Группа изобретений относится к лекарственным средствам, применяемым для лечения профилактики нарушений функций нижних мочевыводящих путей и способ ее применения.
С 2003 г. для клинического определения детрузорной гиперактивности Международным обществом по удержанию мочи (International Continence Society) был предложен термин ГАМП.
ГАМП - синдром, характеризующийся наличием императивных позывов, сопровождающихся или не сопровождающихся ургентным недержанием мочи обычно в сочетании с учащенным мочеиспусканием и ноктурией, при доказанном отсутствии инфекции или других явных патологических изменений.
ГАМП - одно из наиболее частых расстройств мочеиспускания. Так, в Европе приблизительно 17% взрослого населения испытывают симптомы ГАМП.
Кроме императивных позывов, самыми распространенными симптомами ГАМП являются учащенное мочеиспускание (85%), ургентные позывы к мочеиспусканию (54%) и недержание мочи (36%).
Причем как у мужчин, так и у женщин с выраженными симптомами ГАМП отмечается низкий уровень качества жизни и работоспособности, а также тревожность и депрессия. Отмечено, что частота вышеуказанных симптомов увеличивается с возрастом.
К симптоматике ГАМП может приводить гиперактивность детрузора нейрогенного или идиопатического (ненейрогенного) характера.
Нейрогенная детрузорная гиперактивность связана с неврологическими заболеваниями. Нейрогенные нарушения происходят на уровне супраспинальных центров нервной системы и/или проводящих путей спинного мозга.
Нейрогенный ГАМП может встречаться при следующих заболеваниях: травмах и опухолях головного и спинного мозга, воспалительно-дегенеративных заболеваниях позвоночника, рассеянном склерозе, нарушениях мозгового кровообращения, болезни Паркинсона, болезни Альцгеймера и сахарном диабете.
При идиопатической детрузорной гиперактивности причина непроизвольных сокращений детрузора неизвестна.
Несмотря на разнообразие этих заболеваний, все они приводят к принципиально одинаковым, хотя и имеющим специфические оттенки, нарушениям мочеиспускания.
При выявлении ГАМП необходимо проведение терапии для улучшения качества жизни больного путем купирования ургентного и нормализации учащенного мочеиспускания.
Выделяются три основных направления терапии ГАМП: фармакотерапия, поведенческая терапия и биологическая обратная связь, физиотерапия.
Согласно рекомендациям Международного общества по вопросам недержания мочи (ICS - International Continence Society) при лечении больных ГАМП приоритетной является фармакотерапия.
Из уровня техники известно применение биорегуляторных пептидов в клинической практике для направленной коррекции функциональной активности клеток.
Функцию медиаторных межклеточных сигналов на пара- и аутокринном уровнях могут выполнять как секреторные белки - цитокины, так и пептидные регуляторы, выделенные из тканей животных.
Пептидные регуляторы регулируют внутриклеточную экспрессию сигнальных молекул - белков и пептидов, играющих важную роль в паракринной регуляции функции органа.
Данные пептидные регуляторы содержатся в лекарственных препаратах и помогают поддерживать структурный и функциональный гомеостаз клеточных популяций.
При этом механизм биологической активности регуляторных пептидов на молекулярном уровне заключается в следующем.
Полипептиды, взаимодействуя с клеточными рецепторами, запускают каскад внутриклеточных реакций, приводящий к изменению экспрессии белков-сигнальных молекул.
Олигопептиды проникают в ядро клетки через цитоплазму и мембрану ядра. Комплементарное взаимодействие этих пептидов с промоторными зонами генов служит сигналом для транскрипции, трансляции и синтеза белков на рибосомах.
Указанные процессы способствуют изменению функции различных органов и тканей, тем самым обеспечивая необходимый терапевтический эффект.
Применение пептидных биорегуляторов, полученных из тканей животных имеет давнюю историю. Но только в начале двадцатого века были получены научные данные, подтверждающие их биологическую активность при применении пациентами.
Из уровня техники известна фармацевтическая композиция, содержащая выбор аминокислот, содержащихся в наибольшем количестве в исследуемой ткани (см. RU 2161501 29.06.2000 далее - [1])
Данное средство используется для лечения широкого спектра заболеваний, в частности эпифиза и коры головного мозга.
Технический результат решений по патентному документу [1] заключается в выявлении наиболее характерной для конкретного вида ткани животных или человека композиции аминокислот и создании фармацевтической композиции, обладающей строгой специфичностью действия.
Однако, данная известная фармацевтическая композиция содержит искусственные (синтетические) пептиды, обладающие низкими подтвержденными эффективностью и безопасностью.
Из уровня техники известна фармацевтическая композиция, нормализующая тонус мочевого пузыря, содержащая пептиды, полученные из мочевого пузыря телят (см. RU 2302867 22.06.2006 далее - [2]).
Технический результат решений по патентному документу [2] заключается в создании композиции и оптимальной технологии получения пептидов для нее.
Недостатками известного из [2] средства является то, что оно содержит пептиды с молекулярной массой от 74 до 394 Да. При этом на настоящий момент имеются научно доказанные данные о том, что наибольшей активностью при лечении нарушений функций нижних мочевыводящих путей обладают пептиды, полученные из мочевого пузыря крупного рогатого скота животных, имеющих большую молекулярную массу.
Кроме того, известное средство обладает высокой токсичностью за счет использования в нем полипептидов низкомолекулярной фракции в количестве от 70 до 90% и большим количеством нежелательных реакций после его применения.
Технический результат заявленной группы изобретений заключается в:
- снижении гиперрефлекторности при обструкционной гипертрофии мочевого пузыря;
- восстановлении эвакуаторной функции гипертрофированного мочевого пузыря;
- улучшении показателей внутрипузырного давления и эластичности стенок мочевого пузыря;
- нормализации показателей функциональной активности мочевого пузыря (массы, объема мочевого пузыря и их соотношения);
- повышении сократительной активности миоцитов детрузора нормального мочевого пузыря;
- коррекции нарушений мочеиспускания при гиперактивном мочевом пузыре, в том числе на фоне хронического простатита, доброкачественной гиперплазии предстательной железы, климактерического синдрома и других состояний;
- снижении вероятности индуцирования иммунного ответа пациента после введения композиции;
- уменьшении частоты и ургентности мочеиспусканий;
- снижении риска развития прионных болезней (трансмиссивных губчатых энцефалопатий);
- повышении степени очистки лекарственного средства.
Поставленный технический результат достигается в фармацевтической композиции для лечения или профилактики нарушений функций нижних мочевыводящих путей, в частности, для лечения учащенного мочеиспускания и/или недержания мочи, содержащей: пептиды, представляющие собой фрагменты из 10-20 последовательных аминокислотных остатков белка Кальпонин-1 последовательности SEQ ID NO: 1, причем данные фрагменты включают аминокислотную последовательность, приведенную в SEQ ID NO: 2, а также
пептиды, представляющие собой фрагменты из 10-20 последовательных аминокислотных остатков белка Гладкомышечный гамма-актин последовательности SEQ ID NO: 3, причем данные фрагменты включают аминокислотную последовательность, приведенную в SEQ ID NO: 4, а также
пептиды, представляющие собой фрагменты из 10-20 последовательных аминокислотных остатков белка Декорин последовательности SEQ ID NO: 5, причем данные фрагменты включают аминокислотную последовательность, приведенную в SEQ ID NO: 6.
