ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области медицинской химии и области лекарственной терапии, в частности к соединению 2-циано-3,12-диоксоолеан-1,9(11)-диен-17-фенилакриламида и способу их получения; изобретение также относится к их применению в приготовлении противоракового препарата.
УРОВЕНЬ ТЕХНИКИ
Олеаноловая кислота (oleanonic acid, OA) относится к пентациклическим тритерпеноидам природного происхождения, широко распространенным в растительном мире, является активным ингредиентом многих традиционных китайских лекарств и обладает широким спектром биологической активности. Оптимизация структуры на основе OA всегда была одной из горячих точек в исследованиях природной медицинской химии.
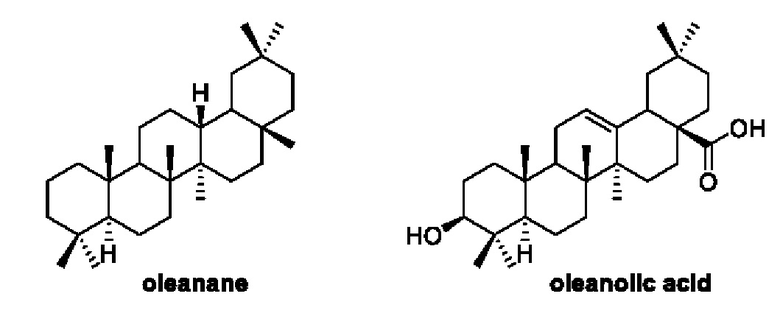
CDDO и его производные (CDDOs) представляют собой полусинтетические производные OA с сильнейшей противоопухолевой и противовоспалительной активностью, обнаруженной на сегодняшний день. В отличие от одноцелевых препаратов, CDDOs полифункциональны, мультитаргетны, действуют на все клеточные сигнальные пути, могут оказывать противоопухолевое действие во множестве процессов, поэтому высокая активность и меньшая резистентность являются преимуществами этих производных. Среди них производное метилового эфира CDDO (2-циан-3,12-диоксолеан-1,9(11)-диен-28-оевая кислота метилового эфира, CDDO-Me), также известное под торговым названием Бардоксолона метила (Bardoxolone Methyl), было испытано в 2006 году в клинических исследованиях по лечению запущенных солидных опухолей и злокачественных лимфом на фазе I (NCT00529438. NCT00508807). Кроме того, в 2008 году проводили утвержденные FDA оценочные исследования CDDO-Me NCT00664027) как препарат для лечения хронической болезни почек (ХБП), вызванной сахарным диабетом типа П. Но, к сожалению, оценка была прекращена в ноябре 2013 года в фазе III клинического исследования из-за высокой летальности, вызванной сердечной недостаточностью. Также сообщалось, что указанное соединение использовалось в клинических исследованиях по легочной артериальной гипертензии (сиротский препарат), утвержденных FDA в 2014 году, в настоящее время находится на фазе III клинического исследования (фаза II NCT02036970; фаза III NCT02657356).
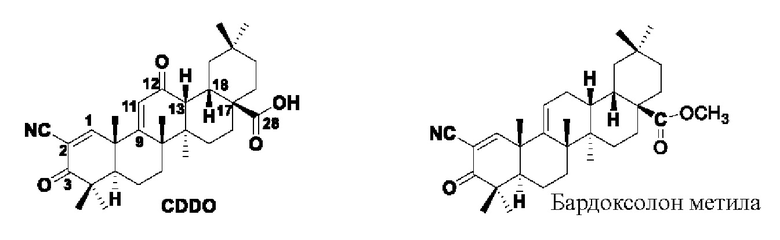
Хотя CDDO-Me обладает хорошей противоопухолевой активностью, его кардиотоксичность все же остается проблемой. Большое практическое значение имело бы сохранение или усиление активности соединений при одновременном снижении их токсичности путем необходимой модификации и реформирования.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение впервые раскрывает соединение 2-циано-3,12-диоксоолеан-1,9(11)-диен-17-фенилакриламида, способ его получения и фармацевтическое применение. Способ получения производного по настоящему изобретению имеет следующие преимущества: сырье дешевое и легкодоступное; реагенты экологически чистые и низкотоксичные; условия мягкие и легко поддающиеся контролю; постобработка удобна и проста; а между тем способ приготовления практичен и выполняем. Согласно результатам фармакологических экспериментов, соединения по настоящему изобретению обладают превосходной противоопухолевой активностью, которая выше или сравнима с противоопухолевой активностью CDDO-Me, а миокардиальная токсичность некоторых соединений значительно снижена. Поэтому указанные соединения могут быть использованы в качестве соединений-кандидатов в противоопухолевые препараты.
Одной из задач настоящего изобретения является разработка соединений 2-циано-3,12-диоксоолеан-1,9(11)-диен-17-фенилакриламида, представленного формулой I:
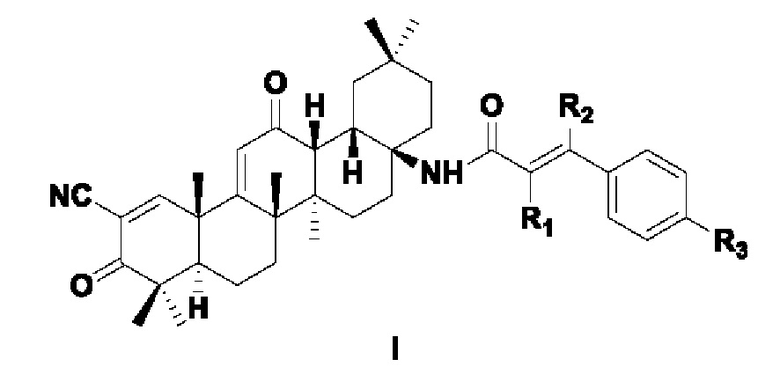
где каждый из R1, R2 и R3 независимо выбран из следующих заместителей: Н, С1-3 алкил, алкокси, галоген, циано, С1-3 алкил, замещенный галогеном или циано, соответственно.
Указанный галоген выбран из F, Cl, Br, I.
Указанная алкоксигруппа представляет собой алкоксигруппу, содержащую 1-3 атома углерода.
В некоторых примерах осуществления настоящего изобретения в указанной формуле I каждый из R1, R2 независимо выбран из Н или циано, соответственно; R3 выбран из Н, С1-3 алкила, алкокси или галогена.
В некоторых примерах осуществления изобретения в указанной формуле I каждый из R1, R2 независимо выбран из Н или циано, соответственно; R3 выбран из F, Cl, Н, метокси.
В некоторых примерах осуществления изобретения в указанной формуле I каждый из R1, R2 в формуле I независимо выбран из Н или циано, соответственно; R3 выбран из Cl, Н.
В некоторых примерах осуществления изобретения в указанной формуле I R1 представляет собой циано, R2 представляет собой Н, a R3 представляет собой Cl или Н.
В другом аспекте настоящее изобретение относится к соединению формулы I, приведенной ниже, или его фармацевтически приемлемой соли:
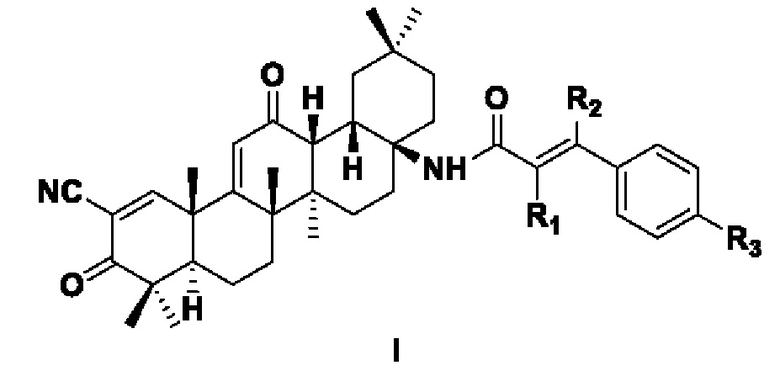
где каждый из R1, R2, R3 независимо выбран из следующих заместителей: Н, С1-6 алкил, С1-6 алкокси, галоген, циано, С1-6 алкил, замещенный галогеном или циано; предпочтительно Н, С1-3 алкил, С1-3 алкокси, галоген, циано, С1-3 алкил, замещенный галогеном или циано.
В некоторых примерах осуществления изобретения в указанной формуле I, каждый из R1, R2 независимо выбран из Н или циано соответственно; R3 выбран из Н, С1-3алкила, алкокси, галогена, С1-3алкила, замещенного галогеном; указанный алкокси представляет собой алкокси, содержащий 1-3 атома углерода; указанный галоген выбран из F, Cl, Br, I.
В некоторых примерах осуществления изобретения в указанной формуле I, R1 представляет собой Н или циано, R2 представляет собой Н или циано, a R3 выбран из Н, метила, метокси, трифторметила, Cl или F.
В некоторых примерах осуществления изобретения в указанной формуле I, R1 представляет собой Н или циано, R2 представляет собой Н или циано, a R3 представляет собой Cl, Н или трифторметил.
В некоторых примерах осуществления соединение формулы I выбрано из следующих соединений:
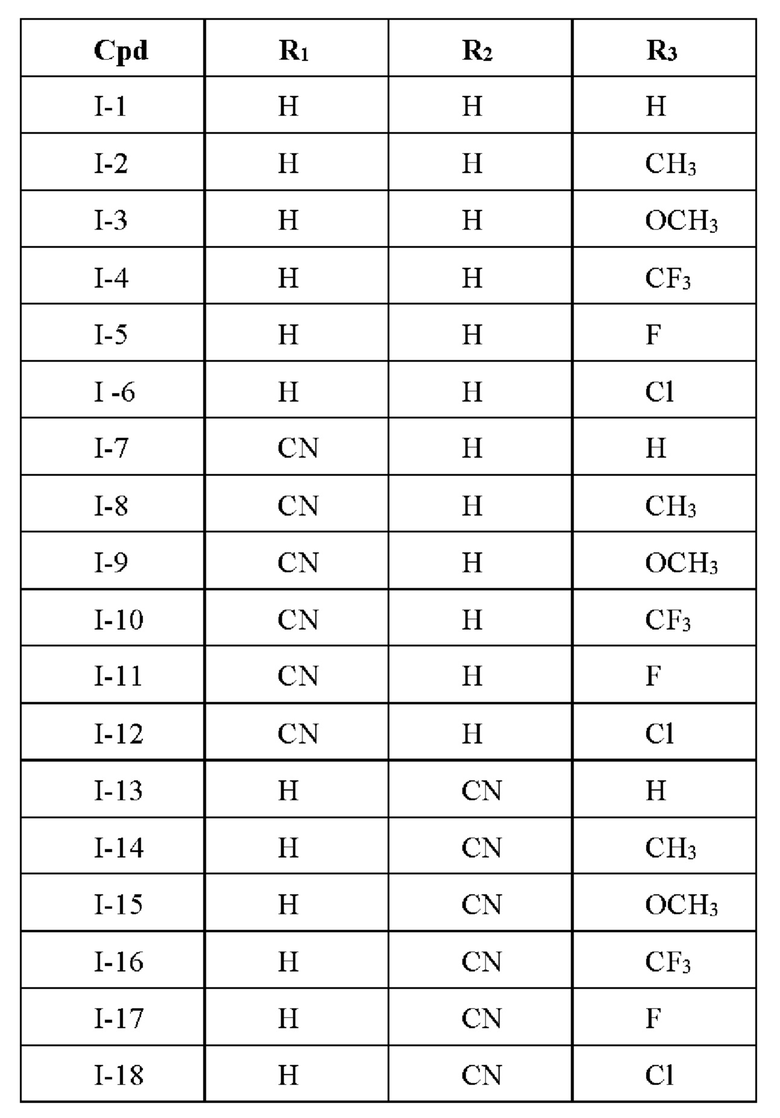
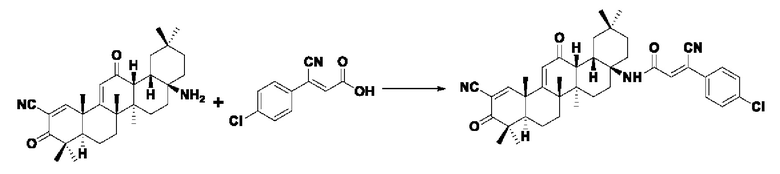
Настоящее изобретение также относится к способу получения указанного соединения, представленного формулой I, отличающемуся тем, что он содержит следующие этапы: 1. Проводили перегруппировку Курциуса CDDO, получено производное (CDDO-NH2), аминированное в положении С-17, со структурой, представленной в формуле II.

