Область техники
Настоящее изобретение относится к гетероциклическому имидазольному соединению, способам его изготовления, а также лекарственным композициям, содержащим его производные, и их применению в качестве лекарств и в качестве ингибиторов поли (АДФ-рибоза) - полимеразы (ПАРП).
Предшествующий уровень техники
Наиболее часто используемыми методами в лечении онкологических заболеваний являются химиотерапия и лучевая терапия. Оба этих метода вызывают разрыв одной или обеих нитей ДНК, а также обладают цитотоксичным эффектом. Целевые опухолевые клетки гибнут из-за повреждений хромосом. Одним из важнейших результатов, который сигнализирует о негативном воздействии на ДНК, является активизация контролирующих клеточный цикл точечных сигналов, цель которой заключается в том, чтобы для защиты клеток остановить их деление в условиях повреждения ДНК, и тем самым избежать повреждения клеток. В большинстве случаев опухолевые клетки, наряду с тем, что показывают повреждения точечных сигналов клеточного цикла, также показывают высокий коэффициент прироста. Таким образом, можно сделать вывод, что в опухолевых клетках существует специальный механизм репарации ДНК, который может очень быстро реагировать и восстанавливать повреждения хромосом, связанные с регулировкой механизма размножения клеток, тем самым позволяя опухолевым клеткам избегать цитотоксического воздействия терапевтических средств и продолжать свое существование.
В клиническом применении терапевтически эффективные концентрации химиотерапевтического препарата или интенсивность излучения могут противодействовать этому механизму репарации ДНК и обеспечивать поражение целевых опухолевых клеток. Однако опухолевые клетки, усиливая свой механизм репарации ДНК, могут выдержать воздействие лечения и выжить даже при смертельных повреждениях ДНК. Чтобы препятствовать развитию невосприимчивости клеток, обычно увеличивают дозировку лекарства или интенсивность излучения, это оказывает негативное воздействие на нормальные структуры рядом с очагом болезни, тем самым процессу лечения сопутствуют серьезные побочные эффекты и возрастает угроза эффективности лечения. Вместе с тем непрерывно повышающаяся невосприимчивость снижает терапевтический эффект. По этой причине можно сделать вывод, что путем регулировки механизма восстановления поврежденных точечных сигналов ДНК можно, используя специфичность опухолевых клеток, повысить цитотоксический эффект лекарства, повреждающего ДНК.
Ферменты ПАРП (поли(АДФ-рибоза)-полимеразы), особенностью которых является активность поли-АДФ-рибозилирования, образуют суперсемейство из 18 видов ядерно-цитоплазматических ферментов. Действие этого поли-АДФ-рибозилирования может регулировать каталитическую активность целевых белков и взаимодействие между белками, а также регулировать и контролировать многие основные биохимические процессы, включая процессы, связанные с репарацией ДНК, клеточной смертью, стабильностью генома.
Фермент ПАРП-1 занимает около 80% от суммарной активности ПАРП в клетке. Он и другой, ближайший ему фермент ПАРП-2 вместе являются представителями семейства ПАРП, обладающими способностью к репарации ДНК. Являясь сенсором и сигнальным белком репарации ДНК, ПАРП-1 может быстро проверять и немедленно соединять поврежденные места ДНК, а после вызывать агрегацию необходимых для репарации ДНК различных белков и затем восстанавливать ДНК. Когда в клетке не хватает ПАРП-1, ПАРП-2 может его заменить в процессе репарации ДНК. Исследования показывают, что по сравнению с нормальной клеткой, в солидной опухоли наблюдается всеобщее усиление белков ПАРП. Кроме того, что касается новообразований (например, рак молочной железы или яичников), в которых наблюдается потеря генов, связанных с репарацией ДНК (например BRC A-1 или BRC A-2), то они демонстрируют крайнюю чувствительность к ингибиторам ПАРП-1, что указывает на потенциальную сферу применения ингибитора ПАРП-1 в качестве самостоятельного препарата в лечении этого вида тройного негативного рака молочной железы. Вместе с тем из-за того, что механизм репарации ДНК запускается, как реакция опухолевой клетки на воздействие химиотерапии и лучевой терапии, то ПАРП-1 рассматривается как значимая мишень в поисках новых способов лечения рака.
Ингибиторы ПАРП, полученные в предыдущих исследованиях, имеют своей моделью никотинамид НАД, который является субстратом для катализа ПАРП, поэтому они разрабатывались как его аналоги. Эти ингибиторы являются конкурирующими ингибиторами НАД, конкурируют с НАД за место в катализе ПАРП, и затем прекращают синтез поли-АДФ-рибозилльной цепи. Без восстановления поли-АДФ-риболизирования ПАРП не может диссоциироваться от поврежденного участка ДНК, что приводит к тому, что другие участвующие в репарации белки не могут проникнуть в поврежденное место и, более того, не могут выполнить процесс репарации. Поэтому под воздействием цитотоксических препаратов или лучевой терапии присутствие ингибитора ПАРП приводит к окончательной гибели опухолевой клетки с поврежденной ДНК.
Кроме того, представляющий собой субстрат для катализа ПАРП и потому постоянно расходующийся, НАД является важным элементом во внутриклеточном процессе синтеза АТФ, поэтому при высоком уровне активности ПАРП количество НАД в клетке заметно снижается, что в свою очередь влияет на синтез АТФ. Если в клетке содержится недостаточное количество АТФ, клетка не может выполнить процесс программируемой клеточной смерти, зависимый от АТФ, а может только запустить специфический процесс апоптоза — некроз. При некрозе высвобождается большое количество факторов воспаления, из-за чего возникает токсическое воздействие на другие органы и ткани. Поэтому игнибиторы ПАРП могут также использоваться для лечения многих заболеваний, связанных с этими механизмами, включая нейродегенеративные заболевания (например, старческое слабоумие, болезнь Хантингтона, болезнь Паркинсона), сахарный диабет, ишемию или осложненные синдромом реперфузии заболевания (например, инфаркт миокарда), заболевания системы кровообращения (например, септический шок), а также воспалительные заболевания (например, хронический ревматизм).
В настоящее время имеется 14 клинически исследуемых ингибиторов ПАРП, в том числе разработанный компанией AstraZeneca ингибитор AZD2281 (структурная формула ниже), для которого в декабре 2014 года от Управления по санитарному надзору за качеством пищевых продуктов и медикаментов США получено разрешение на продажу. Показанием к применению лекарства является чувствительность пациентов с поздней стадией рака яичников к химиотерапии препаратами на основе платины. Соответствующие патентные заявки: W02002036576 и W02006021801.
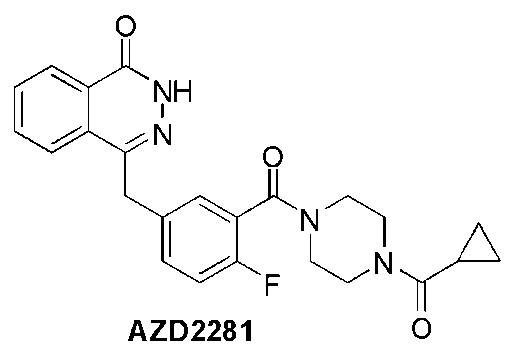
Несмотря на то, что в настоящее время открыт целый ряд ингибиторов ПАРП, по-прежнему важно разрабатывать новые более эффективные соединения, обладающие лучшими фармакокинетическими свойствами и меньшей токсичностью. В настоящем изобретении предложено соединение, которое имеет структуру, описываемую общей формулой (I), а также обнаружено, что соединения, имеющие такую структуру, демонстрируют превосходный результат.
Описание изобретения
Одна из целей настоящего изобретения заключается в предложении нового гетероциклического имидазольного соединения формулы (I) или его фармацевтически приемлемой соли.
Следующая цель настоящего изобретения заключается в предложении способа изготовления указанного гетероциклического имидазольного соединения или его фармацевтически приемлемой соли.
Третья цель настоящего изобретения заключается в предложении промежуточного соединения, получаемого в процессе изготовления указанного гетероциклического имидазольного соединения или его фармацевтически приемлемой соли.
Четвертая цель настоящего изобретения заключается в предложении способа изготовления промежуточного соединения, получаемого в процессе производства указанного гетероциклического имидазольного соединения или его фармацевтически приемлемой соли.
Пятая цель настоящего изобретения заключается в предложении способов применения вышеупомянутого промежуточного соединения в изготовлении указанного гетероциклического имидазольного соединения формулы (I), а также его производных.
Шестая цель настоящего изобретения заключается в предложении лекарственной композиции, активным ингредиентом которого является указанное выше гетероциклическое имидазольное соединение или его фармацевтически приемлемая соль.
Седьмая цель настоящего изобретения заключается в предложении способов применения в лекарствах указанного гетероциклического имидазольного соединения или его фармацевтически приемлемой соли.
Первым предметом настоящего изобретения является гетероциклическое имидазольное соединение, которое представляет собой соединение формулы (I) или его фармацевтически приемлемую соль:

В общей формуле (I):
R представляет собой водород, галоген, (C1-C6) алкил или (C1-C6) галогеналкил;
Одно из обозначений X, Y, Z представляет собой азот, а другие — углеводород, или одно из обозначений X, Y, Z обозначает углеводород, а другие — азот;
M представляет собой азот или CR1, где:
R1 представляет собой водород, кислород, (C1-C6) алкил или (C1-C6) галогеналкил.
