака легкого, рака эндометрия, рака шейки матки, лейкоза, рака мочевого пузыря, неходжкинской лимфомы, глиомы, рака яичников, рака предстательной железы, гепатобилиарного рака, рака пищевода и желудка, саркомы мягких тканей и рака молочной железы.
Вариант осуществления 51. Способ по варианту осуществления 4 9 или 50, где рак имеет мутацию BRAF G466V или BRAF D594G.
Вариант осуществления 52. Способ по любому из вариантов осуществления 26-51, где указанный субъект не подвергался лечению.
Вариант осуществления 53. Способ по любому из вариантов осуществления 26-52, где способ дополнительно включает введение дополнительной противораковой терапии.
Вариант осуществления 54. Способ по варианту осуществления 53, где дополнительная противораковая терапия выбрана из одного или нескольких из хирургического вмешательства, лучевой терапии и противоракового агента.
Вариант осуществления 55. Способ по варианту осуществления 54, где дополнительная противораковая терапия представляет собой противораковый агент.
Вариант осуществления 56. Способ по варианту осуществления 55, где дополнительный противораковый агент выбран из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов SOS1, ингибиторов пути передачи сигнала, ингибиторов контрольных точек, модуляторов пути апоптоза, цитотоксических химиотерапевтических средств, ангиогенез-таргетной терапии и иммуно-таргетных агентов.
Вариант осуществления 57. Способ по варианту осуществления 56, где дополнительный противораковый агент представляет собой ингибитор MEK.
Вариант осуществления 58. Способ по варианту осуществления 57, где ингибитор MEK представляет собой биниметиниб, траметиниб, кобиметиниб, селуметиниб, пимасертиб, рефаметиниб, мирдаметиниб, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамид, 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-дион или его фармацевтически приемлемую соль.
Вариант осуществления 59. Способ по варианту осуществления 58, где ингибитор MEK представляет собой биниметиниб или его фармацевтически приемлемую соль.
Вариант осуществления 60. Способ по варианту осуществления 56, где дополнительный противораковый агент представляет собой ингибитор EGFR.
Вариант осуществления 61. Способ по варианту осуществления 60, где ингибитор EGFR представляет собой цетуксимаб.
Вариант осуществления 62. Способ лечения субъекта с BRAF-ассоциированным метастатическим раком, где субъекта ранее лечили противораковой терапией, где способ включает введение субъекту терапевтически эффективного количества соединения в соответствии с любым из вариантов осуществления 1-23.
Вариант осуществления 63. Способ по варианту осуществления 62, где указанный BRAF-ассоциированный рак имеет мутацию BRAF II класса.
Вариант осуществления 64. Способ по варианту осуществления 63, где указанная мутация II класса представляет собой не-V600 мутацию.
Вариант осуществления 65. Способ по варианту осуществления 62, где указанный BRAF-ассоциированный рак имеет мутацию BRAF I класса.
Вариант осуществления 66. Способ по варианту осуществления 65, где указанный BRAF-ассоциированный рак имеет мутацию BRAF V600E или V600K.
Вариант осуществления 67. Способ по любому из вариантов осуществления 62-66, где указанный субъекта ранее лечили противораковой терапией, которая представляет собой один или несколько противораковых агентов, независимо выбранных из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов SOS1, ингибиторов пути сигнальной трансдукции, ингибиторов контрольных точек, модуляторов пути апоптоза, цитотоксических химиотерапевтических средств, ангиогенез-таргетной терапии и иммуно-таргетных агентов.
Вариант осуществления 68. Способ по варианту осуществления 67, где субъекта ранее лечили ингибитором BRAF.
Вариант осуществления 69. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией ингибитора BRAF и ингибитора MEK.
Вариант осуществления 70. Способ по варианту осуществления 67, где субъекта ранее лечили одним или несколькими ингибиторами контрольных точек.
Вариант осуществления 71. Способ по варианту осуществления 67, где субъекта ранее лечили одним или несколькими ингибиторами PI3K.
Вариант осуществления 72. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией ингибитора BRAF и ингибитора контрольной точки.
Вариант осуществления 73. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией ингибитора BRAF, ингибитора MEK и ингибитора контрольной точки.
Вариант осуществления 74. Способ по варианту 67, в котором субъекта ранее лечили одним или несколькими алкилирующими агентами.
Вариант осуществления 75. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией ингибитора BRAF, ингибитора MEK и ингибитора EGFR.
Вариант осуществления 76. Способ по варианту осуществления 67, где субъекта ранее лечили ингибитором EGFR.
Вариант осуществления 77. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией ингибитора EGFR и одного или нескольких цитотоксических химиотерапевтических агентов.
Вариант осуществления 78. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией ингибитора EGFR и ингибитора BRAF.
Вариант осуществления 79. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией ингибитора MEK и одного или нескольких ингибиторов контрольных точек.
Вариант осуществления 80. Способ по варианту осуществления 67, где субъекта ранее лечили одним или несколькими цитотоксическими химиотерапевтическими агентами.
Вариант осуществления 81. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией терапии антителами и одним или несколькими цитотоксическими химиотерапевтическими агентами.
Вариант осуществления 82. Способ по варианту осуществления 67, где субъекта ранее лечили комбинацией ингибитора EGFR, ингибитора BRAF и одного или нескольких цитотоксических химиотерапевтических агентов.
Вариант осуществления 83. Способ по варианту осуществления 67, где субъекта ранее лечили ингибитором EGFR и одним или несколькими цитотоксическими химиотерапевтическими агентами.
Вариант осуществления 84. Способ по любому из вариантов осуществления 67, 68, 69, 72, 73, 75 и 82, где ингибитор BRAF представляет собой энкорафениб, дабрафениб, вемурафениб или его фармацевтически приемлемую соль.
Вариант осуществления 85. Способ по любому из вариантов осуществления 67, 69, 73, 75 и 79, где ингибитор MEK представляет собой биниметиниб, траметиниб, кобиметиниб или его фармацевтически приемлемую соль.
Вариант осуществления 86. Способ по любому из вариантов осуществления 67, 70, 72, 73 и 79, где ингибитор контрольной точки представляет собой ипилимумаб, ниволумаб или пембролизумаб.
Вариант осуществления 87. Способ по варианту осуществления 67 или 71, где один или несколько ингибиторов PI3K выбраны из бупарлисиба и алпелисиба.
Вариант осуществления 88. Способ по варианту осуществления 67 или 74, где алкилирующий агент выбран из темозоломида, фотемустина, ломустина и кармустина.
Вариант осуществления 89. Способ по любому из вариантов осуществления 67, 75, 76, 77, 78, 82 и 83, где ингибитор EGFR выбран из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба.
Вариант осуществления 90. Способ по любому из вариантов осуществления 67, 77, 80, 81, 82 и 83, где один или несколько цитотоксических химиотерапевтических агентов выбраны из Nordic FLOX (фторурацил, фолиновая кислота и оксалиплатин), оксалиплатина, бевацизумаба, иринотекана, FOLFOXIRI
(оксалиплатин, иринотекан и фторурацил), FOLFIRI (фолиновая кислота, фторурацил и иринотекан) или САРЕОХ (капецитабин и оксалиплатин).
Вариант осуществления 91. Способ по любому из вариантов осуществления 67-90, где субъект является резистентным к указанному предыдущему лечению.
Вариант осуществления 92. Способ по любому из вариантов осуществления 62-91, где у субъекта развились метастазы в головной мозг во время указанного предыдущего лечения.
Вариант осуществления 93. Способ по любому из вариантов осуществления 62-92, где способ дополнительно включает введение дополнительной противораковой терапии.
Вариант осуществления 94. Способ по варианту осуществления 93, где дополнительная противораковая терапия выбрана из одного или нескольких из хирургического вмешательства, лучевой терапии и противоракового агента.
Вариант осуществления 95. Способ по варианту осуществления 94, где дополнительная противораковая терапия представляет собой противораковый агент.
Вариант осуществления 96. Способ по варианту осуществления 95, где дополнительный противораковый агент выбран из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов SOS1, ингибиторов пути передачи сигнала, ингибиторов контрольной точки, модуляторов пути апоптоза, цитотоксических химиотерапевтических средств, ангиогенез-таргетной терапии и иммуно-таргетных агентов.
Вариант осуществления 97. Способ по варианту осуществления 96, где дополнительный противораковый агент представляет собой ингибитор MEK.
Вариант осуществления 98. Способ по варианту осуществления 97, где ингибитор MEK представляет собой биниметиниб, траметиниб, кобиметиниб, селуметиниб, пимасертиб, рефаметиниб, мирдаметиниб, 2-(2-хлор-4-йодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамид, 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3Н,8Н)-дион или его фармацевтически приемлемую соль.
Вариант осуществления 99. Способ по варианту осуществления 95, где ингибитор MEK представляет собой биниметиниб или его фармацевтически приемлемую соль.
Вариант осуществления 100. Способ по варианту осуществления 99, где дополнительный противораковый агент представляет собой ингибитор EGFR.
Вариант осуществления 101. Способ по варианту осуществления 100, где ингибитор EGFR представляет собой цетуксимаб.
Вариант осуществления 102. Способ лечения BRAF-ассоциированной глиомы, где субъекта ранее лечили другой противораковой терапией, где способ включает введение терапевтически эффективного количества соединения в соответствии с любым из вариантов осуществления 1-23.
Вариант осуществления 103. Способ по варианту осуществления 102, где субъекта ранее лечили одним или несколькими цитотоксическими химиотерапевтическими агентами.
Вариант осуществления 104. Способ по варианту осуществления 103, где один или несколько цитотоксических химиотерапевтических агентов выбраны из цисплатина, пеметрекседа, винорелбина и паклитаксела.
Вариант осуществления 105. Способ по варианту осуществления 102, где субъекта ранее лечили алкилирующим агентом.
Вариант осуществления 106. Способ по варианту осуществления 102, где субъекта ранее лечили ингибитором орнитиндекарбоксилазы.
Вариант осуществления 107. Способ в соответствии с вариантом осуществления 102, где субъекта ранее лечили комбинацией алкилирующего агента и ингибитора орнитиндекарбоксилазы.
Вариант осуществления 108. Способ по варианту осуществления 105 или 107, где алкилирующий агент выбран из темозоломида, ломустина и кармустина.
Вариант осуществления 109. Способ по варианту осуществления 106 или 107, где ингибитор орнитиндекарбоксилазы представляет собой рацемический эфлорнитин, D-эфлорнитин или L-эфлорнитин.
Вариант осуществления 110. Способ по варианту осуществления 102, где субъекта ранее лечили ингибитором BRAF.
Вариант осуществления 111. Способ по варианту осуществления 102, где субъекта ранее лечили комбинацией ингибитора BRAF и ингибитора MEK.
Вариант осуществления 112. Способ по варианту осуществления 110 или 111, где ингибитор BRAF представляет собой энкорафениб, дабрафениб, вемурафениб или его фармацевтически приемлемую соль.
Вариант осуществления 113. Способ по варианту осуществления 111, где ингибитор MEK представляет собой биниметиниб, траметиниб, кобиметиниб или его фармацевтически приемлемую соль.
Вариант осуществления 114. Способ по любому из вариантов осуществления 102-113, где противораковая терапия включает хирургическое вмешательство.
Вариант осуществления 115. Способ по любому из вариантов осуществления 102-114, где противораковая терапия включает лучевую терапию.
ПРИМЕРЫ
Следующие примеры иллюстрируют изобретение.
Биологические примеры
Пример А1
Ферментный анализ BRAF V60 0E
Анализ конкурентного замещения сконфигурирован для B-Raf, который отслеживает количество флуоресцентно-меченого «индикатора», связанного с B-Raf через TR-FRET из анти-метки Eu-меченного антитела, также связанного с B-Raf. Для полноразмерного FLAG-меченного B-Raf(V600E), смеси для анализа состоят из 25 мМ K+HEPES, рН 7,4, 10 мМ MgCl2, 0,01% Triton X-100, 1 мМ DTT, 2% ДМСО (из соединения), 50 нМ Tracer 1710 (ThermoFisher, PR917 6A), 0,5 нМ Eu анти-FLAG (М2)-криптатного Ab (Cisbio, 61FG2KLB) и 5 нМ полноразмерных B-Raf(V600E) с N-концевой FLAG-меткой (Origene Technologies, ТР700031). Соединения обычно разводят в ДМСО в пределах 11-точечного диапазона дозирования, созданного с использованием протокола 3-кратного серийного разведения при максимальной дозе 10 мкМ. Анализ проводят в 384-луночных полистироловых не обработанных белых микротитрационных планшетах малого объема (Costar 4512) с конечным объемом 12 мкл. Лунки с низким контролем включают 1 мкМ сильнодействующего ингибитора B-Raf в качестве контроля. Анализы инкубируют при температуре окружающей среды (обычно 22°С) в течение 60 мин, и считывают на микропланшетном ридере PerkinElmer Envision, используя стандартные настройки TRF (АЕх=320 нм, АЕт=615 и 665 нм). Импульсы в установленных соотношениях (6 65 нм/615 нм) преобразуют в долю контроля (РОС) с использованием следующего уравнения:
где
 - средний не ингибированный контроль
- средний не ингибированный контроль
 - средний фон
- средний фон
4-параметрическая логистическая модель соответствует данным РОС для каждого соединения. Исходя из этого соответствия, IC50 оценивают и определяют как концентрацию соединения, при которой кривая наилучшего приближения пересекает 50 РОС. Средние значения IC50 соединений, описанных в настоящем документе, при тестировании в этом анализе представлены в таблице А.
Пример А2
Анализы клеточного ингибирования фосфо-ERK в клетках А375 и Н1755
Клетки А375 и Н1755 получают из Американской коллекции типовых культур (АТСС, Rockville, MD). Клетки А375 поддерживают в среде для выращивания DMEM, содержащей 10% FBS. Клетки Н1755 поддерживают в среде для выращивания RPMI, содержащей 10% FBS.
Клетки собирают в соответствии со стандартными протоколами, подсчитывают и высевают на плоскодонные 9б-луночные планшеты для тканевых культур (Costar #3599) в количестве 2,5×104 клеток/лунку для клеток А375 и 1,5×104 клеток/лунку для клеток Н1755 в 100 мкл/лунку среды для выращивания, содержащей 10% FBS. После инкубации в течение ночи при 37°С с 5% CO2, клетки обрабатывают в течение 2 часов при 37°С, 5% CO2 соединениями, приготовленными в виде 9-точечной серии кратных разведений 1:3,33 с конечной концентрацией соединения в диапазоне от 66 рМ-10 мкМ и постоянной концентрацией ДМСО 0,25%. Контрольные лунки содержат либо только 0,25% ДМСО (не ингибированный контроль), либо 10 мкМ биметиниба (контроль полного ингибирования). Уровни фосфорилированного ERK определяют с использованием протокола In Cell Western: после инкубации с соединением среду для выращивания удаляют, и клетки фиксируют 0,4% формальдегидом в PBS в течение 2 0 минут при комнатной температуре. Клетки пермеабилизируют 100% метанолом в течение 10 минут при комнатной температуре. Планшеты промывают PBS, содержащим 0,05% Tween-2 0, и блокируют в течение 1 часа при комнатной температуре блокирующим буфером LI-COR (LI-COR Biosciences; кат. №927-4 0000). Затем планшеты инкубируют в течение 2 часов при комнатной температуре с 50 мкл анти-фосфо-ERK1/2 (Thr202/Tyr204) в разведении 1:400 (Cell Signaling; кат. №9101) и анти-GAPDH в разведении 1:1000 (Millipore; кат. №МАВ374) в блокирующем буфере LI-COR, содержащем 0,05% Tween-20. Планшеты промывают PBS, содержащим 0,05% Tween-20, затем инкубируют при комнатной температуре в течение 1 часа с 50 мкл анти-кроличьего AlexaFluor 680 в разведении 1:1000 (Life Technologies; кат. №А21109) и анти-мышиного IRDye 800CW в разведении 1:1000 (LI-COR; кат.№92 6-32 210) в блокирующем буфере LI-COR, содержащем 0,05% Tween-20. Планшеты анализируют путем считывания на инфракрасном сканере Odyssey CLx. Для каждой лунки, сигнал фосфо-ERK нормализуют к сигналу GAPDH и преобразовывают в РОС с использованием следующего уравнения:
где
 - средний не ингибированный контроль
- средний не ингибированный контроль
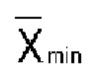 - средний контроль с полным ингибированием
- средний контроль с полным ингибированием
IC50 рассчитывают с использованием 4-параметрической подгонки в программном обеспечении XLfit, и значения представлены в таблице А1.
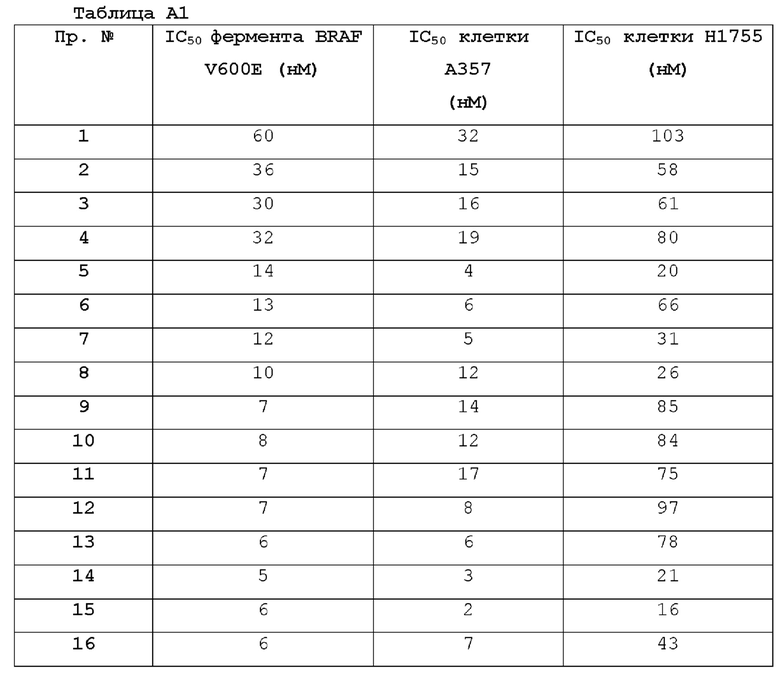
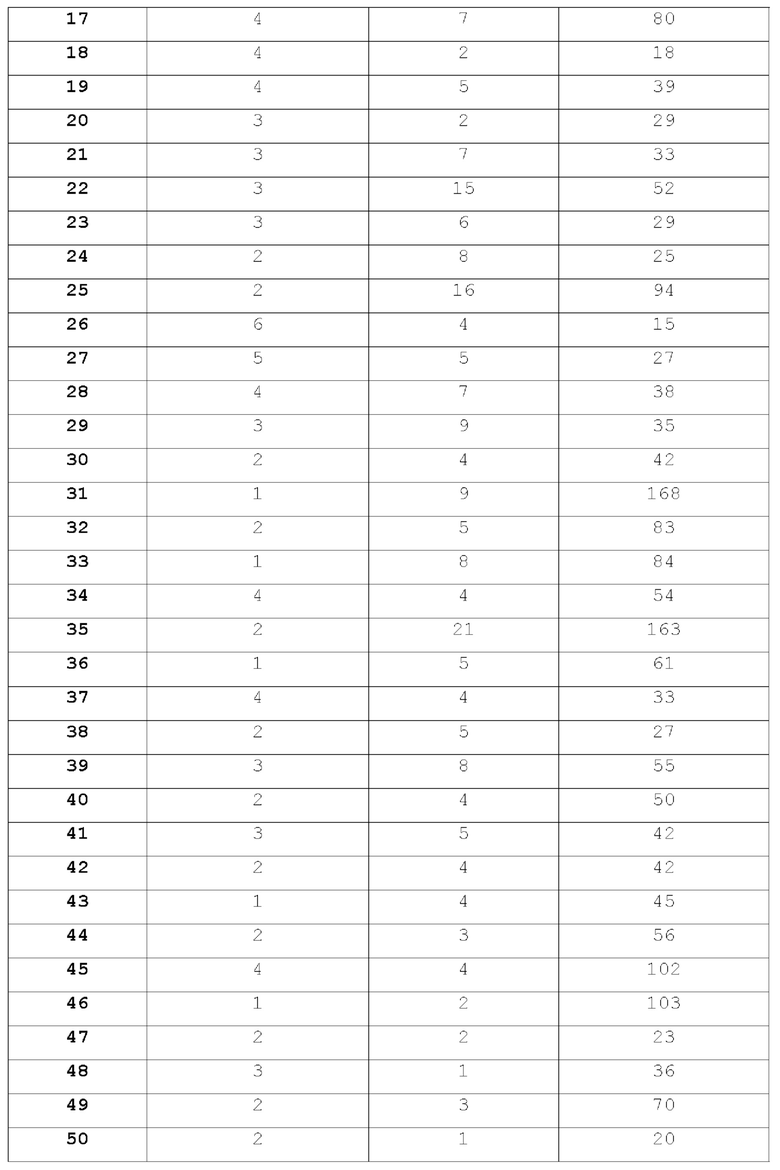
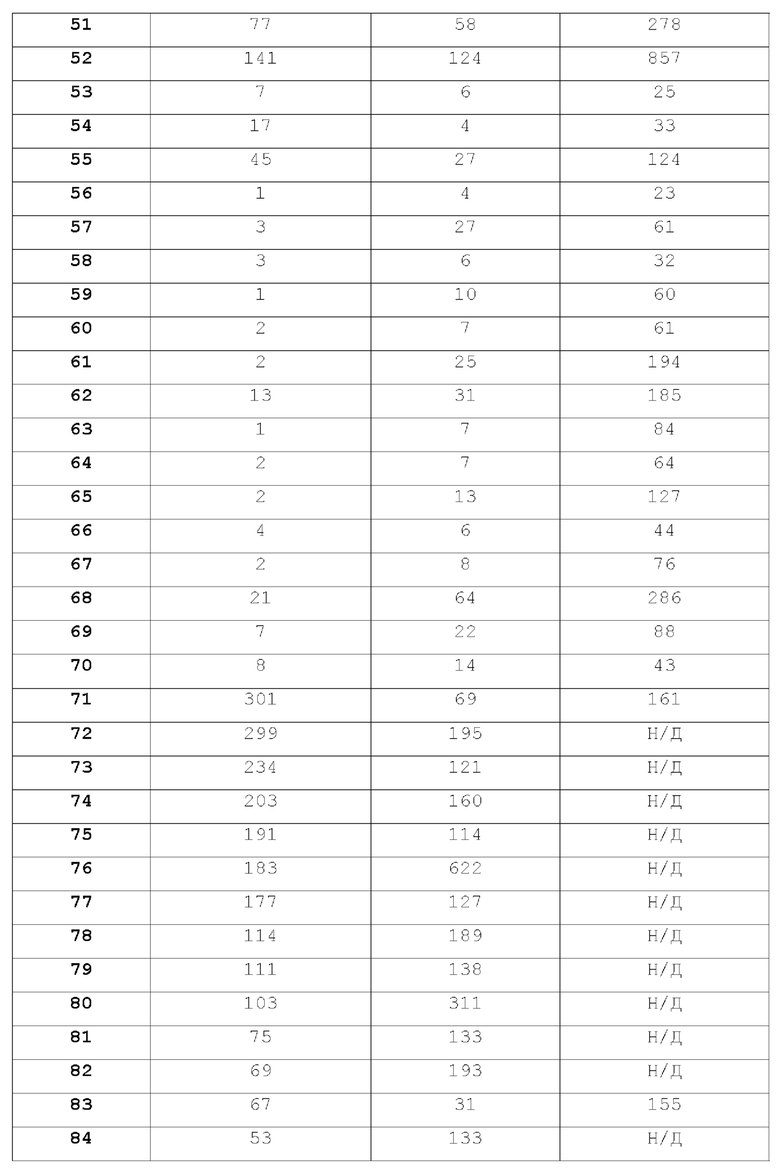
Пример A3
Анализ клеточного ингибирования фосфо-ERK
Соединения примера 13, примера 14 и примера 12 6 оценивают в анализе фосфо-ERK в двух мутантных клеточных линиях BRAF III класса: клетках NCI-H1666 (BRAFG466V) и клетках WM3629 (BRAFD594G/NRASG12D). Клетки NCI-H1666 получают из Американской коллекции типовых культур (АТСС, Rockville, MD), и клетки WM3629 получают от Rockland Immunochemicals (Limerick, PA). Клетки поддерживают в среде для выращивания RPMI, содержащей 10% FBS.
Клетки собирают в соответствии со стандартными протоколами, подсчитывают и высевают на плоскодонные 9б-луночные планшеты для тканевых культур (Costar #3599) по 2,5×104 клеток/лунку в 100 мкл/лунку среды для выращивания, содержащей 10% FBS. После инкубации в течение ночи при 37°С с 5% CO2, клетки обрабатывают в течение 1 часа при 37°С, 5% CO2 ингибиторами, приготовленными в виде серии 9-точечных 1:3,33-кратных разведений с конечными концентрациями соединения 66 рМ-10 мкМ и постоянной концентрацией ДМСО 0,25%. Контрольные лунки содержат либо только 0,25% ДМСО (не ингибированный контроль), либо 10 мкМ биметиниба (контроль полного ингибирования). Уровни фосфорилированного ERK определяют с использованием протокола In Cell Western: после инкубации с соединением, среду для выращивания удаляют и клетки фиксируют 0,4% формальдегидом в PBS в течение 2 0 минут при комнатной температуре. Клетки пермеабилизируют 100% метанолом в течение 10 минут при комнатной температуре. Планшеты промывают PBS, содержащим 0,05% Tween-20, и блокируют в течение 1 часа при комнатной температуре блокирующим буфером LI-COR (LI-COR Biosciences; кат. №927-40000). Затем планшеты инкубируют в течение 2 часов при комнатной температуре с 5 0 мкл анти-фосфо-ERK1/2 (Thr202/Tyr204) в разведении 1:400 (Cell Signaling; кат. №9101) и анти-GAPDH в разведении 1:1000 (Millipore; кат. №МАВ374) в блокирующем буфере LI-COR, содержащем 0,05% Tween-2 0. Планшеты промывают PBS, содержащим 0,05% Tween-2 0, затем инкубируют при комнатной температуре в течение 1 часа с 5 0 мкл анти-кроличьего AlexaFluor 680 в разведении 1:1000 (Life Technologies; кат. №А21109) и анти-мышиного IRDye 800CW в разведении 1:1000 (LI-COR; кат.№926-32210) в блокирующем буфере LI-COR, содержащем 0,05% Tween-2 0. Планшеты анализируют путем считывания на инфракрасном сканере Odyssey CLx. Для каждой лунки, сигнал фосфо-ERK нормализуют к сигналу GAPDH и преобразовывают в РОС с использованием следующего уравнения:
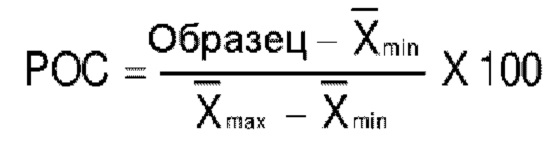
где
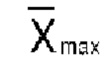 - средний не ингибированный контроль
- средний не ингибированный контроль
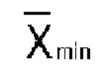 - средний контроль с полным ингибированием
- средний контроль с полным ингибированием
IC50 рассчитывают с использованием 4-параметрической подгонки в программном обеспечении XLfit, и результаты показаны в таблице А2.
Пример А4
Анализ пролиферации
Соединения примеров 13, примеров 14 и примеров 126 оценивают в анализе пролиферации в двух мутантных клеточных линиях BRAF III класса: клетках NCI-H1666 (BRAFG4 66V) и клетках WM3629 (BRAFD594G/NRASG12D). Клетки NCI-H1666 получают из Американской коллекции типовых культур (АТСС, Rockville, MD), и клетки WM3 62 9 получают от Rockland Immunochemicals (Limerick, PA). Клетки поддерживают в среде для выращивания RPMI, содержащей 10% FBS.
Клетки собирают в соответствии со стандартными протоколами, подсчитывают и высевают на плоскодонные 96-луночные планшеты для тканевых культур (Costar #3599) по 2000-5000 клеток/лунку в 100 мкл/лунку среды для выращивания, содержащей 10% FBS. Клетки инкубируют при 37°С с 5% CO2 в течение ночи, затем обрабатывают ингибиторами, приготовленными в виде серии 9-точечных 1:3,33-кратных разведений с конечными концентрациями соединения 66 рМ-10 мкМ и постоянной концентрацией ДМСО 0,25%. Контрольные лунки содержат либо только 0,25% ДМСО. Через 3-5 дней инкубации при 37°С, 5% CO2 жизнеспособность клеток определяют путем добавления 100 мкл реагента CellTiter-Glo® (Promega) в каждую лунку и инкубируют в течение 15 минут при комнатной температуре. Контроли «день 0» определяют путем проведения анализа CellTiter-Glo® на контрольных лунках с ДМСО во время обработки соединением (Контроль «день 0»=0 РОС). Люминесценцию измеряют на планшетном ридере Cytation 5 (BioTek), и значения преобразовывают в РОС с использованием следующего уравнения:
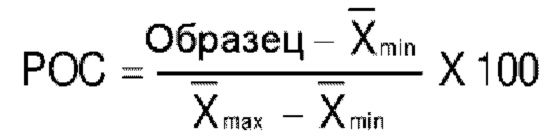
где
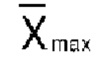 - средний ДМСО контроль
- средний ДМСО контроль
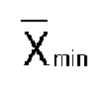 - средний ДМСО контроль в «день 0»
- средний ДМСО контроль в «день 0»
IC50 рассчитывают с использованием 4-параметрической подгонки в программном обеспечении XLfit, и значения показаны в таблице А2.
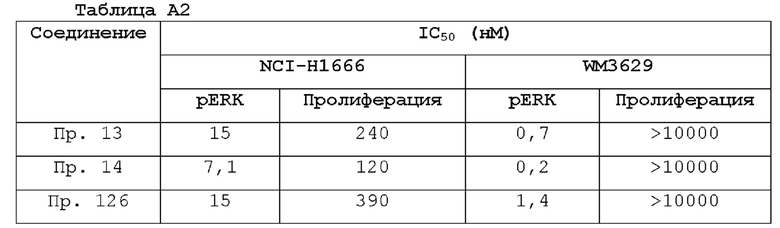
Пример В
Анализ проницаемости MDRl LLC-PK1 и BCRP MDCKII Клетки LLC-PK1 и LLC-PK1, трансфицированные MDR1, культивируют и высевают в соответствии с рекомендациями производителя, за исключением того, что среда для пассажа содержит только 2% фетальной бычьей сыворотки, чтобы увеличить время пассажа до семи дней.
Клетки MDCKII, трансфицированные BCRP, культивируют и высевают в соответствии с рекомендациями производителя. Условия анализа включают наличие и отсутствие BCRP-специфического ингибитора KO143 в концентрации 0,3 мкМ для определения вклада BCRP в значение эффлюкса тестируемого соединения.
Для оценки функциональности эффлюкса Р_gp или BCRP в анализах используют как положительный, так и отрицательный контроль. Исходные растворы для анализа контроля и тестируемого образца готовят в ДМСО для конечных тестируемых концентраций 10 и 1 мкМ, соответственно. Конечная концентрация органических веществ в анализе составляет 1%. Все растворы для дозирования содержат 10 мкМ люцифера желтого для отслеживания целостности монослоя клеток LLC PK1 или MDCKII.
Для апикально-базолатерального определения (А-В) 75 мкл тестируемого образца в транспортном буфере добавляют к апикальной стороне отдельных трансвелл, и в каждую лунку добавляют 250 мкл базолатеральной среды без соединения или люцифера желтого. Для базолатерального и апикального определения (В-А) в каждую лунку добавляют 250 мкл испытуемого образца в транспортном буфере, и в каждую трансвеллу добавляют 7 5 мкл транспортного буфера без соединения или люцифера желтого. Все тесты проводят в трех повторах, и каждое соединение тестируют как на апикально-базолатеральный, так и на базолатерально-апикальный транспорт. Планшеты инкубируют в течение 2 часов на орбитальном шейкере Lab-Line Instruments Titer (VWR, West Chester, PA) при 50 об/мин и 37°C с 5% CO2. Все культуральные планшеты извлекают из инкубатора, и из апикальной и базолатеральной части каждой лунки удаляют по 50 мкл среды и добавляют к 150 мкл 1 мкМ лабеталола в 2:1 ацетонитриле (ацетонитрил): H2O, об./об.
Планшеты считывают с использованием флуорометра Gemini компании Molecular Devices (Sunnyvale, СА) для оценки концентрации желтого люцифера при длинах волн
возбуждения/испускания 425/535 нм. Эти значения принимают, когда обнаруживают, что они составляют менее 2% для апикально-базолатерального и 5% базолатерально-апикального потока через клеточные монослои MDR1-трансфицированных LLC-PK1 или BCRP-трансфицированных MDCKII. Планшеты запечатывают, и содержимое каждой лунки анализируют с помощью ЖХ-МС/МС. Концентрации соединения определяют по отношению площадей пиков соединения к внутреннему стандарту (лабеталолу) по сравнению с дозируемым раствором.
ЖХ-МС анализ
Система ЖХ-МС/МС состоит из автодозатора HTS-PAL (Leap Technologies, Carrboro, NC), HP1200 HPLC (Agilent, Palo Alto, CA) и системы MDSSciex 4000 Q Trap (Applied Biosystems, Foster City, CA). Хроматографическое разделение анализируемого вещества и внутреннего стандарта осуществляют при комнатной температуре с использованием колонки С18 (Kinetics®, 50×300 мм, размер частиц 2,6 мкм, Phenomenex, Torrance, CA) в сочетании с градиентными условиями с использованием подвижных фаз А (вода, содержащая 1% изопропилового спирта и 0,1% муравьиной кислоты) и В (0,1% муравьиной кислоты в ацетонитриле). Общее время работы, включая повторное уравновешивание, для одной инъекции составляет 1,2 минуты. Масс-спектрометрическое определение анализируемых вещества осуществляют с использованием положительного режима ионного распыления. Ответы анализируемого вещества измеряют путем мониторинга множественных реакций (MRM) переходов, уникальных для каждого соединения (протонированный ион-предшественник и выбранные ионы-продукты для каждого тестируемого образца и т/z 329 - т/z 162 для лабеталола, внутреннего стандарта).
Коэффициент проницаемости (Рарр) рассчитывают по следующему уравнению:
Papp=[((Cd*V* (1×106))/(t*0,12см2*C)]
где Cd, V, t и С0 представляют собой определяемую концентрацию (мкМ), объем на стороне дозирования (мл), время инкубации (с) и начальную концентрацию дозирования (мкМ), соответственно. Расчеты Рарр производят для каждого повтора, и затем усредняют. Коэффициенты проницаемости для соединений формулы I приведены в таблице В1. В этом анализе, соединение считается имеющим высокую проницаемость, если проницаемость превышает 8×10-6 см/сек, соединение считается имеющим среднюю проницаемость, если проницаемость составляет от 2×10-6 см/сек до 8×10-6 см/сек, и соединение считается имеющим низкую проницаемость, если проницаемость составляет менее 2×10-6 см/сек.


Коэффициент эффлюкса рассчитывают на основе среднего значения апикально-базолатерального (А-В) Рарр и данных базолатерально-апикального (В-А) Рарр с использованием следующего уравнения:
Коэффициент эффлюкса = Рарр(В-А)/Рарр (А-В)
Коэффициенты эффлюкса для соединений, описанных в настоящем документе, при тестировании в этом анализе представлены в таблице В2.
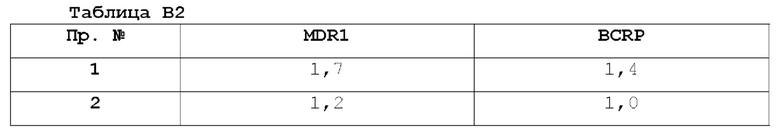
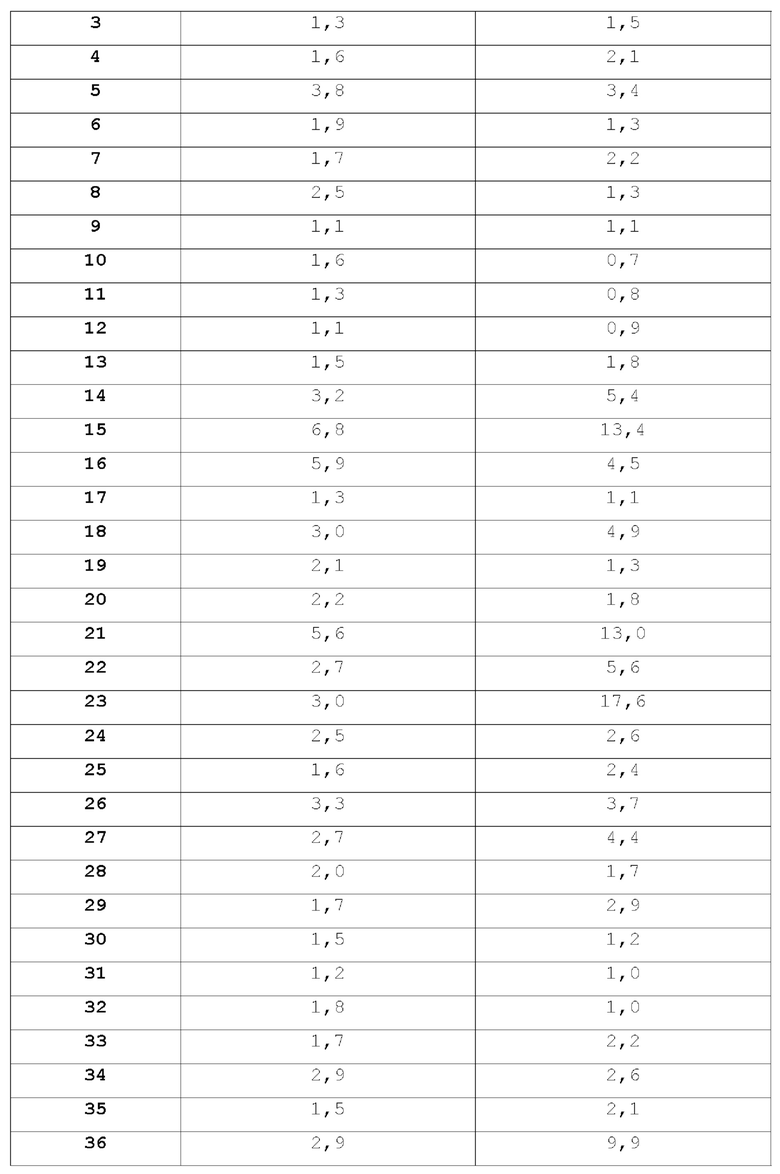
Пример С
РК (отношение свободного мозга к свободной плазме) (мышь) Способность типовых соединений проникать через ВВВ у мышей определяют путем оценки отношения концентраций несвязанного мозга к несвязанной плазме (также называемой свободным мозгом и свободной плазмой) у самцов мышей CD-1.
Уровни соединений в головном мозге получают при пероральном РК введении мышам с типовым временем отбора проб 2, 4, 8, 12 и 24 часа после перорального введения через желудочный зонд в дозе 10 мг/кг. Образцы головного мозга перед анализом хранят при температуре -20±5°С. Концентрации тестируемого соединения в гомогенате мозга мыши определяют тандемной жидкостной хроматографией - масс-спектрометрией (ЖХ-МС/МС) после осаждения белка ацетонитрилом. 12-точечную калибровочную кривую в диапазоне от 0,5 до 10000 нг/мл готовят в двух повторах. Раствор 400 мкг/мл тестируемого соединения в диметилсульфоксиде (ДМСО) серийно разводят (3-кратно) в 100% ДМСО, и затем 2,5 мкл каждого стандартного раствора добавляют к 100 мкл гомогената головного мозга самцов наивных мышей CD-I. Для имитации экстракции на стандартной кривой, ко всем испытуемым образцам добавляют 2,5 мкл ДМСО. В калибровочные и тестируемые образцы гомогената мозга добавляют 10 мкл IS (1 мкг/мл структурного аналога). Гомогенат мозга получают добавлением 0,75 мл 4:1 смеси вода:МеОН к каждому образцу мозга с последующей гомогенизацией в течение 1 минуты с помощью пробирок шариковой мельницы со скоростью 6 м/с с использованием MP Fast Preg-24®. Белки осаждают из 100 мкл образца гомогената головного мозга добавлением 300 мкл ацетонитрила. Образцы перемешивают на вортексе в течение 5 минут и центрифугируют в центрифуге Allegra X-12R (Beckman Coulter, Fullerton, CA; ротор SX4750A) в течение 15 минут при приблизительно 1500×g при 4°С. Аликвоту 100 мкл каждого супернатанта переносят с помощью 550 мкл персонального пипеттора (Apricot Designs, Monrovia, СА) в 96-луночные планшеты и разводят 1:1 водой для ВЭЖХ. Полученные планшеты запечатывают алюминием для анализа ЖХ-МС/МС.
Отношения мозг-плазма рассчитывают, используя концентрацию соединения, измеренную в мозге, деленную на концентрацию соединения, измеренную в плазме. Отношения мозга к плазме всегда получают из одного животного и в один момент времени. Отношения свободного мозга к свободной плазме рассчитывают путем умножения отношения мозга к плазме на свободную фракцию гомогената головного мозга in vitro, деленную на свободную фракцию плазмы in vitro, с использованием следующего уравнения: (В/Р)*(Bfu/Pfu). В Таблице С представлены соотношения свободного мозга к свободной плазме соединений, описанных в настоящем документе.
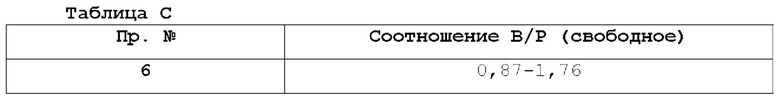
Примеры синтеза
Получение синтетических промежуточных соединения
Промежуточное соединение Р1
6-бром-5-метилхиназолин-4(3Н)-он
6-Амино-3-бром-2-метилбензойную кислоту (10 г, 43 ммоль) и ацетат формамидина (5,4 г, 52 ммоль) растворяют в этаноле (172 мл) в 500-мл колбе с конденсатором с обратным холодильником. Реакционную смесь нагревают до 80°С в течение 16 часов. Смесь охлаждают до температуры окружающей среды и концентрируют в вакууме. Остаток разбавляют водой (300 мл) и энергично перемешивают в течение 60 минут. Полученное твердое вещество выделяют фильтрацией и фильтровальную лепешку промывают водой (500 мл). Твердое вещество сушат под вакуумом с получением 6-бром-5-метилхиназолин-4(3H)-она (6,9 г, 66%) в виде белого твердого вещества. МС (хиад, m/z)=239,0, 241,0 (М+Н).
Промежуточное соединение Р2
6-бром-3,5-диметилхиназолин-4(3Н)-он
6-Бром-5-метилхиназолин-4(3Н)-он (Промежуточное соединение Р1) (11 г, 4 6,0 ммоль), карбонат калия (14,0 г, 101 ммоль) и йодметан (13,1 г, 92,0 ммоль) растворяют в безводном ДМФ (250 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов. Всю реакционную смесь выливают непосредственно в 900 мл воды, и полученную суспензию перемешивают при температуре окружающей среды в течение 30 минут. Твердое вещество собирают фильтрацией и сушат в течение ночи в высоком вакууме с получением 6-бром-3,5-диметилхиназолин-4(3Н)-она (10,1 г, 87%) в виде белого твердого вещества. МС (хиад, m/z)=253,0, 255,0 (М+Н).
Промежуточное соединение Р3
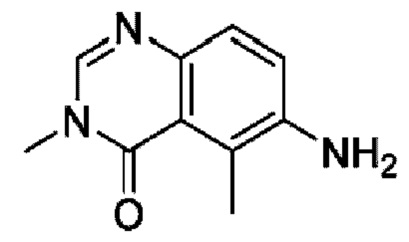
6-Амино-3,5-диметилхиназолин-4(3H)-он
Стадия 1: Получение 6-((4-метоксибензил) амино)-3,5-диметилхиназолин-4(3Н)-она. Раствор (4-метоксифенил)метанамина (1,20 мл, 9,18 ммоль), 6-бром-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение Р2) (2,02 г, 7,98 ммоль), Pd2 (dba)3 (0,365 г, 0,399 ммоль), Xantphos (0,693 г, 1,20 ммоль) и Cs2CO3 (7,80 г, 23,9 ммоль) в толуоле (53,2 мл) помещают в пробирку под давлением и продувают аргоном в течение 10 минут. Реакционный сосуд герметично закрывают и нагревают до 90°С в течение 60 часов. Дополнительный Pd2 (dba)3 (0, 365 г, 0,399 ммоль) и Xantphos (0,693 г, 1,20 ммоль) добавляют и раствор снова продувают аргоном в течение 10 минут, герметично закрывают и нагревают до 90°С в течение еще 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют, концентрируют и очищают колоночной хроматографией, элюируя 5-95% EtOAc/ДХМ с получением 6-((4-метоксибензил)амино)-3,5-диметилхиназолин-4(3H)-она (2,4 г, 97%). МС (хиад, m/z)=310,2 (М+Н).
Стадия 2: Получение 6-амино-3,5-диметилхиназолин-4(3H)-она. Раствор 6((4-метоксибензил)амино)-3,5-диметилхиназолин-4(3H)-она (2,4 г, 7,7 6 ммоль) перемешивают в 5 0 мл ДХМ и 2 5 мл ТФК в течение 2 часов. Раствор концентрируют, и остаток растворяют в 100 мл ДХМ, 10 мл МеОН и энергично перемешивают с 4 г K2CO3 в течение 30 минут.K2CO3 удаляют фильтрацией, и фильтрат концентрируют, и остаток очищают колоночной хроматографией, элюируя 1-10% МеОН/ДХМ (1% NH4OH) с получением 6-амино-3,5-диметилхиназолин-4(3H)-она (1,45 г, 99%). 1Н ЯМР (400 МГц, CDCl3) δ 8,2 (с, 1Н), 7,5 (д, 1Н), 7,1 (д, 1Н), 4,2 (шс, 2Н), 3,6 (с, 3Н), 2,8 (с, 3Н); МС (хиад, m/z)=190,1 (М+Н).
Промежуточное соединение Р4
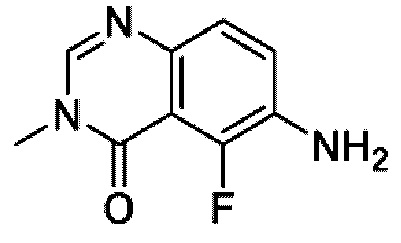
6-Амино-5-фтор-3-метилхиназолин-4(3Н)-он
Стадия 1: Получение 6-бром-5-фторхиназолин-4(3Н)-она. 6-Амино-3-бром-2-фторбензойную кислоту (4,51 г, 19,3 ммоль) растворяют в EtOH (2 00 мл) и затем обрабатывают ацетатом формамидина (6,02 г, 57,8 ммоль) и затем нагревают до 80°С в течение 16 часов. Реакционную смесь обрабатывают дополнительным ацетатом формамидина (3,01 г, 28,9 ммоль) и перемешивают при 80°С в течение дополнительных 4 ч. Реакционную смесь охлаждают до температуры окружающей среды и выливают в воду и затем экстрагируют EtOAc (3х). Объединенные органические слои промывают насыщенным раствором соли (1х) и затем сушат над Na2SO4, фильтруют и концентрируют с получением 6-бром-5-фторхиназолин-4(3Н)-она (4,28 г, 91%). МС (хиад, m/z)=243,0, 245,0 (М+Н).
Стадия 2: Получение 6-бром-5-фтор-3-метилхиназолин-4(3Н)-она. 6-Бром-5-фторхиназолин-4(3Н)-он (4,28 г, 17,6 ммоль) растворяют в ДМФ (70 мл) и затем обрабатывают йодметаном (1,32 мл, 21,1 ммоль), затем карбонатом калия (3,65 г, 2 6,4 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. Реакционную смесь выливают в воду и экстрагируют EtOAc (3х). Объединенные органические слои промывают водой (3х) затем насыщенным раствором соли (1х), затем сушат над Na2SO4, фильтруют, и концентрируют.Полученный остаток очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением 6-бром-5-фтор-3-метилхиназолин-4(3Н)-она (3,33 г, 74%). МС (хиад, m/z)=257,0, 259,0 (М+Н).
