УРОВЕНЬ ТЕХНИКИ
Настоящее изобретение относится к 5-замещенным хиназолинонам для лечения BRAF-ассоциированных заболеваний и нарушений, включая BRAF-ассоциированные опухоли, включая злокачественные и доброкачественные BRAF-ассоциированные опухоли ЦНС и злокачественные экстракраниальные BRAF-ассоциированные опухоли.
Белок BRAF, член семейства серин/треониновых киназ RAF, участвует в каскаде пути подачи сигналов регулируемой Ras-Raf-MEK-внеклеточным сигналом киназы (ERK) или митоген-активированной протеинкиназы (MAPK)/ERK, который влияет на деление и дифференциацию клеток. Мутации в гене BRAF могут привести к неконтролируемому росту и последующему образованию опухоли. Было показано, что BRAF мутирован и/или сверхактивирован при обычных раковых заболеваниях человека, таких как меланома, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников и его метастатические раки, а также первичные опухоли головного мозга. Хотя некоторые ингибиторы BRAF вызывают отличные экстракраниальные реакции, рак все же может развить метастазы в мозг во время или после терапии ингибиторами BRAF (Oliva I.C.G, et al., Annals of Oncology, 29: 1509-1520 (2018)). По оценкам, у 20% всех субъектов с раком развиваются метастазы в головной мозг, при этом большинство метастазов в мозг происходит у пациентов с меланомой, колоректальным раком, раком легких и почечно-клеточной карциномой (Achrol A.S., et al., Nature Reviews (2019), 5:5, pp 1-26). Хотя это наиболее вероятные типы, любой тип рака может распространиться на мозг. Развитие метастазов в головной мозг остается существенным фактором общей смертности от рака у субъектов с прогрессирующей стадией рака, потому что прогноз остается плохим, несмотря на мультимодальное лечение и достижения в области системной терапии, которые включают комбинации хирургического вмешательства, радиационной терапии, химиотерапии, иммунотерапии и/или таргетной терапии.
Кроме того, BRAF был идентифицирован как потенциальная мишень для лечения первичных опухолей головного мозга. О распространенности мутации BRAF-V600E в первичных опухолях головного мозга сообщили Schindler et al. (Acta Neuropathol 121(3):397-405, 2011) на основе анализа 1320 опухолей центральной нервной системы (ЦНС) и Behling et al. (Diagn Pathol 11(1):55, 1-10, 2016), которые проанализировали 969 опухолей ЦНС у детей и взрослых. Эти исследования, в сочетании с другими, сообщают о наличии мутаций BRAF-V600E при различных формах рака, включая папиллярные краниофарингиомы, плеоморфные ксантоастроцитомы (PXA), ганглиоглиомы, астробластомы и другие. (Behling et al., Diagn Pathol 11(1):55, 1-10, 2016; Brastianos et al., Nat Genet 46(2):161-165, 2014; Dougherty et al., Neuro Oncol 12(7):621- 630, 2010; Lehman et al., Neuro Oncol 19(1):31-42, 2017; Mordechai et al., Pediatr Hematol Oncol 32(3):207-211, 2015; Myung et al., Transl Oncol 5(6):430-436, 2012; Schindler et al., Acta Neuropathol 121(3):397-405, 2011).
Гематоэнцефалический барьер (ГЭБ) представляет собой высокоселективный физический, транспортный и метаболический барьер, отделяющий ЦНС от крови. ГЭБ может препятствовать проникновению некоторых лекарственных средств в ткань мозга и является ограничивающим фактором доставки многих периферически вводимых агентов в ЦНС. Многие лекарственные средства, обычно используемые для лечения рака, не могут проникать через ГЭБ. Это означает, что лекарственные средства не могут проникать в мозг и, следовательно, не могут эффективно убивать раковые клетки в головном мозге. Современные методы лечения субъектов с опухолями головного мозга включают хирургическую резекцию, радиационную терапию и/или химиотерапию такими агентами, как темозоломид и/или бевацизумаб. Однако лечение рака головного мозга хирургическим путем не всегда возможно или желательно, например, опухоль может быть недоступна или субъект может быть неспособен выдержать травму при нейрохирургии. Кроме того, известно, что радиационная терапия и лечение цитотоксическими агентами имеют нежелательные побочные эффекты. Например, появляется все больше доказательств того, что использование темозоломида само по себе может вызывать мутации и ухудшать прогноз у значительной части субъектов (B. E. Johnson et al., Science 343: 189-193 (2014)), а на этикетке бевацизумаба есть предупреждения о перфорации желудочно-кишечного тракта, осложнениях при заживлении результатов хирургического вмешательства и ран, а также кровотечении. Ингибиторы киназ полезны для лечения многих периферических видов рака. Однако из-за своих структурных характеристик многие ингибиторы киназ, такие как ингибиторы BRAF (например, вемурафениб и дабрафениб), являются субстратами активных транспортеров, таких как P-гликопротеины (P-gp) или белок резистентности к раку груди (BCRP). Например, сообщалось, что дабрафениб имеет коэффициент эффлюкса MDR1, равный 11,4, коэффициент эффлюкса BCRP, равный 21,0, и общее отношение мозга к плазме, равное 0,023; о свободном соотношении мозга к плазме не сообщалось (Mittapalli, RK, et al., J Pharmacol. Exp Ther 344:655-664, March 2013), и для вемурафениба сообщалось, что коэффициент эффлюкса MDR1 равен 83, a коэффициент эффлюкса BCRP составляет 495, и общее отношение мозга к плазме составляет 0,004; о соотношении свободного мозга к плазме не сообщалось (Mittapalli, RK. et al., J Pharmacol. Exp Ther 342:33-40 (March 2012)
Учитывая, что и P-gp, и BCRP экспрессируются в эндотелиальных клетках, выстилающих кровеносные капилляры головного мозга, активность P-gp и BCRP в ГЭБ играет критическую роль в профилактике распространения большинства киназных ингибиторов в паренхиме мозга. Следовательно, киназные ингибиторы обычно не подходят для лечения опухолей или рака головного мозга, который защищен ГЭБ.
Таким образом, сохраняется потребность в лечении опухолей, несущих мутации BRAF. Кроме того, лечение опухолей ЦНС, в том числе опухолей ЦНС, несущих мутации BRAF, остается неудовлетворенной потребностью.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Следовательно, в настоящем документе представлено соединение формулы I:
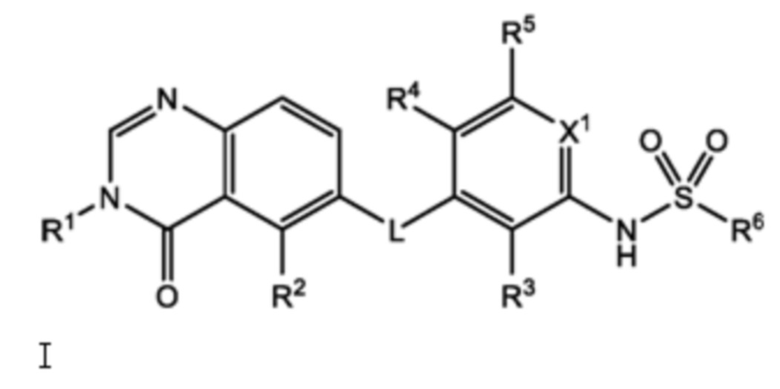
или его фармацевтически приемлемая соль, сольват или полиморф, где L, X1, R1, R2, R3, R4, R5 и R6 такие, как определены в настоящем документе. Определенные соединения формулы I (т.е. соединения формулы II и Formula III как определено в настоящем документе) демонстрируют проникновение в центральную нервную систему (ЦНС) и может быть полезно для лечения BRAF-ассоциированных опухолей ЦНС.
Также в настоящем документе представлена фармацевтическая композиция, содержащая соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, в смеси с фармацевтически приемлемым разбавителем или носителем.
Также в настоящем документе представлен способ лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в таком лечении, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или его фармацевтической композиции, как определено в настоящем документе.
Также в настоящем документе представлен способ ингибирования метастазов, ассоциированных с BRAF-ассоциированной опухолью у субъекта, нуждающегося в таком лечении, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или его фармацевтической композиции, как определено в настоящем документе.
Также в настоящем документе представлен способ ингибирования BRAF киназной активности, in vitro или in vivo, где способ включает контакт клетки с терапевтически эффективным количеством соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или его фармацевтической композиции, как определено в настоящем документе.
Также в настоящем документе представлен способ ингибирования пролиферации клетки, in vitro или in vivo, где способ включает контакт клетки с терапевтически эффективным количеством соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или его фармацевтической композиции, как определено в настоящем документе.
Также в настоящем документе представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф или его фармацевтическая композиция, как определено в настоящем документе, для применения в терапии.
Также в настоящем документе представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф или его фармацевтическая композиция, как определено в настоящем документе, для применения в лечении опухолей.
Также в настоящем документе представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф или его фармацевтическая композиция, как определено в настоящем документе, для применения в ингибировании метастазов, ассоциированных с BRAF-ассоциированной опухолью.
Также в настоящем документе представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, для применения в ингибировании BRAF киназной активности.
Также в настоящем документе представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф или его фармацевтическая композиция, как определено в настоящем документе, для применения в лечении BRAF-ассоциированного заболевания или нарушения (например, BRAF-ассоциированной опухоли).
Также в настоящем документе представлено применение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, как определено в настоящем документе, в производстве лекарственного средства для лечения BRAF-ассоциированной опухоли (например, BRAF-ассоциированной злокачественной опухоли или BRAF-ассоциированной доброкачественной опухоли).
Также в настоящем документе представлено the use соединения формулы I, Formula II or Formula III или его фармацевтически приемлемая соль, сольват или полиморф, как определено в настоящем документе in the manufacture of a medicament for inhibiting metastasis associated with a BRAF-ассоциирован tumor.
Также в настоящем документе представлено использование соединения формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, как определено в настоящем документе, при производстве лекарственного средства для ингибирования активности киназы BRAF.
Также в настоящем документе представлено использование соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, его сольвата или полиморфа, как определено в настоящем документе, при производстве лекарственного средства для лечения BRAF-ассоциированного заболевания или нарушения.
Также в настоящем документе представлен способ лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом, где способ включает (а) определение того, что опухоль ассоциирована с мутацией BRAF; и (b) введение субъекту терапевтического эффективного количества формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или его фармацевтической композиции.
Также в настоящем документе представлена фармацевтическая комбинация для лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом, которая содержит (a) соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф, и (b) дополнительный противораковый агент, где соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль или сольват и дополнительный противораковый агент составлены в виде отдельных композиций или дозировок для отдельного или последовательного применения для лечения BRAF-ассоциированной опухоли, где количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа и дополнительного противоракового агента вместе эффективны при лечении BRAF-ассоциированной опухоли. Также в настоящем документе представлено применение такой комбинации для лечения BRAF-ассоциированной опухоли. также в настоящем документе представлена коммерческая упаковка или продукт, содержащий такую комбинацию в виде комбинированного препарата для раздельного или последовательного применения при лечении BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом.
Также представлены способы лечения субъекта с BRAF-ассоциированной опухолью, которые включают введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа до, во время или после введения другой противораковой терапии (например, хирургического вмешательства, радиотерапии и/или другой противоракового лекарственного средства).
Также в настоящем документе представлен способ получения соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа.
Также в настоящем документе представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, полученный способом получения соединения, как определено в настоящем документе.
Если не указано иное, все технические и научные термины, используемые в данном документе, имеют то же значение, которое обычно понимается специалистом в области, к которой принадлежит это изобретение. В настоящем документе описаны способы и материалы для использования в настоящем изобретении; также могут быть использованы другие подходящие способы и материалы, известные в данной области техники. Материалы, способы и примеры являются только иллюстративными и не предназначены для ограничения. Все публикации, заявки на патенты, патенты, последовательности, записи в базе данных и другие ссылки, упомянутые в данном документе, полностью включены посредством ссылки. В случае противоречия преимущественную силу имеет настоящее описание, включая определения.
Другие особенности и преимущества изобретения будут очевидны из следующего подробного описания и чертежей, а также из формулы изобретения.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
На фигуре 1 показана порошковая рентгеновская дифрактограмма (ПРД) кристаллической формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, Безводной формы А, согласно одному варианту осуществления.
На фигуре 2 показан профиль дифференциальной сканирующей калориметрии (ДСК) N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида Безводной формы А, согласно одному варианту осуществления.
На фигуре 3 показано наложение скана дифференциальной сканирующей калориметрии (ДСК) и скана термогравиметрического анализа (ТГА) N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида формы A.
На фигуре 4 показана порошковая рентгеновская дифрактограмма (ПРД) кристаллической формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы B, согласно одному варианту осуществления.
На фигуре 5 показан профиль дифференциальной сканирующей калориметрии (ДСК) кристаллической формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, безводной формы B, согласно одному варианту осуществления.
На фигуре 6 представлено наложение скана дифференциальной сканирующей калориметрии и скана термогравиметрического анализа N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы B, согласно одному варианту осуществления.
На фигуре 7 показана порошковая рентгеновская дифрактограмма (ПРД) кристаллической формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, моно-анизола формы С, согласно одному варианту осуществления.
На фигуре 8 показано наложение скана дифференциальной сканирующей калориметрии (ДСК) и скана термогравиметрического анализа (ТГА) N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, моно-анизола формы C, согласно одному варианту осуществления.
На фигуре 9 показана порошковая рентгеновская дифрактограмма (ПРД) кристаллической формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, полу-дихлорметана формы D, согласно одному варианту осуществления.
На фигуре 10 показано наложение скана дифференциальной сканирующей калориметрии и скана термогравиметрического анализа N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, полу-дихлорметана формы D, согласно одному варианту осуществления.
На фигуре 11 показана порошковая рентгеновская дифрактограмма (ПРД) кристаллической формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида толуольной формы E, согласно одному варианту осуществления.
На фигуре 12 показано наложение скана дифференциальной сканирующей калориметрии и скана термогравиметрического анализа N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы E, согласно одному варианту осуществления.
На фигуре 13 показана порошковая рентгеновская дифрактограмма (ПРД) кристаллической формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, 1,4-диоксана формы F, согласно одному варианту осуществления.
На фигуре 14 представлено наложение скана дифференциальной сканирующей калориметрии и скана термогравиметрического анализа N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, 1,4-диоксана формы F, согласно одному варианту осуществления.
На фигуре 15 представлен график, показывающий общую биолюминесценцию, нормализованную на мышь, в дни 1-60 перорального введения соединения примера 82 отдельно или в комбинации с биниметинибом в модели A375-люциферазной внутричерепной опухоли у мышей относительно носителя.
На фигуре 16 представлен график Каплана-Мейера в дни 1-60 перорального введения соединения примера 82 отдельно или в комбинации с биниметинибом в модели A375-люциферазной внутричерепной опухоли у мышей относительно носителя, и в течение 30 дней после введения.
На фигуре 17 представлена порошковая рентгеновская дифрактограмма (ПРД) аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, согласно одному варианту осуществления.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем документе представлено соединение формулы I:
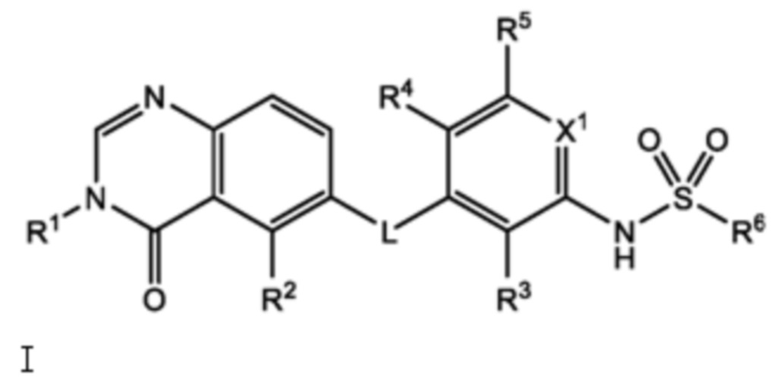
и его фармацевтически приемлемые соли, сольваты и полиморфы, где:
L является O, NH или S;
X1 является CH или N;
R1 является C1-C6 алкилом, C1-C6 дейтероалкилом, C1-C6 фторалкилом, C3-C6 циклоалкилом, (C3-C6 циклоалкил)CH2-, (C1-C6 алкокси)C1-C6 алкилом-, Ar1, Ar1CH2-, hetAr1 или hetCyc1;
Ar1 является фенилом, который необязательно замещен 1-5 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetAr1 является 5-6-членным гетероарильным кольцом, имеющим 1-2 кольцевых атома азота и необязательно замещенным 1-3 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetCyc1 является 4-6-членным насыщенным гетероциклическим кольцом, имеющим один кольцевой атом кислорода;
R2 является метилом, -CD3 или HC≡C-;
R3 является F, Cl, CN или метилом;
R4 является H, F или Cl;
R5 является H, F, Cl или метил;
R6 является C1-C6 алкилом, C1-C6 фторалкилом, (Cyc1)C1-C6 алкилом-, (C1-C3 алкокси)C1-C6 алкилом-, hetAr2, Ar2 или RaRbN-;
Cyc1 является 3-6-членным насыщенным карбоциклическим кольцом;
hetAr2 является 5-6-членным гетероарилом, имеющим 1-2 кольцевых гетероатома, независимо выбранных из N, O и S и необязательно замещенных 1-3 заместителями, независимо выбранными из галогена и C1-C3 алкила;
Ar2 является фенилом, необязательно замещенным 1-5 заместителями, независимо выбранными из галогена и C1-C6 алкила; и
Ra и Rb независимо являются C1-C3 алкилом, или
Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, необязательно замещенное 1-2 галогенами.
Для сложных химических названий, используемых в данном документе, группа заместителя обычно упоминается перед группой, к которой она присоединена. Например, метоксиэтил включает этильную основную цепь с метокси заместителем.
Термин «галоген» означает -F (иногда обозначаемый здесь как «фтор» или «фтористый»), -Cl, -Br и -I.
Используемые здесь термины «C1-C3алкил» и «C1-C6алкил» относятся к насыщенным линейным или разветвленным одновалентным углеводородным радикалам, содержащим от одного до трех или от одного до шести атомов углерода, соответственно. Примеры алкильных групп включают, но не ограничиваются ими, метил, этил, 1-пропил, изопропил, 1-бутил, изобутил, втор-бутил, трет-бутил, 2-метил-2-пропил, пентил, неопентил и гексил.
Термин «C1-C6-фторалкил», используемый в настоящем документе, относится к C1-C6-алкильному радикалу, как определено в настоящем документе, где от одного до трех атомов водорода замещены на от одного до трех атомов фтора, соответственно. Примеры включают, но не ограничиваются ими, фторметил, дифторметил, трифторметил, 2-фторэтил, 2,2-дифторэтил, 2,2,2- и трифторэтил.
Термин «C1-C6 дейтероалкил», используемый в настоящем документе, относится к C1-C6алкильному радикалу, как определено в настоящем документе, который замещен от одного до шести атомов дейтерия. Пример включает, но не ограничивается им, -CD3.
Термины «C1-C3 алкокси» и «C1-C6 алкокси», используемые в настоящем описании, относятся к насыщенным одновалентным алкокси радикалам с линейной или разветвленной цепью, содержащим от одного до трех или от одного до шести атомов углерода, соответственно, где радикал находится на атоме кислорода. Примеры алкоксигрупп включают метокси, этокси, пропокси и изопропокси.
Термины «(C1-C3 алкокси) C1-C6 алкил» и «(C1-C6 алкокси) C1-C6 алкил» в контексте настоящего описания относятся к насыщенным одновалентным радикалам с линейной или разветвленной цепью, содержащим от одного до шести атомов углерода, где один из атомы углерода замещены C1-C3 алкоксигруппой или C1-C6 алкоксигруппой, соответственно, как определено в настоящем документе. Примеры включают метоксиметил (CH3OCH2-) и метоксиэтил (CH3OCH2CH2-).
Используемый здесь термин «C3-C6 циклоалкил» относится к циклопропилу, циклобутилу, циклопентилу или циклогексилу.
Термин «(Cyc1) C1-C6алкил», используемый в настоящем документе, относится к C1-C6алкильному радикалу, как определено в настоящем документе, где один из атомов углерода замещен группой Cyc1, как определено в настоящем документе. Примером является циклопропилметил.
Термин «5-6-членное насыщенное моноциклическое гетероциклическое кольцо» по отношению к кольцу, образованному RaRbN-, где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-6-членное насыщенное моноциклическое гетероциклическое кольцо относится к гетероциклическому кольцу, имеющему один кольцевой атом азота.
Используемый здесь термин «Boc» относится к трет-бутилоксикарбонильной группе, т.е. (CH3)3COC(=O)-.
На протяжении всего описания будет понятно, что количество и природа необязательных групп заместителей будут ограничены в той степени, в которой такие замещения имеют химический смысл.
Термин «соединение», используемый в настоящем документе, включает все стереоизомеры, геометрические изомеры, таутомеры и изотопы изображенных структур. Подразумевается, что соединения, идентифицированные в настоящем документе по названию или структуре как одна конкретная таутомерная форма, включают другие таутомерные формы, если не указано иное.
Термин «таутомер», используемый в настоящем документе, относится к соединениям, структуры которых различаются расположением атомов, но которые существуют в легком и быстром равновесии, и следует понимать, что соединения, представленные в данном документе, могут быть изображены как разные таутомеры, и когда соединения имеют таутомерные формы, предполагается, что все таутомерные формы входят в объем изобретения, и название соединений не исключает какой-либо таутомер.
Следует понимать, что определенные соединения, представленные в настоящем документе, могут содержать один или несколько центров асимметрии и поэтому могут быть получены и выделены в смеси изомеров, такой как рацемическая смесь или в энантиомерно чистой форме.
В одном варианте осуществления формулы I, X1 является CH.
В одном варианте осуществления формулы I, X1 является N.
В одном варианте осуществления формулы I, L является NH.
В одном варианте осуществления формулы I, L является O.
В одном варианте осуществления формулы I, L является S.
В одном варианте осуществления формулы I, R1 является C1-C6 алкилом. Неограничивающие примеры включают метил, этил и изопропил.
В одном варианте осуществления формулы I, R1 является C1-C6 дейтероалкилом. Неограничивающий пример включает -CD3.
В одном варианте осуществления формулы I, R1 является C1-C6 фторалкилом. Неограничивающий пример включает 2,2,2-трифторэтил.
В одном варианте осуществления формулы I, R1 является C3-C6 циклоалкилом. Неограничивающие примеры включают циклопропил, циклобутил и циклопентил.
В одном варианте осуществления формулы I, R1 является (C3-C6 циклоалкил)CH2-. Неограничивающий пример включает циклопропилметил.
В одном варианте осуществления формулы I, R1 является (C1-C6 алкокси)C1-C6 алкилом-. Неограничивающий пример включает метоксиэтил.
В одном варианте осуществления формулы I, R1 является Ar1. В одном варианте осуществления, Ar1 является фенилом, который необязательно замещен 1-3 заместителями, независимо выбранными из галогена и C1-C3 алкила. Неограничивающим примером Ar1 является фенил.
В одном варианте осуществления формулы I, R1 является Ar1CH2-. В одном варианте осуществления, Ar1 часть необязательно замещен 1-2 заместителями, независимо выбранными из галогена и C1-C3 алкила. Неограничивающим примером Ar1CH2- является бензил.
В одном варианте осуществления формулы I, R1 является hetAr1. В одном варианте осуществления, hetAr1 является 5-6-членным гетероарильным кольцом, имеющим 1-2 атома азота и необязательно замещенным 1-2 заместителями, независимо выбранными из галогена и C1-C3 алкила. В одном варианте осуществления, hetAr1 не замещен. Неограничивающим примером является пиридил.
В одном варианте осуществления формулы I, R1 является hetCyc1. Неограничивающий пример включает тетрагидрофуранил.
В одном варианте осуществления формулы I, R2 является метилом.
В одном варианте осуществления формулы I, R2 является -CD3.
В одном варианте осуществления формулы I, R2 является HC≡C-.
В одном варианте осуществления формулы I, R3 является F.
В одном варианте осуществления формулы I, R3 является Cl.
В одном варианте осуществления формулы I, R3 является CN.
В одном варианте осуществления формулы I, R3 является метилом.
В одном варианте осуществления формулы I, R4 является H.
В одном варианте осуществления формулы I, R4 является F.
В одном варианте осуществления формулы I, R4 является Cl.
В одном варианте осуществления формулы I, R5 является H.
В одном варианте осуществления формулы I, R5 является F.
В одном варианте осуществления формулы I, R5 является Cl.
В одном варианте осуществления формулы I, R5 является метилом.
В одном варианте осуществления формулы I, R6 является C1-C6 алкилом. Неограничивающие примеры включают этил, пропил, 2-метилпропил и 1-метилпропил.
В одном варианте осуществления формулы I, R6 является C1-C6 фторалкилом. Неограничивающий пример включает 3-фторпропил.
В одном варианте осуществления формулы I, R6 является (Cyc1)C1-C6 алкилом-. Неограничивающий пример включает циклопропилметил.
В одном варианте осуществления формулы I, R6 является (C1-C3 алкокси)C1-C6 алкилом-. Неограничивающий пример включает 2-метоксиэтил.
В одном варианте осуществления формулы I, R6 является hetAr2. В одном варианте осуществления, hetAr2 является 5-6-членным гетероарилом, имеющим 1-2 кольцевых гетероатома, независимо выбранных из N, O и S и необязательно замещенным 1-2 заместителями, независимо выбранными из галогена и C1-C3 алкила. В одном варианте осуществления, hetAr2 является 5-6-членным гетероарилом, имеющим 1 кольцевой гетероатом, независимо выбранный из N и O и необязательно замещенным 1-2 заместителями, независимо выбранными из галогена и C1-C3 алкила. В одном варианте осуществления, hetAr2 является незамещенным 5-6-членным гетероарилом, имеющим 1 кольцевой гетероатом, выбранный из N и O. Неограничивающие примеры включают пиридил и фуранил.
В одном варианте осуществления формулы I, R6 является Ar2. В одном варианте осуществления, Ar2 является фенилом, необязательно замещенным 1-2 заместителями, независимо выбранными из галогена и C1-C6 алкила. В одном варианте осуществления, Ar2 является фенилом, необязательно замещенным 1-2 галогенами. В одном варианте осуществления, Ar2 является фенилом, необязательно замещенным 1-2 фторами. Неограничивающие примеры включают фенил и 2,4-дифторфенил.
В одном варианте осуществления формулы I, R6 является RaRbN-.
В одном варианте осуществления формулы I, R6 является RaRbN-, где Ra и Rb независимо являются C1-C3 алкилом. Неограничивающим примером является N-этил-N-метиламино.
В одном варианте осуществления формулы I, R6 является RaRbN-, где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, необязательно замещенное 1-2 галогенами. В одном варианте осуществления формулы I, R6 является RaRbN-, где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-членное насыщенное моноциклическое гетероциклическое кольцо, необязательно замещенное 1-2 галогенами. В одном варианте осуществления формулы I, R6 является RaRbN-, где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-членное насыщенное моноциклическое гетероциклическое кольцо, необязательно замещенное 1-2 фторами. В одном варианте осуществления формулы I, R6 является RaRbN-, где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-членное насыщенное моноциклическое гетероциклическое кольцо, необязательно замещенное фтором. Неограничивающим примером является 3-фторпирролидинил.
В одном варианте осуществления формулы I, R6 является C1-C6 алкилом, C1-C6 фторалкилом, (Cyc1)C1-C6 алкилом-, (C1-C3 алкокси)C1-C6 алкилом-, hetAr2 Ar2.
В одном варианте осуществления, X1 является CH и L является NH.
В одном варианте осуществления формулы I, X1 является CH и L является O.
В одном варианте осуществления формулы I, X1 является CH и L является S.
В одном варианте осуществления формулы I, X1 является N и L является NH.
В одном варианте осуществления формулы I, X1 является N и L является O.
В одном варианте осуществления формулы I, X1 является N и L является S.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, и R1, R3, R4, R5 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является F, Cl, CN или метилом, R4 является H, F или Cl, R5 является H, F, Cl или метилом, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является F, R4 является H, F или Cl, R5 является H, F, Cl или метилом, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является F, R4 является H, F или Cl, R5 является H, F, Cl или метилом, R6 является C1-C6 алкилом или C1-C6 фторалкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является F, R4 является H, F или Cl, R5 является H, F, Cl или метилом, R6 является C1-C6 алкилом или C1-C6 фторалкилом, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является Cl, R4 является H или F, R5 является H, F или Cl, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является Cl, R4 является H или F, R5 является H, F или Cl, R6 является C1-C6 алкилом или C1-C6 фторалкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является Cl, R4 является H или F, R5 является H, F или Cl, R6 является C1-C6 алкилом или C1-C6 фторалкилом, и R1 является C1-C6 алкилом или C1-C6 дейтероалкилом.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является CN, R4 является H или F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является CN, R4 является H или F, R5 является H, R6 является C1-C6 алкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является CN, R4 является H или F, R5 является H, R6 является C1-C6 алкилом, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является метилом, R4 является F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является метилом, R4 является F, R5 является H, R6 является C1-C6 алкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является метилом, R3 является метилом, R4 является F, R5 является H, R6 является C1-C6 алкилом, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является HC≡C-, R3 является F, Cl, CN или метилом, R4 является H, F или Cl, R5 является H, F, Cl или метилом, и R1 и R6 такие, как определены для формулы I
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является HC≡C-, R3 является Cl, R4 является F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является HC≡C-, R3 является Cl, R4 является F, R5 является H, R6 является C1-C6 алкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является NH, R2 является HC≡C-, R3 является Cl, R4 является F, R5 является H, R6 является C1-C6 алкилом, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является N, L является NH, R2 является метилом, и R1, R3, R4, R5 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является N, L является NH, R2 является метилом, R3 является F, R4 является F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является N, L является NH, R2 является метилом, R3 является F, R4 является F, R5 является H, R6 является C1-C6 алкилом, C1-C6 фторалкилом или (Cyc1)C1-C6 алкилом-, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является N, L является NH, R2 является метилом, R3 является F, R4 является F, R5 является H, R6 является C1-C6 алкилом, C1-C6 фторалкилом или (Cyc1)C1-C6 алкилом-, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является N, L является NH, R2 является метилом, R3 является Cl, R4 является F или Cl, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является N, L является NH, R2 является метилом, R3 является Cl, R4 является F или Cl, R5 является H, R6 является C1-C6 алкилом, C1-C6 фторалкилом или (Cyc1)C1-C6 алкилом-, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является N, L является NH, R2 является метилом, R3 является Cl, R4 является F или Cl, R5 является H, R6 является C1-C6 алкилом, C1-C6 фторалкилом или (Cyc1)C1-C6 алкилом-, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, и R1, R3, R4, R5 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является F, R4 является H или F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является F, R4 является H или F, R5 является H, R6 является C1-C6 алкилом или C1-C6 фторалкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является F, R4 является H или F, R5 является H, R6 является C1-C6 алкилом или C1-C6 фторалкилом, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является Cl, R4 является F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является Cl, R4 является F, R5 является H, R6 является C1-C6 алкилом или C1-C6 фторалкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является Cl, R4 является F, R5 является H, R6 является C1-C6 алкилом, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является CN, R4 является H или F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является CN, R4 является H или F, R5 является H, R1 является C1-C6 алкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является O, R2 является метилом, R3 является CN, R4 является H или F, R5 является H, R6 является C1-C6 алкилом, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является N, L является O, R2 является метилом, и R1, R3, R4, R5 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является N, L является O, R2 является метилом, R3 является Cl, R4 является H или F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является N, L является O, R2 является метилом, R3 является Cl, R4 является H или F, R5 является H, R6 является C1-C6 алкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является N, L является O, R2 является метилом, R3 является Cl, R4 является H или F, R5 является H, R6 является C1-C6 алкилом, и R1 является метилом.
В одном варианте осуществления формулы I, является X1 является N, L является O, R2 является метилом, R3 является F, R4 является F, R5 является F, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, является X1 является N, L является O, R2 является метилом, R3 является F, R4 является F, R5 является F, R6 является C1-C6 алкилом или фторC1-C6 алкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, является X1 является N, L является O, R2 является метилом, R3 является F, R4 является F, R5 является F, R6 является C1-C6 алкилом или фторC1-C6 алкилом, и R1 является C1-C6 алкилом.
В одном варианте осуществления формулы I, X1 является CH, L является S, R2 является метилом, и R1, R3, R4, R5 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является S, R2 является метилом, R3 является Cl, R4 является F, R5 является H, и R1 и R6 такие, как определены для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является S, R2 является метилом, R3 является Cl, R4 является F, R5 является H, R6 является C1-C6 алкилом, и R1 такой, как определен для формулы I.
В одном варианте осуществления формулы I, X1 является CH, L является S, R2 является метилом, R3 является Cl, R4 является F, R5 является H, R6 является C1-C6 алкилом, и R1 является C1-C6 алкилом.
Любой из вышеупомянутых вариантов осуществления формулы I можно комбинировать друг с другом.
Соединения формулы I включают их фармацевтически приемлемые соли. Кроме того, соединения формулы I также включают другие соли таких соединений, которые не обязательно являются фармацевтически приемлемыми солями, и которые могут быть полезны в качестве промежуточных продуктов для получения и/или очистки соединения формулы I и/или для разделения энантиомеров соединения формулы I.
Термин «фармацевтически приемлемая соль» относится к обычной кислотно-аддитивной или основно-аддитивной соли, которая сохраняет биологическую эффективность и свойства соединений формулы (I), и которые могут быть образованы с подходящими не токсичными органическими или неорганическими кислотами или органическими или неорганическими основаниями. Примеры кислотно-аддитивных солей включают соли, полученные из неорганических кислот, таких как, но не ограниченных ими, хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, сульфаминовая кислота, фосфорная кислота, азотная кислота и перхлорная кислота, полученные из различных органических кислот, таких как, но не ограниченных ими, уксусная кислота, пропионовая кислота, бензойная кислота, гликолевая кислота, фенилуксусная кислота, салициловая кислота, малоновая кислота, малеиновая кислота, олеиновая кислота, памовая кислота, пальмитиновая кислота, бензолсульфоновая кислота, толуолсульфоновая кислота, метансульфоновая кислота, щавелевая кислота, винная кислота, янтарная кислота, лимонная кислота, яблочная кислота, молочная кислота, глутаминовая кислота, фумаровая кислота и подобные. Примерами основно-аддитивных солей являются соли, полученные из гидроксидов аммония, калия, натрия и четвертичного аммония, таких как гидроксид тетраметиламмония. Эти соли часто проявляют более благоприятные свойства растворимости, чем соединения, используемые для их получения, и поэтому более подходят для использования при приготовлении различных фармацевтических составов. В одном варианте осуществления, фармацевтически приемлемые соли формулы I включают кислые соли, такие как трифторацетат.
Кроме того, следует понимать, что соединения формулы I, формулы II и формулы III или их соли могут быть выделены в форме сольватов, и, соответственно, любой такой сольват включен в объем настоящего изобретения.
Термин «сольват» относится к не ковалентным стехиометрическим или не стехиометрическим комбинациям растворителя и растворенного вещества. Термин «гидрат» относится к не ковалентным стехиометрическим или не стехиометрическим комбинациям воды и растворенного вещества. Например, соединения формулы I, формулы II и формулы III и их фармацевтически приемлемые соли и полиморфы могут существовать в не сольватированных, а также в сольватированных формах с фармацевтически приемлемыми растворителями, такими как анизол, дихлорметан, толуол, 1,4-диоксан, вода и подобные.
Соединения формулы I, формулы II и формулы III могут существовать в различных геометрических изомерных формах. Кроме того, некоторые соединения формулы I, формулы II и формулы III могут содержать один или несколько асимметричных центров, таким образом, существовать в стереоизомерных и диастереомерных формах. Термин «стереоизомер» обозначает соединение, которое обладает идентичной молекулярной связностью и множественностью связей, но которое отличается расположением своих атомов в пространстве. Все эти соединения, такие как цис-изомеры, транс-изомеры, диастереомерные смеси, рацематы, нерацемические смеси энантиомеров, по существу чистые и чистые энантиомеры, входят в объем изобретения. В одном варианте осуществления, по существу чистые энантиомеры содержат до 5% масс. соответствующего противоположного энантиомера. В одном варианте осуществления, по существу чистые энантиомеры содержат до 2% масс. соответствующего противоположного энантиомера. В одном варианте осуществления, по существу чистые энантиомеры содержат до 1% масс. соответствующего противоположного энантиомера.
Оптические изомеры могут быть получены путем разделения рацемических смесей известными методами, например, с использованием оптически активной кислоты или основания с образованием диастереоизомерных солей или путем образования ковалентных диастереомеров. Подходящие кислоты включают, например, винную кислоту, диацетилвинную кислоту, дибензоилвинную кислоту, дитолуоилвинную кислоту и камфорсульфоновую кислоту. Смеси диастереоизомеров могут быть разделены на отдельные диастереомеры на основе их физических и/или химических различий способами, известными специалистам в данной области техники, такими как хроматография или фракционная кристаллизация. Затем оптически активные основания или кислоты высвобождают из разделенных диастереоизомерных солей. Различные способы разделения оптических изомеров включают хиральную хроматографию (например, хиральные колонки для ВЭЖХ), необязательно используемую посредством дериватизации с целью максимального разделения энантиомеров. Подходящими колонками для хиральной ВЭЖХ являются колонки Diacel, такие как колонки CHIRALPAK или CHIRALCEL, которые обычно выбирают по желанию. Там, где это применимо, также можно использовать ферментативное разделение, проводимое дериватизацией. Оптически активные соединения формулы II и формулы III также могут быть получены с использованием оптически активных исходных материалов с использованием хирального синтеза без условий реакции рацемизации.
В одном варианте осуществления, соединения формулы I включают соединения примеров 1-80 и их фармацевтически приемлемые соли, сольваты и полиморфы (например, примеры 81-86). В одном варианте осуществления, соединения примеров 1-80 имеют форму свободного основания. В одном варианте осуществления, соединения примеров 1-80 имеют форму кислой соли. В одном варианте осуществления, одно или несколько соединений примеров 1-80 являются трифторацетатом.
Термин «фармацевтически приемлемый» указывает на то, что соединение, его соль или композиция химически и/или токсикологически совместимы с другими ингредиентами, составляющими состав, и/или субъектом, которого ими лечат.
Представленные в настоящем документе соединения могут также содержать не природные пропорции атомных изотопов у одного или нескольких атомов, которые составляют такие соединения. То есть, атом, в частности, когда он упоминается в отношении соединения формулы I, формулы II и формулы III, включает все изотопы и смеси изотопов этого атома, либо встречающиеся в природе, либо полученные синтетическим путем, либо в том виде, в котором распространены в природе, либо в изотопно обогащенной форме. Например, когда упоминается водород, подразумевается, что он относится к 1H, 2H, 3H или их смесям; когда упоминается углерод, подразумевается, что он относится к 11C, 12C, 13C, 14C или их смесям; когда упоминается азот, подразумевается, что он относится к 13N, 14N, 15N или их смесям; когда упоминается кислород, подразумевается, что он относится к 14O, 15О, 16O, 17O, 18O или их смесям; и когда упоминается фтор, подразумевается, что он относится к 18F, 19F или их смесям. Как отмечалось выше, соединения, представленные в настоящем документе, поэтому также включают соединения с одним или несколькими изотопами одного или нескольких атомов и их смеси, включая радиоактивные соединения, где один или несколько не радиоактивных атомов заменены одним из его радиоактивно обогащенных изотопов. Радиомеченые соединения полезны в качестве терапевтических агентов, например, противораковых агентов, исследовательских реактивов, например, аналитических реагентов и диагностических агентов, например, агентов визуализации in vivo. Предполагается, что все изотопные варианты соединений, представленных в настоящем документе, радиоактивные или нет, входят в объем настоящего изобретения.
Соединения формулы I, формулы II и формулы II могут существовать в различных полиморфных формах. Термины «полиморф», «полиморфная форма» и «кристаллическая форма» относятся к различным кристаллическим формам одного соединения. То есть, полиморфами являются отдельные твердые вещества, имеющие одну и ту же молекулярную формулу, однако каждый полиморф может иметь различные физические свойства твердого тела. Следовательно, одно соединение может давать начало множеству полиморфных форм, каждая из которых имеет разные и различные физические свойства твердого тела, такие как разные профили растворимости, скорости растворения, температуры плавления, текучесть и/или разные пики рентгеновской дифракции. Различия в физических свойствах могут влиять на фармацевтические параметры, такие как стабильность при хранении, прессуемость и плотность (которые могут быть важны при составлении и производстве продукта) и скорость растворения (что может быть важным фактором биодоступности). Методики характеризации полиморфных форм включают, но не ограничиваются ими, порошковую рентгеновскую дифрактометрию (ПРД), дифференциальную сканирующую калориметрию (ДСК), термогравиметрический анализ (ТГА), монокристаллическую рентгеновскую дифрактометрию (МРД), вибрационную спектроскопию, например, инфракрасную (ИК) и рамановскую спектроскопию, спектроскопию ядерного магнитного резонанса твердого тела и раствора (ЯМР), оптическую микроскопию, высокотемпературную оптическую микроскопию, сканирующую электронную микроскопию (СЭМ), электронную кристаллографию и количественный анализ, гранулометрический анализ (ГМА), анализ площади поверхности, измерения растворимости, измерения растворения, элементный анализ и анализ Карла Фишера.
В одном варианте осуществления, представленном в настоящем документе, представлены кристаллические формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида или его сольваты.
В одном варианте осуществления, одна или несколько кристаллических форм N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, описанного в настоящем документе, являются безводными.
В одном варианте осуществления, одна или несколько кристаллических форм N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, описанного в настоящем документе, могут быть выделены в виде сольвата. В одном варианте осуществления, кристаллическую форму выделяют в виде сольвата анизола, например сольвата моноанизола. В одном варианте осуществления, кристаллическую форму выделяют в виде сольвата дихлорметана, например, сольвата гемидихлорметана. В одном варианте осуществления, кристаллическую форму выделяют в виде сольвата толуола. В одном варианте осуществления, кристаллическую форму выделяют в виде сольвата 1,4-диоксана.
В одном варианте осуществления, представлены кристаллические формы N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, выбранные из N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида безводной формы А, N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида безводной формы B, N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы C, моно-анизола, N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы D полу-дихлорметана, N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольной формы Е, и N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, 1,4-диоксановой формы F.
В некоторых вариантах осуществления, кристаллические формы могут характеризоваться их порошковыми рентгеновскими дифрактограммами (ПРД). ПРД анализ проводят на приборе PANalytical X’pert pro с датчиком PIXcel (128 каналов), сканируя образцы при 3-35° 2θ. Материал осторожно измельчают для высвобождения любых агломератов и загружают на многолуночный планшет с Mylar полимерной пленкой для поддержки образца. Затем многолуночный планшет помещают в дифрактометр и анализируют с использованием излучения Cu K 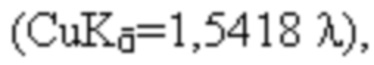 работающего в режиме пропускания (размер шага 0,0130° 2θ, время шага 18,87 с), используя настройки генератора 40 кВ/40 мА. Визуализацию данных и создание изображений производят с помощью настольного приложения HighScore Plus 4,7 (PANalytical, 2017). Приведенные ниже таблицы включают анализ и предоставляют следующие приблизительные данные: 2θ, измеренный в градусах ±0,2 градуса; d, измеренный в Ангстремах ±0,2 Ангстрема; и относительную интенсивность с использованием высоты пика для измерения % высоты (H%) в импульсах в секунду.
работающего в режиме пропускания (размер шага 0,0130° 2θ, время шага 18,87 с), используя настройки генератора 40 кВ/40 мА. Визуализацию данных и создание изображений производят с помощью настольного приложения HighScore Plus 4,7 (PANalytical, 2017). Приведенные ниже таблицы включают анализ и предоставляют следующие приблизительные данные: 2θ, измеренный в градусах ±0,2 градуса; d, измеренный в Ангстремах ±0,2 Ангстрема; и относительную интенсивность с использованием высоты пика для измерения % высоты (H%) в импульсах в секунду.
Должно быть понятно, что 2-тэта значения порошковых рентгеновских дифрактограмм для кристаллических форм N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида могут незначительно отличаться от одного прибора к другому, а также в зависимости от вариаций при подготовке образцов и от партии к партии, и поэтому указанные значения не следует рассматривать как абсолютные. Также будет понятно, что относительные интенсивности пиков могут варьироваться в зависимости от эффектов ориентации, так что интенсивности, показанные на графике ПРД, включенном в настоящий документ, являются иллюстративными и не предназначены для использования для абсолютного сравнения. Соответственно, следует понимать, что фраза «по существу такую же ПРД, как показана на фиг. 1» означает, что для целей сравнения присутствуют, по меньшей мере, 90% пиков, показанных на фиг. 1. Следует понимать, что относительные положения пиков могут отличаться на ±0,2 градуса от положений пиков, показанных на фиг. 1. Также следует понимать, что для целей сравнения разрешена некоторая вариабельность интенсивностей пиков по сравнению с показанными на фиг. 1. То же самое относится к описанию фиг. 4, 7, 9, 11 и 13.
В некоторых вариантах осуществления, кристаллические формы могут быть охарактеризованы их сканами ДСК. Значения теплового перехода измеряют с помощью дифференциальной сканирующей калориметрии («ДСК»). Примерно 1-5 мг материала отвешивают в алюминиевый поддон ДСК и не герметично закрывают алюминиевой крышкой. Чашку для образцов загружают в дифференциальный сканирующий калориметр TA Instruments Discovery DSC 2500, оборудованный кулером RC90. Образец и эталон нагревают до различных температур со скоростью сканирования 10°C/мин и отслеживают полученный ответ теплового потока. Образец повторно охлаждают до 20°C и затем повторно нагревают со скоростью 10°C/мин. В качестве продувочного газа используют азот при скорости потока 50 см3/мин.
В некоторых вариантах осуществления, кристаллические формы могут быть охарактеризованы термогравиметрическим анализом (ТГА)/дифференциальной сканирующей калориметрией (ДСК). Анализ ТГА/ДСК проводят с использованием Приблизительно 5-10 мг материала добавляют в предварительно тарированную открытую алюминиевую чашу и загружают в TA Instruments Discovery SDT 650 Auto-Simultaneous DSC и выдерживают при комнатной температуре. Затем образец нагревают со скоростью 10°C/мин от 30°C до 400°C, в течение этого времени регистрируют изменение веса образца вместе с ответом теплового потока (ДСК). В качестве продувочного газа используют азот при скорости потока 300 см3/мин.
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, безводная форма А
В одном варианте осуществления, представлена кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А, может быть охарактеризована ПРД, как показано на ФИГ. 1.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А, имеет ПРД с, по меньшей мере, 20 характеристическими пиками (2θ градусы ± 0,2), как перечислено в таблице 3.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида безводной формы А имеет ПРД с, по меньшей мере, 8 из характеристических пиков (2θ градусы ± 0,2), перечисленных в таблице 3.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида безводной формы А имеет ПРД с, по меньшей мере, 5 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 3.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида безводной формы А имеет ПРД с, по меньшей мере, 8 характеристическими пиками (2θ градусы ± 0,2), как перечислено в таблице 3A.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида безводной формы А имеет ПРД с, по меньшей мере, 5 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 3B.
В некоторых вариантах осуществления, кристаллический N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид безводной формы А имеет ДСК термограмму, как показано на ФИГ. 2, имеющую температуру начала плавления 162,65 и максимальную температуру плавления 164,39°C.
В некоторых вариантах осуществления, кристаллический N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид безводной формы А имеет ТГА/ДСК профиль, как показан на ФИГ. 3.
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, безводная форма В
В одном варианте осуществления, представлена кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводная форма В, может быть охарактеризована ПРД, как показано на ФИГ. 4.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводная форма В имеет ПРД с, по меньшей мере, 20 характеристическими пиками (2θ градусы ± 0,2), как перечислено в таблице 4.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид безводная форма В имеет ПРД с, по меньшей мере, 8 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 4.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводная форма В, имеет ПРД с, по меньшей мере, 5 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 4.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводная форма В, имеет ПРД с, по меньшей мере, 8 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 4A.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводная форма В, имеет ПРД с, по меньшей мере, 6 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 4B.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводная форма В, имеет ПРД с, по меньшей мере, 5 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 4C.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводная форма В, имеет ПРД с, по меньшей мере, 3 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 4D.
В некоторых вариантах осуществления, кристаллический N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, безводная форма В, имеет ДСК термограмму, как показано на ФИГ. 5, имеющую температуру начала плавления 148,8°C и максимальную температуру плавления 151,39°C.
В некоторых вариантах осуществления, кристаллический N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, безводная форма В, имеет ТГА/ДСК профиль, как показан на ФИГ. 6.
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, форма C моно-анизол
В одном варианте осуществления, представлена кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма С моно-анизол.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма С моно-анизол, может быть охарактеризована ПРД как показано на ФИГ. 7.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма С моно-анизол, имеет ПРД с, по меньшей мере, 20 характеристическими пиками (2θ градусы ± 0,2), как перечислено в таблице 5.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма С моно-анизол, имеет ПРД с, по меньшей мере, 8 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 5.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма С моно-анизол, имеет ПРД с, по меньшей мере, 5 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 5.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма С моно-анизол, имеет ПРД с, по меньшей мере, 8 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 5A.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма С моно-анизол, имеет ПРД с, по меньшей мере, 6 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 5B.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, форма С моно-анизол, имеет ТГА/ДСК профиль, как показан на ФИГ. 8.
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, форма D полу-дихлорметан
В одном варианте осуществления, представлена кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма D полу-дихлорметан.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма D полу-дихлорметан, может быть охарактеризована ПРД, как показано на ФИГ. 9.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма D полу-дихлорметан, имеет ПРД с, по меньшей мере, 20 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 6.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма D полу-дихлорметан, имеет ПРД с, по меньшей мере, 8 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 6.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, форма D полу-дихлорметан, имеет ПРД с, по меньшей мере, 5 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 6.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма D полу-дихлорметан, имеет ПРД с, по меньшей мере, 8 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 6A.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма D полу-дихлорметан, имеет ПРД с, по меньшей мере, 6 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 6B.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма D полу-дихлорметан, имеет ТГА/ДСК профиль, как показан на ФИГ. 10.
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид толуольная форма Е
В одном варианте осуществления, представлена кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольная форма Е.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольная форма Е, может быть охарактеризована ПРД как показано на ФИГ. 11.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольная форма Е, имеет ПРД с, по меньшей мере, 20 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 7.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольная форма Е, имеет ПРД с, по меньшей мере, 8 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 7.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольная форма Е, имеет ПРД с, по меньшей мере, 5 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 7.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольная форма Е, имеет ПРД с, по меньшей мере, 8 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 7A.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольная форма Е, имеет ПРД с, по меньшей мере, 6 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 7B.
В определенных вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольная форма Е, имеет ТГА/ДСК профиль, как показан на ФИГ. 12.
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, форма F 1,4-диоксан
В одном варианте осуществления, представлена кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма F 1,4-диоксан.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма F 1,4-диоксан, может быть охарактеризована ПРД как показано на ФИГ. 13.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма F 1,4-диоксан, имеет ПРД с, по меньшей мере, 20 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 8.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма F 1,4-диоксан, имеет ПРД с, по меньшей мере, 8 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 8.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма F 1,4-диоксан, имеет ПРД с, по меньшей мере, 5 из характеристических пиков (2θ градусы ± 0,2) перечисленных в таблице 8.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма F 1,4-диоксан, имеет ПРД с, по меньшей мере, 8 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 8A.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма F 1,4-диоксан, имеет ПРД с, по меньшей мере, 6 характеристическими пиками (2θ градусы ± 0,2) как перечислено в таблице 8B.
В некоторых вариантах осуществления, кристаллическая форма N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, форма F 1,4-диоксан, имеет ТГА/ДСК профиль, как показан на ФИГ. 14.
В иллюстративных целях на схемах 1-15 показаны общие способы получения соединений, представленных в настоящем документе, а также ключевых промежуточных соединений. Более подробное описание отдельных стадий реакции представлено в разделе «Примеры» ниже. Специалисты в данной области техники поймут, что для синтеза соединений по изобретению можно использовать другие пути синтеза. Хотя конкретные исходные материалы и реагенты изображены на схемах и обсуждаются ниже, другие исходные материалы и реагенты могут быть легко заменены для предоставления множества производных и/или условий реакции. Кроме того, многие соединения, полученные описанными ниже способами, могут быть дополнительно модифицированы в свете этого раскрытия с использованием общепринятой химии, хорошо известной специалистам в данной области техники.
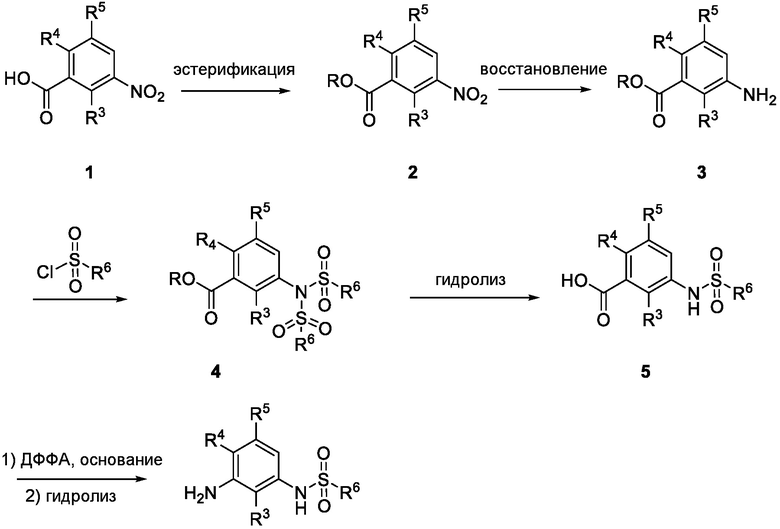
Схема 1
Схема 1 показывает общий способ получения промежуточного соединения 6, которое можно использовать для получения формулы I, где R3, R4, R5 и R6 такие, как определены для формулы I, X1 является CH, и L является NH. Бензойная кислота 1 (где R3, R4 и R5 такие, как определены для формулы I), может быть эстерифицирована с получением соединения 2 (где R является алкилом), например, обработкой триметилсилилдиазометаном в MeOH или в условиях эстерификации Фишера, таких как обработка триметилсилилхлоридом в МеОН. Нитрогруппа промежуточного соединения 2 может быть восстановлена до аминогруппы с использованием стандартных условий, таких как обработка Pd/C и H2 с получением соединения 3. Соединение 3 может быть обработано сульфонилхлоридным реагентом, имеющим формулу R6SO2Cl, где R6 такой, как определен для формулы I, в присутствии основания, такого как NEt3, в органическом растворителе, таком как дихлорметан, с получением соединения 4. Соединение 4 может быть гидролизовано в основных условиях, таких как водный NaOH, в подходящей системе растворителей, такой как ТГФ и/или MeOH, с получением соединения 5. Соединение 5 может быть обработано ДФФА (дифенилфосфоназидом) и основанием, таким как триэтиламин, в подходящем растворителе, таком как ТГФ, с последующим гидролизом с получением соединения 6.
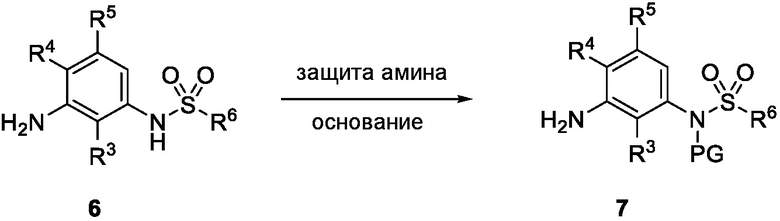
Схема 2
Схема 2 показывает методику получения соединения 7, которое применяют для получения соединения формулы I, где R3, R4, R5 и R6 такие, как описаны для формулы I, X1 является CH, и L является NH. Соединение 6, полученное согласно схеме 1, может быть обработано реагентом, защищающим амин (например, 2-(хлорметокси)этил)триметилсиланом или п-метоксибензилбромид) в присутствии основания (например, гидрида натрия) с получением соединения 7, где PG является аминовой защитной группой (PG) (например, триметилсилилэтоксиметилом или п-метоксибензилом).
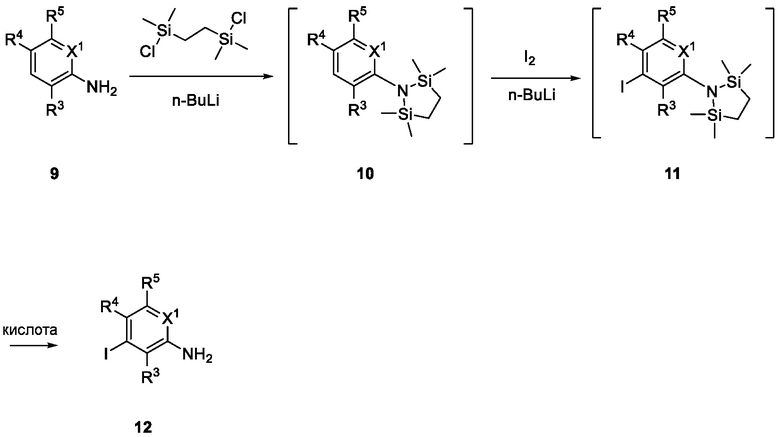
Схема 3
Схема 3 описывает синтез промежуточного соединения 12, которое использует для получения соединения формулы I, где R3, R4 и R5 такие, как определены для формулы I, и X1 является CH или N. Соединение 9 (где X1, R3, R4 и R5 такие, как определены для формулы I) могут быть подвергнуты взаимодействию с 1,2-бис(хлордиметилсилил)этаном в присутствии сильного основания, такого как н-бутиллитий, в подходящем растворителе, таком как ТГФ, при низких температурах, например, -78°C, с получением соединения 1-аза-2,5-дисилациклопентана 10. Соединение 10 может быть подвергнуто взаимодействию с йодом, в присутствии, например, н-бутиллития или сравнимого агента в подходящем растворителе, таком как ТГФ, с получением соединения 11. Соединение 11 может быть лишено защиты взаимодействием с кислотой, такой как HCl, в подходящем растворителе, с получением соединения 12.
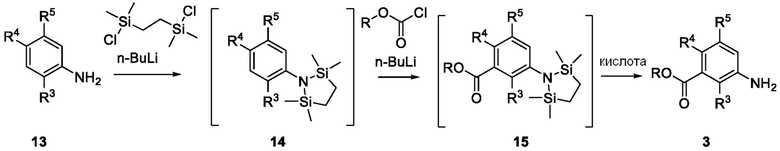
Схема 4
Схема 4 описывает альтернативный синтез соединения 3, где X1 является CH, и где R3, R4 и R5 такие, как определены для формулы I. Соединение 13 (где R3, R4 и R5 такие, как определены для формулы I) может быть подвергнуть взаимодействию с 1,2-бис(хлордиметилсилил)этаном в присутствии сильного основания, такого как н-бутиллитий, в подходящем растворителе, таком как ТГФ, при низких температурах, например, -78°C, с получением соединения 14. Соединение 14 может быть подвергнуто взаимодействию с карбамоилхлоридом, RO(C=O)Cl где R является малым алкилом, таким как метил или этил или бензильной группой, в присутствии, например н-бутиллития, в подходящем растворителе, таком как ТГФ, с получением соединения 15. Соединение 15 может быть лишено защиты взаимодействием с кислотой, такой как HCl, в подходящем растворителе, с получением соединения 3.

Схема 5
Схема 5 описывает синтез промежуточного соединения 18, где R3, R4, R5 и R6 такие, как определены для формулы I и X1 является CH или N. Соединение 12 (полученное согласно схемы 4) может быть подвергнуто взаимодействию с сульфонилхлоридным реагентом, имеющим формулу R6SO2Cl где R6 такой, как определен для формулы I, в присутствии основания, такого как NEt3, в органическом растворителе, таком как дихлорметан с получением соединения 16. Соединение 16 может быть гидролизовано, например, в основных условиях, таких как водный NaOH, в подходящей системе растворителей, такой как ТГФ и/или MeOH, с получением соединения 17. Соединение 17 может быть подвергнуто взаимодействию с реагентом, защищающим аминогруппу, например (2-(хлорметокси)этил)триметилсиланом (SEM-Cl), в стандартных условиях (например, в присутствии основания, такого как гидрид натрия, в подходящем растворителе, таком как ДМФ) с получением соединения 18, где PG является группой защиты амина (например, SEM группой).
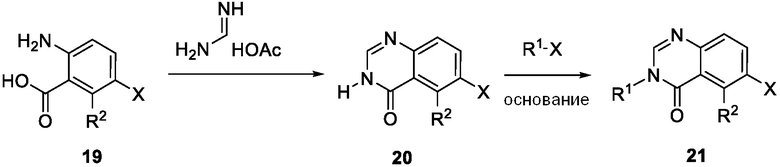
Схема 6
Схема 6 описывает синтез промежуточного соединения 21 где X является галогеном, которое используют для получения соединения формулы I где R1 и R2 такие, как определены для формулы I и X1 является CH. Соединение 19 может быть циклизовано с ацетатом формамидина в органическом растворителе, таком как EtOH, при повышенной температуре с получением соединения 20. Соединение 20 может быть алкилировано с реагентом, имеющим формулу R1X, где R1 такой, как определен для формулы I, и X является галогеном, в присутствии основания, такого как Cs2CO3, в растворителе, таком как ДМФ, с получением соединения 21.
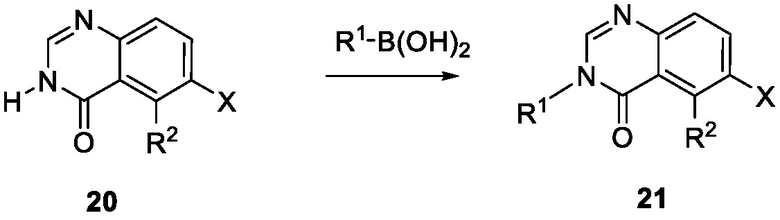
Схема 7
Схема 7 изображает альтернативный путь для синтеза промежуточного соединения 21, где R1 и R2 такие, как определены для формулы I, X1 является CH и X является галогеном, который применяют для получения соединения формулы I. Соединение 20 может быть сопряжено с реагентом на основе бороновой кислоты R1B(OH)2, где R1 является таким, как определен для формулы I, в присутствии катализатора, такого как Cu(OAc)2, и лиганда, такого как пиридин, с получением соединения 21.
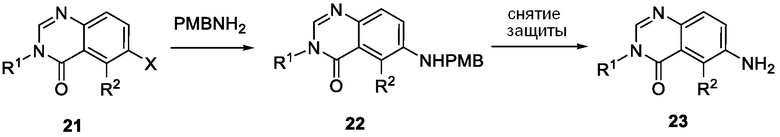
Схема 8
Схема 8 описывает синтез промежуточного соединения 23 где R1 и R2 такие, как определены для формулы I и X1 является CH, которое используют для получения соединения формулы I. Соединение 21 (полученное, например, согласно Схеме 6 или 7) может быть сопряжено с реагентом, имеющим формулу (PG)NH2, где PG является защитной группой амина (такой как п-метоксибензил (PMB)) в присутствии катализатора, которым является палладиевый катализатор (например, Pd2(dba)3), и лиганда (например, Xantphos) с получением соединения 22. Соединение 22 может быть лишено защиты в стандартных условиях, например, с использованием ТФК, с получением соединения 23.
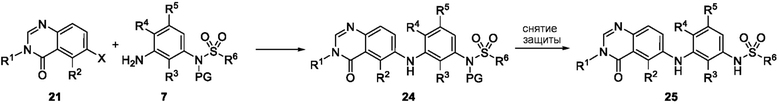
Схема 9
Схема 9 описывает синтез соединения 25, которое является соединением формулы I, где R1, R2, R3, R4, R5 и R6 такие, как определены для формулы I, X1 является CH, и L является NH. Соединение 25 может быть получено сочетанием соединения 21 (полученного, например, согласно Схеме 6 или 7) с соединением 7 (полученным, например, согласно Схеме 2) в присутствии катализатора (например, палладиевого катализатора, например, Pd2(dba)3) и лиганда (например, Xantphos) после снятия защиты в стандартных условиях (например, с ТФК), с получением соединения 25.
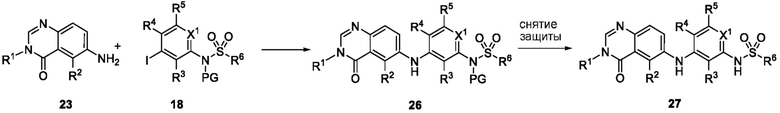
Схема 10
Схема 10 описывает синтез соединения 27, которое является соединением формулы I, где R1, R2, R3, R4, R5 и R6 такие, как определены в настоящем документе, L является NH и X1 является CH или N. Compound 18 (полученное, например, согласно Схеме 9) может быть сопряжено с соединением 23 (полученным, например, согласно Схеме 8) в присутствии катализатора (например, палладиевого катализатора, например, Pd2(dba)3) и лиганда (например, Xantphos) после снятия защиты в стандартных условиях (например, с ТФК), с получением соединения 27.
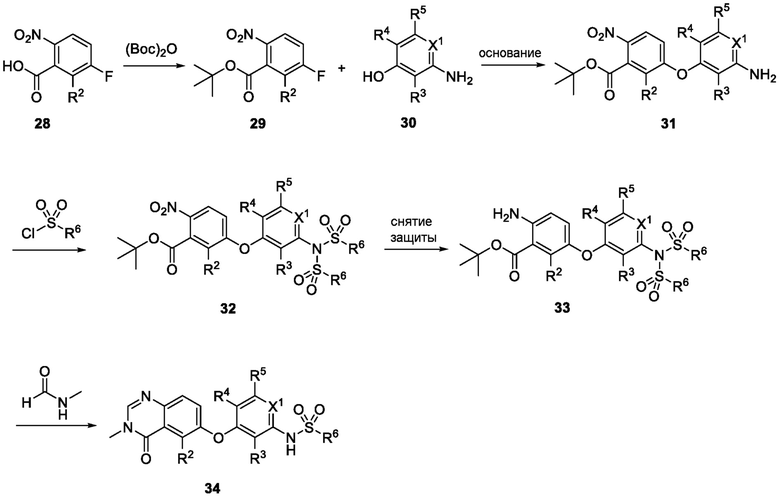
Схема 11
Схема 11 описывает синтез соединения формулы 34, которое является соединением формулы I, где R3, R4, R5 и R6 такие, как определены для формулы I, R1 является метилом, L является O и X1 является CH. Соединение 28 (где R2 такой, как определен для формулы I) может быть подвергнуто взаимодействию с (Boc)2O в смеси органических растворителей, таких как t-BuOH и ДХМ, в присутствии катализатора (например, ДМАП) с получением соединения 29. Соединение 29 может быть сопряжено с соединением 30 в подходящем растворителе, таком как ДМФ, в присутствии основания, такого как Cs2CO3, при повышенной температуре с получением соединения 31. Соединение 31 может быть подвергнуто взаимодействию с сульфонилхлоридным реагентом, имеющим формулу ClSO2R6, в присутствии основания, такого как NEt3, в органическом растворителе, таком как ДХМ, с получением соединения 32. Нитрогруппа соединения 32 может быть восстановлена в стандартных условиях нитро восстановления, таких как обработка Pd/C и H2, с получением соединения 33. Соединение 33 может быть циклизовано с N-метилформамидом в присутствии кислоты, такой как муравьиная кислота, при повышенных температурах, с получением соединения 34.

Схема 12
Схема 12 описывает синтез соединения формулы 34, которое является соединением формулы I, где R3, R4, R5 и R6 такие, как определены для формулы I, R1 является метилом, L является O и X1 является CH. Соединение 28 (где R2 такой, как определен для формулы I) может быть подвергнуто взаимодействию с (Boc)2O в смеси органических растворителей, таких как t-BuOH и ДХМ, в присутствии катализатора (например, ДМАП) с получением соединения 29. Соединение 29 может быть сопряжено с соединением 30 в подходящем растворителе, таком как ДМФ, в присутствии основания, такого как Cs2CO3, при повышенной температуре с получением соединения 31. Нитрогруппа соединения 31 может быть восстановлена в стандартных условиях нитро восстановления, таких как обработка Pd/C и H2 с получением соединения 35. Compound 35 может быть циклизовано с N-метилформамидом в присутствии кислоты, такой как муравьиная кислота, при повышенных температурах, с получением соединения 36. Соединение 36 может быть подвергнуто взаимодействию с сульфонилхлоридным реагентом, имеющим формулу ClSO2R6, в присутствии основания, такого как аминовое основание (например, NEt3), в подходящем органическом растворителе (например, ДХМ) с получением соединения 37. Соединение 37 может быть гидролизовано, например, в основных условиях, таких как водный NaOH, в подходящей системе растворителей, такой как ТГФ и/или MeOH, с получением соединения 34.
Схема 13
Схема 13 описывает синтез соединения формулы 41, которое является соединением формулы I, где R3, R4, R5 и R6 такие, как определены для формулы I, R1 является метилом, R2 является метилом, L является O и X1 является N. Соединение 38, где R1 такой, как определен для формулы I, может быть сопряжено с соединением 39, где X является галогеном, PG является защитной группой амина, и R3, R4 и R5 такие, как определены для формулы I, в подходящем растворителе, таком как ДМФ, в присутствии основания, например, щелочной карбонат (например, Cs2CO3), при повышенной температуре с получением соединения 40. Соединение 40 может быть лишено защиты в стандартных условиях, например с применением ТФК, с получением соединения 41.
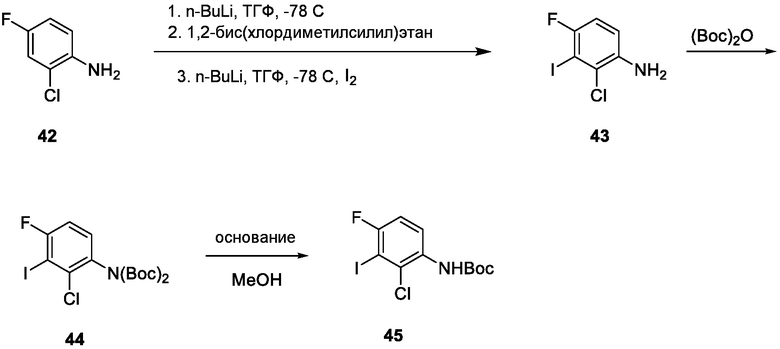
Схема 14
Схема 14 показывает общий способ получения промежуточного соединения 45, которое используют для получения соединений формулы I где R3 является хлором, R4 является фтором и R5 является водородом. Коммерчески доступный 2-хлор-4-фторанилин может быть обработан н-бутиллитием при пониженной температуре, с последующей обработкой 1,2-(бис(хлордиметилсилил)этаном с получением промежуточного 1-(2-хлор-4-фторфенил)-2,2,5,5-тетраметил-1,2,5-азадисилолидина (не показан), который может быть обработан н-бутиллитием с последующей обработкой йодом с получением промежуточного соединения 43. Аминогруппа промежуточного соединения 43 может быть обработана ди-трет-бутилдикарбоната с получением бис-Boc защищенного промежуточного соединения 44. Промежуточное соединение 44 может быть обработано основанием, например, щелочным карбонатом, например, карбонатом калия, с получением Boc-защищенного промежуточного соединения 45.
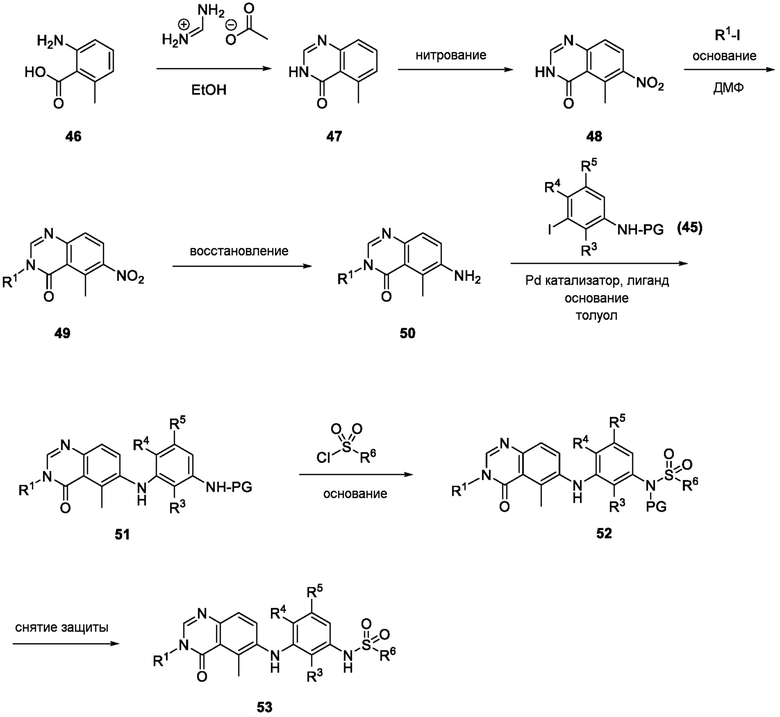
Схема 15
Схема 15 описывает синтез соединения 53, которое является соединением формулы I где X1 является CH, R1 является метилом, и R2, R3, R4, R5 и R6 такие, как определены для формулы I. Коммерчески доступное соединение 46 может быть подвергнуто взаимодействию с ацетатом формамидина при повышенных температурах с получением соединения 47. Соединение 47 может быть нитровано в стандартных условиях, например, в присутствии азотной кислоты и серной кислоты, с получением соединения 48. Соединение 48 может быть алкилировано с MeI в присутствии основания, такого как Cs2CO3, в растворителе, таком как ДМФ, с получением соединения 49. Нитрогруппа соединения 49 может быть восстановлена до аминогруппы с применением стандартных условий, таких как обработка Pd/C и H2 с получением соединения 50. Соединение 50 может быть сопряжено с соединением 45, где PG является защитной группой амина и R3, R4 и R5 такие, как определены для формулы I (полученным, например, как описано на схеме 14) в присутствии катализатора (например, палладиевого катализатора, например, Pd2(dba)3), лиганда (например, Xantphos) и основания (например, щелочного карбоната, например, Cs2CO3) с получением соединения 51. Соединение 51 может быть подвергнуто взаимодействию с сульфонилхлоридом, имеющим формулу R6SO2Cl (где R6 такой, как определен для формулы I), в присутствии основания, такого как NaHMDS, в органическом растворителе, таком как ТГФ, с получением соединения 52. Удаление амино защитной группы соединения 52 в стандартных условиях (например, с ТФК), дает соединение 53.
Синтетические производные, имеющие формулы 21, 23, 24, 26, 31, 32, 33, 35, 36, 37, 40, 45, 49, 50, 51 и 52, как определено на вышеуказанных схемах, представлены как следующие аспекты настоящего изобретения.
В одном варианте осуществления, в настоящем документе представлено соединение, имеющее формулу (23)
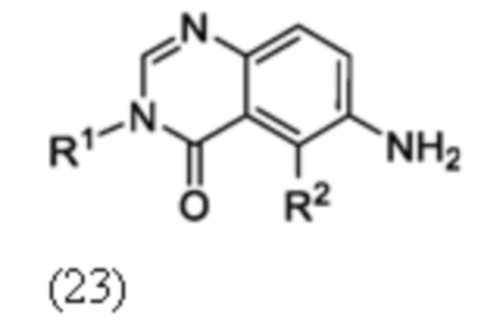
где:
R1 является C1-C6 алкилом, C1-C6 дейтероалкилом, C1-C6 фторалкилом, C3-C6 циклоалкилом, (C3-C6 циклоалкил)CH2-, (C1-C6 алкокси)C1-C6 алкилом-, Ar1, Ar1CH2-, hetAr1 или hetCyc1;
Ar1 является фенилом, который необязательно замещен 1-5 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetAr1 является 5-6-членным гетероарильным кольцом, имеющим 1-2 кольцевых атома азота и необязательно замещенным 1-3 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetCyc1 является 4-6-членным насыщенным гетероциклическим кольцом, имеющим один кольцевой атом кислорода; и
R2 является метилом, -CD3 или HC≡C-.
В одном варианте осуществления формулы (23), R1 является С1-С6 алкилом. В одном варианте осуществления формулы (23), R1 является метилом.
В одном варианте осуществления формулы (23), R1 является С1-С6 алкилом и R2 является метилом. В одном варианте осуществления формулы (23), R1 является метилом и R2 является метилом.
В одном варианте осуществления, в настоящем документе представлено соединение, имеющее формулу (24)
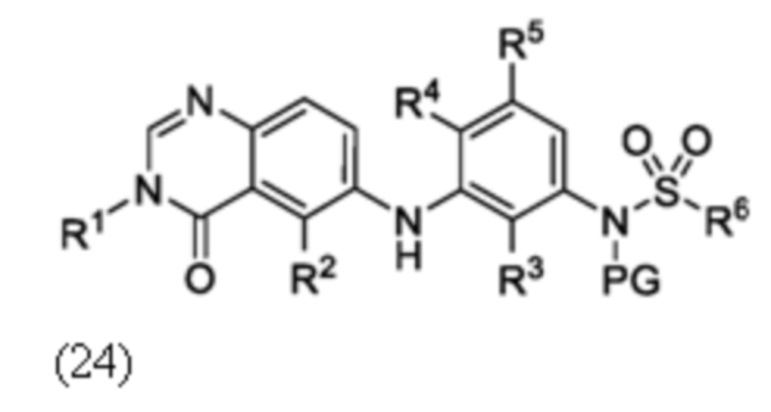
где:
R1 является С1-С6 алкилом, C1-C6 дейтероалкилом, C1-C6 фторалкилом, C3-C6 циклоалкилом, (C3-C6 циклоалкил)CH2-, (C1-C6 алкокси)C1-C6 алкилом-, Ar1, Ar1CH2-, hetAr1 или hetCyc1;
Ar1 является фенилом, который необязательно замещен 1-5 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetAr1 является 5-6-членным гетероарильным кольцом, имеющим 1-2 кольцевых атома азота, и необязательно замещенным 1-3 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetCyc1 является 4-6-членным насыщенным гетероциклическим кольцом, имеющим один кольцевой атом кислорода;
R2 является метилом, -CD3 или HC≡C-;
R3 является F, Cl, CN или метилом;
R4 является H, F или Cl;
R5 является H, F, Cl, метилом;
R6 является С1-С6 алкилом, C1-C6 фторалкилом, (Cyc1)C1-C6 алкилом-, (C1-C3 алкокси)C1-C6 алкилом-, hetAr2, Ar2 или RaRbN-;
Cyc1 является 3-6-членным насыщенным карбоциклическим кольцом;
hetAr2 является 5-6-членным гетероарилом, имеющим 1-2 кольцевых гетероатома, независимо выбранных из N, O и S, и необязательно замещенным 1-3 заместителями, независимо выбранными из галогена и C1-C3 алкила;
Ar2 является фенилом необязательно замещенным 1-5 заместителями, независимо выбранными из галогена и C1-C6 алкила;
Ra и Rb независимо являются C1-C3 алкилом, или
Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, необязательно замещенное фтором; и
PG является защитной группой амина.
В одном варианте осуществления формулы (24), R1 является С1-С6 алкилом.
В одном варианте осуществления формулы (24), R2 является метилом.
В одном варианте осуществления формулы (24), R3 является Cl.
В одном варианте осуществления формулы (24), R4 является F.
В одном варианте осуществления формулы (24), R6 является C1-C6 фторалкилом.
В одном варианте осуществления формулы (24), PG является Boc защитной группой.
В одном варианте осуществления формулы (24), R1 является метилом, R2 является метилом, R3 является Cl, R4 является F и R6 является 3-фторпропилом. В одном варианте осуществления формулы (24), R1 является метилом, R2 является метилом, R3 является Cl, R4 является F, R6 является 3-фторпропилом, и PG является Boc защитной группой.
В одном варианте осуществления формулы (24), R6 является С1-С6 алкилом, C1-C6 фторалкилом, (Cyc1)C1-C6 алкилом-, (C1-C3 алкокси)C1-C6 алкилом-, hetAr2 или Ar2.
В одном варианте осуществления формулы (24), R6 является С1-С6 алкилом или C1-C6 фторалкилом.
Любые из вышеуказанных вариантов осуществления формулы (24) могут быть скомбинированы друг с другом.
В одном варианте осуществления, в настоящем документе представлено соединение, имеющее формулу (45)
где:
R3 является F, Cl, CN или метилом;
R4 является H, F или Cl;
R5 является H, F, Cl, метилом; и
PG является защитной группой амина.
В одном варианте осуществления формулы (45), R3 является Cl.
В одном варианте осуществления формулы (45), R4 является F.
В одном варианте осуществления формулы (45), R5 является H.
В одном варианте осуществления формулы (45), PG является Boc группой.
В одном варианте осуществления формулы (45), R3 является Cl, R4 является F и R5 является F. В одном варианте осуществления формулы (45), R3 является Cl, R4 является F, R5 является F и PG является Boc группой.
Любой из вышеупомянутых вариантов осуществления формулы (45) могут быть скомбинированы друг с другом.
В одном варианте осуществления, в настоящем документе представлено соединение, имеющее формулу (51)