Наиболее предпочтительным является соединение, структура которого находится в соответствии с общей формулой (I), предложенной настоящим изобретением, и в которой:
R представляет собой водород, кислород, метил или трифторметил;
Одно из обозначений X, Y, Z представляет собой азот, а другие — углеводород, или одно из обозначений X, Y, Z обозначает углеводород, а другие — азот;
M представляет собой азот или CR1, где:
R1 представляет собой водород, кислород, метил или трифторметил.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), R представляет собой водород, галоген, (C1-C3) алкоксил или (C1-C3) галогеналкил.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), R представляет собой водород, фтор, метоксил или трифторметил.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), X и Z представляют собой азот, а Y представляет собой углеводород, или X представляет собой азот, а Y и Z — углеводороды, или Z представляет собой азот, а X и Y — углеводороды, или Y представляет собой азот, а X и Z — углеводороды.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), R1 представляет собой водород, кислород, (C1-C6) алкил или (C1-C6) галогеналкил.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), R1 представляет собой водород, кислород, (C1-C3) алкил или (C1-C3) галогеналкил.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), R1 представляет собой водород, кислород, метил или трифторметил.
В одном из предпочтительных вариантов применения настоящего изобретения гетероциклическое имидазольное соединение, описываемое указанной выше формулой (I), представляет собой соединение 4-(3-(пиперазин-1-карбонил)бензил)фталазин-1(2Н)-кетона, а также его фармацевтически приемлемую соль.
Наиболее предпочтительным соединением, которое описывается формулой (I) настоящего изобретения, является соединение, выбранное из нижеследующих соединений (1)–(21): 
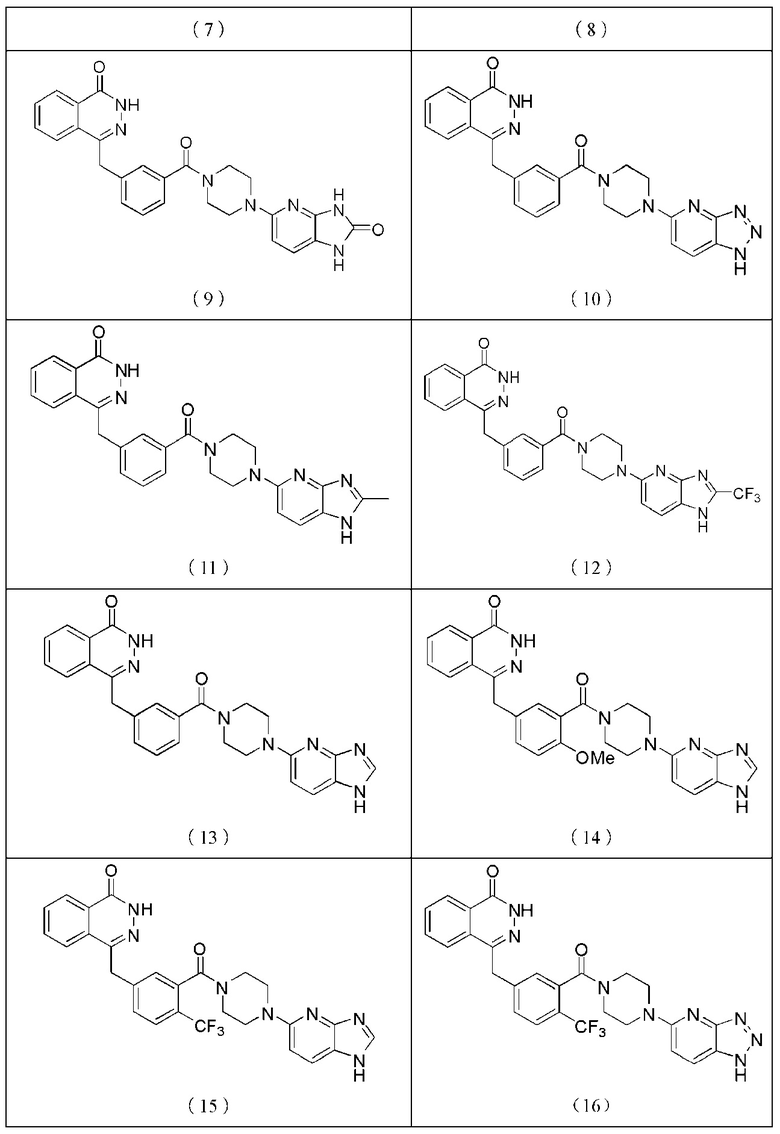
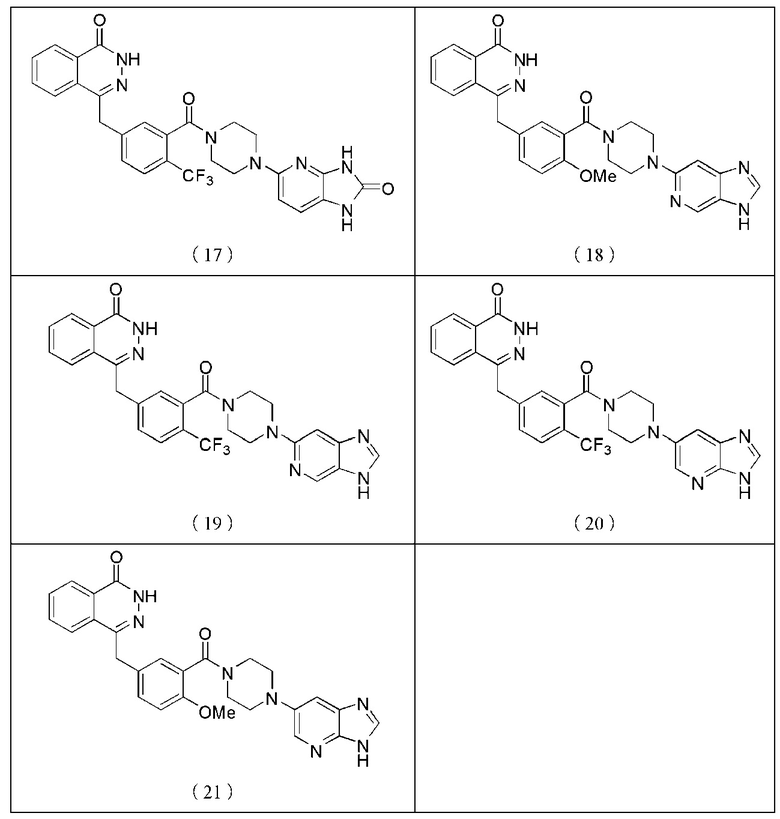
Соединения, описываемые вышеупомянутой формулой (I), представляют собой таутомеры, энантиомеры, диастереомеры, мезомеры, рацематы, а также их смесь.
Соединения, описываемые вышеупомянутой формулой (I), представляют собой фармацевтически приемлемые производные соединения.
Соединения, описываемые вышеупомянутой формулой (I) в настоящем изобретении, могут существовать в виде фармацевтически приемлемых солей. Вторым предметом настоящего изобретения является способ изготовления соединений формулы (I), который описывается следующим уравнением химической реакции:
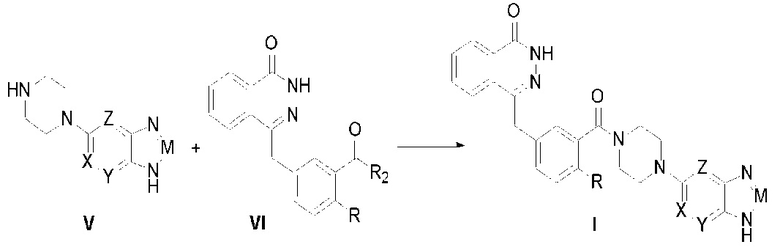
где R, X, Y, Z и M определены как выше, а R2 представляет собой гидроксил, галоген, диимидазол-1-ил; ниже описаны конкретные стадии этой реакции.
Соединение, описываемое общей формулой (I), получают путем реакции конденсации между промежуточным соединением (V) и производным фталазин-карбоновой кислоты (VI).
В одном из вариантов применения настоящего изобретения промежуточное соединение (V) получают следующим способом:
Стадия 1: путем реакции нуклеофильного замещения между монопиперазином и нитрозамещенным гетероциклическим галоидным соединением, содержащим аминогруппу, получают промежуточное соединение (II);
Стадия 2: в промежуточном соединении (II) путем каталитической гидрогенизации восстанавливают нитрогруппу, получая промежуточное соединение (III);
Стадия 3: путем реакции циклизации между промежуточным соединением (III) и ангидридом уксусной кислоты, ангидридом трифторуксусной кислоты, триметилортоформиатом, карбонилдиимидазолом или тринитридными соединениями получают промежуточное соединение (IV);
Стадия 4: из промежуточного соединения (IV) десорбируют защитную группу аминогруппы, получая промежуточное соединение (V);
Уравнения этих реакций представлены ниже:
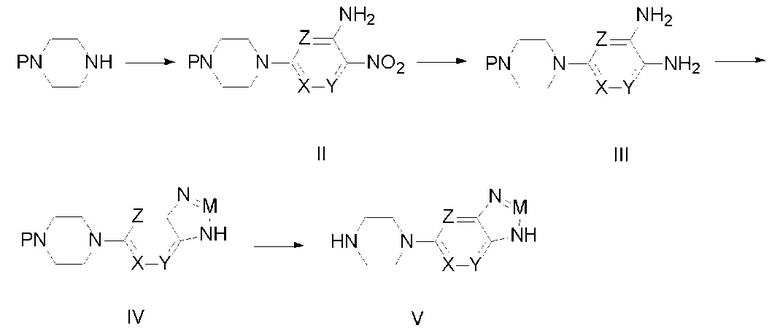
где P представляет собой защитную группу аминогруппы, одно из обозначений X, Y, Z представляет собой азот, а другие — углеводород, или одно из обозначений X, Y, Z обозначает углеводород, а другие — азот;
M представляет собой азот или CR1, где:
R1 представляет собой водород, кислород, метил или трифторметил.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), X и Z представляют собой азот, а Y представляет собой углеводород, или X представляет собой азот, а Y и Z — углеводороды, или Z представляет собой азот, а X и Y — углеводороды, или Y представляет собой азот, а X и Z — углеводороды.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), R1 представляет собой водород, кислород, (C1-C6) алкил или (C1-C6) галогеналкил.