Стадия 3: Получение 6-амино-5-фтор-3-метилхиназолин-4(3Н)-она. 6-Бром-5-фтор-3-метилхиназолин-4(3Н)-он (3,18 г, 12,37 ммоль) растворяют в толуоле (125 мл) и обрабатывают трет-бутил карбаматом (1,59410 г, 13,61 ммоль),
трис(дибензилиденацетон)дипалладием (1,13 г, 1,24 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантеном (1,79 г, 3,09 ммоль) и карбонатом цезия (12,09 г, 37,11 ммоль) и затем продувают аргоном в течение нескольких минут и нагревают до 110°С под баллоном аргона в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (1 л) и перемешивают в течение 15 минут фильтруют через слой Celite® и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (20 мл) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и затем очищают С18 хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным NaHCO3 (1х). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением 6-амино-5-фтор-3-метилхиназолин-4(3Н)-она (1,57 г, 65,7%). 1Н ЯМР (400 МГц, ДМСО) δ 8,05 (с, 1Н), 7,27-7,22 (м, 2Н), 5,51 (шс, 2Н), 3,40 (с, 3Н); МС (хиад, m/z)=194,1 (М+Н).
Промежуточное соединение Р5
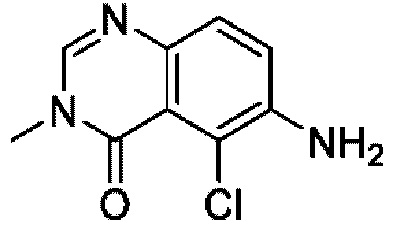
6-Амино-5-хлор-3-метилхиназолин-4(3Н)-она
6-Амино-3-метилхиназолин-4(3Н)-он (3,00 г, 17,1 ммоль) растворяют в ТГФ (170 мл) и затем обрабатывают N-хлорсукцинимидом (2,40 г, 18,0 ммоль) и нагревают до 50°С в течение 16 часов. Реакционную смесь обрабатывают дополнительным N-хлорсукцинимидом (1,14 г, 8,56 ммоль) и перемешивают при 50°С в течение дополнительных 3 часов. Реакционную смесь концентрируют, и полученный остаток разбавляют 1,0 М HCl и экстрагируют ДХМ (3х). Объединенный ДХМ объединенные органические слои промывают 1,0 М HCl (2х) и водный слой нейтрализуют твердым NaHCO3 до примерно рН 7-8, и затем экстрагируют 4:1 ДХМ:ИПС (2х). The объединяют ДХМ:ИПС extracts were сушат над Na2SO4, фильтруют и концентрируют с получением 6-амино-5-хлор-3-метилхиназолин-4(3Н)-она (2,47 г, 69%). 1Н ЯМР (400 МГц, ДМСО) δ 8,10 (с, 1Н), 7,38-7,36 (д, 2Н), 7,29-7,26 (д, 2Н), 5,81 (шс, 2Н), 3,40 (с, 3Н). МС (хиад, m/z)=210,l, 212,1 (М+Н).
Промежуточное соединение Р6
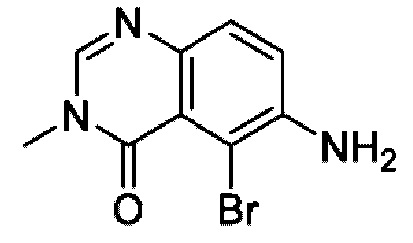
6-Амино-5-бром-3-метил-4а,8а-дигидрохиназолин-4(3Н)-он
6-Амино-3-метилхиназолин-4(3Н)-он (2,00 г, 11,3 ммоль) растворяют в ТГФ (56 мл) и затем обрабатывают N-бромсукцинимидом (2,11 г, 11,9 ммоль) и смесь нагревают до 50°С в течение 60 минут. Реакционную смесь охлаждают до температуры окружающей среды разбавляют этилацетатом (100 мл) и промывают тиосульфатом натрия (насыщенный водный раствор, 100 мл, 1х), затем бикарбонатом натрия (насыщенный водный раствор, 100 мл, 1х). Объединенные органические слои сушат над MgSO4, фильтруют и концентрируют. Остаток очищают С18 хроматографией с обращенной фазой на силикагеле, элюируя водой/ацетонитрилом с получением 6-амино-5-бром-3-метил-4а,8а-дигидрохиназолин-4(3Н)-она (1,01 г, 35%). МС (хиад, m/z)=254,0, 256,0 (М+Н).
Промежуточное соединение Р7
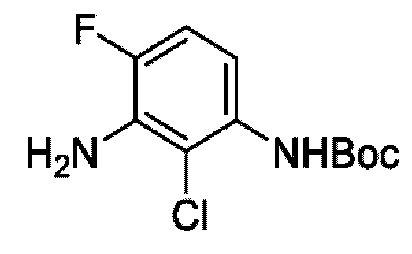
Трет-бутил (3-амино-2-хлор-4-фторфенил)карбамат
Стадия 1: Получение метил 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензоата. Метил 3-амино-2-хлор-6-фторбензоат (5,06 г, 24,9 ммоль) растворяют в ДХМ (250 мл) и охлаждают до 0°С. Реакционную смесь последовательно обрабатывают триэтиламином (10,4 мл, 74,6 ммоль), 4-(диметиламино)пиридином (0,304 г, 2,49 ммоль) и ди-трет-бутилдикарбонатом (13,6 г, 62,1 ммоль) и перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле, элюируя гексаном/ацетоном с получением метил 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензоата (7,55 г, 100%) который применяют сразу на следующей стадии в виде смеси моно/бис-Вос продуктов.
Стадия 2: Получение 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензойной кислоты. Метил 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензоат (7,55 г, 24,9 ммоль) растворяют в 1:1 ТГФ/МеОН (120 мл) и затем обрабатывают 2,0 М водным NaOH (37,3 мл, 74,6 ммоль) и перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют дополнительной водой и экстрагируют Et20 (2×250 мл). Et2O объединенные органические слои объединяют и промывают 1,0 М NaOH (1×50 мл). Объединенные водные слои подкисляют до примерно рН 4 с применением 4,0 М HCl, и затем экстрагируют 4:1 ДХМ/ИПС (2×250 мл). Объединенные органические слои сушат над Na2SO4, фильтруют, и концентрируют с получением 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензойной кислоты (5,53 г, 77%) которую применяют сразу на следующей стадии без очистки.
Стадия 3: Получение трет-бутил (3-амино-2-хлор-4-фторфенил)карбамата.
3-(Трет-бутоксикарбонил)амино)-2-хлор-6-фторбензойную кислоту (5,53 г, 19,1 ммоль) растворяют в ДМФ (100 мл) и последовательно обрабатывают триэтиламином (7,98 мл, 57,27 ммоль) и дифенилфосфорилазидом (6,17 мл, 28,63 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой (20 мл) и нагревают до 80°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют дополнительной водой (50 мл) и затем экстрагируют EtOAc (2×250 мл). Органические экстракты объединяют и промывают водой (3×100 мл) и насыщенным раствором соли (1×50 мл) затем сушат над Na2SO4, фильтруют, и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/ацетоном и затем снова гексаном/МТБЭ с получением трет-бутил (3-амино-2-хлор-4-фторфенил)карбамата (1,05 г, 21%). 1H ЯМР (400 МГц, ДМСО-d6) δ 8,41 (с, 1Н), 6,99-6,94 (м, 1Н), 6,69-6,66 (м, 1Н), 5,34 (с, 2Н), 1,43 (с, 9Н).
Промежуточное соединение Р8
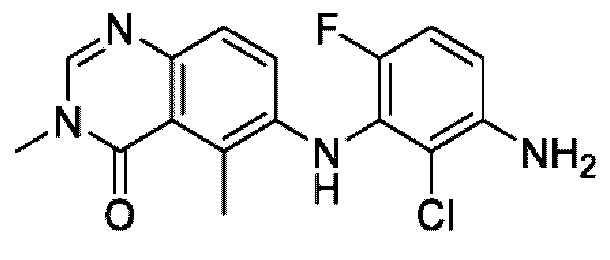
6-((3-Амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3H)-он
Стадия 1: Получение трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)карбамата.
Трет-бутил (3-амино-2-хлор-4-фторфенил)карбамат (56,7 мг, 0,217 ммоль), 6-бром-3,5-диметилхиназолин-4(3Н)-он (50 мг, 0,198 ммоль), карбонат цезия (193 мг, 0,593 ммоль), Xantphos (17,1 мг, 0,0296 ммоль) и трис(дибензилиденацетон)дипалладий (0) (9,05 мг, 0,0099) объединяют в пробирке, затем дегазируют при пониженном давлении и обратно заполняют газообразным аргоном. Толуол (0,98 8 мл) добавляют, и раствор продувают аргоном в течение 5 минут, затем пробирку герметично закрывают и нагревают до 100°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и фильтруют через слой Celite® и очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc с получением трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-карбамата в виде не совсем белого твердого вещества (81 мг, 95%). МС (хиад, m/z)=333,1 (М-Вос).
Стадия 2: Получение 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она.
Трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)карбамат (81 мг, 0,187 ммоль) растворяют в ДХМ (4,7 мл) и обрабатывают трифторуксусной кислотой (1,5 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле, элюируя ДХМ/МеОН и 1% NH4OH с получением 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она в виде светло-желтого твердого вещества (37 мг, 56%). 1H ЯМР (400 МГц, CDCl3) δ 7, 99 (с, 1Н), 7,38 (д, 1Н), 7,02 (д, 1Н), 6,86 (дд, 1Н), 6,51 (дд, 1Н), 3,50 (с, 3Н), 2,90 (с, 3Н); МС (хиад, m/z)=333,1 (М+Н).
Промежуточное соединение Р9
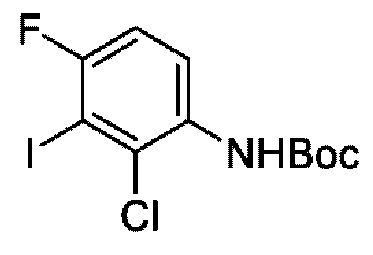
Трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат
Стадия 1: Получение 2-хлор-4-фтор-3-йоданилина.
В 5 л 4-горлой колбе, оборудованной 3 капельными воронками, внутренним датчиком температуры и магнитной мешалкой, 2-хлор-4-фторанилин (82,03 мл, 687,0 ммоль) растворяют в ТГФ (1,5 л) под обратным потоком N2 и охлаждают до -78°С. Реакционную смесь по каплям обрабатывают бутиллитием (2,5 М в гексане) (299,5 мл, 748,8 ммоль) и оставляют перемешиваться при -78°С в течение 15 минут после завершения добавления. Реакционную смесь по каплям обрабатывают ТГФ раствором (500 мл) 1,2-бис(хлордиметилсилил)этана (155,3 г, 721,4 ммоль) и оставляют перемешиваться при -78°С в течение 30 минут после завершения добавления. Реакционную смесь по каплям обрабатывают дополнительным бутиллитием (2,5 М в гексане) (299,5 мл, 748,8 ммоль) и затем ледяную баню удаляют после завершения добавления, и реакционную смесь перемешивают в течение 1 часа. Реакционную смесь охлаждают обратно до -78°С и по каплям обрабатывают дополнительным бутиллитием (2,5 М в гексане) (299,5 мл, 748,8 ммоль) и перемешивают при -78°С в течение 30 минут после завершения добавления. Реакционную смесь по каплям обрабатывают ТГФ раствором (600 мл) йода (249,3 г, 982,4 ммоль) и ледяную баню удаляют, и реакционную смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение 16 часов. Реакционную смесь обрабатывают 1000 мл воды, затем хлористоводородной кислотой (4,0 М водный раствор) (601,1 мл, 2404,5 ммоль) и перемешивают при температуре окружающей среды в течение 1 ч. Реакционную смесь нейтрализуют до примерно рН 8 с применением твердого NaHCO3 и затем обрабатывают тиосульфат натрия (3,0 М водный раствор) (801,5 мл, 2404,5 ммоль) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь переносят в экстракционную воронку, промывая колбу МТБЭ и водой, и затем слои разделяют. Органический слой промывают насыщенным раствором соли (1х) и сушат над Na2SO4, фильтруют, и концентрируют с получением 2-хлор-4-фтор-3-йоданилина (186,49 г, 100%). 1Н ЯМР (400 МГц, ДМСО) δ 6,97-6,93 (м, 1Н), 6,81-6,77 (м, 1Н), 5,41 (шс, 2Н).
Стадия 2: Получение бис-трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата.
В 3 л 1-горлой колбе, 2-хлор-4-фтор-3-йоданилин (186,49 г, 686,99 ммоль) растворяют в ТГФ (2,0 L) и обрабатывают 4-(диметиламино)пиридином (8,39 г, 68,7 ммоль), затем добавляют ди-трет-бутил дикарбонат (314,87 г, 1442,7 ммоль) и затем перемешивают при температуре окружающей среды в течение 1 часа на открытом воздухе с колонкой Vigreux. Реакционную смесь концентрируют досуха. Полученный остаток растворяют в ДХМ (1 л) и разбавляют гексаном (1 л) и перемешивают в течение 15 минут, затем пропускают через тонкий слой диоксида кремния, элюируя дополнительным 1:1 ДХМ:Гексаном (2,5 л). Фильтрат концентрируют досуха и полученное твердое вещество суспендируют в гептане (500 мл) и перемешивают при 80°С в течение 30 минут. Смесь охлаждают до 0°С на ледяной бане и фильтруют, промывают дополнительным охлажденным (0°С) гептаном (500 мл), и собирают светло-рыжевато-коричневое твердое вещество с получением бис-трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (145,5 г, 45%). 1Н ЯМР (400 МГц, ДМСО) δ 7,55-7,51 (м, 1Н), 7,32-7,28 (м, 1Н), 1,33 (с, 18Н).
Стадия 3: Получение трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата.
Бис-трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат (331,7 г, 703,2 ммоль) растворяют в МеОН (1,8 л) и обрабатывают карбонатом калия (106,9 г, 773,5 ммоль), затем нагревают до 65°С в течение 1 часа. Реакционную смесь охлаждают до температуры окружающей среды и выливают в 6,0 л воды и перемешивают в течение 30 минут. Смесь фильтруют, промывают дополнительной водой (1000 мл), и собирают светло-рыжевато-коричневое твердое вещество с получением трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (258,0 г, 99%). 1Н ЯМР (400 МГц, ДМСО) δ 8,82 (с, 1Н), 7,53-7,50 (м, 1Н), 7,24-7,20 (м, 1Н), 1,42 (с, 9Н).
Промежуточное соединение Р10
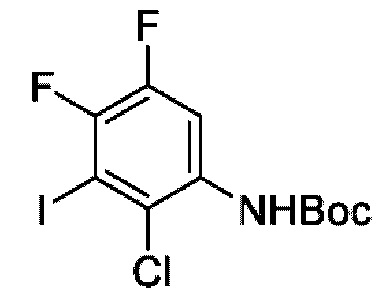
Трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбамат
Стадия 1: Получение 2-хлор-4,5-дифтор-3-йоданилина. Раствор 2-хлор-4,5-дифторанилин (26,0 г, 158,97 ммоль) растворяют в ТГФ (1,0 л) под обратным потоком N2 и охлаждают до -78°С. Реакционную смесь по каплям обрабатывают бутиллитием (2,5 М в гексане) (66,768 мл, 166,92 ммоль) и оставляют перемешиваться при -78°С в течение 15 минут после завершения добавления. Реакционную смесь по каплям обрабатывают ТГФ раствором (250 мл) 1,2-бис(хлордиметилсилил)этана (35,932 г, 166,92 ммоль) и оставляют перемешиваться при -78°С в течение 30 минут после завершения добавления. Реакционную смесь по каплям обрабатывают дополнительным бутиллитием (2,5 М в гексане) (66,768 мл, 166,92 ммоль). Ледяную баню удаляют после завершения добавления и реакционную смесь оставляют перемешиваться в течение 1 часа. Реакционную смесь охлаждают обратно до -78°С и по каплям обрабатывают дополнительным бутиллитием (2,5 М в гексане) (66,768 мл, 166,92 ммоль) и оставляют перемешиваться при -78°С в течение 30 минут после завершения добавления. Реакционную смесь по каплям обрабатывают ТГФ раствором (600 мл) йода (60,522 г, 238,46 ммоль). Ледяную баню удаляют, и реакционную смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение 16 часов. Реакционную смесь обрабатывают 500 мл воды, затем хлористоводородной кислотой (4,0 М водный раствор) (139,10 мл, 556,40 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь нейтрализуют до примерно рН 8 с применением твердого NaHCO3 и затем обрабатывают тиосульфатом натрия (3,0 М водный раствор) (185,47 мл, 556,40 ммоль) и перемешивают при температуре окружающей среды в течение 30 минут. Слои разделяют, и водный слой экстрагируют МТБЭ (1×1000 мл). Объединенные органические слои промывают насыщенным раствором соли (1×500 мл) затем сушат над Na2SO4, фильтруют, и концентрируют с получением 2-хлор-4,5-дифтор-3-йоданилина (46,0 г, 100%). 1Н ЯМР (400 МГц, ДМСО) δ 6,82-6,77 (м, 1Н), 5,68 (шс, 2Н).
Стадия 2: Получение бис-трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбамата.
2-Хлор-4,5-дифтор-3-йоданилин (46,0 г, 159 ммоль) растворяют в ТГФ (1000 мл) и обрабатывают 4-(диметиламино)пиридином (1,94 г, 15,9 ммоль), затем ди-трет-бутилдикарбонатом (72,8 г, 334 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь концентрируют досуха, и полученный остаток растворяют в 1:1 ДХМ:гексаном (500 мл) и перемешивают в течение 15 минут. Смесь пропускают через слой силикагеля, элюируя дополнительным 1:1 ДХМ:Гексаном (1,5 л). Фильтрат концентрируют досуха с получением бис-трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбамата (77,8 г, 100% выход). 1Н ЯМР (400 МГц, ДМСО) δ 7,95-7,90 (м, 1Н), 1,33 (с, 18Н).
Стадия 3: Получение трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбамата.
Бис-трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбамат (77,8 г, 159 ммоль) растворяют в МеОН (650 мл) и обрабатывают карбонатом калия (24,2 г, 175 ммоль), и реакционную смесь нагревают до 65°С в течение 1,5 часов. Реакционную смесь охлаждают до температуры окружающей среды, затем выливают в воду (2 л) и перемешивают в течение 15 минут. Реакционную смесь фильтруют, промывают дополнительной водой, и собирают полученное коричневое твердое вещество. Твердое вещество сушат в высоком вакууме в течение 16 часов и затем суспендируют в гептане (250 мл) и перемешивают в течение 15 минут. Суспензию фильтруют, и твердое вещество промывают дополнительный гептан с получением трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбамата (34,9 г, 56,4%). 1Н ЯМР (400 МГц, ДМСО) δ 8,94 (шс, 1Н), 7,76-7,71 (м, 1Н), 1,43 (с, 9Н).
Промежуточное соединение Р11
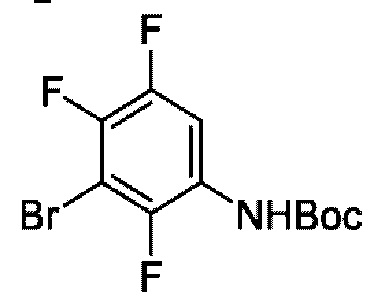
Трет-бутил (3-бром-2,4,5-трифторфенил)карбамат
Стадия А: Получение бис-трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбамата.
К раствору 3-бром-2,4,5-трифторанилина (3,0 г, 13 ммоль) в ТГФ (88 мл, 13 ммоль) добавляют ДМАП (0,16 г, 1,3 ммоль) и ди-трет-бутил дикарбонат (6,7 г, 31 ммоль), и раствор перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь концентрируют и очищают хроматографией на силикагеле (50% ДХМ/гексан) с получением бис-трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбамата, который применяют непосредственно на следующей стадии.
Стадия B: Получение трет-бутил (3-бром-2,4,5-трифторфенил)карбамата.
К раствору бис-трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (5,7 г, 13 ммоль) в метаноле (67 мл) добавляют карбонат калия (2,03 г, 14,7 ммоль), и реакционную смесь нагревают до 65°С в течение 3 часов. Раствор охлаждают до комнатной температуры и концентрируют. Полученное твердое вещество разбавляют водой, перемешивают в течение 15 минут, и затем фильтруют, промывают водой, и сушат в течение ночи с получением трет-бутил (3-бром-2,4,5-трифторфенил)карбамата (4,15 г, 95%). 1H ЯМР (400 МГц, ДМСО) δ 9,4 (шс, 1Н), 7,82 (м, 1Н), 1,46, (с, 9Н).
Промежуточное соединение Р12
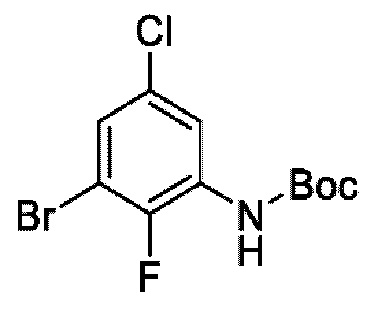
Трет-бутил (3-бром-5-хлор-2-фторфенил)карбамат
3-Бром-5-хлор-2-фторанилин (2,43 г, 10,8 ммоль) растворяют в ТГФ (110 мл) и обрабатывают 4-(диметиламино)пиридином (0,132 г, 1,08 ммоль), затем ди-трет-бутил дикарбонатом (4,96 г, 22,7 ммоль) и смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь концентрируют досуха, и полученный остаток растворяют в МеОН (100 мл) и обрабатывают карбонатом калия (2,99 г, 21,7 ммоль) и нагревают при 70°С в течение 30 минут. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Остаток растворяют в ДХМ и промывают водой (1х), затем сушат над Na2SO4, фильтруют, и концентрируют.Полученный остаток очищают хроматографией на силикагеле (Гексан/EtOAc) с получением трет-бутил (3-бром-5-хлор-2-фторфенил)карбамата (2,92 г, 83%). 1H ЯМР (400 МГц, ДМСО) δ 9,45 (шс, 1Н), 7,81-7,79 (м, 1Н), 7,54-7,52 (м, 1Н), 1,47 (с, 9Н).
Промежуточное соединение Р13
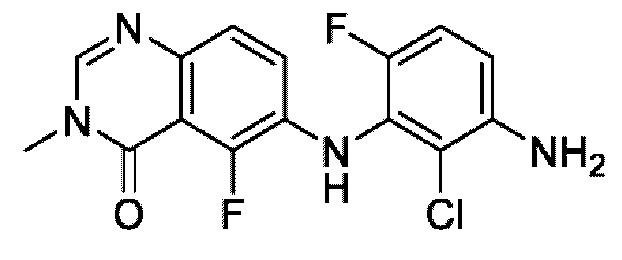
6-((3-Амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-он
6-Амино-5-фтор-3-метилхиназолин-4(3Н)-он (Промежуточное соединение Р4; 1,50 г, 7,76 ммоль) растворяют в толуоле (78 мл) и обрабатывают трет-бутил (2-хлор-4-фтор-3-йодфенил)карбаматом (Промежуточное соединение Р9; 3,02 г, 8,15 ммоль), трис(дибензилиденацетон)диппалладием (0) (0,36 г, 0,39 ммоль), Xantphos (0,67 г, 1,16 ммоль) и карбонатом цезия (5,06 г, 15,53 ммоль). Реакционную смесь продувают аргоном в течение нескольких минут и затем нагревают при 110°С в течение 24 часов под баллоном аргона. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (1 л) и перемешивают в течение 15 минут, затем фильтруют через слой Celite® и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (20 мл) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и очищают С18 хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным NaHCO3 (1х) и затем сушат над Na2SO4, фильтруют, и концентрируют с получением 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (1,26 г, 48%). 1Н ЯМР (400 МГц, ДМСО) δ 8,14 (с, 1Н), 7,78 (с, 1Н), 7,31-7,29 (д, 1Н), 7,05-7,00 (т, 1Н), 6,94-6,89 (т, 1Н), 6,70-6,67 (м, 1Н), 5,28 (шс, 2Н), 3,43 (с, 3Н). МС (хиад, m/z)=337,1, 339,1 (М+Н).
Промежуточное соединение Р14
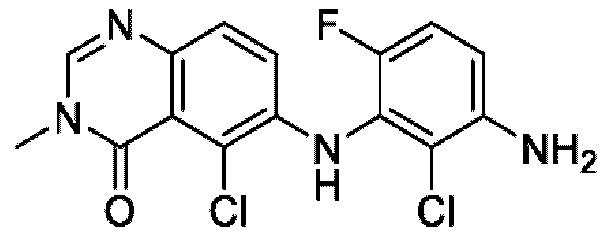
6-((3-Амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-он
6-Амино-5-хлор-3-метилхиназолин-4(3Н)-он (2,35 г, 11,2101 ммоль) растворяют в толуоле (110 мл) и обрабатывают трет-бутил (2-хлор-4-фтор-3-йодфенил)карбаматом (4,37 г, 11,77 ммоль), трис(дибензилиденацетон)диппалладием (0) (0,51 г, 0,56 ммоль), Xantphos (0,97 г, 1,68 ммоль), и карбонатом цезия (7,30 г, 22,42 ммоль). Реакционную смесь продувают аргоном в течение нескольких минут и затем нагревают, перемешивают при 110°С под баллоном аргона в течение 60 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (1 л) и перемешивают в течение 15 минут, затем фильтруют через слой Celite® и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ/Ацетон). Выделенный продукт растворяют в 1:1 ДХМ:ТФК (20 мл) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и затем очищают С18 хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным NaHCO3 (1х) и затем сушат над Na2SO4, фильтруют, и концентрируют с получением 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (2,67 г, 67%). 1Н ЯМР (400 МГц, ДМСО) δ 8,16 (с, 1Н), 7,47 (с, 1Н), 7,41-7,39 (д, 1Н), 7,08-7,03 (т, 1Н), 6,76-6,72 (м, 2Н), 5,31 (с, 2Н), 3,41 (с, 3Н). МС (хиад, m/z)=353,0, 355,0 (М+Н).
Промежуточное соединение Р15
6-((3-Амино-2-хлор-6-фторфенил)амино)-5-бром-3-метилхиназолин-4(3Н)-он
6-Амино-5-бром-3-метилхиназолин-4(3Н)-он (800 мг, 3,15 ммоль) растворяют в толуоле (32 мл) и обрабатывают трет-бутил (2-хлор-4-фтор-3-йодфенил)карбаматом (1,23 г, 3,31 ммоль), трис(дибензилиденацетон)диппалладием (0) (144 мг, 0,16 ммоль), Xantphos (273 мг, 0,47 ммоль) и карбонатом цезия (2,05 г, 6,30 ммоль). Реакционную смесь продувают аргоном в течение нескольких минут и затем нагревают и перемешивают при 110°С под баллоном аргона в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (100 мл) и перемешивают в течение 15 минут, затем фильтруют через слой Celite® и концентрируют. Полученный остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода, 0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным водным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с MgSO4, фильтруют, и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (20 мл) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и промывают насыщенным водным NaHCO3 с получением 6-((3-Амино-2-хлор-6-фторфенил)амино)-5-бром-3-метилхиназолин-4(3Н)-она (1,2 г, 96%). МС (хиад, m/z)=397,0, 399,0 (М+Н).
Промежуточное соединение Р16
6-((3-Амино-2-хлор-5,6-дифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-он
6-Амино-5-хлор-3-метилхиназолин-4(3Н)-он (1,00 г, 4,77 ммоль) растворяют в толуоле (48 мл) и обрабатывают трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбаматом (1,95 г, 5,01 ммоль), трис(дибензилиденацетон)диппалладием (0) (218 мг, 0,24 ммоль), Xantphos (414 мг, 0,72 ммоль) и карбонатом цезия (3,89 г, 11,9 ммоль). Реакционную смесь продувают аргоном в течение нескольких минут и перемешивают при 110°С под баллоном аргона в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (100 мл) и перемешивают в течение 15 минут, затем фильтруют через слой Celite® и концентрируют. Полученный остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода, 0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным водным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с MgSO4, фильтруют, и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (35 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют, растворяют в ДХМ (75 мл) и промывают насыщенным водным NaHCO3. Органический остаток в ДХМ концентрируют и очищают хроматографией на силикагеле (55-90% EtOAc/Гексан) с получением с получением 6-((3-Амино-2-хлор-5,6-дифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (390 мг, 22%). МС (хиад, m/z)=371,0, 373,1 (М+Н).
Промежуточное соединение Р17
6-((3-Амино-2-хлор-5,6-дифторфенил)амино)-3,5-диметилхиназолин-4(3Н)-он
6-Амино-3,5-диметилхиназолин-4(3Н)-он (2,0 г, 11,0 ммоль) растворяют в толуоле (106 мл) и обрабатывают трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбаматом (4,9 г, 13,0 ммоль), трис(дибензилиденацетон)диппалладием (0) (0,97 г, 1,1 ммоль), Xantphos (1,5 г, 2,6 ммоль) и карбонатом цезия (6,9 г, 21,0 ммоль). Реакционную смесь продувают аргоном в течение нескольких минут и затем перемешивают при 110°С под баллоном аргона в течение 72 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (500 мл) и перемешивают в течение 15 минут, затем фильтруют через слой Celite® и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ-EtOAc). Полученный остаток растворяют в 1:1 ДХМ:ТФК (22 мл) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и обрабатывают 500 мл NaHCO3 и перемешивают в течение 30 минут при температуре окружающей среды. Смесь экстрагируют 4:1 ДХМ:ИПС. Органический слой отделяют, сушат над безводным Na2SO4, фильтруют, и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (ДХМ/Ацетон) с получением 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она (1,2 г, 32%) в виде рыжевато-коричневого твердого вещества. МС (хиад, m/z)=351,0, 353,0 (М+Н).
Промежуточное соединение Р18
6-((3-Амино-2-хлор-5;6-дифторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-он
6-Амино-5-фтор-3-метилхиназолин-4(3Н)-он (1170 мг, 6,05648 ммоль) растворяют в толуоле (61 мл) и обрабатывают трет-бутил (2-хлор-4,5-дифтор-3-йодфенил)карбаматом (2477,37 мг, 6,35931 ммоль), трис(дибензилиденацетон)диппалладием (554,613 мг, 0,605648 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантеном (876,119 мг, 1,51412 ммоль) и карбонатом цезия (3946,63 мг, 12,1130 ммоль). Реакционную смесь продувают аргоном в течение нескольких минут и нагревают до 110°С в течение 16 часов под баллоном аргона. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ и перемешивают в течение 15 минут, затем фильтруют через слой Celite® и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ/Ацетон) и затем полученный остаток растворяют в 1:1 ДХМ:ТФК (20 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют и разбавляют 4:1 ДХМ:ИПС и промывают насыщенным NaHCO3 (1х) затем сушат над Na2SO4, фильтруют, и концентрируют с получением 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (1,21 г, 56%). МС (хиад, m/z)=355,0, 357,0 (М+Н).
Промежуточное соединение Р19
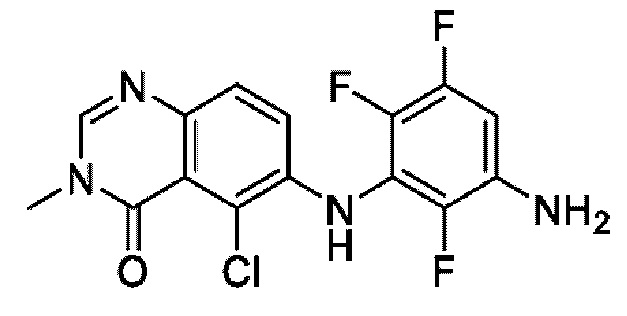
6-((3-Амино-2,5,6-трифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-он
Стадия А: Получение трет-бутил (3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)карбамата.
Раствор 6-амино-5-хлор-3-метилхиназолин-4(3Н)-она (200 мг, 0,95 ммоль), трет-бутил (3-бром-2,4,5-трифторфенил)карбамата (327 мг, 1,00 ммоль), Pd2(dba)3 (43,7 мг, 0,0477 ммоль), Xantphos (82,8 мг, 0,143 ммоль) и карбоната цезия (777 мг, 2,39 ммоль) в толуоле (9,5, мл) продувают аргоном в течение 5 минут и затем нагревают до 110°С под азотом в течение 16 часов. Реакцию охлаждают до температуры окружающей среды, фильтруют через Celite®, концентрируют, и очищают хроматографией на силикагеле (5-95% ДХМ/EtOAc) с получением трет-бутил (3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)карбамата (87 мг, 20,0%). МС (хиад, m/z)=455,1 (М+Н).
Стадия В: Получение 6-((3-амино-2,5,6-трифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она.
Раствор трет-бутил (3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)карбамата (87 мг, 0,19 ммоль) в дихлорметане (960 мкл) и ТФК (960 мкл) перемешивают при температуре окружающей среды в течение 1 часа. Раствор разделяют между дихлорметаном и насыщенным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют, и очищают хроматографией на силикагеле (5-95% EtOAc/ДХМ) с получением 6-((3-амино-2,5,6-трифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (65 мг, 96%). МС (хиад, m/z)=355,1 (М+Н).
Промежуточное соединение Р20
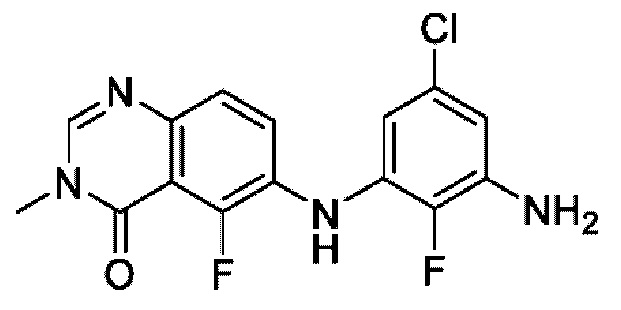
6-((3-амино-5-хлор-2-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-он
Стадия А: Получение трет-бутил (5-хлор-2-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)карбамата.
Раствор 6-амино-5-фтор-3-метилхиназолин-4(3Н)-она (300 мг, 1,6 ммоль), трет-бутил (3-бром-5-хлор-2-фторфенил)карбамата (529 мг, 1,63 ммоль), Pd2 (dba)3 (71,1 мг, 0,0776 ммоль), Xantphos (135 мг, 0,233 ммоль) и карбоната цезия (1265 мг, 3,88 ммоль) в толуоле (15,5 мл) продувают аргоном в течение 5 минут и затем нагревают до 110°С под азотом в течение 16 часов. Реакцию охлаждают до температуры окружающей среды, фильтруют через Celite®, концентрируют, и очищают хроматографией на силикагеле (1-15% МеОН/ДХМ, 1% NH4OH) с получением трет-бутил (5-хлор-2-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)карбамата (189 мг, 27,9%). МС (хиад, m/z)=437,1 (М+Н).
Стадия В: Получение 6-((3-амино-5-хлор-2-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она.
Раствор трет-бутил (5-хлор-2-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)карбамата (189 мг, 0,433 ммоль) в дихлорметане (1082 мкл) и ТФК (1082 мкл) перемешивают при температуре окружающей среды в течение 30 минут. Раствор концентрируют и разделяют между ДХМ и насыщенным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют, и очищают хроматографией на силикагеле (1-15% МеОН/ДХМ, 1% NH4OH) с получением 6-((3-амино-5-хлор-2-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (70 мг, 48,0%). МС (хиад, m/z)=337,1 (М+Н).
Промежуточное соединение Р21
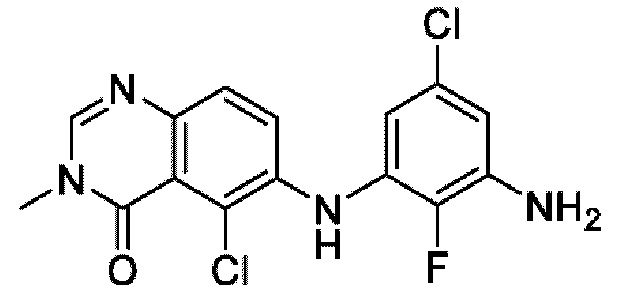
6-((3-амино-5-хлор-2-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-он
Стадия А: Получение трет-бутил (5-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)карбамата. Раствор 6-амино-5-хлор-3-метилхиназолин-4(3Н)-он (820 мг, 2,35 ммоль), трет-бутил (3-бром-5-хлор-2-фторфенил)карбамат (800 мг, 2,46 ммоль), Pd2 (dba)3 (107 мг, 0,117 ммоль), Xantphos (204 мг, 0,352 ммоль), и карбонат цезия (1912 мг, 5,87 ммоль) в толуоле (23,5 мл) продувают аргоном в течение 5 минут и затем нагревают до 110°С под азотом в течение 16 часов. Реакцию охлаждают до температуры окружающей среды, фильтруют через Celite®, концентрируют, и очищают хроматографией на силикагеле (5-95% ДХМ/EtOAc) с получением трет-бутил (5-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)карбамата (525 мг, 49,3%). МС (хиад, m/z)=453,1 (М+Н).
Стадия В: Получение 6-((3-амино-5-хлор-2-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она.
Раствор трет-бутил (5-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)карбамата (525 мг, 1,16 ммоль) в дихлорметане (2895 мкл) и ТФК (2895 мкл) перемешивают при температуре окружающей среды в течение 1 часа. Раствор концентрируют и разделяют между дихлорметаном и насыщенным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют, и очищают хроматографией на силикагеле (5-95% EtOAc/ДХМ) с получением 6-((3-амино-5-хлор-2-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (260 мг, 63,6%). МС (хиад, т/т.)=353,1 (М+Н).
Промежуточное соединение Р22
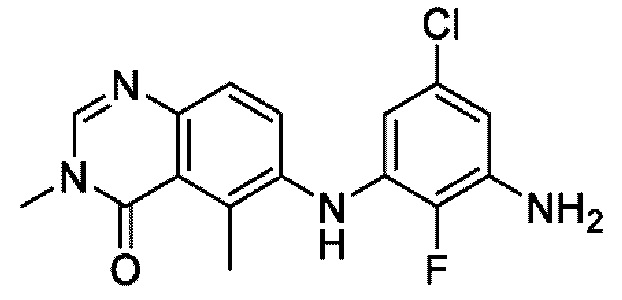
6-((3-амино-5-хлор-2-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-он
6-Амино-3,5-диметилхиназолин-4(3Н)-он (500 мг, 2,64 ммоль) растворяют в толуоле (26 мл) и обрабатывают трет-бутил (3-бром-5-хлор-2-фторфенил)карбаматом (1,03 г, 3,17 ммоль), трис(дибензилиденацетон)диппалладием (0) (241,98 мг, 0,26 ммоль), Xantphos (382,25 мг, 0,66 ммоль) и карбонатом цезия (1,721 г, 5,28 ммоль). Реакционную смесь продувают аргоном в течение нескольких минут и затем нагревают до 110°С под баллоном аргона в течение 24 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (500 мл) и перемешивают в течение 15 минут, затем фильтруют через слой Celite® и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ/EtOAc) и затем растворяют в 1:1 ДХМ:ТФК (10 мл) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и обрабатывают 500 мл NaHCO3 и перемешивают в течение 30 минут при температуре окружающей среды. Смесь экстрагируют 4:1 ДХМ:ИПС, и органический слой отделяют, сушат над безводным Na2SO4, фильтруют, и концентрируют с получением 6-((3-амино-5-хлор-2-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она (0,8 г, 90%) в виде желтого твердого вещества. МС (хиад, m/z)=333, 1, 335,1 (М+Н).
Промежуточное соединение Р23
2-Хлор-4-фтор-3-метоксианилин
Стадия 1: Получение 2-хлор-4-фтор-3-метоксибензальдегида.
N,N,N'-триметилэтилендиамин (1,77 мл, 13,6 ммоль) растворяют в ТГФ (50 мл) и охлаждают до -42°С под обратным потоком азота, затем обрабатывают п-бутиллитием (2,5 М в гексане, 5,45 мл, 13,6 ммоль) и оставляют перемешиваться при -42°С в течение 30 минут. Реакционную смесь охлаждают до -78°С и обрабатывают 50 мл ТГФ раствором 4-фтор-3-метоксибензальдегида (2,0 г, 13,0 ммоль) и затем нагревают до -42°С и перемешивают в течение 30 минут. Реакционную смесь охлаждают до -78°С и обрабатывают n-бутиллитием (2,5 М в гексане, 5,45 мл, 13,6 ммоль) и затем нагревают до -42°С и оставляют перемешиваться в течение 1 ч. Реакционную смесь быстро переносят через канюлю в 50 мл ТГФ раствор гексахлорэтана (6,14 г, 26,0 ммоль) при температуре окружающей среды, и перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь обрабатывают 4,0 М HCl и экстрагируют Et2O (2×250 мл). Органические фазы объединяют и промывают 1,0 М NaOH (1×100 мл), 1,0 М HCl (1×100 мл), и насыщенным раствором соли (1×50 мл) и затем сушат над Na2SO4, фильтруют, и концентрируют. Остаток очищают С18 хроматографией с обращенной фазой (вода/ацетонитрил с 0,1% ТФК) и фракции, содержащие желаемый продукт, объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным NaHCO3 (1×100 мл), сушат над Na2SO4, фильтруют, и концентрируют с получением 2-хлор-4-фтор-3-метоксибензальдегида (1,12 г, 46%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 10,26 (с, 1Н), 7,70-7,67 (м, 1Н), 7,52-7,48 (т, 1Н), 3,94 (с, 3Н).
Стадия 2: Получение 2-хлор-4-фтор-3-метоксибензойной кислоты.
2-Хлор-4-фтор-3-метоксибензальдегид (1,07 г, 5,67 ммоль) растворяют в ацетонитриле (57 мл) и обрабатывают водным 1,0 М двухосновным раствором фосфата натрия (8,51 мл, 8,51 ммоль) и затем охлаждают до 0°С. Реакционную смесь обрабатывают водным 35% масс, раствором перекиси водорода (0,732 мл, 8,51 ммоль) затем по каплям добавляют водный 1,0 М раствор хлорита натрия (8,51 мл, 8,51 ммоль) и затем оставляют нагреваться до температуры окружающей среды и перемешивают в течение 16 часов. Реакционную смесь обрабатывают 3,0 М раствором тиосульфата натрия и разбавляют 1,0 М NaOH, затем промывают Et2O (2×250 мл). Водный слой подкисляют до примерно рН 2 с применением 4,0 М HCl и экстрагируют 4:1 ДХМ:ИПС (2×250 мл). Органические экстракты объединяют и сушат над Na2SO4, фильтруют, и концентрируют с получением 2-хлор-4-фтор-3-метоксибензойной кислоты (1,16 г, 99%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 13,45 (шс, 1Н), 7,59-7,56 (м, 1Н), 7,40-7,36 (т, 1H), 3,89 (с, 3Н).
Стадия 3: Получение 2-хлор-4-фтор-3-метоксианилина.
2-Хлор-4-фтор-3-метоксибензойную кислоту (1,16 г, 5,670 ммоль) растворяют в ДМФ (57 мл) и обрабатывают триэтиламином (2,37 мл, 17,01 ммоль) затем дифенилфосфорилазидом (1,83 мл, 8,51 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают 10 мл воды и нагревают до 8 0°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют EtOAc (2×250 мл). Органические фазы объединяют и промывают водой (3×100 мл) и насыщенным раствором соли (1×50 мл), сушат над Na2SO4, фильтруют, и концентрируют.Остаток очищают хроматографией на силикагеле (Гексан/EtOAc) с получением 2-хлор-4-фтор-3-метоксианилина (640,9 мг, 64%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 6,98-6,94 (т, 1Н), 6,52-6,49 (м, 1Н), 5,24 (шс, 2Н), 3,82 (с, 3Н).
Промежуточное соединение Р24
3-Амино-2-хлор-6-фторфенол
К раствору 2-хлор-4-фтор-3-метоксианилина (400 мг, 2,2 ммоль) в ДХМ (10 мл) добавляют BBr3 (4,5 мл, 1,0 М в ДХМ, 4,5 ммоль) при 0°С, и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь гасят метанолом (3 мл) и затем выливают в холодную воду (75 мл) и экстрагируют этилацетатом (100 мл × 2). Органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной хроматографией на силикагеле (10% этилацетат в гексане) с получением 3-амино-2-хлор-6-фторфенола в виде не совсем белого твердого вещества (220 мг, 60%). 1Н ЯМР (400 МГц, ДМСО-d6) δ 9,87 (шс, 1Н), 6,83 (дд, 1Н), 6,22-60,18 (м, 1Н), 5,03 (шс, 2Н). МС (m/z)=159,8 (М-Н).
Промежуточное соединение Р25
3-Фтор-2-метил-6-нитробензойная кислота
К охлажденному на льду раствору 3-фтор-2-метилбензойной кислоты (5 г, 32,4 ммоль) в концентрированной H2SO4 (50 мл) медленно добавляют концентрированную HNO3 (3,5 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 6 часов. Реакционную смесь выливают в ледяную воду, и твердое вещество собирают фильтрацией, промывают водой (2×50 мл) и сушат при пониженном давлении с получением 3-фтор-2-метил-6-нитробензойной кислоты в виде не совсем белого твердого вещества (3,0 г, 47%). 1Н ЯМР (400 МГц, MeOD) δ 8,11-8,07 (м, 1Н), 7,34 (т, 1Н), 2,32 (с, 3Н); МС (m/z)=197,6 (М-Н).
Промежуточное соединение Р26
Трет-бутил 3-фтор-2-метил-6-нитробензоат
К перемешиваемому раствору 3-фтор-2-метил-6-нитробензойной кислоты (800 мг, 4,02 ммоль) в смеси трет-BuOH и ДХМ (1:1, 5 мл) добавляют Boc2O (1,38 мл, 6,03 ммоль), затем ДМАП (147 мг, 1,2 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов в атмосфере азота. Реакционную смесь концентрируют, и неочищенный остаток гасят водой (150 мл) и экстрагируют этилацетатом (2×150 мл). Объединенные органические слои промывают водой (2×100 мл), насыщенным раствором соли (100 мл), сушат над безводным сульфатом натрия, фильтруют, и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной хроматографией на силикагеле с применением 15-20% этилацетата в гексане с получением трет-бутил 3-фтор-2-метил-6-нитробензоата в виде бесцветной жидкости (600 мг, 60%). 1Н ЯМР (400 МГц, CDCl3) δ 8,10-8,07 (дд, 1Н), 7,34 (т, 1Н), 2,30 (с, 3Н), 1,60 (с, 9Н).
Промежуточное соединение Р27
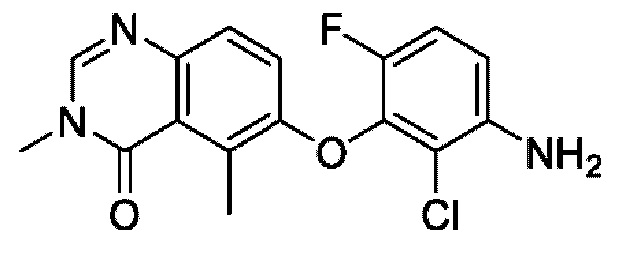
6-(3-Амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-он
Стадия 1: Получение метил 3-фтор-2-метил-6-нитробензоата. Метил 3-фтор-2-метилбензоат (4,17 г, 24,8 ммоль) растворяют в серной кислоте (24,1 мл, 248 ммоль), и смесь охлаждают до 0°С.
Туда медленно добавляют азотную кислоту (1,36 мл, 29,8 ммоль), и смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение 1 часа. Реакционную смесь выливают в 500 мл ледяной воды и перемешивают в течение 15 минут. Смесь экстрагируют EtOAc (2х), объединенные органические слои промывают насыщенным NaHCO3 (2х), насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют, и концентрируют.Неочищенный продукт очищают хроматографией с обращенной фазой (5->95% вода-АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным NaHCO3 (1х). Объединенные органические слои сушат над Na2SO4, фильтруют, и концентрируют с получением метил 3-фтор-2-метил-6-нитробензоата (3,80 г, 72%).
Стадия 2: Получение метил 3-(3-амино-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата.
К раствору метил 3-фтор-2-метил-6-нитробензоата (686 мг, 3,22 ммоль) и 3-амино-2-хлор-6-фторфенола (572 мг, 3,54 ммоль) в ДМСО (15 мл) добавляют карбонат калия (1112 мг, 8,05 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют EtOAc и водой. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией, элюируя 10-50% EtOAc в гексане с получением метил 3-(3-амино-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата (950 мг, 83%).
Стадия 3: Получение метил 3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата.