2. Соединение формулы I получено посредством реакции конденсации соединения формулы II со соединением формулы Ш (соединение фенилакриловой кислоты):
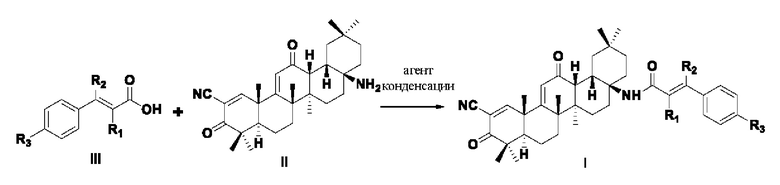
Указанный агент конденсации представляет собой один или несколько агентов, выбранных из дициклогексилкарбодиимида, N,N-диизопропилкарбодиимида, 1-этил-(3-диметиламинопропил) карбодиимида, бензотриазол-1-
илокситрис(диметиламино)фосфония гексафторфосфата или бензотриазол-1-илокситрипирролидин гексафторфосфата;
Растворитель представляет собой один или комбинацию более из следующих, но не ограничиваясь ими: из N,N-диметилформамида, ацетона, ацетонитрила, толуола, бензола, диметилбензола, 1,4-диоксана, этилацетата, дихлорметана, хлороформа, тетрагидрофурана или эфира; предпочтительная температура реакции составляет 0°С~60°С.
Еще одной задачей настоящего изобретения является разработка фармацевтической композиции, содержащей эффективную дозу соединения по настоящему изобретению или его фармацевтически приемлемой соли и один или несколько фармацевтически приемлемых фармацевтических эксципиентов. Соединения по настоящему изобретению могут быть приготовлены в виде различных дозированных форм, таких как таблетки, капсулы, гранулы, жидкие препараты и т.д., отдельно или с одним или несколькими фармацевтически приемлемыми носителями для клинического перорального, инъекционного или местного введения. В этих различных составах соединение по настоящему изобретению может присутствовать в количестве 0,1%-99,9%. Дозировка соединения по настоящему изобретению может составлять 0,00I-10000 мг/кг/0,3 дня, и ее можно соответствующим образом корректировать в соответствии с клиническими потребностями.
Термин "фармацевтически приемлемая соль", используемый в настоящем изобретении, в настоящем изобретении относится к фармакологически приемлемой соли присоединения кислоты или основания, или их соединения с растворителем.
Еще другой задачей настоящего изобретения является разработка применения таких соединений или их фармацевтически приемлемых солей и их фармацевтических композиций для получения противоопухолевых препаратов. Указанные опухоли выбираются из рака легкого, рака печени, рака толстой кишки, рака поджелудочной железы, рака молочной железы, рака предстательной железы, рака головного мозга, рака яичников, рака шейки матки, рака яичек, рака почки, рака головы и шеи, лимфомы, меланомы или лейкемии. В частности, Указанные опухоли выбираются из рака легкого, рака печени или рака молочной железы. Предпочтительно рак легких. Ряд тестов на опухолевые клетки показывает, что соединения по настоящему изобретению обладают противоопухолевой активностью широкого спектра действия со значениями IC50 в наномолярном и микромолярном диапазоне, сравнимыми с активностью положительного контрольного препарата CDDO-Me. Среди них токсичность соединений I-7 и I-12 значительно менее токсичны для эмбриональных кардиомиоцитов крысы Н9С2 (значения IC50 3,176±1,74 мкМ и 3,143±1,53 мкМ, соответственно), чем CDDO-Me (IC50 0,308±0,01 мкМ).
Одной из задач настоящего изобретения является применением указанных соединений или их фармацевтически приемлемых солей и их фармацевтических композиций для получения противоопухолевых препаратов. Указанные опухоли выбираются из рака легких, рака печени, асцитной опухоли, метастазов в мозг, рака толстой кишки, рака поджелудочной железы, рака груди, рака простаты, рака мозга, рака яичников, рака шейки матки, рака яичек, рака почек, рака головы и шеи, лимфомы, меланомы или лейкемии; предпочтительно из рака легких, рака печени, рака груди, асцитной опухоли, рака поджелудочной железы, метастазов в мозг; более предпочтительно из немелкоклеточного рака легких, рака печени, рака груди, асцитной опухоли. Ряд тестов на опухолевые клетки показывает, что соединения по настоящем изобретению обладают противоопухолевой активностью широкого спектра действия со значениями IC50 в наномолярном и микромолярном диапазоне, сравнимыми с активностью положительного контрольного препарата CDDO-Me. Среди них токсичность соединений I-7 и I-12 на эмбриональные кардиомиоциты крысы Н9С2 (значения IC50 3,176±1,74 мкМ и 3,143±1,53 мкМ, соответственно) значительно ниже, чем у CDDO-Me (IC50 0,308±0,01 мкМ); кроме того, соединения I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, I-12, I-13, I-14, I-16, I-18 проявили более низкую токсичность для эпителиальных клеток почечных канальцев человека HK-2.
Дальнейшей задачей настоящего изобретения является разработка указанных соединений или фармацевтически приемлемых их солей и фармацевтических композиций, которые используются против опухолей. Указанные опухоли выбираются из рака легких, рака печени, асцитной опухоли, метастазов в мозг, рака толстой кишки, рака поджелудочной железы, рака груди, рака простаты, рака мозга, рака яичников, рака шейки матки, рака яичек, рака почек, рака головы и шеи, лимфомы, меланомы или лейкемии; предпочтительно из рака легких, рака печени, рака груди, асцитной опухоли, рака поджелудочной железы, метастазов в мозг; более предпочтительно из немелкоклеточного рака легких, рака печени, рака груди, асцитной опухоли. Ряд тестов на опухолевые клетки показал, что соединения по настоящем изобретению обладают противоопухолевой активностью широкого спектра действия со значениями IC50 в наномолярном и микромолярном диапазоне, сравнимыми с активностью положительного контрольного препарата CDDO-Me. Среди них токсичность соединений I-7 и I-12 на эмбриональные кардиомиоциты крысы Н9С2 (значения IC50 3,176±1,74 мкМ и 3,143±1,53 мкМ, соответственно) значительно ниже, чем у CDDO-Me (IC50 0,308±0,01 мкМ); более того, соединения I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, I-12, I-13, I-14, I-16 и I-18 проявили более низкую токсичность для эпителиальных клеток почечных канальцев человека HK-2.
Другой задачей настоящего изобретения является разработка способа лечения опухолей, включающего введение субъекту или пациенту терапевтически эффективного количества указанных соединений или их фармацевтически приемлемых солей и их фармацевтических композиций. Указанные опухоли выбираются из рака легких, рака печени, асцитной опухоли, метастазов в мозг, рака толстой кишки, рака поджелудочной железы, рака груди, рака простаты, рака мозга, рака яичников, рака шейки матки, рака яичек, рака почек, рака головы и шеи, лимфомы, меланомы или лейкемии; предпочтительно из рака легких, рака печени, рака груди, асцитной опухоли, рака поджелудочной железы, метастазов в мозг; более предпочтительно из немелкоклеточного рака легких, рака печени, рака груди, асцитной опухоли. Ряд тестов на опухолевые клетки показал, что соединения по настоящем изобретению обладают противоопухолевой активностью широкого спектра действия со значениями IC50 в наномолярном и микромолярном диапазоне, сравнимыми с активностью положительного контрольного препарата CDDO-Me. Среди них токсичность соединений I-7 и I-12 на эмбриональные кардиомиоциты крысы Н9С2 (значения IC50 3,176±1,74 мкМ и 3,143±1,53 мкМ, соответственно) значительно ниже, чем у CDDO-Me (IC50 0,308±0,01 мкМ); более того, соединения I-1, I-2, I-3, I-4, I-5, I-6, I-7, I-8, I-9, I-12, I-13, I-14, I-16 и I-18 проявили более низкую токсичность для эпителиальных клеток почечных канальцев человека HK-2.
Хотя предпочтительные примеры осуществления изобретения были показаны и описаны в настоящем документе, специалистам в данной области будет очевидно, что такие примеры осуществления представлены только в качестве примера. Многие изменения, вариации и альтернативы, которые, не отходя от сущности и объема изобретения, теперь придут на ум специалистам в данной области. Следует понимать, что для реализации настоящего изобретения могут быть использованы различные альтернативные примеры осуществления изобретения, описанные в настоящем документе. Прилагаемая формула изобретения предназначена для ограничения объема настоящего изобретения и, таким образом, охватывает способы и конструкции, и их эквивалентные формы в пределах объема правовой охраны данного изобретения, определенного формулой изобретения.
Некоторые химические термины
Если не указано иное, все технические и научные термины, используемые в настоящем документе, имеют такое же значение, которое обычно понимается специалистами в данной области техники. Если не указано иное, все патенты, патентные заявки и опубликованные материалы, цитируемые в настоящем документе, включены в настоящий документ во всей полноте посредством ссылки.
Следует понимать, что вышеизложенное общее описание и последующее подробное изложение являются лишь примерами и пояснениями, и не ограничивают ни один из заявленных объектов изобретения, в настоящей заявке использование единственного числа включает множественное, если специально не указано иное. Следует отметить, что используемое в настоящем описании и прилагаемой формуле изобретения указания на единственное число предполагает указание и на множественное число, если только контекст четко не указывает на обратное. Следует также отметить, что применение «или» означает «и/или», если не указано иное. Кроме того, использование термина «включая», а также других его форм, таких как «включает», «включают» и «включали», не является ограничивающим.