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), R1 представляет собой водород, кислород, (C1-C3) алкил или (C1-C3) галогеналкил
В одном из вариантов применения настоящего изобретения в соединении, описываемом общей формулой (I), R1 представляет собой водород, кислород, метил или трифторметил.
Предпочтительно, чтобы указанное выше производное соединение фталазин-карбоновой кислоты (VI) было следующим:
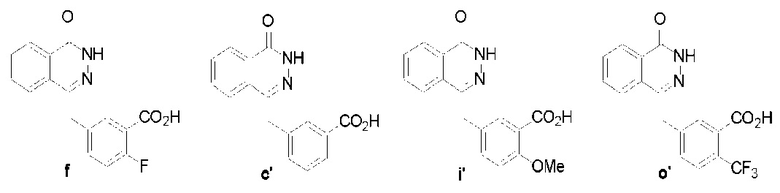
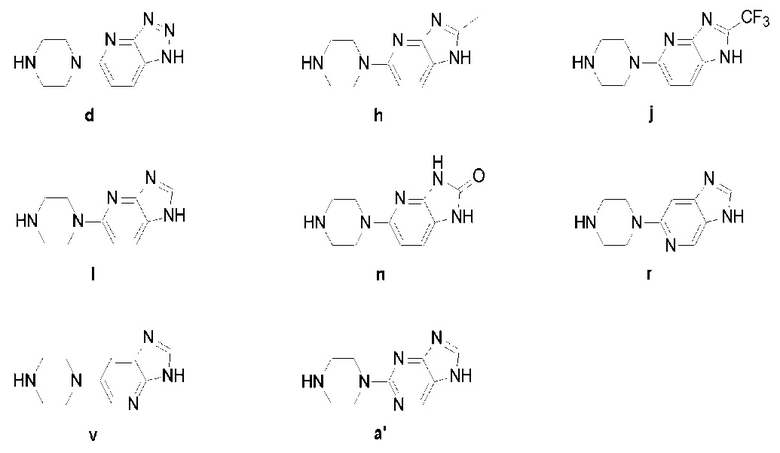
Предпочтительно, чтобы указанное выше промежуточное соединение (V) было следующим:
В одном из вариантов реализации настоящего изобретения конденсирующее средство в упомянутой реакции конденсации получается из 1,1-карбонилимидазола, 1-этил-(3-диметиламинопропил)карбодиамина гидрохлорида, 2-(7-азобензотриазол)-N,N,N',N'-тетраметилмочевины гексафторфосфата, бензодинитрозол-N,N,N',N'-тетраметилмочевины гексафторфосфата.
В одном из вариантов реализации настоящего изобретения растворитель в упомянутой реакции конденсации получается из дихлорметана, этилацетата, диметилсульфоксида, тетрагидрофурана, диметилформамида, диметилацетамида, N-диметилпирролинкетона, ацетона.
В одном из вариантов реализации настоящего изобретения в упомянутую реакцию конденсации добавляют неорганическое основание или алкалоид.
В одном из вариантов реализации настоящего изобретения упомянутый алкалоид получается из триэтиламина, диэтиламина, диизопропилэтиламина, пиперидина.
В одном из вариантов применения настоящего изобретения путем реакции циклизации между промежуточным соединением (III) и нитритом натрия, ангидридом уксусной кислоты, ангидридом трифторуксусной кислоты, триметилортоформиатом или тринитридными соединениями получают промежуточное соединение (IV);
В одном из вариантов применения настоящего изобретения путем реакции циклизации между промежуточным соединением (III) и ангидридом уксусной кислоты, ангидридом трифторуксусной кислоты, триметилортоформиатом или азидом натрия получают промежуточное соединение (IV);
Третьим предметом настоящего изобретения является промежуточное соединение, которое может использоваться в изготовлении гетероциклического имидазольного соединения, описываемого указанной выше общей формулой (I).
Это промежуточное соединение описывается структурной формулой (V), представленной ниже:
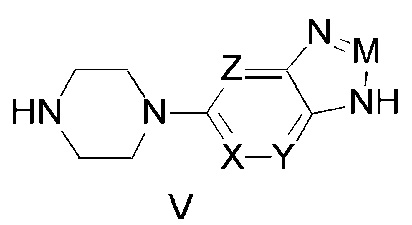
где в промежуточном соединении (V):
одно из обозначений X, Y, Z представляет собой азот, а другие — углеводород, или одно из обозначений X, Y, Z обозначает углеводород, а другие — азот;
M представляет собой азот или CR1, где:
R1 представляет собой водород, кислород, алкил, алкоксил или галогеналкил.
В одном из вариантов применения настоящего изобретения X и Z представляют собой азот, а Y представляет собой углеводород, или X представляет собой азот, а Y и Z — углеводороды, или Z представляет собой азот, а X и Y — углеводороды, или Y представляет собой азот, а X и Z — углеводороды.
В одном из вариантов применения настоящего изобретения R1 представляет собой водород, кислород, (C1-C6) алкил или (C1-C6) галогеналкил.
В одном из вариантов применения настоящего изобретения R1 представляет собой водород, кислород, (C1-C3) алкил или (C1-C3) галогеналкил.
В одном из вариантов применения настоящего изобретения R1 представляет собой водород, кислород, метил или трифторметил.
Предпочтительно, чтобы указанное промежуточное соединение (V) было следующим:
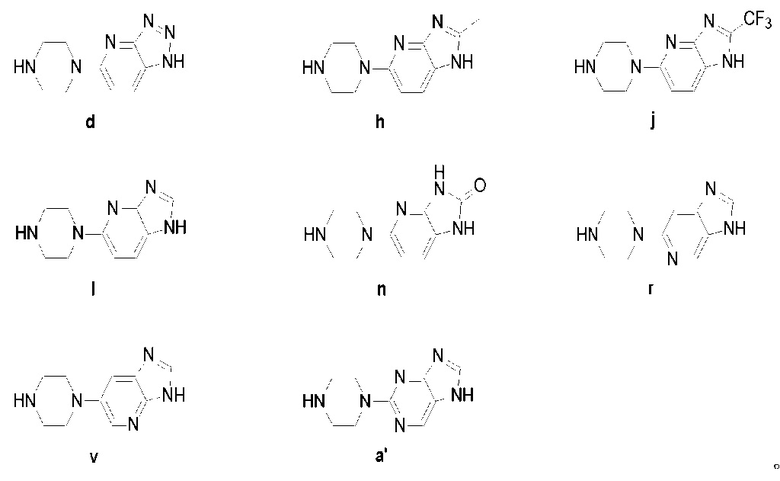
Способ изготовления промежуточного соединения (V) является четвертым предметом настоящего изобретения. Промежуточное соединение (V) получают следующим образом:
Стадия 1: путем реакции нуклеофильного замещения между монопиперазином и нитрозамещенным гетероциклическим галоидным соединением, содержащим аминогруппу, получают промежуточное соединение (II);
Стадия 2: в промежуточном соединении (II) путем каталитической гидрогенизации восстанавливают нитрогруппу, получая промежуточное соединение (III);
Стадия 3: путем реакции циклизации между промежуточным соединением (III) и нитритом натрия, ангидридом уксусной кислоты, или ангидридом трифторуксусной кислоты, или триметилортоформиатом, или тринитридным соединением получают промежуточное соединение (IV);
Стадия 4: из промежуточного соединения (IV) удаляют защитную группу аминогруппы, получая промежуточное соединение (V);
Формулы этих реакций представлены ниже:
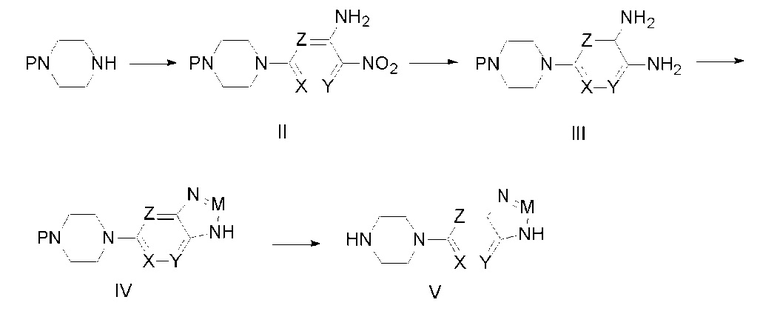
где P представляет собой защитную группу аминогруппы, одно из обозначений X, Y, Z представляет собой азот, а другие — углеводород, или одно из обозначений X, Y, Z обозначает углеводород, а другие — азот; M представляет собой азот или CR1, где: R1 представляет собой водород, оксогруппу, метил или трифторметил.
В одном из вариантов применения настоящего изобретения путем реакции циклизации между промежуточным соединением (III) и ангидридом уксусной кислоты, ангидридом трифторуксусной кислоты, триметилортоформиатом или азидом натрия получают промежуточное соединение (IV);
Промежуточное соединение (V), являющееся пятой целью настоящего изобретения, применяется в изготовлении соединения, описываемого упомянутой выше формулой (I), или его фармацевтически приемлемой соли.