где:
R1 является С1-С6 алкилом, C1-C6 дейтероалкилом, C1-C6 фторалкилом, C3-C6 циклоалкилом, (C3-C6 циклоалкил)CH2-, (C1-C6 алкокси)C1-C6 алкилом-, Ar1, Ar1CH2-, hetAr1 или hetCyc1;
Ar1 является фенилом который необязательно замещен 1-5 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetAr1 является 5-6-членным гетероарильным кольцом, имеющим 1-2 кольцевых атома азота, и необязательно замещенным 1-3 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetCyc1 является 4-6-членным насыщенным гетероциклическим кольцом, имеющим один кольцевой атом кислорода;
R3 является F, Cl, CN или метилом;
R4 является H, F или Cl;
R5 является H, F, Cl, метилом; и
PG является защитной группой амина.
В одном варианте осуществления формулы (51), R1 является С1-С6 алкилом. В одном варианте осуществления формулы (51), R1 является метилом.
В одном варианте осуществления формулы (51), R3 является Cl.
В одном варианте осуществления формулы (51), R4 является F.
В одном варианте осуществления формулы (51), R5 является H.
В одном варианте осуществления формулы (51), PG является Boc защитной группой.
В одном варианте осуществления формулы (51), R1 является С1-С6 алкилом, R3 является Cl, R4 является F и R5 является H. В одном варианте осуществления формулы (51), R1 является С1-С6 алкилом, R3 является Cl, R4 является F, R5 является H и PG является Boc группой. В одном варианте осуществления формулы (51), R1 является метилом, R3 является Cl, R4 является F, и R5 является H. В одном варианте осуществления формулы (51), R1 является метилом, R3 является Cl, R4 является F, R5 является H и PG является Boc группой.
Любой из вышеупомянутых вариантов осуществления формулы (51) могут быть скомбинированы друг с другом.
Также в настоящем документе представлен способ получения соединения формулы I или его фармацевтически приемлемая соль или сольват, который включает:
(a) для соединения формулы I, где X1 является CH, L является NH, и R1, R2, R3, R4, R5 и R6 такие, как определены для формулы I, сочетание соединения, имеющего формулу (21):
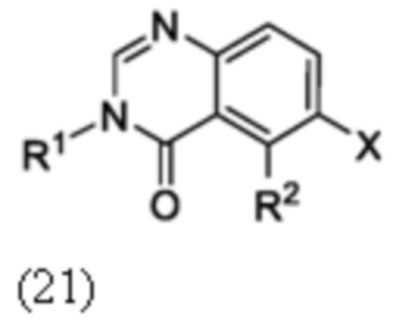
где R1 и R2 такие, как определены для формулы I и X является галогеном, с соединением, имеющим формулу (7):
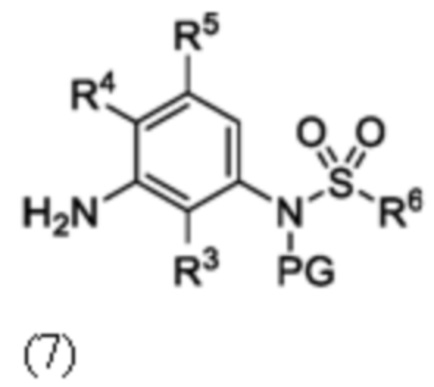
где R3, R4, R5 и R6 такие, как определены для формулы I, и PG является защитной группой амина, в присутствии катализатора и лиганда, после удаления аминовой защитной группой; или
(b) для соединения формулы I где L является NH, и X1, R1, R2, R3, R4, R5 и R6 такие, как определены для формулы I, сочетание соединения, имеющего формулу (23):
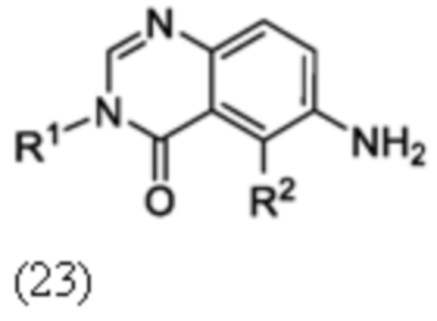
где R1 и R2 такие, как определены для формулы I, с соединением, имеющим формулу (18)
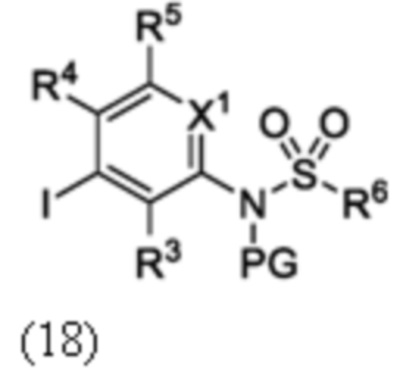
где X1, R3, R4, R5 и R6 такие, как определены для формулы I, и PG является защитной группой амина, в присутствии катализатора и лиганда, с последующим удалением аминовой защитной группы; или
(c) для соединения формулы I, где L является O и X1, R1, R2, R3, R4, R5 и R6 такие, как определены для формулы I, циклизацию соединения, имеющего формулу (33):
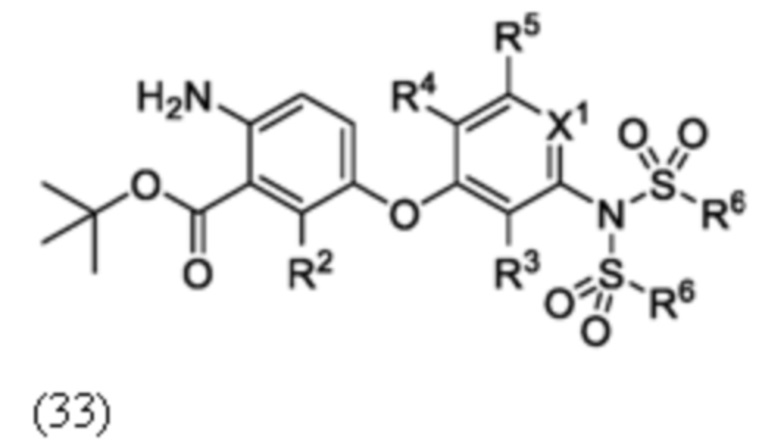
где X1, R2, R3, R4, R5 и R6 такие, как определены для формулы I, с N-метилформамидом в присутствии кислоты; или
(d) для соединения формулы I, где R1 является метилом, L является O, X1 является CH, R3, R4, R5 и R6 такие, как определены для формулы I, сочетание соединения, имеющего формулу (36)
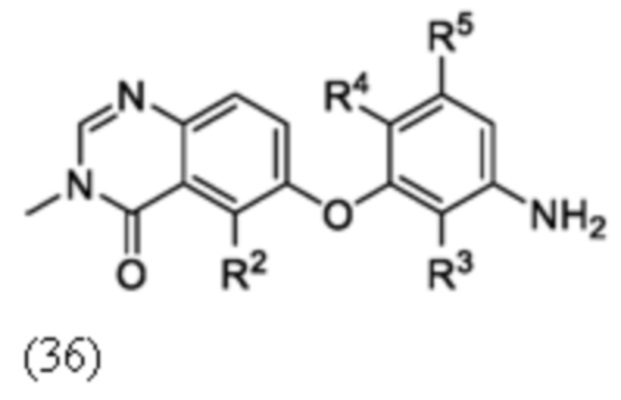
с реагентом, имеющим формулу R6SO2Cl в присутствии аминового основания, с последующим гидролизом; или
(e) для соединения формулы I, где L является O, R2 является метилом, X1 является N, и R1, R3, R4, R5 и R6 такие, как определены для формулы I, сочетание соединения, имеющего формулу (38)
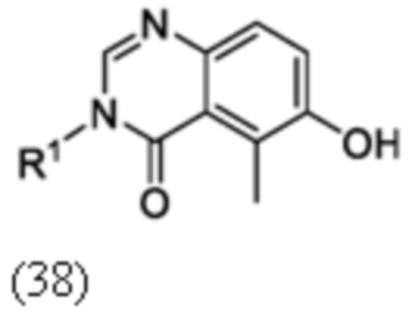
где R1 такой, как определен для формулы I, с соединением, имеющим формулу (39)
где X является галогеном, PG является защитной группой амина, и R3, R4, R5 и R6 такие, как определены для формулы I, в присутствии щелочного карбоната, с последующим удалением аминовой защитной группы; или
(f) для соединения формулы I где X1 является CH, R1 является метилом, и R2, R3, R4, R5 и R6 такие, как определены для формулы I, взаимодействие соединения, имеющего формулу (51)
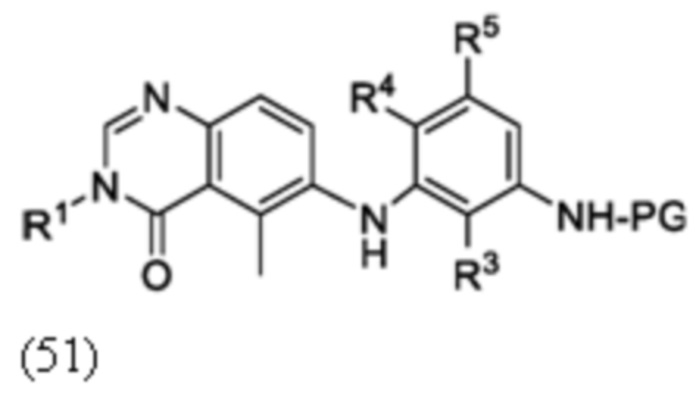
где X1 является CH, R1 является метилом, и R2, R3, R4, R5 и R6 такие, как определены для формулы I и PG является защитной группой амина, с соединением, имеющим формулу:
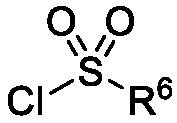
где R6 такой, как определен для формулы I, в присутствии основания, с получением соединения, имеющего формулу (52)
с последующим удалением аминовой защитной группы; и
необязательно, получение фармацевтически приемлемой соли или сольвата.
Используемый здесь термин «аминовая защитная группа» относится к производным групп, обычно используемых для блокирования или защиты аминогруппы, в то время как реакции проводят с другими функциональными группами соединения. Примеры подходящих защитных групп для использования в любом из способов, описанных в настоящем документе, включают карбаматы, амиды, алкильные и арильные группы, имины, а также многие производные N-гетероатома, которые могут быть удалены для регенерации желаемой группы амина. Неограничивающими примерами аминовых защитных групп являются трет-бутилоксикарбонил («Boc»), 2-триметилсилилэтоксиметил (SEM) и п-метоксибензил (PMB). Дополнительные примеры этих групп и других защитных групп можно найти в T. W. Greene, et al., Greene’s Protective Groups in Organic Synthesis. New York: Wiley Interscience, 2006.
Соединения формулы I полезны для лечения заболеваний и нарушений, которые можно лечить с помощью ингибитора киназы BRAF, таких как заболевания и нарушения, ассоциированные с BRAF, например, пролиферативные нарушения, такие как рак, включая солидные опухоли. Способность тестируемых соединений действовать как ингибиторы BRAF может быть продемонстрирована с помощью анализа, описанного в примере A. Значения IC50 показаны в таблице A.
В некоторых вариантах осуществления, определенные соединения формулы I или их фармацевтически приемлемая соль, сольват или полиморф, т.е. соединения формулы II или их фармацевтически приемлемая соль, сольват или полиморф, и формулы III или их фармацевтически приемлемая соль, сольват или полиморф, как описано ниже в настоящем документе, демонстрируют удивительную способность проникать в мозг и/или ЦНС. Такие соединения способны пересекать ГЭБ и ингибировать киназу BRAF в головном мозге и/или других структурах ЦНС. В некоторых вариантах осуществления, соединения, представленные в настоящем документе, способны пересекать ГЭБ в терапевтически эффективном количестве. Например, лечение субъекта с раком (например, BRAF-ассоциированным раком, таким как BRAF-ассоциированный рак ЦНС) может включать введение (например, пероральное введение) формулы I или его фармацевтически приемлемой соли, сольвата или полиморфа (т. е., соединения формулы II и формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа) субъекту. Соответственно, в некоторых вариантах осуществления соединения, представленные в настоящем документе, применяют для лечения рака ЦНС.
Используемые здесь термины «лечить» или «лечение» относятся к терапевтическим или паллиативным мерам. Благоприятные или желаемые клинические результаты включают, но не ограничены ими, облегчение, полное или частичное, симптомов, связанных с заболеванием или нарушением или состоянием, уменьшение степени заболевания, стабилизацию (т.е. не ухудшение) состояния заболевания, задержки или замедления прогрессирования заболевания, улучшения или временного смягчения болезненного состояния (например, одного или нескольких симптомов заболевания) и ремиссии (частичной или полной), обнаруживаемой или не обнаруживаемой. Однако «лечить» или «лечение» также может включать терапевтические меры (например, ингибирование киназы BRAF в BRAF-ассоциированной опухоли), которые временно ухудшают внешний вид и/или симптомы у субъекта. Используемые в настоящем документе термины «лечение» и «лечить» при обращении, например, к лечению рака, не предназначены для использования в качестве абсолютных терминов. Например, «лечение рака» и «лечить рак», используемые в клинических условиях, предназначены для включения получения полезных или желаемых клинических результатов и могут включать улучшение состояния субъекта, страдающего раком. Благоприятные или желаемые клинические результаты включают, но не ограничены ими, одно или несколько из следующего: уменьшение пролиферации (или разрушение) неопластических или раковых клеток, ингибирование метастазирования неопластических клеток, уменьшение метастазов у субъекта, уменьшение или уменьшение размера опухоли, изменение скорости роста одной или нескольких опухолей у субъекта, увеличение периода ремиссии у субъекта (например, по сравнению с одним или несколькими показателями у субъекта, имеющего аналогичный рак, не получающего лечение или другое лечение или по сравнению с одним или несколькими показателями у того же субъекта до лечения), уменьшение симптомов, вызванных заболеванием, повышение качества жизни тех, кто страдает от заболевания (например, оцененного с помощью FACT-G или EORTC-QLQC30), уменьшение дозы других лекарственных средств, необходимых для лечения заболевания, замедление прогрессирования заболевания и/или продление выживаемости субъектов, страдающих заболеванием. «Лечение» также может означать продление выживаемости по сравнению с ожидаемой выживаемостью, если не получал лечения, например, увеличение общей выживаемости (OS) по сравнению с субъектом, не получающим лечение, как описано в настоящем документе, и/или увеличение выживаемости без прогрессирования (PFS) по сравнению с субъектом, не получающим лечение, как описано в настоящем документе.
Используемый в настоящем документе термин «субъект» относится к любому животному, включая млекопитающих, таких как мыши, крысы, другие грызуны, кролики, собаки, кошки, свиньи, крупный рогатый скот, овцы, лошади, приматы и люди. В некоторых вариантах осуществления, субъектом является человек. В некоторых вариантах осуществления, субъект испытал и/или продемонстрировал, по меньшей мере, один симптом заболевания или нарушения, которое необходимо лечить и/или предотвращать. В некоторых вариантах осуществления, у субъекта идентифицирована или диагностирована опухоль с мутацией BRAF (BRAF-ассоциированная опухоль) (например, по данным одобренного регулирующим органом, например, одобренного FDA, анализа или набора). В некоторых вариантах осуществления, у субъекта имеется опухоль, положительная в отношении мутации BRAF (например, по данным анализа или набора, одобренного регулирующим органом). Субъект может быть субъектом, опухоль которого имеет мутацию BRAF (например, если опухоль идентифицирована как таковая по данным утвержденного регулирующим органом, например, утвержденного FDA набора или анализа). В некоторых вариантах осуществления, у субъекта подозревают BRAF-ассоциированную опухоль. В некоторых вариантах осуществления, субъект имеет историю болезни, указывающую, что субъект имеет опухоль, имеющую мутацию BRAF (и, необязательно, история болезни указывает, что субъекта следует лечить любой из композиций, представленных в настоящем документе). В некоторых вариантах осуществления, субъектом является человек. В некоторых вариантах осуществления, субъектом-человеком является субъект детского возраста.
Используемый в настоящем документе термин «субъект детского возраста» относится к субъекту в возрасте до 21 года на момент постановки диагноза или лечения. Термин «детский возраст» можно далее разделить на различные субпопуляции, включая: новорожденных (от рождения до первого месяца жизни); младенцев (от 1 месяца до двух лет); детей (от двух лет до 12 лет); и подростков (от 12 до 21 года (вплоть до, но не включая, двадцать второй день рождения)). Berhman RE, Kliegman R, Arvin AM, Nelson WE, Nelson Textbook of Pediatrics, 15th Ed. Philadelphia: W.B. Saunders Company, 1996; Rudolph AM, et al. Rudolph’s Pediatrics, 21st Ed. New York: McGraw-Hill, 2002; и Avery MD, First LR. Pediatric Medicine, 2nd Ed. Baltimore: Williams & Wilkins; 1994. В некоторых вариантах осуществления, субъект детского возраста имеет возраст от рождения до первых 28 дней жизни, возраст от 29 дней до менее двух лет, возраст от двух лет до менее 12 лет или возраст от 12 лет до 21 года (вплоть, но не включая, двадцать второй день рождения). В некоторых вариантах осуществления, субъект детского возраста имеет возраст от рождения до первых 28 дней жизни, возраста от 29 дней до менее 1 года, возраст от одного месяца до менее четырех месяцев, возраст от трех месяцев до менее семи месяцев, возраст от шести месяцев до менее 1 года, возраста от 1 года до менее 2 лет, возраст от 2 лет до менее 3 лет, возрасте от 2 лет до 7 лет, возраст от 3 до 5 лет, возраст от 5 до 10 лет, возраст от 6 до 13 лет, возраст от 10 до 15 лет или возраст от 15 до 22 лет.
В некоторых вариантах осуществления, соединения формулы I или их фармацевтически приемлемые соли или сольваты полезны для профилактики заболеваний и нарушений, как определено в настоящем документе. Используемый в настоящем документе термин «профилактика» означает профилактику начала, рецидива или распространения, полностью или частично, заболевания или состояния, как описано в настоящем документе.
Термин «BRAF-ассоциированное» по отношению к заболеванию или нарушению в контексте настоящего описания относится к заболеваниям или нарушениям, связанным с одной или несколькими мутациями BRAF или имеющим одну или несколько мутаций BRAF. Неограничивающие примеры BRAF-ассоциированного заболевания или нарушения, включают, например, BRAF- ассоциированные опухоли.
Фраза «мутация BRAF» относится к генетической мутации (например, хромосомной транслокации, которая приводит к одной или нескольким мутациям в гене BRAF, что приводит к экспрессии белка BRAF с одной или несколькими точечными мутациями по сравнению с BRAF дикого типа. Неограничивающие примеры мутаций BRAF включают мутации BRAF V600, например, V600E, V600K, V600R и V600S.
Термин «дикий тип» описывает нуклеиновую кислоту (например, ген BRAF или мРНК BRAF), которая обычно обнаруживается у субъекта, у которого нет заболевания или нарушения, связанного с эталонной нуклеиновой кислотой или белком.
Термин «BRAF дикого типа» описывает нуклеиновую кислоту BRAF (например, ген BRAF или мРНК BRAF) или белок BRAF, который обнаружен у субъекта, у которого нет BRAF-ассоциированного заболевания, например, BRAF-ассоциированного рака (и, необязательно, также не имеет повышенного риска развития BRAF-ассоциированного заболевания и/или не имеет подозрений на наличие BRAF-ассоциированного заболевания) или обнаруживается в клетке или ткани субъекта, у которого нет BRAF-ассоциированного заболевания, например, BRAF-ассоциированного рака (и, необязательно, также не имеет повышенного риска развития BRAF-ассоциированной болезни и/или не подозревается в наличии BRAF-ассоциированной болезни).
Используемый в настоящем документе термин «опухоль» относится к аномальному росту ткани, который возникает в результате неконтролируемой, как правило, быстрой клеточной пролиферации. Опухоль может быть доброкачественной опухолью (не раковой) или злокачественной опухолью (т.е. раком). Опухоль может быть солидной опухолью или гемобластозом (т.е. гематологической опухолью, также известной как рак крови).
Используемый в настоящем документе термин «BRAF-ассоциированная опухоль», относится к опухолям, связанным с мутацией BRAF или имеющим мутацию BRAF, например, мутацию BRAF V600, например, мутацию BRAF V600E, V600K, V600R или V600S. BRAF-ассоциированные опухоли, включают как доброкачественные BRAF-ассоциированные опухоли, так и злокачественные BRAF-ассоциированные опухоли (т.е. BRAF-ассоциированные раки).
Термин «BRAF-ассоциированный рак» в контексте настоящего описания относится к раковым заболеваниям, связанным с мутацией BRAF или имеющим мутацию BRAF, например, мутации BRAF V600, например, мутации BRAF V600E, V600K, V600R или V600S. В настоящем документе описаны неограничивающие примеры BRAF-ассоциированного рака.
Термин «регулирующий орган» относится к агентству страны по разрешению медицинского использования фармацевтических агентов в стране. Например, неограничивающим примером регулирующего органа является U.S. Food and Drug Administration (FDA).
В настоящем документе представлен способ лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в таком лечении, где способ включает введение субъекту терапевтического количества эффективного количества формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или его фармацевтической композиции. Например, в настоящем документе представлены способы лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в таком лечении, где способ включает а) обнаружение мутации BRAF в образце, взятом у субъекта; и b) введение терапевтически эффективного количества формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, мутацией BRAF является V600E и/или V600K и/или V600D и/или V600R. В некоторых вариантах осуществления, мутацией BRAF является V600E. В некоторых вариантах осуществления, мутацией BRAF является V600K.
В некоторых вариантах осуществления любого из способов применения, описанных в настоящем документе, BRAF-ассоциированная опухоль, представляет собой солидную опухоль. В некоторых вариантах осуществления, опухоль является внутричерепной. В некоторых вариантах осуществления, опухоль является экстракраниальной. В некоторых вариантах осуществления любого из способов применения, описанных в настоящем документе, BRAF-ассоциированной опухолью является злокачественная BRAF-ассоциированная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления любого из способов применения, описанных в настоящем документе, раком является меланома, рак толстой кишки, колоректальный рак, рак легкого (например, мелкоклеточный рак легкого или немелкоклеточный рак легкого), рак щитовидной железы (например, папиллярный рак щитовидной железы, медуллярный рак щитовидной железы, дифференцированный рак щитовидной железы, рецидивирующий рак щитовидной железы или рефрактерный дифференцированный рак щитовидной железы), рак груди, рак яичников, рак ЦНС, рак кости, рак заднего прохода, анального канала или аноректума, рак глаза рак желчных протоков, протоковая карцинома in situ, рак печени, желчного пузыря или плевры, рак рта, рак полости рта, рак губы, рак ротоглотки, рак носа, носовой полости или среднего уха, рак вульвы, рак пищевода, рак шейки матки, карциноидная опухоль желудочно-кишечного тракта, рак подглоточника, рак почки, рак гортани, рак печени, рак легких, меланома, рак носоглотки, рак периферической нервной системы (например, нейробластома), рак яичников, рак поджелудочной железы, брюшины, сальника, рак брыжейки, рак глотки, рак простаты, рак почек (например, почечно-клеточная карцинома (RCC)), рак тонкой кишки, рак мягких тканей, рак желудка, рак яичек, рак матки, рак мочеточника или рак мочевого пузыря. В одном варианте осуществления, BRAF-ассоциированным раком является рак ЦНС, меланома, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников или нейробластома. В некоторых вариантах осуществления, BRAF-ассоциированным раком является внечерепной рак, выбранный из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого, рака яичников и нейробластомы. В некоторых вариантах осуществления, BRAF-ассоциированным раком является меланома. В некоторых вариантах осуществления, BRAF-ассоциированным раком является рак толстой кишки. В некоторых вариантах осуществления, BRAF-ассоциированным раком является рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным раком является немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным раком является рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированный рак представляет собой нейробластому. В некоторых вариантах осуществления, BRAF-ассоциированным раком является внутричерепной рак (рак мозга). В некоторых вариантах осуществления, BRAF-ассоциированным раком является рак ЦНС.
Термин «метастаз» является известным в данной области техники термином, который относится к распространению раковых клеток из места, где они впервые образовались (первичный сайт), к одному или нескольким другим сайтам у субъекта (одному или нескольким вторичным сайтам). При метастазировании раковые клетки отрываются от исходной (первичной) опухоли, путешествуют по кровеносной или лимфатической системе и образуют новую опухоль (метастатическую опухоль) в других органах или тканях тела. Новая метастатическая опухоль включает те же или аналогичные раковые клетки, что и первичная опухоль. На вторичном сайте опухолевые клетки могут пролиферировать и начать рост или колонизацию вторичной опухоли на этом удаленном сайте.
Термин «метастатический рак» (также известный как «вторичный рак»), используемый в настоящем документе, относится к типу рака, который возникает в одном типе ткани, но затем распространяется на одну или несколько тканей за пределами происхождения (первичного) рака. Метастатический рак головного мозга относится к раку в головном мозге, то есть к раку, который возник в ткани, отличной от мозга, и метастазировал в мозг.
В одном варианте осуществления, BRAF-ассоциированной опухолью является злокачественная BRAF-ассоциированная опухоль ЦНС (т.е. BRAF-ассоциированный рак ЦНС). Термин «рак ЦНС» или «рак ЦНС» или используемый в настоящем документе взаимозаменяемо относится к раку (т.е. злокачественной опухоли) ЦНС, включая рак головного мозга (также известный как внутричерепные опухоли), рак спинного мозга и рак мозговых оболочек, окружающих головной и спинной мозг. Термин «BRAF-ассоциированный рак ЦНС» относится к раку ЦНС, связанному с мутацией BRAF или имеющему мутацию BRAF. Раки ЦНС включают метастатический рак головного мозга и злокачественные первичные опухоли головного мозга.
В одном варианте осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак головного мозга. BRAF-ассоциированный метастатический рак головного мозга может быть результатом любого рака, описанного в настоящем документе, где у субъекта развился, по меньшей мере, один метастаз в головной мозг. В одном варианте осуществления, BRAF-ассоциированным метастатическим раком головного мозга является метастатическая меланома, метастатический колоректальный рак или метастатический немелкоклеточный рак легкого. В одном варианте осуществления, BRAF-ассоциированным метастатическим раком мозга является метастатическая меланома. В одном варианте осуществления, BRAF-ассоциированным метастатическим раком мозга является метастатический колоректальный рак. В одном варианте осуществления, BRAF-ассоциированным метастатическим раком головного мозга является метастатический немелкоклеточный рак легкого. В одном варианте осуществления, BRAF-ассоциированным метастатическим раком мозга является метастатический рак яичников. В одном варианте осуществления метастатическим раком мозга является метастатический рак щитовидной железы. В одном варианте осуществления, BRAF-ассоциированным метастатическим раком мозга является рак почки. В одном варианте осуществления, раком является BRAF-ассоциированный метастатический рак с, по меньшей мере, одним метастазом в мозг (т.е. метастатический рак головного мозга). В одном варианте осуществления, раком является BRAF-ассоциированная метастатическая меланома с, по меньшей мере, одним метастазом в мозг. В одном варианте осуществления, раком является BRAF-ассоциированный метастатический колоректальный рак с, по меньшей мере, одним метастазом в мозг. В одном варианте осуществления, раком является BRAF-ассоциированный метастатический немелкоклеточный рак легкого с, по меньшей мере, одним метастазом в мозг. В одном варианте осуществления, раком является BRAF-ассоциированный метастатический рак яичников с, по меньшей мере, одним метастазом в мозг. В одном варианте осуществления, раком является BRAF-ассоциированный метастатический рак щитовидной железы с, по меньшей мере, одним метастазом в мозг. В одном варианте осуществления, раком является BRAF-ассоциированная нейробластома с, по меньшей мере, одним метастазом в мозг.
Лептоменингеальными метастазами (лептоменингеальной болезнью (LMD)) является подмножество метастазов ЦНС, которые растут в слизистой оболочке головного мозга или позвоночника и/или в спинномозговой жидкости (СМЖ) или лептоменингеальный карциноматоз. У млекопитающих мозговые оболочки представляют собой твердую мозговую оболочку, паутинную оболочку и мягкую мозговую оболочку. СМЖ располагается в субарахноидальном пространстве между паутинной оболочкой и мягкой мозговой оболочкой. Вместе паутинную оболочку и мягкую мозговую оболочку иногда называют лептоменингами. Когда LMD возникает в лептоменингах и/или СМЖ, окружающих спинной мозг, ее можно назвать «внечерепной LMD». Когда LMD возникает в лептоменингах и/или СМЖ головного мозга, ее можно назвать «внутричерепной LMD». Поскольку раковые клетки LMD могут находиться во взвешенном состоянии в спинномозговой жидкости, они могут быстро распространяться по ЦНС. В результате у LMD плохой прогноз, выживаемость обычно измеряется месяцами. В одном варианте осуществления, метастатический раком является BRAF-ассоциированная LMD. В одном варианте осуществления, метастатическим раком является внутричерепная BRAF-ассоциированная LMD. В одном варианте осуществления, метастатическим раком является внечерепная BRAF-ассоциированная LMD. BRAF-ассоциированными раками с наиболее высокой частотой лептоменингеальных метастазов являются рак легких и меланома. В одном варианте осуществления, BRAF-ассоциированной LMD является LMD, полученная из метастазов меланомы (т.е. LMD является метастатической меланомой). В одном варианте осуществления, BRAF-ассоциированной LMD является LMD, полученная из метастазов колоректального рака (т.е. LMD является метастатическим колоректальным раком). В одном варианте осуществления, BRAF-ассоциированной LMD является LMD, полученная из метастазов немелкоклеточного рака легкого (т.е. LMD является метастатическим немелкоклеточным раком легкого).
В одном варианте осуществления, раком является BRAF-ассоциированный рак, имеющий высокий риск метастазирования. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является рак, имеющий мутации BRAF V600E, V600K, V600R и/или V600S. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является меланома, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников или нейробластома. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является меланома, колоректальный рак, рак щитовидной железы, немелкоклеточный рак легкого, рак яичников или нейробластома, каждый из которых имеет мутацию BRAF V600E, V600K, V600R и/или V600S. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является меланома. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является меланома, имеющая мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является колоректальный рак. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является колоректальный рак, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является рак щитовидной железы. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является рак щитовидной железы, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является немелкоклеточный рак легкого. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является немелкоклеточный рак легкого, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является рак яичников. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является рак яичника, имеющий мутацию BRAF V600E или мутацию BRAF V600K. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является нейробластома. В одном варианте осуществления, BRAF-ассоциированным раком, имеющим высокий риск метастазирования, является нейробластома, имеющая мутацию BRAF V600E или мутацию BRAF V600K.
В одном варианте осуществления, BRAF-ассоциированной опухолью ЦНС является первичная BRAF-ассоциированная опухоль головного мозга. Первичными опухолями головного мозга являются опухоли, которые начинаются в головном мозге или позвоночнике и известны под общим названием глиомы. Термин «глиома» используется для описания опухолей, которые возникают в глиальных клетках, присутствующих в ЦНС. Согласно классификации опухолей головного мозга ВОЗ, глиомы классифицируются по активности клеток и агрессивности по шкале, включающей степень I (доброкачественные опухоли ЦНС) и степени II - IV (злокачественные опухоли ЦНС):
Глиома I степени (пилоцитарная астроцитома): обычно возникает у детей в мозжечке или стволе мозга, а иногда и в полушариях головного мозга, и растет медленно. I степень может встречаться у взрослых. Несмотря на то, что они доброкачественные (степень I по классификации ВОЗ), трудности с лечением этого заболевания делают их рост злокачественным по поведению с высоким уровнем заболеваемости (Rostami, Acta Neurochir (Wien). 2017; 159(11): 2217-2221).
Глиома II степени (глиомы низкой степени злокачественности): включает астроцитому, олигодендроглиому и смешанную олигоастроцитому. Глиомы II степени обычно возникают у молодых людей (20-50 лет) и чаще всего встречаются в полушариях головного мозга. Из-за инфильтративной природы этих опухолей могут возникать рецидивы. Некоторые глиомы II степени рецидивируют и развиваются в более агрессивные опухоли (III или IV степени).
Глиома III степени (злокачественная глиома): включает анапластическую астроцитому, анапластическую олигодендроглиому и анапластическую смешанную олигоастроцитому. Опухоли III степени агрессивны, злокачественные опухоли высокой степени злокачественности проникают в близлежащие ткани головного мозга с выступами в виде щупалец, что затрудняет полное хирургическое удаление.
Глиомы IV степени: включает мультиформную глиобластому (GBM) и глиосаркому; (GBM) является злокачественной глиомой. GBM является самой агрессивной и самой распространенной первичной опухолью головного мозга. Мультиформная глиобластома обычно быстро распространяется и проникает в другие части мозга с выступами в виде щупалец, что затрудняет полное хирургическое удаление. Глиосаркома является злокачественным раком, который определяется как глиобластома, состоящая из глиоматозного и саркоматозного компонентов.
В одном варианте осуществления, BRAF-ассоциированной первичной опухолью головного мозга является глиома.
Доброкачественные первичные опухоли головного мозга могут вызвать сильную боль, необратимое повреждение головного мозга и смерть, а в некоторых случаях могут стать злокачественными. Неограничивающие примеры доброкачественных первичных опухолей головного мозга включают глиомы I степени, папиллярные краниофарингиомы, менингиому (включая рабдоидную менингиому), атипичные тератоидные/рабдоидные опухоли, дизэмбриопластическую нейроэпителиальную опухоль (DNT), пилоцитарную астроцитому, олигодендроглиому, смешанную олигоастроцитому, анапластическую астроцитому, анапластическую олигодендроглиому, анапластическую смешанную олигоастроцитому, диффузную астроцитому, эпендимому, плеоморфную ксантоастроцитому (PXA), ганглиоглиому, глиосаркому или анапластическую ганглиоглиому. В одном варианте осуществления, BRAF-ассоциированной опухолью является доброкачественная первичная опухоль головного мозга.
В одном варианте осуществления BRAF-ассоциированным раком является рак периферической нервной системы. В одном варианте осуществления, раком периферической нервной системы является нейробластома. В одном варианте осуществления, раком является BRAF-ассоциированный рак.
Было обнаружено, что подмножество соединений формулы I, то есть соединения формулы II или их фармацевтически приемлемые соли, сольваты или полиморфы, проявляют неожиданную пенетрантность мозга и/или ЦНС. Такие соединения способны пересекать ГЭБ и ингибировать киназу BRAF в головном мозге и/или других структурах ЦНС. В некоторых вариантах осуществления, соединения формулы II или их фармацевтически приемлемые соли или сольваты способны пересекать ГЭБ в терапевтически эффективном количестве.
В одном варианте осуществления, в настоящем документе представлены соединения формулы II
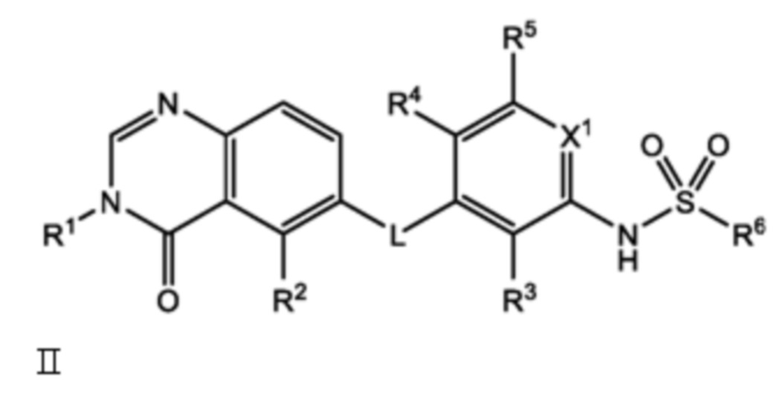
и их фармацевтически приемлемые соли, сольваты или полиморфы, где:
L является NH или O или S;
X1 является CH или N;
R1 является С1-С6 алкилом, C1-C6 дейтероалкилом, C1-C6 фторалкилом, C3-C6 циклоалкилом, (C3-C6 циклоалкил)CH2-, Ar1, Ar1CH2- или hetCyc1;
Ar1 является фенилом который необязательно замещен 1-5 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetCyc1 является 4-6-членным насыщенным гетероциклическим кольцом, имеющим один кольцевой атом кислорода;
R2 является метилом;
R3 является F или Cl;
R4 является H, F или Cl;
R5 является H, F, Cl или метилом;
R6 является С1-С6 алкилом, C1-C6 фторалкилом, (Cyc1)C1-C6 алкилом-, (C1-C3 алкокси)C1-C6 алкилом-, Ar2 или RaRbN-;
Cyc1 является 3-6-членным насыщенным карбоциклическим кольцом;
Ar2 является фенилом необязательно замещенным 1-5 заместителями, независимо выбранными из галогена и C1-C3 алкила; и
Ra и Rb независимо являются C1-C6 алкилом, или
Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, необязательно замещенное 1-2 галогенами;
и их фармацевтически приемлемые соли.
В одном варианте осуществления формулы II, R1 является С1-С6 алкилом. В одном варианте осуществления, формулы II, R1 является метилом, этилом или изопропилом. В одном варианте осуществления, формулы II, R1 является метилом.
В одном варианте осуществления формулы II, R1 является C1-C6 дейтероалкилом. в одном варианте осуществления формулы II, R1 является -CD3.
В одном варианте осуществления формулы II, R1 является C1-C6 фторалкилом. В одном варианте осуществления формулы II, R1 является 2,2,2-трифторэтилом.
В одном варианте осуществления формулы II, R1 является C3-C6 циклоалкилом. В одном варианте осуществления формулы II, R1 является циклопропилом, циклобутилом или циклопентилом.
В одном варианте осуществления формулы II, R1 является (C3-C6 циклоалкил)CH2-. В одном варианте осуществления формулы II, R1 является циклопропилметилом.
В одном варианте осуществления формулы II, R1 является Ar1. В одном варианте осуществления формулы II, R1 является фенилом.
В одном варианте осуществления формулы II, R1 является Ar1CH2-. В одном варианте осуществления формулы II, R1 является бензилом.
В одном варианте осуществления формулы II, R1 является hetCyc1. В одном варианте осуществления формулы II, R1 является тетрагидрофуранилом.
В одном варианте осуществления формулы II, R3 является F.
В одном варианте осуществления формулы II, R3 является Cl.
В одном варианте осуществления формулы II, R4 является H.
В одном варианте осуществления формулы II, R4 является F.
В одном варианте осуществления формулы II, R4 является Cl.
В одном варианте осуществления формулы II, R5 является H.
В одном варианте осуществления формулы II, R5 является F.
В одном варианте осуществления формулы II, R5 является Cl
В одном варианте осуществления формулы II, R5 является метилом.
В одном варианте осуществления формулы II, R6 является С1-С6 алкилом. В одном варианте осуществления формулы II, R6 является этилом, пропилом, 2-метилпроп-1-илом или 1-метилпроп-1-илом.
В одном варианте осуществления формулы II, R6 является C1-C6 фторалкилом. В одном варианте осуществления формулы II, R6 является 3-фторпроп-1-илом.
В одном варианте осуществления формулы II, R6 является (Cyc1)C1-C6 алкилом-. В одном варианте осуществления формулы II, R6 является циклопропилметилом.
В одном варианте осуществления формулы II, R6 является (C1-C3 алкокси)C1-C6 алкилом-. В одном варианте осуществления формулы II, R6 является (2-метокси)этилом.
В одном варианте осуществления формулы II, R6 является Ar2. В одном варианте осуществления формулы II, R6 является фенилом необязательно замещенным 1-3 галогенами. В одном варианте осуществления формулы II, R6 является фенилом необязательно замещенным 1-3 атомами фтора. В одном варианте осуществления формулы II, R6 является фенилом необязательно замещенным одним или двумя атомами фтора. В одном варианте осуществления формулы II, R6 является фенилом или 2,5-дифторфенилом.
В одном варианте осуществления формулы II, R6 является RaRbN-.
В одном варианте осуществления формулы II, R6 является RaRbN- где Ra и Rb независимо являются C1-C6 алкилом. В одном варианте осуществления формулы II, R6 является N-метил-N-этиламино-.
В одном варианте осуществления формулы II, R6 является RaRbN- где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, необязательно замещенное 1-2 галогенами. В одном варианте осуществления формулы II, R6 является RaRbN-, где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-членное насыщенное моноциклическое гетероциклическое кольцо, необязательно замещенное 1-2 галогенами. В одном варианте осуществления формулы II, R6 является RaRbN-, где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-членное насыщенное моноциклическое гетероциклическое кольцо, необязательно замещенное 1-2 атомами фтора. В одном варианте осуществления формулы II, R6 является RaRbN-, где Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-членное насыщенное моноциклическое гетероциклическое кольцо, необязательно замещенное фтором. В одном варианте осуществления формулы II, R6 является 3-фторпирролидинилом.
В одном варианте осуществления формулы II, R6 является С1-С6 алкилом, C1-C6 фторалкилом, (Cyc1)C1-C6 алкилом-, (C1-C3 алкокси)C1-C6 алкилом- или Ar2.
Любой из вышеупомянутых вариантов осуществления формулы II могут быть скомбинированы друг с другом.
В одном варианте осуществления, соединения формулы II включают соединения формулы II-A
и их фармацевтически приемлемые соли, сольваты или полиморфы, где:
L является NH или O или S;
X1 является CH или N;
R1 является С1-С6 алкилом, C1-C6 дейтероалкилом, C1-C6 фторалкилом, C3-C6 циклоалкилом, (C3-C6 циклоалкил)CH2-, Ar1, Ar1CH2- или hetCyc1;
Ar1 является фенилом который необязательно замещен 1-5 заместителями, независимо выбранными из галогена и C1-C3 алкила;
hetCyc1 является 4-6-членным насыщенным гетероциклическим кольцом, имеющим один кольцевой атом кислорода;
R2 является метилом;
R3 является F или Cl;
R4 является H, F или Cl;
R5 является H, F, Cl или метилом;
R6 является С1-С6 алкилом, C1-C6 фторалкилом, (Cyc1)C1-C6 алкилом-, (C1-C3 алкокси)C1-C6 алкилом-, Ar2 или RaRbN-;
Cyc1 является 3-6-членным насыщенным карбоциклическим кольцом;
Ar2 является фенилом необязательно замещенным 1-5 заместителями, независимо выбранными из галогена и C1-C3 алкила; и
Ra и Rb независимо являются C1-C6 алкилом, или
Ra и Rb вместе с атомом азота, к которому они присоединены, образуют 5-6-членное насыщенное гетероциклическое кольцо, необязательно замещенное 1-2 галогенами;
где:
если L является NH, X1 является CH и R3 является F, то R4 является F или H, R5 является H, Cl или метилом и R6 является С1-С6 алкилом, (Cyc1)C1-C6 алкилом-, Ar2 или RaRbN-;
если L1 является NH, X1 является CH, R3 является Cl, R4 является F и R5 является F, то R6 является С1-С6 алкилом, (Cyc1)C1-C6 алкилом-, Ar2 или RaRbN-, и
если L1 является NH и X1 является N, то R3 является Cl, R4 является Cl и R5 является H.
насыщенным моноциклическим гетероциклическим насыщенным моноциклическим гетероциклическим насыщенным моноциклическим гетероциклическим насыщенным моноциклическим гетероциклическим.
В одном варианте осуществления, соединения формулы II выбирают из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82, имеющих следующие структуры, соответственно:
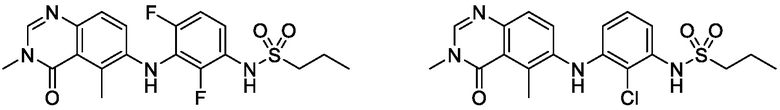
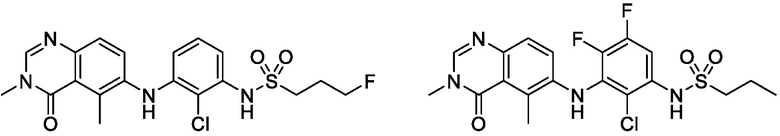
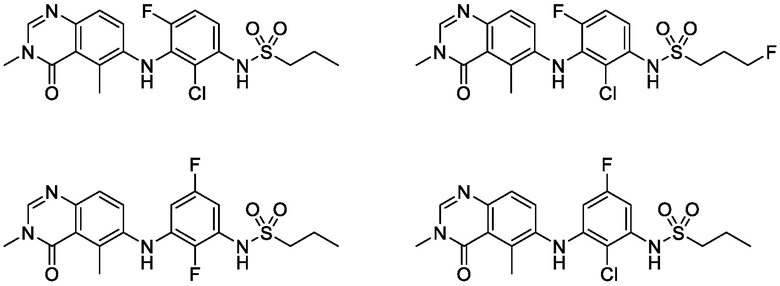
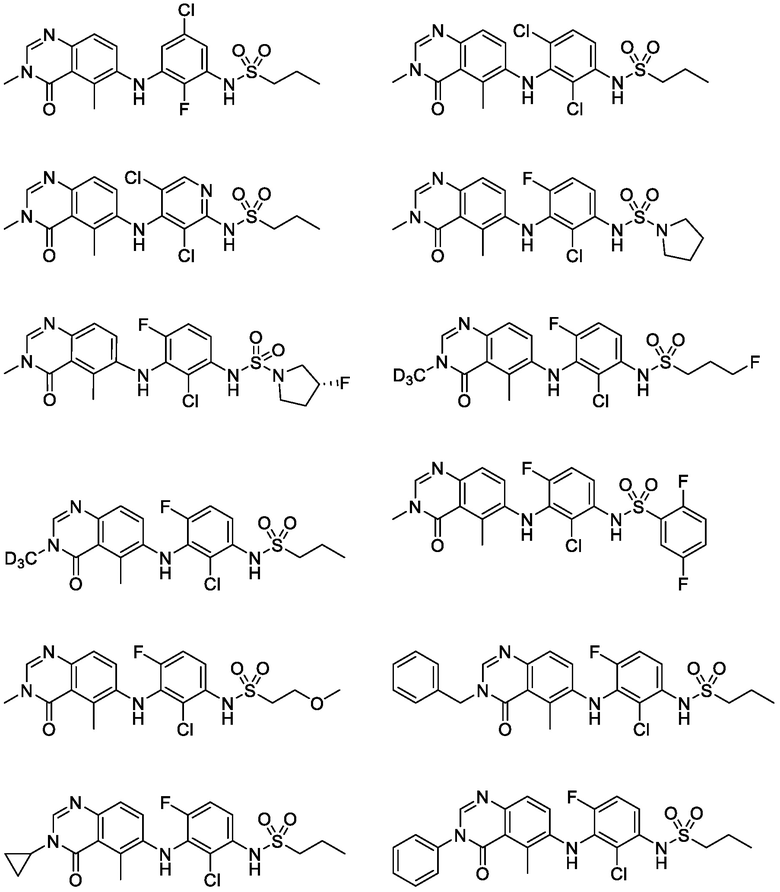
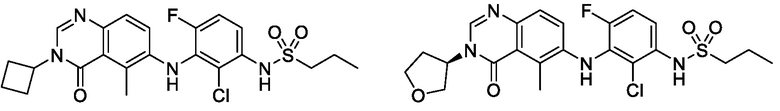
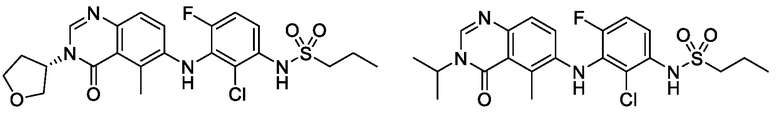
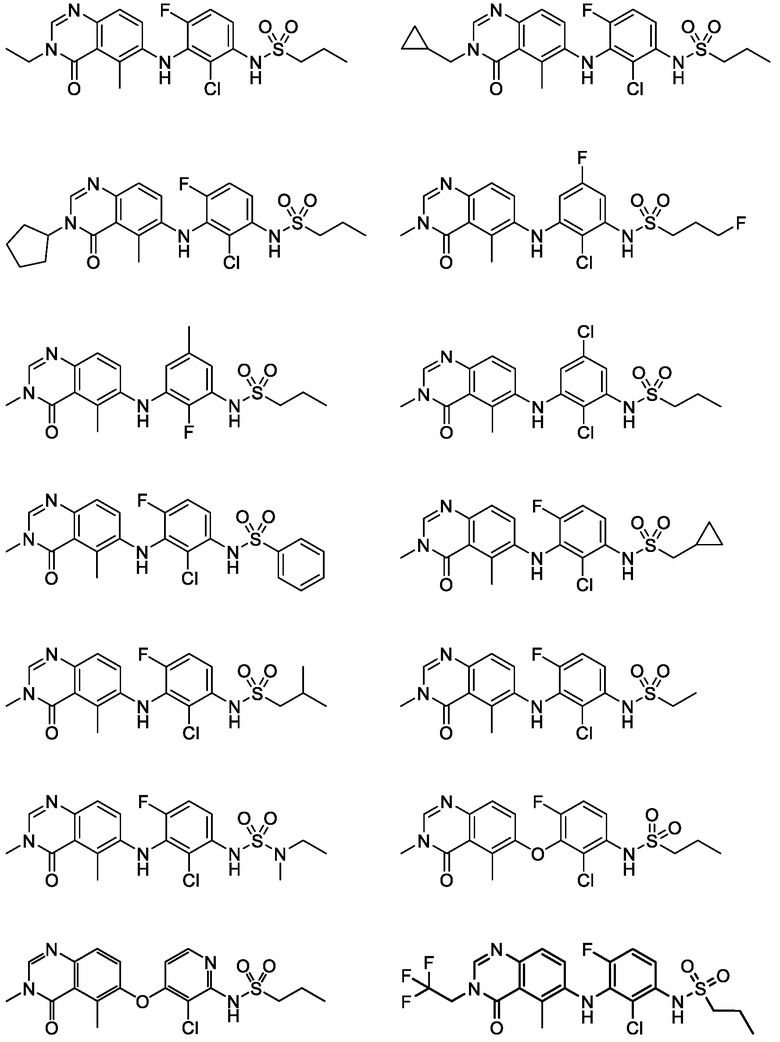
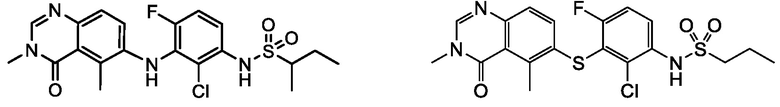
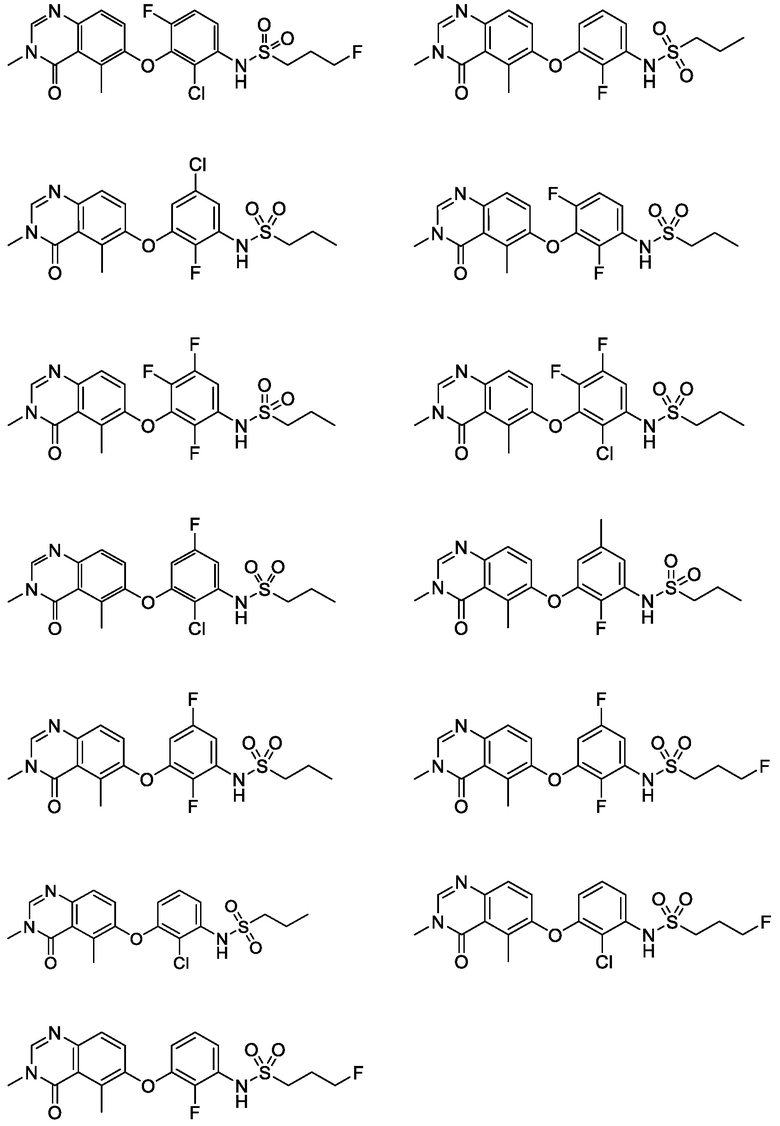
и их фармацевтически приемлемые соли и сольваты, и
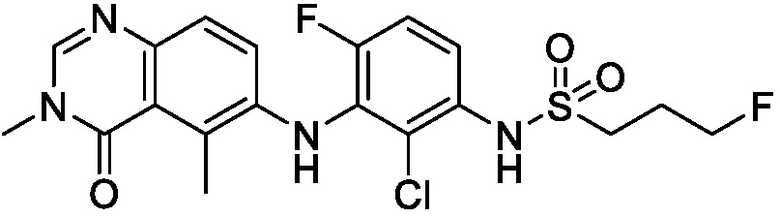 безводная форма В.
безводная форма В.
В одном варианте осуществления, соединения примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 имеют форму свободного основания. В одном варианте осуществления, соединения примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 имеют форму кислой соли. В одном варианте осуществления, соединения примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 являются трифторацетатами.
Было обнаружено, что конкретная подгруппа соединений формулы II, то есть соединения формулы III или их фармацевтически приемлемые соли, сольваты или полиморфы, обладают особенно неожиданной пенетрантностью в ЦНС. В одном варианте осуществления, соединения формулы III обладают особенно неожиданной пенетрантностью в мозг.
В одном варианте осуществления, в настоящем документе представлены соединения формулы III, имеющие формулу:
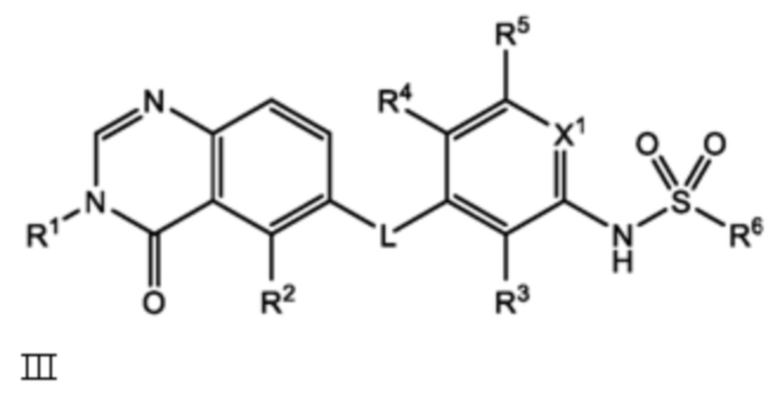
и их фармацевтически приемлемые соли, сольваты или полиморфы, где:
L является NH или O;
X1 является CH или N;
R1 является С1-С6 алкилом или C1-C6 дейтероалкилом;
R2 является метилом;
R3 является F или Cl;
R4 является H или F;
R5 является H или F; и
R6 C1-C6 алкилом или C1-C6 фторалкилом.
В одном варианте осуществления формулы III, L является NH.
В одном варианте осуществления формулы III, L является O.
В одном варианте осуществления формулы III, R1 является С1-С6 алкилом. В одном варианте осуществления формулы III, R1 является метилом.
В одном варианте осуществления формулы III, R1 является C1-C6 дейтероалкилом. В одном варианте осуществления формулы III, R1 является -CD3.
В одном варианте осуществления формулы III, R3 является F.
В одном варианте осуществления формулы III, R3 является Cl.
В одном варианте осуществления формулы III, R4 является H.
В одном варианте осуществления формулы III, R4 является F.
В одном варианте осуществления формулы III, R5 является H.
В одном варианте осуществления формулы III, R5 является F.
В одном варианте осуществления формулы III, R6 является С1-С6 алкилом. В одном варианте осуществления формулы III, R6 является этилом. В одном варианте осуществления формулы III, R6 является пропилом.
В одном варианте осуществления формулы III, R6 является C1-C6 фторалкилом. В одном варианте осуществления формулы III, R6 является 3-фторпропилом.
Любой из вышеупомянутых вариантов осуществления формулы III могут быть скомбинированы друг с другом.
В одном варианте осуществления, соединения формулы III включают соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемые соли и сольваты, и пример 82, имеющие следующие структуры, соответственно:
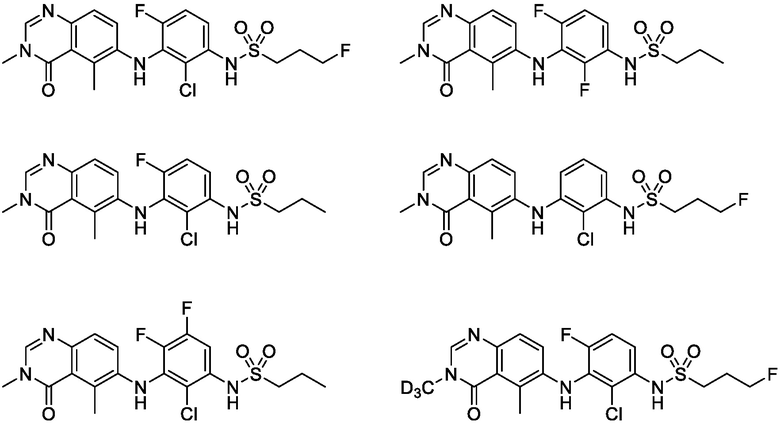
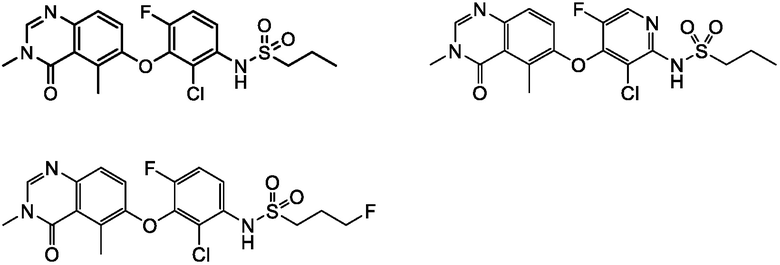
и их фармацевтически приемлемые соли и сольваты, и
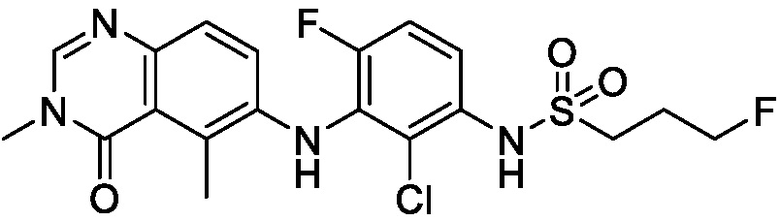 безводная форма В.
безводная форма В.
В одном варианте осуществления, соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 имеют форму свободного основания. В одном варианте осуществления, соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 имеют форму кислой соли. В одном варианте осуществления, соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 являются трифторацетатами. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82.
В одном варианте осуществления, соединения формулы III включают соединения формулы III-A, имеющие формулу:
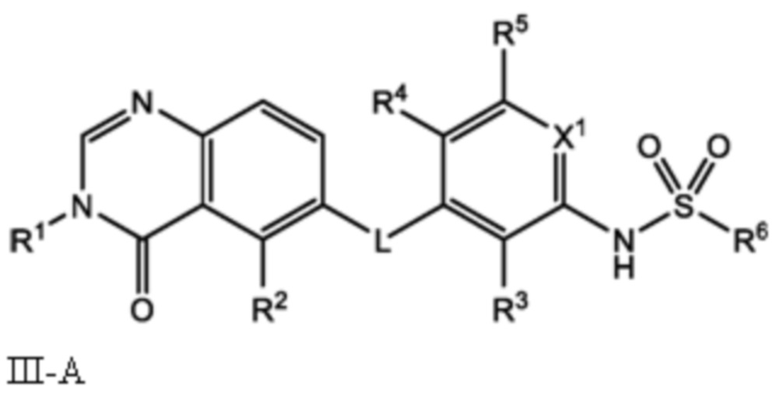
и их фармацевтически приемлемые соли, сольваты или полиморфы, где:
L является NH или O;
X1 является CH или N;
R1 является С1-С6 алкилом или C1-C6 дейтероалкилом;
R2 является метилом;
R3 является F или Cl;
R4 является H или F;
R5 является H или F; и
R6 C1-C6 алкилом или C1-C6 фторалкилом;
где если R3 является F и R4 является F, то R5 является H и R6 является С1-С6 алкилом, и
если R3 является Cl, R4 является F и R5 является F, то R6 является С1-С6 алкилом.
Соответственно, соединения формулы II или формулы III и их фармацевтически приемлемые соли и соли, описанные в настоящем документе, также могут быть использованы для лечения BRAF-ассоциированных опухолей ЦНС. Например, лечение субъекта с BRAF-ассоциированной опухолью ЦНС может включать введение (например, пероральное введение) формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, субъекту. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС имеет мутацию BRAF V600. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС имеет мутацию BRAF V600E и/или V600K и/или V600D и/или V600R. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС имеет мутацию BRAF V600E. В некоторых вариантах осуществления, BRAF-ассоциированный рак ЦНС имеет мутацию BRAF V600K. В некоторых вариантах осуществления, субъекта ранее лечили одним или несколькими другими видами противораковой терапии, например, противораковым средством, хирургическим вмешательством и/или радиационной терапией, например, как описано ниже. В некоторых вариантах осуществления, субъекта лечат соединением формулы II или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом в сочетании с одним или несколькими другими видами противораковой терапии, например, противораковым агентом, хирургическим вмешательством и/или радиационной терапией, например, как описано ниже. В некоторых вариантах осуществления, субъекта лечат одним или несколькими способами лечения рака, например, противораковым средством, хирургическим вмешательством и/или радиационной терапией после введения формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, например, как описано ниже.
В некоторых вариантах осуществления любого из способов, описанных в настоящем документе, опухолью является BRAF-ассоциированная опухоль ЦНС, и способ включает введение соединения формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является опухоль ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью ЦНС является злокачественная опухоль ЦНС (рак ЦНС). В некоторых вариантах осуществления, злокачественной опухолью ЦНС является метастатический рак ЦНС. В некоторых вариантах осуществления, метастатический рак ЦНС выбран из метастатической меланомы, метастатического колоректального рака, метастатического немелкоклеточного рака легкого, метастатического рака щитовидной железы и метастатического рака яичников. В некоторых вариантах осуществления, метастатическим раком ЦНС является метастатическая меланома. В некоторых вариантах осуществления, метастатическим раком ЦНС является колоректальный рак. В некоторых вариантах осуществления, метастатическим раком ЦНС является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, метастатическим раком ЦНС является метастатический рак щитовидной железы. В некоторых вариантах осуществления, метастатическим раком ЦНС является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью ЦНС является LMD. В некоторых вариантах осуществления, LMD является внутричерепной. В некоторых вариантах осуществления, LMD является внечерепной. В некоторых вариантах осуществления, LMD является метастатической меланомой. В некоторых вариантах осуществления, LMD выбран из метастатической меланомы, метастатического колоректального рака и метастатического немелкоклеточного рака легкого. В некоторых вариантах осуществления, LMD является метастатический колоректальный рак. В некоторых вариантах осуществления, LMD является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга. В некоторых вариантах осуществления, первичной опухолью головного мозга является глиома 2 степени. В некоторых вариантах осуществления, первичной опухолью головного мозга является глиома 3 степени. В некоторых вариантах осуществления, первичной опухолью головного мозга является глиома 4 степени. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью ЦНС является доброкачественная опухоль. В некоторых вариантах осуществления, доброкачественной опухолью ЦНС является папиллярная краниофарингиома, менингиома (включая рабдоидную менингиому), атипичная тератоидная/рабдоидная опухоль или дисэмбриопластическая нейроэпителиальная опухоль (DNT). В некоторых вариантах осуществления, соединением является соединение формулы III. В некоторых вариантах осуществления, соединением является соединение формулы III. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82.
Способность определять, может ли соединение подходить для лечения рака ЦНС, может быть определена, например, через определение того, является ли соединение субстратом транспортера эффлюкса и/или измерение проницаемости клеток и/или измерение содержания соотношения свободной крови к свободной плазме, как описано в настоящем документе.
В некоторых вариантах осуществления, соединения формулы II или их фармацевтически приемлемая соль, сольват или полиморф или формулы III или их фармацевтически приемлемая соль, сольват или полиморф, демонстрируют высокую проницаемость в клетки. Способы определения проницаемости соединения формулы II или III или его фармацевтически приемлемой соли, сольвата или полиморфа могут быть определены в соответствии с анализом, описанным в примере B, и коэффициенты проницаемости для соединения формулы I, II и III представлены в таблице B1.
Определенные соединения формулы I или их фармацевтически приемлемая соль, сольват или полиморф, т.е. соединения формулы II или их фармацевтически приемлемые соли или сольваты или соединения формулы III или их фармацевтически приемлемые соли или сольваты демонстрируют низкий эффлюкс. Способы in vitro оценки того, являются ли соединения формулы II или их фармацевтически приемлемые соли или сольваты или формула III или их фармацевтически приемлемые соли или сольваты субстратами для транспортеров эффлюкса P-гликопротеина (P-gp или белок мультилекарственной резистентности 1 (MDR1)) и белок резистентности к раку груди (BCRP) описаны в примере B, и коэффициенты эффлюкса соединений формулы II представлены в таблице B3. В одном варианте осуществления, соединения формулы II и их фармацевтически приемлемые соли и сольваты или формулы III и их фармацевтически приемлемые соли и сольваты имеют коэффициент эффлюкса ≤3,0 при тестировании на клетках, экспрессирующих P-gp. В одном варианте осуществления, соединения формулы II и их фармацевтически приемлемые соли и сольваты, имеющие коэффициент эффлюкса ≤3,0 при тестировании в клетках, экспрессирующих P-gp, выбраны из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59,61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80, и их фармацевтически приемлемых солей и сольватов, и примера 81-86. В одном варианте осуществления, соединения формулы II и их фармацевтически приемлемые соли и сольваты или формулы III и их фармацевтически приемлемые соли и сольваты, имеют коэффициент эффлюкса ≤3,0 при тестировании в клетках, экспрессирующих P-gp, и коэффициент эффлюкса ≤4,5 при тестировании в клетках, экспрессирующих BCRP. В одном варианте осуществления, соединения формулы II и их фармацевтически приемлемые соли и сольваты или формулы III и их фармацевтически приемлемые соли и сольваты, имеющие коэффициент эффлюкса ≤3,0 при тестировании на клетках, экспрессирующих P-gp, и коэффициент эффлюкса ≤3,0 при тестировании в клетках, экспрессирующих BCRP, выбраны из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 81-86.
В вариантах осуществления, соединения формулы II и их фармацевтически приемлемые соли и сольваты или формулы III и их фармацевтически приемлемые соли и сольваты, демонстрируют отношение мозг (несвязанный)/плазма (несвязанная) от среднего до высокого (т.е. свободное отношение мозг/плазма от среднего до высокого). Способность соединения формулы II и их фармацевтически приемлемых солей и сольватов или формулы III или их фармацевтически приемлемой соли, сольвата или полиморфа, проникать через ГЭБ субъекта (например, человека) может быть определена на подходящей модели на животных (например, грызунах, например, мышах). Например, способность некоторых соединений формулы III проникать через ГЭБ у мышей определяют путем оценки соотношения несвязанного мозга к несвязанной плазме (свободный B/P) у мышей, например, как описано в Примере C, и соотношения свободного мозга к свободной плазме представлены в Таблице C2. Соотношение свободного мозга к свободной плазме более 0,3 свидетельствует о значительной степени свободного проникновения в ЦНС.
Соответственно, в некоторых вариантах осуществления, способы по настоящему изобретению включают способы лечения BRAF-ассоциированного рака ЦНС у субъекта, нуждающегося в этом. В одном варианте осуществления, способ включает введение соединения формулы III и его фармацевтически приемлемых солей и сольватов или его фармацевтически приемлемой композиции, как описано в настоящем документе, так что, по меньшей мере, часть соединения формулы III проникает через ГЭБ, как продемонстрировано на подходящей животной модели. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,3 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,35 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,4 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,45 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,5 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, отношение мозг/плазма всего количества лекарственного средства составляет, по меньшей мере, приблизительно 0,55 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарства составляет, по меньшей мере, приблизительно 0,6 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,65 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,7 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления соотношение мозг/плазма всего лекарственного средства составляет по меньшей мере, приблизительно 0,75 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,8 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, отношение мозг/плазма всего количества лекарственного средства составляет, по меньшей мере, приблизительно 0,85 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,9 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 0,95 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 1,0 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарства составляет, по меньшей мере, приблизительно 1,0 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, отношение мозг/плазма всего количества лекарственного средства составляет, по меньшей мере, приблизительно 1,1 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, соотношение мозг/плазма всего лекарственного средства составляет, по меньшей мере, приблизительно 1,2 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, отношение мозг/плазма всего количества лекарственного средства составляет, по меньшей мере, приблизительно 1,3 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, отношение мозг/плазма всего количества лекарственного средства составляет, по меньшей мере, приблизительно 1,4 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, отношение мозг/плазма общего количества лекарственного средства составляет, по меньшей мере, приблизительно 1,5 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, отношение мозг/плазма общего количества лекарственного средства составляет, по меньшей мере, приблизительно 1,6 после введения (например, перорального или внутривенного введения) субъекту. В некоторых вариантах осуществления, отношение мозг/плазма общего количества лекарственного средства составляет, по меньшей мере, приблизительно 1,7 после введения (например, перорального или внутривенного введения) субъекту. Следует отметить, что доля соединения, которое проникает через ГЭБ, рассчитывается на основе площади под кривой зависимости концентрации от времени для данного периода времени (AUC0-t) в мозге по сравнению с плазмой. Соответственно, доля представляют собой соотношение концентраций. То есть, если (AUC0-24ч) для соединения составляет 20 нг/мл в мозге и 80 нг/мл в плазме, то доля соединения, проникающего через ГЭБ, составляет 20% (20 нг/мл в мозге деленные на общую концентрацию (20 нг/мл+80 нг/мл)) (то есть отношение мозга к плазме, равное 0,20). В некоторых вариантах осуществления, доля рассчитывается на основе площади под кривой зависимости концентрации от времени для периода времени от t=0 (время дозирования) до последней точки концентрации, поддающейся количественной оценке, т.е. (AUC0-last).
Мутации в гене BRAF были идентифицированы при злокачественных меланомах, папиллярных карциномах щитовидной железы, колоректальных карциномах, немелкоклеточных карциномах легких (NSCLC), карциномах яичников и их метастатических опухолях, а также в первичных опухолях головного мозга (Davies H., et al., Nature 417(6892):949-954, 2002). Например, мутации BRAF наблюдались во многих метастатических опухолях ЦНС, включая метастазы меланомы в мозг (Flaherty KT, et al., Nat Rev Cancer (2012) 12(5):349-61), метастазы колоректального рака в мозг и метастазы немелкоклеточного рака легкого в мозг (Berghoff, AS, Preusser M., Curr Opin Neurol (2014) 27(6):689-696), папиллярный рак щитовидной железы (Kim, WW et al., J Otolaryngol Head Neck Surg. 2018; 47:4, 1-6) и рак яичников (Grisham RN., et al., Cancer. 2013;119:548-554).
Мутации BRAF также наблюдались в злокачественных первичных опухолях головного мозга, включая глиомы IV степени, например, глиобластомы и глиосаркомы, анапластические астроцитомы (опухоли высокой степени злокачественности) и анапластические ганглиоглиомы III степени по ВОЗ (Berghoff, AS, Preusser M., Curr Opin Neurol (2014) 27(6):689-696); Schindler et al. (Acta Neuropathol 121(3):397-405, 2011); Behling et al. (Diagn Pathol 11(1):55, 2016)) в детской и взрослой популяциях.
BRAF мутации наблюдались также при доброкачественных первичных опухолях головного мозга, например в астроцитомах II степени по ВОЗ, плеоморфных ксантоастроцитомах II степени по ВОЗ (PXA), плеоморфных ксантоастроцитомах с анаплазией, пилоцитарной астроцитоме (PA), папиллярных краниофарингиомах, ганглиолгиомах, астробластомах, пилоцитарных астроцитомах, атипичных тератоидных/рабдоидных опухолях, рабдоидных менингиомах (Berghoff, AS, Preusser M., Curr Opin Neurol (2014) 27(6):689-696; Schindler et al. (Acta Neuropathol 121(3):397-405, 2011); Behling et al. (Diagn Pathol 11(1):55, 2016); (Behling et al., Diagn Pathol 11(1):55, 2016; Brastianos et al., Nat Genet 46(2):161-165, 2014; Dougherty et al., Neuro Oncol 12(7):621- 630, 2010; Lehman et al., Neuro Oncol 19(1):31-42, 2017; Mordechai et al., Pediatr Hematol Oncol 32(3):207-211, 2015; Myung et al., Transl Oncol 5(6):430-436, 2012; Schindler et al., Acta Neuropathol 121(3):397-405, 2011)) в детской и взрослой популяции.
Мутации BRAF также были обнаружены в рецидивирующих нейробластомах (Eleveld, TF, et al., Nat Genet 47(8):864-871, 2015). Нейробластомой является детская опухоль периферической нервной системы. Большинство субъектов с нейробластомой имеют опухоли, которые изначально отвечают на химиотерапию, но у значительной части субъектов будут возникать резистентные к терапии рецидивы.
Соответственно, также в настоящем документе представлен способ лечения субъекта, который диагностирован или идентифицирован как имеющий BRAF-ассоциированную опухоль, например, любую из типовых BRAF-ассоциированных опухолей, описанных в настоящем документе, включающий введение субъекту терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или его фармацевтической композиция, как определено в настоящем документе. В некоторых вариантах осуществления, субъект, который был идентифицирован или диагностирован как имеющий BRAF-ассоциированную опухоль с помощью одобренного регулирующим органом, например, одобренного FDA, теста или анализа для выявления мутации BRAF у субъекта или образца биопсии от субъекта или при выполнении любого из неограничивающих примеров анализов, описанных в настоящем документе. В некоторых вариантах осуществления, тест или анализ представлен в виде набора. Например, BRAF-ассоциированная опухоль может быть раком, который включает одну или несколько мутаций BRAF (например, V600E и/или V600K). В некоторых вариантах осуществления, соединение формулы I выбрано из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления BRAF-ассоциированным раком ЦНС является BRAF-ассоциированный метастатический рак, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы II или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединение формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъект является взрослым субъектом. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Также представлены способы лечения опухоли у субъекта, нуждающегося в этом, включающие: (а) обнаружение BRAF-ассоциированной опухоли у субъекта; и (b) введение субъекту терапевтического эффективного количества формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или его фармацевтической композиции. В некоторых вариантах осуществления этих способов, опухолью является BRAF-ассоциированная доброкачественная опухоль. В некоторых вариантах осуществления этих способов, опухолью является BRAF-ассоциированная злокачественная опухоль. В некоторых вариантах осуществления этих способов, опухолью является BRAF-ассоциированная злокачественная опухоль (например, любая из BRAF-ассоциированных злокачественных опухолей, описанных в настоящем документе), и способ дополнительно включает введение субъекту одной или нескольких дополнительных противораковых терапий, например, хирургического вмешательства (например, по меньшей мере, частичной резекции опухоли) и/или радиационной терапии и/или противоракового агента. В некоторых вариантах осуществления этих способов, опухолью является BRAF-ассоциированная доброкачественная опухоль, например, BRAF-ассоциированная доброкачественная опухоль ЦНС, и способ дополнительно включает введение субъекту одной или нескольких дополнительных противораковых терапий, например, хирургического вмешательства (например, по меньшей мере, частичной резекции опухоли) и/или радиационной терапии и/или противоракового агента. В некоторых вариантах осуществления, у субъекта установлено наличие BRAF-ассоциированной опухоли с помощью использования одобренного регулирующим органом, например, одобренного FDA, теста или анализа для идентификации мутации BRAF у субъекта или в образце биопсии от субъекта (например, биопсии ткани или жидкости) или через выполнение любого из неограничивающих примеров анализов, описанных в настоящем документе. В некоторых вариантах осуществления, тест или анализ представлен в виде набора. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает введение соединения формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является BRAF-ассоциированный метастатический рак, и способ включает введение соединения формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает введение соединения формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает введение соединения формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы III. В некоторых вариантах осуществления, соединением является соединение формулы III. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и его фармацевтически приемлемых солей или сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Также представлены способы лечения субъекта, имеющего BRAF-ассоциированную опухоль, которые включают выполнение анализа образца, полученного от субъекта, для определения того, что у субъекта имеется опухоль с мутацией BRAF, и введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или его фармацевтической композиции субъекту, у которого определена мутация BRAF. В некоторых вариантах осуществления этих способов, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак), и способ дополнительно включает введение субъекту одного или нескольких других видов противораковой терапии, например, хирургического вмешательства (например, по меньшей мере, частичной резекции опухоли) и/или радиационной терапии и/или лечения противораковым агентом. В некоторых вариантах осуществления этих способов, субъекта ранее лечили другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В некоторых вариантах осуществления, субъектом является субъект с подозрением на наличие BRAF-ассоциированной опухоли, субъект, у которого имеется один или несколько симптомов BRAF-ассоциированной опухоли или субъект, имеющий повышенный риск развития BRAF-ассоциированной опухоли. В некоторых вариантах осуществления, в анализе используют секвенирование следующего поколения, пиросеквенирование, иммуногистохимию или Break apart FISH анализ. В некоторых вариантах осуществления, анализом является анализ, одобренный регулирующим органом, например, набор, одобренный FDA. В некоторых вариантах осуществления, анализом является жидкостная биопсия. В некоторых вариантах осуществления, биопсией является биопсия ткани. В некоторых вариантах осуществления, раком является рак ЦНС, и биопсией является жидкостная биопсия (например, СМЖ). В некоторых вариантах осуществления, раком является рак ЦНС, и биопсией является биопсия ткани (например, образец опухоли, полученный во время традиционной хирургической операции или биопсия стереотаксической иглой, например, стереотаксическая пункционная биопсия под контролем КТ или МРТ). В настоящем документе описаны дополнительные неограничивающие анализы, которые можно использовать в этих способах. Дополнительные анализы также известны в данной области техники. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является BRAF-ассоциированный метастатический рак, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Таже представлено соединение формулы I, формулы II или формулы III или его фармацевтический приемлемая соль, сольват или полиморф или его фармацевтическая композиция для применения в лечении BRAF-ассоциированной опухоли у субъекта, идентифицированного или диагностированного как имеющего BRAF-ассоциированную опухоль через стадию выполнения анализа (например, in vitro анализа) образца, полученного от субъекта, для определения того, что субъект имеет мутацию BRAF, где наличие BRAF мутации указывает на то, что субъект имеет BRAF-ассоциированную опухоль. Также представлено применение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа для изготовления лекарственного средства для лечения BRAF-ассоциированной опухоли у субъекта, идентифицированного или диагностированного как имеющего BRAF-ассоциированную опухоль через стадию выполнения анализа образца, полученного от субъекта, для определения того, что субъект имеет мутацию BRAF, где наличие BRAF мутации указывает на то, что субъект имеет BRAF-ассоциированную опухоль. Некоторые варианты осуществления любого из способов или применений, описанных в настоящем документе, дополнительно включают запись в истории болезни субъекта (например, на машиночитаемом носителе) о том, что у субъекта определено наличие мутации BRAF при выполнении анализа, и ему следует вводить соединение формула I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф или его фармацевтическую композицию. В некоторых вариантах осуществления, анализ использует секвенирование следующего поколения, пиросеквенирование, иммуногистохимию или Break apart FISH анализ. В некоторых вариантах осуществления, анализом является анализ, утвержденный регулирующим органом, например, набор, утвержденный FDA. В некоторых вариантах осуществления, анализом является жидкостная биопсия. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является злокачественная BRAF-ассоциированная опухоль (т.е., BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является BRAF-ассоциированный метастатический рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная РМР или внечерепная РМР. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС. В некоторых вариантах осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Также представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, для использования при лечении BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом или у субъекта, у которого выявлено или диагностировано наличие BRAF-ассоциированной опухоли. Также представлено использование соединения формулы I, формулы II или формулы III или его эффективно приемлемой соли, сольвата или полиморфа для производства лекарственного средства для лечения BRAF-ассоциированной опухоли у субъекта, у которого идентифицирована или диагностирована BRAF-ассоциированная опухоль. В некоторых вариантах осуществления, субъект идентифицируется или диагностируется как имеющий BRAF-ассоциированную опухоль с помощью одобренного регулирующим органом, например, одобренного FDA, набора для идентификации мутации BRAF у субъекта или в образце биопсии от субъекта. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является злокачественная BRAF-ассоциированная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает использование соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак, и способ включает использование формулы II или его фармацевтически приемлемая соль, сольват или полиморф или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает использование соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает использование соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или ее фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, у субъекта идентифицирована или диагностирована опухоль с мутацией BRAF. В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, у субъекта есть опухоль, положительная в отношении мутации BRAF. В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, субъектом может быть субъект с опухолью (опухолями), положительной по мутации BRAF. В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, субъектом может быть субъект, опухоль (опухоли) которого имеет мутацию BRAF. В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, у субъекта подозревается BRAF-ассоциированная опухоль. В некоторых вариантах осуществления, в настоящем документе представлены способы лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в таком лечении, где способ включает а) обнаружение мутации BRAF; и b) введение терапевтически эффективного количества формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, мутация BRAF определяется с использованием одобренного регулирующим органом, например, одобренного FDA, анализа или набора. В некоторых вариантах осуществления, мутацией BRAF является BRAF V600E или BRAF V600K. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС. В некоторых вариантах осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, субъект имеет историю болезни, указывающую, что у субъекта есть опухоль, которая имеет одну или несколько мутаций BRAF. В некоторых вариантах осуществления, клиническая история болезни указывает, что субъекта следует лечить одним или несколькими соединениями формулы I, формулы II или формулы III или их фармацевтически приемлемыми солями или сольватами или композициями, представленными в настоящем документе. В некоторых вариантах осуществления, мутация BRAF определяется с использованием одобренного регулирующим органом, например, одобренного FDA анализа или набора. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак, ЦНС. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Также представлены способы лечения субъекта, которые включают введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа субъекту, имеющему клиническую историю болезни, которая указывает на то, что субъект имеет мутацию BRAF. Также представлено использование соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа для производства лекарственного средства для лечения BRAF-ассоциированной опухоли у субъекта, имеющего клиническую историю болезни, которая указывает на наличие у субъекта мутация BRAF. Некоторые варианты осуществления этих способов и использования могут дополнительно включать: стадию проведения анализа (например, анализа in vitro) образца, полученного от субъекта, для определения, есть ли у субъекта мутация BRAF, и записи информации в файл с историей болезни субъекта (например, на машиночитаемый носитель), что у субъекта обнаружена мутация BRAF. В некоторых вариантах осуществления, анализом является анализ in vitro. Например, анализ, который использует секвенирование следующего поколения, иммуногистохимию или Break apart FISH анализ. В некоторых вариантах осуществления, анализом является одобренный регулирующим органом, например, одобренный FDA, набор. В некоторых вариантах осуществления, анализом является биопсию жидкости. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Также в настоящем документе представлен способ лечения субъекта. В некоторых вариантах осуществления, способ включает выполнение анализа образца, полученного от субъекта, чтобы определить, есть ли у субъекта мутация BRAF. В некоторых таких вариантах осуществления, способ также включает введение субъекту, у которого определена мутация BRAF, терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, способ включает определение того, что у субъекта есть мутация BRAF, с помощью анализа, проведенного на образце, полученном от субъекта. В таких вариантах осуществления, способ также включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. Некоторые варианты осуществления, этих способов дополнительно включают введение субъекту другой противораковой терапии. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Также представлены способы (например, способы in vitro) выбора лечения для субъекта, у которого идентифицирована или диагностирована BRAF-ассоциированная опухоль. Некоторые варианты осуществления могут дополнительно включать введение выбранного лечения субъекту, у которого идентифицирована или диагностирована BRAF-ассоциированная опухоль. Например, выбранное лечение может включать введение терапевтически эффективного количества формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. Некоторые варианты осуществления, могут дополнительно включать стадию проведения анализа образца, полученного от субъекта, для определения того, что субъект имеет мутацию BRAF, и идентификации и диагностики субъекта, у которого определена мутация BRAF, как имеющего BRAF-ассоциированную опухоль. В некоторых вариантах осуществления, субъект был идентифицирован или диагностирован как имеющий BRAF-ассоциированную опухоль с помощью использования одобренного регулирующим органом, например, одобренного FDA, набора для выявления мутации BRAF у субъекта или образца биопсии от субъекта. В некоторых вариантах осуществления, анализом является анализ in vitro. Например, анализ, который использует секвенирование следующего поколения, иммуногистохимию или Break apart FISH анализ. В некоторых вариантах осуществления, анализом является одобренный регулирующим органом, например, одобренный FDA, набор. В некоторых вариантах осуществления, анализом является жидкостная биопсия. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Также в настоящем документе представлены способы выбора лечения субъекта, где способы включают в себя стадию проведения анализа образца, полученного от субъекта, для определения наличия у субъекта мутации BRAF, и идентификации или диагностики субъекта, определенного как имеющего мутацию BRAF как имеющего BRAF-ассоциированную опухоль. Некоторые варианты осуществления дополнительно включают введение выбранного лечения субъекту, у которого идентифицирована или диагностирована BRAF-ассоциированная опухоль. Например, выбранное лечение может включать введение терапевтически эффективного количества формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа субъекту, идентифицированному или диагностированному как имеющий BRAF-ассоциированную опухоль. В некоторых вариантах осуществления, анализом является анализ in vitro. Например, анализ, который использует секвенирование следующего поколения, иммуногистохимию или Break apart FISH анализ. В некоторых вариантах осуществления, анализом является одобренный регулирующим органом, например, одобренный FDA, набор. В некоторых вариантах осуществления, анализом является биопсию жидкости. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль, (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и пример 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
Также представлены способы выбора субъекта для лечения, где способы включают выбор, идентификацию или диагностику субъекта, имеющего BRAF-ассоциированная опухоль, и выбор субъекта для лечения, включая введение терапевтически эффективного количества соединения формулы I, формулы II. или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, идентификация или диагностика у субъекта BRAF-ассоциированной опухоли может включать стадию выполнения анализа образца, полученного от субъекта, для определения того, что у субъекта есть мутация BRAF, и идентификацию или диагностику субъекта, определенного как имеющего мутацию BRAF как имеющего BRAF-ассоциированную опухоль. В некоторых вариантах осуществления, способ выбора субъекта для лечения можно использовать как часть клинического исследования, которое включает введение различных способов лечения BRAF-ассоциированной опухоли. В некоторых вариантах осуществления, анализом является анализ in vitro. Например, анализ, который использует секвенирование следующего поколения, иммуногистохимию или Break apart FISH анализ. В некоторых вариантах осуществления, анализом является одобренный регулирующим органом, например, одобренный FDA, набор. В некоторых вариантах осуществления, анализом является жидкостная биопсия. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является BRAF-ассоциированная злокачественная опухоль (т.е. BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является метастатический BRAF-ассоциированный рак, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль головного мозга, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, соединением является соединение формулы III или его приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением формулы III является пример 1 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 4 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 7 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 9 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 10 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 23 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 55 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 63 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 67 или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединением формулы III является пример 82. В некоторых вариантах осуществления, субъектом является взрослый субъект. В некоторых вариантах осуществления, субъектом является субъект детского возраста.
В некоторых вариантах осуществления любого из способов или применений, описанных в настоящем документе, анализ, используемый для определения наличия у субъекта мутации BRAF с использованием образца, взятого у субъекта, может включать, например, секвенирование следующего поколения, иммуногистохимию, флуоресцентную микроскопию, Break apart FISH анализ, Саузерн блоттинг, Вестерн блоттинг, FACS анализ, Нозерн блоттинг и амплификацию на основе ПЦР (например, РВ-ПЦР и количественную РВ-ПЦР в реальном времени). Как хорошо известно в данной области техники, анализы обычно проводят, например, по меньшей мере, с одним меченым зондом нуклеиновой кислоты или, по меньшей мере, с одним меченым антителом или его антигенсвязывающим фрагментом. Анализы могут использовать другие методы обнаружения, известные в данной области техники, для обнаружения мутации BRAF. В некоторых вариантах осуществления, образцом является биологический образец или образец биопсии (например, образец биопсии, залитый парафином) от субъекта. В некоторых вариантах осуществления, субъектом является субъект с подозрением на наличие BRAF-ассоциированной опухоли, субъект, имеющий один или несколько симптомов BRAF-ассоциированной опухоли и/или субъект, который имеет повышенный риск развития BRAF-ассоциированной опухоли.
В некоторых вариантах осуществления, биопсией является биопсия опухоли (например, образец опухоли, полученный во время традиционной хирургической операции или биопсия стереотаксической иглой, например, стереотаксическая пункционная биопсия под контролем КТ или МРТ). Способы биопсии ткани могут использоваться для определения общей опухолевой массы и/или мутации BRAF.
В некоторых вариантах осуществления, мутация BRAF может быть идентифицирована с использованием жидкостной биопсии (по другому называемой жидкостной биопсией или жидкофазной биопсией). См., например, Karachialiou et al., “Real-time liquid biopsies become a reality in cancer treatment”, Ann. Transl. Med., 3(3):36, 2016. Методы жидкостной биопсии можно использовать для определения общей опухолевой массы и/или мутации BRAF. Жидкостные биопсии могут быть выполнены на биологических образцах, полученных относительно легко от субъекта (например, с помощью простого забора крови), и обычно менее инвазивны, чем традиционные способы, используемые для выявления опухолевой массы и/или мутации BRAF. В некоторых вариантах осуществления, жидкостная биопсия может использоваться для обнаружения наличия мутации BRAF на более ранней стадии, чем традиционные способы. В некоторых вариантах осуществления, биологический образец для использования в жидкостной биопсии может включать СМЖ, кровь, плазму, мочу, слюну, мокроту, бронхо-альвеолярный лаваж, желчь, лимфатическую жидкость, кистозную жидкость, стул, асцит и их комбинации. В некоторых вариантах осуществления, жидкостная биопсия может использоваться для обнаружения циркулирующих опухолевых клеток (CTC). В некоторых вариантах осуществления, жидкостная биопсия может использоваться для обнаружения внеклеточной ДНК. В некоторых вариантах осуществления, внеклеточной ДНК, обнаруженной с помощью жидкостной биопсии, является циркулирующая опухолевая ДНК (цоДНК), полученная из опухолевых клеток. Анализ цоДНК (например, с использованием методов чувствительного обнаружения, таких как, помимо прочего, секвенирование следующего поколения (NGS), традиционная ПЦР, цифровая ПЦР или анализ на микрочипах), можно использовать для идентификации мутации BRAF.
В некоторых вариантах осуществления, мутация BRAF, идентифицированная с помощью жидкостной биопсии, также присутствует в раковой клетке, которая присутствует у субъекта (например, в опухоли). В некоторых вариантах осуществления, любой из типов мутаций BRAF можно обнаружить с помощью жидкостной биопсии. В некоторых вариантах осуществления, генетическая мутация, идентифицированная с помощью жидкостной биопсии, может использоваться для идентификации субъекта как кандидата для конкретного лечения. Например, обнаружение мутации BRAF у субъекта может указывать на то, что субъект будет отвечать на лечение, которое включает введение формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа.
«Опухолевая масса», также называемая «опухолевой нагрузкой», относится к общему количеству опухолевого материала, распределенного по всему телу. Опухолевая масса относится к общему количеству раковых клеток или общему размеру опухоли (опухолей) по всему телу, включая лимфатические узлы и костный мозг(?) (в оригинале bone narrow, но кажется, что это опечатка). Опухолевую массу можно определить различными методами, известными в данной области техники, такими как, например, измерение размеров опухоли (опухолей) при удалении из субъекта, например, с помощью штангенциркуля, или при нахождении в теле с использованием методов визуализации, например, магнитно-резонансной томографии (МРТ), компьютерной томографии (КТ), мультидетекторной компьютерной томографии (МДКТ), позитронно-эмиссионной томографии (ПЭТ), рентгена, ультразвука или сканирования костей.
Термин «размер опухоли» относится к общему размеру опухоли, который можно измерить как длину и ширину опухоли. Размер опухоли может быть определен различными способами, известными в данной области техники, такими как, например, измерение размеров опухоли (опухолей) после удаления из субъекта, например, с помощью штангенциркуля или во время нахождения в теле с использованием методов визуализации, например, МРТ, сканирования костей, УЗИ или КТ.
Жидкостная биопсия может выполняться несколько раз в течение курса диагностики, курса мониторинга и/или курса лечения для определения одного или нескольких клинически значимых параметров, включая, без ограничения, прогрессирование заболевания или эффективность лечения после введение лечения субъекту. Например, первая жидкостная биопсия может быть выполнена в первый момент времени, и вторая жидкостная биопсия может быть выполнена во второй момент времени во время курса диагностики, курса мониторинга и/или курса лечения. В некоторых вариантах осуществления, первая временная точка может быть временной точкой до постановки диагноза у субъекта с заболеванием (например, когда субъект здоров), и вторая временная точка может быть временной точкой после того, как у субъекта развилось заболевание (например, вторая временная точка может использоваться для диагностики субъекта с заболеванием). В некоторых вариантах осуществления, первая временная точка может быть временной точкой до постановки диагноза у субъекта с заболеванием (например, когда субъект здоров), после которой субъект находится под наблюдением, и вторая временная точка может быть временной точкой после мониторинга субъекта. В некоторых вариантах осуществления, первая временная точка может быть временной точкой после постановки диагноза субъекту с заболеванием, после которой пациенту назначается лечение, и вторая временная точка может быть временной точкой после того, как лечение проведено; в таких случаях, вторая временная точка может использоваться для оценки эффективности лечения (например, если генетическая мутация (мутации), обнаруженная в первой временной точке, имеет пониженную распространенность или не поддается обнаружению). В некоторых вариантах осуществления, лечение, которое будет назначено субъекту, может включать соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф.
В одном варианте осуществления, соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, может использоваться отдельно или в комбинации с одной или несколькими различными формами лечения для лечения субъекта со злокачественной опухолью. Например, соединение формулы I, формула II или формула III или их фармацевтически приемлемые соли или сольваты также могут использоваться в комбинации с одной или несколькими дополнительными противораковыми терапиями, например хирургическим вмешательством, радиационной терапией и/или противораковым агентом, который действует таким же или другим механизмом действия. В одном варианте осуществления, лечение субъекта, имеющего BRAF-ассоциированную злокачественную опухоль, соединением формулы I, формулы II или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом в комбинации с одним или несколькими дополнительными методами лечения, например, хирургическим вмешательством, радиационной терапией и/или противораковым агентом, могут иметь повышенную терапевтическую эффективность по сравнению с лечением того же субъекта или аналогичного субъекта соединением формулы I, формулы II или формулы III или фармацевтически приемлемой солью или сольватом в качестве монотерапии.
Соответственно, в одном варианте осуществления, в настоящем документе представлены способы лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), которые включают: введение субъекту (i) терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли или сольвата в качестве монотерапии, или (ii) терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в сочетании с одним или несколькими дополнительными противораковыми терапиями. В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят вторую противораковую терапию в течение указанного периода времени. В одном варианте осуществления, второй противораковой терапией является второй противораковый агент.
Также в настоящем документе представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, для использования в сочетании с дополнительной противораковой терапией. В настоящем документе также представлена дополнительная противораковая терапия для использования в комбинации с соединением формулы I, формулы II или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом.
Также в настоящем документе представлено соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, для использования при лечении BRAF-ассоциированной опухоли путем совместного введения с дополнительной противораковой терапией. В настоящем документе также представлена дополнительная противораковая терапия для лечения BRAF-ассоциированной опухоли путем совместного введения с соединением формулы I, формулы II или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом.
В некоторых вариантах осуществления, субъекту вводят одну или несколько противораковых терапий, отличных от соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, перед введением соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, одна или несколько противораковых терапий, выбранных из хирургии и/или радиационной терапии, и/или противоракового агента, который действует по тому же самому или по другому механизму действия. Например, в некоторых вариантах осуществления, субъект, нуждающийся в этом, может быть подвергнут, по меньшей мере, частичной резекции опухоли перед введением соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, лечение, по меньшей мере, частичной резекции опухоли уменьшает размер опухоли (например, опухолевую массу) до введения одной или нескольких доз формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, субъект, нуждающийся в этом, может пройти радиационную терапию перед введением формулы I, формулы II или формулы III или ее фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, субъект, нуждающийся в этом, может пройти лечение одним или несколькими противораковыми агентами, отличными от соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, перед введением соединения формулы I, формула II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, у субъекта имеется рак, резистентный или рефрактерный к предыдущей терапии.
Соответственно, в некоторых вариантах осуществления, в настоящем документе представлены способы лечения субъекта, имеющего BRAF-ассоциированную опухоль, включающие (i) введение одной или нескольких противораковых терапий указанному субъекту в течение периода времени, и (ii) после (i), введение (a) соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, в качестве монотерапии, или (b) соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, в сочетании с одной или несколькими дополнительными противораковыми терапиями.
В некоторых вариантах осуществления, соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф, можно вводить до введения одной или нескольких противораковых терапий (например, хирургического вмешательства, радиационной терапии и/или противоракового агента, который действует по тому же или другому механизму действия) для лечения субъекта с опухолью. Например, в некоторых вариантах осуществления, субъект, нуждающийся в этом, может подвергнуться, по меньшей мере, частичной резекции опухоли после введения соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, субъект, нуждающийся в этом, может пройти радиационную терапию после введения соединения формулы I, формулы II или формулы III или ее фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, субъект, который в этом нуждается, может пройти лечение соединением формулы I, формулы II или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом перед введением соединения одного или нескольких противораковых агентов, отличных от соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82.
Соответственно, в некоторых вариантах осуществления, в настоящем документе представлены способы лечения субъекта, страдающего BRAF-ассоциированной опухолью, включающие (i) введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, и (ii) после указанного периода времени, введение одной или нескольких противораковых терапий. Например, субъекту, нуждающемуся в этом, можно ввести одну или несколько доз соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, а затем провести, по меньшей мере, частичную резекцию опухоли. В некоторых вариантах осуществления, лечение одной или несколькими дозами соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом уменьшает размер опухоли (например, опухолевую массу) до, по меньшей мере, частичной резекции опухоли. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82.
В некоторых вариантах осуществления любого из описанных выше способов, дополнительной противораковой терапией является хирургическое вмешательство, радиационная терапия и/или противораковый агент, который действует по тому же самому или по другому механизму действия.
Неограничивающие примеры дополнительных противораковых агентов, которые могут применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, включают дополнительные ингибиторы киназ, отличные от соединений формулы I, формулы II или формулы III или их фармацевтически приемлемой соли, сольвата или полиморфа, включая ингибиторы MEK, ингибиторы BRAF (например, ингибиторы BRAF, отличные от соединения формулы I, II или III), ингибиторы EGFR, ингибиторы HER2 и/или HER3, ингибиторы SHP2, ингибиторы Axl, ингибиторы PI3K, ингибиторы SOS1, ингибиторы пути сигнальной трансдукции, ингибиторы контрольных точек, модуляторы пути апоптоза, цитотоксические химиотерапевтические средства, таргетные терапии ангиогенеза, и иммунотаргетные агенты, включая иммунотерапию.
В одном варианте осуществления, противораковым агентом, который может применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, является таргетный терапевтический агент. Термин «таргетный терапевтический агент», используемый в настоящем документе, относится к молекуле, которая блокирует рост раковых клеток путем вмешательства в конкретные таргетные молекулы, необходимые для канцерогенеза и роста опухоли, а не простым вмешательством во все быстро делящиеся клетки (например, как при традиционной цитотоксической химиотерапии), и включает, но не ограничивается ими, терапевтические агенты, таргетирующие рецепторную тирозинкиназу, ингибиторы пути сигнальной трансдукции (например, ингибиторы пути Ras-Raf-MEK-ERK, ингибиторы пути PI3K-Akt-mTOR-S6K («ингибиторы PI3K»)) и модуляторы пути апоптоза.
В некоторых вариантах осуществления, противораковый агент, который может применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, является ингибитором MEK. Неограничивающие примеры ингибиторов MEK включают биниметиниб, траметиниб, кобиметиниб, селуметиниб, пимасертиб, рефаметиниб, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфениламино)бензамид (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамид (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-дион (TAK-733). Дополнительные примеры ингибиторов MEK включают соединения, описанные в WO 03/077914, WO 2005/023759, WO 2005/051301, US 7,517,994, US 7,732,616, WO 2005/051906, WO 2005/051302, WO 2005/051300 и WO 2007/044084. В некоторых вариантах осуществления, ингибитором MEK является биниметиниб.
В некоторых вариантах осуществления, противораковым агентом, который может быть использован в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, является другой ингибитор BRAF, отличный от соединения формулы I, формулы II или формулы III. Неограничивающие примеры других ингибиторов BRAF включают энкорафениб, дабрафениб и вемурафениб, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамид (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамид (PLX8394). Дополнительные примеры ингибиторов BRAF известны в данной области техники.
В некоторых вариантах осуществления, противораковым агентом, который можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, является ингибитор EGFR. Неограничивающие примеры ингибиторов EGFR включают цетуксимаб (Erbitux®), панитумумаб (Vectibix®), осимертиниб (меректиниб, Tagrisso®), эрлотиниб (Tarceva®), гефитиниб (Iressa®), нецитумумаб (Portrazza™), нератиниб (Portrazza™), лапатиниб (Tykerb®), вандетаниб (Caprelsa®), бригатиниб (Alunbrig®) и ингибиторы EGFR, описанные в публикациях РСТ №№ WO 2019/071351 и WO 2017/117680, обе которые полностью включены в настоящий документ посредством ссылки. Дополнительные примеры ингибиторов EGFR известны в данной области техники.
В некоторых вариантах осуществления, противораковым агентом, который может применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, является ингибитор HER2 и/или HER3. Неограничивающие примеры ингибиторов HER2 и/или HER3 включают лапатиниб, канертиниб, (E)-2-метокси-N-(3-(4-(3-метил-4-(6-метилпиридин-3-илокси)фениламино)хиназолин-6-ил)аллил)ацетамид (CP-724714), сапитиниб, 7-[[4-[(3-этинилфенил)амино]-7-метокси-6-хиназолинил]окси]-N-гидрокси-гептанамид (CUDC-101), мубритиниб, 6-[4-[(4-этилпиперазин-1-ил)метил]фенил]-N-[(1R)-1-фенилэтил]-7H-пирроло[2,3-d]пиримидин-4-амин (AEE788), ирбинитиниб (тукатиниб), позиотиниб, N-[4-[1-[4-(4-ацетил-1-пиперазинил)циклогексил]-4-амино-3-пиразоло[3,4-d]пиримидинил]-2-метоксифенил]-1-метил-2-индолкарбоксамид (KIN001-111), 7-циклопентил-5-(4-феноксифенил)-7H-пирроло[2,3-d]пиримидин-4-иламин (KIN001-051), 6,7-диметокси-N-(4-феноксифенил)хиназолин-4-амин (KIN001-30), дазатиниб и босутиниб.
В некоторых вариантах осуществления, противораковым агентом, который может применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, является ингибитор SHP2. Неограничивающие примеры ингибиторов SHP2 включают 6-(4-амино-4-метилпиперидин-1-ил)-3-(2,3-дихлорфенил)пиразин-2-амин (SHP099), [3-[(3S,4S)-4-амино-3-метил-2-окса-8-азаспиро[4,5]декан-8-ил]-6-(2,3-дихлорфенил)-5-метилпиразин-2-ил]метанол (RMC-4550) RMC-4630, TNO155 и соединения, описанные в WO 2015/107493, WO 2015/107494, WO 2015/107495, WO 2019/075265, PCT/US2019/056786 и PCT/IB2020/053019.
В некоторых вариантах осуществления, соединение формулы I, II или III или его практически приемлемая соль, сольват или полиморф вводят в комбинации с ингибитором MEK и ингибитором SHP2, например, любым из ингибиторов MEK и ингибиторов SHP2, описанных в настоящем документе. В некоторых вариантах осуществления, соединение формулы I, II или III или его приемлемую соль, сольват или полиморф вводят в комбинации с ингибитором MEK, которым является биниметиниб, и ингибитором SHP2, например, ингибиторами SHP2, описанными в настоящем документе.
В некоторых вариантах осуществления, противораковым агентом, который может применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, является ингибитор Axl. Неограничивающие примеры ингибиторов Axl включают бемцентиниб, YW327.6S2 (моноклональное антитело), GL2I.T (рецептор-ловушку), 2-(5-хлор-2-(4-((4-метилпиперазин-1-ил)метил)фениламино)пиримидин-4-иламино)-N, N-диметилбензолсульфонамид (TP-0903), 3-[2-[[3-фтор-4-(4-метил-1-пиперазинил)фенил]амино]-5-метил-7H-пирроло[2,3-d]пиримидин-4-ил]-бензолацетонитрил (SGI-7079), гильтеритиниб, босутиниб, кабозантиниб, сунитиниб, форетиниб, амуватиниб, глезатиниб, N-(4-((2-амино-3-хлорпиридин-4-ил)окси)-3-фторфенил)-4-этокси-1-(4-фторфенил)-2-оксо-1,2-дигидропиридин-3-карбоксамид (BMS777607), мерестиниб, (Z)-3-((3-((4-(морфолинометил)-1H-пиррол-2-ил)метилен)-2-оксоиндолин-5-ил)метил)тиазолидин-2,4-дион (S49076), и соединения, описанные в WO 2020/047184.
В некоторых вариантах осуществления, противораковым агентом, который можно применять в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом к любому из описанных выше способов, является ингибитор SOS1. Неограничивающие примеры ингибиторов SOS1 включают ингибиторы, описанные в публикации РСТ № WO 2018/115380, которая полностью включена в настоящий документ посредством ссылки.
В некоторых вариантах осуществления, противораковым агентом, который можно применять в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом к любому из описанных выше способов, является ингибитор PI3K. Неограничивающие примеры включают бупарлисиб (BKM120), альпелисиб (BYL719), самотолисиб (LY3023414), 8-[(1R)-1-[(3,5-дифторфенил)амино]этил]-N, N-диметил-2-(морфолин-4-ил)-4-оксо-4H-хромен-6-карбоксамид (AZD8186), теналисиб (RP6530), гидрохлорид воксталисиба (SAR-245409), гедатолисиб (PF-05212384), панулисиб (P-7170), таселисиб (GDC-0032), транс-2-амино-8-[4-(2-гидроксиэтокси)циклогексил]-6-(6-метоксипиридин-3-ил)-4-метилпиридо[2,3-d]пиримидин-7(8H)-он (PF-04691502), дувелисиб (ABBV-954), ацетат N2-[4-оксо-4-[4-(4-оксо-8-фенил-4H-1-бензопиран-2-ил)морфолин-4-иум-4-илметокси]бутирил]-L-аргинил-глицил-L-аспартил-L-серина (SF-1126), пиктилизиб (GDC-0941), 2-метил-1-[2-метил-3-(трифторметил)бензил]-6-(морфолин-4-ил)-1H-бензимидазол-4-карбоновая кислота (GSK2636771), иделалисиб (GS-1101), тозилат умбралисиба (TGR-1202), пиктилисиб (GDC-0941), гидрохлорид копанлисиба (BAY 84-1236), дактолисиб (BEZ-235), 1-(4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)пиразин-2-ил]-1-этил-1H-1,2,4-триазол-3-ил]пиперидин-1-ил)-3-гидроксипропан-1-он (AZD-8835), 5-[6,6-диметил-4-(морфолин-4-ил)-8,9-дигидро-6H-[1,4]оксазино[4,3-e]пурин-2-ил]пиримидин-2-амин (GDC-0084), эверолимус, рапамицин, перифозин, сиролимус и темсиролимус.
В некоторых вариантах осуществления, противораковым агентом, который можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом по любому из описанных выше способов, является иммунотерапия. Термин «иммунотерапия» относится к агенту, который модулирует иммунную систему. В некоторых вариантах осуществления, иммунотерапия может увеличивать экспрессию и/или активность регулятора иммунной системы. В некоторых вариантах осуществления, иммунотерапия может снижать экспрессию и/или активность регулятора иммунной системы. В некоторых вариантах осуществления, иммунотерапия может задействовать и/или усилить активность иммунной клетки.
В некоторых вариантах осуществления, иммунотерапия, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является терапия антителами (например, моноклональным антителом, конъюгированным антителом). В некоторых вариантах осуществления, терапией антителом является бевацизумаб (Mvasti™, Avastin®), трастузумаб (Herceptin®), авелумаб (Bavencio®), ритуксимаб (MabThera™, Rituxan®), эдреколомаб (Panorex), даратумуаб (Darzalex®), оларатумаб (Lartruvo™), офатумумаб (Arzerra®), алемтузумаб (Campath®), цетуксимаб (Erbitux®), ореговомаб, пембролизумаб (Keytruda®), динутиксимаб (Unituxin®), обинутузумаб (Gazyva®, 206), тремелимумаб (CP-675,206), рамуцирумаб (Cyramza®), ублитуксимаб (TG-1101), панитумумаб (Vectibix®), элотузумаб (Empliciti™), нецитумумаб (Portrazza™), цирмтузумаб (UC-961), ибритумомаб (Zevalin®), изатуксимаб (SAR650984) нимотузумаб, фрезолимумаб (GC1008), лирилумаб (INN), могамулизумаб (Poteligeo®), фиклатузумаб (AV-299), деносумаб (Xgeva®), ганитумаб, урелумаб, пидилизумаб, аматуксимаб, блинатумомаб (AMG103; Blincyto®) или мидостаурин (Rydapt).
В некоторых вариантах осуществления, иммунотерапия, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является конъюгат антитело-лекарственное средство. В некоторых вариантах осуществления, конъюгатом антитело-лекарственное средство является гемтузумаб озогамицин (Mylotarg™), инотузумаб озогамицин (Besponsa®), брентуксимаб ведотин (Adcetris®), адо-трастузумаб эмтанзин (TDM-1; Kadcyla®), мирветуксимаб соравтанзин (IMGN853) или анетумаб равтанзин.
В некоторых вариантах осуществления, иммунотерапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, включает токсин. В некоторых вариантах осуществления, иммунотерапией является денилейкин дифтитокс (Ontak®).
В некоторых вариантах осуществления, иммунотерапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является цитокиновая терапия. В некоторых вариантах осуществления, цитокиновой терапией является терапия интерлейкином 2 (IL-2), терапия интерфероном альфа (IFNα), терапия гранулоцитарным колониестимулирующим фактором (G-CSF), терапия интерлейкином 12 (IL-12), терапия интерлейкином 15 (IL-15), терапия интерлейкином 7 (IL-7) или терапия эритропоэтином-альфа (EPO). В некоторых вариантах осуществления, терапией с использованием IL-2 является альдеслейкин (Proleukin®). В некоторых вариантах осуществления, терапией IFNα является IntronA® (Роферон-A®). В некоторых вариантах осуществления, терапией G-CSF является филграстим (Neupogen®).
В некоторых вариантах осуществления, иммунотерапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является ингибитор иммунной контрольной точки. В некоторых вариантах осуществления, иммунотерапия включает один или несколько ингибиторов иммунных контрольных точек. В некоторых вариантах осуществления, ингибитором иммунной контрольной точки является ингибитор CTLA-4, ингибитор PD-1 или ингибитор PD-L1. В некоторых вариантах осуществления, ингибитором CTLA-4 является ипилимумаб (Yervoy®) или тремелимумаб (CP-675,206). В некоторых вариантах осуществления, ингибитором PD-1 является пембролизумаб (Keytruda®), ниволумаб (Opdivo®) и RN888. В некоторых вариантах осуществления, ингибитором PD-L1 является атезолизумаб (Tecentriq®), авелумаб (Bavencio®) или дурвалумаб (Imfinzi™).
В некоторых вариантах осуществления, иммунотерапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является иммунотерапия на основе мРНК. В некоторых вариантах осуществления, иммунотерапией на основе мРНК является CV9104 (см., например, Rausch et al. (2014) Human Vaccine Immunother 10(11): 3146-52; и Kubler et al. (2015) J. Immunother Cancer 3:26).
В некоторых вариантах осуществления, иммунотерапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является терапия онколитическим вирусом. В некоторых вариантах осуществления, терапией онколитическим вирусом является талимоген альхерпарепвек (T-VEC; Imlygic®).
В некоторых вариантах осуществления, иммунотерапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является противораковая вакцина. В некоторых вариантах осуществления, противораковой вакциной является вакцина против вируса папилломы человека (HPV). В некоторых вариантах осуществления, вакциной против HPV является Gardasil®, Gardasil9® или Cervarix®. В некоторых вариантах осуществления, противораковой вакциной является вакцина против вируса гепатита B (HBV). В некоторых вариантах осуществления, вакциной против HBV является Engerix-B®, Recombivax HB® или GI-13020 (Tarmogen®). В некоторых вариантах осуществления, противораковой вакциной является Twinrix® или Pediarix®. В некоторых вариантах осуществления, противораковой вакциной является BiovaxID®, Oncophage®, GVAX, ADXS11-001, ALVAC-CEA, PROSTVAC®, Rindopepimut®, CimaVax-EGF, лапулейцел-T (APC8024; Neuvenge™), GRNVAC1, GRNVAC2, GRN-1201, гепкортеспенлизимут-L (Hepko-V5), DCVAX®, SCIB1, BMT CTN 1401, PrCa VBIR, PANVAC, ProstAtak®, DPX-Survivac или виагенпуматуцел-L (HS-110).
В некоторых вариантах осуществления, иммунотерапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является пептидная вакцина. В некоторых вариантах осуществления, пептидной вакциной является нелипепимут-S (E75) (NeuVax™), IMA901 или SurVaxM (SVN53-67). В некоторых вариантах осуществления, противораковой вакциной является иммуногенная персональная неоантигенная вакцина (см., например, Ott et al. (2017) Nature 547: 217-221; Sahin et al. (2017) Nature 547: 222-226). В некоторых вариантах осуществления, противораковой вакциной является RGSH4K или NEO-PV-01. В некоторых вариантах осуществления, противораковой вакциной является вакцина на основе ДНК. В некоторых вариантах осуществления, вакциной на основе ДНК является ДНК вакцина маммаглобин-A (см., например, Kim et al. (2016) OncoImmunology 5(2): e1069940).
В некоторых вариантах осуществления, иммунотерапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является клеточная иммунотерапия (например, адоптивная T-клеточная терапия, терапия дендритными клетками, терапия естественными киллерами). В некоторых вариантах осуществления, клеточной иммунотерапией является сипулейцел-Т (APC8015; Provenge™; Plosker (2011) Drugs 71(1): 101-108). В некоторых вариантах осуществления, клеточная иммунотерапия включает клетки, экспрессирующие химерный антигенный рецептор (CAR). В некоторых вариантах осуществления, клеточной иммунотерапией является CAR-T-клеточная терапия. В некоторых вариантах осуществления, CAR-T клеточной терапией является тисагенлеклейцел (Kymriah™).
В некоторых вариантах осуществления, противораковым агентом, который может применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в любом из описанных выше способов, является цитотоксический химиотерапевтический агент. Неограничивающие примеры цитотоксических химиотерапевтических агентов включают триоксид мышьяка, блеомицин, кабазитаксел, капецитабин, карбоплатин, цисплатин, циклофосфамид, цитарабин, дакарбазин, даунорубицин, доцетаксел, доксорубицин, этопозид, 5-фторурацил, фолиновую кислоту, гемцитабин, иринотекан, ломустин, метотрексат, митомицин C, оксалиплатин, паклитаксел, пеметрексед, темозоломид и винкристин и их комбинации, например, Nordic FLOX (фторурацил, фолиновая кислота и оксалиплатин), FOLFOXIRI (оксалиплатин, иринотекан и фторурацил), FOLFIRI (фолиновая кислота, фторурацил и иринотекан) или CAPEOX (капецитабин и оксалиплатин).
В некоторых вариантах осуществления, противораковым агентом, который может применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, является таргетная терапия ангиогенеза. Неограничивающие примеры таргетной терапии ангиогенеза, включают афлиберцепт и бевацизумаб.
В некоторых вариантах осуществления, противораковый агент, который может применяться в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом в соответствии с любым из описанных выше способов, включает модуляторы пути апоптоза (например, обатаклакс).
В некоторых вариантах осуществления, противораковой терапией, которую можно применять в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом, по любому из описанных выше способов, является радиационная терапия. Неограничивающие примеры радиационной терапии включают внешнюю радиационную терапию (например, внешнюю радиационную терапию с использованием киловольтных рентгеновских лучей или мегавольтных рентгеновских лучей) или внутреннюю радиационную терапию. Внутренняя радиационная терапия (также называемая брахитерапией) может включать, например, внутреннюю радиационную терапию низкими дозами или внутреннюю радиационную терапию высокими дозами. Внутренняя радиационная терапия низкими дозами включает, например, введение небольших радиоактивных гранул (также называемых семенами) в раковую ткань субъекта или рядом с ней. Внутренняя радиационная терапия высокими дозами включает, например, введение тонкой трубки (например, катетера) или имплантата в злокачественную ткань субъекта или рядом с ней, и подачу высокой дозы облучения на тонкую трубку или имплантат с использованием радиационного аппарата. Способы проведения радиационной терапии у субъекта, страдающего раком, известны в данной области техники. В вариантах реализации, где опухолью является опухоль ЦНС, радиационная терапия может включать радиационную терапию всего мозга (WBRT) или стереотаксическую радиохирургию (SRS), такую как Cyberknife®, XKnife®, Gamma Knife® или ExacTrac®.
В некоторых вариантах осуществления, противораковой терапией, которую можно использовать в комбинации с соединением формулы I, II или III или его фармацевтически приемлемой солью, сольватом или полиморфом, по любому из описанных выше способов, является хирургическое вмешательство. Неограничивающие примеры хирургического вмешательства включают, например, открытую операцию или минимально инвазивную операцию. Хирургия может включать, например, по меньшей мере, частичную резекцию опухоли, удаление всей опухоли, циторедукцию опухоли или удаление опухоли, которая вызывает боль или давление у субъекта. Способы выполнения открытой хирургии и минимально инвазивной хирургии у субъекта, страдающего раком, известны в данной области техники.
В некоторых вариантах осуществления, дополнительная терапия включает любую из перечисленных выше терапий или противораковых агентов, которые являются стандартами лечения рака, когда рак имеет мутацию BRAF.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят ингибитор MEK (например, любой из ингибиторов MEK, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, ингибитором MEK является биниметиниб. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят ингибитор BRAF (например, любой из ингибиторов BRAF, описанных в настоящем документе, включая второе соединение формулы I, II или III или его фармацевтическое приемлемую соль, сольват или полиморф) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 или его фармацевтически приемлемая соль и сольват и примеры 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль и сольват. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят ингибитор EGFR (например, любой из ингибиторов EGFR, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемые соли и сольваты, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82. В одном варианте осуществления, опухолью является рак легкого.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят ингибитор HER2 и/или HER3 в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из Примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят ингибитор Axl (например, любой из ингибиторов Axl, описанных в настоящем документе, включая соединения формулы I и соединения, отличные от соединения формулы I) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят ингибитор SOS1 (например, любой из ингибиторов SOS1, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят ингибитор передачи сигнала (например, любой из ингибиторов передачи сигнала, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят ингибитор контрольной точки (например, любой из ингибиторов контрольной точки, описанный в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят модулятор пути апоптоза (например, любой из модуляторов пути апоптоза, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из Примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят цитотоксический химиотерапевтический агент (например, любой из цитотоксических химиотерапевтических агентов, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединение формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение периода времени, когда субъекту вводят таргетную терапию ангиогенеза (например, любую из таргетных терапий ангиогенеза, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф, и пример 82. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов и примера 82.
В одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль (например, любую из BRAF-ассоциированных опухолей, описанных в настоящем документе), включающий введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение определенного периода времени, когда субъекту вводят иммунотаргетный агент (например, любой из иммунотаргетных агентов, описанных в настоящем документе) в течение указанного периода времени. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 11, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбрано из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов и примера 82.
Также в настоящем документе представлена фармацевтическая комбинация для лечения BRAF-ассоциированной опухоли у субъекта, нуждающегося в этом, которая включает (а) соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф, и (b) по меньшей мере, один дополнительный противораковый агент (например, любой из иллюстративных дополнительных противораковых агентов, описанных в настоящем документе или известных в данной области техники), где соединение формулы I, формула II или формула III или его фармацевтически приемлемую соль, сольват или полиморф, и, по меньшей мере, один дополнительный противораковый агент готовят отдельно для одновременного, раздельного или последовательного применения для лечения опухоли, где количества соединения формулы I, формулы II или формулы III или их фармацевтически приемлемой соли или сольвата, и дополнительного противоракового агента вместе эффективны при лечении опухоли; (ii) использование такой комбинации для приготовления лекарственного средства для лечения опухоли; и (iii) коммерческая упаковка или продукт, содержащий такую комбинацию, как комбинированный препарат для одновременного, раздельного или последовательного использования; и к способу лечения опухоли у пациента, нуждающегося в этом.
Используемый в настоящем документе термин «фармацевтическая комбинация» относится к не фиксированной комбинации активных ингредиентов. Термин «не фиксированная комбинация» означает, что соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, и, по меньшей мере, один дополнительный противораковый агент составляются в виде отдельных композиций или дозировок, таких, которые могут вводиться нуждающемуся в этом субъекту одновременно или раздельно с варьируемыми временными интервалами, где такое введение обеспечивает эффективные уровни двух или нескольких соединений в организме субъекта. Они также применимы к коктейльным терапиям, например, при приеме трех или нескольких активных ингредиентов. Точно так же термин «комбинация», относящийся к соединению формулы I, формулы II или формулы III, используемому с комбинацией другого противоракового агента, относится к не фиксированной комбинации.
Соответственно, также в настоящем документе представлен способ лечения BRAF-ассоциированной опухоли, включающий введение субъекту, нуждающемуся в этом, фармацевтической комбинации для лечения указанной опухоли, который включает (а) соединение формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, и (b) дополнительное противораковое средство для одновременного, раздельного или последовательного применения для лечения опухоли, где количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, и дополнительное противораковое средство вместе эффективны в лечении опухоли. В одном варианте осуществления, BRAF-ассоциированной опухолью является злокачественная опухоль, и дополнительным противораковым агентом является противораковый агент, например, любой из противораковых агентов, описанных в настоящем документе. В некоторых вариантах осуществления, соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль или сольват, и дополнительный противораковый агент вводят одновременно в виде отдельных доз. В некоторых вариантах осуществления, соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль или сольват, и дополнительный противораковый агент вводят в виде отдельных доз, последовательно в любом порядке, например, ежедневных или прерывистых доз, в совместно терапевтически эффективных количествах. Дополнительные противораковые агенты могут быть введены с одной или несколькими дозами соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа или их фармацевтической композицией, как часть одной и той же или в разных дозированных формах, одинаковыми или разными путями введения, и/или по одинаковой или разным схемам введения согласно стандартной фармацевтической практике, известной специалисту в данной области техники. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является злокачественная BRAF-ассоциированная опухоль (т.е., BRAF-ассоциированный рак). В некоторых вариантах осуществления, BRAF-ассоциированным раком является BRAF-ассоциированный рак ЦНС, и фармацевтическая комбинация содержит соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является BRAF-ассоциированный метастатический рак, и фармацевтическая комбинация содержит соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатическая меланома, и фармацевтическая комбинация содержит соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический колоректальный рак, и фармацевтическая комбинация содержит соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический немелкоклеточный рак легкого, и фармацевтическая комбинация включает соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак щитовидной железы, и фармацевтическая комбинация содержит соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является метастатический рак яичников, и фармацевтическая комбинация содержит соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированным метастатическим раком является внутричерепная LMD или внечерепная LMD, и фармацевтическая комбинация содержит соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированным раком ЦНС является первичная опухоль мозга, и фармацевтическая комбинация включает соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф, или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль ЦНС, и фармацевтическая комбинация содержит соединение формулы II или его фармацевтически приемлемую соль, сольват или полиморф или формулы III или его фармацевтически приемлемую соль, сольват или полиморф. В некоторых вариантах осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединением формулы I является соединение, выбранное из примеров 1-80 и их фармацевтически приемлемых солей и сольватов, и примеров 81-86. В одном варианте осуществления, соединением является соединение формулы II или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы II выбрано из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67. 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80, и их фармацевтически приемлемые соли и сольваты, и пример 82. В одном варианте осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В одном варианте осуществления, соединение формулы III выбирают из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей или сольватов, и примера 82.
В некоторых вариантах осуществления любого из способов, описанных в настоящем документе, у субъекта есть BRAF-ассоциированная опухоль (например, доброкачественная, злокачественная или метастатическая опухоль), где субъекта лечили предыдущей терапией или стандартной терапией (например, лечили одним или несколькими противораковыми агентами, отличными от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа, и/или радиационной терапией и/или хирургией), где указанная BRAF-ассоциированная опухоль стала резистентной или рефрактерной к указанной предшествующей терапии. В некоторых вариантах осуществления, у субъекта имеется BRAF-ассоциированная опухоль (например, местнораспространенная или метастатическая опухоль), которая не имеет стандартной терапии. В одном варианте осуществления, способ включает введение соединения формулы I, выбранного из примеров 1-80, и их фармацевтически приемлемых солей и сольватов и примеров 81-86. В одном варианте осуществления, способ включает введение соединения формулы II или его приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, способ включает введение соединения формулы II, выбранного из примеров 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80 и их фармацевтически приемлемых солей и сольватов, и примера 82. В одном варианте осуществления, способ включает введение соединения формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, способ включает введение соединения формулы III, выбранного из примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67, и его фармацевтически приемлемых солей или сольватов, и примера 82.
Соответственно, в одном варианте осуществления, в настоящем документе представлен способ лечения субъекта, имеющего BRAF-ассоциированную опухоль, где субъекта ранее лечили одним или несколькими противораковыми терапиями (например, противораковым агентом, радиационной терапией и/или хирургическим вмешательством), где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, II III или его фармацевтически приемлемой соли, сольвата или полиморфа.
Различные опухоли с мутацией BRAF не обязательно имеют общие механизмы резистентности к ингибиторам BRAF, и относительно первичных опухолей головного мозга имеется ограниченное количество исследований, описывающих вовлеченные механизмы резистентности. Yao, TW et al. (Oncotarget 8(1):583-595, 2017) и Ywo, Z., et al. (Cancer Cell 28(3):370-383, 2015) обнаружили гиперактивацию рецепторов эпидермального фактора роста (EGFR) в клеточных линиях глиомы, резистентных к ингибиторам BRAF, и лечение ингибиторами EGFR вместе с ингибитором BRAF дало многообещающие результаты по преодолению резистентности к ингибитору BRAF в клетках взрослой и детской глиомы. Для клеток рака щитовидной железы, резистентность к ингибитору BRAF в значительной степени обусловлена передачей сигналов HER3, общего молекулярного пути, вовлеченного в глиомагенез; поэтому добавление ингибиторов HER киназы к лечению ингибитором BRAF также может быть полезным при опухолях головного мозга (Montero-Conde et al., Cancer Discov 3(5):520-533, 2013). Кроме того, Yao, TW et al. (Oncotarget 8(1):583-595, 2017) обнаружили повышенную экспрессию и активность Axl в отношении резистентных к ингибитору BRAF клеток при подавлении глиомы BRAF-V600E с использованием ингибиторов Axl. В том же исследовании, повторная сенсибилизация резистентных к ингибитору BRAF клеток после отмены ингибитора BRAF в течение 48 часов предполагает, что периодическое использование ингибитора BRAF может снизить резистентность к ингибитору BRAF (Yao et al. (Oncotarget 8(1):583-595, 2017). WO 2013/070996, которая полностью включена в настоящий документ, описывает способ лечения пролиферативного заболевания (например, рака, например, меланомы), включающий подавление резистентности к лечению ингибитором BRAF энкорафенибом путем введения энкорафениба согласно схеме периодического дозирования. Ma, XH et al. (J Clin Invest 124(3):1406-1417, 2014) предположили, что активация аутофагии, вызванной стрессом эндоплазматического ретикулума, является механизмом резистентности к ингибитору BRAF, и Levy, JM et al. (Cancer Discov 4(7):773-780, 2014) показали клиническое и рентгенологическое улучшение при добавлении ингибитора аутофагии хлороквина у субъекта с ганглиоглиомой ствола головного мозга, которая ранее прогрессировала на лечении вемурафенибом.
В некоторых вариантах осуществления, субъект, страдающий метастатической меланомой (например, метастатической меланома с мутацией BRAF), получал лечение ингибитором BRAF (например, ингибитором BRAF, отличным от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида, (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатической меланомой (например, метастатической меланомой с мутацией BRAF), получал лечение ингибитором BRAF (например, отличным от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа) и ингибитором MEK до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида, и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил)]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), и ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, и ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиметиниба. В одном варианте осуществления, субъекта ранее лечили энкорафенибом и биниметинибом. В одном варианте осуществления, субъекта ранее лечили дабрафенибом и траметинибом. В одном варианте осуществления, субъекта ранее лечили вемурафенибом и кобиметинибом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатической меланомой (например, метастатической меланомой с мутацией BRAF), получал лечение одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой соль, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами контрольной точки, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатической меланомой (например, метастатической меланомой с мутацией BRAF), получал лечение одним или несколькими ингибиторами PI3K перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами PI3K, выбранными из бупарлисиба (BKM120), альпелисиба (BYL719), самотолисиба (LY3023414), 8-[(1R)-1-[(3,5-дифторфенил)амино]этил]-N, N-диметил-2-(морфолин-4-ил)-4-оксо-4H-хромен-6-карбоксамида (AZD8186), теналисиба (RP6530), гидрохлорида вокталисиба (SAR-245409), гедатолисиба (PF-05212384), панулисиба (P-7170), тазелисиба (GDC-0032), транс-2-амино-8-[4-(2-гидроксиэтокси)циклогексил]-6-(6-метоксиэтидин-3-ил)-4-метилпиридо[2,3-d]пиримидин-7(8H)-она (PF-04691502), дувелисиба (ABBV-954), N2-[4-оксо-4-[4-(4-оксо-8-фенил-4H-1-бензопиран-2-ил)морфолин-4-иум-4-илметокси]бутирил]-L-аргинил-глицил-L-аспартил-L-серинацетата (SF-1126), пиктилизиба (GDC- 0941), 2-метил-1-[2-метил-3-(трифторметил)бензил]-6-(морфолин-4-ил)-1H-бензимидазол-4-карбоновой кислоты (GSK2636771), иделалисиба (GS-1101), тозилата умбралисиба (TGR-1202), пиктилизиба (GDC-0941), гидрохлорида копанлисиба (BAY 84-1236), дактолизиба (BEZ-235), 1-(4-[5-[5-амино-6-(5-трет-бутил-1,3,4-оксадиазол-2-ил)писразин-2-ил]-1-этил-1H-1,2,4-триазол-3-ил]пиперидин-1-ил)-3-гидроксипропан-1-она (AZD-8835), 5-[6,6-диметил-4-(морфолин-4-ил)-8,9-дигидро-6H-[1,4]оксазино[4,3-е]пурин-2-ил]пиримидин-2-амина (GDC-0084), эверолимуса, рапамицина, перифозина, сиролимуса и темсиролимуса. В одном варианте осуществления, субъекта ранее лечили бупарлизибом или альпелисибом, отдельно или в комбинации. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатической меланомой (например, метастатической меланомой с мутацией BRAF), получал лечение ингибитором BRAF, отличным от соединения формулы I, формулы II, формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил)]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) и одним или несколькими ингибиторами контрольной точки, независимо выбранными из ипилимумаба, ниволумаба и пембролизумаба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатической меланомой (например, метастатической меланомой с мутацией BRAF), получал лечение ингибитором BRAF (например, отличным от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа), ингибитором MEK и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1) до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулой III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733) и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиметиниба, и одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелизумаба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатической меланомой (например, метастатическая меланома, мутантная по BRAF), получал лечение одним или несколькими алкилирующими агентами перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулой III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили одним или несколькими алкилирующими агентами, выбранными из темозоломида, фотемустина, ломустина и кармустина. В одном варианте осуществления, субъекта ранее лечили темозоломидом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение ингибитором BRAF, отличным от соединения формулы I, формулы II, формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, ингибитором MEK и ингибитором EGFR перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720), и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733), и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиметиниба, и ингибитором EGFR, выбранным из цетуксимаба и панитумумаба. В одном варианте осуществления, субъекта ранее лечили энкорафенибом, биниметинибом и цетуксимабом. В одном варианте осуществления, субъекта ранее лечили дабрафенибом, траметинибом и панитумумабом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение ингибитором EGFR перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394). В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение цетуксимабом или панитумумабом перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получал лечение ингибитором EGFR и одним или несколькими цитотоксическими химиотерапевтическими агентами до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и одним или несколькими цитотоксическими химиотерапевтическими агентами. В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба или панитумумаба и одним или несколькими цитотоксическими химиотерапевтическими агентами, такими как Nordic FLOX (фторурацил, фолиновая кислота и оксалиплатин) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение ингибитором EGFR и ингибитором BRAF перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394). В одном варианте осуществления, субъекта ранее лечили ингибитором EGFR, выбранным из цетуксимаба и панитумумаба, и ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба. В одном варианте осуществления, субъекта ранее лечили энкорафенибом и цетуксимабом. В одном варианте осуществления, субъекта ранее лечили вемурафенибом и панитумумабом. В одном варианте осуществления, субъекта ранее лечили дабрафенибом и панитумумабом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение ингибитором MEK и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA- 4, ингибитором PD-1 и/или ингибитором PD-L1) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодофениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733 ), и одним или несколькими ингибиторами контрольной точки (например, любым из ингибиторов контрольной точки, описанных в настоящем документе, например, ингибитора CTLA-4, ингибитора PD-1 и/или ингибитора PD-L1). В одном варианте осуществления, субъекта ранее лечили ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиметиниба, и одним или несколькими ингибиторами контрольных точек, независимо выбранных из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK, которым является биниметиниб, и ингибиторами контрольной точки ниволумабом и ипилимумабом. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK биниметинибом и ингибитором контрольной точки пембролизумабом. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK биниметинибом и ингибитором контрольной точки авелумабом. В одном варианте осуществления, субъекта ранее лечили ингибитором MEK траметинибом и ингибиторами контрольной точки ниволумабом и ипилимумабом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) перед обработкой соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, формулой III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами контрольной точки, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, субъекта ранее лечили ниволумабом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение одним или несколькими цитотоксическими химиотерапевтическими агентами перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение оксалиплатином, иринотеканом, FOLFOXIRI (оксалиплатин, иринотекан и фторурацил), FOLFIRI (фолиновая кислота и фторотеурацил) или CAPEOX (капецитабин и оксалиплатин) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение терапией антителами и одним или несколькими цитотоксическими химиотерапевтическими агентами перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение терапией антителами, которыми является бевацизумаб, и одним или несколькими цитотоксическими химиотерапевтическими агентами. В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение бевацизумабом и иринотеканом, бевацизумабом и FOLFOXIRI (оксалиплатином, иринотеканом и фурацилом) или бевацизумабом и FOLFIRI (фолиновой кислотой, фторурацилом и иринотеканом) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение ингибитором EGFR, ингибитором BRAF, отличным от соединения формулы I, формулы II или формулы III, и одним или несколькими цитотоксическими химиотерапевтическими агентами до лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В некоторых вариантах осуществления, субъект, имеющий метастатический колоректальный BRAF-ассоциированный рак (например, метастатический колоректальный рак с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, ингибитором BRAF, выбранным из ингибитора BRAF, выбранного из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) и одним или несколькими цитотоксическими химиотерапевтическими агентами. В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба и панитумумаба, ингибитором BRAF, которым является вемурафениб, и цитотоксическим химиотерапевтическим агентом, которым является иринотекан. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, мутантным метастатическим колоректальным раком, связанным с BRAF), получал лечение ингибитором EGFR и одним или несколькими цитотоксическими химиотерапевтическими агентами перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатический колоректальный рак с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба, и одним или несколькими цитотоксическими химиотерапевтическими агентами. В некоторых вариантах осуществления, субъект, имеющий BRAF-ассоциированный метастатический колоректальный рак (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение ингибитором EGFR, выбранным из цетуксимаба и панитумумаба, и цитотоксическим химиотерапевтическим агентом, которым является иринотекан или FOLFIRI (фолиновая кислота, фторурацил и иринотекан). В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал хирургическое лечение перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом или формулой III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получил хирургическое лечение с последующим лечением ингибитором BRAF, отличным от соединения формулы I, формулы II, формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, ингибитором MEK и ингибитором EGFR перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее подвергали хирургическому вмешательству и ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3).-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамид (PLX8394), ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733), и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вьетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее подвергали хирургическому лечению и ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиметиниба, и ингибитором EGFR, выбранным из цетуксимаба и панитумумаба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий BRAF-ассоциированным метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение радиационной терапией (например, радиационной терапия всего мозга или стереотаксической радиохирургией) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатическим колоректальным раком (например, метастатическим колоректальным раком с мутацией BRAF), получал лечение радиационной терапией (например, радиационной терапией всего мозга или стереотаксической радиохирургией) с последующим лечением ингибитором BRAF, ингибитором MEK и ингибитором EGFR до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили радиационной терапией и ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3)-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-иодофениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733), и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее подвергали хирургическому лечению и ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиметиниба, и ингибитором EGFR, выбранным из цетуксимаба и панитумумаба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатическим немелкоклеточным раком легкого (например, метастатическим немелкоклеточным раком легкого с мутацией BRAF), получал лечение одним или несколькими ингибиторами EGFR перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами EGFR, независимо выбранными из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили эрлотинибом. В одном варианте осуществления, субъекта ранее лечили гефитинибом. В одном варианте осуществления, субъекта ранее лечили эрлотинибом и гефитинибом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, страдающий метастатическим немелкоклеточным раком легкого (например, метастатическим немелкоклеточным раком легкого с мутацией BRAF), получал лечение ингибитором BRAF, отличным от соединения формулы I, формулы II, формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733), и ингибитором EGFR, выбранным из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из вемурафениба, дабрафениба и энкорафениба, и ингибитором EGFR, выбранным из цетуксимаба и панитумумаба, до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В некоторых вариантах осуществления, субъект, имеющий метастатический рак щитовидной железы (например, метастатическим раком щитовидной железы с мутацией BRAF) получал лечение с ингибитором BRAF (например, ингибитором BRAF, отличным от соединения формулы I, формулы II, формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1Н-пирроло[2,3-Ь]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1Н-пирроло[2,3-Ь]пиридин3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), ингибитора МЕК, выбранного из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2-(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-йодфениламин)бензамида (PD-325901), 2-(2-хлор-4-йодфениламин)-N-(циклопропилметоксите)-3,4-дифторбензамида (CI-1040) и 3-[2-(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин4,7(3H,8Н)-диона (TAK-733), и ингибитора EGFR, выбранного из цетуксимаба, панитумумаба, осимертиниба, эрлотиниба, гефитиниба, нецитумумаба, нератиниба, лапатиниба, вандетаниба и бригатиниба. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из вемурафениба, дабрафениба и энкорафениба перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, у субъекта развились метастазы в мозг во время указанного предыдущего лечения.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную LMD и его ранее лечили ингибитором BRAF (например, ингибитором BRAF, отличным от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа) и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394) и одним или несколькими ингибиторами контрольной точки, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную LMD и его ранее лечили ингибитором BRAF (например, ингибитором BRAF, отличным от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа), ингибитором MEK и одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1) до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733), и ингибитором контрольной точки (например, любым из ингибиторов контрольной точки, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или ингибитором PD-L1). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиметиниба, и одним или несколькими ингибиторами контрольных точек, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелизумаба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную LMD и его ранее лечили одним или несколькими ингибиторами контрольных точек (например, любым из ингибиторов контрольных точек, описанных в настоящем документе, например, ингибитором CTLA-4, ингибитором PD-1 и/или или ингибитором PD-L1) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили одним или несколькими ингибиторами контрольной точки, независимо выбранными из ипилимумаба, ниволумаба, пембролизумаба и авелумаба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и его ранее лечили хирургическим путем перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и его ранее лечили радиационной терапией (например, радиационной терапией всего мозга или стереотаксической радиохирургией) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и его ранее лечили одним или несколькими цитотоксическими химиотерапевтическими агентами перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили одним или несколькими цитотоксическими химиотерапевтическими агентами, независимо выбранными из цисплатина, пеметрекседа, винорелбина и паклитаксела. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и ранее его лечили ингибитором орнитиндекарбоксилазы перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект ранее получал лечение ингибитором орнитиндекарбоксилазы, которым является эфлорнитин (в виде рацемата или D- или L-энантиомера). В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и его ранее лечили алкилирующим агентом перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект ранее получал лечение алкилирующим агентом, выбранным из темозоломида, ломустина и кармустина. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и его ранее лечили алкилирующим агентом и ингибитором орнитиндекарбоксилазы перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект ранее получал лечение алкилирующим агентом, выбранным из темозоломида, ломустина и кармустина, и ингибитором орнитиндекарбоксилазы, которым является эфлорнитин (в виде рацемата или D- или L-энантиомера). В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и его ранее лечили радиационной терапией (например, радиационной терапией всего мозга или стереотаксической радиохирургией) и алкилирующим агентом перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект ранее получал лечение радиационной терапией (например, радиационной терапией всего мозга или стереотаксической радиохирургией) и алкилирующим агентом, выбранным из темозоломида, ломустина и кармустина. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и ранее получал терапию антителами перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом или формулой III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект ранее получал лечение антителом, которым является бевацизумаб. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и его ранее лечили хирургическим вмешательством и радиационной терапией перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом или формулой III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и ранее получал хирургическое лечение, радиационную терапию и алкилирующий агент перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили хирургическим вмешательством, радиационной терапией (например, радиационной терапией всего мозга или стереотаксической радиохирургией) и алкилирующим агентом, выбранным из темозоломида, ломустина и кармустина. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и ранее его лечили ингибитором BRAF (например, ингибитором BRAF, отличным от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа) до лечения соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект ранее получал лечение ингибитором BRAF, выбранным из N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720), вемурафениба, дабрафениба, энкорафениба и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3)-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамид (PLX8394). В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную глиому, и ранее его лечили ингибитором BRAF (например, ингибитором BRAF, отличным от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа) и ингибитором MEK перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъект ранее получал лечение ингибитором BRAF, выбранным из N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720), вемурафениба, дабрафениба, энкорафениба и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3)-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394), и ингибитором MEK, выбранным из биниметиниба, траметиниба, кобиметиниба, селуметиниба, пимасертиба, рефаметиниба, N-[2(R),3-дигидроксипропокси]-3,4-дифтор-2-(2-фтор-4-иодфениламино)бензамида (PD-325901), 2-(2-хлор-4-иодфениламино)-N-(циклопропилметокси)-3,4-дифторбензамида (CI-1040) и 3-[2(R),3-дигидроксипропил]-6-фтор-5-(2-фтор-4-йодфениламино)-8-метилпиридо[2,3-d]пиримидин-4,7(3H,8H)-диона (TAK-733). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба, и ингибитором MEK, выбранным из биниметиниба, траметиниба и кобиметиниба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, глиомой является глиома 2-й, 3-й или 4-й степени.
В одном варианте осуществления, субъект имеет BRAF-ассоциированную ганглиоглиому ствола мозга, и его ранее лечили ингибитором BRAF (например, ингибитором BRAF, отличным от соединения формулы I, II или III или его фармацевтически приемлемой соли, сольвата или полиморфа) перед лечением соединением формулы II или его фармацевтически приемлемой солью, сольватом или полиморфом, или формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом. В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба, вемурафениба, N-[3-(5-хлор-1H-пирроло[2,3-b]пиридин-3-илкарбонил)-2,4-дифторфенил]пропан-1-сульфонамида (PLX4720) и (3R)-N-(3-[[5-(2-циклопропилпиримидин-5-ил)-1H-пирроло[2,3-b]пиридин-3-ил]карбонил]-2,4-дифторфенил)-3-фторпирролидин-1-сульфонамида (PLX8394). В одном варианте осуществления, субъекта ранее лечили ингибитором BRAF, выбранным из энкорафениба, дабрафениба и вемурафениба. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению.
Также в настоящем документе представлен способ лечения заболевания или расстройства, опосредованного BRAF, у субъекта, нуждающегося в таком лечении, где способ включает введение субъекту терапевтического эффективного количества соединения формулы I, формулы II, формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, или его фармацевтической композиции. Заболевание или нарушение, опосредованное BRAF, может включать любое заболевание, нарушение или состояние, которое прямо или косвенно связано с мутацией BRAF. В некоторых вариантах осуществления, заболеванием является рак (например, BRAF-ассоциированный рак). В некоторых вариантах осуществления, раком является любой из видов BRAF-ассоциированных рака или опухолей, описанных в настоящем документе. В некоторых вариантах осуществления, раком является рак ЦНС, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, раком ЦНС является метастатический рак. В некоторых вариантах осуществления, раком ЦНС является первичная опухоль головного мозга. В некоторых вариантах осуществления, соединением является соединение формулы III или его фармацевтически приемлемая соль, сольват или полиморф. В некоторых вариантах осуществления, соединение формулы III выбрано из соединения примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67 и их фармацевтически приемлемых солей и сольватов, и примера 82.
Хотя генетическая основа туморогенеза может различаться для разных типов рака, клеточные и молекулярные механизмы, необходимые для метастазирования, по-видимому, схожи для всех типов солидных опухолей. Во время метастатического каскада, раковые клетки теряют реакцию ингибирования роста, претерпевают изменения в адгезивности и вырабатывают ферменты, которые могут разрушать компоненты внеклеточного матрикса. Это приводит к отделению опухолевых клеток от исходной опухоли, инфильтрации в кровоток через новообразованную сосудистую сеть, миграции и пропотевания опухолевых клеток в благоприятные отдаленные участки, где они могут образовывать колонии. Ряд генов был идентифицирован как промоторы или супрессоры метастазирования.
Соответственно, в настоящем документе также представлены способы лечения, ингибирования, профилактики, помощи в профилактике или уменьшения симптомов метастазирования BRAF-ассоциированного рака у субъекта, нуждающегося в этом, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, или его фармацевтической композиции. В некоторых вариантах осуществления, соединение формулы I, формула II или формула III или ее фармацевтически приемлемую соль, сольват или полиморф используют в комбинации с другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли), и/или радиационной терапией, и/или лечения противораковым агентом. В одном варианте осуществления, рак является метастатический рак с метастазами в мозг, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, раком является метастатическая меланома с метастазами в мозг. В одном варианте осуществления, раком является метастатический колоректальный рак с метастазами в мозг. В одном варианте осуществления, раком является метастатический немелкоклеточный рак легкого с метастазами в мозг. В одном варианте осуществления, раком является метастатический рак яичников с метастазами в мозг. В одном варианте осуществления, раком является метастатический рак щитовидной железы с метастазами в мозг. В одном варианте осуществления, раком является нейробластома с метастазами в мозг, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, субъекта ранее лечили другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, субъекта лечат соединением формулы I, формулы II, формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом, в комбинации с другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом.
Также в настоящем документе представлены способы ингибирования метастазов у субъекта, нуждающегося в этом, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, его фармацевтической композиции. В некоторых вариантах осуществления, соединение формулы I, формула II или формула III или его фармацевтически приемлемую соль, сольват или полиморф, используют в комбинации с другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, раком является метастатический рак с метастазами в мозг, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, раком является метастатическая меланома с метастазами в мозг. В одном варианте осуществления, раком является метастатический колоректальный рак с метастазами в мозг. В одном варианте осуществления, раком является метастатический немелкоклеточный рак легкого с метастазами в мозг. В одном варианте осуществления, раком является метастатический рак яичников с метастазами в мозг. В одном варианте осуществления, раком является метастатический рак щитовидной железы с метастазами в мозг. В одном варианте осуществления, раком является нейробластома с метастазами в мозг, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, субъекта ранее лечили другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, субъекта лечат соединением формулы I, формулы II, формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом в комбинации с другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, противораковым лечением является противораковый агент. В одном варианте осуществления, противораковый агент выбран из одного или нескольких ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов SOS1, ингибиторов сигнальной трансдукции, ингибиторов контрольных точек, модуляторов пути апоптоз, цитотоксических химиотерапевтических агентов, таргетных терапий ангиогенеза и иммунотаргетных агентов. В одном варианте осуществления, противораковым агентом является ингибитор МЕК. В одном варианте осуществления, ингибитор МЕК выбран из биниметиниба, траметиниба и кобиметиниба. В одном варианте осуществления, ингибитором МЕК является биниметиниб.
Используемый в настоящем документе термин «лечение метастазов» означает уменьшение размера, прогрессирования и/или дальнейшего распространения одного или нескольких метастазов.
В настоящем документе также представлены способы ингибирования метастазов у субъекта, нуждающегося в этом, где способ включает введение субъекту терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, или его фармацевтической композиции. В некоторых вариантах осуществления, соединение формулы I, формулы II или формулы III или ее фармацевтически приемлемой соли, сольвата или полиморфа, используют в комбинации с другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, раком является метастатический рак с метастазами в мозг, и способ включает введение соединения формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, раком является метастатическая меланома с метастазами в мозг. В одном варианте осуществления, раком является метастатический колоректальный рак с метастазами в мозг. В одном варианте осуществления, раком является метастатический немелкоклеточный рак легкого с метастазами в мозг. В одном варианте осуществления, раком является метастатический рак яичников с метастазами в мозг. В одном варианте осуществления, раком является метастатический рак щитовидной железы с метастазами в мозг. В одном варианте осуществления, раком является нейробластома с метастазами в мозг, и способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, субъекта ранее лечили другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, субъекта лечат соединением формулы I, формулы II, формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом в комбинации с другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, противораковой терапией является противораковый агент. В одном варианте осуществления, противораковый агент выбирают из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов SOS1, ингибиторов пути передачи сигнала, ингибиторов контрольных точек, модуляторов пути апоптоза, цитотоксических химиотерапевтических агентов, таргетных терапий ангиогенеза и иммунотаргетных агентов. В одном варианте осуществления, противораковым агентом является ингибитор MEK. В одном варианте осуществления, ингибитор MEK выбран из биниметиниба, траметиниба и кобиметиниба. В одном варианте осуществления, ингибитором MEK является биниметиниб.
Используемый в настоящем документе термин «ингибирование метастазов» означает уменьшение возникновения (или повторения) одного или нескольких метастазов, профилактику возникновения (или повторного возникновения) одного или нескольких метастазов или уменьшение распространения одного или нескольких метастазов.
Также представлены способы снижения риска развития одного или нескольких метастазов или одного или нескольких дополнительных метастазов у субъекта, имеющего BRAF-ассоциированный рак, которые включают: выбор, идентификацию или диагностику субъекта как больного BRAF-ассоциированным раком и введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа субъекту, выбранному, идентифицированному или диагностированному как имеющий BRAF-ассоциированный рак. Также предусмотрены способы снижения риска развития одного или нескольких метастазов или одного или нескольких дополнительных метастазов у субъекта, страдающего раком, ассоциированным с BRAF, который включает введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, субъекту, имеющему BRAF-ассоциированный рак. Снижение риска развития одного или нескольких метастазов или одного или нескольких дополнительных метастазов у субъекта, имеющего BRAF-ассоциированный рак, можно сравнить с риском развития одного или нескольких метастазов или одного или нескольких дополнительных метастазов у субъекта до лечения, или по сравнению с субъектом или популяцией субъектов, страдающих подобным или тем же самым BRAF-ассоциированным раком, которые не получали лечения или другого лечения.
Фраза «риск развития одного или нескольких метастазов» означает риск того, что у субъекта или субъекта, имеющего первичную опухоль, разовьется дополнительная опухоль (например, солидная опухоль) в месте, удаленном от первичной опухоли у субъекта или субъекта в течение установленного периода времени, где дополнительная опухоль включает такие же или аналогичные раковые клетки, что и первичная опухоль. В настоящем документе описаны способы снижения риска развития одного или нескольких метастазов у субъекта или субъекта, страдающего раком.
Фраза «риск развития дополнительных метастазов» означает риск того, что у субъекта или субъекта, имеющего первичную опухоль и одну или несколько дополнительных опухолей на участках, удаленных от первичной опухоли (где одна или несколько дополнительных опухолей включают такие же или аналогичные раковые клетки, что и первичная опухоль), разовьется одна или несколько дополнительных опухолей, удаленных от первичной опухоли, где дополнительные опухоли включают те же или аналогичные раковые клетки, что и первичная опухоль. В настоящем документе описаны способы снижения риска развития дополнительных метастазов.
Также в настоящем документе представлен способ лечения BRAF-ассоциированной опухоли, метастазов BRAF-ассоциированной опухоли или их комбинации у субъекта, нуждающегося в этом, где способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, субъекту. В одном варианте осуществления, у субъекта есть, по меньшей мере, один метастаз или существует риск развития, по меньшей мере, одного метастаза. В одном варианте осуществления, субъект имеет, по меньшей мере, один метастаз, где способ включает введение соединения формулы II или его фармацевтически приемлемой соли, сольвата или полиморфа, или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В одном варианте осуществления, у субъекта есть риск развития, по меньшей мере, одного метастаза. В одном варианте осуществления, субъект подвержен риску развития, по меньшей мере, одного метастаза, где указанный субъект имеет рак, выбранный из меланомы, колоректального рака, рака щитовидной железы, немелкоклеточного рака легкого или рака яичников. В одном варианте осуществления, раком является BRAF мутантный рак, например, BRAF мутантный рак V600, например, BRAF V600E, BRAF V600K, BRAF V600D или BRAF V600R. В одном варианте осуществления, субъекта ранее лечили другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, субъект стал невосприимчивым к указанному предыдущему лечению. В одном варианте осуществления, субъекта лечат соединением формулы I, формулы II, формулы III или его фармацевтически приемлемой солью, сольватом или полиморфом, в комбинации с другим противораковым лечением, например, хирургическим вмешательством (например, по меньшей мере, частичной резекцией опухоли) и/или радиационной терапией и/или лечением противораковым агентом. В одном варианте осуществления, противораковой терапией является противораковый агент, выбранный из ингибиторов MEK, ингибиторов BRAF, ингибиторов EGFR, ингибиторов SOS1, ингибиторов HER2 и/или HER3, ингибиторов Axl, ингибиторов PI3K, ингибиторов пути передачи сигнала, ингибиторов контрольных точек, модуляторов пути апоптоза, цитотоксических химиотерапевтических агентов, таргетной терапии ангиогенеза, и иммунотаргетного агента. В одном варианте осуществления, противораковым агентом является ингибитор MEK. В одном варианте осуществления, ингибитор MEK выбран из биниметиниба, траметиниба и кобиметиниба.
В некоторых вариантах осуществления, субъекту вводят один или несколько агентов для ослабления побочных эффектов лечения (например, один или несколько из кортикостероидов, антагонистов серотонина, антагонистов дофамина, ингибиторов NK-1, каннабиноидов, противотревожных лекарственных средств (например, лоразепама или диазепама), антибиотиков, противогрибковых средств, колониестимулирующего фактора, железных добавок, Прокрита, эпоэтина альфа, дарбэпоэтина альфа, противорвотных агентов, диуретиков, НПВП, анальгетиков, метотрексата, антидиуретиков, пробиотиков, препаратов для регулировки артериального давления, агентов против тошноты, слабительных и т. д.).
В одном варианте осуществления, BRAF-ассоциированной опухолью является доброкачественная опухоль, и соединение формулы I, формулы II или формулы III или его фармацевтически приемлемая соль, сольват или полиморф, может использоваться отдельно или в комбинации с одним или несколькими разными формами лечения для лечения субъекта с доброкачественной опухолью.
В некоторых вариантах осуществления, субъект имеет опухоль ЦНС, и ему вводят один или несколько агентов для облегчения одного или нескольких симптомов, связанных с опухолью ЦНС, включая, помимо прочего, судороги, тошноту, головные боли, нечеткость зрения, потерю зрения, потерю равновесия, изменения мелкой моторики и сонливость. Примеры таких агентов для облегчения одного или нескольких симптомов, связанных с опухолью ЦНС, включают кортикостероиды, противосудорожные препараты (например, каннабидиол, габапентин или прегабалин), обезболивающие (например, НПВП, ацетаминофен) и агенты против тошноты.
Также представлен способ ингибирования активности BRAF киназы в клетке млекопитающего, включающий контакт клетки с эффективным количеством соединения формулы I, формулы II или формулы III. В некоторых вариантах осуществления, контакт происходит in vitro. В некоторых вариантах осуществления, контакт происходит in vivo. В некоторых вариантах осуществления, контакт осуществляется in vivo, где способ включает введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа субъекту, имеющему клетку, обладающую активностью BRAF киназы. В некоторых вариантах осуществления, клеткой является раковая клетка. В некоторых вариантах осуществления, раковой клеткой является любая злокачественная опухоль, как описано в настоящем документе. В некоторых вариантах осуществления, раковой клеткой является BRAF-ассоциированная злокачественная клетка. В некоторых вариантах осуществления, клеткой является клетка мозга (например, нервная клетка или глиальная клетка).
Также представлен способ ингибирования активности BRAF киназы в клетке млекопитающего, включающий контакт клетки с эффективным количеством соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, контакт происходит in vitro. В некоторых вариантах осуществления, контакт происходит in vivo. В некоторых вариантах осуществления, контакт осуществляется in vivo, где способ включает введение терапевтически эффективного количества соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа, млекопитающему, имеющему клетку, обладающую активностью BRAF киназы. В некоторых вариантах осуществления, клеткой млекопитающего является раковая клетка млекопитающего. В некоторых вариантах осуществления, раковой клеткой млекопитающего является любой рак, как описано в настоящем документе. В некоторых вариантах осуществления, раковой клеткой млекопитающего является BRAF-ассоциированная раковая клетка. В некоторых вариантах осуществления, клеткой млекопитающего является клетка мозга.
Используемый в настоящем документе термин «контакт» относится к объединению указанных фрагментов в системе in vitro или в системе in vivo. Например, «контакт» BRAF киназы с соединением, представленным в настоящем документе, включает контакт клетки, содержащей BRAF киназу, с соединением, представленным в настоящем документе, а также, например, введение соединения в настоящем документе в образец, содержащий клеточный или очищенный препарат, содержащий BRAF киназу.
Также в настоящем документе представлен способ ингибирования пролиферации клеток in vitro или in vivo, где способ включает контакт клетки с терапевтическими эффективным количеством соединения формулы I, формула II или формула III или его фармацевтически приемлемой солью, сольватом или полиморфом или его фармацевтической композиции, как определено в настоящем документе.
Используемый в настоящем документе термин «терапевтически эффективное количество» соединения, его фармацевтической композиции или его фармацевтической комбинации включает количество, достаточное для достижения любого одного или нескольких полезных или желаемых результатов. Для профилактического использования полезные или желаемые результаты включают устранение или снижение риска, уменьшение тяжести или отсрочку начала заболевания, включая биохимические, гистологические и/или поведенческие симптомы заболевания, его осложнения и промежуточные патологические фенотипы, проявляющиеся во время развития заболевания. Для терапевтического использования полезные или желаемые результаты включают предоставление терапевтического эффекта, который может включать уменьшение размера опухоли, ингибирование (например, замедление до некоторой степени, предпочтительно, остановку) прогрессирования опухоли, ингибирование (например, замедление до некоторой степени, предпочтительно, остановку) роста опухоли, ингибирование (например, замедление до некоторой степени, предпочтительно, остановку) инвазивности опухоли и/или ингибирование (например, замедление до некоторой степени, предпочтительно, остановку) метастазирования опухоли. Специалист в данной области техники понимает, что прогрессирование опухоли у людей может быть определено множеством способов. Например, размер опухоли близко к коже можно измерить, установив ширину и глубину опухоли с помощью штангенциркуля, а затем рассчитав объем опухоли. Менее доступные опухоли, такие как рак легких и ЦНС, можно измерить, наблюдая изображения, полученные при сканировании с помощью магнитно-резонансной томографии (МРТ). Опухоли ЦНС, такие как опухоли головного мозга, можно измерить с помощью комбинации МРТ-сканирования и мониторинга неврологических функций. Рост опухоли головного мозга обычно связан со снижением неврологической работоспособности. Оказание терапевтического эффекта также включает продление выживаемости субъекта или субъекта сверх ожидаемого при отсутствии лечения, и/или облегчение до некоторой степени (или, предпочтительно, устранение) одного или нескольких признаков или симптомов, ассоциированных с раком. В одном варианте осуществления, лечение субъекта или субъекта соединением или комбинацией по изобретению продлевает выживаемость сверх ожидаемого в отсутствие лечения на 1 или более месяцев, например, на 3 или более месяцев, например, на 6 или более месяцев, например, на 1 год или более, например, на 2 года или более, например, на 3 года или более, например, на 5 лет или более, например, на 10 лет или более. Оказание терапевтического эффекта также включает уменьшение количества раковых клеток. Оказание терапевтического эффекта также включает уничтожение раковых клеток. Оказание терапевтического эффекта также включает уменьшение массы опухоли. Оказание терапевтического эффекта также включает в себя переход рака в стадию ремиссии. Терапевтически эффективное количество можно вводить за одно или несколько введений. Для целей настоящего изобретения, дозированным терапевтически эффективным количеством соединения или его фармацевтической композиции является количество, достаточное для осуществления профилактического или терапевтического лечения прямо или косвенно. Как понятно в клиническом контексте, дозировка терапевтически эффективного количества соединения или его фармацевтической композиции может быть достигнута в сочетании с другой терапией. Таким образом, «терапевтически эффективное количество» можно рассматривать в контексте введения одной или нескольких терапий (например, одного или нескольких противораковых агентов), и можно считать, что один агент вводится в терапевтически эффективном количестве, если в сочетании с одним или несколькими другими агентами, может быть или достигается желаемый результат. Что касается лечения рака, терапевтически эффективное количество может также относиться к тому количеству, которое имеет эффект (1) уменьшения размера опухоли, (2) ингибирования (то есть замедления до некоторой степени, предпочтительно, остановки) появление метастазов опухоли, (3) ингибирования до некоторой степени (то есть, замедления до некоторой степени, предпочтительно, остановки) роста опухоли или инвазивности опухоли, и/или (4) ослабления до некоторой степени (или, предпочтительно, устранения) одного или нескольких признаков или симптомов, ассоциированных с раком. Терапевтическая или фармакологическая эффективность доз и схем введения также может быть охарактеризована как способность индуцировать, усиливать, поддерживать или продлевать контроль заболевания и/или общую выживаемость у субъектов с этими специфическими опухолями, которую можно измерить как продление времени до прогрессирования заболевания.
В одном варианте осуществления, субъект, леченный в соответствии с любым из способов, описанных в настоящем документе, может быть оценен в соответствии с одним или несколькими стандартными критериями оценки ответа, известными в данной области техники, включая RECIST (Response Evaluation Criteria in Solid Tumors, например, RECIST версии 1.0, RECIST версии 1.1 и модифицированный RECIST 1.1 (mRECIST 1.1)), RANO-BM (Response Assessment in Neuro-Oncology Brain Metastases), Macdonald, RANO-LMD и NANO (Neurologic Assessment in Neuro-Oncology). В одном варианте осуществления любого из указанных критериев, опухоль оценивают с помощью исследования визуализации (например, МРТ, КТ, МДКТ или ПЭТ). В одном варианте осуществления, ответ на лечение оценивают в соответствии с RECIST версии 1.1, где: полный ответ (CR) определен как полное исчезновение всех опухолевых поражений; частичный ответ (PR) определен как уменьшение суммы измерений опухоли, по меньшей мере, на 30%; прогрессирующее заболевание (PD) определено как, по меньшей мере, 20% увеличение суммы измерений опухоли (где развитие новых поражений или существенное прогрессирование поражений вне мишени также определяется как PD), где увеличение, по меньшей мере, 5 мм от исходного уровня оценивается как PD; и стабильное заболевание (SD) определено как отсутствие достаточного сокращения, чтобы соответствовать критериям PR, или достаточного увеличения, чтобы соответствовать критериям PD, принимая в качестве эталона наименьшую сумму диаметров во время лечения. В одном варианте осуществления, оценки включают внутричерепной ответ (оцениваемый согласно модифицированному RECIST с использованием МРТ гадолиниевым усилением), экстракраниальный ответ, общую частоту ответа, частоту контроля заболевания (DCR), продолжительность ответа (DOR), выживаемость без прогрессирования (PFS) и общую выживаемость (OS).
В одном варианте осуществления, у субъекта имеется опухоль ЦНС и, по меньшей мере, одна поддающаяся измерению внутричерепная опухоль. В одном варианте осуществления, по меньшей мере, одну измеримую внутричерепную опухоль измеряют с помощью МРТ или КТ.
«Измеримая» опухоль (опухолевое поражение) означает опухоль, которая может быть точно измерена, по меньшей мере, в одном измерении (самый длинный диаметр в плоскости измерения не регистрируется) с минимальным размером: 10 мм по КТ сканированию (толщина среза КТ сканирования не более 5 мм); измерение штангенциркулем 10 мм при клиническом осмотре; 20 мм на рентгенограмме грудной клетки.
При использовании в качестве фармацевтических агентов, соединения формулы I, формулы II или формулы III, включая их фармацевтически приемлемые соли или сольваты, можно вводить в форме фармацевтических композиций. Эти композиции могут быть приготовлены способом, хорошо известным в области фармацевтики, и могут вводиться различными путями, в зависимости от того, желательно ли местное или системное лечение, и от области, подлежащей лечению. Введение может быть местным (включая трансдермальное, эпидермальное, офтальмологическое и на слизистые оболочки, включая интраназальное, вагинальное и ректальное введение), легочным (например, путем ингаляции или инсуффляции порошков или аэрозолей, в том числе с помощью небулайзера; интратрахеально или интраназально), пероральным или парентеральным. Пероральное введение может включать дозированную форму, составленную для введения один или два раза в день (BID). Парентеральное введение включает внутривенное, внутриартериальное, подкожное, внутрибрюшинное внутримышечное введение или инъекцию или инфузию; или внутричерепное, например, интратекальное или внутрижелудочковое введение. Парентеральное введение может осуществляться в виде однократной болюсной дозы или, например, с помощью перфузионного насоса непрерывного действия. Фармацевтические композиции и составы для местного применения могут включать трансдермальные пластыри, мази, лосьоны, кремы, гели, капли, суппозитории, спреи, жидкости и порошки. Могут быть необходимы или желательны обычные фармацевтические носители, водные, порошковые или масляные основы, загустители и т.п.
Также в настоящем документе представлены фармацевтические композиции, которые содержат в качестве активного ингредиента соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф в комбинации с одним или несколькими фармацевтически приемлемыми носителями (эксципиентами). Например, фармацевтическую композицию получают с использованием соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа. В некоторых вариантах осуществления, композиция подходит для местного введения. При составлении композиций, представленных в настоящем документе, активный ингредиент обычно смешивают с эксципиентом, разбавляют эксципиентом или заключают в такой носитель, например, в форме капсулы, саше, бумаги или другого контейнера. Когда эксципиент служит разбавителем, он может быть твердым, полутвердым или жидким материалом, который действует как носитель, носитель или среда для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, пастилок, саше, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (в твердой или в жидкой среде), мазей, содержащих, например, до 10% масс. активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных растворов для инъекций и порошков в стерильной упаковке. В некоторых вариантах осуществления, композиция составлена для перорального введения. В некоторых вариантах осуществления, композицией является твердый пероральный состав. В некоторых вариантах осуществления, композиция составлена в виде таблетки или капсулы.
Далее в настоящем документе представлены фармацевтические композиции, содержащие соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф с фармацевтически приемлемым носителем. Фармацевтические композиции, содержащие соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф в качестве активного ингредиента, можно получить тщательным смешиванием соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа с фармацевтическим носителем согласно общепринятым методикам приготовления фармацевтических смесей. Носитель может принимать самые разные формы в зависимости от желаемого пути введения (например, перорального, парентерального). В некоторых вариантах осуществления, композицией является твердая пероральная композиция.
Подходящие фармацевтически приемлемые носители хорошо известны в данной области техники. Описание некоторых из этих фармацевтически приемлемых носителей можно найти в The Handbook of Pharmaceutical Excipients, опубликованном American Pharmaceutical Association и Pharmaceutical Society of Great Britain.
Способы составления фармацевтических композиций описаны в многочисленных публикациях, таких Pharmaceutical Dosage Forms: Tablets, Second Edition, Revised and Expanded, Volumes 1-3, edited by Lieberman et al; Pharmaceutical Dosage Forms: Parenteral Medications, Volumes 1-2, edited by Avis et al; and Pharmaceutical Dosage Forms: Disperse Systems, Volumes 1-2, edited by Lieberman et al; published by Marcel Dekker, Inc.
При приготовлении композиций в пероральной дозированной форме можно использовать любую из обычных фармацевтических сред. Таким образом, для жидких пероральных препаратов, таких как суспензии, эликсиры и растворы, подходящие носители и добавки включают воду, гликоли, масла, спирты, ароматизаторы, консерванты, стабилизаторы, красители и подобные; для твердых пероральных препаратов, таких как порошки, капсулы и таблетки, подходящие носители и добавки включают крахмалы, сахара, разбавители, гранулирующие агенты, лубриканты, связующие агенты, разрыхлители и подобные. Подходящие связующие агенты включают, без ограничения, крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, натуральные и синтетические камеди, такие как гуммиарабик, трагакант или олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и подобные. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и подобные. Твердые пероральные препараты также могут быть покрыты такими веществами, как сахар, или иметь энтеросолюбильное покрытие, чтобы модулировать основной участок абсорбции. Для парентерального введения носитель обычно состоит из стерильной воды, и для повышения растворимости или консервации могут быть добавлены другие ингредиенты. Суспензии или растворы для инъекций также могут быть приготовлены с использованием водных носителей вместе с соответствующими добавками. Фармацевтические композиции в настоящем документе будут содержать на единицу дозировки, например, таблетку, капсулу, порошок, инъекцию, чайную ложку и подобные, количество активного ингредиента, необходимое для доставки терапевтически эффективного количества, как описано в настоящем документе.
Композиции, содержащие соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф, могут быть составлены в виде стандартной дозированная формы, где каждая доза содержит от примерно 5 до примерно 1000 мг (1 г), чаще от примерно 100 мг до примерно 500 мг активного ингредиента. Термин «стандартная дозированная форма» относится к физически дискретным единицам, подходящим в качестве единичных доз для людей и других субъектов, где каждая единица содержит заранее определенное количество активного продукта (т.е. соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф), рассчитанное для получения желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим эксципиентом.
В некоторых вариантах осуществления, композиция, представленная в настоящем документе, содержит от примерно 5 мг до примерно 50 мг активного ингредиента. Специалист в данной области техники поймет, что включены соединения или композиции, содержащие от примерно 5 мг до примерно 10 мг, от примерно 10 мг до примерно 15 мг, от примерно 15 мг до примерно 20 мг, от примерно 20 мг до примерно 25 мг, от примерно 25 мг до примерно 30 мг, от примерно 30 мг до примерно 35 мг, от примерно 35 мг до примерно 40 мг, от примерно 40 мг до примерно 45 мг или от примерно 45 мг до примерно 50 мг активного ингредиента.
В некоторых вариантах осуществления, композиция, представленная в настоящем документе, содержит от примерно 50 мг до примерно 500 мг активного ингредиента. Специалист в данной области техники поймет, что включены соединения или композиции, содержащие от примерно 50 мг до примерно 100 мг, от примерно 100 мг до примерно 150 мг, от примерно 150 мг до примерно 200 мг, от примерно 200 мг до примерно 250 мг, от примерно 250 мг до примерно 300 мг, от примерно 300 мг до примерно 300 мг, от примерно 350 мг до примерно 400 мг или от примерно 450 мг до примерно 500 мг активного ингредиента. В некоторых вариантах осуществления, композиция, представленная в настоящем документе, содержит примерно 10 мг, примерно 20 мг, примерно 80 мг или примерно 160 мг активного ингредиента.
В некоторых вариантах осуществления, композиция, представленная в настоящем документе, содержит от примерно 500 мг до примерно 1000 мг активного ингредиента. Специалист в настоящей области техники поймет, что включены соединения или композиции, содержащие от примерно 500 мг до примерно 550 мг, от примерно 550 мг до примерно 600 мг, от примерно 600 мг до примерно 650 мг, от примерно 650 мг до примерно 700 мг, примерно 700 мг до примерно 750 мг, от примерно 750 мг до примерно 800 мг, от примерно 800 мг до примерно 850 мг, от примерно 850 мг до примерно 900 мг, от примерно 900 мг до примерно 950 мг или от примерно 950 мг до примерно 1000 мг активного ингредиента.
Суточная дозировка соединения формулы I, формулы II или формулы III или его приемлемой соли, сольвата или полиморфа может варьироваться в широком диапазоне от 1,0 до 10000 мг на взрослого человека в сутки или выше, или в любом другом диапазоне. Для перорального применения, композиции предпочтительно представлены в форме таблеток, содержащих 0,01, 0,05, 0,1, 0,5, 1,0, 2,5, 5,0, 10,0, 15,0, 25,0, 50,0, 100, 150, 160, 200, 250 и 500 миллиграммов активного ингредиента для симптоматической корректировки дозировки для субъекта, подлежащего лечению. Терапевтически эффективное количество лекарственного средства обычно доставляется на уровне дозировки от примерно 0,1 мг/кг до примерно 1000 мг/кг массы тела в сутки или в любом диапазоне в этих пределах. Предпочтительно, диапазон составляет от примерно 0,5 до примерно 500 мг/кг массы тела в сутки или любой диапазон в этих пределах. Более предпочтительно, от примерно 1,0 до примерно 250 мг/кг массы тела в сутки или в этих пределах. Более предпочтительно, от примерно 0,1 до примерно 100 мг/кг массы тела в сутки или в этих пределах. В одном примере, диапазон может составлять от примерно 0,1 до примерно 50,0 мг/кг массы тела в сутки или любое количество или диапазон в этих пределах. В другом примере, диапазон может составлять от примерно 0,1 до примерно 15,0 мг/кг массы тела в сутки или любой диапазон в этих пределах. В еще одном примере, диапазон может составлять от примерно 0,5 до примерно 7,5 мг/кг массы тела в сутки или любое количество в этих пределах. Фармацевтические композиции, содержащие соединение формулы I, формулы II или формулы III или его фармацевтически приемлемую соль, сольват или полиморф, можно вводить по схеме от 1 до 4 раз в день или в виде однократной суточной дозы.
Активное соединение может быть эффективным в широком диапазоне доз и обычно вводится в терапевтически эффективном количестве. Оптимальные дозировки для введения могут быть легко определены специалистами в данной области техники. Следовательно, будет понятно, что количество фактически вводимого соединения обычно определяется врачом и будет варьироваться в зависимости от соответствующих обстоятельств, включая способ введения, фактически вводимое соединение, силу препарата, состояние, подлежащее лечению, и прогрессирование болезненного состояния. Кроме того, факторы, связанные с конкретным субъектом, которого лечат, включая ответ субъекта, возраст, вес, диету, время введения и тяжесть симптомов субъекта, могут вызвать необходимость корректировки дозировок.
В некоторых вариантах осуществления, соединения, представленные в настоящем документе, могут быть введены в количестве от примерно 1 мг/кг до примерно 100 мг/кг. В некоторых вариантах осуществления, соединение, представленное в настоящем документе, может быть введено в количестве от примерно 1 мг/кг до примерно 20 мг/кг, от примерно 5 мг/кг до примерно 50 мг/кг, от примерно 10 мг/кг до примерно 40 мг/кг, от примерно 15 мг/кг до примерно 45 мг/кг, от примерно 20 мг/кг до примерно 60 мг/кг или от примерно 40 мг/кг до примерно 70 мг/кг. Например, примерно 5 мг/кг, примерно 10 мг/кг, примерно 15 мг/кг, примерно 20 мг/кг, примерно 25 мг/кг, примерно 30 мг/кг, примерно 35 мг/кг, примерно 40 мг/кг, примерно 45 мг/кг, примерно 50 мг/кг, примерно 55 мг/кг, примерно 60 мг/кг, примерно 65 мг/кг, примерно 70 мг/кг, примерно 75 мг/кг, примерно 80 мг/кг, примерно 85 мг/кг, примерно 90 мг/кг, примерно 95 мг/кг или примерно 100 мг/кг. В некоторых вариантах осуществления, таким введением может быть введение один раз в день (QD) или два раза в день (BID). В некоторых вариантах осуществления, такое введение может происходить по схеме прерывистого дозирования. Например, в одном варианте осуществления, схема дозирования включает введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа один раз в день в течение периода 1, 2, 3, 4, 5 или 6 недель, за которыми следует период в 1, 2, 3, 4, 5 или 6 недель без лечения и повторение цикла, пока субъект лечится указанным соединением. В одном варианте осуществления, схема дозирования включает введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение 4 недель с последующим периодом в две недели без лечения и повторением цикла, пока субъект лечится указанным соединением. В одном варианте осуществления, схема дозирования включает введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение 1 недели с последующим периодом в 1 неделю без лечения и повторением цикла, пока субъект лечится указанным соединением. В одном варианте осуществления, схема дозирования включает введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение 2 недель с последующим периодом 1 или 2 недели без лечения и повторение цикла, пока субъект лечится указанным соединением. В одном варианте осуществления, схема дозирования включает введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение 3 недель с последующим периодом в 1, 2 или 3 недели без лечения, и повторение цикла, пока субъект лечится указанным соединением. В одном варианте осуществления, схема дозирования включает введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение 4 недель с последующим периодом в 1, 2, 3 или 4 недели без лечения, и повторение цикла, пока субъект лечится указанным соединением. В одном варианте осуществления, схема дозирования включает введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение 5 недель с последующим периодом 1, 2, 3, 4 или 5 недель без лечения, и повторение цикла, пока субъект лечится указанным соединением. В одном варианте осуществления, схема дозирования включает введение соединения формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа в течение 6 недель с последующим периодом 1, 2, 3, 4, 5. или 6 недель без лечения, и повторение цикла, пока субъект лечится указанным соединением. В любой из указанных периодических схем дозирования, введение формулы I, формулы II или формулы III или его фармацевтически приемлемой соли, сольвата или полиморфа составляет один раз в сутки (QD).
Специалист в данной области поймет, что испытания как in vivo, так и in vitro с использованием подходящих, известных и общепринятых моделей клеток и/или животных позволяют прогнозировать способность тестируемого соединения лечить или предотвращать данное нарушение.
Специалист в данной области также поймет, что клинические испытания на людях, включая первые испытания на людях, испытания в диапазоне доз и испытания эффективности, на здоровых субъектах и/или лицах, страдающих данным нарушением, могут быть завершены в соответствии со способами, хорошо известными в клинической и медицинской области.
В настоящем документе представлены фармацевтические наборы, полезные, например, при лечении BRAF-ассоциированных заболеваний или нарушений, таких как рак, которые включают один или несколько контейнеров, содержащих фармацевтическую композицию, содержащую терапевтически эффективное количество соединения, представленного в настоящем документе. Такие наборы могут дополнительно включать, если желательно, один или несколько различных традиционных фармацевтических компонентов набора, таких как, например, контейнеры с одним или несколькими фармацевтически приемлемыми носителями, дополнительными контейнерами и т. д., что будет очевидно для специалистов в данной области техники. В набор также могут быть включены инструкции в виде вкладышей или этикеток с указанием количества компонентов для введения, руководства по применению и/или руководства по смешиванию компонентов.
ПРИМЕРЫ
Следующие примеры иллюстрируют изобретение.
Биологические примеры
Пример A
Ферментный анализ BRAF V600E и V600K
Для B-Raf был сконфигурирован конкурентный анализ замещения, который отслеживает количество флуоресцентно-меченого «индикатора», связанного с B-Raf через TR-FRET из антитела, меченного анти-меткой Eu антитела, также связанного с B-Raf. Для полноразмерного FLAG-меченного B-Raf (V600E) аналитические смеси состоят из 25 мМ K+HEPES, pH 7,4, 10 мМ MgCl2, 0,01% Triton X-100, 1 мМ DTT, 2% ДМСО (из соединения), 50 нМ Tracer 1710 (ThermoFisher, PR9176A), 0,5 нМ Eu анти-FLAG (M2)-криптата Ab (Cisbio, 61FG2KLB) и 5 нМ полноразмерного B-Raf FLAG-меченный на N-конце (V600E) (Origene Technologies, TP700031). При анализе B-Raf (V600K) делают следующие замены: 15 нМ Tracer 178 (ThermoFisher, PV5593), 2 нМ Eu-анти-GST Ab (ThermoFisher, PV5595) и 5 нМ GST-меченный на N-конце B-Raf (V600K) (331-конец, SignalChem, B08-12DG). Соединения обычно разводят ДМСО в пределах 11-точечного диапазона дозирования, созданного с использованием протокола 3-кратного серийного разведения при верхней дозе 10 мкМ. Анализ проводят в 384-луночных полистирольных, необработанных, белых микротитровальных планшетах с низким объемом (Costar 4512) в конечном объеме 12 мкл. Лунки с низким контролем включают 1 мкМ эффективного ингибитора B-Raf в качестве контроля. Анализы инкубируют при температуре окружающей среды (обычно 22°C) в течение 60 мин, и затем считывают на микропланшетном ридере PerkinElmer EnVision, используя стандартные настройки TRF (λEx=320 нм, λEm=615 и 665 нм). Соотнесенные числа (665 нм/615 нм) преобразованы в долю от контроля (POC), используя следующее уравнение:
где:
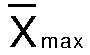 средние неингибированные контроли
средние неингибированные контроли
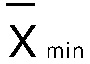 средний фон
средний фон
4-параметрическая логистическая модель соответствует данным POC для каждого соединения. Исходя из этой подгонки, оценивают IC50 и определяют как концентрацию соединения, при которой наиболее подходящая кривая пересекает 50 POC. Усредненные значения IC50 соединений, описанных в настоящем документе, при тестировании в этом анализе, представлены в Таблице A.
(нМ)
(нМ)
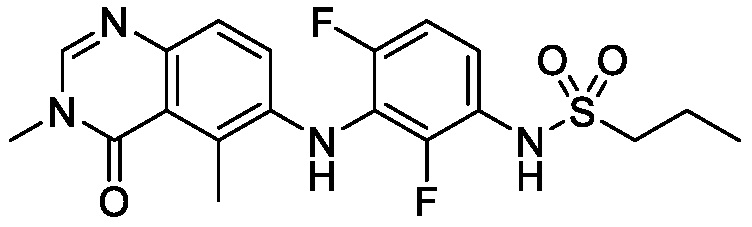
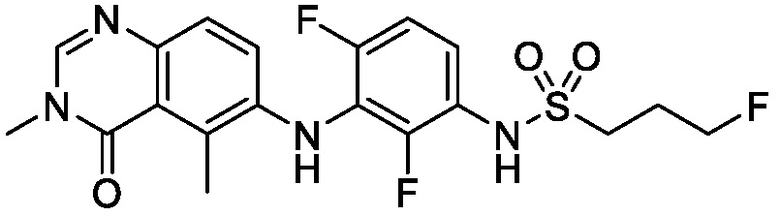
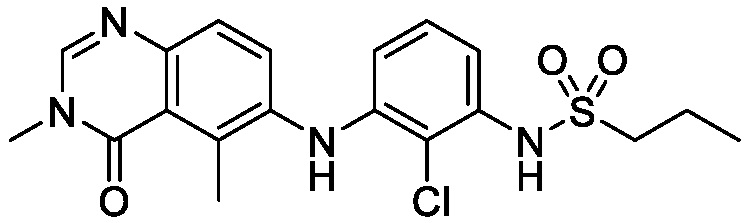
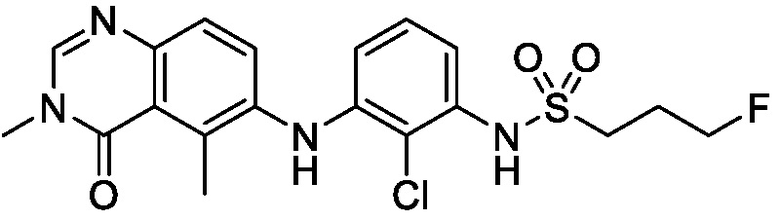
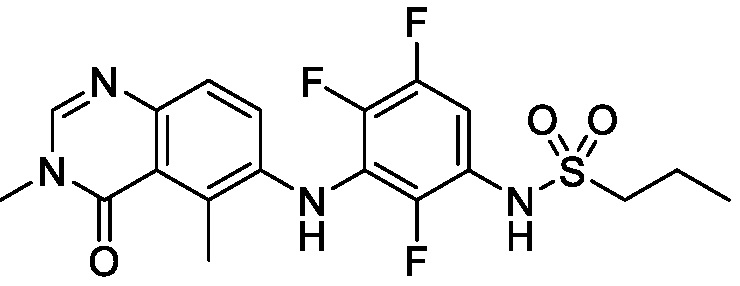
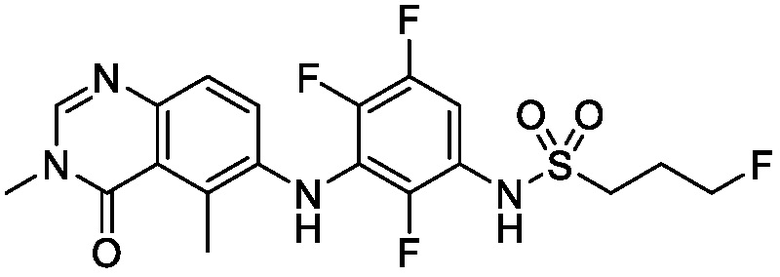
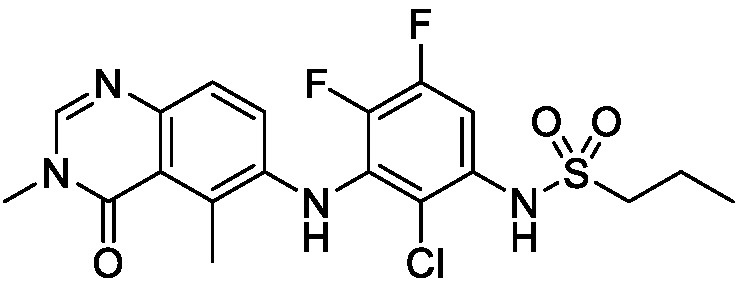
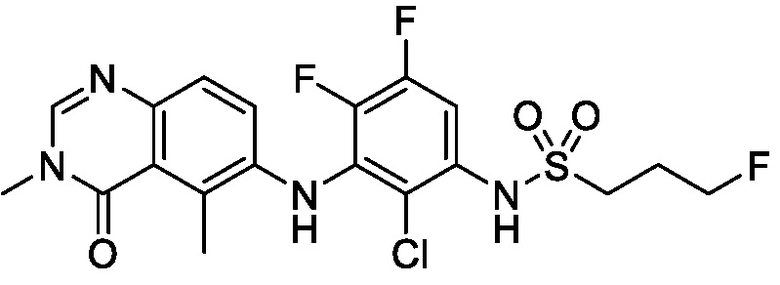
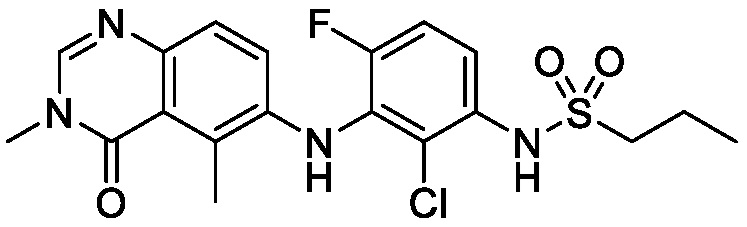
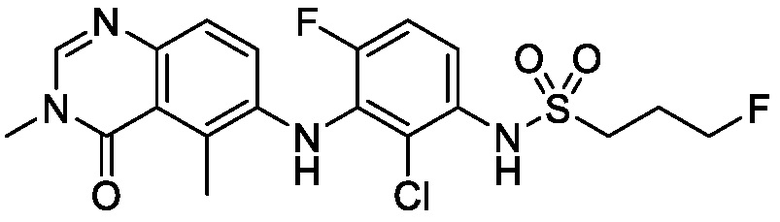
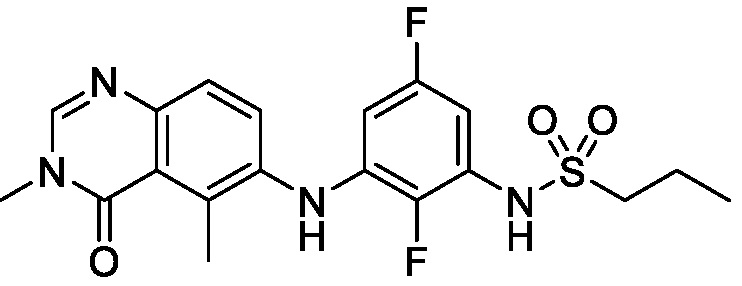
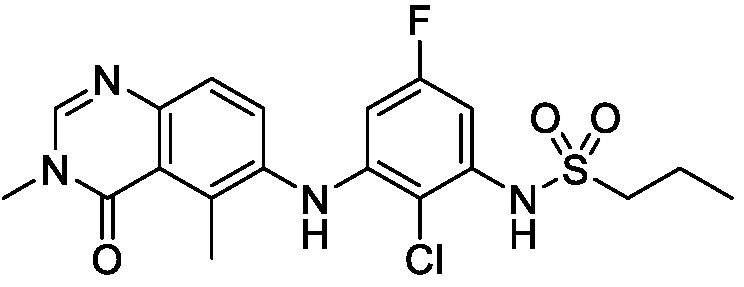
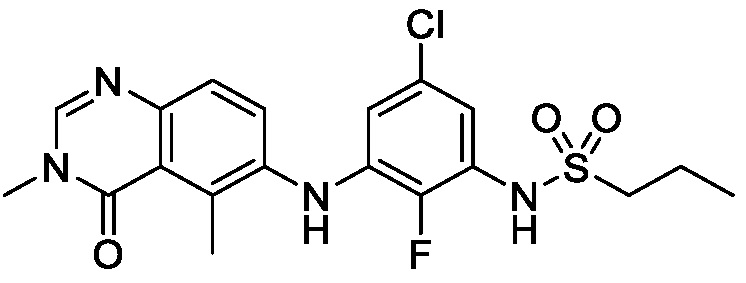