Технический результат заявленной группы изобретений также заключается в том, что композиция вводится пациенту один или более раз вместе с носителем.
Технический результат заявленной группы изобретений также заключается в том, что композиция может вводится пациенту парентерально или внутримышечно.
Технический результат заявленной группы изобретений также заключается в применении фармацевтической композиции в фармацевтически эффективном количестве для восстановлении эвакуаторной функции мочевого пузыря, улучшения показателей внутрипузырного давления и эластичности стенок мочевого пузыря, нормализации показателей функциональной активности мочевого пузыря (массы, объема мочевого пузыря и их соотношения) при обструкционной гипертрофии мочевого пузыря).
Технический результат заявленной группы изобретений также заключается в применении фармацевтической композиции в фармацевтически эффективном количестве для повышения сократительной активности миоцитов детрузора при гипоактивном мочевом пузыре.
Технический результат заявленной группы изобретений также заключается в применении фармацевтической композиции в фармацевтически эффективном количестве для лечения учащенного мочеиспускания и/или недержания мочи.
Технический результат заявленной группы изобретений также заключается в применении фармацевтической композиции в фармацевтически эффективном количестве у пациентов с гиперактивным мочевым пузырем.
Аминокислотные последовательности белков приведены в Таблице 1.
Графические материалы.
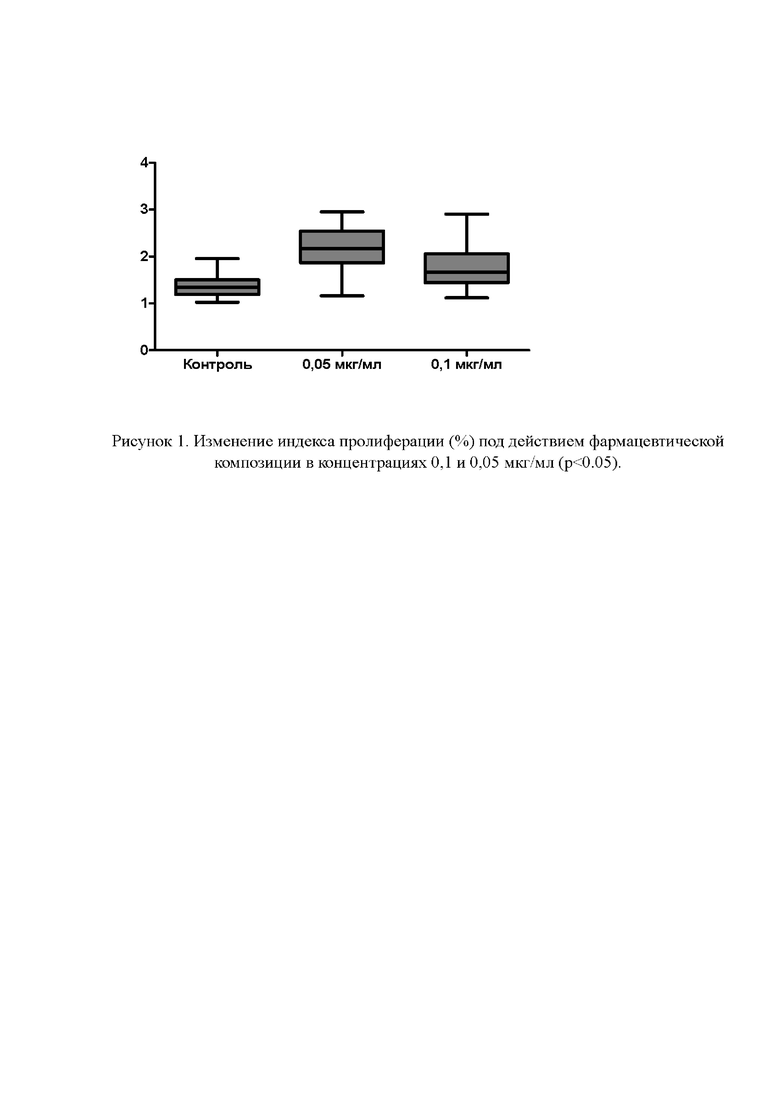
Рисунок 1. Изменение индекса пролиферации (%) под действием фармацевтической композиции в концентрациях 0,1 и 0,05 мкг/мл (р<0.05).
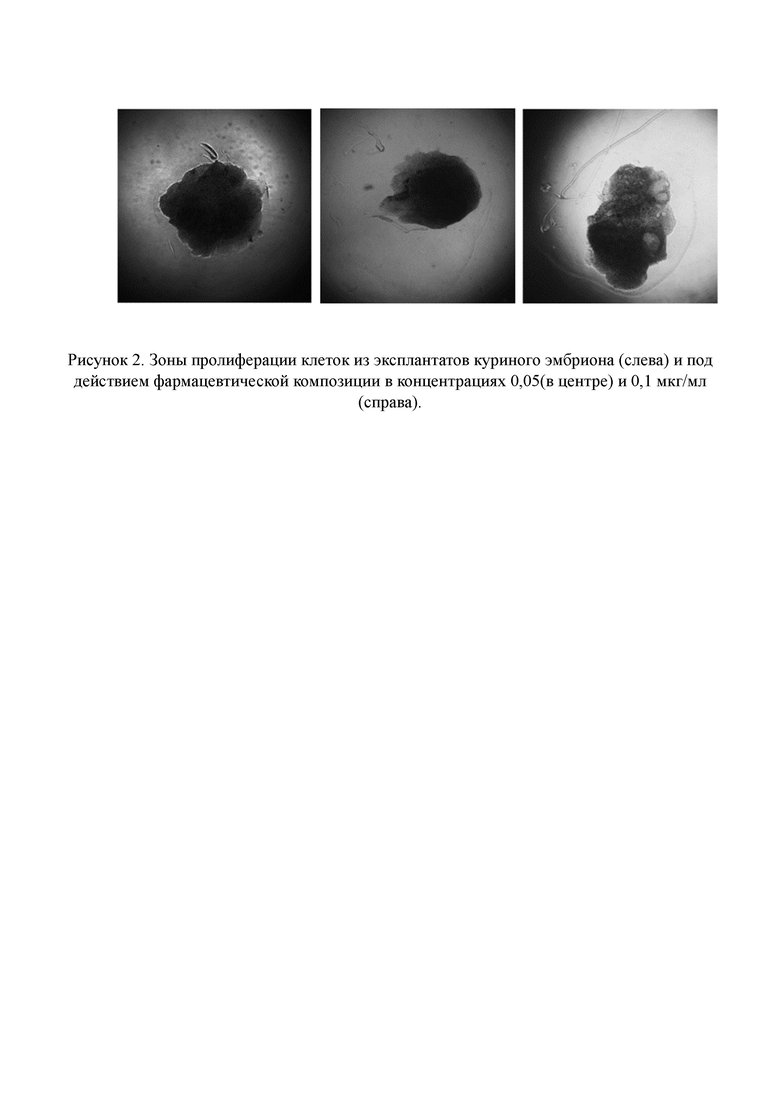
Рисунок 2. Зоны пролиферации клеток из эксплантатов куриного эмбриона (слева) и под действием фармацевтической композиции в концентрациях 0,05 (в центре) и 0,1 мкг/мл (справа).