Лекарственная композиция, являющаяся шестой целью настоящего изобретения, включает соединение формулы (I), содержащее терапевтически эффективную дозу активного компонента этой композиции или его фармацевтически приемлемую соль и один или несколько видов фармацевтических носителей, эксципиент и/или разжижающее средство.
Упомянутые лекарственные комплексы можно изготовить в форме таблеток, капсул, водных и масляных суспензий, растворимых порошков, гранул, лекарственных пастил, эмульсий, сиропов, кремов, мазей, суппозиториев или растворов для инъекций.
В упомянутых лекарственных комплексах соединения, описываемые общей формулой (I), существуют в свободном виде.
Одним из способов реализации, являющихся седьмой целью настоящего изобретения, является применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарств от заболеваний, которые лечатся торможением активности ПАРП.
Одним из способов реализации, являющихся седьмой целью настоящего изобретения, является применение упомянутой лекарственной композиции в изготовлении лекарств от заболеваний, которые лечатся торможением активности ПАРП.
Заболеваниями, которые лечатся торможением активности ПАРП, могут быть заболевания сосудов, септический шок, ишемические повреждения, нейротоксичность, геморрагический шок, воспалительное заболевание, рассеянный склероз, нейродегенеративные заболевания или сахарный диабет. Cantoni и др. (Biochim. Biophys. Acta, 1989, 1014: 1-7) и Liaudet и др. (Proc. Natl. Acad. Sci. U.S.A., 97(3), 2000, 97(3): 10203-10208) предоставили исследования связи упомянутых выше заболеваний с активностью ПАРП.
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение соединения, описываемого общей формулой (I), в изготовлении поддерживающих лекарственных средств, используемых при лечении опухолей.
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение соединения, описываемого общей формулой (I), или его фармацевтически приемлемых солей в изготовлении поддерживающих лекарственных средств, используемых при лечении опухолей.
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение указанных лекарственных составов в изготовлении поддерживающих лекарственных средств, используемых при лечении рака.
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение соединения, описываемого общей формулой (I), в изготовлении лекарственных препаратов, используемых в противораковой химиотерапии или для усиления химиорадиотерапии.
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение соединения, описываемого общей формулой (I), или его фармацевтически приемлемых солей в изготовлении лекарственных препаратов, используемых в противораковой химиотерапии или для усиления химиорадиотерапии.
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение указанных лекарственных составов в изготовлении лекарственных препаратов, используемых в противораковой химиотерапии или для усиления химиорадиотерапии.
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение соединения, описываемого общей формулой (I), в изготовлении лекарств для индивидульного лечения рака, дефектного по репарации двухнитевых разрывов (ДНР) ДНК, зависимой от гомологической рекомбинации (ГР).
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение соединения, описываемого общей формулой (I), или его фармацевтически приемлемой соли в изготовлении лекарств для индивидульного лечения рака, дефектного по репарации двухнитевых разрывов (ДНР) ДНК, зависимой от гомологической рекомбинации (ГР).
Одним из способов применения, которые являются седьмой целью настоящего изобретения, является применение упомянутой лекарственной композиции в изготовлении лекарств для индивидульного лечения рака, дефектного по репарации двухнитевых разрывов (ДНР) ДНК, зависимой от гомологической рекомбинации (ГР).
Предпочтительно, чтобы пути упомянутой репарации двухнитевых разрывов ДНК, зависимой от гомологической рекомбинации (ГР), были дефектными.
В том числе, предпочтительно, чтобы упомянутый рак содержал один или несколько видов раковых клеток, у которых снижена или утрачена способность к репарации двухнитевых разрывов ДНК, зависимой от гомологической рекомбинации (ГР), по сравнению с нормальными клетками.
Предпочтительно, чтобы упомянутый рак имел дефектный или мутантный фенотип BRCA-1 или BRCA-2. Более того, наиболее предпочтительно, чтобы упомянутый рак был дефектным или мутантным раком BRCA-1 и/или BRCA-2.
Предпочтительно, чтобы упомянутый рак был раком молочной железы, раком яичников, раком поджелудочной железы, раком предстательной железы, раком прямой кишки, раком ободочной кишки, раком печени.
Для того, чтобы проверить уровень эффективности воздействия соединения, предложенного настоящим изобретением, на ферменты ПАРП, при определении активности воздействия каждого соединения настоящего изобретения на ферменты ПАРП использовались испытания активности ферментов на биохимическом уровне
ПАРП — это вид ферментов посттрансляционной модификации, который может быть активирован повреждением ДНК. ПАРП присутствует в каталитических процессах организма, главным образом, в некоторых процессах поли-АДФ-рибозилирования, зависящих от НАД (никотинамидадениндинуклеотид), его субстрат включает преимущественно некоторые нуклеопротеины внутри ПАРП, например, одни из них — гистоны. В настоящем изобретении путем измерения ПАРП под действием НАД в процессе поли-АДФ-рибозилировании гистонов, содержащихся на 96-луночном культуральном планшете, измеряется активность ПАРП, и, соответственно, измеряется активность ПАРП после ингибирующего воздействия, тем самым оценивается степень подавления активности ПАРП видом соединений, предложенных настоящим изобретением.
Способы реализации
Данные ниже примеры соединения используются для дальнейшего описания настоящего изобретения, однако эти примеры отнюдь не ограничивают сферу применения настоящего изобретения.
В примерах реализации настоящего изобретения экспериментальные методы, для которых не указаны конкретные условия, используются в соответствии с нормальными условиями, или условиями, рекомендуемыми компанией-производителем сырья или товара. Реагенты, для которых не указывается конкретный источник, закупаются в обычном порядке.
Ниже даются значения терминов, используемых в описании изобретения или в разделе формулы изобретения, если не заявлено обратное.
В настоящем изобретении термин «(С1-С6) алкил» означает линейный или разветвленный насыщенный одновалентный алкил, содержащий от 1 до 6 атомов углерода. Примеры подобных радикалов включают (но не ограничиваются) метил, этил, пропил, изопропил, н-бутил, изобутил и трет-бутил.
Термин «(С1-С6) галогеналкил» означает линейный или разветвленный насыщенный одновалентный алкил, содержащий от 1 до 6 атомов углерода, в котором атомы водорода частично или полностью замещены атомами галогена.
Термин «(С1-С6) алкоксил» означает линейный или разветвленный насыщенный одновалентный алкил, содержащий от 1 до 6 атомов углерода, связанный с атомом кислорода. Примеры подобных радикалов включают (но не ограничиваются) метоксил, этоксил, пропоксил, изопропоксил, н-бутоксил, изобутоксил и трет-бутоксил.
Термин «энантиомеры» означает стереоизомеры, представляющие собой зеркальные отражения друг друга.
Термин «энантиомеры» означает стереоизомеры, молекулы которых имеют один или несколько хиральных центров и не являются зеркальными отражениями друг друга
Термин «конформер» означает изомер, являющийся результатом ограниченного вращения органической молекулы вокруг одинарной химической связи.
Термин «таутомерия» означает явление, при котором два вида функциональных изомеров каких-либо органических структур легко переходят друг в друга и при этом между ними устанавливается равновесие, изомеры, соответствующие этому явления, называются таутомерами.
Термин «мезомер» означает оптически неактивное соединение, образованное молекулами, которые содержат несимметричные атомы, но обладают факторами симметрии.
Термин «рацемат» означает эквимолярную смесь оптически активных хиральных молекул и их энантиомеров.
Термин «метаболит и его прекурсор» означает получаемое в метаболическом процессе или расходуемое вещество; термин «пролекарство» означает соединение, которое получают из лекарственного вещества путем изменения его химической структуры, вне живого организма или человеческого тела это соединение неактивно, а внутри организма превращается в первоначальное вещество и действует как лекарство.
Термин «производное» означает сравнительно сложный продукт, в котором атомы или радикалы первоначального соединения замещены другими атомами или радикалами.
Термин «терапевтически эффективная доза» означает любое количество, которое производит необходимую биологическую реакцию.
Термин «галоген» означает F (фтор), Cl (хлор), Br (бром), I (йод).
Термин «лекарственная композиция» означает смесь одного или двух соединений, описываемых настоящим изобретением, с другими химическими веществами, например фармацевтически приемлемым носителем. Цель лекарственной композиции заключается в том, чтобы способствовать процессу усвоения лекарства.
Термин «фармацевтический носитель» означает неактивный компонент в лекарственной копозиции, который не оказывает явного возбуждения на организм и не мешает биологической активности и свойствам принимаемого соединения, например: карбонат кальция, фосфат кальция, различные углеводы (например, лактоза, маннит), крахмал, циклодекстрин, стеарат магния, целлюлоза, карбонат магния, полимеры акриловой или метилакриловой кислоты, гели, вода, полиэтиленгликоль, пропиленгликоль, этиленгликоль, касторовое масло, гидрированное или полиэтоксилированное гидрированное касторовое масло, кунжутное, кукурузное, арахисовое масла и другие вещества.
В указанных выше лекарственных композициях помимо фармацевтически приемлемых носителей также могут содержаться адъюванты, обычно применяемые в фармакологии, например: антибактериальные, противогрибковые, противомикробные средства, консерванты, пигменты, солюбилизаторы, загустители, поверхностно-активные вещества, комплексообразующие агенты, протеины, аминокислоты, жиры, сахариды, витамины, минералы, микроэлементы, подсластители, химические красители, ароматизаторы или их сочетания.
