Изобретение относится к технологии углеродных наноматериалов, конкретно, к технологии получения углеродных наноразмерных частиц, в том числе, растворимых в воде.
Обычно наночастицами считаются частицы, размер которых менее 100 нм. Если их размер находится в диапазоне 2-20 нм, такие частицы называют квантовыми точками, хотя в некоторых источниках квантовыми точками называют и углеродные частицы размером до 50 нм. Четкой общепринятой границы размеров квантовых точек в настоящее время нет. Характерным свойством углеродных частиц размером 2-10 нм является флуоресценция, причем, чем меньше частица, тем короче длина волны флуоресценции. Чисто ориентировочно, на основании публикаций в научно-технической литературе, можно оценивать, что голубой цвет флуоресценции соответствует размеру частиц (области делокализации электронов) 2-4 нм, зеленый - 5-7 нм, желтый 8-10 нм, белый - смесь фракций разного размера. Но это очень условная оценка, спектр флуоресценции зависит не только от размера области делокализации электронов, но и от структуры. В большинстве случаев углеродные наночастицы указанных размеров имеют графеновую структуру, но нередко они называются графеновыми квантовыми точками без приведения доказательств структуры. Также, в большинстве случаев углеродные (графеновые) наночастицы (квантовые точки) имеют функциональные группы (обычно кислород- или азот-содержащие), которые придают этим частицам те или иные свойства, например, растворимость в воде или органических растворителях, способность реагировать с теми или иными молекулами, способность подвергаться вторичной функционализации. Элементное содержание кислорода или азота в таких квантовых точках может быть весьма велико (до 10-30%).
Эффективность углеродных наночастиц в тех или иных приложениях в общем не имеет прямой связи с их размером или какими-либо другими параметрами. Для одних применений лучше подходят частицы малого размера, например, 2-10 нм, и с одним составом поверхностных групп, для других применений лучше более крупные наночастицы, например 30-50 нм, и с другой природой поверхностных групп. Для одних применений требуется узкий диапазон распределения частиц по размерам, для других применений это не существенно.
Существуют два принципиально разных подхода к получению углеродных наночастиц:
- «Снизу вверх», то есть, синтез углеродных частиц из углеродсодержащих молекул путем их карбонизации в соответствующих условиях, например, гидротермальной или сольвотермальной карбонизации, или простой термообработки смеси молекулярных предшественников.
- «Сверху вниз», то есть, дробление крупных углеродных структур на наноразмерные частицы, преимущественно химическими методами. Наиболее часто такое дробление достигается окислительной обработкой углеродсодержащего сырья (графита, угля, кокса) в тех или иных условиях.
Рассмотрим известные методы получения углеродных наночастиц. Поскольку публикация по данной теме очень много, далее мы выберем те способы, которые являются характерными для той или иной группы методов.
Первая группа методов, «снизу вверх», состоит в том, что раствор тех или иных органических молекул (чаще всего лимонная кислота, глюкоза) подвергают гидротермальной карбонизации в автоклаве под давлением, обычно при температуре 180-220°С. Часто в исходный раствор органических молекул, способных к гидротермальной карбонизации, добавляют те или иные вещества, содержащие гетероатомы, например, азот- или серу-содержащие соединения, тогда полученные квантовые точки содержат эти гетероатомы в качестве функциональных групп. По данному направлению в научно-технической литературе есть тысячи публикаций, рассматривать все их здесь нет возможности. Достаточно понимать вышеуказанную общую технологическую схему. Состав исходных веществ и условия карбонизации (например, гидротермальной) подбирают так, чтобы получить углеродные квантовые точки с теми или иными параметрами. В качестве примера по данной технологии синтеза углеродных наночастиц можно сослаться на обзорные публикации:
[Linlin Shi, Boyang Wang, Siyu Lu. Efficient bottom-up synthesis of graphene quantum dots at an atomically precise level // Matter, Volume 6, Issue 3, 1 March 2023, Pages 728-760].
[Philippe Pierrat, Jean-Jacques Gaumet. Graphene quantum dots: Emerging organic materials with remarkable and tunable luminescence features // Tetrahedron Letters Volume 61, Issue 49, 3 December 2020, 152554].
[Xiliu Zhang, Changbo Wei, Ye Li, Dongsheng Yu. Shining luminescent graphene quantum dots: Synthesis, physicochemical properties, and biomedical applications // TrAC Trends in Analytical Chemistry Volume 116, July 2019, Pages 109-121].
[Dibyendu Ghosh, Krishnendu Sarkar, Pooja Devi, Ki-Hyun Kim, Praveen Kumar. Current and future perspectives of carbon and graphene quantum dots: From synthesis to strategy for building optoelectronic and energy devices // Renewable and Sustainable Energy Reviews, Volume 135, 2021, 110391].
[Manila Ozhukil Valappil, Vijayamohanan K. Pillai, Subbiah Alwarappan. Spotlighting graphene quantum dots and beyond: Synthesis, properties and sensing applications // Applied Materials Today, Volume 9, December 2017, Pages 350-371].
Рассмотренный метод синтеза углеродных квантовых точек «снизу вверх» позволяет варьировать свойства получаемых квантовых точек в широких пределах путем варьирования состава исходных веществ и условий гидротермальной карбонизации. Однако, существенным недостатком данного метода является то, что, как правило, гидротермальная и сольвотермальная карбонизация органических молекул идет с образованием множества побочных продуктов, и выделение углеродных наночастиц, в частности квантовых точек, с заданными параметрами из полученной реакционной смеси представляет собой сложную задачу. Эта задача решается с применением диализа, ультрафильтрации, электрофореза, хроматографии, ультрацентрифугирования. Все эти методики занимают длительное время, требуют сложного оборудования, а их производительность мала для массовой наработки квантовых точек.
Для массовой наработки углеродных (графеновых) квантовых точек более приемлемыми являются способы «сверху вниз», то есть, химическое дробление углеродных (графеновых) структур на мелкие фрагменты. Рассмотрим подробно эти способы.
В работе
[Shanli Yang, Yingru Li, Shaofei Wang, Jingsong Xu, Lang Shao, Tao Gai, Hao Tang, Yiming Ren, Mingfu Chu, Bianyuan Xia. A novel synthesis of graphene quantum dots via thermal treatment of crude graphite oxide in a dry and alkaline condition, and their application in uranyl detection // Heliyon Volume 6, Issue 9, September 2020, e04533]
описан способ получения графеновых квантовых точек из оксида графена. Согласно этому способу, вначале получали оксид графена путем окисления графита перманганатом калия в концентрированной серной кислоте по методу Хаммерса. Затем сухой оксид графена перерабатывали следующим образом. Сначала КОН (20 г) растворяли в 25 мл смеси этанол/вода (этанол/вода = 4:1) с образованием сильнощелочного раствора. Порошок сухого оксида графена (5 г) добавляли в этот раствор медленно при перемешивании, чтобы избежать сильного кипения. Получилась своего рода черная и вязкая паста. Эту черную пасту высушили при 80°C, и получили твердое вещество черного цвета. Вещество поместили в муфельную печь, нагрели до 300°С со скоростью 10°С/мин и выдержали при этой температуре в течение 4 ч в условиях окружающей атмосферы. Затем полученный продукт промывали деионизированной водой, при этом графеновые квантовые точки переходили в фильтрат вместе с другими водорастворимыми примесями. Фильтрат очищали диализом в течение двух недель с использованием диализной установки для удаления оставшихся примесей. Диализ проводили через мембрану, задерживающую частицы с молекулярной массой более 1000 г/моль, меньшие частицы удалялись через эту мембрану. Таким образом была получена водная дисперсия графеновых квантовых точек. Наконец, водную дисперсию этих квантовых точек дополнительно концентрировали для достижения желаемой концентрации с помощью роторного испарителя.
По своей структуре полученные квантовые точки представляют собой мелкие фрагменты графена размером 5-14 нм с большим количеством кислородных (гидроксильных и карбоксильных) групп по периферии частиц. Благодаря этим полярным группам и достигается растворимость полученных частиц в воде (преимущественно, при щелочном рН). Хотя об этом прямо в цитируемой работе не говорится, можно предположить, что в результате промывки диализом графеновые частицы содержат карбоксильные группы, в которых протоны замещены на ионы калия. Иначе вряд ли может быть, учитывая, что отмывку продукта диализом проводили, судя по описанию, нейтральной водой. Поскольку в исходном щелочном растворе карбоксильные группы находились в виде калиевых солей, без подкисления промывной воды маловероятно, чтобы ионы калия могли уйти.
Описанный способ имеет следующие недостатки. Во-первых, процесс получения оксида графена окислением графита по Хаммерсу достаточно трудоемкий и затратный, требует большого количества реагентов и времени. Так, для окисления графита перманганатом калия по Хаммерсу на 1 масс. ч. Графита требуется затратить 55 масс. ч. концентрированной серной кислоты и 3 масс. ч. перманганата калия. Во-вторых, описанный процесс переработки оксида графена в квантовые точки тоже весьма трудоемкий и продолжительный. Одна только промывка диализом занимает две недели. Кроме того, при высушивании спиртовой пасты, содержащей гидроксид калия и оксид графена, пары спирта или уходят в атмосферу, или же, если синтез масштабировать, требуется специальная установка для сушки с улавливанием паров спирта. Также, и концентрирование готового раствора квантовых точек в роторном испарителе занимает много времени.