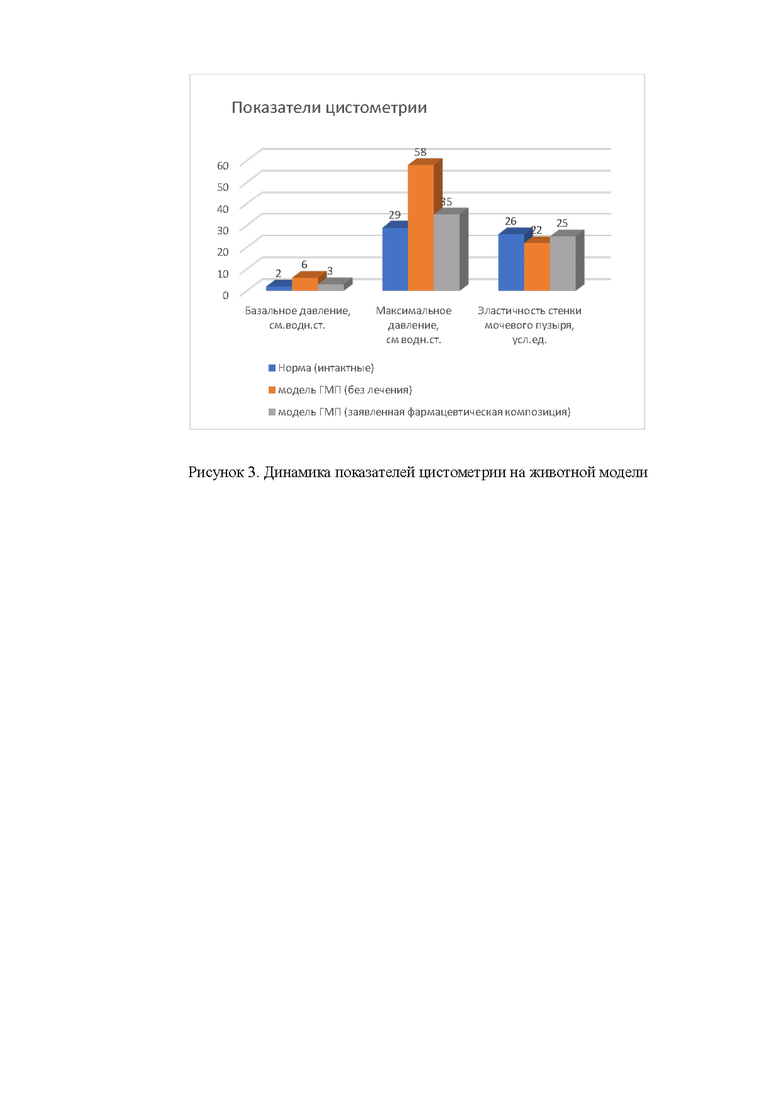
Рисунок 3. Динамика показателей цистометрии на животной модели.
Рисунок 4. Динамика показателей функциональной активности мочевого пузыря на животной модели.
Таблица 1. Аминокислотные последовательности белков/фрагментов белков.
Таблица 2. Результаты оценки фильтратов.
Таблица 3. Аминокислотные последовательности фрагментов белка SEQ ID NO: 1
Таблица 4. Аминокислотные последовательности фрагментов белка SEQ ID NO: 3
Таблица 5. Аминокислотные последовательности фрагментов белка SEQ ID NO: 5
Сущность изобретения
Оценка биологической активности заявленной фармацевтической композиции.
Оценка биологической активности заявленной фармацевтической композиции проводилась на тканях куриных эмбрионов в концентрациях 0,5 и 0,1 мкг/мл.
Биологическую активность оценивали по влиянию препарата на развитие выселяющихся пролиферирующих и мигрирующих клеток в зоне роста эксплантата в органотипической культуре мышечной ткани куриного эмбриона.
Исследование проводили с помощью инвертированного светового микроскопа.
Для количественной оценки влияния препарата на рост эксплантатов ткани применяли морфометрический метод.
Использовали приборы и оборудование: ламинарный бокс с вертикальным воздушным потоком NUAIRE (США); шейкер лабораторный орбитальный термостатируемый BIOSAN PST-HL60 (Латвия); CO2-инкубатор NUAIRE (США); микроскоп инвертированный Leica DM IL LED (Германия), фотоаппарат Canon 5D MkII; программное обеспечение ImageJ v1.53.
Применяли материалы и реактивы: Хенкса раствор без фенолового красного 450 мл (ПанЭко, кат. №Р020п, РУ № ФСР 2012/12980); питательную среду MEM (Minimum Essention Medium Eagle, Corning, кат. №10-010-CV); фетальную бычью сыворотку (HyClone, кат. № SV30160.03); раствор глюкозы 5%, стерильный (Мосфарм); гентамицин (Lonza, кат. №17-518Z); вода деонизованная, стерильная, 450 мл (ПанЭко, Кат. №: Р009п); поли-L-лизина гидробромид 25 мг («ПанЭко», кат. № Ф086); чашки Петри 35×10 mm (SPL Lifesciences, кат. №26035 на ПанЭко); планшет 96-луночный (Thermo Fisher Scientific, Nunc 167008); инструменты для глазной хирургии (ПТО «Медтехника»); куриные эмбрионы со сроком инкубации 16-17 суток.
Использовали 96-луночный планшет покрытый поли-L-лизином.
На дно лунки 96-луночного планшета (150 мкл/лунка) наносили раствор поли-L-лизина гидробромида (рабочая концентрация 25 мкг/мл) для полного покрытия рабочей поверхности и инкубировали 10 минут при комнатной температуре.
После чего раствор полимера отбирали и дважды промывали дистиллированной водой, оставляли на 1 ч до полного высыхания с открытой крышкой в стерильных условиях ламинарного бокса.
Интенсивность роста ткани оценивали по величине индекса пролиферации, который рассчитывался в %, как отношение площади всего эксплантата с зоной роста клеток к исходной площади эксплантата:
Δ%=(Sэкспл+зона/ Sэкспл)×100%
Для сравнения эффективности препаратов определяли относительную пролиферацию, полученную при делении ИП препарата на ИП контроля и выраженный в процентах.
Для приготовления раствора фармацевтической композиции использовали ее сухой лиофилизат - 5 мг и 5 мл стерильной апирогенной воды.