К раствору метил 3-(3-амино-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата (683,4 мг, 1,927 ммоль) в диоксане (17,8 мл) добавляют ТЭА (805,6 мкл, 5,780 ммоль), ДМАП (23,54 мг, 0,1927 ммоль) и (Вос)2O (1261 мг, 5,780 ммоль), и реакционную смесь нагревают до 100°С в течение 1 часа. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (5->50% Гексан-EtOAc) с получением 3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата (970 мг, 91%).
Стадия 4: Получение метил 6-амино-3-(3-(бис(трет-бутоксикарбонил)амино-2-хлор-6-фторфенокси)-2-метилбензоата.
К раствору метил 3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата (319 мг, 0,575 ммоль) в 1:1 ТГФ/насыщенном водном NH4Cl (10 мл) добавляют порошок железа (321 мг, 5,75 ммоль) и реакционную смесь нагревают до 65°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют 20% ИПС в ДХМ и воде. Твердое вещество удаляют фильтрацией, и фильтрат промывают водой (1х), насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют, и концентрируют с получением 6-амино-3-(3-(бис(трет-бутоксикарбонил)амино-2-хлор-6-фторфенокси)-2-метилбензоата (260 мг). Неочищенный продукт применяют непосредственно на следующей стадии. МС (m/z)=525,2 (М+Н).
Стадия 5: Получение трет-бутил (трет-бутоксикарбонил)(2-хлор-4-фтор-3-((5-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата.
Раствор метил 6-амино-3-(3-(бис(трет-бутоксикарбонил)амино-2-хлор-6-фторфенокси)-2-метилбензоата (260 мг, 0,495 ммоль) и ацетата формамидина (61,9 мг, 0,594 ммоль) в EtOH (4 мл) нагревают до 70°С в течение 24 часов. Реакционную смесь концентрируют, и остаток очищают колоночной хроматографией, элюируя 25-100% EtOAc в гексане с получением трет-бутил (трет-бутоксикарбонил)(2-хлор-4-фтор-3-((5-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата (0,244 г, 95%). МС (m/z)=520,1 (М+Н).
Стадия 6: Получение трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата.
К раствору трет-бутил (трет-бутоксикарбонил)(2-хлор-4-фтор-3-((5-метил-4-оксо-3,4 -дигидрохиназолин-6-ил)окси)фенил)карбамата (0,244 г, 0,469 ммоль) в ДМФ добавляют K2CO3 (0,0973 г, 0,704 ммоль) затем Mel (0,0439 мл, 0,704 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют водой и экстрагируют EtOAc. Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата. Неочищенный продукт применяют на следующей стадии как есть. МС (m/z)=534,2 (М+Н).
Стадия 7: Получение 6-(3-амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-она.
К раствору трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (0,251 г, 0,470 ммоль) в ДХМ (4 мл) добавляют ТФК (4 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь концентрируют, и остаток очищают колоночной хроматографией, элюируя 50-100% EtOAc в гексане с получением 6-(3-амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-она (0,144 г, 92%). МС (m/z)=334,1 (М+Н).
Промежуточное соединение Р28
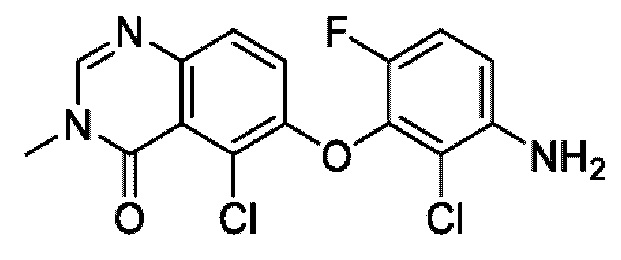
6-(3-Амино-2-хлор-6-фторфенокси)-5-хлор-3-метил-3, 4-дигидрохиназолин-4-он
Стадия 1: Получение метил 2-хлор-3-фторбензоата. К раствору 2-хлор-3-фторбензойной кислоты (3 г, 17,24 ммоль) в метаноле (60 мл) добавляют концентрированную H2SO4 (0,5 мл), и реакционную смесь нагревают при 60°С в атмосфере азота в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют при пониженном давлении. Реакционную смесь разделяют между этилацетатом (100 мл) и насыщенным водным NaHCO3 (100 мл). Органическую фазу отделяют и промывают насыщенным раствором соли (100 мл), сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении с получением метил 2-хлор-3-фторбензоата в виде желтой жидкости (2,9 g) которую применяют на следующей стадии без дальнейшей очистки 1Н ЯМР (400 МГц, ДМСО-d6) δ 7,70-7,62 (м, 2Н), 7,56-7,48 (м, 1Н), 3,88 (с, 3Н).
Стадия 2: Получение метил 2-хлор-3-фтор-6-нитробензоата.
К охлажденному на льду раствору метил 2-хлор-3-фторбензоата (0,531 г, 2,82 ммоль) в концентрированной H2SO4 (5 мл) добавляют HNO3 (0,233 мл, 3,52 ммоль, 70% масс./масс.). Реакционную смесь удаляют с ледяной бани и перемешивают при температуре окружающей среды в течение 4 часов. Реакционную смесь выливают в ледяную воду (50 мл) и добавляют твердый Na2CO3 для доведения раствора до рН 2. Водную фазу экстрагируют ДХМ (3×25 мл) и объединенные органические фазы концентрируют с получением желтого масла, которое очищают хроматографией с обращенной фазой (5-50% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и экстрагируют ДХМ (3×25 мл). Органические фазы объединяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением метил 2-хлор-3-фтор-6-нитробензоата (411 мг, 59,4%) в виде прозрачного желтого масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ (8,41-8,36, м, 1Н), (7,91-7,84, м, 1Н), (3,96, с, 3Н).
Стадия 3: Получение метил 3-(3-амино-2-хлор-6-фторфенокси)-2-хлор-6-нитробензоата.
К раствору метил 2-хлор-3-фтор-6-нитробензоата (85 мг, 0,364 ммоль) в ДМСО (1,8 мл) добавляют твердый K2CO3 (126 мг, 0,910 ммоль) и 3-амино-2-хлор-6-фторфенол (64,7 мг, 0,400 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов затем разделяют между этилацетатом (25 мл) и водой (15 мл). Органическую фазу отделяют и промывают насыщенным раствором соли (15 мл), затем сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением коричневого твердого вещества, которое очищают хроматографией на силикагеле (0-50% гексан/этилацетат) с получением метил 3-(3-амино-2-хлор-6-фторфенокси)-2-хлор-6-нитробензоата (100 мг, 73,3%) в виде желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,06 (д, 1Н), 7,02 (т, 1Н), 6,76-6,68 (м, 2Н),), 4,09 (шс, 2Н), 4,07 (с, 3Н).
Стадия 4: Получение метил 3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-хлор-6-нитробензоата.
К раствору метил 3-(3-амино-2-хлор-6-фторфенокси)-2-хлор-6-нитробензоата (100 мг, 0,267 ммоль) в 1,4-диоксане (2,6 мл) добавляют триэтиламин (0,112 мл, 0,800 ммоль), N,N-диметилпиридин-4-амин (3,26 мг, 0,0267 ммоль) и ди-трет-бутилдикарбонат (175 мг, 0,800 ммоль). Реакционную смесь нагревают при 100°С в течение 1 часа затем охлаждают до температуры окружающей среды и разделяют между этилацетатом (25 мл) и водой (15 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (0-40% гексан/этилацетат) с получением метил 3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-хлор-6-нитробензоата (128 мг, 84%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,05 (д, 1Н), 7,26-7,22 (м, 2Н), 6,63-6,59 (м, 1Н), 4,07 (с, 3Н), 1,43 (с, 18Н).
Стадия 5: Получение метил 6-амино-3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-хлорбензоата.
К раствору метил 3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-хлор-6-нитробензоата (105 мг, 0,182 ммоль) в тетрагидрофуране (2 мл) добавляют порошок железа (102 мг, 1,82 ммоль) и насыщенный водный NH4Cl (2 мл). Реакционную смесь нагревают при 65°С в течение 16 часов. Реакцию охлаждают до температуры окружающей среды и разделяют между этилацетатом (25 мл) и водой (25 мл), затем фильтруют. Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением метил 6-амино-3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-хлорбензоата в виде желтого масла (99 мг), которое применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 6: Получение трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата.
К раствору метил 6-амино-3-(3-(бис(трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-хлорбензоата (99 мг, 0,182 ммоль) в этаноле (1 мл) добавляют ацетат формамидина (20,0 мг, 0,193 ммоль). Реакционную смесь герметично закрывают и нагревают при 100°С в микроволновом реакторе в течение 2 часов. Реакцию охлаждают до температуры окружающей среды затем концентрируют с получением смеси трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата и трет-бутил (2-хлор-3-((5-хлор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата, которую применяют непосредственно на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=540,l (М+Н)
Стадия 7: Получение трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата.
К смеси трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата и трет-бутил (2-хлор-3-((5-хлор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (99 мг, 0,183 ммоль, рассчитано с применением молекулярной массы трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата) в N,N-диметилформамиде (1 мл) добавляют K2CO3 (38,0 мг, 0,275 ммоль) и йодметан (12,5 мкл, 0,202 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов затем разделяют между водой (10 мл) и этилацетатом (10 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением желтого масла, которое очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1%ТФК) с получением 40 мг смеси продукта, содержащей трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат и трет-бутил (2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат (40 мг) в виде прозрачного масла, которое используют на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=554,1 (М+Н).
Стадия 8: Получение 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она.
Смесь трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата и трет-бутил (2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (40 мг) растворяют в ДХМ (1 мл) и трифторуксусной кислоте (1 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа затем концентрируют с получением белого твердого вещества, которое очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции разбавляют насыщенным водным NaHCO3 (15 мл) и экстрагируют ДХМ (3×15 мл). Органические фазы объединяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-он (29,2 мг, 37% со стадии 5) в виде рыжевато-коричневого твердого вещества. МС (хиад, m/z)=354,0 (М+Н)
Промежуточное соединение Р29
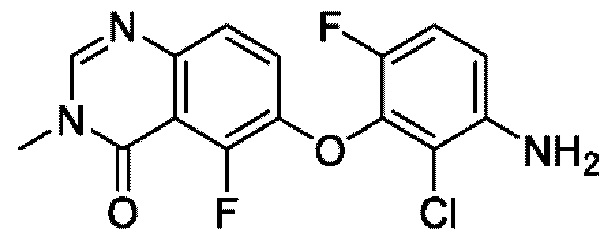
6-(3-Амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-он
Стадия 1: Получение метил 2,3-дифторбензоата. 2,3-дифторбензойную кислоту (913,1 мг, 5,775 ммоль) растворяют в МеОН (58 мл) затем обрабатывают серной кислотой (140,2 мкл, 1,444 ммоль) и смесь нагревают до 70°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Остаток разбавляют ДХМ и промывают насыщенным NaHCO3 (1х) затем сушат над Na2SO4, фильтруют, и концентрируют с получением метил 2,3-дифторбензоата (714,4 мг, 71,9%). 1Н ЯМР (400 МГц, ДМСО) δ 7,74-7,65 (м, 2Н), 7,35-7,29 (м, 1Н), 3,85 (с, 3Н).
Стадия 2: Получение метил 2,3-дифтор-6-нитробензоатв. К холодному (0°С) раствору метил 2,3-дифторбензоата (714,4 мг, 4,150 ммоль) в серной кислоте (4030 мкл, 41,50 ммоль) добавляют азотную кислоту (297,8 мкл, 4,565 ммоль) по каплям. Реакционную смесь перемешивают при 0°С в течение 15 минут и затем при температуре окружающей среды в течение 16 часов. Реакционную смесь выливают в ледяную воду (100 мл) и перемешивают в течение 15 минут, затем нейтрализуют до примерно рН 8 с применением твердого Na2CO3. Реакционную смесь экстрагируют EtOAc (2х), и объединенные органические слои промывают водой (1х), насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют и концентрируют. Полученный остаток очищают хроматографией на силикагеле (Гексан/ацетон) с получением 1:1 смеси метил 2,3-дифтор-6-нитробензоат и метил 2,3-дифтор-5-нитробензоата (665,6 мг, 74%), которую применяют непосредственно на следующей стадии без очистки.
Стадия 3: Получение метил 3-(3-амино-2-хлор-6-фторфенокси)-2-фтор-6-нитробензоата. Смесь метил 2,3-дифтор-6-нитробензоата и метил 2,3-дифтор-5-нитробензоата (665,6 мг, 3,065 ммоль) растворяют в ДМСО (12 мл) и обрабатывают 3-амино-2-хлор-6-фторфенолом (544,8 мг, 3,372 ммоль), затем карбонатом калия (635,5 мг, 4,598 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь выливают в воду и экстрагируют EtOAc (2х). Объединенные органические слои промывают водой (3х),насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют, и концентрируют с получением 1:1 смеси метил 3-(3-амино-2-хлор-6-фторфенокси)-2-фтор-6-нитробензоата и метил 2-(3-((бис-трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-3-фтор-5-нитробензоата (860,9 мг, 78%) которую применяют непосредственно на следующей стадии без очистки.
Стадия 4: Получение метил 3-(3-((бис-трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-фтор-6-нитробензоат. 1:1 смесь метил 3-(3-амино-2-хлор-6-фторфенокси)-2-фтор-6-нитробензоата и метил 2-(3-((бис-трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-3-фтор-5-нитробензоата (860,9 мг, 2,400 ммоль) растворяют в ТГФ (24 мл) и обрабатывают 4-(диметиламино)пиридином (2 9,32 мг, 0,2 4 00 ммоль), затем ди-трет-бутил дикарбонатом (1100 мг, 5,040 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 60 часов. Реакционную смесь концентрируют и очищают хроматографией на силикагеле (Гексан/EtOAc) с получением метил 3-(3-((бис-трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-фтор-6-нитробензоата (476,9 мг, 36%). 1Н ЯМР (400 МГц, ДМСО) δ 8,16-8,13 (дд, 1Н), 7,63-7,61 (м, 2Н), 7,03-6,99 (т, 1Н), 3,94 (с, 3Н), 1,33 (с, 18Н).
Стадия 5: Получение метил 6-амино-3-(3-((бис-трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-фторбензоата.
Метил 3-(3-((бис-трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-фтор-6-нитробензоат (461,7 мг, 0,8261 ммоль) растворяют в 1:1 ТГФ: насыщенным NH4Cl (16,5 мл, 0,05 М) и затем обрабатывают железом (922,6 мг, 16,52 ммоль) и реакционную смесь нагревают до 65°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют EtOAc. Органический слой промывают водой (1х), насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют и концентрируют с получением смеси метил 6-амино-3-(3-((бис-трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-фторбензоата и метил 6-амино-3-(3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-фторбензоата (395,1 мг, 90%).
Стадия 6: Получение бис-трет-бутил
(2-хлор-4-фтор-3-((5-фтор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата. Смесь метил 6-амино-3-(3-((бис-трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-фторбензоата и метил 6-амино-3-(3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторфенокси)-2-фторбензоата (395,1 мг, 0,7470 ммоль) растворяют в EtOH (15 мл) и обрабатывают ацетатом формамидина (388,8 мг, 3,735 ммоль) и реакционную смесь нагревают до 100°С в течение 8 часов микроволнами. Реакционную смесь концентрируют и остаток разбавляют EtOAc. Объединенные органические слои промывают водой (1х), насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют, и концентрируют с получением смеси бис-трет-бутил (2-хлор-4-фтор-3-((5-фтор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата и трет-бутил (2-хлор-4-фтор-3-((5-фтор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата (342,4 мг, 88%).
Стадия 7: Получение 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она. Смесь бис-трет-бутил (2-хлор-4-фтор-3-((5-фтор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата и трет-бутил (2-хлор-4-фтор-3-((5-фтор-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата (342,4 мг, 0,6535 ммоль) растворяют в ДМФ (6,5 мл) и обрабатывают карбонатом калия (117,4 мг, 0,8496 ммоль) затем йодметаном (44,75 мкл, 0,7189 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь разбавляют водой и экстрагируют EtOAc (2х). Объединенные органические слои промывают водой (3х), насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют, и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (5,0 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют и очищают С18 хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Объединенные желаемые фракции затем разделяют между 4:1 ДХМ:ИПС и насыщенным NaHCO3 (1х). Объединенные органические слои сушат над Na2SO4, фильтруют, и концентрируют с получением 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (206,1 мг, 93%). МС (хиад, m/z)=338,1, 340,1 (М+Н).
Промежуточное соединение Р30
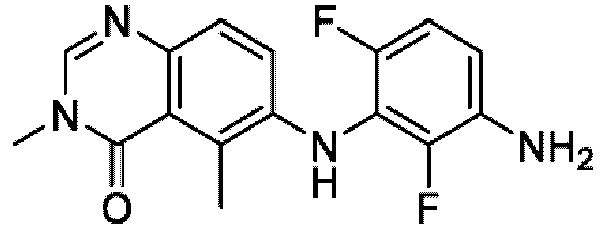
6-((3-амино-2,6-дифторфенил)амино)-3,5-диметилхиназолин-4(3Н)-он
Стадия 1: Получение ди-трет-бутил (3-бром-2,4-дифторфенил)дикарбамата. 3-Бром-2,4-дифторанилин (375 мг, 1,80 ммоль) перемешивают при температуре окружающей среды с ди-трет-бутилдикарбонатом (826 мг, 3,79 ммоль) и ДМАП (44,1 мг, 0,361 ммоль) в ТГФ (9,014 мл) в течение 48 часов. Реакционную смесь разбавляют этилацетатом и промывают насыщенным водным NH4Cl. Объединенные органические слои сушат над сульфатом магния, фильтруют и концентрируют. Продукт применяют непосредственно на следующей стадии без очистки.
Стадия 2: Получение трет-бутил (3-бром-2,4-дифторфенил)карбамата. Ди-трет-бутил (3-бром-2,4-дифторфенил)дикарбамат (735 мг, 1,80 ммоль) растворяют в МеОН (6,0 мл) и добавляют карбонат калия (113 мкл, 1,98 ммоль), и реакцию нагревают до 65°С в течение 1 часа. Реакционную смесь охлаждают до температуры окружающей среды и затем фильтруют через слой Celite и концентрируют. Неочищенный продукт очищают хроматографией с нормальной фазой (0-20% гексан/EtOAc) с получением трет-бутил (3-бром-2,4-дифторфенил)карбамата в виде белого твердого вещества (447 мг, 81% за 2-стадии).
Стадия 3: Получение 6-((3-амино-2,6-дифторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она. 6-Амино-3,5-диметилхиназолин-4(3Н)-он (50 мг, 0,26 ммоль), трет-бутил (3-бром-2,4-дифторфенил)карбамат (81 мг, 0,26 ммоль), карбонат цезия (172 мг, 0,53 ммоль), Pd2 (dba)3 (24 мг, 0,026 ммоль) и Xantphos (38 мг, 0,066 ммоль) растворяют в толуоле (2,6 мл) и затем нагревают до 110°С в течение 24 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют через Celite, концентрируют, и затем восстанавливают в 1:1 ДХМ/ТФК (5 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 15 минут. Летучие вещества удаляют в вакууме, и неочищенный продукт очищают хроматографией с обращенной фазой (0-30% MeCN/H2O, 0,1% ТФК). Полученный продукт разбавляют ДХМ, промывают насыщ. водным NaHCO3, и концентрируют с получением 6-((3-амино-2,6-дифторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она(32 мг, 38%). МС (хиад, m/z)=317,1 (М+Н).
Промежуточное соединение Р31
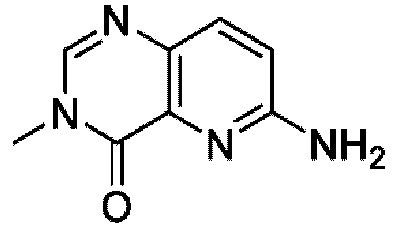
6-амино-3-метилпиридо[3,2-d]пиримидин-4(3Н)-он
Стадия 1: 6-хлор-3-метилпиридо[3,2-d]пиримидин-4(3Н)-она. К раствору 6-хлорпиридо[3,2-d]пиримидин-4(3Н)-она (100 мг, 0,551 ммоль) и йодметана (44 мкл, 0,71 ммоль) в N,N-диметилформамиде (1,1 мл) добавляют карбонат калия (114 мг, 0,826 ммоль) и перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь затем разделяют между этилацетатом и водой, и органический слой промывают насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют, и концентрируют. Полученный остаток затем очищают хроматографией на силикагеле (5-95% EtOAc/ДХМ) с получением 6-хлор-3-метилпиридо[3,2-d]пиримидин-4(3Н)-она (67 мг, 62% выход). МС (хиад, m/z)=196,1 (М+Н).
Стадия 2: 6-((4-метоксибензил)амино)-3-метилпиридо[3,2-d]пиримидин-4(3Н)-она. Раствор 6-хлор-3-метилпиридо[3,2-d]пиримидин-4(3Н)-она (25 мг, 0,128 ммоль), (4-метоксифенил)метанамина (33 мкл, 0,25 ммоль), и основания Хюнига (33 мкл, 0,19 ммоль) в ДМСО (426 мкл) нагревают при 90°С в течение 48 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ и водой. Органический слой промывают насыщенным раствором соли (1х), сушат над Na2SO4, фильтруют, и концентрируют. Полученный остаток очищают хроматографией на силикагеле (1-10% МеОН/ДХМ, 1% NH4OH) с получением неочищенного 6-((4-метоксибензил)амино)-3-метилпиридо[3,2-d]пиримидин-4(3Н)-она (25 мг, 66% выход). МС (хиад, m/z)=297,1 (М+Н).
Стадия 3: 6-амино-3-метилпиридо[3,2-d]пиримидин-4(3Н)-она. Неочищенный 6-((4-метоксибензил)амино)-3-метилпиридо[3,2-d]пиримидин-4(3Н)-он (25 мг, 0,084 ммоль) перемешивают в ТФК (422 мкл) и дихлорметане (422 мкл) в течение 3 часов при температуре окружающей среды. Реакционную смесь затем концентрируют, растворяют в ТФК (422 мкл) и затем нагревают в герметично закрытой пробирке при 60°С в течение 3 дней. Реакционную смесь затем концентрируют и растворяют в 1 мл метанола и 1 мл ДХМ и затем пропускают через бикарбонатную основную смолу. Фильтрат затем концентрируют и очищают хроматографией на силикагеле (1-15% МеОН/ДХМ, 1% NH4OH) с получением 6-амино-3-метилпиридо[3,2-d]пиримидин-4(3Н)-она (7,0 мг, 47% выход). МС (хиад, m/z)=177,1 (М+Н).
Промежуточное соединение Р32
трет-Бутил_(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат
Стадия 1: Получение трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата. 6-(3-Амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-он (1,5 г, 4,5 ммоль) растворяют в ТГФ (22 мл) затем обрабатывают ди-трет-бутилдикарбонатом (2,2 г, 9,9 ммоль) и N,N-диметилпиридин-4-амином (55 мг, 0,45 ммоль). Реакционную смесь нагревают до 45°С в течение 12 часов. Реакционную смесь концентрируют и очищают колоночной хроматографией на силикагеле (0-20% ДХМ/EtOAc) с получением трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (2 г, 83%) в виде белого твердого вещества. ЯМР (400 МГц, (CD3)2SO) δ 8,28 (с, 1Н), 7,55-7,54 (д, 1Н), 7,53 (с, 1Н), 7,47-7,41 (д, 1Н), 6,86-6,78 (д, 1Н), 3,46 (с, 3Н), 2,92 (с, 3Н), 1,36 (с, 18Н); МС (хиад, m/z)=534,2, 536,2 (М+Н).
Стадия 2: Получение трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата. Трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат (2 г, 3,7 ммоль) растворяют в МеОН (19 мл) и обрабатывают карбонатом калия (0,62 г, 4,5 ммоль) затем нагревают до 60°С в течение 12 часов. Реакционную смесь охлаждают до температуры окружающей среды, выливают в воду, обрабатывают ультразвуком и фильтруют. Полученное твердое вещество очищают колоночной хроматографией на силикагеле (0-50% ДХМ/Ацетон) с получением трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (1,5 г, 98%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) δ 8, 89 (с, 1Н), 8,27 (с, 1Н), 7,53-7,40 (м, 3Н), 6,97-6,92 (д, 1Н), 3,46 (с, 3Н), 2,91 (с, 3Н), 1,45 (с, 9Н); МС (хиад, m/z)=434,1, 436,1 (М+Н).
Промежуточное соединение Р33
трет-Бутил_(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат
Стадия 1: Получение трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата. 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-он (1,5 г, 4,2 ммоль) растворяют в ТГФ (21 мл) и обрабатывают ди-трет-бутилдикарбонатом (2,0 г, 9,3 ммоль) и N,N-диметилпиридин-4-амином (52 мг, 0,43 ммоль) затем нагревают до 45°С в течение 12 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют и очищают колоночной хроматографией на силикагеле (0-20% ДХМ/EtOAc) с получением промежуточного соединения трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (2 г, 85%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) δ 8,36 (с, 1Н), 7,62-7,54 (м, 3Н), 7,02-6,96 (д, 1Н), 3,46 (с, 3Н), 1,36 (с, 18Н); МС (хиад, m/z)=554,2, 556,2 (М+Н).
Стадия 2: Получение трет-бутил (2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата. Трет-бутил (трет-бутоксикарбонил)(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат (2 г, 3,6 ммоль) растворяют в МеОН (18 мл) и обрабатывают карбонатом калия (0,60 г, 4,3 ммоль) затем нагревают до 60°С в течение 12 часов. Реакцию охлаждают до температуры окружающей среды, выливают в воду, обрабатывают ультразвуком и фильтруют, и полученное твердое вещество очищают колоночной хроматографией на силикагеле (0-50% ДХМ/Ацетон) с получением трет-бутил (2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (1,6 г, 100%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) δ 8,92 (с, 1Н), 8,35 (с, 1Н), 7,59-7,53 (м, 2Н), 7,49-7,43 (т, 1Н), 7,15-7,11 (м, 1Н), 3,46 (с, 3Н), 1,46 (с, 9Н); МС (хиад, m/z)=454,2, 456,2 (М+Н).
Промежуточное соединение Р34
трет-Бутил_(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамат
Стадия 1: Получение трет-бутил (трет-бутоксикарбонил)(2-хлор-4-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата. 6-(3-Амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-он (1,5 г, 4,4 ммоль) растворяют в ТГФ (22 мл) и обрабатывают ди-трет-бутилдикарбонатом (2,1 г, 9,8 ммоль) и N,N-диметилпиридин-4-амином (54 мг, 0,44 ммоль) затем нагревают до 45°С в течение 12 часов. Реакцию концентрируют и очищают колоночной хроматографией на силикагеле (0-20% ДХМ/EtOAc) до трет-бутил (трет-бутоксикарбонил)(2-хлор-4-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата (1,8 г, 75%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) δ 8,34 (с, 1Н), 7,57-7,53 (д, 2Н), 7,49-7,46 (м, 1Н), 7,23-7,17 (т, 1Н), 3,45 (с, 3Н), 1,35 (с, 18Н); МС (хиад, m/z)=538,1, 540,1 (М+Н).
Стадия 2: Получение трет-бутил (2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата. Трет-бутил (трет-бутоксикарбонил)(2-хлор-4-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамат (1,8 г, 3,35 ммоль) растворяют в МеОН (16,7 мл) и обрабатывают карбонатом калия (0,55 г, 4,02 ммоль) и реакцию нагревают до 60°С в течение 12 часов. Реакцию охлаждают до температуры окружающей среды, выливают в воду, обрабатывают ультразвуком, и фильтруют, и полученное твердое вещество очищают колоночной хроматографией на силикагеле (0-50% ДХМ/Ацетон) с получением трет-бутил (2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамата (1,16 г, 80%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) δ 8,92 (с, 1Н), 8,33 (с, 1Н), 7,56-7,51 (м, 1Н), 7,48-7,41 (м, 2Н), 7,31-7,25 (т, 1Н), 3,46 (с, 3Н), 1,45 (с, 9Н); МС (хиад, m/z)=438,1, 440,1.
Получение синтетических примеров
Пример 1
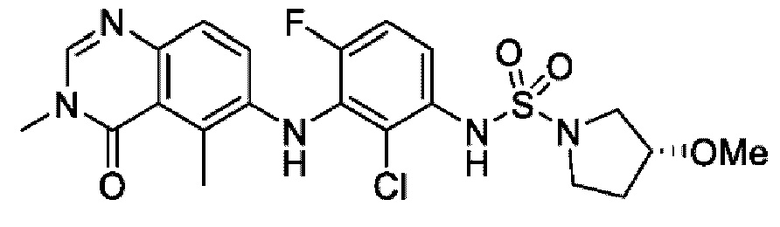
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксипирролидин-1-сульфонамид
Стадия 1: Получение (R)-3-метоксипирролидин-1-сульфонилхлорида. Суспензию (R)-3-метоксипирролидина гидрохлорида (198 мг, 1,44 ммоль) и N,N-диизопропилэтиламина (376 мкл, 2,16 ммоль) перемешивают в ДХМ (6 мл) при температуре окружающей среды до полного растворения смеси. Реакционную смесь охлаждают до 0°С и обрабатывают сульфурилхлоридом (349 мкл, 4,32 ммоль). Холодную баню удаляют через 30 минут, и смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют дополнительным ДХМ и промывают 1,0 М HCl (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют, и концентрируют с получением неочищенного (R)-3-метоксипирролидин-1-сульфонилхлорида (212 мг, 74%), который применяют на следующей стадии как есть.
Стадия 2: Получение (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксипирролидин-1-сульфонамида. Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она (50 мг, 0,15 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (212 мг, 1,0 ммоль) в пиридине (0,8 мл) герметично закрывают и нагревают при 65°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Неочищенный продукт очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют, и концентрируют. Продукт затем очищают колоночной хроматографией на силикагеле (50-100% EtOAc/гексан) с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксипирролидин-1-сульфонамида (40 мг, 54%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,92 (с, 1Н), 7,44-7,39 (м, 2Н), 7,08-7,02 (м, 2Н), 6,88 (с, 1Н), 5,57 (с, 1Н), 3,96-3,92 (м, 1Н), 3,54 (с, 3Н), 3,50-3,41 (м, 4Н), 3,25 (с, 3Н), 2,96 (с, 3Н), 2,09-2,03 (м, 1Н), 1,99-1,90 (м, 1Н). МС (хиад, m/z)=496, 1 (М+Н).
Пример 2
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-этил-N-метил)-сульфамида трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (35 мг, 0,10 ммоль) и N-этил-N-метилсульфамоилхлорида (47 мг, 0,30 ммоль) в пиридине (0,50 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, фильтруют и концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-этил-N-метил)-сульфамида трифторацетата (22 мг, 48%). 1H ЯМР (400 МГц, CDCl3) δ 8,67 (с, 1Н), 7,65 (д, 1Н), 7,56-7,52 (м, 1Н), 7,17 (т, 1Н), 7,01-6,98 (м, 1Н), 6,75 (с, 1Н), 6,55 (с, 1Н), 3,69 (с, 3Н), 3,31-3,26 (м, 2Н), 2,87 (с, 3Н), 1,15 (т, 1Н). МС (хиад, m/z)=474,1 (М+Н).
Пример 3
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N,N-диметил)сульфамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08494 ммоль) и диметилсульфамоила хлорида (120 мг, 0,8494 ммоль) в пиридине (0,8 мл) нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют, и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc), затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют, и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N,N-диметил)-сульфамида (13,6 мг, 35%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO): δ 9,47 (с, 1Н), 8,21 (с, 1Н), 7,76 (с, 1Н), 7,53-7,33 (м, 3Н), 6,78-6,72 (м, 1Н), 3,44 (с, 3Н), 2,73 (с, 6Н); МС (хиад, m/z)=460,1, 462,1 (М+Н).
Пример 4
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-(N-этил-N-метил)-сульфамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (28 мг, 0,0832 ммоль) и этил(метил)сульфамоилхлорида (106 мг, 0,673 ммоль) в пиридине (0,8 мл) нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют, и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc) затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют, и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-(N-этил-N-метил)-сульфамида (28 мг, 74%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) δ 9,33 (с, 1Н), 8,15 (с, 1Н), 8,02 (с, 1Н), 7,39-7,24 (м, 3Н), 7,01-6,93 (т, 1Н), 3,41 (с, 3Н), 3,13-3,06 (м, 2Н), 2,72 (с, 3Н), 1,02-0,98 (т, 3Н); МС (хиад, m/z)=458,1, 460,1 (М+Н).
Пример 5
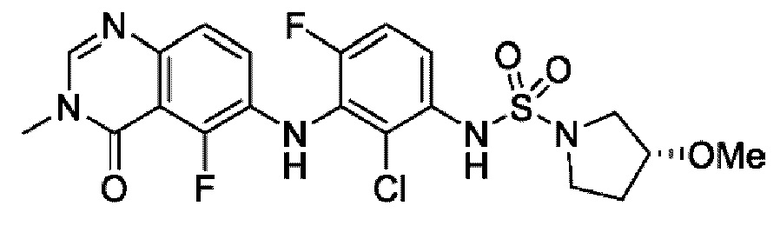
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксипирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (70 мг, 0,21 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (166 мг, 0,83 ммоль) в пиридине (2 мл) герметично закрывают и нагревают при 65°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Неочищенный продукт очищают колоночной хроматографией на силикагеле (30-100% EtOAc/гексан) с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-(N-изопропил-N-метил)-сульфамида (68 мг, 65%). 1H ЯМР (400 МГц, CDCl3) δ 7,89 (с, 1Н), 7,54-7,51 (м, 1Н), 7,40-7,38 (дд, 1Н), 7,14-7,09 (т, 1Н), 7,06-6,99 (м, 1Н), 6,90 (с, 1Н), 5,91 (с, 1Н), 3,96-3,93 (м, 1Н), 3,57 (с, 3Н), 3,52-3,41 (м, 4Н), 3,25 (с, 3Н), 2,12-2,05 (м, 1Н), 2,00-1,91 (м, 1Н). МС (хиад, m/z)=500,1 (М+Н).
Пример 6
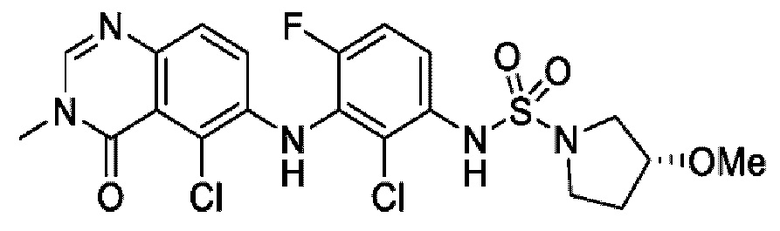
(R)-N-(2-хлор-4-фтор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксипирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (70 мг, 0,20 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (158 мг, 0,79 ммоль) в пиридине (2 мл) герметично закрывают и нагревают при 65°С в течение 60 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Неочищенный продукт очищают колоночной хроматографией на силикагеле (30-100% EtOAc/гексан) с получением (R)-N-(2-хлор-4-фтор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксипирролидин-1-сульфонамида (59 мг, 58%). 1Н ЯМР (400 МГц, CDCl3) δ 7,91 (с, 1Н), 7,58-7,54 (м, 1Н), 7,50-7,48 (д, 1Н), 7,15-7,11 (т, 1Н), 6,97-6,94 (м, 1Н), 6,91 (с, 1Н), 6,44 (с, 1Н), 3,96-3,93 (м, 1Н), 3,56 (с, 3Н), 3,52-3,41 (м, 4Н), 3,25 (с, 3Н), 2,11-2,05 (м, 1Н), 1,99-1,90 (м, 1Н). МС (хиад, m/z)=516,1 (М+Н).
Пример 7
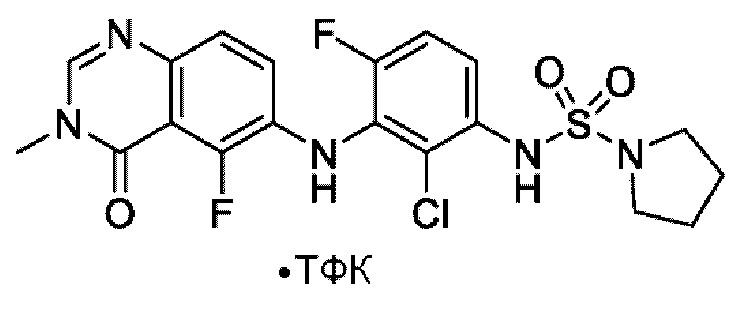
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-он (150 мг, 0,45 ммоль) и пирролидин-1-сульфонилхлорида (190 мг, 1,10 ммоль) в пиридине (2,2 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида трифторацетата (161 мг, 77%). 1Н ЯМР (400 МГц, CDCl3) δ 8,32 (с, 1Н), 7,56-7,53 (м, 1Н), 7,49-7,46 (м, 1Н), 7,15 (т, 1Н), 7,09-7,03 (м, 1Н), 6,75 (с, 1Н), 5,98 (с, 1Н), 3,64 (с, 3Н), 3,37-3,34 (м, 4Н), 1,91-1,88 (м, 4Н). МС (хиад, m/z)=470,1 (М+Н).
Пример 8
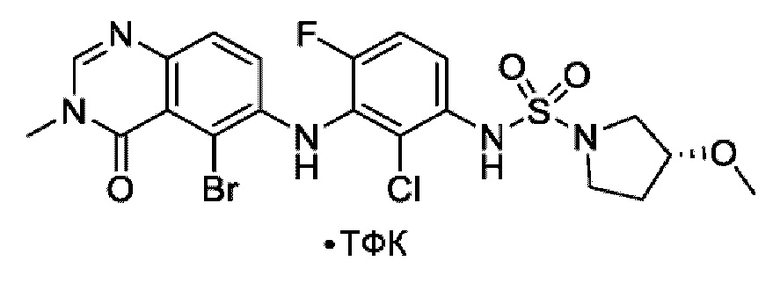
(R)-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-метоксипирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-бром-3-метилхиназолин-4(3Н)-она (35 мг, 0,088 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (44 мг, 0,22 ммоль) в пиридине (0,44 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-метоксипирролидин-1-сульфонамида трифторацетата (20 мг, 40%). 1Н ЯМР (400 МГц, CDCl3) δ 8,18 (с, 1Н), 7,61-7,57 (м, 1Н), 7,15 (т, 1Н), 7,00-6,95 (м, 2Н), 6,93 (с, 1Н), 6,66 (с, 1Н), 3,97-3,95 (м, 1Н), 3,62 (с, 3Н), 3,53-3,43 (м, 2Н), 3,27 (с, 3Н), 2,07-1,93 (м, 4Н). МС (хиад, m/z)=560,0, 562,0 (М+Н).
Пример 9
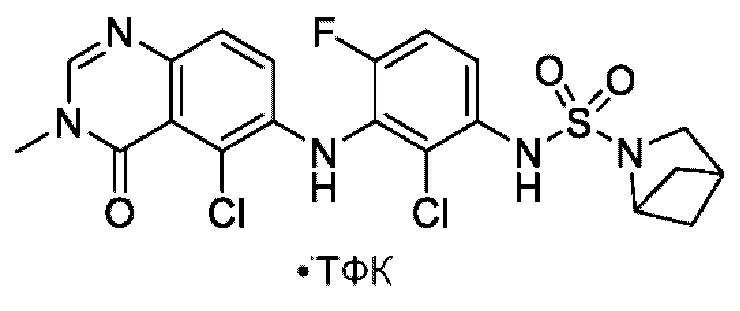
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-азабицикло[2,1,1]гексан-2-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (30 мг, 0,085 ммоль) и 2-азабицикло[2,1,1]гексан-2-сульфонилхлорида (46 мг, 0,26 ммоль) в пиридине (0,43 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuS04. Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида трифторацетата (21 мг, 50%). 1Н ЯМР (400 МГц, CDCL3) δ 8,46 (с, 1Н), 7,64-7,61 (м, 2Н), 7,16 (т, 1Н), 7,00-6,97 (м, 1Н), 6,82 (с, 1Н), 6,51 (с, 1Н), 4,27-4,24 (м, 1Н), 3,53 (с, 3Н), 3,40 (с, 2Н), 2,92-2,88 (м, 1Н), 1,96-1,94 (м, 2Н), 1,55-1,53 (м, 2Н). МС (хиад, m/z)=498,1 (М+Н).
Пример 10
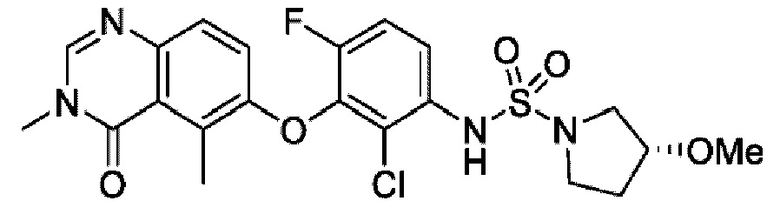
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксипирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-она (70 мг, 0,21 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (167 мг, 0,84 ммоль) в пиридине (2 мл) герметично закрывают и нагревают при 65°С в течение 60 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Неочищенный продукт очищают колоночной хроматографией на силикагеле (30-100% EtOAc/гексан) с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксипирролидин-1-сульфонамида (55 мг, 53%). 1Н ЯМР (400 МГц, CDCl3) δ 7,94 (с, 1Н), 7,58-7,55 (м, 1Н), 7,43 (д, 1Н), 7,15-7,11 (т, 1Н), 6,93-6,91 (м, 2Н), 3,96-3,93 (м, 1Н), 3,55 (с, 3Н), 3,51-3,40 (м, 4Н), 3,26 (с, 3Н), 3,01 (с, 3Н), 2,12-2,05 (м, 1Н), 1,99-1,90 (м, 1Н). МС (хиад, m/z)=497,1 (М+Н).
Пример 11
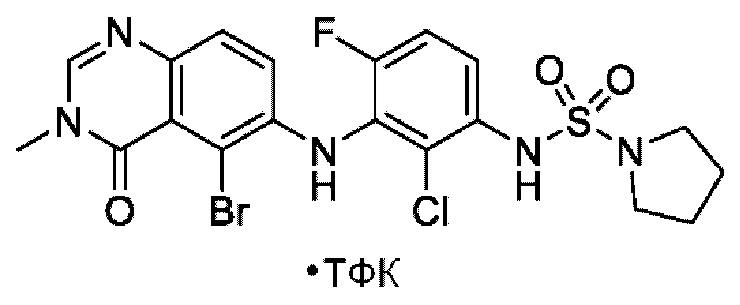
N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)пирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-бром-3-метилхиназолин-4(3Н)-она (250 мг, 0,63 ммоль) и пирролидин-1-сульфонилхлорида (320 мг, 1,89 ммоль) в пиридине (3,1 мл) герметично закрывают и нагревают при 70°С в течение 18 часов.
Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)пирролидин-1-сульфонамида трифторацетата (202 мг, 60%). 1Н ЯМР (400 МГц, CDCl3) δ 7,98 (с, 1Н), 7,58-7,54 (м, 1Н), 7,51-7,49 (м, 1Н), 7,14 (т, 1Н), 6,93-6,90 (м, 1Н), 6,64 (с, 1Н), 3,57 (с, 3Н), 3,36-3,31 (м, 4Н), 1,90-1,86 (м, 4Н). МС (хиад, m/z)=530,0, 532,0 (М+Н).
Пример 12
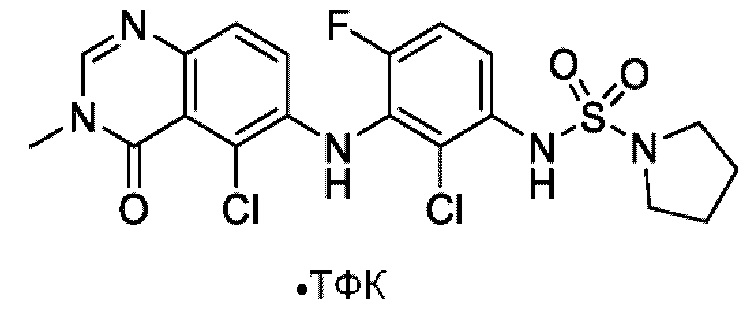
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (150 мг, 0,42 ммоль) и пирролидин-1-сульфонилхлорида (180 мг, 1,06 ммоль) в пиридине (2,1 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамида трифторацетата (179 мг, 87%). 1Н ЯМР (400 МГц, CDCl3) δ 8,59 (с, 1Н), 7,64-7,58 (м, 2Н), 7,17 (т, 1Н), 7,01-6,97 (м, 1Н), 6,77 (с, 1Н), 6,54 (с, 1Н), 3,68 (с, 1Н), 3,38-3,34 (м, 4Н), 1,92-1,88 (м, 4Н). МС (хиад, m/z)=486,1 (М+Н).
Пример 13
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,089 ммоль) и 2-азабицикло[2,1,1]гексан-2-сульфонилхлорида (49 мг, 0,27 ммоль) в пиридине (0,45 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида трифторацетата (18 мг, 41%). 1Н ЯМР (400 МГц, CDCl3) δ 8,25 (с, 1Н), 7,60-7,56 (м, 1Н), 7,47-7,56 (м, 1Н), 7,14 (т, 1Н), 7,08-7,03 (м, 1Н), 6,80 (с, 1Н), 5,96 (с, 1Н), 4,26-4,24 (м, 1Н), 3,63 (с, 3Н), 3,40 (с, 2Н), 2,91-2,88 (м, 1Н), 1,96-1,93 (м, 2Н), 1,54-1,53 (м, 2Н). МС (хиад, m/z)=482,1 (М+Н).
Пример 14
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамид и кристаллическая форма (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида, форма А
Способ А получения (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида.
6-((3-Амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-он (Промежуточное соединение Р13; 139,5 мг, 0,4143 ммоль) растворяют в пиридине (4,1 мл) и обрабатывают (3R)-3-фторпирролидин-1-сульфонилхлоридом (388,6 мг, 2,071 ммоль), и реакционную смесь нагревают до 60°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и затем разбавляют EtOAc и промывают 10% лимонной кислотой (2х) затем насыщенным раствором соли (1х) затем сушат над Na2SO4, фильтруют, и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ/Ацетон), затем С18 хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Объединенные желаемые фракции затем разделяют между 4:1 ДХМ : ИПС и насыщенным NaHCO3 (1х) затем сушат над Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в ДХМ и промывают 1,0 М NaOH (2х), и затем объединенные водные слои экстрагируют ДХМ (1х). Водный слой подкисляют до примерно рН 2 с применением 4,0 М HCl и затем экстрагируют 4:1 ДХМ : ИПС (2х) и затем сушат над Na2SO4, фильтруют и концентрируют с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида (74,8 мг, 37%). 1Н ЯМР (400 МГц, (CD3)2SO) δ 9,54 (с, 1Н), 8,18 (с, 1Н), 8,07 (с, 1Н), 7,42-7,38 (м, 1Н), 7,35-7,30 (м, 2Н), 7,03-6,99 (т, 1Н), 5,39-5,26 (м, 1Н), 3,50-3,39 (м, 6Н), 3,35-3,28 (м, 1Н), 2,15-2,00 (м, 2Н). МС (хиад, m/z)=488,1, 490,1 (М+Н).
Способ В получения (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида.
Стадия 1: Получение трет-бутил (R)-(2-хлор-4-фтор-3-йодфенил)((3-фторпирролидин-1-ил)сульфонил)карбамата. Трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат (Промежуточное соединение Р9; 20 г, 54 ммоль) растворяют в ТГФ (269 мл) и охлаждают до 0°С. Реакционную смесь обрабатывают гидридом натрия (4,3 г, 108 ммоль, 60% масс., в минеральном масле) порциями, и затем ледяную баню удаляют и смесь перемешивают в течение 15 минут. Реакционную смесь обрабатывают (R)-3-фторпирролидин-1-сульфонилхлоридом (20 г, 108 ммоль) и затем нагревают до 50°С в течение 12 часов. Реакционную смесь охлаждают до температуры окружающей среды и медленно выливают в перемешиваемую колбу с ледяной водой (500 мл) и затем экстрагируют EtOAc (3×100 мл). Органические экстракты промывают насыщенным раствором соли (1×50 мл), сушат над Na2SO4, фильтруют, и концентрируют. Остаток очищают хроматографией на силикагеле (50-100% Гексан/ДХМ). Желаемые фракции объединяют и концентрируют, и затем полученное твердое вещество обрабатывают ультразвуком с охлажденным МеОН, фильтруют, и промывают минимальным МеОН с получением трет-бутил (R)-(2-хлор-4-фтор-3-йодфенил)((3-фторпирролидин-1-ил)сульфонил)карбамата (21 г, 75%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,43-7,38 (м, 1Н), 7,07-7,03 (м, 1Н), 5,39-5,23 (м, 1Н), 4,02-3,72 (м, 4Н), 2,36-2,03 (м, 2Н), 1,39 (с, 9Н).