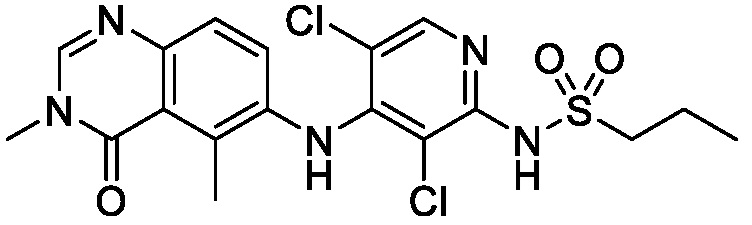
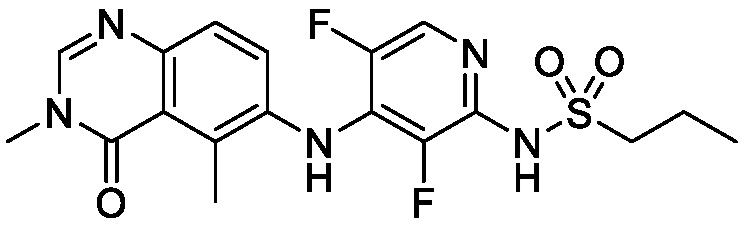
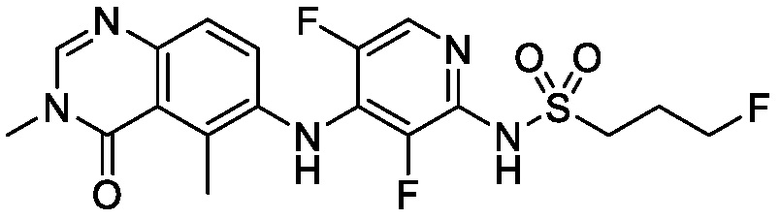
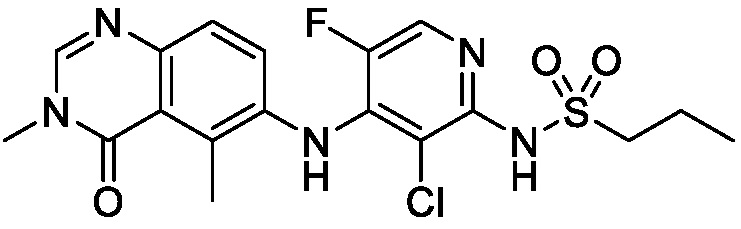
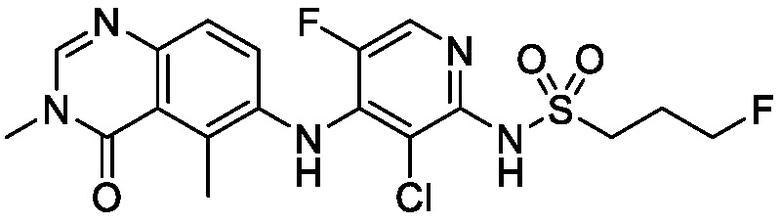
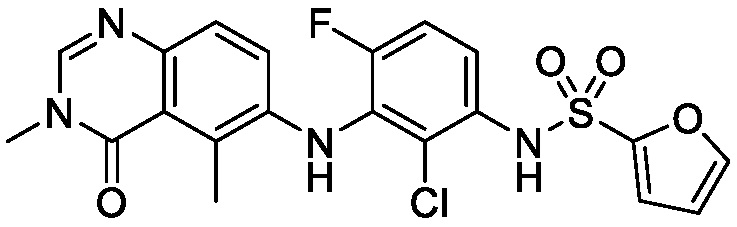
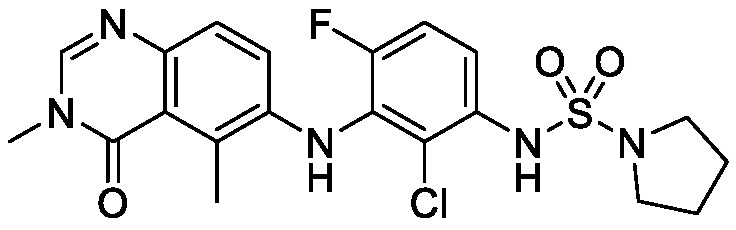
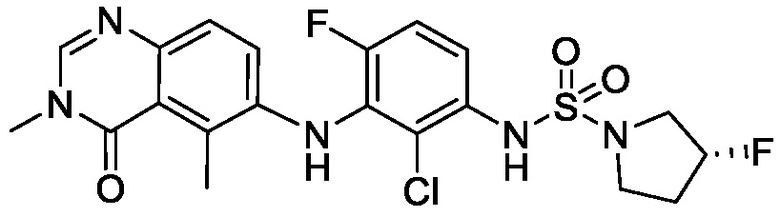
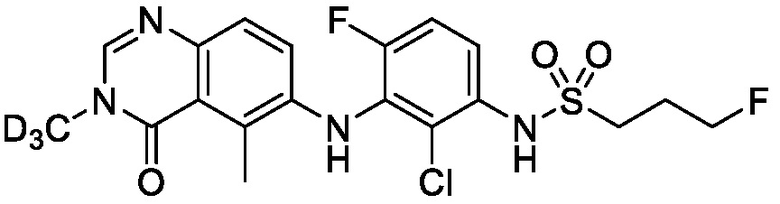

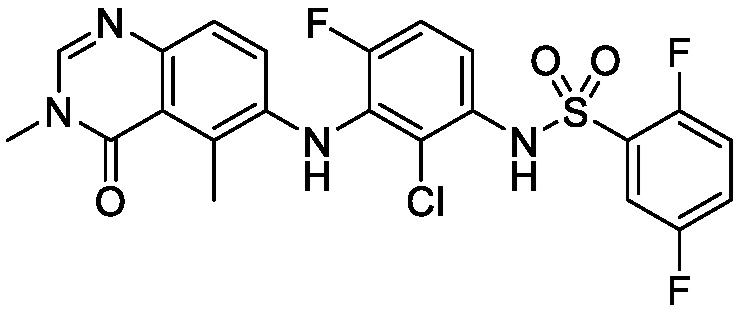
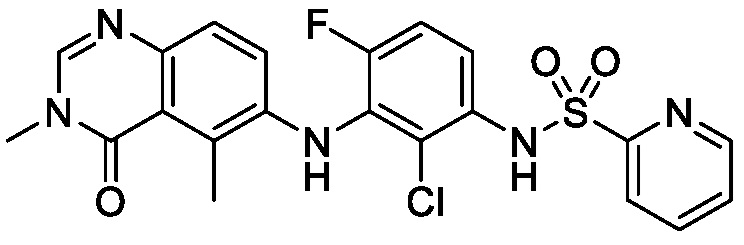

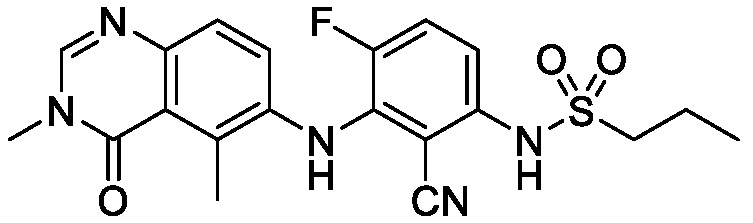
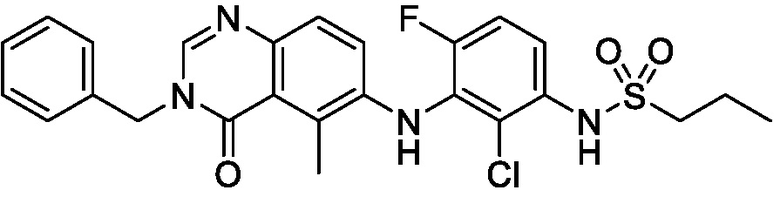
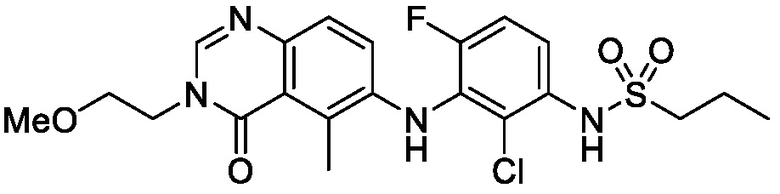

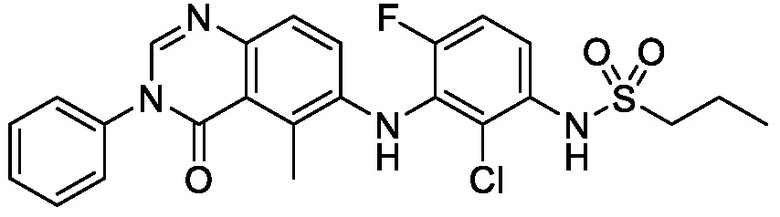
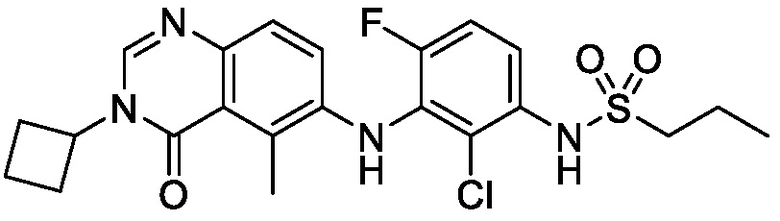
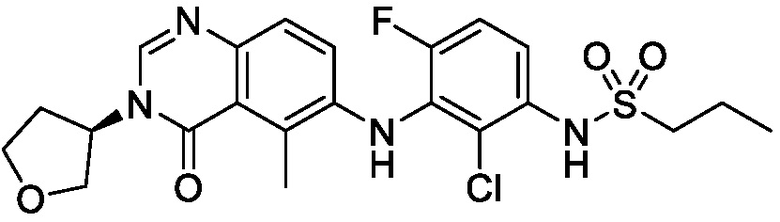
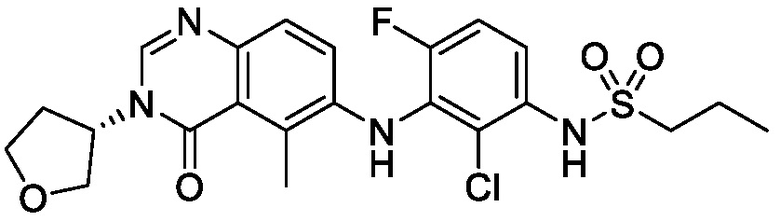
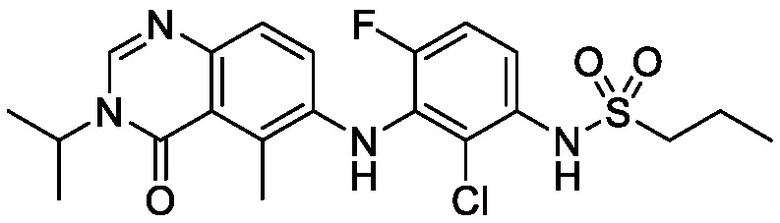
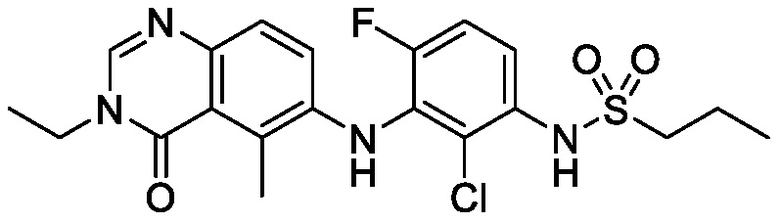
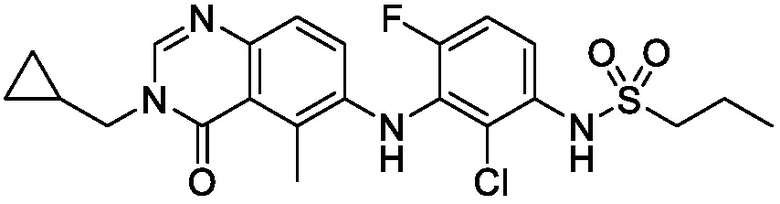
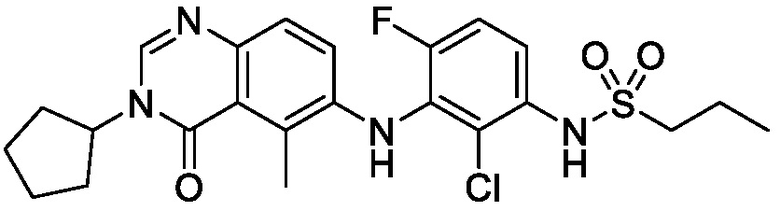

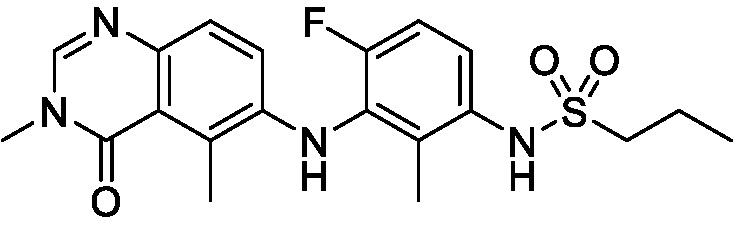
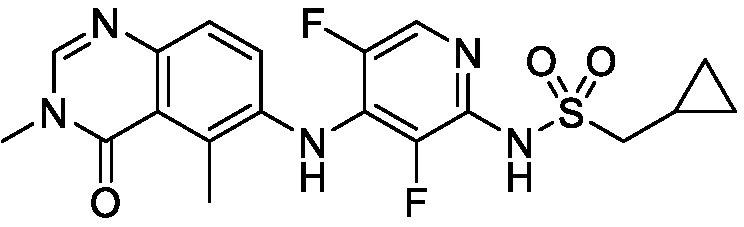
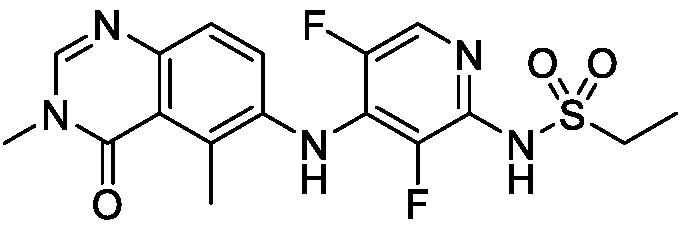
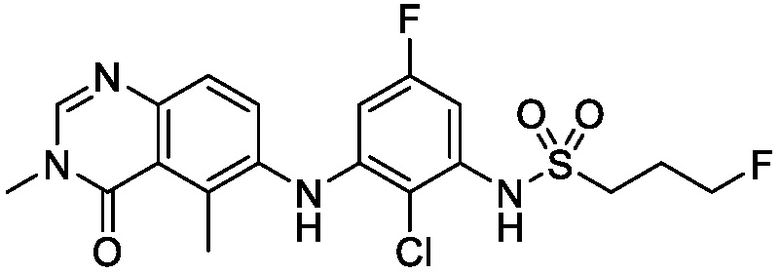
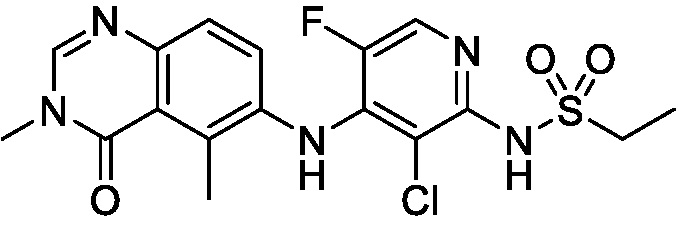
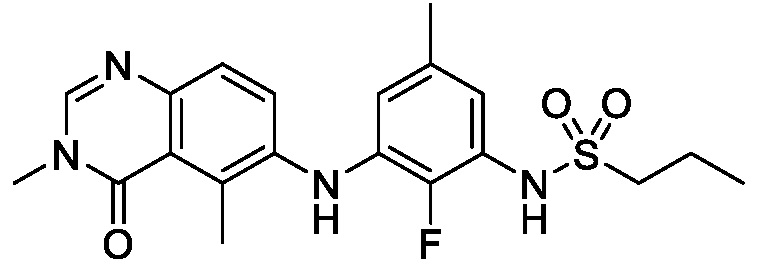
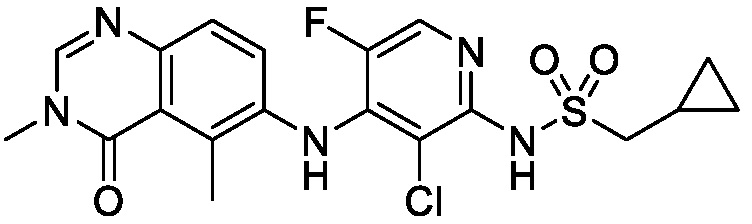
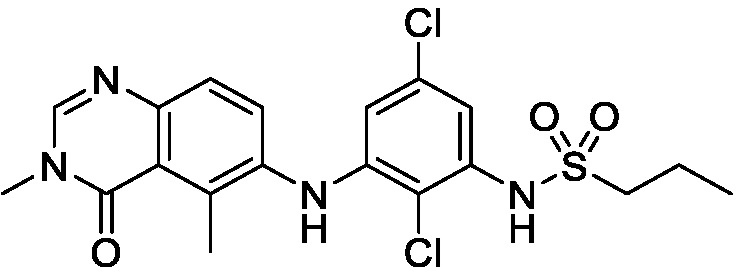
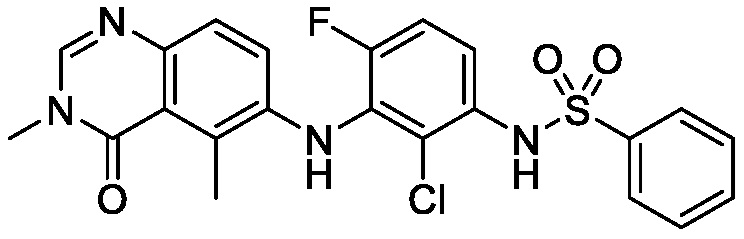
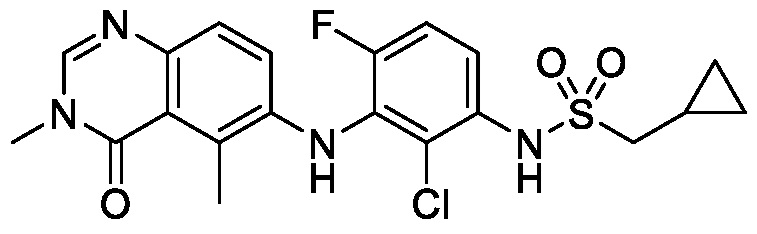
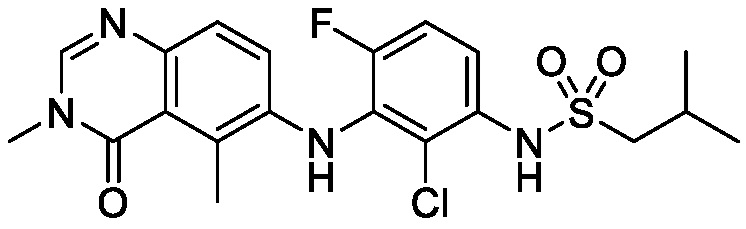
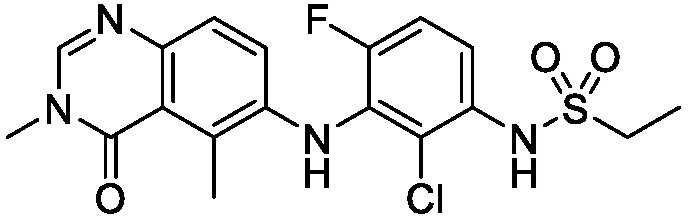
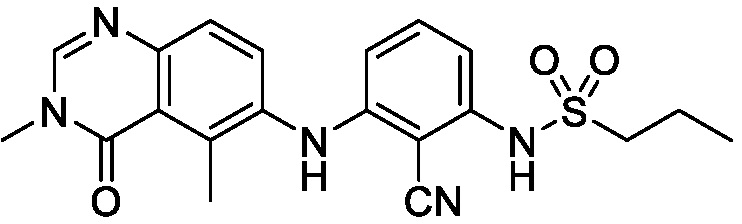
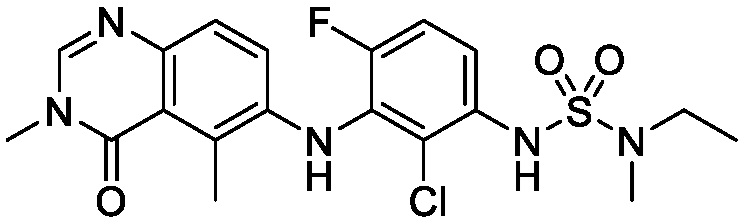
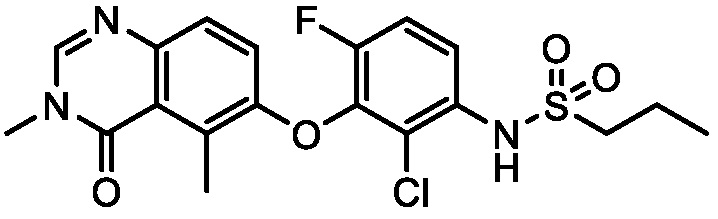
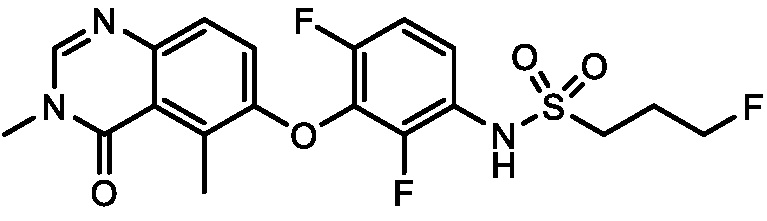
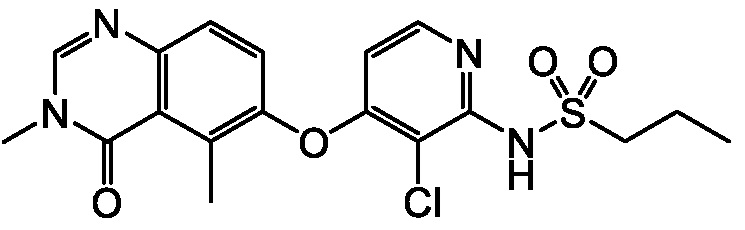
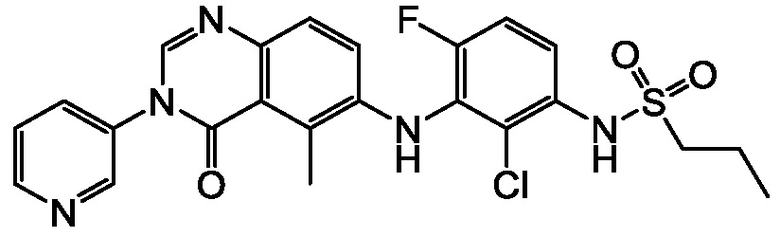
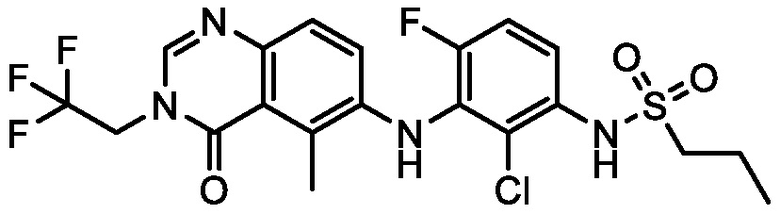
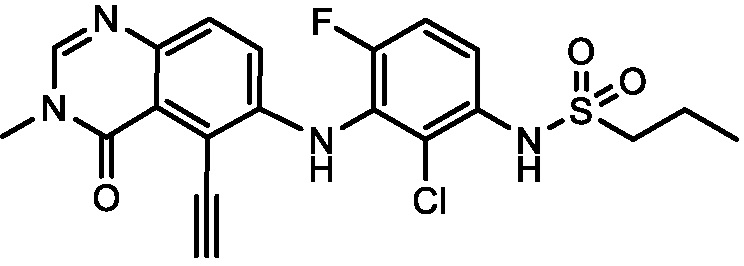
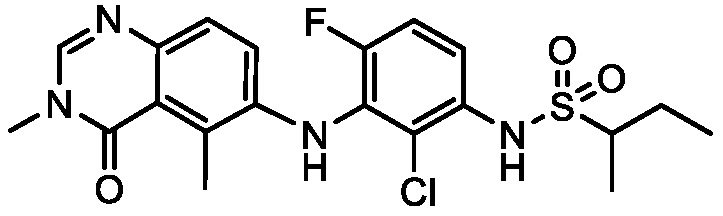
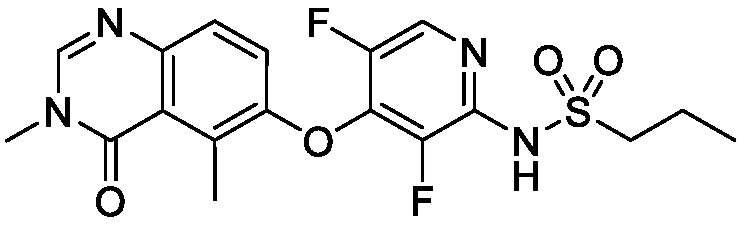
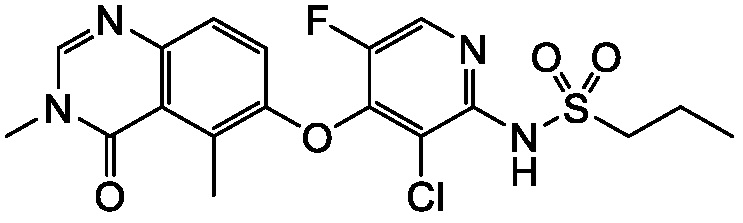
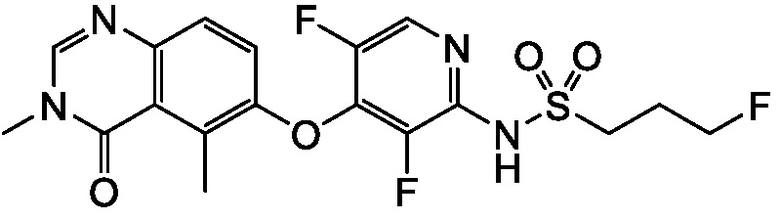