В работе
[Su Zhang, Jiayao Zhu, Yan Qing, Chengwei Fan, Luxiang Wang, Yudai Huang, Rui Sheng, Yong Guo Dianzeng Jia, Tao Wang, Yanliang Pan, Yan Lv, Huaihe Song. Construction of hierarchical porous carbon nanosheets from template-assisted assembly of coal-based graphene quantum dots for high performance supercapacitor electrodes // Materials Today Energy 6 (2017) 36-45]. Согласно этому способу, 5 г тонкоизмельченного битуминозного угля помещают в трехгорлую стеклянную колбу. Затем медленно при перемешивании добавляют 25 мл 67%-ной азотной кислоты и 75 мл 98%-ной серной кислоты (в какой последовательности, не указано). Реакционную смесь нагревают до 80°С на водяной бане и кипятят при этой температуре (имеется в виду, с обратным холодильником) на протяжении 10 часов. После охлаждения до комнатной температуры смесь разбавляют 1 л воды и продолжают перемешивание еще 12 часов. Полученные квантовые точки отделяют центрифугированием (5 мин 4000 об/мин). Чтобы удалить избыток кислот, продукт очищают диализом (отсечка 3500 Дальтон), пока рН диализной воды станет равен 7. The primary coal-based GQDs were separated by centrifugation (4000 rpm, 5 min). Далее снова подвергают центрифугированию 6 мин 10000 об/мин. Полученные углеродные квантовые точки высушивают в сушильном шкафу при 80°С. Размер полученных квантовых точек составляет несколько нанометров. Далее для получения композита этот продукт диспергируют под действием ультразвука в этаноле.
Общим существенным признаком с заявляемым изобретением является наличие стадии окисления углеродного материала сильным окислителем в среде концентрированной серной кислоты. По данному признаку рассмотренный способ совпадает с другими вариантами этого способа, описанными в трех публикациях, цитируемых далее, и имеет те же недостатки, которые также будут рассмотрены далее.
В работах
[Jin Wang, Mingming Chen, Chengyang Wang, Jiuzhou Wang, Jiaming Zheng. Preparation of mesoporous carbons from amphiphilic carbonaceous material for high-performance electric double-layer capacitors // Journal of Power Sources 196 (2011) 550-558]
[Jiu-zhou Wang, Li-qun Wang, Ming-ming Chen, Cheng-yang Wang, Cui Zhang, Fei He. Nanoporous carbons from oxidized green needle coke for use in high performance supercapacitors // New Carbon Materials, 2015, 30(2): 141-149]
[Jiuzhou Wang, Mingming Chen, Chengyang Wang, Jin Wang, Jiaming Zheng. A facile method to prepare carbon aerogels from amphiphilic carbon material // Materials Letters 68 (2012) 446-449]
описано получение так называемого «амфифильного» углерода, представляющего собой наноразмерные частицы, содержащие преимущественно углерод и значительное количество кислородных групп, в том числе, гидроксильных, карбоксильных. По существу, в этих публикациях описан один и тот же способ, который по совокупности технологических операций наиболее близок к заявляемому изобретению и является прототипом. Этот способ включает окисление кокса смесью концентрированных азотной и серной кислот. В качестве исходного кокса использовали сырой игольчатый кос, полученный при температуре коксования около 500°С, без прокаливания при высокой температуре. Этот кокс окисляли в смеси концентрированных азотной и серной кислот в объемном соотношении 3:7. Таким образом, этот способ отличается от описанного в рассмотренной выше публикации только исходным сырьем (кокс вместо битуминозного угля). Что касается температуры окисления, возможны варианты. Однако, температуру окисления нельзя рассматривать как существенный признак, потому что температурный интервал реакции окисления в данной системе может быть выбран исходя из известных каждому квалифицированному специалисту данных о свойствах системы азотная кислота - серная кислота. В первой из цитированных работ, как следует из приведенной там ссылки на первоисточник [D. Tateishi, K. Esumi, H. Honda, H. Oda. Preparation of carbonaceous gel beads // Carbon 30 (1992) 942-944], окисление проводили в течение 3 ч при 80°С, и ограничение по температуре понятно, потому что выше этой температуры азотная кислота начинает кипеть и интенсивно испаряться. Во второй публикации температура окисления комнатная, но время окисления больше. Независимо от температурного режима окисления, получили «амфифильный углерод», который обладал способностью растворяться в воде при щелочном рН, и не растворялся при нейтральном и кислом рН. Растворимость этого углерода объяснялась наличием большого количества кислородных групп (гидроксильных, карбоксильных), которые ионизируются при щелочном рН и тем самым создают электростатическое отталкивание между частицами в водном растворе, препятствуя агломерации. Размер этих частиц, как следует из цитированных работ, 30-50 нм в нативном виде и около 25 нм после прокаливания сухого геля при 500°С.
Таким образом, по существу все рассмотренные выше публикации описывают один и тот же способ, главным существенным признаком которого, совпадающим с заявляемым изобретением, является окисление углеродного материала (кокса или угля) в смеси концентрированных азотной и серной кислот, температурный режим может варьироваться, и после окисления разбавление реакционной смеси водой. Затем следует выделение углеродных наночастиц из полученной реакционной смеси тем или иным известным методом (например, диализ, ультрафильтрация, ультрацентрифугирование).
Недостатком рассмотренного способа является агрессивность и токсичность применяемого окислителя - концентрированной (дымящей) азотной кислоты. Кроме того, что пары азотной кислоты очень токсичны, в процессе окисления она восстанавливается углеродом и выделяется очень токсичный диоксид азота. Кроме того, недостатком является очень большой расход кислот в пересчете на массу исходного кокса.
Данных о механизме окисления в этой системе в литературе мы не нашли. Однако, в результате наших собственных опытов выяснилось следующее. Окисление сырого кокса смесью концентрированных азотной и серной кислот в указанном в прототипе соотношении протекает с сильным выделением тепла, вследствие чего необходим точный температурный контроль реакционной смеси. Если допустить даже локальный перегрев реакционной смеси за счет выделяющегося в реакции тепла, то реакция самоускоряется с выделением большого количества газообразного диоксида азота, реакционная смесь вспенивается и выбрасывается из колбы. Такое поведение данной системы создает большие сложности при масштабировании синтеза. Также, диоксид азота является очень токсичным веществом. Далее, даже если удалось поддерживать температуру в процессе окисления в заданном диапазоне (не превышая 80°С), так что в процессе окисления не происходило существенного выделения диоксида азота, при разбавлении реакционной смеси водой диоксид азота все равно выделяется в большом количестве, и при масштабировании синтеза это тоже серьезная проблема. Далее реакционную смесь после завершения окисления разбавляют водой, окисленный кокс отделяют центрифугированием, промывают водой до рН3, и высушивают. Как показали наши опыты, промывка водой до рН3 в данном способе существенна. Если пытаться промывать окисленный кокс водой до нейтрального рН, на фильтре или центрифугированием, по мере приближения рН к нейтральному продукт начинает переходить в коллоидный раствор, который проходит через фильтр или плохо осаждается в центрифуге. Для того, чтобы устранить образование коллоида, необходимо вести промывку до слабокислого рН.
Можно предположить, что окисление сырого кокса указанной смесью кислот происходит по следующему механизму:
HNO3 + H2SO4 → NO2+HSO4- + H2O (эта реакция образования бисульфата нитрония известна в нитрующей смеси при нитровании органических соединений)
NO2+HSO4- → NO+HSO4- + [O] (бисульфат нитрозония известен)
Кокс + [O] → окисленный кокс (собственно окисление кокса)
NO+HSO4- + H2O → NO + NO2 + H2SO4 (гидролиз бисульфата нитрозония при разбавлении реакционной смеси водой)
2NO + O2 (воздух) → 2NO2
Таким образом, труднопреодолимыми недостатками рассмотренного способа является также сложность контроля температурного режима в процессе окисления кокса и, как уже отмечалось выше, выделение очень токсичного диоксида азота, не говоря уже об опасности работы с концентрированной азотной кислотой, пары которой также очень токсичны.
Задачей заявляемого изобретения является устранение недостатков прототипа путем выбора соответствующего окислителя, режимов окисления и последующей обработки, и способа выделения и очистки конечного продукта.
Поставленная задача решается тем, что в способе получения углеродных наночастиц, включающем обработку кокса окислителем в серной кислоте, в качестве окислителя используют персульфат аммония, в качестве серной кислоты - серную кислоту с концентрацией от 98 до 100%, причем, после обработки кокса окислителем проводят обработку окисленного кокса водным раствором травящего реагента, а в качестве последнего используют перекись водорода при температуре обработки от 90 до 100°С, или гидроксид калия при температуре обработки от 160 до 250°С.
Здесь необходимы пояснения. В качестве исходного кокса необходимо, как и в способе-прототипе, использовать кокс, полученный при температуре коксования около 500°С («сырой кокс»). Кокс, прокаленный при высокой температуре (графитизированный), как показали проведенные опыты, не пригоден для реализации заявляемого изобретения. Это обусловлено тем, что структура сырого кокса состоит из наноразмерных фрагментов графеновых слоев, соединенных прослойками аморфного углерода. Последний, как наименее химически стабильный, вытравливается окислителем в первую очередь, в результате чего остаются наночастицы с графеновой структурой. Если же брать графитизированный кокс, вся его структура преимущественно графеновая и окислительной фрагментации на наночастицы в данных условиях она не поддается.
Массовое соотношение персульфата аммония к коксу может варьироваться, однако, оптимальным, как показали проведенные опыты, является массовое соотношение 4:1. Выбор такого массового соотношения обоснован тем, что реакция окисления кокса персульфатом аммония в серной кислоте вначале сопровождается сильным выделением тепла, но тепловой эффект падает по мере увеличения количества персульфата аммония и становится малозаметным при достижении указанного массового соотношения. Это свидетельствует о том, что при данном количестве персульфата аммония достигается практически полное в данных условиях окисление кокса. Однако, технический результат изобретения достигается и при больших количествах персульфата аммония.