Стадия 2: Получение трет-бутил (R)-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)((3-фторпирролидин-1-ил)сульфонил)карбамата. 6-Амино-5-фтор-3-метилхиназолин-4(3Н)-он (15,7 г, 81 ммоль), трет-бутил (R)-(2-хлор-4-фтор-3-йодфенил)(3-фторпирролидин-1-ил)сульфонил)карбамат (38,5 г, 73,7 ммоль), три(дибензилиденацетон)дипалладий (6,74 г, 7,37 ммоль), (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфан) (10,7 г, 18,4 ммоль) и карбонат цезия (72,0 г, 221 ммоль) суспендируют в толуоле (491 мл). Реакционную смесь продувают аргоном в течение 15 минут и затем нагревают до 100°С под баллоном аргона в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, растворяют в 4:1 ДХМ : ИПС (500 мл) и EtOAc (200 мл) и фильтруют через слой Celite. Celite промывают дополнительным 4:1 ДХМ : ИПС (2×50 мл), и фильтрат концентрируют с получением неочищенного трет-бутил (R)-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)((3-фторпирролидин-1-ил)сульфонил)карбамата (43,3 г, 100%), который применяют непосредственно на следующей стадии. МС (хиад, m/z)=588,1, 590,1 (М+Н).
Стадия 3: Получение (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида. трет-Бутил (R)-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)((3-фторпирролидин-1-ил)сульфонил)карбамат (43,3 г, 73,6 ммоль) растворяют в 1:1 ДХМ : ТФК (150 мл) и перемешивают при температуре окружающей среды в течение 2 часов. Смесь концентрируют, растворяют в 4:1 ДХМ : ИПС и промывают NaHCO3 (2×100 мл) и насыщенным раствором соли (1×50 мл). Органическую фазу промывают 1 N NaOH (3×100 мл), и объединенные водные слои экстрагируют ДХМ (2×50 мл). NaOH водный слой подкисляют 4N HCl до рН 1 и экстрагируют 4:1 ДХМ : ИПС (3×100 мл). ДХМ : ИПС органическую фазу промывают NaHCO3 (1×100 мл) и насыщенным раствором соли (1×50 мл), сушат над Na2SO4, фильтруют, и концентрируют. Полученный остаток очищают хроматографией на силикагеле (0-100% ДХМ/EtOAc) с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида (29,7 г, 83%). 1Н ЯМР (400 МГц, CDCl3) δ 7,90 (с, 1Н), 7,55-7,51 (м, 1Н), 7,40-7,38 (м, 1Н), 7,15-7,11 (т, 1Н), 7,05-7,00 (м, 1Н), 6,79 (с, 1Н), 5,92 (с, 1Н), 5,30-5,15 (м, 1Н), 3,69-3,58 (м, 5Н), 3,52-3,45 (м, 2Н), 2,31-2,21 (м, 1Н), 2,13-1,94 (м, 1Н). МС (хиад, m/z) =488,1, 490,1 (М+Н).
Способ С. Получение кристаллической формы (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамид, формы A. (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамид, полученный способом А или способом В (63,7 г, 131 ммоль) обрабатывают EtOAc (76 мл, 0,83 мг/мл) и смесь перемешивают до растворения. Реакционную смесь перемешивают в течение 1 часа до отверждения, и затем дополнительный EtOAc добавляют для возобновления перемешивания, и смесь оставляют перемешиваться в течение 12 часов при температуре окружающей среды. Раствор разбавляют охлажденным EtOAc (100 мл), фильтруют, и промывают дополнительным охлажденным EtOAc (25 мл) с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида, формы А (52,3 г, 82%) в виде белого твердого вещества, которое характеризуют порошковой рентгеновской дифракцией. Порошковую рентгеновскую дифракцию проводят с применением дифрактометра Rigaku MiniFlex 6G, оборудованного источником излучения Cu. Дифрагированное излучение определяют датчиком D/teX Ultra2. Напряжение и силу тока рентгеновской трубки устанавливают на 40 кВ и 15 мА, соответственно. Данные собирают в угломере MiniFlex при длине волны Cu от 3,0 до 45,0°2-Тэта с применением ширины шага 0,01° и скорости шага 3,00°/мин. Коробку с входной щелью устанавливают на 1,25° и щель, ограничивающую длину, устанавливают на 10 мм. Образец вращают при 10 об/мин во время сбора. Образцы получают, помещая их в силиконовый держатель для образца с низким фоном.
Данные собирают и анализируют с использованием программного обеспечения SmartLab Studio II (версия 4.3.147.0). Программное обеспечение автоматически идентифицирует пики в файле данных PXRD с использованием способа второй производной с последующим ручным выбором пиков. Как правило, выбирают пики с относительной интенсивностью ≥3%. Типичная ошибка, связанная с положением пика кристаллического материала, по данным PXRD, указанная в USP, составляет до +/- 0,2° 2-тета (USP-941). На фигуре 1 показана PXRD картина кристаллической структуры (3R)-N-{2-хлор-4-фтор-3-[(5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино]фенил}-3-фторпирролидин-1-сульфонамида формы А, и в таблице D представлены пики PXRD кристаллической картины кристаллической формы (3R)-N-{2-хлор-4-фтор-3-[(5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино]фенил}-3-фторпирролидин-1-сульфонамида формы А.
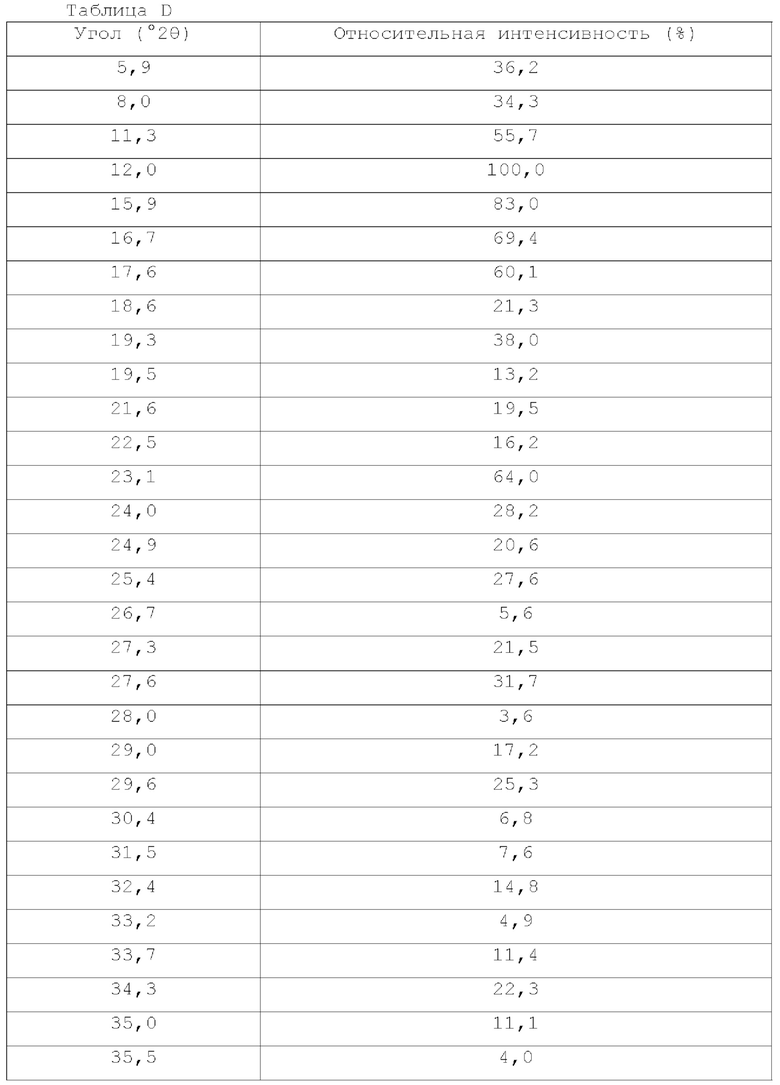
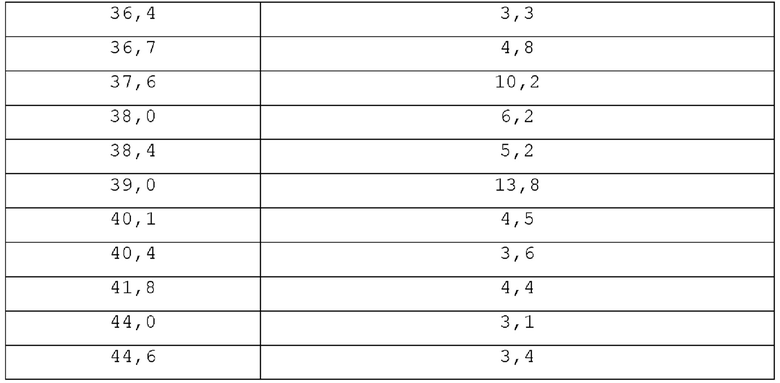
Пример 15
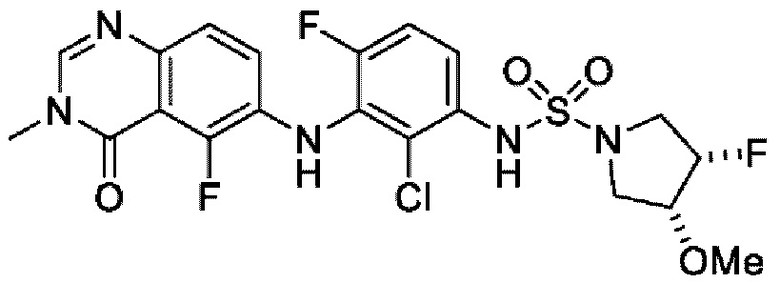
цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-4-метоксипирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,0891 ммоль) и цис-3-фтор-4-метоксипирролидин-1-сульфонилхлорида (97 мг, 0,445 ммоль) в пиридине (0,8 мл) нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc), затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-4-метоксипирролидин-1-сульфонамида (11,2 мг, 24%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,90 (с, 1Н), 7,54-7,46 (м, 1Н), 7,43-7,36 (д, 1Н), 7,18-7,09 (т, 1Н), 7,09-6,99 (м, 1Н), 6,76 (с, 1Н), 5,92 (с, 1Н), 5,23-4,97 (м, 1Н), 3,97-3,82 (м, 1Н), 3,82-3,74 (м, 1Н), 3,70-3,67 (д, 1Н), 3,62-3,58 (м, 1Н), 3,57 (с, 3Н), 3,44 (с, 3Н), 3,35-3,26 (м, 1Н); МС (хиад, m/z)=518,1, 520,1 (М+Н).
Пример 16
цис-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-фтор-4-метоксипирролидин-1-сульфонамид трифторацетат
Стадия 1: Получение цис-3-фтор-4-метоксипирролидин-1-сульфонилхлорида. Суспензию цис-3-фтор-4-метоксипирролидина гидрохлорида (0,834 г, 5,36 ммоль) и N, N-диизопропилэтиламина (1,40 мл, 8,04 ммоль) перемешивают в ДХМ (13 мл) при температуре окружающей среды до полного растворения смеси. Реакционную смесь охлаждают до 0°С и обрабатывают сульфурилхлоридом (1,08 мл, 13,4 ммоль). Холодную баню удаляют через 30 минут, и смесь перемешивают при температуре окружающей среды в течение 18 часов. Реакционную смесь разбавляют дополнительным ДХМ и промывают 1,0 М HCl (1х). Органический слой отделяют, сушат над безводным MgSO4, фильтруют и концентрируют с получением неочищенного цис-3-фтор-4-метоксипирролидин-1-сульфонилхлорида, который используют на следующей стадии как есть.
Стадия 2: Получение цис-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-фтор-4-метоксипирролидин-1-сульфонамида трифторацетата. Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-бром-3-метилхиназолин-4(3Н)-она (35 мг, 0,088 ммоль) и цис-3-фтор-4-метоксипирролидин-1-сульфонилхлорида (48 мг, 0,22 ммоль) в пиридине (0,44 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением цис-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-фтор-4-метоксипирролидин-1-сульфонамида трифторацетата (3,0 мг, 6%). 1H ЯМР (400 МГц, CDCl3) δ 8,17 (с, 1Н), 7,59-7,56 (м, 2Н), 7,16 (т, 1Н), 6,97-6,94 (м, 1Н), 6,78 (с, 1Н), 6,65 (с, 1Н), 5,20-5,04 (м, 1Н), 3,96-3,86 (м, 1Н), 3,81-3,77 (м, 1Н), 3,68-3,67 (м, 1Н), 3,61 (с, 3Н), 3,45 (м, 3Н), 3,32-3,27 (м, 1Н), 2,10-2,05 (м, 2Н). МС (хиад, m/z)=578,0, 580,0 (М+Н).
Пример 17
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-азабицикло[3,1,0]гексан-3-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (35 мг, 0,099 ммоль) и 3-азабицикло[3,1,0]гексан-3-сульфонилхлорида (72 мг, 0,40 ммоль) в пиридине (0,8 мл) герметично закрывают и нагревают при 65°С в течение 60 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Неочищенный продукт очищают колоночной хроматографией на силикагеле (50-100% EtOAc/гексан), затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным NaHCO3. Объединенные органические слои отделяют, сушат над безводным Na2SO4, фильтруют, и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида (34 мг, 69%). 1Н ЯМР (400 МГц, CDCl3) δ 7,93 (с, 1Н), 7,51-7,47 (м, 2Н), 7,17-7,12 (т, 1Н), 6,96-6,93 (м, 1Н), 6,78 (с, 1Н), 6,46 (с, 1Н), 3,57 (с, 3Н), 3,49 (с, 1Н), 3,47 (с, 1Н), 3,40-3,37 (м, 2Н), 1,53-1,50 (м, 2Н), 0,64-0,59 (м, 1Н), 0,21-0,18 (м, 1Н). МС (хиад, m/z)=498,1 (М+Н).
Пример 18
цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-4-метоксипирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (30 мг, 0,0849 ммоль) и цис-3-фтор-4-метоксипирролидин-1-сульфонилхлорида (108 мг, 0,496 ммоль) в пиридине (0,8 мл) нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют, и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc) с получением цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-4-метоксипирролидин-1-сульфонамида (45,4 мг, 21%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,93 (с, 1Н), 7,58-7,53 (м, 1Н), 7,53-7,48 (д, 1Н), 7,18-7,12 (т, 1Н), 6,99-6,92 (м, 1Н), 6,78 (с, 1Н), 6,45 (с, 1Н), 5,20-5,02 (м, 1Н), 3,96-3,85 (м, 1Н), 3,82-3,76 (м, 1Н), 3,71-3,66 (д, 1Н), 3,63-3,59 (т, 1Н), 3,57 (с, 3Н), 3,44 (с, 3Н), 3,35-3,25 (м, 1Н); МС (хиад m/z)=534,1, 536,1 (М+Н).
Пример 19
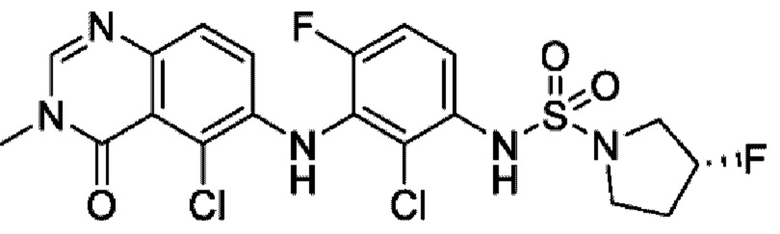
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид
6-((3-Амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-он (152,6 мг, 0,4321 ммоль) растворяют в пиридине (1,8 мл), затем обрабатывают (3R)-3-фторпирролидин-1-сульфонилхлоридом (121,6 мг, 0,6481 ммоль) и нагревают до 60°С в течение 16 часов. Реакционную смесь обрабатывают дополнительным (3R)-3-фторпирролидин-1-сульфонилхлоридом (121,6 мг, 0,6481 ммоль) и перемешивают при 60°С в течение дополнительных 3 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют EtOAc и промывают 10% лимонной кислотой (2х) и насыщенным раствором соли (1х), затем сушат над Na2SO4, фильтруют, и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамида (93,9 мг, 43%). 1H ЯМР (400 МГц, (CD3)2SO) δ 9,56 (с, 1Н), 8,18 (с, 1Н), 7,72 (с, 1Н), 7,50-7,46 (м, 1Н), 7,41-7,33 (м, 2Н), 6,73-6,70 (дд, 1Н), 5,36-5,23 (м, 1Н), 3,47-3,30 (м, 7Н), 2,12-1,97 (м, 2Н). МС (хиад, m/z)=504,0, 506,0 (М+Н).
Пример 20
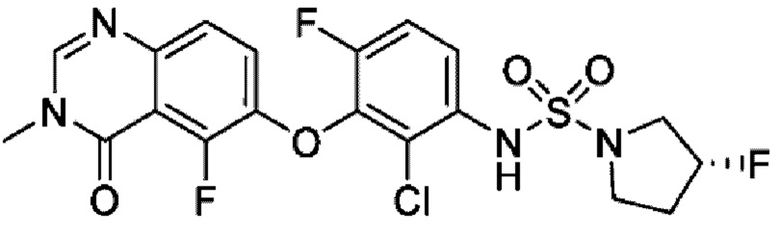
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторпирролидин-1-сульфонамид
К раствору 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,089 ммоль) в пиридине (0,8 мл) добавляют (R)-3-фторпирролидин-1-сульфонилхлорид (67 мг, 0,36 ммоль), и реакционную смесь герметично закрывают и перемешивают при 65°С в течение 48 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Неочищенный продукт очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК), затем колоночной хроматографией на силикагеле (50-100% EtOAc/гексан) с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторпирролидин-1-сульфонамида (28 мг, 64%). 1Н ЯМР (400 МГц, CDCl3) δ 7,96 (с, 1Н), 7,59-7,55 (м, 1Н), 7,43-7,40 (дд, 1Н), 7,24-7,20 (т, 1Н), 7,14-7,09 (т, 1Н), 6,80 (с, 1Н), 5,29-5,14 (м, 1Н), 3,67-3,43 (м, 7Н), 2,30-2,20 (м, 1Н), 2,11-1,92 (м, 1Н). МС (хиад, m/z)=489, 1 (М+Н).
Пример 21
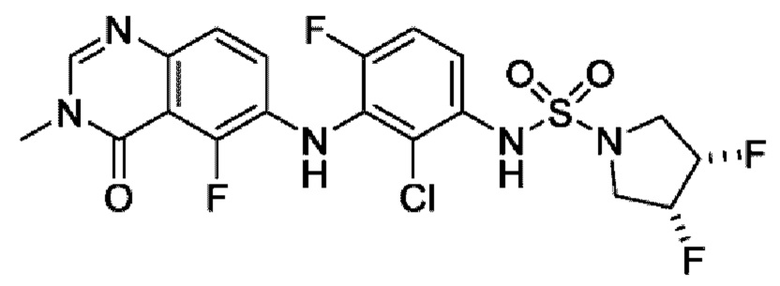
цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,4-дифторпирролидин-1-сульфонамид
Стадия 1: Получение цис-3,4-дифторпирролидин-1-сульфонилхлорида. Суспензию цис-3,4-дифторпирролидина HCl (1,0 г, 6,97 ммоль) и N, N-диизопропилэтиламина (376 мкл, 2,16 ммоль) перемешивают в ДХМ (70 мл) при температуре окружающей среды до полного растворения смеси. Реакционную смесь охлаждают до 0°С и обрабатывают сульфурилхлоридом (1,69 мл, 20,9 ммоль). Ледяную баню удаляют через 30 минут, и смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют дополнительным ДХМ и промывают 1,0 М HCl (3х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением неочищенного цис-3,4-дифторпирролидин-1-сульфонилхлорида (1/21 г, 85%) который применяют непосредственно на следующей стадии.
Стадия 2: Получение цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,4-дифторпирролидин-1-сульфонамида. Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,0891 ммоль) и цис-3,4-дифторпирролидин-1-сульфонилхлорида (92 мг, 0,45 ммоль) в пиридине (0,8 мл) нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc), затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,4-дифторпирролидин-1-сульфонамида (22 мг, 49%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 9,70 (с, 1Н), 8,18 (с, 1Н), 8,05 (с, 1Н), 7,40-7,24 (м, 3Н), 7,05-6,96 (т, 1Н), 5,41-5,15 (м, 2Н), 3,70-3,56 (м, 2Н), 3,48-3,37 (м, 6Н); МС (хиад, m/z)=506,1, 508,1 (М+Н).
Пример 22

цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,4-дифторпирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (30 мг, 0,085 ммоль) и цис-3,4-дифторпирролидин-1-сульфонилхлорида (87 мг, 0,42 ммоль) в пиридине (0,8 мл) нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc), затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,4-дифторпирролидин-1-сульфонамида (22 мг, 50%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) δ 9,73 (с, 1Н), 8,18 (с, 1Н), 7,73 (с, 1Н), 7,49-7,32 (м, 3Н), 6,77-6,70 (м, 1Н), 5,38-5,16 (м, 2Н), 3,68-3,55 (м, 2Н), 3,46-3,33 (м, 5Н); МС (хиад, m/z)=522,0, 524,0 (М+Н).
Пример 23
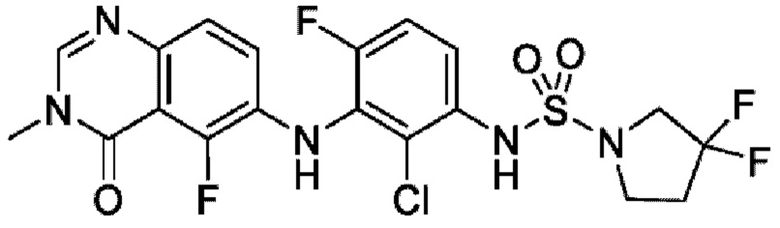
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,3-дифторпирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,089 ммоль) и 3,3-дифторпирролидин-1-сульфонилхлорида (55 мг, 0,27 ммоль) в пиридине (0,5 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 (15 мл) и ДХМ (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,3-дифторпирролидин-1-сульфонамида (8 мг, 18%) в виде белого твердого вещества 1Н ЯМР (400 МГц, CDCl3) δ 7,91 (с, 1Н), 7,51-7,46 (м, 1Н), 7,42-7,38 (м, 1Н), 7,15 (т, 1Н), 7,00 (м, 1Н), 6,75 (с, 1Н), 3,71-3,54 (м, 7Н), 2,44-2,31 (м, 2Н); МС (хиад, m/z)=506,1 (М+Н).
Пример 24
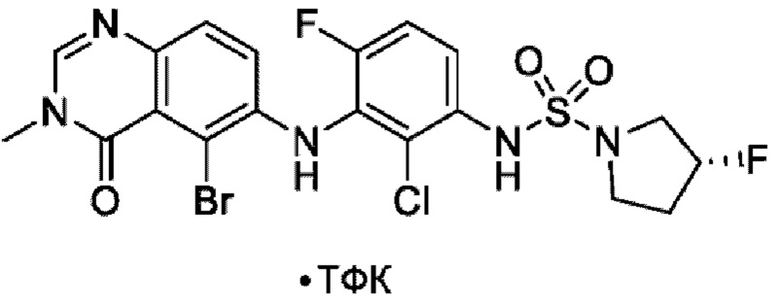
(R)-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-фторпирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-бром-3-метилхиназолин-4(3Н)-она (35 мг, 0,088 ммоль) и (R)-3-фторпирролидин-1-сульфонилхлорида (41 мг, 0,22 ммоль) в пиридине (0,44 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)-3-фторпирролидин-1-сульфонамида трифторацетата (3,0 мг, 6%). 1Н ЯМР (400 МГц, CDCl3) δ 8,09 (с, 1Н), 7,61-7,52 (м, 2Н), 7,16 (т, 1Н), 6,95-6,92 (м, 1Н), 6,78 (с, 1Н), 6,63 (с, 1Н), 5,30-5,15 (м, 1Н), 3,66-3,60 (м, 1Н), 3,60 (с, 3Н), 3,53-3,45 (м, 1Н), 2,35-2,21 (м, 2Н), 2,14-2,00 (м, 2Н). МС (хиад, m/z)=548,0, 550,0 (М+Н).
Пример 25
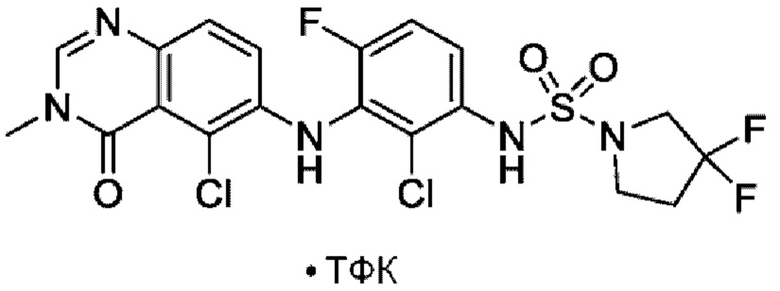
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторпирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (35 мг, 0,099 ммоль) и 3,3-дифторпирролидин-1-сульфонилхлорида (61 мг, 0,30 ммоль) в пиридине (0,50 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторпирролидин-1-сульфонамида трифторацетата (21 мг, 40%). 1H ЯМР (400 МГц, CDCl3) δ 8,14 (с, 1Н), 7,57-7,55 (м, 2Н), 7,18 (т, 1Н), 6,98-6,95 (м, 1Н), 6,77 (с, 1Н), 6,49 (с, 1Н), 3,67-3,63 (м, 2Н), 3,61 (с, 3Н), 3,59-3,54 (м, 2Н), 2,47-2,33 (м, 2Н). МС (хиад, m/z)=522,0, 524,0 (М+Н).
Пример 26
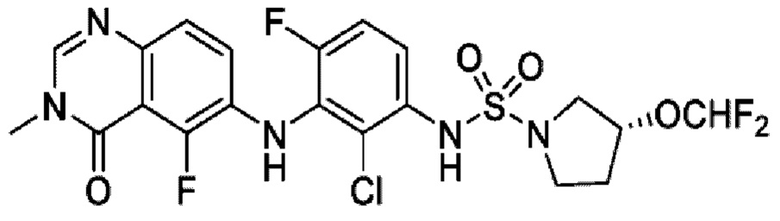
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(дифторметокси)пирролидин-1-сульфонамид
Стадия 1: Получение (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорида. К охлажденному на льду раствору (R)-3-(дифторметокси)пирролидина (107 мг, 0,780 ммоль) и N-этил-N-изопропилпропан-2-амина (204 мкл, 1,17 ммоль) в ДХМ (6 мл) добавляют сульфурилдихлорид (189 мкл, 2,34 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 18 часов. Реакционную смесь разбавляют ДХМ (20 мл) и промывают водным 1М HCl (10 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорида, который применяют непосредственно на следующей стадии без дальнейшей очистки. 1H ЯМР (400 МГц, CDCl3) δ 6,47-6,09 (м, 1Н), 5,00-4,95 (м, 1Н), 3,73-3,58 (м, 4Н), 2,30-2,20 (м, 2Н).
Стадия 2: Получение (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(дифторметокси)пирролидин-1-сульфонамида. Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (50 мг, 0,149 ммоль) и (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорида (174,9 мг, 0,742 ммоль) в пиридине (1 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают хроматографией на силикагеле (гексан/этилацетат 0-100%), затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 (15 мл) и ДХМ (30 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(дифторметокси)пирролидин-1-сульфонамида (28,02 мг, 35%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,99 (с, 1Н), 7,55-7,49 (м, 1Н), 7,43-7,38 (м, 1Н), 7,13 (т, 1Н), 7,08-7,01 (м, 1Н), 6,77 (с, 1Н), 4,87-4,80 (м, 1Н), 3,62-3,43 (м, 7Н), 2,17-2,08 (м, 2Н); МС (хиад m/z)=536,1 (М+Н).
Пример 27
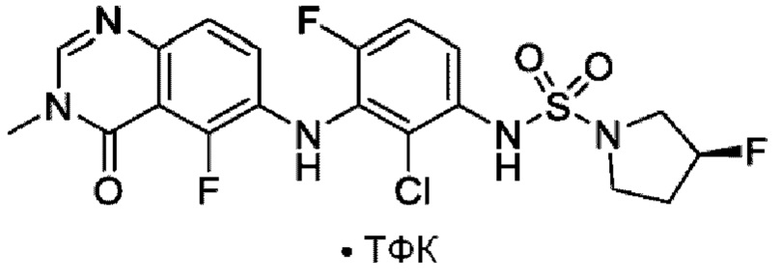
(S)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,09 ммоль) и (S)-3-фторпирролидин-1-сульфонилхлорида (50 мг, 0,3 ммоль) в пиридине (445 мкл, 0,0891 ммоль) нагревают до 70°С в герметично закрытой пробирке в течение 24 часов. Раствор охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт лиофилизируют с получением (S)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида трифторацетата (18,6 мг, 42,8%). 1Н ЯМР (400 МГц, (CD3)2SO) δ 9,54 (с, 1Н), 8,21 (с, 1Н), 8,09 (с, 1Н), 7,42-7,30 (м, 2Н), 7,03-6,99 (т, 1Н), 5,32 (д, 1Н), 3,50-3,30 (м, 7Н), 2,16-1,96 (м, 2Н); МС (хиад m/z)=488,1 (М+Н).
Пример 28
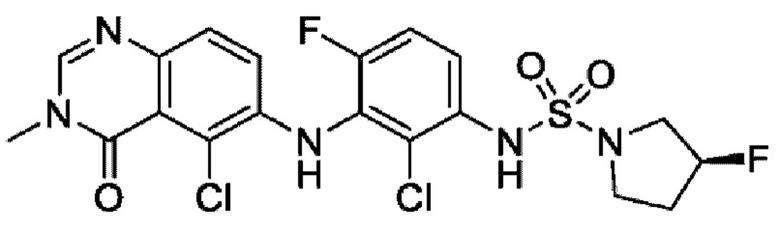
(S)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (30 мг, 0,09 ммоль) и (S)-3-фторпирролидин-1-сульфонилхлорида (48 мг, 0,25 ммоль) в пиридине (425 мкл, 0,085 ммоль) нагревают до 70°С в герметично закрытой пробирке в течение 24 часов. Раствор охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией на силикагеле (1-10% МеОН/ДХМ, 1% NH4OH) с получением (S)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамида (14 мг, 33%). 1Н ЯМР (400 МГц, CD3OD) δ 8,44 (с, 1Н), 7,67-7,63 (м, 1Н), 7,43 (д, 1Н), 7,28-7,23 (т, 1Н), 6,92-6,89 (м, 1Н), 5,31-5,15 (м, 1Н), 3,61-3,39 (м, 7Н), 2,22-2,00 (м, 2Н); МС (хиад m/z)=504,1 (М+Н).
Пример 29
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (52 мг, 0,1472 ммоль) и (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорида (173,5 мг, 0,7362 ммоль) в пиридине (0,800 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (гексан/этилацетат 0-100%), затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 (15 мл) и ДХМ (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамида (27,34 мг, 34%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,93 (с, 1Н), 7,59-7,53 (м, 1Н), 7,50 (д, 1Н), 7,15 (т, 1Н), 6,98-6,94 (м, 1Н), 6,77 (с, 1Н), 6,46 (с, 1Н), 6,35-5,97 (м, 1Н), 4,88-4,81(м, 1Н), 3,63-3,44 (м, 7Н), 2,17-2,10 (м, 2Н); МС (хиад, m/z)=552,0 (М+Н).
Пример 30
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-азабицикло[3,1,0]гексан-3-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,09 ммоль) и 3-азабицикло[3,1,0]гексан-3-сульфонилхлорида (48 мг, 0,27 ммоль) в пиридине (445 мкл, 0,0891 ммоль) нагревают до 70°С в герметично закрытой пробирке в течение 24 часов. Раствор охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт лиофилизируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида трифторацетат (23,5 мг, 54,7%). 1Н ЯМР (400 МГц, (CD3)2SO) δ 9,46 (с, 1Н), 8,19 (с, 1Н), 8,08 (с, 1Н), 7,38-7,30 (м, 3Н), 7,03-6,98 (т, 1Н), 3,44 (с, 3Н), 3,29 (с, 4Н), 1,56-1,52 (м, 2Н), 0,60-0,54 (м, 1Н), 0,16-0,13 (м, 1Н); МС (хиад, m/z)=482,1 (М+Н).
Пример 31
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (100 мг, 0,2694 ммоль) и пирролидин-1-сульфонилхлорида (114,2 мг, 0,6735 ммоль) в пиридине (1,35 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (15 мл) и 10% водным CuSO4 (15 мл). Органическую фазу отделяют, сушат над Mg2SO4, фильтруют и концентрируют. Реакционную смесь очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции собирают и лиофилизируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пирролидин-1-сульфонамида трифторацетата (73,5 мг, 54%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,34 (с, 1Н), 7,61 (д, 1Н), 7,49-7,45 (м, 1Н), 7,06-7,03 (м, 1Н), 6,85 (с, 1Н), 6,54 (с, 1Н), 3,64 (с, 3Н), 3,40-3,36 (м, 4Н), 1,95-1,92 (м, 4Н). МС (хиад, m/z)=504,0 (М+Н).
Пример 32
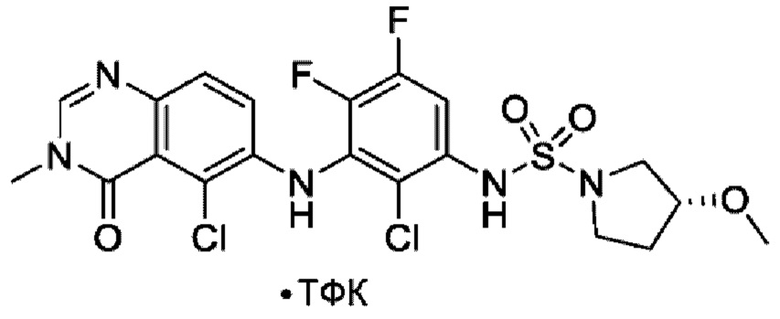
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-метоксипирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (100 мг, 0,269 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (134,5 мг, 0,6735 ммоль) в пиридине (1,35 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-метоксипирролидин-1-сульфонамида трифторацетата (144 мг, 60%). 1Н ЯМР (400 МГц, CDCl3) δ 8,34 (с, 1Н), 7,60 (д, 1Н), 7,53-7,48 (м, 1Н), 7,07-7,04 (м, 2Н), 6,54 (с, 1Н), 4,00-3,98 (м, 1Н), 3,64 (с, 3Н), 3,56-3,43 (м, 4Н), 3,29 (с, 3Н), 2,18-2,12 (м, 1Н), 2,04-1,94 (м, 1Н). МС (хиад, m/z)=534,1 (М+Н).
Пример 33
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фторпирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (100 мг, 0,269 ммоль) и (R)-3-фторпирролидин-1-сульфонилхлорида (126,4 мг, 0,6735 ммоль) в пиридине (1,35 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фторпирролидин-1-сульфонамида трифторацетата (141 мг, 43%). 1Н ЯМР (400 МГц, CDCl3) δ 8,78 (с, 1Н), 7,66 (д, 1Н), 7,58-7,53 (м, 1Н), 7,07-7,05 (м, 1Н), 6,89 (с, 1Н), 6,57 (с, 1Н), 5,33-5,18 (м, 1Н), 3,71 (с, 3Н), 3,68-3,62 (м, 2Н), 3,55-3,46 (м, 2Н), 2,36-2,26 (м, 1Н), 2,18-1,99 (м, 1Н). МС (хиад, m/z)=522,1 (М+Н).
Пример 34
(R)-N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-3-метоксипирролидин-1-сульфонамид
К раствору 6-((3-амино-2,5,6-трифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (15 мг, 0,042 ммоль) в пиридине (211 мкл, 0,042 ммоль) добавляют (R)-3-метоксипирролидин-1-сульфонилхлорид (21 мг, 0,11 ммоль), и реакционную смесь нагревают в герметично закрытой пробирке до 70°С в течение 16 часов. Раствор охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией на силикагеле (1-15% МеОН/ДХМ, 1% NH4OH) с получением (R)-N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-3-метоксипирролидин-1-сульфонамида (12 мг, 55%). 1Н ЯМР (400 МГц, CDCl3) δ 7,95 (с, 1Н), 7,54 (д, 1Н), 7,40-7,33 (м, 1Н), 7,14-7,10 (м, 1Н), 7,04 (шс, 1Н), 6,32 (шс, 1Н), 3,58 (с, 3Н), 3,55-3,37 (м, 5Н), 3,29 (с, 3Н), 1,99-1,93 (м, 2Н); МС (хиад, m/z)=518,1 (М+Н).
Пример 35
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фторпирролидин-1-сульфонамид
Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она (125 мг, 0,35 ммоль) и (R)-3-фторпирролидин-1-сульфонилхлорида (334,3 мг, 1,78 ммоль) в пиридине (3,6 мл) нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc), затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным водным NaHCO3 (1х), и органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фторпирролидин-1-сульфонамида (50 мг, 28%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,95 (с, 1Н), 7,49-7,46 (д, 1Н), 7,32-7,27 (м, 1Н), 7,15-7,13 (м, 1Н), 6,81 (с, 1Н), 5,65 (с, 1Н), 5,32-5,16 (м, 1Н), 3,71-3,47 (м, 7Н), 2,96 (с, 3Н), 2,34-1,98 (м, 2Н); МС (хиад, m/z)=502,1, 504,1 (М+Н).
Пример 36
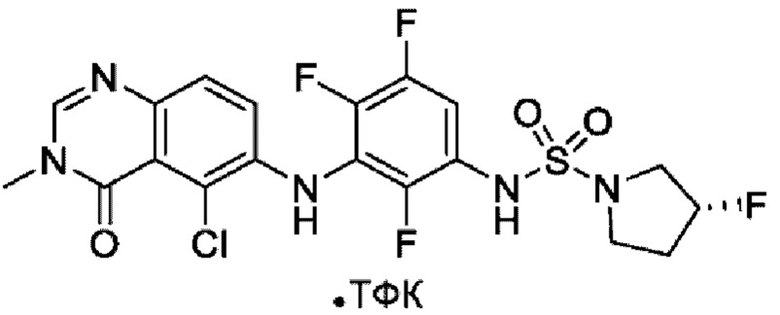
(R)-N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-3-фторпирролидин-1-сульфонамид трифторацетат
К раствору 6-((3-амино-2,5,6-трифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (13 мг, 0,037 ммоль) в пиридине (183 мкл, 0,037 ммоль) добавляют (R)-3-фторпирролидин-1-сульфонилхлорид (17 мг, 0,092 ммоль), и реакционную смесь нагревают в герметично закрытой пробирке до 70°С в течение 6 часов. Раствор охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт лиофилизируют с получением (R)-N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-3-фторпирролидин-1-сульфонамида трифторацетата (5 мг, 27% выход). 1Н ЯМР (400 МГц, CDCl3) δ 8,55 (с, 1Н), 7,64 (д, 1Н), 7,45-7,38 (м, 1Н), 7,16-7,12 (м, 1Н), 6,65 (шс, 1Н), 6,40 (с, 1Н), 5,25 (д, 1Н), 3,69 (с, 3Н), 3,66-3,44 (м, 4Н), 2,35-2,00 (м, 2Н); МС (хиад, m/z)=506,1 (М+Н).
Пример 37
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксипирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (96 мг, 0,2711 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (270,6 мг, 1,355 ммоль) в пиридине (2,2 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды, затем разделяют между ДХМ (75 мл) и 10% водным CuSO4 (75 мл). Органическую фазу сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (гексан/этилацетат 0-100%), затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 (30 мл) и ДХМ (15×3 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксипирролидин-1-сульфонамида (38,53 мг, 27%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,99 (с, 1Н), 7,61-7,56 (м, 1Н), 7,50 (д, 1Н), 7,14 (т, 1Н), 7,04-7,00 (м, 2Н), 3,97-3,92 (м, 1Н), 3,57 (с, 3Н), 3,52-3,37 (м, 4Н), 3,25 (с, 3Н), 2,13-2,04 (м, 1Н), 2,00-1,87 (м, 1Н); МС (хиад, m/z)=517,0 (М+Н).
Пример 38
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (75 мг, 0,2118 ммоль) и (R)-3-фторпирролидин-1-сульфонилхлорида (119,2 мг, 0,6353 ммоль) в пиридине (1,8 мл) нагревают при 70°С в течение 48 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (50 мл) и 10% водным CuSO4 (50 мл). Органическую фазу сушат над Na2SO4, фильтруют затем концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (гексан/этилацетат 0-100%) затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 (15 мл) и ДХМ (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамида (39,6 мг, 37%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,63-7,58k (м, 1Н), 7,50 (д, 1Н), 7,15 (т, 1Н), 7,03-6,99 (д, 1Н), 6,89 (с, 1Н), 5,30-5,13 (м, 1Н), 3,68-3,42 (м, 7Н), 2,32-2,18 (м, 1Н), 2,13-1,93 (м, 1Н); МС (хиад, m/z)=505,0 (М+Н).
Пример 39
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (50 мг, 0,141 ммоль) и пирролидин-1-сульфонилхлорида (120 мг, 0,706 ммоль) в пиридине (1,1 мл) перемешивают при 70°С в течение 48 часов. Реакционную смесь охлаждают до температуры окружающей среды, затем разделяют между ДХМ (50 мл) и 10% водным CuSO4 (50 мл). Органическую фазу сушат над Na2SO4, фильтруют затем концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (гексан/этилацетат 0-100%), затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 (15 мл) и ДХМ (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пирролидин-1-сульфонамида (45,6 мг, 66%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,60-7,55 (м, 1Н), 7,49 (д, 1Н), 7,14 (т, 1Н), 7,00 (д, 1Н), 6,93 (с, 1Н), 3,57 (с, 3Н), 3,36-3,26 (м, 4Н), 1,90-1,80 (м, 4Н); МС (хиад, m/z)=487,0 (М+Н).
Пример 40
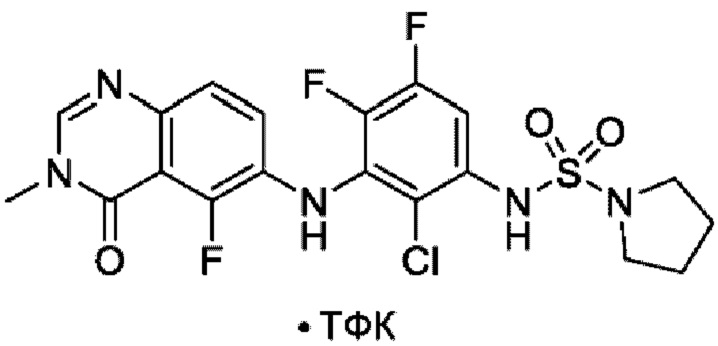
N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (35 мг, 0,09867 ммоль) и пирролидин-1-сульфонилхлорида (41,84 мг, 0,2467 ммоль) в пиридине (0,49 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида трифторацетата (32,4 мг, 67%). 1Н ЯМР (400 МГц, CDCl3) δ 8,55 (с, 1Н), 7,54 (д, 1Н), 7,46-7,41 (м, 1Н), 7,19-7,13 (м, 1Н), 6,85 (с, 1Н), 6,05 (с, 1Н), 3,68 (с, 3Н), 3,40-3,36 (м, 4Н), 1,95-1,92 (м, 4Н). МС (хиад, m/z)=488,1 (М+Н).
Пример 41
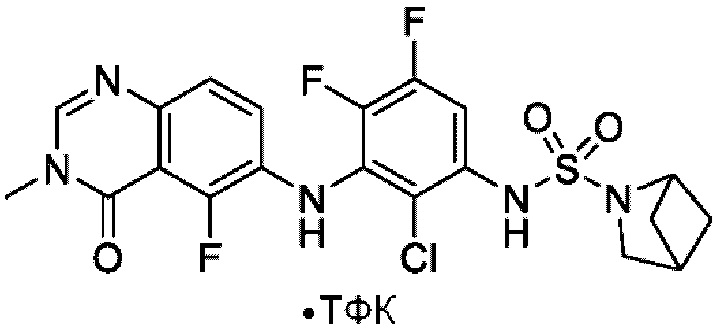
N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (35 мг, 0,09867 ммоль) и 2-азабицикло[2,1,1]гексан-2-сульфонилхлорида (44,80 мг, 0,2467 ммоль) в пиридине (0,49 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида трифторацетата (16,4 мг, 33%). 1Н ЯМР (400 МГц, CDCl3) δ 8,39 (с, 1Н), 7,52-7,46 (м, 2Н), 7,17-7,11 (м, 1Н), 6,88 (с, 1Н), 6,02 (с, 1Н), 4,27 (д, 1Н), 3,65 (с, 3Н), 3,41 (с, 2Н), 2,93-2,90 (м, 1Н), 1,99-1,95 (м, 2Н), 1,56-1,55 (м, 2Н). МС (хиад, m/z)=500,1 (М+Н).
Пример 42
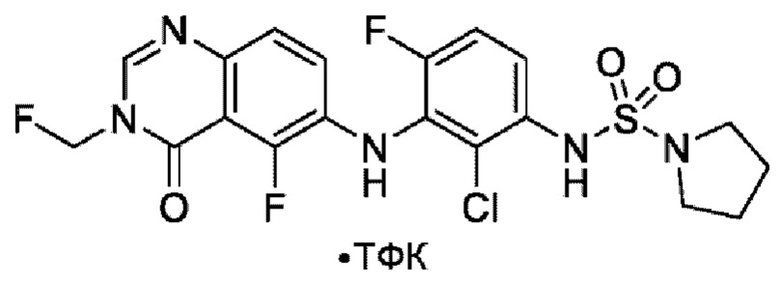
N-(2-хлор-4-фтор-3-((5-фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамид трифторацетат
Стадия 1: Получение 6-бром-5-фтор-3-(фторметил)хиназолин-4(3Н)-она. 6-Бром-5-фторхиназолин-4(3Н)-он (0,512 г, 2,107 ммоль) растворяют в N, N-диметилформамиде (5,26 мл) и обрабатывают йодфторметаном (0,1566 мл, 2,317 ммоль) и карбонатом калия (0,4367 г, 3,160 ммоль). Реакцию выдерживают при температуре окружающей среды в течение 16 часов. Неочищенную реакционную смесь разбавляют ДХМ (30 мл) и фильтруют через Celite®. Фильтрат промывают водой (20 мл). Органический слой собирают, сушат над MgSO4 и концентрируют с получением 6-бром-5-фтор-3-(фторметил)хиназолин-4(3Н)-она, который применяют на следующей стадии без очистки.
Стадия 2: Получение трет-бутил (5-фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)карбамата. 6-Бром-5-фтор-3-(фторметил)хиназолин-4(3Н)-он (500 мг, 1,82 ммоль) растворяют в толуоле (18 мл) и обрабатывают трет-бутилкарбаматом (224 мг, 1,91 ммоль), трис(дибензилиденацетон)диппалладием (0) (83,2 мг, 0,0909 ммоль), Xantphos (158 мг, 0,273 ммоль) и карбонатом цезия (1777 мг, 5,45 ммоль). Раствор продувают аргоном в течение нескольких минут и затем нагревают и перемешивают при 110°С в атмосфере аргона в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (30 мл) и перемешивают в течение 15 минут затем фильтруют через Celite® и концентрируют. Полученный остаток очищают хроматографией на силикагеле (55-95% EtOAc/Гексан) с получением (5-фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)карбамата (270 мг, 47% за две стадии). МС (хиад, m/z)=312,1 (М+Н).
Стадия 3: Получение 6-амино-5-фтор-3-(фторметил)хиназолин-4(3Н)-она. (5-Фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)карбамат (267 мг, 0,858 ммоль) растворяют в 10 мл 1:1 раствора ТФК/ДХМ, и реакционную смесь перемешивают при температуре окружающей среды в течение 15 минут. Растворитель удаляют в вакууме. Полученный остаток растворяют в 4:1 ДХМ/ИПС (10 мл) и промывают насыщенным водным NaHCO3 (10 мл, х3). Органический слой сушат над MgSO4, фильтруют и концентрируют с получением 6-амино-5-фтор-3-(фторметил)хиназолин-4(3Н)-она, который используют без дальнейшей очистки. МС (хиад, m/z)=212,1 (М+Н).