Определение стандартных химических терминов можно найти в работах, на которые сделана ссылка, включая, без ограничения, Carey and Sundberg "Advanced Organic Chemistry 4thEd." Vols. A (2000) and В (2001), Plenum Press, New York. Если не указано иное, используются стандартные способы масс-спектроскопии, ядерного магнитного резонанса (NMR), высокоэффективной жидкостной хроматографии (HPLC), инфракрасного (IR) спектроскопического анализа и UV/Vis-спектроскопии, и фармакологии, известные в области техники. Если не указаны специальные определения, номенклатура, используемая в настоящей заявке, является известной в области техники. Стандартные Способ могут использоваться для химического синтеза, химических анализов, получения, изготовления и доставки фармацевтических средств и лечения пациентов. Реакции и Способ очистки могут быть проведены, например, с применением Набор реагентов согласно инструкциям изготовителя, или так, как это обычно выполняется в данной области техники, или как описано в настоящей заявке. Вышеизложенные методики и процедуры могут в целом выполняться обычными способами, известными в данной области техники, и как описано в различных общих и более конкретных источниках, цитируемых и обсуждаемых в описании данного изобретения. Во всем описании группы и их заместители могут быть выбраны специалистом в данной области таким образом, чтобы получить стабильные фрагменты и соединения.
Когда заместитель описывается обычной химической формулой, написанной слева направо, заместитель также включает химически эквивалентный заместитель, полученный при записи структурной формулы справа налево. Например, СН2О эквивалентен ОСН2.
Терминал "соединение" в настоящей заявке может содержать все стереоизомеры, геометрические изомеры, изомеры взаимопревращений и изотопы. Соединения по настоящей заявке могут быть асимметричными, например, иметь один или более стереоизомеров. Если не указано иное, включены все стереоизомеры, такие как энантиомеры и диастереоизомеры. Соединения по настоящей заявке, содержащие асимметрично замещенные атомы углерода, могут быть выделены в оптически активной чистой форме или в рацемической форме. Оптически активная чистая форма может быть выделена из рацемической смеси или синтезирована с использованием хирального сырья или хиральных реагентов. Соединения по настоящей заявке также включают взаимно изменяющуюся изомерную форму. Взаимные изомерные формы возникают в результате обмена одной связи с соседней двойной связью и вместе с миграцией протона. Соединения по настоящей заявке также включают все атомы изотопа, либо в промежуточном соединении, либо в конечном соединении. Атомы изотопов включают атомы, имеющие одинаковый атомный номер, но разные массовые числа. Например, изотопы водорода включают тритий и дейтерий. То есть, соединения по настоящей заявке включают соединения, в которых часть или весь водород (Н) заменен тритием (Т) и/или дейтерием (D); также соединения, в которых часть или весь 12С заменен 13С и/или 14С; и соединения, в которых другие изотопы (например, N, О, Р, S) заменены друг на друга, например, 14N и 15N, 18О и 17О; 31Р и 32Р; 35S и 36S, и т.д. Соединения, описанные в настоящем документе, могут содержать один или более стереоцентров, и каждый центр может существовать в R или S-конфигурации или их комбинациях. Подобным образом, соединения, представленные в настоящем описании, могут содержать одну или более двойных связей, и каждая может существовать в Е (транс) или Z (цис)-конфигурации или их комбинациях. Представление одного конкретного стереоизомера, региоизомера, диастереомера, энантиомера или эпимера следует понимать как включающее все возможные стереоизомеры, региоизомеры, диастереомеры, энантиомеры или эпимеры и их смеси. Таким образом, соединения, представленные в настоящем описании, включают все отдельные конфигурационные стереоизомерные, региоизомерные, диастереомерные, энантиомерные и эпимерные формы, также как их соответствующие смеси. Методики обращения или оставления без изменений определенного стереоцентра и методики для разделения смесей стереоизомеров хорошо известны в данной области техники, и специалист в данной области техники будет в состоянии выбрать соответствующий метод для конкретной ситуации.
Термин "необязательный/произвольный " или "необязательно/произвольно", что в дальнейшем описанное явление или состояние может или не может возникнуть и что описание включает случаи, когда указанное явление или состояние происходит, и случаи, когда не происходит.
Как используется в настоящем документе, C1-3 означает, что данная часть содержит 1-3 атома углерода, т.е. группа содержит 1 атом углерода, 2 атома углерода, 3 атома углерода. Так, например, "C1-С4 алкил" означает алкильную группу с 1-4 атомами углерода, т.е. указанная алкильная группа выбирается из метила, этила, пропила, изопропила, н-бутила, изобутила, сек-бутила и трет-бутила.
Используемый в настоящей заявке термин "алкил", отдельно или в комбинации, относится к алифатическому углеводороду с необязательно замещенными прямыми цепями или необязательно замещенными разветвленными цепями. Предпочтительно, "алкил" в настоящей заявке может иметь I-20 атомов углерода примерно, например, 1-10 атомов углерода, I-8 атомов углерода, или I-6 атомов углерода, или I-4 атомов углерода, или 1-3 атомов углерода. Примеры алкильных групп в настоящем документе включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил и т.д. Определенные группы, такие как "алкил", когда появляется числовой диапазон, например, "C1-С6 алкил" или "C1-6 алкил", относятся к алкильным группам, которые состоят из 1 атома углерода, 2 атомов углерода, 3 атомов углерода. Термин "алкил" относится к алкильным группам, которые могут состоять из 1 атома углерода, 2 атомов углерода, 3 атомов углерода, 4 атомов углерода, 5 атомов углерода или 6 атомов углерода, и включает случаи, когда числовой диапазон не указан.
Как используется в данном документе, "алкил" включает алкильные группы в сочетании с другими группами, например, алкил в алкокси.
Используемый в настоящей заявке термин "галоген", отдельно или в комбинации, выбран из F, Cl, Br, I.
Используемые в настоящей заявке термины "галогенированный" или "галогензамещенный", по отдельности или в сочетании, означают, что один или несколько атомов водорода опционально замещенной группы (например, алкил, алкенил и алкинил) замещены атомами фтора, хлора, брома, йода или их комбинацией. В некоторых примерах осуществления изобретения два или более атомов водорода замещаются атомами галогена, которые идентичны друг другу (например, дифторметил, трифторметил); в других примерах осуществления изобретения два или более атомов водорода замещаются атомами галогена, которые не идентичны Друг Другу (например, I-хлор-I-фтор-I-йодоэтил). Неограничивающими примерами галоалкила являются фторметил и бромэтил. Неограничивающими примерами галоалкенила являются бромовинилы. Неограничивающими примерами галоалкинилов являются хлорэтинилы.
Используемые в настоящем описании термины "лечить", "лечащий" или "лечение" и другие грамматические эквиваленты включают облегчение, смягчение или уменьшение интенсивности симптомов заболевания или состояния, предотвращение дополнительных симптомов, уменьшение интенсивности или предотвращение лежащих в основе метаболических причин симптомов, ингибирование заболевания или состояния, например, прекращение развития заболевания или состояния, облегчение заболевания или состояния, вызывание ремиссии заболевания или состояния, облегчение состояния, вызванного заболеванием или состоянием, или прекращение симптомов заболевания или состояния и предназначены для включения профилактики. Термины дополнительно включают достижение терапевтического эффекта и/или профилактического эффекта. Под терапевтическим эффектом подразумевается устранение или уменьшение интенсивности основного нарушения, подлежащего лечению. Кроме того, терапевтический эффект достигается при устранении или уменьшении интенсивности одного или более физиологических симптомов, связанных с основным нарушением, так что у пациента наблюдается улучшение несмотря на то, что пациент все еще может страдать от основного нарушения. Для профилактического эффекта композиции могут вводиться пациенту с риском развития конкретного заболевания или пациенту, жалующемуся на один или более физиологических симптомов заболевания, даже если диагноз данного заболевания может быть не поставлен.
Как используется в настоящем документе, термины "эффективное количество", "терапевтически эффективное количество" или "фармацевтически эффективное количество" относятся к количеству одного или более активных веществ (таких как соединение по настоящей заявке), которое при введении достаточно для обеспечения некоторого облегчения одного или нескольких нежелательных физиологический изменений или нарушений подлежащего лечению. Результатом может быть уменьшение и/или ремиссия признаков, симптомов или причин заболевания, или любое другое желаемое изменение в биологической системе. Например, "эффективное количество" для терапевтических целей - это количество композиции, включающей раскрытые в настоящем документе соединения, необходимое для обеспечения клинически значимого облегчения состояния. Эффективное количество, подходящее для применения в каждом конкретном случае, может быть определено с помощью таких методов, как тесты на эскалацию дозы.
Используемый в настоящем описании термин "приемлемый " применительно к составу, композиции или ингредиенту означает не имеющий постоянного негативного воздействия на общее состояние здоровья объекта, подлежащего лечению.
Термин "фармацевтически приемлемый", используемый здесь, относится к веществу (такому как носитель или разбавитель), которое не влияет на биологическую активность или свойства соединений по настоящей заявке и является относительно нетоксичным, т.е. вещество может вводить индивидуумам, не вызывая неблагоприятных эффектов биологической реакции или взаимодействуя нежелательным образом с любым компонентом, содержащимся в композиции.
Используемый в настоящем описании термин "фармацевтическая композиция" относится к биологически активному соединению, необязательно смешанному, по меньшей мере, с одним фармацевтически приемлемым химическим компонентом, таким как, но не ограничиваясь ими, носители, стабилизаторы, разбавители, диспергирующие агенты, суспендирующие агенты, загустители и/или эксципиенты.
Термин "носитель", используемый в настоящем документе, относится к относительно нетоксичному веществу, которое облегчает введение соединений по настоящей заявке в клетки или ткани.
Используемый здесь термин "фармацевтически приемлемые соли " относится к солям, которые сохраняют биологическую потенцию свободной кислоты и свободного основания указанного соединения и не оказывает неблагоприятного биологического или иного воздействия. Соединения по настоящей заявке также включают фармацевтически приемлемые соли. Фармацевтически приемлемые соли относится к форме, в которой основной радикал в исходном соединении превращается в соль. Фармацевтически приемлемые соли включают без ограничения, соли основного радикала, такие как неорганические или органические соли аминовой (амино) группы. Фармацевтически приемлемые соли по настоящей заявке могут быть синтезированы из исходного соединения, т.е. основные группы в исходном соединении реагируют с 1-4 эквивалентами кислоты в системе растворителя. Подходящие соли перечислены в Remingtong's Pharmaceutical Scicences, 17 edth, Mack Publishing Company, Easton, Pa., 1985, p. 1418 и Journal of Pharmaceutical Science, 66, 2 (1977).