Настоящим изобретением открыто некоторое соединение, а также применение этого соединения в качестве ингиботора поли(АДФ-рибоза)-полимеразы. Технические специалисты этой области могут заимствовать содержание этого изобретения и применять его, подходящим образом улучшая технические параметры. Особо следует подчеркнуть, что любые подобные заменители или преобразования будут совершенно очевидны для специалистов этой области, поэтому считается, что они учтены в настоящем изобретении. Методы настоящего изобретения а также его применение уже описаны посредством примеров реализации, которые дали достаточно хорошие результаты. Очевидно, что специалисты смогут, внося в рамках настоящего изобретения модификации или целесообразные изменения и сочетания, реализовывать и применять технологии настоящего изобретения.
Представленные ниже комбинированные примеры реализации еще больше поясняют настоящее изобретение.
Примеры изготовления
Структурная формула соединения положительно определена путем ядерного магнитного резонанса (ЯМР) или масс-спектрометрии (МС). Сдвиг (S) ЯМР задан единицами в миллионных долях, 10-6 (ppm). Растворитель, применяемый для измерений, представляет собой дейтерированный метанол, дейтерированный хлороформ, в качестве внутреннего стандарта используется тетраметилсилан.
При масс-спектрометрии использовалась жидкость и масс-спектрограф (производитель: компания «Симадзу», серийный номер: LCMS-2020).
Можно использовать начальное сырье, уже известное по настоящему изобретению, или его можно комбинировать в соответствии с уже известными методами данной области, или можно напрямую закупить необходимые коммерческие продукты.
Пример реализации 1
Соединение (1): получение 4-(3-(4-(1H-[1,2,3]триазол[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
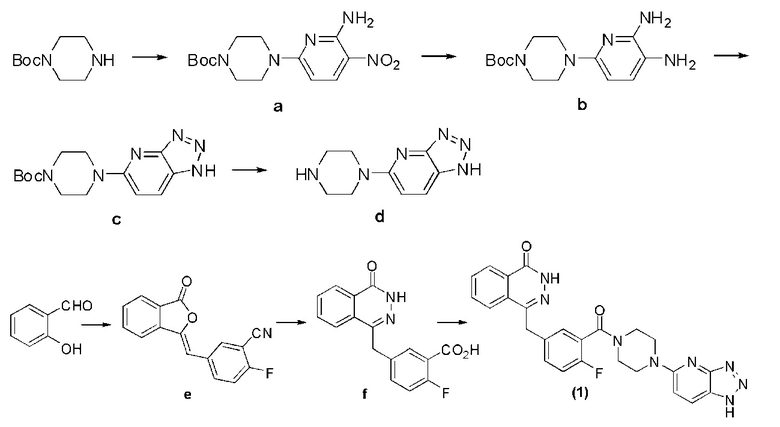
Стадия 1: получение 4-(6-амино-5-нитропиридин-2-ил)пиперазин-1-терт бутилкарбоната.
В диметилформамид (10 мл), содержащий защитный пиперазин моно-трет-бутилоксикарбонила (1,86 г, 10 ммоль), добавляют 6-хлор-3-нитро-2-аминопиридин (1,91 г, 11 ммоль) и диизопропилэтиламин (1,55 г, 12 ммоль), реакция проходит при комнатной температуре, через 8 часов путем декомпрессии удаляют растворитель, оставшееся вещество подвергают флэш-хроматографии (отношение дихлорметана к метанолу составляет 50 к 1) и получают твердое соединение а белого цвета: 4-(6-амино-5-5-нитропиперидин-2-ил)пиперазин-1-трет бутилкарбонат (2,72 г, коэффициент выработки 84%). MS (ESI) m/z: [M+H]+= 324.
Стадия 2: получение 4-(5,6-диаминопиридин-2-ил)пиперазин -1-трет бутилкарбоната.
10%-ный карбид палладия (259 мг) добавляют в раствор метанола (20 мл) с содержащимся в нем соединением а (2,59 г, 8 ммоль), в течение 7 часов при нормальной температуре протекает гидрирование, затем проводят фильтрацию. Оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 10 к 1) и получают твердое вещество b желтого цвета: 4-(5,6-диаминопиперидин-2-ил)пиперазин-1-трет бутилкарбонат (2,25 г, коэффициент выработки 93%). MS (ESI) m/z: [M+H]+=294.
Стадия 3: получение 4-(1H-[1,2,3]триазол[4,5-b]пиридин-5-ил)пиперазин-1-трет бутилкарбоната.
В раствор уксусной кислоты (30 мл), содержащий соединение b (1,76 г, 6 ммоль), добавляют нитрит натрия (0,42 г, 6 ммоль), нагревают до флегмы, после 8 часов реакции охлаждают, путем декомпрессии удаляют растворитель, оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 10 к 1) и получают твердое вещество c желтого цвета: 4-(1H-[1,2,3]триазол[4,5-b]пиридин-5-ил)пиперазин-1-трет бутилкарбонат (1,64 г, коэффициент выработки 90%). MS (ESI) m/z: [M+H]+=305.
Стадия 4: получение (пиперазин-1-ил)-1H-[1,2,3]триазол[4,5-b]пиперидина.
В раствор дихлорметана (10 мл), содержащий соединение c (1,52 г, 5 ммоль), добавляют трифторуксусную кислоту (2,28 г, 20 ммоль), реакция протекает при нормальной температуре 8 часов, затем путем декомпрессии удаляют растворитель, оставшееся вещество растворяют в дихлорметане (20 мл), добавляют гидрокарбонат натрия до уровня pH=8, растворитель удаляют путем выпаривания, оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 10 к 1) и получают твердое соединение d желтого цвета: 5-(пиперазин-1-ил)-1H-[1,2,3]триазол[4,5-b]пиперидин (0,87 г, коэффициент выработки 86%). MS (ESI) m/z: [M+H]+=205.
Стадия 5: получение 2-фтор-4-((3-оксоизобензофуран-1-(3H)-иден)метил)фенилциана.
В ледяной ванне в раствор абсолютного метанола (1 л), содержащий метилат натрия (61,8 г, 1,14 моль), медленно вводят диметилэфир ортофосфористой кислоты (97 мл, 1,06 моль). Температуру в реакционной системе поддерживают на уровне не выше 5°C, в течение 20 минут медленно капают 2-карбоксил-бензальдегид (135 г, 0,9 моль). В описанной выше реакционной системе постепенно повышают температуру до уровня нормальной, а также в течение 30 минут медленно капают метилсульфокислоту (81,6 мл, 1,26 моль). Растворитель удаляют путем декомпрессии, затем оставшееся вещество разбавляют водой (600 мл), а также с помощью дихлорметана (500 мл) трижды проводят экстракцию. Используя воду (100 мл) вместе с органической фазой, дважды производят экстракцию, органическая фаза дегидрируется с помощью ангидрида сульфата магния. Путем декомпрессии удаляют растворитель и получают твердое соединение желтого цвета: 3-оксо-1,3-дигидробензоизофуран-1-ил-диметилфосфит. Это соединение, не подвергая очистке, используют в следующей одноступенчатой реакции. В раствор тетрагидрофурана (330 мл), содержащий полученное в предыдущей реакции неочищенное соединение 3-оксо-1,3-дигидробензоизофуран-1-ил-диметилфосфита, добавляют 2-фтор-5-формилфенилциан (20,9 г, 0,14 моль), температуру реакционной системы снижают до 15°C, в течение 30 минут медленно капают триэтиламин (19,5 мл, 0,14 моль). Температура описанной выше реакционной системы постепенно повышается до нормальной, путем декомпрессии удаляют растворитель, оставшееся вещество промешивают водой (250 мл), отфильтровывают и получают твердое соединение e белого цвета: 2-фтор-4-((3-оксоизобензофуран-1-(3H)-иден)метил)фенилциан (37,2 г, коэффициент выработки 96%).
Стадия 6: получение 2-фтор-5-((4-оксо-3,4-дигидрофталазин-1-ил)метилбензойной кислоты.
В водный раствор (200 мл), содержащий соединение е (37 г, 0,14 моль), добавляют 13N раствор гидроксида натрия (50 мл), повышают температуру до 90°C, перемешивают 1 час. Снижают температуру в описанной выше реакционной системе до 70°C, затем добавляют гидразин-гидрат (100 мл, 2 моля) и перемешивают в течение 18 часов, поддерживая температуру на уровне 70°C. Реакционный раствор охлаждают до нормальной температуры, используя 8N соляной кислоты, регулируют водородный показатель описанной выше реакционной системы до уровня pH=4, фильтруют, фильтрованный осадок последовательно промывают водой (60 мл) дважды и диэтиловым эфиром (50 мл) трижды. Затем вакуумной сушкой получают твердое соединение f белого цвета: 2-фтор-5-((4-оксо-3,4-дигидрофталазин-1-ил)метилбензойная кислота (30,1 г, коэффициент выработки 77%). MS (ESI) m/z: [M+H]+=299.
Стадия 7: получение 4-(3-(4-(1H-[1,2,3]триазол[4,5-b]пиридин-5-ил)пиперазин-1-карбонил )-4- фторбензил)фталазин-1(дигидро)-кетона.