Стадия 4: Получение трет-бутил (2-хлор-4-фтор-3-((5-фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)карбамата. 6-Амино-5-фтор-3-(фторметил)хиназолин-4(3Н)-он (200 мг, 0,947 ммоль) растворяют в толуоле (9 мл) и обрабатывают трет-бутил (2-хлор-4-фтор-3-йодфенил)карбаматом (370 мг, 0,994 ммоль), трис(дибензилиденацетон)диппалладием (0) (43,4 мг, 0,0474 ммоль), Xantphos (82,2 мг, 0,142 ммоль) и карбонатом цезия (926 мг, 2,84 ммоль). Раствор продувают аргоном в течение нескольких минут и затем нагревают и перемешивают при 110°С в атмосфере аргона в течение 6 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и перемешивают в течение 15 минут, затем фильтруют через Celite и концентрируют. Полученный остаток очищают хроматографией на силикагеле (25-75% EtOAc/Гексан) с получением трет-бутил (2-хлор-4-фтор-3-((5-фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)карбамата (332 мг, 75% за две стадии). МС (хиад, m/z)=455,1 (М+Н).
Стадия 5: Получение 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-(фторметил)хиназолин-4(3Н)-она. Трет-бутил (2-хлор-4-фтор-3-((5-фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)карбамат (322 мг, 0,708 ммоль) растворяют в 10 мл 1:1 раствора ТФК/ДХМ, и реакционную смесь перемешивают при температуре окружающей среды в течение 15 минут. Растворитель удаляют в вакууме. Полученный остаток растворяют в 4:1 ДХМ/ИПС (10 мл) и промывают насыщенным водным NaHCO3 (10 мл, х3). Органический слой сушат над MgSO4, фильтруют и концентрируют с получением 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-(фторметил)хиназолин-4(3Н)-она (96 мг, 38%) который используют без дальнейшей очистки. МС (хиад, m/z)=355,1 (М+Н).
Стадия 6: Получение N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)пирролидин-1-сульфонамид трифторацетата. Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-(фторметил)хиназолин-4(3Н)-она (20 мг, 0,05638 ммоль) и пирролидин-1-сульфонилхлорида (23,91 мг, 0,1410 ммоль) в пиридине (0,28 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-4-фтор-3-((5-фтор-3-(фторметил)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида трифторацетата (15,4 мг, 56%). 1Н ЯМР (400 МГц, CDCl3) δ 8,28 (с, 1Н), 7,58-7,54 (м, 1Н), 7,48-7,46 (м, 1Н), 7,16 (т, 1Н), 7,09-7,04 (м, 1Н), 6,77 (с, 1Н), 6,05 (д, 2Н), 6,00 (с, 1Н), 3,37-3,34 (м, 4Н), 1,91-1,88 (м, 4Н). МС (хиад, m/z)=488,1 (М+Н).
Пример 43
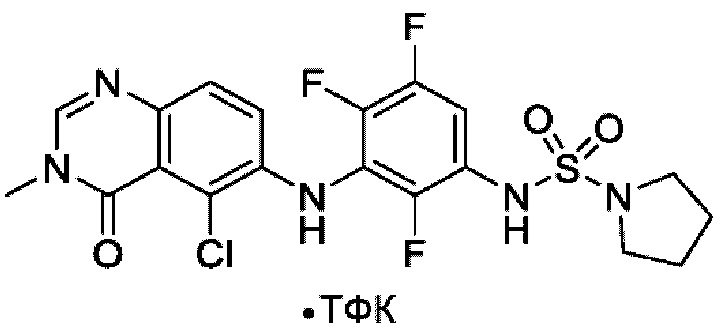
N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)пирролидин-1-сульфонамид трифторацетат
К раствору 6-((3-амино-2,5,6-трифторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (13 мг, 0,037 ммоль) в пиридине (183 мкл, 0,0366 ммоль) добавляют пирролидин-1-сульфонилхлорид (10,5 мкл, 0,0916 ммоль), и реакционную смесь нагревают в герметично закрытой пробирке до 70°С в течение 5 часов. Раствор концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт лиофилизируют с получением N-(3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)пирролидин-1-сульфонамида трифторацетата (9 мг, 50,3%). 1H ЯМР (400 МГц, CDCl3) δ 8,60 (с, 1Н), 7,67 (д, 1Н), 7,39-7,33 (м, 1Н), 7,16-7,12 (м, 1Н), 6,58 (шс, 1Н), 6,42 (с, 1Н), 3,69 (с, 3Н), 3,40-3,36 (м, 4Н), 1,96-1,92 (м, 4Н); МС (хиад, m/z)=488,1 (М+Н).
Пример 44
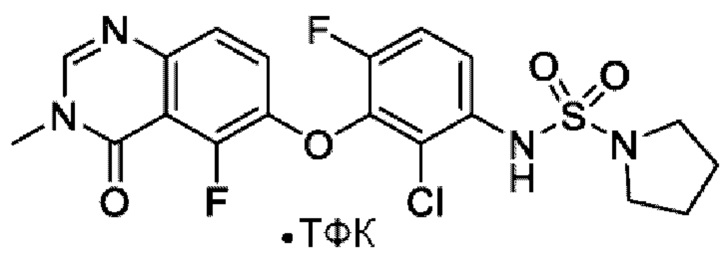
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пирролидин-1-сульфонамид трифторацетат
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (85 мг, 0,2517 ммоль) и пирролидин-1-сульфонилхлорида (106,7 мг, 0,6292 ммоль) в пиридине (1,26 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пирролидин-1-сульфонамида трифторацетата (119 мг, 68%). 1Н ЯМР (400 МГц, CDCl3) δ 8,31 (с, 1Н), 7,60-4,56 (м, 1Н), 7,31-7,27 (м, 1Н), 7,14 (т, 1Н), 6,77 (с, 1Н), 3,63 (с, 3Н), 3,35-3,32 (м, 4Н), 1,91-1,87 (м, 4Н). МС (хиад, m/z)=471,1 (М+Н).
Пример 45
N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)фенил)-(N-этил-N-метил)-сульфамид трифторацетат
Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08457 ммоль) и N-этил-N-метилсульфамоилхлорида (33,32 мг, 0,2114 ммоль) в пиридине (0,42 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)фенил) - (N-этил-N-метил)-сульфамида трифторацетата (26,1 мг, 65%). 1Н ЯМР (400 МГц, CDCl3) δ 8,38 (с, 1Н), 7,52-7,50 (д, 1Н), 7,39-7,34 (м, 1Н), 7,18-7,12 (м, 1Н), 6,78 (с, 1Н), 6,03 (с, 1Н), 3,66 (с, 3Н), 3,34-3,29 (м, 2Н), 2,88 (с, 3Н), 1,20-1,16 (т, 3Н); МС (хиад, m/z)=476,1 (М+Н).
Пример 46
(R)-N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамид трифторацетат. Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08457 ммоль) и (R)-3-фторпирролидин-1-сульфонилхлорида (39,67 мг, 0,2114 ммоль) в пиридине (0,42 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида трифторацетата (24,1 мг, 56%). 1Н ЯМР (400 МГц, CDCl3) δ 8,37 (с, 1Н), 7,52-7,45 (м, 2Н), 7,16-7,11 (м, 1Н), 6,85 (с, 1Н), 6,01 (с, 1Н), 5,32-5,17 (м, 1Н), 3,68-3,62 (м, 5Н), 3,55-3,47 (м, 2Н), 2,35-2,25 (м, 1Н), 2,18-1,98 (м, 1Н). МС (хиад, m/z)=506,1 (М+Н).
Пример 47
(R)-N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксипирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-5,6-дифторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08457 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (42,21 мг, 0,2114 ммоль) в пиридине (0,42 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-4,5-дифтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксипирролидин-1-сульфонамида трифторацетата (16,4 мг, 37%). 1Н ЯМР (400 МГц, CDCl3) δ 8,42 (с, 1Н), 7,53-7,44 (м, 2Н), 7,17-7,12 (м, 1Н), 7,03 (с, 1Н), 6,03 (с, 1Н), 4,00-3,98 (м, 1Н), 3,66 (с, 3Н), 3,59-3,43 (м, 4Н), 3,29 (с, 3Н), 2,18-2,12 (м, 1Н), 2,03-1,94 (м, 1Н). МС (хиад, m/z)=518,1 (М+Н).
Пример 48
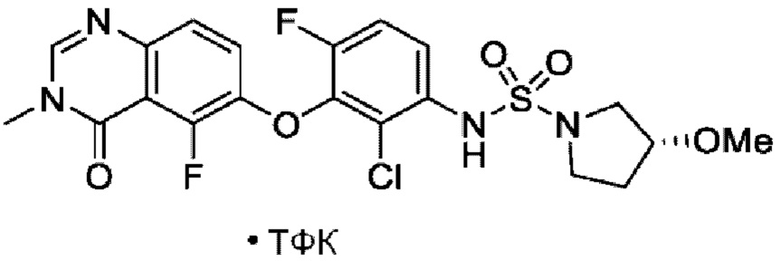
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-метоксипирролидин-1-сульфонамид трифторацетат
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (102 мг, 0,3020 ммоль) и (R)-3-метоксипирролидин-1-сульфонилхлорида (150,8 мг, 0,7551 ммоль) в пиридине (1,51 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4 (15 мл). Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-метоксипирролидин-1-сульфонамида трифторацетата (78,3 мг, 52%). 1Н ЯМР (400 МГц, CDCl3) δ 8,16 (с, 1Н), 7,60-7,57 (м, 1Н), 7,48-7,45 (м, 1Н), 7,27-7,23 (м, 1Н), 7,13 (т, 1Н), 6,94 (с, 1Н), 3,97-3,94 (м, 1Н), 3,61 (с, 3Н), 3,52-3,40 (м, 4Н), 3,27 (с, 3Н) 2,13-2,07 (м, 1Н), 2,00-1,91 (м, 1Н). МС (хиад, m/z)=501,1 (М+Н).
Пример 49
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамид
К раствору 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (15 мг, 0,044 ммоль) в пиридине (222 мкл, 0,044 ммоль) добавляют 2-азабицикло[2,1,1]гексан-2-сульфонилхлорид (24 мг, 0,13 ммоль), и реакционную смесь нагревают в герметично закрытой пробирке до 70°С в течение 16 часов. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт разделяют между дихлорметаном и насыщенным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида (7 мг, 33%). 1Н ЯМР (400 МГц, CDCl3) δ 7,98 (с, 1Н), 7,60-7,56 (м, 1Н), 7,44-7,42 (м, 1Н), 7,27-7,14 (м, 1Н), 7,14-7,10 (т, 1Н), 6,84 (шс, 1Н), 4,24-7,22 (м, 1Н), 3,58 (с, 3Н), 2,89-2,86 (м, 1Н), 2,35 (с, 2Н), 1,94-1,91 (м, 2Н), 1,52-1,50 (м, 2Н); МС (хиад, m/z)=483,1 (М+Н).
Пример 50
N-(2-хлор-3-((5-циано-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид трифторацетат
N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)пирролидин-1-сульфонамид 2,2,2-трифторацетат (60 мг, 0,09305 ммоль) и цианид меди(I) (33,34 мг, 0,3722 ммоль) в N, N-диметилформамиде (0,93 мл) герметично закрывают в пробирке и нагревают до 100°С в течение 4 часов. Реакцию оставляют охлаждаться до температуры окружающей среды. Неочищенный раствор фильтруют через 0,45 мкм шприцевый фильтр и экстрагируют между H2O и 4:1 ДХМ/ИПС. Объединенные органические слои собирают, сушат над MgSO4 и концентрируют. Неочищенную смесь очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК) с получением N-(2-хлор-3-((5-циано-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамида трифторацетата (10,8 мг, 24%). 1Н ЯМР (400 МГц, CDCl3) δ 8,21 (с, 1Н), 7,78 (д, 1Н), 7,68-7,65 (м, 1Н), 7,19 (т, 1Н), 6,78 (с, 1Н), 6,69 (с, 1Н), 3,68 (с, 3Н), 3,37-3,34 (м, 4Н), 1,92-1,89 (м, 4Н). МС (хиад, m/z)=477,1 (М+Н).
Пример 51

N-(2-хлор-4-фтор-3-((3-метил-4-оксо-5-винил-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамид трифторацетат
N-(3-((5-бром-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-хлор-4-фторфенил)пирролидин-1-сульфонамид 2,2,2-трифторацетат (100 мг, 0,155 ммоль) и карбонат цезия (126 мг, 0,388 ммоль) растворяют в 3,2 мл диоксана и 1,0 мл воды. К раствору добавляют пинаколовый циклический эфир винилбороновой кислоты (65,8 мкл, 0,388 ммоль) и продукт присоединения дихлор [1,1'-бис(дифенилфосфино)ферроцен]палладия и дихлорметана (3,80 мг, 0,00465 ммоль). Раствор нагревают до 80°С в течение 16 часов. Реакцию оставляют охлаждаться до температуры окружающей среды. Неочищенный раствор фильтруют через 0,45 мкм шприцевый фильтр и экстрагируют между H2O (5 мл) и ДХМ (5 мл, х2). Объединенные органические слои собирают, сушат над MgSO4 и концентрируют. Неочищенную смесь очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК) с получением N-(2-хлор-4-фтор-3-((3-метил-4-оксо-5-винил-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида трифторацетата (29,0 мг, 39%). 1H ЯМР (400 МГц, CDCl3) δ 8,66 (с, 1Н), 7,63 (д, 1Н), 7,56-7,52 (м, 1Н), 7,41-7,33 (м, 1Н), 7,14 (т, 1Н), 7,00-6,97 (м, 1Н), 6,74 (с, 1Н), 6,63 (с, 1Н), 5,87-5,84 (м, 1Н), 5,66-5,61 (м, 1Н), 3,65 (с, 3Н), 3,37-3,34 (м, 4Н), 1,91-1,88 (м, 4Н). МС (хиад, m/z)=478,1 (М+Н).
Пример 52

N-(2-хлор-3-((5-этил-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид трифторацетат
К раствору N-(2-хлор-4-фтор-3-((3-метил-4-оксо-5-винил-3,4-дигидрохиназолин-6-ил)амино)фенил)пирролидин-1-сульфонамида 2,2,2-трифторацетата (11 мг, 0,0186 ммоль) в метаноле (372 мкл) добавляют палладий на угле (10% масс., 8 мг, 0,00752 ммоль) и реакционную смесь продувают Ar в течение 5 минут, затем перемешивают при температуре окружающей среды под баллоном водорода в течение 30 минут. Реакционную смесь фильтруют через 0,45 мкм шприцевый фильтр, и неочищенный раствор очищают хроматографией с обращенной фазой (5-95 H2O/АЦН, 0,1% ТФК) с получением N-(2-хлор-3-((5-этил-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамида трифторацетата (8,8 мг, 99%). 1Н ЯМР (400 МГц, CDCl3) δ 8,71 (с, 1Н), 7,59 (д, 1Н), 7,50-7,46 (м, 1Н), 7,13 (т, 1Н), 7,05-7,02 (м, 1Н), 6,74 (с, 1Н), 5,84 (с, 1Н), 3,67 (с, 3Н), 3,54-3,48 (м, 2Н), 3,38-3,35 (м, 4Н), 1,92-1,89 (м, 4Н), 1,37 (т, 2Н). МС (хиад, m/z)=480,2 (М+Н).
Пример 53
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метокси-d3)пирролидин-1-сульфонамид трифторацетат
Стадия 1: Получение (3R)-3-(метокси-d3)пирролидина гидрохлорида. Трет-бутил (R)-3-гидроксипирролидин-1-карбоксилат (1,00 г, 5,34 ммоль) растворяют в N, N-диметилформамиде (26,7 мл, 5,34 ммоль) и охлаждают до 0°С. Гидрид натрия (0,214 г, 5,34 ммоль) добавляют, и полученный раствор нагревают до температуры окружающей среды и выдерживают в течение 10 минут. Йодметан-d3 (0,349 мл, 5,61 ммоль) добавляют к реакции и перемешивают при температуре окружающей среды в течение одного часа. Реакцию обрабатывают добавлением 20 мл H2O. Раствор экстрагируют ДХМ (2×20 мл) и органический слой собирают и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (0-10% МеОН в ДХМ). Желаемые фракции собирают и концентрируют. Полученный пирролидин растворяют в ДХМ (10 мл) и добавляют 3 мл 5N HCl в изопропиловом спирте. Реакцию перемешивают при температуре окружающей среды в течение ночи, затем летучие вещества концентрируют с получением желаемого продукта (605 мг, 81%), который используют как есть на следующей стадии.
Стадия 2: Получение (3R)-3-(метокси-d3)пирролидин-1-сульфонилхлорида. Суспензию (3R)-3-(метокси-d3)пирролидина гидрохлорида (0,448 г, 4,30 ммоль) и N-этил-N-изопропилпропан-2-амина (1,12 мл, 6,45 ммоль) перемешивают в ДХМ (14,3 мл) при температуре окружающей среды до полного растворения смеси. Реакционную смесь охлаждают до 0°С и обрабатывают сульфурилхлоридом (0,70 мл, 8,60 ммоль). Смесь нагревают до температуры окружающей среды через 30 минут и оставляют перемешиваться в течение 18 часов. Реакционную смесь разбавляют дополнительным ДХМ (20 мл) и промывают 1,0 М HCl (20 мл). Органический слой отделяют, сушат над безводным MgSO4, фильтруют и концентрируют с получением неочищенного (3R)-3-(метокси-d3)пирролидин-1-сульфонилхлорида, который используют на следующей стадии как есть.
Стадия 3: Получение (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метокси-d3)пирролидин-1-сульфонамида трифторацетата. Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (25 мг, 0,07079 ммоль) и (R)-3-(метокси-d3) пирролидин-1-сульфонилхлорида (28,69 мг, 0,1416 ммоль) в пиридине (0,35 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метокси-d3)пирролидин-1-сульфонамида трифторацетата (18,7 мг, 51%). 1Н ЯМР (400 МГц, CDCl3) δ 8,22 (с, 1Н), 7,61-7,52 (м, 2Н), 7,15 (т, 1Н), 7,00-6,97 (м, 1Н), 6,93 (с, 1Н), 6,49 (с, 1Н), 3,97-3,95 (м, 1Н), 3,62 (с, 3Н), 3,53-3,42 (м, 4Н), 2,14-2,07 (м, 1Н), 2,01-1,92 (м, 1Н). МС (хиад, m/z)=519,1 (М+Н).
Пример 54
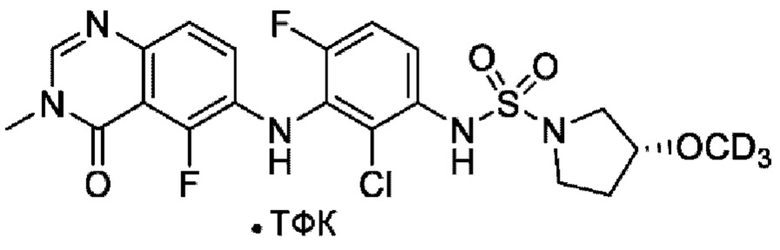
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(метокси-d3)пирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (25 мг, 0,07424 ммоль) и (R)-3-(метокси-d3)пирролидин-1-сульфонилхлорида (30,09 мг, 0,1485 ммоль) в пиридине (0,37 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, фильтруют и концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(метокси-d3)пирролидин-1-сульфонамида трифторацетата (16,5 мг, 44%). 1Н ЯМР (400 МГц, CDCl3) δ 8,31 (с, 1Н), 7,58-7,55 (м, 1Н), 7,48-7,45 (м, 1Н), 7,14 (т, 1Н), 7,09-7,04 (м, 1Н), 6,92 (с, 1Н), 5,97 (с, 1Н), 3,97-3,95 (м, 1Н), 3,64 (с, 3Н), 3,53-3,42 (м, 4Н), 2,13-2,07 (м, 1Н), 2,01-1,92 (м, 1Н); МС (хиад, m/z)=503,1 (М+Н).
Пример 55
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метокси-d3)пирролидин-1-сульфонамид трифторацетат
Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она (25 мг, 0,07513 ммоль) и (R)-3-(метокси-d3)пирролидин-1-сульфонилхлорида (30,45 мг, 0,1503 ммоль) в пиридине (0,37 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют, и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метокси-d3)пирролидин-1-сульфонамида трифторацетата (13,0 мг, 35%). 1Н ЯМР (400 МГц, CDCl3) δ 8,66 (с, 1Н), 7,57 (д, 1Н), 7,51-7,48 (м, 1Н), 7,11 (т, 1Н), 7,09-7,06 (м, 1Н), 6,89 (с, 1Н), 5,66 (с, 1Н), 3,98-3,95 (м, 1Н), 3,66 (с, 3Н), 3,53-3,43 (м, 4Н), 2,96 (с, 3Н), 2,13-2,07 (м, 1Н), 2,02-1,93 (м, 1Н); МС (хиад, m/z)=499,2 (М+Н).
Пример 56
(R)-N-(5-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-3-фторпирролидин-1-сульфонамид трифторацетат
К раствору 6-((3-амино-5-хлор-2-фторфенил)амино)-5-хлор-3-метилхиназолин-4(3Н)-она (20 мг, 0,057 ммоль) в пиридине (283 мкл, 0,057 ммоль) добавляют (R)-3-фторпирролидин-1-сульфонилхлорид (27 мг, 0,14 ммоль), и реакционную смесь нагревают в герметично закрытой пробирке при 70°С в течение 16 часов. Раствор охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт лиофилизируют с получением (R)-N-(5-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-3-фторпирролидин-1-сульфонамида трифторацетата (11 мг, 39%). 1Н ЯМР (400 МГц, CDCl3) δ 8,51 (с, 1Н), 7,70-7,61 (м, 2Н), 7,34-7,32 (м, 1Н), 7,03-7,00 (м, 1Н), 6,72 (шс, 1Н), 6,55 (шс, 1Н), 5,25 (д, 1Н), 3,73-3,49 (м, 7Н), 2,36-2,00 (м, 2Н); МС (хиад, m/z)=504,1 (М+Н).
Пример 57
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамид трифторацетат
К раствору 6-(3-амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-она (30 мг, 0,090 ммоль) в пиридине (449 мкл) добавляют (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорид (64 мг, 0,27 ммоль), и реакционную смесь нагревают в герметично закрытой пробирке при 70°С в течение 16 часов. Раствор охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт лиофилизируют с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамида трифторацетата (10 мг, 21%). 1Н ЯМР (400 МГц, CDCl3) δ 8,59 (с, 1Н), 7,61-7,54 (м, 2Н), 7,20-7,15 (т, 1Н), 6,98 (д, 1Н), 6,81 (шс, 1Н), 6,45-6,00 (м, 1Н), 5,08-4,84 (м, 1Н), 3,85-3,45 (м, 7Н), 3,01 (с, 3Н), 2,28-2,12 (м, 2Н); МС (хиад, m/z)=533,1 (М+Н).
Пример 58
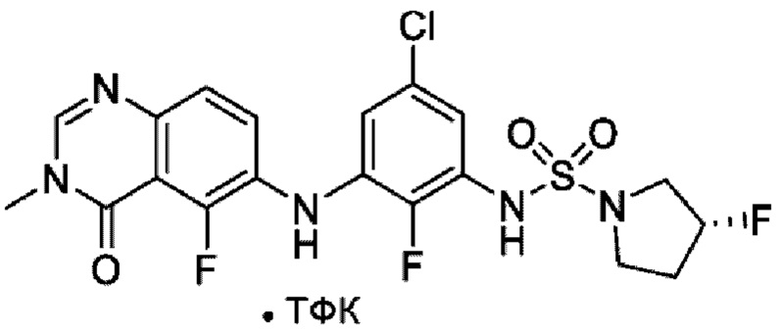
(R)-N-(5-хлор-2-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамид трифторацетат
К раствору 6-((3-амино-5-хлор-2-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (20 мг, 0,059 ммоль) в пиридине (297 мкл, 0,059 ммоль) добавляют (R)-3-фторпирролидин-1-сульфонилхлорид (28 мг, 0,15 ммоль), и реакционную смесь нагревают в герметично закрытой пробирке при 70°С в течение 16 часов. Раствор охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт лиофилизируют с получением (R)-N-(5-хлор-2-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпирролидин-1-сульфонамида трифторацетата (12 мг, 41%). 1Н ЯМР (400 МГц, CDCl3) δ 8,67 (с, 1Н), 7,71-7,61 (м, 2Н), 7,30-7,27 (м, 1Н), 6,98-6,95 (м, 1Н), 6,72 (шс, 1Н), 5,40-5,20 (м, 1Н), 4,00-3,50 (м, 7Н), 2,25-2,00 (м, 2Н); МС (хиад, m/z)=488,1 (М+Н).
Пример 59
цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3,4-дифторпирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (25 мг, 0,071 ммоль) и цис-3,4-дифторпирролидин-1-сульфонилхлорида (73 мг, 0,35 ммоль) в пиридине (0,5 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды затем разделяют между ДХМ (25 мл) и насыщенным водным CuSO4 (25 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (0-100% EtOAc/гексан), затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции разбавляют насыщенным водным NaHCO3 (15 мл) затем экстрагируют ДХМ (30 мл). Органические фазы объединяют и сушат над Na2SO4, фильтруют и концентрируют с получением цис-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3,4-дифторпирролидин-1-сульфонамида (18 мг, 49%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,61-7,56 (м, 1Н), 7,53 (д, 1Н), 7,19 (т, 1Н), 7,03 (д, 1Н), 6,80 (с, 1Н), 5,21-4,98 (м, 2Н), 3,82-3,67 (м, 2Н), 3,65-3,48 (м, 5Н); МС (хиад, m/z)=523,0 (М+Н).
Пример 60
(R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамид
Стадия 1: Получение (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорида. К охлажденному на льду раствору (R)-3-(дифторметокси)пирролидина (107 мг, 0,780 ммоль) и N-этил-N-изопропилпропан-2-амина (204 мкл, 1,17 ммоль) в ДХМ (6 мл) добавляют сульфурилдихлорид (189 мкл, 2,34 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 18 часов. Реакционную смесь разбавляют ДХМ (20 мл) и промывают водным 1М HCl (10 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорида, который применяют непосредственно на следующей стадии без дальнейшей очистки. 1Н ЯМР (400 МГц, CDCl3) δ 6,47-6,09 (м, 1Н), 5,00-4,95 (м, 1Н), 3,73-3,58 (м, 4Н), 2,30-2,20 (м, 2Н).
Стадия 2: Получение (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамида. Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (56 мг, 0,158 ммоль) и (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорида (186 мг, 0,791 ммоль) в пиридине (0,800 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (гексан/этилацетат 0-100%) затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Фракции, содержащие желаемый продукт, объединяют и разделяют между насыщенным водным NaHCO3 (15 мл) и ДХМ (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением (R)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(дифторметокси)пирролидин-1-сульфонамида (13,9 мг, 16%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,63-7,58 (м, 1Н), 7,53 (д, 1Н), 7,17 (т, 1Н), 7,03 (д, 1Н), 6,78 (с, 1Н), 6,39-5,98 (м, 1Н), 4,88-4,81 (м, 1Н), 3,63-3,41 (м, 6Н), 2,21-2,04 (м, 3Н); МС (хиад, m/z)=553,0 (М+Н).
Пример 61
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3,3-дифторпирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (51 мг, 0,144 ммоль) и 3,3-дифторпирролидин-1-сульфонилхлорида (148 мг, 0,720 ммоль) в пиридине (0,500 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (гексан/этилацетат 0-100%), затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 (15 мл) и ДХМ (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3,3-дифторпирролидин-1-сульфонамида (21,2 мг, 28%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 8,05 (с, 1Н), 7,60-7,52 (м, 2Н), 7,20-7,15 (т, 1Н), 7,06-7,03 (д, 1Н), 3,67-3,52 (м, 7Н), 2,42-2,32 (м, 2Н); МС (хиад, m/z)=523,0 (М+Н).
Пример 62
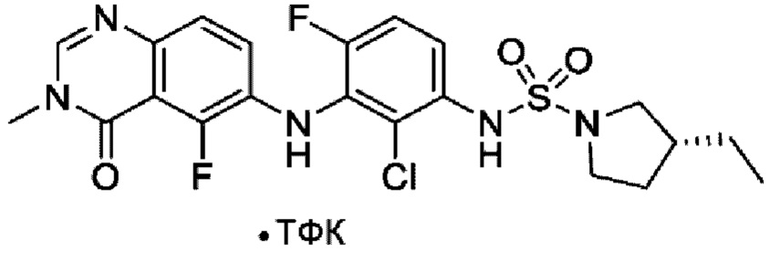
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-этилпирролидин-1-сульфонамид трифторацетат
Стадия 1: Получение (3R)-3-этилпирролидин-1-сульфонилхлорида. Суспензию (R)-3-этилпирролидина гидрохлорида (150 мг, 1,11 ммоль) и N-этил-N-изопропилпропан-2-амина (0,289 мл, 1,66 ммоль) перемешивают в ДХМ (2,7 мл) при температуре окружающей среды до полного растворения смеси. Реакционную смесь охлаждают до 0°С и обрабатывают сульфурилхлоридом (0,179 мл, 2,11 ммоль). Смесь нагревают до температуры окружающей среды через 30 минут и оставляют перемешиваться в течение 18 часов. Реакционную смесь разбавляют дополнительный ДХМ (5 мл) и промывают 1,0 М HCl (5 мл). Органический слой отделяют, сушат над безводным MgSO4, фильтруют и концентрируют с получением неочищенного (3R)-3-этилпирролидин-1-сульфонилхлорида, который используют на следующей стадии как есть.
Стадия 2: Получение (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-этилпирролидин-1-сульфонамида трифторацетата. Раствор 6-((3-амино-2-хлор-6-фторфенил)амино)-5-фтор-3-метилхиназолин-4(3Н)-она (20 мг, 0,05940 ммоль) и (R)-3-этилпирролидин-1-сульфонилхлорида (23,48 мг, 0,1188 ммоль) в пиридине (0,30 мл) герметично закрывают и нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ (15 мл) и промывают 10% водным CuSO4. Экстракт ДХМ сушат над MgSO4, концентрируют и остаток очищают хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-этилпирролидин-1-сульфонамида трифторацетата (18,1 мг, 61%). 1Н ЯМР (400 МГц, CDCl3) δ 8,28 (с, 1Н), 7,56-7,52 (м, 1Н), 7,48-7,45 (м, 1Н), 7,15 (т, 1Н), 7,08-7,03 (м, 1Н), 6,75 (с, 1Н), 5,97 (с, 1Н), 3,64 (с, 3Н), 3,55-3,43 (м, 2Н), 3,34-3,28 (м, 1Н), 2,94-2,89 (м, 1Н), 2,11-2,00 (м, 2Н), 1,40-1,32 (м, 2Н), 0,89 (т, 3Н). МС (хиад, m/z)=498,2 (М+Н).
Пример 63
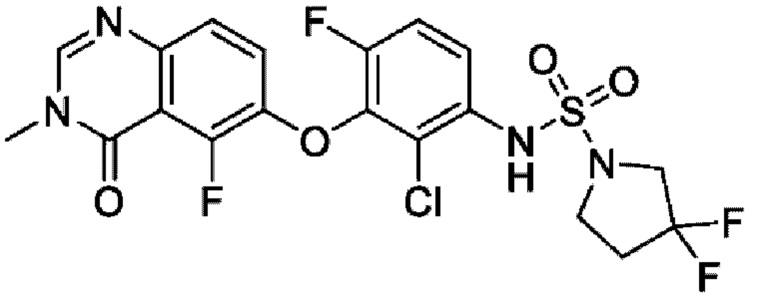
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3,3-дифторпирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08 ммоль) и 3,3-дифторпирролидин-1-сульфонилхлорида (91,3 мг, 0,44 ммоль) растворяют в пиридине (444 мкл, 0,08 ммоль) и нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc) затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным водным NaHCO3 (1х), и органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3,3-дифторпирролидин-1-сульфонамида (18,1 мг, 40%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,98 (с, 1Н), 7,58-7,51 (м, 1Н), 7,48-7,41 (м, 1Н), 7,30-7,21 (м, 1Н), 7,20-7,10 (т, 1Н), 6,75 (с, 1Н), 3,69-3,61 (т, 2Н), 3,60-3,53 (м, 5Н), 2,45-2,30 (м, 2Н); МС (хиад, m/z)=507,1, 509,1 (М+Н).
Пример 64
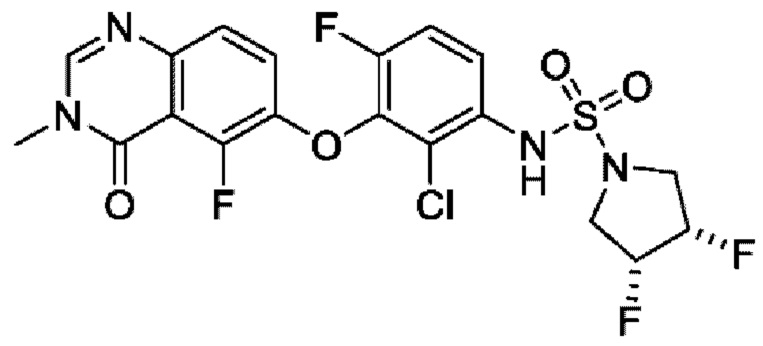
цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3,4-дифторпирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08 ммоль) и цис-3,4-дифторпирролидин-1-сульфонилхлорида (91,3 мг, 0,44 ммоль) растворяют в пиридине (444 мкл, 0,08 ммоль) и нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc) затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным водным NaHCO3 (1х) и органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением цис-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3,4-дифторпирролидин-1-сульфонамида (10,7 мг, 24%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,97 (с, 1Н), 7,57-7,51 (м, 1Н), 7,47-7,43 (м, 1Н), 7,31-7,27 (д, 1Н), 7,18-7,11 (т, 1Н), 6,76 (с, 1Н), 5,21-4,98 (м, 2Н), 3,77-3,53 (м, 7Н); МС (хиад, m/z)=507,1, 509,1 (М+Н).
Пример 65
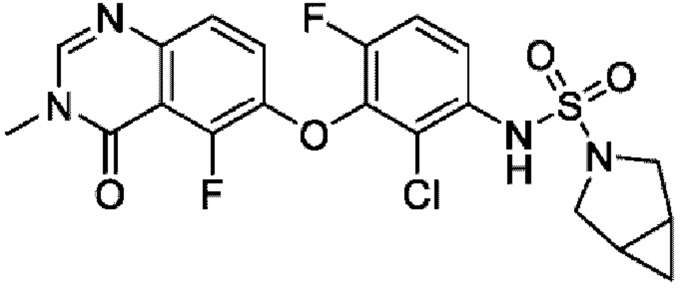
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-азабицикло[3,1,0]гексан-3-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08 ммоль) и 3-азабицикло[3,1,0]гексан-3-сульфонилхлорида (81 мг, 0,44 ммоль) растворяют в пиридине (444 мкл, 0,08 ммоль) и нагревают при 70°С в течение 32 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc) затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным водным NaHCO3 (1х), и органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида (31 мг, 72%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,97 (с, 1Н), 7,53-7,47 (м, 1Н), 7,46-7,42 (м, 1Н), 7,25-7,21 (д, 1Н), 7,15-7,10 (т, 1Н), 6,76 (с, 1Н), 3,57 (с, 3Н), 3,51-3,44 (д, 2Н), 3,42-3,35 (д, 2Н), 0,93-0,81 (м, 2Н), 0,68-0,59 (м, 1Н), 0,23-0,16 (м, 1Н); МС (хиад, m/z)=483,0, 485,0 (М+Н).
Пример 66
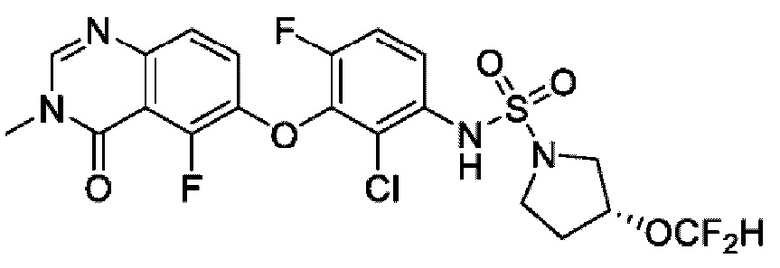
(R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-(дифторметокси)пирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08 ммоль) и (R)-3-(дифторметокси)пирролидин-1-сульфонилхлорида (105 мг, 0,44 ммоль) растворяют в пиридине (444 мкл, 0,08 ммоль) и нагревают при 70°С в течение 24 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют этилацетатом и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (ДХМ/EtOAc), затем хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ : ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением (R)-N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-(дифторметокси)пирролидин-1-сульфонамида (3,5 мг, 7%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ 7,97 (с, 1Н), 7,61-7,55 (м, 1Н), 7,48-7,43 (м, 1Н), 7,30-7,27 (д, 1Н), 7,17-7,09 (т, 1Н), 6,77 (с, 1Н), 6,40-6,00 (т, 1Н), 4,88-4,82 (д, 1Н), 3,59-3,45 (м, 7Н), 2,16-2,10 (м, 2Н); МС (хиад, m/z)=537,0, 539,0 (М+Н).
Пример 67
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-2-азабицикло[2,1,11 гексан-2-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (50 мг и 0,1412 ммоль), 2-азабицикло[2,1,1]гексан-2-сульфонилхлорида (128,2 мг, 0,7059 ммоль) в пиридине (0,700 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (гексан/этилацетат 0-100%), затем хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 (15 мл) и ДХМ (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-2-азабицикло[2,1,1]гексан-2-сульфонамида (18,4 мг, 26%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,99 (с, 1Н), 7,64-7,58 (м, 1Н), 7,53 (д, 1Н), 7,16 (т, 1Н), 7,03 (д, 1Н), 6,82 (с, 1Н), 4,27-4,22 (м, 1Н), 3,58 (с, 3Н),3,38 (с, 2Н), 2,92-2,86 (м, 1Н), 1,98-1,89 (м, 2Н), 1,56-1,50 (м, 2Н); МС (хиад, m/z) = 499,1 (М+Н).
Пример 68
6-(2-Хлор-3-{[этил(метил)сульфамоил]амино}-6-фторфенокси)-3,5-диметил-3,4 дигидрохиназолин-4-он
К раствору 6-(3-амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-она (160,0 мг, 0,48 ммоль) в ДХМ (10 мл) добавляют последовательно пиридин (0,19 мл, 2,39 ммоль), этил (метил) сульфамоилхлорид (378 мг, 2,39 ммоль) и ДМАП (5,8 мг, 0,048 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 48 часов под азотом. Реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (2×50 мл). Объединенные органические слои промывают водой (2×25 мл) и насыщенным раствором соли (25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией с обращенной фазой (30-95% АЦН/вода с 20 мМ бикарбоната аммония) с получением 6-(2-хлор-3-{[этил(метил)сульфамоил]амино}-6-фторфенокси)-3,5-диметил-3,4-дигидрохиназолин-4-она в виде бледно-белого твердого вещества (23 мг, 10%), 1H ЯМР (400 МГц, Метанол-d4) δ 8,18 (с, 1Н), 7,56 (дд, 1Н), 7,41 (д, 1Н), 7,28 (т, 1Н), 6,96 (д, 1Н), 3,54 (с, 3Н), 3,21 (м, 2Н), 2,97 (с, 3Н), 2,82 (с, 3Н), 1,09 (т, 3Н); МС (m/z) = 455,1, 457,1 (М+Н).
Пример 69
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пирролидин-1-сульфонамид
К раствору 6-(3-амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-она (130,0 мг, 0,39 ммоль) в ДХМ (5 мл) добавляют Et3N (0,75 мл, 5,83 ммоль) и пирролидин-1-сульфонилхлорид (790 мг, 4,67 ммоль), и смесь перемешивают в течение 16 часов при температуре окружающей среды под азотом. Реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (2×50 мл). Объединенные органические слои промывают водой (2×25 мл) и насыщенным раствором соли (25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией с обращенной фазой (30-95% АЦН/вода с 20 мМ бикарбоната аммония) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пирролидин-1-сульфонамида в виде белого твердого вещества (23,4 мг, 13%). 1H ЯМР (400 МГц, Метанол-d4) δ 8,18 (с, 1Н), 7,60 (дд, 1Н), 7,41 (д, 1Н), 7,28 (т, 1Н), 6,96 (д, 1Н), 3,54 (с, 3Н), 3,30 (м, 4Н), 2,97 (с, 3Н), 1,85 (м, 4Н); МС (m/z) = 467,3, 469,3 (М+Н).
Пример 70
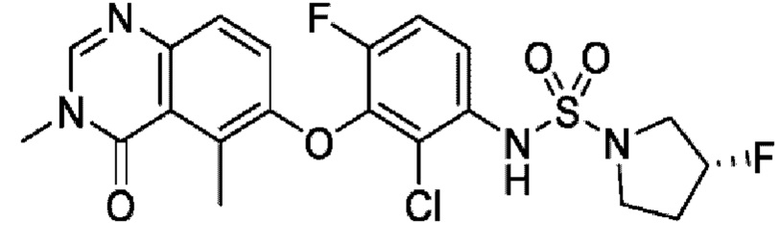
(R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамид
К раствору 6-(3-амино-2-хлор-6-фторфенокси)-3,5-диметилхиназолин-4(3Н)-она (119 мг, 0,357 ммоль) в пиридине (2 мл) добавляют (R)-3-фторпирролидин-1-сульфонилхлорид (147 мг, 0,784 ммоль), и реакционную смесь перемешивают при 65°С в течение 48 часов. Реакционную смесь концентрируют, и неочищенный продукт очищают колоночной хроматографией с обращенной фазой (5-95% MeCN/вода с 0,1% ТФК) с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамида (77 мг, 45%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,95 (с, 1Н), 7,59-7,56 (м, 1Н), 7,41 (д, 1Н), 7,16-7,12 (т, 1Н), 6,91-6,89 (м, 2Н), 5,29-5,14 (м, 1Н), 3,68-3,44 (м, 7Н), 3,00 (с, 3Н), 2,31-2,21 (м, 1Н), 2,13-1,94 (м, 1Н). МС (m/z) = 485,1, 487,1 (М+Н).
Соединения в Таблице 1 получают с применением методики, подобной описанным для синтеза (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксипирролидин-1-сульфонамида (Пример 1) с применением следующих модификаций: на стадии I, замена (R)-3-метоксипирролидина гидрохлорида подходящим амином или гидрохлоридом амина, и на стадии 2, замена 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-она и/или (R)-3-метоксипирролидин-1-сульфонилхлорида подходящим хиназолин-4(3Н)-оном и/или продуктом сульфонилхлорида со стадии 1.