Если не указано иное, соли в данной заявке относятся к кислотным солям, образованным с органическими кислотами/неорганическими кислотами, и основным солям, образованным с органическими основаниями/неорганическими основаниями. Кроме того, амфотерные ионы (внутренние соли) образуются, когда основная функциональная группа общего соединения представляет собой пиридин или имидазол (но не ограничиваясь пиридином или имидазолом), а кислотная функциональная группа представляет собой карбоновую кислоту (но не ограничиваясь карбоновой кислотой), амфотерные ионы также включены в соли в данной заявке.
ПОДРОБНОЕ ОПИСАНИЕ ПРИМЕРОВ РЕАЛИЗАЦИИ
Для того чтобы более четко проиллюстрировать настоящее изобретение, здесь приводится ряд примеров. Эти примеры являются иллюстративными и не должны рассматриваться как ограничивающие настоящее изобретение.
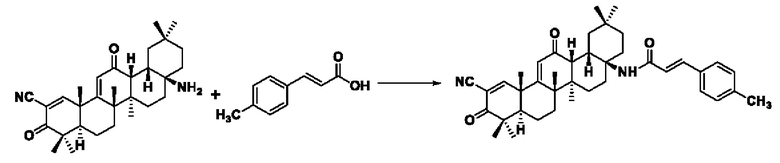
Пример 1: Синтез I-1
Растворяли 0,080 г (0,170 ммоль) CDDO-NH2 в 5 мл дихлорметана, добавляли 0,046 г (0,260 ммоль) 3-фенилакрил, перемешивали, добавляли 0,177 г бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфат (РуВор, 0,340 ммоль), N,N-диизопропилэтиламин 0,074 мл (0,425 ммоль), реакция была завершена при комнатной температуре в течение 12 часов. После обнаружения с помощью TLC того, что реакция завершена полностью, добавляли 5 мл воды и 5 мл дихлорметана для экстракции и разделения, водный слой экстрагировали с помощью 5 мл дихлорметана, объединяли органические слои. Органический слой промывали 5 мл насыщенного раствора хлорида натрия и сушили над безводным сульфатом натрия. После вакуумной фильтрации, ротационного испарения, колоночной хроматографии органического слоя получали 0,036 г соединения в виде желто-белого твердого вещества с выходом 44,9%.
ESI-MS: 593.4 [М+Н]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.89 (3H, s), 1.04 (3H, s), 1.05 (3H, s), 1.17 (3H, s), 1.26 (3H, s), 1.42 (3H, s), 1.47 (3H, s), 6.01 (1H, s), 6.53 (1H, d,J=15.60 Hz), 7.35 (3H, m), 7.48 (2H, m), 7.60 (1H, d,J=15.54 Hz), 8.08 (1H, s).

Пример 2: Синтез I-2
Синтез производился в соответствии с способом получения соединения I-1, заменяли 3-(4-метилфенил)акрил на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,024 г целевого соединения I-2 в виде бледно-желтого твердого вещества, с выходом 28,8%.
ESI-MS: 607.4 [М+Н]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.90 (3H, s), 1.05 (6H, s), 1.17 (3H, s), 1.27 (3H, s), 1.41 (3H, s), 1.47 (3H, s), 2.36 (3H, s), 6.01 (1H, s), 6.43 (1H, d,J=12.60 Hz), 7.19 (2H, s), 7.38 (2H, s), 7.57 (1H, d,J=15.27 Hz), 8.06 (1H, s).
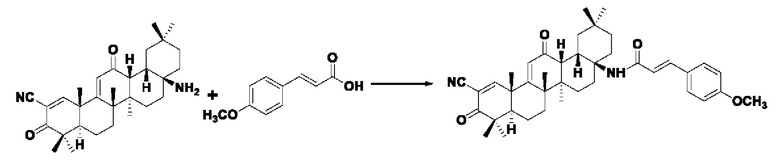
Пример 3: Синтез I-3
Синтез производился в соответствии с способом получения соединения I-1, заменяли 3-(4-метоксифенил)акрил на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,035 г целевого соединения I-3 в виде желто-белого твердого вещества с выходом 41,2%.
ESI-MS: 623.4 [M+H]+, 645.4 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.92 (3H, s), 1.06 (6H, s), 1.18 (3H, s), 1.27 (3H, s), 1.42 (3H, s), 1.48 (3H, s), 3.84 (3H, s), 6.01 (1H, s), 6.90 (1H, d,J=8.49 Hz), 7.45 (1H, d,J=8.43 Hz), 7.58 (4H, m), 8.05 (1H, s).
Пример 4: Синтез I-4
Синтез производился в соответствии с способом получения соединения I-1, заменяли 3-(4-трифторметилфенил)акрил на 3-фенилакрил в качестве реакционного реагента при неизменных условиях было получено 0,018 г целевого соединения I-4 в виде бледно-желтого твердого вещества с выходом 20,4%.
ESI-MS: 661.4 [М+Н]+, 683.4 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.91 (3H, s), 1.06 (3H, s), 1.07 (3H, s), 1.18 (3H, s), 1.27 (3H, s), 1.44 (3H, s), 1.49 (3H, s), 6.03 (1H, s), 6.54 (1H, d,J=15.30 Hz), 7.61 (4H, m), 7.63 (1H, d,J=12.96 Hz), 8.09 (1H, s).

Пример 5: Синтез I-5
Синтез производился в соответствии с способом получения соединения I-1, заменяли 3-(4-фторфенил)акрил на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,019 г целевого соединения I-5 в виде бледно-желтого твердого вещества, с выходом 23,4%.
ESI-MS: 611.4 [М+Н]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.89 (3H,s), 1.05 (3H,s), 1.06 (3H, s), 1.17(3H, s), 1.26 (3H, s), 1.45 (3H, s), 1.49 (3H, s), 6.01 (1H, s), 6.55 (1H, d, J=8.58 Hz), 7.59 (2H, s), 7.64 (2H, s), 7.63 (1H, d,J=8.61 Hz), 8.09 (1H, s).
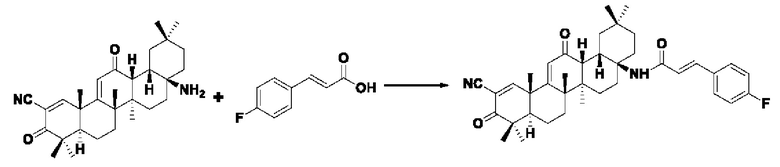
Пример 6: Синтез I-6
Синтез производился в соответствии с способом получения соединения I-1, заменяли 3-(4-хлорфенил)акрил на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,025 г целевого соединения I-6 в виде желто-белого твердого вещества, с выходом 29,1%.
ESI-MS: 627.3 [М+Н]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.90 (3H, s), 1.05 (3H, s), 1.06 (3H, s), 1.18 (3H, s), 1.27 (3H, s), 1.44 (3H, s), 1.49 (3H, s), 6.03 (1H, s), 6.53 (1H, d, J=8.66 Hz), 7.62 (2H, s), 7.68 (2H, s), 7.61 (1H, d,J=8.64 Hz), 8.09 (1H, s).
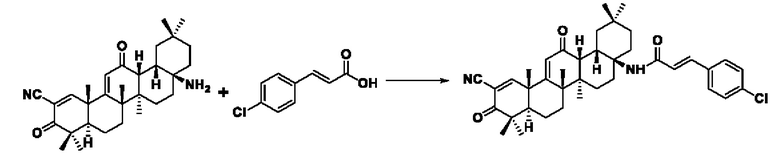
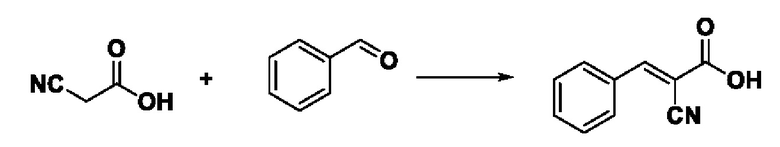
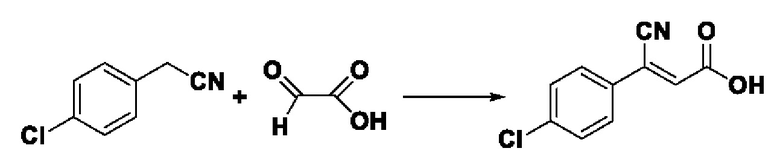
Пример 7: Синтез промежуточного соединения III-1 2-циано-3-фенилакрила Синтез промежуточного соединения III-1 2-циано-3-фенилакриловой кислоты
Растворяли 0,425 г (5 ммоль) цианоуксусной кислоты и 0,477 г (4,5 ммоль) бензальдегида в 20 мл толуола, добавляли 0,058 г (0,75 ммоль) ацетата аммония, нагревают проточным способом, реакция была завершена в течение 6 7 ч, завершение реакции определяли с помощью TLC. После охлаждения до комнатной температуры производили фильтрацию, фильтровальную лепешку сушили в вакууме, получали 0,643 г промежуточного соединения Ш-1 в виде белого твердого вещества с выходом 82,4%.
ESI-MS: 174.0 [М+Н]+.
Формула реакции выглядит следующим образом.
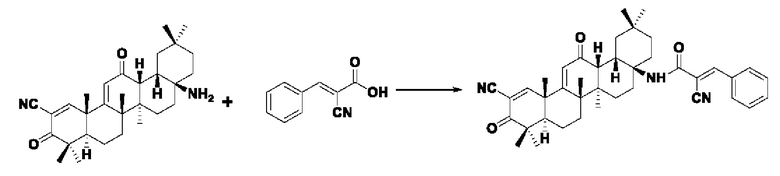
Пример 8: Синтез I-7
Синтез производился в соответствии с способом получения соединения I-1, заменяли 2-циано-3-фенилакрил на 3-фенилакрил в качестве реакционного реагента при прочих неизменных условиях было получено 0,026 г целевого соединения I-7, бледно-желтого твердого вещества, с выходом 30,7%.
ESI-MS: 618.4 [М+Н]+, 640.4 [M+Na]+, 616.4 [М-Н]-
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.93 (3H, s), 1.06 (3H, s), 1.13 (3H, s), 1.18 (3H, s), 1.27 (3H, s), 1.34 (3H, s), 1.49 (3H, s), 6.01 (1H, s), 7.35 (3H, m), 7.48 (2H, m), 8.06 (1H, s), 8.21 (1H, s).
Пример 9: Синтез промежуточного соединения III-2 2-циано-3-(4-метилфенил) акрила
Синтез производился в соответствии с способом синтеза промежуточного соединения III-1 2-циано-3-фенилакрил, заменяли 4-метилбензальдегида на бензальдегид, другие условия оставались неизменными, было получено 0,727 г промежуточного соединения III-2 с выходом 86,3%.
ESI-MS: 188.0 [М+Н]+.
Формула реакции выглядит следующим образом.