Что касается выбора концентрации серной кислоты. Как показали проведенные опыты, наиболее эффективно окисление кокса персульфатом аммония протекает в среде 100%-ной серной кислоты. При этом привес окисленного кокса по сравнению с исходным достигает 28-30% за счет присоединения окисных групп. В этих условиях реакция окисления кокса практически не сопровождается побочной реакцией разложения перекисных соединений с выделением газообразного кислорода. Если брать 98%-ную серную кислоту, эффективность окисления кокса несколько снижается, привес за счет присоединения окисных групп составляет около 20-21%. Но при этом уже становится заметной побочная реакция разложения перекисных соединений с выделением газообразного кислорода, что и снижает долю окислителя, вступающему в реакцию окисления кокса. Однако, технический результат заявляемого изобретения при применении 98%-ной серной кислоты все еще достигается. Если же в качестве серной кислоты брать обычную концентрированную серную кислоту, содержащую, согласно ГОСТ 4204-77, 93,6-95,6% H2SO4 (остальное вода), эффективность окисления кокса персульфатом аммония резко снижается, а доля побочной реакции разложения перекисных соединений с выделением газообразного кислорода резко возрастает. Это связано с тем, что при наличии в системе воды происходит гидролиз пероксодисерной кислоты до пероксомоносерной, которая значительно менее стабильна, а затем и до свободной перекиси водорода, которая способна каталитически разлагаться при контакте с углями.
Следует отметить, что применение как растворов перекиси водорода, так и гидроксида калия в качестве травящих реагентов для графеновых материалов описано в многочисленных публикациях, однако, в сочетании с окислением углеродного материала персульфатом аммония в серной кислоте травление этими реагентами ранее не было известно.
Если в качестве травящего реагента применяют перекись водорода, массовое соотношение перекиси водорода к окисленному коксу берут от 4:1 до 5:1 в расчете на безводные компоненты, а после обработки окисленного кокса перекисью водорода полученный раствор выпаривают. В таком варианте специальных операций по выделению углеродных наночастиц из реакционной смеси не требуется, потому что при выпаривании не прореагировавшая перекись водорода, которая может содержаться в реакционной смеси, испаряется вместе с водой. Что касается времени обработки окисленного кокса раствором перекиси водорода, оно может варьироваться в широких пределах, но оптимальным временем является около 12 часов. Что касается температуры обработки окисленного кокса раствором перекиси водорода, как показали проведенные опыты, травление окисленного кокса перекисью водорода протекает с приемлемой скоростью в диапазоне температур от 90 до 100°С (при атмосферном давлении). В литературе есть примеры травления графеновых материалов перекисью водорода и при более высоких температурах, до 160-180°С, однако при этом требуется проводить процесс в автоклаве, что создает проблему при масштабировании процесса. Кроме того, при проведении этого процесса в автоклаве возникает еще проблема со стравливанием избыточного давления газообразного кислорода, который в большем или меньшем количестве выделяется при побочной реакции разложения перекиси водорода. Учитывая сказанное, представляется оптимальным проводить заявляемый процесс при атмосферном давлении в указанном диапазоне температур.
Что касается температуры выпаривания раствора после обработки окисленного кокса перекисью водорода, она не должна быть слишком высокой, потому что при высокой температуре происходит спекание углеродных наночастиц и они теряют растворимость в воде. Практически, как показали проведенные опыты, наиболее удобно выпаривание раствора может быть проведено в вентилируемом сушильном шкафу при температуре 60°С, при этом раствор следует налить в широкий полипропиленовый поддон. Однако, для ускорения процесса может быть применено выпаривание под вакуумом.
Если в качестве травящего реагента применяется раствор гидроксида калия, избыточный гидроксид калия (а также, образующийся в результате реакции травления карбонат калия) остаются в реакционной смеси и простым выпариванием раствора от них невозможно избавиться. Потому при применении в качестве травящего реагента гидроксида калия возможны два варианта дальнейшей обработки реакционной смеси, а именно.
Первый вариант, реакционную смесь растворяют в воде или разбавляют водой, и осаждают углеродные наночастицы добавлением минеральной кислоты до кислого рН. В кислой среде углеродные наночастицы нерастворимы и образуют достаточно крупные агрегаты, которые могут быть отфильтрованы через обычный фильтр с размером пор порядка нескольких мкм. Осажденные наночастицы отделяют фильтрацией, с последующей промывкой водой, подкисленной азотной кислотой до рН 2,5-3 (0,3 мл 65%-ной азотной кислоты на 1 л дистиллированной воды). Подкисление промывной воды необходимо для того, чтобы предотвратить переход наночастиц в коллоидный раствор и проскок через фильтр.
Вообще говоря, гидроксид калия и карбонат калия, присутствующие в реакционной смеси после травления окисленного кокса гидроксидом калия, можно удалить из раствора и обработкой сильнокислой катионообменной смолой в Н-форме. При этом анионы гидроксила и карбоната быстро и необратимо нейтрализуются смолой, а катионы калия переходят из раствора в гранулы смолы. Такой вариант обработки реакционной смеси тоже может быть применен, однако, простое осаждение наночастиц кислотой и промывка слабокислой водой удобнее и экономичнее.
Что касается температуры обработки окисленного кокса раствором гидроксида калия. Как показали проведенные опыты, эту обработку наиболее целесообразно проводить в интервале температур от 160 до 250°С. Выбор данного температурного интервала обусловлен следующим. Ниже 160°С травящее действие раствора гидроксида калия недостаточно эффективно, реакция сильно замедляется и диспергирование окисленного кокса на наноразмерные частицы не происходит или происходит в недостаточной степени. Выше же 250°С возможно спекание углеродных наночастиц.
Далее изобретение подтверждается примерами реализации.
Для реализации изобретения применяли следующие исходные вещества.
Сырой нефтяной игольчатый кокс производства АО «Газпромнефть-ОНПЗ», полученный из нефтяного сырья при температуре коксования около 500°С. Этот кокс не проводил электрического тока (сопротивление кусков более 2 МОм). Перед дальнейшими операциями кокс был измельчен в лопастной мельнице ударного типа до прохождения через проволочное сито из нержавеющей стали с просветом ячеек 0,2 мм.
Следует отметить, что кокс, прокаленный при высокой температуре (графитизированный), для реализации настоящего изобретения не пригоден.
Персульфат аммония использовали марки ЧДА.
Для получения 100%-ной серной кислоты концентрированную серную кислоту марки ХЧ (около 95% H2SO4) закрепляли 24%-ным олеумом. Точку эквивалентности контролировали по дымлению в контакте с влажным воздухом. Если дымления не было, значит, концентрация серной кислоты меньше 100%. Тогда добавляли еще немного олеума до появления слабого дымка. Согласно
[А. Гордон, Р. Форд. Спутник химика. Физико-химические свойства, методики, библиография. М., «Мир», 1976, 510 с. - С. 442]
такой простой метод позволяет контролировать отклонение состава от 100%-ной серной кислоты с точностью ±0,02%.
Гидроксид калия брали марки ЧДА в виде гранул, содержащих 85% КОН (остальное вода).
Остальные материалы и оборудование были стандартными лабораторными.
Распределение частиц по размерам определяли с помощью прибора Nicomp 380 ZLS. Этот прибор определяет размер частиц по динамическому рассеянию света. Раствор углеродных наночастиц готовили в водном аммиаке (0,25 моль/л аммиака). При этом концентрацию наночастиц выбирали в пределах 0,1-0,5% масс. в зависимости от интенсивности окраски (для сильно окрашенных образцов брали меньшие концентрации).
Рентгеновские дифрактограммы записывали с помощью прибора ARL Equinox 1000 («Thermo Scientific Instruments Group», USA), излучение 1,54056 А, CuKα.
Изобретение иллюстрируется фигурами графических изображений:
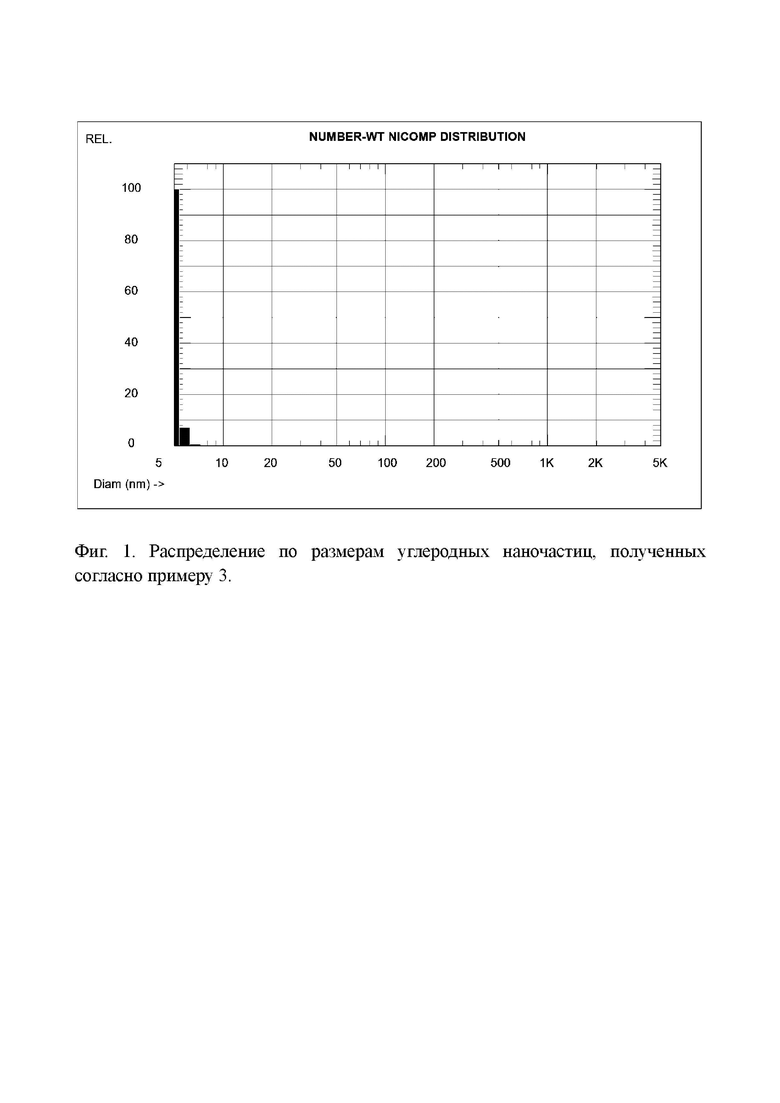
Фиг. 1 - Распределение по размерам углеродных наночастиц, полученных согласно примеру 3.