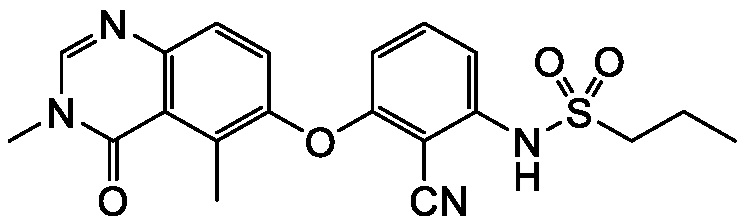
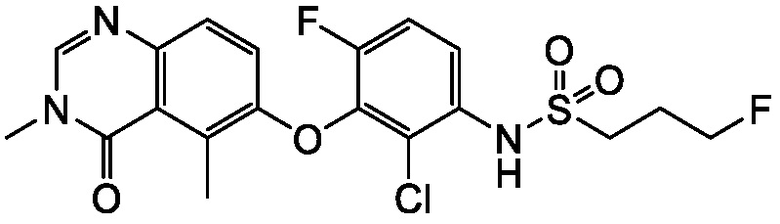
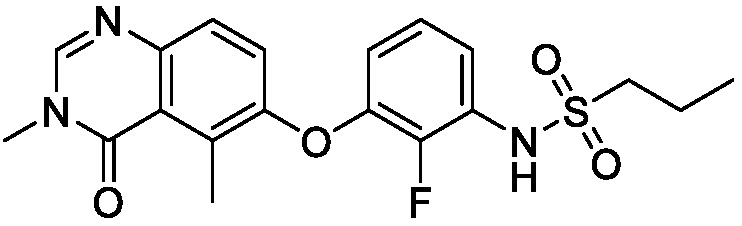
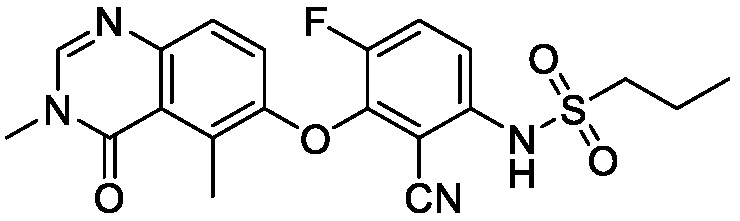
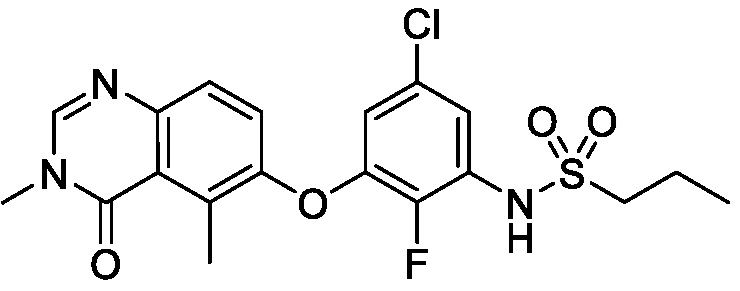
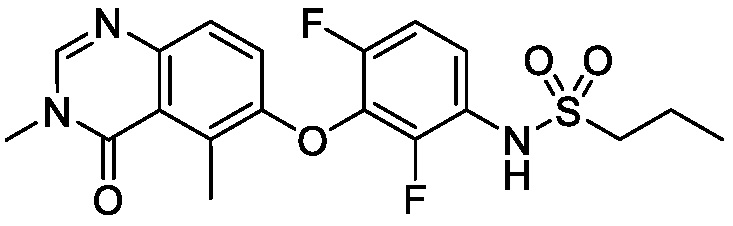
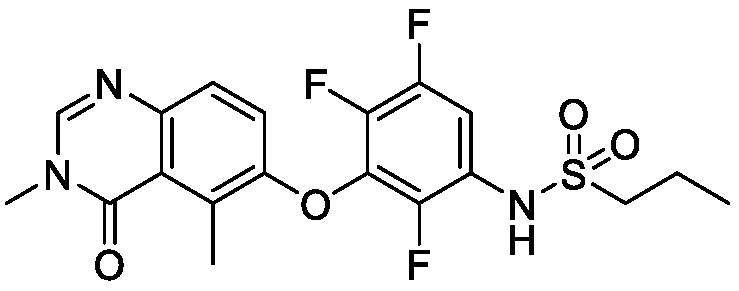
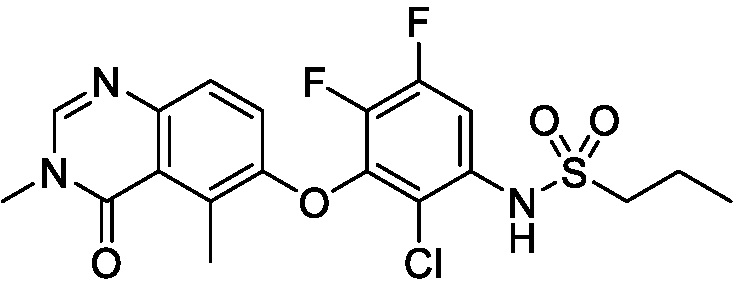
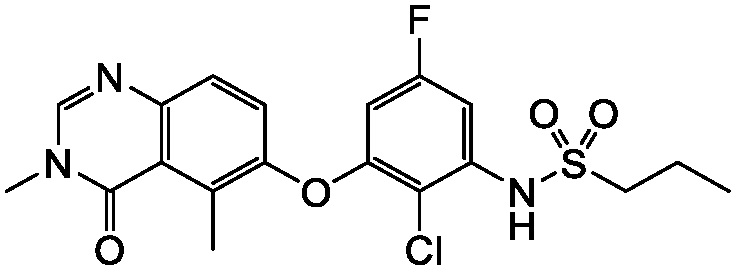
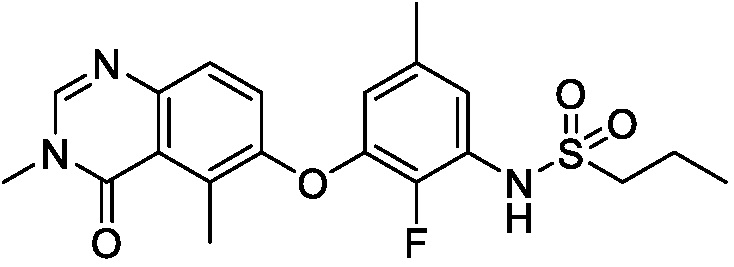
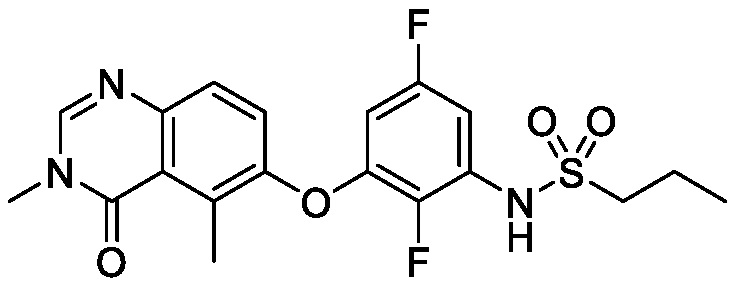
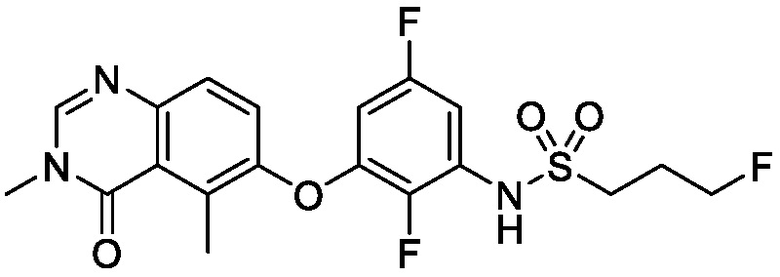
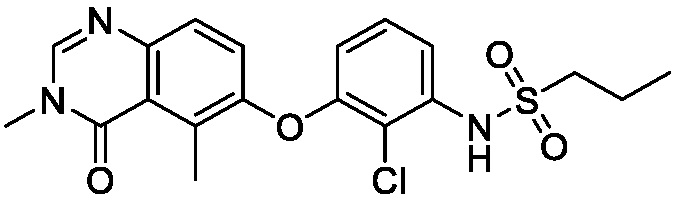
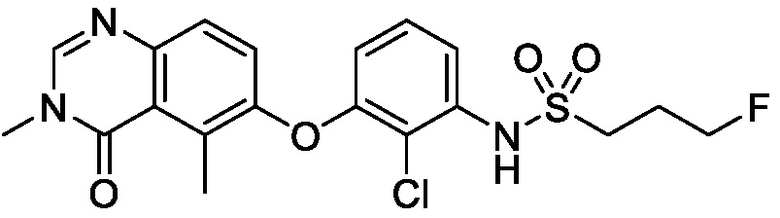
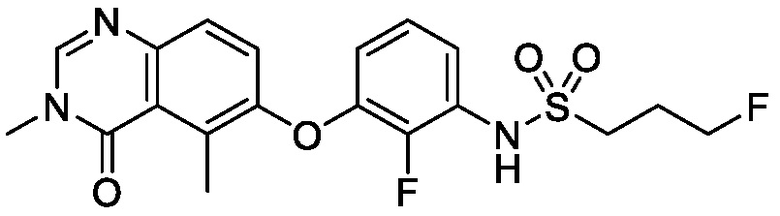
Пример B
Анализ проницаемости MDR1 LLC-PK1 и BCRP MDCKII
Клетки LLC-PK1 и MDR1 трансфицированные LLC-PK1 культивируют и высевают в соответствии с рекомендациями производителя, за исключением того, что среда для пассажа содержит только 2% фетальной телячьей сыворотки, чтобы продлить время пассажа до семи дней.
BCRP трансфицированные клетки MDCKII культивируют и высевают в соответствии с рекомендациями производителя. Условия анализа включены с и без BCRP-специфического ингибитора, KO143, в концентрации 0,3 мкМ, чтобы установить вклад BCRP в величину эффлюкса тестируемого соединения.
Как положительный, так и отрицательный контроль используют для оценки функциональности эффлюкса P-gp или BCRP в анализах. Исходные растворы для анализа контроля и исследуемого препарата готовят в ДМСО до конечных концентраций 10 и 1 мкМ, соответственно. Конечная концентрация органических веществ в анализе составляет 1%. Все дозируемые растворы содержат 10 мкМ люцифера желтого для контроля целостности монослоя клеток LLC PK1 или MDCKII.
Для апикально-базолатерального определения (A - B), 75 мкл тестируемого продукта в транспортном буфере добавляют к апикальной стороне индивидуальных трансвелов, и 250 мкл базолатеральной среды без соединения или люцифера желтого добавляют в каждую лунку. Для базолатерально-апикального определения (B - A), 250 мкл тестируемого продукта в транспортном буфере добавляют в каждую лунку, и 75 мкл транспортного буфера без соединения или люцифера желтого добавляют в каждый трансвел. Все тесты проводят в трех экземплярах, и каждое соединение тестируют для определения и апикально-базолатерального, и базолатерально-апикального транспорта. Планшеты инкубируют в течение 2 часов на орбитальном шейкере Lab-Line Instruments Titer Orbital Shaker (VWR, West Chester, PA) при 50 об/мин и 37°C с 5% CO2. Все культуральные планшеты удаляют из инкубатора, и 50 мкл среды удаляют из апикальной и базолатеральной части каждой лунки и добавляют к 150 мкл 1 мкМ лабеталола в 2:1 ацетонитриле (ацетонитрил): H2O, об./об.
Планшеты считывают с использованием флуориметра Molecular Devices (Sunnyvale, CA) Gemini Fluorometer для оценки концентраций люцифера желтого при длинах волн возбуждения/испускания 425/535 нм. Эти значения принимают, когда они составляют менее 2% для апикально-базолатерального и 5% для базолатерально-апикального потока через монослои клеток LLC-PK1, трансфицированных MDR1, или MDCKII, трансфицированных BCRP. Планшеты герметично закрывают, и содержимое каждой лунки анализируют с помощью ЖХ-МС/МС. Концентрации соединения определяют по отношению площадей пиков соединения к внутреннему стандарту (лабеталолу) по сравнению с дозируемым раствором.
ЖХ-МС анализ
Система ЖХ-МС/МС состоит из автосэмплера HTS-PAL (Leap Technologies, Carrboro, NC), HP1200 HPLC (Agilent, Palo Alto, CA) и системы ловушек MDS Sciex 4000 Q Trap system (Applied Biosystems, Foster City, CA). Хроматографическое разделение аналита и внутреннего стандарта достигается при комнатной температуре с использованием колонки C18 (Kinetics®, 50×300 мм, размер частиц 2,6 мкм, Phenomenex, Torrance, CA) в сочетании с градиентными условиями с использованием подвижных фаз A (вода, содержащая 1% изопропилового спирта и 0,1% муравьиной кислоты) и B (0,1% муравьиной кислоты в ацетонитриле). Общее время прогона, включая повторное уравновешивание, для одного впрыска составляет 1,2 минуты. Масс-спектрометрическое обнаружение аналитов проводят с использованием положительного режима ионного распыления. Ответы аналитов измеряют посредством множественного мониторинга реакций (MRM) переходов, уникальных для каждого соединения (протонированный ион-предшественник и выбранные ионы продукта для каждого исследуемого продукта и m/z 329 до m/z 162 для лабеталола, внутреннего стандарта).
Коэффициент проницаемости (Papp) рассчитывают из следующего уравнения:
Papp=[((Cd*V*(1×106))/(t*0,12cm2*C)]
где Cd, V, t и C0 являются определенной концентрацией (мкМ), объемом на стороне дозирования (мл), временем инкубации (с) и начальной концентрацией дозирования (мкМ), соответственно. Расчеты для Papp проводят для каждого повтора, и затем усредняют. Коэффициенты проницаемости для соединения формулы I представлены в таблице B1. В этом анализе соединение определяется как имеющее высокую проницаемость, если проницаемость превышает 8×10-6 см/сек, соединение определяется как имеющее среднюю проницаемость, если проницаемость составляет от 2×10-6 см/сек до 8×10-6 см/сек, и соединение определяется как имеющее низкую проницаемость, если проницаемость составляет менее 2×10-6 см/сек.
(*10-6 см/с)
Н/Д: не доступно
Коэффициент эффлюкса рассчитывают из средних апикально-базолатеральных (A-B) Papp данных и базолатерально-апикальных (B-A) Papp данных:
Коэффициент эффлюкса=Papp(B-A)/Papp(A-B)
Коэффициенты эффлюкса для типовых соединений описаны в WO 2012/118492, более конкретно, для соединений примеров 34 и 37, при тестировании в этом анализе, показаны в Таблице B2, и коэффициенты эффлюкса для соединения формулы I (включая соединения формулы II и III) при тестировании в этом анализе представлены в Таблице B3. Соединения формулы II (примеры 1, 3, 4, 7, 9, 10, 11, 12, 13, 14, 15, 21, 22, 23, 24, 25, 27, 29, 31, 32, 33, 34, 35, 36, 37, 38, 39, 44, 46, 48, 49, 50, 51, 52, 54, 55, 57, 59, 61, 65, 67, 68, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79 и 80) и формулы III (примеры 1, 4, 7, 9, 10, 23, 55, 63 и 67) демонстрируют тенденцию к пониженным коэффициентам эффлюкса в обоих MDR1 и BCRP анализах по сравнению с типовыми соединениями в WO 2012/118492, что указывает на то, что соединения формул II и III будут иметь повышенную проницаемость по сравнению с типовыми соединениями из WO 2012/118492.
Пример 34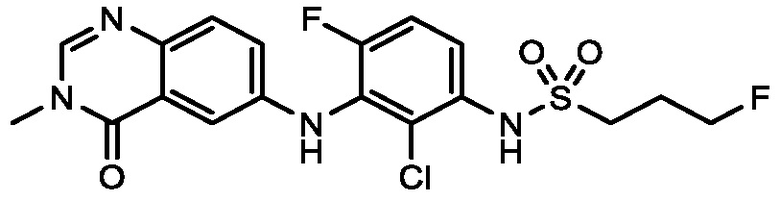
Пример 37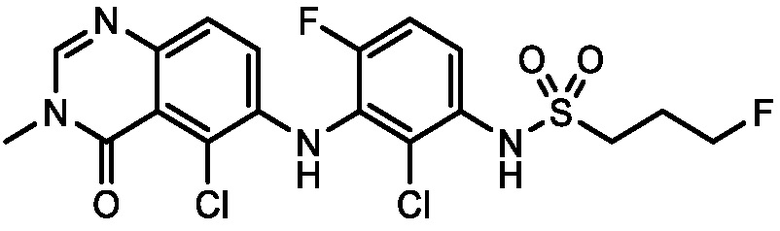
Н/Д: не доступно
Пример C
ФК (соотношение свободного мозга к свободной плазме) (мышь)
Способность типовых соединений проникать через ГЭБ у мышей определяют через оценку соотношения концентраций несвязанного мозга и несвязанной плазмы (также называемого соотношением концентрация свободного мозга к свободной плазме) у самцов мышей CD-1.
Уровни соединений в головном мозге получают из ФК при пероральном дозировании мышам с типовым временем отбора проб через 2, 4, 8, 12 и 24 часа после перорального введения через зонд в дозе 10 мг/кг. Перед анализом образцы мозга хранят при -20±5°C. Концентрации тестируемого соединения в гомогенате мозга мыши определяют методом жидкостной хроматографии и тандемной масс-спектрометрии (ЖХ-МС/МС) после осаждения белка ацетонитрилом. Калибровочную кривую по 12 точкам в диапазоне от 0,5 до 10000 нг/мл получают в двух экземплярах. Раствор 400 мкг/мл тестируемого соединения в диметилсульфоксиде (ДМСО) серийно разводят (3-кратно) в 100% ДМСО, и затем 2,5 мкл каждого стандартного раствора добавляют к 100 мкл гомогената мозга самцов наивных мышей CD-1. Для имитации экстракции на стандартной кривой во все исследуемые образцы добавляют 2,5 мкл ДМСО. В калибровочные и тестовые образцы гомогената головного мозга добавляют 10 мкл IS (1 мкг/мл структурного аналога). Гомогенат головного мозга получают добавлением 0,75 мл 4:1 смеси вода:МеОН к каждому образцу головного мозга с последующей гомогенизацией в течение 1 минуты с помощью шариковых мельниц при 6 м/с использованием MP Fast Prep-24®. Белки осаждают из 100 мкл образца гомогената мозга добавлением 300 мкл ацетонитрила. Образцы перемешивают на вихревой мешалке в течение 5 минут и центрифугируют в центрифуге Allegra X-12R (Beckman Coulter, Fullerton, CA; SX4750A ротор) в течение 15 минут при приблизительно 1500 x g при 4°C. 100 мкл аликвоту каждого супернатанта переносят с помощью 550 мкл Personal Pipettor (Apricot Designs, Monrovia, CA) в 96-луночные планшеты и разбавляют 1:1 водой для ВЭЖХ. Полученные планшеты герметизируют алюминием для анализа ЖХ-МС/МС.
Отношения мозга к плазме рассчитывают с применением концентрации соединения, измеренной в головном мозге, деленной на концентрацию соединения, измеренную в плазме. Соотношения мозга к плазме всегда создают для одного животного и одного момента времени. Соотношение свободного мозга к свободной плазме рассчитывают путем умножения отношения мозга к плазме на фракцию, свободную от гомогената мозга in vitro, деленную на фракцию, свободную от плазмы in vitro, с использованием следующего уравнения: (B/P)*(Bfu/Pfu).
Таблица C1 предоставляет соотношения свободного мозга к свободной плазме соединений примеров 34 и 37, описанных в WO 2012/118492, при испытании в анализе из примера C. Таблица C2 предоставляет соотношения свободного мозга к свободной плазме соединений примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67, описанные в настоящем документе. Данные в Таблице C1 и Таблице C2 демонстрируют неожиданное улучшение соотношений свободного мозга к свободной плазме для соединений примеров 1, 4, 7, 9, 10, 23, 55, 63 и 67.
Пример 34
Пример 37
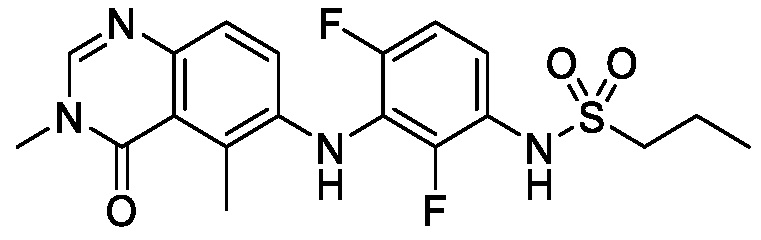
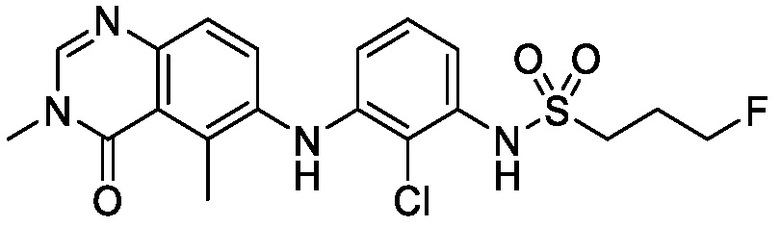
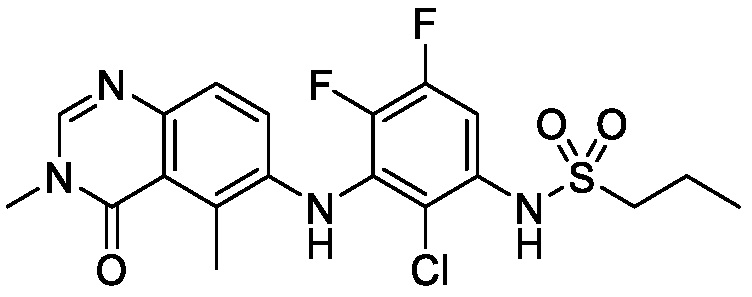

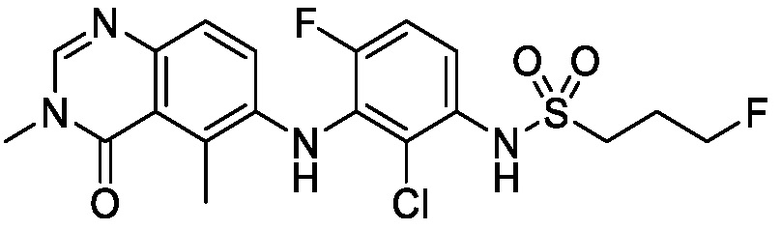
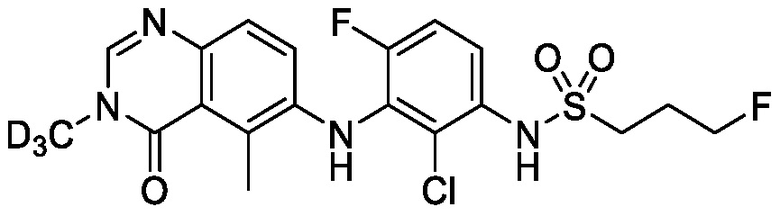
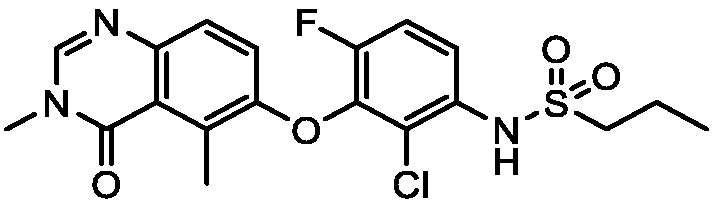
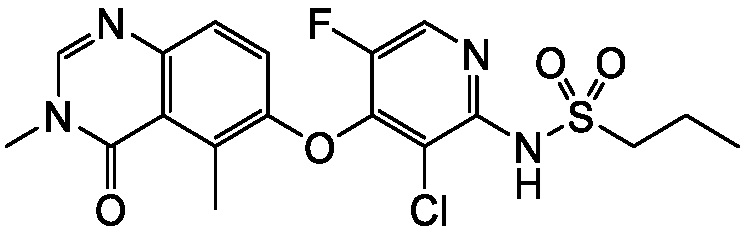
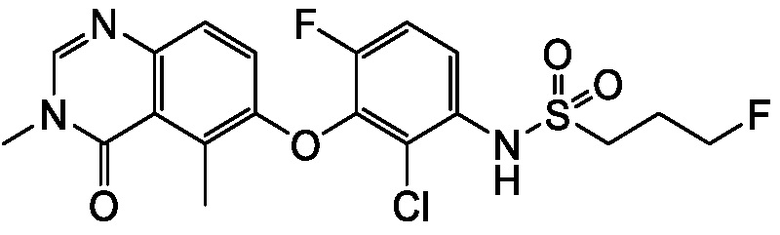
Пример D
Внутричерепная эффективность А375-люциферазы и выживаемость после лечения N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамидом, безводной формой В (Пример 82)
Самок мышей nu/nu NCr от Envigo (Indianapolis, IN) получают в возрасте 5-6 недель и размещают в группах по 5. После минимального периода акклиматизации в течение одной недели мыши получают ортотопические (в.ч.) внутричерепные инъекции 3,0×103 A375-люциферазных опухолевых клеток, суспендированных в объеме 10 мкл 0,9% стерильного фосфатно-солевого буфера Дульбекко (DPBS) с использованием подхода «сверху вниз» (Laursen and Belknap, 1986). Через семь дней после имплантации клеток (день 1) животных произвольно распределяют и сортируют на пять групп (n=10/группу). Мышам, несущим ксенотрансплантаты A375-люциферазной внутричерепной опухоли, вводят тестируемые соединения N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, безводную форму В (пример 82) и ингибитор MEK биниметиниб либо в виде отдельных агентов, либо в комбинации.
Тестируемые соединения дают в объеме дозы 10 мл/кг, и носители подбирают для каждой группы, чтобы контролировать стресс от дозирования, т.е. все группы получают дозу, вводимую дважды в день (BID) через желудочный зонд (PO) в соответствующих объемах доз и по схеме, показанной в таблице 9. Рост опухоли контролируют путем измерения активности люциферазы два раза в неделю. Общий поток на мышь может варьироваться на порядок в день 1 начала исследования (начало дозирования). Общий поток нормализуют для каждой мыши, принимая первое измерение потока на 2 день за 100%. Потерю массы тела как индикатор прогрессирования опухоли определяют ежедневно в течение интервала дозирования, а затем по мере необходимости (по меньшей мере, один раз в неделю). Выживаемость оценивают до 90 дня (30 дней после прекращения приема препарата).
Дозы и схемы, использованные в этом исследовании, хорошо переносятся во всех группах с максимальной потерей веса тела <5% от введения соединения и отсутствием смертей, связанных с введением исследуемого продукта.
Соединение примера 82, вводимое в виде единственного агента, является высокоэффективным в этой модели при обеих используемых дозах. Пример 82, вводимый два раза в день (BID) в течение 60 дней подряд через желудочный зонд (ПО), оказывает значительное влияние на рост опухоли, как показано на Фигуре 15.
Соединение Примера 82 и биниметиниб, вводимые в комбинации, являются значительно более эффективными, чем только соединение Примера 82, как показано на ФИГ. 15. Комбинация значительно увеличивает ингибирование роста опухоли и улучшает выживаемость в мышиной модели метастатической меланомы V600E, как показано на ФИГ. 16 и как суммировано в Таблице 10.
(к-во животных в день 90)
Примеры синтеза
Получение промежуточных соединений для синтеза
Промежуточное соединение P1
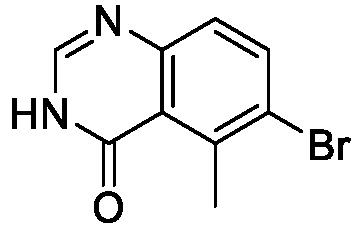
6-бром-5-метилхиназолин - 4(3 H )-он
6-Амино-3-бром-2-метилбензойную кислоту (10 г, 43 ммоль) и ацетат формамидина (5,4 г, 52 ммоль) растворяют в этаноле (172 мл) в 500 мл колбе с конденсатором с обратным холодильником. Реакционную смесь нагревают до 80°C в течение 16 часов. Смесь охлаждают до температуры окружающей среды и концентрируют в вакууме. Остаток разбавляют водой (300 мл) и энергично перемешивают в течение 60 минут. Полученное твердое вещество выделяют фильтрацией и фильтровальную лепешку промывают водой (500 мл). Твердые вещества сушат под вакуумом с получением 6-бром-5-метилхиназолин-4(3H)-она (6,9 г, 66%) в виде белого твердого вещества. МС (хиад, m/z)=239,0, 241,0 (M+H).
Промежуточное соединение P2
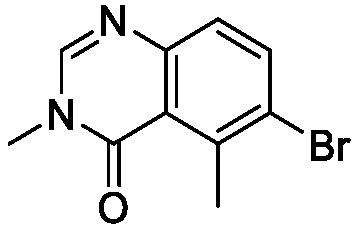
6-бром-3,5-диметилхиназолин - 4(3 H )-он
6-Бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (11 г, 46,0 ммоль), карбонат калия (14,0 г, 101 ммоль) и йодметан (13,1 г, 92,0 ммоль) растворяют в безводном ДМФ (250 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов. Всю реакционную смесь выливают непосредственно в 900 мл воды и полученную суспензию перемешивают при температуре окружающей среды в течение 30 минут. Твердые вещества собирают фильтрацией и сушат в течение ночи в высоком вакууме с получением 6-бром-3,5-диметилхиназолин-4(3H)-она (10,1 г, 87%) в виде белого твердого вещества. МС (хиад, m/z)=253,0, 255,0 (M+H).
Промежуточное соединение P3
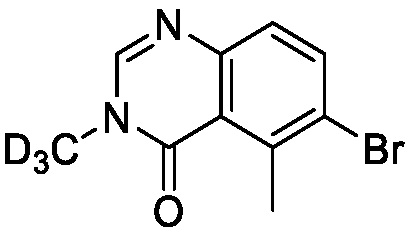
6-бром-5-метил-3-(метил-d3)хиназолин - 4(3H)-он
В 50 мл колбу добавляют 6-бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (3,37 г, 14,1 ммоль), ДМФ (94,0 мл), карбонат калия (4,29 г, 31,0 ммоль) и йодметан-d3 (1,75 мл, 28,2 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь разбавляют водой и экстрагируют EtOAc. Органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают перекристаллизацией из EtOAc с получением 6-бром-5-метил-3-(метил-d3)хиназолин-4(3H)-она (2,94 г, 81%) в виде твердого вещества. МС (хиад, m/z)=256,0, 258,0 (M+H).
Промежуточное соединение P4
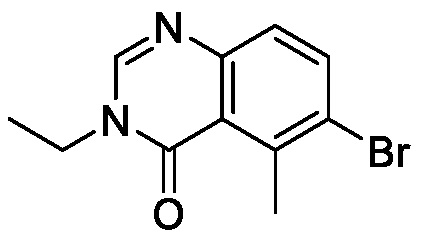
6-бром-3-этил-5-метилхиназолин - 4(3 H )-он
В 10 мл колбу добавляют 6-бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (0,100 г, 0,418 ммоль), йодэтан (0,101 мл, 1,25 ммоль), карбонат цезия (0,204 г, 0,627 ммоль) и ДМФ (4 мл). Колбу герметично закрывают резиновой мембраной и смесь перемешивают в течение ночи при температуре окружающей среды. Смесь разбавляют водой и этилацетатом. Водный слой экстрагируют EtOAc, и объединенные органические экстракты промывают насыщенным раствором соли (2x), сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают колоночной хроматографией, элюируя 30% - 70% EtOAc/гексаном с получением 6-бром-3-этил-5-метилхиназолин-4(3H)-она (0,060 г, 53%) в виде белого твердого вещества. МС (хиад, m/z)=267,0, 269,0 (M+H).
Промежуточное соединение P5
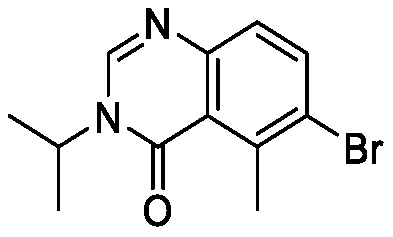
6-бром-3-изопропил-5-метилхиназолин - 4(3 H )-он
В 10 мл колбу добавляют 6-бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (0,100 г, 0,418 ммоль), 2-йодпропан (0,125 мл, 1,25 ммоль), карбонат цезия (0,204 г, 0,627 ммоль) и ДМФ (4 мл). Колбу герметично закрывают резиновой мембраной, и смесь перемешивают в течение ночи при температуре окружающей среды. Смесь разбавляют водой и этилацетатом. Водный слой экстрагируют EtOAc, и объединенные органические экстракты промывают насыщенным раствором соли (2x), сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают колоночной хроматографией, элюируя 30% - 70% EtOAc/гексаном с получением 6-бром-3-изопропил-5-метилхиназолин-4(3H)-она (0,0716 г, 61%) в виде белого твердого вещества. МС (хиад, m/z)=281,0, 283,0 (M+H).
Промежуточное соединение P6
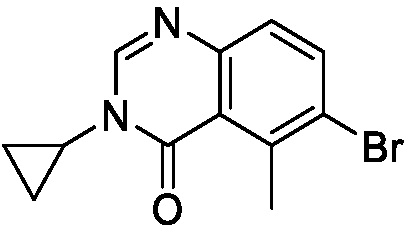
6-бром-3-циклопропил-5-метилхиназолин - 4(3 H )-он
В 10 мл колбу добавляют циклопропилбороновую кислоту (0,036 г, 0,42 ммоль), 6-бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (0,05 г, 0,21 ммоль), карбонат цезия (0,14 г, 0,42 ммоль) и диоксан (4 мл). К суспензии добавляют 1,10-фенантролин (0,04 г, 0,2 ммоль) и затем ацетат меди(II) (0,038 г, 0,21 ммоль). Смесь нагревают на открытом воздухе при 80°C в течение 40 часов и затем охлаждают до комнатной температуры, гасят насыщенным водным NH4Cl, и разбавляют этилацетатом. Водный слой экстрагируют EtOAc (4x) и объединенные органические экстракты сушат над Na2SO4 и концентрируют в вакууме. Неочищенный продукт очищают колоночной хроматографией, элюируя 30% - 70% EtOAc/гексаном с получением 6-бром-3-циклопропил-5-метилхиназолин-4(3H)-она (0,02 г, 34%) в виде белого твердого вещества. МС (хиад, m/z)=279,0, 281,0 (M+H).
Промежуточное соединение P7
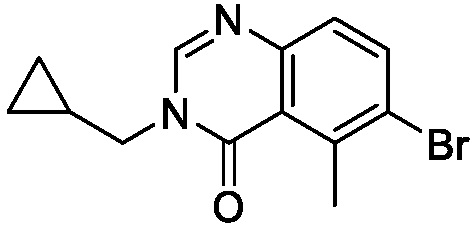
6-бром-3-(циклопропилметил)-5-метилхиназолин - 4(3 H )-он
К раствору 6-бром-5-метилхиназолин-4(3H)-она (Промежуточное соединение P1) (0,100 г, 0,418 ммоль) в ДМФ (1,67 мл) добавляют K2CO3 (0,116 г, 0,837 ммоль), затем (бромметил)циклопропан (0,102 г, 0,753 ммоль). Смесь перемешивают в течение 6 часов при температуре окружающей среды. Смесь разбавляют насыщенным водным NH4Cl и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют до твердого вещества. Неочищенный продукт очищают колоночной хроматографией, элюируя 0-20% EtOAc/CH2Cl2 с получением 6-бром-3-(циклопропилметил)-5-метилхиназолин-4(3H)-она (0,108 г, 88%) в виде белого твердого вещества. МС (хиад, m/z)=293,0, 295,0 (M+H).
Промежуточное соединение P8

6-бром-3-циклобутил-5-метилхиназолин - 4(3 H )-он
В 10 мл колбу добавляют бромциклобутан (0,118 мл, 1,26 ммоль), 6-бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (0,100 г, 0,418 ммоль), карбонат цезия (0,204 г, 0,627 ммоль) и ДМФ (2,1 мл). Сосуд герметично закрывают резиновой мембраной, помещают в атмосферу N2, перемешивают при температуре окружающей среды в течение 17 часов и затем при 70°C в течение 24 часов. Смесь охлаждают до комнатной температуры, разбавляют этилацетатом, и промывают насыщенным раствором соли. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают колоночной хроматографией, элюируя 30% - 70% EtOAc/гексаном с получением 6-бром-3-циклобутил-5-метилхиназолин-4(3H)-она (0,047 г, 38%) в виде белого твердого вещества. МС (хиад, m/z)=293,1, 295,1 (M+H).
Промежуточное соединение P9
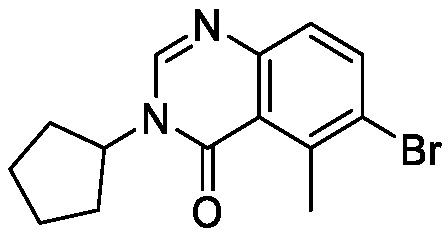
6-бром-3-циклопентил-5-метилхиназолин - 4(3 H )-он
В круглодонную колбу, оборудованную лопастной мешалкой, загружают 6-бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (0,10 г, 0,42 ммоль), сухой ДМА (4 мл), карбонат цезия (0,28 г, 0,84 ммоль) и циклопентил йодид (0,12 г, 0,63 ммоль). Смесь перемешивают при температуре окружающей среды в течение 16 часов. Смесь разбавляют водой и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной хроматографией, элюируя 10% - 75% EtOAc/гексаном с получением 6-бром-3-циклопентил-5-метилхиназолин-4(3H)-она (0,045 г, 35%). МС (хиад, m/z)=307,0, 309,0 (M+H).
Промежуточное соединение P10
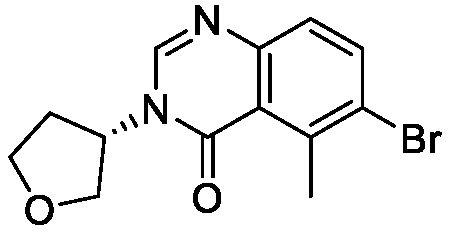
( S )-6-бром-5-метил-3-(тетрагидрофуран - 3-ил)хиназолин - 4(3 H )-он
В круглодонную колбу, оборудованную лопастной мешалкой и входом для азота, загружают 6-бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (0,10 г, 0,418 ммоль), сухой ДМФ (4 мл), (R)-3-йодтетрагидрофуран (0,124 г, 0,627 ммоль) и карбонат цезия (0,273 г, 0,837 ммоль). Эту смесь перемешивают при температуре окружающей среды в течение 12 часов. Через 12 часов, добавляют еще 1,0 экв. (R)-3-йодтетрагидрофурана (0,124 г, 0,627 ммоль) и добавляют 2,0 экв. карбоната цезия (0,273 г, 0,837 ммоль) и смесь нагревают до 70°C в течение еще 24 часов. Смесь разбавляют водой и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной хроматографией, элюируя 5% - 95% EtOAc/гексаном с получением (S)-6-бром-5-метил-3-(тетрагидрофуран-3-ил)хиназолин-4(3H)-она (0,054 г, 41%) в виде белого твердого вещества. МС (хиад, m/z)=309,0, 311,0 (M+H).
Промежуточное соединение P11
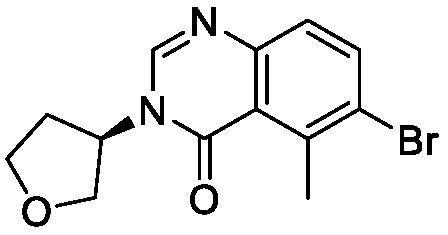
( R )-6-бром-5-метил-3-(тетрагидрофуран - 3-ил)хиназолин - 4(3 H )-он
В круглодонную колбу, оборудованную лопастной мешалкой и входом для азота, загружают 6-бром-5-метилхиназолин-4(3H)-он (Промежуточное соединение P1) (0,10 г, 0,42 ммоль), сухой ДМФ (4 мл), (S)-3-йодтетрагидрофуран (0,12 г, 0,63 ммоль) и карбонат цезия (0,27 г, 0,84 ммоль). Эту смесь перемешивают при комнатной температуре в течение 12 часов в атмосфере азота. Через 12 часов, добавляют еще 1,5 экв. (S)-3-йодтетрагидрофурана (0,12 г, 0,63 ммоль) и 2,0 экв. карбоната цезия (0,27 г, 0,84 ммоль) и смесь нагревают до 60°C в течение еще 24 часов. Смесь разбавляют водой и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной хроматографией, элюируя 5% - 85% EtOAc/гексаном с получением (R)-6-бром-5-метил-3-(тетрагидрофуран-3-ил)хиназолин-4(3H)-она (0,045 г, 35%) в виде белого твердого вещества. МС (хиад, m/z)=309,0, 311,0 (M+H).
Промежуточное соединение P12
6-бром-3-(2-метоксиэтил)-5-метилхиназолин - 4(3 H )-он
К суспензии 6-бром-5-метилхиназолин-4(3H)-она (Промежуточное соединение P1) (0,100 г, 0,4183 ммоль) в ДМФ (1,67 мл) добавляют K2CO3 (0,1214 г, 0,8784 ммоль) затем 2-бромэтилметиловый эфир (0,0786 мл, 0,8366 ммоль). Смесь перемешивают в течение 18 часов при температуре окружающей среды. Смесь разбавляют насыщенным водным NH4Cl и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют до твердого вещества. Неочищенный продукт очищают колоночной хроматографией, элюируя 0-30% EtOAc/CH2Cl2 с получением 6-бром-3-(2-метоксиэтил)-5-метилхиназолин-4(3H)-она (0,105 г, 85%) в виде белого твердого вещества. МС (хиад, m/z)=297,0, 299,0 (M+H).
Промежуточное соединение P13
6-бром-5-метил-3-фенилхиназолин - 4(3 H )-он
В пробирку под давлением, оборудованную лопастной мешалкой, загружают 6-бром-5-метилхиназолин-4(3H)-оном (Промежуточное соединение P1) (0,050 г, 0,21 ммоль), фенилбороновую кислоту (0,033 г, 0,27 ммоль), ДХМ (2 мл), пиридин (0,036 г, 0,46 ммоль) и ацетат меди (II) (0,084 г, 0,46 ммоль). Пробирку накрывают резиновой мембраной, которую протыкают иглой, чтобы обеспечить приток воздуха, и смесь перемешивают при температуре окружающей среды в течение 48 часов. Смесь разбавляют ДХМ и фильтруют через GF/F фильтровальную бумагу, и фильтрат концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной хроматографией, элюируя 90% EtOAc/гексаном с получением 6-бром-5-метил-3-фенилхиназолин-4(3H)-она (0,020 г, 30%) в виде белого твердого вещества. МС (хиад, m/z)=315,0, 317,0 (M+H).
Промежуточное соединение P14
3-бензил-6-бром-5-метилхиназолин - 4(3 H )-он
К раствору 6-бром-5-метилхиназолин-4(3H)-она (Промежуточное соединение P1) (100 мг, 0,4183 ммоль) в ДМФ (1,673 мл) добавляют карбонат калия (121,4 мг, 0,8784 ммоль) и бензилбромид (74,63 мкл, 0,6274 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 18 часов. Смесь разбавляют a насыщенным водным раствором NH4Cl и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле, элюируя 0-30% CH2Cl2/EtOAc с получением 3-бензил-6-бром-5-метилхиназолин-4(3H)-она (110 мг, 79%) в виде белого твердого вещества. МС (хиад, m/z)=329,0, 331,0 (M+H).
Промежуточное соединение P15
6-амино-3,5-диметилхиназолин - 4(3 H )-он
Стадия 1: Получение 6-((4-метоксибензил)амино)-3,5-диметилхиназолин-4(3H)-она. Раствор (4-метоксифенил)метанамина (1,20 мл, 9,18 ммоль), 6-бром-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P2) (2,02 г, 7,98 ммоль), Pd2(dba)3 (0,365 г, 0,399 ммоль), Xantphos (0,693 г, 1,20 ммоль), и Cs2CO3 (7,80 г, 23,9 ммоль) в толуоле (53,2 мл) помещают в пробирку под давлением и продувают аргоном в течение 10 минут. Реакционный сосуд герметично закрывают и нагревают до 90°C в течение 60 часов. Добавляют дополнительный Pd2(dba)3 (0,365 г, 0,399 ммоль) и Xantphos (0,693 г, 1,20 ммоль) и раствор снова продувают аргоном в течение 10 минут, герметично закрывают и нагревают до 90°C в течение еще 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют, концентрируют, и очищают колоночной хроматографией, элюируя 5-95% EtOAc/ДХМ с получением 6-((4-метоксибензил)амино)-3,5-диметилхиназолин-4(3H)-она (2,4 г, 97%). МС (хиад, m/z)=310,2 (M+H).
Стадия 2: Получение 6-амино-3,5-диметилхиназолин-4(3H)-она. Раствор 6-((4-метоксибензил)амино)-3,5-диметилхиназолин-4(3H)-она (2,4 г, 7,76 ммоль) перемешивают в 50 мл ДХМ и 25 мл ТФК в течение 2 часов. Раствор концентрируют, и остаток растворяют в 100 мл ДХМ, 10 мл MeOH, и энергично перемешивают с 4 г K2CO3 в течение 30 минут. K2CO3 удаляют фильтрацией, и фильтрат концентрируют и остаток очищают колоночной хроматографией, элюируя 1-10% MeOH/ДХМ (1% NH4OH) с получением 6-амино-3,5-диметилхиназолин-4(3H)-она (1,45 г, 99%). 1H ЯМР (400 MГц, CDCl3) δ 8,2 (с, 1H), 7,5 (д, 1H), 7,1 (д, 1H), 4,2 (шс, 2H), 3,6 (с, 3H), 2,8 (с, 3H); МС (хиад, m/z)=190,1 (M+H).
Промежуточное соединение P16
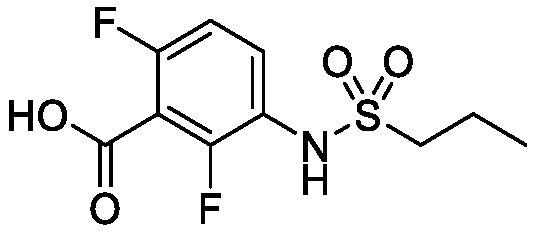
2,6-Дифтор-3-(пропилсульфонамидо)бензойная кислота
Стадия 1: Получение метил 2,6-дифтор-3-нитробензоата. В 1 л колбу загружают 2,6-дифтор-3-нитробензойную кислоту (17,0 г, 83,7 ммоль) и MeOH (170 мл). Колбу помещают на баню с ледяной водой, и капельную воронку, загруженную раствором триметилсилила (TMS) диазометана в гексане (2M, 209 мл, 419 ммоль) присоединяют к колбе. Раствор TMS диазометана медленно добавляют в реакционную колбу в течение 2 часов. Большой избыток реагента требуется для того, чтобы реакция завершилась, что определяют прекращением выделения N2 при дальнейшей добавлении реагента. Раствор концентрируют в вакууме с получением неочищенного метил 2,6-дифтор-3-нитробензоата в виде твердого вещества (18,2 г). Продукт используют без дальнейшей очистки.
Стадия 2: Получение метил 3-амино-2,6-дифторбензоата. В 1 л колбу с загруженным метил 2,6-дифтор-3-нитробензоатом (18,2 г, 83,8 ммоль) добавляют 10% масс. Pd на активированном угле (4,46 г, 4,19 ммоль) в атмосфере азота. В колбу добавляют EtOH (350 мл), и водород пропускают через смесь в течение 15 минут. Реакционную смесь перемешивают под двумя баллонами с водородом в течение ночи. Баллоны повторно загружают H2 (г) и смесь перемешивают дополнительные 4 часа. После потребления исходного материала и промежуточного соединения гидроксиламина по данным ТСХ, азот пропускают через реакционную смесь. Смесь фильтруют через стекловолоконную фильтровальную (GF/F) бумагу дважды. Раствор концентрируют с получением неочищенного метил 3-амино-2,6-дифторбензоата в виде масла (15,66 г). Продукт используют без дальнейшей очистки.
Стадия 3: Получение метил 2,6-дифтор-3-(N-(пропилсульфонил)пропилсульфонамидо)бензоата. Пропан-1-сульфонилхлорид (23,46 мл, 209,3 ммоль) медленно добавляют к охлажденному раствору метил 3-амино-2,6-дифторбензоата (15,66 г, 83,7 ммоль) и триэтиламина (35,00 мл, 251,1 ммоль) в CH2Cl2 (175 мл). Реакционную смесь перемешивают в течение 1 часа при комнатной температуре. Воду (300 мл) добавляют, и органический слой отделяют, промывают водой (2×300 мл) и насыщенным раствором соли (200 мл), затем сушат (Na2SO4), фильтруют и концентрируют до масла. Неочищенный продукт очищают колоночной хроматографией, элюируя 15% EtOAc/гексаном с получением метил 2,6-дифтор-3-(N-(пропилсульфонил)пропилсульфонамидо)бензоата в виде твердого вещества (24,4 г, 73% в течение 3 стадий). 1H ЯМР (400 MГц, CDCl3) δ 7,52-7,45 (м, 1H), 7,08-7,02 (м, 1H), 3,97 (с, 3H), 3,68-3,59 (м, 2H), 3,53-3,45 (м, 2H), 2,02-1,89 (м, 4H), 1,10 (т, J=7,4 Гц, 6H).
Стадия 4: Получение 2,6-дифтор-3-(пропилсульфонамидо)бензойную кислоту. A 1N водный раствор NaOH (150 мл, 150 ммоль) добавляют к раствору метил 2,6-дифтор-3-(N-(пропилсульфонил)пропилсульфонамидо)бензоата (20,0 г, 50,1 ммоль) в 4:1 ТГФ/MeOH (250 мл). Реакционную смесь перемешивают при комнатной температуре в течение 12 часов. Органические растворители концентрируют в вакууме до половины объема (температура водяной бани 35°C). 1N HCl (150 мл) медленно добавляют к смеси, и полученное твердое вещество фильтруют и промывают водой (4×50 мл). Продукт промывают Et2O (4×15 мл) с получением 2,6-дифтор-3-(пропилсульфонамидо)бензойной кислоты (10,7 г, 77%) в виде твердого вещества. 1H ЯМР (400 MГц, (CD3)2SO) δ 9,74 (с, 1H), 7,57-7,50 (м, 1H), 7,23-7,17 (м, 1H), 3,11-3,06 (м, 2H), 1,79-1,69 (м, 2H), 0,98 (т, J=7,4 Гц, 3H); МС (хиад, m/z)=278,0 (M-H).
Промежуточное соединение P17
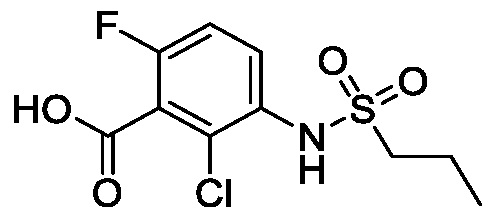
2-Хлор-6-фтор-3-(пропилсульфонамидо)бензойная кислота
Стадия 1: Получение 1-(2-хлор-4-фторфенил)-2,5-диметил-1H-пиррола. В 20 л 4-горлую круглодонную колбу добавляют раствор 2-хлор-4-фторбензоламина (1300 г, 8,82 моль) в толуоле (10 л), 4-метилбензолсульфоновую кислоту (3,1 г, 17,84 ммоль), и гексан-2,5-дион (1222,5 г, 10,62 моль). Полученный раствор нагревают до кипения с обратным холодильником в течение 1 часа, затем охлаждают до температуры окружающей среды. pH раствора доводят до примерно 8 водным карбонатом натрия (1M). Полученную смесь промывают водой (5000 мл) и концентрируют в вакууме. Неочищенный продукт очищают дистилляцией при 140°C с получением 1-(2-хлор-4-фторфенил)-2,5-диметил-1H-пиррола (1700 г, 85%) в виде твердого вещества.
Стадия 2: Получение метил 2-хлор-3-(2,5-диметил-1H-пиррол-1-ил)-6-фторбензоата. В 5000 мл 4-горлую круглодонную колбу, продутую и выдерживаемую в инертной атмосфере азота, помещают раствор 1-(2-хлор-4-фторфенил)-2,5-диметил-1H-пиррола (390 г, 1,65 моль) в тетрагидрофуране (2000 мл). Реакционный раствор охлаждают до -78°C. Раствор n-BuLi (2,5M в гексане, 728 мл, 1,82 моль) добавляют по каплям при перемешивании в реакционный сосуд в течение 80 минут, и метилхлорформиат (215,5 г, 2,27 моль) добавляют по каплям при перемешивании в течение 90 минут. Реакционный раствор затем перемешивают в течение 60 минут при -78°C и гасят добавлением NH4Cl/воды (1000 мл). Полученный раствор экстрагируют этилацетатом (1500 мл). Органические слои объединяют, промывают водой (1500 мл) и водным хлоридом натрия (1500 мл), сушат над безводным MgSO4, и концентрируют в вакууме с получением метил 2-хлор-3-(2,5-диметил-1H-пиррол-1-ил)-6-фторбензоата в виде масла (неочищенное, 566,7 г), которое применяют без дальнейшей очистки.
Стадия 3: Получение метил 3-амино-2-хлор-6-фторбензоата. Раствор метил 2-хлор-3-(2,5-диметил-1H-пиррол-1-ил)-6-фторбензоата (1500 г, 5,05 моль) в EtOH/H2O (7500/2500 мл), NH2OH·HCl (5520 г, 79,20 моль) и триэтиламина (2140 г, 20,98 моль) помещают в 25 л 4-горлые круглодонные колбы. Полученный раствор нагревают до кипения с обратным холодильником в течение 18 часов на масляной бане, охлаждают до комнатной температуры, концентрируют, и экстрагируют этилацетатом (3×3000 мл). Органические экстракты объединяют, сушат над безводным Na2SO4, и концентрируют в вакууме. Остаток очищают с применением колонке с силикагелем, элюируя Et2O/EtOAc (20:1-10:1) с получением метил 3-амино-2-хлор-6-фторбензоата в виде масла (980 г, 95%).
Стадия 4: Получение метил 2-хлор-6-фтор-3-(пропилсульфонамидо)бензоата. Раствор метил 3-амино-2-хлор-6-фторбензоата (980 г, 4,76 моль) в дихлорметане (8000 мл) помещают в 20 л 4-горлую круглодонную колбу. Триэтиламин (1454 г, 14,25 моль) добавляют по каплям в течение 80 минут в этот реакционный сосуд при перемешивании при 0°C, затем добавляют пропан-1-сульфонилхлорид (1725 г, 11,94 моль). Полученный раствор перемешивают при комнатной температуре в течение 2 часов, затем разбавляют водой (1000 мл). Органический слой промывают гидрохлоридом (1000 мл) и водой (1000 мл), сушат над Na2SO4, и концентрируют с получением метил 2-хлор-6-фтор-3-(пропилсульфонамидо)бензоата в виде твердого вещества (1500 г, 97%).
Стадия 5: Получение 2-хлор-6-фтор-3-(пропилсульфонамидо)бензойной кислоты. Раствор метил 2-хлор-6-фтор-3-(пропилсульфонамидо)бензоата (1500 г, 4,61 моль) в ТГФ/H2O (3000 мл/3000 мл) и гидроксида калия (1000 г, 17,68 моль) помещают в 10 л 4-горлую круглодонную колбу. Полученный раствор кипятят с обратным холодильником в течение 2 часов, охлаждают до комнатной температуры и экстрагируют этилацетатом (3×2000 мл). Водные слои объединяют, и pH доводят до 2 гидрохлоридом (2M). Полученный раствор экстрагируют дихлорметаном (2×3000 мл). Органические слои объединяют, сушат над безводным сульфатом натрия и концентрируют с получением 2-хлор-6-фтор-3-(пропилсульфонамидо)бензойной кислоты в виде твердого вещества (517,5 г, 37%). 1H ЯМР (400 MГц, CDCl3): δ 7,86-7,83 (д, 1H, J=14,4Гц), 7,20-7,16 (д, 1H, J=17,2 Гц), 6,81 (с, 1H), 3,11-3,07 (м, 2H, J=15,6 Гц), 1,93-1,86 (м, 2H, J=30,8 Гц), 1,1,0-1,06 (м, 3H, J=15,2Гц); МС (хиад, m/z)=296,1 (M+H).
Промежуточное соединение P18
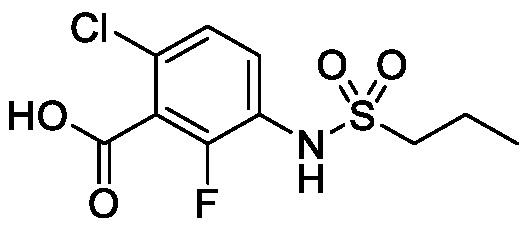
6-Хлор-2-фтор-3-(пропилсульфонамидо)бензойная кислота
Стадия 1: Получение бензил 3-амино-6-хлор-2-фторбензоата. В высушенную пламенем круглодонную колбу, оборудованную лопастной мешалкой и резиновой мембраной, загружают 4-хлор-2-фторанилин (5,00 г, 34,35 ммоль) и безводный ТГФ (170 мл). Этот раствор охлаждают до -78°C, и n-BuLi (2,5M в гексане, 14,7 мл, 36,75 ммоль) добавляют в течение 15 минут. Эту смесь перемешивают при -78°C в течение 20 минут, и затем ТГФ раствор (25 мл) 1,2-бис(хлордиметилсилил)этана (7,76 г, 36,1 ммоль) добавляют в течение 10 минут к реакционной смеси. Реакционную смесь перемешивают при -78°C в течение 1 часа, и затем медленно добавляют n-BuLi (2,5 M в гексане, 15,11 мл, 37,79 ммоль). Смесь нагревают до комнатной температуры в течение одного часа, затем снова охлаждают до -78°C. Медленно добавляют третью партию n-BuLi (2,5M в гексане, 15,66 мл, 39,16 ммоль), и смесь перемешивают при -78°C в течение 75 минут. Медленно добавляют бензилхлорформиат (7,40 г, 41,22 ммоль), и смесь перемешивают при -78°C в течение одного часа. Охлаждающую баню удаляют, и смесь нагревают до температуры окружающей среды в течение 30 минут и затем гасят водой (70 мл) и концентрируют HCl (25 мл). Смесь продолжают нагревать до комнатной температуры. Смесь экстрагируют EtOAc. Экстракты дважды промывают насыщенным раствором NaHCO3, один раз водой, сушат над сульфатом натрия и концентрируют. Остаток очищают колоночной хроматографией на (30% этилацетате/гексане) с получением бензил 3-амино-6-хлор-2-фторбензоата (4,3 г, 45%) в виде масла. 1H ЯМР ((CD3)2SO, 400 MГц) δ 7,37-7,48 (м, 5H), 7,07 (дд, 1H, J=8, 2), 6,87 (т, 1H, J=8), 5,61 (шс, 2H), 5,40 (с, 2H).
Стадия 2: Получение бензил 6-хлор-2-фтор-3-(N-(пропилсульфонил)пропилсульфонамидо)бензоата. Бензил 3-амино-6-хлор-2-фторбензоат (4,3 г, 15,37 ммоль) растворяют в сухом дихлорметане (270 мл). Триэтиламин (5,36 мл, 38,43 ммоль) добавляют, и смесь охлаждают до 0°C. Пропан-1-сульфонил хлорид (3,63 мл, 32,3 ммоль) добавляют через шприц, и выпадает осадок. После завершения добавления, смесь нагревают до комнатной температуры, и исходный продукт потребляется по данным ТСХ (3:1 гексан:этилацетатом). Смесь разбавляют дихлорметаном (200 мл), промывают 2M водным HCl (2×100 мл), насыщенным водным раствором NaHCO3, сушат над сульфатом натрия и концентрируют. Остаток очищают колоночной хроматографией (40% этилацетат/гексан) с получением бензил 6-хлор-2-фтор-3-(N-(пропилсульфонил)пропилсульфонамидо)бензоата (5,5 г, 72%) в виде масла, которое медленно отверждается при выстаивании. ЯМР (CDCl3, 400 MГц) δ 7,28-7,45 (м, 7H), 5,42 (с, 2H), 3,58-3,66 (м, 4H), 3,43-3,52 (м, 4H), 1,08 (т, 6H, J=8).
Стадия 3: Получение 6-хлор-2-фтор-3-(пропилсульфонамидо)бензойной кислоты. Бензил 6-хлор-2-фтор-3-(N-(пропилсульфонил)пропилсульфонамидо) бензоат (5,4 г, 10,98 ммоль) растворяют в ТГФ (100 мл) и 1M водном KOH (100 мл). Смесь кипятят с обратным холодильником в течение 16 часов и затем охлаждают до комнатной температуры. Смесь подкисляют до pH примерно 2 2M водным HCl и экстрагируют EtOAc (2x). Экстракты промывают водой, сушат над сульфатом натрия и концентрируют до твердого вещества, которое растирают с гексаном/Et2O с получением 6-хлор-2-фтор-3-(пропилсульфонамидо)бензойной кислоты (2,2 г, 68%) в виде твердого вещества. 1H ЯМР ((CD3)2SO, 400 MГц) δ 9,93 (с, 1H), 7,49 (т, 1H, J=8), 7,38 (дд, 1H, J=8, 2), 3,11-3,16 (м, 2H), 1,68-1,78 (м, 2H), 0,97 (т, 3H, J=8).
Промежуточное соединение P19
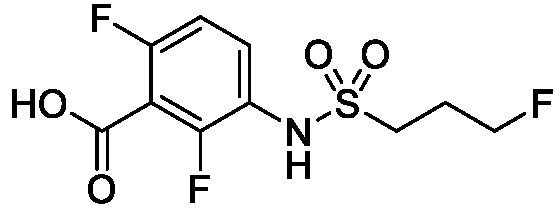
2,6-Дифтор-3-(3-фторпропилсульфонамидо)бензойная кислота
Стадия 1: Получение метил 2,6-дифтор-3-(N-(3-фторпропилсульфонил)-3-фторпропилсульфонамидо)бензоата. Метил 2,6-дифтор-3-(N-(3-фторпропилсульфонил)-3-фторпропилсульфонамидо)бензоат получают по методике, описанной для Промежуточного соединения 16, Стадия 3, заменяя 3-фторпропилсульфонилхлоридом пропан-1-сульфонилхлорид. 1H ЯМР (400 MГц, (CD3)2SO) δ 8,05-7,99 (м, 1H), 7,44 (т, 1H), 4,62 (т, 2H), 4,50 (т, 2H), 3,93 (с, 3H), 3,89-3,74 (м, 4H), 2,26-2,11 (м, 4H).
Стадия 2: Получение 2,6-Дифтор-3-(3-фторпропилсульфонамидо)бензойной кислоты. 2,6-Дифтор-3-(3-фторпропилсульфонамидо)бензойную кислоту получают по методике, описанной для Промежуточного соединения P16, Стадия 4, заменяя метил 2,6-дифтор-3-(N-(3-фторпропилсульфонил)-3-фторпропилсульфонамидо)бензоатом метил 2,6-дифтор-3-(N-(пропилсульфонил)-пропил-сульфонамидо)бензоат. 1H ЯМР (400 MГц, CD3OD) δ 7,64 (м, 1H), 7,07 (м, 1H), 4,58 (м, 1H), 4,47 (м, 1H), 3,22 (м, 2H), 2,26-2,12 (м, 2H); МС (хиад, m/z)=296,1 (M-H).
Промежуточное соединение P20
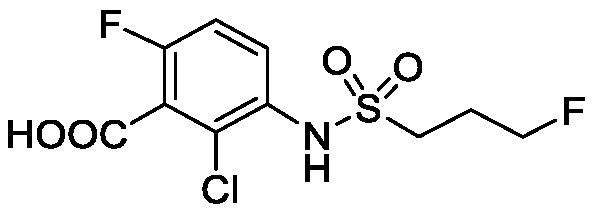
2-Хлор-6-фтор-3-(3-фторпропилсульфонамидо)бензойная кислота
Стадия 1: Получение метил 2-хлор-6-фтор-3-(3-фтор-N-(3-фторпропилсульфонил)пропилсульфонамидо)бензоата. Метил 2-хлор-6-фтор-3-(3-фтор-N-(3-фторпропилсульфонил)пропилсульфонамидо)бензоат получают по методике, описанной для Промежуточного соединения P16, Стадия 3, заменяя метил 3-амино-2-хлор-6-фторбензоатом метил 3-амино-2,6-дифторбензоат и 3-фторпропилсульфонилхлоридом пропан-1-сульфонил хлорид.
Стадия 2: Получение 2-Хлор-6-фтор-3-(3-фторпропилсульфонамидо)бензойной кислоты. 2-Хлор-6-фтор-3-(3-фторпропилсульфонамидо)бензойную кислоту (93% за 2 стадии) получают по методике, описанной для Промежуточного соединения P16, Стадия 4, заменяя метил 2-хлор-6-фтор-3-(3-фтор-N-(3-фторпропилсульфонил)пропилсульфонамидо)бензоатом бензил 6-хлор-2-фтор-3-(N-(пропилсульфонил)пропилсульфонамидо)бензоат. 1H ЯМР (400 MГц, CD3OD) δ 7,63 (м, 1H), 7,19 (м, 1H), 4,56 (м, 1H), 4,44 (м, 1H), 3,21 (м, 2H), 2,25-2,12 (м, 2H); МС (хиад, m/z)=312,1, 314,1 (M-H).
Промежуточное соединение P21
N- (3-Амино-2,4-дифторфенил)пропан-1-сульфонамид
Триэтиламин (4,68 мл, 33,59 ммоль) и дифенилфосфорилазид (3,73 мл, 16,79 ммоль) добавляют к раствору 2,6-дифтор-3-(пропилсульфонамидо)бензойной кислоты (4,078 г, 14,6 ммоль) в ТГФ (60 мл). Реакционную смесь перемешивают при комнатной температуре в течение 3 часов и затем нагревают до 80°C в течение 2 часов. Добавляют воду (10 мл), и смесь перемешивают при 80°C в течение 15 часов. Реакционную смесь разбавляют EtOAc (300 мл), и органический слой промывают насыщенным водным раствором NaHCO3 и насыщенным раствором соли. Растворитель удаляют при пониженном давлении, и остаток очищают колоночной хроматографией на силикагеле элюируя 30/70 EtOAc/гексаном с получением N-(3-амино-2,4-дифторфенил)пропан-1-сульфонамида (2,03 г, 55%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,32 (с, 1H), 6,90-6,80 (м, 1H), 6,51 (тд, J=8,7, 5,5, 1H), 5,28 (с, 2H), 3,05-2,96 (м, 2H), 1,82-1,64 (м, 2H), 1,01-0,90 (м, 3H); МС (хиад, m/z)=251,1 (M+H).
Промежуточное соединение P22
N- (3-Амино-4-хлор-2-фторфенил)пропан-1-сульфонамид
Триэтиламин (1,84 мл, 13,2 ммоль) и дифенилфосфорилазид (1,43 мл, 6,61 ммоль) добавляют к раствору 6-хлор-2-фтор-3-(пропилсульфонамидо)бензойной кислоты (1,70 г, 5,75 ммоль) в ТГФ (23 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа, нагревают до 70°C и перемешивают в течение 1 часа. Добавляют воду (6 мл), после чего реакционную смесь перемешивают снова при 70°C в течение 3 часов. Смесь охлаждают до комнатной температуры, добавляют этилацетат, и слои разделяют. Органическую фазу сушат с сульфатом натрия, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают флэш-хроматографией на силикагеле, элюируя 0-50% EtOAc/гептаном с получением N-(3-амино-4-хлор-2-фторфенил)пропан-1-сульфонамида (1,01 г, 66%) в виде твердого вещества. 1H ЯМР (500 MГц, (CD3)2SO) δ 9,54 (с, 1H), 7,02 (д, 1H), 6,58 (т, 1H), 5,50 (с, 2H), 3,09-2,95 (т, 2H), 1,81-1,64 (м, 2H), 0,96 (т, 3H); МС (хиад, m/z)=267,1 (M+H).
Промежуточное соединение P23
N- (3-Амино-2-хлор-4-фторфенил)пропан-1-сульфонамид
Указанное в заголовке соединение получают с применением методики, описанной в Промежуточном соединении P21 с применением 2-хлор-6-фтор-3-(пропилсульфонамидо)бензойной кислоты вместо 2,6-дифтор-3-(пропилсульфонамидо)бензойной кислоты в качестве исходного материала. 1H ЯМР (400 MГц, (CD3)2SO) δ 9,20 (с, 1H), 7,28-6,99 (м, 1H), 6,63 (тд, J=8,7, 5,5, 1H), 5,45 (с, 2H), 3,07-2,99 (м, 2H), 1,88-1,69 (м, 2H), 1,03-0,95 (м, 3H); МС (хиад, m/z)=267,1 (M+H).
Промежуточное соединение P24
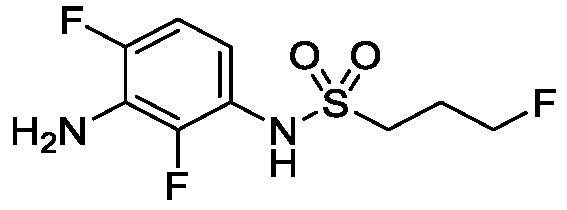
N- (3-Амино-2,4-дифторфенил)-3-фторпропан-1-сульфонамид
Триэтиламин (0,682 мл, 4,89 ммоль) и дифенилфосфорилазид (0,547 мл, 2,45 ммоль) добавляют к раствору 2,6-дифтор-3-(3-фторпропилсульфонамидо)бензойной кислоты (0,485 г, 1,63 ммоль) в ДМФ (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют воду (1,18 мл, 65,2 ммоль), и смесь перемешивают при 80°C в течение 15 часов. Реакционную смесь разбавляют этилацетатом (100 мл), и органический слой промывают насыщенным водным NaHCO3, сушат над сульфатом натрия и концентрируют. Неочищенный продукт очищают с применением хроматографии на силикагеле, элюируя гексаном/этилацетатом (4:1) с получением N-(3-амино-2,4-дифторфенил)-3-фторпропан-1-сульфонамида (0,34 г, 77%). 1H ЯМР (400 MГц, (CD3)2SO) δ 9,49 (шс, 1H), 6,87 (м, 1H), 6,52 (м, 1H), 5,33 (шс, 2H), 4,60 (м, 1H), 4,48 (м, 1H), 3,14 (м, 2H), 2,16-2,03 (м, 2H); МС (хиад, m/z)=267,1 (M-H).
Промежуточное соединение P25
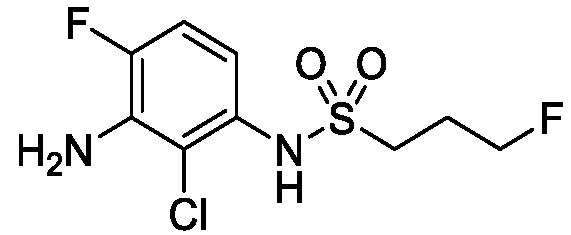
N- (3-Амино-2-хлор-4-фторфенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение метил 3-амино-2-хлор-6-фторбензоата. 2-Хлор-4-фтор анилин (0,82 мл, 6,87 ммоль) растворяют в ТГФ (54 мл) и охлаждают до -78°C в обратном потоке азота. Реакционную смесь медленно обрабатывают н-бутиллитием (2,5 M в гексане, 2,94 мл, 7,35 ммоль) и затем перемешивают при -78°C в течение 15 минут после завершения добавления. Реакционную смесь обрабатывают по каплям 15 мл ТГФ раствора 1,2-бис(хлордиметилсилил)этана (1,55 г, 7,21 ммоль) и перемешивают при -78°C в течение 15 минут после завершения добавления. Реакционную смесь медленно обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 2,94 мл, 7,35 ммоль) и ледяную баню удаляют после завершения добавления, и реакционную смесь нагревают в течение 30 минут. Реакционную смесь обратно охлаждают до -78°C и медленно обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 2,94 мл, 7,35 ммоль) и перемешивают при -78°C в течение 30 минут, затем обрабатывают по каплям метилхлорформиатом (0,63 мл, 7,83 ммоль). После завершения добавления, ледяную баню удаляют и реакционную смесь перемешивают в течение 1 часа. Реакционную смесь обрабатывают 4,0 M HCl и перемешивают при температуре окружающей среды в течение 30 минут и затем реакционную смесь нейтрализуют до примерно pH 8 с применением твердого NaHCO3. Реакционную смесь экстрагируют EtOAc (2x) и органические слои объединяют и промывают водой и насыщенным раствором соли и затем сушат над Na2SO4, фильтруют, и концентрируют. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением метил 3-амино-2-хлор-6-фторбензоата (1,23 г, 88%). 1H ЯМР (400 MГц, ДМСО-d6) δ 7,08 (т, 1H), 6,95 (т, 1H), 5,51 (с, 2H), 3,89 (с, 3H).
Стадия 2: Получение 2-хлор-6-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)-сульфонамидо)бензоата. В 250 мл колбу добавляют метил 3-амино-2-хлор-6-фторбензоат (5,09 г, 25,0 ммоль), дихлорметан (125 мл) и триэтиламин (10,5 мл, 75,0 ммоль) под азотом. Раствор охлаждают до 0°C и обрабатывают 3-фторпропан-1-сульфонилхлоридом (6,25 мл, 52,5 ммоль) в течение 20 минут. Реакционную смесь перемешивают в течение 15 часов при температуре окружающей среды после завершения добавления. Реакционную смесь концентрируют и восстанавливают EtOAc, который промывают 0,1N HCl водным раствором, затем насыщенным раствором соли. Органический слой сушат над Na2SO4 и концентрируют, затем очищают хроматографией на силикагеле (EtOAc/Гексан) с получением метил 2-хлор-6-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)-пропил)сульфонамидо)бензоата в виде коричневой жидкости (10,1 г, 90%). 1H ЯМР (400 MГц, CDCl3) δ 7,49-7,52 (дд, 1H), 7,16 (т, 1H), 4,63 (т, 2H), 4,53 (т, 2H), 3,99 (с, 3H), 3,74-3,85 (м, 4H), 2,29-2,39 (м, 4H).
Стадия 3: Получение 2-хлор-6-фтор-3-((3-фторпропил)сульфонамидо)бензойной кислоты. В 250 мл колбу добавляют метил 2-хлор-6-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)-пропил)сульфонамидо)бензоат (4,0 г, 8,85 ммоль), тетрагидрофуран (59,0 мл), метанол (9,84 мл) и 2,0 M гидроксида калия (26,6 мл, 53,1 ммоль). Реакционную смесь перемешивают при 50°C в течение 14 часов. Реакционную смесь концентрируют и остаток растворяют в EtOAc и pH доводят до примерно 2 1M водным раствором HCl. Водный слой экстрагируют EtOAc, и объединенные органические слои промывают насыщенным раствором соли. Органический слой сушат над Na2SO4 и концентрируют с получением 2-хлор-6-фтор-3-((3-фторпропил)сульфонамидо)бензойной кислоты в виде коричневого твердого вещества (2,15 г, 77%). 1H ЯМР (400 MГц, CDCl3) δ 7,58-7,61 (дд, 1H), 7,03 (т, 1H), 4,51 (т, 1H), 4,42 (т, 1H), 3,13 (т, 2H), 2,10-2,21 (м, 2H); МС (хиад, m/z)=312,1 (M-H).
Стадия 4: Получение N-(3-амино-2-хлор-4-фторфенил)-3-фторпропан-1-сульфонамида. В 100 мл колбу добавляют 2-хлор-6-фтор-3-((3-фторпропил)сульфонамидо)бензойную кислоту (2,15 г, 6,854 ммоль), ДМФ (11,42 мл) и триэтиламин (2,866 мл, 20,56 ммоль) под азотом. Дифенилфосфорилазид (2,216 мл, 10,28 ммоль) добавляют, и реакционную смесь перемешивают при температуре окружающей среды в течение 90 минут, в это время добавляют воду (18,28 мл, 6,854 ммоль) и реакционную смесь нагревают до 80°C в течение 15 часов. Реакционную смесь разбавляют EtOAc и промывают насыщенным водным NaHCO3. Водный слой экстрагируют EtOAc (x4) и объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают с применением хроматографии на силикагеле, элюируя EtOAc/гексаном с получением N-(3-амино-2-хлор-4-фторфенил)-3-фторпропан-1-сульфонамида (0,91 г, 47%). 1H ЯМР (400 MГц, CDCl3) δ 6,94-7,02 (м, 2H), 6,55 (шс, 1H), 4,58 (т, 1H), 4,48 (т, 1H), 4,22 (шс, 2H), 3,22 (т, 2H), 2,16-2,27 (м, 2H); МС (хиад, m/z)=283,0 (M-H).
Промежуточное соединение P26
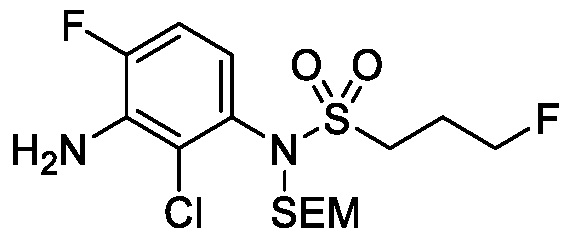
N- (3-амино-2-хлор-4-фторфенил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
В 100 мл колбу добавляют N-(3-амино-2-хлор-4-фторфенил)-3-фторпропан-1-сульфонамид (Промежуточное соединение P25) (2,04 г, 7,17 ммоль) и ДМФ (28,7 мл) под азотом. Раствор охлаждают до 0°C и обрабатывают 60% гидридом натрия в минеральном масле (0,387 г, 9,67 ммоль). Реакционную смесь перемешивают при 0°C в течение 15 минут, затем 2-(триметилсилил)этоксиметил хлорид (1,53 мл, 8,60 ммоль) добавляют по каплям в течение 5 минут. Реакционную смесь отслеживают ТСХ до тех пор, пока все исходные продукты не будут израсходованы. Реакционную смесь разбавляют EtOAc и водой. Водный слой экстрагируют EtOAc, и объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4 и концентрируют. Неочищенный продукт очищают с применением хроматографии на силикагеле, элюируя EtOAc/гексаном с получением N-(3-амино-2-хлор-4-фторфенил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)-пропан-1-сульфонамида (2,83 г, 95%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 6,99-6,94 (дд, 1H), 6,88-6,84 (дд, 1H), 5,24 (д, 1H), 4,68 (д, 1H), 4,62 (т, 1H), 4,50 (т, 1H), 4,22 (с, 2H), 3,69-3,58 (м, 2H), 3,29 (дд, 2H), 2,34-2,21 (м, 2H), 0,94-0,88 (м, 2H), 0,00 (с, 9H).
Промежуточное соединение P27
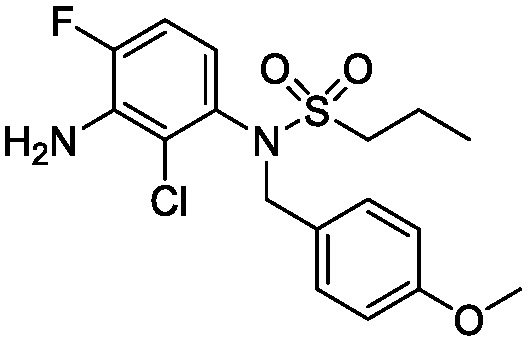
N- (3-Амино-2-хлор-4-фторфенил)- N- (4-метоксибензил)пропан-1-сульфонамид
N-(3-Амино-2-хлор-4-фторфенил)пропан-1-сульфонамид (75 г, 280 ммоль) растворяют в ДМФ (200 мл). 60% суспензию гидрида натрия в минеральном масле (11,85 г, 296 ммоль) добавляют несколькими порциями в течение 15 минут. Реакционную смесь перемешивают при комнатной температуре в течение 90 минут и нагревают до 40°C в течение двух часов. Эту гомогенную смесь охлаждают до 0°C, и п-метоксибензилхлорид (40,03 мл, 295,25 ммоль) добавляют в течение 5 минут. Реакционную смесь перемешивают и нагревают до комнатной температуры. Через 14 часов, реакционную смесь выливают в разбавленный раствор хлорида аммония (1750 мл) и водный слой декантируют для удаления масла. Это масло три раза растирают с водой (2 л). Оставшийся продукт переносят в 1 л стакан, разбавляют водой (800 мл), обрабатывают ультразвуком в течение 30 минут и затем перемешивают при комнатной температуре в течение 1 часа. Полученное твердое вещество собирают фильтрацией и сушат лиофилизацией с получением N-(3-амино-2-хлор-4-фторфенил)-N-(4-метоксибензил)пропан-1-сульфонамида (111,9 г, 99%). 1H ЯМР (500 MГц, (CD3)2SO) δ=7,11 (д, J=8,6, 2H), 6,96 (дд, J=10,6, 8,8, 1H), 6,81 (т, J=5,7, 2H), 6,51 (дд, J=8,7, 5,1, 1H), 5,42 (с, 2H), 4,71 (д, J=14,4, 1H), 4,57 (д, J=14,4, 1H), 3,70 (с, 3H), 3,21 (тд, J=6,7, 1,4, 2H), 1,77 (дд, J=15,3, 7,5, 2H), 1,00 (т, J=7,4, 3H).
Промежуточное соединение P28
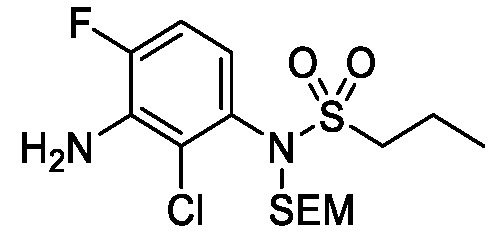
N- (3-амино-2-хлор-4-фторфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
К раствору N-(3-амино-2-хлор-4-фторфенил)пропан-1-сульфонамида (Промежуточное соединение P27) (10,0 г, 37,5 ммоль) в ДМФ (187 мл) при 0°C добавляют 60% суспензию гидрида натрия в минеральном масле (1,65 г, 41,2 ммоль) и (2-(хлорметокси)этил)триметилсилан (6,58 мл, 39,4 ммоль) и раствор перемешивают при температуре окружающей среды в течение 4 часов. Реакционную смесь гасят водой и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют в вакууме. Неочищенный продукт очищают хроматографией на силикагеле, элюируя 5% - 35% EtOAc/гексаном с получением N-(3-амино-2-хлор-4-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (12,7 г, 85%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 6,97 (м, 1H), 6,87 (м, 1H), 5,23 (д, 1H), 4,69 (д, 1H), 4,2 (с, 2H), 3,65 (м, 2H), 3,1 (т, 2H), 1,92 (м, 2H), 1,05 (т, 3H), 0,91 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P29
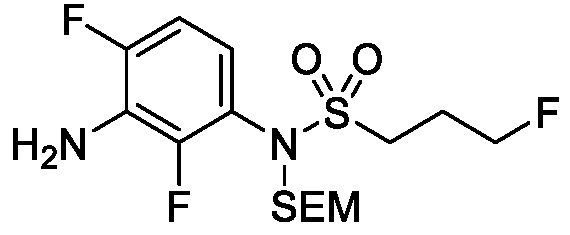
N- (3-амино-2,4-дифторфенил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
В 10 мл колбу добавляют N-(3-амино-2,4-дифторфенил)-3-фторпропан-1-сульфонамид (Промежуточное соединение P24) (204 мг, 0,764 ммоль) и ДМФ (3,0 мл) под азотом. Раствор охлаждают до 0°C и обрабатывают 60% гидридом натрия в минеральном масле (41,3 мг, 1,03 ммоль). Реакционную смесь перемешивают при 0°C в течение 15 минут, затем 2-(триметилсилил)этоксиметил хлорид (0,163 мл, 0,917 ммоль) добавляют по каплям в течение 5 минут. Реакционную смесь отслеживают ТСХ до тех пор, пока все исходные продукты не будут израсходованы. Реакционную смесь разбавляют EtOAc и водой. Водный слой экстрагируют EtOAc и объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают с применением хроматографии на силикагеле, элюируя EtOAc/гексаном с получением N-(3-амино-2,4-дифторфенил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида (59,3 мг, 20%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ (ч/млн) 6,73-6,86 (м, 2H), 4,96 (с, 2H), 4,61 (т, 1H), 4,50 (т, 1H), 3,83 (с, 2H), 3,65 (т, 2H), 3,26 (т, 2H), 2,18-2,31 (м, 2H), 0,92 (т, 2H), 0,01 (с, 9H).
Промежуточное соединение P30
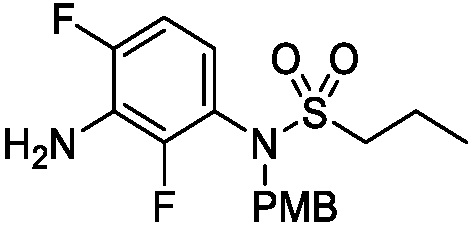
N- (3-амино-2,4-дифторфенил)- N- (4-метоксибензил)пропан-1-сульфонамид
К раствору N-(3-амино-2,4-дифторфенил)пропан-1-сульфонамида (1,05 г, 4,196 ммоль) в ДМФ (20 мл) при 0°C добавляют 60% суспензию гидрида натрия в минеральном масле (0,1678 г, 4,196 ммоль) и смесь перемешивают при 0°C в течение 15 минут. К реакционной смеси добавляют 1-(хлорметил)-4-метоксибензол (0,5694 мл, 4,196 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Раствор гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексан с получением N-(3-амино-2,4-дифторфенил)-N-(4-метоксибензил)пропан-1-сульфонамида (0,789 г, 51%). 1H ЯМР (400 MГц, CDCl3) δ 7,14 (д, 2H), 6,78 (д, 2H), 6,67 (м, 1H), 6,42 (м, 1H), 4,69 (с, 2H), 3,77 (с, 3H), 3,06 (т, 2H), 1,92 (м, 2H), 1,06 (т, 3H).
Промежуточное соединение P31
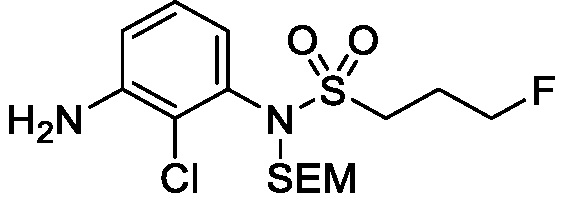
N- (3-амино-2-хлорфенил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-нитрофенил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида. Раствор 2-хлор-3-нитроанилина (1,0 г, 5,79 ммоль) в CH2Cl2 (29,0 мл) обрабатывают триэтиламином (1,01 мл, 7,24 ммоль), затем 3-фтор-пропан-1-сульфонилхлоридом (1,47 мл, 11,6 ммоль) и смесь перемешивают при температуре окружающей среды в течение 2 часов. Добавляют дополнительный триэтиламин (1,01 мл, 7,24 ммоль) и 3-фтор-пропан-1-сульфонилхлорид (1,47 мл, 11,6 ммоль) и смесь перемешивают при температуре окружающей среды в течение 30 минут. Добавляют 50 мл 0,5 N HCl раствора и слои смешивают и отделяют. Органический слой сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-нитрофенил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (2,44 г, 99%) и используют без дальнейшей очистки.
Стадия 2: Получение N-(2-хлор-3-нитрофенил)-3-фторпропан-1-сульфонамида. К раствору N-(2-хлор-3-нитрофенил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (2,44 г, 5,80 ммоль) в ТГФ (11,6 мл) и MeOH (3,87 мл) добавляют NaOH (5,80 мл, 11,6 ммоль, 2,0M водным), и смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят добавлением 1N HCl (10 мл) и смесь концентрируют для удаления MeOH. Смесь экстрагируют EtOAc, и объединенные экстракты сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией, элюируя 5% EtOAc/ДХМ с получением N-(2-хлор-3-нитрофенил)-3-фторпропан-1-сульфонамида (0,933 г, 54%) в виде желтого твердого вещества.
Стадия 3: Получение N-(3-амино-2-хлорфенил)-3-фторпропан-1-сульфонамида. К раствору N-(2-хлор-3-нитрофенил)-3-фторпропан-1-сульфонамида (0,933 г, 3,14 ммоль) в EtOH (15,7 мл) и воде (3,93 мл) добавляют порошок железа (1,76 г, 31,4 ммоль) одной порцией, затем NH4Cl (0,168 г, 3,14 ммоль). Смесь нагревают до 80°C в течение 2,5 часов. Смесь фильтруют через Celite®, промывают MeOH, и фильтрат концентрируют. Неочищенный остаток растворяют в EtOAc, затем промывают водой и сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией, элюируя 5% - 40% EtOAc/гексаном с получением N-(3-амино-2-хлорфенил)-3-фторпропан-1-сульфонамида в виде оранжевого масла (0,711 г, 84%).
Стадия 4: Получение N-(3-амино-2-хлорфенил)-3-фтор-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. Раствор N-(3-амино-2-хлорфенил)-3-фторпропан-1-сульфонамида (0,679 г, 2,55 ммоль) в ДМФ (10,2 мл) охлаждают до 0°C на бане лед/вода. Раствор обрабатывают 60% гидридом натрия в минеральном масле (0,137 г, 3,44 ммоль) и смесь перемешивают в течение 15 минут. Затем 2-(триметилсилил)этоксиметил хлорид (0,541 мл, 3,05 ммоль) добавляют по каплям, и смесь перемешивают в течение дополнительного 1 часа при 0°C. Реакционную смесь гасят добавлением насыщенным водным раствором NH4Cl, и смесь нагревают до температуры окружающей среды и экстрагируют EtOAc. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией, элюируя 10% - 30% EtOAc/гексаном с получением N-(3-амино-2-хлорфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,726 г, 71%) в виде густого бледно-желтого масла. 1H ЯМР (400 MГц, CDCl3) δ 7,09 (т, 1H); 6,90 (дд, 1H); 6,80 (дд, 1H); 5,26 (шм, 1H); 4,74 (шм, 1H); 4,62 (т, 1H); 4,51 (т, 1H); 4,18 (шс, 2H); 3,65 (шм, 2H); 3,31 (м, 2H); 2,29 (м, 2H); 0,92 (м, 2H); 0,01 (с, 9H).
Промежуточное соединение P32
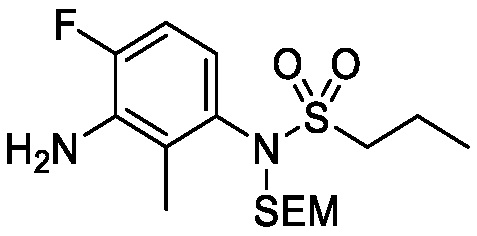
N- (3-амино-4-фтор-2-метилфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 6-фтор-2-метил-3-(пропилсульфонамидо)бензойной кислоты. К раствору метил 3-амино-6-фтор-2-метилбензоата (900 мг, 4,913 ммоль) и триэтиламина (2,05 мл, 14,74 ммоль) в дихлорметане (19,6 мл) добавляют пропан-1-сульфонилхлорид (1,38 мл, 12,28 ммоль), и смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят насыщенным водным NaHCO3 и экстрагируют ДХМ. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Остаток растворяют в тетрагидрофуране (19,6 мл) и метаноле (4,9 мл) и обрабатывают 2,0 M гидроксида калия (14,7 мл, 29,48 ммоль) при 50°C в течение 60 часов. Раствор концентрируют и промывают Et2O. Водный слой доводят до примерно pH 2 концентрированной HCl и экстрагируют 4:1 ДХМ/ИПА. Органические слои сушат с Na2SO4, фильтруют и концентрируют с получением 6-фтор-2-метил-3-(пропилсульфонамидо)бензойной кислоты (1,13 г, 84%). 1H ЯМР (400 MГц, ДМСО) δ 9,2 (с, 1H), 7,35 (м, 1H), 7,15 (т, 1H), 3,08 (м, 2H), 2,3 (с, 3H), 1,73 (м, 2H), 0,99 (т, 3H).
Стадия 2: Получение N-(3-амино-4-фтор-2-метилфенил)пропан-1-сульфонамида. К раствору 6-фтор-2-метил-3-(пропилсульфонамидо)бензойной кислоты (1,13 г, 4,105 ммоль) в ДМФ (41,05 мл) добавляют триэтиламин (1,716 мл, 12,31 ммоль) и дифенилфосфорилазид (1,33 мл, 6,157 ммоль). Раствор перемешивают при температуре окружающей среды в течение 1 часа. Затем добавляют воду (5,86 мл, 4,105 ммоль) и раствор нагревают до 80°C в течение ночи. Раствор гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают колоночной хроматографией, элюируя 10:90 EtOAc/гексан с получением N-(3-амино-4-фтор-2-метилфенил)пропан-1-сульфонамида (0,88 г, 87%). МС (хиад, m/z)=247,1 [M+H].
Стадия 3: К раствору N-(3-амино-4-фтор-2-метилфенил)пропан-1-сульфонамида (880 мг, 3,57 ммоль) в ДМФ (23,8 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 193 мг, 4,82 ммоль) и (2-(хлорметокси)этил)триметилсилан (717 мкл, 4,29 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают 60% суспензией гидрида натрия в минеральном масле, элюируя 10% - 50% EtOAc/гексаном с получением N-(3-амино-4-фтор-2-метилфенил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида (590 мг, 44%). 1H ЯМР (400 MГц, CDCl3) δ 6,87 (м, 1H), 6,78 (м, 1H), 5,2 (д, 1H), 4,48 (д, 1H), 3,58 (т, 2H), 3,14 (т, 2H), 2,19 (с, 3H), 1,89 (м, 2H), 1,15 (т, 3H), 0,94 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P33
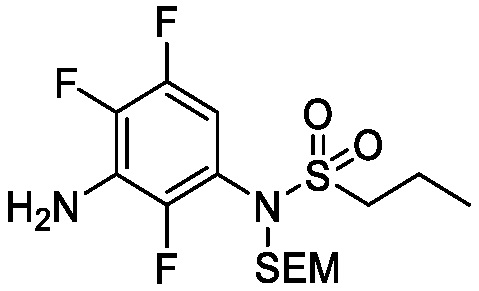
N- (3-амино-2,4,5-трифторфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение метил 3-амино-2,5,6-трифторбензоата. 2,4,5-Трифторанилин (3,00 г, 20,394 ммоль) растворяют в ТГФ (100 мл) и охлаждают до -78°C в обратном потоке N2. Реакционную смесь медленно обрабатывают н-бутиллитием (2,5 M в гексане, 8,97 мл, 22,434 ммоль) и перемешивают при -78°C в течение 15 минут. Реакционную смесь обрабатывают по каплям ТГФ раствором (50 мл) 1,2-бис(хлордиметилсилил)этана (4,61 г, 21,414 ммоль) и перемешивают при -78°C в течение 15 минут после завершения добавления. Реакционную смесь медленно обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 8,97 мл, 22,434 ммоль). Ледяную баню удаляют и реакционную смесь нагревают в течение 30 минут. Реакционную смесь обратно охлаждают до -78°C и медленно обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 8,97 мл, 22,434 ммоль) и перемешивают при -78°C в течение 30 минут. Реакционную смесь обрабатывают по каплям метилхлорформиатом (2,36 мл, 30,591 ммоль). Холодную баню удаляют, и смесь перемешивают в течение 1 часа. Реакционную смесь гасят водой и подкисляют примерно pH 1 с применением 4,0 M HCl и перемешивают при комнатной температуре в течение 30 минут. Реакционную смесь доводят до примерно pH 8 с применением твердого NaHCO3 и экстрагируют EtOAc (2x). Объединенные органические экстракты промывают водой, насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле, элюируя 5:95 гексан/этилацетатом, затем С18 хроматографией с обращенной фазой, элюируя 5:95 водой/ацетонитрилом с 0,1% ТФК. Продукт разделяют между ДХМ и насыщенным водным NaHCO3. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением метил 3-амино-2,5,6-трифторбензоата (2,34 г, 56%). 1H ЯМР (400 MГц, ДМСО) δ 6,88 (м, 1H), 5,58 (с, 2H), 3,87 (с, 3H).
Стадия 2: Получение метил 2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)бензоата. К раствору метил 3-амино-2,5,6-трифторбензоата (2,32 г, 11,31 ммоль) и триэтиламина (4,729 мл, 33,93 ммоль) в дихлорметане (45,24 мл) добавляют пропан-1-сульфонилхлорид (3,170 мл, 28,27 ммоль), и смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением метил 2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)бензоата (3,08 г, 65%). 1H ЯМР (400 MГц, CDCl3) δ 7,37 (м, 1H), 3,99 (с, 3H), 3,63 (м, 2H), 3,49 (м, 2H), 1,94 (м, 4H), 1,1 (т, 6H).
Стадия 3: Получение 2,3,6-трифтор-5-(пропилсульфонамидо)бензойной кислоты. К раствору метил 2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)бензоата (3,08 г, 7,38 ммоль) в метаноле (8,20 мл) и тетрагидрофуране (49,2 мл) добавляют 2,0 M гидроксида калия (22,1 мл, 44,3 ммоль), и смесь нагревают до 50°C в течение ночи. ТГФ/MeOH удаляют под вакуумом, и остаток разбавляют водой и промывают простым эфиром. Водный слой доводят до примерно pH 2 концентрированной HCl и экстрагируют 4:1 ДХМ/ИПА. Органические слои сушат с Na2SO4, фильтруют и концентрируют с получением 2,3,6-трифтор-5-(пропилсульфонамидо)бензойной кислоты (1,6 г, 73%). МС (хиад, m/z)=296,1 [M-H].
Стадия 4: Получение N-(3-амино-2,4,5-трифторфенил)пропан-1-сульфонамида. К раствору 2,3,6-трифтор-5-(пропилсульфонамидо)бензойной кислоты (1,6 г, 5,383 ммоль) в ДМФ (53,83 мл) добавляют триэтиламин (2,251 мл, 16,15 ммоль) и дифенилфосфорилазид (1,740 мл, 8,074 ммоль). Раствор перемешивают при температуре окружающей среды в течение 1 часа. Затем добавляют воду (7,690 мл, 5,383 ммоль), и реакционную смесь нагревают до 80°C в течение 24 часов. Раствор гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 50% EtOAc/гексаном с получением N-(3-амино-2,4,5-трифторфенил)пропан-1-сульфонамида (0,733 г, 51%). МС (хиад, m/z)=267,0 [M-H].
Стадия 5: Получение N-(3-амино-2,4,5-трифторфенил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида. К раствору N-(3-амино-2,4,5-трифторфенил)пропан-1-сульфонамида (733 мг, 2,73 ммоль) в ДМФ (13,7 мл) при 0°C добавляют 60% суспензию гидрида натрия в минеральном масле (148 мг, 3,69 ммоль) и (2-(хлорметокси)этил)триметилсилан (582 мкл, 3,28 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(3-амино-2,4,5-трифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (1,02 г, 94%). 1H ЯМР (400 MГц, CDCl3) δ 6,66 (м, 1H), 4,93 (с, 2H), 3,97 (шс, 2H), 3,65 (м, 2H), 3,07 (м, 2H), 1,88 (м, 2H), 1,04 (т, 3H), 0,91 (м, 2H), 0,02 (с, 9H).
Промежуточное соединение P34
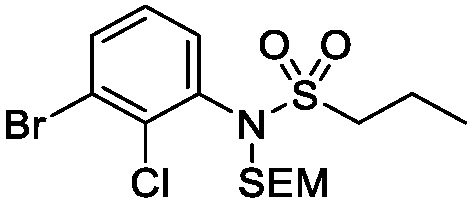
N- (3-бром-2-хлорфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-бром-2-хлорфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. 3-бром-2-хлоранилин (2,026 г, 9,813 ммоль) растворяют в ДХМ (20 мл) и обрабатывают триэтиламином (4,103 мл, 29,44 ммоль). 1-Пропансульфонилхлорид (2,420 мл, 21,59 ммоль) добавляют, и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Через 16 часов добавляют дополнительный триэтиламин (2,05 мл, 14,72 ммоль, 1,5 экв.) и 1-пропансульфонилхлорид (1,21 мл, 10,79 ммоль, 1,1 экв.) и реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь разбавляют ДХМ (20 мл) и промывают насыщенным водным раствором NaHCO3 (50 мл). Органическую фазу отделяют и сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-бром-2-хлорфенил)-N-(пропилсульфонил)-пропан-1-сульфонамида (4,1 г, 99%). Продукт применяют непосредственно на стадии 2 без дальнейшей очистки.
Стадия 2: Получение N-(3-бром-2-хлорфенил)пропан-1-сульфонамида. N-(3-бром-2-хлорфенил)-N-(пропилсульфонил)пропан-1-сульфонамид (4,1 г, 9,81 ммоль) растворяют в ТГФ (50 мл) и обрабатывают 1,0 M гидроксидом натрия (19,63 мл, 19,63 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь разбавляют водой и доводят до примерно pH 4 с применением 4,0 M HCl. Водную фазу экстрагируют ДХМ (2×50 мл). Органические фазы объединяют, сушат над Na2SO4, фильтруют и концентрируют до коричневого масла. Неочищенный продукт очищают с применением хроматографии на силикагеле, элюируя 14% EtOAc/86% гексаном с получением N-(3-бром-2-хлорфенил)пропан-1-сульфонамида в виде желтого твердого вещества (2,90 г, 95%). 1H ЯМР (400 MГц, CDCl3) δ 7,66 (дд, 1H) , 7,43 (дд, 1H), 7,16 (т, 1H), 6,85 (с, 1H), 3,11-3,05 (м, 2H), 1,91-1,81 (м, 2H), 1,04 (т, 3H).
Стадия 3: Получение N-(3-бром-2-хлорфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. N-(3-бром-2-хлорфенил)пропан-1-сульфонамид (1,063 г, 3,40 ммоль) растворяют в ДМФ (18 мл) и охлаждают на бане лед/вода в течение 10 минут под аргоном. К реакционной смеси добавляют 60% суспензию гидрида натрия в минеральном масле (0,1428 г, 3,57 ммоль) и реакционную смесь перемешивают в течение примерно 15 минут до прекращения выделения газа. (2-(Хлорметокси)этил)триметилсилан (0,724 мл, 4,08 ммоль) добавляют, и реакционную смесь нагревают до температуры окружающей среды под аргоном в течение 45 минут. Раствор гасят H2O (50 мл) и продукт экстрагируют EtOAc (50 мл). Органическую фазу промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(3-бром-2-хлорфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида в виде светло-желтого масла (1,38 г, 92%). 1H ЯМР (400 MГц, CDCl3) δ 7,56 (дд, 1H), 7,47 (дд, 1H), 7,19 (т, 1H), 5,22 (д,1H), 4,78 (д, 1H), 3,67 (д, 2H), 3,13-3,06 (м, 2H), 1,98-1,84 (м, 2H), 1,05 (т, 3H), 0,99-0,81(м, 2H), 0,01 (с, 9H).
Промежуточное соединение P35