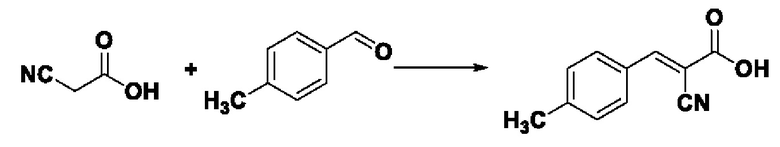
Пример 10: Синтез I-8
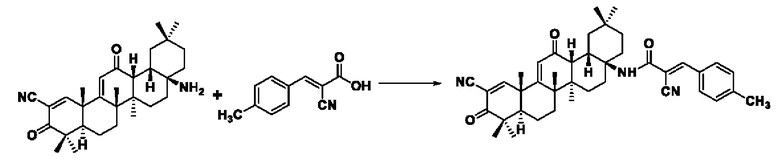
Синтез производился в соответствии с способом I-1, заменяли 2-циано-3-(4-метилфенил)акрил на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,031 г целевого соединения I-8, бледно-желтого твердого вещества, с выходом 35,9%.
ESI-MS: 632.4 [М+Н]+, 654.4 [M+Na]+, 630.4 [М-Н]-
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.93 (3H, s), 1.06 (3H, s), 1.13 (3H, s), 1.18 (3H, s), 1.27 (3H, s), 1.34 (3H, s), 1.49 (3H, s), 2.43 (3H, s), 6.01 (1H, s), 6.99 (2H, d, J=8.48 Hz), 7.92 (2H, d, J=8.61 Hz), 8.06 (1H, s), 8.21 (1H, s).
Пример 11: Синтез промежуточного соединения II I-32-циано-3-(4-метоксифенил) акрила
Синтез производился в соответствии с способом синтеза промежуточного соединения III-1 2-циано-3-фенилакрила, заменяли 4-метоксилбензальдегид на бензальдегид, другие условия оставались неизменными, было получено 0,794 г промежуточного соединения III-3 с выходом 86,8%.
ESI-MS: 204.0 [М+Н]+
Формула реакции выглядит следующим образом.
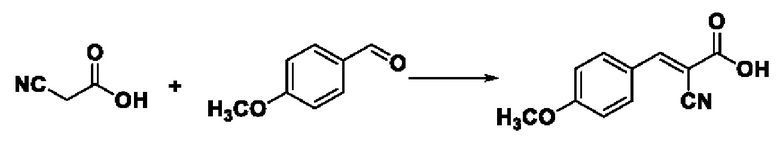
Пример 12: Синтез I-9
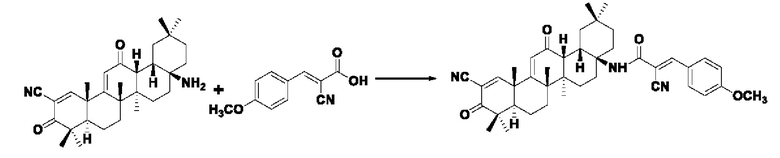
Синтез производился в соответствии с способом I-1, заменяли2-циано-3-(4-метоксифенил)акрил на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,030 г целевого соединения I-9 в виде желтого твердого вещества с выходом 33,7%.
ESI-MS: 648.4 [М+Н]+, 670.4 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.93 (3H, s), 1.06 (3H, s), 1.12 (3H, s), 1.17 (3H, s), 1.27 (3H, s), 1.34 (3H, s), 1.49 (3H, s), 3.11 (3H, s), 6.01 (1H, s), 6.99 (2H, d, J=8.49 Hz), 7.92 (2H, d,J=8.67 Hz), 8.06 (1H, s), 8.21 (1H, s).
Пример 13: Синтез промежуточного соединения III-4 2-циано-3-(4-трифторметилфенил)акрила
Синтез производился в соответствии с способом синтеза промежуточного соединения III-1 2-циано-3-фенилакрила, заменяли 4-трифторметилбензальдегид на бензальдегид, другие условия оставались неизменными, было получено 0,872 г промежуточного соединения III-4 с выходом 80,4%.
ESI-MS: 242.0 [М+Н]+.
Формула реакции выглядит следующим образом.
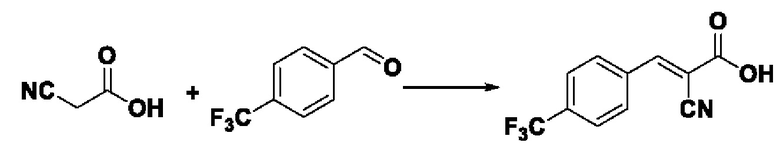
Пример 14: Синтез I-10
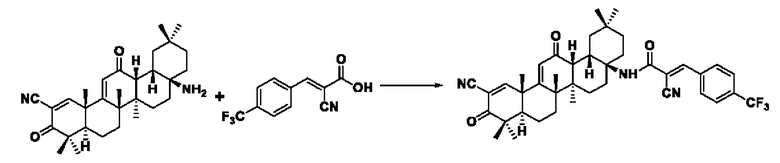
Синтез производился в соответствии с способом I-1, заменяли 2-циано-3-(4-трифторметилфенил)акрил на 3-фенилакрил в качестве реакционного реагента, а другие условия оставались неизменными, было получено 0,031 г целевого соединения I-10, белое твердое вещество, с выходом 32,9%.
ESI-MS: 686.4 [М+Н]+, 684.4 [М-Н]-
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.91 (3H, s), 1.02 (3H, s), 1.08 (3H, s), 1.17 (3H, s), 1.27 (3H, s), 1.44 (3H, s), 1.50 (3H, s), 6.05 (1H, s), 7.68 (2H, d,J=8.07 Hz), 7.88 (2H, d,J=8.01 Hz), 8.09 (1H, s).
Пример 15: Синтез промежуточного соединения III-5 2-циано-3-(4-фторфенил) акрила
Синтез производился в соответствии с способом синтеза промежуточного соединения III-1 2-циано-3-фенилакрила, заменяли 4-фторбензальдегид на бензальдегид, другие условия оставались неизменными, было получено 0,683 г промежуточного соединения III-5 с выходом 79,4%.
ESI-MS: 192.0 [М+Н]+.
Формула реакции выглядит следующим образом.
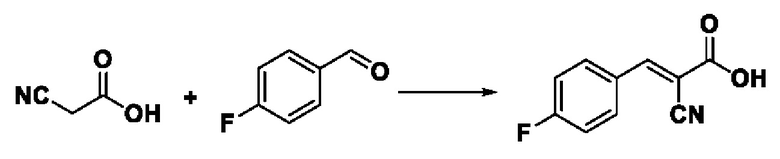
Пример 16: Синтез I-11
Синтез производился в соответствии с способом I-1, заменяли 2-циано-3-(4-фторфенил)акрил на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,022 г целевого соединения I-11 в виде бледно-желтого твердого вещества с выходом 25,4%.
ESI-MS: 636.4 [М+Н]+, 658.4 [М+Na]+, 635.4 [М-Н]-.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.94 (3H, s), 1.07 (3H, s), 1.13 (3H, s), 1.18 (3H, s), 1.27 (3H, s), 1.44 (3H, s), 1.50 (3H, s), 6.05 (1H, s), 7.21 (4H, m), 8.06 (1H, s), 8.26 (1H, s).
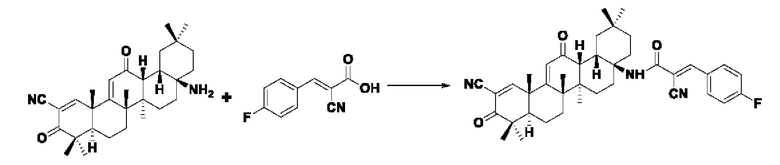
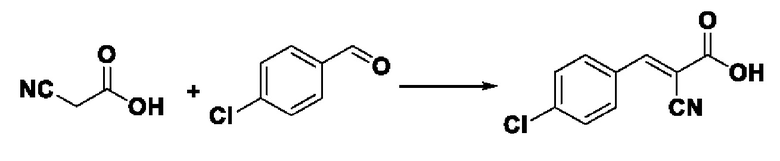
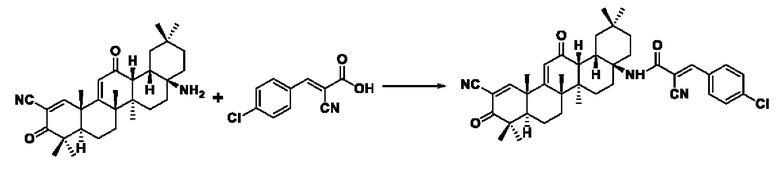
Пример 17: Синтез промежуточного соединения II I -6 2-циано-3-(4-хлорфенил) акрила
Синтез производился в соответствии с способом синтеза промежуточного продукта III-1 2-циано-3-фенилакрил, заменяли 4-хлорбензальдегид на бензальдегид, другие условия оставались неизменными, было получено 0,712 г промежуточного продукта III-6 с выходом 76,2%.
ESI-MS: 208.0 [М+Н]+.
Формула реакции выглядит следующим образом.
Пример 18: Синтез I-12
Синтез производился в соответствии с способом I-1, заменяли 2-циано-3-(4-хлорфенил)акрил на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,033 г целевого соединения I-12 в виде желто-белого твердого вещества, с выходом 37,9%.
ESI-MS: 652.3 [М+Н]+, 674.3 [M+Naf, 650.3 [М-Н]-
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.92 (3H, s), 1.02 (3H, s), 1.08 (3H, s), 1.17 (3H, s), 1.27 (3H, s), 1.44 (3H, s), 1.50 (3H, s), 6.05 (1H, s), 7.42 (2H, d,J=8.47 Hz), 7.82 (2H, d,J=8.51 Hz), 8.12(1H, s).
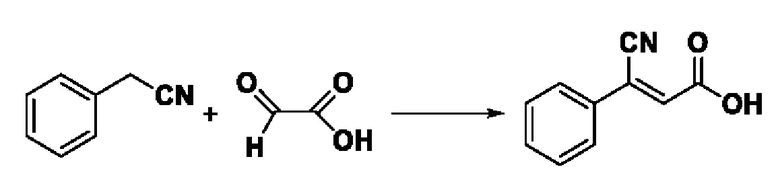
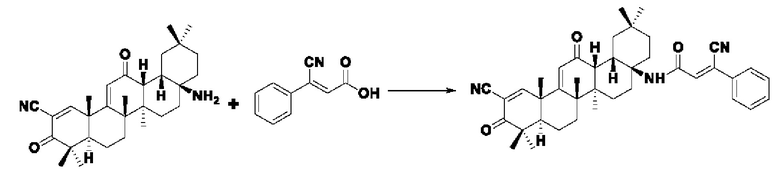
Пример 19: Синтез промежуточного соединения III-7 3-циано-3-фенилакрила
Растворяли 0,735 г (5 ммоль) бензола ацетонитрила в 50 мл метанола, добавляли 0,464 г (5 ммоль) гликолевой кислоты, 1,035 г (7,5 ммоль) карбоната калия и нагревали проточным способом, а конечную точку реакции показывали по результату проверки с помощью TLC. После экстракции фильтровальную лепешку промывали дихлорметаном, растворяли в воде и доводили до рН=4 добавлением разбавленной соляной кислоты, после чего выпадало белое твердое вещество. Объединили органический слой экстрагировали этилацетатом (30 мл × 2),и высушили над безводным сульфатом натрия. После вакуумной фильтрации высушили в печи при постоянной температуре, было получено 0,679 г промежуточного соединения III-7 в виде белого твердого вещества с выходом 78,5%.