Воду и лиофилизат тщательно перемешивали, далее готовили разведения в конечных концентрациях - 0,1 и 0,05 мкг/мл в полной питательной среде.
Все эксперименты были выполнены в 2 повторах на не менее чем 15 эксплантатах в каждом повторе.
Культивирование эксплантатов в течение 72 ч группе вызывает прирост Δ%=136.3% от площади эксплантатов.
Фармацевтическая композиция в исследованных концентрациях 0,1 и 0,05 мкг/мл обладает статистически значимым стимулирующим действием на выход клеток из эксплантатов и вызывает формирование зоны роста по отношению к контролю (р<0.05).
Наибольшая зона роста формировалась под действием фармацевтической композиции в концентрации 0,05 мкг/мл: дозозависимый прирост по отношению к контролю составил 56,8%, и 29,0% для концентрации 0,1 мкг/мл (см. Рисунок 1, 2).
Таким образом, индекс площади эксплантатов органотипических тканей, полученных от КЭ и инкубируемых в среде с добавлением раствора фармацевтической композиции значимо больше индекса площади эксплантатов в контроле (среда без добавления раствора исследуемой фармацевтической композиции). Что свидетельствует о высокой биологической активности фармацевтической композиции.
Оценка биологической активности заявленной фармацевтической композиции.
Проводились исследования сократительной активности клеток изолированной мышцы детрузора in vitro («в пробирке») на нормальном и гипертрофированном мочевом пузыре, а также исследования на модели гиперактивного мочевого пузыря (ГМП)* у лабораторных животных (крыс) с использованием цистометрии.
Изучение влияния на гладкомышечные клетки детрузора мочевого пузыря in vitro.
Исследовалось влияние фармацевтической композиции на сократительную активность клеток детрузора в экспериментальной модели in vitro (см. рис. 3).
Исследование проводили на изолированной мышце сжимателя мочевого пузыря (детрузора) с измерением степени ее сокращения на нормальном и гипертрофированном мочевом пузыре. Горизонтальные полоски детрузора помещали между фиксированным стержнем и датчиком силы в специальную ванночку. В раствор Кребса, в который помещали полоски мышцы, добавляли фармацевтическую композицию в концентрациях 8, 16 и 32 мкг/мл, затем регистрировали сократительную активность мышечных волокон детрузора с помощью преобразователя давления, присоединенного к полиграфу.
Установлено, что фармацевтическая композиция оказывает дозозависимое регулирующее действие на функцию детрузора. На препаратах нормального мочевого пузыря сократительная активность мышц повышается на 82,1-167,3% (при разных дозировках), а на препаратах патологического (гипертрофированного) мочевого пузыря исходно повышенная сократительная активность миоцитов детрузора подавляется под действием фармацевтической композиции, соответственно, на 12,7%, на 33,4% и на 43,8% при дозировках 8, 16 и 32 мкг/мл.
В дальнейших экспериментах концентрация 32 мкг/мл была принята за терапевтически эффективную дозу - 5 мг в пересчете на человеческую дозировку.
Результаты исследования свидетельствуют о стимулирующем действии фармацевтической композиции на тонус гладких мышц нормального мочевого пузыря и ингибирующем действии на тонус мышц гипертрофированного мочевого пузыря в условиях in vitro.
Исследование на модели гиперактивности мочевого пузыря у лабораторных животных с использованием цистометрии.
Для формирования ГМП у животных (крыс) использовали модель гипертрофии мочевого пузыря* (см. рис. 4). Гипертрофия мочевого пузыря вызывается созданием препятствия в мочеиспускательном канале (модель обструкционной гипертрофии МП, которая провоцирует у животных наступление дисфункции детрузора).
*Для моделирования детрузорной гиперактивности (ГМП) в доклинических исследованиях применяется модель обструкционной гипертрофии МП: создается частичная непроходимость уретры. Для этого мочеиспускательный канал перевязывают лавсановой лигатурой в присутствии находящегося в просвете катетера диаметром 1 мм (операция под наркозом). Лигатуру накладывают суммарно на 6 недель.
Начиная с 4-й недели после наложения лигатуры проводился курс терапии фармацевтической композиции по 1 введению в/м 1 раз в сут 10 дней, затем наступал период ожидания (10 дней после окончания курса).
После периода ожидания (через 6 недель от начала формирования гипертрофии мочевого пузыря) лигатуру снимали и проводили цистометрию. Измеряли максимальный объем мочевого пузыря, базальное давление, внутрипузырное давление при максимальном наполнении, эластичность стенки мочевого пузыря (рассчитывается как отношение объема наполнения к внутрипузырному давлению).
Результаты цистометрии показали, что при формировании гипертрофии мочевого пузыря у крыс нарастает дисфункция детрузора: значительно увеличивается базальное и максимальное внутрипузырное давление при достоверном снижении показателя эластичности стенки мочевого пузыря. Введение Везустена приводило к достоверному улучшению показателей: снижению внутрипузырного давления и повышению эластичности стенки мочевого пузыря по сравнению с показателями контрольной группы; в результате показатели цистометрии животных после терапии фармацевтической композиции практически не отличались от показателей здоровых животных, см рисунок «Динамика показателей цистометрии на животной модели».
Далее оценивали показатели функциональной активности мочевого пузыря: массу, объем мочевого пузыря и их соотношение, остаточную мочу, отношение остаточной мочи к объему мочевого пузыря.
При формировании обструкции значительно (почти в 6 раз) увеличилась масса и почти в 7 раз - объем мочевого пузыря; объем остаточной мочи составил 64,6% от объема мочевого пузыря (рис. 2).
В результате терапии препаратом фармацевтической композиции показатели функциональной активности* мочевого пузыря приблизились к показателям здоровых животных:
масса и объем мочевого пузыря снизились в 3,3-4,5 раза по сравнению с показателями контрольной группы (группы без лечения), рис. 2;
объем остаточной мочи** не превышал 11% от объема мочевого пузыря (по сравнению 0% у здоровых животных и 64,6% в контрольной группе).
*масса, объем мочевого пузыря и их соотношение
**объем остаточной мочи (ООМ) на этой модели коррелирует с выраженностью дисфункции детрузора, повышение ООМ свидетельствует о нарушении эвакуаторной функции МП.
Сущность изобретения.
Исследование безопасности и переносимости возрастающих доз заявленной композиции при однократном и последующем многократном внутримышечном введении у здоровых добровольцев.
Исследование проводилось как двойное-слепое, рандомизированное, плацебо-контролируемое исследование безопасности и переносимости возрастающих доз заявленной композиции при однократном и последующем многократном внутримышечном введении у здоровых добровольцев.
Всего в исследовании было скринировано 45 добровольцев. В исследование были рандомизированы 32 добровольца. Из них в когорты однократного дозирования были рандомизированы по 6 добровольцев в группы заявленной фармацевтической композиции 5 мг однократно и фармацевтической композиции 10 мг однократно. В группу плацебо однократно были рандомизированы 4 добровольца.