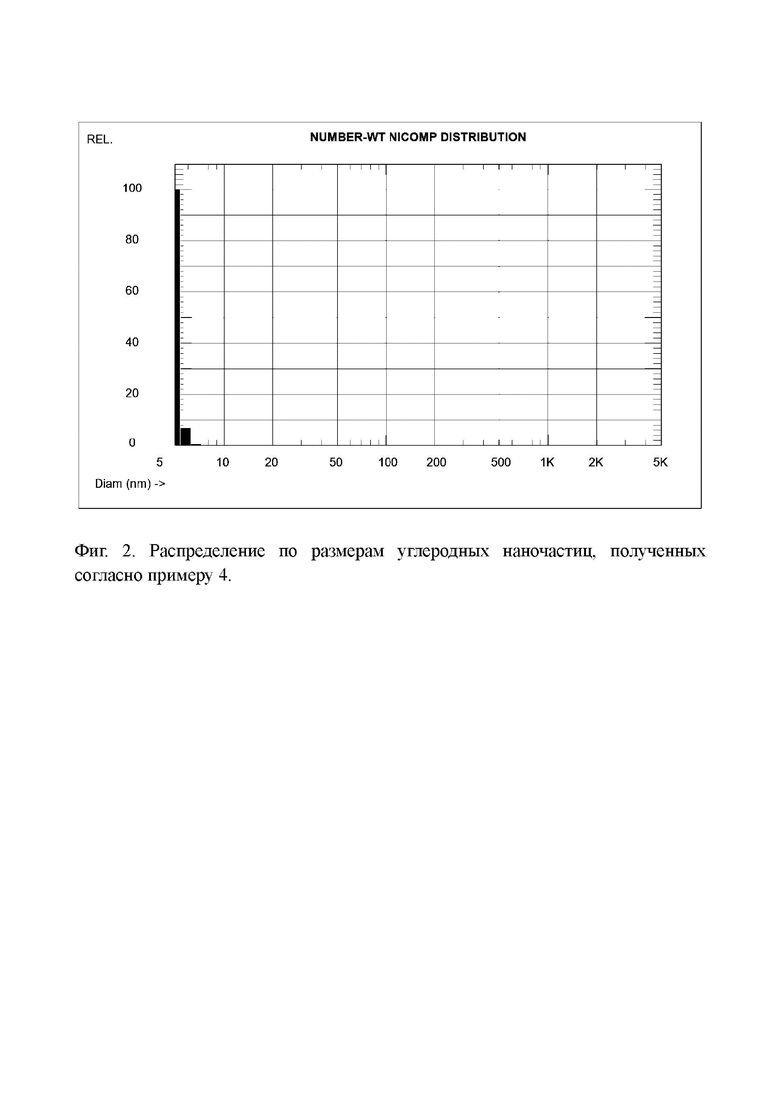
Фиг. 2. Распределение по размерам углеродных наночастиц, полученных согласно примеру 4.
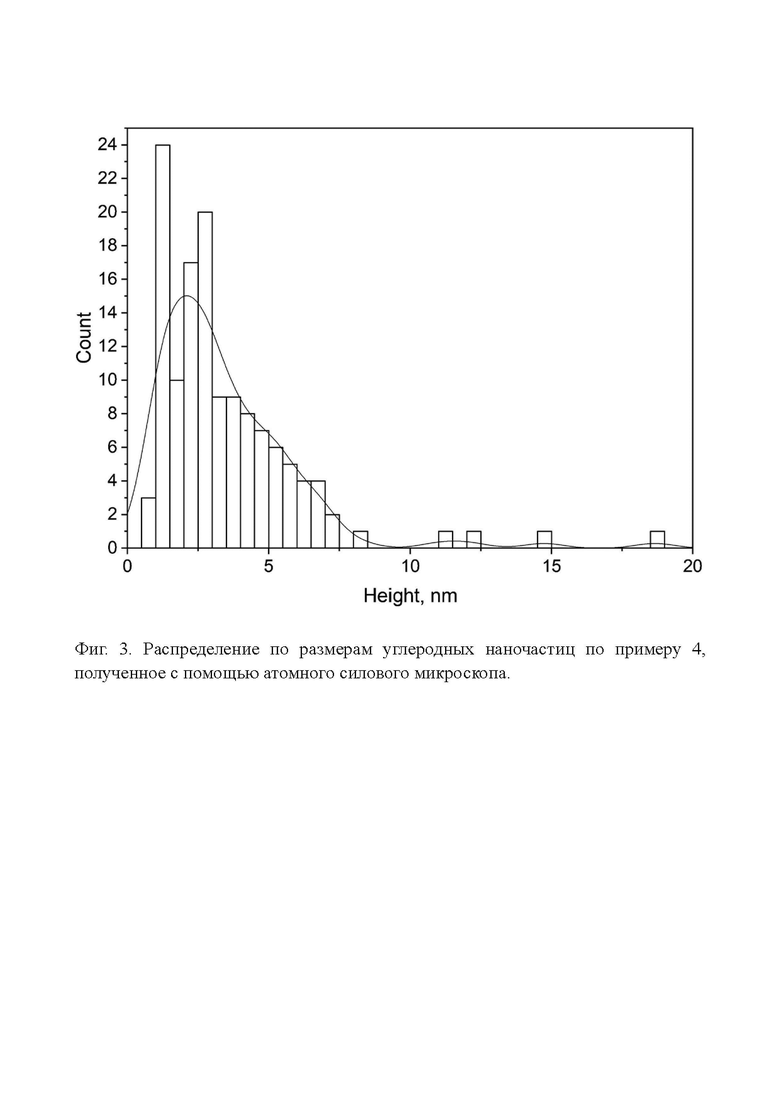
Фиг. 3. Распределение по размерам углеродных наночастиц по примеру 4, полученное с помощью атомного силового микроскопа.
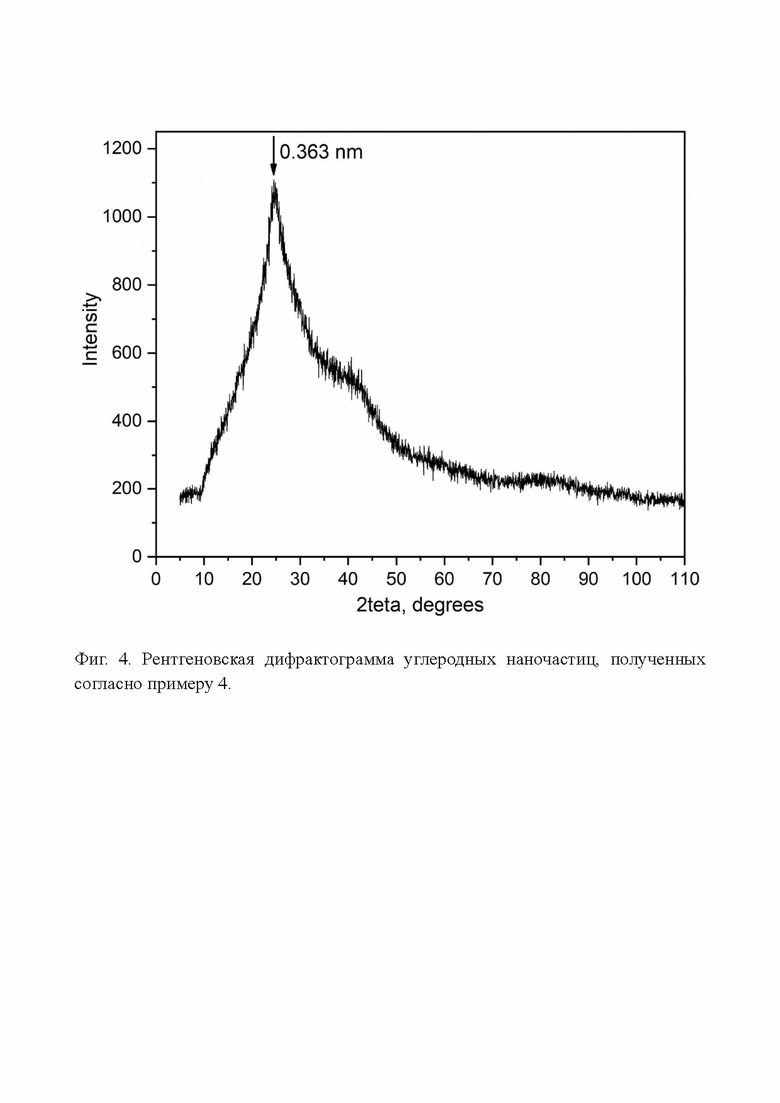
Фиг. 4. Рентгеновская дифрактограмма углеродных наночастиц, полученных согласно примеру 4.