ESI-MS: 174.0 [М+Н]+.
Формула реакции выглядит следующим образом.
Пример 20: Синтез I-13
Синтез производился в соответствии с способом получения соединения I-1, заменяли промежуточное соединение III-7 на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,030 г целевого соединения I-13 в виде бледно-желто-белого твердого вещества, с выходом 36,2%.
ESI-MS: 618.4 [М+Н]+, 640.4 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.91 (3H, s), 1.04 (3H, s), 1.06 (3H, s), 1.18 (3H, s), 1.34 (3H, s), 1.42 (3H, s), 1.47 (3H, s), 6.00 (1H, s), 7.03 (1H, s), 7.47 (2H, m), 7.69 (3H, m), 8.04 (1H, s).
Пример 21: Синтез промежуточного соединения III-8 3-циано-3-(4-метилфенил) акрила
Синтез производился в соответствии с способом синтеза промежуточного продукта III-7 3-циано-3-фенилакрил, заменяли 4-метилфенилацетонитрила на фенилацетонитрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,733 г промежуточного продукта III-8 с выходом 78,2%.
ESI-MS: 188.0 [М+Н]+.
Формула реакции выглядит следующим образом.
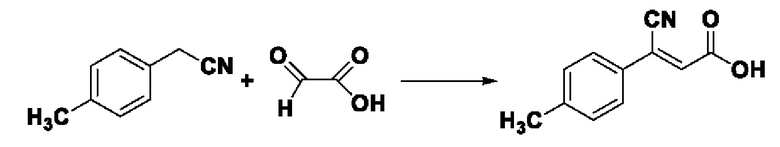
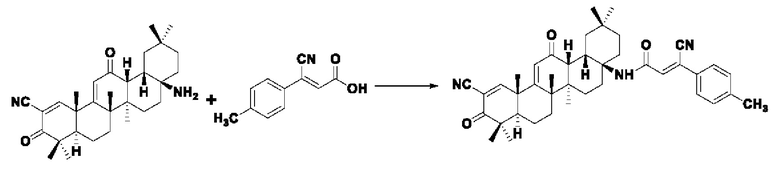
Пример 22: Синтез I-14
Синтез производился в соответствии с способом получения соединения I-1, заменяли промежуточное соединение III-8 на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,035 г целевого соединения I-14 в виде желто-белого твердого вещества с выходом 40,7%.
ESI-MS: 632.4 [М+Н]+, 654.4 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.90 (3H, s), 1.03 (3H, s), 1.06 (3H, s), 1.18 (3H, s), 1.27 (3H, s), 1.42 (3H, s), 1.47 (3H, s), 2.40 (3H, s), 6.00 (1H, s), 7.01 (1H, s), 7.26 (2H, d, J=8.16 Hz), 7.68 (2H, d,J=7.7 Hz), 8.04 (1H, s).
Пример 23: Синтез промежуточного соединения III-9 3-циано-3-(4-метоксифенил) акрила
Синтез производился в соответствии с способом синтеза промежуточного продукта Ш-7 3-циано-3-фенилакрила, заменяли 4-метоксилфенилацетонитрил на фенилацетонитрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,757 г промежуточного продукта III-9 с выходом 74,5%.
ESI-MS: 204.0 [М+Н]+.
Формула реакции выглядит следующим образом.
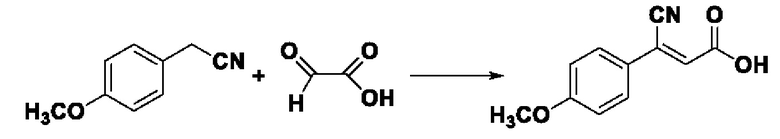
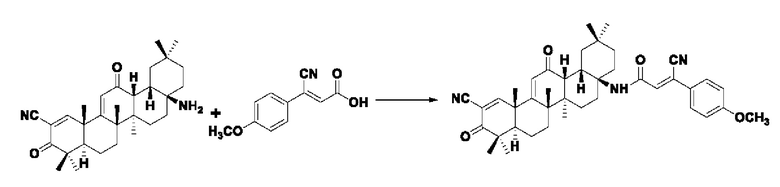
Пример 24: Синтез I-15
Синтез производился в соответствии с способом получения соединения I-1, заменяли промежуточное соединение III-9 на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,031 г целевого соединения I-15 в виде бледно-желто-коричневого твердого вещества с выходом 35,4%.
ESI-MS: 648.4 [М+Н]+, 670.4 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.89 (3H, s), 0.99 (3H, s), 1.04 (3H, s), 1.17 (3H, s), 1.27 (3H, s), 1.40 (3H, s), 1.45 (3H, s), 3.85 (3H, s), 6.00 (1H, s), 6.94 (2H, d, J=8.76 Hz), 7.03 (1H, s), 7.65 (2H, d, J=8.85 Hz), 8.06 (1H, s).
Пример 25: Синтез промежуточного соединения III-10 3-циано-3-(4-трифторметилфенил)акрила
Синтез производился в соответствии с способом синтеза промежуточного продукта III-7 3-циано-3-фенилакрила, заменяли 4-трифторметилфенилацетонитрил на фенилацетонитрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,833 г промежуточного продукта III-10 с выходом 69,1%.
ESI-MS: 242.0 [М+Н]+.
Формула реакции выглядит следующим образом.
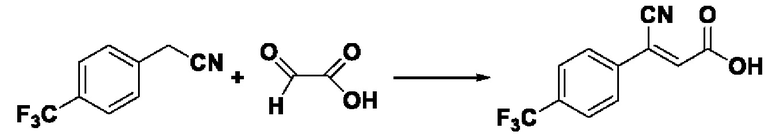
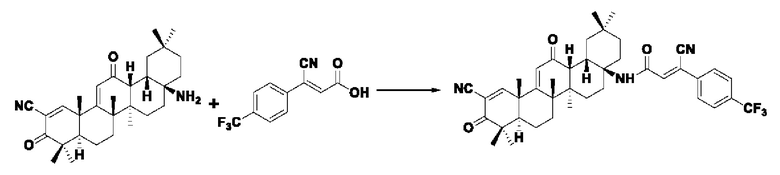
Пример 26: Синтез I-16
Синтез производился в соответствии с способом получения соединения I-1, заменяли промежуточного соединения III-10 на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,056 г целевого соединения I-16 в виде бледно-желто-коричневого твердого вещества с выходом 60,2%.
ESI-MS: 686.4 [М+Н]+, 708.4 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.89 (3H, s), 1.00 (3H, s), 1.06 (3H, s), 1.18 (3H, s), 1.28 (3H, s), 1.42 (3H, s), 1.47 (3H, s), 6.04 (1H, s), 7.15 (1H, s), 7.73 (2H, d, J=8.13 Hz), 7.82 (2H, d,J=7.89 Hz), 8.09 (1H, s).
Пример 27: Синтез промежуточного соединения III-11 3-циано-3-(4-фторфенил) акрила
Синтез производился в соответствии с способом синтеза промежуточного продукта III-7 3-циано-3-фенилакрил, заменяли 4-фторфенилацетонитрил на фенилацетонитрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,956 г промежуточного продукта III-11 с выходом 81,9%.
ESI-MS: 192.0 [М+Н]+.
Формула реакции выглядит следующим образом.
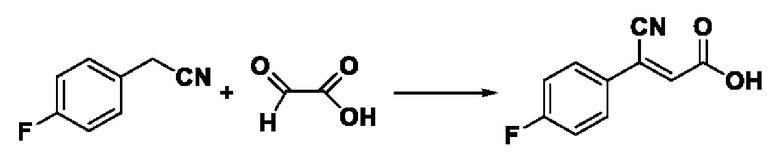
Пример 28: Синтез I-17
Синтез производился в соответствии с способом получения соединения I-1, заменяли промежуточное соединение III-11 на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,023 г целевого соединения I-17в виде желто-белого твердого вещества с выходом 27,0%.
ESI-MS: 636.4 [М+Н]+, 658.4 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.90 (3H, s), 1.01 (3H, s), 1.06 (3H, s), 1.18 (3H, s), 1.34 (3H, s), 1.43 (3H, s), 1.49 (3H, s), 6.04 (1H, s), 6.96 (1H, s), 7.15 (2H, d, J=8.49 Hz), 7.70 (2H, d, J=8.76 Hz), 8.09 (1H, s).
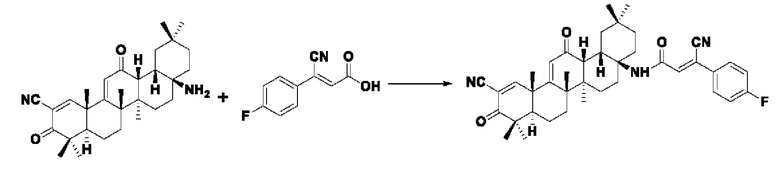
Пример 29: Синтез промежуточного соединения III-12 3-циано-3-(4-хлорфенил) акрила
Синтез производился в соответствии с способом синтеза промежуточного продукта III-7 3-циано-3-фенилакрила, заменяли 4-хлорфенилацетонитрил на фенилацетонитрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,843 г промежуточного продукта III-12 с выходом 81,2%.
ESI-MS: 208.0 [М+Н]+.
Формула реакции выглядит следующим образом.
Пример 30: Синтез I-18
Синтез производился в соответствии с способом получения соединения I-1, заменяли промежуточное соединение III-12 на 3-фенилакрил в качестве реакционного реагента, другие условия оставались неизменными, было получено 0,027 г целевого соединения 1-18 в виде бледного желто-коричневого твердого вещества с выходом 30,3%.
ESI-MS: 652.3 [М+Н]+, 674.3 [M+Na]+.
1H-NMR (300 MHz, CDCl3, TMS), δ ppm: 0.88 (3H, s), 0.96 (3H, s), 1.04 (3H, s), 1.17 (3H, s), 1.26 (3H, s), 1.40 (3H, s), 1.45 (3H, s), 6.02 (1H, s), 7.23 (1H, s), 7.40 (2H, d, J=8.40 Hz), 7.64 (2H, d,J=8.43 Hz), 8.09 (1H, s).
Пример 31: Фармакологические эксперименты с соединениями
Противоопухолевую активность соединений по настоящему изобретению определяли способом метилтиазолилтетразолия (МТТ), метил бардоксолона (CDDO-Me) выбирали в качестве положительного контрольного препарата.