N- (3-бром-2,5-дихлорфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-бром-2,5-дихлорфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. Триэтиламин (1,794 мл, 12,87 ммоль) добавляют к 3-бром-2,5-дихлоранилину (1,0 г, 4,151 ммоль) и пропан-1-сульфонилхлориду (1,070 мл, 9,547 ммоль) в ДХМ (30 мл), и смесь перемешивают при температуре окружающей среды в течение 60 часов. Реакционную смесь гасят водой (100 мл) и водный слой экстрагируют ДХМ (2×40 мл). Органические слои объединяют, промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-бром-2,5-дихлорфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,87 г, 99%), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(3-бром-2,5-дихлорфенил)пропан-1-сульфонамида. К раствору N-(3-бром-2,5-дихлорфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,87 г, 4,13 ммоль, 1,92 M) в ацетонитриле (40 мл) добавляют моногидрат карбоната натрия (15 мл, 28,9 ммоль) и реакционную смесь нагревают до 80°C в течение 6 часов. Реакционную смесь концентрируют для удаления ацетонитрила и добавляют 10% водную лимонную кислоту для доведения рН до примерно 3. Продукт экстрагируют ДХМ (3×50 мл) и органические слои объединяют, сушат над Na2SO4, фильтруют и концентрируют с получением темно-коричневого твердого вещества, которое очищают хроматографией на силикагеле, элюируя 20% EtOAc/гексаном с получением N-(3-бром-2,5-дихлорфенил)пропан-1-сульфонамида в виде светло-коричневого твердого вещества (700 мг, 49%). МС (хиад, m/z)=343,9 (M-H).
Стадия 3: Получение N-(3-бром-2,5-дихлорфенил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида. N-(3-бром-2,5-дихлорфенил)пропан-1-сульфонамид (645 мг, 1,859 ммоль) растворяют в ДМФ (9,2 мл) под аргоном и смесь охлаждают на бане лед/вода. К реакционной смеси добавляют (81,77 мг, 2,044 ммоль) и реакционную смесь перемешивают при 0°C в течение 30 минут. (2-(Хлорметокси)этил)триметилсилан (395,6 мкл, 2,230 ммоль) добавляют, и реакционную смесь нагревают до температуры окружающей среды в течение ночи. Раствор гасят H2O (50 мл), и продукт экстрагируют EtOAc (50 мл). Органическую фазу промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (5-50% EtOAc/гексан) с получением N-(3-бром-2,5-дихлорфенил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида в виде желтого масла (575,8 мг, 65%). 1H ЯМР (400 MГц, CDCl3) δ 7,68 (д, 1H), 7,47 (д, 1H), 5,21 (д, 1H), 4,74 (д, 1H), 3,67 (д, 2H), 3,14-3,05 (м, 2H), 1,98-1,85 (м, 2H), 1,06 (т, 3H), 0,99-0,82 (м, 2H), 0,02 (с, 9H).
Промежуточное соединение P36
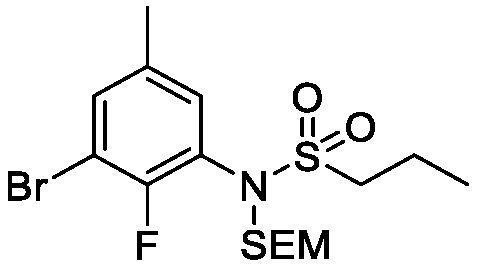
N- (3-бром-2-фтор-5-метилфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-бром-2-фтор-5-метилфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. Триэтиламин (1059 мкл, 7,596 ммоль) добавляют к раствору 3-бром-2-фтор-5-метиланилина (500 мг, 2,450 ммоль) и пропан-1-сульфонилхлорида (631,8 мкл, 5,636 ммоль) в ДХМ (24,5 мл), и смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь гасят 100 мл воды, и водный слой экстрагируют ДХМ (2×40 мл). Объединенные органические экстракты промывают насыщенным раствором соли, сушат с Na2SO4 и концентрируют с получением N-(3-бром-2-фтор-5-метилфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,01 г, 99%) в виде оранжевого твердого вещества, которое применяют непосредственно на следующей стадии.
Стадия 2: Получение N-(3-бром-2-фтор-5-метилфенил)пропан-1-сульфонамида. К раствору N-(3-бром-2-фтор-5-метилфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,01 г, 2,40 ммоль) в ацетонитриле (25 мл) добавляют карбонат натрия (15 мл, 18,16 ммоль, 1,2M), и реакционную смесь нагревают до 80°C в течение одного часа. Реакционную смесь концентрируют для удаления ацетонитрила, и 10% водную лимонную кислоту добавляют для доведения рН до примерно 3. Продукт экстрагируют ДХМ (3×50 мл) и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением темно-коричневого твердого вещества, которое очищают хроматографией на силикагеле, элюируя 10% EtOAc/ДХМ, с получением N-(3-бром-2-фтор-5-метилфенил)пропан-1-сульфонамида в виде белого твердого вещества (650,9 мг, 81%). 1H ЯМР (400 MГц, CDCl3) δ 7,37-7,32 (м, 1H), 7,17-7,12 (м, 1H), 6,48 (с, 1H), 3,12-3,05 (м, 2H), 2,31 (с, 3H), 1,93-1,81 (м, 2H), 1,05 (т, 3H).
Стадия 3: Получение N-(3-бром-2-фтор-5-метилфенил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. N-(3-бром-2-фтор-5-метилфенил)пропан-1-сульфонамид (524 мг, 1,689 ммоль) растворяют в ДМФ (8,45 мл) под аргоном и смесь охлаждают на бане лед/вода. К реакционной смеси добавляют 60% суспензию гидрида натрия в минеральном масле (74,32 мг, 1,858 ммоль) и реакционную смесь перемешивают при 0°C в течение 30 минут. (2-(Хлорметокси)этил)триметилсилан (359,5 мкл, 2,027 ммоль) добавляют, и реакционную смесь нагревают до температуры окружающей среды в течение 1 часа. Реакционную смесь гасят 10% водной лимонной кислотой (50 мл), и водный слой экстрагируют EtOAc (50 мл). Органическую фазу промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением желтого масла, которое очищают хроматографией на силикагеле, элюируя 20% EtOAc/гексаном, с получением N-(3-бром-2-фтор-5-метилфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида в виде белых кристаллов (588,4 мг, 79%). 1H ЯМР (400 MГц, CDCl3) δ 7,40-7,36 (м, 1H), 7,20-7,16 (м, 1H), 4,98 (с, 2H), 3,72-3,66 (м, 2H), 3,10-3,03 (м, 2H), 2,33 (с, 3H), 1,95-1,83 (м, 2H), 1,06 (т, 3H), 0,95-0,88 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P37
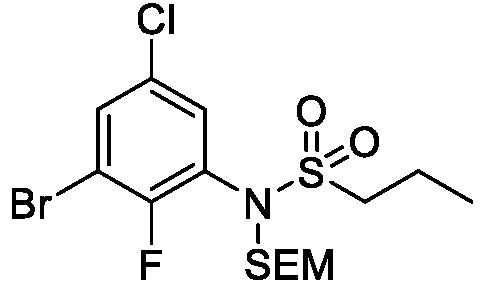
N- (3-бром-5-хлор-2-фторфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-бром-5-хлор-2-фторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. Триэтиламин (1733 мкл, 12,43 ммоль) добавляют к 3-бром-5-хлор-2-фторанилину (900 мг, 4,010 ммоль) и пропан-1-сульфонилхлориду (1034 мкл, 9,222 ммоль) в ДХМ (30 мл) и смесь перемешивают при комнатной температуре в течение 16 часов. Реакционную смесь гасят 100 мл воды, и водный слой экстрагируют ДХМ (2×40 мл). Органические слои объединяют, промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(3-бром-5-хлор-2-фторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,7 г, 95%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(3-бром-5-хлор-2-фторфенил)пропан-1-сульфонамида. К раствору N-(3-бром-5-хлор-2-фторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,7 г, 4,01 ммоль) в ацетонитриле (30 мл) добавляют карбонат натрия (15 мл, 28,5 ммоль, 1,9 M), и реакционную смесь нагревают до 80°C в течение 1 часа. Реакционную смесь концентрируют, и pH доводят до примерно 3 10% водной лимонной кислотой (50 мл) и EtOAc (50 мл). Слои разделяют, и водный слой экстрагируют EtOAc (3×50 мл). Органические слои объединяют, промывают насыщенным раствором соли (25 мл), сушат над Na2SO4, фильтруют и концентрируют с получением светло-коричневого твердого вещества, которое очищают хроматографией на силикагеле, элюируя гексаном/EtOAc (9:1) с получением N-(3-бром-5-хлор-2-фторфенил)пропан-1-сульфонамид в виде белого твердого вещества (1,3 г, 97%). МС (хиад, m/z)=327,9, 329,9 (M-H).
Стадия 3: Получение N-(3-бром-5-хлор-2-фторфенил)-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида. N-(3-бром-5-хлор-2-фторфенил)пропан-1-сульфонамид (1 г, 3,025 ммоль) растворяют в ДМФ (15 мл) под аргоном и охлаждают на ледяной бане в течение 5 минут. К реакционной смеси добавляют 60% суспензию гидрида натрия в минеральном масле (0,1331 г, 3,327 ммоль) и реакционную смесь перемешивают при 0°C в течение 30 минут. (2-(хлорметокси)этил)триметилсилан (0,644 мл, 3,630 ммоль) добавляют, и ледяную баню удаляют и реакционную смесь нагревают до температуры окружающей среды в течение 1 часа. Реакционную смесь гасят добавлением воды (50 мл) и продукт экстрагируют EtOAc (3×50 мл). Органические фазы объединяют, промывают насыщенным раствором соли (50 мл), сушат над Na2SO4, фильтруют и концентрируют с получением коричневого масла, которое очищают хроматографией на силикагеле, элюируя гексаном/EtOAc (9:1), с получением N-(3-бром-5-хлор-2-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида в виде прозрачного масла (751,9 мг, 54%). 1H ЯМР (400 MГц, CDCl3) δ 7,60-7,56 (м, 1H), 7,43-7,39 (м, 1H), 4,97 (с, 2H), 3,71-3,63 (м, 2H), 3,12-3,04 (м, 2H), 1,95-1,83 (м, 2H), 1,07 (т, 3H), 0,96-0,88 (м, 2H), 0,02 (с, 9H).
Промежуточное соединение P38
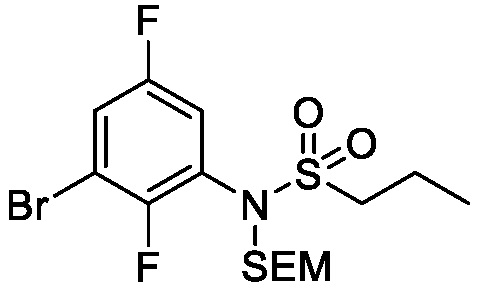
N- (3-бром-2,5-дифторфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-бром-2,5-дифторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. Триэтиламин (1,768 мл, 12,68 ммоль) добавляют к раствору 3-бром-2,5-дифторанилин гидрохлорида (1 г, 4,091 ммоль) и пропан-1-сульфонилхлорида (0,963 мл, 8,59 ммоль) в ДХМ (41 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов, затем гасят водой (100 мл). Слои разделяют, и водный слой экстрагируют ДХМ (2×40 мл). Органические слои объединяют, промывают насыщенным раствором соли (50 мл) и сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-бром-2,5-дифторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,6 г, 93%), который применяют непосредственно на следующей стадии.
Стадия 2: Получение N-(3-бром-2,5-дифторфенил)пропан-1-сульфонамида. К раствору N-(3-бром-2,5-дифторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,6 г, 3,81 ммоль) в ацетонитриле (30 мл) добавляют карбонат натрия (15 мл, 26,6 ммоль, 1,77 M) и реакционную смесь нагревают до 80°C в течение 1 часа. Реакционную смесь концентрируют для удаления ацетонитрила, pH доводят до примерно 3 10% водной лимонной кислотой (50 мл) и EtOAc (50 мл). Слои разделяют, и водный слой экстрагируют EtOAc (3×50 мл). Органические слои объединяют, промывают насыщенным раствором соли и сушат над Na2SO4, фильтруют и концентрируют с получением светло-коричневого твердого вещества, которое очищают на силикагеле, элюируя гексаном/EtOAc (9:1), с получением N-(3-бром-2,5-дифторфенил)пропан-1-сульфонамида в виде белого твердого вещества (844 мг, 71%). МС (хиад, m/z)=311,9, 313,9 (M-H).
Стадия 3: Получение N-(3-бром-2,5-дифторфенил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. N-(3-бром-2,5-дифторфенил)пропан-1-сульфонамид (700 мг, 2,228 ммоль) растворяют в ДМФ (11 мл) под аргоном и охлаждают на бане лед/вода в течение 5 минут. 60% суспензию гидрида натрия в минеральном масле (93,58 мг, 2,340 ммоль) добавляют, и реакционную смесь перемешивают до прекращения выделения газа. (2-(Хлорметокси)этил)триметилсилан (474,3 мкл, 2,674 ммоль) добавляют, и реакционную смесь нагревают до температуры окружающей среды в течение 16 часов. Реакционную смесь гасят водой (50 мл) и EtOAc (50 мл). Органическую фазу промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением в виде коричневого твердого вещества. Твердое вещество очищают хроматографией на силикагеле, элюируя гексаном/EtOAc (1:1) с получением N-(3-бром-2,5-дифторфенил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида в виде белых кристаллов (801,4 мг, 81%). 1H ЯМР (400 MГц, CDCl3) δ 7,37-7,32 (м, 1H), 7,21-7,16 (м, 1H), 4,98 (с, 2H), 3,70-3,64 (м, 2H), 3,11-3,05 (м, 2H), 1,94-1,84 (м, 2H), 1,07 (т, 3H), 0,95-0,88 (м, 2H), 0,02 (с, 9H).
Промежуточное соединение P39

N- (3-бром-2-хлор-5-фторфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-бром-2-хлор-5-фторфенил)пропан-1-сульфонамида. 3-Бром-2-хлор-5-фторанилин (2,38 г, 10,60 ммоль) растворяют в ДХМ (105 мл) и затем последовательно обрабатывают триэтиламином (4,434 мл, 31,81 ммоль) и 1-пропансульфонилхлоридом (2,615 мл, 23,33 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1,5 часов. Реакционную смесь разбавляют ДХМ (250 мл), промывают насыщенным NaHCO3, сушат над Na2SO4, фильтруют и концентрируют досуха. Остаток растворяют в ТГФ (50 мл) и обрабатывают гидроксидом калия (15,91 мл, 31,81 ммоль) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь подкисляют примерно pH 4 с применением 4,0 M HCl и затем экстрагируют ДХМ (2×200 мл). Органические слои объединяют и сушат над Na2SO4, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением N-(3-бром-2-хлор-5-фторфенил)пропан-1-сульфонамида (3,14 г, 90%). 1H ЯМР (500 MГц, (CD3)2SO) δ=9,83 (с, 1H), 7,67-7,64 (дд, 1H), 7,40-7,36 (дд, 1H), 3,23-3,19 (м, 2H), 1,79-1,69 (м, 2H), 0,99-0,95 (т, 3H).
Стадия 2: Получение N-(3-бром-2-хлор-5-фторфенил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. N-(3-бром-2-хлор-5-фторфенил)пропан-1-сульфонамид (3,14 г, 9,50 ммоль) растворяют в ДМФ (48 мл), охлаждают на бане лед/вода. 60% суспензию гидрида натрия в минеральном масле (0,570 г, 14,2 ммоль) добавляют, и реакционную смесь перемешивают до прекращения выделения газа. 2-(Триметилсилил)этоксиметилхлорид (2,02 мл, 11,4 ммоль) добавляют, и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 16 часов. Реакционную смесь обрабатывают водой (50 мл) и экстрагируют EtOAc (2×250 мл), и затем объединенные органические слои промывают водой (3×100 мл) и насыщенным раствором соли (1×50 мл) и затем сушат над Na2SO4, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-бром-2-хлор-5-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (4,00 г, 91%). 1H ЯМР (500 MГц, (CD3)2SO) δ=7,96-7,93 (дд, 1H), 7,63-7,60 (дд, 1H), 5,15-5,10 (м, 1H), 4,83-4,78 (м, 1H), 3,68-3,60 (м, 2H), 3,37-3,28 (м, 2H), 1,83-1,73 (м, 2H), 1,03-1,00 (т, 3H), 0,88-0,84 (т, 2H), 0,00 (с, 9H).
Промежуточное соединение P40
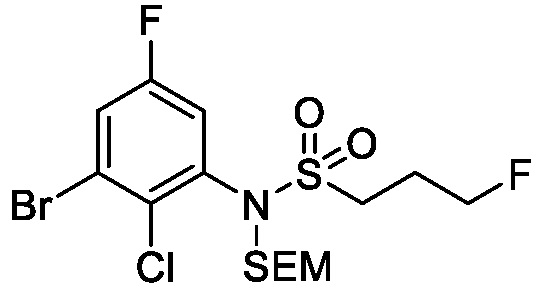
N- (3-бром-2-хлор-5-фторфенил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-бром-2-хлор-5-фторфенил)-3-фторпропан-1-сульфонамида. 3-Бром-2-хлор-5-фторанилин (1,10 г, 4,90 ммоль) растворяют в ДХМ (50 мл) и затем последовательно обрабатывают триэтиламином (2,05 мл, 14,7 ммоль) и 3-фторпропан-1-сульфонилхлоридом (1,28 мл, 10,8 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь разбавляют дополнительным ДХМ (100 мл) и промывают насыщенным NaHCO3 (1×100 мл) и затем сушат над Na2SO4, фильтруют и концентрируют досуха. Остаток растворяют в ТГФ (50 мл) и обрабатывают 2,0 M гидроксида калия (7,35 мл, 14,7 ммоль) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь подкисляют примерно pH 4 с применением 4,0 M HCl, и затем экстрагируют ДХМ (2×200 мл) и затем объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-бром-2-хлор-5-фторфенил)-3-фторпропан-1-сульфонамида (1,29 г, 76%). 1H ЯМР (500 MГц, (CD3)2SO) δ=10,00 (с, 1H), 7,70-7,67 (дд, 1H), 7,41-7,38 (дд, 1H), 4,61-4,58 (т, 1H), 4,49-4,47 (т, 1H), 3,35-3,31 (м, 2H), 2,18-2,05 (м, 2H).
Стадия 2: Получение N-(3-бром-2-хлор-5-фторфенил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида. N-(3-бром-2-хлор-5-фторфенил)-3-фторпропан-1-сульфонамид (1,29 г, 3,70 ммоль) растворяют в ДМФ (19 мл), охлаждают до 0°C и затем последовательно обрабатывают гидридом натрия (60% суспензия в минеральном масле, 0,222 г, 5,55 ммоль) и 2-(триметилсилил)этоксиметилхлоридом (0,788 мл, 4,44 ммоль). Реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 16 часов. Реакционную смесь обрабатывают водой (50 мл) и экстрагируют EtOAc (2×200 мл) и затем объединенные органические слои промывают водой (3×100 мл) и насыщенным раствором соли (1×50 мл) и затем сушат над Na2SO4, фильтруют и концентрируют досуха. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением N-(3-бром-2-хлор-5-фторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (1,24 г, 70%). 1H ЯМР (500 MГц, (CD3)2SO) δ=7,98-7,95 (дд, 1H), 7,66-7,63 (дд, 1H), 5,17-5,14 (м, 1H), 4,83-4,80 (м, 1H), 4,65-4,62 (т, 1H), 4,53-4,50 (т, 1H), 3,69-3,60 (м, 2H), 3,51-3,42 (м, 2H), 2,20-2,10 (м, 2H), 0,89-0,85 (м, 2H), 0,00 (с, 9H).
Промежуточное соединение P41
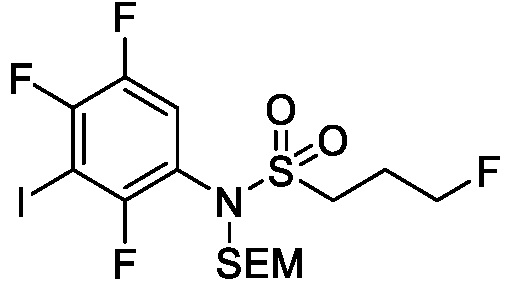
3-фтор- N- (2,4,5-трифтор-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 2,4,5-трифтор-3-йоданилина. Раствор 2,4,5-трифторанилина (3,0 г, 20,394 ммоль) в 150 мл ТГФ охлаждают до -78°C под азотом и раствор медленно обрабатывают н-бутиллитием (2,5 M в гексане, 8,97 мл, 22,43 ммоль) и перемешивают при -78°C в течение 15 минут. Затем раствор 1,2-бис(хлордиметилсилил)этана (4,61 г, 21,4 ммоль) в 50 мл ТГФ медленно добавляют в реакционную смесь и перемешивают в течение 15 минут. Затем медленно добавляют н-бутиллитий (2,5 M в гексане, 8,97 мл, 22,43 ммоль), и реакционную смесь удаляют из бани -78°C и нагревают в течение 30 минут. Реакционную смесь обратно охлаждают до -78°C и медленно добавляют н-бутиллитий (2,5 M в гексане, 8,97 мл, 22,43 ммоль) и перемешивают при -78°C в течение 30 минут. Затем медленно добавляют раствор йода (7,764 г, 30,59 ммоль) в 50 мл ТГФ, и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Раствор гасят ~10 мл 4M HCl и перемешивают в течение 30 минут. Затем добавляют 20 мл насыщенного тиосульфата натрия, и раствор доводят до примерно pH 8 твердым NaHCO3 и перемешивают в течение 15 минут. Раствор гасят водой и экстрагируют EtOAc. Органические слои промывают водой, насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают двуокисью кремния, элюируя 10% - 50% EtOAc/гексан с получением 2,4,5-трифтор-3-йоданилина (5,1 г, 92%). 1H ЯМР (400 MГц, CDCl3) δ 6,66 (м, 1H), 3,78 (шс, 2H).
Стадия 2: Получение 3-фтор-N-(2,4,5-трифтор-3-йодфенил)пропан-1-сульфонамида. К раствору 2,4,5-трифтор-3-йоданилина (2,5 г, 9,16 ммоль) в дихлорметане (45,8 мл) при 0°C добавляют 3-фторпропан-1-сульфонилхлорид (2,29 мл, 19,2 ммоль) и триэтиламин (3,83 мл, 27,5 ммоль), и смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Остаток растворяют в ацетонитриле (45,8 мл) и обрабатывают 2,0 M Na2CO3 (36,6 мл, 73,3 ммоль) при 60°C в течение 45 минут. pH доводят до примерно 3 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением 3-фтор-N-(2,4,5-трифтор-3-йодфенил)пропан-1-сульфонамида (2,72 г, 75%). МС (хиад, m/z)=395,9 [M-H].
Стадия 3: Получение 3-фтор-N-(2,4,5-трифтор-3-йодфенил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. К раствору 3-фтор-N-(2,4,5-трифтор-3-йодфенил)-пропан-1-сульфонамида (2,72 г, 6,85 ммоль) в ДМФ (34,2 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 0,370 г, 9,25 ммоль) и (2-(хлорметокси)этил)триметилсилан (1,46 мл, 8,22 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексан с получением 3-фтор-N-(2,4,5-трифтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (3,11 г, 86%). 1H ЯМР (400 MГц, CDCl3) δ 7,39 (м, 1H), 4,95 (с, 2H), 4,62 (т, 1H), 4,51 (т, 1H), 3,66 (т, 2H), 3,24 (т, 2H), 2,25 (м, 2H), 0,93 (т, 2H), 0,03 (с, 9H).
Промежуточное соединение P42
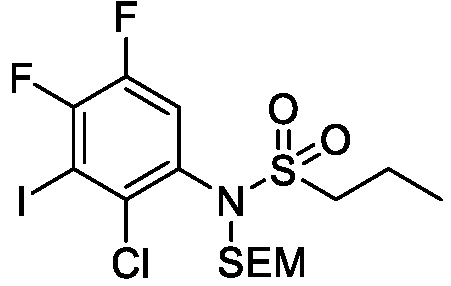
N- (2-хлор-4,5-дифтор-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 2-хлор-4,5-дифтор-3-йоданилина. 2-Хлор-4,5-дифторанилин (5,08 г, 31,06 ммоль) растворяют в ТГФ (200 мл) и охлаждают до -78°C в обратном потоке N2. Реакционную смесь медленно обрабатывают н-бутиллитием (2,5 M в гексане, 13,667 мл, 34,167 ммоль) и перемешивают при -78°C в течение 15 минут. Реакционную смесь обрабатывают по каплям ТГФ раствором (50 мл) 1,2-бис(хлордиметилсилил)этана (7,021 г, 32,61 ммоль) и перемешивают при -78°C в течение 15 минут после завершения добавления. Реакционную смесь обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 13,667 мл, 34,167 ммоль). Ледяную баню удаляют, и реакционную смесь нагревают в течение 30 минут. Реакционную смесь обратно охлаждают до -78°C и медленно обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 13,667 мл, 34,167 ммоль) и перемешивают при -78°C в течение 30 минут. Реакционную смесь обрабатывают по каплям ТГФ раствором (100 мл) йода (11,825 г, 46,591 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Раствор гасят 4M HCl to примерно pH 1 и перемешивают в течение 30 минут. Раствор доводят до примерно pH 8 твердым NaHCO3 и перемешивают в течение 15 минут. Реакционную смесь обрабатывают 3,0 M тиосульфатом натрия и экстрагируют EtOAc (2x). Органические слои промывают водой, насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (5-50% EtOAc/гексан) с получением 2-хлор-4,5-дифтор-3-йоданилина (4,86 г, 54%). 1H ЯМР (400 MГц, ДМСО) δ 6,83 (м, 1H) 5,75 (с, 2H).
Стадия 2: Получение N-(2-хлор-4,5-дифтор-3-йодфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. К раствору 2-хлор-4,5-дифтор-3-йоданилина (2,1 г, 7,255 ммоль) в дихлорметане (36,28 мл) при 0°C добавляют пропан-1-сульфонилхлорид (1,708 мл, 15,24 ммоль) и триэтиламин (3,034 мл, 21,77 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4,5-дифтор-3-йодфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (3,1 г, 85%), который применяют без очистки.
Стадия 3: Получение N-(2-хлор-4,5-дифтор-3-йодфенил)пропан-1-сульфонамида. Раствор N-(2-хлор-4,5-дифтор-3-йодфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (3,1 г, 6,2 ммоль) в ацетонитриле (31 мл) и 2.0 M карбонате натрия (25 мл, 49 ммоль) нагревают до 60°C в течение 90 минут. Раствор доводят до примерно pH 4 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 1% - 20% EtOAc/ДХМ с получением N-(2-хлор-4,5-дифтор-3-йодфенил)пропан-1-сульфонамида (0,91 г, 37%). МС (хиад, m/z)=393,9 [M-H].
Стадия 4: Получение N-(2-хлор-4,5-дифтор-3-йодфенил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. К раствору N-(2-хлор-4,5-дифтор-3-йодфенил)пропан-1-сульфонамида (910 мг, 2,30 ммоль) в ДМФ (11,5 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 124,2 мг, 3,106 ммоль) и (2-(хлорметокси)этил)триметилсилан (489,6 мкл, 2,760 ммоль), и реакционную смесь перемешивают при 0°C в течение 1 часа. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексан с получением N-(2-хлор-4,5-дифтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (1118 мг, 92%). 1H ЯМР (400 MГц, CDCl3) δ 7,44 (м, 1H), 5,21 (д, 1H), 4,78 (д, 1H), 3,7 (м, 2H), 3,05 (м, 2H), 1,95 (м, 2H), 1,16 (т, 3H), 1,0 (м, 2H) 0,03 (с, 9H).
Промежуточное соединение P44
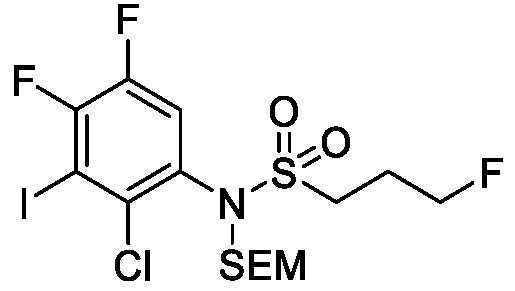
N- (2-хлор-4,5-дифтор-3-йодфенил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 2-хлор-4,5-дифтор-3-йоданилина. 2-Хлор-4,5-дифторанилин (5,08 г, 31,060 ммоль) растворяют в ТГФ (200 мл) и охлаждают до -78°C в обратном потоке азота. Реакционную смесь медленно обрабатывают н-бутиллитием (2,5 M в гексане, 13,667 мл, 34,167 ммоль) и перемешивают при -78°C в течение 15 минут. Реакционную смесь обрабатывают по каплям ТГФ раствором (50 мл) 1,2-бис(хлордиметилсилил)этана (7,021 г, 32,614 ммоль) и перемешивают при -78°C в течение 15 минут после завершения добавления. Реакционную смесь обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 13,667 мл, 34,167 ммоль) Ледяную баню удаляют, и реакционную смесь нагревают в течение 30 минут. Реакционную смесь обратно охлаждают до -78°C и медленно обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 13,667 мл, 34,167 ммоль) и перемешивают при -78°C в течение 30 минут. Реакционную смесь обрабатывают по каплям ТГФ раствором (100 мл) йода (11,825 г, 46,591 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Раствор гасят 4M HCl до примерно pH 1 и перемешивают в течение 30 минут. Реакционную смесь доводят до примерно pH 8 с применением твердого NaHCO3. Реакционную смесь обрабатывают 3,0 M тиосульфатом натрия и экстрагируют EtOAc (2x). Органические слои промывают водой, насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением 2-хлор-4,5-дифтор-3-йоданилина (4,86 г, 54%). 1H ЯМР (400 MГц, ДМСО) δ 6,83 (м, 1H) 5,75 (с, 2H).
Стадия 2: Получение N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фтор-N-((3-фторпропил)-сульфонил)пропан-1-сульфонамида. К раствору 2-хлор-4,5-дифтор-3-йоданилина (2,1 г, 7,26 ммоль) в дихлорметане (36,3 мл) при 0°C добавляют 3-фторпропан-1-сульфонилхлорид (1,81 мл, 15,2 ммоль) и триэтиламин (3,03 мл, 21,8 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 45 минут. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фтор-N-((3-фторпропил)-сульфонил)пропан-1-сульфонамида (3,8 г, 97%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 3: Получение N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фторпропан-1-сульфонамида. Раствор N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фтор-N-((3-фторпропил)сульфонил)-пропан-1-сульфонамида (3,8 г, 7,07 ммоль) в ацетонитриле (35,3 мл) и 2,0 M карбонате натрия (28,3 мл, 56,5 ммоль) нагревают до 60°C в течение 1 часа. Раствор доводят до примерно pH 4 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают двуокисью кремния, элюируя 1% - 20% EtOAc/ДХМ с получением N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фторпропан-1-сульфонамида (2,89 г, 99%). МС (хиад, m/z)=411,9 [M-H].
Стадия 4: Получение N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида. К раствору N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фторпропан-1-сульфонамида (2,89 г, 6,99 ммоль) в ДМФ (34,9 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 0,377 г, 9,43 ммоль) и (2-(хлорметокси)этил)триметилсилан (1,49 мл, 8,39 ммоль), и реакционную смесь перемешивают при 0°C в течение 45 минут. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (3,46 г, 91%). 1H ЯМР (400 MГц, CDCl3) δ 7,48 (м, 1H), 5,21 (д, 1H), 4,78 (д, 1H), 4,65 (т, 1H), 4,58 (т, 1H), 3,7 (м, 2H), 3,3 (т, 2H), 2,3 (м, 2H), 0,95 (м, 2H), 0,02 (с, 9H).
Промежуточное соединение P45
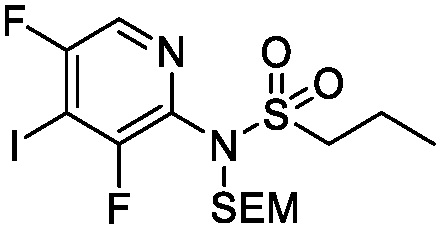
N- (3,5-дифтор-4-йодпиридин - 2-ил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 3,5-дифтор-4-йодпиридин-2-амина. 2-Амино-3,5-дифторпиридин (7,07 г, 54,3 ммоль) растворяют в ТГФ (250 мл) и охлаждают до -78°C. Реакционную смесь обрабатывают 2,5 M раствором н-бутиллития в гексане (54,3 мл, 136 ммоль) и перемешивают при -78°C в течение 1 часа после завершения добавления. Реакционную смесь обрабатывают по каплям 50 мл ТГФ раствором йода (41,4 г, 163 ммоль) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят 10% тиосульфатом натрия и экстрагируют EtOAc (2x). Органические слои промывают водой и насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 5% - 60% гексаном/ацетоном) с получением 3,5-дифтор-4-йодпиридин-2-амина (8,5g, 61%). МС (хиад, m/z)=257,0 [M+H].
Стадия 2: Получение N-(3,5-дифтор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида. Триэтиламин (1,134 мл, 8,138 ммоль) добавляют к 3,5-дифтор-4-йодпиридин-2-амину (0,992 г, 3,875 ммоль) и пропан-1-сульфонилхлориду (0,912 мл, 8,138 ммоль) в ДХМ (38,75 мл), и смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические экстракты промывают насыщенным раствором соли и сушат с Na2SO4 и концентрируют с получением N-(3,5-дифтор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,8 г, 99%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 3: Получение N-(3,5-дифтор-4-йодпиридин-2-ил)пропан-1-сульфонамида. Раствор N-(3,5-дифтор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида (1,8 г, 3,84 ммоль) в ацетонитриле (19,2 мл) и 2,0 M карбоната натрия (13,5 мл, 26,9 ммоль) нагревают до 80°C в течение 1 часа. Раствор доводят до примерно pH 3 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 5% - 95% EtOAc/ДХМ с получением N-(3,5-дифтор-4-йодпиридин-2-ил)пропан-1-сульфонамида (0,664 г, 48%). МС (хиад, m/z)=363,0 [M+H].
Стадия 4: Получение N-(3,5-дифтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. К раствору N-(3,5-дифтор-4-йодпиридин-2-ил)пропан-1-сульфонамида (661 мг, 1,83 ммоль) в ДМФ (9,1 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (98,6 мг, 2,46 ммоль) и (2-(хлорметокси)этил)триметилсилане (388 мкл, 2,19 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(3,5-дифтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (808 мг, 90%) в виде масла. 1H ЯМР (400 MГц, CDCl3) δ 8,07 (с, 1H), 5,10 (с, 2H), 3,68 (т, 2H), 3,34 (т, 2H), 1,95 (м, 2H) 1,09 (т, 2H), 0,89 (т, 3H), 0,02 (с, 9H); МС (хиад, m/z)=493,1 [M+H].
Промежуточное соединение P46
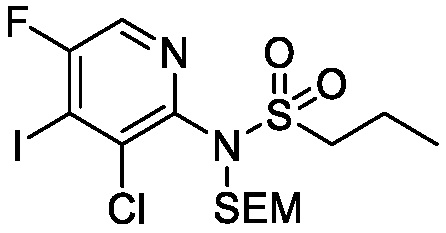
N- (3,5-дифтор-4-йодпиридин - 2-ил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 3-хлор-5-фтор-4-йодпиридин-2-амина. В 500 мл колбу добавляют 3-хлор-5-фторпиридин-2-амин (5,67 г, 38,7 ммоль) и тетрагидрофуран (193 мл). Раствор охлаждают до -78°C и обрабатывают добавлением по каплям 2,5 M раствора н-бутиллития в гексане (38,7 мл, 96,7 ммоль) и перемешивают при -78°C в течение 1 часа. Раствор йода (29,5 г, 116 ммоль) в тетрагидрофуране (59,3 мл) добавляют по каплям из капельной воронки. Реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 30 минут. Реакционную смесь гасят насыщенным водным Na2S2O3 и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 0% - 95% ацетон/ДХМ с получением 3-хлор-5-фтор-4-йодпиридин-2-амина (5,7g, 54%). МС (хиад, m/z)=272,9 [M+H].
Стадия 2: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида. К раствору 3-хлор-5-фтор-4-йодпиридин-2-амина (4,0 г, 14,68 ммоль) и пропан-1-сульфонилхлорида (3,456 мл, 30,83 ммоль) в дихлорметане (73,41 мл) добавляют триэтиламин (4,297 мл, 30,83 ммоль), и смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида (7 г, 98%), который применяют непосредственно на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=485,0 [M+H].
Стадия 3: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)пропан-1-сульфонамида. Раствор N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида (7,0 г, 14 ммоль) в ацетонитриле (72 мл) и 2,0 M Na2CO3 (51 мл, 101 ммоль) перемешивают при температуре окружающей среды в течение 60 часов и затем нагревают до 70°C в течение 90 минут. Раствор доводят до примерно pH 3 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Реакционную смесь Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают двуокисью кремния, элюируя 5% - 75% EtOAc/гексаном с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)пропан-1-сульфонамида (4,3 г, 79%). МС (хиад, m/z)=378,9 [M+H].
Стадия 4: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида. К раствору N-(3-хлор-5-фтор-4-йодпиридин-2-ил)пропан-1-сульфонамида (4,30 г, 11,4 ммоль) в ДМФ (56,8 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (0,613 г, 15,3 ммоль) и (2-(хлорметокси)этил)триметилсилан (2,42 мл, 13,6 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 1 часа. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (4,08 г, 71%) в виде масла. 1H ЯМР (400 MГц, CDCl3) δ 8,13 (с, 1H), 5,06 (с, 2H), 3,68 (т, 2H), 3,38 (т, 2H), 1,95 (м, 2H) 1,09 (т, 2H), 0,89 (т, 3H), 0,02 (с, 9H); МС (хиад, m/z)=509,1 [M+H].
Промежуточное соединение P47
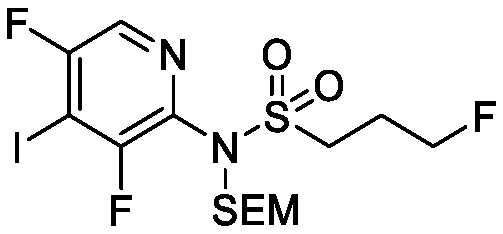
N- (3,5-дифтор-4-йодпиридин - 2-ил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 3,5-дифтор-4-йодпиридин-2-амина. 2-Амино-3,5-дифторпиридин (7,07 г, 54,3 ммоль) растворяют в ТГФ (250 мл) и охлаждают до -78°C. Реакционную смесь обрабатывают 2,5 M раствором н-бутиллития в гексане (54,3 мл, 136 ммоль) и перемешивают при -78°C в течение 1 часа после завершения добавления. Реакционную смесь обрабатывают по каплям 50 мл ТГФ раствора йода (41,4 г, 163 ммоль) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят 10% тиосульфатом натрия и экстрагируют EtOAc (2x). Органические слои промывают водой и насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 5% - 60% гексаном/ацетоном с получением 3,5-дифтор-4-йодпиридин-2-амина (8,5 г, 61%). МС (хиад, m/z)=257,0 [M+H].
Стадия 2: Получение N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)-сульфонил)пропан-1-сульфонамида. К раствору 3,5-дифтор-4-йодпиридин-2-амина (1,40 г, 5,47 ммоль) и 3-фторпропан-1-сульфонилхлорида (1,43 мл, 12,0 ммоль) в дихлорметане (27,3 мл) добавляют триэтиламин (2,29 мл, 16,4 ммоль) и смесь перемешивают при температуре окружающей среды в течение 45 минут. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (2,7 г, 98%) который применяют непосредственно на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=504,9 [M+H].
Стадия 3: Получение N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида. Раствор N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (2,7 г, 5,4 ммоль) в ацетонитриле (27 мл) и 2,0 M карбонате натрия (21 мл, 43 ммоль) нагревают до 60°C в течение 90 минут. Раствор доводят до примерно pH 4 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 1% - 20% EtOAc/ДХМ с получением N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида (1,6 г, 79%). МС (хиад, m/z)=380,9 [M+H].
Стадия 4: Получение N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида. К раствору N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида (1,6 г, 4,209 ммоль) в ДМФ (21,05 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 0,2273 г, 5,682 ммоль) и затем (2-(хлорметокси)этил)триметилсилан (0,8959 мл, 5,051 ммоль) и реакционную смесь перемешивают при 0°C в течение 30 минут. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (1,97 г, 92%) в виде масла. 1H ЯМР (400 MГц, CDCl3) δ 8,08 (с, 1H), 5,10 (с, 2H), 4,66 (т, 1H), 4,54 (т, 1H), 3,68 (т, 2H), 3,55 (т, 2H), 2,34 (м, 2H) 1,09 (т, 2H), 0,89 (т, 3H) 0,01 (с, 9H); МС (хиад, m/z)=511,0 [M+H].
Промежуточное соединение P48
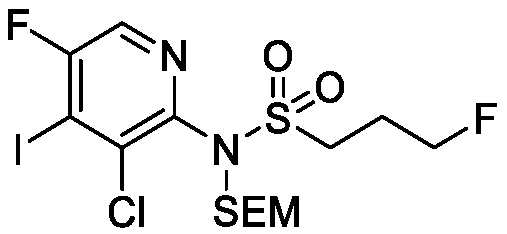
N- (3-хлор-5-фтор-4-йодпиридин - 2-ил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 3-хлор-5-фтор-4-йодпиридин-2-амина. В 500 мл колбу добавляют 3-хлор-5-фторпиридин-2-амин (5,67 г, 38,7 ммоль) и тетрагидрофуран (193 мл). Раствор охлаждают до -78°C и обрабатывают добавлением по каплям 2,5 M раствора н-бутиллития в гексане (38,7 мл, 96,7 ммоль) и перемешивают при -78°C в течение 1 часа. Затем раствор йода (29,5 г, 116 ммоль) в тетрагидрофуране (59,3 мл) добавляют по каплям из капельной воронки, и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 30 минут. Реакционную смесь гасят насыщенным водным Na2S2O3 и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 0-95% ацетон/ДХМ) с получением 3-хлор-5-фтор-4-йодпиридин-2-амина (5,7 г, 54%). МС (хиад, m/z)=272,9 [M+H].
Стадия 2: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)-сульфонил)пропан-1-сульфонамида. К раствору 3-хлор-5-фтор-4-йодпиридин-2-амин (1,28 г, 4,698 ммоль) и 3-фторпропан-1-сульфонилхлорида (1,362 мл, 11,75 ммоль) в дихлорметане (23,49 мл) добавляют триэтиламин (1,637 мл, 11,75 ммоль) и смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь гасят водой и экстрагируют ДХМ. Реакционную смесь Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (1,85 г, 76%) который применяют непосредственно на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=520,9 [M+H].
Стадия 3: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида. Раствор N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)сульфонил)-пропан-1-сульфонамида (1,85 г, 3,55 ммоль) в ацетонитриле (17,8 мл) и 2,0 M Na2CO3 (12,4 мл, 24,9 ммоль) нагревают до 60°C в течение 90 минут. Раствор доводят до примерно pH 3 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле, элюируя 5% - 95% EtOAc/гексаном с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида (1,01 г, 72%). МС (хиад, m/z)=396,9 [M+H].
Стадия 4: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида. К раствору N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида (1,01 г, 2,55 ммоль) в ДМФ (12,7 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 0,138 г, 3,44 ммоль) и (2-(хлорметокси)этил)триметилсилан (0,542 мл, 3,06 ммоль) и реакционную смесь перемешивают при 0°C в течение 2 часов. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,912 г, 68%) в виде масла. 1H ЯМР (400 MГц, CDCl3) δ 8,14 (с, 1H), 5,06 (с, 2H), 4,65 (т, 1H), 4,54, (т, 1H), 3,66 (т, 2H), 3,59 (т, 2H), 2,34 (м, 2H) 1,09 (т, 2H), 0,89 (т, 3H) 0,01 (с, 9H); МС (хиад, m/z)=526,9 [M+H].
Промежуточное соединение P49
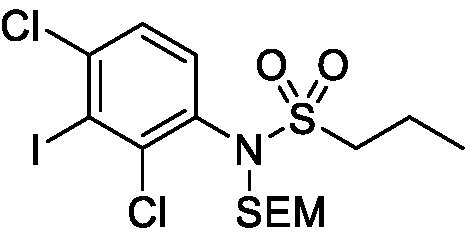
N- (2,4-дихлор-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 2,4-дихлор-3-йоданилина. Раствор 2,4-дихлоранилина (2,0 г, 12,345 ммоль) в 80 мл ТГФ охлаждают до -78°C под азотом, и раствор медленно обрабатывают н-бутиллитием (2,5 M в гексане, 5,432 мл, 13,579 ммоль) и перемешивают при -78°C в течение 15 минут. Затем раствор 1,2-бис(хлордиметилсилил)этан (2,790 г, 12,962 ммоль) в 30 мл ТГФ добавляют к реакционной смеси, и смесь перемешивают при -78°C в течение 15 минут. Медленно добавляют н-бутиллитий (2,5 M в гексане, 5,432 мл, 13,579 ммоль), и реакционную смесь удаляют из бани -78°C и нагревают в течение 30 минут. Реакционную смесь обратно охлаждают до -78°C и медленно добавляют н-бутиллитий (2,5 M в гексане, 5,432 мл, 13,579 ммоль) и перемешивают при -78°C в течение 20 минут. Затем медленно добавляют раствор йода (4,70 г, 18,517 ммоль) в 30 мл ТГФ, и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Раствор гасят ~10 мл 4M HCl и перемешивают в течение 30 минут. Затем 20 мл насыщенного тиосульфата натрия добавляют и раствор доводят до примерно pH 8 твердым NaHCO3 и перемешивают в течение 15 минут. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают водой, насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле, элюируя 10% - 50% EtOAc/гексаном с получением 2,4-дихлор-3-йоданилина (1,35 г, 38%). МС (хиад, m/z)=287,9 [M+H].
Стадия 2: Получение N-(2,4-дихлор-3-йодфенил)пропан-1-сульфонамида. К раствору 2,4-дихлор-3-йоданилина (1,35 г, 4,69 ммоль) в дихлорметане (23,4 мл) при 0°C добавляют триэтиламин (1,96 мл, 14,1 ммоль) и пропан-1-сульфонилхлорид (1,11 мл, 9,85 ммоль), и смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь гасят водой и экстрагируют ДХМ. Объединенные органические слои концентрируют и остаток растворяют в ацетонитрил (23,4 мл), обрабатывают 2,0 M Na2CO3 (18,8 мл, 37,5 ммоль) и перемешивают при 60°C в течение 16 часов. Раствор доводят до примерно pH 5 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют с получением N-(2,4-дихлор-3-йодфенил)пропан-1-сульфонамида (1,8 г, 97%). МС (хиад, m/z)=391,9 [M-H].
Стадия 3: Получение N-(2,4-дихлор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида. К раствору N-(2,4-дихлор-3-йодфенил)пропан-1-сульфонамида (1,8 г, 4,57 ммоль) в ДМФ (30,5 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 0,247 г, 6,17 ммоль) и затем (2-(хлорметокси)этил)триметилсилан (0,917 мл, 5,48 ммоль), и раствор перемешивают при температуре окружающей среды в течение 45 минут. Реакционную смесь гасят водой и экстрагируют EtOAc. Объединенные органические слои концентрируют и остаток очищают хроматографией на силикагеле, элюируя 10% - 50% EtOAc/гексаном с получением N-(2,4-дихлор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (2,3 г, 96%). 1H ЯМР (400 MГц, CDCl3) δ 7,45 (с, 1H), 7,25 (с, 1H), 5,21 (д, 1H), 4,77 (д, 1H), 3,68 (м, 2H), 3,09 (т, 2H), 1,91 (м, 2H), 1,05 (т, 3H), 0,91 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P50
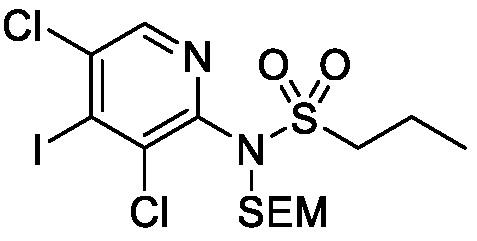
N- (3,5-дихлор-4-йодпиридин - 2-ил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 3,5-дихлор-4-йодпиридин-2-амина. Раствор 3,5-дихлорпиридин-2-амина (2,9 г, 17,791 ммоль) в 150 мл ТГФ охлаждают до -78°C под N2 и раствор медленно обрабатывают н-бутиллитием (2,5 M в гексане, 7,83 мл, 19,570 ммоль) и перемешивают при -78°C в течение 15 минут. Затем раствор 1,2-бис(хлордиметилсилил)этана (4,021 г, 18,681 ммоль) в 50 мл ТГФ медленно добавляют к реакционной смеси и смесь перемешивают при -78°C в течение 15 минут. Затем медленно добавляют н-бутиллитий (2,5 M в гексане, 7,83 мл, 19,570 ммоль), и реакционную смесь удаляют из бани -78°C и нагревают в течение 30 минут. Реакционную смесь обратно охлаждают до -78°C и медленно добавляют н-бутиллитий (2,5 M в гексане, 7,83 мл, 19,570 ммоль), и смесь перемешивают при -78°C в течение 20 минут. Раствор йода (6,773 г, 26,687 ммоль) в 50 мл ТГФ медленно добавляют, и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Раствор гасят примерно 10 мл 4M HCl и перемешивают в течение 30 минут. Затем добавляют 20 мл насыщенного тиосульфата натрия и раствор доводят до примерно pH 8 твердым NaHCO3 и перемешивают в течение 15 минут. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают водой, насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (10-50% EtOAc/гексан) с получением 3,5-дихлор-4-йодпиридин-2-амина (3,35 г, 65%). МС (хиад, m/z)=288,9 [M+H].
Стадия 2: Получение N-(3,5-дихлор-4-йодпиридин-2-ил)пропан-1-сульфонамида. К раствору 3,5-дихлор-4-йодпиридин-2-амина (3,35 г, 11,6 ммоль) в дихлорметане (58,0 мл) при 0°C добавляют пропан-1-сульфонилхлорид (2,73 мл, 24,4 ммоль) и триэтиламин (4,85 мл, 34,8 ммоль), и смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь гасят водой и экстрагируют ДХМ. Объединенные органические слои концентрируют и остаток растворяют в ацетонитриле (58,0 мл) и обрабатывают 2,0 M Na2CO3 (46,4 мл, 92,8 ммоль) при 60°C в течение 4 часов. Раствор доводят до pH ~5 10% лимонной кислотой. Реакционную смесь гасят водой и экстрагируют ДХМ. Объединенные органические слои концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(3,5-дихлор-4-йодпиридин-2-ил)пропан-1-сульфонамида (1,85 г, 40%). МС (хиад, m/z)=394,9 [M+H].
Стадия 3: Получение N-(3,5-дихлор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. К раствору N-(3,5-дихлор-4-йодпиридин-2-ил)пропан-1-сульфонамида (1,85 г, 4,68 ммоль) в ДМФ (23,4 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 0,253 г, 6,32 ммоль) и (2-(хлорметокси)этил)триметилсилан (0,997 мл, 5,62 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь гасят водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и остаток очищают хроматографией на силикагеле, элюируя 5% - 50% EtOAc/гексаном с получением N-(3,5-дихлор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (1,94 г, 79%). МС (хиад, m/z)=525,0 [M+H].
Промежуточное соединение P51
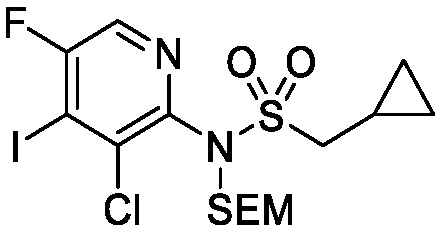
N- (3-хлор-5-фтор-4-йодпиридин - 2-ил)-1-циклопропил- N- ((2-(триметилсилил)этокси)-метил)метансульфонамид
Стадия 1: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-1-циклопропил-N-((циклопропил-метил)сульфонил)метансульфонамида. Триэтиламин (1,586 мл, 11,38 ммоль) добавляют к раствору 3-хлор-5-фтор-4-йодпиридин-2-амина (1 г, 3,670 ммоль) и циклопропил-метансульфонилхлорид (0,9008 мл, 7,708 ммоль) в ДХМ (37 мл) при 0°C. Реакционную смесь перемешивают при 0°C в течение 10 минут, затем нагревают до 40°C в течение 16 часов. Реакционную смесь гасят 100 мл воды и экстрагируют ДХМ (2×40 мл). Органические слои объединяют, промывают насыщенным раствором соли и сушат с Na2SO4. Раствор фильтруют и концентрируют с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-1-циклопропил-N-((циклопропилметил)-сульфонил)метансульфонамида (1,68 г, 90%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-1-циклопропилметан-сульфонамида. Раствор N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-1-циклопропил-N-((циклопропилметил)сульфонил)метансульфонамида (1,68 г, 3,30 ммоль) в ацетонитриле (40 мл) и моногидрате карбоната натрия (15 мл, 23,1 ммоль,1,54 M) нагревают до 80°C в течение одного часа. Реакционную смесь концентрируют для удаления ацетонитрила, и pH was доводят до примерно 3 10% лимонной кислотой в воде. Продукт экстрагируют ДХМ (3×50 мл) и органические экстракты объединяют, сушат над сульфатом натрия, фильтруют и концентрируют с получением темно-коричневого твердого вещества, которое очищают хроматографией на силикагеле, элюируя 90% ДХМ/10% EtOAc с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-1-циклопропилметансульфонамида (400 мг, 31%). МС (хиад, m/z)=390,9 (M+H).
Стадия 3: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-1-циклопропил-N-((2-(триметил-силил)этокси)метил)метансульфонамида. N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-1-цикло-пропилметансульфонамид (400 мг, 1,024 ммоль) растворяют в ДМФ (5,1 мл). Раствор охлаждают до 0°C с применением ледяной бани и добавляют гидрид натрия (60% суспензия в минеральном масле, 43,01 мг, 1,075 ммоль). Реакционную смесь перемешивают при 0°C в течение 15 мин и (2-(хлорметокси)этил)триметилсилан (218,0 мкл, 1,229 ммоль) добавляют по каплям. Реакционную смесь перемешивают при температуре окружающей среды в течение 2 часов и затем гасят водой и экстрагируют EtOAc. Органическую фазу сушат над Na2SO4, фильтруют и концентрируют. Неочищенную смесь очищают гель-хроматографией на двуокиси кремния, элюируя 0-100% EtOAc/Hex с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-1-циклопропил-N-((2-(триметилсилил)этокси)метил)-метансульфонамида в виде желтого масла (317,3 мг, 59%). МС (хиад, m/z)=521,0 (M+H).
Промежуточное соединение P52
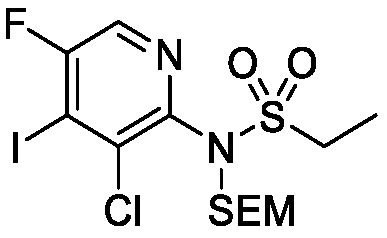
N- (3-хлор-5-фтор-4-йодпиридин - 2-ил)- N- ((2-(триметилсилил)этокси)метил)этан-сульфонамид
Стадия 1: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-(этилсульфонил)этан-сульфонамида. Триэтиламин (1,586 мл, 11,38 ммоль) добавляют к 3-хлор-5-фтор-4-йодпиридин-2-амину (1 г, 3,670 ммоль) и этансульфонилхлориду (0,7303 мл, 7,708 ммоль) в ДХМ (37 мл) при 0°C. Реакционную смесь перемешивают при 0°C в течение 10 минут и затем нагревают до температуры окружающей среды в течение ночи. Реакционную смесь гасят водой (100 мл) и водный слой экстрагируют ДХМ (2×40 мл). Органические слои объединяют и промывают насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-(этилсульфонил)этансульфонамида (1,5 г, 89%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)этансульфонамида. К раствору N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-(этилсульфонил)этансульфонамида (1,5 г, 3,29 ммоль) в ацетонитриле (30 мл) добавляют 15 мл водный раствор моногидрата карбоната натрия (2,9 г, 23,0 ммоль), и реакционную смесь нагревают до 80°C в течение 1 часа. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют для удаления ацетонитрила, и затем обрабатывают 10% водным раствором лимонной кислоты (50 мл), затем EtOAc (50 мл). Слои разделяют, и водный слой экстрагируют дополнительным EtOAc, и органические экстракты объединяют и промывают насыщенным раствором соли и сушат над Na2SO4, фильтруют и концентрируют с получением светло-коричневого твердого вещества, которое очищают гель-хроматографией на двуокиси кремния (ДХМ/EtOAc) с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)этансульфонамида (623 мг, 41%). МС (хиад, m/z)=364,8 (M+H).
Стадия 3: Получение N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)-метил)этансульфонамида. N-(3-хлор-5-фтор-4-йодпиридин-2-ил)этансульфонамид (623 мг, 1,71 ммоль) растворяют в ДМФ (11 мл) и охлаждают на бане с ледяной водой в течение 5 минут в атмосфере аргона. Гидрид натрия (60% суспензия в минеральном масле, 75,19 мг, 1,88 ммоль) добавляют к раствору, и реакционную смесь перемешивают до прекращения выделения газа и затем обрабатывают (2-(хлорметокси)этил)триметилсиланом (363,7 мкл, 2,05 ммоль). Реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 74 часов, затем нагревают до 40°C в течение 6 часов. Реакционную смесь гасят водой (50 мл) и затем экстрагируют EtOAc (50 мл). Органическую фазу промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Неочищенную реакционную смесь очищают гель-хроматографией на двуокиси кремния, элюируя гексаном/EtOAc с получением N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)-метил)этансульфонамида в виде желтого масла (751,8 мг, 89%). МС (хиад, m/z)=495,0 (M+H).
Промежуточное соединение P53
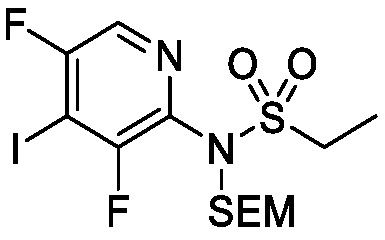
N- (3,5-дифтор-4-йодпиридин - 2-ил)- N- ((2-(триметилсилил)этокси)метил)этансульфонамид
Стадия 1: Получение N-(3,5-дифтор-4-йодпиридин-2-ил)-N-(этилсульфонил)этансульфонамида. К раствору 3,5-дифтор-4-йодпиридин-2-амина (400 мг, 1,563 ммоль) и этансульфонилхлорида (0,370 мл, 3,906 ммоль) в дихлорметане (7,81 мл) добавляют триэтиламин (0,653 мл, 4,688 ммоль), и смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь разбавляют EtOAc и затем промывают водой затем насыщенным раствором соли. Органические экстракты объединяют и сушат над Na2SO4, фильтруют и концентрируют с получением N-(3,5-дифтор-4-йодпиридин-2-ил)-N-(этилсульфонил)этансульфонамида (0,500 г, 73%), который применяют непосредственно на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=440,9 (M+H).
Стадия 2: Получение N-(3,5-фтор-4-йодпиридин-2-ил)этансульфонамида. Раствор N-(3,5-дифтор-4-йодпиридин-2-ил)-N-(этилсульфонил)этансульфонамида (0,50 г, 1,14 ммоль) в ацетонитриле (5,68 мл) и 2,0 M водном карбонате натрия (4,5 мл, 9,09 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3,5-фтор-4-йодпиридин-2-ил)этансульфонамида (0,364 г, 92%). МС (хиад, m/z)=349,0 (M+H).
Стадия 3: Получение N-(3,5-дифтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)-метил)этансульфонамида. К раствору N-(3,5-дифтор-4-йодпиридин-2-ил)этан-сульфонамида (0,364 г, 1,05 ммоль) в ДМФ (4,2 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (56,5 мг, 1,41 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,223 мл, 1,25 ммоль), и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3,5-дифтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)этан-сульфонамида (0,343 г, 69%) в виде масла. 1H ЯМР (400 MГц, CDCl3) δ 8,06 (с, 1H), 5,09 (с, 2H), 3,69-3,65 (дд, 2H), 3,37 (q, 2H), 1,46 (т, 3H), 0,86-0,90 (дд, 2H), 0,01 (с, 9H).
Промежуточное соединение P54
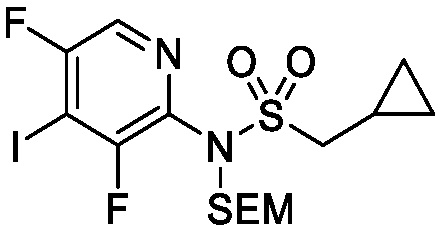
1-циклопропил- N- (3,5-дифтор-4-йодпиридин - 2-ил)- N- ((2-(триметилсилил)этокси)метил)-метансульфонамид
Стадия 1: Получение 1-циклопропил-N-((циклопропилметил)сульфонил)-N-3,5-дифтор-4-йодпиридин-2-ил)метансульфонамида. К раствору 3,5-дифтор-4-йодпиридин-2-амина (500 мг, 1,95 ммоль) и циклопропилметансульфонилхлорида (0,656 мл, 5,86 ммоль) в дихлорметане (9,8 мл) добавляют триэтиламин (1,089 мл, 7,81 ммоль) и смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь разбавляют EtOAc и промывают водой, затем насыщенным раствором соли. Объединенные органические слои сушат с Na2SO4, фильтруют и концентрируют с получением 1-циклопропил-N-((циклопропилметил)сульфонил)-N-3,5-дифтор-4-йодпиридин-2-ил)метансульфонамида (0,915 г, 95%), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение 1-циклопропил-N-(3,5-фтор-4-йодпиридин-2-ил)метансульфонамида. Раствор 1-циклопропил-N-((циклопропилметил)сульфонил)-N-3,5-дифтор-4-йодпиридин-2-ил)метансульфонамида (0,915 г, 1,86 ммоль) в ацетонитриле (9,3 мл) и 2,0 M водном карбонате натрия (7,4 мл, 14,9 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением 1-циклопропил-N-(3,5-фтор-4-йодпиридин-2-ил)метансульфонамида (0,680 г, 98%). МС (хиад, m/z)=372,9 (M-H).
Стадия 3: Получение 1-циклопропил-N-(3,5-дифтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)-этокси)метил)метансульфонамида. К раствору 1-циклопропил-N-(3,5-фтор-4-йодпиридин-2-ил)метансульфонамида (0,680 г, 1,82 ммоль) в ДМФ (7,2 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (98,1 мг, 2,45 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,387 мл, 2,18 ммоль), и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои, сушат над Na2SO4, фильтруют, концентрируют. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением 1-циклопропил-N-(3,5-дифтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)-метансульфонамида (0,682 г, 74%) в виде масла. 1H ЯМР (400 MГц, CDCl3) δ 8,04 (с, 1H), 5,13 (с, 2H), 3,73-3,68 (дд, 2H), 3,24-3,23 (д, 2H), 1,27-1,24 (м, 1H), 0,91-0,86 (дд, 2H), 0,74-0,69 (м, 2H), 0,52-0,48 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P55
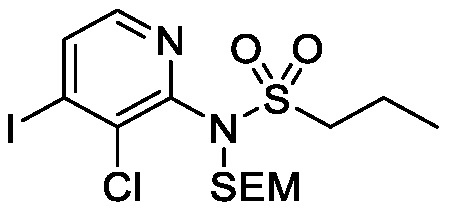
N- (3-хлор-4-йодпиридин - 2-ил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-хлор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида. К раствору 3-хлор-йодпиридин-2-амина (243 мг, 0,953 ммоль) и 1-пропансульфонилхлорида (0,246 мл, 2,19 ммоль) в дихлорметане (4,77 мл) добавляют триэтиламин (0,399 мл, 2,86 ммоль) и смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь разбавляют EtOAc и промывают водой, затем насыщенным раствором соли и затем органические слои сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-хлор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида (0,189 г, 43%), который применяют непосредственно на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=466,9 (M+H).
Стадия 2: Получение N-(3-хлор-4-йодпиридин-2-ил)пропан-1-сульфонамида. Раствор N-(3-хлор-4-йодпиридин-2-ил)-N-(пропилсульфонил)пропан-1-сульфонамида (0,189 г, 0,405 ммоль) в ацетонитриле (2,03 мл) и 2,0 M водном карбонате натрия (1,6 мл, 3,24 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-хлор-4-йодпиридин-2-ил)пропан-1-сульфонамида (0,121 г, 83%). МС (хиад, m/z)=360,9 (M+H).
Стадия 3: Получение N-(3-хлор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида. К раствору N-(3-хлор-4-йодпиридин-2-ил)пропан-1-сульфонамида (0,121 г, 0,336 ммоль) в ДМФ (1,3 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (16,1 мг, 0,403 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,080 мл, 0,453 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc и затем объединенные органические экстракты промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-хлор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,13 г, 79%) в виде масла. 1H ЯМР (400 MГц, CDCl3) δ 8,00 (д, 1H), 7,80 (д, 1H), 5,09 (с, 2H), 3,67-3,63 (дд, 2H), 3,45-3,41 (дд, 2H), 2,01-1,91 (м, 2H), 1,08 (т, 3H), 0,91-0,86 (дд, 2H), 0,01 (с, 9H).
Промежуточное соединение P56

N- (3-хлор-4-йодпиридин - 2-ил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-хлор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)сульфонил)-пропан-1-сульфонамида. К раствору 3-хлор-йодпиридин-2-амина (256 мг, 1,01 ммоль) и 3-фторпропан-1-сульфонилхлорида (0,275 мл, 2,31 ммоль) в дихлорметане (5,03 мл) добавляют триэтиламин (0,421 мл, 3,02 ммоль), и смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь разбавляют EtOAc и промывают водой затем насыщенным раствором соли. Органические экстракты сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-хлор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (0,433 г, 86%) который применяют непосредственно на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=502,9 (M+H).
Стадия 2: Получение N-(3-хлор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида. Раствор N-(3-хлор-4-йодпиридин-2-ил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (0,433 г, 0,861 ммоль) в ацетонитриле (4,31 мл) и 2,0 M водном карбонате натрия (3,45 мл, 6,89 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат с Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-хлор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида (0,292 г, 90%). МС (хиад, m/z)=379,0 (M+H).
Стадия 3: Получение N-(3-хлор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. К раствору N-(3-хлор-4-йодпиридин-2-ил)-3-фторпропан-1-сульфонамида (0,292 г, 0,771 ммоль) в ДМФ (3,09 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (41,6 мг, 1,04 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,164 мл, 0,926 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-хлор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида (0,200 г, 51%) в виде масла. 1H ЯМР (400 MГц, CDCl3) δ 8,00 (д, 1H), 7,81 (д, 1H), 5,09 (с, 2H), 4,65 (т, 1H), 4,53 (т, 1H), 3,66-3,61 (дд, 4H), 2,41-2,27 (м, 2H), 0,90-0,86 (дд, 2H), 0,01 (с, 9H).
Промежуточное соединение P57
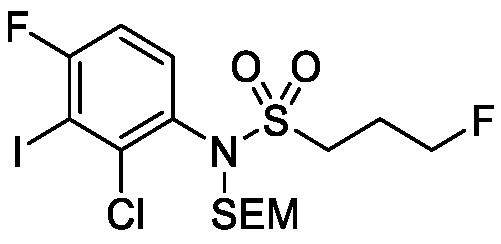
N- (2-хлор-4-фтор-3-йодфенил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение 2-хлор-4-фтор-3-йоданилина. 2-Хлор-4-фторанилин (2,44 г, 16,76 ммоль) растворяют в ТГФ (100 мл) и охлаждают до -78°C в обратном потоке N2. Реакционную смесь медленно обрабатывают н-бутиллитием (2,5 M в гексане, 7,38 мл, 18,44 ммоль) и затем перемешивают при -78°C в течение 15 минут после завершения добавления. Реакционную смесь обрабатывают по каплям 50 мл ТГФ раствора 1,2-бис(хлордиметилсилил)этана (3,79 г, 17,60 ммоль) и перемешивают при -78°C в течение 15 минут после завершения добавления. Реакционную смесь медленно обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 7,38 мл, 18,44 ммоль) и ледяную баню удаляют после завершения добавления и реакционную смесь нагревают в течение 30 минут. Реакционную смесь обратно охлаждают до -78°C и медленно обрабатывают дополнительным н-бутиллитием (2,5 M в гексане, 7,38 мл, 18,44 ммоль) и перемешивают при -78°C в течение 30 минут затем обрабатывают по каплям 50 мл ТГФ раствором йода (6,38 г, 25,14 ммоль). После завершения добавления ледяную баню удаляют, и реакционную смесь перемешивают в течение 1 часа. Реакционную смесь обрабатывают водой и затем подкисляют примерно pH 1 с применением 4,0 M HCl и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь нейтрализуют до примерно pH 8 с применением твердого NaHCO3 и затем обрабатывают водным раствором 3,0 M тиосульфата натрия. Реакционную смесь экстрагируют EtOAc (2×250 мл). Органические слои объединяют и промывают водой (1×100 мл) и насыщенным раствором соли (1×100 мл) и затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) затем С18 хроматографией с обращенной фазой, элюируя водой/ацетонитрилом с 0,1% ТФК. Желаемые фракции объединяют и разделяют между 4:1 ДХМ/ИПА и насыщенным NaHCO3 (1×100 мл). Органические слои сушат над Na2SO4, фильтруют и концентрируют с получением 2-хлор-4-фтор-3-йоданилина (2,70 г, 59%). 1H ЯМР (400 MГц, ДМСО-d6) δ 6,99-6,95 (м, 1H), 6,83-6,79 (м, 1H), 5,44 (с, 2H).
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)-3-фтор-N-((3-фторпропил)сульфонил)-пропан-1-сульфонамида. К раствору 2-хлор-4-фтор-3-йоданилина (1,7 г, 6,262 ммоль) и 3-фторпропан-1-сульфонилхлорида (1,815 мл, 15,66 ммоль) в дихлорметане (31,31 мл) добавляют триэтиламин (2,182 мл, 15,66 ммоль) и смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют ДХМ (2x) и затем объединенные органические экстракты промывают насыщенным раствором соли. Органические экстракты сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (3,0 г, 92%), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил)-3-фторпропан-1-сульфонамида. Раствор N-(2-хлор-4-фтор-3-йодфенил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида (3,0 г, 5,8 ммоль) в ацетонитриле (29 мл) и 2,0 M водном карбонате натрия (20 мл, 40 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и затем подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют ДХМ (2x), и объединенные органические слои промывают насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, гексан/EtOAc, с получением N-(2-хлор-4-фтор-3-йодфенил)-3-фторпропан-1-сульфонамида (2,1 г, 92%). МС (хиад, m/z)=393,9 (M-H).
Стадия 4: Получение N-(2-хлор-4-фтор-3-йодфенил)-3-фтор-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)-3-фторпропан-1-сульфонамида (2,1 г, 5,31 ммоль) в ДМФ (26,5 мл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 0,287 г, 7,17 ммоль), затем (2-(хлорметокси)этил)триметилсилан (1,13 мл, 6,37 ммоль) и реакционную смесь перемешивают при 0°C в течение 30 минут. Реакционную смесь обрабатывают водой и экстрагируют EtOAc и затем объединенные органические слои промывают водой, затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, гексан/EtOAc, с получением N-(2-хлор-4-фтор-3-йодфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (2,7 г, 97%). 1H ЯМР (500 MГц, CDCl3) δ 7,52 (дд, 1H), 7,07 (дд, 1H), 5,21 (д, 1H), 4,71 (д, 1H), 4,62 (т, 1H), 4,52 (т, 1H), 3,73 (м, 1H), 3,59 (м, 1H), 3,28 (т, 2H), 2,28 (м, 2H), 0,88, (т, 2H), 0,02 (с, 9H).
Промежуточное соединение P58
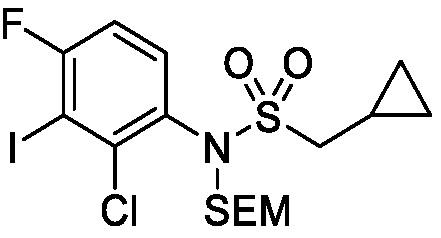
N- (2-хлор-4-фтор-3-йодфенил)-1-циклопропил- N- ((2-(триметилсилил)этокси)метил)-метансульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-1-циклопропил-N-((циклопропил-метил)сульфонил)метансульфонамида. К раствору 2-хлор-4-фтор-3-йоданалина (150 мг, 0,553 ммоль) и циклопропанметансульфонилхлорида (0,186 мл, 1,66 ммоль) в дихлорметане (2,76 мл) добавляют триэтиламин (0,308 мл, 2,21 ммоль), и смесь перемешивают при температуре окружающей среды в течение ночи. Реакционную смесь обрабатывают водой и экстрагируют EtOAc (2x). Объединенные органические слои промывают насыщенным раствором соли и сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-1-циклопропил-N-((циклопропилметил)сульфонил)метансульфонамида (0,281 г, 99%), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)-1-циклопропилметансульфонамида. Раствор N-(2-хлор-4-фтор-3-йодфенил)-1-циклопропил-N-((циклопропилметил)сульфонил)-метансульфонамида (0,281 г, 0,553 ммоль) в ацетонитриле (2,77 мл) и 2,0 M водном карбонате натрия (2,21 мл, 4,43 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и затем подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением N-(2-хлор-4-фтор-3-йодфенил)-1-циклопропилметансульфонамида (0,192 г, 89%). 1H ЯМР (400 MГц, CDCl3) δ 7,76-7,69 (м, 1H), 7,06-6,99 (м, 1H), 3,03 (д, 2H), 1,14-1,06 (м, 1H), 0,71-0,63 (м, 2H), 0,33-0,25 (м, 2H).
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил)-1-циклопропил-N-((2-(триметилсилил)-этокси)метил)метансульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)-1-циклопропилметансульфонамида (0,192 г, 0,493 ммоль) в ДМФ (1,9 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (26,6 мг, 0,665 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,105 мл, 0,591 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением N-(2-хлор-4-фтор-3-йодфенил)-1-циклопропил-N-((2-(триметил-силил)этокси)метил)метансульфонамида (0,16 г, 63%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,51 (д, 1H), 7,05 (д, 1H), 5,23 (д, 1H), 4,73 (д, 1H), 3,77-3,54 (м, 2H), 3,12-2,97 (м, 2H), 1,28-1,18 (м, 1H), 0,95-0,85 (м, 2H), 0,74-0,69 (м, 2H), 0,44-0,40 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P59
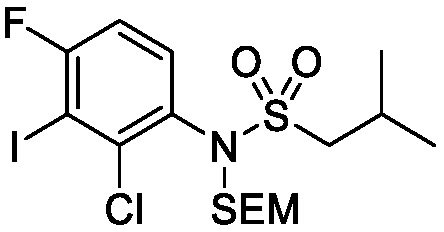
N- (2-хлор-4-фтор-3-йодфенил)-2-метил- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-(изобутилсульфонил)-2-метилпропан-1-сульфонамида. К раствору 2-хлор-4-фтор-3-йоданалина (152 мг, 0,560 ммоль) и 2-метилпропан-1-сульфонилхлорида (0,183 мл, 1,4 ммоль) в дихлорметане (2,8 мл) добавляют триэтиламин (0,234 мл, 1,68 ммоль) и реакционную смесь нагревают до 50°C в течение 15 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют, затем обрабатывают водой и экстрагируют EtOAc. Объединенные органические экстракты промывают насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-N-(изобутилсульфонил)-2-метилпропан-1-сульфонамида (0,274 г, 96%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)-2-метилпропан-1-сульфонамида. Раствор N-(2-хлор-4-фтор-3-йодфенил)-N-(изобутилсульфонил)-2-метилпропан-1-сульфонамида (0,274 г, 0,536 ммоль) в ацетонитриле (2,68 мл) и 2,0 M водном карбонате натрия (2,14 мл, 4,28 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и затем подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические экстракты промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(2-хлор-4-фтор-3-йодфенил)-2-метилпропан-1-сульфонамида (0,138 г, 63%). 1H ЯМР (400 MГц, CDCl3) δ 7,72-7,67 (м, 1H), 7,09-7,02 (м, 1H), 6,67 (шс, 1H), 2,94 (д, 2H), 2,35-2,23 (м, 1H), 1,09 (д, 6H).
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил-2-метил-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)-2-метилпропан-1-сульфонамида (0,138 г, 0,352 ммоль) в ДМФ (1,4 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (19,0 мг, 0,476 ммоль). Через 15 минут, реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,075 мл, 0,423 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением N-(2-хлор-4-фтор-3-йодфенил-2-метил-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,113 г, 61%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,52-7,49 (дд, 1H), 7,07-7,04 (дд, 1H), 5,21 (д, 1H), 4,70 (д, 1H), 3,76-3,55 (м, 2H), 3,01 (д, 2H), 2,38-2,28 (м, 1H), 1,10 (д, 6H), 0,95-0,85 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P60
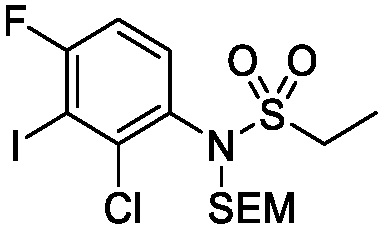
N- (2-хлор-4-фтор-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)этансульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-(этилсульфонил)этилсульфонамида. К раствору 2-хлор-4-фтор-3-йоданилина (150 мг, 0,553 ммоль) и этансульфонилхлорида (0,157 мл, 1,66 ммоль) в дихлорметане (2,8 мл) добавляют триэтиламин (0,308 мл, 2,21 ммоль), и смесь перемешивают при температуре окружающей среды в течение 15 часов. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические экстракты промывают насыщенным раствором соли затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-N-(этилсульфонил)этилсульфонамида (0,250 г, 99%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)этансульфонамида. Раствор N-(2-хлор-4-фтор-3-йодфенил)-N-(этилсульфонил)этиансульфонамида (0,250 г, 0,45 ммоль) в ацетонитриле (2,3 мл) и 2,0 M водном карбонате натрия (1,8 мл, 3,6 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и затем подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(2-хлор-4-фтор-3-йодфенил)этансульфонамида (0,158 г, 63%). 1H ЯМР (400 MГц, CDCl3) δ 7,73-7,68 (м, 1H), 7,07-7,01 (м, 1H), 3,10 (кв, 2H), 1,37 (т, 3H).
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил-N-((2-(триметилсилил)этокси)-метил)этансульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)-этансульфонамида (0,158 г, 0,435 ммоль) в ДМФ (1,7 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (23,5 мг, 0,587 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,093 мл, 0,521 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(2-хлор-4-фтор-3-йодфенил-N-((2-(триметилсилил)этокси)метил)этансульфонамида (0,166 г, 77%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,53-7,49 (дд, 1H), 7,07-7,04 (дд, 1H), 5,20 (д, 1H), 4,74 (д, 1H), 3,78-3,71 (м, 1H), 3,63-3,56 (м, 1H), 3,14 (кв, 2H), 1,45 (т, 3H), 0,97-0,84 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P61
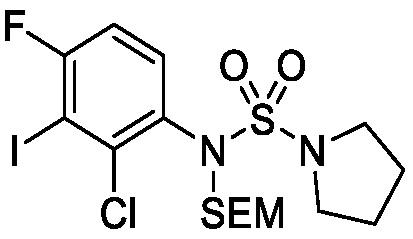
N- (2-хлор-4-фтор-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)пирролидин-1-сульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-(пирролидин-1-илсульфонил)пирролидин-1-сульфонамида. К раствору 2-хлор-4-фтор-3-йоданалина (150 мг, 0,553 ммоль) в пиридине (2,8 мл) добавляют пирролидин-1-сульфонилхлорид (0,070 мл, 0,608 ммоль), и смесь перемешивают при температуре окружающей среды в течение 15 часов. Реакционную смесь обрабатывают водой и экстрагируют EtOAc. Органические слои промывают насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-N-(пирролидин-1-илсульфонил)пирролидин-1-сульфонамида (0,290 г, 98%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)пирролидин-1-сульфонамида. Раствор N-(2-хлор-4-фтор-3-йодфенил)-N-(пирролидин-1-илсульфонил)пирролидин-1-сульфонамида (0,290 г, 0,539 ммоль) в ацетонитриле (2,7 мл) и 2,0 M водном карбонате натрия (2,2 мл, 4,3 ммоль) нагревают до 60°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и затем подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(2-хлор-4-фтор-3-йодфенил)пирролидин-1-сульфонамида (0,093 г, 42%).
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)-пирролидин-1-сульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)пирролидин-1-сульфонамида (0,093 г, 0,229 ммоль) в ДМФ (0,92 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (12,4 мг, 0,309 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,049 мл, 0,275 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc, с получением N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пирролидин-1-сульфонамида (0,087 г, 71%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,68-7,64 (дд, 1H), 7,05-7,02 (дд, 1H), 5,19 (д, 1H), 4,68 (д, 1H), 3,78-3,72 (м, 1H), 3,64-3,58 (м, 1H), 3,37 (д, 2H), 3,21 (д, 2H), 1,89-1,83 (м, 4H), 0,93-0,88 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P62
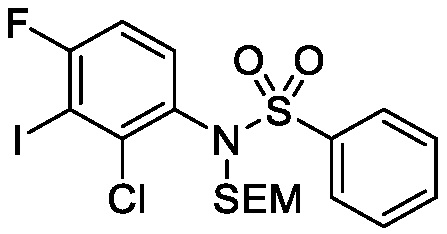
N- (2-хлор-4-фтор-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)бензолсульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-(фенилсульфонила). К раствору 2-хлор-4-фтор-3-йоданилина (152 мг, 0,560 ммоль) в тетрагидрофуране (1,4 мл) и пиридине (1,4 мл) добавляют фенилсульфонилхлорид (0,079 мл, 0,62 ммоль), и смесь перемешивают при температуре окружающей среды в течение 15 часов. Реакционную смесь концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-N-(фенилсульфонил)бензолсульфонамида (0,240 г, 104%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)-бензолсульфонамида. Раствор N-(2-хлор-4-фтор-3-йодфенил)-N-(фенилсульфонил)бензолсульфонамида (0,240 г, 0,45 ммоль) в 4:1 ТГФ/MeOH (2,3 мл) обрабатывают 2,0 M водным раствором KOH (1,3 мл, 2,25 ммоль) и смесь нагревают до 60°C в течение 1 часа. Реакционную смесь охлаждают до температуры окружающей среды и затем подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-бензолсульфонамида (0,220 г, 95%), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)-метил)бензолсульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)-бензолсульфонамида (0,220 г, 0,534 ммоль) в ДМФ (2,1 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (28,9 мг, 0,722 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,114 мл, 0,641 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат с Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, гексан/EtOAc, с получением N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (0,180 г, 62%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,74-7,71 (м, 2H), 7,61-7,56 (м, 1H), 7,49-7,44 (м, 2H), 7,23-7,19 (дд, 1H), 6,99-6,94 (дд, 1H), 5,32 (д, 1H), 4,73 (д, 1H), 3,70-3,58 (м, 2H), 0,92-0,82 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P63
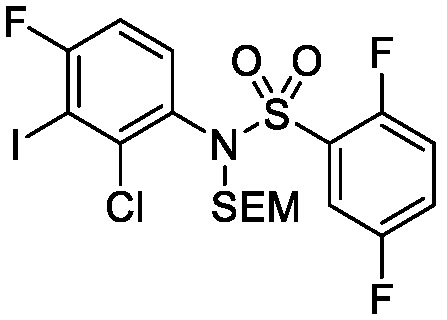
N- (2-хлор-4-фтор-3-йодфенил)-2,5-дифтор- N- ((2-(триметилсилил)этокси)метил)бензолсульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-((2,5-дифторфенил)сульфонил)-2,5-дифторбензолсульфонамида. К раствору 2-хлор-4-фтор-3-йоданилина (150 мг, 0,553 ммоль) в тетрагидрофуране (1,4 мл) и пиридине (1,4 мл) добавляют 2,5-дифторбензолсульфонилхлорид (0,082 мл, 0,608 ммоль) и смесь перемешивают при температуре окружающей среды в течение 15 часов. Реакционную смесь концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-N-((2,5-дифторфенил)сульфонил)-2,5-дифторбензолсульфонамида (0,253 г, 73%) который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)-2,5-дифторбензолсульфонамида. Раствор N-(2-хлор-4-фтор-3-йодфенил)-N-((2,5-дифторфенил)сульфонил)-2,5-дифтор-бензолсульфонамида (0,253 г, 0,41 ммоль) в 4:1 ТГФ/MeOH (2,03 мл) обрабатывают 2,0 M водным раствором КОН (1,03 мл, 2,05 ммоль), и реакционную смесь нагревают до 60°C в течение 1 часа. Реакционную смесь охлаждают до температуры окружающей среды и затем подкисляют примерно pH 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-2,5-дифторбензолсульфонамида (0,173 г, 70%), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил)-2,5-дифтор-N-((2-(триметилсилил)-этокси)метил)бензолсульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)-2,5-дифторбензолсульфонамида (0,173 г, 0,387 ммоль) в ДМФ (1,5 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (20,9 мг, 0,522 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,082 мл, 0,464 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, гексан/EtOAc с получением N-(2-хлор-4-фтор-3-йодфенил)-2,5-дифтор-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида (0,118 г, 53%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,41-7,34 (м, 2H), 7,29-7,22 (м, 1H), 7,21-7,14 (м, 1H), 7,05-6,99 (м, 1H), 5,43 (д, 1H), 4,91 (д, 1H), 3,82-3,63 (м, 2H), 0,98-0,83 (м, 2H), 0,02 (с, 9H).
Промежуточное соединение P64
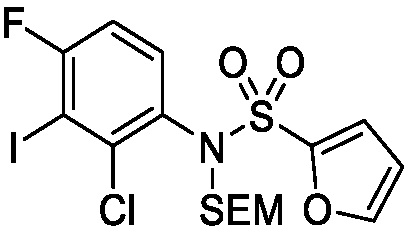
N- (2-хлор-4-фтор-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)фуран - 2-сульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-(фуран-2-илсульфонил)фуран-2-сульфонамида. К раствору 2-хлор-4-фтор-3-йоданалина (150 мг, 0,553 ммоль) в дихлорметане (2,8 мл) и триэтиламине (0,231 мл, 1,66 ммоль) добавляют фуран-2-сульфонилхлорид (0,193 мг, 1,16 ммоль) и смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-N-(фуран-2-илсульфонил)фуран-2-сульфонамида (0,061 г, 21%), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)фуран-2-сульфонамида. Раствор N-(2-хлор-4-фтор-3-йодфенил)-N-(фуран-2-илсульфонил)фуран-2-сульфонамида (0,061 г, 0,115 ммоль) в 4:1 ТГФ/MeOH (0,692 мл) обрабатывают 2,0 M водным раствором КОН (0,290 мл, 0,577 ммоль) и смесь перемешивают при температуре окружающей среды в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и затем подкисляют примерно pH примерно 3 10% водной лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc, и объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)фуран-2-сульфонамида (0,035 г, 76%), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил-N-((2-(триметилсилил)этокси)-метил)фуран-2-сульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)фуран-2-сульфонамида (0,035 г, 0,0879 ммоль) в ДМФ (0,35 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (4,75 мг, 0,119 ммоль). Через 15 минут реакционную смесь обрабатывают (2-(хлорметокси)этил)триметилсиланом (0,019 мл, 0,105 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc, и затем объединенные органические слои промывают водой затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(2-хлор-4-фтор-3-йодфенил-N-((2-(триметилсилил)этокси)метил)фуран-2-сульфонамида (0,032 г, 68%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,60-7,59 (дд, 1H), 7,30-7,26 (дд, 1H), 7,03-6,99 (дд, 1H), 6,91-6,90 (дд, 1H), 6,51-6,50 (дд, 1H), 5,30 (д, 1H), 4,87 (д, 1H), 3,76-3,63 (м, 2H), 0,96-0,82 (м, 2H), 0,01 (с, 9H).
Промежуточное соединение P65
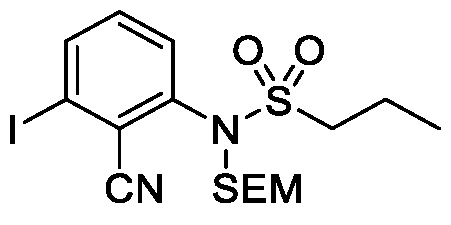
N- (2-циано-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-циано-3-йодфенил)пропан-1-сульфонамида. К раствору пропан-1-сульфонамида (46 мг, 0,37 ммоль) в ДМФ (810 мкл) добавляют гидрид натрия (60% суспензия в минеральном масле, 15 мг, 0,37 ммоль) и смесь нагревают до 40°C в течение 15 минут. Реакционную смесь обрабатывают 2-фтор-6-йодбензонитрилом (40 мг, 0,16 ммоль) и нагревают до 90°C в течение 2 часов и затем перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь подкисляют примерно pH 3 10% водной лимонной кислотой и экстрагируют EtOAc (2x), и объединенные органические слои промывают водой и насыщенным раствором соли затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(2-циано-3-йодфенил)пропан-1-сульфонамида (51 мг, 90%). 1H ЯМР (400 MГц, CDCl3) δ 7,76 (д, 1H), 7,68 (д, 1H), 7,28 (т, 1H), 6,89 (шс, 1H), 3,17 (т, 2H), 1,91 (м, 2H), 1,07 (т, 3H).
Стадия 2: Получение N-(2-циано-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. К раствору N-(2-циано-3-йодфенил)пропан-1-сульфонамида (50 мг, 0,143 ммоль) в ДМФ (952 мкл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 7,71 мг, 0,193 ммоль) и (2-(хлорметокси)этил)триметилсилан (28,7 мкл, 0,171 ммоль) и реакционную смесь перемешивают при 0°C в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc (2x), и затем объединенные органические слои промывают водой затем насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc) с получением N-(2-циано-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (50 мг, 73%). 1H ЯМР (400 MГц, CDCl3) δ 7,93 (д, 1H), 7,56 (д, 1H), 7,33 (т, 1H), 5,08 (с, 2H), 3,70 (т, 2H), 3,16 (т, 2H), 1,94 (м, 2H), 1,08 (т, 3H), 0,92 (т, 2H), 0,01 (с, 9H).
Промежуточное соединение P66
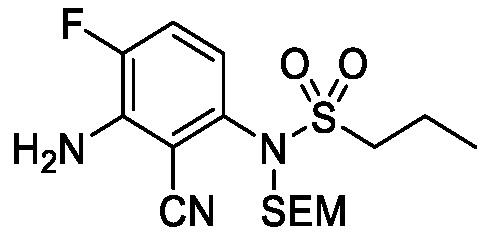
N- (3-амино-2-циано-4-фторфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-амино-2-циано-4-фторфенил)пропан-1-сульфонамида. К раствору пропан-1-сульфонамида (232 мг, 1,88 ммоль) в ДМСО (2 мл) добавляют гидрид натрия (60% суспензия в минеральном масле, 82,4 мг, 2,06 ммоль) и смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь обрабатывают 2 мл ДМСО раствором 2-амино-3,6-дифторбензонитрила (138 мг, 0,895 ммоль) и затем реакционную смесь нагревают до 100°C в течение 16 часов и затем при 120°C в течение 24 часов. Реакционную смесь охлаждают до температуры окружающей среды и обрабатывают 0,5 M водным NaOH и затем экстрагируют EtOAc (2x). Водный слой подкисляют примерно pH 4 и экстрагируют EtOAc (2x), и затем объединенные органические слои промывают насыщенным раствором соли затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-амино-2-циано-4-фторфенил)пропан-1-сульфонамида (26 мг, 11%). 1H ЯМР (400 MГц, CDCl3) δ 7,15 (дд, 1H), 6,92 (дд, 1H), 6,54 (шс, 1H), 4,58 (шс, 2H), 3,12 (т, 2H), 1,91 (м, 2H), 1,06 (т, 3H).
Стадия 2: Получение N-(3-амино-2-циано-4-фторфенил)-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида. К раствору N-(3-амино-2-циано-4-фторфенил)пропан-1-сульфонамида (25 мг, 0,0972 ммоль) в ДМФ (648 мкл) при 0°C добавляют гидрид натрия (60% суспензия в минеральном масле, 4,28 мг, 0,107 ммоль) и затем (2-(хлорметокси)этил)триметилсилан (17,1 мкл, 0,102 ммоль), и реакционную смесь перемешивают при 0°C в течение 30 минут Реакционную смесь обрабатывают водой и экстрагируют EtOAc (2x), и затем объединенные органические слои промывают водой затем насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-амино-2-циано-4-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (16 мг, 43%). 1H ЯМР (400 MГц, CDCl3) δ 7,17 (дд, 1H), 6,81 (дд, 1H), 5,01 (с, 2H), 4,60 (шс, 2H), 3,69 (т, 2H), 3,12 (т, 2H), 1,92 (м, 2H), 1,06 (т, 3H), 0,93 (т, 2H), 0,02 (с, 9H).
Промежуточное соединение P67
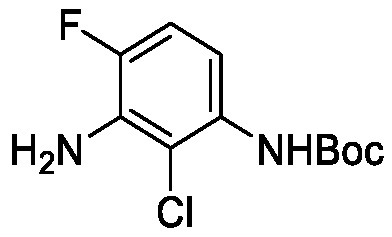
Трет-бутил (3-амино-2-хлор-4-фторфенил)карбамат
Стадия 1: Получение метил 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензоата. Метил 3-амино-2-хлор-6-фторбензоат (5,06 г, 24,9 ммоль) растворяют в ДХМ (250 мл) и охлаждают до 0°C. Реакционную смесь последовательно обрабатывают триэтиламином (10,4 мл, 74,6 ммоль), 4-(диметиламино)пиридинлом (0,304 г, 2,49 ммоль) и затем ди-трет-бутилдикарбонатом (13,6 г, 62,1 ммоль) и перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле, элюируя гексаном/ацетоном с получением метил 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензоата (7,55 г, 100%), который используют непосредственно на следующей стадии в виде смеси моно/бис-Вос продуктов.
Стадия 2: Получение 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензойной кислоты. Метил 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензоат (7,55 г, 24,9 ммоль) растворяют в 1:1 ТГФ/MeOH (120 мл) и затем обрабатывают 2,0 M водным NaOH (37,3 мл, 74,6 ммоль) и перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют дополнительной водой и экстрагируют Et2O (2×250 мл). Et2O органические слои объединяют и промывают 1,0 M NaOH (1×50 мл) и затем объединенные водные слои подкисляют примерно pH 4 с применением 4,0 M HCl и затем экстрагируют 4:1 ДХМ/ИПА (2×250 мл). ДХМ/ИПА органические слои объединяют и сушат над Na2SO4, фильтруют и концентрируют с получением 3-((трет-бутоксикарбонил)амино)-2-хлор-6-фторбензойной кислоты (5,53 г, 77%), которую применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 3: Получение трет-бутил (3-амино-2-хлор-4-фторфенил)карбамата. 3-((Трет-бутоксикарбонил)амино)-2-хлор-6-фторбензойную кислоту (5,53 г, 19,1 ммоль) растворяют в ДМФ (100 мл) и последовательно обрабатывают триэтиламином (7,98 мл, 57,27 ммоль), затем дифенилфосфорилазидом (6,17 мл, 28,63 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой (20 мл) и нагревают до 80°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют дополнительной водой (50 мл) и затем экстрагируют EtOAc (2×250 мл). Органические экстракты объединяют и промывают водой (3×100 мл) и насыщенным раствором соли (1×50 мл), затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/ацетоном и затем снова гексаном/МТБЭ с получением трет-бутил (3-амино-2-хлор-4-фторфенил)карбамата (1,05 г, 21%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,41 (с, 1H), 6,99-6,94 (м, 1H), 6,69-6,66 (м, 1H), 5,34 (с, 2H), 1,43 (с, 9H).
Промежуточное соединение P68
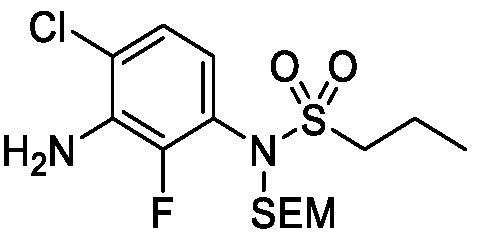
N- (3-амино-4-хлор-2-фторфенил)- N- ((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
К раствору N-(3-амино-4-хлор-2-фторфенил)пропан-1-сульфонамида (3,0 г, 11,2 ммоль) в ДМФ (45,0 мл) при 0°C добавляют 60% гидрид натрия в виде суспензии в минеральном масле (607 мг, 15,2 ммоль). Через 15 минут реакционную смесь обрабатывают по каплям (2-(хлорметокси)этил)триметилсиланом (2,4 мл, 13,5 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают водой и экстрагируют EtOAc (2x) и затем объединенные органические слои промывают водой затем насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc с получением N-(3-амино-4-хлор-2-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (3,85 г, 86%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,08 (д, 1H), 6,75 (дд, 1H), 4,97 (с, 2H), 4,16 (с, 2H), 3,65 (дд, 2H), 3,08 (дд, 2H), 1,93-1,83 (м, 2H), 1,05 (т, 3H), 0,91 (дд, 2H), 0,01 (с, 9H).
Промежуточное соединение P69