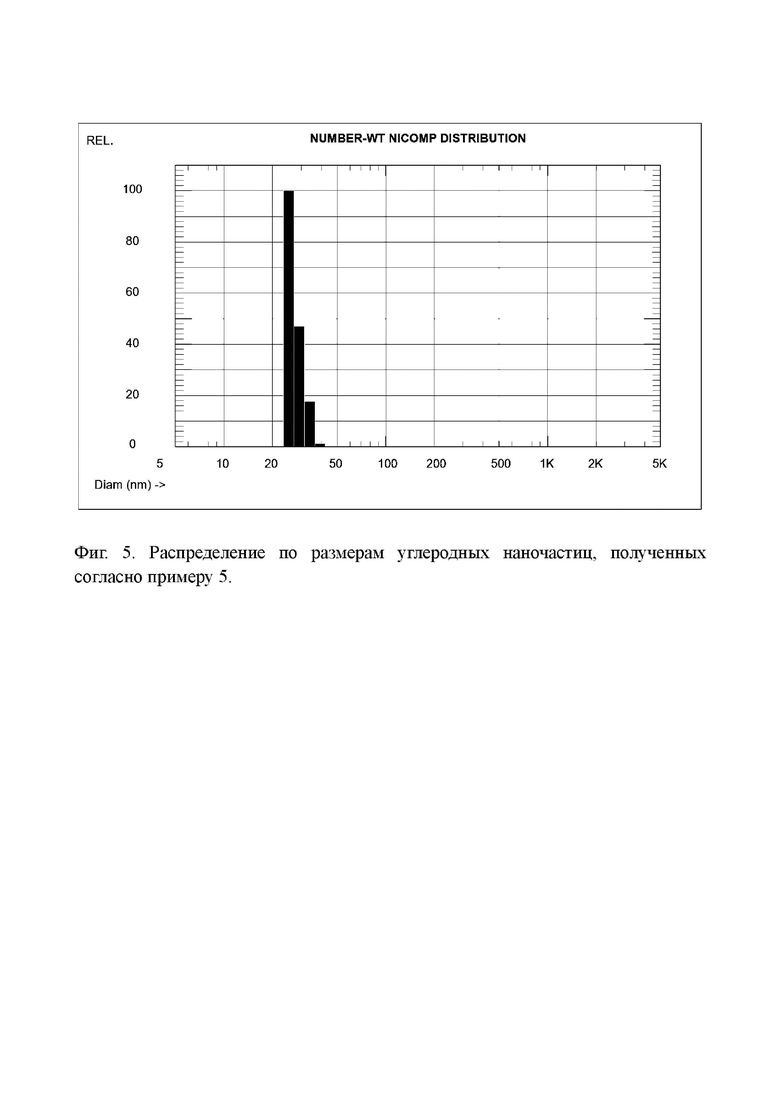
Фиг. 5. Распределение по размерам углеродных наночастиц, полученных согласно примеру 5.
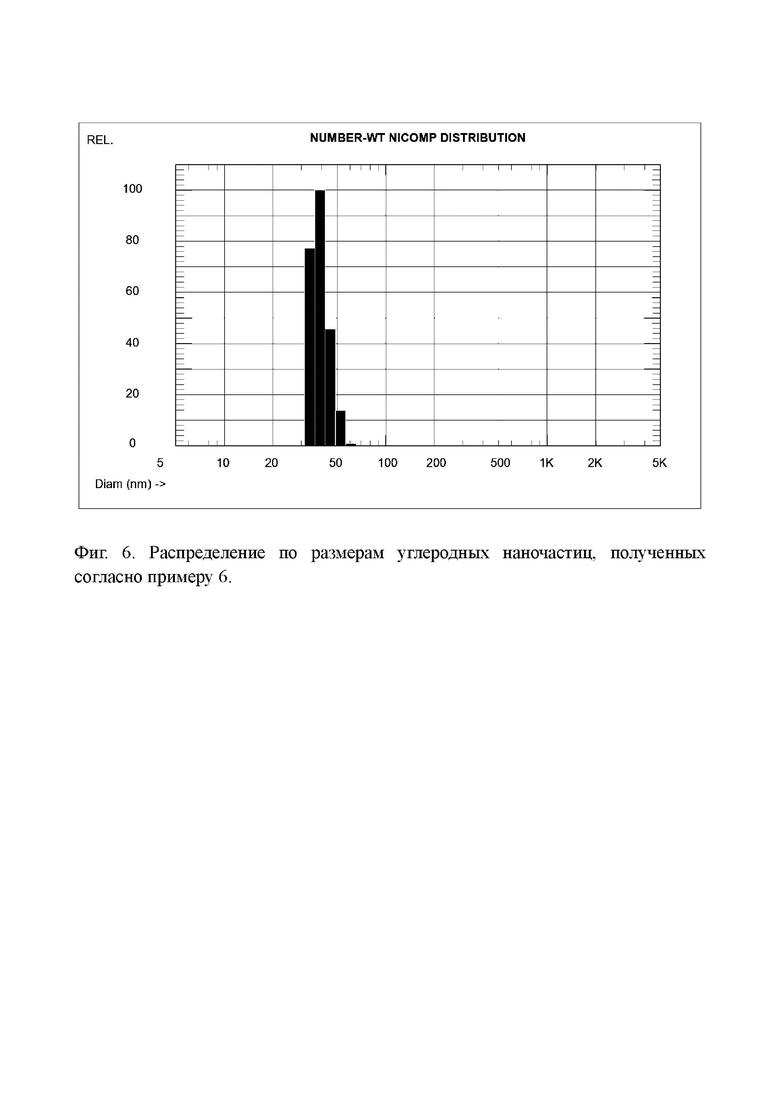
Фиг. 6. Распределение по размерам углеродных наночастиц, полученных согласно примеру 6.
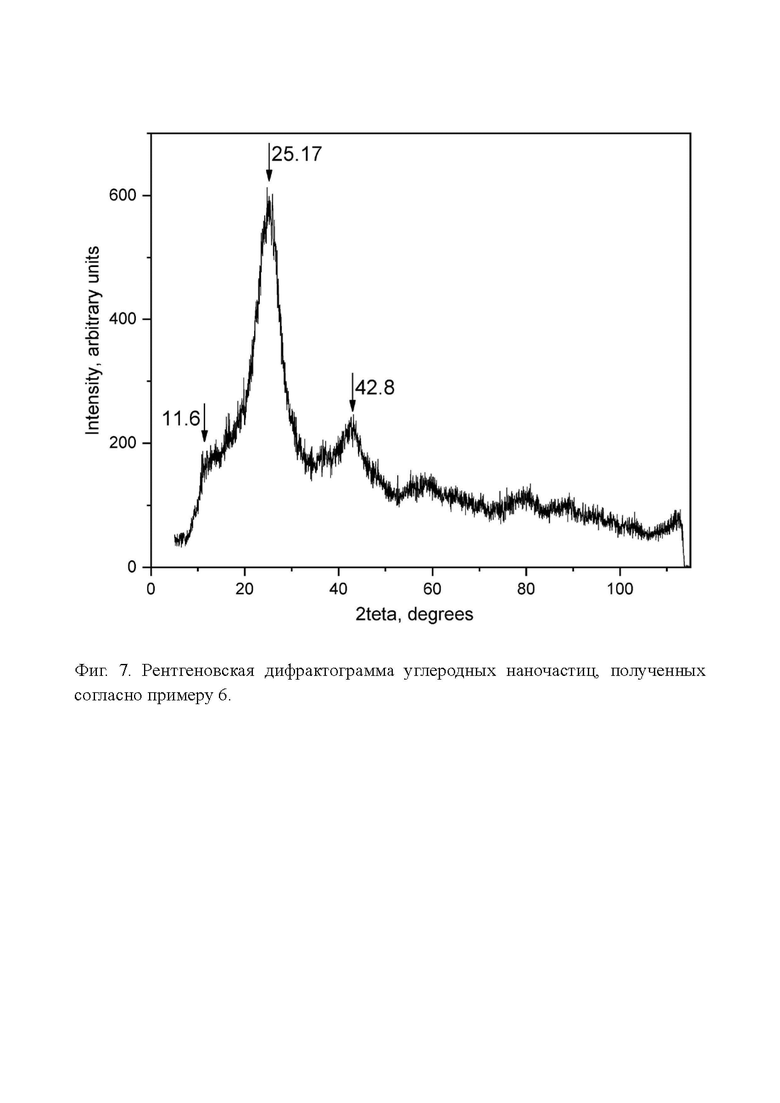
Фиг. 7. Рентгеновская дифрактограмма углеродных наночастиц, полученных согласно примеру 6.