В раствор диметилформамида (5 мл), который содержит соединение f (50 мг, 0,17 ммоль), добавляют соединение d (49 мг, 0,24 ммоль), 2-(7-азабензотриазол)-N,N,N′,N′-тетраметилкарбамид-гексафторфосфорную кислоту (77 мг, 0,2 ммоль) и триэтиламин (70 мг, 0,7 ммоль) и оставляют перемешиваться при комнатной температуре в течение ночи. Удаляют растворитель выпариванием, оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 10 к 1) и получают твердое соединение белого цвета (1): 4-(3-(4-(1H-[1,2,3]триазол[4,5-b]пиридин-5-ил)пиперазин-1-карбонил )-4- фторбензил)фталазин-1(дигидро)-кетон (16 мг, коэффициент выработки 20%). MS (ESI) m/z: [M+H]+=485. 1H NMR (300 MHz, DMSO-d6): δ 12,57 (s, 1H), 8,24-8,12 (m, 2H), 7,96-7,74 (m, 1H), 7,89-7,81 (m, 3H), 7,43-7,38 (m, 2H), 7,26-7,21 (m, 1H), 7,05-6,99 (m, 1H), 4,32 (s, 2H), 3,73 (br, 6H), 3,57 (br, 2H).
Пример реализации 2
Соединение (2): получение 4-(4-фтор-3-(4-(2-метил-1H-имидазо-[4,5-b]-пиридин-5-ил)-пиперазин-1-карбонил)-бензил)-фталазин-1-(дигидро)-кетон, ниже дано конкретное уравнение химической реакции:
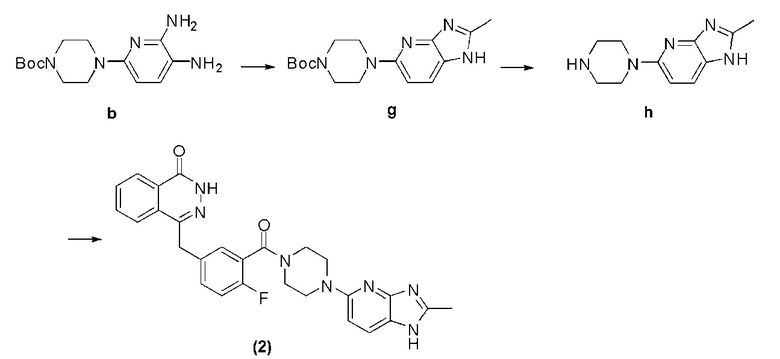
Стадия 1: получение 4-(2-метил-1H-имидазо-[4,5-b]-пиридин-5-ил)-пиперазин-1-трет-бутилкарбоната.
В раствор уксусной кислоты (30 мл), содержащий соединение b (1,47 г, 5 ммоль), добавляют ангидрид уксусной кислоты (0,56 г, 5,5 ммоль), температуру повышают до флегмы и после 8 часов протекания реакции охлаждают. Путем декомпрессии удаляют растворитель, оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 10 к 1) и получают твердое соединение g желтого цвета: 4-(2-метил-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-трет бутилкарбонат (0,73 г, коэффициент выработки 46%). MS (ESI) m/z: [M+H]+=318.
Стадия 2: получение 2-метил-5-(пиперазин-1-ил)-1H-имидазо[4,5-b]пиридина.
Используется метод, аналогичный методу получения соединения d на стадии 4 в примере 1: путем реакции десорбции защитной группы между соединением g и трифторуксусной кислотой получают соединение h: 2-метил -5-(пиперазин-1-ил)-1H-имидазо[4,5-b]пиридин (320 мг, коэффициент выработки 82%). MS (ESI) m/z: [M+H]+=218.
Стадия 3: получение 4-(4-фтор-3-(4-(2-метил-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения d на стадии 7 в примере 1: путем реакции конденсации между соединением h и соединением f получают соединение (2): 4-(4-фтор-3-(4-(2-метил-1H-имидазо-[4,5-b]-пиридин-5-ил)-пиперазин-1-карбонил)-бензил)-фталазин-1-(дигидро)-кетон (26 мг, коэффициент выработки 32%). MS (ESI) m/z: [M+H]+=498. 1H NMR (300 MHz, DMSO-d6): δ 12,55 (s, 1H), 8,23-8,12 (m, 2H), 7,96-7,75 (m, 1H), 7,89-7,80 (m, 3H), 7,44-7,38 (m, 2H), 7,27-7,22 (m, 1H), 7,06-6,98 (m, 1H), 4,33 (s, 2H), 3,72 (br, 4H), 3,56 (br, 4H), 2,63 (s, 3H).
Пример реализации 3
Соединение (3): получение 4-(4-фтор-3-(4-(2-трифторметил)-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетон, ниже дано конкретное уравнение химической реакции:
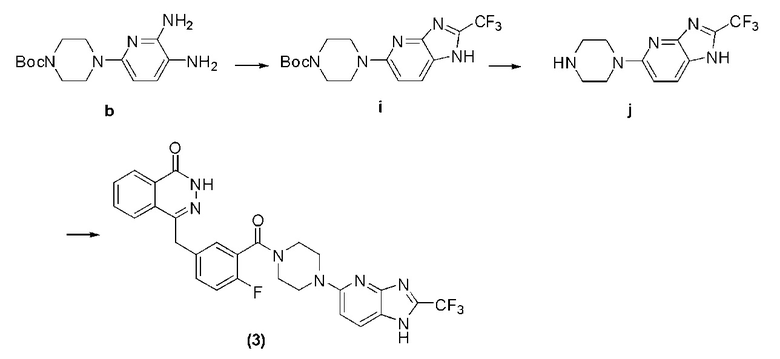
Стадия 1: получение 4-(2-трифторметил-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-трет бутилкарбоната.
В раствор трифторуксусной кислоты (30 мл), содержащий соединение b (1,47 г, 5 ммоль), добавляют ангидрид уксусной кислоты (1,16 г, 5,5 ммоль), повышают температуру до флегмы, после 8 часов протекания реакции температуру охлаждают, путем декомпрессии удаляют растворитель, оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 10 к 1) и получают твердое соединение i светло-желтого цвета: 4-(2-трифторметил-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-трет бутилкарбонат (0,69 г, коэффициент выработки 37%). MS (ESI) m/z: [M+H]+=372.
Стадия 2: получение 5-(пиперазин-1-ил)-2-трифторметил-1H-имидазо[4,5-b]пиридина.
Используется метод, аналогичный методу получения соединения d на стадии 4 в примере 1: путем реакции десорбции защитной группы между соединением i и трифторуксусной кислотой получают соединение j: 5-(пиперазин-1-ил)-2-трифторметил-1H-имидазо[4,5-b]пиридин (269 мг, коэффициент выработки 78%). MS (ESI) m/z: [M+H]+=272.
Стадия 3: получение 4-(4-фтор-3-(4-(2-трифторметил)-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере 1: путем реакции конденсации между соединением j и соединением f получают соединение (3): 4-(4-фтор-3-(4-(2-метил-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетон (38 мг, коэффициент выработки 41%). MS (ESI) m/z: [M+H]+=552. 1H NMR (300 MHz, DMSO-d6): δ 12,59 (br, 1H), 8,25 (d, 1H, J=8,1 Hz), 7,98-7,89 (m, 3H), 7,87-7,80 (m, 2H), 7,45-7,38 (m, 2H), 7,26-7,20 (m, 1H), 6,92 (d, 1H, J=9,0 Hz), 4,33 (s, 2H), 3,73 (br, 2H), 3,63 (br, 2H), 3,46 (br, 4H).
Пример реализации 4
Соединение (4): получение 4-(3-(4-(lH-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетон, ниже дано конкретное уравнение химической реакции:

Стадия 1: получение 4-(1H-имидазо-[4,5-b]-пиридин-5-ил)-пиперазин-1-трет бутилкарбоната.
В раствор триметилортоформиата (1,47 г, 5 ммоль), содержащий соединение b (6 г), добавляют толуолсульфоновую кислоту (86 мг, 0,5 ммоль), температуру повышают до флегмы, после 8 часов протекания реакции охлаждают, путем декомпрессии удаляют растворитель, оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 10 к 1) и получают твердое соединение k светло-желтого цвета: 4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-трет бутилкарбонат (0,73 г, коэффициент выработки 48%). MS (ESI) m/z: [M+H]+=304.
Стадия 2: получение 5-(пиперазин-1-ил)-1H-имидазо[4,5-b]пиридина.
Используется метод, аналогичный методу получения соединения d на стадии 4 в примере 1: путем реакции десорбции защитной группы между соединением k и трифторуксусной кислотой получают соединение l: 5-(пиперазин-1-ил)-1H-имидазо[4,5-b]пиридин (307 мг, коэффициент выработки 73%). MS (ESI) m/z: [M+H]+=204.
Стадия 3: получение 4-(3-(4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 1 в примере 1: путем реакции конденсации между соединением l и соединением f получают соединение (4): 4-(3-(4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетон (25 мг, коэффициент выработки 31%). MS (ESI) m/z: [M+H]+=484. 1H NMR (300 MHz, DMSО-d6): δ 12,61 (br, 1H), 8,27-8,24 (m, 1H), 8,16 (s, 1H), 8,00-7,97 (m, 1H), 7,93-7,82 (m, 4H), 7,45-7,39 (m, 2H), 7,28-7,22 (m, 1H), 6,83-6,80 (m, 1H), 4,34 (s, 2H), 3,73 (br, 2H), 3,58 (br, 2H), 3,42 (br, 4H).