В когорты многократного дозирования были рандомизированы по 6 добровольцев в группы в группу заявленной фармацевтической композиции 5 мг многократно» и заявленной фармацевтической композиции 10 мг многократно и 4 добровольца в группу плацебо многократно.
Все рандомизированные добровольцы вошли в популяцию для анализа безопасности.
Все рандомизированные в когорты однократного и многократного дозирования добровольцы завершили исследование в соответствии с протоколом.
В ходе данного исследования заявленная фармацевтическая композиция продемонстрировала благоприятный профиль безопасности, общую переносимость и хорошую местную переносимость. Серьезных нежелательных явлений (далее - СНЯ) и случаев смерти в ходе исследования зарегистрировано не было.
Всего на фоне введения исследуемого препарата у 11/32 (34.4%) добровольцев было зарегистрировано 26 нежелательных явления (далее НЯ) в системно-органных классах «Общие расстройства и нарушения в месте введения» (General disorders and administration site conditions) и «Лабораторные и инструментальные исследования» (Investigations).
НЯ со степенью тяжести III и IV, отнесенных к категории связанных с введением исследуемого препарата, также как и не связанных с введением исследуемого препарата, в исследовании зарегистрировано не было.
Все зарегистрированные НЯ соответствовали I или II степени тяжести по СТСАЕ (версия 4.03).
НЯ, связанных с введением исследуемого препарата и приведших к досрочному завершению исследования, в исследовании зарегистрировано не было.
Таким образом, в ходе исследования заявленная фармацевтическая композиция продемонстрировала благоприятный профиль безопасности, общую переносимость и хорошую переносимость в выбранном диапазоне доз и режимах введения.
Оценка биологической активности заявленной фармацевтической композиции.
Для оценки биологической активности заявленной фармацевтической композиции в форме раствора для внутримышечного введения 5 мг было проведено многоцентровое, двойное слепое, плацебо-контролируемое рандомизированное исследование в параллельных группах у пациентов с гиперактивным мочевым пузырем (ГАМП).
Пациенты, успешно прошедшие скрининг и удовлетворяющие критериям включения/невключения, были рандомизированы в 2 группы в соотношении 1:1.
Группа А (n=75): пациенты получали исследуемую фармацевтическую композицию, внутримышечно, в дозе 5 мг, 3 раза в неделю, в период лечения было введено 10 доз препарата.
Группа В (n=75): пациенты получали плацебо внутримышечно 3 раза в неделю, в период лечения введено 10 доз плацебо.
Дни введения в группах А и В: 1, 3, 5, 8, 10, 12, 15, 17, 19, 22.
Допускалось отклонение в ±1 день при посещении клинического центра для введения препарата/плацебо.
Если пациент пропускал визит для введения исследуемого препарата/плацебо, то эта доза считалась пропущенной и ее принимать не следовало.
Пациент исключался из исследования в случае пропуска суммарно между визитами более 3 доз препарата на протяжении всего периода лечения.
В рамках настоящего исследования для пациентов была сохранена возможность использования при необходимости базовой терапии ГАМП.
В ходе проведения исследования было скринировано 152 пациента, из них 150 рандомизированы, на этапе скрининга выбыло 2 пациента в связи с несоответствием критериям включения/невключения в исследование, преждевременно (после рандомизации) выбывших пациентов не было.
В популяцию ITT и РР были включены все 150 рандомизированных пациентов.
Пациенты мужского и женского пола в возрасте от 18 до 70 лет, соответствующие критериям включения/невключения в исследование.
У всех участвующих в исследовании пациентов был верифицированный диагноз ГАМП (учащенное мочеиспускание, ургентные позывы с или без потери мочи) в течение 3 месяцев и более.
Максимальная продолжительность участия пациента в исследовании составила не более 91 дня.
В исследовании было выявлено, что среднее изменение (уменьшение) количества баллов (за сутки) на ВФОТ по сравнению с Визитом Скрининга (Исходно) в группе заявленной фармацевтической композиции составило 10,018±9,195 баллов и в группе Плацебо 5,089±6,427, при этом выявлены статистически значимые различия между группами по данному показателю (р=0,0007), нижняя граница 95% доверительного интервала для разности средних групп А и В составила 2,369.
На основании вышеуказанных данных был сделан вывод о превосходящей эффективности заявленной фармацевтической композиции по сравнению с плацебо в отношении снижения тяжести симптомов ГАМП по шкале TUFS, граница превосходящей эффективности 2,36 балла (р=0.022).
Доля пациентов, у которых на ВФОТ наблюдалось ≥50% уменьшение количества эпизодов недержания мочи (3+4 степень выраженность позывов по PPIUS) за 24 часа по сравнению с исходным уровнем.
В качестве второй первичной переменной эффективности в исследовании проводили оценку по числу пациентов, у которых наблюдался терапевтический ответ (далее - ТО), который определяли, как уменьшение количества эпизодов недержания мочи (3+4 степень выраженность позывов по PPIUS) на 50% и более за 24 часа на ВФОТ по сравнению с исходным уровнем.
При проведении анализа были выявлены статистически значимые различия между группами по доле пациентов с ТО на ВФОТ. Рассчитана нижняя граница 95% доверительного интервала (далее - ДИ) для разности долей пациентов, у которых был достигнут ТО, при этом выявлена превосходящая эффективность заявленной фармацевтической композиции по сравнению с плацебо в отношении доли пациентов с терапевтическим ответом, который определялся как уменьшение количества эпизодов недержания мочи (3+4 степень выраженность позывов по PPIUS) за 24 часа ВФОТ на 50% и более по сравнению с исходным уровнем.
Нижняя граница доверительного интервала для разности долей составила 0,0850, установлено, что граница превосходящей эффективности составила 8% (р=0,0244), при проведении точечной оценки разность долей составила 0,2400 или 24%.
Таким образом, сделан вывод о превосходящей эффективности заявленной фармацевтической композиции по сравнению с плацебо в отношении уменьшения количества эпизодов недержания мочи (3+4 степень выраженность позывов по PPIUS) за 24 часа ВФОТ на 50% и более по сравнению с исходным уровнем.
В исследовании также было установлено, что в результате проведенного межгруппового сравнения среднего изменения степени тяжести симптомов ГАМП по шкале TUFS на визите промежуточной оценки терапии (ВПОТ) по сравнению с исходным уровнем, были выявлены статистически значимые различия между группами пациентов, получавших заявленной фармацевтической композиции и плацебо (р=0,0092), проявляющиеся в снижении степени тяжести симптомов ГАМП на фоне введения заявленной фармацевтической композиции.
В исследовании также было установлено, что в результате проведенного межгруппового сравнения среднего изменения количества эпизодов недержания мочи (3+4 степень выраженности позывов по PPIUS) на визите финальной оценки терапии (ВФОТ) по сравнению с исходным уровнем, были выявлены статистически значимые различия между группами пациентов, получавших заявленную фармацевтическую композицию и плацебо (р=0,001), проявляющиеся в снижении количества эпизодов недержания мочи на фоне введения заявленной фармацевтической композиции.