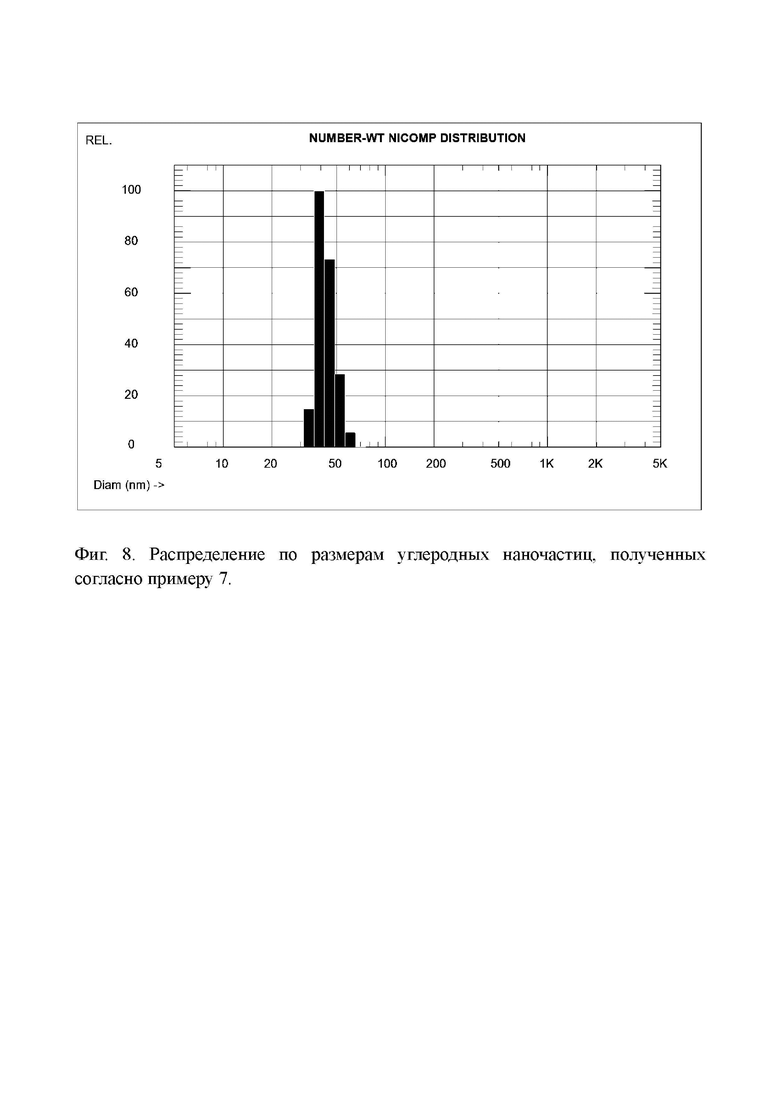
Фиг. 8. Распределение по размерам углеродных наночастиц, полученных согласно примеру 7.
Пример 1. Окисление кокса персульфатом аммония в 100%-ной серной кислоте.
В 2-литровый стакан из термостойкого стекла, закрепленный в охлаждающей водяной бане и снабженный фторопластовой мешалкой, влили 260 мл (475 г) 100%-ной серной кислоты. Внутри реакционной смеси был погружен термодатчик. Включили мешалку на 150 об/мин и загрузили 50 г сырого игольчатого кокса, измельченного в порошок с размером частиц менее 0,2 мм. Температура смеси самопроизвольно поднялась на 8 градусов. Значит, какая-то реакция серной кислоты с коксом медленно происходит даже без персульфата. Поскольку при этом в начальный момент был заметен запах диоксида серы, можно полагать, что эта первичная реакция, протекающая в небольшой степени, состоит в восстановлении серной кислоты до диоксида серы. Далее запах диоксида серы исчез. Небольшими порциями в течение часа всыпали в стакан 200 г персульфата аммония с такой скоростью, чтобы температура внутри реакционной смеси была в пределах 40-50°С. В конце экзотермический эффект сильно ослаб, реакционная смесь, вначале жидкая, загустела, однако, еще поддавалась перемешиванию мешалкой. После добавления всего персульфата аммония продолжали перемешивание еще в течение часа, при этом температуру смеси поддерживали теплой баней около 30°С. Затем поменяли воду в бане на холодную и при перемешивании медленно, вначале очень медленно, влили 1,25 л воды. Вначале сильный экзотермический эффект. Воду добавляли с такой скоростью, чтобы температура смеси была в пределах 50-60°С. В конце увеличили скорость мешалки до 200 об/мин. После прибавления всей воды разобрали установку, накрыли теплый стакан и оставили до завтра. Сверху присутствует небольшое количество пены с пузырьками газа. По всей видимости, это кислород за счет разложения избыточного персульфата аммония. Тот факт, что в конце добавления персульфата аммония экзотермический эффект почти исчез, говорит, что данное количество персульфата аммония (4:1) достаточно для полного окисления сырого игольчатого кокса и больше персульфата аммония давать нет смысла. После отстаивания суспензию перенесли на фильтр (воронка Бюхнера с фильтрующим слоем из нетканого полипропиленового материала Спанбонд), после стекания жидкости осадок несколько раз промыли на фильтре водой, подкисленной азотной кислотой до рН 2,5-3 (1,5 мл 65%-ной азотной кислоты на 5 л воды). Подкисление промывной воды необходимо для предотвращения ухода продукта в коллоид, что происходит при отмывке окисленного кокса водой до нейтрального рН. Затем продукт откачали на фильтре под вакуумом для удаления излишка жидкости и высушили в сушильном шкафу при 60°С до постоянной массы, которая составляла 65,0 г. Незначительное количество азотной кислоты, присутствующей во влажном продукте, испаряется вместе с водой во время сушки. Таким образом, за счет присоединения окисных групп масса кокса увеличилась на 30% по сравнению с исходной. При этом, существенного окисления кокса до летучих веществ (углекислого газа) не происходило, судя по тому, что в процессе окисления не наблюдалось газовыделения. Также, в этих условиях не происходило и заметного выделения газообразного кислорода за счет разложения перекисных соединений.
Полученный окисленный игольчатый кокс представлял собой непрочные ссохшиеся комки, которые для удобства последующих операций измельчили до прохождения через сито 0,2 мм.
Отходы производства по описанной методике представляют собой водный раствор серной кислоты и сульфата аммония с незначительной примесью азотной кислоты. Этот раствор можно нейтрализовать аммиаком с получением раствора сульфата аммония (с незначительной примесью нитрата аммония), который можно применять в качестве удобрения как в виде раствора, так и высушенного продукта.
Пример 2. Окисление кокса персульфатом аммония в 98%-ной серной кислоте.
В отличие от 100%-ной серной кислоты, которая отсутствует в продаже (замерзает при +10°С и потому не удобна для транспортировки, особенно в холодное время года), 98%-ная серная кислота имеется в продаже. Окисление кокса персульфатом аммония в среде 98%-ной серной кислоты протекает несколько менее эффективно (что видно по немного меньшему привесу окисленного кокса за счет окисных групп), однако вполне приемлемо для реализации заявляемого изобретения.
Окисление кокса персульфатом аммония в 98%-ной серной кислоте и все последующие операции проводили таким же образом, как описано в примере 1, но вместо 100%-ной серной кислоты взяли 98%-ную, количество которой было меньше и составляло 200 г на 50 г сырого игольчатого кокса. Возможность использования меньшего количества кислоты обусловлена тем, что при окислении в 98%-ной кислоте частицы кокса меньше разбухают и вязкость реакционной смеси меньше, чем в 100%-ной кислоте, что дает возможность обеспечить приемлемую для перемешивания механической мешалкой густоту реакционной смеси при меньшем количестве серной кислоты. Температурный режим в процессе окисления был таким же, как и в примере 1. После всех операций и высушивания получили 60,7 г окисленного кокса, то есть, масса кокса после окисления увеличилась на 21,4%. В этом примере привес за счет окисных групп был меньше, чем в примере 1. Это говорит, что окисление в 98%-ной серной кислоте происходит несколько менее эффективно, чем в 100%-ной, но и такой окисленный кокс пригоден для реализации заявляемого изобретения.
Пример 3. Получение углеродных наночастиц из кокса, окисленного в 100%-ной серной кислоте, методом травления перекисью водорода.
В 3-литровый стеклянный реактор, снабженный крышкой с отверстием для вала мешалки, фторопластовой мешалкой и водяной баней, поместили 130 г окисленного кокса, полученного согласно примеру 1 (что соответствует 100 г исходного не окисленного кокса), и добавили 1150 мл воды и 218 мл 50%-ной перекиси водорода (плотность 1,195 г/см3). Включили мешалку на 200 об/мин и нагрев водяной бани. Когда вода в водяной бане закипела, с этого момента выдержали реакционную смесь к кипящей водяной бане в течение 4 часов, после чего добавили еще 218 мл 50%-ной перекиси водорода и выдержали реакционную смесь при перемешивании в кипящей водяной бане еще 3 часа, после чего выключили нагрев и мешалку и оставили до завтра. На следующий день разобрали установку. Реакционная смесь представляла собой гелеобразную массу. Перемешивая вручную, добавили постепенно 200 мл воды. Образовалась густая, но жидкая масса. Добавили к ней 435 мл 50%-ной перекиси водорода, снова собрали установку, как вначале, и выдержали при перемешивании в течение 7 часов в кипящей водяной бане, затем все выключили и оставили остывать.
Таким образом, суммарное соотношение безводной перекиси водорода к окисленному коксу составляет 4:1.
Здесь необходимы пояснения. Порционное добавление перекиси водорода проводили с целью лучшего контроля процесса, потому что в ходе процесса происходит частичное разложение перекиси водорода с выделением газообразного кислорода, и при однократной загрузке всей перекиси водорода за счет слишком сильного газовыделения реакционная смесь могла бы вылезти из реактора в виде пены. Что касается температурного режима, то датчик, помещенный внутри реактора, показывал в первые часы температуру около 90°С, но по мере разложения перекиси водорода и ослабления газовыделения температура реакционной смеси приближалась к температуре кипящей бани (100°С). Меньшая температура вначале процесса была обусловлена уносом из реакционной смеси водяных паров вместе с газообразным кислородом, что и вызывало понижение температуры за счет теплоты испарения воды.
После остывания получили 440 г темно-красно-коричневой жидкости без осадка, представляющей собой раствор углеродных наночастиц. Этот раствор распределили в два плоских полипропиленовых поддона с размером дна 23*35 см и высушили в вентилируемом сушильном шкафу при температуре 60°С. Получили 85,0 г сухих углеродных наночастиц в виде черного твердого вещества, хорошо растворимого в воде и в водном растворе аммиака.