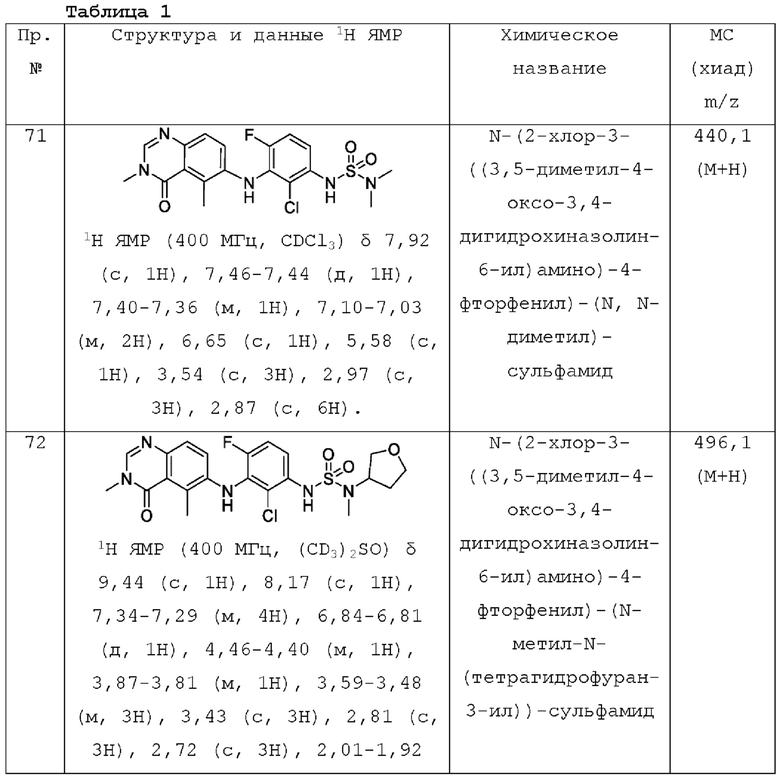
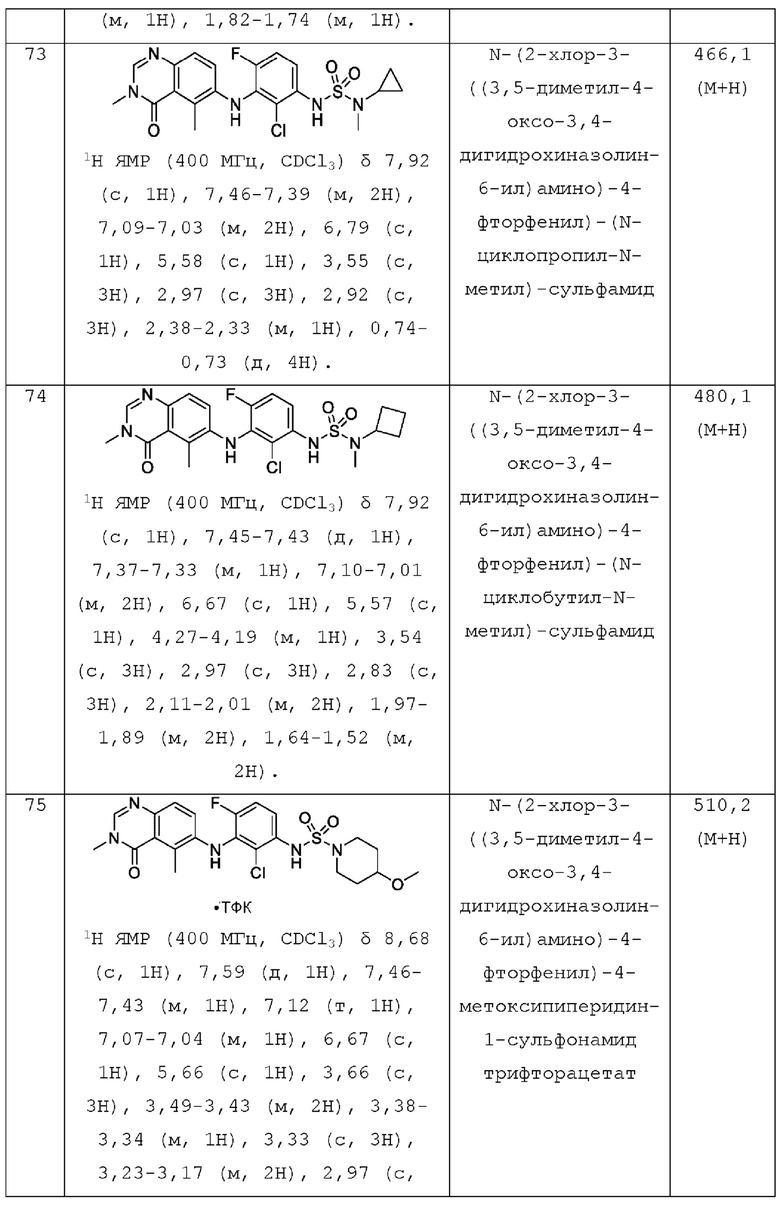
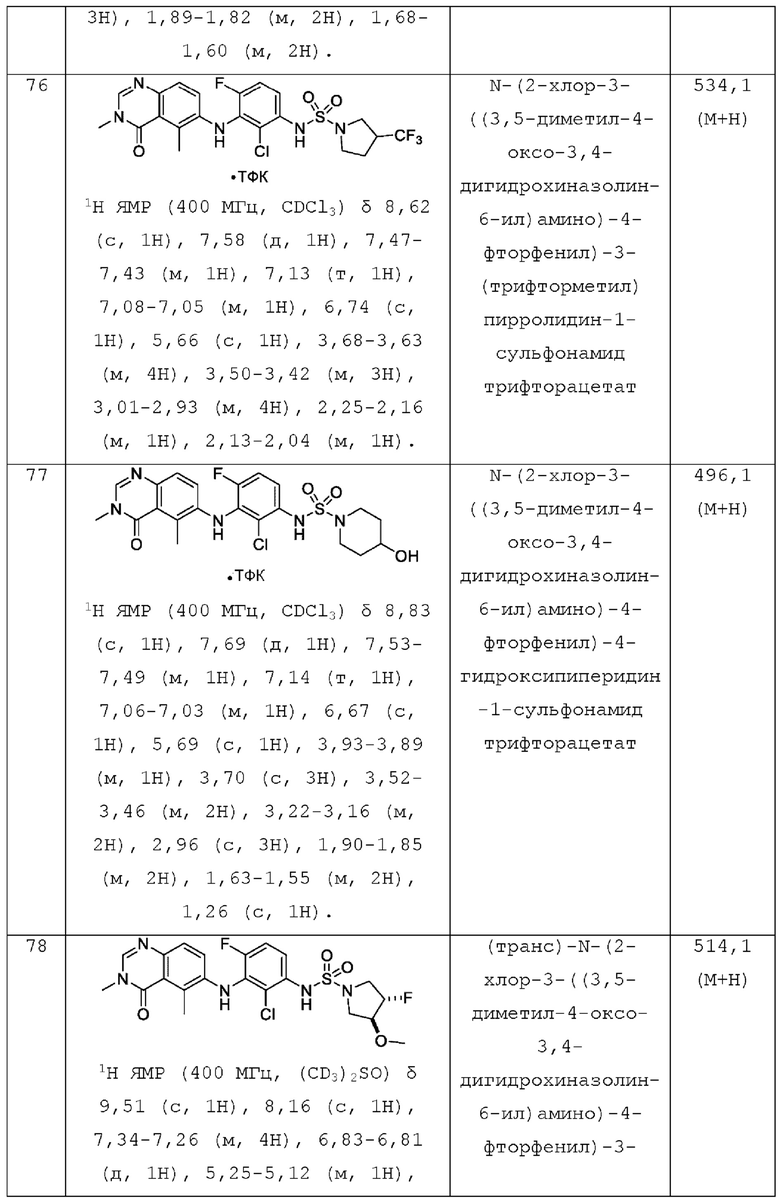
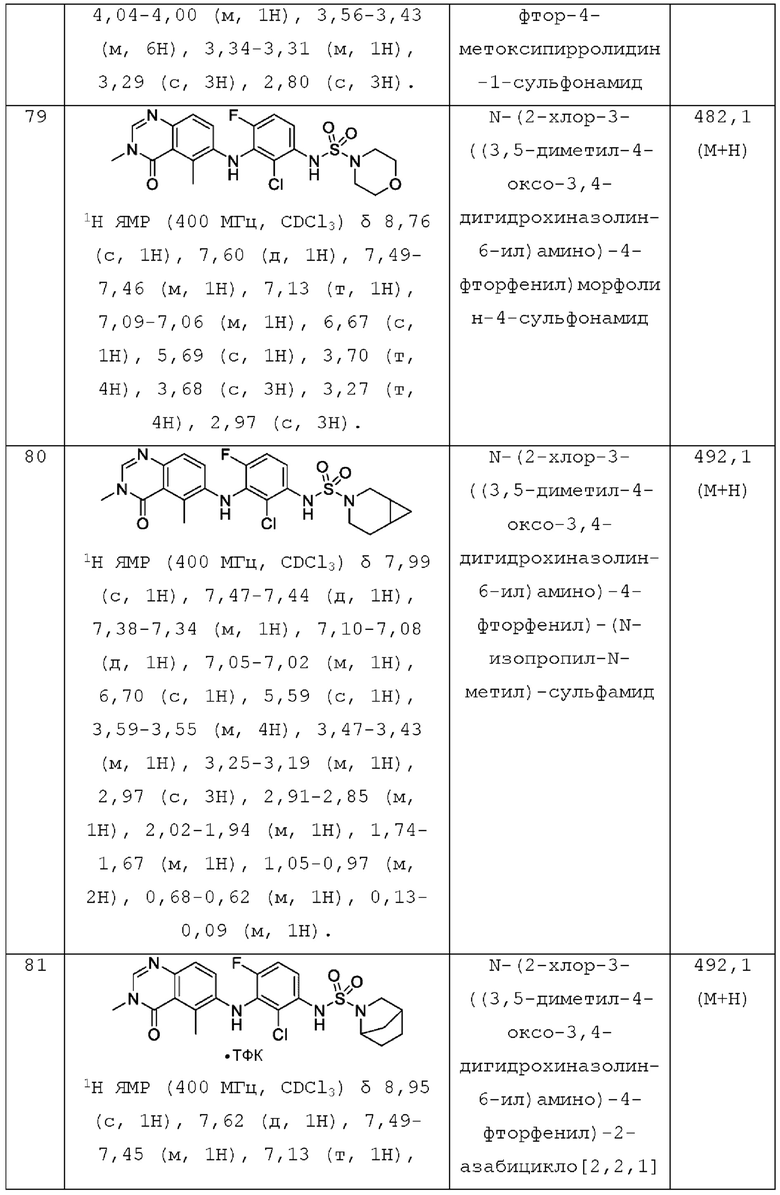
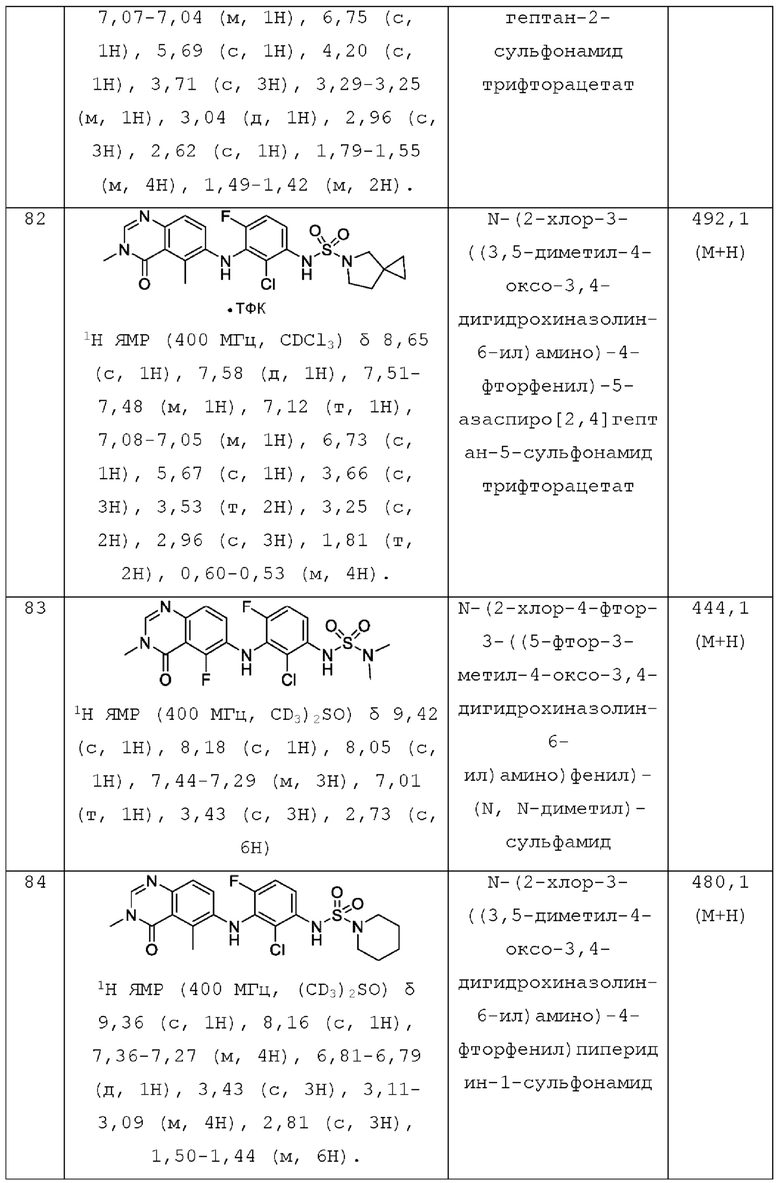
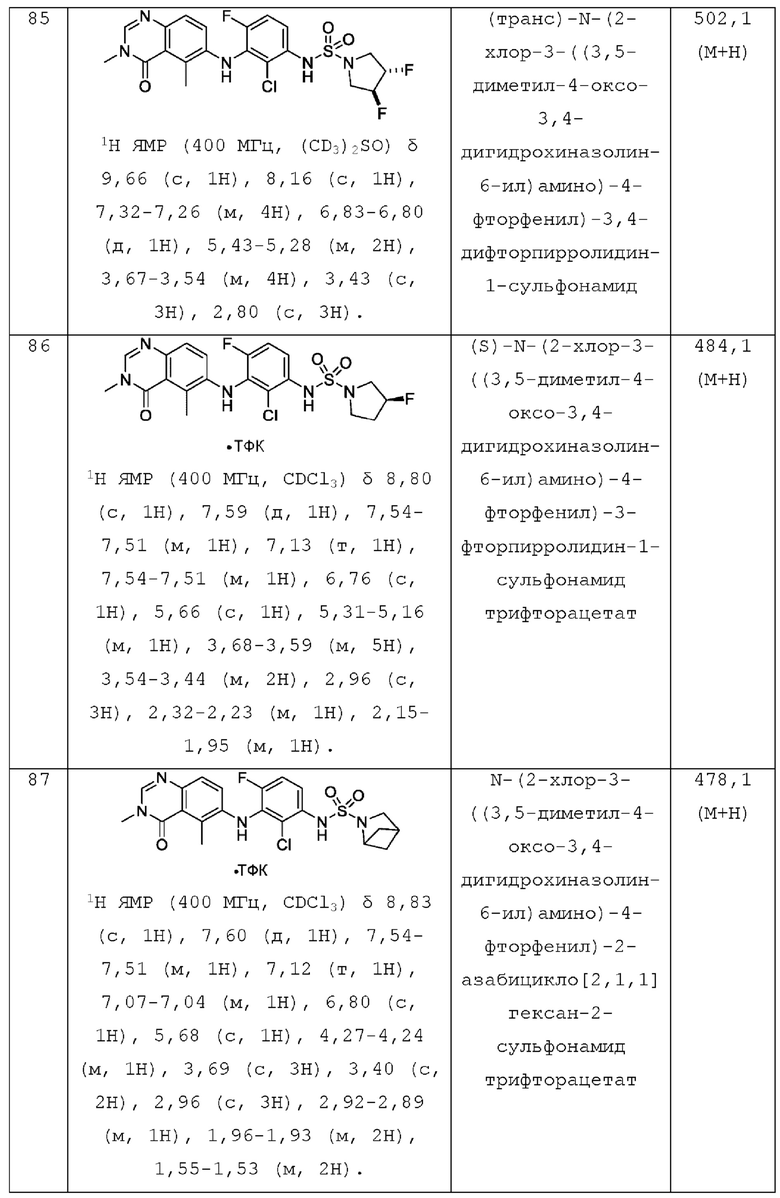
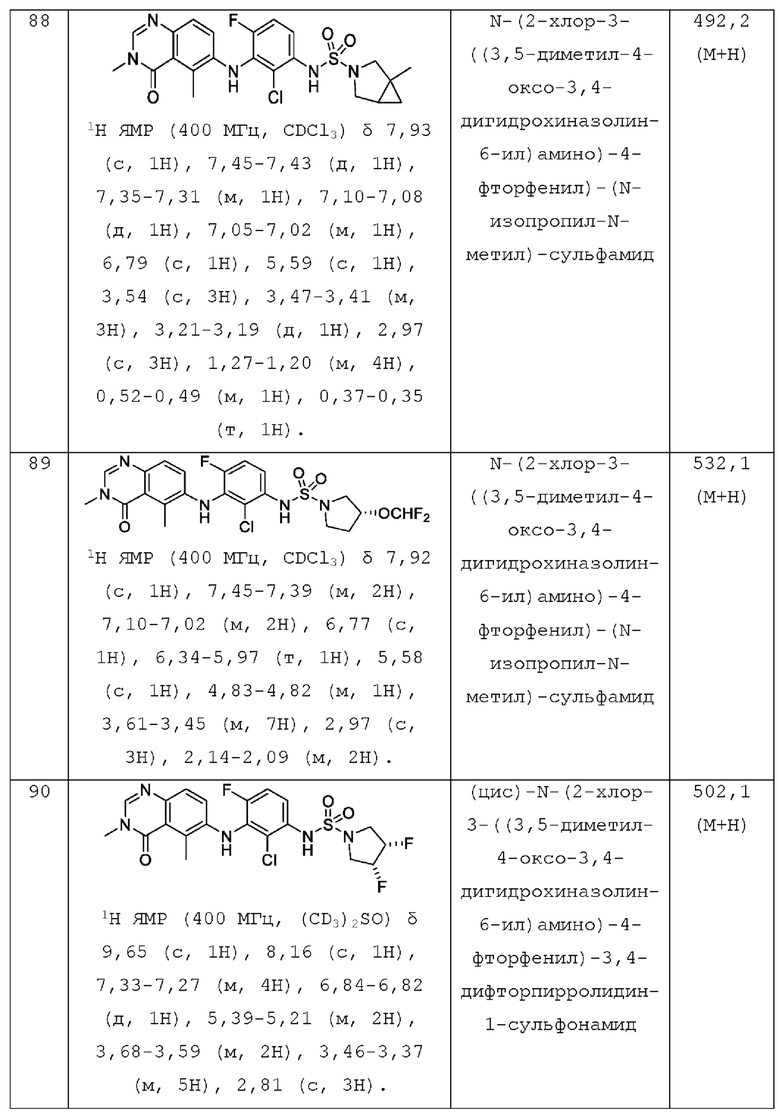
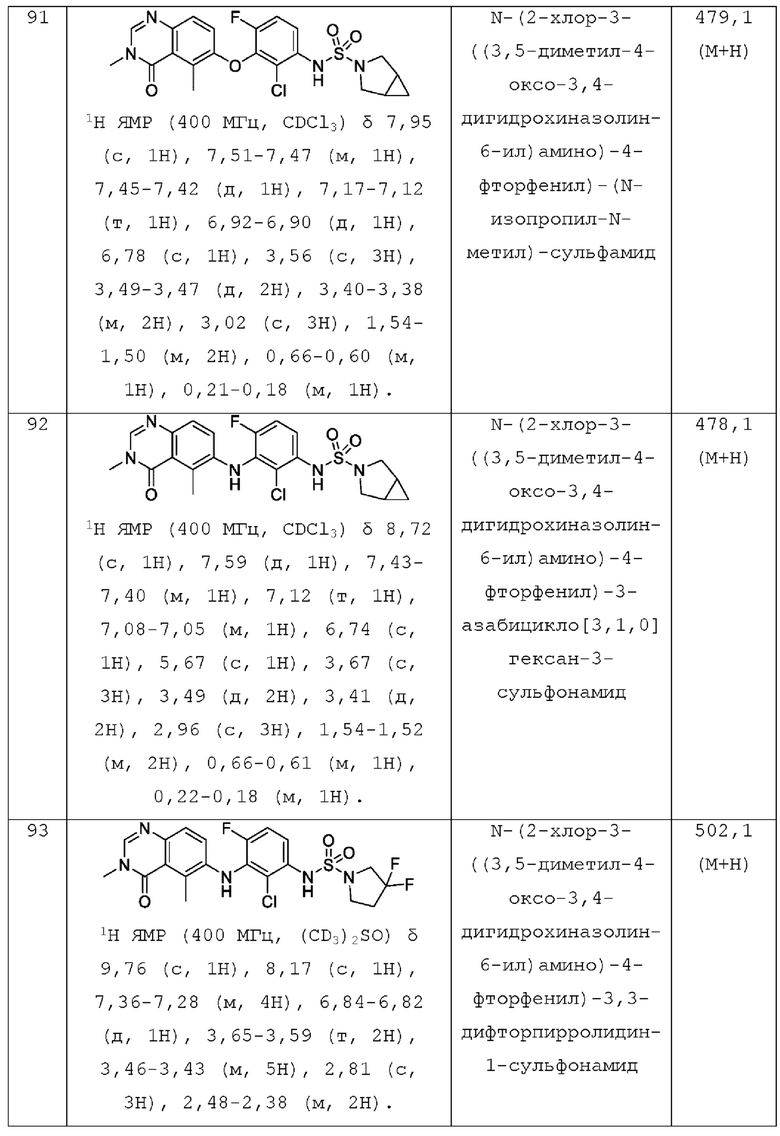
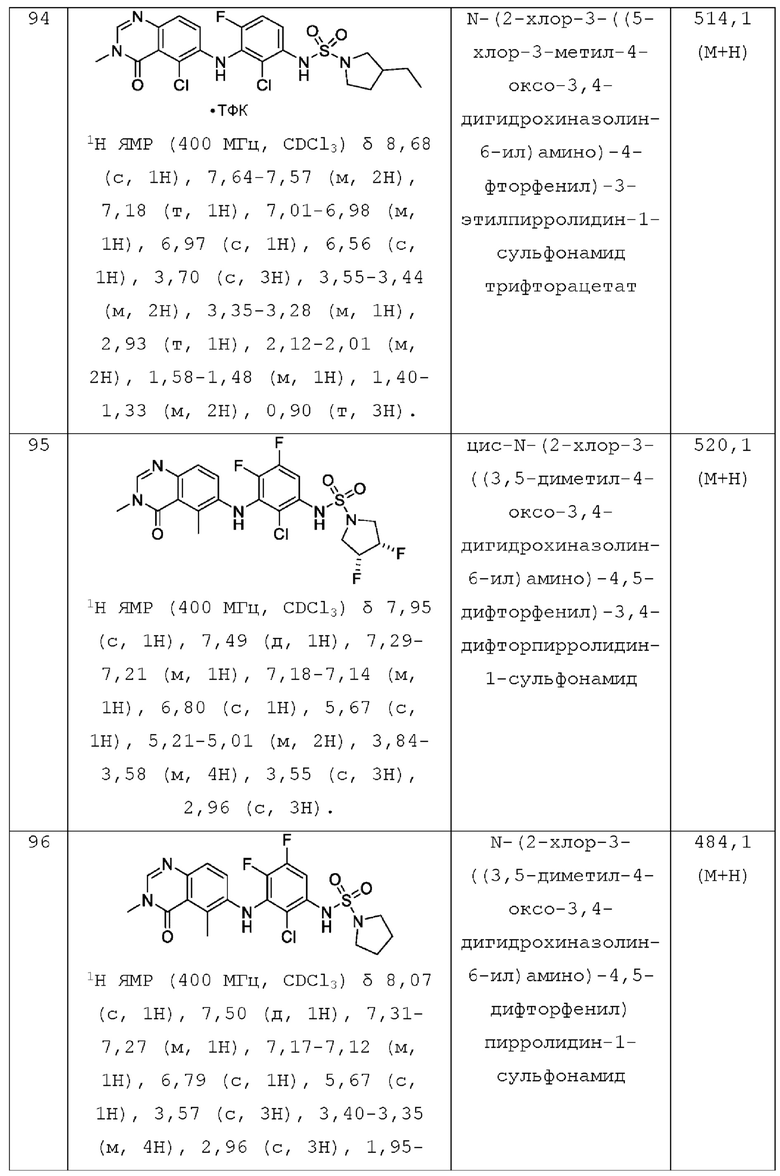
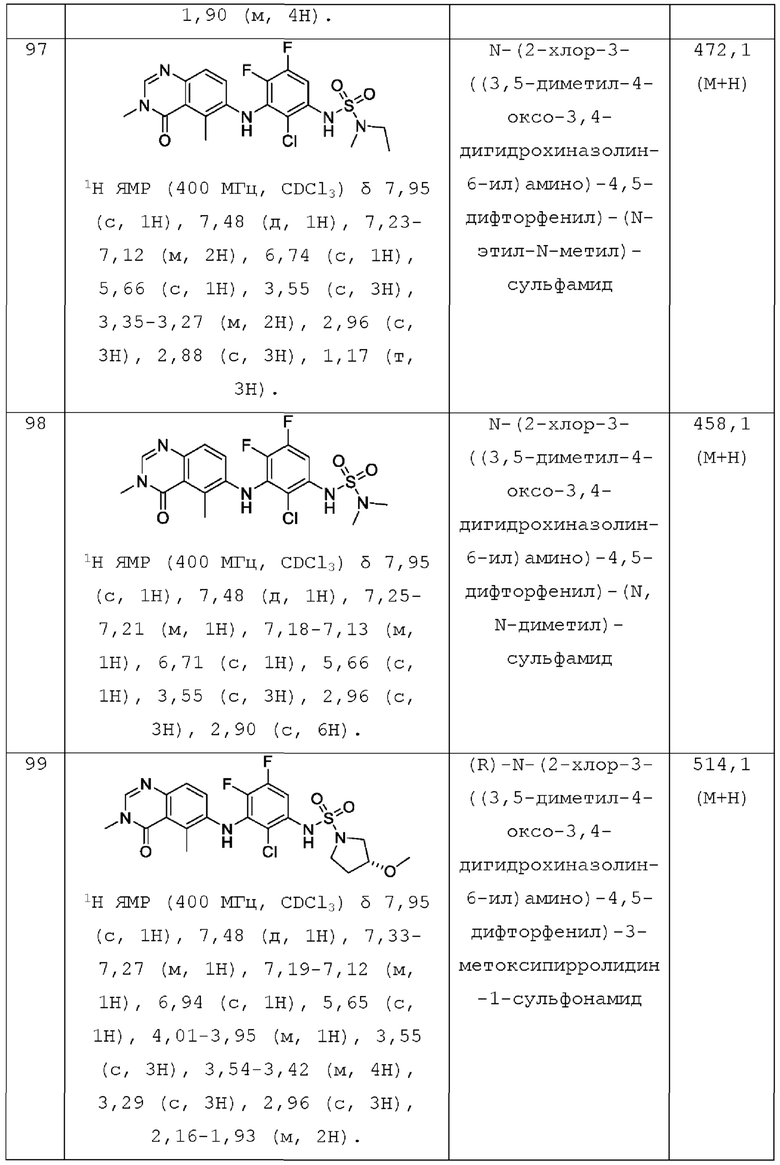
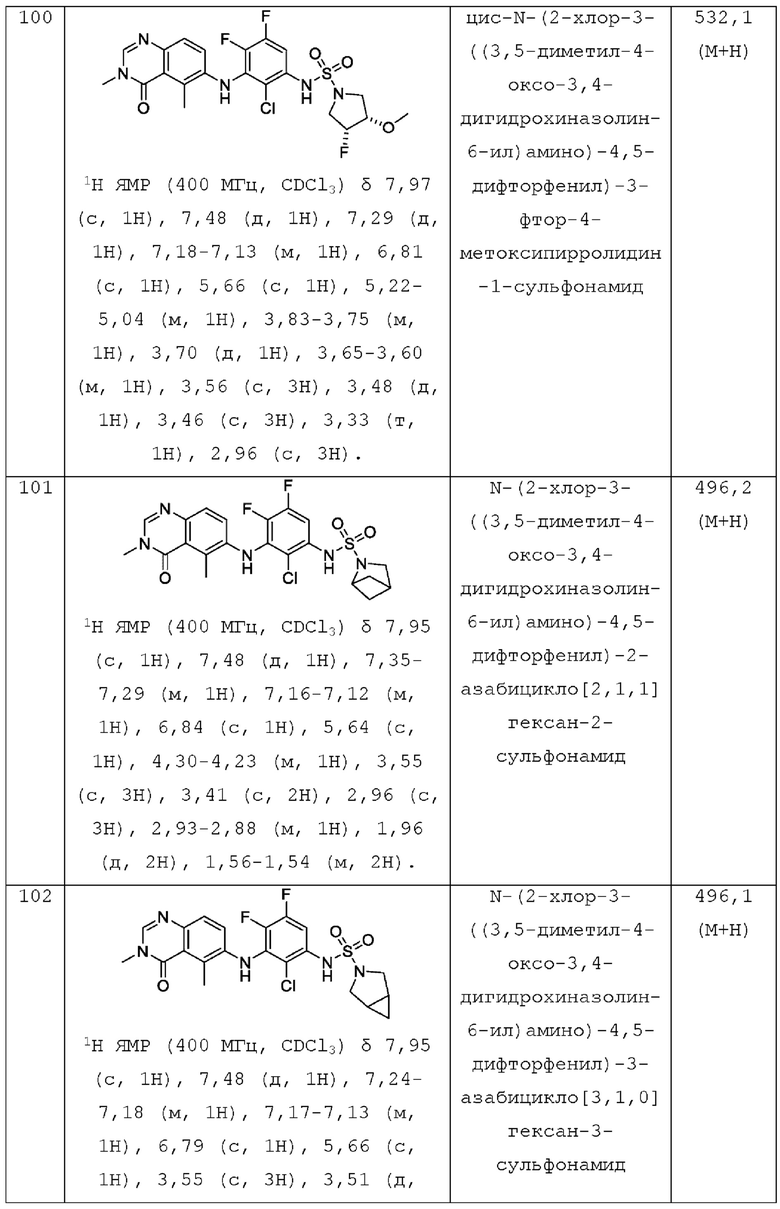
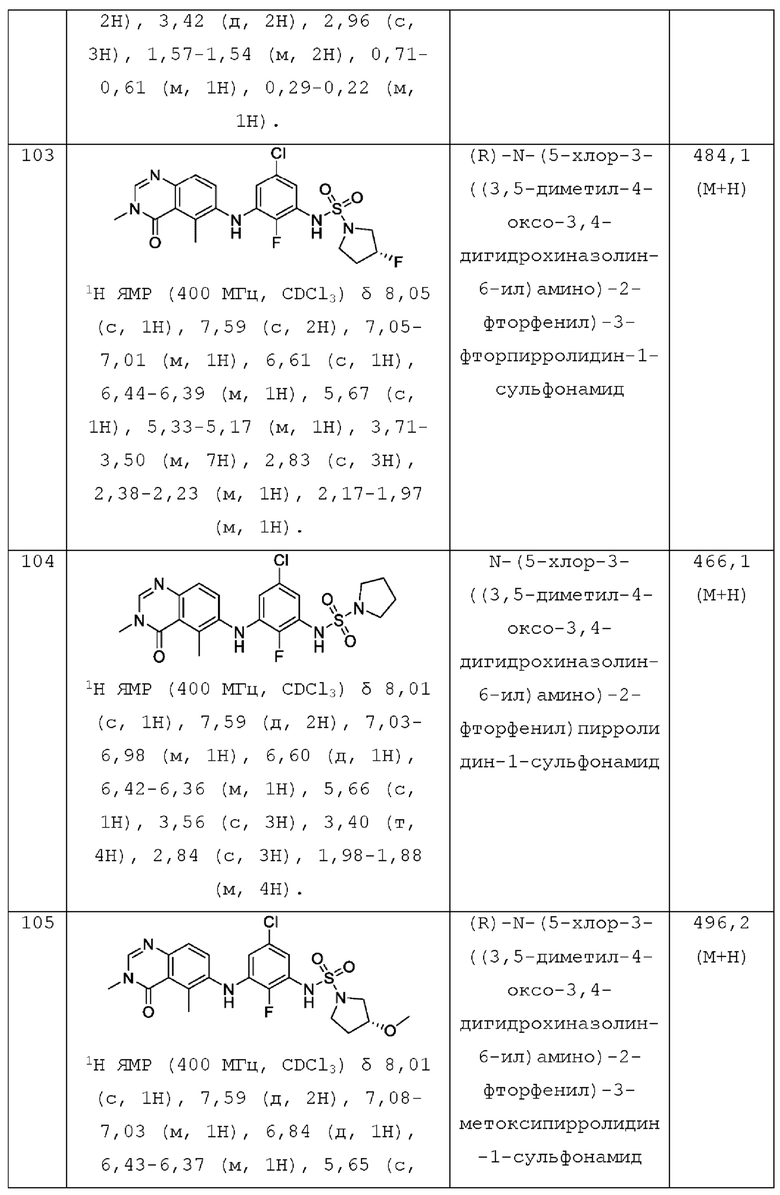
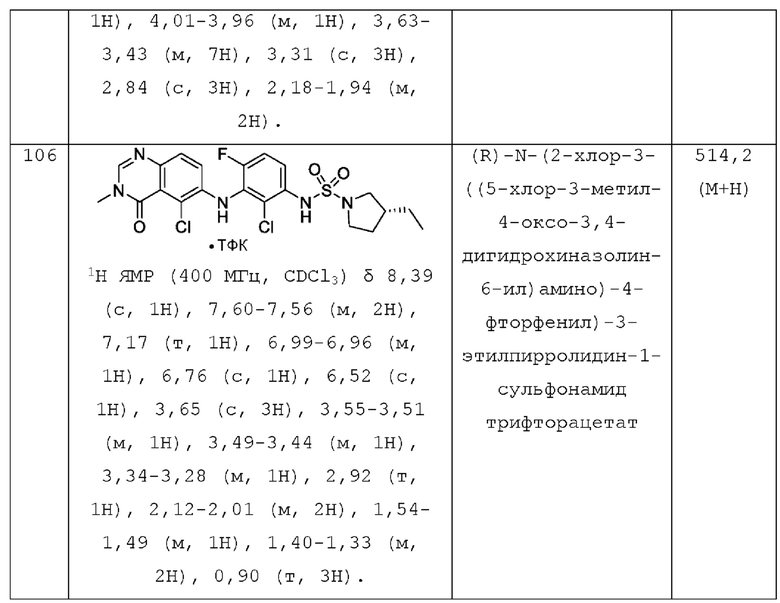
Пример 107
(S)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (51 мг, 0,144 ммоль) и (S)-3-фторпирролидин-1-сульфонилхлорида (135 мг, 0,720 ммоль) в пиридине (0,50 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл), и органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют. Реакционную смесь очищают хроматографией на силикагеле (0-100% гексан/этилацетат), затем хроматографией с обращенной фазой (5-95% MeCN/вода с 0,1% ТФК). Фракции, содержащие желаемый продукт, объединяют и разделяют между насыщенным водным NaHCO3 и ДХМ (30 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют с получением (S)-N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторпирролидин-1-сульфонамида (23,5 мг, 32,3% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,64-7,59 (м, 1Н), 7,52 (д, 1Н), 7,16 (т, 1Н), 7,03 (д, 1Н), 6,79 (с, 1Н), 5,30-5,14 (м, 1Н), 3,70-3,56 (м, 5Н), 3,53-3,43 (м, 2Н), 2,33-2,21 (м, 1Н), 2,15-1,93 (м, 1Н); МС (хиад, m/z) = 505, 0, 507,0 (М+Н).
Пример 108
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-азабицикло[3,1,0]гексан-3-сульфонамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (30 мг, 0,085 ммоль) и 3-азабицикло[3,1,0]гексан-3-сульфонилхлорида (46 мг, 0,25 ммоль) в пиридине (565 мкл, 0,085 ммоль) нагревают до 70°С в течение 16 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт разделяют между дихлорметаном и насыщенным NaHCO3. Органический слой промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением неочищенного продукта. Неочищенный продукт очищают хроматографией на силикагеле (5-95% EtOAc/ДХМ), и продукт разделяют между дихлорметаном и насыщенным NaHCO3. Органический слой промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-азабицикло[3,1,0]гексан-3-сульфонамида (13 мг, 31% выход). 1H ЯМР (400 МГц, CDCl3) δ 9,0 (с, 1Н), 7,55-7,51 (м, 2Н), 7,18-7,14 (т, 1Н), 7,03 (д, 1Н), 6,80 (с, 1Н), 3,57 (с, 3Н), 3,49-3,46 (м, 2Н), 3,40-3,36 (м, 2Н), 1,54-1,50 (м, 2Н), 0,66-0,60 (м, 1Н), 0,22-0,18 (м, 1Н); МС (хиад, m/z) = 499,1, 501,1 (М+Н).
Пример 109
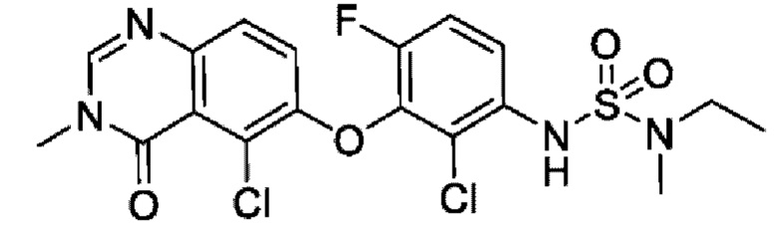
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-(N-этил-N-метил)-сульфамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (50 мг, 0,141 ммоль) и (N-этил-N-метил)сульфамоилхлорида (86,9 мкл, 0,706 ммоль) в пиридине (0,5 мл) нагревают при 70°С в течение 8 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (0-100% гексан/EtOAc), затем ВЭЖХ хроматографией с обращенной фазой (5-95% MeCN/вода с 0,1% ТФК). Желаемые фракции разделяют между насыщенным водным NaHCO3 и ДХМ (20 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-(N-этил-N-метил)-сульфамида (32,3 мг, 48,1% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,56-7,49 (м, 2Н), 7,16 (т, 1Н), 7,03 (д, 1Н), 6,72 (с, 1Н), 3,58 (с, 3Н), 3,26 (м, 2Н), 2,85 (с, 3Н), 1,13 (т, 3Н); МС (хиад, m/z) = 475,1, 477,1 (М+Н).
Пример 110
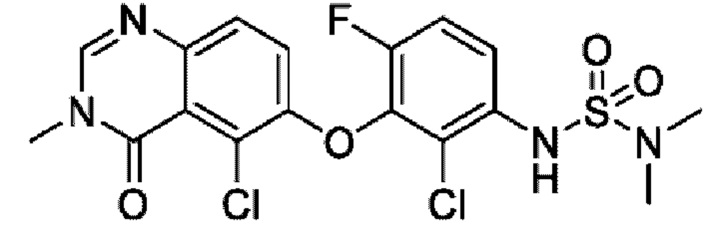
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-(N,N-диметил)-сульфамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-хлор-3-метилхиназолин-4(3Н)-она (50 мг, 0,1412 ммоль) и диметилсульфамоилхлорида (75,30 мкл, 0,7059 ммоль) в пиридине (0,5 мл) нагревают при 70°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между ДХМ (25 мл) и 10% водным CuSO4 (25 мл). Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь очищают хроматографией на силикагеле (0-100% EtOAc/гексан), затем ВЭЖХ хроматографией с обращенной фазой (5-95% АЦН/вода с 0,1% ТФК). Желаемые фракции разделяют между насыщенным водным NaHCO3 и ДХМ (20 мл) и органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-(N,N-диметил)-сульфамида (28,84 мг, 44,28% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,60-7,51 (м, 2Н), 7,16 (т, 1Н), 7,04 (д, 1Н), 6,68 (с, 1Н), 3,58 (с, 3Н), 2,87 (с, 6Н); МС (хиад, m/z) = 461,1, 463,1 (М+Н).
Пример 111
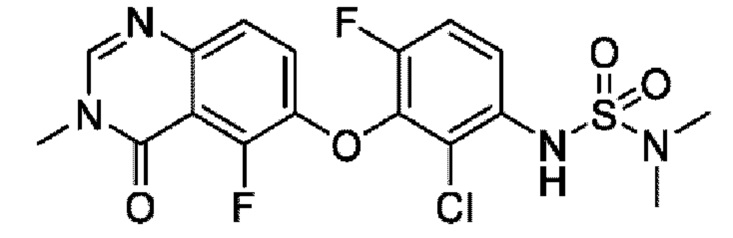
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-(N,N-диметил)-сульфамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08 ммоль) и (N,N-диметил)сульфамоилхлорида (47,7 мкл, 0,44 ммоль) в пиридине (0,88 мл) нагревают при 60°С в течение 3 дней в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют EtOAc и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), затем сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (30-100% EtOAc/ДХМ) с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-(N,N-диметил)-сульфамида (21,3 мг, 54%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,97 (с, 1Н), 7,56-7,51 (м, 1Н), 7,48-7,43 (м, 1Н), 7,29-7,22 (м, 1Н), 7,17-7,08 (т, 1Н), 6,67 (с, 1Н), 3,57 (с, 3Н), 2,86 (с, 6Н); МС (хиад, m/z) = 445,0, 447,1 (М+Н).
Пример 112

N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-(N-этил-N-метил)-сульфамид
Раствор 6-(3-амино-2-хлор-6-фторфенокси)-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,08 ммоль) и (N-этил-N-метил)сульфамоилхлорида (70 мг, 0,44 ммоль) в пиридине (0,88 мл) нагревают при 60°С в течение 3 дней в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют EtOAc и промывают 10% лимонной кислотой (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (30-100% EtOAc/ДХМ), затем хроматографией с обращенной фазой (5-95% MeCN/H2O с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-(N-этил-N-метил)-сульфамида (7,1 мг, 17%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 7,97 (с, 1Н), 7,53-7,48 (м, 1Н), 7,47-4,42 (д, 1Н), 7,25-7,20 (м, 1Н), 7,16-7,08 (т, 1Н), 6,70 (с, 1Н), 3,57 (с, 3Н), 3,31-3,22 (м, 2Н), 2,85 (с, 3Н), 1,15-1,10 (т, 3Н); МС (хиад, m/z) = 459,0, 461,0 (М+Н).
Пример 113
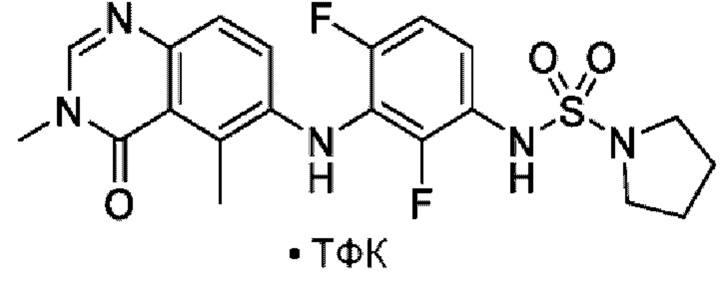
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)пирролидин-1-сульфонамид трифторацетат
6-((3-амино-2,6-дифторфенил)амино)-3,5-диметилхиназолин-4(3Н)-он (22 мг, 0,06955 ммоль), пирролидин-1-сульфонилхлорид (19,93 мкл, 0,1739 ммоль) и пиридин (347,8 мкл, 0,06955 ммоль) добавляют в 3 мл пробирку, и реакцию герметично закрывают и нагревают до 70°С в течение 16 часов. Реакцию охлаждают до температуры окружающей среды и разделяют между ДХМ и насыщенным водным CuSO4. Органический слой концентрируют и полученный остаток очищают ВЭЖХ с обращенной фазой (5-95% MeCN/H2O, 0,1% ТФК). Желаемые фракции лиофилизируют с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)пирролидин-1-сульфонамида трифторацетата (23 мг, 74% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,56 (с, 1Н), 7,58 (д, 1Н), 7,36-7,30 (м, 1Н), 7,16-7,13 (м, 1Н), 7,02-6,97 (м, 1Н), 6,43 (с, 1Н), 5,48 (с, 1Н), 3,64 (с, 3Н), 3,37-3,30 (м, 4Н), 2,95 (с, 3Н), 1,93-1,88 (м, 4Н). МС (хиад, m/z) = 450,1 (М+Н).
Пример 114
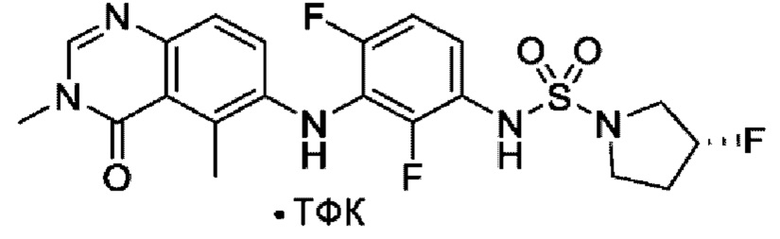
(R)-N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамид трифторацетат
6-((3-Амино-2,6-дифторфенил)амино)-3,5-диметилхиназолин-4(3Н)-он (8,0 мг, 0,0253 ммоль), (R)-3-фторпирролидин-1-сульфонилхлорид (11,9 мг, 0,0632 ммоль) и пиридин (126 мкл, 0,0253 ммоль) добавляют в 3 мл пробирку, и реакцию герметично закрывают и нагревают до 70°С в течение 16 часов. Реакцию охлаждают до температуры окружающей среды и разделяют между ДХМ и насыщенным водным CuSO4. Органический слой концентрируют и неочищенный продукт очищают ВЭЖХ с обращенной фазой (5-95% MeCN/Н2О, 0,1% ТФК). Желаемые фракции лиофилизируют с получением (R)-N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида трифторацетата (4 мг, 32% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,66 (с, 1Н), 7,59 (д, 1Н), 7,40-7,35 (м, 1Н), 7,18-7,15 (м, 1Н), 7,02-6,97 (м, 1Н), 6,44 (с, 1Н), 5,48 (с, 1Н), 5,31-5,17 (м, 1Н), 3,66 (с, 3Н), 3,49-3,10 (м, 2Н), 2,94 (с, 3Н), 2,32-2,22 (м, 2Н), 2,16-1,98 (м, 2Н). МС (хиад, m/z) = 468,1 (М+Н).
Пример 115
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамид
Трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат (30 мг, 0,07 ммоль) растворяют в ТГФ (0,7 мл), охлаждают до 0°С и обрабатывают гидридом натрия (5,5 мг, 0,14 ммоль). Реакцию перемешивают в течение 10 минут и затем обрабатывают 3-(метоксиметил)азетидин-1-сульфонилхлоридом (28 мг, 0,14 ммоль) и затем нагревают до 50°С в течение 12 часов. Реакционную смесь охлаждают до температуры окружающей среды и выливают в 20 мл ледяной воды, и водный слой экстрагируют ДХМ (3×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют, разбавляют ДХМ, промывают насыщенным NaHCO3 (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией с обращенной фазой (5-95% вода/АЦН с 0,1% ТФК) и желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамида (17 мг, 49%) в виде белого твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 9,72 (с, 1Н), 8,28 (с, 1Н), 7,55-7,43 (м, 3Н), 6,98-6,94 (д, 1Н), 3,90-3,83 (т, 2Н), 3,61-3,55 (м, 2Н), 3,46 (с, 3Н), 3,43-3,40 (д, 2Н), 3,23 (с, 3Н), 2,91 (с, 3Н), 2,81-2,70 (м, 1Н); МС (хиад, m/z) = 497,1, 499,1 (М+Н).
Пример 116
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторазетидин-1-сульфонамид
Трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат (30 мг, 0,07 ммоль) растворяют в ТГФ (0,7 мл), охлаждают до 0°С и обрабатывают гидридом натрия (5,5 мг, 0,14 ммоль). Реакцию перемешивают в течение 10 минут и затем обрабатывают 3-фторазетидин-1-сульфонилхлоридом (24 мг, 0,14 ммоль) и нагревают до 50°С в течение 12 часов. Реакционную смесь охлаждают до температуры окружающей среды и выливают в 20 мл ледяной воды. Водный слой экстрагируют 4:1 ДХМ/ИПС (3×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакцию концентрируют, разбавляют ДХМ, промывают насыщенным NaHCO3 (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией с обращенной фазой (5-95% вода/АЦН с 0,1% ТФК) и желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3. Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторазетидин-1-сульфонамида (13 мг, 40%) в виде белого твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 9,96 (с, 1Н), 8,28 (с, 1Н), 7,52-7,42 (м, 3Н), 6,99-6,94 (д, 1Н), 5,46-5,24 (м, 1Н), 4,20-4,08 (м, 2Н), 4,01-3,89 (м, 2Н), 3,46 (с, 3Н), 2,91 (с, 3Н); МС (хиад, m/z) = 471,0, 473,0 (М+Н).
Пример 117
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксиазетидин-1-сульфонамид
Трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамат (30 мг, 0,07 ммоль) растворяют в ТГФ (0,7 мл), охлаждают до 0°С и обрабатывают гидридом натрия (5,5 мг, 0,14 ммоль). Реакционную смесь перемешивают в течение 10 минут и обрабатывают 3-метоксиазетидин-1-сульфонилхлоридом (26 мг, 0,14 ммоль) и нагревают до 50°С в течение 24 часов. Реакционную смесь охлаждают до температуры окружающей среды и выливают в 20 мл ледяной воды. Водный слой экстрагируют ДХМ (2×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакцию концентрируют, разбавляют ДХМ, промывают насыщенным NaHCO3 (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией с обращенной фазой (5-95% вода/АЦН с 0,1% ТФК), и желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксиазетидин-1-сульфонамида (13 мг, 39%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 9,82 (с, 1Н), 8,28 (с, 1Н), 7,54-7,41 (м, 3Н), 7,00-6,94 (д, 1Н), 4,18-4,10 (м, 1Н), 4,00-3,95 (м, 2Н), 3,73-3,67 (м, 2Н), 3,46 (с, 3Н), 3,18 (с, 3Н), 2,91 (с, 3Н); МС (хиад, m/z) = 483,1, 485,1 (М+Н).
Пример 118
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамид
Раствор трет-бутил (2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (25 мг, 0,055 ммоль) и гидрида натрия (4,4 мг, 0,11 ммоль) в ТГФ (0,5 мл) перемешивают при 0°С в течение 10 минут. Добавляют 3-(метоксиметил)азетидин-1-сульфонилхлорид (22 мг, 0,11 ммоль), и реакционную смесь нагревают до температуры окружающей среды нагревают при 50°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между водой (25 мл) и 4:1 ДХМ:ИПС (25 мл). Органическую фазу сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Реакционную смесь разбавляют ДХМ (5 мл) и добавляют ТФК (5 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 30 минут, затем концентрируют и очищают ВЭЖХ хроматографией с обращенной фазой (5-95% ацетонитрил:Н2О с 0,1% ТФК). Фракции, содержащие желаемый продукт, объединяют и разделяют между насыщенным водным NaHCO3 и ДХМ (25 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамида (7,7 мг, 27% выход) в виде белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,61-7,52 (м, 2Н), 7,17 (т, 1Н), 7,05 (д, 1Н), 6,67 (с, 1Н), 4,01 (т, 2Н), 3,82-3,77 (м, 2Н), 3,58 (с, 3Н), 3,47 (д, 2Н), 3,35 (с, 3Н), 2,88-2,76 (м, 1Н); МС (хиад, m/z) = 517,1, 519,1 (М+Н).
Пример 119
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксиазетидин-1-сульфонамид
Раствор трет-бутил (2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (0,674 мл, 0,0674 ммоль) в ТГФ (0,7 мл) и гидрида натрия (5,39 мг, 0,135 ммоль) перемешивают при 0°С в течение 10 минут. Добавляют 3-метоксиазетидин-1-сульфонилхлорид (25,0 мг, 0,135 ммоль), и реакционную смесь нагревают до температуры окружающей среды затем нагревают до 50°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между водой (25 мл) и 4:1 ДХМ:ИПС (25 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют с получением неочищенной смеси, которую берут в ДХМ (5 мл) и трифторуксусной кислоте (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют и очищают ВЭЖХ хроматографией с обращенной фазой (5-95% ацетонитрил:Н2О с 0,1% ТФК). Желаемые фракции разделяют между насыщ. водным NaHCO3 и ДХМ (25 мл), и органическую фазу сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-метоксиазетидин-1-сульфонамида (8,3 мг, 24,5% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,17 (с, 1Н), 7,61-7,54 (м, 2Н), 7,18 (т, 1Н), 7,07 (д, 1Н), 6,67 (с, 1Н), 4,19-4,04 (м, 3Н), 3,93-3,87 (м, 2Н), 3,61 (с, 3Н), 3,27 (с, 3Н); МС (хиад, m/z) = 503,1, 505,1 (М+Н).
Пример 120
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил) окси)-4-фторфенил)-3-фторазетидин-1-сульфонамид
Раствор трет-бутил (2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)карбамата (0,605 мл, 0,0605 ммоль) в ТГФ (0,6 мл) и гидрида натрия (4,84 мг, 0,121 ммоль) перемешивают при 0°С в течение 10 минут, затем добавляют 3-фторазетидин-1-сульфонилхлорид (21,0 мг, 0,121 ммоль), и реакционную смесь оставляют нагреваться до температуры окружающей среды затем нагревают до 50°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между водой (25 мл) и 4:1 ДХМ:ИПС (25 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении и реакционную смесь берут в ДХМ (5 мл) и ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют и очищают ВЭЖХ хроматографией с обращенной фазой (5-95% ацетонитрил:Н2О с 0,1% ТФК). Желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 и ДХМ (25 мл), и органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)-3-фторазетидин-1-сульфонамида (7,8 мг, 26,2% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,00 (с, 1Н), 7,59-7,51 (м, 2Н), 7,18 (т, 1Н), 7,05 (д, 1Н), 6,68 (с, 1Н), 5,35-5,14 (м, 1Н), 4,26-4,08 (м, 4Н), 3,59 (с, 3Н); МС (хиад, m/z) = 491,0, 493,0 (М+Н).