Пример реализации 5
Соединение (5): получение 4-(4-фтор-3-(4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
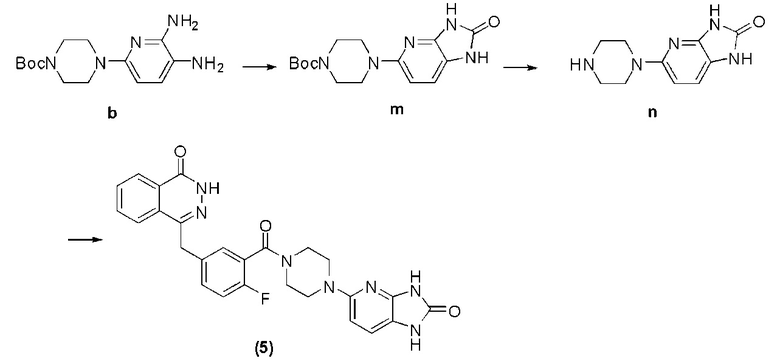
Стадия 1: получение 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-трет бутилкарбоната.
В раствор ангидридного тетрагидрофурана (20 мл), содержащий соединение b (1,47 г, 5 ммоль), добавляют карбонилдиимидазол (10 ммоль), температуру повышают до флегмы, после 8 часов протекания реакции охлаждают, путем декомпрессии удаляют растворитель, оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 10 к 1) и получают твердое соединение m желтого цвета: 4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-трет бутилкарбонат (1,24 г, коэффициент выработки 78%). MS (ESI) m/z: [M+H]+=320.
Стадия 2: получение 5-(пиперазин-1-ил)-1H-имидазо[4,5-b]пиридин-2(3H)-кетона.
Используется метод, аналогичный методу получения соединения d на стадии 4 в примере 1: путем реакции десорбции защитной группы между соединением m и трифторуксусной кислотой получают соединение n: 5-(пиперазин-1-ил)-1H-имидазо[4,5-b]пиридин-2(3H)-кетон (331 мг, коэффициент выработки 79%). MS (ESI) m/z: [M+H]+=220.
Стадия 3: получение 4-(4-фтор-3-(4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением n и соединением f получают соединение (5): 4-(4-фтор-3-(4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетон (32 мг, коэффициент выработки 36%). MS (ESI) m/z: [M+H]+=500. 1H NMR (300 MHz, DMSО-d6): δ 12,58 (br, 1H), 10,97 (br, 1H), 10,39 (br, 1H), 8,28-8,26 (m, 1H), 7,99-7,96 (m, 1H), 7,92-7,81 (m, 2H), 7,46-7,42 (m, 1H), 7,39-7,37 (m, 1H), 7,27-7,20 (m, 1H), 7,11 (d, 1H, J=8,4Hz), 6,36 (d, 1H, J=8,4Hz), 4,33 (s, 2H), 3,73 (br, 2H), 3,40 (br, 2H), 3,26-3,21 (br, 4H).
Пример реализации 6
Соединение (6): получение 4-(3-(4-(3H-имидазо[4,5-c]пиридин-6-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
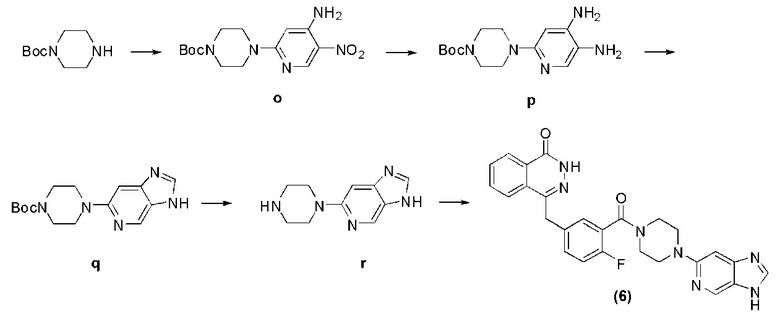
Стадия 1: получение 4-(4-амино-5-нитропиридин-2-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения a на стадии 1 в примере реализации 1, путем реакции нуклеофильного замещения между защитным пиперазином моно-трет-оксокарбонила и 2-хлор-5-нитро-4-аминопиридином получают соединение o: 4-(4-амино-5-нитропиридин-2-ил)пиперазин-1-трет бутилкарбонат (1,1 г, коэффициент выработки 86%). MS (ESI) m/z: [M+H]+=324.
Стадия 2: получение 4-(4,5-диаминопиридин-2-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения b на стадии 2 в примере реализации 1, путем реакции каталитической гидрогенизации из соединения o получают соединение p: 4-(4,5-диаминопиридин-2-ил)пиперазин-1-трет бутилкарбонат (0,9 г, коэффициент выработки 97%). MS (ESI) m/z: [M+H]+=294.
Стадия 3: получение 4-(3H-имидазо[4,5-c]пиридин-6-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения k на стадии 1 в примере реализации 4, путем реакции циклизации между соединением p и триметилортоформиатом получают соединение q: 4-(3H-имидазо[4,5-c]пиридин-6-ил)пиперазин-1-трет бутилкарбонат (0,6 г, коэффициент выработки 82%). MS (ESI) m/z: [M+H]+=304.
Стадия 4: получение 6-(пиперазин-1-ил)-3H-имидазо[4,5-c]пиридина.
Используется метод, аналогичный методу получения соединения d на стадии 4 в примере 1: путем реакции десорбции защитной группы между соединением m и трифторуксусной кислотой получают соединение r: 6-(пиперазин-1-ил)-3H-имидазо[4,5-c]пиридин (279 мг, коэффициент выработки 75%). MS (ESI) m/z: [M+H]+=204.
Стадия 5: получение 4-(3-(4-(3H-имидазо[4,5-c]пиридин-6-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1, путем реакции конденсации между соединением n и соединением f получают соединение (6): 4-(3-(4-(3H-имидазо[4,5-c]пиридин-6-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетон (16 мг, коэффициент выработки 20%). MS (ESI) m/z: [M+H]+=484. 1H NMR (300MHz, DMSO-d6): δ 12,57 (s, 1H), 12,35 (s, 1H), 8,54 (s, 1H), 8,25 (d, 1H, J=7,8 Hz), 8,09 (s, 1H), 7,98-7,80 (m, 3H), 7,42-7,37 (m, 2H), 7,26-7,20 (m, 2H), 6,76 (s, 1H), 4,33 (s, 2H), 3,75 (br, 2H), 3,50 (br, 2H), 3,39 (br, 4H).
Пример реализации 7
Соединение (7): получение 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:

Стадия 1: получение 4-(5-амино-6-нитропиридин-3-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения a на стадии 1 в примере реализации 1, путем реакции нуклеофильного замещения между защитным пиперазином моно-трет-оксобутила и 5-бром-2-нитро-3-аминопиридином получают соединение s: 4-(5-амино-6-нитропиридин-3-ил)пиперазин-1-трет бутилкарбонат (0,7 г, коэффициент выработки 82%). MS (ESI) m/z: [M+H]+=324.
Стадия 2: получение 4-(5,6-диаминопиридин-3-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения b на стадии 2 в примере реализации 1, путем реакции каталитической гидрогенизации из соединения s, получают соединение t: 4-(5,6-диаминопиридин-3-ил)пиперазин-1-трет бутилкарбонат (0,52 г, коэффициент выработки 91%). MS (ESI) m/z: [M+H]+=294.
Стадия 3: получение 4-(3H-имидазо[4,5-c]пиридин-6-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения k на стадии 1 в примере реализации 4, путем реакции циклизации между соединением t и триметилортоформиатом получают соединение u: 4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-трет бутилкарбонат (0,36 г, коэффициент выработки 73%). MS (ESI) m/z: [M+H]+=304.
Стадия 4: получение 6-(пиперазин-1-ил)-3H-имидазо[4,5-b]пиридина.
Используется метод, аналогичный методу получения соединения d на стадии 4 в примере 1: путем реакции десорбции защитной группы между соединением u и трифторуксусной кислотой получают соединение v: 6-(пиперазин-1-ил)-3H-имидазо[4,5-b]пиридин (126 мг, коэффициент выработки 82%). MS (ESI) m/z: [M+H]+=204.
Стадия 5: получение 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением v и соединением f получают соединение (7): 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-фторбензил)фталазин-1(дигидро)-кетон (16 мг, коэффициент выработки 22%). MS(ESI): m/z 484 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,59 (s, 1H), 8,25-8,20 (m, 3H), 7,98-7,79 (m, 3H), 7,51-7,45 (m, 1H), 7,42-7,37 (m, 3H), 7,26-7,20 (m, 1H), 4,33 (s, 2H), 4,33 (s, 2H ), 3,78 (br, 2H), 3,55-3,47 (m, 2H), 3,19-3,14 (m, 2H), 3,03 (br, 2H).
Пример реализации 8
Соединение (8): получение 4-(3-(4-(7H-пурин-2-ил)пиперазин-1-карбонил)-4- фторбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
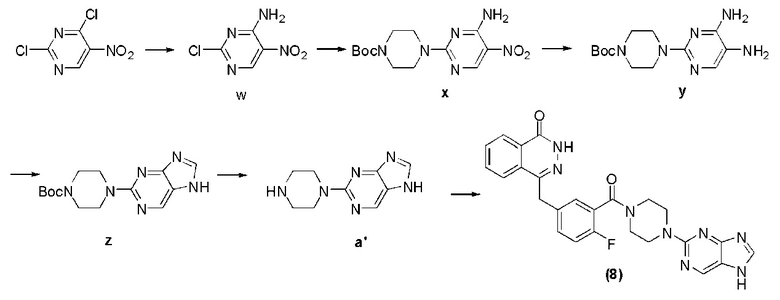
Стадия 1: получение 2-хлор-5-нитро-4-аминопиримидина.