В исследовании также было установлено, что в результате проведенного межгруппового сравнения среднего изменения количества эпизодов недержания мочи (3+4 степень выраженности позывов по PPIUS) на визите промежуточной оценки терапии (ВПОТ) по сравнению с исходным уровнем, были выявлены статистически значимые различия между группами пациентов, получавших заявленную фармацевтическую композицию и плацебо (р=0,0108), проявляющиеся в снижении количества эпизодов недержания мочи на фоне введения заявленной фармацевтической композиции.
В исследовании также было установлено, что в результате проведенного межгруппового сравнения среднего изменения количества мочеиспускания на визите финальной оценки терапии (ВФОТ) по сравнению с исходным уровнем, были выявлены статистически значимые различия между группами пациентов, получавших препарат Везустим и плацебо (р=0,0021), проявляющиеся в снижении количества мочеиспусканий на фоне введения заявленной фармацевтической композиции.
В исследовании также было установлено, что в результате проведенного межгруппового сравнения среднего изменения количества мочеиспускания на визите промежуточной оценки терапии (ВПОТ) по сравнению с исходным уровнем, были выявлены статистически значимые различия между группами пациентов, получавших заявленную фармацевтическую композицию и плацебо (р=0,0229), проявляющиеся в снижении количества мочеиспусканий на фоне введения заявленной фармацевтической композиции.
В исследовании также было установлено, что в результате проведенного межгруппового сравнения среднего изменения количества баллов по основному опроснику OAB-q на визите финальной оценки терапии (ВФОТ) по сравнению с исходным уровнем, были выявлены статистически значимые различия между группами пациентов, получавших заявленную фармацевтическую композицию и плацебо (р=0,0002), проявляющиеся в снижении степени беспокойства вследствие симптомов ГАМП.
В исследовании также было установлено, что в результате проведенного межгруппового сравнения среднего изменения количества баллов по дополнительному опроснику OAB-q на визите финальной оценки терапии (ВФОТ) по сравнению с исходным уровнем, были выявлены статистически значимые различия между группами пациентов, получавших заявленную фармацевтическую композицию и плацебо (р=0,0032), проявляющиеся в снижении степени беспокойства вследствие симптомов ГАМП.
Нежелательные явления
За время проведения настоящего клинического исследования всего было зарегистрировано 169 НЯ (76 НЯ в группе пациентов, получавших заявленную фармацевтическую композицию и 93 НЯ в группе пациентов, получавших плацебо).
Всего НЯ были выявлены у 72 пациентов (у 32 пациентов в группе приема заявленной фармацевтической композиции и у 40 пациентов в группе плацебо).
При оценке числа пациентов, у которых были зарегистрированы НЯ, статистически значимых различий между группами выявлено не было (значение р=0,253).
За время исследования случаев развития СНЯ зарегистрировано не было.
Наиболее часто встречающимися НЯ были отклонения лабораторных показателей (клинического и биохимического анализов крови, общего анализа мочи).
По мнению исследователей, эти отклонения не имеют связи или имеют сомнительную связь с применением препаратов.
За время проведения исследования зарегистрировано 13 НЯ в месте инъекции: 6 НЯ в группе заявленной фармацевтической композиции и 7 НЯ в группе плацебо, все НЯ были легкой степени тяжести и не потребовали дополнительных действий от персонала, разрешились самостоятельно.
По результатам проведенного исследования у пациентов с (ГАМП) установлено, что заявленная фармацевтическая композиция при применении по схеме 3 раза в неделю в дозе 5 мг обладает клинической эффективностью и снижает степень тяжести симптомов ГАМП по шкале TUFS, а также оказывает положительное влияние на уменьшение количества эпизодов недержания мочи (3+4 степень выраженности позывов по PPIUS) на 50% и более.
В рамках данного исследования было также продемонстрировано положительное влияние заявленной фармацевтической композиции на уменьшение среднего значения количества баллов по основному и дополнительному опроснику OAB-q, что свидетельствует о том, что наличие симптомов ГАМП стало в меньшей степени беспокоить пациентов, получавших исследуемый препарат.
В рамках данного исследования было доказано значимое снижение общего количества мочеиспусканий у пациентов, получавших исследуемую заявленную фармацевтическую композицию в сравнении с плацебо.
Заявленная фармацевтическая композиция имела благоприятный профиль безопасности, поскольку в исследовании не наблюдалось статистически значимых различий между группами композиции и плацебо в отношении показателей безопасности, оцениваемых в исследовании.
Определение количества и идентификация аминокислотных остатков белка в заявленной фармацевтической композиции.
К настоящему моменту известно большое количество методов определения аминокислотных остатков белка в растворе.
Эти методы, обычно, используются для определения общего содержания белка, и в основе принципа измерения лежит взаимодействие с веществами, содержащими пептидные связи.
Для количественного определения аминокислотных остатков белка в составе фармацевтической композиции использовали методику Waddell W.
Данная методика позволила определить содержание аминокислотных остатков белка в заявленной фармацевтической композиции методом спектрофотометрии в УФ области по разности значений оптической плотности на двух длинах волн (215 и 225 нм).
Кроме того, для количественного определения аминокислотных остатков белка в растворе и лиофилизате предложенной фармацевтической композиции был использован метод спектрофотометрии на длине волны 280 нм (по ароматическим аминокислотам в составе).
Для проведения вышеуказанного анализа также были применены методы Лоури и спектрофлуориметрии.
Для анализа использовались соответствующие наборы, например: Thermo Fischer кат №23225.
В качестве стандарта в вышеуказанных аналитических методах был использован бычий сывороточный альбумин. (Protein assay technical handbook, Thermo Ficher Scientific, 2017 https://assets.thermofisher.com/TFS-Assets/LSG/brochures/protein-assay-technical-handook.pdf далее [3]).
Данные методы являются общеизвестными, широко используемыми методами исследовательского и технологического контроля.
Для идентификации аминокислотных остатков белка и установления их последовательности в заявленной группе изобретений использовался метод ВЭЖХ МС-МС (см. Tandem Mass Spectrometry for Peptide and Protein Sequence Analysis. J.J. Coon, J. E.P. Syka, J. Shabanowitz, D.F. Hunt. BioTechniques, Volume 38, Issue 4. Apr 2005. Pages 507-655 далее [4]).
На сегодняшний день тандемная масс-спектрометрия является общепринятым методом, позволяющим устанавливать покомпонентную аминокислотную последовательность, в том числе и в сложных смесях, содержащих тысячи индивидуальных аминокислотных остатков белка.
Дальнейшее отнесение индивидуальных аминокислотных остатков белка к белку-предшественнику выполняется программными средствами с использованием международных протеомных баз, таких как UniProt и др (см.; Orbitrap Mass Spectrometry. R. Zubarev, A. Makarov. Anal. Chem. 2013, 85, 11, 5288-5296 далее [5]).