Пример 121
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-(метоксиметил)азетидин-1-сульфонамид
Трет-бутил (2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамат (25 мг, 0,06 ммоль) растворяют в ТГФ (0,6 мл), охлаждают до 0°С и обрабатывают гидридом натрия (4,6 мг, 0,11 ммоль). Реакцию перемешивают в течение 10 минут и обрабатывают 3-(метоксиметил)азетидин-1-сульфонилхлоридом (23 мг, 0,11 ммоль) и нагревают до 50°С в течение 12 часов. Реакцию охлаждают до температуры окружающей среды и выливают в 20 мл ледяной воды. Водный слой экстрагируют 4:1 ДХМ/ИПС (3×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют, разбавляют ДХМ, промывают насыщенным NaHCO3 (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (0-100% ДХМ/EtOAc), затем хроматографией с обращенной фазой (5-95% водой/ацетонитрилом с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х) и органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-(метоксиметил)азетидин-1-сульфонамида (10 мг, 35%) в виде не совсем белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 8,02 (с, 1Н), 7,58-7,54 (м, 1Н), 7,47-7,43 (м, 1Н), 7,29-7,22 (м, 1Н), 7,17-7,11 (т, 1Н), 6,67 (с, 1Н), 4,04-3,97 (т, 2Н), 3,82-3,76 (м, 2Н), 3,58 (с, 3Н), 3,49-3,45 (д, 2Н), 3,35 (с, 3Н), 2,88-2,77 (м, 1Н); МС (хиад, m/z) = 501,0, 503,1 (М+Н).
Пример 122
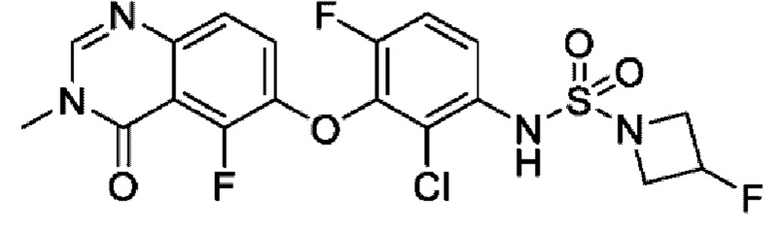
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторазетидин-1-сульфонамид
Трет-бутил (2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамат (30 мг, 0,07 ммоль) растворяют в ТГФ (0,6 мл), охлаждают до 0°С и обрабатывают гидридом натрия (5,5 мг, 0,14 ммоль). Реакцию перемешивают в течение 10 минут и затем обрабатывают 3-фторазетидин-1-сульфонилхлоридом (24 мг, 0,14 ммоль). Реакционную смесь нагревают до 50°С в течение 12 часов. Реакцию охлаждают до температуры окружающей среды и выливают в 20 мл ледяной воды, и водный слой экстрагируют ДХМ (3×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакцию концентрируют, разбавляют ДХМ, промывают насыщенным NaHCO3 (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (0-100% ДХМ/EtOAc) затем хроматографией с обращенной фазой (5-95% водой/ацетонитрилом с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х) и органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторазетидин-1-сульфонамида (14 мг, 43%) в виде белого твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 9,99 (с, 1Н), 8,33 (с, 1Н), 7,55-7,43 (м, 3Н), 7,36-7,29 (т, 1Н), 5,45-5,25 (д, 1Н), 4,20-4,08 (м, 2Н), 3,99-3,88 (м, 2Н), 3,46 (с, 3Н); МС (хиад, m/z) = 475,1, 477,1 (М+Н).
Пример 123
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-метоксиазетидин-1-сульфонамид
Трет-бутил (2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)карбамат (30 мг, 0,07 ммоль) растворяют в ТГФ (0,6 мл), охлаждают до 0°С и обрабатывают гидридом натрия (5,5 мг, 0,14 ммоль). Реакцию перемешивают в течение 10 минут и затем обрабатывают 3-метоксиазетидин-1-сульфонилхлоридом (25 мг, 0,14 ммоль) и нагревают до 50°С в течение 12 часов. Реакцию охлаждают до температуры окружающей среды и выливают в 20 мл ледяной воды. Водный слой экстрагируют ДХМ (3×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в 1:1 ДХМ:ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакцию концентрируют, разбавляют ДХМ, промывают насыщенным NaHCO3 (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (0-100% ДХМ/EtOAc) затем хроматографией с обращенной фазой (5-95% вода/ацетонитрил с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-метоксиазетидин-1-сульфонамида (14 мг, 42%) в виде белого твердого вещества. 1H ЯМР (400 МГц, (CD3)2SO) δ 9,85 (с, 1Н), 8,33 (с, 1Н), 7,55-7,43 (м, 3Н), 7,35-7,28 (т, 1Н), 4,18-4,10 (м, 1Н), 4,00-3,93 (м, 2Н), 3,73-3,66 (м, 2Н), 3,46 (с, 3Н), 3,17 (с, 3Н); МС (хиад, m/z) = 487,1, 489,1 (М+Н).
Пример 124
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)азетидин-1-сульфонамид трифторацетат
Стадия 1: Трет-бутил (азетидин-1-илсульфонил)(2-хлор-4-фтор-3-йодфенил)карбамат. К раствору трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (75 мг, 0,20 ммоль) в тетрагидрофуране (2,0 мл) при 0°С добавляют гидрид натрия (60% в минеральном масле, 12 мг, 0,30 ммоль) и перемешивают в течение 10 минут. Азетидин-1-сульфонилхлорид (63 мг, 0,40 ммоль) добавляют, и раствор нагревают до 50°С в течение 4 часов, при этом реакция останавливается. Раствор разделяют между дихлорметаном и насыщенным NaHCO3 и органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% ДХМ/гексан, затем промывают 80% EtOAc/гексаном) с получением трет-бутил (азетидин-1-илсульфонил) (2-хлор-4-фтор-3-йодфенил)карбамат (64 мг, 65% выход).
Стадия 2: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)азетидин-1-сульфонамид трифторацетат. Раствор 6-амино-3,5-диметилхиназолин-4(3Н)-она (25 мг, 0,13 ммоль), трет-бутил (азетидин-1-илсульфонил)(2-хлор-4-фтор-3-йодфенил) карбамата (64 мг, 0,13 ммоль), трис(дибензилиденацетон)дипалладия (6 мг, 0,006 ммоль), Xantphos (11 мг, 0,019 ммоль) и карбоната цезия (86 мг, 0,26 ммоль) в толуоле (1320 мкл) продувают аргоном и нагревают до 110°С в течение ночи в герметично закрытой пробирке. На следующее утро, раствор фильтруют через Celite®, концентрируют и затем перемешивают в 1 мл ДХМ и 1 мл ТФК в течение 1 часа. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК), и продукт лиофилизируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)азетидин-1-сульфонамида трифторацетата (22 мг, 36% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,74 (с, 1Н), 7,60 (д, 1Н), 7,51-7,47 (м, 1Н), 7,16-7,11 (т, 1Н), 7,10-7,06 (м, 1Н), 6,60 (шс, 1Н), 5,68 (шс, 1Н), 4,02-3,98 (т, 4Н), 3,67 (с, 3Н), 2,96 (с, 3Н), 2,29-2,22 (м, 2Н); МС (хиад, m/z) = 452,1, 454,1 (М+Н).
Пример 125
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид
Стадия 1: Трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-фторазетидин-1-ил)сульфонил)карбамат. К раствору трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (100 мг, 0,269 ммоль) в тетрагидрофуране (1790 мкл) при 0°С добавляют гидрид натрия (60% в минеральном масле, 16 мг, 0,40 ммоль) и перемешивают в течение 10 минут. 3-Фторазетидин-1-сульфонилхлорид (70 мг, 0,40 ммоль) добавляют, и раствор нагревают до 50°С в течение 5 часов. Раствор затем разделяют между дихлорметаном и насыщенным NaHCO3 и затем органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% EtOAc/гексан) с получением трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-фторазетидин-1-ил)сульфонил)карбамата (60 мг, 44% выход).
Стадия 2: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид. Раствор 6-амино-3,5-диметилхиназолин-4(3Н)-она (33 мг, 0,17 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-фторазетидин-1-ил)сульфонил)карбамата (60 мг, 0,11 ммоль), трис(дибензилиденацетон)дипалладия (10 мг, 0,011 ммоль), Xantphos (17 мг, 0,029 ммоль) и карбоната цезия (76 мг, 0,23 ммоль) в толуоле (790 мкл) продувают аргоном и нагревают до 110°С в течение ночи в герметично закрытой пробирке. Раствор фильтруют через Celite®, концентрируют и фильтрат перемешивают в 1 мл of ДХМ и 1 мл ТФК в течение 30 минут. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК) и продукт разделяют между ДХМ и насыщенным NaHCO3. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамида (32 мг, 58% выход). 1H ЯМР (400 МГц, CDCl3) δ 7,93 (с, 1Н), 7,45 (д, 1Н), 7,40-7,37 (м, 1Н), 7,12-7,04 (м, 2Н), 6,69 (шс, 1Н), 5,61 (шс, 1Н), 5,33-5,15 (м, 1Н), 4,25-4,08 (м, 4Н), 3,54 (с, 3Н), 2,98 (с, 3Н); МС (хиад, m/z) = 470,1, 472,1 (М+Н).
Пример 126
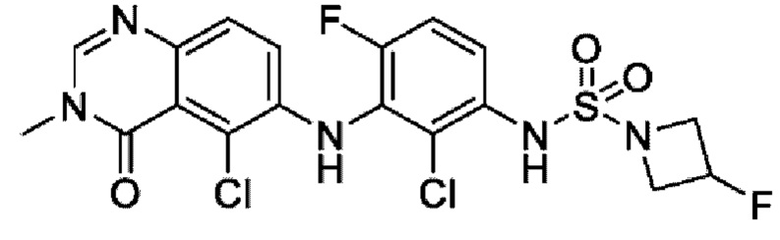
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид
Раствор 6-амино-5-хлор-3-метилхиназолин-4(3Н)-она (90 мг, 0,42 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3-фторазетидин-1-ил)сульфонил)карбамата (218 мг, 0,429 ммоль), трис(дибензилиденацетон)дипалладия (39 мг, 0,042 ммоль), Xantphos (62 мг, 0,10 ммоль) и карбоната цезия (279 мг, 0,858 ммоль) в толуоле (2860 мкл) продувают аргоном и нагревают до 110°С в течение ночи в герметично закрытой пробирке. Раствор фильтруют через Celite®, концентрируют и остаток перемешивают в 1 мл ДХМ и 1 мл ТФК в течение 1 часа. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК), и продукт разделяют между ДХМ и насыщенным NaHCO3. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамида (78 мг, 37% выход). 1H ЯМР (400 МГц, CDCl3) δ 7,94 (с, 1Н), 7,56-7,52 (м, 1Н), 7,51 (д, 1Н), 7,19-7,14 (т, 1Н), 6,99-6,95 (м, 1Н), 6,72 (шс, 1Н), 6,47 (шс, 1Н), 5,35-5,15 (м, 1Н), 4,25-4,10 (м, 4Н), 3,57 (с, 3Н); МС (хиад, m/z) = 490,1, 492,1 (М+Н).
Пример 127
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторазетидин-1-сульфонамид трифторацетат
Раствор 6-амино-5-фтор-3-метилхиназолин-4(3Н)-она (12 мг, 0,062 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3-фторазетидин-1-ил)сульфонил)карбамата (31 мг, 0,062 ммоль), трис(дибензилиденацетон)дипалладия (2 мг, 0,003 ммоль), Xantphos (5 мг, 0,009 ммоль) и карбоната цезия (40 мг, 0,12 ммоль) в толуоле (620 мкл) продувают аргоном и нагревают до 110°С в герметично закрытой пробирке в течение ночи. Раствор фильтруют через Celite, концентрируют и остаток перемешивают в 1 мл ДХМ и 1 мл ТФК в течение 1 часа. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК) и продукт лиофилизируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторазетидин-1-сульфонамида трифторацетата (4 мг, 13% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,49 (с, 1Н), 7,56-7,49 (м, 2Н), 7,20-7,15 (т, 1Н), 7,12-7,06 (м, 1Н), 6,68 (шс, 1Н), 6,01 (шс, 1Н), 5,34-5,16 (м, 1Н), 4,26-4,09 (м, 4Н), 3,67 (с, 3Н); МС (хиад, m/z) = 475,0, 477,0 (М+Н).
Пример 128
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксиазетидин-1-сульфонамид трифторацетат
Стадия 1: трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3-метоксиазетидин-1-ил)сульфонил)карбамат. К раствору трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (40 мг, 0,11 ммоль) в тетрагидрофуране (1070 мкл) при 0°С добавляют гидридом натрия (60% в минеральном масле, 6 мг, 0,2 ммоль) и перемешивают в течение 10 минут. 3-Метоксиазетидин-1-сульфонилхлорид (40 мг, 0,22 ммоль) добавляют, и раствор нагревают до 50°С в течение 2 часов. Раствор разделяют между дихлорметаном и насыщенным NaHCO3 и органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% ДХМ/гексан, затем промывают 80% EtOAc/гексаном) с получением трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3-метоксиазетидин-1-ил)сульфонил)карбамата (49 мг, 87% выход).
Стадия 2: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксиазетидин-1-сульфонамид трифторацетат. Раствор 6-амино-3,5-диметилхиназолин-4(3Н)-она (18 мг, 0,095 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3-метоксиазетидин-1-ил)сульфонил)карбамата (49 мг, 0,095 ммоль), трис(дибензилиденацетон)дипалладия (4 мг, 0,004 ммоль), Xantphos (8 мг, 0,01 ммоль) и карбоната цезия (61 мг, 0,19 ммоль) в толуоле (950 мкл) продувают аргоном и нагревают до 110°С в герметично закрытой пробирке в течение 2 часов. Раствор фильтруют через Celite, концентрируют и остаток перемешивают в 1 мл ДХМ и 1 мл ТФК в течение 1 часа. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК) и продукт лиофилизируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксиазетидин-1-сульфонамида трифторацетата (12 мг, 26% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,68 (с, 1Н), 7,58 (д, 1Н), 7,50-7,46 (м, 1Н), 7,11-7,06 (м, 2Н), 6,63 (шс, 1Н), 5,66 (шс, 1Н), 4,19-4,13 (м, 1Н), 4,11-4,07 (м, 2Н), 3,93-3,89 (м, 2Н), 3,66 (с, 3Н), 3,26 (с, 3Н), 2,97 (с, 3Н); МС (хиад, m/z) = 482,2, 484,1 (М+Н).
Пример 129
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метоксиазетидин-1-сульфонамид
6-Амино-5-хлор-3-метилхиназолин-4(3Н)-он (70 мг, 0,334 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-метоксиазетидин-1-ил)сульфонил)карбамат (183 мг, 0,351 ммоль), карбонат цезия (218 мг, 0,668 ммоль), трис(дибензилиденацетон)дипалладий(0) (30,6 мг, 0,0334 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфан) (48,3 мг, 0,0835 ммоль) растворяют в толуоле (3,3 мл) и нагревают при 110°С в течение ночи в герметично закрытой пробирке. Реакционную смесь оставляют охлаждаться до температуры окружающей среды, и неочищенную реакционную смесь разбавляют ДХМ и фильтруют через Celite. Растворитель концентрируют, затем восстанавливают в 1:1 ДХМ:ТФК (10 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Неочищенную реакционную смесь концентрируют и очищают хроматографией с обращенной фазой (5-95%, MeCN/Н2О, 0,1% ТФК). Желаемый продукт экстрагируют ДХМ и насыщенным водным NaHCO3 и слой ДХМ концентрируют с получением желаемого продукта (57 мг, 34% выход). 1H ЯМР (400 МГц, CDCl3) δ 7,99 (с, 1Н), 7,58-7,51 (м, 2Н), 7,16 (т, 1Н), 6,99-6,97 (м, 1Н), 6,67 (с, 1Н), 6,47 (с, 1Н), 4,18-4,13 (м, 1Н), 4,08 (т, 2Н), 3,92-3,89 (м, 2Н), 3,58 (с, 3Н), 3,27 (с, 3Н). МС (хиад, m/z) = 502,1, 504,1 (М+Н).
Пример 130
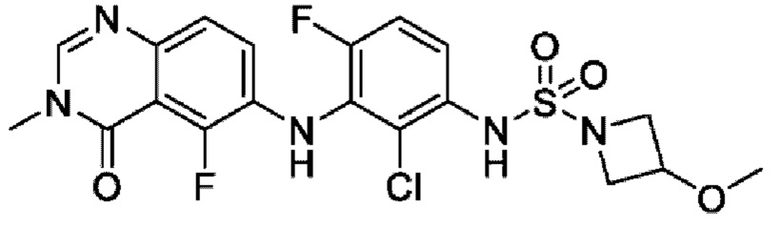
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-метоксиазетидин-1-сульфонамид
6-Амино-5-фтор-3-метилхиназолин-4(3Н)-он (84,44 мг, 0,4371 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-метоксиазетидин-1-ил)сульфонил)карбамат (239 мг, 0,4590 ммоль), карбонат цезия (284,8 мг, 0,8742 ммоль), трис(дибензилиденацетон)дипалладий(0) (40,03 мг, 0,04371 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфан) (63,23 мг, 0,1093 ммоль) растворяют в толуоле (4,4 мл) и нагревают при 110°С в течение ночи в герметично закрытой пробирке. Реакционную смесь оставляют охлаждаться до температуры окружающей среды и неочищенную реакционную смесь разбавляют ДХМ и фильтруют через Celite. Растворитель концентрируют, затем восстанавливают в 1:1 ДХМ:ТФК (10 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Неочищенную реакционную смесь концентрируют и очищают хроматографией с обращенной фазой (5-95%, MeCN/Н2О, 0,1% ТФК). Желаемый продукт экстрагируют ДХМ и насыщенным водным NaHCO3 и слой ДХМ концентрируют с получением желаемого продукта (98 мг, 46% выход). 1H ЯМР (400 МГц, CDCl3) δ 7,92 (с, 1Н), 7,53-7,50 (м, 1Н), 7,41-7,39 (м, 1Н) 7,16-7,11 (м, 1Н), 7,07-7,02 (м, 1Н), 6,68 (с, 1Н), 5,90 (с, 1Н), 4,14 (т, 1Н), 4,07 (т, 2Н), 3,92-3,89 (м, 2Н), 3,58 (с, 3Н), 3,27 (с, 3Н). МС (хиад, m/z) = 486,1, 488,1 (М+Н).
Пример 131
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамид
Стадия 1: 3-(метоксиметил)азетидин-1-сульфонилхлорид. К раствору 3-(метоксиметил)азетидина гидрохлорида (200 мг, 1,45 ммоль) в ДХМ (3630 мкл) добавляют сульфурилдихлорид (290 мкл, 3,6 ммоль) и основание Хюнига (380 мкл, 2,2 ммоль) по каплям, и реакцию перемешивают при температуре окружающей среды в течение 48 часов. Реакционную смесь разделяют между ДХМ и 1 N HCl, и слой ДХМ сушат над Na2SO4, фильтруют и концентрируют с получением 3-(метоксиметил)азетидин-1-сульфонилхлорида (273 мг, 94,1% выход).
Стадия 2: трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-(метоксиметил)азетидин-1-ил)сульфонил)карбамат. К раствору трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (300 мг, 0,807 ммоль) в тетрагидрофуране (5380 мкл) при 0°С добавляют гидрид натрия (60% в минеральном масле, 48 мг, 1,2 ммоль) и перемешивают в течение 10 минут. 3-(Метоксиметил)азетидин-1-сульфонилхлорид (242 мг, 1,21 ммоль) добавляют, и раствор нагревают до 50°С в течение 16 часов в герметично закрытой пробирке. Раствор разделяют между ДХМ и насыщенным NaHCO3 и органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% EtOAc/гексан) с получением неочищенного продукта. Неочищенный продукт концентрируют и повторно очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт разделяют между ДХМ и насыщенным NaHCO3 и органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3-(метоксиметил)азетидин-1-ил)сульфонил)карбамата (232 мг, 53,7% выход).
Стадия 3: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамид. Раствор 6-амино-3,5-диметилхиназолин-4(3Н)-она (83 мг, 0,43 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-(метоксиметил)азетидин-1-ил)сульфонил)карбамата (234 мг, 0,438 ммоль), трис(дибензилиденацетон)дипалладия (40 мг, 0,043 ммоль), Xantphos (63 мг, 0,10 ммоль) и карбоната цезия (285 мг, 0,877 ммоль) в толуоле (2920 мкл) продувают аргоном и нагревают до 110°С в течение 16 часов в герметично закрытой пробирке. Раствор фильтруют через Celite®, концентрируют и остаток перемешивают в 1 мл ДХМ и 1 мл ТФК в течение 1 часа. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт разделяют между ДХМ и насыщенным NaHCO3 и органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамида (165 мг, 75,8% выход). 1H ЯМР (400 МГц, CDCl3) δ 7,93 (с, 1Н), 7,46-7,39 (м, 2Н), 7,12-7,04 (м, 2Н), 6,68 (шс, 1Н), 5,60 (шс, 1Н), 4,03-3,99 (т, 2Н), 3,80-3,76 (м, 2Н), 3,54 (с, 3Н), 3,48 (д, 2Н), 3,34 (с, 3Н), 2,97 (с, 3Н), 2,85-2,77 (м, 1Н); МС (хиад, m/z) = 496,2, 498,2 (М+Н).
Пример 132
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамид трифторацетат
Раствор 6-амино-5-хлор-3-метилхиназолин-4(3Н)-она (20 мг, 0,095 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-(метоксиметил)азетидин-1-ил)сульфонил)карбамата (76 мг, 0,14 ммоль), трис(дибензилиденацетон)дипалладия (4 мг, 0,004 ммоль), Xantphos (8 мг, 0,01 ммоль) и карбоната цезия (6 мг, 0,1 ммоль) в толуоле (950 мкл) продувают аргоном и нагревают до 110°С в герметично закрытой пробирке в течение 16 часов. Раствор фильтруют через Celite®, концентрируют и остаток перемешивают в 1 мл ДХМ и 1 мл ТФК в течение 1 часа. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК) и продукт лиофилизируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)азетидин-1-сульфонамида трифторацетата (2,0 мг, 4,1% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,41 (с, 1Н), 7,61-7,57 (м, 2Н), 7,20-7,15 (т, 1Н), 7,02-6,99 (м, 1Н), 6,67 (шс, 1Н), 6,53 (шс, 1Н), 4,04-4,00 (т, 1Н), 3,82-3,78 (м, 1Н), 3,65 (с, 3Н), 3,49 (д, 2Н), 3,35 (с, 3Н), 2,87-2,79 (м, 1Н); МС (хиад, m/z) = 516,1, 518,1 (М+Н).
Пример 133
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(метоксиметил)азетидин-1-сульфонамид трифторацетат
Раствор 6-амино-5-фтор-3-метилхиназолин-4(3Н)-она (20 мг, 0,10 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-(метоксиметил)азетидин-1-ил)сульфонил)карбамата (83 мг, 0,15 ммоль), трис(дибензилиденацетон)дипалладия (4 мг, 0,005 ммоль), Xantphos (8 мг, 0,02 ммоль) и карбоната цезия (67 мг, 0,20 ммоль) в толуоле (1030 мкл) продувают аргоном и нагревают до 110°С в герметично закрытой пробирке в течение 16 часов. Раствор фильтруют через Celite®, концентрируют и остаток перемешивают в 1 мл ДХМ и 1 мл ТФК в течение 1 часа. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК) и продукт лиофилизируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(метоксиметил)азетидин-1-сульфонамида трифторацетата (4,0 мг, 7,7% выход). 1H ЯМР (400 МГц, CDCl3) δ 8,48 (с, 1Н), 7,58-7,54 (м, 1Н), 7,50 (д, 1Н), 7,19-7,14 (т, 1Н), 7,11-7,06 (м, 1Н), 6,67 (шс, 1Н), 6,01 (шс, 1Н), 4,04-4,00 (т, 2Н), 3,82-3,78 (м, 2Н), 3,67 (с, 3Н), 3,50-3,48 (д, 2Н), 3,35 (с, 3Н), 2,87-2,79 (м, 1Н); МС (хиад, m/z) = 500,1, 502,1 (М+Н).
Пример 134
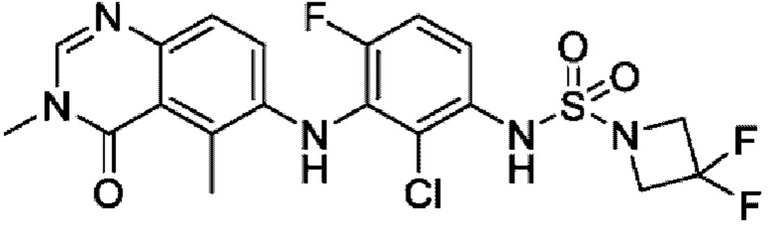
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторазетидин-1-сульфонамид
Стадия 1: Трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3,3-дифторазетидин-1-ил)сульфонил)карбамат. Трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат (553,3 мг, 1,489 ммоль) растворяют в ТГФ (15 мл) и обрабатывают гидридом натрия (60% в минеральном масле, 178,7 мг, 4,467 ммоль) и перемешивают при температуре окружающей среды в течение 15 минут. Реакционную смесь обрабатывают 3,3-дифторазетидин-1-сульфонилхлоридом (855,8 мг, 4,467 ммоль) и нагревают до 60°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и обрабатывают водой и экстрагируют EtOAc (2x) и the объединяют organic layer was промывают водой (3х), насыщенным раствором соли (1х) и сушат над Na2SO4, фильтруют и концентрируют. Полученный остаток очищают хроматографией на силикагеле (Гексан/EtOAc) с получением трет-бутил (2-хлор-4-фтор-3-йодфенил)((3,3-дифторазетидин-1-ил)сульфонил)карбамата (311,3 мг, 39,7% выход). 1H ЯМР (400 МГц, (CD3)2SO) δ 7,68-7,64 (м, 1Н), 7,39-7,35 (м, 1Н), 4,74-4,67 (т, 2Н), 4,46-4,40 (т, 2Н), 1,38 (с, 9Н).
Стадия 2: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторазетидин-1-сульфонамид. 6-Амино-3,5-диметилхиназолин-4(3Н)-он (31,5 мг, 0,166475 ммоль) растворяют в толуоле (1,7 мл) и обрабатывают трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3,3-дифторазетидин-1-ил)сульфонил)карбаматом (96,4493 мг, 0,183122 ммоль), трис(дибензилиденацетон)диппалладием (15,2446 мг, 0,0166475 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантеном (24,0819 мг, 0,0416187 ммоль) и карбонатом цезия (162,722 мг, 0,499424 ммоль), затем продувают аргоном, герметично закрывают и нагревают до 110°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ затем промывают водой (1х) и сушат над Na2SO4, фильтруют и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ/Ацетон). Полученный остаток растворяют в 1:1 ДХМ:ТФК (2,0 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют и очищают С18 хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК), и желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 и 4:1 ДХМ:ИПС. Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторазетидин-1-сульфонамида (33,0 мг, 40,6% выход). 1H ЯМР (400 МГц, (CD3)2SO) δ 10,04 (с, 1Н), 8,17 (с, 1Н), 7,35-7,31 (м, 4Н), 6,86-6,84 (дд, 1Н), 4,36-4,30 (т, 4Н), 3,43 (с, 3Н), 2,81 (с, 3Н). МС (хиад, m/z) = 488,1, 490,1 (М+Н).
Пример 135
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторазетидин-1-сульфонамид
6-Амино-5-хлор-3-метилхиназолин-4(3Н)-он (31,4 мг, 0,149786 ммоль) растворяют в толуоле (1,5 мл) и обрабатывают трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3,3-дифторазетидин-1-ил)сульфонил)карбаматом (86,7802 мг, 0,164764 ммоль), трис(дибензилиденацетон)диппалладием (13,7164 мг, 0,0149786 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантеном (21,6677 мг, 0,0374464 ммоль) и карбонатом цезия (146,409 мг, 0,449357 ммоль), затем продувают аргоном, герметично закрывают и нагревают до 110°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ и промывают водой (1х) затем сушат над Na2SO4, фильтруют и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ/Ацетон). Полученный остаток растворяют в 1:1 ДХМ:ТФК (2,0 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют и очищают С18 хроматографией с обращенной фазой (вода/АЦН с 0,1% ТФК) и желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 и 4:1 ДХМ:ИПС. Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3,3-дифторазетидин-1-сульфонамида (29,0 мг, 38,1% выход). 1H ЯМР (400 МГц, (CD3)2SO) δ 10,14 (с, 1Н), 8,21 (с, 1Н), 7,79 (с, 1Н), 7,53-7,49 (м, 1Н), 7,45-7,38 (м, 2Н), 6,79-6,76 (дд, 1Н), 4,36-4,30 (т, 4Н), 3,45 (с, 3Н). МС (хиад, m/z) = 508,0, 510,0 (М+Н).
Пример 136
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,3-дифторазетидин-1-сульфонамид
6-Амино-5-фтор-3-метилхиназолин-4(3Н)-он (32,1 мг, 0,166165 ммоль) растворяют в толуоле (1,7 мл) и обрабатывают трет-бутил
(2-хлор-4-фтор-3-йодфенил)((3,3-дифторазетидин-1-ил)сульфонил)карбаматом (96,27 мг, 0,183 ммоль), трис(дибензилиденацетон)диппалладием (15,2163 мг, 0,0166 ммоль), 4,5-бис(дифенилфосфино)-9,9-диметилксантеном (24,0371 мг, 0,0415413 ммоль) и карбонатом цезия (162,419 мг, 0,498 ммоль), затем продувают аргоном, герметично закрывают и нагревают до 110°С в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют ДХМ и промывают водой (1х) затем сушат над Na2SO4, фильтруют и концентрируют. Полученный остаток очищают хроматографией на силикагеле (ДХМ/Ацетон). Полученный остаток растворяют в 1:1 ДХМ:ТФК (2,0 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют и очищают С18 хроматографией с обращенной фазой (водой/ацетонитрилом с 0,1% ТФК) и желаемые фракции объединяют и разделяют между насыщенным водным NaHCO3 и 4:1 ДХМ:ИПС. Органическую фазу отделяют, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3,3-дифторазетидин-1-сульфонамида (22,6 мг, 27,7% выход). 1H ЯМР (400 МГц, (CD3)2SO) δ 10,09 (с, 1Н), 8,19 (с, 1Н), 8,10 (с, 1Н), 7,40-7,32 (м, 3Н), 7,07-7,02 (т, 1Н), 4,37-4,31 (т, 4Н), 3,44 (с, 3Н). МС (хиад, m/z) = 492,1, 494,1 (М+Н).
Пример 137
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((дифторметокси)метил)азетидин-1-сульфонамид трифторацетат
Стадия 1: Получение 3-((дифторметокси)метил)азетидин-1-сульфонилхлорида. 3-((дифторметокси)метил)азетидин (200 мг, 1,46 ммоль) перемешивают с N-этил-N-изопропилпропан-2-амином (379 мкл, 2,19 ммоль) при температуре окружающей среды в дихлорметане (3646 мкл) в течение 5 минут. Реакционную смесь охлаждают до -10°С и затем к реакции по каплям добавляют сульфурилдихлорид (295 мкл, 3,65 ммоль) в виде чистой жидкости. Реакцию оставляют нагреваться до температуры окружающей среды и перемешивают в течение 16 часов. Реакционную смесь разбавляют ДХМ и промывают 1N водн. HCl. Слой ДХМ сушат над MgSO4, концентрируют и используют без очистки непосредственно на следующей стадии (345 мг, 100%).
Стадия 2: Получение трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-((дифторметокси)-метил)азетидин-1-ил)сульфонил)карбамата. Трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат (105 мг, 0,283 ммоль) растворяют в тетрагидрофуране (1413 мкл) и охлаждают до 0°С. Гидрид натрия (60% в минеральном масле, 17,0 мг, 0,424 ммоль) добавляют, и полученный раствор оставляют нагреваться до температуры окружающей среды, затем добавляют 3-((дифторметокси)-метил)азетидин-1-сульфонилхлорид (99,9 мг, 0,424 ммоль) и реакционную смесь нагревают до 60°С в течение 16 часов. Неочищенную реакционную смесь охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией на силикагеле (Гексан/EtOAc) с получением трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-((дифторметокси)-метил)азетидин-1-ил)сульфонил)карбамата (98 мг, 61%).
Стадия 3: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((дифторметокси)метил)азетидин-1-сульфонамид трифторацетат. 6-амино-3,5-диметилхиназолин-4(3Н)-он (31 мг, 0,164 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-((дифторметокси)метил)азетидин-1-ил)сульфонил)карбамат (98,2 мг, 0,172 ммоль), (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфан) (14,2 мг, 0,0246 ммоль), карбонат цезия (107 мг, 0,328 ммоль) и трис(дибензилиденацетон)диппалладий(0) (7,50 мг, 0,00819 ммоль) растворяют в толуоле (1,6 мл), герметично закрывают и нагревают до 110°С в течение 16 часов. Реакционную смесь оставляют охлаждаться до температуры окружающей среды, фильтруют через Celite, концентрируют и остаток восстанавливают в 1:1 ДХМ:ТФК (2 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Летучие вещества удаляют в вакууме, и неочищенный продукт очищают хроматографией с обращенной фазой (5-95% MeCN/H2O, 0,1% ТФК). Желаемые фракции лиофилизируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((дифторметокси)метил)азетидин-1-сульфонамида трифторацетата (38 мг, 44%). 1H ЯМР (400 МГц, CDCl3) δ 8,58 (с, 1Н), 7,57 (д, 1Н), 7,48-7,44 (м, 1Н), 7,13 (т, 1Н), 7,09-7,06 (м, 1Н), 6,62 (с, 1Н), 6,41-6,04 (м, 1Н), 5,66 (с, 1Н), 4,05 (т, 2Н), 3,98-3,97 (д, 2Н), 3,83-3,79 (м, 2Н), 3,65 (с, 3Н), 2,97 (с, 3Н), 2,93-2,87 (м, 1Н). МС (хиад, m/z) = 532,1, 534,1 (М+Н).
Пример 138
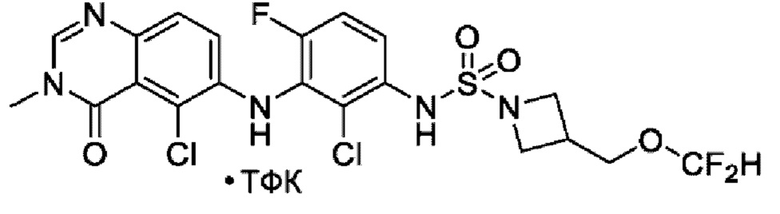
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((дифторметокси)метил)азетидин-1-сульфонамид трифторацетат
6-Амино-5-хлор-3-метилхиназолин-4(3Н)-он (28,1 мг, 0,1340 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-((дифторметокси)метил)азетидин-1-ил)сульфонил)карбамат (80,33 мг, 0,1407 ммоль), карбонат цезия (87,35 мг, 0,2681 ммоль), трис(дибензилиденацетон)дипалладий(0) (6,137 мг, 0,006702 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис-(дифенилфосфан) (9,307 мг, 0,01609 ммоль) растворяют в толуоле (1,3 мл), герметично закрывают и нагревают до 110°С в течение 16 часов. Реакционную смесь оставляют охлаждаться до температуры окружающей среды, фильтруют через Celite, концентрируют и остаток восстанавливают в 1:1 ДХМ:ТФК (2 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Летучие вещества удаляют в вакууме, и неочищенный продукт очищают хроматографией с обращенной фазой (5-95% MeCN/H2O, 0,1% ТФК). Желаемые фракции лиофилизируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-((дифторметокси)метил)азетидин-1-сульфонамида трифторацетата (33 мг, 44%). 1H ЯМР (400 МГц, CDCl3) δ 8,50 (с, 1Н), 7,61 (д, 1Н), 7,60-7,57 (м, 1Н), 7,19 (т, 1Н), 7,02-6,99 (м, 1Н), 6,66 (с, 1Н), 6,53 (с, 1Н), 6,41-6,04 (м, 1Н), 4,05 (т, 2Н), 3,99-3,97 (д, 2Н), 3,83-3,80 (м, 2Н), 3,67 (с, 3Н), 2,96-2,86 (м, 1Н). МС (хиад, m/z) = 552,1, 554,1 (М+Н).
Пример 139
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-((дифторметокси)метил)азетидин-1-сульфонамид трифторацетат
6-Амино-5-фтор-3-метилхиназолин-4(3Н)-он (25,9 мг, 0,1341 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-((дифторметокси)метил)азетидин-1-ил)сульфонил)карбамат (80,35 мг, 0,1408 ммоль), карбонат цезия (87,37 мг, 0,2681 ммоль), трис(дибензилиденацетон)дипалладий(0) (6,139 мг, 0,006704 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфан) (9,309 мг, 0,01609 ммоль) растворяют в толуоле (1,3 мл), герметично закрывают и нагревают до 110°С в течение 16 часов. Реакционную смесь оставляют охлаждаться до температуры окружающей среды, фильтруют через Celite, концентрируют и остаток восстанавливают в 1:1 ДХМ/ТФК (2 мл). Реакцию перемешивают при температуре окружающей среды в течение 30 минут и летучие вещества удаляют в вакууме. Неочищенный продукт очищают хроматографией с обращенной фазой (5-95% MeCN/Н2О, 0,1% ТФК). Желаемые фракции лиофилизируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-((дифторметокси)метил)азетидин-1-сульфонамида трифторацетата (39 мг, 55%). 1H ЯМР (400 МГц, CDCl3) δ 8,54 (с, 1Н), 7,57-7,50 (м, 2Н), 7,17 (т, 1Н), 7,12-7,07 (м, 1Н), 6,65 (с, 1Н), 6,41-6,04 (м, 1Н), 6,01 (с, 1Н), 4,05 (т, 2Н), 3,98-3,97 (д, 2Н), 3,83-3,79 (м, 2Н), 3,68 (с, 3Н), 2,96-2,85 (м, 1Н). МС (хиад, m/z) = 536,1, 538,1 (М+Н).
Пример 140
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)-3-метилазетидин-1-сульфонамид трифторацетат
Стадия 1: 3-(метоксиметил)-3-метилазетидин-1-сульфонилхлорид. К раствору 3-(метоксиметил)-3-метилазетидина гидрохлорида (230 мг, 1,52 ммоль) и N-этил-N-изопропилпропан-2-амина (400 мкл, 2 ммоль) в дихлорметане (5060 мкл) при 0°С добавляют сульфурилдихлорид (300 мкл, 3,8 ммоль), и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 16 часов. Раствор разбавляют ДХМ и промывают 1 N HCl (1х) и слой ДХМ сушат над Na2SO4, фильтруют и концентрируют с получением 3-(метоксиметил)-3-метилазетидин-1-сульфонилхлорида (240 мг, 74% выход).
Стадия 2: трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3-(метоксиметил)-3-метилазетидин-1-ил)сульфонил)карбамат. К раствору трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат (25 мг, 0,067 ммоль) в тетрагидрофуране (340 мкл) при 0°С добавляют гидрид натрия (60% в минеральном масле, 4,0 мг, 0,10 ммоль) и перемешивают в течение 10 минут. 3-(Метоксиметил)-3-метилазетидин-1-сульфонилхлорид (22 мг, 0,10 ммоль) добавляют, и раствор нагревают до 50°С в герметично закрытой пробирке в течение 16 часов. Раствор разделяют между дихлорметаном и насыщенным NaHCO3 и органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% EtOAc/гексан) с получением трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-(метоксиметил)-3-метилазетидин-1-ил)сульфонил)карбамата (34 мг, 92% выход).
Стадия 3: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)-3-метилазетидин-1-сульфонамид трифторацетат.Раствор 6-амино-3,5-диметилхиназолин-4(3Н)-она (12 мг, 0,063 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-(метоксиметил)-3-метилазетидин-1-ил)сульфонил)карбамата (34 мг, 0,063 ммоль), трис(дибензилиденацетон)дипалладий (5 мг, 0,00 6 ммоль), Xantphos (9 мг, 0,01 ммоль) и карбонат цезия (41 мг, 0,12 ммоль) в толуоле (420 мкл) продувают аргоном и нагревают до 110°С в течение 16 часов в герметично закрытой пробирке. Раствор фильтруют через Celite®, концентрируют и перемешивают в 1 мл ДХМ и 1 мл ТФК в течение 1 часа. Раствор концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК) и продукт лиофилизируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(метоксиметил)-3-метилазетидин-1-сульфонамида трифторацетата (6,0 мг, 19% выход). 1Н ЯМР (400 МГц, CDCl3) (8,61 (с, 1Н), 7,57 (д, 1Н), 7,51-7,47 (м, 1Н), 7,15-7,06 (м, 2Н), 6,68 (шс, 1Н), 5,67 (шс, 1Н), 3,87 (д, 2Н), 3,65 (с, 3Н), 3,60 (д, 2Н), 3,36 (с, 3Н), 3,32 (с, 2Н), 2,97 (с, 3Н), 1,28 (с, 3Н); МС (хиад, m/z) = 510,2, 512,2 (М+Н).
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-этоксиазетидин-1-сульфонамид
Стадия 1: трет-бутил 3-этоксиазетидин-1-карбоксилат.
Раствор трет-бутил 3-гидроксиазетидин-1-карбоксилат (63,5 мл, 12,7 ммоль) в 1:1 ДМФ:ТГФ (60 мл) и гидрид натрия (0,7 62 г, 19,1 ммоль) перемешивают в ледяной бане в течение 10 минут. Йодэтан (3,05 мл, 3 8,1 ммоль) добавляют, и реакционную смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение 18 часов. Реакционную смесь разбавляют водой (100 мл) и экстрагируют EtOAc (100 мл). Органическую фазу сушат над Na2SO4, фильтруют и концентрируют с получением трет-бутил 3-этоксиазетидин-1-карбоксилата (2,56 г, 100% выход), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: 3-этоксиазетидин гидрохлорид. Раствор трет-бутил 3-этоксиазетидин-1-карбоксилат (2,50 г, 12,4 ммоль) в 1:1:3 ТГФ:ДМФ:EtOAc (150 мл) перемешивают при температуре окружающей среды при добавлении раствора 5N HCl (74,5 мл, 373 ммоль) в ИПС. Реакционную смесь перемешивают при температуре окружающей среды, затем концентрируют с получением 3-этоксиазетидина гидрохлорида (1,3 г, 76,1% выход) в виде красного масла, которое используют на следующих стадиях без дальнейшей очистки. 1Н ЯМР (4 00 МГц, D4-Метанол) (4,48-4,40 (м, 1Н), 4,33-4,25 (м, 2Н), 4,01-3,93 (м, 2Н), 3,53 (м, 2Н), 1,23 (т, 3Н).
Стадия 3: 3-этоксиазетидин-1-сульфонилхлорид. Раствор 3-этоксиазетидина гидрохлорида (500 мг, 3,63 ммоль) в ДХМ (30 мл) охлаждают до 0°С и обрабатывают N-этил-N-изопропилпропан-2-амином (94 9 мкл, 5,4 5 ммоль), затем сульфурилдихлоридом (881 мкл, 10,9 ммоль). Реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 18 часов затем разбавляют ДХМ (70 мл) и промывают 1N HCl (2×50 мл). Органические фазы объединяют и промывают насыщенным раствором соли (25 мл), затем сушат над Na2SO4, фильтруют и концентрируют с получением 3-этоксиазетидин-1-сульфонилхлорида (700 мг, 96,5% выход) в виде желтого масла, которое используют без дальнейшей очистки. 1Н ЯМР (400 МГц, CDCl3) (4,36-4,30 (м, 1Н), 4,29-4,22 (м, 2Н), 4,04-3,99 (м, 2Н), 3,47 (м, 2Н), 1,24 (т, 3Н).
Стадия 4: трет-бутил (2-хлор-4-фтор-3-йодфенил) ((3-этоксиазетидин-1-ил)сульфонил)карбамат. Раствор трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат (961 мкл, 0,673 ммоль) в ТГФ (1 мл) обрабатывают гидридом натрия (53,8 мг, 1,35 ммоль, 60% дисперсия в минеральном масле) и перемешивают при 0°С в течение 30 минут.3-этоксиазетидин-1-сульфонилхлорид (269 мг, 1,35 ммоль) добавляют, и реакционную смесь нагревают до температуры окружающей среды и нагревают при 50°С в течение 96 часов. Реакционную смесь разбавляют водой (50 мл) и насыщенным водным NaHCO3 (50 мл), и водный слой экстрагируют 4:1 ДХМ ИПС (3×25 мл). Органические фазы объединяют и сушат над Na2SO4, фильтруют и концентрируют с получением коричневого масла, которое очищают хроматографией на силикагеле (0-50% гексан: EtOAc) с получением трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-этоксиазетидин-1-ил) сульфонил) карбамата (279 мг, 77,5% выход). 1Н ЯМР (400 МГц, CDCl3) (7, 40-7, 35 (м, 1Н), 7,07-7,01 (м, 1Н), 4, 40-4, 28 (м, 3Н), 4,26-4,18 (м, 2Н), 3,46 (кв, 2Н), 1,43 (с, 9Н), 1,23 (т, 3Н).
Стадия 5: N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-этоксиазетидин-1-сульфонамид. Раствор 6-амино-5-хлор-3-метилхиназолин-4(3Н)-она (30 мг, 0,143 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-этоксиазетидин-1-ил)сульфонил)карбамата (84,2 мг, 0,157 ммоль), Pd2(dba)3 (13,1 мг, 0,0143 ммоль), карбоната цезия (140 мг, 0,429 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфана) (20,7 мг, 0,0358 ммоль) в толуоле (0,9 мл) продувают аргоном в течение 5 минут затем нагревают при 100°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды, затем разделяют между водой (25 мл) и EtOAc (50 мл). Органическую фазу отделяют и промывают насыщенным раствором соли (25 мл) затем сушат над Na2SO4, фильтруют и концентрируют до коричневого масла. Неочищенную реакционную смесь растворяют в ДХМ (25 мл) и ТФК (10 мл) и перемешивают в течение 2 0 минут при температуре окружающей среды затем концентрируют с получением коричневого масла, которое очищают ВЭЖХ (5-95% ацетонитрил: H2O с 0,1%ТФК). Желаемые фракции объединяют и рН доводят до 9 насыщенным водным NaHCO3. Водную фазу экстрагируют ДХМ (2×50 мл), и органические фазы объединяют, сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-этоксиазетидин-1-сульфонамида (39,4 мг, 53,3% выход) в виде бежевого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) (7,93(с, 1Н), 7, 58-7, 48 (м, 2Н), 7,15 (т, 1Н), 7,01-6,95 (м, 1Н), 6,68 (с, 1Н), 6,45 (с, 1Н), 4,27-4,19 (м, 1Н), 4,10-4,04 (м, 2Н), 3,95-3,88 (м, 2Н), 3,57 (с, 3Н), 3,41 (м, 2Н), 1,19 (т, 3Н); МС (хиад, m/z) = 516,1, 518,1 (М+Н).