Инструменты: сверхчистый стол (SW-CJ-1FD, AIRTECH, ООО сучжоуская компания воздушной техники Аньтай), СО2- инкубатор постояннойтемпературы(3111, Thermo, США), инвертированный биомикроскоп (IX71, OLYMPUS, Япония), микропланшетный ридер ELISA (модель 680, BIO-RAD, США), шейкер платформы (Kylin-bell lab Instruments), автоклав (YXO.SG41.280, ООО шанхайская компания Хуасянь) и центрифуга (SIGMA).
Реактивы: DMEM (GIBCO), фетальная бычья сыворотка (GIBCO), трипсин (SIGMA) и DMSO (SIGMA)
Клеточные штаммы: клеточный штамм немелкоклеточного рака легкого человека А549, клеточный штамм рака печени человека HepG2, клеточный штамм рака молочной железы человека MCF-7, эпителиальные клетки почечных канальцев человека HK-2, эмбриональные кардиомиоциты крысы Н9С2 (все предоставлены ОАО Цзянской биотехнической компанией Кайцзи). Компания с ограниченной ответственностью.). I
Способ: криоконсервированные штаммы клеток реанимировали и культивировали в инкубаторе СО2 при постоянной температуре 37°С, жидкость меняли один раз в день. Посев проводили, когда клеточные штаммы находились в хорошем состоянии в экспоненциальной фазе. Добавляли 1 мл 0,25% раствора трипсина, переварили в течение 1-2 мин. Состояние клеток наблюдали под микроскопом, и когда прилипшие клетки становились круглыми и уменьшались, аспирировали раствор для переваривания, добавляли 1-2 мл среды DMEM, содержащей 10% фетальной бычьей сыворотки, для получения клеточной суспензии, на которой проводили подсчет клеток. Количество необходимой клеточной суспензии рассчитывалосьисходя из 5×104 клеток на лунку и общего количества лунок. Клеточную суспензию инокулировали на 96-луночный планшет в объеме 100 мкл/лунку среду блокировали PBS, после чего инкубировали в СО2 - инкубаторе при постоянной температуре 37°С в течение 24 часов.
Испытуемый препарат, положительный контрольный препарат CDDO-Me и пустая контрольная группа DMSO были приготовлены в среде DMEM до конечной концентрации 5 мкМ/лунку, каждый препарат был помещен в 3 повторные лунки, после чего инкубировали в течение 48 ч. Реагент МТТ добавляли в 96-луночный планшет в количестве 10 мкл/лунку для дальнейшей инкубации в течение 4 ч. Среду в планшете аспирировали и в каждую лунку добавляли 100 мкл DMSO, после чего встряхивали на платформенном шейкере в течение 10 мин, чтобы кристаллы растворились. Поглощение в каждой лунке проверяли с помощью микропланшетного ридера ELISA при длине волны 570 нм, и скорость ингибирования клеток рассчитывали по формуле, приведенной ниже. Среднее значение трех результатов предварительного скрининга принимали за окончательный показатель ингибирования. Соединения с показателем ингибирования предварительного скрининга более 60% подвергались градиентному скринингу концентрации (5-кратное разведение) для расчета значений IC50 тестируемого препарата (рассчитанных с помощью программного обеспечения graphpad), и результаты трех повторных экспериментов были окончательными значениями IC50 тестируемых соединений.
Степень ингибирования клеток (%)=[(значение OD контрольного контроля - значение OD группы дозирования) / значение OD контрольной группы] × 100%
=
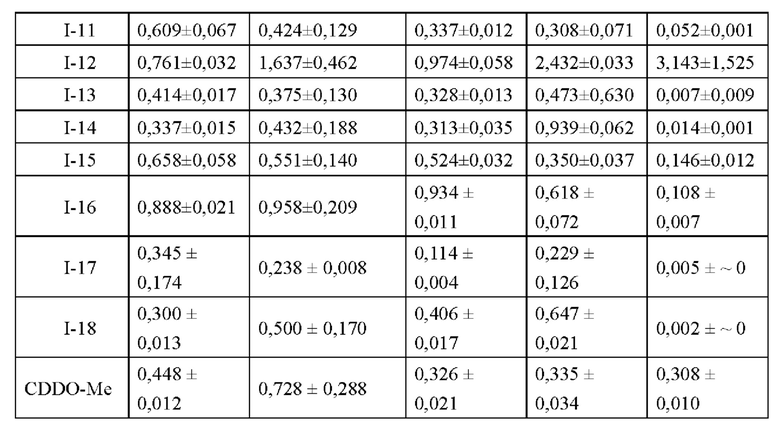
Как видно из таблицы 1, большинство соединений по настоящему изобретению обладают большей или сравнимой ингибирующей активностью в отношении кардиомиоцитов Н9С2, чем CDDO-Me. Соединения I-1, I-2, I-3, I-4, I-5, I -6, I-7, I-8, I-9, I-12, I-13, I-14, I-16 и I-18 проявляли более низкую токсичность в отношении эпителиальных клеток почечных канальцев HK-2 человека. В частности, соединения I-7 и I-12 обладали относительно небольшой ингибирующей активностью в отношении нормальных кардиомиоцитов Н9С2 (значения IC50 3,176±1,74 мкМ и 3,143±1,53 мкМ соответственно), примерно 1/10 ингибирующей активности CDDO-Me. (IC50 0,308±0,01 мкМ), демонстрировали относительно низкую миокардиальную токсичность.
Пример 32. Ингибирующее действие соединений по настоящему изобретению на опухоли у мышей с опухолями.
1) Экспериментальные животные: 72 самки мышей ICR в возрасте от 6 до 8 недель; экспериментальные клетки: клетки асцитной опухоли S180.
8 животных содержались в нормальной группе, способ разработки моделей на остальных мышах заключается в следующем:
2) Пролиферация и пассаж асцитных опухолевых клеток S180: Клетки асцитной опухоли S180, криоконсервированные были взяты при -80°С, были разморожены на водяной бане при 37°С, помещены в 12 мл центрифужную пробирку, в которую было добавлено соответствующее количество физиологического раствора, и центрифугированы при 1200 об/мин в течение 5 мин. Супернатант отбрасывали, затем клетки ресуспендировали в 300 мкл физиологического раствора и внутрибрюшинно вводили мыши ICR. По мере вздутия брюшной полости мыши проводили внутрибрюшинную инъекцию клеток второй мыши ICR, и клетки были готовы к инокуляции, когда брюшная полость перестала вздуваться.
3) Инокуляция опухолевых клеток S180 в подмышечную впадину мышей: клетки S180, культивированные до 2 пассажей, центрифугировали и добавляли физраствор для получения концентрации 1×107 клеток/мл. Клетки в объеме 100 мкл вводили подкожно в подмышечную впадину мышей. Была разработана модель опухолей на мышах.
Модельные мыши были разделены на группу опухолей, группу соединения I-7 в дозе 5 мг/кг, группу соединения I-7 в дозе 15 мг/кг, группу соединения I-12 в дозе 5 мг/кг, группу соединения I-12 в дозе 15 мг/кг, группу соединения I-16 в дозе 5 мг/кг, группу соединения I-16 в дозе 15 мг/кг и группу CDDO-Me в дозе 15 мг/кг.
В каждой группе было по 8 мышей.
4) Дозировка: лекарственные препараты вводились внутрижелудочно по мере роста опухоли.
Соединение I-7 в дозе 5 мг/кг вводили группе соединения I-7 в дозе 5 мг/кг;
соединение I-7 в дозе 15 мг/кг вводили группе соединения I-7 в дозе 15 мг/кг;
соединение I-12 в дозе 5 мг/кг вводили группе соединения I-12 в дозе 5 мг/кг;
соединение I-12 в дозе 15 мг/кг вводили группе соединения I-12 в дозе 15 мг/кг;
соединение I-16 в дозе5 мг/кг вводили группе соединения I-16 в дозе 5 мг/кг;
соединение I-16 в дозе 15 мг/кг вводили группе соединения I-16 в дозе 15 мг/кг; и
CDDO-Me в дозе 5 мг/кг вводили группе CDDO-Me 15 в дозе мг/кг.
В Нормальной группе и группе модели вводили равный объем 0,5% кроскармеллозы натрия.
Введение проводили в течение 7 дней подряд. Мышей вскрывали на следующий день после введения последней дозы.
Показатели эксперимента: измеряли массу тела, массу селезенки и массу опухоли. Селезенки измельчали на льду с последующим лизисом эритроцитов и инкубацией с антителами FITC-CD11b, LY6C и LY6G в течение 15 минут, а затем тестировали на CD11b+LY6Chi+и CD11b+LY6G+с помощью проточной цитометрии.
Экспериментальные результаты представлены в таблицах 2 и 3.

Клетки-супрессоры миелоидного происхождени (MDSC) представляют собой группу незрелых клеток миелоидного происхождения. MDSC могут дифференцироваться в зрелые макрофаги, гранулоциты и дендритные клетки в нормальных условиях, но будут агрегироваться и активироваться в патологических условиях. В опухолях MDSC могут подавлять Т-клеточные иммунные ответы, а также могут взаимодействовать с другими иммунными клетками и вместе способствовать формированию иммуносупрессии в микроокружении опухоли. Кроме того, MDSC также могут способствовать пролиферации и метастазированию опухоли неиммунными путями, такими как разрушение внеклеточного матрикса, стимулирование ангиогенеза и т.д. Индоламин 2,3-диоксигеназа 1 (IDOl) как один из ключевых функциональных маркеров MDSC может катализировать триптофан, метаболизм в микроокружении опухоли, что приводит к высвобождению растворимого кинуренина и его нижестоящих метаболитов и вызывает иммунную толерантность Treg и антигенпрезентирующих клеток, что приводит к ускользанию опухоли от иммунного ответа. Как сообщалось в некоторых исследованиях, MDSCs вовлечены в прогрессирование рака поджелудочной железы, рака молочной железы, метастазов в головной мозг и т.д. и имеют большую исследовательскую ценность в качестве потенциальной терапевтической мишени и надежного прогностического маркера.
Указанные примеры по настоящему изобретению показали, что соединения по настоящему изобретению могут ингибировать экспрессию MDSC и улучшать микроокружение опухоли, что свидетельствует об их терапевтическом действии на рак легких, рак печени, рак поджелудочной железы, рак молочной железы, асцитную опухоль, метастазы в головной мозг, и т.п.