Алгоритмы отнесения идентифицированных пептидных последовательностей к белкам-предшественникам учитывает длину белка, количество уникальных пептидов, идентифицированных в пробе для данного белка, вероятность ложного определения последовательности пептида и другие факторы (см. Rune, Matthiesen (2006). Mass Spectrometry Data Analysis in Proteomics || Protein Identification by Tandem Mass Spectrometry and Sequence Database Searching., 10.1385/1597452750(), 87-120. doi:10.1385/1-59745-275-0:87 далее [6]).
Результатом масс-спектрометрического анализа аминокислотной смеси является перечень идентифицированных пептидов и белков-предшественников (для пептидов с длиной аминокислотной последовательности более 6 аминокислот), а также всевозможные аналитические характеристики, включая интенсивность пика каждого идентифицированного пептида, соотношение заряда к массе для молекулярного иона, его заряд и др.
Первый способ получения заявленной фармацевтической композиции.
Общеизвестно, что «прионные болезни» или трансмиссивные губчатые энцефалопатии, относящиеся к группе нейродегенеративных заболеваний людей и животных. Данное заболевание вызывается возбудителями - прионами (белками) с аномальной третичной структурой и молекулярной массой 27-36 кДа.
Поэтому при производстве заявленной фармацевтической композиции, содержащей аминокислотные остатки белка, одной из основных технологических задач являлось исключение попадания в нее любых веществ с молекулярной массой выше 10 кДа, включая прионные белки.
В качестве сырья для получения заявленной фармацевтической композиции использовали мочевой пузырь крупного рогатого скота.
Указанное сырье замораживали и выдерживали в течение 2 недель при температуре (-19)÷(-21)°С.
После чего замороженное сырье размораживали при комнатной температуре в течение приблизительно 20 часов.
Затем замороженные мочевые пузыри крупного рогатого скота измельчали и загружали в среду экстрагирования - 3% уксусную кислоту в воде.
Объемное соотношение сырья и экстрагента составляло 1:2.
Экстрагирование проводили при температуре 11÷13°С.
Экстракцию проводили, в течение 10 минут при постоянном перемешивании.
После получения однородной взвеси в нее добавляли 1% раствор хлористого цинка в объемном соотношении 1:50.
Таким образом, при загрузке сырья в количестве 50 кг, экстракционная смесь содержала по массе: 2% уксусная кислота, 0,02% хлористый цинк.
Смесь перемешивали по 1 ч через каждые 4 ч отстаивания в течение 48 часов.
После этого проводили декантирование экстракционной массы мочевых пузырей крупного рогатого скота на декантере (декантерной центрифуге).
Затем проводили фильтрацию на глубинном фильтре, задерживающем твердые частицы размером свыше 3 микрон.
Полученный фильтрат передавали на стадию ультрафильтрации на полых волокнах с номинальным отсечением по молекулярной массе (рейтингом) 13 кДа.
Полученный пермеат сушили в лиофильной сушке в течение 3-5 суток и получали ~ 1,6 кг субстанции полипептидов.
Далее 1,6 кг субстанции полипептидов мочевых пузырей крупного рогатого скота растворяли в дистиллированной воде при комнатной температуре и постоянном перемешивании.
Фильтровали полученный раствор через фильтр 0,22 мкм.
Полученный раствор подвергали ультрафильтрационной очистке на установке при трансмембранном давлении не более 2,0 кгс/см2 через материал с рейтингом 10 кДа.
Далее разводили раствор до концентрации 5,5 мг/мл полипептидов.
Затем в раствор добавляли глицин до его конечной концентрации 20 мг/мл при рН 5,0÷6,6. После чего раствор подвергали стерилизующей фильтрации под давлением не более 2,0 кгс/см2, разливали во флаконы 5 мг и получали приблизительно 90000 флаконов готовой лекарственной формы «раствор».
Для получения лекарственной формы «лиофилизат» раствор фармацевтической композиции после ее розлива во флаконы подвергают лиофилизации в течение 72 часов при давлении не более 0,05 атм.
Второй способ получения заявленной фармацевтической композиции.
В качестве исходного сырья использовали 100 кг мочевых пузырей крупного рогатого скота.
Вышеуказанное сырье замораживали и выдерживали при температуре (-19)÷(-21)°С.
После чего сырье измельчали и загружали в среду экстрагирования, а именно: 3% уксусную кислоту в воде.
Объемное соотношение сырья и экстрагента составляло 1:2, при температуре экстрагирования - 15÷20°С.
Экстракцию проводили при постоянном перемешивании не менее 10 мин., после получения однородной взвеси в нее добавляли 1% раствор хлористого цинка в объемном соотношении 1:50.
Таким образом экстракционная смесь содержала по массе: 2% уксусная кислота, 0,02% хлористый цинк.
Смесь перемешивали по 1 ч через каждые 4 ч отстаивания в течение 48 ч.
Затем экстракт отделяли от балластных веществ сепарированием на центрифужном сепараторе.
К экстракту добавляли ацетон в объемном соотношении 1:4, выдерживали при температуре не выше 5°С не менее 4 ч.
После формирования осадка декантировали надосадочную жидкость.
Полученный осадок, содержащий активное вещество, промывали на нутч-фильтре двукратными объемами охлажденного до температуры 7÷16°С ацетона до получения осадка светло-серого цвета.
Удаляли ацетон, например сушкой при атмосферном давлении и комнатной температуре в вытяжном шкафу при периодическом перемешивании.
Полученные ~3 кг сухой субстанции имели потерю в массе при высушивании не более 10,0%.
Затем 3 кг сухой субстанции полипептидов мочевых пузырей крупного рогатого скота, полученной изложенным выше способом, растворяли в дистиллированной воде при комнатной температуре и постоянном перемешивании.
Отделяли осадок нерастворимых веществ центрифугированием, и надосадочную жидкость фильтровали через фильтр 0,22 мкм.
Полученный раствор подвергали ультрафильтрационной очистке на установке при трансмембранном давлении не более 2,0 кгс/см2 через материалы с рейтингом 10 кДа.
Разводили раствор до концентрации 5,5 мг/мл полипептидов.
В раствор добавляли глицин до его конечной концентрации 20 мг/мл при рН 5,0÷6,6, раствор подвергали стерилизующей фильтрации под давлением не более 2,0 кгс/см2, разливали в объеме, соответствующем дозировке 5 мг на флакон.
В результате получали около 90000 флаконов готовой фармацевтической композиции в форме «раствор».
В другом случае раствор после ультрафильтрационной очистки, полученный изложенным выше способом, разводили до концентрации 11 мг/мл полипептидов.
В раствор добавляли глицин до его конечной концентрации 40 мг/мл при рН 5,0÷6,6, раствор подвергали стерилизующей фильтрации под давлением не более 2,0 кгс/см2.
Затем раствор разливали во флаконы в дозировке 10 мг.
Таким образом было получено около 45000 флаконов готовой фармацевтической композиции в форме «раствор».