6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин - 4(3 H )-он
Стадия 1: Получение трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)карбамата. Трет-бутил (3-амино-2-хлор-4-фторфенил)карбамат (56,7 мг, 0,217 ммоль), 6-бром-3,5диметилхиназолин-4(3H)-он (50 мг, 0,198 ммоль), карбонат цезия (193 мг, 0,593 ммоль), Xantphos (17,1 мг, 0,0296 ммоль) и трис(дибензилиденацетон)дипалладий (0) (9,05 мг, 0,0099) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Добавляют толуол (0,988 мл), и раствор продувают аргоном в течение 5 минут, затем пробирку герметично закрывают и нагревают до 100°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и фильтруют через слой Celite® и очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc с получением трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-карбамата в виде беловатого твердого вещества (81 мг, 95%). МС (хиад, m/z)=333,1 (M-Boc).
Стадия 2: Получение 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3H)-она. Трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)карбамат (81 мг, 0,187 ммоль) растворяют в ДХМ (4,7 мл) и обрабатывают трифторуксусной кислотой (1,5 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле, элюируя ДХМ/MeOH и 1% NH4OH с получением 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3H)-она в виде светло-желтого твердого вещества (37 мг, 56%). 1H ЯМР (400 MГц, CDCl3) δ 7,99 (с, 1H), 7,38 (д, 1H), 7,02 (д, 1H), 6,86 (дд, 1H), 6,51 (дд, 1H), 3,50 (с, 3H), 2,90 (с, 3H); МС (хиад, m/z)=333,1 (M+H).
Промежуточное соединение P70
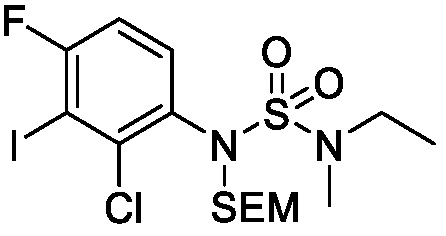
N- (2-хлор-4-фтор-3-йодфенил)- N- ((2-(триметилсилил)этокси)метил)- N- этил- N- метил-1-сульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-этил-N-метил-1-сульфонамида. К раствору 2-хлор-4-фтор-3-йоданилина (145 мг, 0,534 ммоль) в пиридине (2671 мкл) добавляют этил(метил)сульфамоилхлорид (177 мг, 1,12 ммоль), и реакционную смесь перемешивают в течение ночи при температуре окружающей среды. Реакционную смесь обрабатывают 10% водной лимонной кислотой и экстрагируют EtOAc (2x), и объединенные органические слои промывают водой и насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc и затем выделенный продукт растворяют в ацетонитриле (2671 мкл) и 2,0 M водным Na2CO3 (2671 мкл, 5,34 ммоль) и нагревают до 65°C в течение 2 часов. Реакционную смесь очищают С18 хроматографией с обращенной фазой, элюируя H2O/ацетонитрилом с 0,1% ТФК, и выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3 затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-N-этил-N-метил-1-сульфонамида (21 мг, 10%). 1H ЯМР (400 MГц, CDCl3) δ 7,64 (дд, 1H), 7,02 (дд, 1H), 6,71 (шс, 1H), 3,22 (кв, 2H), 2,81 (с, 3H), 1,01 (т, 3H).
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)-N-этил-N-метил-1-сульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)-N-этил-N-метил-1-сульфонамида (21 мг, 0,0535 ммоль) в ДМФ (357 мкл) при 0°C добавляют 60% гидрид натрия в минеральном масле (2,57 мг, 0,0642 ммоль) и затем (2-(хлорметокси)этил)триметилсилан (9,39 мкл, 0,0562 ммоль), и раствор перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь обрабатывают водой и экстрагируют EtOAc (2x), и затем объединенные органические слои промывают водой затем насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc) с получением N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)-N-этил-N-метил-1-сульфонамида (27,5 мг, 98%). 1H ЯМР (400 MГц, CDCl3) δ 7,66 (дд, 1H), 7,04 (дд, 1H), 5,19 (д, 1H), 4,68 (д, 1H), 3,64 (т, 2H), 3,14 (1, 2H), 2,80 (с, 3H), 0,92 (м, 5H), 0,01 (с, 9H).
Промежуточное соединение P71

(R)- N- (2-хлор-4-фтор-3-йодфенил)-3-фтор- N- ((2-(триметилсилил)этокси)метил)-пирролидин-1-сульфонамид
Стадия 1: Получение (R)-N-(2-хлор-4-фтор-3-йодфенил)-3-фторпирролидин-1-сульфонамида. К раствору 2-хлор-4-фтор-3-йоданилина (197 мг, 0,726 ммоль) в пиридине (3629 мкл) добавляют (R)-3-фторпирролидин-1-сульфонилхлорид (286 мг, 1,52 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 48 часов. Реакционную смесь обрабатывают 10% водной лимонной кислотой и экстрагируют EtOAc (2x), и объединенные органические слои промывают водой и насыщенным раствором соли затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают С18 хроматографией с обращенной фазой, элюируя H2O/ацетонитрилом с 0,1% ТФК, и выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют с получением (R)-N-(2-хлор-4-фтор-3-йодфенил)-3-фторпирролидин-1-сульфонамида (200 мг, 43%). 1H ЯМР (400 MГц, CDCl3) δ 7,68 (дд, 1H), 7,02 (дд, 1H), 6,78 (шс, 1H), 3,59 (м, 3H), 3,47 (м, 2H), 2,24 (м, 1H), 2,05 (м, 1H).
Стадия 2: Получение (R)-N-(2-хлор-4-фтор-3-йодфенил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пирролидин-1-сульфонамида. К раствору (R)-N-(2-хлор-4-фтор-3-йодфенил)-3-фторпирролидин-1-сульфонамида (132 мг, 0,312 ммоль) в ДМФ (2082 мкл) при 0°C добавляют 60% гидрид натрия в минеральном масле (15,0 мг, 0,375 ммоль) и затем (2-(хлорметокси)этил)триметилсилан (54,8 мкл, 0,328 ммоль), и раствор перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь обрабатывают водой и экстрагируют EtOAc (2x), и затем объединенные органические слои промывают водой затем насыщенным раствором соли и затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле, элюируя гексаном/EtOAc и затем повторно очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют с получением (R)-N-(2-хлор-4-фтор-3-йодфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пирролидин-1-сульфонамида (25,2 мг, 15%). 1H ЯМР (400 MГц, CDCl3) δ 7,64 (м, 1H), 7,04 (дд, 1H), 5,29 (м, 1H), 5,18 (м, 1H), 3,64 (м, 4H), 3,49 (м, 3H), 2,24 (м, 1H), 2,09 (м, 1H), 0,91 (т, 2H), 0,01 (с, 9H).
Промежуточное соединение P72
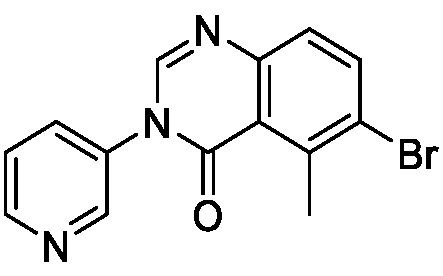
6-бром-5-метил-3-(пиридин-3-ил)хиназолин-4(3H)-он
Пиридин-3-илбороновую кислоту (51,4 мг, 0,418 ммоль), ацетат меди(II) (38,0 мг, 0,209 ммоль) и 15 мг измельченных молекулярных сит 4 ангстрема суспендируют в безводном ДХМ (3 мл). Суспензию перемешивают в течение 5 минут при температуре окружающей среды под баллоном кислорода. 6-Бром-5-метилхиназолин-4(3H)-он (50 мг, 0,209 ммоль) и пиридин (33,8 мкл, 0,418 ммоль) добавляют, и реакционную смесь перемешивают при 40°C в течение 42 часов под баллоном кислорода. Реакционную смесь разбавляют ДХМ (15 мл) и твердые вещества удаляют фильтрацией и промывают ДХМ (10 мл). Органический фильтрат промывают водным насыщенным бикарбонатом натрия (3×25 мл). Водные промывки объединяют и экстрагируют ДХМ:ИПА (4:1) (2×10 мл). Органические фазы объединяют и сушат над сульфатом натрия, фильтруют и концентрируют с получением зеленого масла, которое очищают хроматографией на силикагеле, элюируя ДХМ и MeOH (0%-10% MeOH, с 1% NH4OH) с получением 6-бром-5-метил-3-(пиридин-3-ил)хиназолин-4(3H)-она (38,7 мг, 59%) в виде белого твердого вещества. МС (хиад, m/z)=316,0, 318,0 (M+H).
Промежуточное соединение P73
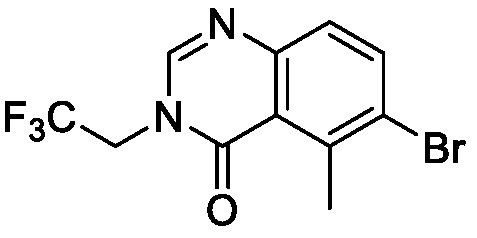
6-бром-5-метил-3-(2,2,2-трифторэтил)хиназолин-4(3H)-он
К раствору 6-бром-5-метилхиназолин-4(3H)-она (50 мг, 0,2091 ммоль) в ДМФ (2,1 мл) добавляют K2CO3 (115,6 мг, 0,8366 ммоль), и реакционную смесь перемешивают под аргоном при температуре окружающей среды в течение 5 минут. 2,2,2-Трифторэтил трифторметансульфонат (33,14 мкл, 0,2301 ммоль) добавляют, и реакционную смесь перемешивают в течение ночи при температуре окружающей среды. Реакционную смесь гасят водой (15 мл) и экстрагируют EtOAc (3×15 мл). Органические фазы объединяют, сушат над сульфатом натрия и концентрируют с получением белого твердого вещества, которое очищают хроматографией на силикагеле, элюируя гексаном и EtOAc (0-30% EtOAc) с получением 6-бром-5-метил-3-(2,2,2-трифторэтил)хиназолин-4(3H)-она (60 мг, 89%) в виде воскообразного белого твердого вещества. МС (хиад, m/z)=321,0, 323,0 (M+H).
Промежуточное соединение P74
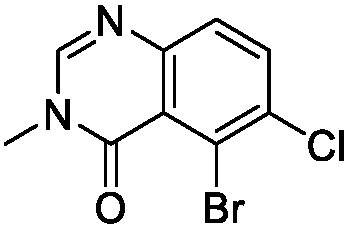
5-бром-6-хлор-3-метилхиназолин-4(3H)-он
Стадия 1: Получение 5-бром-6-хлорхиназолин-4(3H)-она. Раствор 6-амино-2-бром-3-хлорбензойной кислоты (1,21 г, 4,831 ммоль), формамида (0,288 мл, 7,246 ммоль) и POCl3 (2,25 мл, 24,15 ммоль) нагревают до 95°C в течение 2 часов. Реакционную смесь охлаждают до температуры окружающей среды и затем концентрируют для удаления excess POCl3, и остаток осторожно гасят водой. Полученную желтую суспензию перемешивают при температуре окружающей среды в течение 30 минут, и твердые вещества собирают фильтрацией, промывают водой и сушат в вакууме с получением 5-бром-6-хлорхиназолин-4(3H)-она (1,20 г, 96%), который применяют непосредственно на следующей стадии без дальнейшей очистки. МС (хиад, m/z)=259,0, 261,0 (M+H).
Стадия 2: Получение 5-бром-6-хлор-3-метилхиназолин-4(3H)-она. К раствору 5-бром-6-хлорхиназолин-4(3H)-она (1,2 г, 4,62 ммоль) в ДМФ (20 мл) добавляют карбонат калия (1,41 г, 10,2 ммоль), затем йодметан (0,576 мл, 9,25 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют 100 мл воды и экстрагируют EtOAc (x4). Органические фазы объединяют, промывают насыщенным раствором соли, сушат над Na2SO4, затем концентрируют. Неочищенный продукт очищают колоночной хроматографией, элюируя 10-50% ацетоном в ДХМ с получением 5-бром-6-хлор-3-метилхиназолин-4(3H)-она (0,78 г, 62%). МС (хиад, m/z)=274,9, 276,9 (M+H).
Промежуточное соединение P75
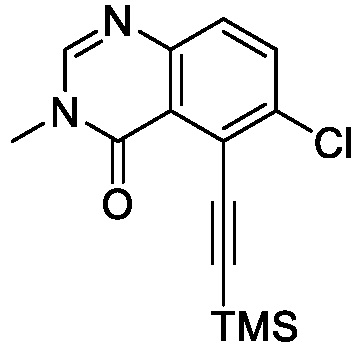
6-хлор-3-метил-5-((триметилсилил)этинил)хиназолин-4(3H)-он
5-Бром-6-хлор-3-метилхиназолин-4(3H)-он (276,5 мг, 1,011 ммоль) растворяют в диоксане (10 мл) и обрабатывают триметилсилилацетиленом (171,44 мкл, 1,213 ммоль), хлоридом бис(трифенилфосфин)палладия(II) (141,91 мг, 0,202 ммоль), йодида меди (I) (38,506 мг, 0,202 ммоль) и триэтиламина (563,62 мкл, 4,044 ммоль), и затем продувают аргоном в течение 5 минут, герметично закрывают и нагревают до 70°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией на силикагеле (гексан/ацетон), затем С18 хроматографией с обращенной фазой (вода-ацетонитрил с 0,1% ТФК). Желаемые фракции затем концентрируют досуха и полученный остаток растворяют в 4:1 ДХМ:ИПА и промывают насыщенным NaHCO3 (1x). Органические фракции объединяют и сушат над Na2SO4, фильтруют и концентрируют с получением 6-хлор-3-метил-5-((триметилсилил)этинил)хиназолин-4(3H)-оном (115,4 мг, 39%). МС (хиад, m/z)=291,1, 293,1 (M-H).
Промежуточное соединение P76
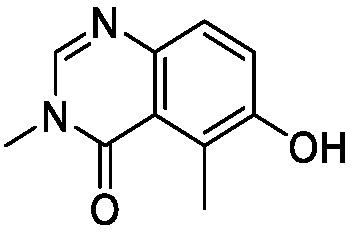
6-гидрокси-3,5-диметилхиназолин-4(3H)-он
Стадия 1: Получение 3,5-диметил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-4(3H)-она. В пробирку добавляют 6-бром-3,5-диметилхиназолин-4(3H)-он (2,70 г, 10,7 ммоль), бис(пинаколато)диборон (3,25 г, 12,8 ммоль), KOAc (3,14 г, 32,0 ммоль) и ДМСО (26,7 мл). Суспензию продувают барботированием Ar в течение 5 минут, затем добавляют аддукт дихлорметана и дихлор[1,1'-бис(дифенилфосфино)ферроцен]палладия (II) (0,263 г, 0,320 ммоль) одной порцией и пробирку герметично закрывают. Смесь нагревают до 90°C в течение 15 часов. Смесь охлаждают до температуры окружающей среды, разбавляют водой (150 мл) и перемешивают в течение 15 минут. Полученное твердое вещество выделяют вакуумной фильтрацией и промывают дополнительной водой. Полученное твердое вещество растворяют в EtOAc (150 мл), и раствор сушат над Na2SO4. Смесь затем фильтруют непосредственно через слой двуокиси кремния и желаемый продукт элюируют EtOAc. Фильтрат концентрируют с получением 3,5-диметил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-4(3H)-она в виде рыжевато-коричневого твердого вещества (2,48 г, 77%). МС (хиад, m/z)=301,1 (M+H).
Стадия 2: Получение 6-гидрокси-3,5-диметилхиназолин-4(3H)-она. Раствор 3,5-диметил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)хиназолин-4(3H)-она (2,48 г, 8,26 ммоль) в ТГФ (41,3 мл) охлаждают до 0°C в бане лед/вода, затем NaOH (2,0 M водным, 20,7 мл, 41,3 ммоль) добавляют по каплям. Медленно добавляют перекись водорода (35% масс., водную, 5,68 мл, 66,1 ммоль). Реакционную смесь перемешивают при 0°C в течение 2,5 часов и затем гасят добавлением тиосульфатом натрия (3,0 M водным, 24,8 мл, 74,4 ммоль), и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 1 часа. Реакционную смесь разбавляют 0,1 N водным NaOH и промывают МТБЭ (2x). Водный слой доводят до примерно pH 4 твердым лимонной кислотой. Полученное твердое вещество выделяют вакуумной фильтрацией, промывают дополнительной водой и затем сушат в течение ночи под вакуумом при температуре окружающей среды с получением 6-гидрокси-3,5-диметилхиназолин-4(3H)-она в виде беловатого твердого вещества (1,49 г, 94%). МС (хиад, m/z)=191,1 (M+H).
Промежуточное соединение P77
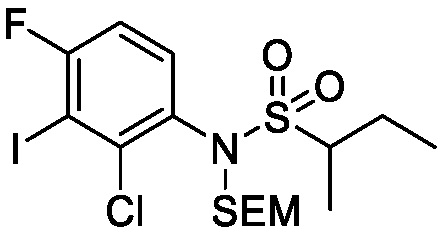
N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)бутан-2-сульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)бутан-2-сульфонамида. К раствору 2-хлор-4-фтор-3-йоданилина (250 мг, 0,921 ммоль) в дихлорметане (4,6 мл) при 0°C добавляют триэтиламин (385 мкл, 2,76 ммоль) и бутан-2-сульфонилхлорид (303 мг, 1,93 ммоль), и реакционную смесь перемешивают в течение ночи при температуре окружающей среды. Реакционную смесь разбавляют ДХМ и промывают водой (1x) и насыщенным раствором соли (1x). Органический слой сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (10-75% EtOAc/гексан) с получением N-(2-хлор-4-фтор-3-йодфенил)бутан-2-сульфонамида (250 мг, 52%). 1H ЯМР (400 MГц, CDCl3) δ 7,75 (дд, 1H), 7,02 (дд, 1H), 2,99 (м, 1H), 1,37 (д, 3H), 1,14 (м, 2H), 1,01 (т, 3H).
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)бутан-2-сульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)бутан-2-сульфонамида (188 мг, 0,480 ммоль) в N, N-диметилформамиде (32 мл) при 0°C добавляют 60% гидрид натрия в минеральном масле (23,0 мг, 0,576 ммоль), затем (2-(хлорметокси)этил)триметилсилан (84,3 мкл, 0,504 ммоль). Ледяную баню удаляют и раствор перемешивают при температуре окружающей среды в течение 30 минут. Раствор затем разбавляют EtOAc и гасят водой. Органический слой промывают водой (1x) и насыщенным раствором соли (1x), сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)бутан-2-сульфонамида (185 мг, 52%). 1H ЯМР (400 MГц, CDCl3) δ 7,51 (дд, 1H), 7,05 (дд, 1H), 5,16 (дд, 1H), 4,81 (дд, 1H), 3,69 (м, 2H), 2,94 (м, 1H), 1,40 (дд, 3H), 1,13 (м, 2H), 1,03 (м, 3H), 0,89 (м, 2H), 0,02 (с, 9H).
Промежуточное соединение P78
2-хлор-4-фтор-3-метоксианилин
Стадия 1: Получение 2-хлор-4-фтор-3-метоксибензальдегида. N, N,N'-триметилэтилендиамин (1,77 мл, 13,6 ммоль) растворяют в ТГФ (50 мл) и охлаждают до -42°C в обратном потоке азота, затем обрабатывают н-бутиллитием (2,5 M в гексане, 5,45 мл, 13,6 ммоль) и перемешивают при -42°C в течение 30 минут после завершения добавления. Реакционную смесь затем охлаждают до -78°C и обрабатывают 50 мл ТГФ раствора 4-фтор-3-метоксибензальдегида (2,0 г, 13,0 ммоль) и затем нагревают до -42°C и перемешивают в течение 30 минут. Реакционную смесь охлаждают до -78°C и обрабатывают н-бутиллитием (2,5 M в гексане, 5,45 мл, 13,6 ммоль) и затем нагревают до -42°C и перемешивают в течение 1 ч. Реакционную смесь быстро переносят через канюлю в 50 мл ТГФ раствор гексахлорэтана (6,14 г, 26,0 ммоль) при температуре окружающей среды и перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь обрабатывают 4,0 M HCl и экстрагируют Et2O (2×250 мл). Органические фазы объединяют и промывают 1,0 M NaOH (1×100 мл), 1,0 M HCl (1×100 мл) и насыщенным раствором соли (1×50 мл) и затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают С18 хроматографией с обращенной фазой (вода/ацетонитрил с 0,1% ТФК) и фракции, содержащие желаемый продукт, объединяют и разделяют между 4:1 ДХМ:ИПА и насыщенным NaHCO3 (1×100 мл), сушат над Na2SO4, фильтруют и концентрируют с получением 2-хлор-4-фтор-3-метоксибензальдегида (1,12 г, 46%). 1H ЯМР (400 MГц, ДМСО-d6) δ 10,26 (с, 1H), 7,70-7,67 (м, 1H), 7,52-7,48 (т, 1H), 3,94 (с, 3H).
Стадия 2: Получение 2-хлор-4-фтор-3-метоксибензойной кислоты. 2-Хлор-4-фтор-3-метоксибензальдегид (1,07 г, 5,67 ммоль) растворяют в ацетонитриле (57 мл) и обрабатывают водным 1,0 M двуосновным раствором фосфата натрия (8,51 мл, 8,51 ммоль) и затем охлаждают до 0°C. Реакционную смесь обрабатывают водным 35% масс. раствором перекиси водорода (0,732 мл, 8,51 ммоль), затем по каплям добавляют водный 1,0 M раствор хлорита натрия (8,51 мл, 8,51 ммоль) и затем нагревают до температуры окружающей среды и перемешивают в течение 16 часов. Реакционную смесь обрабатывают 3,0 M раствором тиосульфата натрия и разбавляют 1,0 M NaOH, затем промывают Et2O (2×250 мл). Водный слой подкисляют до примерно pH 2 с применением 4,0 M HCl и экстрагируют 4:1 ДХМ:ИПА (2×250 мл). Органические экстракты объединяют и сушат над Na2SO4, фильтруют и концентрируют с получением 2-хлор-4-фтор-3-метоксибензойной кислоты (1,16 г, 99%). 1H ЯМР (400 MГц, ДМСО-d6) δ 13,45 (шс, 1H), 7,59-7,56 (м, 1H), 7,40-7,36 (т, 1H), 3,89 (с, 3H).
Стадия 3: Получение 2-хлор-4-фтор-3-метоксианилина. 2-Хлор-4-фтор-3-метоксибензойную кислоту (1,16 г, 5,670 ммоль) растворяют в ДМФ (57 мл) и обрабатывают триэтиламином (2,37 мл, 17,01 ммоль), затем дифенилфосфорилазидом (1,83 мл, 8,51 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь обрабатывают 10 мл воды и нагревают до 80°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют EtOAc (2×250 мл). Органические фазы объединяют и промывают водой (3×100 мл) и насыщенным раствором соли (1×50 мл), сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (гексан/EtOAc) с получением 2-хлор-4-фтор-3-метоксианилина (640,9 мг, 64%). 1H ЯМР (400 MГц, ДМСО-d6) δ 6,98-6,94 (т, 1H), 6,52-6,49 (м, 1H), 5,24 (шс, 2H), 3,82 (с, 3H).
Промежуточное соединение P79
3-амино-2-хлор-6-фторфенол
К раствору 2-хлор-4-фтор-3-метоксианилина (400 мг, 2,2 ммоль) в ДХМ (10 мл) добавляют BBr3 (4,5 мл, 1,0 M в ДХМ, 4,5 ммоль) при 0°C и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь гасят метанолом (3 мл) и затем выливают в холодную воду (75 мл) и экстрагируют этилацетатом (100 мл x 2). Органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% этилацетат в гексане) с получением 3-амино-2-хлор-6-фторфенола в виде беловатого твердого вещества (220 мг, 60%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,87 (шс, 1H), 6,83 (дд, 1H), 6,22-60,18 (м, 1H), 5,03 (шс, 2H). МС (m/z)=159,8 (M-H).
Промежуточное соединение P80
3-амино-2,6-дифторфенол
Стадия 1: Получение 2-(2,4-дифтор-3-метоксифенил)изоиндолин-1,3-диона. Смесь 2,4-дифтор-3-метоксианилина (4,89 г, 30,7 ммоль), изобензофуран-1,3-диона (4,55 г, 30,7 ммоль) и триэтиламина (4,28 мл, 30,7 ммоль) в толуоле (76,8 мл) нагревают до кипения с обратным холодильником с ловушкой Дина-Старка в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Остаток разбавляют EtOAc и гасят водой. Органическую фазу промывают водой (1x) и насыщенным раствором соли (1x), сушат над Na2SO4, фильтруют и концентрируют и остаток очищают хроматографией на силикагеле (100% ДХМ) с получением 2-(2,4-дифтор-3-метоксифенил)изоиндолин-1,3-диона (8,0 г, 90%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,0 (м, 2H), 7,95 (м, 2H), 7,35 (м, 2H), 3,98 (с, 3H).
Стадия 2: Получение 2-(2,4-дифтор-3-гидроксифенил)изоиндолин-1,3-диона. К раствору 2-(2,4-дифтор-3-метоксифенил)изоиндолин-1,3-диона (7,6 г, 26 ммоль) в дихлорметане (131 мл) при 0°C добавляют 1,0 M трибромборан (34 мл, 34 ммоль), и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 4 часов. Раствор выливают в лед и перемешивают в течение 2 часов. Полученные твердые вещества собирают фильтрацией и сушат в вакууме с получением 2-(2,4-дифтор-3-гидроксифенил)изоиндолин-1,3-диона (6,2 г, 86%). МС (хиад, m/z)=274,1 [M-H].
Стадия 3: Получение 3-амино-2,6-дифторфенола. Раствор 2-(2,4-дифтор-3-гидроксифенил)изоиндолин-1,3-диона (200 мг, 0,727 ммоль) и 1,0 M гидразина (1453 мкл, 1,45 ммоль) в метаноле (1453 мкл) перемешивают при температуре окружающей среды в течение ночи. Раствор концентрируют, и остаток очищают хроматографией на силикагеле (1-20% MeOH/ДХМ, 1% NH4OH) с получением 3-амино-2,6-дифторфенола (91 мг, 86%). 1H ЯМР (400 MГц, CD3OD) δ 6,62 (м, 1H), 6,24 (м, 1H); МС (хиад, m/z)=146,1 [M+H].
Промежуточное соединение P81
Трет- бутил 3-фтор-2-метил-6-нитробензоат
К перемешиваемому раствору 3-фтор-2-метил-6-нитробензойной кислоты (800 мг, 4,02 ммоль) в смеси трет-BuOH и ДХМ (1:1, 5 мл) добавляют Boc2O (1,38 мл, 6,03 ммоль) затем ДМАП (147 мг, 1,2 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов в атмосфере азота. Реакционную смесь концентрируют и неочищенный остаток гасят водой (150 мл) и экстрагируют этилацетатом (2×150 мл). Объединенные органические слои промывают водой (2×100 мл), насыщенным раствором соли (100 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с применением 15-20% этилацетата в гексане с получением трет-бутил 3-фтор-2-метил-6-нитробензоата в виде бесцветной жидкости (600 мг, 60%). 1H ЯМР (400 MГц, CDCl3) δ 8,10-8,07 (дд, 1H), 7,34 (т, 1H), 2,30 (с, 3H), 1,60 (с, 9H).
Промежуточное соединение P82
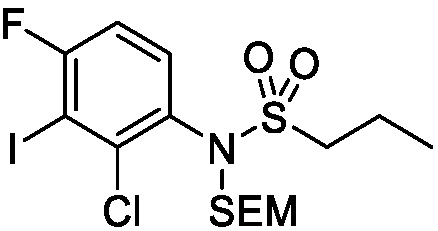
N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. К раствору 2-хлор-4-фтор-3-йоданилина (2,0 г, 7,4 ммоль) в ДХМ (50 мл) при 0°C добавляют триэтиламин (2,6 мл, 18,5 ммоль) и пропан 1-сульфонилхлорид (2,1 мл, 18,5 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов под азотом. Реакционную смесь выливают в воду и экстрагируют ДХМ (3x 20 мл). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с применением 20% этилацетата в гексане с получением N-(2-хлор-4-фтор-3-йодфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (2,5 г, 70%) в виде коричневого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,48-7,39 (м, 1H), 7,12-7,03 (м, 1H), 3,73-3,61 (м, 2H), 3,62-3,49 (м, 2H), 2,06-1,88 (м, 4H), 1,09 (т, J=7,4 Гц, 6H).
Стадия 2: Получение N-(2-хлор-4-фтор-3-йодфенил)пропан-1-сульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (8,0 г, 16,59 ммоль) в ацетонитриле (100 мл) добавляют бикарбонат натрия (17,5 г, 170 ммоль) и 25 мл воды, и реакционную смесь нагревают при 70°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, подкисляют примерно pH 1 насыщенным раствором лимонной кислоты и экстрагируют этилацетатом (3×50 мл). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния, элюируя 20% этилацетатом в гексане с получением N-(2-хлор-4-фтор-3-йодфенил)пропан-1-сульфонамида (5,0 г, 80%) в виде коричневого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,74-7,66 (м, 1H), 7,09-7,00 (м, 1H), 6,67 (с, 1H), 3,08-2,99 (м, 2H), 1,92-1,78 (м, 2H), 1,03 (т, J=7,5 Гц, 3H); МС (m/z)=377,9 (M+H).
Стадия 3: Получение N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. К раствору N-(2-хлор-4-фтор-3-йодфенил)пропан-1-сульфонамида (2,0 г, 7,38 ммоль) в ДМФ (10 мл) при 0°C добавляют 60% гидрид натрия в минеральном масле (396 мг, 9,88 ммоль) порциями, и реакционную смесь перемешивают при 0°C в течение 30 минут. Туда добавляют 2-(триметилсилил)этоксиметилхлорид (1,5 мл, 8,85 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь выливают в холодную воду и экстрагируют этилацетатом (100 мл x 3). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с применением 5% этилацетатом в гексане с получением N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (3,2 г, 85%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,67-7,58 (м, 1H), 7,41-7,30 (м, 1H), 5,09 (д, J=11,1 Гц, 1H), 4,71 (д, J=11,1 Гц, 1H), 3,60 (кв, J=7,7 Гц, 2H), 3,31-3,15 (м, 2H), 1,75 (кв, J=7,5 Гц, 2H), 0,99 (т, J=7,4 Гц, 3H), 0,83 (т, J=8,2 Гц, 2H), -0,02 (с, 9H).
Промежуточное соединение P83
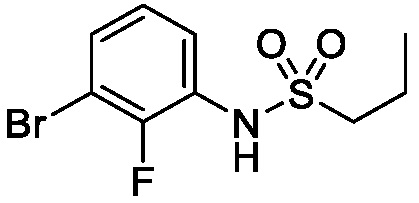
N-(3-бром-2-фторфенил)пропан-1-сульфонамид
К раствору 3-бром-2-фторанилина (1,0 г, 5,26 ммоль) в пиридине (10 мл) добавляют пропан-1-сульфонилхлорид (5,92 мл, 52,62 ммоль), и реакционную смесь перемешивают при 60°C в течение 3 часов в атмосфере аргона. Реакционную смесь охлаждают до температуры окружающей среды, и растворитель удаляют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (5% этилацетатом в гексане) с получением N-(3-бром-2-фторфенил)пропан-1-сульфонамида в виде бледно-желтого твердого вещества (950 мг, 60%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,86 (с, 1H), 7,57-7,48 (м, 1H), 7,46-7,37 (м, 1H), 7,19-7,10 (м, 1H), 3,16-3,08 (м, 2H), 1,80-1,66 (м, 2H), 0,97 (т, J=7,4 Гц, 3H).
Промежуточное соединение P84
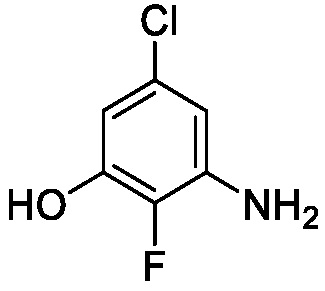
3-амино-5-хлор-2-фторфенол
Стадия 1: Получение 5-хлор-2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина. Раствор 3-бром-5-хлор-2-фторанилина (184 мг, 0,82 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (271 мг, 1,07 ммоль), PdCl2(dppf)-dcm (18 мг, 0,025 ммоль) и ацетата калия (161 мг, 1,64 ммоль) в N, N-диметилформамиде (1949 мкл) продувают аргоном в течение 10 минут и нагревают до 100°C под баллоном аргона в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют EtOAc (3 x). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (5-50% EtOAc/гексан) с получением 5-хлор-2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (169 мг, 76%).
Стадия 2: Получение 3-амино-5-хлор-2-фторфенола. К раствору 5-хлор-2-фтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (134 мг, 0,494 ммоль) в тетрагидрофуране (4935 мкл) при 0°C добавляют 2,0 M гидроксид натрия (1234 мкл, 2,47 ммоль) и 35% перекись водорода (340 мкл, 3,95 ммоль), и реакционную смесь перемешивают при 0°C в течение 30 минут. Реакционную смесь гасят 5 мл насыщенного тиосульфата натрия и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь подкисляют примерно pH 5 10% лимонной кислотой, разбавляют водой и экстрагируют EtOAc (3x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% EtOAc/гексан) с получением 3-амино-5-хлор-2-фторфенола (63 мг, 79%). 1H ЯМР (400 MГц, CDCl3) δ 6,39 (м, 1H), 6,33 (м, 1H), 5,02 (м, 1H), 3,79 (шс, 2H).
Промежуточное соединение P85
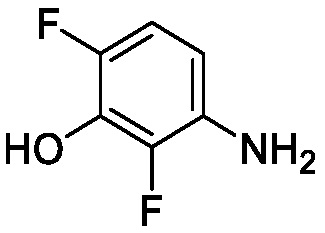
3-амино-2,6-дифторфенол
Стадия 1: Получение 2-(2,4-дифтор-3-метоксифенил)изоиндолин-1,3-диона. Смесь 2,4-дифтор-3-метоксианилина (4,4 г, 28 ммоль), изобензофуран-1,3-диона (4,1 г, 28 ммоль) и триэтиламина (3,9 мл, 28 ммоль) в 69 мл толуола нагревают до кипения с обратным холодильником с ловушкой Дина-Старка в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют EtOAc (3 x). Объединенные органические слои промывают водой, насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией с обращенной фазой (5-95% АЦН/вода, 0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным водным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют с получением 2-(2,4-дифтор-3-метоксифенил)изоиндолин-1,3-диона (6,3 г, 79%). 1H ЯМР (400 MГц, CDCl3) δ 7,02 (м, 4H), 6,69 (м, 1H), 6,38 (м, 1H), 3,98 (с, 3H).
Стадия 2: Получение 2-(2,4-дифтор-3-гидроксифенил)изоиндолин-1,3-диона. К раствору 2-(2,4-дифтор-3-метоксифенил)изоиндолин-1,3-диона (6,1 г, 21,1 ммоль) в 70 мл дихлорметана при 0°C добавляют 1,0 M трибромборан (27,4 мл, 27,4 ммоль) в ДХМ, и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 16 часов. Раствор выливают в лед, и полученные твердые вещества собирают фильтрацией и сушат в вакууме с получением 2-(2,4-дифтор-3-гидроксифенил)изоиндолин-1,3-диона (4,29 г, 74%). 1H ЯМР (400 MГц, CDCl3) δ 7,97 (м, 2H), 7,82 (м, 2H), 7,05 (м, 1H), 6,88 (м, 1H).
Стадия 3: Получение 3-амино-2,6-дифторфенола. Раствор 2-(2,4-дифтор-3-гидроксифенил)изоиндолин-1,3-диона (4,29 г, 15,6 ммоль) и 1,0 M гидразина (31,2 мл, 31,2 ммоль) перемешивают в метаноле (31,2 мл) при температуре окружающей среды в течение 16 часов. Реакционную смесь фильтруют через Целит, Целит промывают метанолом, и фильтрат концентрируют при пониженном давлении. Остаток доводят до примерно pH 5 лимонной кислотой в воде и экстрагируют EtOAc (3 x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% EtOAc/гексан) с получением 3-амино-2,6-дифторфенола (2,11 г, 93%). МС (хиад, m/z)=146,1 (M+H).
Промежуточное соединение P86
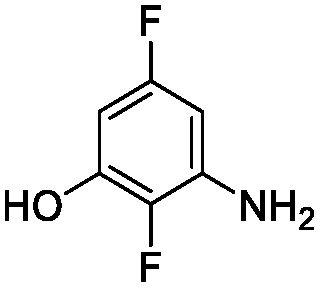
3-амино-2,5-дифторфенол
Раствор 2,5-дифтор-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (0,500 г, 1,960 ммоль) в ТГФ (9,80 мл) охлаждают до 0°C в бане лед/вода и 2,0 M NaOH (4,90 мл, 9,801 ммоль) добавляют по каплям. Туда медленно добавляют 35% масс. перекись водорода (1,35 мл, 15,68 ммоль), и смесь перемешивают при 0°C в течение 2,5 часов. Реакционную смесь гасят 3,0 M тиосульфатом натрия (5,88 мл, 17,64 ммоль), и смесь нагревают до температуры окружающей среды и перемешивают в течение 0,5 часа. Смесь разбавляют 0,1N NaOH и смесь экстрагируют МТБЭ (2x). Водный слой обрабатывают твердым лимонной кислотой до примерно pH 4-5. Смесь экстрагируют EtOAc (3x) и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением 3-амино-2,5-дифторфенола (0,236 г, 82%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,75 (с, 1H), 5,97 (ддд, J=10,6, 6,3, 3,1 Гц, 1H), 5,87 (ддд, J=9,4, 6,3, 3,1 Гц, 1H), 5,28 (шс, 2H).
Промежуточное соединение P87
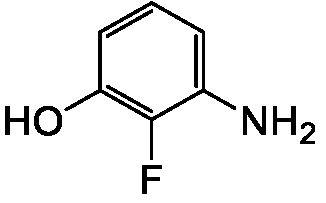
3-амино-2-фторфенол
К раствору 2-фтор-3-метоксианилина (2 г, 14,18 ммоль) в ДХМ (15 мл) добавляют BBr3 (29 мл, 1M в ДХМ, 28,36 ммоль) при 0°C, и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь гасят метанолом (30 мл) и концентрируют. Остаток выливают в ледяную воду (20 мл) и экстрагируют этилацетатом (2×200 мл). Объединенные органические слои промывают водой (2×100 мл) и насыщенным раствором соли (100 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением 3-амино-2-фторфенола (1,6 г, 88%) в виде коричневого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 9,28 (с-ш, 1H), 6,60 (т, J=8,1 Гц, 1H), 6,18 (т, J=7,9 Гц, 1H), 6,11 (т, J=7,9 Гц, 1H), 4,93 (с-ш, 2H). МС (хиад, m/z)=126,0 (M-H).
Промежуточное соединение P88
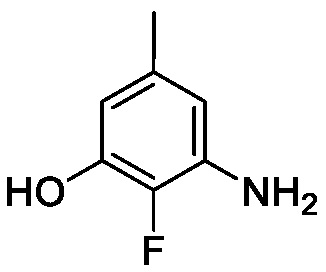
3-амино-2-фтор-5-метилфенол
Стадия 1: Получение трет-бутил(2-фтор-5-метилфенокси)диметилсилана. К раствору 2-фтор-5-метилфенола (12 г, 95,23 ммоль) в N, N-диметилформамиде (30 мл) добавляют 1H-имидазол (9,71 г, 142,85 ммоль), и реакционную смесь охлаждают до 0°C. Туда добавляют трет-бутил диметилсилилхлорид (21,42 г, 142,85 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют холодной водой и экстрагируют этилацетатом (3×100 мл). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (5% EtOAc/гексан) с получением трет-бутил(2-фтор-5-метилфенокси)диметилсилана в виде бесцветного масла (21,3 г, 94%). 1H ЯМР (400 MГц, CDCl3) δ 6,88-6,93 (м, 1H), 6,67-6,72 (м, 2H), 2,25 (с, 3H), 1,00 (с, 9H), 0,19 (с, 6H).
Стадия 2: Получение 3-((трет-бутилдиметилсилил)окси)-2-фтор-5-метилбензальдегида. К перемешиваемому раствору трет-бутил(2-фтор-5-метилфенокси)диметилсилана (5,0 г, 20,83 ммоль) в ТГФ (100 мл) добавляют N, N,N′,N′′,N′′-пентаметилдиэтилентриамин (PMDTA, 2,6 мл, 12,5 ммоль), и реакционную смесь охлаждают до -78°C. Туда добавляют n-BuLi (1,6 M в гексане, 14 мл, 22,91 ммоль) по каплям и реакционную смесь перемешивают при -35°C в течение 1 часа. Реакционную смесь охлаждают до -78°C и N, N-диметилформамид (9,7 мл, 125 ммоль) добавляют по каплям. Реакционную смесь медленно нагревают до комнатной температуры, перемешивают в течение 1 часа и гасят насыщенным водным раствором хлорида аммония (70 мл). Полученную смесь экстрагируют этилацетатом (3×100 мл) и органический слой промывают холодной водой (2×20 мл). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением 3-((трет-бутилдиметилсилил)окси)-2-фтор-5-метилбензальдегида в виде бесцветной вязкой жидкости (3,5 г, 63%). 1H ЯМР (400 MГц, ДМСО-d6) δ 10,15 (с, 1H), 7,15-7,20 (м, 2H), 2,29 (с, 3H), 0,97 (с, 9H), 0,19 (с, 6H).
Стадия 3: Получение 2-фтор-3-гидрокси-5-метилбензойной кислоты. К раствору 3-((трет-бутилдиметилсилил)окси)-2-фтор-5-метилбензальдегида (11,0 г, 41,04 ммоль) в ацетонитриле ( 40 мл) добавляют насыщенный водный раствор двухосновного фосфата натрия (9,6 г, 61,56 ммоль) и реакционную смесь охлаждают до 0°C. Туда добавляют водную перекись водорода (30% масс/масс; 6,9 мл, 61,56 ммоль) затем по каплям добавляют насыщенный водный раствор хлорита натрия (5,54 г, 61,56 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь обрабатывают насыщенным водным раствором тиосульфата натрия (100 мл), подщелачивают до примерно pH 9 с 2N NaOH (50 мл) и экстрагируют этилацетатом (3×30 мл). Водный слой подкисляют 4N раствором HCl (50 мл) и экстрагируют этилацетатом (3×200 мл). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют с получением неочищенной 2-фтор-3-гидрокси-5-метилбензойной кислоты в виде коричневого твердого вещества (4,5 г, 64%), которое используют как таковое на следующей стадии. 1H ЯМР (400 MГц, ДМСО-d6) δ 13,00 (с-ш, 1H), 9,93 (с-ш, 1H), 7,01 (д, J=5,2 Гц, 1H), 6,94 (д, J=8 Гц, 1H), 2,21 (с, 3H); МС (m/z)=168,9 (M-H).
Стадия 4: Получение метил 2-фтор-3-метокси-5-метилбензоата. К раствору 2-фтор-3-гидрокси-5-метилбензойной кислоты (4,5 г, 26,47 ммоль) в N, N-диметилформамиде (10 мл) при 0°C добавляют карбонат калия (14,61 г, 105,88 ммоль) и метилйодид (9,9 мл, 158,82 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 3 часов. Реакционную смесь разбавляют водой (30 мл) и экстрагируют этилацетатом (3×100 мл). Объединенные органические слои промывают холодной водой (2×30 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением метил 2-фтор-3-метокси-5-метилбензоата в виде коричневой жидкости (2,75 г, 52%). 1H ЯМР (400 MГц, ДМСО-d6) δ 7,22-7,24 (м, 1H), 6,91 (д, J=8 Гц, 1H), 3,89, (с, 3H). 3,86 (с, 3H), 2,31 (с, 3H); МС (m/z)=199,3 (M+H).
Стадия 5: Получение 2-фтор-3-метокси-5-метилбензойной кислоты. К раствору метил 2-фтор-3-метокси-5-метилбензоата (4,0 г, 20,20 ммоль) в ТГФ и воды (4:1, 50 мл) добавляют моногидрат гидроксида лития (3,8 г, 90,90 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 3 часов. Реакционную смесь охлаждают до 0°C, гасят водным 1N HCl до примерно pH 1 и экстрагируют этилацетатом (3×100 мл). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют с получением неочищенной 2-фтор-3-метокси-5-метилбензойной кислоты в виде белого твердого вещества (3,0 г, 80%), которое используют как таковое на следующей стадии. 1H ЯМР (400 MГц, ДМСО-d6) δ 13,12 (с-ш, 1H), 7,14-7,21 (м, 2H), 3,83 (с, 3H), 1,90 (с, 3H); МС (m/z)=183,2 (M-1).
Стадия 6: Получение трет-бутил (2-фтор-3-метокси-5-метилфенил)карбамата. К перемешиваемому раствору 2-фтор-3-метокси-5-метилбензойной кислоты (3,0 г, 16,30 ммоль) в смеси трет-BuOH/толуол (1:1, 60 мл) добавляют N, N-диизопропилэтиламин (DIPEA, 4,5 мл, 26,08 ммоль) и смесь охлаждают до 0°C. Туда добавляют дифенилфосфорный азид (ДФФА, 5,25 мл, 24,45 ммоль), и реакционную смесь нагревают до температуры окружающей среды и затем кипятят с обратным холодильником при 110°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой (20 мл) и экстрагируют этилацетатом (3×100 мл). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением трет-бутил (2-фтор-3-метокси-5-метилфенил)карбамата в виде бесцветного твердого вещества (1,7 г, 41%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,80 (с-ш, 1H), 6,95 (д, J=8 Гц, 1H), 6,69 (д, J=8 Гц, 1H), 3,78 (с, 3H), 2,23 (с, 3H), 1,44 (с, 9H).
Стадия 7: Получение 3-амино-2-фтор-5-метилфенола. К раствору трет-бутил (2-фтор-3-метокси-5-метилфенил)карбамата (1,7 г, 6,66 ммоль) в ДХМ (10 мл) при 0°C добавляют трибромид бора (17 мл, 1M в ДХМ, 16,66 ммоль) и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь гасят метанолом (30 мл) и затем концентрируют. Остаток обрабатывают насыщенным водным бикарбонатом натрия до примерно pH 5 и экстрагируют этилацетатом (2×200 мл). Объединенные органические слои промывают водой (100 мл), сушат над безводным Na2SO4, фильтруют и концентрируют с получением неочищенного 3-амино-2-фтор-5-метилфенола в виде коричневого твердого вещества (1,2 г, неочищенный), которое используют как есть без очистки. МС (m/z)=140,0 (M-H).
Промежуточное соединение P89
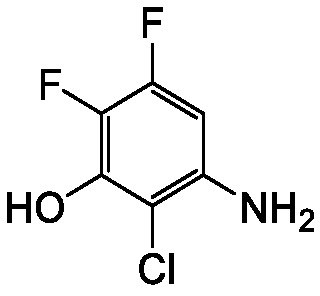
3-амино-2-хлор-5,6-дифторфенол
Стадия 1: Получение 3,4-дифтор-2-метоксианилина. К суспензии Fe-порошка (26 г, 465,61 ммоль) в метаноле (70 мл) и воде (30 мл) добавляют концентрированную HCl (4 мл) по каплям и смесь нагревают до 75°C. Туда добавляют раствор 1,2-дифтор-3-метокси-4-нитробензола (22 г, 116,40 ммоль) в метаноле (30 мл) по каплям в течение 30 минут, и смесь перемешивают при 75°C в течение 4 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют через Целит и Целит промывают ДХМ (2×250 мл). Фильтрат концентрируют и неочищенный остаток разбавляют водой (250 мл) и экстрагируют ДХМ (2×500 мл). Органический слой промывают водой (2×100 мл) и насыщенным раствором соли (100 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (20% EtOAc/гексан) с получением 3,4-дифтор-2-метоксианилина в виде светло-желтой жидкости (15 г, 81%). 1H ЯМР (400 MГц, ДМСО-d6) δ 6,83 (кв, J=9,0 Гц, 1H), 6,47-6,38 (м, 1H), 5,01 (с-ш, 2H), 3,78 (с, 3H).
Стадия 2: Получение 6-бром-3,4-дифтор-2-метоксианилина. К раствору 3,4-дифтор-2-метоксианилина (15,0 г, 94,3 ммоль) в ДХМ (50 мл) добавляют раствор N-бромсукцинимида (16,6 г, 94,3 ммоль) в ДМФ (10 мл), и реакционную смесь перемешивают при 0°C в течение 30 минут. Реакционную смесь концентрируют, и остаток разбавляют водой и экстрагируют этилацетатом (2×500 мл). Объединенные органические слои промывают насыщенным Na2S2O3 (4×150 мл), водой (3×75 мл) и насыщенным раствором соли (2×100 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией на амине двуокиси кремния (5% EtOAc/гексан) с получением 6-бром-3,4-дифтор-2-метоксианилина (10,0 г, 44%). 1H ЯМР (400 MГц, ДМСО-d6) δ 7,35 (дд, J=7,9, 10,2 Гц, 1H), 5,14 (с-ш, 2H), 3,83 (с, 3H); МС (m/z) =238,0, 240,0 [M+H].
Стадия 3: Получение 1-бром-2-хлор-4,5-дифтор-3-метоксибензола. К смеси безводного хлорида меди (1,01 г, 7,56 ммоль) и 6-бром-3,4-дифтор-2-метоксианилина (1,2 г, 5,04 ммоль) в безводном ацетонитриле (10 мл) добавляют трет-бутилнитрит (1,04 г, 10,08 ммоль) по каплям при 55°C в течение 5 минут. Реакционную смесь перемешивают при 55°C в течение еще 10 минут и гасят охлажденной 10% водной HCl (10 мл) и экстрагируют этилацетатом (2×100 мл). Объединенные органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (гексан) с получением 1-бром-2-хлор-4,5-дифтор-3-метоксибензола в виде светло-желтой жидкости (1,0 г, 77%). 1H ЯМР (400 MГц, ДМСО-d6) δ 7,84 (т, J=8,8 Гц, 1H), 3,98 (с, 3H).
Стадия 4: Получение 2-хлор-4,5-дифтор-3-метоксианилина. К раствору 1-бром-2-хлор-4,5-дифтор-3-метоксибензола (6,0 г, 23,34 ммоль), имина бензофенона (8,46 г, 46,69 ммоль) и NaOtBu (3,37 г, 35,01 ммоль) в толуоле (15 мл) добавляют Pd2(dba)3 (1,07 г, 1,17 ммоль) и BINAP (1,45 г, 2,34 ммоль) и реакционную смесь опрыскивают Ar в течение 15 минут, герметично закрывают и затем нагревают при 110°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют при пониженном давлении. Остаток разбавляют водой и экстрагируют этилацетатом (2×250 мл). Объединенные органические слои промывают водой (2×75 мл) и насыщенным раствором соли (2×50 мл), сушат над безводным Na2SO4, фильтруют и концентрируют с получением неочищенного N-(2-хлор-4,5-дифтор-3-метоксифенил)-1,1-дифенилметанимина (7,0 г), который применяют непосредственно на следующей стадии без дальнейшей очистки.
Стадия 5: Получение 2-хлор-4,5-дифтор-3-метоксианилина. К раствору N-(2-хлор-4,5-дифтор-3-метоксифенил)-1,1-дифенилметанимина (7,0 г) in ТГФ (10 мл) добавляют 1N HCl (10 мл, 10 ммоль) по каплям, и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и остаток растворяют в воде и pH доводят до примерно 7 твердым NaHCO3. Водный слой экстрагируют этилацетатом (2×250 мл) и объединенные органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией на амине двуокиси кремния (10% EtOAc/гексан) с получением 2-хлор-4,5-дифтор-3-метоксианилина в виде бледно-желтой жидкости (1,85 г, 22%). 1H ЯМР (400 MГц, ДМСО-d6) δ 6,51 (дд, J=7,3, 12,8 Гц, 1H), 5,55 (с-ш, 2H), 3,88 (с, 3H).
Стадия 6: Получение 3-амино-2-хлор-5,6-дифторфенола. К раствору 2-хлор-4,5-дифтор-3-метоксианилина (2,2 г, 9,24 ммоль) в ДХМ (30 мл) добавляют раствор 1N BBr3 в ДХМ (28,0 мл, 27,73 ммоль) при 0°C. Реакционную смесь перемешивают при 0°C в течение 1 часа и затем при 25°C в течение 16 часов под Ar. Реакционную смесь концентрируют, и остаток растворяют в MeOH при 0°C и pH доводят до примерно 7 насыщенным NaHCO3. Реакционную смесь концентрируют и водный слой экстрагируют этилацетатом (2×250 мл). Объединенные органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным Na2SO4, фильтруют и концентрируют с получением 3-амино-2-хлор-5,6-дифторфенола в виде бледно-коричневого твердого вещества (1,1 г, 66%). 1H ЯМР (400 MГц, ДМСО-d6) δ 5,96 (дд, J=6,9, 12,8 Гц, 1H), 5,03 (с-ш, 2H). МС (m/z) =177,8 [M-H].
Промежуточное соединение P90

3-фтор-2-метил-6-нитробензойная кислота
К ледяному раствору 3-фтор-2-метилбензойной кислоты (5 г, 32,4 ммоль) в концентрированной H2SO4 (50 мл) медленно добавляют концентрированную HNO3 (3,5 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 6 часов. Реакционную смесь выливают в ледяную воду и твердое вещество собирают фильтрацией, промывают водой (2x 50 мл) и сушат при пониженном давлении с получением 3-фтор-2-метил-6-нитробензойной кислоты в виде беловатого твердого вещества (3,0 г, 47%). 1H ЯМР (400 MГц, MeOD) δ 8,11-8,07 (м, 1H), 7,34 (т, 1H), 2,32 (с, 3H); МС (m/z)=197,6 (M-H).
Промежуточное соединение P91
3-амино-2,5,6-трифторфенол
Стадия 1: Получение трет-бутил (2,4,5-трифтор-3-метоксифенил)карбамата. К раствору 2,4,5-трифтор-3-метоксибензойной кислоты (5,0 г, 24,27 ммоль) в смеси толуола:трет-бутанола (1:1, 20 мл) добавляют N, N-диизопропилэтиламин (5,7 мл, 31,55 ммоль) и смесь охлаждают до 0°C. Туда добавляют дифенилфосфорилазид (7,8 мл, 36,4 ммоль), и реакционную смесь нагревают до температуры окружающей среды и затем перемешивают при 100°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют этилацетатом (3×50 мл). Объединенные органические слои сушат с Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением трет-бутил (2,4,5-трифтор-3-метоксифенил)карбамата в виде бесцветного твердого вещества (5,0 г, 74%). 1H ЯМР (400 MГц, CDCl3) δ 7,73 (с-ш, 1H), 6,60 (с, 1H), 4,01 (с, 3H), 1,51 (с, 9H); МС (m/z)=278,0 (M+H).
Стадия 2: Получение 2,4,5-трифтор-3-метоксианилина. К раствору трет-бутил (2,4,5-трифтор-3-метоксифенил)карбамата (5,0 г, 18,0 ммоль) в 1,4-диоксане (20 мл) добавляют 4N HCl в диоксане (22,5 мл, 90,25 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь концентрируют, и pH доводят до примерно 7 насыщенным водным бикарбонатом натрия. Водный слой экстрагируют этилацетатом (3×100 мл), и объединенные органические слои сушат с Na2SO4, фильтруют и концентрируют с получением 2,4,5-трифтор-3-метоксианилина в виде коричневатого полутвердого вещества (2,5 г, 60%) которое применяют непосредственно на следующей стадии. 1H ЯМР (400 MГц, CDCl3) δ 6,33-6,21 (м, 1H), 3,99 (с, 3H), 3,66 (с-ш, 2H); МС (m/z)=175,9 (M-H).
Стадия 3: Получение 3-амино-2,5,6-трифторфенола. К перемешиваемому раствору 2,4,5-трифтор-3-метоксианилина (2,5 г, 17,73 ммоль) в дихлорметане (20 мл) добавляют 1,0 M трибромид бора в ДХМ (35,5 мл, 35,46 ммоль) при 0°C. Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь гасят метанолом (50 мл), концентрируют при пониженном давлении и pH доводят до примерно 6 насыщенным раствором бикарбоната натрия (10 мл). Водный слой экстрагируют этилацетатом (2×50 мл), и объединенные органические слои сушат с Na2SO4, фильтруют и концентрируют с получением 3-амино-2,5,6-трифторфенола в виде коричневого твердого вещества (1,75 г, 61%), которое применяют непосредственно на следующей стадии. 1H ЯМР (400 MГц, ДМСО-d6) δ 10,27 (с-ш, 1H), 6,36-5,85 (м, 1H), 5,14 (с-ш, 2H); МС (m/z)=161,9 (M-H).
Получение синтетических примеров
Пример 1
N- (3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-2,4-дифторфенил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4-дифторфенил)-N-(4-метоксибензил)пропан-1-сульфонамида. N-(3-амино-2,4-дифторфенил)-N-(4-метоксибензил)пропан-1-сульфонамид (83,7 мг, 0,226 ммоль), 6-бром-3,5-диметилхиназолин-4(3H)-он (52 мг, 0,205 ммоль), карбонат цезия (201 мг, 0,616 ммоль), Xantphos (17,8 мг, 0,0308 ммоль) и трис(дибензилиденацетон)дипалладий (0) (9,41 мг, 0,0103) объединяют в пробирке затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (1,03 мл) добавляют, и раствор продувают аргоном в течение 5 минут, затем пробирку герметично закрывают и нагревают до 90°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и фильтруют через слой Celite® и очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4-дифторфенил)-N-(4-метоксибензил)пропан-1-сульфонамида в виде белого твердого вещества (65 мг, 58%). МС (хиад, m/z)=543,2 (M+H).
Стадия 2: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4-дифторфенил)пропан-1-сульфонамида. N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4-дифторфенил)-N-(4-метоксибензил)пропан-1-сульфонамид (65 мг, 0,12 ммоль) растворяют в ДХМ (3,0 мл) и обрабатывают трифторуксусной кислотой (1,0 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и остаток очищают хроматографией на силикагеле, элюируя ДХМ/MeOH с 1% NH4OH с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)пропан-1-сульфонамида в виде белого твердого вещества (37,5 мг, 43%). 1H ЯМР (400 MГц, CDCl3) δ 7,94 (с, 1H), 7,45 (д, 1H), 7,29-7,23 (м, 1H), 7,16-7,12 (м, 1H), 6,98 (т, 1H), 6,62 (шс, 1H), 5,41 (шс, 1H), 3,54 (с, 3H), 3,07 (дд, 2H), 2,95 (с, 3H), 1,92-1,83 (м, 2H), 1,05 (т, 3H); МС (хиад, m/z)=423,1 (M+H).
Пример 2
N- (3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-2,4-дифторфенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида. N-(3-амино-2,4-дифторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамид (34,6 мг, 0,087 ммоль), 6-бром-3,5-диметилхиназолин-4(3H)-он (20 мг, 0,079 ммоль), карбонат цезия (77 мг, 0,237 ммоль), Xantphos (6,9 мг, 0,012 ммоль) и трис(дибензилиденацетон)дипалладий (0) (3,62 мг, 0,00395) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (0,395 мл) добавляют, и раствор продувают аргоном в течение 5 мин, затем пробирку герметично закрывают и нагревают до 90°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и фильтруют через слой Celite® и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)-N-((2-(триметилсилил)-этокси)метил))пропан-1-сульфонамида в виде белого твердого вещества (35,6 мг, 79%). МС (хиад, m/z)=571,2 (M+H).
Стадия 2: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)3-фторпропан-1-сульфонамида. N-(3-((3,5-диметил-4-оксо-3,4-дигидро-хиназолин-6-ил)амино-2,4-дифторфенил)-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамид (35,6 мг, 0,0624 ммоль) растворяют в ДХМ (1,5 мл) и обрабатывают трифторуксусной кислотой (0,5 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле (ДХМ/MeOH с 1% NH4OH) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4-дифторфенил)3-фторпропан-1-сульфонамида в виде белого твердого вещества (16,0 мг, 46%). 1H ЯМР (400 MГц, CDCl3) δ 7,90 (с, 1H), 7,42 (д, 1H), 7,17-7,22 (м, 1H), 7,09-7,12 (м, 1H), 6,92-6,97 (м, 1H), 5,49 (шс, 1H), 4,57 (т, 1H), 4,45 (т, 1H), 3,51 (с, 3H), 3,21 (дд, 2H), 2,91 (с, 3H), 2,15-2,27 (м, 2H). МС (хиад, m/z)=441,1 (M+H).
Пример 3
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)фенил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3-бром-2-хлорфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (41 мг, 0,093 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (18 мг, 0,093 ммоль), трис(дибензилиденацетон)дипалладия (0) (4,2 мг, 0,0046 ммоль), Xantphos (8,0 мг, 0,014 ммоль) и карбоната цезия (90 мг, 0,28 ммоль) в толуоле (617 мкл) продувают аргоном в течение 10 минут, затем герметично закрывают и нагревают в пробирке до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды и очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. МС (хиад, m/z)=551,2 [M+H].
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пропан-1-сульфонамида. Раствор N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида перемешивают в 1 мл ДХМ и 0,2 мл трифторуксусной кислоты в течение 45 минут. Реакционную смесь концентрируют и растворяют в 1 мл ТГФ и 1 мл насыщенного NaHCO3 и перемешивают в течение 30 минут. Реакционную смесь разбавляют дополнительным насыщенным NaHCO3 и экстрагируют ДХМ (2x), и затем объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли. Органические слои сушат над Na2SO4, затем фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пропан-1-сульфонамида (17 мг, 44% за 2 стадии). 1H ЯМР (400 MГц, CDCl3) δ 8,01 (с, 1H), 7,62 (д, 1H), 7,57 (д, 1H), 7,15 (д, 1H), 7,07 (т, 1H), 6,78 (с-ш, 1H), 6,47 (д, 1H), 5,98 (с-ш, 1H), 3,57 (с, 3H), 3,14 (т, 2H), 2,82 (с, 3H), 1,90 (м, 2H), 1,06 (т, 3H). МС (хиад, m/z)=421,1 [M+H].
Пример 4
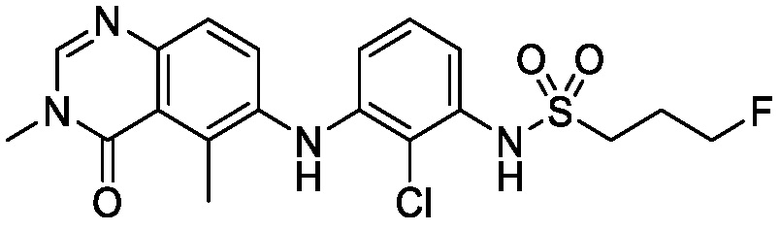
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)фенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Суспензию N-(3-амино-2-хлорфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (0,045 г, 0,11 ммоль), 6-бром-3,5-диметилхиназолин-4(3H)-она (0,026 г, 0,10 ммоль), трис(дибензилиденацетон)дипалладия (0) (0,0047 г, 0,0051 ммоль), Xantphos (0,0089 г, 0,015 ммоль) и карбоната цезия (0,10 г, 0,31 ммоль) в толуоле (1,0 мл) опрыскивают барботированием аргона в течение 5 минут. Пробирку герметично закрывают, и смесь нагревают at 90°C в течение ночи. Смесь охлаждают до температуры окружающей среды и разбавляют насыщенным водным раствором NH4Cl и смесь экстрагируют EtOAc (3x). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией, элюируя ДХМ/EtOAc с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида в виде бледно-желтой твердой пены (0,055 г, 94%). МС (хиад, m/z)=569,2 [M+H].
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпропан-1-сульфонамида. В пробирку, содержащую N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамид (0,055 г, 0,097 ммоль), добавляют CH2Cl2 (0,483 мл) и раствор обрабатывают трифторуксусной кислотой (0,185 мл, 2,42 ммоль). Раствор перемешивают при температуре окружающей среды в течение 2 часов. Смесь добавляют в насыщенный водный раствор NaHCO3, и смесь экстрагируют CHCl3. Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией (ДХМ/MeOH) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпропан-1-сульфонамида в виде беловатого твердого вещества (0,026 г, 58%). 1H ЯМР (400 MГц, CDCl3) δ 8,00 (с, 1H); 7,59 (м, 2H); 7,15 (дд, 1H); 7,08 (т, 1H); 6,80 (шс, 1H); 6,49 (дд, 1H); 5,98 (шс, 1H); 4,63 (т, 1H); 4,51 (т, 1H); 3,56 (с, 3H); 3,32 (м, 2H); 2,83 (с, 3H); 2,27 (м, 2H); МС (хиад, m/z)=439,0 [M+H].
Пример 5
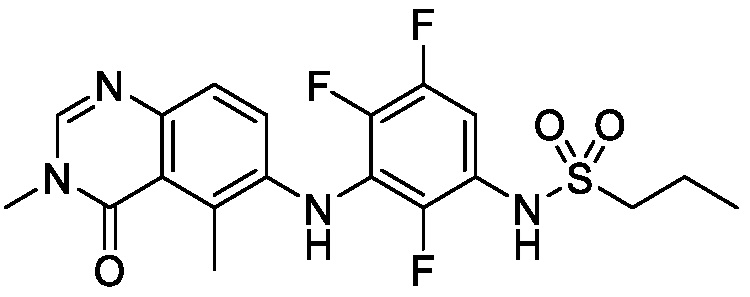
N- (3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-2,4,5-трифторфенил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(2,4,5-трифтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид (105 мг, 0,206 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (39,0 мг, 0,206 ммоль), трис(дибензилиденацетон)дипалладия (0) (9,44 мг, 0,0103 ммоль), Xantphos (17,9 мг, 0,0309 ммоль) и карбоната цезия (201 мг, 0,618 ммоль) в толуоле (1374 мкл) продувают аргоном в течение 10 минут и затем нагревают в герметично закрытой колбе до 90°C в течение 3 часов. Реакционную смесь охлаждают до температуры окружающей среды и затем очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (59 мг, 50%). МС (хиад, m/z)=571,2 [M+H].
Стадия 2: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)пропан-1-сульфонамида. Раствор N-(3-((3,5-диметил-4-оксо-3,4-дигидро-хиназолин-6-ил)амино)-2,4,5-трифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (96 мг, 0,168 ммоль) в дихлорметане (1 мл) и трифторуксусной кислоте (0,5 мл) перемешивают в течение 30 минут при температуре окружающей среды. Раствор концентрируют, и затем растворяют в тетрагидрофуране (1 мл) и насыщенном NaHCO3 (1 мл) и перемешивают при температуре окружающей среды в течение 20 минут. Реакционную смесь разбавляют дополнительным насыщенным NaHCO3 и экстрагируют ДХМ (2x), и затем объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Органические слои очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc, с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)пропан-1-сульфонамида (47,5 мг, 64%). 1H ЯМР (400 MГц, CDCl3) δ 9,37 (с, 1H), 7,71 (д, 1H), 7,27 (м, 2H), 6,52 (с-ш, 1H), 5,63 (шс, 1H), 3,79 (с, 3H), 3,16 (т, 2H), 2,94 (с, 3H), 1,90 (м, 2H), 1,09 (т, 3H); МС (хиад, m/z)=441,1 [M+H].
Пример 6
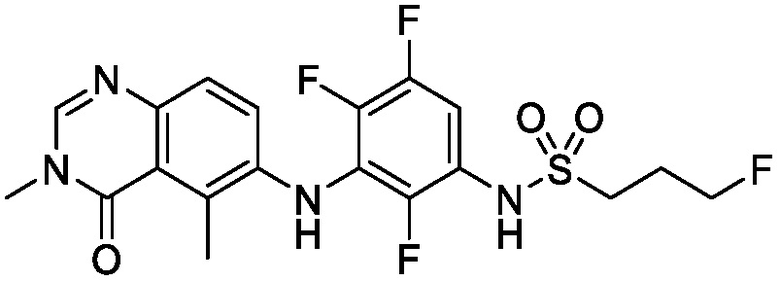
N- (3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-2,4,5-трифторфенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4,5-трифторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида. 3-Фтор-N-(2,4,5-трифтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамид (39 мг, 0,074 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-он (Промежуточное соединение P15) (14 мг, 0,074 ммоль), карбонат цезия (72 мг, 0,22 ммоль), Xantphos (9,0 мг, 0,016 ммоль) и трис(дибензилиденацетон)дипалладий (0) (4,7 мг, 0,0052) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (0,395 мл) добавляют, и раствор продувают аргоном в течение 5 минут затем пробирку герметично закрывают и нагревают до 110°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и фильтруют через слой Celite® и очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc, с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4,5-трифторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида в виде белого твердого вещества (28,9 мг, 66%). МС (хиад, m/z)=589,2 (M+H).
Стадия 2: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-3-фторпропан-1-сульфонамида. N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4,5-трифторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамид (28,9 мг, 0,0491 ммоль) растворяют в ДХМ (1,2 мл) и обрабатывают трифторуксусной кислотой (0,41 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и остаток очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,4,5-трифторфенил)-3-фторпропан-1-сульфонамида в виде белого твердого вещества (8,20 мг, 24%). 1H ЯМР (400 MГц, CDCl3) δ 7,89 (с, 1H), 7,35 (д, 1H), 7,13 (д, 1H), 6,98-7,05 (м, 1H), 6,02 (шс, 1H), 4,51 (т, 1H), 4,39 (т, 1H), 3,46 (с, 3H), 3,15 (дд, 2H), 2,82 (с, 3H), 2,06-2,19 (м, 2H); МС (хиад, m/z)=459,1 (M+H).
Пример 7
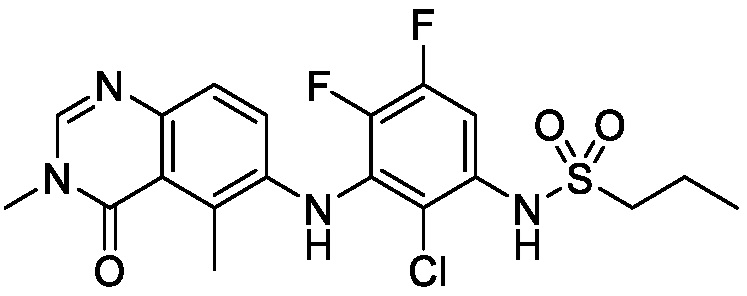
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4,5-дифторфенил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(2-хлор-4,5-дифтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (39 мг, 0,074 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (14 мг, 0,074 ммоль), трис(дибензилиденацетон)дипалладия (0) (3,4 мг, 0,0037 ммоль), Xantphos (6,4 мг, 0,011 ммоль) и карбоната цезия (72 мг, 0,22 ммоль) в толуоле (494 мкл) продувают аргоном в течение 10 минут и затем нагревают в герметично закрытой пробирке до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды и очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc, с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (33 мг, 75%). МС (хиад, m/z)=587,2 [M+H].
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пропан-1-сульфонамида. Раствор N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (33 мг, 0,567 ммоль) в 1 мл ДХМ и 0,2 мл трифторуксусной кислоты перемешивают при температуре окружающей среды в течение 1 часа. Раствор концентрируют и растворяют в 1 мл ТГФ и 1 мл насыщенного NaHCO3 и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь разбавляют дополнительным насыщенным NaHCO3 и экстрагируют ДХМ (2x), и затем объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)пропан-1-сульфонамида (15 мг, 44%). 1H ЯМР (400 MГц, CDCl3) δ 7,96 (с, 1H), 7,48 (д, 1H), 7,31 (дд, 1H), 7,18 (дд, 1H), 6,72 (шс, 1H), 5,69 (шс, 1H), 3,56 (с, 3H), 3,12 (т, 2H), 2,96 (с, 3H), 1,88 (м, 2H), 1,07 (т, 3H); МС (хиад, m/z)=457,1 [M+H].
Пример 8
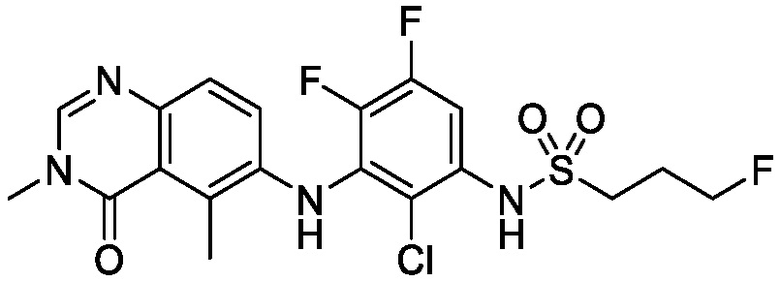
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4,5-дифторфенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида. N-(2-хлор-4,5-дифтор-3-йодфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамид (52 мг, 0,095 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-он (Промежуточное соединение P15) (18 мг, 0,095 ммоль), карбонат цезия (93 мг, 0,29 ммоль), Xantphos (14,0 мг, 0,024 ммоль) и трис(дибензилиденацетон)дипалладий (0) (7,4 мг, 0,0081) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (0,475 мл) добавляют, и раствор продувают аргоном в течение 5 минут затем пробирку герметично закрывают и нагревают до 110°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и фильтруют через слой Celite®, затем очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида в виде белого твердого вещества (34,5 мг, 60%). МС (хиад, m/z)=605,2 (M+H).
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фторпропан-1-сульфонамида. N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фтор-N-((2-(триметилсилил)этокси)-метил))пропан-1-сульфонамид (34,5 мг, 0,057 ммоль) растворяют в ДХМ (1,4 мл) и обрабатывают трифторуксусной кислотой (0,48 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют, и остаток очищают хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4,5-дифторфенил)-3-фторпропан-1-сульфонамида в виде белого твердого вещества (20,5 мг, 45%). 1H ЯМР (400 MГц, CDCl3) δ 7,95 (с, 1H), 7,49 (д, 1H), 7,30 (д, 1H), 7,17 (дд, 1H), 6,72 (шс, 1H), 5,69 (шс, 1H), 4,63 (т, 1H), 4,51 (т, 1H), 3,55 (с, 3H), 3,29 (дд, 2H), 2,95 (с, 3H), 2,18-2,32 (м, 2H); МС (хиад, m/z)=475,1 (M+H).
Пример 9
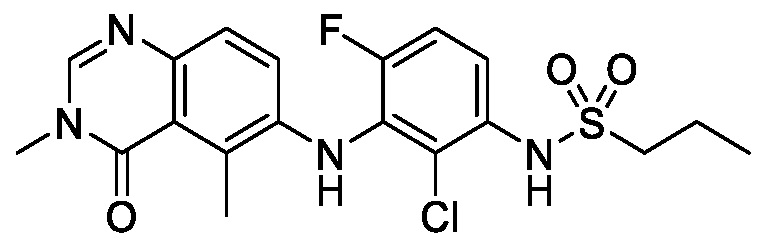
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-(4-метоксибензил)пропан-1-сульфонамида. Раствор N-(3-амино-2-хлор-4-фторфенил)-N-(4-метоксибензил)пропан-1-сульфонамида (42 мг, 0,11 ммоль), 6-бром-3,5-диметилхиназолин-4(3H)-она (27 мг, 0,11 ммоль), трис(дибензилиденацетон)дипалладия (0) (5,0 мг, 0,0054 ммоль), Xantphos (9,4 мг, 0,016 ммоль) и карбоната цезия (106 мг, 0,33 ммоль) в толуоле (724 мкл) продувают аргоном в течение 10 минут и затем нагревают в герметично закрытой пробирке до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-(4-метоксибензил)пропан-1-сульфонамида (43 мг, 70%). МС (хиад, m/z)=559,2 [M+H].
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пропан-1-сульфонамида. Раствор N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-(4-метоксибензил)пропан-1-сульфонамида (43 мг, 0,077 ммоль) в 1 мл ДХМ и 0,2 мл трифторуксусной кислоты перемешивают при температуре окружающей среды в течение 3 часов. Реакционную смесь концентрируют, и остаток очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пропан-1-сульфонамида (23 мг, 48%). 1H ЯМР (400 MГц, CDCl3) δ 7,94 (с, 1H), 7,44 (д, 1H), 7,41 (дд, 1H), 7,11 (д, 1H), 7,08 (д, 1H), 6,76 (шс, 1H), 5,61 (шс, 1H), 3,55 (с, 3H), 3,10 (т, 2H), 2,97 (с, 3H), 1,88 (м, 2H), 1,05 (т, 3H); МС (хиад, m/z)=439,1 [M+H].
Пример 10
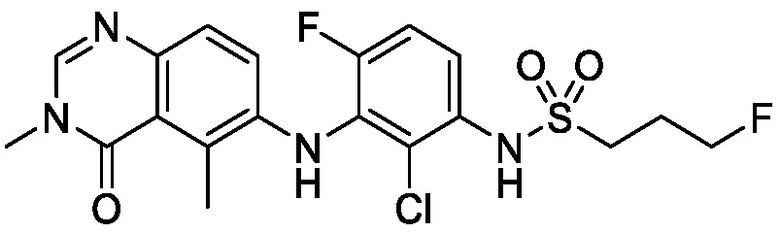
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид
Способ A
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3-амино-2-хлор-4-фторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (Промежуточное соединение P26; 150 мг, 0,361 ммоль), 6-бром-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P2; 91,5 мг, 0,361 ммоль), трис(дибензилиденацетон)дипалладия (0) (16,6 мг, 0,0181 ммоль), Xantphos (31,4 мг, 0,0542 ммоль) и карбоната цезия (353 мг, 1,08 ммоль) в толуоле (2410 мкл) продувают аргоном в течение 10 минут и затем нагревают в герметично закрытой пробирке до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (200 мг, 94%). МС (хиад, m/z)=587,2 [M+H].
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида. Раствор N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида (200 мг, 0,34 ммоль) перемешивают в 2 мл ДХМ и 0,5 мл трифторуксусной кислоты при температуре окружающей среды в течение 45 минут. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле (ДХМ/MeOH с 1% NH4OH) и С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК). Фракции, содержащие желаемый продукт, растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли. Органические слои сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида (65 мг, 39%). 1H ЯМР (400 MГц, CDCl3) δ 7,93 (с, 1H), 7,44 (д, 1H), 7,41 (дд, 1H), 7,12 (д, 1H), 7,08 (д, 1H), 6,79 (шс, 1H), 5,61 (шс, 1H), 4,61 (т, 1H), 4,50 (т, 1H), 3,55 (с, 3H), 3,28 (т, 2H), 2,98 (с, 3H), 2,25 (т, 2H); МС (хиад, m/z)=457,1 [M+H].
Способ B
Стадия A: Получение 5-метилхиназолин-4(3H)-она. Раствор 2-амино-6-метилбензойной кислоты (74,7 г, 494 ммоль) и ацетата формамидина (61,7 г, 593 ммоль) в этаноле (1,2 л) нагревают при кипении с обратным холодильником в атмосфере азота в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды с получением густой суспензии. Твердые вещества выделяют фильтрацией и промывают этанолом (500 мл) и сушат в вакуумной печи при 50°C в течение 18 часов. Фильтрат концентрируют до 500 мл, затем разбавляют водой (1 л) и выдерживают при температуре окружающей среды в течение 18 часов с получением густой суспензии. Твердые вещества выделяют фильтрацией и сушат в вакуумной печи при 50°C в течение 18 часов. Твердые вещества после первой фильтрации и второй фильтрации объединяют с получением 5-метилхиназолин-4(3H)-она (63,3 г, 80%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 12,01 (шс, 1H), 8,00 (с, 1H), 7,63 (т, 1H), 7,47 (д, 1H), 7,26 (д, 1H), 2,78 (с, 3H); МС (хиад, m/z)=161,2 (M+H).
Стадия B: Получение 5-метил-6-нитрохиназолин-4(3H)-она. Раствор 5-метилхиназолин-4(3H)-она (50 г, 312,2 ммоль) в концентрированной серной кислоте (555 мл) охлаждают до внутренней температуры 10°C с внешней баней с ледяной водой. К реакционной смеси добавляют раствор азотной кислоты (5M раствор в концентрированной серной кислоте, 68,67 мл, 343,4 ммоль). Скорость добавления контролирует для сохранения внутренней температуры ниже 30°C. Реакционную смесь перемешивают в течение 20 минут и затем разбавляют ледяной водой (5,5 л) и охлаждают с внешней баней с ледяной водой до достижения внутренней температуры реакционной смеси <25°C. pH реакционной смеси доводят до 4 медленным добавлением 10 M водного NaOH (2 л) с получением густой желтой суспензии. Твердые вещества выделяют фильтрацией и промывают водой (1 л) с получением фильтровальной лепешки. Фильтровальную лепешку суспендируют в метаноле (7 л) и этилацетате (500 мл), затем нагревают до 55°C с получением оранжевого раствора. При охлаждении до температуры окружающей среды в течение 18 часов образуется суспензия желтых кристаллов. Кристаллы фильтруют, промывают метанолом (1 л) и сушат в печи с высоким вакуумом при 50°C в течение 18 часов с получением 5-метил-6-нитрохиназолин-4(3H)-она (59,94 г, 94%) в виде желтых кристаллов. 1H ЯМР (400 MГц, ДМСО-d6) δ 12,50 (шс, 1H), 8,18 (с, 1H), 8,13 (д, 1H), 7,63 (д, 1H), 2,82 (с, 3 H); МС (хиад, m/z)=206,1 (M+H).
Стадия C: Получение 3,5-диметил-6-нитрохиназолин-4(3H)-она. Раствор 5-метил-6-нитрохиназолин-4(3H)-она (59,94 г, 292,1 ммоль) в ДМФ (0,2 M, 1,5 л) охлаждают на внешней бане с ледяной водой. К реакционной смеси добавляют K2CO3 (80,75 г, 584,3 ммоль) и йодметан (27,28 мл, 438,2 ммоль). Реакционную смесь перемешивают в течение 3 часов при температуре окружающей среды затем разбавляют водой (7,5 л) с получением густой желтой суспензии, которую перемешивают в течение 1 часа при температуре окружающей среды. Твердые вещества выделяют фильтрацией, промывают водой (1 л) и сушат под вакуумом в течение 18 часов с получением 3,5-диметил-6-нитрохиназолин-4(3H)-она (45,85 г, 72%) в виде тускло-желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,49 (с, 1H), 8,13 (д, 1H), 7,63 (д, 1H), 3,47 (с, 3H), 2,81 (с, 3H); МС (хиад, m/z)=220,1 (M+H)
Стадия D: Получение 6-амино-3,5-диметилхиназолин-4(3H)-она: Суспензию 3,5-диметил-6-нитрохиназолин-4(3H)-она (45,85 г, 209,2 ммоль) и палладия на угле (11,13 г, 5,229 ммоль) в метаноле (3 л) опрыскивают азотом в течение 30 минут и затем помещают под баллон с водородом. Реакционную смесь перемешивают при температуре окружающей среды в течение 18 часов, затем опрыскивают азотом и фильтруют через слой диатомовой земли. Фильтрат концентрируют с получением 6-амино-3,5-диметилхиназолин-4(3H)-она (37,59 г, 95%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,02 (с, 1H), 7,26 (д, 1H), 7,13 (д, 1H), 5,28 (шс, 2H), 3,39 (с, 3H), 2,62 (с, 3H); МС (хиад, m/z)=190,1 (M+H)
Стадия E: Получение 2-хлор-4-фтор-3-йоданилина. В 5-л 4-горлой колбе, оборудованной 3 капельными воронками, внутренним температурным зондом и магнитной лопастной мешалкой, 2-хлор-4-фторанилин (82,03 мл, 687,00 ммоль) растворяют в ТГФ (1,5 л) в обратном потоке N2 и охлаждают до -78°C. Реакционную смесь затем обрабатывают по каплям н-бутиллитием (2,5 M в гексане) (299,53 мл, 748,82 ммоль) и перемешивают при -78°C в течение 15 минут после завершения добавления. Реакционную смесь обрабатывают по каплям ТГФ раствором (500 мл) 1,2-бис(хлордиметилсилил)этана (155,28 г, 721,35 ммоль) и перемешивают при -78°C в течение 30 минут после завершения добавления. Реакционную смесь обрабатывают по каплям дополнительным н-бутиллитием (2,5 M в гексане) (299,53 мл, 748,82 ммоль) и затем ледяную баню удаляют после завершения добавления, и реакционную смесь перемешивают в течение 1 часа. Реакционную смесь затем обратно охлаждают до -78°C и обрабатывают по каплям дополнительным н-бутиллитием (2,5 M в гексане) (299,53 мл, 748,82 ммоль) и перемешивают при -78°C в течение 30 минут после завершения добавления. Реакционную смесь обрабатывают по каплям ТГФ раствором (600 мл) йода (249,34 г, 982,40 ммоль), и ледяную баню удаляют и реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 16 часов. Реакционную смесь обрабатывают 1 л воды, затем хлористоводородной кислотой (4,0 M водным раствором) (601,12 мл, 2404,5 ммоль) и перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь нейтрализуют до примерно pH 8 с применением твердого NaHCO3 и затем обрабатывают тиосульфатом натрия (3,0 M водным раствором) (801,49 мл, 2404,5 ммоль) и перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь переносят в 6 л экстракционную воронку, колбу промывают МТБЭ и водой и слои разделяют. Водный слой экстрагируют МТБЭ (1x) и органические слои объединяют, промывают насыщенным раствором соли (1x), сушат над Na2SO4, фильтруют и концентрируют с получением 2-хлор-4-фтор-3-йоданилина (186,49 г, 100%) который применяют непосредственно на следующей стадии. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,08-7,04 (м, 1H), 6,91-6,88 (м, 1H), 5,52 (шс, 2H). МС (хиад, m/z)=271,9, 273,9 (M+H).
Стадия F: Получение бис-трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата. В 3 л 1-горлой колбе, 2-хлор-4-фтор-3-йоданилин (186,49 г, 687,0 ммоль) растворяют в ТГФ (2,0 л) и обрабатывают 4-(диметиламино)пиридином (8,393 г, 68,7 ммоль), затем добавляют ди-трет-бутил дикарбонат (314,87 г, 1442,7 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа на открытом воздухе с применением колонки Vigreux. Реакционную смесь затем концентрируют досуха и полученное твердое вещество суспендируют в гептане (1 л) и перемешивают в течение 30 минут. Полученные твердые вещества собирают фильтрацией, промывают дополнительным гептаном (250 мл x 2) с получением бис-трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (150,7 г, 47%) в виде светлого рыжевато-коричневого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 7,55-7,51 (м, 1H), 7,32-7,27 (м, 1H), 1,33 (с, 18H).
Стадия G: Получение Трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата. Бис-трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамат (150,7 г, 319,5 ммоль) растворяют в MeOH (800 мл) и обрабатывают карбонатом калия (48,57 г, 351,4 ммоль) и реакционную смесь нагревают до 65°C в течение 1 часа. Реакционную смесь затем охлаждают до температуры окружающей среды и выливают в 3,0 л воды и перемешивают в течение 30 минут. Полученные твердые вещества собирают фильтрацией и промывают дополнительной водой (1 л). Твердые вещества затем растворяют в ДХМ (1 л), разбавляют гексаном (1 л) и фильтруют через слой геля двуокиси кремния (500 г), элюируя дополнительным 1:1 ДХМ:гексаном (4,0 л). Фракции, содержащие желаемый продукт, концентрируют с получением трет-бутил (2-хлор-4-фтор-3-йодфенил)карбамата (101,0 г, 85%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,82 (шс, 1H), 7,53-7,49 (м, 1H), 7,27-7,20 (м, 1H), 1,42 (с, 9H).
Стадия H: Получение трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)карбамата. В 3 л колбу, оборудованную конденсатором с обратным холодильником и магнитной лопастной мешалкой, 6-амино-3,5-диметилхиназолин-4(3H)-он (50,5 г, 266,9 ммоль) растворяют в толуоле (1065 мл) и обрабатывают трет-бутил (2-хлор-4-фтор-3-йодфенил)карбаматом (109,1 г, 293,6 ммоль), трис(дибензилиденацетон)дипалладием (0) (2,44 г, 2,67 ммоль), Xantphos (3,86 г, 6,67 ммоль) и карбонатом цезия (173,9 г, 533,78 ммоль). Реакционную смесь продувают аргоном в течение 15 минут и затем нагревают до 110°C под баллоном аргона в течение 24 часов. Реакционную смесь охлаждают до температуры окружающей среды и затем разбавляют ДХМ (2,0 л) и перемешивают в течение 30 минут. Реакционную смесь фильтруют через Celite® и фильтрат концентрируют. Полученное твердое вещество растворяют в ДХМ (2,5 л) и затем выливают в перемешиваемый раствор гептана (12,5 л) и перемешивают при температуре окружающей среды в течение 30 минут. Полученные твердые вещества собирают фильтрацией, промывают дополнительным гептаном (1,5 л) с получением трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)карбамата (95,3 г, 83%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,70 (с, 1H), 8,15 (с, 1H), 7,36-7,23 (м, 4H), 6,81-6,79 (м, 1H), 3,43 (с, 3H), 2,81 (с, 3H), 1,45 (с, 9H); МС (хиад, m/z)=433,2, 435,2 (M+H).
Стадия I: Получение трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)((3-фторпропил)сульфонил)карбамата. В 5 л 3-горлой колбе, оборудованной 2 капельными воронками, внутренним температурным зондом, магнитной лопастной мешалкой и входом для азота, трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)карбамат (95,3 г, 220 ммоль) растворяют в ТГФ (2,2 л) и охлаждают до 5°C в обратном потоке N2. Реакционную смесь обрабатывают по каплям 1,0 M раствором бис(триметилсилил)амида натрия в ТГФ (209 мл, 209 ммоль) и затем перемешивают в течение 15 минут после завершения добавления. Реакционную смесь обрабатывают по каплям 150 мл ТГФ раствора 3-фторпропан-1-сульфонилхлорида (33,6 г, 209 ммоль) и ледяную баню затем удаляют, и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют с получением трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)((3-фторпропил)сульфонил)карбамата (123 г, 100%) который применяют непосредственно на следующей стадии. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,17 (с, 1H), 7,53 (с, 1H), 7,40 (с, 1H), 7,39 (с, 1H), 7,33-7,31 (д, 1H), 6,80-6,78 (д, 1H), 4,67-4,64 (т, 1H), 4,55-4,52 (т, 1H), 4,00-3,93 (м, 1H), 3,85-3,77 (м, 1H), 3,43 (с, 3H), 2,80 (с, 3H), 2,25-2,19 (м, 2H), 1,41 (с, 9H). МС (m/z)=557,2, 559,2 (M+H).
Стадия J: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида. К раствору трет-бутил (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)карбамата (123 г, 221,0 ммоль) в ДХМ (500 мл) добавляют стабильный поток 500 мл трифторуксусной кислоте и смесь перемешивают при температуре окружающей среды в течение 1 часа после завершения добавления. Реакционную смесь концентрируют, и полученный остаток растворяют в ДХМ (1500 мл), обрабатывают 1500 мл воды и нейтрализуют до примерно pH 7-8 с применением твердой NaHCO3. Раствор затем фильтруют, и слои разделяют и водный слой NaHCO3 экстрагируют ДХМ (1x). Объединенные органические слои промывают насыщенным NaHCO3 (1x) и затем 1,0 M NaOH (3x). Объединенные водные слои NaOH промывают ДХМ (1x) и водный слой NaOH переносят в колбу, и разбавляют 4:1 ДХМ:ИПА (1000 мл) и подкисляют примерно pH 2 с применением 4,0 M HCl. Слои разделяют, и водный слой HCl экстрагируют 4:1 ДХМ:ИПА (2x). Объединенные слои 4:1 ДХМ:ИПА сушат над Na2SO4, фильтруют и концентрируют. Полученный остаток растворяют в ДХМ (2000 мл) и перемешивают с SiliaMetS тиолом (100 граммов) при температуре окружающей среды в течение 16 часов. Смесь загружают в слой геля двуокиси кремния (500 г) и элюируют EtOAc (8,0 л). Фильтрат концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида (78,9 г, 78,4%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,64 (шс, 1H), 8,16 (с, 1H), 7,34-7,25 (м, 4H), 6,88-6,85 (д, 1H), 4,62-4,59 (т, 1H), 4,50-4,47 (т, 1H), 3,43 (с, 3H), 3,25-3,21 (м, 2H), 2,81 (с, 3H), 2,19-2,06 (м, 2H). МС (m/z)=457,1, 459,1 (M+H).
Пример 11
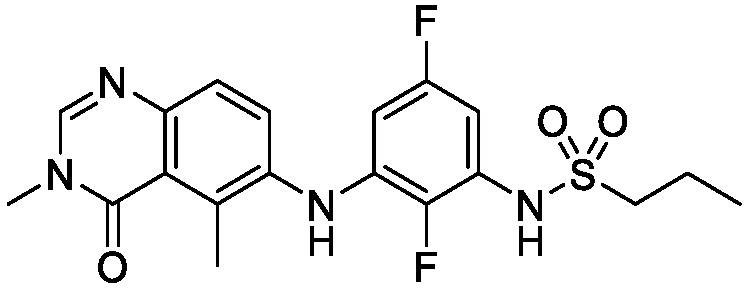
N- (3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-2,5-дифторфенил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3-бром-2,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (72,6 мг, 0,163 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (30,9 мг, 0,163 ммоль), трис(дибензилиденацетон)дипалладия (0) (10,5 мг, 0,0114 ммоль), Xantphos (19,8 мг, 0,0343 ммоль) и карбоната цезия (160 мг, 0,490 ммоль) в толуоле (4 мл) продувают аргоном в течение 5 минут и затем нагревают при 100°C в течение 18 часов в 15 мл герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, затем разбавляют водой (25 мл) и 4:1 ДХМ:ИПА (25 мл). Слои разделяют, и органическую фазу сушат над Na2SO4, фильтруют и концентрируют с получением коричневого масла, которое очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (62,9 мг, 70%) в виде беловатой пены. МС (хиад, m/z)=553,2 (M+H)
Стадия 2: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)пропан-1-сульфонамида. N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид (62,9 мг, 0,114 ммоль) растворяют в ДХМ (5 мл) и чистой трифторэтановой кислоте (3507 мкл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1,5 часов. Реакционную смесь концентрируют, и затем очищают хроматографией на силикагеле (ДХМ/EtOAc) затем С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и затем выделенный продукт растворяют в 4:1 ДХМ:ИПА и промывают насыщенным NaHCO3 затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)пропан-1-сульфонамида (13,6 мг, 28%). 1H ЯМР (400 MГц, CDCl3) δ 8,01 (с, 1H), 7,64-7,56 (м, 1H), 6,82-6,75 (м, 1H), 6,55 (с, 1H), 6,19-6,13 (м, 1H), 5,74 (с, 1H), 3,56 (с, 3H), 3,21-3,14 (м, 2H), 2,84 (с, 3H), 1,97-1,86 (м, 2H), 1,09 (т, 3H); МС (хиад, m/z)=423,1 (M+H).
Пример 12
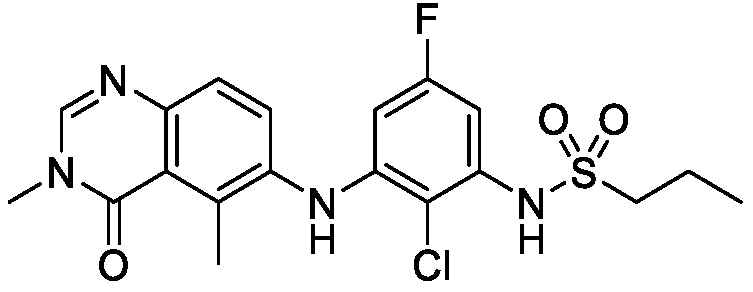
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-5-фторфенил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3-бром-2-хлор-5-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (47 мг, 0,102 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (19,3 мг, 0,102 ммоль), трис(дибензилиденацетон)дипалладия (0) (7,00 мг, 0,00765 ммоль), Xantphos (13,3 мг, 0,0229 ммоль) и карбоната цезия (99,7 мг, 0,306 ммоль) в толуоле (4 мл) продувают аргоном в течение 10 минут и затем нагревают до 110°C в течение ночи под баллоном аргона. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (ДХМ/ацетон) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторфенил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (38 мг, 66%). МС (хиад, m/z)=569,2 (M+H).
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторфенил)пропан-1-сульфонамида. Раствор N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,038 г, 0,067 ммоль) в трифторуксусной кислоте (4 мл) перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют, и остаток обрабатывают ТГФ и насыщенным NaHCO3 (4 мл, 1:1) в течение 30 минут. Реакционную смесь разделяют между этилацетатом и водой. Органические слои отделяют, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (ДХМ/ацетон) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторфенил)пропан-1-сульфонамида (13 мг, 44%). 1H ЯМР (400 MГц, CDCl3) δ 8,03 (с, 1H), 7,60 (с, 2H), 6,91 (дд, 1H), 6,86 (шс, 1H), 6,09-6,05 (м, 2H), 3,56 (с, 3H), 3,19-3,14 (м, 2H), 2,81 (с, 3H), 1,95-1,85 (м, 2H), 1,07 (т, 3H); МС (хиад, m/z)=439,1 (M+H).
Пример 13
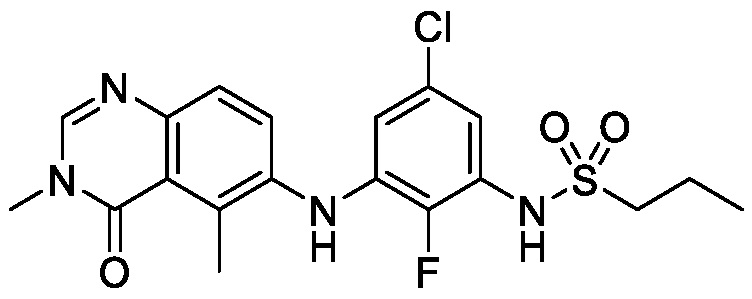
N- (5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-2-фторфенил)пропан-1-сульфонамид
Стадия 1: Получение N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3-бром-5-хлор-2-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (75,2 мг, 0,163 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (29,9 мг, 0,158 ммоль), трис(дибензилиденацетон)дипалладия (0) (10,1 мг, 0,0111 ммоль), Xantphos (19,2 мг, 0,0332 ммоль) и карбоната цезия (154 мг, 0,474 ммоль) в толуоле (1,5 мл) продувают аргоном в течение 5 минут и затем нагревают до 100°C в течение 18 часов под аргоном в 15 мл герметично закрытой пробирке. Реакционную смесь разбавляют водой (25 мл) и 4:1 ДХМ:ИПА (25 мл). Слои разделяют, и органическую фазу сушат над Na2SO4, фильтруют и концентрируют затем очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (60,0 мг, 67%) в виде желтого масла. МС (хиад, m/z)=569,2 (M+H).
Стадия 2: Получение N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)пропан-1-сульфонамида. N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид (60 мг, 0,105 ммоль) растворяют в ДХМ (5 мл) и трифторуксусной кислоте (3249 мкл) при 0°C, затем нагревают до температуры окружающей среды и перемешивают в течение 30 минут. Растворитель удаляют в вакууме и желтое масло растворяют в ДХМ (25 мл) и перемешивают с насыщенным водным раствором NaHCO3 (15 мл) в течение 30 минут. Слои разделяют, и органическую фазу сушат над Na2SO4, фильтруют и концентрируют затем очищают гель-хроматографией на двуокиси кремния (ДХМ/EtOAc) с получением N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2-фторфенил)-пропан-1-сульфонамида (13,6 мг, 29%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 8,01 (с, 1H), 7,62-7,56 (м, 2H), 7,04 (дд, 1H), 6,52 (д, 1H), 6,42 (дд, 1H), 5,69 (д, 1H), 3,56 (с, 3H), 3,21-3,14 (м, 2H), 2,84 (с, 3H), 1,97-1,86 (м, 2H), 1,09 (т, 3H); МС (хиад, m/z)=439,0 (M+H).
Пример 14
N- (2,4-дихлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)фенил)пропан-1-сульфонамид
Стадия 1: Получение N-(2,4-дихлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(2,4-дихлор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (50 мг, 0,095 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (18 мг, 0,095 ммоль), трис(дибензилиденацетон)дипалладия (0) (4,4 мг, 0,0048 ммоль), Xantphos (8,3 мг, 0,014 ммоль) и карбоната цезия (93 мг, 0,29 ммоль) в толуоле (636 мкл) продувают аргоном в течение 10 минут и затем герметично закрывают и нагревают в пробирке до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(2,4-дихлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (45 мг, 80%). МС (хиад, m/z)=585,1 [M+H].
Стадия 2: Получение N-(2,4-дихлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пропан-1-сульфонамида. Раствор N-(2,4-дихлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (45 мг, 0,077 ммоль) в 1 мл ДХМ и 0,2 мл трифторуксусной кислоты перемешивают при температуре окружающей среды в течение 30 минут. Раствор концентрируют и затем растворяют в 1 мл ТГФ и 1 мл насыщенного NaHCO3 и перемешивают при температуре окружающей среды в течение 20 минут. Реакционную смесь разбавляют дополнительной водой и экстрагируют ДХМ (2x), и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (ДХМ/MeOH) с получением N-(2,4-дихлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)пропан-1-сульфонамида (23 мг, 53%). 1H ЯМР (400 MГц, CDCl3) δ 7,92 (с, 1H), 7,48 (д, 1H), 7,41 (т, 1H), 6,88 (д, 1H), 6,79 (шс, 1H), 5,72 (шс, 1H), 3,55 (с, 3H), 3,11 (т, 2H), 3,00 (с, 3H), 1,88 (м, 2H), 1,04 (т, 3H); МС (хиад, m/z)=455,1 [M+H].
Пример 15
N- (3,5-дихлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)пиридин - 2-ил)пропан-1-сульфонамид
Стадия 1: Получение N-(3,5-дихлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)пиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3,5-дихлор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (40 мг, 0,076 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (14 мг, 0,076 ммоль), трис(дибензилиденацетон)дипалладия (0) (3,5 мг, 0,0038 ммоль), Xantphos (6,6 мг, 0,011 ммоль) и карбоната цезия (74 мг, 0,23 ммоль) в толуоле (508 мкл) продувают аргоном в течение 10 минут и затем герметично закрывают и нагревают в пробирке до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(3,5-дихлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)пиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (20 мг, 44%). МС (хиад, m/z)=586,1 [M+H].
Стадия 2: Получение N-(3,5-дихлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)пиридин-2-ил)пропан-1-сульфонамида. Раствор N-(3,5-дихлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)пиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (20 мг, 0,034 ммоль) в 1 мл ДХМ и 0,2 мл трифторуксусной кислоты перемешивают при температуре окружающей среды в течение 45 минут. Раствор концентрируют, и остаток очищают хроматографией на силикагеле (ДХМ/MeOH с 1% NH4OH) с получением N-(3,5-дихлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)пиридин-2-ил)пропан-1-сульфонамида (13 мг, 37%). 1H ЯМР (400 MГц, CDCl3) δ 8,12 (с, 1H), 8,01 (с, 1H), 7,50 (д, 1H), 7,24 (д, 1H), 7,21 (шс, 1H), 6,25 (шс, 1H), 3,66 (т, 2H), 3,56 (с, 3H), 2,91 (с, 3H), 1,92 (м, 2H), 1,09 (т, 3H); МС (хиад, m/z)=456,1 [M+H].
Пример 16
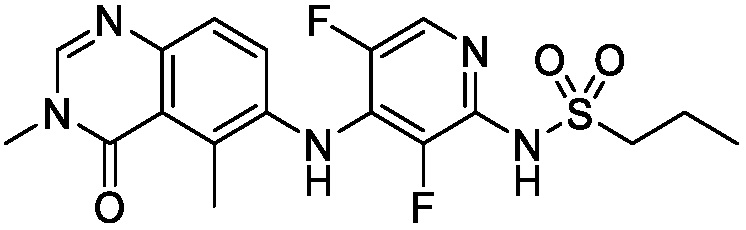
N- (4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-3,5-дифторпиридин - 2-ил)пропан-1-сульфонамид
Стадия 1: Получение N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3,5-дифтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (38 мг, 0,077 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (15 мг, 0,077 ммоль), трис(дибензилиденацетон)дипалладия (0) (3,5 мг, 0,0039 ммоль), Xantphos (6,7 мг, 0,012 ммоль) и карбоната цезия (75 мг, 0,23 ммоль) в толуоле (514 мкл) продувают аргоном в течение 10 минут и затем герметично закрывают и нагревают в пробирке до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (35 мг, 81%). МС (хиад, m/z)=554,2 [M+H].
Стадия 2: Получение N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)пропан-1-сульфонамида. Раствор N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (35 мг, 0,063 ммоль) в 1 мл ДХМ и 0,2 мл трифторуксусной кислоты перемешивают при температуре окружающей среды в течение 45 минут. Реакционную смесь концентрируют, и остаток очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК), и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3 затем насыщенным раствором соли затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)пропан-1-сульфонамида (21 мг, 64%). 1H ЯМР (400 MГц, CDCl3) δ 8,04 (с, 1H), 7,94 (с, 1H), 7,48 (д, 1H), 7,41 (д, 1H), 5,93 (с, 1H), 3,62 (т, 2H), 3,54 (с, 3H), 2,89 (с, 3H), 1,93 (м, 2H), 1,08 (т, 3H); МС (хиад, m/z)=424,1 [M+H].
Пример 17
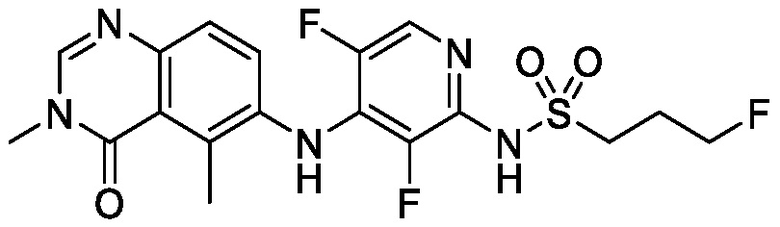
N- (4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-3,5-дифторпиридин - 2-ил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (44 мг, 0,086 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (16 мг, 0,086 ммоль), трис(дибензилиденацетон)дипалладия (0) (3,9 мг, 0,0043 ммоль), Xantphos (7,5 мг, 0,013 ммоль) и карбоната цезия (84 мг, 0,26 ммоль) в толуоле (575 мкл) продувают аргоном в течение 10 минут и затем герметично закрывают и нагревают в пробирке до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды, концентрируют и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)-метил)пропан-1-сульфонамида (37 мг, 76%). МС (хиад, m/z)=572,2 [M+H].
Стадия 2: Получение N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)-3-фторпропан-1-сульфонамида. Раствор N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пропан-1-сульфонамида (37 мг, 0,065 ммоль) в 1 мл ДХМ и 0,2 мл трифторуксусной кислоты перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле (ДХМ/MeOH с 1% NH4OH) с получением N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-3,5-дифторпиридин-2-ил)-3-фторпропан-1-сульфонамида (18 мг, 47%). 1H ЯМР (400 MГц, CDCl3) δ 8,02 (с, 1H), 7,94 (с, 1H), 7,52 (д, 1H), 7,42 (д, 1H), 5,92 (шс, 1H), 4,66 (т, 1H), 4,54 (т, 1H), 3,80 (т, 2H), 3,55 (с, 3H), 2,90 (с, 3H), 2,31 (м, 2H); МС (хиад, m/z)=442,1 [M+H].
Пример 18
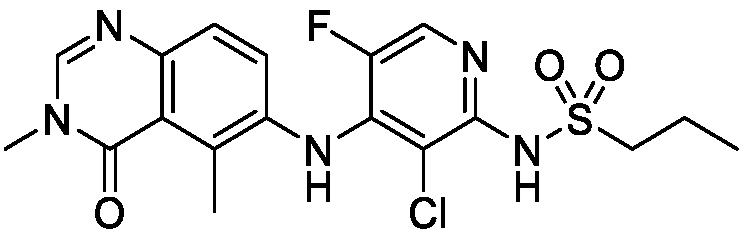
N- (3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-5-фторпиридин - 2-ил)пропан-1-сульфонамид
Стадия 1: Получение N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (105 мг, 0,206 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (39,0 мг, 0,206 ммоль), трис(дибензилиденацетон)дипалладия (0) (9,45 мг, 0,0103 ммоль), Xantphos (17,9 мг, 0,0310 ммоль) и карбоната цезия (202 мг, 0,619 ммоль) в толуоле (1376 мкл) продувают аргоном в течение 10 минут и затем нагревают в герметично закрытой колбе до 90°C в течение ночи. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют, концентрируют и затем очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (111 мг, 94%). МС (хиад, m/z)=570,2 [M+H].
Стадия 2: Получение N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)пропан-1-сульфонамида. Раствор N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)-пропан-1-сульфонамида (111 мг, 0,19 ммоль) в 1 мл ДХМ и 0,5 мл трифторуксусной кислоты перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь концентрируют и растворяют в ДХМ и промывают насыщенным водным NaHCO3 и затем сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)пропан-1-сульфонамида (71 мг, 78%). 1H ЯМР (400 MГц, CDCl3) δ 8,04 (с, 1H), 7,96 (д, 1H), 7,54 (д, 1H), 7,40 (д, 1H), 6,11 (шс, 1H), 3,66 (т, 2H), 3,56 (с, 3H), 2,89 (с, 3H), 1,95 (м, 2H), 1,09 (т, 3H); МС (хиад, m/z)=440,1 [M+H].
Пример 19
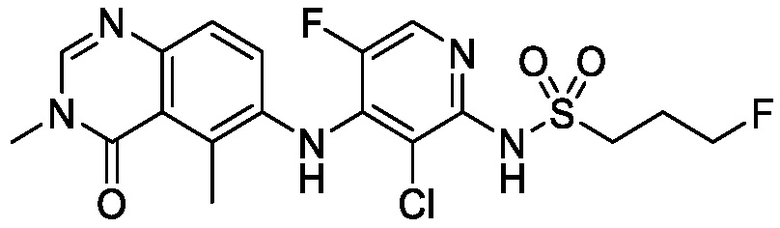
N- (3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-5-фторпиридин - 2-ил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида. N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамид (53 мг, 0,101 ммоль), 6-амино-3,5диметилхиназолин-4(3H)-он (17,4 мг, 0,092 ммоль), карбонат цезия (90 мг, 0,276 ммоль), Xantphos (8,0 мг, 0,0138 ммоль) и трис(дибензилиденацетон)дипалладий (0) (4,2 мг, 0,0046) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (0,46 мл) добавляют, и раствор продувают аргоном в течение 5 минут затем пробирку герметично закрывают и нагревают до 100°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют через слой Celite® и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида в виде белого твердого вещества (34,5 мг, 63%). МС (хиад, m/z)=588,2 (M+H).
Стадия 2: Получение N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)-3-фторпропан-1-сульфонамида. N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)-метил))пропан-1-сульфонамид (34,5 мг, 0,057 ммоль) растворяют в ДХМ (1,4 мл) и обрабатывают трифторуксусной кислотой (0,48 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и остаток очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3 затем насыщенным раствором соли затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-5-фторпиридин-2-ил)-3-фторпропан-1-сульфонамида в виде белого твердого вещества (12,1 мг, 29%). 1H ЯМР (400 MГц, CDCl3) δ 7,94 (с, 1H), 7,76 (д, 1H), 7,37 (д, 1H), 7,31 (дд, 1H), 4,52 (т, 1H), 4,41 (т, 1H), 3,87 (с, 3H), 3,64 (т, 2H), 3,43 (с, 3H), 2,22-2,09 (м, 2H); МС (хиад, m/z)=458,0 (M+H).
Пример 20
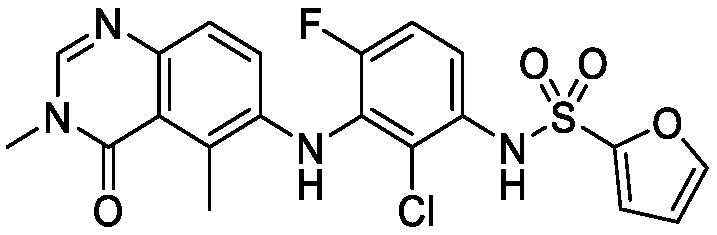
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)фуран - 2-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-((2-(триметилсилил)этокси)метил)фуран-2-сульфонамида. N-(2-хлор-4-фтор-3-йодфенил)-N-((2-триметилсилил)этокси)метил)фуран-2-сульфонамид (31,2 мг, 0,0587 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-он (Промежуточное соединение P15) (10 мг, 0,0534 ммоль), карбонат цезия (52 мг, 0,16 ммоль), Xantphos (4,6 мг, 0,00801 ммоль) и трис(дибензилиденацетон)дипалладий (0) (2,4 мг, 0,00267) объединяют в пробирке затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (0,356 мл) добавляют, и раствор продувают аргоном в течение 5 минут затем пробирку герметично закрывают и нагревают до 90°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды затем фильтруют через слой Celite® и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-((2-(триметил-силил)этокси)метил)фуран-2-сульфонамида в виде белого твердого вещества (15,3 мг, 48%). МС (хиад, m/z)=593,1 (M+H).
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)фуран-2-сульфонамида. N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-((2-(триметилсилил)этокси)метил)фуран-2-сульфонамид (15,3 мг, 0,0258 ммоль) растворяют в ДХМ (0,65 мл) и обрабатывают трифторуксусной кислотой (0,215 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле, элюируя ДХМ/MeOH с 1% NH4OH с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)фуран-2-сульфонамида в виде белого твердого вещества (8,4 мг, 32%). 1H ЯМР (400 MГц, CDCl3) δ 7,93 (с, 1H), 7,55 (кв, 1H), 7,40-7,36 (м, 2H), 7,07 (дд, 2H), 7,00 (дд, 1H), 6,89 (с, 1H), 6,48 (дд, 1H), 5,51 (шс, 1H), 3,54 (с, 3H), 2,93 (с, 3H); МС (хиад, m/z)=463,0 (M+H).
Пример 21
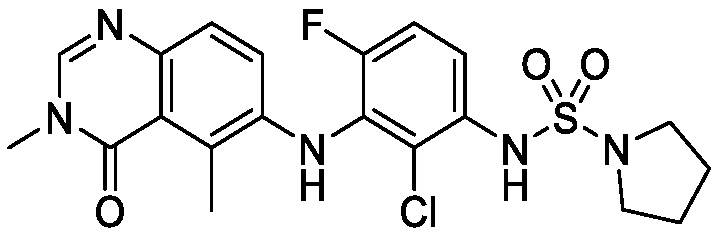
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-4-фторфенил)-N-((2-триметилсилил)этокси)метил)пирролидин-1-сульфонамида. N-(2-хлор-4-фтор-3-йодфенил)-N-((2-триметилсилил)этокси)метил)пирролидин-1-сульфонамид (52 мг, 0,097 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-он (Промежуточное соединение P15) (16,6 мг, 0,088 ммоль), карбонат цезия (86 мг, 0,263 ммоль), Xantphos (7,61 мг, 0,0132 ммоль) и трис(дибензилиденацетон)дипалладий (0) (4,0 мг, 0,00439) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (0,585 мл) добавляют, и раствор продувают аргоном в течение 5 мин, затем пробирку герметично закрывают и нагревают до 90°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют через слой Celite® и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-4-фторфенил)-N-((2-триметилсилил)-этокси)метил)пирролидин-1-сульфонамида в виде белого твердого вещества (36 мг, 69%). МС (хиад, m/z)=596,2 (M+H).
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамида. N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-4-фторфенил)-N-((2-триметилсилил)этокси)метил)пирролидин-1-сульфонамид (36 мг, 0,0604 ммоль) растворяют в ДХМ (1,5 мл) и обрабатывают трифторуксусной кислотой (0,500 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют и остаток очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3 затем насыщенным раствором соли затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пирролидин-1-сульфонамида в виде белого твердого вещества (4,2 мг, 10%). 1H ЯМР (400 MГц, CDCl3) δ 7,93 (с, 1H), 7,44 (д, 1H), 7,41 (дд, 1H), 7,08 (д, 1H), 7,03 (дд, 1H), 6,73 (с, 1H), 5,58 (шс, 1H), 3,54 (с, 3H), 3,36-3,33 (м, 4H), 2,97 (с, 3H), 1,89-1,86 (м, 4H); МС (хиад, m/z)=466,1 (M+H).
Пример 22
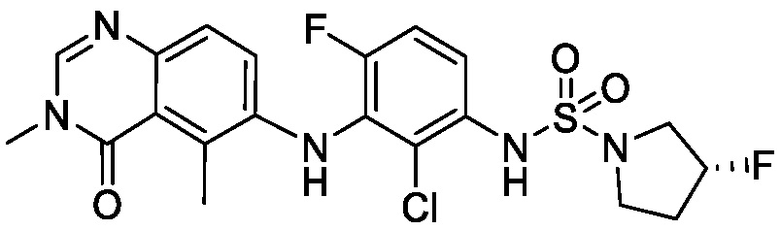
(R)- N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамид
Стадия 1: Получение (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пирролидин-1-сульфонамида. Раствор (R)-N-(2-хлор-4-фтор-3-йодфенил)-3-фтор-N-((2-(триметил-силил)этокси)метил)пирролидин-1-сульфонамида (25 мг, 0,045 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) (8,6 мг, 0,045 ммоль), трис(дибензилиденацетон)дипалладия (0) (2,1 мг, 0,0023 ммоль), Xantphos (3,9 мг, 0,0068 ммоль) и карбоната цезия (44 мг, 0,14 ммоль) в толуоле (301 мкл) продувают аргоном в течение 10 минут и затем герметично закрывают в пробирке и нагревают до 90°C в течение 24 часов. Реакционную смесь охлаждают до температуры окружающей среды, затем фильтруют через Целит® и концентрируют с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пирролидин-1-сульфонамида (25 мг, 90%), который используют на следующей стадией без очистки. МС (хиад, m/z)=614,2 (M+H).
Стадия 2: Получение (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамида. (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фтор-N-((2-(триметилсилил)-этокси)метил)пирролидин-1-сульфонамид (25 мг, 0,041 ммоль) перемешивают в 0,2 мл трифторуксусной кислоты и 0,2 мл ДХМ в течение 30 минут. Реакционную смесь концентрируют и растворяют в 0,2 мл ТГФ и затем обрабатывают 0,2 мл насыщенного NaHCO3 и перемешивают при температуре окружающей среды в течение 20 минут. Реакционную смесь разбавляют водой и экстрагируют ДХМ (2x), и затем объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Органические слои очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют с получением (R)-N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпирролидин-1-сульфонамида (13 мг, 59%). 1H ЯМР (400 MГц, CDCl3) δ 7,92 (с, 1H), 7,42 (м, 2H), 7,06 (м, 2H), 6,75 (шс, 1H), 5,57 (шс, 1H), 3,63 (м, 3H), 3,55 (с, 3H), 3,49 (м, 2H), 2,98 (с, 3H), 2,26 (м, 1H), 2,09 (м, 1H); МС (хиад, m/z)=484,1 [M+H].
Пример 23
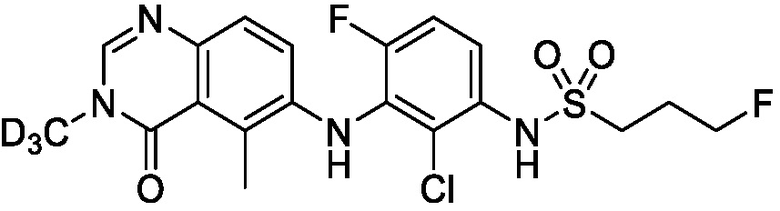
N- (2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-фенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида. N-(3-амино-2-хлор-4-фторфенил)-3-фтор-N-((2-(триметилсилил)этокси)-метил))пропан-1-сульфонамид (533 мг, 1,283 ммоль), 6-бром-5-метил-3-(метил-d3)хиназолин-4(3H)-он (298,8 мг, 1,167 ммоль), карбонат цезия (1140 мг, 3,5 ммоль), Xantphos (101 мг, 0,175 ммоль) и трис(дибензилиденацетон)дипалладий (0) (53,4 мг, 0,0583) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (5,83 мл) добавляют и раствор продувают аргоном в течение 5 минут затем пробирку герметично закрывают и нагревают до 100°C в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют через слой Celite® и очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc с получением N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамида в виде белого твердого вещества (670 мг, 97%). МС (хиад, m/z)=590,2 (M+H).
Стадия 2: Получение N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпропан-1-сульфонамида. N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фтор-N-((2-(триметилсилил)этокси)метил))пропан-1-сульфонамид (670 мг, 1,135 ммоль) растворяют в ДХМ (28 мл) и обрабатывают трифторуксусной кислотой (9,5 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь гасят водным раствора насыщенным NaHCO3 и экстрагируют EtOAc (3x). Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК), и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-3-фторпропан-1-сульфонамида в виде белого твердого вещества (319 мг, 60%) 1H ЯМР (400 MГц, CDCl3) δ 7,92 (с, 1H), 7,46 (д, 1H), 7,40 (дд, 1H), 7,12 (д, 1H), 7,07 (дд, 1H), 6,65 (шс, 1H), 5,60 (шс, 1H), 4,62 (т, 1H), 4,50 (т, 1H), 3,27 (дд, 2H), 2,98 (с, 3H), 2,31-2,18 (м, 2H); МС (хиад, m/z)=460,1 (M+H).
Пример 24
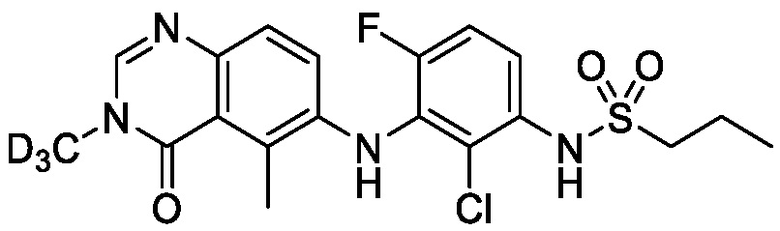
N- (2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)фенил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-(4-метоксибензил)пропан-1-сульфонамида. N-(3-амино-2-хлор-4-фторфенил)-N-(метоксибензил)пропан-1-сульфонамид (116 мг, 0,301 ммоль), 6-бром-5-метил-3-(метил-d3)хиназолин-4(3H)-он (70 мг, 0,273 ммоль), карбонат цезия (267 мг, 0,820 ммоль), Xantphos (23,7 мг, 0,0410 ммоль) и трис(дибензилиденацетон)дипалладий (0) (12,5 мг, 0,0137) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (1,37 мл) добавляют и раствор продувают аргоном в течение 5 минут, затем пробирку герметично закрывают и нагревают до 90°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, затем фильтруют через слой Celite® и очищают хроматографией на силикагеле, элюируя ДХМ/EtOAc с получением N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-(4-метоксибензил)пропан-1-сульфонамида в виде белого твердого вещества (144 мг, 94%). МС (хиад, m/z)=562,2 (M+H).
Стадия 2: Получение N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-пропан-1-сульфонамида. N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-N-(4-метоксибензил)пропан-1-сульфонамид (144 мг, 0,256 ммоль) растворяют в ДХМ (6,4 мл) и обрабатывают трифторуксусной кислотой (2,1 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь гасят водным раствором насыщенного NaHCO3 и экстрагируют EtOAc (3x). Объединенные органические фракции сушат над Na2SO4, фильтруют и концентрируют. Неочищенную смесь очищают хроматографией на силикагеле (ДХМ/MeOH с 1% NH4OH) с получением N-(2-хлор-4-фтор-3-((5-метил-3-(метил-d3)-4-оксо-3,4-дигидрохиназолин-6-ил)амино)фенил)-пропан-1-сульфонамида в виде желтого твердого вещества (66 мг, 55%) 1H ЯМР (400 MГц, CDCl3) δ 7,92 (с, 1H), 7,45 (д, 1H), 7,41 (дд, 1H), 7,11 (д, 1H), 7,06 (дд, 1H), 6,62 (шс, 1H), 5,60 (шс, 1H), 3,10 (дд, 2H), 2,98 (с, 3H), 1,93-1,83 (м, 2H), 1,05 (т, 3H); МС (хиад, m/z)=442,1 (M+H).
Пример 25
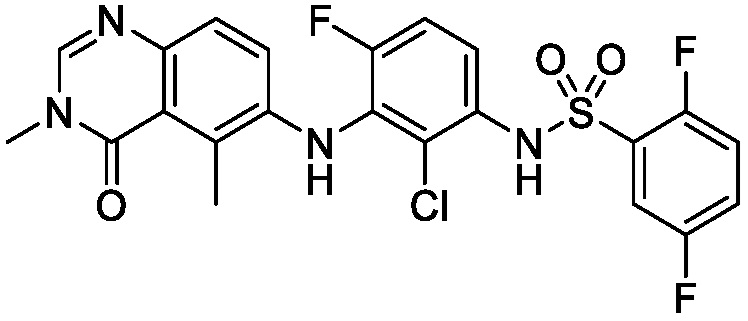
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)-2,5-дифторбензолсульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2,5-дифтор-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида. N-(2-хлор-4-фтор-3-йодфенил)-2,5-дифтор-N-((2-триметилсилил)этокси)метил)бензолсульфонамид (63 мг, 0,109 ммоль), 6-амино-3,5-диметилхиназолин-4(3H)-он (Промежуточное соединение P15) (18,8 мг, 0,0994 ммоль), карбонат цезия (97 мг, 0,298 ммоль), Xantphos (8,6 мг, 0,0149 ммоль) и трис(дибензилиденацетон)дипалладий (0) (4,6 мг, 0,00497) объединяют в пробирке, затем дегазируют при пониженном давлении и повторно заполняют аргоном. Толуол (0,662 мл) добавляют, и раствор продувают аргоном в течение 5 минут затем пробирку герметично закрывают и нагревают до 90°C в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды фильтруют через слой Celite® и очищают хроматографией на силикагеле (ДХМ/EtOAc) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2,5-дифтор-N-((2-(триметилсилил)этокси)метил)бензолсульфонамида в виде белого твердого вещества (15,1 мг, 24%). МС (хиад, m/z)=639,2 (M+H).
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2,5-дифторбензолсульфонамида. N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2,5-дифтор-N-((2-(триметилсилил)этокси)метил)бензол-сульфонамид (15,1 мг, 0,0236 ммоль) растворяют в ДХМ (0,47 мл) и обрабатывают трифторуксусной кислотой (0,197 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 ч. Реакционную смесь концентрируют, и остаток очищают хроматографией на силикагеле, элюируя ДХМ/MeOH с 1% NH4OH с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2,5-дифторбензолсульфонамида в виде беловатого твердого вещества (2,4 мг, 5,0%). 1H ЯМР (400 MГц, CDCl3) δ 7,86 (с, 1H), 7,45-7,40 (м, 1H), 7,29 (д, 1H), 7,22-7,14 (м, 2H), 7,11-7,04 (м, 1H), 6,95 (т, 1H), 6,82-6,77 (м, 1H), 5,63 (шс, 1H), 3,47 (с, 3H), 2,81 (с, 3H); МС (хиад, m/z)=509,1 (M+H).
Пример 26
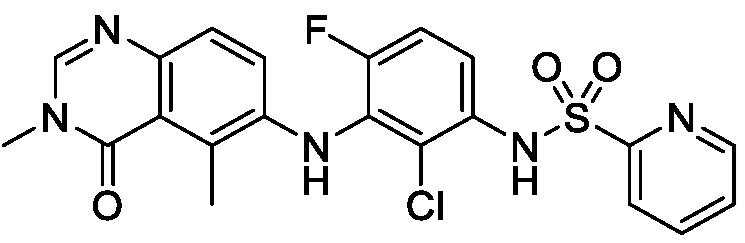
N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)пиридин-2-сульфонамид
К раствору 6-((амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3H)-она (18 мг, 0,054 ммоль) и триэтиламина (0,030 мл, 0,216 ммоль) в ДХМ (0,540 мл) добавляют пиридин-2-сульфонилхлорид (20,2 мг, 0,114 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов, затем гасят водой. Водный слой экстрагируют EtOAc (3x) и объединенные органические слои промывают насыщенным раствором соли (1x), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК) и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3, затем насыщенным раствором соли, затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пиридин-2-сульфонамида в виде белого твердого вещества (2,75 мг, 11%). 1H ЯМР (400 MГц, CD3OD) δ 8,63 (д, 1H), 8,11 (с, 1H), 8,01 (т, 1H), 7,92-7,89 (м, 1H), 7,62-7,59 (м, 1H), 7,36 (дд, 1H), 7,32 (д, 1H), 7,14 (т, 1H), 6,77 (д, 1H), 3,52 (с, 3H), 2,83 (с, 3H); МС (хиад, m/z)=474,1 (M+H).
Пример 27