Определение распределения частиц по размерам в растворе этого вещества дало средний размер частиц 5,1 нм (Фиг. 1).
Что касается элементного состава. Данные, полученные для образца по примеру 3 методом энергодисперсионной спектроскопии в сканирующем электронном микроскопе:
Углерод =45,1% масс.
Кислород =48,6% масс.
Сера =6,3% масс.
Обращает на себя внимание присутствие заметного количества серы. Вероятно, она присутствует в виде сульфо-групп, которые присоединились к коксу в процессе окисления кокса персульфатом аммония в 100%-ной серной кислоте (которая сама по себе обладает сульфирующим действием). Если для образца, полученного по примеру 3, приписать соответствующую часть кислорода сульфо-группам, получим следующий пересчитанный элементный состав:
Углерод =45,1% масс.
Кислород =39,2% масс.
Сульфо-группы =15,7% масс.
Таким образом, по соотношению кислорода и углерода полученные наночастицы близки к оксиду графена, однако, имеют существенно другую структуру.
Следует отметить, что и наночастицы, описанные в способе-прототипе, также имеют в своем составе сульфо-группы [Jiuzhou Wang, Mingming Chen, Chengyang Wang, Jin Wang, Jiaming Zheng. A facile method to prepare carbon aerogels from amphiphilic carbon material // Materials Letters 68 (2012) 446-449].
Обилие поверхностных функциональных групп благоприятно для различных применений полученных углеродных наночастиц.
Пример 4. Получение углеродных наночастиц из кокса, окисленного в 100%-ной серной кислоте, методом травления перекисью водорода.
Повторили синтез по примеру 3, но суммарное количество перекиси водорода к окисленному коксу взяли 5:1 вместо 4:1, как было в примере 3. То есть, количество перекиси водорода, прибавляемой на каждой стадии, в данном примере увеличили в 1,25 раза. В результате получили такой же темно-красно-коричневый раствор углеродных наночастиц, при высушивании которого получили сухие углеродные наночастицы в виде черного твердого вещества массой 82,0 г.
Определение распределения наночастиц по размерам в растворе этого вещества дало средний размер частиц тоже 5,1 нм (Фиг. 2). Таким образом, увеличение массы перекиси водорода от 4:1 до 5:1 к окисленному коксу не привело к заметному изменению среднего размера наночастиц.
Данные распределения по размерам для наночастиц, полученных по примеру 4, подтверждаются также данными, полученными с помощью атомного силового микроскопа (Фиг. 3). Видно, что почти все частицы находятся в диапазоне размеров 2-8 нм.
Что касается структуры полученных углеродных наночастиц. Углеродные наночастицы в сухом виде, полученные по примеру 4, на рентгеновской дифрактограмме показывают очень широкую полосу (Фиг. 4) с пиком, соответствующим межплоскостному расстоянию 0,363 нм (для графита 0,3354 нм). Причем, в отличие от оксида графена, между графеновыми слоями этих частиц нет интеркалированных ионов или молекул. Для оксида графена характерна дифрактограмма с одним относительно узким пиком, соответствующим межслоевому расстоянию в пределах 0,7-1,4 нм в зависимости от условий синтеза.
Пример 5. Получение углеродных наночастиц из кокса, окисленного в 98%-ной серной кислоте, методом травления перекисью водорода.
Повторили синтез по примеру 4 (перекись водорода : окисленный кокс =5:1), но в качестве окисленного кокса взяли кокс, окисленный в 98%-ной серной кислоте согласно примеру 2. При этом количество окисленного кокса взяли такое, которое соответствует 100 г исходного не окисленного кокса, то есть, 121,4 г. В результате всех операций получили сухие углеродные наночастицы в виде черного твердого вещества массой 71,5 г.
Определение распределения частиц по размерам в растворе этого вещества дало средний размер частиц 27,3 нм (Фиг. 5). Таким образом, при применении кокса, окисленного персульфатом аммония в среде 98%-ной серной кислоты, средний размер частиц значительно увеличивается по сравнению с коксом, окисленным в 100%-ной серной кислоте. Однако, этот размер все еще находится в диапазоне наноразмеров и такие наночастицы тоже могут найти применение. Следует отметить, что для углеродных наноматериалов не существует каких-то универсальных критериев качества. Для одних применений лучше частицы с одной структурой и размером, для других применений оптимальные параметры могу быть другими.
Пример 6. Получение углеродных наночастиц из окисленного кокса методом травления гидроксидом калия при 200°С.
В ванне из фторопласта-4 растворили 200 г гранулированного гидроксида калия (содержащего согласно сертификату качества 86% КОН, остальное вода) в 200 мл воды. При растворении КОН происходило сильное саморазогревание. Когда раствор КОН остыл, добавили 65 г окисленного кокса, полученного согласно примеру 1, и тщательно перемешали. При этом наблюдалось небольшое саморазогревание. Ванну закрыли фторопластовой крышкой не герметично, но так, чтобы пары воды могли выходить, но не было прямого воздухообмена с наружным воздухом. Этот момент важен из следующих соображений. В таблице показана зависимость температуры кипения растворов КОН при атмосферном давлении от концентрации КОН:
Из этой таблицы следует, что водные растворы КОН указанного состава находятся в равновесии с водяным паром при атмосферном давлении и указанных температурах. Если мы будем нагревать разбавленный раствор КОН до какой-то из указанных температур, то избыточная вода будет испаряться, пока не будет достигнута указанная равновесная концентрация КОН в растворе. Но это при условии, что пары воды могут выходить из системы, не создавая избыточного давления, и в то же время воздухообмен с окружающей атмосферой будет сведен к минимуму. Если допустить прямой воздухообмен, вода из раствора будет испаряться и дальше. Это плохо для реализации заявляемого изобретения, потому что в таком случае концентрация КОН в растворе никак не контролируется. Практически, как показали проведенные опыты, условия, близкие к равновесным, можно легко создать, закрыв ванну плотно, но не герметично прилегающей крышкой.
Закрытую крышкой ванну с реакционной смесью поместили в печку, включили печку на 200°С, и по достижению этой температуры выдержали 6 часов при этой температуре. Затем выключили печку и оставили остывать. На следующий день извлекли и вскрыли ванну. Находящийся в ней продукт (твердый темно-коричневый пористый корж) перенесли в 10-литровое пластиковое ведро и растворили в воде. Смыли водой остатки из ванны, добавили в ведро воду до общего объема 6 л и тщательно перемешали. Получили темно-коричневый раствор, в котором содержалось небольшое количество осадка. Закрытое крышкой ведро оставили отстаиваться несколько дней для осаждения осадка.
Затем сифоном медленно отобрали верхние 5 литров раствора, чтобы не взмучивать осадок, и профильтровали этот раствор через фильтр из микроволокнистого полипропилена (фильтрующий материал ПП-190 производства ООО «Технофильтр»). Далее постепенно переносили на фильтр остальной продукт, стараясь первые порции раствора переносить на фильтр по возможности без осадка, и фильтровали, откачивая водоструйным насосом, Фильтрация шла медленно, очень тонкий осадок забивал фильтр и создавал плотный, плохо фильтрующийся слой. Удалось отобрать только 0,5 л фильтрата, остальное пришлось выбросить из-за очень медленной скорости фильтрации. Оставшуюся суспензию можно было бы отцентрифугировать, но имеющаяся лабораторная центрифуга была слишком малого объема для переработки раствора за приемлемое время. Тем не менее, при наличии большой центрифуги можно было бы заменить отстаивание исходного раствора центрифугированием, что сэкономило бы время. Таким образом, из 6 л исходного раствора с осадком получили 5,5 л профильтрованного раствора без осадка. По расчету, здесь содержится КОН =200*0,86*5,5/6 =157,7 г 100%-ного КОН или (/56,11=) 2,81 моль КОН. К этому раствору в пластиковом ведре медленно, при перемешивании, прибавили раствор 250 мл 65%-ной азотной кислоты (ее концентрация согласно справочным данным 14,3 моль/л) в 500 мл воды. Таким образом, азотной кислоты прибавлено 3,58 моль, что на 27% больше, чем теоретически необходимое количество для нейтрализации соединений калия. В процессе добавления азотной кислоты наблюдалось выделение газа, по запаху углекислого, что свидетельствует об образовании в реакционной смеси при спекании КОН с окисленным коксом некоторого количества карбоната калия.
Из подкисленного раствора выпал осадок углеродных наночастиц, которые в кислой среде теряют растворимость и образуют агрегаты, поддающиеся фильтрации. На следующий день сифоном отобрали бесцветную жидкость над осадком, насколько это было возможно, и суспензию фильтровали через такой же фильтр с фильтрующим слоем ПП-190 из микроволокнистого полипропилена, при слабом вакууме (относительное разрежение 1-2 метра водяного столба). В данном случае применять для ускорения фильтрации более сильный вакуум не желательно, потому что агрегаты наночастиц слабые и могут начать проходить через фильтр в виде коллоида. Осадок на фильтре промывали водой, подкисленной азотной кислотой до рН 2,5-3 (1,5 мл 65%-ной азотной кислоты на 5 л деионизированной воды). Промывку проводили до тех пор, пока рН промывной воды, выходящей из фильтра, сравнялась с рН промывной воды, подаваемой на фильтр. Получили слой осадка, который осторожно сняли с фильтра шпателем из нержавеющей стали и перенесли в герметичный полипропиленовый контейнер. Примечание: осадок при механическом перемешивании легко переходит в коллоидную жидкость, в связи с чем механическое воздействие на него во время промывки не допускается.