Кроме того, следует отметить, что соединения, выбранные в примере 32, являются репрезентативными примерами целевых соединений, описанных здесь, и не имеют целью ограничить настоящее изобретение.
| название | год | авторы | номер документа |
|---|---|---|---|
| Соединение-антагонист PD-L1 | 2020 |
|
RU2823231C1 |
| ХИМИЧЕСКИЙ СИНТЕЗ И ПРОТИВООПУХОЛЕВЫЙ И ПРОТИВОМЕТАСТАТИЧЕСКИЙ ЭФФЕКТЫ КОНЪЮГАТА ДВОЙНОГО ДЕЙСТВИЯ | 2011 |
|
RU2729419C2 |
| Производные бензимидазол-2-пиперазина, полезные в качестве ингибитора поли(АДФ-рибоза)-полимеразы (PARP) | 2014 |
|
RU2649002C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2016 |
|
RU2686314C1 |
| ПРОИЗВОДНЫЕ ИЗОХИНОЛИНОНА, ПОЛЕЗНЫЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2015 |
|
RU2690853C2 |
| БЕНЗОПРОИЗВОДНЫЕ С ШЕСТИЧЛЕННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА DPP-4 И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2702644C2 |
| СЛОЖНОЭФИРНОЕ ПРОИЗВОДНОЕ 2-АМИНО-БИЦИКЛО[3.1.0]ГЕКСАН-2,6-ДИКАРБОНОВОЙ КИСЛОТЫ | 2004 |
|
RU2409557C2 |
| ПРОИЗВОДНЫЕ 1-(3-АМИНОФЕНИЛ)-6,8-ДИМЕТИЛ-5-(4-ИОД-2-ФТОР-ФЕНИЛАМИНО)-3-ЦИКЛОПРОПИЛ-1H,6H-ПИРИДО[4,3-d]ПИРИМИДИН-2,4,7-ТРИОНА В КАЧЕСТВЕ ИНГИБИТОРОВ МЕК1/2 | 2015 |
|
RU2605400C1 |
| НОВЫЕ ПРОИЗВОДНЫЕ БЕНЗОДИАЗЕПИНА | 2019 |
|
RU2799340C2 |
| Способ получения ингибитора ацетилхолинэстеразы (AChE) и ингибитор, полученный способом | 2021 |
|
RU2779555C1 |
Настоящее изобретение относится к области медицинской химии и фармакотерапии, в частности к производному 2-циано-3,12-диоксоолеан-1,9(11)-диен-17-фенилакриламида формулы I или его фармацевтически приемлемой соли. В формуле I: каждый из R1, R2 независимо выбран из Н или циано; R3 выбран из следующих заместителей: Н, C1-6 алкил, С1-6 алкокси, галоген, циано, С1-6 алкил, замещенный галогеном или циано. Также предложены способ получения соединения формулы I, содержащая его фармацевтическая композиция и его применение в приготовлении препарата для лечения опухоли. Предложенные соединения обладают превосходной противоопухолевой активностью и могут быть использованы в качестве соединений-кандидатов в противоопухолевые препараты. 5 н. и 12 з.п. ф-лы, 3 табл., 32 пр.
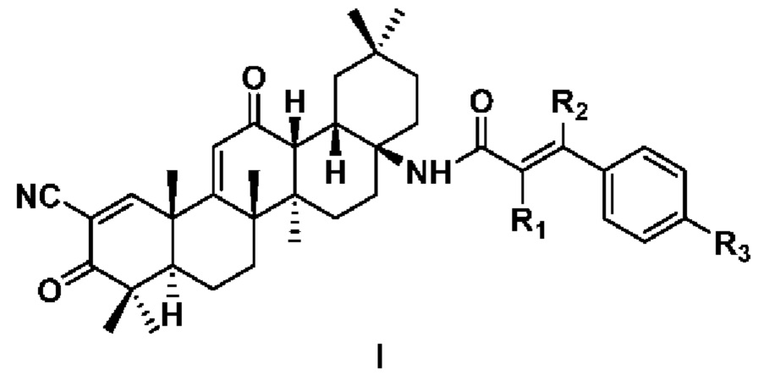
1. Соединение формулы I или его фармацевтически приемлемая соль 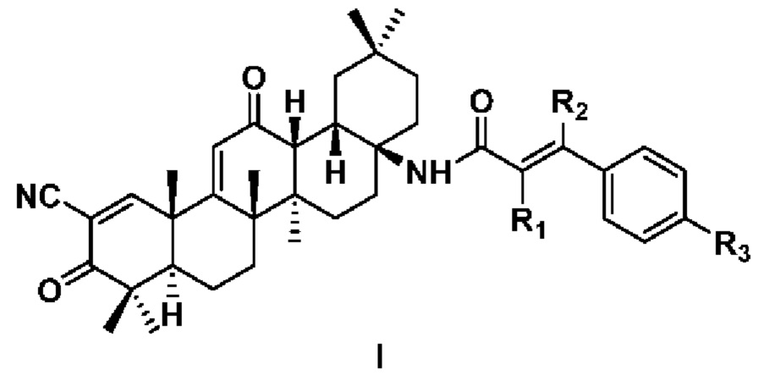
где каждый из R1, R2 независимо выбран из Н или циано; и
R3 выбран из следующих заместителей: Н, C1-6 алкил, С1-6 алкокси, галоген, циано, С1-6 алкил, замещенный галогеном или циано.
2. Соединение или его фармацевтически приемлемая соль по п. 1, где каждый из R1, R2 независимо выбран из Н или циано; и
R3 выбран из: Н, C1-3 алкила, C1-3 алкокси, галогена, C1-3 алкила, замещенного галогеном или циано.
3. Соединение или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что каждый из R1 и R2 независимо выбран из Н или циано; и
R3 выбран из Н, C1-3 алкила, C1-3 алкокси, галогена, C1-3 алкила, замещенного галогеном; и галоген выбран из F, Cl, Br, I.
4. Соединение или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что R1 представляет собой Н или циано; R2 представляет собой Н или цианогруппу и R3 выбран из Н, метила, метоксила, трифторметила, Cl или F.
5. Соединение или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что R1 представляет собой Н или циано; R2 представляет собой Н или цианогруппу и R3 представляет собой Cl, Н или трифторметил.
6. Соединение или его фармацевтически приемлемая соль по п. 1, отличающиеся тем, что соединение выбрано из:
3-фенил-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил) акриламид, 3-(4-метилфенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-(4-метоксифенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-(4-трифторметилфенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-(4-фторфенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-(4-хлорфенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
2-циано-3-фенил-N-(2-циано-3,12-диоксолан-1,9(11)-диен-19-ил)акриламид,
2-циано-3-(4-метилфенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
2-циано-3-(4-метоксифенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
2-циано-3-(4-трифторметилфенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
2-циано-3-(4-фторфенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
2-циано-3-(4-хлорфенил)-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-фенил-3-циано-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-(4-метилфенил)-3-циано-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-(4-метоксифенил)-3-циано-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-(4-трифторметилфенил)-3-циано-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид,
3-(4-фторфенил)-3-циано-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид и
3-(4-хлорфенил)-3-циано-N-(2-циано-3,12-диоксоолеан-1,9(11)-диен-19-ил)акриламид.
7. Способ синтеза соединения формулы I по любому из пп. 1-6, отличающийся тем, что способ синтеза заключается в следующем:
получается соединение формулы I посредством реакции конденсации соединения формулы II с соединением формулы III
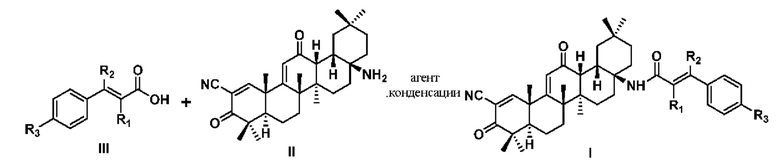
где каждый из R1, R2 и R3 независимо определен в любом из пп. 1-6, а агент конденсации представляет собой один или несколько агентов, выбранных из дициклогексилкарбодиимида, N,N-диизопропилкарбодиимида, 1-этил-(3-диметиламинопропил)карбодиимида. бензотриазол-1-окситрис(диметиламино)фосфония гексафторфосфат или бензотриазол-1-илокситрипирролидин гексафторфосфат.
8. Способ синтеза по п. 7, отличающийся тем, что способ синтеза описанной формулы II заключается в следующем:
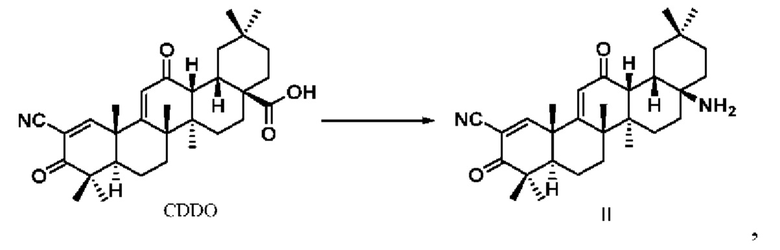
получается производное, аминированное в положении С-17 путем перегруппировки Курциуса CDDO, то есть соединение формулы II.
9. Фармацевтическая композиция, обладающая противоопухолевой активностью и содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-6 и один или несколько фармацевтически приемлемых фармацевтических адъювантов.
10. Применение соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или фармацевтической композиции по п. 9 в приготовлении препарата для лечения опухоли.
11. Применение по п. 10, отличающееся тем, что указанные опухоли выбраны из рака легкого, рака печени, асцитной опухоли, метастазов в головной мозг, рака толстой кишки, рака поджелудочной железы, рака молочной железы, рака предстательной железы, рака головного мозга, рака яичников, рака шейки матки, рака яичка, рака почки, рака головы и шеи, лимфомы, меланомы или лейкемии.
12. Применение по п. 10, отличающееся тем, что указанная опухоль выбрана из рака легкого, рака печени, рака молочной железы, асцитной опухоли, рака поджелудочной железы, метастазов в головной мозг.
13. Применение по п. 10, отличающееся тем, что указанная опухоль выбрана из немелкоклеточного рака легкого, рака печени, рака молочной железы и асцитной опухоли.
14. Способ лечения опухоли, содержащий введение субъекту или пациенту в терапевтически эффективной дозе соединения или его фармацевтически приемлемой соли по любому из пп. 1-6 или фармацевтической композиции по п. 9.
15. Способ по п. 14, отличающийся тем, что указанные опухоли выбраны из рака легкого, рака печени, асцитной опухоли, метастазов в головной мозг, рака толстой кишки, рака поджелудочной железы, рака молочной железы, рака предстательной железы, рака головного мозга, рака яичников, рака шейки матки, рака яичка, рака почки, рака головы и шеи, лимфомы, меланомы или лейкемии.
16. Способ по п. 14, отличающийся тем, что указанная опухоль выбрана из рака легкого, рака печени, рака молочной железы, асцитной опухоли, рака поджелудочной железы, метастазов в головной мозг.
17. Способ по п. 14, отличающийся тем, что указанная опухоль выбрана из немелкоклеточного рака легкого, рака печени, рака молочной железы и асцитной опухоли.
| EA 201001555 A1, 30.06.2011 | |||
| WO 2009146216 A2, 03.12.2009 | |||
| CN 106632576 A, 10.05.2017 | |||
| WO 2015027206 A1, 26.02.2015 | |||
| WO 2018089539 A1, 17.05.2018. |