N- (2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)-2-метоксиэтан-1-сульфонамид
Стадия 1: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-метокси-N-((2-метоксиэтил)сульфонил)этан-1-сульфонамида. К раствору 6-((3-амино-2-хлор-6-фторфенил)амино)-3,5-диметилхиназолин-4(3H)-она (15,3 мг, 0,046 ммоль) и триэтиламина (0,026 мл, 0,184 ммоль) в ДХМ (0,115 мл) при 0°C добавляют 2-метоксиэтилсульфонилхлорид (0,011 мл, 0,0966 ммоль). Реакционную смесь нагревают до температуры окружающей среды и перемешивают в течение 16 часов, затем гасят водой. Водный слой экстрагируют EtOAc (3x), и объединенные органические слои промывают насыщенным раствором соли (1x), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле, элюируя ДХМ/MeOH с 1% NH4OH с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-метокси-N-((2-метоксиэтил)сульфонил)этан-1-сульфонамида (18 мг, 68%). МС (хиад, m/z)=577,1 (M+H).
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-метоксиэтан-1-сульфонамида. N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-метокси-N-((2-метоксиэтил)сульфонил)этан-1-сульфонамид (18 мг, 0,033 ммоль) растворяют в MeCN (0,165 мл) и обрабатывают 2,0 M водным раствором Na2CO3 (0,132 мл, 0,263 ммоль), затем нагревают до 60°C в течение 1 часа. Реакционную смесь охлаждают до температуры окружающей среды и затем обрабатывают водой и экстрагируют EtOAc (3x). Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (ДХМ/MeOH с 1% NH4OH) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-2-метоксиэтан-1-сульфонамида в виде белого твердого вещества (1,9 мг, 13%). 1H ЯМР (400 MГц, CDCl3) δ 7,92 (с, 1H), 7,47-7,44 (м, 2H), 7,11-7,05 (м, 2H), 6,96 (шс, 1H), 5,60 (шс, 1H), 3,83 (т, 2H), 3,55 (с, 3H), 3,40-3,38 (м, 2H), 3,35 (с, 3H), 2,97 (с, 3H); МС (хиад, m/z)=455,1 (M+H).
Пример 28
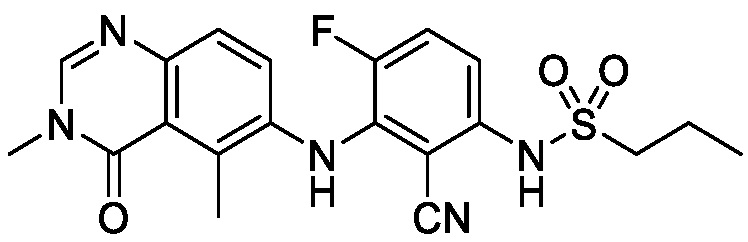
N- (2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин - 6-ил)амино)-4-фторфенил)пропан-1-сульфонамид
Стадия 1: Получение N-(2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. Раствор N-(3-амино-2-циано-4-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (16 мг, 0,041 ммоль), 6-бром-3,5-диметилхиназолин-4(3H)-она (10 мг, 0,041 ммоль), трис(дибензилиденацетон)дипалладия (0) (1,9 мг, 0,0021 ммоль), Xantphos (3,6 мг, 0,0062 ммоль) и карбоната цезия (40 мг, 0,12 ммоль) в толуоле (275 мкл) продувают аргоном в течение 10 минут и затем герметично закрывают и нагревают в пробирке до 90°C в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и затем разбавляют водой и экстрагируют ДХМ (2x), и объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют, концентрируют. Остаток берут далее в виде неочищенного N-(2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида без очистки. МС (хиад, m/z)=560,2 [M+H].
Стадия 2: Получение N-(2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пропан-1-сульфонамида. Раствор N-(2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамид перемешивают в 0,5 мл ДХМ и 0,2 мл трифторуксусной кислоты в течение 30 минут. Реакционную смесь концентрируют, и остаток очищают С18 хроматографией с обращенной фазой (H2O/ацетонитрил с 0,1% ТФК), и затем выделенный продукт растворяют в ДХМ и промывают насыщенным NaHCO3 затем насыщенным раствором соли затем сушат над Na2SO4, фильтруют и концентрируют с получением N-(2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)пропан-1-сульфонамида (6,4 мг, 36%). 1H ЯМР (400 MГц, CDCl3) δ 8,01 (с, 1H), 7,55 (д, 1H), 7,40 (д, 1H), 7,26 (дд, 1H), 7,18 (дд, 1H), 6,58 (шс, 1H), 5,94 (шс, 1H), 3,56 (с, 3H), 3,13 (м, 2H), 2,91 (с, 3H), 1,88 (м, 2H), 1,06 (т, 3H); МС (хиад, m/z)=430,2 [M+H].
Соединения в Таблице 1 получают с применением способа, аналогичного тому, который описан для синтеза N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4-дифторфенил)пропан-1-сульфонамида (Пример 1) с применением следующих модификаций: на стадии 1, заменяя 6-бром-3,5диметилхиназолин-4(3H)-он и/или N-(3-амино-2,4-дифторфенил)-N-(4-метоксибензил)пропан-1-сульфонамид на подходящий 3,5-замещенный 6-бром-хиназолин-4(3H)-он и SEM или PMB-защищенный анилин; и на стадии 2, заменяя N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4-дифторфенил)-N-(4-метоксибензил)пропан-1-сульфонамид на подходящий SEM или PMB-защищенный продукт сочетания со стадии 1. Соединения в таблице 1, которые являются солями ТФК, выделяют при очистке неочищенного продукта С18 хроматографией с обращенной фазой, элюируя H2O/ацетонитрилом с 0,1% ТФК, затем концентрации фракций, содержащих желаемый продукт.
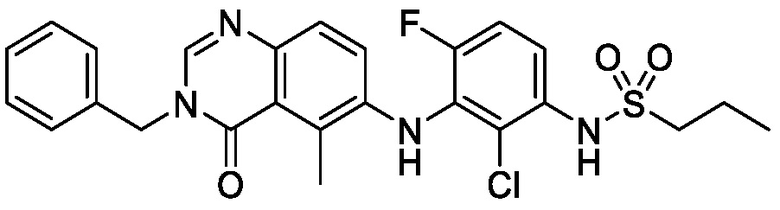
(M+H)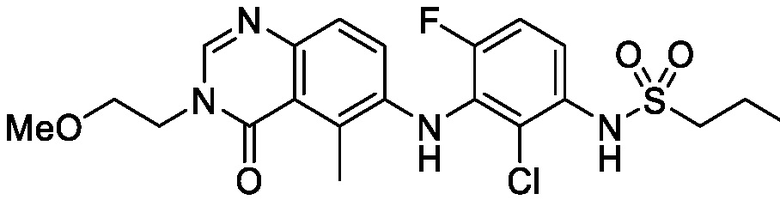
(M+H)
(M+H)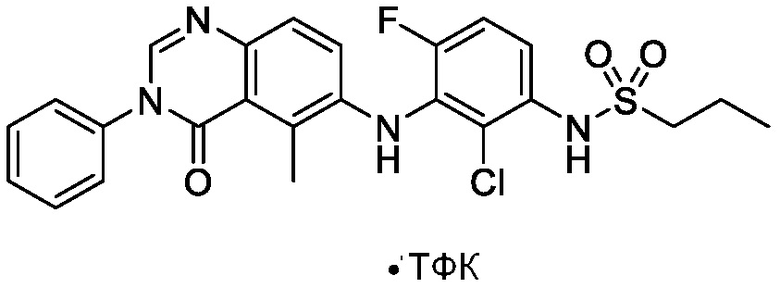
(M+H)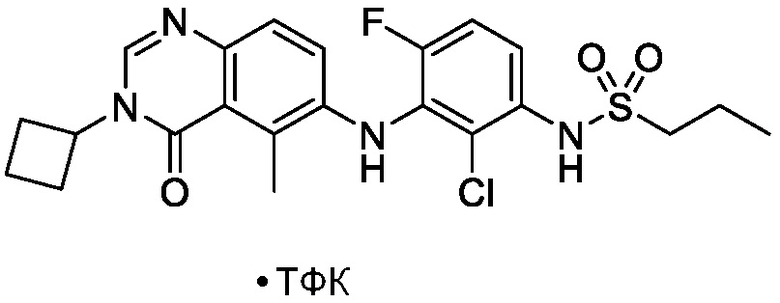
(M+H)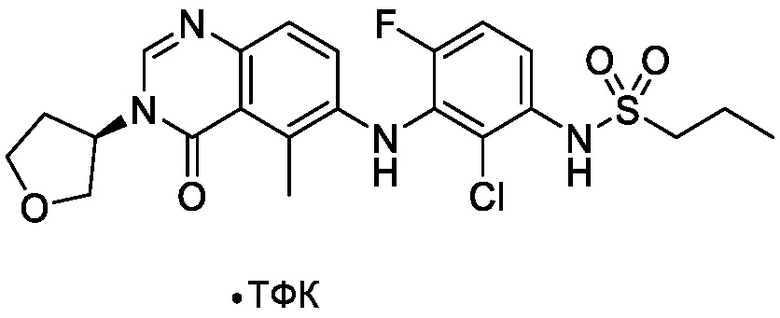
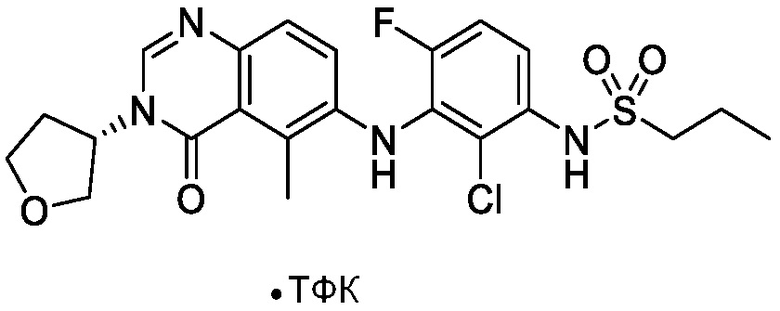
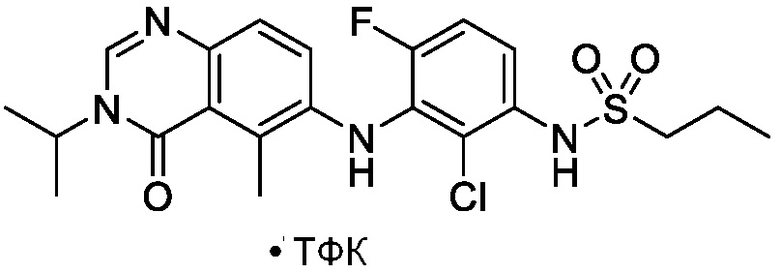

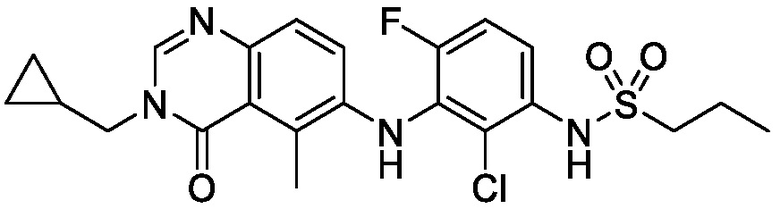
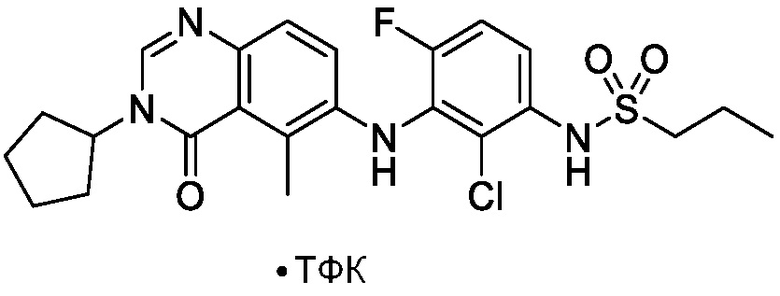
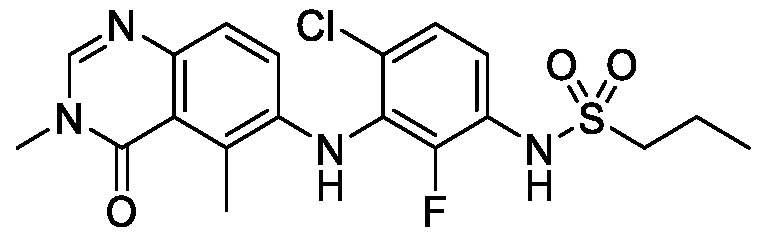
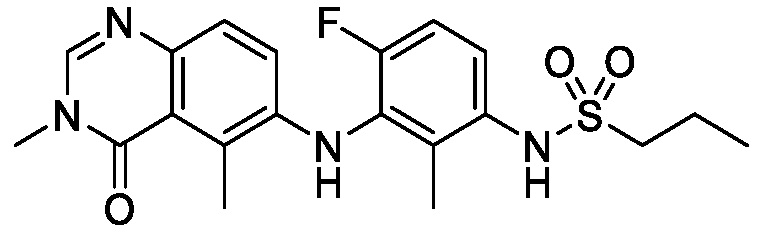
Соединения в Таблице 2 получают с применением способа, аналогичного тому, который описан для синтеза N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)пропан-1-сульфонамида (Пример 11) с применением следующих модификаций: на стадии замена N-(3-бром-2,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида и/или 6-амино-3,5-диметилхиназолин-4(3H)-она (Промежуточное соединение P15) на подходящий 3,5-замещенный 6-аминохиназолин-4(3H)-он и SEM или PMB-замещенный арил- или гетероарилгалогенид; и на стадии 2 замена N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (Пример 11, Стадия 1) на подходящий SEM или PMB-защищенный продукт сочетания.
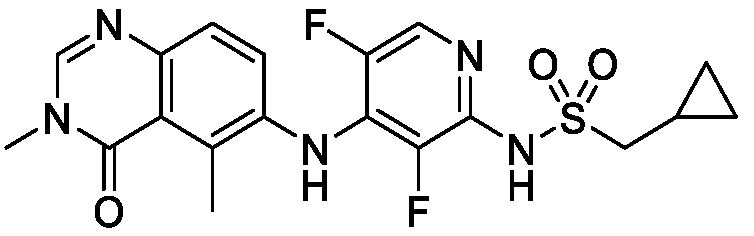
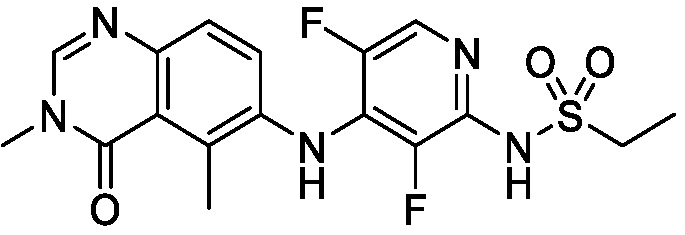
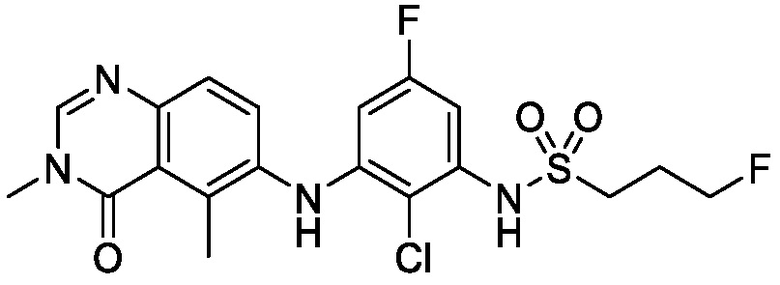
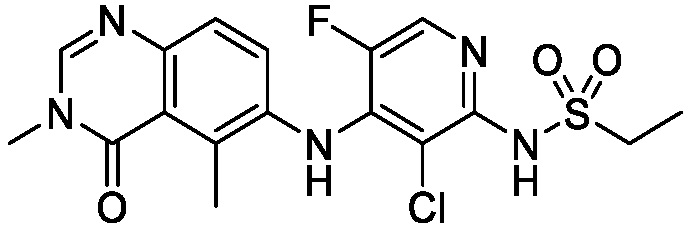
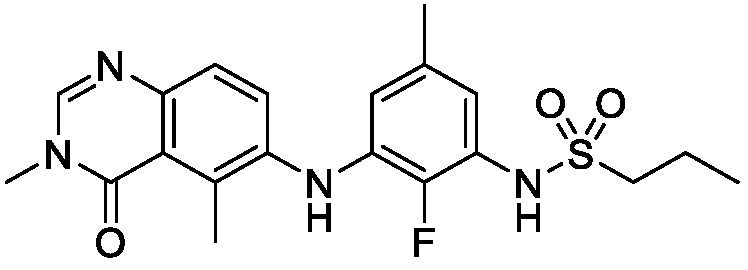
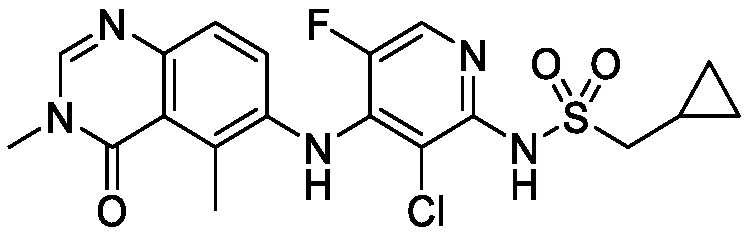
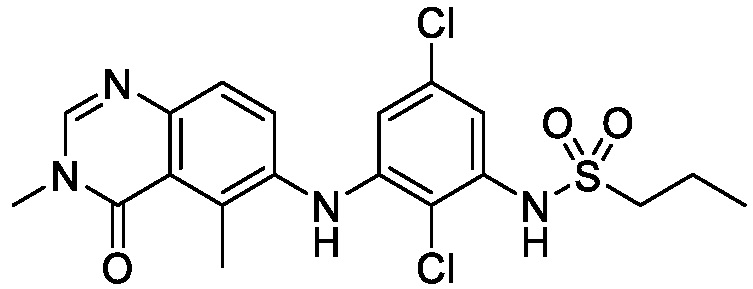
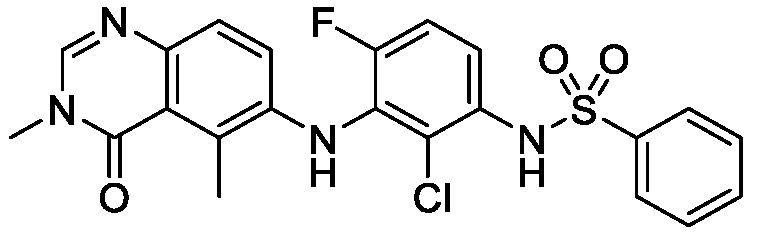
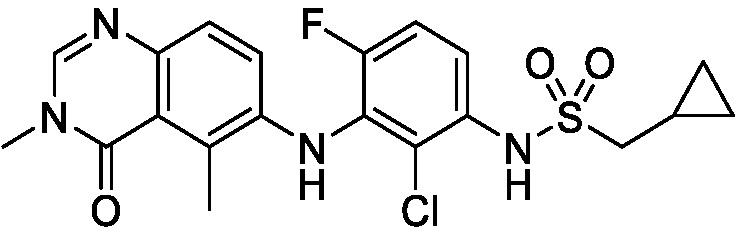
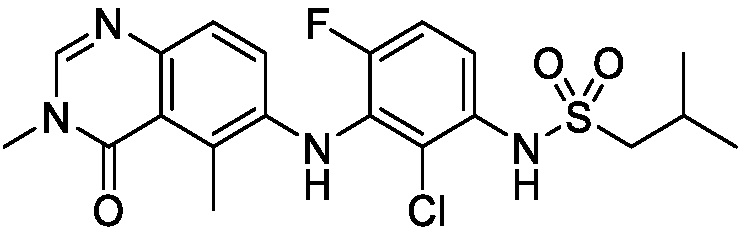
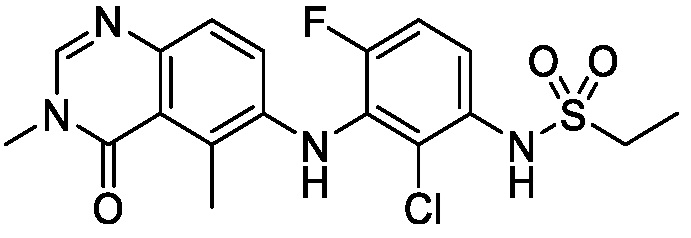
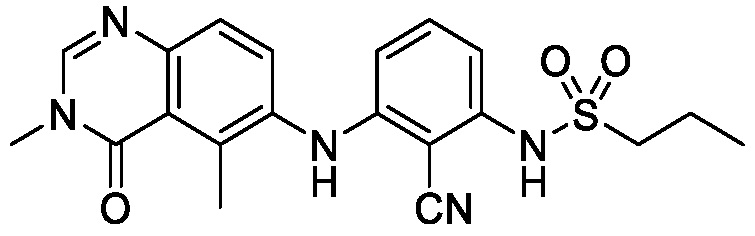
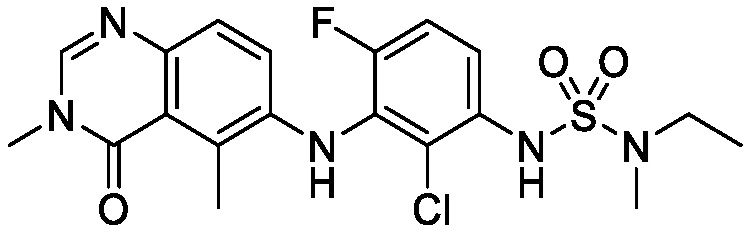
Пример 55
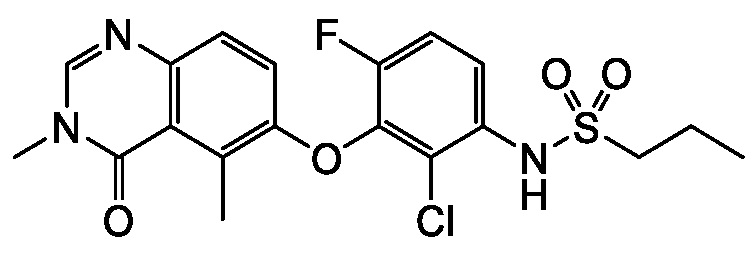
N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-yl)окси]-4-фторфенил}пропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата. К раствору 3-амино-2-хлор-6-фторфенола (100 мг, 0,62 ммоль) в ДМФ (3,0 мл) добавляют Cs2CO3 (505 мг, 1,55 ммоль), затем трет-бутил 3-фтор-2-метил -6-нитробензоат (136 мг, 0,68 ммоль) и реакционную смесь нагревают при 100°C в течение 2 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, гасят водой и экстрагируют этилацетатом (2×100 мл). Органические слои промывают водой (2×50 мл), насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (50% этилацетатом в гексане) с получением трет-бутил 3-(3-амино-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата в виде светло-желтого твердого вещества (140 мг, 56%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,04 (д, 1H), 7,21 (т, 1H), 6,79-6,73 (м, 2H), 5,57 (шс, 2H), 2,37 (с, 3H), 1,58 (с, 9H); МС (m/z)=394,9 (M-H).
Стадия 2: Получение трет-бутил 3-{2-хлор-6-фтор-3-[N-(пропан-1-сульфонил)пропан-1-сульфонамидо]-фенокси}-2-метил-6-нитробензоата. К перемешиваемому раствору трет-бутил 3-(3-амино-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата (140,0 мг, 0,35 ммоль) в дихлорметане (6 мл) добавляют триэтиламин (0,15 мл, 1,06 ммоль) под азотом. Раствор охлаждают до 0°C и обрабатывают пропан-1-сульфонилхлоридом (0,09 мл, 0,74 ммоль) и перемешивают в течение 6 часов при температуре окружающей среды. Реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (2×100 мл). Органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (50% этилацетатом в гексане) с получением трет-бутил 3-{2-хлор-6-фтор-3-[N-(пропан-1-сульфонил)пропан-1-сульфонамидо]-фенокси}-2-метил-6-нитробензоата в виде светло-желтого твердого вещества (160 мг, 60%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,10 (д, 1H), 7,88-7,84 (м, 1H), 7,70 (т, 1H), 6,75 (д, 1H), 3,77-3,76 (м, 4H), 2,40 (с, 3H), 1,87-1,80 (м, 4H), 1,58 (с, 9H), 1,01 (т, 6H).
Стадия 3: Получение трет-бутил-6-амино-3-{2-хлор-6-фтор-3-[N-(пропан-1-сульфонил)пропан-1-сульфонамидо]фенокси}-2-метилбензоата. К раствору трет-бутил-3-{2-хлор-6-фтор-3-[N-(пропан-1-сульфонил)пропан-1-сульфонамидо]фенокси}-2-метил-6-нитробензоата (160 мг, 0,23 ммоль) в смеси метанола (3 мл) и воды (0,5 мл) при температуре окружающей среды добавляют хлорид аммония (70 мг, 1,3 ммоль), затем порошок Fe (147 мг, 2,6 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь фильтруют через Celite®, промывают ДХМ (2×30 мл) и концентрируют при пониженном давлении. Остаток разбавляют водой (30 мл) и экстрагируют ДХМ (2×100 мл). Органический слой промывают водой (2×50 мл) и насыщенным раствором соли (100 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (5-10% этилацетатом в гексане) с получением трет-бутил-6-амино-3-{2-хлор-6-фтор-3-[N-(пропан-1-сульфонил)пропан-1-сульфонамидо]фенокси}-2-метилбензоата в виде светло-желтого твердого вещества (80 мг, 53%). 1H ЯМР (400 MГц, ДМСО-d6) δ 7,69-7,66 (дд, 1H), 7,52 (т, 1H), 6,52 (д, 1H), 6,33 (д, 1H), 5,16 (шс, 2H), 3,78-3,63 (м, 4H), 2,30 (с, 3H), 1,85-1,83 (м, 4H), 1,56 (с, 9H), 1,01 (т, 6H).
Стадия 4: Получение N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-4-фторфенил}пропан-1-сульфонамида. К раствору трет-бутил-6-амино-3-{2-хлор-6-фтор-3-[N-(пропан-1-сульфонил)пропан-1-сульфонамидо]фенокси}-2-метилбензоата (55 мг, 0,12 ммоль) в N-метилформамиде (1,5 мл) добавляют муравьиную кислоту (0,004 мл, 0,11 ммоль), и реакционную смесь нагревают до 180°C в течение 2 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют этилацетатом (2×80 мл). Органический слой промывают водой (2×50 мл), насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (50% этилацетатом в гексане) затем растирают с диэтиловым эфиром с получением N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-4-фторфенил}пропан-1-сульфонамида в виде беловатого твердого вещества (25 мг, 56%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,66 (шс, 1H), 8,27 (с, 1H), 7,50-7,43 (м, 3H), 7,00 (д, 1H), 3,45 (с, 3H), 3,13 (т, 2H), 2,90 (с, 3H), 1,78-1,72 (м, 2H), 0,97 (т, 3H). МС (m/z)=440,1 [M+H].
Пример 56
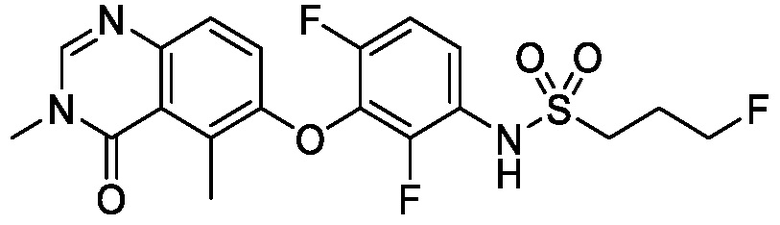
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4-дифторфенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2,6-дифторфенокси)-2-метил-6-нитробензоата. К раствору 3-амино-2,6-дифторфенола (70 мг, 0,482 ммоль) в N, N-диметилформамиде (2,4 мл) добавляют карбонат цезия (393 мг, 1,21 ммоль) и трет-бутил 3-фтор-2-метил-6-нитробензоат (135 мг, 0,531 ммоль), и реакционную смесь нагревают до 100°C под азотом в течение 2 часов. Раствор охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют EtOAc. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (5-95% EtOAc/гексан) с получением трет-бутил 3-(3-амино-2,6-дифторфенокси)-2-метил-6-нитробензоата (134 мг, 73%). 1H ЯМР (400 MГц, CDCl3) δ 7,94 (д, 1H), 6,85 (м, 1H), 6,67 (м, 2H), 2,44 (с, 3H), 1,64 (с, 9H).
Стадия 2: Получение трет-бутил 3-(2,6-дифтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата. К раствору трет-бутил 3-(3-амино-2,6-дифторфенокси)-2-метил-6-нитробензоата (134 мг, 0,352 ммоль) в дихлорметане (3,5 мл) при 0°C добавляют триэтиламин (147 мкл, 1,06 ммоль) и 3-фторпропан-1-сульфонилхлорид (92,2 мкл, 0,775 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь разбавляют водой и экстрагируют ДХМ. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (5-95% EtOAc/гексан) с получением трет-бутил 3-(2,6-дифтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата (208 мг, 94%). 1H ЯМР (400 MГц, CDCl3) δ 7,98 (д, 1H), 7,35 (м, 1H), 7,18 (м, 1H), 6,63 (д, 1H), 4,64 (т, 2H), 4,51 (т, 2H), 3,73 (м, 4H), 2,46 (с, 3H), 2,32 (м, 4H), 1,65 (с, 9H).
Стадия 3: Получение 6-амино-3-(2,6-дифтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата. Раствор трет-бутил 3-(2,6-дифтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата (170 мг, 0,270 ммоль) и 10% палладия на угле (28,8 мг, 0,0270 ммоль) в метаноле (2,7 мл) перемешивают при температуре окружающей среды под баллоном водорода в течение 8 часов. Раствор фильтруют через Celite® и концентрируют с получением трет-бутил 6-амино-3-(2,6-дифтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата (122 мг, 75%). 1H ЯМР (400 MГц, CDCl3) δ 7,18 (м, 1H), 7,06 (м, 1H), 6,54 (д, 1H), 6,46 (д, 1H), 4,62 (т, 2H), 4,51 (т, 2H), 3,72 (м, 4H), 2,42 (с, 3H), 2,32 (м, 4H), 1,61 (с, 9H); МС (хиад, m/z)=599,2 (M+H).
Стадия 4: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4-дифторфенил)-3-фторпропан-1-сульфонамида. Раствор трет-бутил 6-амино-3-(2,6-дифтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата (34 мг, 0,057 ммоль) в N-метилформамиде (568 мкл) и муравьиной кислоте (2,9 мкл, 0,062 ммоль) нагревают до 180°C в течение 90 минут. Реакционную смесь охлаждают до температуры окружающей среды и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Продукт затем разделяют между ДХМ и насыщенным водным NaHCO3. Органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4-дифторфенил)-3-фторпропан-1-сульфонамида (7 мг, 28%). 1H ЯМР (400 MГц, CDCl3) δ 7,98 (с, 1H), 7,42 (м, 2H), 7,04 (м, 2H), 6,78 (шс, 1H), 4,61 (т, 1H), 4,49 (т, 1H), 3,55 (с, 3H), 3,26 (т, 2H), 2,98 (с, 3H), 2,22 (м, 2H); МС (хиад, m/z)=442,2 (M+H).
Пример 57
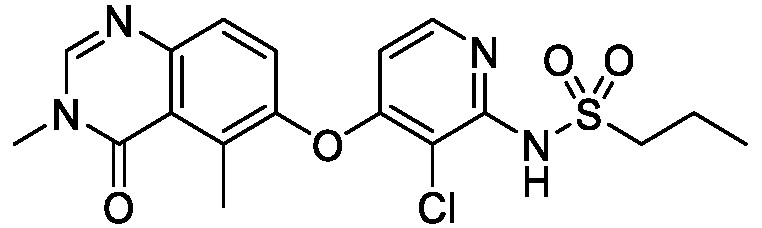
N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)пиридин-2-ил)пропан-1-сульфонамид
Стадия 1: Получение 6-((2,3-дихлорпиридин-4-ил)окси)-3,5-диметилхиназолин-4(3H)-она. В пробирку добавляют 6-гидрокси-3,5-диметилхиназолин-4(3H)-он (0,050 г, 0,26 ммоль), 2,3-дихлор-4-йодпиридин (0,086 г, 0,32 ммоль) и ДМФ (1,3 мл). Полученный раствор обрабатывают карбонатом цезия (0,13 г, 0,39 ммоль), и реакционную смесь нагревают до 100°C и перемешивают в течение 7 часов. Смесь охлаждают до температуры окружающей среды и разбавляют водой (3 мл). Полученное твердое вещество собирают вакуумной фильтрацией, промывают дополнительной водой и сушат в вакууме. Неочищенный продукт очищают колоночной хроматографией (50-100% EtOAc/ДХМ) с получением 6-((2,3-дихлорпиридин-4-ил)окси)-3,5-диметилхиназолин-4(3H)-она в виде белого твердого вещества (0,061 г, 69%). МС (хиад, m/z)=336,0, 338,0 (M+H).
Стадия 2: Получение N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)пиридин-2-ил)пропан-1-сульфонамида. В пробирку добавляют 6-((2,3-дихлорпиридин-4-ил)окси)-3,5-диметилхиназолин-4(3H)-он (0,060 г, 0,18 ммоль), пропан-1-сульфонамид (0,033 г, 0,27 ммоль), трис(дибензилиденацетон)дипалладий (0) (0,016 г, 0,018 ммоль), Xantphos (0,026 г, 0,045 ммоль) и карбонат цезия (0,17 г, 0,54 ммоль). Добавляют диоксан (1,8 мл) и смесь продувают барботированием аргона в течение 10 минут. Пробирку герметично закрывают и смесь нагревают при 90°C в течение 14 часов, и затем при 110°C в течение еще 8 часов. Смесь затем охлаждают до температуры окружающей среды, разбавляют насыщенным водным раствором NH4Cl и экстрагируют 5% ИПА/CHCl3 (3x). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт затем очищают колоночной гель-хроматографией на двуокиси кремния (0-4% MeOH/ДХМ) с получением N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)пиридин-2-ил)пропан-1-сульфонамида в виде беловатого твердого вещества (0,054 г, 68%). МС (хиад, m/z)=423,1, 425,1 (M+H).
Следующие соединения получают с применением способа, аналогичного тому, который описан для синтеза N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино-2,4-дифторфенил)пропан-1-сульфонамида (Пример 1)
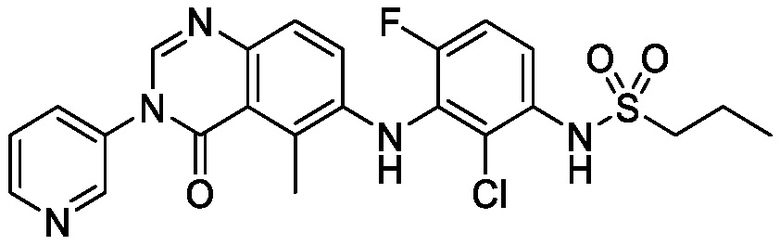
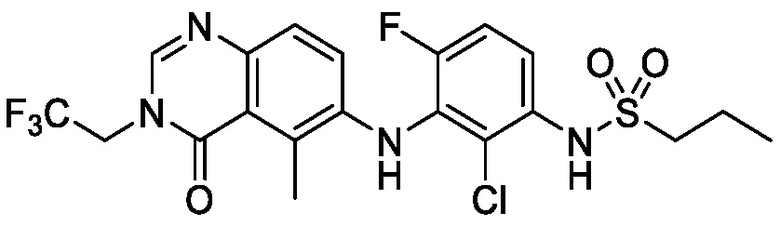
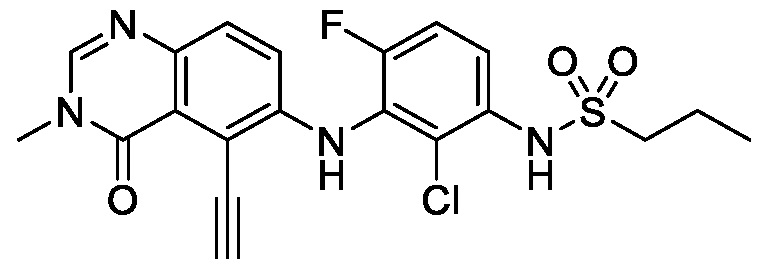
Следующие соединения получают с применением способа, аналогичного тому, который описан для синтеза N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-2,5-дифторфенил)пропан-1-сульфонамида (Пример 11).
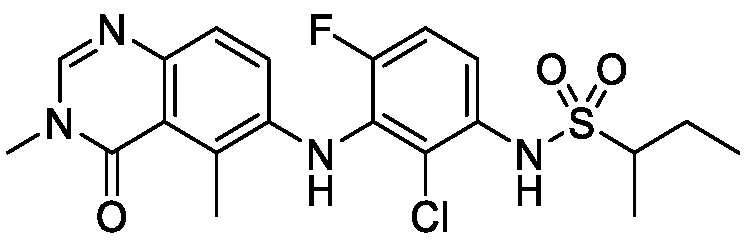
Пример 62
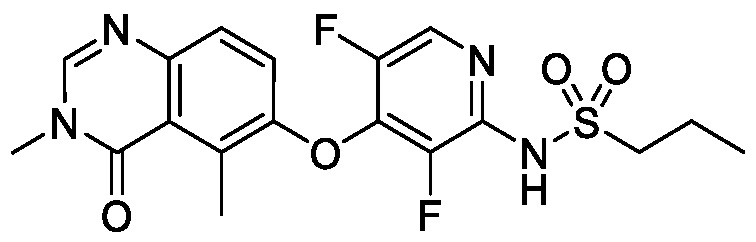
N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)пропан-1-сульфонамид
Стадия 1: Получение N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. К раствору 6-гидрокси-3,5-диметилхиназолин-4(3H)-она (0,040 г, 0,21 ммоль) и N-(3,5-дифтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,12 г, 0,25 ммоль) в ДМФ (2,1 мл) добавляют карбонат цезия (0,14 г, 0,42 ммоль), и смесь нагревают до 100°C в течение 1 часа. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между этилацетатом и водой. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией с обращенной фазой, элюируя 5-95% ацетонитрилом в воде (0,1% ТФК). Неочищенный продукт растворяют в ДХМ/ИПА (4:1), промывают насыщенным NaHCO3, насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением с получением N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,073 г, 63%). МС (хиад, m/z)=555,2 (M+H).
Стадия 2: Получение N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)пропан-1-сульфонамида. Раствор N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,073 г, 0,13 ммоль) в ТФК (4 мл) перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь концентрируют, и неочищенный продукт очищают хроматографией с обращенной фазой, элюируя 5-95% ацетонитрилом в воде (0,1% ТФК). Выделенный продукт растворяют в ДХМ/ИПА (4:1), промывают насыщенным NaHCO3, насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением с получением N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)пропан-1-сульфонамида (15 мг, 27%). МС (хиад, m/z)=425,1 (M+H).
Пример 63
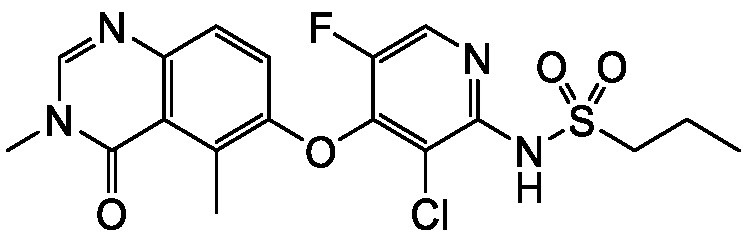
N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторпиридин-2-ил)пропан-1-сульфонамид
К раствору 6-гидрокси-3,5-диметилхиназолин-4(3H)-она (0,043 г, 0,23 ммоль) и N-(3-хлор-5-фтор-4-йодпиридин-2-ил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,14 г, 0,27 ммоль) в ДМФ (2,3 мл) добавляют карбонат цезия (0,15 г, 0,45 ммоль) и смесь нагревают до 100°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между этилацетатом и водой. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией с обращенной фазой, элюируя 5-95% ацетонитрилом в воде (0,1% ТФК). Выделенный продукт растворяют в ДХМ/ИПА (4:1), промывают насыщенным NaHCO3, насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением с получением N-(3-хлор-4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторпиридин-2-ил)пропан-1-сульфонамида (15 мг, 15%). МС (хиад, m/z)=441,1, 443,1 (M+H).
Пример 64
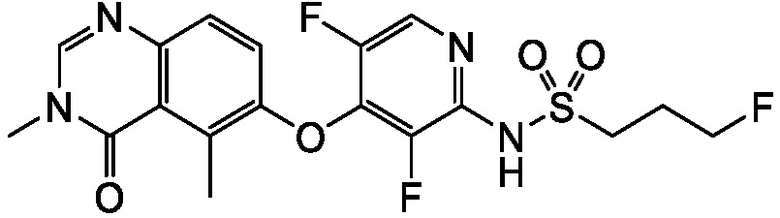
N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида. К раствору 6-гидрокси-3,5-диметилхиназолин-4(3H)-она (0,050 г, 0,26 ммоль), N-(3,5-дифтор-4-йодпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,15 г, 0,29 ммоль) в ДМФ (2,6 мл) добавляют карбонат цезия (0,17 г, 0,53 ммоль), и реакционную смесь перемешивают при 100°C в течение 3 часов. Реакционную смесь охлаждают до температуры окружающей среды и разделяют между этилацетатом и водой. Объединенные органические слои промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают хроматографией с обращенной фазой, элюируя 5-95% ацетонитрилом в воде (0,1% ТФК). Выделенный продукт растворяют в ДХМ/ИПА (4:1), промывают насыщенным NaHCO3, насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,029 г, 19%). МС (хиад, m/z)=573,2 (M+H).
Стадия 2: Получение N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-3-фторпропан-1-сульфонамида. Раствор N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-3-фтор-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (0,029 г, 0,051 ммоль) в ТФК (4 мл) перемешивают при температуре окружающей среды в течение 2 часов. Реакционную смесь концентрируют и очищают хроматографией с обращенной фазой (5-95% ацетонитрил в воде, 0,1% ТФК). Выделенный продукт растворяют в ДХМ/ИПА (4:1), промывают насыщенным NaHCO3, насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют с получением N-(4-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,5-дифторпиридин-2-ил)-3-фторпропан-1-сульфонамида (3 мг, 13%). МС (хиад, m/z)=443,1 (M+H).
Пример 65
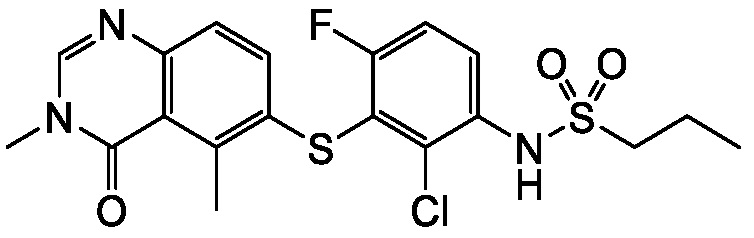
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)тио)-4-фторфенил)пропан-1-сульфонамид
Стадия 1: Получение 2-этилгексил 3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)тио)пропаноат. К раствору 6-бром-3,5-диметилхиназолин-4(3H)-она (600,0 мг, 2,37 ммоль), 2-этилгексил 3-меркаптопропаноата (1,06 мл, 4,74 ммоль) и N, N-диизопропилэтиламина (0,83 мл, 4,74 ммоль) в диоксане (6 мл) добавляют Xantphos (411,50 мг, 0,71 ммоль) и Pd(OAc)2 (160,00 мг, 0,71 ммоль). Реакционную смесь дегазируют аргоном в течение 10 минут, герметично закрывают и затем нагревают при 140°C в течение 48 часов. После завершения реакции, реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (2×100 мл). Органические слои промывают водой (2×50 мл), насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (60% этилацетатом в гексане) с получением 2-этилгексил 3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)тио)пропаноата в виде желтой жидкости (800 мг, 86%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,31 (с, 1H), 7,79 (д, J=8,6 Гц, 1H), 7,48 (д, J=8,6 Гц, 1H), 3,92 (д, J=5,8 Гц, 2H), 3,43 (с, 3H), 3,19 (т, J=6,8 Гц, 2H), 2,89 (с, 3H), 2,60 (т, J=6,7 Гц, 2H), 1,54-1,44 (м, 1H), 1,34-1,19 (м, 8H), 0,87-0,78 (м, 6H); МС (хиад, m/z)=390,8 (M+H).
Стадия 2: Получение 6-меркапто-3,5-диметилхиназолин-4(3H)-она. К раствору 2-этилгексил-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)тио)пропаноата (280,0 мг, 0,72 ммоль) в ТГФ (3 мл) при 0°C добавляют этоксид натрия (53,7 мг, 0,79 ммоль), и реакционную смесь перемешивают при 0°C в атмосфере азота в течение 30 минут. После завершения реакции, растворитель выпаривают и твердое вещество промывают ДХМ. Полученное твердое вещество затем растворяют в воде и добавляют 2N HCl для доведения рН до примерно 7. Осадок собирают фильтрацией и сушат в вакууме с получением 6-меркапто-3,5-диметилхиназолин-4(3H)-она в виде светло-желтого твердого вещества (130 мг, 87%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,27 (с, 1H), 7,80 (д, J=8,5 Гц, 1H), 7,38 (д, J=8,6 Гц, 1H), 5,61 (с, 1H), 3,43 (с, 3H), 2,80 (с, 3H); МС (хиад, m/z)=207,0 (M+H).
Стадия 3: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)тио)-4-фторфенил)пропан-1-сульфонамида. К раствору 6-меркапто-3,5-диметилхиназолин-4(3H)-она (54,00 мг, 0,26 ммоль), N-(2-хлор-4-фтор-3-йодфенил)-N-((2-(триметилсилил)этокси)метил)пропан-1-сульфонамида (132,72 мг, 0,26 ммоль) и трикалийфосфата (111,14 мг, 0,52 ммоль) в ДМА (1,0 мл) добавляют DMEDA (0,01 мл, 0,11 ммоль) и CuI (9,97 мг, 0,05 ммоль). Реакционную смесь дегазируют Ar в течение 10 минут, герметично закрывают и нагревают при 130°C в течение 16 часов. После завершения реакции, реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (2×25 мл). Органические слои промывают водой (2×10 мл), насыщенным раствором соли (10 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (40% этилацетатом в гексане) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)тио)-4-фторфенил)пропан-1-сульфонамида в виде светло-коричневого твердого вещества (14 мг, 12%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,63 (с, 1H), 8,32 (с, 1H), 7,68-7,57 (м, 1H), 7,41 (т, J=8,9 Гц, 2H), 7,21 (д, J=8,7 Гц, 1H), 3,44 (с, 3H), 3,15-3,06 (м, 2H), 2,98 (с, 3H), 1,79-1,65 (м, 2H), 0,94 (т, J=7,4 Гц, 3H); МС (хиад, m/z)=456,1 (M+H).
Пример 66