Получили 268,5 г влажного продукта, в котором содержалось 9,66% сухого вещества (высушивание пробы при 60°С). Таким образом, в этой влажной пасте (которая превратилась в густую жидкость при перемешивании) содержится 25,94 г сухого вещества. Чтобы оценить выход углеродных наночастиц по отношению к исходному коксу, следует отметить, что мы отобрали 5,5 л раствора из исходных 6 л. То есть, если бы удалось полностью отделить раствор от небольшого количества осадка, выход сухих наночастиц составил бы 25,94*6/5,5 =28,30 г, что составляет 43,5% от массы окисленного кокса, или 56,6% от массы исходного не окисленного кокса.
Полученный влажный продукт далее использовали в виде водной пасты без высушивания, потому что в результате высушивания происходит сильная агрегация наночастиц.
Распределение частиц по размерам в растворе этого продукта показано на Фиг. 6. Средний размер частиц составляет 39,4 нм.
Таким образом, травление окисленного кокса раствором гидроксида калия дает наночастицы большего размера, чем при травлении перекисью водорода. Однако, для определенных применений, описание которых не относится к заявляемому изобретению, эти наночастицы показывают хорошие результаты.
Элементный анализ продукта по примеру 5 методом энергодисперсионной спектроскопии в сканирующем электронном микроскопе дал следующие результаты (в атомных %):
Углерод =81,13 ат%
Кислород =18,08 ат%
Сера =0,79 ат%.
Вероятно, сера присутствует в виде сульфоновых групп, присоединившихся к углероду в процессе окисления персульфатом аммония в 100%-ной серной кислоте.
На фиг. 7 показана рентгеновская дифрактограмма образца по примеру 6. Виден широкий пик с наиболее интенсивным максимумом при 2teta=25,17°, который соответствует межплоскостному расстоянию 0,356 нм, что немного больше, чем межслоевое расстояние графита (0,3354 нм). Также просматривается небольшое плечо, 2teta=11,6°, соответствующее межплоскостному расстоянию около 0,76 нм. На основании этих данных можно предположить, что материал неоднороден. Судя по меньшей ширине основного пика по сравнению с Фиг. 5 по примеру 4, наночастицы, полученные в результате травления окисленного кокса гидроксидом калия, большего размера, чем при травлении перекисью водорода. Это согласуется с вышеприведенными данными по распределению частиц по размерам.
Пример 7. Получение углеродных наночастиц из окисленного кокса методом травления гидроксидом калия при 250°С.
Повторили синтез углеродных наночастиц как в примере 6, но теперь температуру обработки гидроксидом калия увеличили до 250°С. Все остальные операции проводили в тех же условиях и с теми же количествами реагентов. В результате получили 282,0 г водной пасты углеродных наночастиц, в которой массовое содержание сухого вещества (высушенного при 60°С) составляло 10,55%. Соответственно, выход сухих наночастиц составлял 29,75 г, что равно 59,5% от массы исходного (не окисленного) кокса.
Распределение частиц по размерам показано на Фиг. 8, средний размер частиц равен 43,1 нм.
Проведенные опыты показали, что превышение температуры травления гидроксидом калия выше 250°С приводит к значительному уменьшению выхода растворимых наночастиц, предположительно, вследствие их спекания. С другой стороны, уменьшение температуры травления окисленного кокса ниже 160°С приводит к значительному увеличению размеров частиц, причем, размер становится больше 100 нм, то есть, такие частицы не могут называться наноразмерными. Таким образом, технический результат достигается в интервале температур обработки окисленного кокса гидроксидом калия от 160 до 250°С, причем, оптимальная температура около 200°С.
Углеродные наночастицы, полученные согласно заявляемому изобретению, могут применяться в качестве:
- эффективных адсорбентов органических и неорганических веществ, в том числе органических красителей, соединений тяжелых металлов, радионуклидов;
- добавок в полимеры;
- диспергаторов для углеродных нанотрубок;
- получения нанокомпозиционных материалов;
- получения углеродных аэрогелей;
- после соответствующей обработки, в качестве компонентов электродных материалов химических источников тока.
Возможны и другие применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ активации кокса | 2024 |
|
RU2832352C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДА ГРАФЕНА | 2018 |
|
RU2709594C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАФЕНА | 2016 |
|
RU2657504C2 |
| СПОСОБ МОДИФИЦИРОВАНИЯ УГЛЕРОДНЫХ НАНОТРУБОК | 2012 |
|
RU2528985C2 |
| Способ получения малослойных форм восстановленного оксида графена из графита однореакторным методом | 2024 |
|
RU2829356C1 |
| Способ получения оксида графена | 2022 |
|
RU2796672C2 |
| Способ получения электропроводящего гидрофильного аэрогеля на основе композита из графена и углеродных нанотрубок | 2017 |
|
RU2662484C2 |
| НАНОКОМПОЗИТНЫЙ ДИСПЕРСНЫЙ МАГНИТНЫЙ МАТЕРИАЛ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2010 |
|
RU2426188C1 |
| СПОСОБ ПОЛУЧЕНИЯ АКТИВИРОВАННОГО ВЫСОКОДИСПЕРСНОГО ПРЕПАРАТА ГРАФИТА ДЛЯ ПОКРЫТИЙ НА УЛЬТРАТОНКИХ СТЕКЛЯННЫХ ВОЛОКНАХ | 2014 |
|
RU2583099C1 |
| Способ получения углеродных точек из прекурсора бересты березы | 2020 |
|
RU2727388C1 |
Изобретение относится к химической промышленности, нанотехнологии и охране окружающей среды. Сначала обрабатывают кокс окислителем - персульфатом аммония, в серной кислоте с концентрацией от 98 до 100%. Затем окисленный кокс обрабатывают водным раствором травящего реагента. В качестве травящего реагента используют перекись водорода при температуре обработки от 90 до 100°С и массовом соотношении перекиси водорода к окисленному коксу от 4:1 до 5:1 в расчете на безводные компоненты, после чего полученный раствор выпаривают. В качестве травящего реагента в альтернативном варианте используют гидроксид калия при температуре обработки от 160 до 250°С с последующим растворением полученной реакционной смеси в воде, осаждением углеродных наночастиц добавлением минеральной кислоты до кислого рН, отделением их фильтрацией, промывкой водой и подкисленной азотной кислотой до рН 2,5-3. Предложенный способ получения углеродных наночастиц легко масштабируется и исключает выделение азотной кислоты и токсичных оксидов азота. 2 з.п. ф-лы, 8 ил., 1 табл., 7 пр.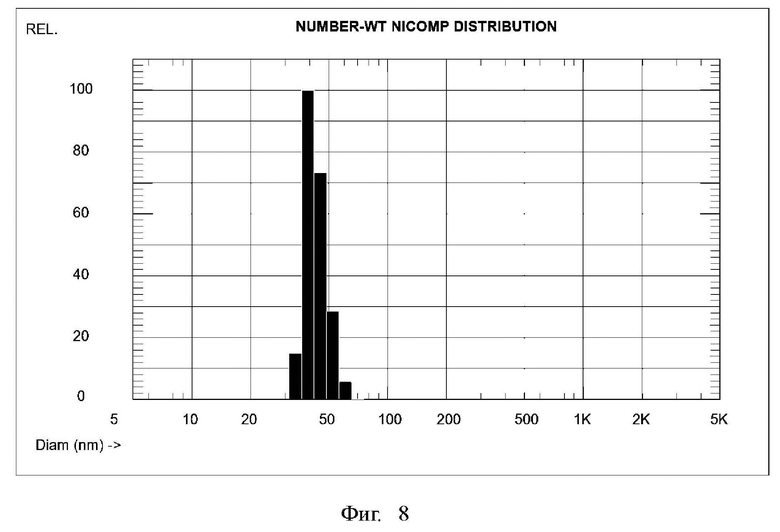
1. Способ получения углеродных наночастиц, включающий обработку кокса окислителем в серной кислоте, отличающийся тем, что в качестве окислителя используют персульфат аммония, в качестве серной кислоты - серную кислоту с концентрацией от 98 до 100%, причем после обработки кокса окислителем проводят обработку окисленного кокса водным раствором травящего реагента, а в качестве последнего используют перекись водорода при температуре обработки от 90 до 100°С или гидроксид калия при температуре обработки от 160 до 250°С.
2. Способ по п. 1, отличающийся тем, что массовое соотношение перекиси водорода к окисленному коксу берут от 4:1 до 5:1 в расчете на безводные компоненты, а после обработки окисленного кокса перекисью водорода полученный раствор выпаривают.
3. Способ по п. 1, отличающийся тем, что реакционную смесь после обработки окисленного кокса раствором гидроксида калия растворяют в воде, осаждают углеродные наночастицы добавлением минеральной кислоты до кислого рН и отделяют наночастицы фильтрацией с последующей промывкой водой, подкисленной азотной кислотой до рН 2,5-3.
| JIUZHOU WANG et al., A facile method to prepare carbon aerogels from amphiphilic carbon material, Mater | |||
| Lett., 2012, v | |||
| Способ получения смеси хлоргидратов опийных алкалоидов (пантопона) из опийных вытяжек с любым содержанием морфия | 1921 |
|
SU68A1 |
| Способ пропитывания дерева | 1921 |
|
SU446A1 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАФЕНА | 2016 |
|
RU2657504C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГРАФЕНОВОГО МАТЕРИАЛА | 2018 |
|
RU2693755C1 |
| ТКАЧЕВ С.В., Восстановленный оксид графена: получение, строение, свойства, Автореферат диссертации на соискание ученой степени кандидата химических наук, | |||