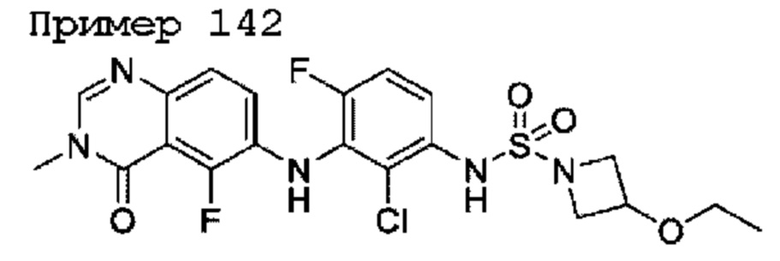
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-этоксиазетидин-1-сульфонамид
Раствор 6-амино-5-фтор-3-метилхиназолин-4(3Н)-она (30 мг, 0,155 ммоль), трет-бутил (2-хлор-4-фтор-3-йодфенил)((3-этоксиазетидин-1-ил)сульфонил)карбамата (91,4 мг, 0,171 ммоль), Pd2(dba)3 (14,2 мг, 0,0155 ммоль), карбоната цезия (152 мг, 0,466 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфана) (22,5 мг, 0,0388 ммоль) в толуоле (0,9 мл) продувают аргоном в течение 5 минут, затем нагревают при 100°С в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды затем разделяют между водой (25 мл) и EtOAc (50 мл). Органическую фазу отделяют и промывают насыщенным раствором соли (25 мл), затем сушат над Na2SO4, фильтруют и концентрируют до коричневого масла. Неочищенную реакционную смесь растворяют в ДХМ (25 мл) и ТФК (10 мл) и перемешивают в течение 2 0 минут при температуре окружающей среды затем концентрируют с получением коричневого масла, которое очищают ВЭЖХ (5-95% ацетонитрил: Н2О с 0,1%ТФК). Желаемые фракции объединяют и рН доводят до 9 насыщенным водным NaHCO3. Водную фазу экстрагируют ДХМ (2×50 мл), и органические фазы объединяют, сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-этоксиазетидин-1-сульфонамида (32,9 мг, 42,4% выход) в виде бежевого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) (7, 90 (с, 1Н), 7, 54-7, 48 (м, 1Н), 7, 43-7, 38 (м, 1Н), 7,14 (т, 1Н), 7,08-7,02 (м, 1Н), 6,64 (с, 1Н), 5,92 (с, 1Н), 4,27-4,19 (м, 1Н), 4,10-4,03 (м, 2Н), 3, 94-3, 89 (м, 2Н), 3,57 (с, 3Н), 3,42 (м, 2Н), 1,19 (т, 3Н); МС (хиад, m/z)=500,1, 502,1 (М+Н).
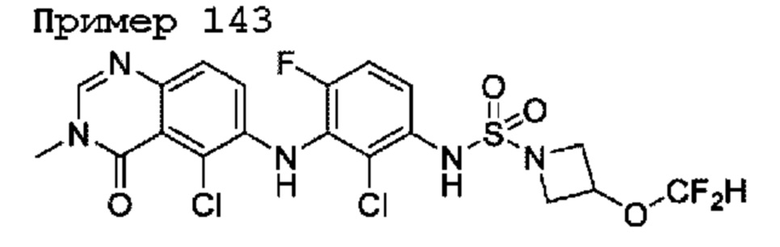
N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(дифторметокси)азетидин-1-сульфонамид
Стадия 1: Получение 3-(дифторметокси) азетидин-1-сульфонилхлорида. 3-(дифторметокси)азетидин гидрохлорид (513 мг, 3,21 ммоль) суспендируют в ДХМ (15,7 мл) и туда добавляют N,N-диизопропилэтиламин (818,7 мкл, 4,70 ммоль) и перемешивают при температуре окружающей среды до растворения. Реакционную смесь охлаждают до 0°С и обрабатывают сульфурилдихлоридом (0,77 мл, 9,64 ммоль) по каплям и перемешивают при температуре окружающей среды в течение 12 часов. Реакционную смесь разбавляют ДХМ, промывают 10% HCl (3×25 мл), насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют с получением 3-(дифторметокси)азетидин-1-сульфонилхлорида (366 мг, 51%) в виде желтой жидкости которое используют на следующей стадии без очистки. 1Н ЯМР (400 МГц, CDCl3) (6,48-6,08 (т, 1Н), 5,08-5,01 (м, 1Н), 4,41-4,35 (м, 2Н), 4,21-4,16 (м, 2Н).
Стадия 2: Получение трет-бутил (2-хлор-4-фтор-3-йодфенил)(3-(дифторметокси)азетидин-1-ил)сульфонил)карбамата. Трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат (307 мг, 0,826 ммоль) растворяют в ТГФ (4,1 мл), охлаждают до 0°С и обрабатывают гидридом натрия (66,1 мг, 1,65 ммоль). Реакцию перемешивают при температуре окружающей среды в течение 15 минут и обрабатывают 3-(дифторметокси)азетидин-1-сульфонилхлоридом (366 мг, 1,65 ммоль), и затем нагревают до 50°С в течение 12 часов. Реакционную смесь охлаждают до температуры окружающей среды и выливают в 2 0 мл ледяной воды. Водный слой экстрагируют EtOAc (2×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией с обращенной фазой (5-95% вода/АЦН с 0,1% ТФК) и желаемые фракции объединяют, разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х). Органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением трет-бутил (2-хлор-4-фтор-3-йодфенил)(3-(дифторметокси)азетидин-1-ил)сульфонил)карбамата (228 мг, 50%) в виде не совсем белого полутвердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) (7,62-7,57 (м, 1Н), 7,38-7,33 (м, 1Н), 6,97-6,60 (т, 1Н), 5,07-5,00 (м, 1Н), 4,50-4,44 (м, 2Н), 4,27-4,18 (м, 2Н), 1,39 (с, 9Н).
Стадия 3: Получение N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(дифторметокси)азетидин-1-сульфонамида. Трет-бутил (2-хлор-4-фтор-3-йодфенил)(3-(дифторметокси)азетидин-1-ил)сульфонил)карбамат (58,43 мг, 0,11 ммоль), 6-амино-5-хлор-3-метилхиназолин-4(3Н)-он (22 мг, 0,105 ммоль), карбонат цезия (68,4 мг, 0,21 ммоль), трис(дибензилиденацетон)диппалладий (9,6 мг, 0,011 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфан) (15,2 мг, 0,026 ммоль) суспендируют в толуоле (1,05 мл), и реакцию продувают аргоном в течение 15 минут, герметично закрывают и нагревают до 110°С в течение 12 часов. Неочищенную реакцию охлаждают до температуры окружающей среды, затем разбавляют ДХМ и фильтруют через короткий слой Celite(и концентрируют. Остаток растворяют в 1:1 ДХМ:ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 30 минут. Реакцию концентрируют, разбавляют ДХМ, промывают насыщенным NaHCO3 (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (0-100% ДХМ/EtOAc), затем хроматографией с обращенной фазой (5-95% вода/ацетонитрил с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ:ИПС и насыщенным водным NaHCO3 (1х), и органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N- (2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-(дифторметокси)азетидин-1-сульфонамида (26 мг, 46%) в виде светло-коричневого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) (9, 86 (с, 1Н), 8,21 (с, 1Н), 7,77 (с, 1Н), 7, 54-7, 36 (м, 3Н), 6, 93-6,53 (м, 2Н), 4, 97-4, 88 (м, 1Н), 4,15-4,08 (м, 2Н), 3,90-3,82 (м, 2Н), 3,44 (с, 3Н); МС (хиад, m/z)=539, 0, 541, 0, 543,0
(М+Н).
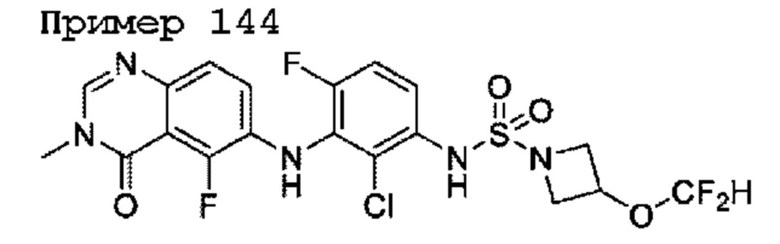
N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(дифторметокси)азетидин-1-сульфонамид
Трет-бутил (2-хлор-4-фтор-3-йодфенил)(3-(дифторметокси)азетидин-1-ил)сульфонил)карбамат (61,0 мг, 0,11 ммоль), 6-амино-5-фтор-3-метилхиназолин-4(3Н)-он (21 мг, 0,11 ммоль), карбонат цезия (71,0 мг, 0,22 ммоль), трис(дибензилиденацетон)диппалладий (10,0 мг, 0,011 ммоль) и (9,9-диметил-9Н-ксантен-4,5-диил)бис(дифенилфосфан) (16 мг, 0,026 ммоль) суспендируют в толуоле (1,08 мл), и реакцию продувают аргоном в течение 15 минут, герметично закрывают и нагревают до 110°С в течение 12 часов. Неочищенную реакцию охлаждают до температуры окружающей среды, затем разбавляют ДХМ и фильтруют через короткий слой Celite(и концентрируют. Остаток растворяют в 1:1 ДХМ:ТФК (5 мл) и перемешивают при температуре окружающей среды в течение 3 0 минут. Реакцию концентрируют, разбавляют ДХМ, промывают насыщенным NaHCO3 (3×50 мл) и насыщенным раствором соли (1×25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Остаток очищают колоночной хроматографией на силикагеле (0-100% ДХМ/EtOAc) затем хроматографией с обращенной фазой (5-95% вода/АЦН с 0,1% ТФК). Желаемые фракции объединяют и разделяют между 4:1 ДХМ: ИПС и насыщенным водным NaHCO3 (1х) и органический слой отделяют, сушат над безводным Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-фтор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-(дифторметокси)азетидин-1-сульфонамида (20 мг, 35%) в виде не совсем белого твердого вещества. 1Н ЯМР (400 МГц, (CD3)2SO) δ 9,81 (с, 1Н), 8,18 (с, 1Н), 8,08 (с, 1Н), 7,43-7,29 (м, 3Н), 7, 08-7, 00 (т, 1Н), 6, 93-6,53 (т, 1Н), 4, 97-4, 89 (м, 1Н), 4,15-4,08 (м, 2Н), 3, 89-3, 82 (м, 2Н), 3,43 (с, 3Н); МС (хиад, m/z) = 522,0, 524,0 (М+Н).

N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-3-(метоксиметил)азетидин-1-сульфонамид трифторацетат
Стадия 1: трет-бутил 3-фтор-3-(метоксиметил)азетидин-1-карбоксилат. К раствору трет-бутил 3-фтор-3-(гидроксиметил)азетидин-1-карбоксилата (600 мг, 2,92 ммоль) в тетрагидрофуране (14,6 мл) при 0°С добавляют гидрид натрия (60% в минеральном масле, 175 мг, 4,39 ммоль) и перемешивают при температуре окружающей среды в течение 10 минут. Йодметан (3 64 мкл, 5,85 ммоль) добавляют и перемешивают при температуре окружающей среды в течение 45 минут. Раствор разделяют между насыщенным NaHCO3 и ДХМ, и органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют, затем очищают хроматографией на силикагеле (5-75% EtOAc/гексан) с получением трет-бутил 3-фтор-3-(метоксиметил)азетидин-1-карбоксилата (631 мг, 98,4% выход).
Стадия 2: 3-фтор-3-(метоксиметил)азетидин гидрохлорид. Раствор трет-бутил 3-фтор-3-(метоксиметил)азетидин-1-карбоксилата (631 мг, 2,8 8 ммоль) в 4 М HCl в диоксане (5,76 мл) перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь концентрируют с получением 3-фтор-3-(метоксиметил)азетидина гидрохлорида (440 мг, 98% выход).
Стадия 3: 3-фтор-3-(метоксиметил) азетидин-1-сульфонилхлорид. К раствору 3-фтор-3-(метоксиметил)азетидина гидрохлорида (440 мг, 2,8 3 ммоль) и N-этил-N-изопропилпропан-2-амина (740 мкл, 4,2 4 ммоль) в дихлорметане (9,42 мл) при 0°С добавляют сульфурилдихлорид (570 мкл, 7,07 ммоль) и нагревают до температуры окружающей среды и перемешивают в течение 16 часов. Раствор разбавляют дополнительным ДХМ и промывают 1 N HCl (1х), затем сушат над Na2SO4, фильтруют и концентрируют с получением 3-фтор-3-(метоксиметил)азетидин-1-сульфонилхлорида (601 мг, 97,7% выход).
Стадия 4: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-3-(метоксиметил)азетидин-1-сульфонамид трифторацетат. 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-он (25 мг, 0,075 ммоль), 3-фтор-3-(метоксиметил)азетидин-1-сульфонилхлорид (16 мг, 0,075 ммоль) и бис((трифторметил)сульфонил)амид кальция (45 мг, 0,075 ммоль) в толуоле (190 мкл) нагревают до 100°С в течение 16 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, затем фильтруют, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-3-(метоксиметил)азетидин-1-сульфонамида трифторацетата (6,0 мг, 16% выход). 1Н ЯМР (400 МГц, CDCl3) δ 8, 82 (с, 1Н), 7,60 (д, 1Н), 7,53-7,49 (м, 1Н), 7,18-7,14 (т, 1Н), 7,11-7,08 (м, 1Н), 6,69 (шс, 1Н), 5,73 (шс, 1Н), 4,16-4,02 (м, 4Н), 3,73 (с, 3Н), 3,65 (д, 2Н), 3,43 (с, 3Н), 2,95 (с, 3Н); МС (хиад, m/z) = 514,1, 516,1 (М+Н).
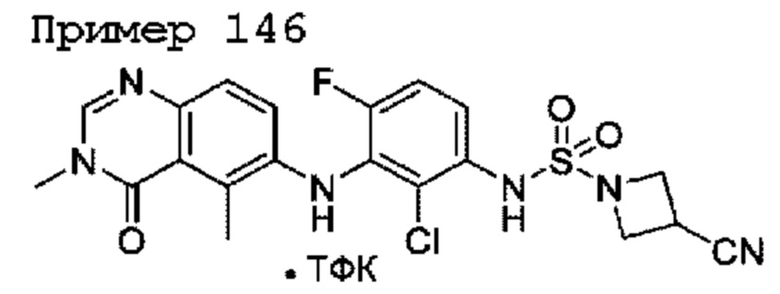
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-цианоазетидин-1-сульфонамид трифторацетат
6-((3-Амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-он (25 мг, 0,0751 ммоль), 3-цианоазетидин-1-сульфонилхлорид (13,6 мг, 0,0751 ммоль) и бис((трифторметил)сульфонил)амид кальция (45,1 мг, 0,0 751 ммоль) суспендируют в толуоле (150 мкл) в пробирке, герметично закрывают и нагревают при 100°С в течение 16 часов. Реакцию оставляют охлаждаться до температуры окружающей среды и концентрируют в вакууме. Неочищенную реакционную смесь очищают хроматографией с обращенной фаз MeCN/H2O, 0, 1% ТФК). Желаемые фракции объединяют и лиофилизируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-цианоазетидин-1-сульфонамида трифторацетата (18 мг, 50% выход). 1Н ЯМР (400 МГц, CDCl3) δ 8,67 (с, 1Н), 7,61-7,58 (м, 1Н), 7,15 (т, 1Н), 7,12-7,09 (м, 1Н), 6,63 (с, 1Н), 5,69 (с, 1Н), 4,27-4,18 (м, 4Н), 3,66 (с, 3Н), 3,50-3, 43 (м, 1Н), 2,97 (с, 3Н). МС (хиад, m/z) = 477,1, 479,1 (М+Н).
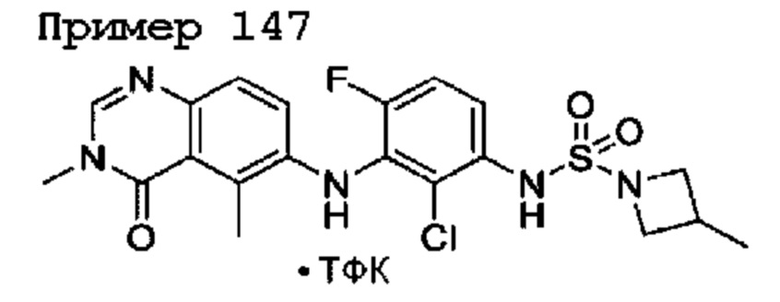
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метилазетидин-1-сульфонамид трифторацетат
Стадия 1: Получение 3-метилазетидин-1-сульфонилхлорид. 3-Метилазетидин гидрохлорид (75 мг, 0,697 ммоль) перемешивают с N-этил-N-изопропилпропан-2-амином (181 мкл, 1,05 ммоль) при температуре окружающей среды в дихлорметане (1743 мкл) в течение 5 минут. Реакционную смесь охлаждают до -10°С (лед/ацетон) и по каплям добавляют сульфурилдихлорид (141 мкл, 1,74 ммоль) в виде чистой жидкости. Реакционную смесь оставляют нагреваться до температуры окружающей среды и перемешивают в течение 16 часов. Раствор разбавляют дополнительный ДХМ и промывают 1 N HCl (1х), затем сушат над Na2SO4, фильтруют и концентрируют с получением 3-метилазетидин-1-сульфонилхлорида (118 мг, 100% выход), который применяют непосредственно на следующей стадии.
Стадия 2: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-метилазетидин-1-сульфонамид трифторацетат. 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3Н)-он (2 0 мг, 0,0 601 ммоль), 3-метилазетидин-1-сульфонилхлорид (10,2 мг, 0,0601 ммоль) и бис((трифторметил)сульфонил)амид кальция (36,1 мг, 0,0 601 ммоль) суспендируют в толуоле (12 0 мкл) в пробирке, герметично закрывают и нагревают при 100°С в течение 16 часов. Реакцию оставляют охлаждаться до температуры окружающей среды и концентрируют в вакууме. Неочищенную реакционную смесь очищают хроматографией с обращенной фазой (5-95% MeCN/H2O, 0,1% ТФК). Желаемые фракции объединяют и лиофилизируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-цианоазетидин-1-сульфонамида трифторацетата (9,4 мг, 33% выход). 1Н ЯМР (400 МГц, CDCl3) δ 8,95 (с, 1Н), 7,63 (д, 1Н), 7,55-7,51 (м, 1Н), 7,16 (т, 1Н), 7,11-7,08 (м, 1Н), 6,62 (с, 1Н), 5,73 (с, 1Н), 4,03 (т, 2Н), 3,73 (с, 3Н), 3,61 (т, 2Н), 2,96 (с, 3Н), 2,74-2,65 (м, 1Н) 1,23 (д, 3Н). МС (хиад, m/z) = 466,l, 468, 1 (М+Н).
Следующие соединения также получают в соответствии с методиками, описанными в настоящем документе.
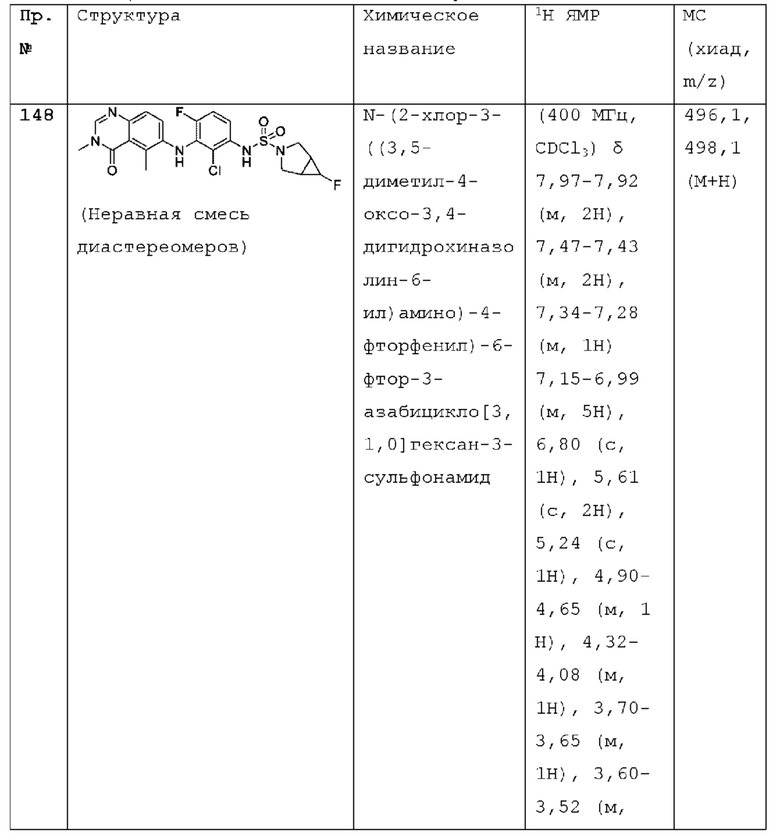

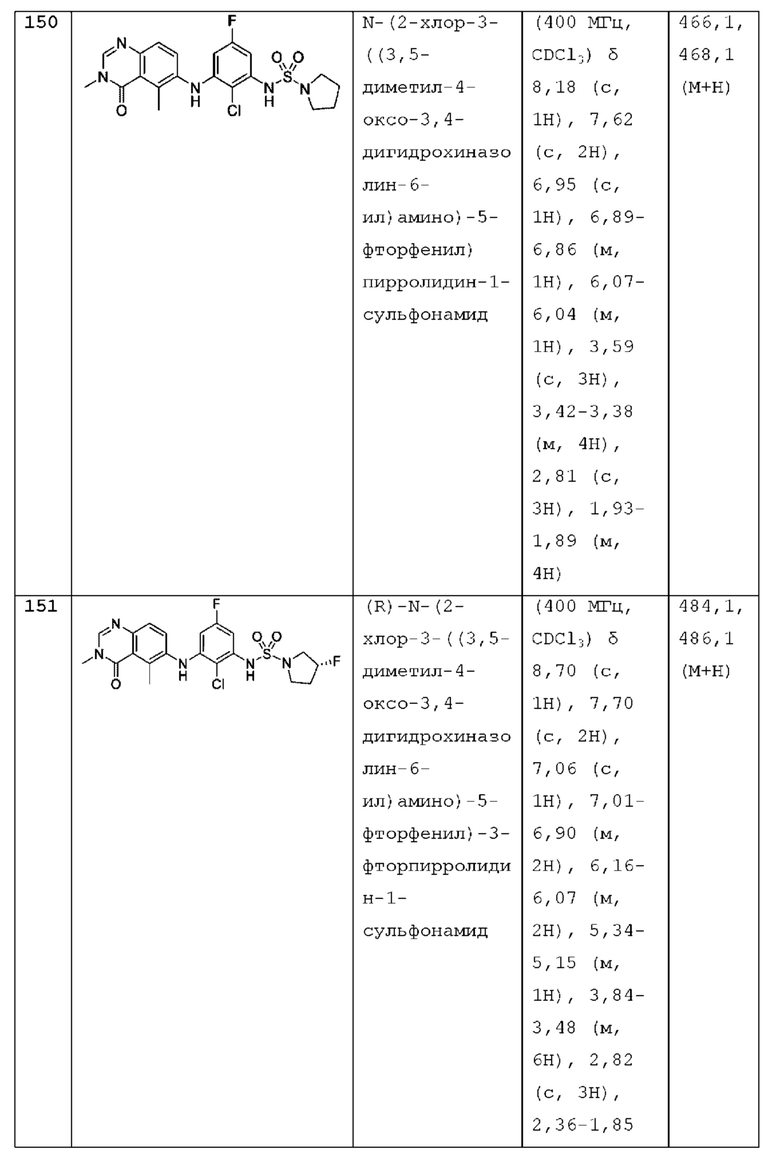
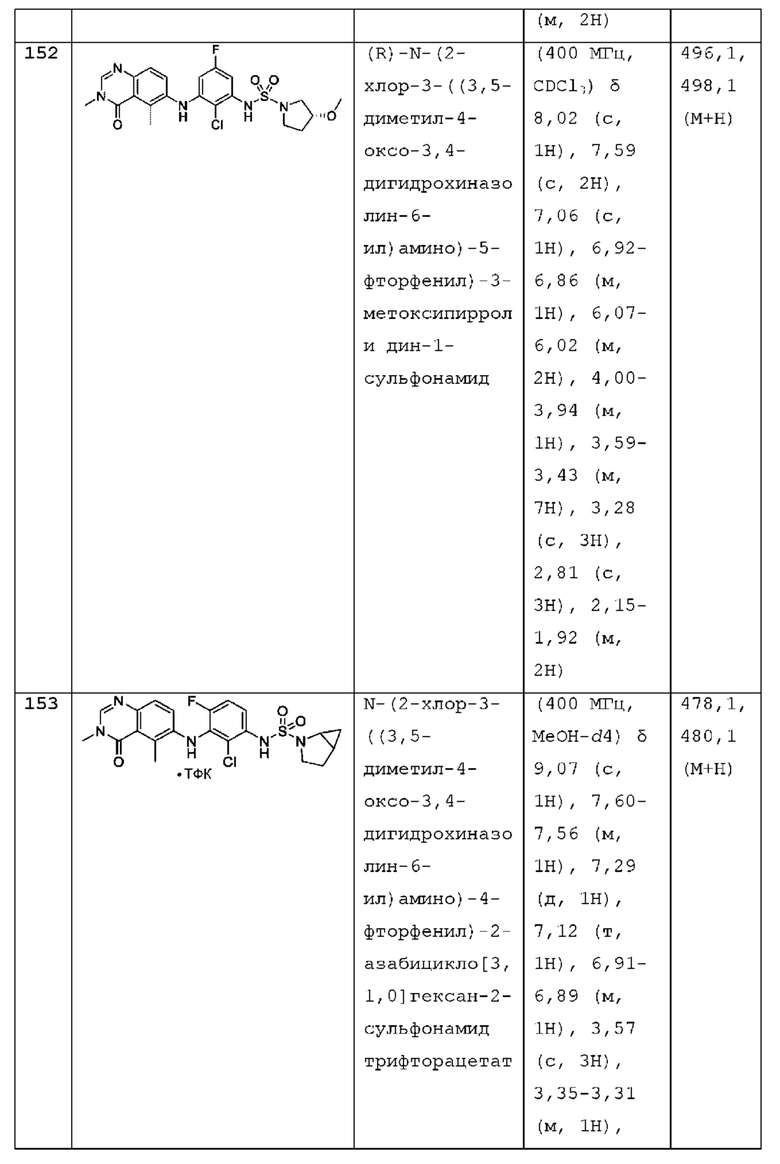
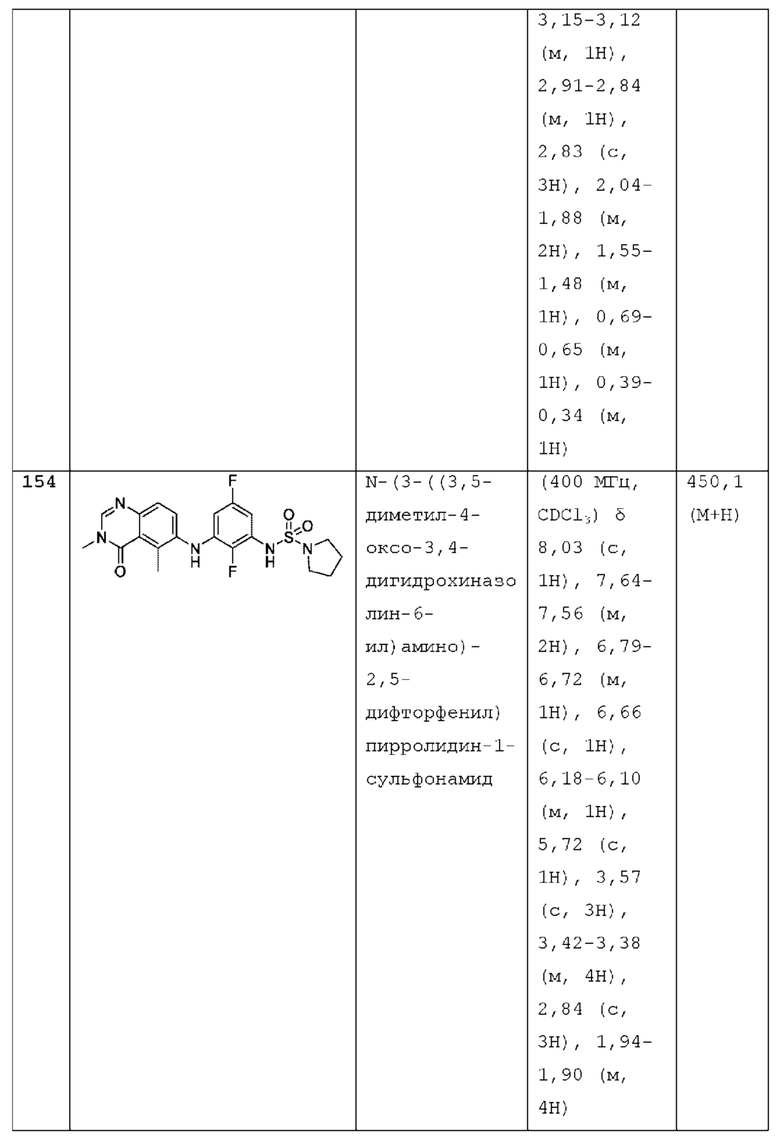

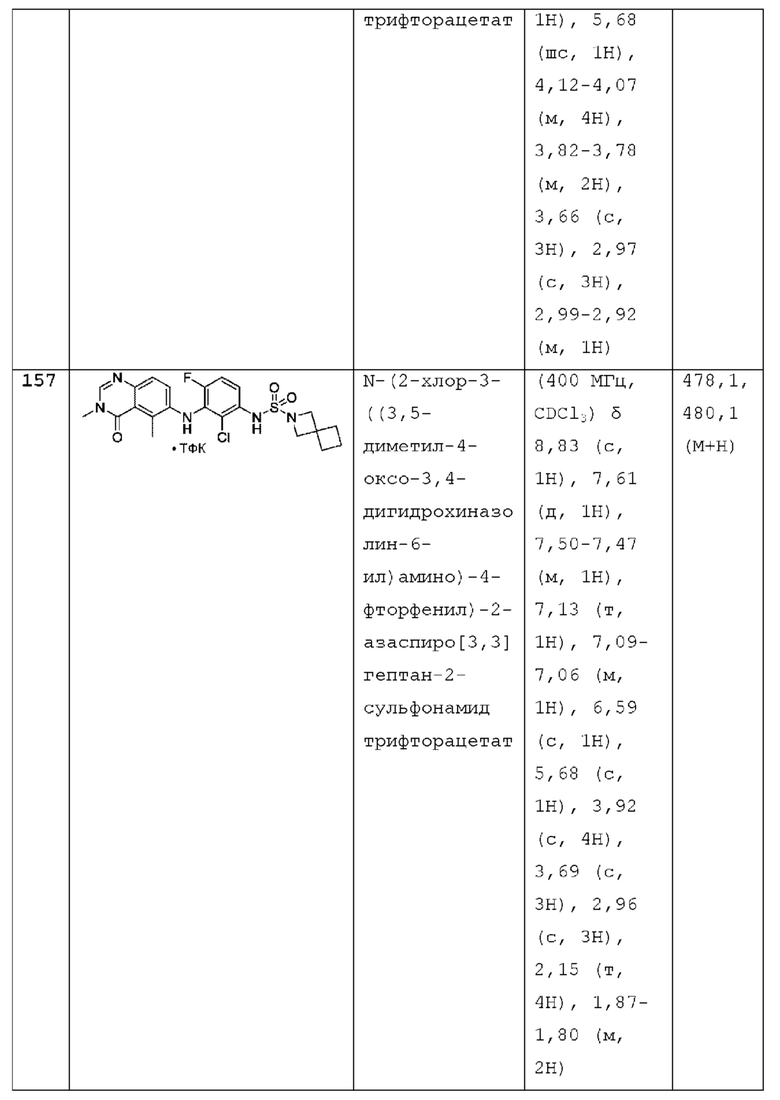
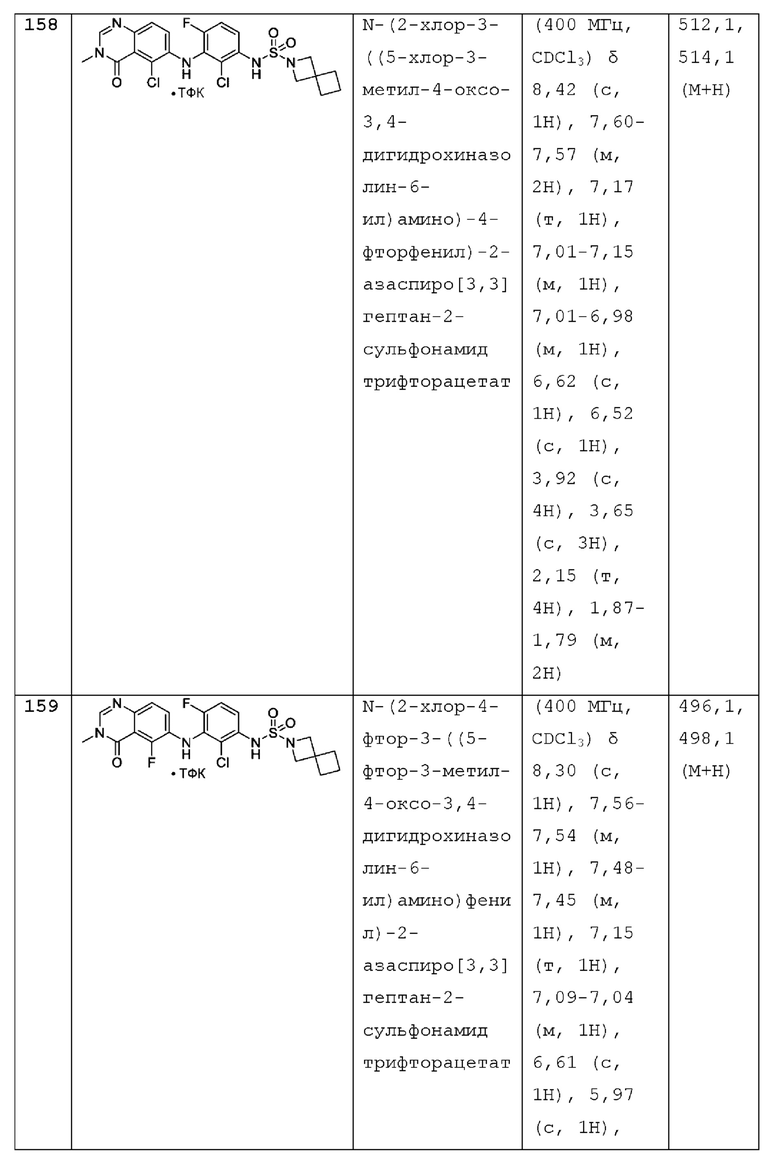
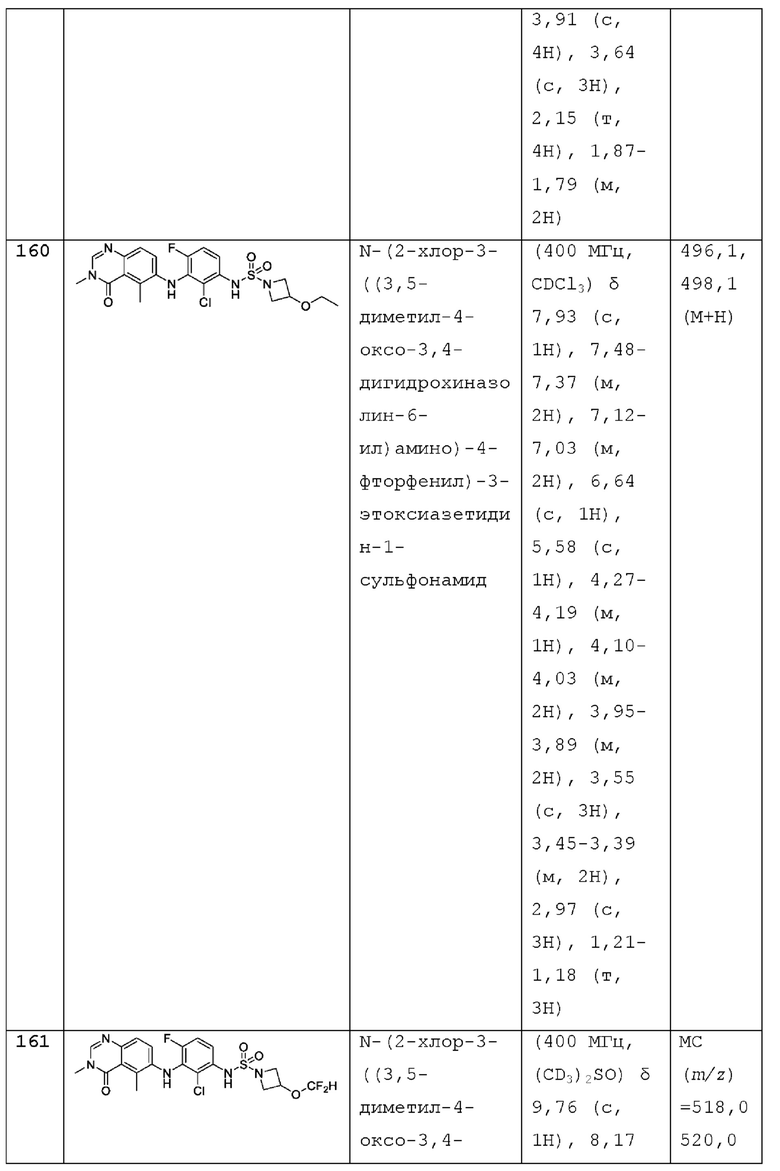
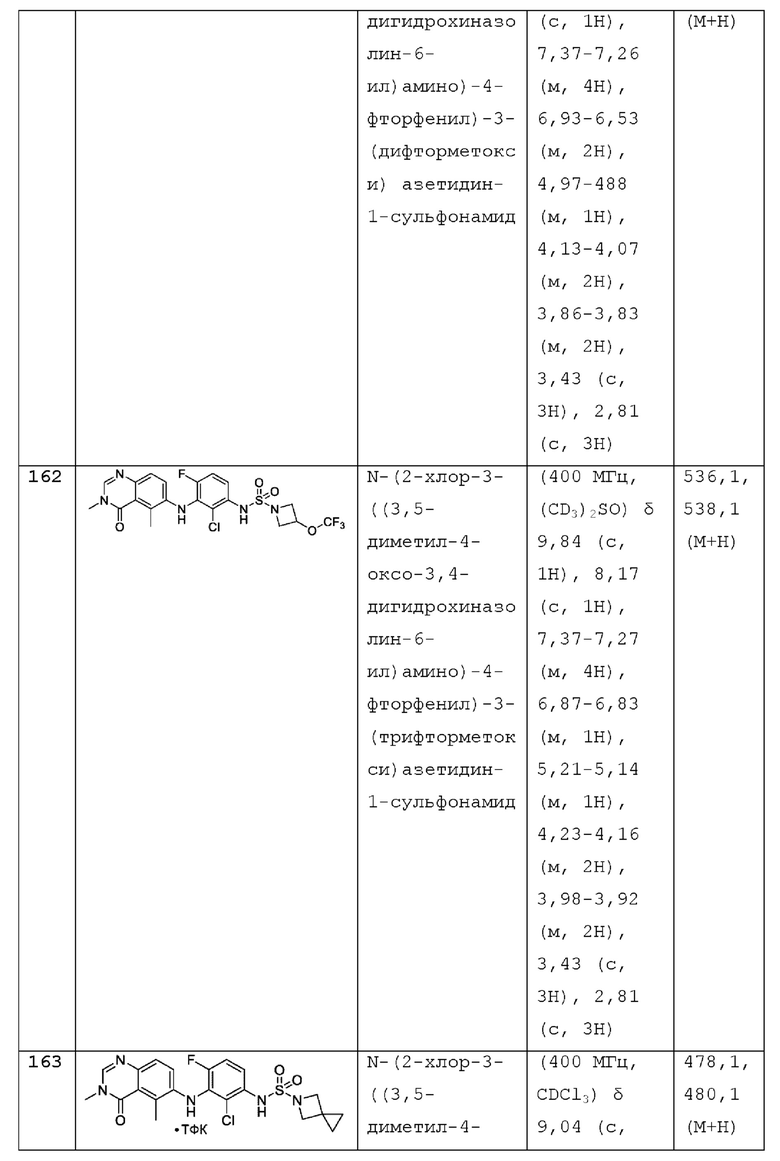


| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ХИНАЗОЛИН-4-ОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2020 |
|
RU2797606C1 |
| 3,4-ДИГИДРО-2,7-НАФТИРИДИН-1,6(2H,7H)-ДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ MEK | 2022 |
|
RU2826000C1 |
| НОВЫЕ ИНГИБИТОРЫ BRAF КАК «РАЗРУШИТЕЛИ ПАРАДОКСА» | 2020 |
|
RU2825870C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2019 |
|
RU2821941C2 |
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛИЛЬНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2020 |
|
RU2811612C2 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ИМИНОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2018 |
|
RU2801302C2 |
| СПИРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2016 |
|
RU2745069C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛО[3,4-b]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ТАМ И МЕТ | 2019 |
|
RU2773611C1 |
| МОДУЛЯТОРЫ MAS-СВЯЗАННОГО РЕЦЕПТОРА G-БЕЛКА X2 И СВЯЗАННЫЕ С НИМИ ПРОДУКТЫ И СПОСОБЫ | 2021 |
|
RU2841536C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ МЕТИЛХИНАЗОЛИНОНА | 2020 |
|
RU2802968C1 |
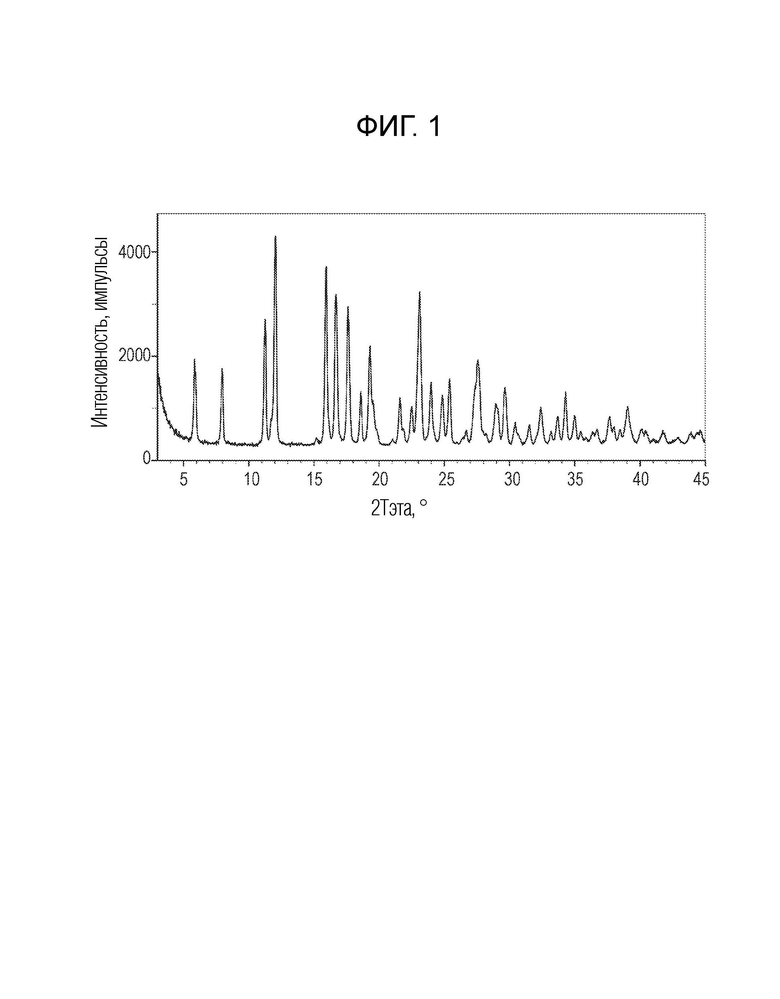
Изобретение относится к соединению, представляющему собой N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид формулы I, или его фармацевтически приемлемой соли, фармацевтической композиции на его основе, а также способу лечения BRAF-ассоциированной опухоли с их использованием. 4 н. и 21 з.п. ф-ы, 1 ил., 140 пр.
1. Соединение, которое представляет собой N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид, имеющий структуру
или его фармацевтически приемлемую соль.
2. Соединение, которое представляет собой N-(2-хлор-3-((5-хлор-3-метил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторазетидин-1-сульфонамид, имеющий структуру
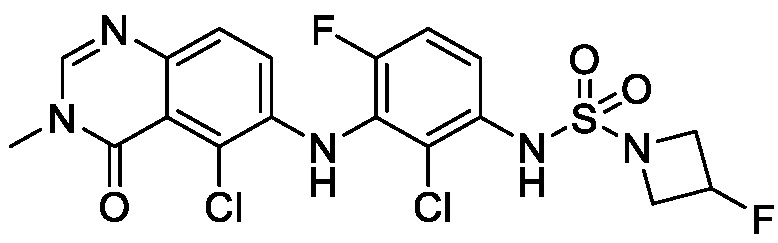 .
.
3. Фармацевтическая композиция для лечения BRAF-ассоциированной опухоли, содержащая терапевтически эффективное количество соединения по п. 1 или 2 или его фармацевтически приемлемой соли и один или несколько фармацевтически приемлемых носителей.
4. Способ лечения BRAF-ассоциированной опухоли у нуждающегося в этом субъекта, включающий введение субъекту терапевтически эффективного количества соединения по п. 1 или 2 или его фармацевтически приемлемой соли.
5. Способ по п. 4, отличающийся тем, что указанная BRAF-ассоциированная опухоль имеет мутацию BRAF II класса.
6. Способ по п. 5, отличающийся тем, что указанная мутация BRAF II класса представляет собой не-V600 мутацию BRAF.
7. Способ по п. 6, отличающийся тем, что указанная не-V600 мутация BRAF представляет собой BRAF G469A или G469R.
8. Способ по п. 5, отличающийся тем, что указанная мутация BRAF II класса представляет собой вариант сплайсинга BRAF V600E.
9. Способ по п. 8, отличающийся тем, что указанный вариант сплайсинга BRAF V600E представляет собой p61BRAF (V600E).
10. Способ по любому из пп. 4-9, отличающийся тем, что указанная BRAF-ассоциированная опухоль представляет собой рак, выбранный из рака легкого, меланомы, колоректального рака, рака молочной железы, рака поджелудочной железы, рака щитовидной железы, рака предстательной железы, аденоидно-кистозной карциномы, рака аппендикса, рака тонкой кишки, плоскоклеточной карциномы головы и шеи, ангиосаркомы, рака мочевого пузыря, новообразования плазматических клеток, гепатопанкреатобилиарной карциномы, карциномы яичника, нейроэндокринного рака, холангиокарциномы и рака ЦНС.
11. Способ по любому из пп. 4-10, отличающийся тем, что указанный рак представляет собой метастатический рак.
12. Способ по п. 11, отличающийся тем, что указанный рак представляет собой метастатический рак ЦНС.
13. Способ по любому из пп. 4-9, отличающийся тем, что указанная BRAF-ассоциированная опухоль представляет собой первичную опухоль головного мозга.
14. Способ по п. 13, отличающийся тем, что указанная первичная опухоль головного мозга представляет собой глиому 2 степени, глиому 3 степени или глиому 4 степени.
15. Способ по п. 4, отличающийся тем, что указанная BRAF-ассоциированная опухоль имеет мутацию BRAF I класса.
16. Способ по п. 15, отличающийся тем, что указанная мутация BRAF I класса представляет собой BRAF V600E или BRAF V600K.
17. Способ по п. 15 или 16, отличающийся тем, что указанная BRAF-ассоциированная опухоль выбрана из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого, рака яичников, почечно-клеточной карциномы и их метастатического рака и первичных опухолей головного мозга.
18. Способ по любому из пп. 4-17, дополнительно включающий введение дополнительной противораковой терапии.
19. Способ по п. 18, отличающийся тем, что дополнительная противораковая терапия выбрана из одного или нескольких из хирургического вмешательства, лучевой терапии и противоракового агента.
20. Способ по п. 19, отличающийся тем, что дополнительная противораковая терапия представляет собой противораковый агент.
21. Способ по п. 20, отличающийся тем, что дополнительный противораковый агент выбран из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов SOS1, ингибиторов пути передачи сигнала, ингибиторов контрольных точек, модуляторов пути апоптоза, цитотоксических химиотерапевтических средств, ангиогенез-таргетной терапии и иммуно-таргетных агентов.
22. Способ по п. 21, отличающийся тем, что дополнительный противораковый агент представляет собой ингибитор MEK.
23. Способ по п. 22, отличающийся тем, что ингибитор МЕК представляет собой биниметиниб или его фармацевтически приемлемую соль.
24. Способ по п. 21, отличающийся тем, что дополнительный противораковый агент представляет собой ингибитор EGFR.
25. Способ по п. 24, отличающийся тем, что ингибитор EGFR представляет собой цетуксимаб.
| WO 2012118492 A1, 07.09.2012 | |||
| WO 2008079903 A1, 03.07.2008 | |||
| WO 2019060611 A1, 28.03.2019 | |||
| КРИСТАЛЛИЧЕСКАЯ ФОРМА N-(3-(5-(4-ХЛОРОФЕНИЛ)-1Н-ПИРАЗОЛО[3,4-В]ПИРИДИН-3-КАРБОНИЛ)-2,4-ДИФТОРОФЕНИЛ) ПРОПАН-1-СУЛЬФОНАМИДА, АКТИВНЫЙ КОМПОНЕНТ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2018 |
|
RU2678455C1 |