Для получения лекарственной формы «лиофилизат» раствор фармацевтической композиции после розлива во флаконы подвергают лиофилизации в течение 72 часов при давлении не более 0,05 атм.
По итогам проведенных экспериментов в качестве оптимальной технологии для приготовления фармацевтической композиции в инъекционной форме была выбрана мембрана с рейтингом 10 кДа.
При этом мембрана с рейтингом 13 кДа может быть успешно использована при производстве других форм выпуска препарата, в которых фактор иммуногенности не является существенным, например, свечей или пероральных форм. Результаты оценки фильтратов приведены в Таблице 2.
Таблица 1. Аминокислотные последовательности белков/фрагментов белков.
последовательности
HNHHPHNYYNSA
QMWISKPEYDEAGPSIVHRKCF
GNYK
Таблица. 2 Результаты оценки фильтратов
примесей
Таблица 3. Аминокислотные последовательности фрагментов белка SEQ ID NO: 1
Таблица 4. Аминокислотные последовательности фрагментов белка SEQ ID NO: 3
Таблица 5. Таблица 5. Аминокислотные последовательности фрагментов белка SEQ ID NO: 5
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ И/ИЛИ ПРОФИЛАКТИКИ СИМПТОМОВ НИЖНИХ МОЧЕВЫХ ПУТЕЙ | 2020 |
|
RU2808426C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ОСНОВЕ ПЕПТИДА, НОРМАЛИЗУЮЩЕГО МОЧЕИСПУСКАНИЕ, И СПОСОБ ЕЕ ПРИМЕНЕНИЯ | 2007 |
|
RU2367467C2 |
| СПОСОБ ЛЕЧЕНИЯ ГИПЕРАКТИВНОГО МОЧЕВОГО ПУЗЫРЯ | 2017 |
|
RU2644304C1 |
| ЛЕЧЕНИЕ ИНТЕРСТИЦИАЛЬНОГО ЦИСТИТА | 2008 |
|
RU2570559C2 |
| Способ лечения синдрома гиперактивного мочевого пузыря | 2022 |
|
RU2792534C1 |
| ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЯ | 2006 |
|
RU2435610C2 |
| СПОСОБ КОМБИНИРОВАННОГО НЕИНВАЗИВНОГО ЛЕЧЕНИЯ ГИПЕРАКТИВНОСТИ ДЕТРУЗОРА У ПАЦИЕНТОВ ПОСЛЕ ТРАНСУРЕТРАЛЬНОЙ РЕЗЕКЦИИ ПРЕДСТАТЕЛЬНОЙ ЖЕЛЕЗЫ | 2023 |
|
RU2804515C1 |
| СПОСОБ ПРОГНОЗИРОВАНИЯ ФОРМИРОВАНИЯ ОСЛОЖНЕНИЙ ПРИ ГИПЕРАКТИВНОМ МОЧЕВОМ ПУЗЫРЕ У ДЕТЕЙ | 2012 |
|
RU2497455C1 |
| СПОСОБ ЛЕЧЕНИЯ ДЕТЕЙ С НЕЙРОГЕННОЙ ДИСФУНКЦИЕЙ МОЧЕВОГО ПУЗЫРЯ ПРИ СНИЖЕНИИ АКТИВНОСТИ ПОЗЫВА К МОЧЕИСПУСКАНИЮ МЕТОДОМ БИОЛОГИЧЕСКИ ОБРАТНОЙ СВЯЗИ | 2010 |
|
RU2452531C1 |
| УСОВЕРШЕНСТВОВАННАЯ СХЕМА ИНЪЕКЦИИ В МОЧЕВОЙ ПУЗЫРЬ ДЛЯ ВВЕДЕНИЯ БОТУЛОТОКСИНОВ | 2017 |
|
RU2741966C2 |

Группа изобретений относится к фармацевтической композиции для лечения гиперактивного мочевого пузыря у пациентов, содержащей в эффективном количестве: пептиды, состоящие из фрагмента из 10-20 последовательных аминокислотных остатков белка Кальпонин-1 последовательности SEQ ID NO: 1, причем данный фрагмент включает аминокислотную последовательность, приведенную в SEQ ID NO: 2, а также пептиды, состоящие из фрагмента из 10-20 последовательных аминокислотных остатков белка Гладкомышечный гамма-актин последовательности SEQ ID NO: 3, причем данный фрагмент включает аминокислотную последовательность, приведенную в SEQ ID NO: 4, а также пептиды, состоящие из фрагмента из 10-20 последовательных аминокислотных остатков белка Декорин последовательности SEQ ID NO: 5, причем данный фрагмент включает аминокислотную последовательность, приведенную в SEQ ID NO: 6, а также относится к применению фармацевтической композиции в эффективном количестве для лечения учащенного мочеиспускания и/или недержания мочи у пациентов с гиперактивным мочевым пузырем, и также относится к применению фармацевтической композиции в эффективном количестве для лечения гиперактивного мочевого пузыря у пациентов. 3 н. и 3 з.п. ф-лы, 4 ил., 5 табл.
1. Фармацевтическая композиция для лечения гиперактивного мочевого пузыря у пациентов, содержащая в эффективном количестве:
пептиды, состоящие из фрагмента из 10-20 последовательных аминокислотных остатков белка Кальпонин-1 последовательности SEQ ID NO: 1, причем данный фрагмент включает аминокислотную последовательность, приведенную в SEQ ID NO: 2, а также
пептиды, состоящие из фрагмента из 10-20 последовательных аминокислотных остатков белка Гладкомышечный гамма-актин последовательности SEQ ID NO: 3, причем данный фрагмент включает аминокислотную последовательность, приведенную в SEQ ID NO: 4, а также
пептиды, состоящие из фрагмента из 10-20 последовательных аминокислотных остатков белка Декорин последовательности SEQ ID NO: 5, причем данный фрагмент включает аминокислотную последовательность, приведенную в SEQ ID NO: 6.
2. Композиция по п. 1, отличающаяся тем, что содержит носитель.
3. Композиция по п. 1, отличающаяся тем, что композиция предназначена для парентерального введения.
4. Композиция по п. 3, отличающаяся тем, что композиция предназначена для внутримышечного введения.
5. Применение фармацевтической композиции по п. 1 в эффективном количестве для лечения учащенного мочеиспускания и/или недержания мочи у пациентов с гиперактивным мочевым пузырем.
6. Применение фармацевтической композиции по п. 1 в эффективном количестве для лечения гиперактивного мочевого пузыря у пациентов.
| Ioan Scarneciu et al., Overactive bladder: A review and update / Exp Ther Med,2021, Vol.22, N.6, pp.1444 | |||
| СРЕДСТВО, НОРМАЛИЗУЮЩЕЕ ТОНУС МОЧЕВОГО ПУЗЫРЯ, И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2006 |
|
RU2302867C1 |
| WO 2019090263 A1, 09.05.2019 | |||
| СПОСОБ ОБРАБОТКИ АРАХИСА | 1994 |
|
RU2057465C1 |
| WO 2002068649 A2, 06.09.2002. | |||