N-{2-Циано-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]фенил}пропан-1-сульфонамид
Стадия 1: Получение 2-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-6-фторбензонитрилк. К холодному (0°C) раствору 6-гидрокси-3,5-диметил-3H-хиназолин-4-она (233 мг, 1,22 ммоль) в ДМФ (4 мл) добавляют 60% NaH в минеральном масле (49,0 мг, 1,22 ммоль), и реакционную смесь перемешивают при 0°C в течение 30 минут. Туда добавляют 2,6-дифтор-бензонитрил (170,41 мг, 1,22 ммоль) и смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь гасят ледяной водой и экстрагируют этилацетатом (2 × 100 мл). Органический слой промывают насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с применением 50% этилацетата/гексана с получением 2-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-6-фторбензонитрила (151 мг, 39%) в виде беловатого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,39 (с, 1H), 7,70-7,58 (м, 3H), 7,22 (т, J=8,7 Гц, 1H), 6,53 (д, J=8,7 Гц, 1H), 3,47 (с, 3H), 2,66 (с, 3H).
Стадия 2: Получение N-{2-циано-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]фенил}пропан-1-сульфонамида. К раствору амида пропан-1-сульфоновой кислоты (120,2 мг, 0,98 ммоль) в NMP (5 мл) в герметично закрытой пробирке добавляют NaH (60% в минеральном масле, 39,1 мг, 0,98 ммоль), и реакционную смесь перемешивают при 60°C в течение 40 минут. Туда добавляют 2-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-6-фторбензонитрил (151,0 мг, 0,49 ммоль), и реакционную смесь перемешивают при 130°C в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды и разбавляют 0,5M раствором NaOH. Водный слой промывают этилацетатом (2 ×15 мл) и затем подкисляют примерно pH 5 6N HCl. Водный слой экстрагируют этилацетатом (3 х 15 мл), объединенные органические слои промывают водой и насыщенным раствором соли, сушат над безводным Na2SO4, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают хроматографией с обращенной фазой (20-95% ацетонитрил/вода, 20 мМ NH4CO3) с получением N-{2-циано-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]фенил}пропан-1-сульфонамида (100 мг, 49%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 10,24 (с, 1H), 8,37 (с, 1H), 7,64-7,54 (м, 2H), 7,54-7,44 (м, 1H), 7,17 (д, J=8,3 Гц, 1H), 6,43 (с, 1H), 3,47 (с, 3H), 3,20 (м, 2H), 2,66 (с, 3H), 1,80 (кв, J=7,7 Гц, 2H), 1,01 (т, J=7,4 Гц, 3H); МС (m/z)=413,3 (M+H).
Пример 67
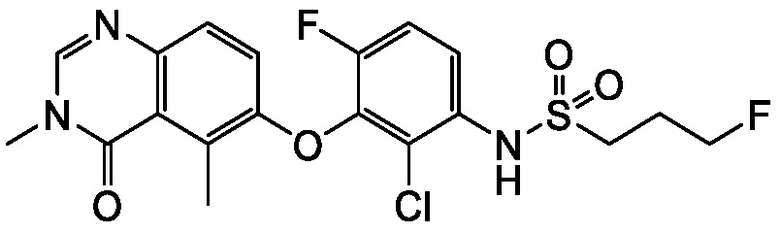
N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-4-фторфенил}-3-фторпропан-1-сульфонамид
Стадия 1: Получение трет-бутил-3-{2-хлор-6-фтор-3-[N-(3-фторпропансульфонил)-3-фторпропансульфонамидо]фенокси}2-метил-6-нитробензоата. К раствору трет-бутил 3-(3-амино-2-хлор-6-фторфенокси)-2-метил-6-нитробензоата (500,0 мг, 1,26 ммоль) в дихлорметане (15 мл) добавляют триэтиламин (0,53 мл, 3,78 ммоль) под азотом. Раствор охлаждают до 0°C и обрабатывают 3-фторпропан-1-сульфонилхлоридом (505 мг, 3,15 ммоль) и реакционную смесь перемешивают в течение 6 часов при температуре окружающей среды. После завершения реакции, реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (2×100 мл). Органические слои промывают водой (2×50 мл), насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (50% этилацетатом в гексане) с получением трет-бутил-3-{2-хлор-6-фтор-3-[N-(3-фторпропансульфонил)-3-фторпропансульфонамидо]фенокси}2-метил-6-нитробензоата в виде светло-желтого твердого вещества (600 мг, 73%). 1H ЯМР (400 MГц, CDCl3) δ 7,96 (д, J=9,1 Гц, 1H), 7,47-7,39 (м, 1H), 7,29 (т, J=8,8 Гц, 1H), 6,50 (д, J=9,2 Гц, 1H), 4,64 (т, J=5,6 Гц, 2H), 4,52 (т, J=5,6 Гц, 2H), 3,89-3,71 (м, 4H), 2,46 (с, 3H), 2,50-2,26 (м, 4H), 1,65 (с, 9H).
Стадия 2: Получение трет-бутил-6-амино-3-{2-хлор-6-фтор-3-[N-(3-фторпропансульфонил)-3-фторпропансульфонамидо]фенокси}-2-метилбензоата. К раствору трет-бутил-3-{2-хлор-6-фтор-3-[N-(3-фторпропансульфонил)-3-фторпропансульфонамидо]фенокси}-2-метил-6-нитробензоата (600 мг, 0,93 ммоль) в смеси метанола (10 мл) и воды (3 мл) при температуре окружающей среды добавляют хлорид аммония (250 мг, 4,6 ммоль) и порошок железа (520 мг, 9,3 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. После завершения реакции, реакционную смесь фильтруют через Celite®, промывают ДХМ (2×30 мл) и концентрируют при пониженном давлении. Неочищенный остаток разбавляют водой (30 мл) и экстрагируют ДХМ (2×100 мл). Органический слой промывают водой (2×50 мл), насыщенным раствором соли (100 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (5-10% этилацетатом в гексане) с получением трет-бутил-6-амино-3-{2-хлор-6-фтор-3-[N-(3-фторпропансульфонил)-3-фторпропансульфонамидо]фенокси}-2-метилбензоата в виде коричневого липкого твердого вещества (300 мг, 53%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,82-9,70 (м, 1H), 7,79-7,70 (м, 1H), 7,56 (т, J=9,5 Гц, 1H), 7,46-7,34 (м, 1H), 6,51 (т, J=9,1 Гц, 1H), 6,35 (т, J=8,3 Гц, 1H), 5,22-5,08 (м, 2H), 4,67-4,56 (м, 2H), 4,55-4,44 (м, 2H), 3,96-3,75 (м, 2H), 3,29-3,20 (м, 1H), 2,38-2,23 (м, 4H), 2,27-2,16 (м, 1H), 2,20-2,03 (м, 1H), 1,56 (с, 9H).
Стадия 3: Получение N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-4-фторфенил}-3-фторпропан-1-сульфонамида. К раствору трет-бутил-6-амино-3-{2-хлор-6-фтор-3-[N-(3-фторпропансульфонил)-3-фторпропансульфонамидо]фенокси}-2-метилбензоата (250 мг, 0,82 ммоль) в N-метилформамиде (4 мл) добавляют муравьиную кислоту (0,031 мл, 0,82 ммоль), и реакционную смесь нагревают до 180°C в течение 2 часов. После завершения реакции, реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют этилацетатом (2×80 мл). Органический слой промывают водой (2×50 мл), насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (50% этилацетатом в гексане) затем растирают с диэтиловым эфиром с получением N-{2-хлор-3-[(3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси]-4-фторфенил}-3-фторпропан-1-сульфонамида в виде беловатого твердого вещества (70 мг, 20%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,82 (с, 1H), 8,28 (с, 1H), 7,54-7,40 (м, 3H), 7,00 (д, J=9,0 Гц, 1H), 4,60 (т, J=6,0 Гц, 1H), 4,49 (т, J=6,0 Гц, 1H), 3,46 (с, 3H), 3,27 (т, J=7,9 Гц, 2H), 2,91 (с, 3H), 2,22-2,04 (м, 2H); МС (хиад, m/z)=458,1 (M+H).
Пример 68
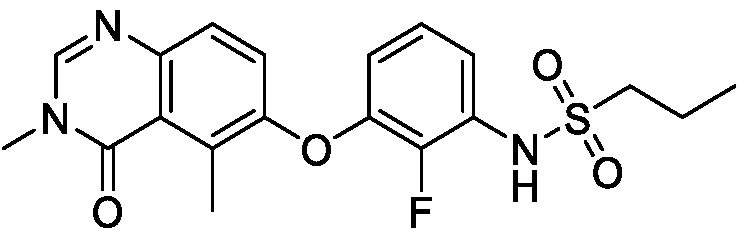
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фторфенил)пропан-1-сульфонамид
Раствор N-(3-бром-2-фторфенил)пропан-1-сульфонамида (1,2 г, 4,05 ммоль), 6-гидрокси-3,5-диметилхиназолин-4(3H)-она (1,2 г, 6,07 ммоль), K3PO4 (2,58 г, 12,15 ммоль), пиколиновой кислоты (498 мг, 4,05 ммоль) и CuI (772 мг, 4,05 ммоль) в ДМСО (12 мл) дегазируют в течение 15 минут продуванием аргоном. Реакционную смесь герметично закрывают и нагревают до 150°C в течение 16 часов. После завершения реакции, реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (2×100 мл). Органические слои промывают водой (2×50 мл), насыщенным раствором соли (100 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают хроматографией с обращенной фазой (30-95% ацетонитрил/вода, 20 мМ NH4HCO3) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фторфенил)пропан-1-сульфонамида (66 мг, 4%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,80 (с, 1H), 8,33 (с, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,38 (д, J=8,8 Гц, 1H), 7,17 (т, J=7,6 Гц, 1H), 7,09 (т, J=8,3 Гц, 1H), 6,69 (т, J=7,7 Гц, 1H), 3,46 (с, 3H), 3,18-3,09 (м, 2H), 2,74 (с, 3H), 1,81-1,69 (м, 2H), 0,98 (т, J=7,5 Гц, 3H); МС (m/z)=406,3 (M+H).
Пример 69
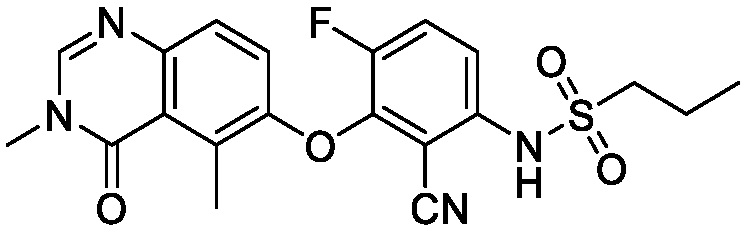
N-(2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пропан-1-сульфонамид
Стадия 1: Получение 2-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,6-дифторбензонитрила. К перемешиваемому раствору 6-гидрокси-3,5-диметилхиназолин-4(3H)-она (250 мг, 1,31 ммоль) в ДМФ (5,0 мл) добавляют 60% NaH в минеральном масле (62 мг, 1,579 ммоль)) при 0°C и реакционную смесь перемешивают в течение 30 минут при температуре окружающей среды. Затем добавляют 2,3,6-трифторбензонитрил (206,69 мг, 1,31 ммоль), и реакционную смесь перемешивают в течение 2 часов при температуре окружающей среды. После завершения реакции, реакционную смесь охлаждают до 5°C и реакционную смесь гасят насыщенным водным раствора NH4Cl (15 мл), разбавляют водой и экстрагируют ДХМ (2×50 мл). Органические слои промывают водой (2×25 мл), насыщенным раствором соли (25 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (30% этилацетатом в гексане) с получением 2-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,6-дифторбензонитрила в виде беловатого твердого вещества (300 мг, 77%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,32 (с, 1H), 7,95-7,83 (м, 1H), 7,54-7,43 (м, 2H), 7,41 (д, J=9,0 Гц, 1H), 3,46 (с, 3H), 2,87 (с, 3H).
Стадия 2: Получение N-(2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пропан-1-сульфонамида. К перемешиваемому раствору пропан-1-сульфонамида (210 мг, 1,71 ммоль) в ДМФ (5,0 мл) добавляют 60% NaH в минеральном масле (92 мг, 2,28 ммоль)) при 0°C и реакционную смесь перемешивают в течение 1 часа при температуре окружающей среды. 2-(3,5-Диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-3,6-дифторбензонитрил (350 мг, 1,06 ммоль) добавляют, и реакционную смесь перемешивают в течение 2 часов при 100°C. Реакционную смесь охлаждают до 5°C, и реакционную смесь гасят насыщенным водным раствора NH4Cl (20 мл), разбавляют водой и экстрагируют ДХМ (3×50 мл). Органические слои промывают водой (2×30 мл), насыщенным раствором соли (30 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают хроматографией с обращенной фазой (10-95% ацетонитрил/вода, 20 мМ NH4HCO3) с получением N-(2-циано-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4-фторфенил)пропан-1-сульфонамида в виде белого твердого вещества (70 мг, 15%). 1H ЯМР (400 MГц, ДМСО-d6) δ 10,24 (с, 1H), 8,37 (с, 1H), 7,81-7,75 (м, 1H), 7,50-7,47 (м, 1H), 7,42-7,38 (м, 1H), 7,25 (д, J=8,3 Гц, 1H), 3,47 (с, 3H), 3,20 (м, 2H), 2,90 (с, 3H), 1,80 (кв, J=7,7 Гц, 2H), 1,01 (т, J=7,4 Гц, 3H); МС (m/z)=431,3 (M+H).
Пример 70
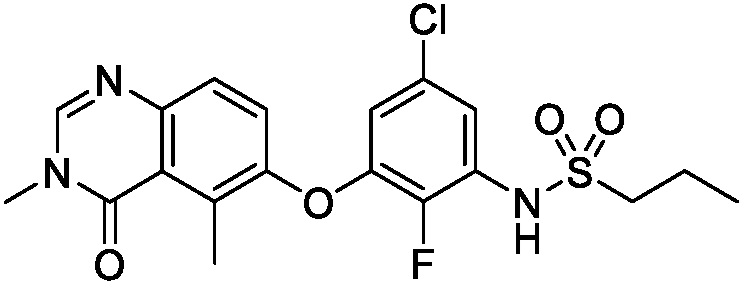
N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фторфенил)пропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-5-хлор-2-фторфенокси)-2-метил-6-нитробензоата. К раствору 3-амино-5-хлор-2-фторфенола (61 мг, 0,38 ммоль) в N, N-диметилформамиде (1888 мкл) добавляют карбонат цезия (308 мг, 0,94 ммоль) и трет-бутил 3-фтор-2-метил-6-нитробензоат (106 мг, 0,42 ммоль), и реакционную смесь нагревают до 100°C под азотом в течение 90 минут. Раствор охлаждают до комнатной температуры, разбавляют водой и экстрагируют EtOAc (3 x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (5-95% EtOAc/гексан) и затем хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным водным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют с получением трет-бутил 3-(3-амино-5-хлор-2-фторфенокси)-2-метил-6-нитробензоата (90 мг, 60%). 1H ЯМР (400 MГц, CDCl3) δ 7,98 (д, 1H), 6,78 (д, 1H), 6,66 (дд, 1H), 6,43 (дд, 1H), 2,39 (с, 3H), 1,64 (с, 9H).
Стадия 2: Получение трет-бутил 6-амино-3-(3-амино-5-хлор-2-фторфенокси)-2-метилбензоата. Раствор трет-бутил 3-(3-амино-5-хлор-2-фторфенокси)-2-метил-6-нитробензоата (47 мг, 0,12 ммоль), порошка цинка (77 мг, 1,2 ммоль) и NH4Cl (127 мг, 2,4 ммоль) в метаноле (592 мкл) перемешивают при температуре окружающей среды в течение 30 минут. Раствор фильтруют через Целит, Целит промывают MeOH и фильтрат концентрируют. Неочищенный продукт очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным водным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют с получением трет-бутил 6-амино-3-(3-амино-5-хлор-2-фторфенокси)-2-метилбензоата (20 мг, 46%). МС (хиад, m/z)=367,1 (M+H).
Стадия 3: Получение 6-(3-амино-5-хлор-2-фторфенокси)-3,5-диметилхиназолин-4(3H)-она. Раствор трет-бутил 6-амино-3-(3-амино-5-хлор-2-фторфенокси)-2-метилбензоата (20 мг, 0,055 ммоль) в N-метилформамиде (545 мкл, 0,055 ммоль) и муравьиной кислоте (2,8 мкл, 0,060 ммоль) нагревают до 180°C в течение 16 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют насыщенным водным NaHCO3 и экстрагируют ДХМ (3x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% EtOAc/ДХМ) с получением 6-(3-амино-5-хлор-2-фторфенокси)-3,5-диметилхиназолин-4(3H)-она (7,2 мг, 40%). МС (хиад, m/z)=334,0, 336,0 (M+H).
Стадия 4: Получение N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фторфенил)пропан-1-сульфонамида. К раствору 6-(3-амино-5-хлор-2-фторфенокси)-3,5-диметилхиназолин-4(3H)-она (7,2 мг, 0,02157 ммоль) в дихлорметане (215,7 мкл) при 0°C добавляют триэтиламин (9,021 мкл, 0,06472 ммоль) и пропан-1-сульфонилхлорида (5,342 мкл, 0,04746 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. Реакционную смесь разбавляют водой и экстрагируют ДХМ (3x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют и концентрируют. Остаток растворяют в ацетонитрил (107,9 мкл) и нагревают до 60°C с 2 M Na2CO3 (107,9 мкл, 0,02157 ммоль) в течение 1 часа. Раствор доводят до примерно pH 5 10% лимонной кислотой, разбавляют водой и экстрагируют ДХМ (3x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией с обращенной фазой (5-95% MeCN/вода, 0,1% ТФК). Выделенный продукт растворяют в ДХМ и промывают насыщенным водным NaHCO3. Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют с получением N-(5-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фторфенил)пропан-1-сульфонамида (4,5 мг, 47%). 1H ЯМР (400 MГц, CDCl3) δ 8,02 (с, 1H), 7,58 (д, 1H), 7,33 (м, 2H), 6,41 (м, 1H), 3,57 (с, 3H), 3,18 (т, 2H), 2,80 (с, 3H), 1,92 (м, 2H), 1,08 (т, 3H).
Пример 71
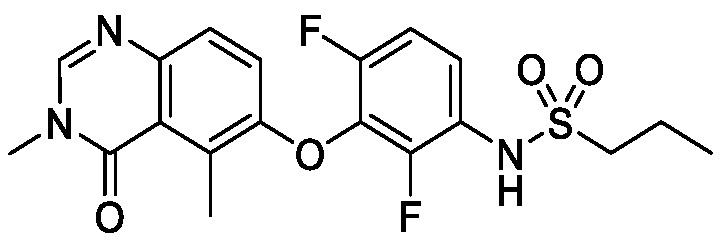
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4-дифторфенил)пропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2,6-дифторфенокси)-2-метил-6-нитробензоата. К раствору 3-амино-2,6-дифторфенола (271 мг, 1,87 ммоль) в N, N-диметилформамиде (9338 мкл) добавляют карбонат цезия (1521 мг, 4,67 ммоль) и трет-бутил 3-фтор-2-метил-6-нитробензоат (524 мг, 2,05 ммоль), и реакционную смесь нагревают до 100°C под азотом в течение 90 минут. Раствор охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют EtOAc (3x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-50% EtOAc/гексан) с получением трет-бутил 3-(3-амино-2,6-дифторфенокси)-2-метил-6-нитробензоата (681 мг, 96%). 1H ЯМР (400 MГц, CDCl3) δ 7,96 (д, 1H), 6,86 (м, 1H), 6,65 (м, 2H), 2,45 (с, 3H), 1,65 (с, 9H).
Стадия 2: Получение трет-бутил 6-амино-3-(3-амино-2,6-дифторфенокси)-2-метилбензоата. Раствор трет-бутил 3-(3-амино-2,6-дифторфенокси)-2-метил-6-нитробензоата (681 мг, 1,79 ммоль) и 10% палладия на угле (191 мг, 0,179 ммоль) в метаноле (11936 мкл, 1,79 ммоль) продувают аргоном в течение 10 минут и затем перемешивают под баллоном водорода в течение 90 минут. Палладий удаляют фильтрацией, и фильтрат концентрируют с получением трет-бутил 6-амино-3-(3-амино-2,6-дифторфенокси)-2-метилбензоата (529 мг, 84%). МС (хиад, m/z)=351,1 (M+H).
Стадия 3: Получение 6-(3-амино-2,6-дифторфенокси)-3,5-диметилхиназолин-4(3H)-она. Раствор трет-бутил 6-амино-3-(3-амино-2-фтор-6-метилфенокси)-2-метилбензоата (150 мг, 0,433 ммоль) в N-метилформамиде (4330 мкл, 0,433 ммоль) нагревают до 180°C в течение 3 часов. Раствор охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют EtOAc (3x). Органические фазы концентрируют и нагревают до 90°C в растворе 3 мл диоксана и 3 мл 6 M NaOH в течение 16 часов. Раствор охлаждают до температуры окружающей среды и доводят до примерно pH 5 10% лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют ДХМ (3x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% EtOAc/ДХМ) с получением 6-(3-амино-2,6-дифторфенокси)-3,5-диметилхиназолин-4(3H)-она (79 мг, 58%). МС (хиад, m/z)=318,1 (M+H).
Стадия 4: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4-дифторфенил)пропан-1-сульфонамида. К раствору 6-(3-амино-2,6-дифторфенокси)-3,5-диметилхиназолин-4(3H)-она (76 мг, 0,2395 ммоль) в дихлорметане (2395 мкл) добавляют триэтиламин (100,2 мкл, 0,7186 ммоль) и пропан-1-сульфонилхлорид (59,31 мкл, 0,5270 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 30 минут. Реакционную смесь разбавляют водой и экстрагируют ДХМ (3x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют. Остаток растворяют в 1 мл MeCN и 1 мл 2 M Na2CO3 и нагревают до 80°C в течение 30 минут. Раствор охлаждают до температуры окружающей среды и доводят до примерно pH 5 10% лимонной кислотой. Реакционную смесь разбавляют водой и экстрагируют EtOAc (3x). Объединенные органические слои промывают насыщенным раствором соли, сушат с Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (5-95% EtOAc/ДХМ) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4-дифторфенил)пропан-1-сульфонамида (55 мг, 54%). 1H ЯМР (400 MГц, CDCl3) δ 7,97 (с, 1H), 7,42 (м, 2H), 7,03 (м, 2H), 6,48 (шс, 1H), 3,55 (с, 3H), 3,09 (м, 2H), 2,99 (с, 3H), 1,88 (м, 2H), 1,05 (т, 3H). МС (хиад, m/z)=424,1 (M+H).
Пример 72
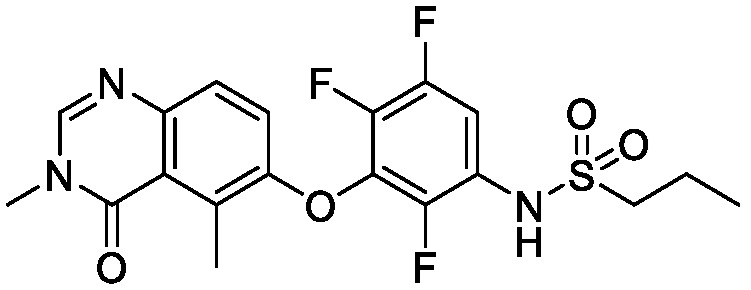
N -(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4,5-трифторфенил)пропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2,5,6-трифторфенокси)-2-метил-6-нитробензоата. К раствору 3-амино-2,5,6-трифторфенола (1,0 г, 6,1 ммоль) в ДМФ (5 мл) добавляют карбонат калия (2,54 г, 18,4 ммоль) и раствор трет-бутил 3-фтор-2-метил-6-нитробензоата (1,87 г, 7,36 ммоль) в ДМФ (3 мл) и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь выливают в ледяную воду (20 мл) и экстрагируют этилацетатом (3×50 мл). Объединенные органические слои сушат над Na2SO4, фильтруют, концентрируют и очищают хроматографией на силикагеле (10% EtOAc/гексан) с получением трет-бутил 3-(3-амино-2,5,6-трифторфенокси)-2-метил-6-нитробензоата в виде коричневого твердого вещества (1,0 г, 41%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,05 (д, J=9,1 Гц, 1H), 7,04 (д, J=9,1 Гц, 1H), 6,80-6,68 (м, 1H), 5,66 (с-ш, 2H), 2,37 (с, 3H), 1,58 (с, 9H).
Стадия 2: Получение трет-бутил (2-метил-6-нитро-3-(2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)фенокси)бензоата. К раствору трет-бутил 3-(3-амино-2,5,6-трифторфенокси)-2-метил-6-нитробензоата (1,0 г, 2,5 ммоль) в дихлорметане (10 мл) при 0°C добавляют триэтиламин (1,0 мл, 7,5 ммоль) и пропан сульфонилхлорид (0,75 мл, 6,2 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 6 часов. Реакционную смесь гасят холодной водой (10 мл) и экстрагируют дихлорметаном (3×30 мл). Объединенные органические слои промывают водой (10 мл), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (20% EtOAc/гексан) с получением трет-бутил (2-метил-6-нитро-3-(2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)фенокси)бензоата в виде коричневого полутвердого вещества (1,0 г, 65%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,15-8,04 (м, 2H), 7,12 (дд, J=3,6, 9,7 Гц, 1H), 3,78-3,56 (м, 4H), 2,40 (с, 3H), 1,92-1,67 (м, 4H), 1,58 (с, 9H), 1,05-0,95 (м, 6H).
Стадия 3: Получение трет-бутил 6-амино-2-метил-3-(2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)фенокси)бензоата. К раствору трет-бутил 2-метил-6-нитро-3-(2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)фенокси)бензоата (1,0 г, 1,63 ммоль) в метаноле (50 мл) добавляют 10% Pd на угле (50% влажная подложка) (500 мг, 50% загрузка) и смесь продувают аргоном в течение 10 минут. Реакционную смесь помещают под баллон H2 и перемешивают при температуре окружающей среды в течение 3 дней. Реакционную смесь фильтруют через слой Целита, и фильтрат концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (30% EtOAc/гексан) с получением трет-бутил 6-амино-2-метил-3-(2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)фенокси)бензоата в виде коричневой липкой жидкости (300 мг, 31%). 1H ЯМР (400 MГц, ДМСО-d6) δ 7,94-7,83 (м, 1H), 6,66 (д, J=8,9 Гц, 1H), 6,55 (д, J=9,0 Гц, 1H), 5,27 (с-ш, 2H), 3,75-3,55 (м, 4H), 2,29 (с, 3H), 1,90-1,70 (м, 4H), 1,56 (с, 9H), 1,00 (т, J=7,4 Гц, 6H); МС (m/z)=581,2 (M+H).
Стадия 4: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4,5-трифторфенил)пропан-1-сульфонамида. К раствору трет-бутил 6-амино-2-метил-3-(2,3,6-трифтор-5-(N-(пропилсульфонил)пропилсульфонамидо)фенокси)бензоата (400 мг, 0,086 ммоль) в N-метилформамиде (2 мл) добавляют муравьиную кислоту (0,5 мл). Реакционную пробирку промывают аргоном в течение 10 минут, затем герметично закрывают и реакционную смесь перемешивают при 180°C в течение 3 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют этилацетатом (3×20 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией с обращенной фазой (30-100% АЦН/вода с 10 мМ бикарбоната аммония) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,4,5-трифторфенил)пропан-1-сульфонамида в виде беловатого твердого вещества (99 мг, 32%), 1H ЯМР (400 MГц, ДМСО-d6) δ 10,02 (с-ш, 1H), 8,30 (с, 1H), 7,50-7,36 (м, 2H), 7,34 (д, J=9,0 Гц, 1H), 3,46 (с, 3H), 3,19-3,11 (м, 2H), 2,88 (с, 3H), 1,70 (кв, J=7,6 Гц, 2H), 0,99-0,89 (м, 3H); МС (m/z)=442,4 (M+H).
Пример 73
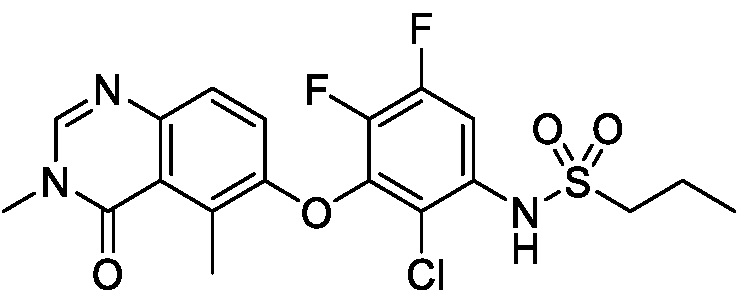
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4,5-дифторфенил)пропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2-хлор-5,6-дифторфенокси)-2-метил-6-нитробензоата. К раствору 3-амино-2-хлор-5,6-дифторфенола (550 мг, 3,07 ммоль) в ДМФ (10,0 мл) добавляют K2CO3 (848,00 мг, 6,15 ммоль) затем трет-бутил 3-фтор-2-метил-6-нитробензоат (783 мг, 3,07 ммоль), и реакционную смесь нагревают при 90°C в течение 16 часов в герметично закрытой пробирке. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют этилацетатом (4×100 мл). Объединенные органические слои промывают водой (4×50 мл) и насыщенным раствором соли (2×25 мл), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением трет-бутил 3-(3-амино-2-хлор-5,6-дифторфенокси)-2-метил-6-нитробензоата (450 мг, 35%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,03 (д, J=9,2 Гц, 1H), 6,90 (д, J=9,2 Гц, 1H), 6,78 (дд, J=7,4, 12,9 Гц, 1H), 5,88 (с-ш, 2H), 2,38 (с, 3H), 1,58 (с, 9H); МС (m/z)=412,5 (M-H).
Стадия 2: Получение трет-бутил-3-(2-хлор-5,6-дифтор-3-пропилсульфонамидо)фенокси)-2-метил-6-нитробензоата. К раствору трет-бутил 3-(3-амино-2-хлор-5,6-дифторфенокси)-2-метил-6-нитробензоата (500 мг, 1,21 ммоль) в дихлорметане (10 мл) при 0°C добавляют триэтиламин (2,40 мл, 18,12 ммоль), затем пропан-1-сульфонил хлорид (0,80 мл, 8,45 ммоль), и реакционную смесь перемешивают в течение 1 часов при температуре окружающей среды. Реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (3×100 мл). Объединенные органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным Na2SO4, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением трет-бутил-3-(2-хлор-5,6-дифтор-3-пропилсульфонамидо)фенокси)-2-метил-6-нитробензоата в виде светло-желтого твердого вещества (600 мг, 95%). МС (m/z)=518,7 [M-H].
Стадия 3: Получение трет-бутил-6-амино-3-(2-хлор-5,6-дифтор-3-(пропилсульфонамидо)фенокси)-2-(метил)бензоата. К раствору трет-бутил-3-(2-хлор-5,6-дифтор-3-пропилсульфонамидо)фенокси)-2-метил-6-нитробензоата (300 мг, 0,57 ммоль) в смеси метанола (10 мл) и воды (5 мл) добавляют хлорид аммония (153,00 мг, 2,89 ммоль), затем порошок Fe (224,00 мг, 8,67 ммоль), и реакционную смесь перемешивают при 75°C в течение 4 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют через Целит, Целит промывают ДХМ (2×50 мл) и фильтрат концентрируют при пониженном давлении. Неочищенный остаток разбавляют водой (50 мл) и экстрагируют ДХМ (2×100 мл). Органический слой промывают водой (2×50 мл) и насыщенным раствором соли (100 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (20% EtOAc/гексан) с получением трет-бутил-6-амино-3-(2-хлор-5,6-дифтор-3-(пропилсульфонамидо)фенокси)-2-(метил)бензоата в виде белого твердого вещества (200 мг, 72%). МС (m/z)=488,9 [M-H].
Стадия 4: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4,5-дифторфенил)пропан-1-сульфонамида. К раствору трет-бутил-6-амино-3-(2-хлор-5,6-дифтор-3-(пропилсульфонамидо)фенокси)-2-(метил)бензоата (300 мг, 0,61 ммоль) в N-метилформамиде (3,0 мл) добавляют муравьиную кислоту (каталитическое количество), и реакционную смесь нагревают до 180°C в течение 2 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют этилацетатом (3×100 мл). Органический слой промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (50% EtOAc/гексан), затем растирают с диэтиловым эфиром с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-4,5-дифторфенил)пропан-1-сульфонамида в виде беловатого твердого вещества (70 мг, 25%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,85 (с-ш, 1H), 8,29 (с, 1H), 7,53 (дд, J=7,5, 11,7 Гц, 1H), 7,44 (д, J=9,0 Гц, 1H), 7,17 (д, J=8,9 Гц, 1H), 3,46 (с, 3H), 3,22-3,17 (м, 2H), 2,90 (с, 3H), 1,75 (кв, J=7,6 Гц, 2H), 0,97 (т, J=7,4 Гц, 3H); МС (m/z)=458,4, 460,3 [M+H].
Пример 74
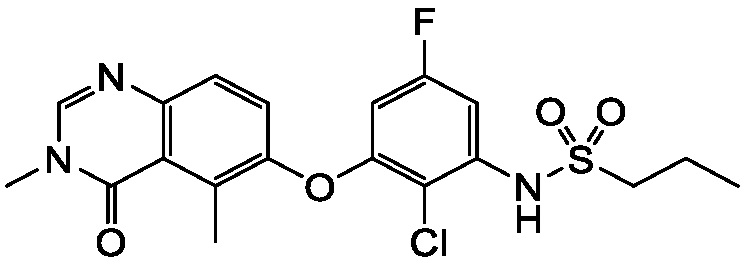
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторфенил)пропан-1-сульфонамид
Стадия 1: Получение 6-(3-амино-2-хлор-5-фторфенокси)-3,5-диметилхиназолин-4(3H)-она. К перемешиваемому раствору 6-гидрокси-3,5-диметилхиназолин-4(3H)-она (1,0 г, 5,25 ммоль), 3-бром-2-хлор-5-фторанилина (1,77 г, 7,88 ммоль) и трифосфата калия (3,3 г, 15,77 ммоль) в ДМСО (10 мл) в герметично закрытой пробирке добавляют пиколиновую кислоту (65 мг, 0,52 ммоль) и CuI (301 мг, 1,57 ммоль). Реакционную смесь продувают аргоном в течение 10 минут, герметично закрывают и нагревают при 140°C в течение 18 часов. Реакционную смесь охлаждают до температуры окружающей среды, фильтруют через слой Целита, и Целит промывают этилацетатом. Фильтрат разбавляют водой и экстрагируют этилацетатом (2×100 мл). Объединенные органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией в течение амин двуокисью кремния (50% EtOAc/гексан) с получением 6-(3-амино-2-хлор-5-фторфенокси)-3,5-диметилхиназолин-4(3H)-она (170 мг, 10%) в виде коричневого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,32 (с, 1H), 7,53 (д, J=8,8 Гц, 1H), 7,38 (д, J=8,8 Гц, 1H), 6,35 (дд, J=2,9, 10,9 Гц, 1H), 5,91 (с-ш, 2H), 5,67 (дд, J=2,8, 9,9 Гц, 1H), 3,45 (с, 3H), 2,67 (с, 3H); МС (m/z)=334,1 (M+H).
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. К раствору 6-(3-амино-2-хлор-5-фторфенокси)-3,5-диметилхиназолин-4(3H)-она (170 мг, 0,51 ммоль) в ДХМ (10 мл) добавляют триэтиламин (0,3 мл, 2,24 ммоль) затем пропан-1-сульфонилхлорид (0,21 мл, 1,87 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 6 часов в атмосфере азота. Реакционную смесь разбавляют водой и экстрагируют этилацетатом (2×50 мл). Объединенные органические слои промывают водой (2×25 мл) и насыщенным раствором соли (25 мл), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией над амином двуокиси кремния (50% EtOAc/гексан) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (116 мг, 42%) в виде желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,37 (с, 1H), 7,59 (д, J=8,8 Гц, 1H), 7,53 (дд, J=2,9, 8,5 Гц, 1H), 7,48 (д, J=8,8 Гц, 1H), 6,85 (дд, J=2,9, 9,5 Гц, 1H), 3,87-3,75 (м, 2H), 3,74-3,62 (м, 2H), 3,47 (с, 3H), 2,67 (с, 3H), 1,94-1,74 (м, 4H), 1,04 (т, J=7,4 Гц, 6H); МС (m/z)=546,2 (M+H).
Стадия 3: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторфенил)пропан-1-сульфонамида. К перемешиваемому раствору N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (7) (116 мг, 0,212 ммоль) в смеси ацетонитрила (12 мл) и воды (4 мл) добавляют бикарбонат натрия (139 мг, 1,66 ммоль), и реакционную смесь нагревают при 80°C в течение 3 часов. Реакционную смесь охлаждают до температуры окружающей среды и концентрируют. Остаток разбавляют водой и экстрагируют этилацетатом (3x 20 мл). Объединенные органические слои промывают водой (2×20 мл) и насыщенным раствором соли (20 мл), сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией с обращенной фазой (20-100% АЦН/вода с 10 мМ бикарбоната аммония) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-5-фторфенил)пропан-1-сульфонамида (14 мг, 15%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 9,78 (с-ш, 1H), 8,35 (с, 1H), 7,56 (д, J=8,8 Гц, 1H), 7,45 (д, J=8,9 Гц, 1H), 7,10 (дд, J=2,9, 10,2 Гц, 1H), 6,49-6,41 (м, 1H), 3,46 (с, 3H), 3,23 (т, J=7,7 Гц, 2H), 2,68 (с, 3H), 1,76 (p, J=7,5 Гц, 2H), 0,99 (т, J=7,4 Гц, 3H); МС (m/z)=440,3, 442,3 (M+H).
Пример 75
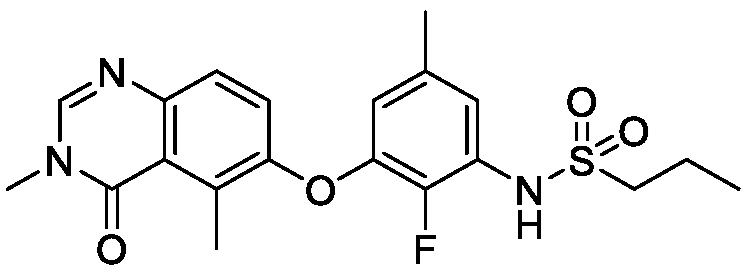
N -(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фтор-5-метилфенил)пропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2-фтор-5-метилфенокси)-2-метил-6-нитробензоата. К раствору 3-амино-2-фтор-5-метилфенола (1,2 г, неочищенный) в ДМФ (7 мл) добавляют K2CO3 (4,69 г, 34 ммоль), затем раствор трет-бутил 3-фтор-2-метил-6-нитробензоата (2,6 г, 10,21 ммоль) в ДМФ (3 мл), и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. Реакционную смесь выливают в ледяную воду и экстрагируют этилацетатом (3×100 мл). Органический слой сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением трет-бутил 3-(3-амино-2-фтор-5-метилфенокси)-2-метил-6-нитробензоата в виде коричневого полутвердого вещества (1,5 г, 47%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,06 (д, J=8 Гц, 1H), 6,84 (д, J=12 Гц, 1H), 6,50 (д, J=4 Гц, 1H), 6,23 (д, J=4 Гц, 1H), 5,37(с-ш, 2H), 2,31 (с, 3H), 2,15 (с, 3H), 1,57 (с, 9H).
Стадия 2: Получение трет-бутил 6-амино-3-(3-амино-2-фтор-5-метилфенокси)-2-метилбензоата. К раствору трет-бутил 3-(3-амино-2-фтор-5-метилфенокси)-2-метил-6-нитробензоата (1,5 г, 4,29 ммоль) в метаноле-воде (1:1; 60 мл) при температуре окружающей среды добавляют хлорид аммония (1,13 г, 21,49 ммоль) и порошок Fe (2,39 г, 42,98 ммоль) и реакционную смесь перемешивают при 80°C в течение 2 часов. Реакционную смесь фильтруют через слой Целита, Целит промывают дихлорметаном и фильтрат концентрируют. Остаток экстрагируют дихлорметаном (3×50 мл) и объединенные органические слои промывают водой, сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (20% EtOAc/гексан) с получением трет-бутил 6-амино-3-(3-амино-2-фтор-5-метилфенокси)-2-метилбензоата в виде белого твердого вещества (800 мг, 54%). ЯМР (400 MГц, ДМСО-d6) δ 6,69-6,83 (м, 1H), 6,60-6,65 (м, 1H), 6,21 (д, J=4 Гц, 1H), 5,60 (J=8 Гц, 1H), 5,35 (с-ш, 1H), 5,13 (с-ш, 1H), 2,10 (с, 3H), 2,01 (с, 3H), 1,54 (с, 9H); МС (m/z)=347,3 (M+H).
Стадия 3: Получение 6-(3-амино-2-фтор-5-метилфенокси)-3,5-диметилхиназолин-4(3H)-она. Раствор трет-бутил 6-амино-3-(3-амино-2-фтор-5-метилфенокси)-2-метилбензоата (800 мг, 2,31 ммоль) в N-метилформамиде (2 мл) перемешивают при 180°C в герметично закрытой пробирке под аргоном в течение 3 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой и экстрагируют этилацетатом (3×30 мл). Объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного 6-(3-амино-2-фтор-5-метилфенокси)-3,5-диметилхиназолин-4(3H)-она (500 мг), который применяют на следующей стадии без дальнейшей очистки. МС (m/z)=314,2 (M+H).
Стадия 4: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фтор-5-метилфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. К раствору 6-(3-амино-2-фтор-5-метилфенокси)-3,5-диметилхиназолин-4(3H)-она (500 мг, неочищенный) в дихлорметане (10 мл) при 0°C добавляют триэтиламин (0,67 мл, 4,79 ммоль) и 1-пропансульфонилхлорид (0,47 мл, 3,99 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 6 часов. Реакционную смесь гасят водой (10 мл) и экстрагируют дихлорметаном (3x 20 мл). Объединенные органические слои промывают водой (20 мл), сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фтор-5-метилфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (900 мг), который применяют на следующей стадии без дальнейшей очистки. МС (m/z)=526,2 (M+H).
Стадия 5: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фтор-5-метилфенил)пропан-1-сульфонамида. К раствору N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фтор-5-метилфенил)-N-(пропилсульфонил)пропан-1-сульфонамида (900 мг, неочищенный) в 1:1 смеси ацетонитрила/воды (40 мл) добавляют карбонат натрия (1,26 г, 12 ммоль), и реакционную смесь перемешивают при 80°C в течение 2 часов. Реакционную смесь охлаждают до температуры окружающей среды и pH доводят примерно 7 10% водной лимонной кислотой раствор. Водный слой экстрагируют этилацетатом (3×20 мл), и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной хроматографией с обращенной фазой (30-95% АЦН/вода с 20 мМ бикарбоната аммония) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фтор-5-метилфенил)пропан-1-сульфонамида в виде беловатого твердого вещества (70 мг, 10%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,71 (с-ш, 1H), 8,32 (с, 1H), 7,52 (д, J=8,8 Гц, 1H), 7,36 (д, J=8,8 Гц, 1H), 6,96 (д, J=5,7 Гц, 1H), 6,50 (д, J=6,5 Гц, 1H), 3,45 (с, 3H), 3,12 (т, J=7,7 Гц, 2H) 2,73 (с, 3H), 2,18 (с, 3H), 1,73 (м, 2H), 0,97 (т, J=7,4 Гц, 3H); МС (m/z)=420,2 (M+H).
Пример 76
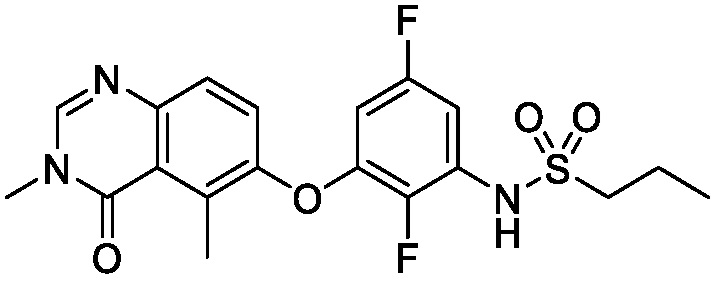
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)пропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2,5-дифторфенокси)-2-метил-6-нитробензоата. К раствору 3-амино-2,5-дифторфенола (0,100 г, 0,689 ммоль) и трет-бутил 3-фтор-2-метил-6-нитробензоата (0,193 г, 0,758 ммоль) в ДМФ (2,76 мл) добавляют карбонат цезия (0,561 г, 1,72 ммоль) и смесь нагревают до 100°C и перемешивают в течение 2 часов. Смесь охлаждают до температуры окружающей среды и разбавляют водой. Смесь экстрагируют EtOAc (3x) и объединенные органические слои сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (10-20% EtOAc/гексан) с получением трет-бутил 3-(3-амино-2,5-дифторфенокси)-2-метил-6-нитробензоата в виде желтого твердого вещества (0,254 г, 96%). 1H ЯМР (400 MГц, CDCl3) δ 7,97 (д, J=9,0 Гц, 1H), 6,79 (д, J=9,0 Гц, 1H), 6,39 (ддд, J=9,4, 6,3, 2,7 Гц, 1H), 6,17 (ддд, J=8,6, 5,5, 2,7 Гц, 1H), 3,99 (шс, 2H), 2,39 (с, 3H), 1,65 (с, 9H).
Стадия 2: Получение трет-бутил 6-амино-3-(3-амино-2,5-дифторфенокси)-2-метилбензоата. К раствору трет-бутил 3-(3-амино-2,5-дифторфенокси)-2-метил-6-нитробензоата (0,246 г, 0,647 ммоль) в EtOAc (3,23 мл) и MeOH (3,23 мл) добавляют палладий (0,0688 г, 0,0323 ммоль, Pd/C, 10% масс., тип Degussa). Систему продувают H2 газом, и затем перемешивают в атмосфере H2 (давление баллона) в течение 3 часов. Смесь фильтруют через нейлоновый фильтр, и твердое вещество промывают дополнительным MeOH и EtOAc. Фильтрат концентрируют с получением трет-бутил 6-амино-3-(3-амино-2,5-дифторфенокси)-2-метилбензоата (0,230 г, 99%) в виде густого желтого масла. 1H ЯМР (400 MГц, CDCl3) δ 6,87 (д, J=8,6 Гц, 1H), 6,53 (д, J=8,6 Гц, 1H), 6,13 (ддд, J=9,4, 6,3, 3,1 Гц, 1H), 5,73 (ддд, J=9,7, 6,3, 2,7 Гц, 1H), 2,25 (с, 3H), 1,60 (с, 9H).
Стадия 3: Получение 6-(3-амино-2,5-дифторфенокси)-3,5-диметилхиназолин-4(3H)-она. Смесь трет-бутил 6-амино-3-(3-амино-2,5-дифторфенокси)-2-метилбензоата (0,227 г, 0,648 ммоль) и метилформамида (1,89 мл, 32,4 ммоль) нагревают до 150°C, где ее перемешивают в течение 22 часов. Смесь охлаждают до температуры окружающей среды и затем разбавляют водой (15 мл), что дает мутную рыжевато-коричневую суспензию. Смесь разбавляют 5 мл насыщенного водного раствора NaHCO3 и затем экстрагируют EtOAc (3x). Объединенные органические экстракты промывают насыщенным раствором соли и сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (20-50% EtOAc/ДХМ) с получением 6-(3-амино-2,5-дифторфенокси)-3,5-диметилхиназолин-4(3H)-она (0,138 г, 67%) в виде кремового твердого вещества. МС (хиад, m/z)=318,1 (M+H).
Стадия 4: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида. В пробирку добавляют 6-(3-амино-2,5-дифторфенокси)-3,5-диметилхиназолин-4(3H)-он (0,040 г, 0,1261 ммоль) и CH2Cl2 (1,26 мл) и суспензию охлаждают до 0°C. Добавляют триэтиламин (0,053 мл, 0,378 ммоль), затем 1-пропансульфонил хлорид (0,031 мл, 0,277 ммоль) и смесь перемешивают при 0°C в течение 1 часа. Смесь разбавляют насыщенным водным раствором NH4Cl и экстрагируют EtOAc (3x). Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-N-(пропилсульфонил)пропан-1-сульфонамида в виде бежевой пены, которую используют на стадии 5 без очистки. МС (хиад, m/z)=530,1 (M+H).
Стадия 5: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)пропан-1-сульфонамида. В пробирку, содержащую N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-N-(пропилсульфонил)пропан-1-сульфонамид (0,067 г, 0,13 ммоль), добавляют CH3CN (0,63 мл) затем 2M Na2CO3 (0,63 мл, 1,3 ммоль). Смесь нагревают до 40°C и перемешивают в течение 3 часов и затем нагревают до 50°C, где ее перемешивают в течение дополнительного 3 часов. Смесь охлаждают до температуры окружающей среды, разбавляют водой и pH доводят до примерно 5 твердым лимонной кислотой (0,24 г, 1,3 ммоль). Смесь экстрагируют EtOAc (3x), и объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (10-30% EtOAc/ДХМ) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)пропан-1-сульфонамида (0,043 г, 78%) в виде белого твердого вещества. МС (хиад, m/z)=424,1 (M+H).
Пример 77
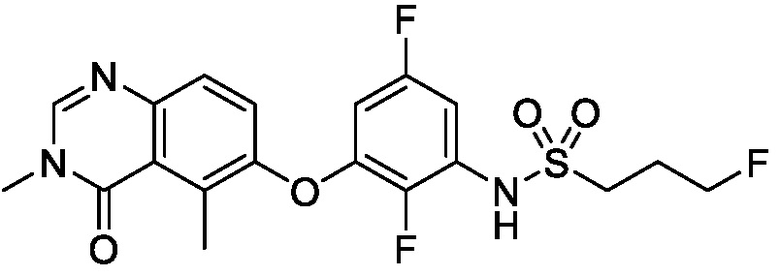
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида. В пробирку добавляют 6-(3-амино-2,5-дифторфенокси)-3,5-диметилхиназолин-4(3H)-он (0,040 г, 0,126 ммоль) и CH2Cl2 (1,26 мл) и суспензию охлаждают до 0°C. Добавляют триэтиламин (0,053 мл, 0,378 ммоль), затем 3-фторпропан-1-сульфонилхлорид (0,035 мл, 0,277 ммоль) и смесь перемешивают при 0°C в течение 1 часа. Смесь разбавляют насыщенным водным раствором NH4Cl и экстрагируют EtOAc (3x). Объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют с получением неочищенного N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамида в виде бежевой пены, которую применяют непосредственно на стадии 2. МС (хиад, m/z)=566,1 (M+H).
Стадия 2: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-3-фторпропан-1-сульфонамида. В пробирку, содержащую неочищенный N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-3-фтор-N-((3-фторпропил)сульфонил)пропан-1-сульфонамид (0,071 г, 0,126 ммоль), добавляют CH3CN (0,628 мл) затем 2M Na2CO3 (0,628 мл, 1,26 ммоль). Смесь нагревают до 50°C, где ее перемешивают в течение 1 часа. Смесь охлаждают до температуры окружающей среды и разбавляют водой и pH доводят до примерно 5 твердой лимонной кислотой (0,241 г, 1,26 ммоль). Смесь экстрагируют EtOAc (3x) и объединенные органические экстракты сушат над Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией на силикагеле (10-30% EtOAc/ДХМ) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2,5-дифторфенил)-3-фторпропан-1-сульфонамида (0,046 г, 82%) в виде беловатого твердого вещества. МС (хиад, m/z)=442,1 (M+H).
Пример 78
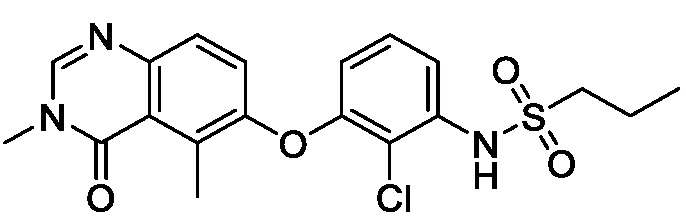
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пропан-1-сульфонамид
Стадия 1: Получение 6-(3-амино-2-хлорфенокси)-3,5-диметилхиназолин-4(3H)-она. Раствор 6-бром-3,5-диметилхиназолин-4(3H)-она (300,0 мг, 1,18 ммоль), 3-амино-2-хлорфенола (339,1 мг, 2,37 ммоль), трикалийфосфата (1,0 г, 4,74 ммоль), пиколиновой кислоты (43,76 мг, 0,36 ммоль) и CuI (112,6 мг, 0,59 ммоль) в ДМСО (6 мл) опрыскивают Ar в течение пяти минут. Реакционную смесь герметично закрывают и нагревают при 130°C в течение 48 часов. После завершения реакции, реакционную смесь концентрируют при пониженном давлении, разбавляют водой и экстрагируют этилацетатом (2×100 мл). Объединенные органические слои промывают водой (2×50 мл) и насыщенным раствором соли (50 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (60% этилацетатом в гексане) с получением 6-(3-амино-2-хлорфенокси)-3,5-диметилхиназолин-4(3H)-она в виде светло-коричневого твердого вещества (260 мг, 69%). 1H ЯМР (400 MГц, ДМСО-d6) δ 8,31 (с, 1H), 7,51 (д, J=8,7 Гц, 1H), 7,32-7,25 (м, 1H), 6,92 (т, J=8,1 Гц, 1H), 6,58-6,54 (м, 1H), 5,92-5,88 (м, 1H), 5,57 (с, 2H), 3,45 (с, 3H), 2,70 (с, 3H); МС (m/z)=316,0 (M+H).
Стадия 2: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-N-(пропилсульфонил)пропан-1-сульфонамида и N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пропан-1-сульфонамида. К раствору 6-(3-амино-2-хлорфенокси)-3,5-диметилхиназолин-4(3H)-она (200 мг, 0,64 ммоль) в ДХМ (5,0 мл) добавляют триэтиламин (1,26 мл, 9,52 ммоль), затем пропан-1-сульфонилхлорид (0,42 мл, 4,44 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа в атмосфере азота. После завершения реакции, реакционную смесь разбавляют водой и экстрагируют этилацетатом (2×250 мл). Объединенные органические слои промывают водой (2×100 мл) и насыщенным раствором соли (100 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (40% этилацетатом в гексане) с получением смеси N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-N-(пропилсульфонил)пропан-1-сульфонамида и N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пропан-1-сульфонамида в виде бледно белого твердого вещества (200 мг). МС (m/z)=528,2, 422,4 (M+H).
Стадия 3: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пропан-1-сульфонамида. К раствору N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-N-(пропилсульфонил)пропан-1-сульфонамида и N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пропан-1-сульфонамида (240 мг) в MeOH добавляют гидроксид натрия (36,43 мг, 0,91 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 1 часа. После завершения реакции, реакционную смесь выпаривают досуха при пониженном давлении. Реакционную смесь разбавляют водой и экстрагируют этилацетатом (2×50 мл) и объединенные органические слои промывают водой (2×25 мл) и насыщенным раствором соли (25 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (80% этилацетатом в гексане) с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)пропан-1-сульфонамид в виде белого твердого вещества (70,0 мг, 36%). 1H ЯМР (400 MГц, ДМСО-d6) δ 9,57 (с, 1H), 8,34 (с, 1H), 7,55 (д, J=8,7 Гц, 1H), 7,36 (д, J=8,8 Гц, 1H), 7,25 (д, J=5,0 Гц, 2H), 6,65-6,57 (м, 1H), 3,46 (с, 3H), 3,19-3,14 (м, 2H), 2,69 (с, 3H), 1,78 (кв, J=7,5 Гц, 2H), 0,99 (т, J=7,4 Гц, 3H); МС (m/z)=422,4 (M+H).
Пример 79
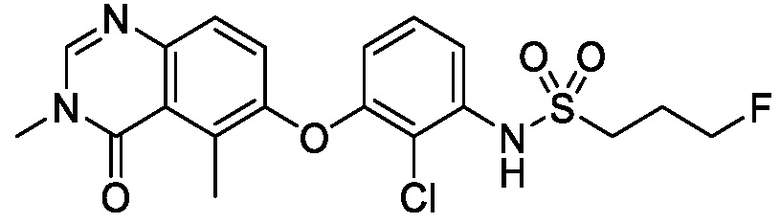
N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2-хлорфенокси)-2-метил-6-нитробензоата. К перемешиваемому раствору 3-амино-2-хлорфенола (500 мг, 3,84 ммоль) в ДМФ (10 мл) добавляют карбонат цезия (2,84 г, 8,74 ммоль) и трет-бутил 3-фтор-2-метил-6-нитробензоат (934 мг, 3,84 ммоль). Реакционную смесь нагревают при 120°C в течение 2 часов. После завершения реакции, реакционную смесь выливают в ледяную воду (30 мл) и экстрагируют этилацетатом (3×40 мл). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с применением 10% этилацетатом в гексане с получением трет-бутил 3-(3-амино-2-хлорфенокси)-2-метил-6-нитробензоата (800 мг, 60%) в виде желтой жидкости. 1H ЯМР (400 MГц, CDCl3) δ 7,92 (д, J=9,2 Гц, 1H), 7,08 (т, J=8,1 Гц, 1H), 6,71-6,60 (м, 2H), 6,44 (д, J=9,2 Гц, 1H), 5,29 (с, 1H), 4,22 (с, 2H), 2,41 (с, 3H), 1,64 (с, 9H).
Стадия 2: Получение трет-бутил 3-(2-хлор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата. К перемешиваемому раствору трет-бутил 3-(3-амино-2-хлорфенокси)-2-метил-6-нитробензоата (300 мг, 0,794 ммоль) в ДХМ (15 мл) при 0°C последовательно добавляют триэтиламин (0,330 мл, 2,38 ммоль) и 3-фторпропансульфонилхлорид (0,317 мг, 1,98 ммоль). Реакционную смесь перемешивают при температуре окружающей среды в течение 6 часов и растворитель выпаривают при пониженном давлении. Остаток разбавляют водой (10 мл) и экстрагируют ДХМ (3x 20 мл). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с применением 5% этилацетата в гексане с получением трет-бутил 3-(2-хлор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата (400 мг, 80%) в виде коричневой жидкости. 1H ЯМР (400 MГц, CDCl3) δ 7,97 (д, J=9,1 Гц, 1H), 7,47-7,34 (м, 2H), 7,22-7,18 (м, 1H), 6,64 (д, J=9,1 Гц, 1H), 4,64 (т, J=5,6 Гц, 2H), 4,53 (т, J=5,7 Гц, 2H), 3,91-3,73 (м, 4H), 2,44-2,26 (м, 4H), 2,03 (с, 3H), 1,64 (с, 9H).
Стадия 3: Получение трет-бутил 6-амино-3-(2-хлор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата. К перемешиваемому раствору трет-бутил 3-(2-хлор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата (1,0 г, 1,59 ммоль) в смеси метанол/вода (2:1) при температуре окружающей среды добавляют хлорид аммония (426 мг, 7,97 ммоль) и порошок железа (890 мг, 15,94 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов. После завершения реакции, реакционную смесь фильтруют через Целит, Целит промывают ДХМ и фильтрат концентрируют при пониженном давлении. Остаток разбавляют водой (10 мл) и экстрагируют ДХМ (3x 20 мл). Объединенные органические слои сушат над безводным сульфатом натрия, фильтруют и концентрируют при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с применением 10% этилацетата в гексане с получением трет-бутил 6-амино-3-(2-хлор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата (650 мг, 68%) в виде белого твердого вещества. 1H ЯМР (400 MГц, CDCl3) δ 7,25 (с, 1H), 7,17 (т, J=8,2 Гц, 1H), 7,08 (дд, J=1,5, 7,9 Гц, 1H), 6,86 (д, J=8,7 Гц, 1H), 6,74 (дд, J=1,5, 8,3 Гц, 1H), 6,55 (д, J=8,7 Гц, 1H), 4,72 (с, 2H), 4,65 (т, J=5,7 Гц, 2H), 4,53 (т, J=5,7 Гц, 2H), 3,93-3,75 (м, 4H), 2,46-2,28 (м, 4H), 2,21 (с, 3H), 1,59 (с, 9H).
Стадия 4: Получение N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторпропан-1-сульфонамида. Смесь трет-бутил 6-амино-3-(2-хлор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата (350 мг, 0,60 ммоль), N-метилформамида (1 мл) и муравьиной кислоты (0,5 мл) нагревают до 180°C в герметично закрытой пробирке в течение 2 часов. После завершения реакции, реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой (30 мл) и экстрагируют ДХМ (2×50 мл). Объединенные органические слои промывают водой (2×25 мл) и насыщенным раствором соли (25 мл), сушат над безводным сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с применением 50% этилацетатом в гексане с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)фенил)-3-фторпропан-1-сульфонамида (60 мг, 23%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 9,72 (с, 1H), 8,33 (с, 1H), 7,55 (д, J=8,8 Гц, 1H), 7,36 (д, J=8,8 Гц, 1H), 7,31-7,21 (м, 2H), 6,67-6,59 (м, 1H), 4,62 (т, J=5,9 Гц, 1H), 4,50 (т, J=6,0 Гц, 1H), 3,46 (с, 3H), 3,30-3,25 (м, 2H), 2,69 (с, 3H), 2,24-2,06 (м, 2H); МС (m/z)=440,3 (M+H).
Пример 80
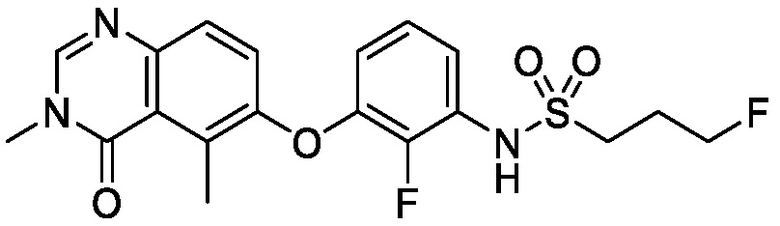
N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фторфенил)-3-фторпропан-1-сульфонамид
Стадия 1: Получение трет-бутил 3-(3-амино-2-фторфенокси)-2-метил-6-нитробензоата. К перемешиваемому раствору 3-амино-2-фторфенола (1 г, 6,21 ммоль) в ДМФ (5 мл) добавляют K2CO3 (3,26 г, 23,62 ммоль) и раствор трет-бутил 3-фтор-2-метил-6-нитробензоата (2,4 г, 7,45 ммоль) в ДМФ (3 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 часов, выливают в ледяную воду и экстрагируют этилацетатом (3×20 мл). Объединенные органические слои сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением трет-бутил 3-(3-амино-2-фторфенокси)-2-метил-6-нитробензоата (2,5 г, 87%) в виде светло-желтого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,07 (д, J=9,2 Гц, 1H), 6,99-6,90 (м, 1H), 6,84 (д, J=9,2 Гц, 1H), 6,76-6,67 (м, 1H), 6,47-6,37 (м, 1H), 5,47 (с-ш, 2H), 2,33 (с, 3H), 1,58 (с, 9H) ; МС (хиад, m/z)=361,3 (M-H).
Стадия 2: Получение трет-бутил 3-(2-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата. К раствору трет-бутил 3-(3-амино-2-фторфенокси)-2-метил-6-нитробензоата (2,5 г, 6,86 ммоль) в ДХМ (15 мл) при 0°C добавляют триэтиламин (3,4 мл, 23,9 ммоль) затем 3-фторпропансульфонилхлорид (2,74 г, 17,17 ммоль), и реакционную смесь перемешивают при температуре окружающей среды в течение 6 часов в атмосфере азота. Реакционную смесь гасят водой (10 мл) и экстрагируют ДХМ (2×20 мл). Объединенные органические слои промывают водой (10 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния (10% EtOAc/гексан) с получением трет-бутил 3-(2-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата (2,2 г, 52%) в виде коричневого полутвердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 8,10 (д, J=9,1 Гц, 1H), 7,72-7,57 (м, 2H), 7,45 (т, J=8,2 Гц, 1H), 6,88 (д, J=9,0 Гц, 1H), 4,62 (т, J=5,7 Гц, 2H), 4,50 (т, J=5,7 Гц, 2H), 3,91-3,72 (м, 4H), 2,36 (с, 3H), 2,29-2,05 (м, 4H), 1,58 (с, 9H).
Стадия 3: Получение трет-бутил 6-амино-3-(2-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата. К раствору трет-бутил 3-(2-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метил-6-нитробензоата (1,0 г, 1,63 ммоль) в MeOH (10 мл) добавляют 10% Pd/C (примерно 50% влажный) (500,0 мг, 50% масс/масс) и реакционную смесь продувают аргоном в течение 10 минут. Реакционную смесь перемешивают под баллоном водорода при температуре окружающей среды в течение 12 часов, фильтруют через Целит и фильтрат концентрируют. Неочищенный продукт очищают колоночной гель-хроматографией на двуокиси кремния с получением трет-бутил 6-амино-3-(2-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата (700 мг, 73%) в виде бесцветной смолистой жидкости. МС (хиад, m/z)=581,3 (M+H).
Стадия 4: Получение N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фторфенил)-3-фторпропан-1-сульфонамида. Раствор трет-бутил 6-амино-3-(2-фтор-3-((3-фтор-N-((3-фторпропил)сульфонил)пропил)сульфонамидо)фенокси)-2-метилбензоата (400 мг, 0,689 ммоль), N-метилформамида (2 мл) и муравьиной кислоты (10 мл) нагревают при 180°C в герметично закрытой пробирке в течение 2 часов. Реакционную смесь охлаждают до температуры окружающей среды, разбавляют водой (30 мл) и экстрагируют ДХМ (2×50 мл). Объединенные органические слои промывают водой (2×25 мл) и насыщенным раствором соли (25 мл), сушат над безводным Na2SO4, фильтруют и концентрируют. Неочищенный продукт очищают хроматографией с обращенной фазой (20-95% MeCN/вода, 20 мМ бикарбоната аммония) с получением N-(3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)окси)-2-фторфенил)-3-фторпропан-1-сульфонамида (155 мг, 53%) в виде белого твердого вещества. 1H ЯМР (400 MГц, ДМСО-d6) δ 9,93 (с, 1H), 8,32 (с, 1H), 7,53 (д, J=8,9 Гц, 1H), 7,38 (д, J=8,9 Гц, 1H), 7,18 (т, J=7,4 Гц, 1H), 7,11 (т, J=8,2 Гц, 1H), 6,71 (т, J=8,0 Гц, 1H), 4,61 (т, J=5,9 Гц, 1H), 4,49 (т, J=5,9 Гц, 1H), 3,46 (с, 3H), 3,30-3,25 (м, 2H), 2,74 (с, 3H), 2,21-2,03 (м, 2H). МС (m/z)=424,4 (M+H).
Пример 81
Способы получения N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А
Способ 1A: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид форму С растворяют в 20% воде/EtOH (12 об.) и нагревают до кипения с обратным холодильником. Раствор медленно по каплям переносят в воду (10 об.). Суспензию перемешивают в течение 2 ч, затем фильтруют и сушат с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А. ПРД показана на ФИГ. 1 и отнесение пиков показано в таблице 3. типовая термограмма ДСК показана на ФИГ. 2.
Способ 2A: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид форму С берут в EtOH (44 об.) и нагревают до 78°C до достижения полного растворения. Быстро добавляют воду (73 об.) и суспензию охлаждают до 22°C и хранят при 5°C в течение 15 ч. Твердые вещества фильтруют и сушат с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А.
Способ 3A: Аморфный N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид получают растворением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной форма В в 1,4-диоксане и затем лиофилизацией и повторением этого процесса до тех пор, пока продукт не будет признан полностью аморфным по ПРД (фигура 17). К аморфному N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамиду (20 мг) добавляют 2 капли сверхнасыщенного ТГФ раствора и 2×2,8 мм гранул шаровой мельницы. Образец загружают в шаровую мельницу и встряхивают с 3×60 секундными интервалами с 10-секундной паузой между интервалами. Выделенные влажные твердые вещества сушат с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А.
Способ 4A: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид форму F сушат в вакуумной печи в течение 12 ч при 40°C с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А.
Способ 5A: Насыщенный раствор аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в 1,4-диоксане хранят при 2-8°C в течение 72 часов. Полученные твердые вещества фильтруют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы А.
Пример 82
Способы получения N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В
Способ 1B: В этой методике, S=коэффициент масштабирования (г/г). N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид (либо в аморфной форме, либо в форме A или форме С) растворяют в этаноле (9,4 S) и деионизированной воде (3,0 S) при 65-75°C. Раствор доочищают на фильтре при 65-75°C. Реактор промывают вперед горячим (65-75°C) раствором этанола (1,3 S) и воды (0,4 S). Воду (5,6 S) добавляют к фильтрованному раствору при 65-75°C в течение 30-120 минут, и температуру понижают до 60-65°C. Раствор засевают N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамидом, формой B (0,001S) и температуру понижают до 20-25°C в течение 4-8 ч. Суспензию выстаивают 10-24 ч при 20-25°C. Твердые вещества выделяют центрифугированием, промывают смесью холодного этанола (0,7 S) и деионизированной воды (0,9 S), чистой деионизированной водой (2,5 S) и сушат при 28-32°C в течение минимум 16 ч с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В. ПРД показана на ФИГ. 4 и отнесение пиков показано в таблице 4. Типовая термограмма ДСК показана на ФИГ. 5, имеющая максимальную температуру плавления примерно 151,39°C. ФИГ. 6 является наложением скана дифференциальной сканирующей калориметрии и скана термогравиметрического анализа N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В. ТГА/ДСК анализ обнаружил незначительную потерю массы 0,2% масс. во время эндотермического события, наблюдаемого на следе ДСК, которое имеет начало при 148°C и пик при 152°C, что является плавлением безводной формы В. Потеря массы наиболее вероятно происходит из-за движения образца во время плавления. Никакой другой потери массы или событий не наблюдают до тех пор, пока продукт не начинает разлагаться при приблизительно 250°C.
Способ 2B: К аморфному N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамиду (20 мг; получен, как описано в примере 81, Способ 3A) добавляют 2 капли сверхнасыщенного 2-этоксиэтанола, хлороформ, 2-пропанол, этанол, этанол/воду, этилацетат, гептан, изопропилацетат, метанол, 2-метил ТГФ, метилизобутилкетон или раствор трифторэтанола и 2×2,8 мм гранулы шаровой мельницы. Образцы загружают в шаровую мельницу и встряхивают в течение 3×60 секунд интервалов с 10 секундной паузой между интервалами, и полученные твердые вещества выделяют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
Способ 3B: N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид, форму D, сушат в вакуумной печи в течение 12 ч при 40°C с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
Способ 4B: Насыщенный раствор аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в трифторэтаноле хранят при 2-8°C в течение 72 часов. Полученные твердые вещества фильтруют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
Способ 5B: Насыщенный раствор аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в трифторэтаноле хранят при -10°C в течение 72 часов. Полученные твердые вещества фильтруют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
Способ 6B: Насыщенный раствор аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в 2-этоксиэтаноле, уксусной кислоте, ацетоне, анизоле, бензиловом спирте, хлороформе, дихлорметане, 2-пропаноле, этаноле, этаноле/воде, этилацетате, гептане, изопропилацетате, метаноле, 2-метил ТГФ, метилизобутилкетоне, трет-бутилметиловом эфире, трифторэтаноле или воде подвергают тепловому циклу от 20°C до 40°C в 4-часовых циклах в течение 48 часов при перемешивании. Твердые вещества выделяют и сушат с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
Способ 7B: Насыщенный раствор аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в 2-этоксиэтаноле, N, N’-диметилформамиде, N, N’-диметилацетамиде или трифторэтаноле переносят в 2 мл пробирки. Затем пробирки открывают, и растворитель выпаривают при температуре окружающей среды. Полученный твердый продукт выделяют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
Способ 8B: Трет-бутилметиловый эфир добавляют по каплям при перемешивании к насыщенному раствору аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в N, N’-диметилформамиде. Суспензию затем открывают и выпаривают при температуре окружающей среды. Полученные твердые вещества восстанавливают с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид безводная форма В.
Способ 9B: Трет-бутилметиловый эфир добавляют по каплям при перемешивании к насыщенному раствору аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в N, N’-диметилацетамиде. Суспензию затем открывают и выпаривают при температуре окружающей среды. Полученные твердые вещества восстанавливают с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
Способ 10B: Воду добавляют по каплям при перемешивании к насыщенному раствору аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в ацетоне. Суспензию затем открывают и выпаривают при температуре окружающей среды. Полученные твердые вещества восстанавливают с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, безводной формы В.
Пример 83
Способы получения N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы С моно-анизола
К аморфному N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида (20 мг; получен как описано в примере 81, способ 3A) добавляют 2 капли сверхнасыщенного раствора анизола и 2×2,8 мм гранулы шаровой мельницы. Образцы загружают в шаровую мельницу и встряхивают в течение 3×60 секундных интервалов с 10 секундной паузой между интервалами, и полученные твердые вещества выделяют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы С моно-анизола. ПРД показана на ФИГ. 7 и отнесение пиков показано в таблице 5. ФИГ. 8 представляет наложение скана дифференциальной сканирующей калориметрии (ДСК) и скана термогравиметрического анализа (ТГА) N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида формы С моно-анизола. ТГА/ДСК анализ показал 18,4% потерю массы от 92°C до 122°C. Это равно 0,95 экв. анизола. Из-за небольшого количества материала, доступного для анализа (0,2 мг) и времени выдержки в условиях окружающей среды перед анализом, количество анизола, присутствующего на термограмме, может быть неточно представлено, но эта информация, по-видимому, позволяет предположить, что форма С является сольватом моно-анизола. При высыхании форма С моно-анизол превращается в слабо кристаллическую форму B.
Пример 84
Способы получения N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамиды формы D сольвата полу-дихлорметана
Способ 1D: К аморфному N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамиду (20 мг; получен как описано в примере 81, Способ 3A) добавляют 2 капли сверхнасыщенного раствора дихлорметана и 2×2,8 мм гранулы шаровой мельницы. Образец загружают в шаровую мельницу и встряхивают в течение 3×60 секундных интервалов с 10 секундной паузой между интервалами, и полученные твердые вещества выделяют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида формы D полу-дихлорметана. ПРД показана на ФИГ. 9 и отнесение пиков показано в таблице 6. ФИГ. 10 представляет наложение скана дифференциальной сканирующей калориметрии (ДСК) и скана термогравиметрического анализа N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида формы D полу-дихлорметана. Термогравиметрический анализ (ТГА) формы D показал 10,2% потерю массы между 61°C и 88°C. Это эквивалентно 0,61 экв. дихлорметана. Дифференциальный тепловой анализ показал незначительное эндотермическое событие с наступлением при 147°C и пиком при 150°C. Затем возникает другое незначительное эндотермическое событие с наступлением при 164°C и пиком при 165°C. Это согласуется с формой Form A (наступление при 162°C и пик при 165°C). При сушке, форма D полу-дихлорметан превращается в кристаллическую форму B.
Способ 2D: Аморфный N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамид растворяют в дихлорметане. Раствор концентрируют, и твердые вещества выделяют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида формы D полу-дихлорметана.
Пример 85
Способы получения N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы E, сольвата моно-толуола
Способ 1E: К аморфному N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамиду (20 мг; получен как описано в примере 81, Способ 3A) добавляют 2 капли сверхнасыщенного толуольного раствора N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида и 2×2,8 мм гранулы шаровой мельницы. Образец загружают в шаровую мельницу и встряхивают в течение 3×60 секундных интервалов с 10 секундной паузой между интервалами, и полученные влажные твердые вещества выделяют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, толуольной формы Е. ПРД показана на ФИГ. 11 и отнесение пиков показано в таблице 7. ФИГ. 12 представляет наложение скана дифференциальной сканирующей калориметрии (ДСК) и скана термогравиметрического анализа N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы E. Термогравиметрический анализ (ТГА) показал 17,1% потерю массы от 87 до 128°C. Это эквивалентно 1,02 экв. толуола. Во время этой потери массы наблюдается изменение в базовой линии. Наблюдают событие с наступлением при 93°C и пиком при 103°C. После этого, наблюдают эндотермическое событие с наступлением при 164°C и пиком при 165°C. Это согласуется с формой A (наступление при 162°C и пиком при 165°C).
Способ 2E: Насыщенный раствор аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в толуоле подвергают тепловому циклу от 20°C до 40°C в 4-часовых циклах в течение 48 часов при перемешивании. Полученные твердые вещества выделяют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида толуольной формы Е.
Пример 86
Способы получения N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида формы F 1,4 диоксана
Способ 1F: Насыщенный раствор аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в 1,4-диоксане хранят при 2-8°C в течение 72 часов. Полученные твердые вещества фильтруют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида формы F 1,4-диоксана. ПРД показана на ФИГ. 13 и отнесение пиков показано в таблице 8. ФИГ. 14 представляет наложение скана дифференциальной сканирующей калориметрии (ДСК) и скана термогравиметрического анализа N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы F 1,4-диоксана. ТГА/ДСК анализ формы F показал 23,7% потерю массы от 49 до 89°C, что равно 1,6 эквивалента 1,4-диоксана. Эта потеря массы по видимому представляет десольватирование N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида формы F 1,4-диоксана до N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы A, та как эндотермическое событие плавления формы A наблюдается на следе ДСК с наступлением при 161°C и пиком при 164°C.
Способ 2F: Насыщенный раствор аморфного N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида в 1,4-диоксане хранят при -10°C в течение 72 часов. Полученные твердые вещества фильтруют с получением N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида, формы F, 1,4 диоксана.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ 4-ОКСО-3,4-ДИГИДРОХИНАЗОЛИНОНА ДЛЯ ЛЕЧЕНИЯ BRAF-АССОЦИИРОВАННЫХ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ | 2021 |
|
RU2814662C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКОГО ИМИНОПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2018 |
|
RU2801302C2 |
| 3,4-ДИГИДРО-2,7-НАФТИРИДИН-1,6(2H,7H)-ДИОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ MEK | 2022 |
|
RU2826000C1 |
| НОВЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ КАК ИНГИБИТОРЫ BRD4 | 2016 |
|
RU2721120C2 |
| ПРОИЗВОДНЫЕ БЕНЗИЗОКСАЗОЛСУЛЬФОНАМИДА | 2020 |
|
RU2813356C2 |
| ХИНОЛИНИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2803116C2 |
| СОЕДИНЕНИЯ ПИРАЗОЛО[3,4-b]ПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ ТАМ И МЕТ | 2019 |
|
RU2773611C1 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ПИРРОЛАМИДОПИРИДОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2809596C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2801647C2 |
| Ингибиторы рецептора эпидермального фактора роста | 2022 |
|
RU2821531C2 |
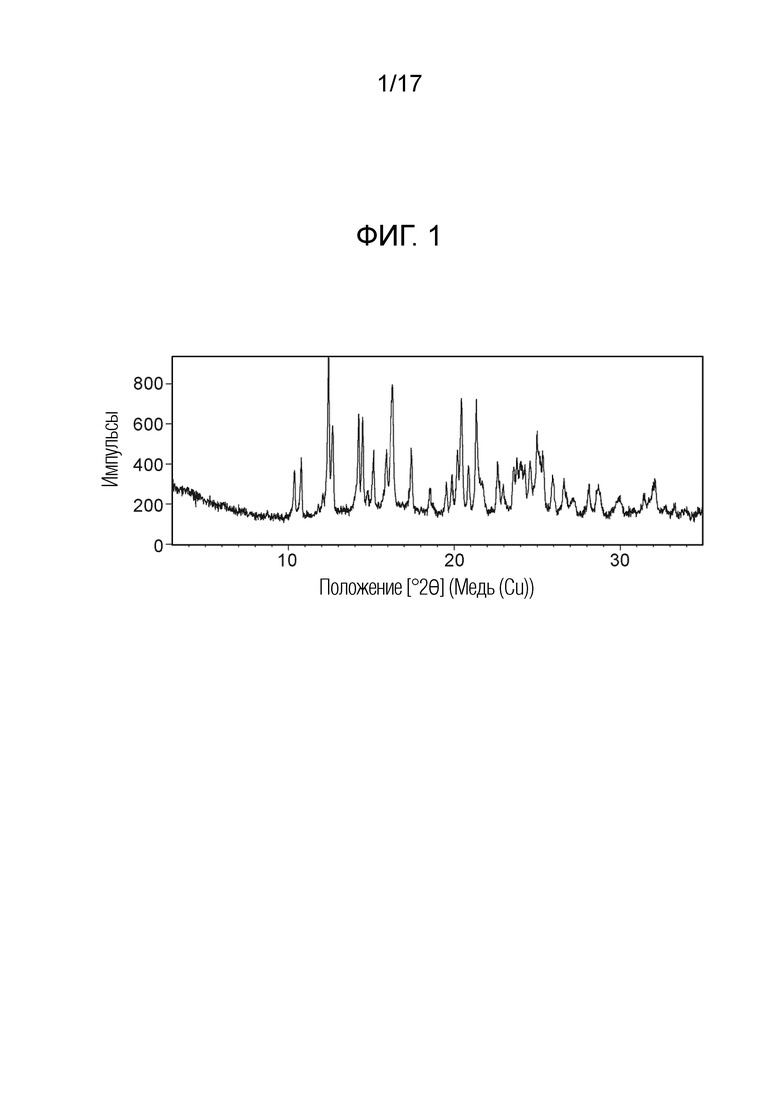
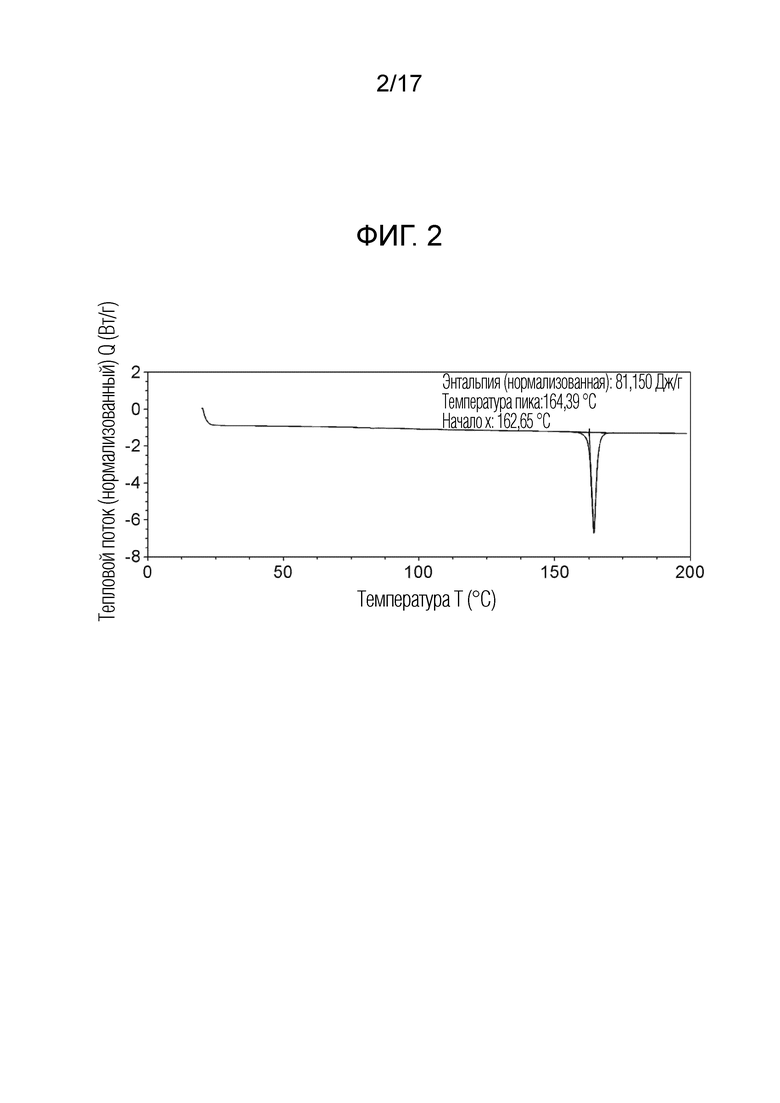

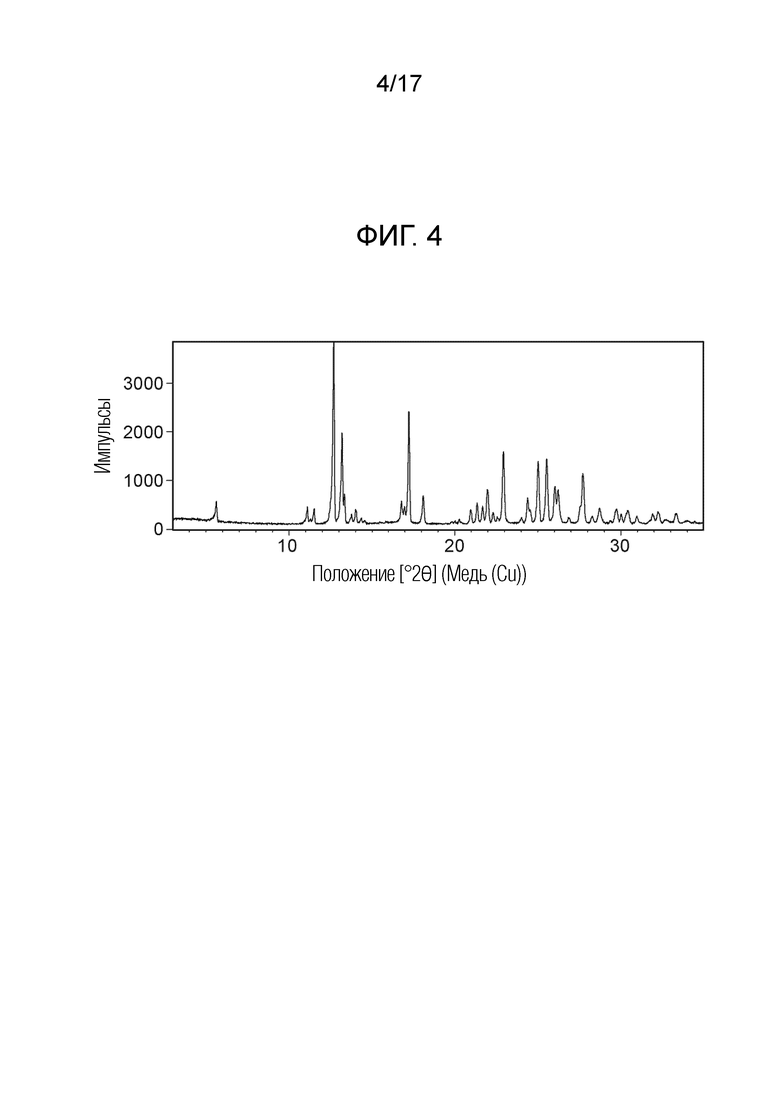
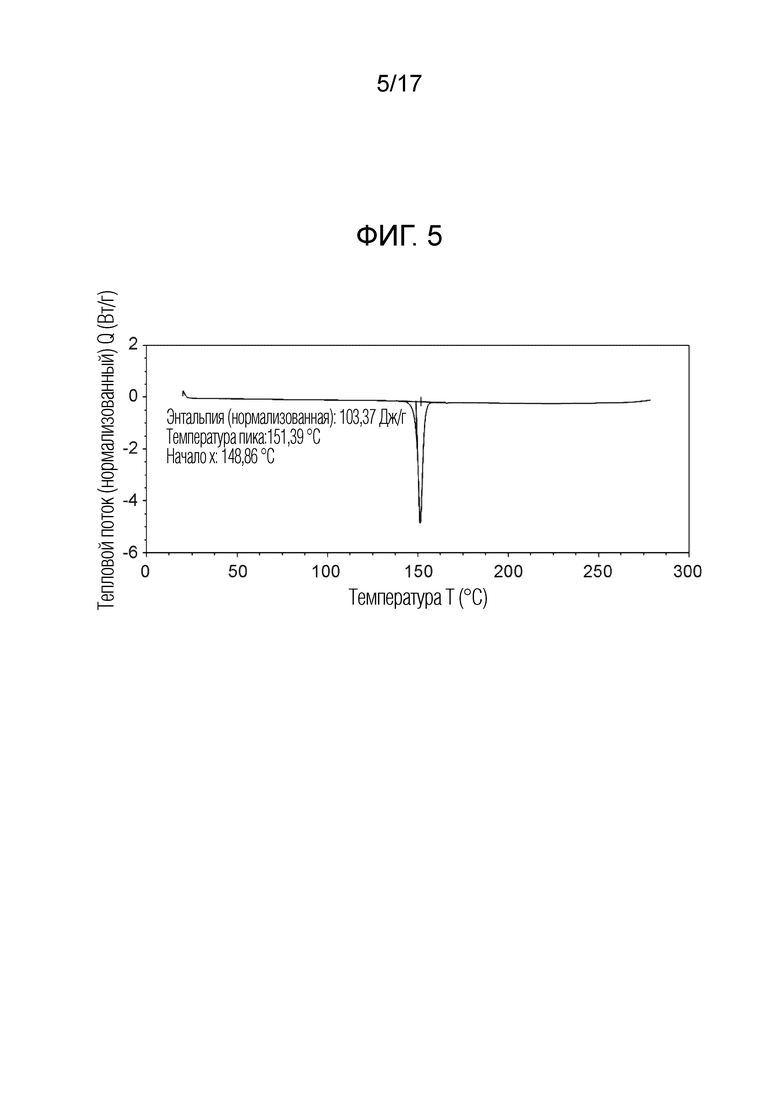
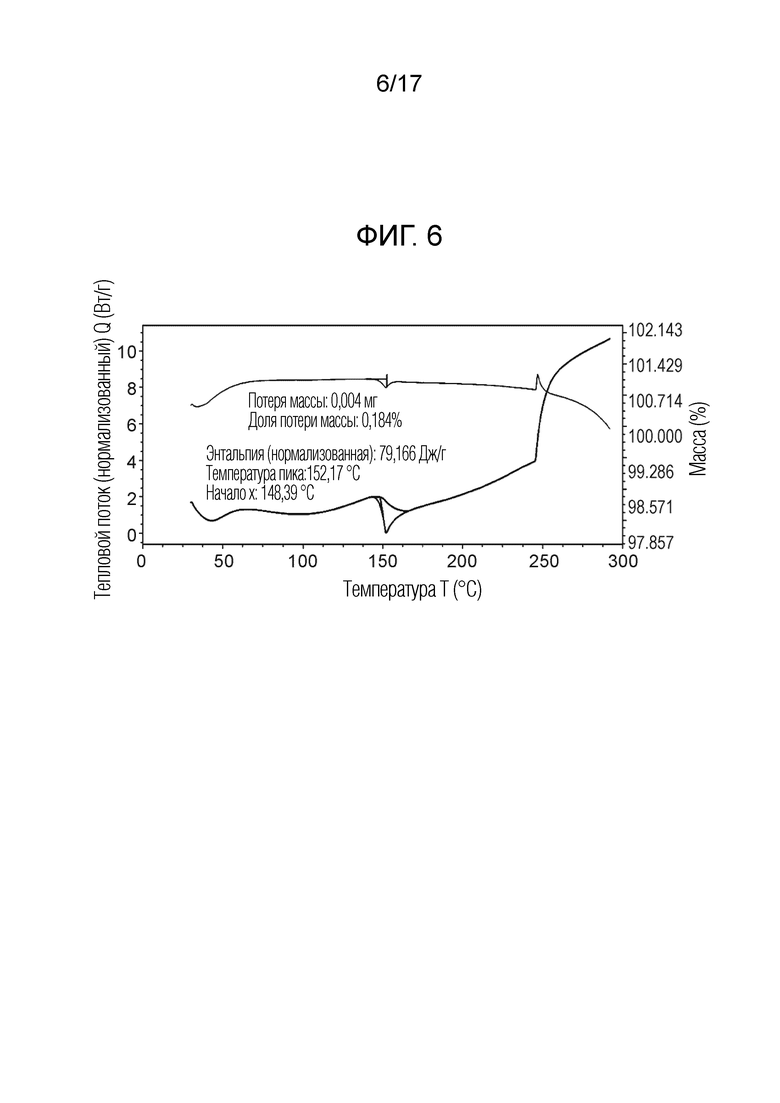
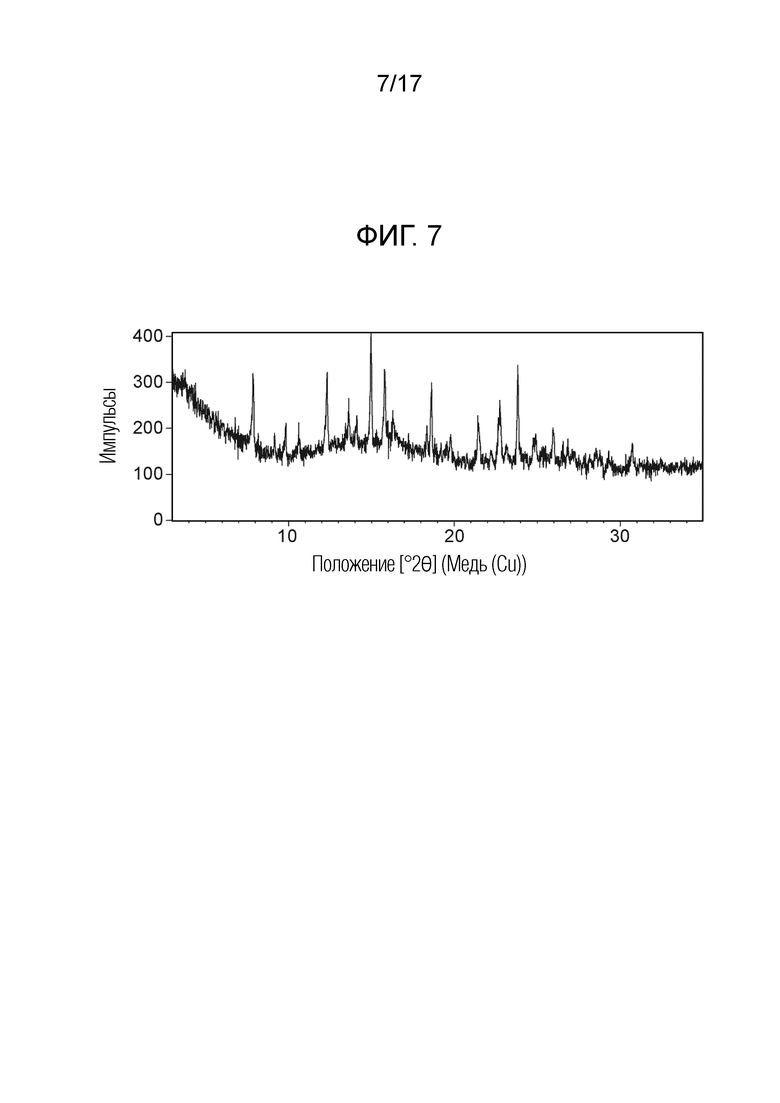
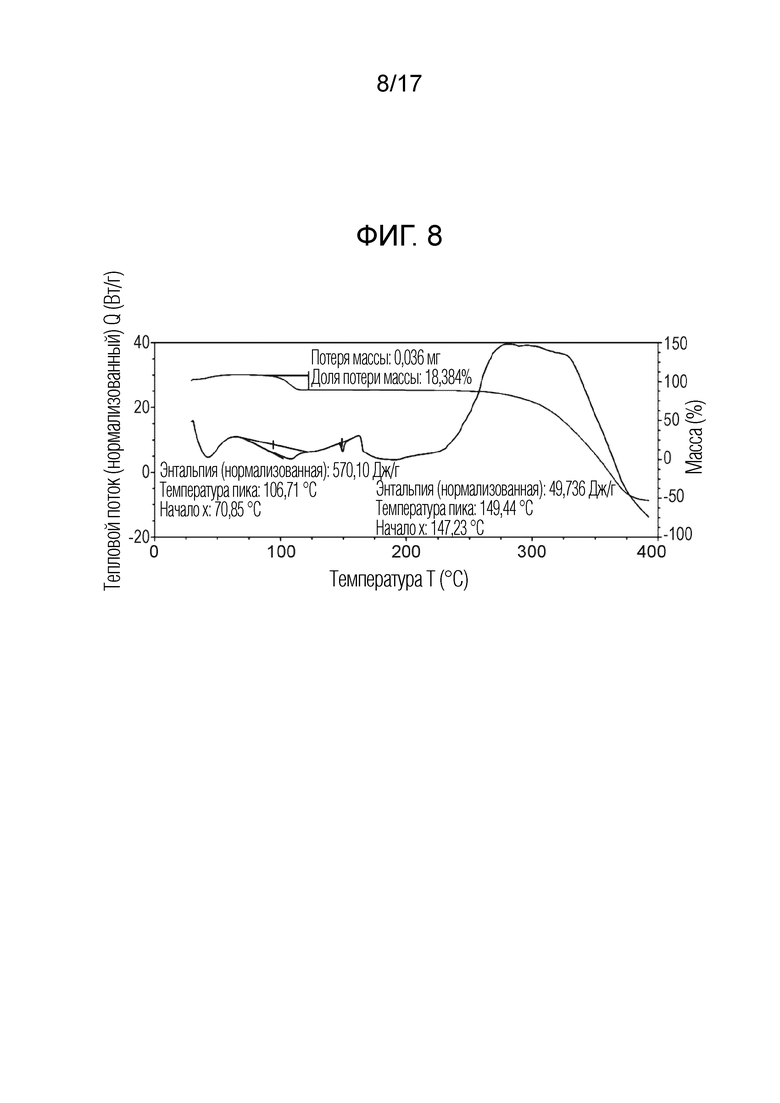
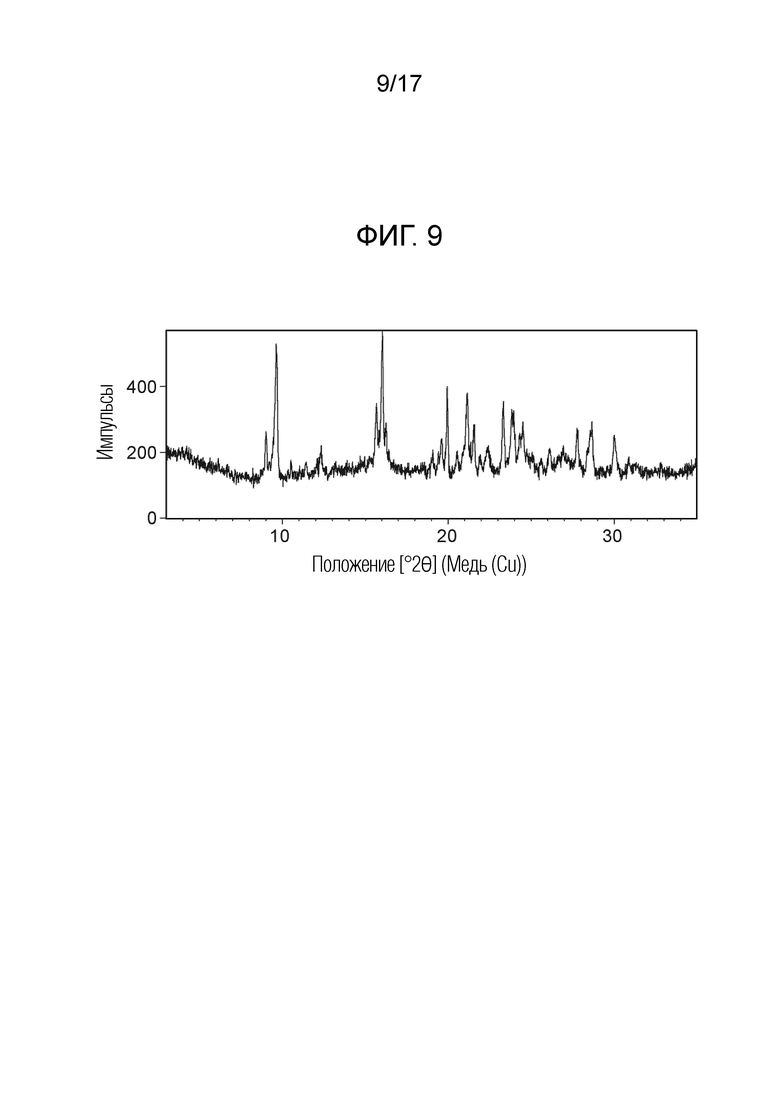
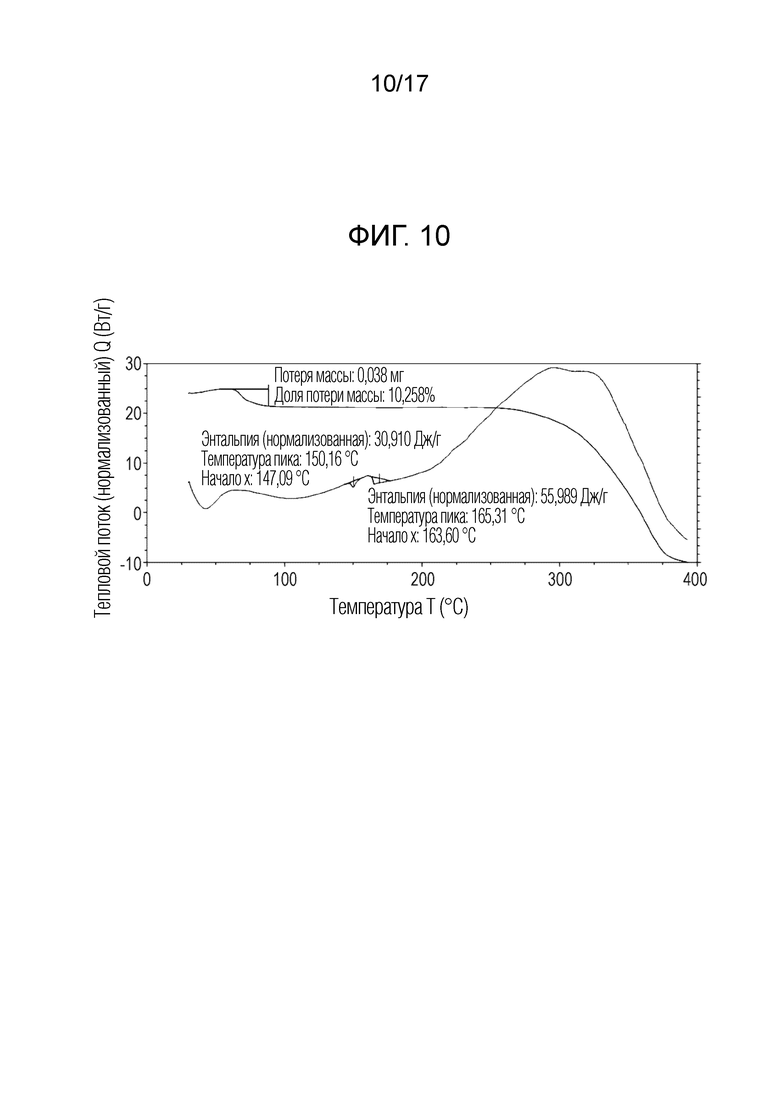
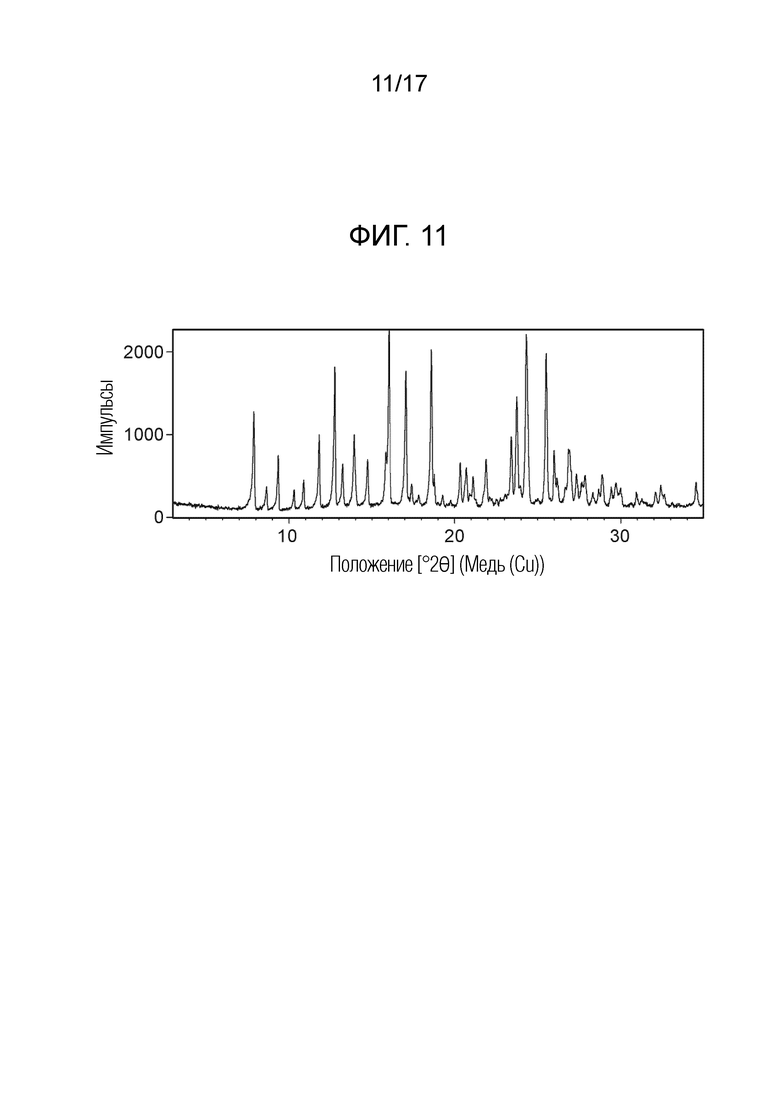
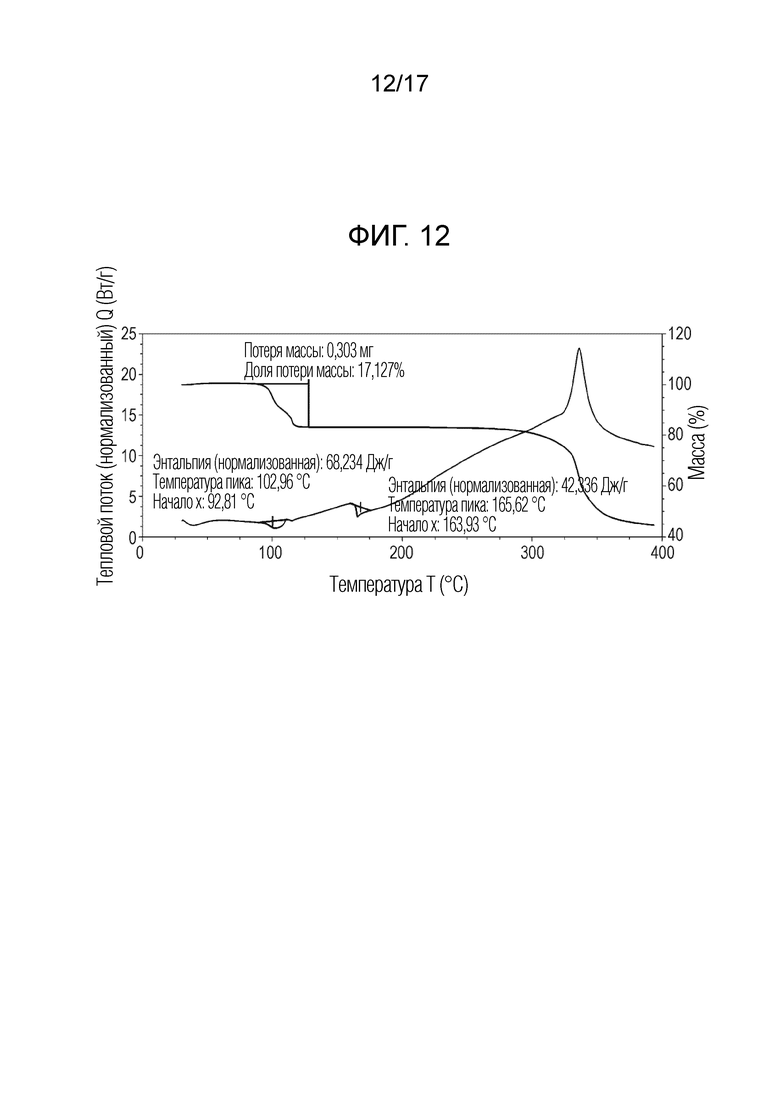
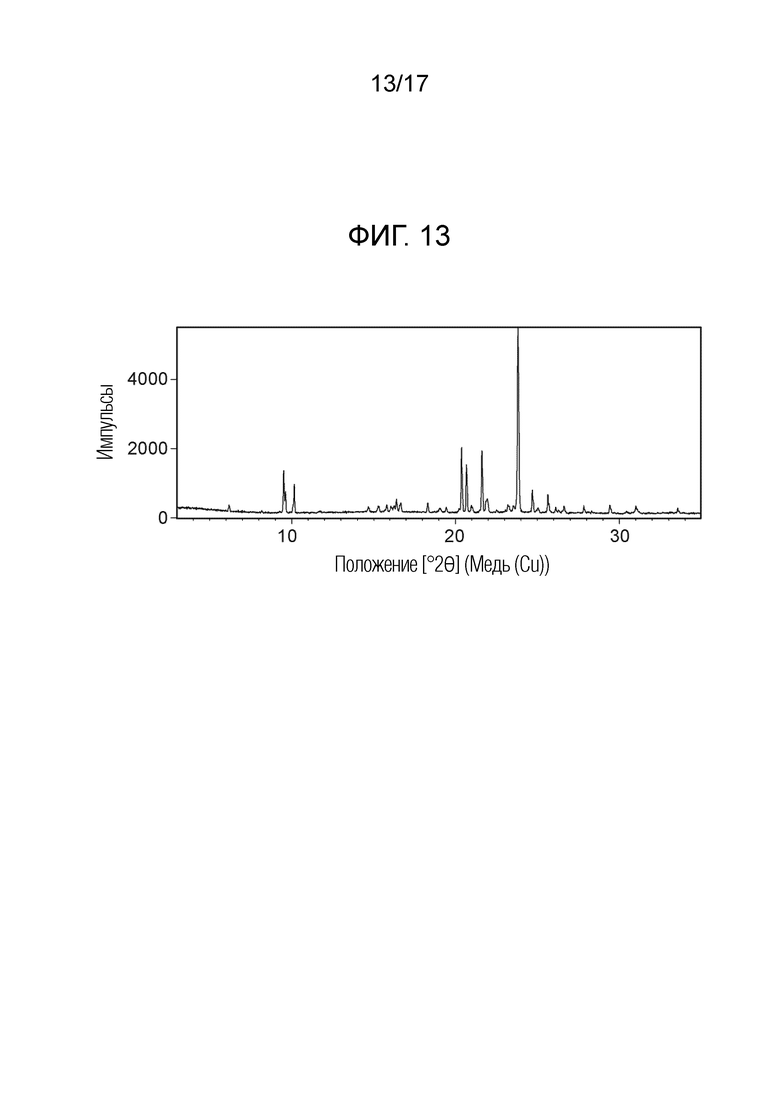
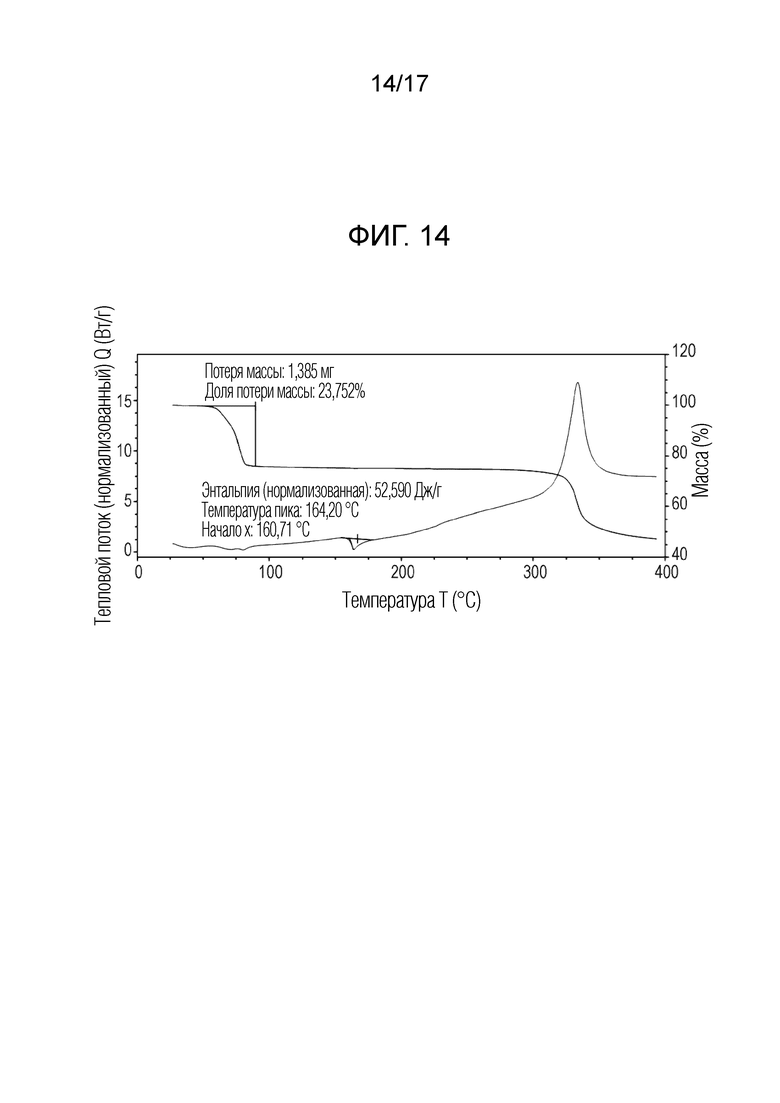
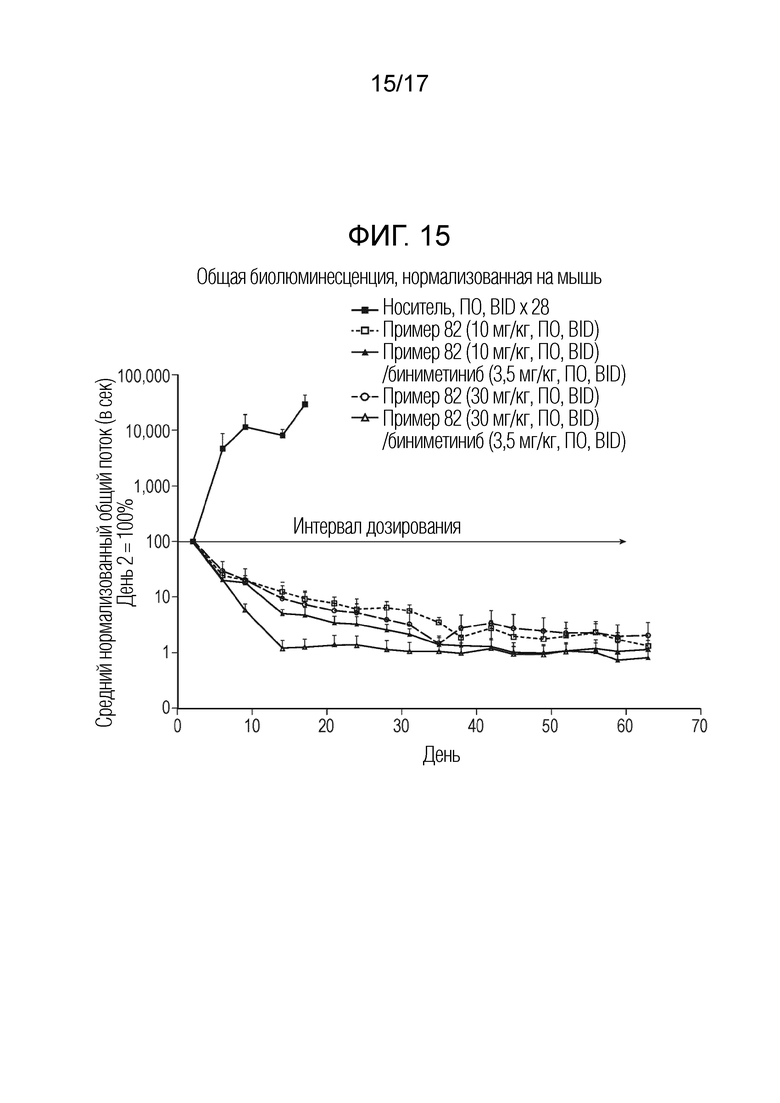
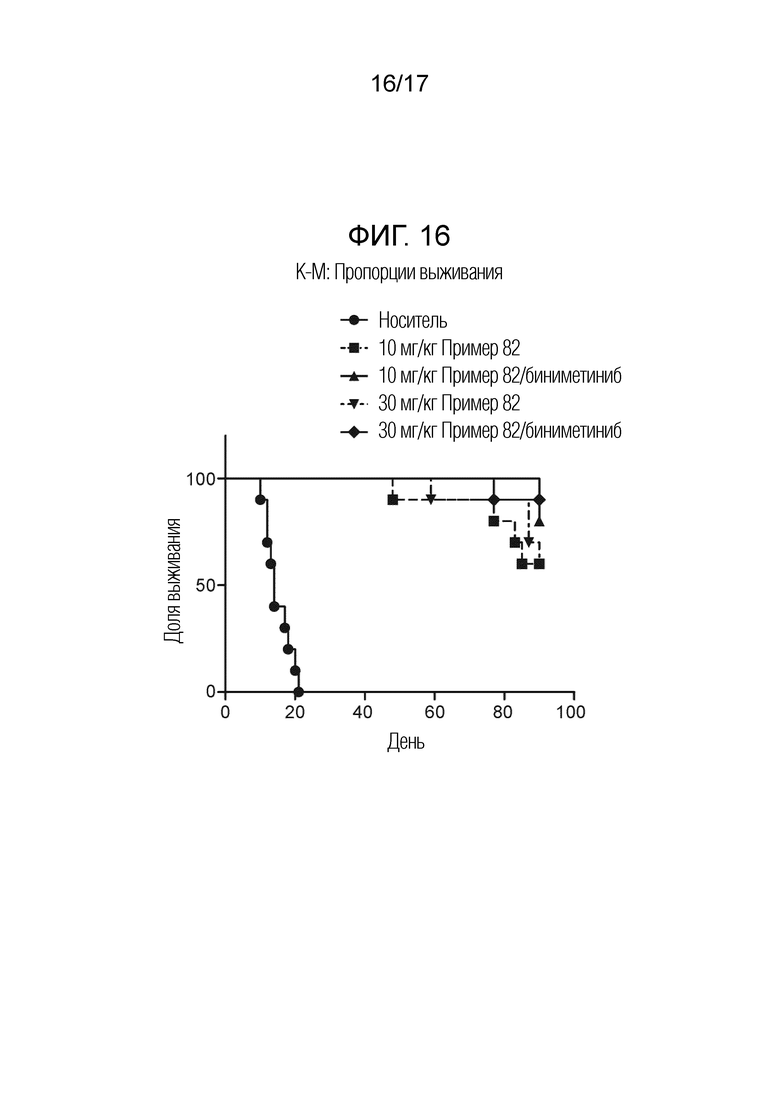
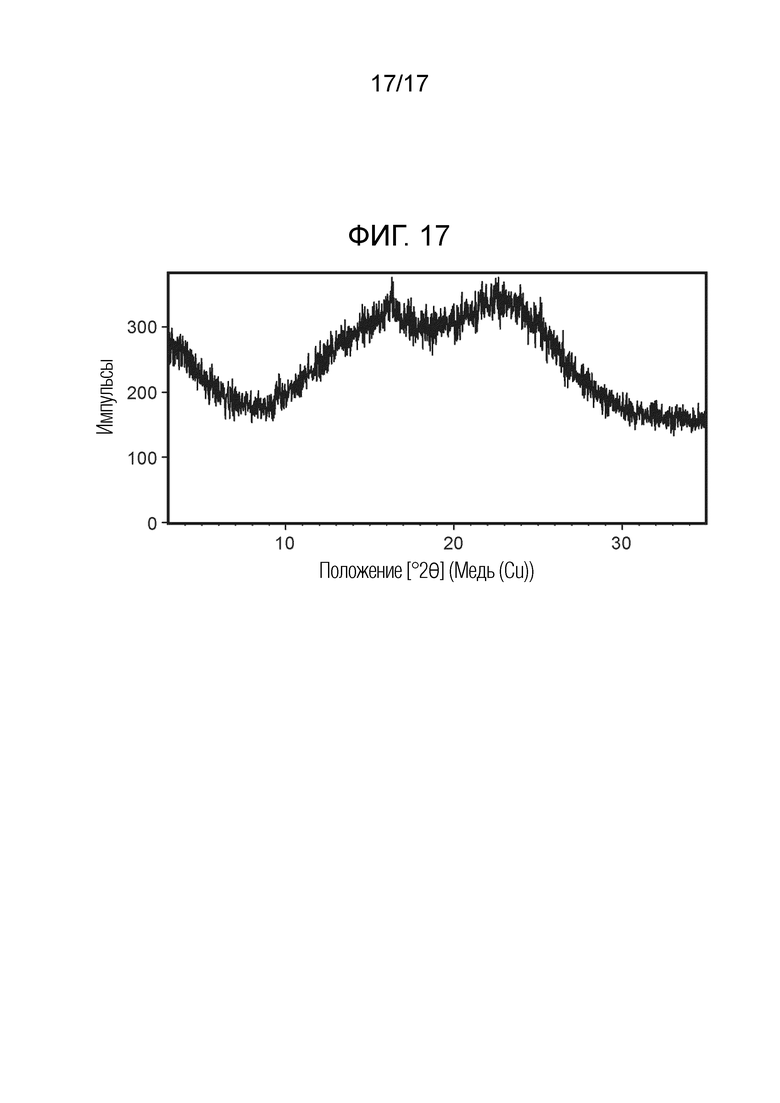
Изобретение относится к области органической химии, а именно к соединению формулы III или к его фармацевтически приемлемой соли, где L является NH или O; X1 является CH или N; R1 является С1-С6 алкилом или C1-C6 дейтероалкилом; R2 является метилом; R3 является F или Cl; R4 является H или F; R5 является H или F; и R6 является C1-C6 алкилом или C1-C6 фторалкилом. Также изобретение относится к безводным формам А и В N-(2-хлор-3-((3,5-диметил-4-оксо-3,4-дигидрохиназолин-6-ил)амино)-4-фторфенил)-3-фторпропан-1-сульфонамида и фармацевтической композиции на основе указанных соединений. Технический результат: эффективное лечение BRAF-ассоциированной опухоли. 5 н. и 1 з.п. ф-лы, 17 ил., 31 табл., 90 пр.
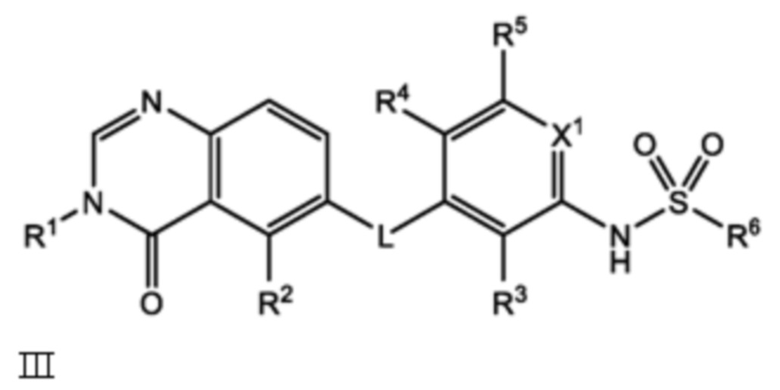
1. Соединение формулы III
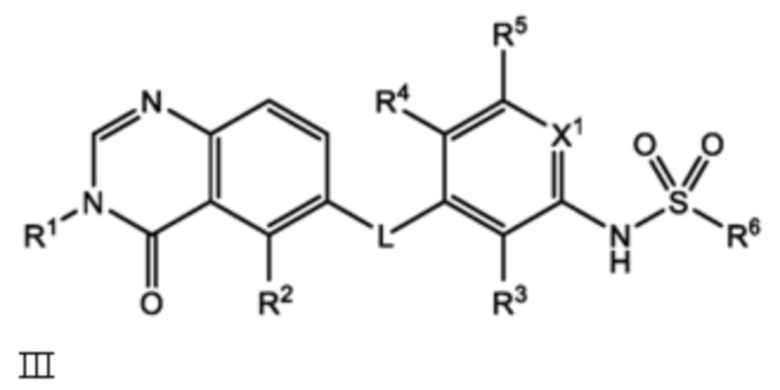
или его фармацевтически приемлемая соль, где:
L является NH или O;
X1 является CH или N;
R1 является С1-С6 алкилом или C1-C6 дейтероалкилом;
R2 является метилом;
R3 является F или Cl;
R4 является H или F;
R5 является H или F; и
R6 является C1-C6 алкилом или C1-C6 фторалкилом.
2. Соединение по п. 1, выбранное из:
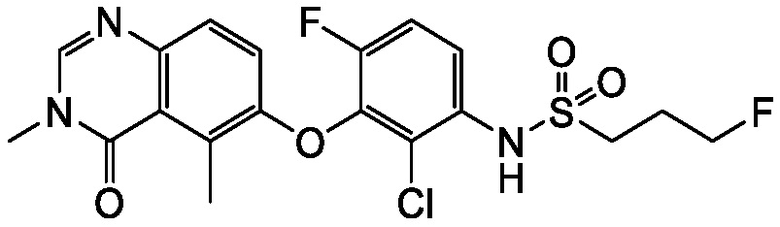 ,
,
или его фармацевтически приемлемая соль.
3. Соединение, которым является
 ,
,
или его фармацевтически приемлемая соль.
4. Соединение, которым является
 , безводная форма В, имеющая ПРД, содержащую пики при значениях 2θ 17,2, 22,9, 25,5, 26,2 и 27,7 (±0,2° 2θ).
, безводная форма В, имеющая ПРД, содержащую пики при значениях 2θ 17,2, 22,9, 25,5, 26,2 и 27,7 (±0,2° 2θ).
5. Соединение, которым является
, безводная форма А, имеющая ПРД, содержащую пики при значениях 2θ 10,4, 10,8, 12,3, 14,2, 14,5, 16,3, 20,5 и 23,6 (±0,2° 2θ).
6. Фармацевтическая композиция для лечения BRAF-ассоциированной опухоли, содержащая эффективное количество соединения по любому из пп. 1-5 в смеси с фармацевтически приемлемым разбавителем или носителем.
| WO 2012118492 A1, 07.09.2012 | |||
| WENGLOWSKY STEVE ET AL | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, AMSTERDAM, NL, 24(8), 13.03.2014, с.1923-1927 | |||
| WO 2019060611 A1, 28.03.2019 | |||
| Способ получения алюминиевого сплава | 1929 |
|
SU19350A1 |