В тетрагидрофуран (10 мл), содержащий 2,4-дихлор-нитропиримидин (500 мг, 2,5 ммоль), добавляют гидрокарбонат натрия (238 мг, 2,8 ммоль) и аммиачную воду (0,3 мЛ), после 2 часов протекания реакции при 55°C путем декомпрессии удаляют растворитель, оставшееся вещество подвергают флэш-хроматографии (соотношение дихлорметана к метанолу составляет 100 к 1) и получают твердое соединение w белого цвета: 2-хлор-5-нитро-4-аминопиримидин (0,47 г, коэффициент выработки 84%). MS (ESI) m/z: [M+H]+=175.
Стадия 2: получение 4-(5-амино-6-нитропиримидин-2-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения a на стадии 1 в примере реализации 1, путем реакции нуклеофильного замещения между защитным пиперазином моно-трет оксобутила и соединением w получают соединение x: 4-(5-амино-6-нитропиримидин-2-ил)пиперазин-1-трет бутилкарбонат (0,61 г, коэффициент выработки 87%). MS (ESI) m/z: [M+H]+=325.
Стадия 3: получение 4-(4,5-диаминопиридин-2-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения b на стадии 2 в примере реализации 1, путем реакции каталитической гидрогенизации из соединения x, получают соединение z: 4-(4,5-диаминопиридин-2-ил)пиперазин-1-трет бутилкарбонат (0,26 г, коэффициент выработки 76%). MS (ESI) m/z: [M+H]+=295.
Стадия 4: получение 4-(7H-пурин-2-ил)пиперазин-1-трет бутилкарбоната.
Используется метод, аналогичный методу получения соединения k на стадии 1 в примере реализации 4, путем реакции циклизации между соединением y и триметилортоформиатом получают соединение z: 4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-трет бутилкарбонат (0,36 г, коэффициент выработки 73%). MS (ESI) m/z: [M+H]+=305.
Стадия 5: получение 2-(пиперазин-1-ил)-7H-пурина.
Используется метод, аналогичный методу получения соединения d на стадии 4 в примере 1: путем реакции десорбции защитной группы между соединением z и трифторуксусной кислотой получают соединение a′: 2-(пиперазин-1-ил)-7H-пурин (141 мг, коэффициент выработки 74%). MS (ESI) m/z: [M+H]+=205.
Стадия 6: получение 4-(3-(4-(7H-пурин-2-ил)пиперазин-1-карбонил)фторбензил)фталазил-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением a и соединением f получают соединение (8): 4-(3-(4-(7H-пурин-2-ил)пиперазин-1-карбонил)-4-фторбензил)фталазил-1(дигидро)-кетон (88 мг, коэффициент выработки 74%). MS (ESI) m/z: 485 [M+l]+. 1NMR (300MHz, DMSO-d6): δ 12,78 (s, 1H), 12,57 (s, 1H), 8,72 (s, 1H), 8,26-8,24 (m, 1H), 8,12 (s, 1H), 7,98-7,96 (m, 1H), 7,91-7,87 (m, 1H), 7,84-7,80 (m, 1H), 7,45-7,41 (m, 1H), 7,39-7,37 (m, 1H), 7,25-7,21 (m, 1H), 4,32 (s, 2H), 3,81-3,79 (m, 2H), 3,72-3,65 (m, 4H), 3,28-3,26 (m, 2H).
Пример реализации 9
Соединение (9): получение 4-(3-(4-(2-карбонил-2,3-дигидро-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
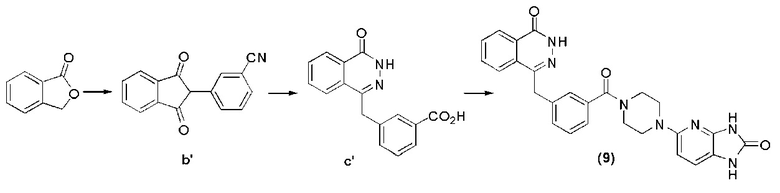
Стадия 1: получение 3-(1,3-диоксо-2,3-дигидро-1H-инден-2-ил)фенилциана.
В ледяной ванне в раствор этилпропионата (200 мл), содержащий изобензофуран-1(3H)-кетон (51 г, 0,38 моль) и трицианобензальдегид (52 г, 0,39 моль), в течение 40 минут постепенно добавляют метиловый раствор метоксида натрия (320 мл). В реакционной системе поддерживается температура ниже 30°C, затем в описанной выше реакционной системе медленно повышают температуру до нормальной и нагревают до флегмы в течение 1 часа, вводят метанол (100 мл) и производят перемешивание в режиме потока в течение 1 часа. Описанную выше реакционную систему охлаждают до нормальной температуры, путем декомпрессии удаляют растворитель, затем оставшееся вещество разводят водой (1 л) и отфильтровывают. Фильтрационный кек трижды промывают диэтиловым эфиром (200 мл), затем это соединение окисляют с помощью уксусной кислоты (110 мл) и отфильтровывают. Фильтрационный кек промывают водой (100 мл) и получают твердое соединение b′ красного цвета: 3-(1,3-диоксо-2,3-дигидро-1H-инден-2-ил)фенилциан (69 г, коэффициент выработки 94%).
Стадия 2: получение 3-((4-оксо-3,4-дигидрофталазил-1-ил)метил)бензойной кислоты.
Используется метод, аналогичный методу получения соединения f на стадии 6 в примере реализации 1, путем реакции гидролиза из соединения b получают соединение c′: 3-((4-оксо-3,4-дигидрофталазил-1-ил)метил)бензойная кислота (28 г, коэффициент выработки 55%). MS (ESI) m/z: 281 [M+l]+.
Стадия 3: получение 4-(3-(4-(2-карбонил-2,3-диоксо-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением n и соединением f получают соединение (9): 4-(3-(4-(2-карбонил-2,3-диоксо-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетон (37 мг, коэффициент выработки 46%). MS (ESI) m/z: 482 [M+l]+. 1H NMR (300 MHz, DMSО-d6): δ 12,58 (br, 1H ), 10,95 (br, 1H), 10,37 (m, 1H), 8,27-8,24 (m, 1H), 7,97-7,80 (m, 3H), 7,42-7,35 (m, 3H ), 7,26-7,23 (m, 1H), 7,11-7,09 (m, 1H), 6,34 (d, 1H, J=8,7 Hz), 4,35 (s, 2H), 3,69-3,47 (m, 4H), 3,24-3,14 (m, 4H).
Пример реализации 10
Соединение (10): получение 4-(3-(4-(lH-[1,2,3]-триазол[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
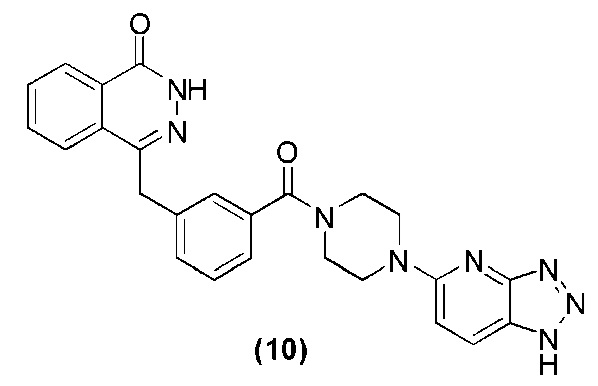
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением c и соединением d получают соединение (10): 4-(3-(4-(lH-[1,2,3]триазол[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-бензил)фталазин-1(дигидро)-кетон (41 мг, коэффициент выработки 52%). MS (ESI) m/z: 467 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,53 (s, 1H), 8,21-8,10 (m, 2H), 7,93-7,71 (m, 1H), 7,87-7,80 (m, 3H), 7,41-7,35 (m, 3H), 7,24-7,20 (m, 1H), 7,02-6,96 (m, 1H), 4,30 (s, 2H), 3,71 (br, 6H), 3,55 (br, 2H).
Пример реализации 11
Соединение (11): получение 4-(3-(4-(2-метил-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
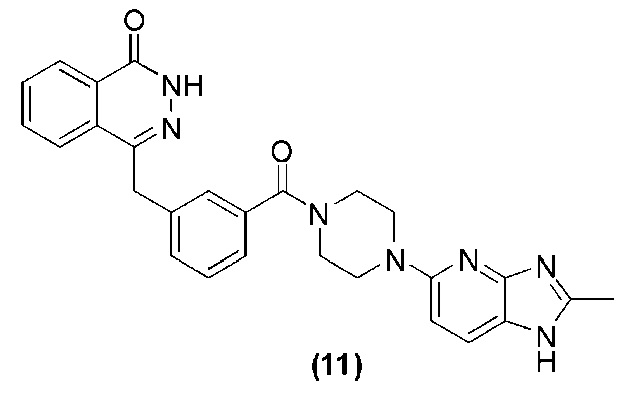
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением c и соединением h получают соединение (11): 4-(3-(4-(2-метил-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетон (034 мг, коэффициент выработки 45%). MS (ESI) m/z: 480 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,52 (s, 1H), 8,21-8,10 (m, 2H), 7,94-7,72 (m, 1H), 7,87-7,77 (m, 3H), 7,41-7,34 (m, 3H), 7,26-7,21 (m, 1H), 7,03-6,97 (m, 1H), 4,31 (s, 2H), 3,71 (br, 4H), 3,52 (br, 4H), 2,61 (s, 3H).
Пример реализации 12
Соединение (12): получение 4-(3-(4-(2-трифторметил)-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетон, ниже дано конкретное уравнение химической реакции:
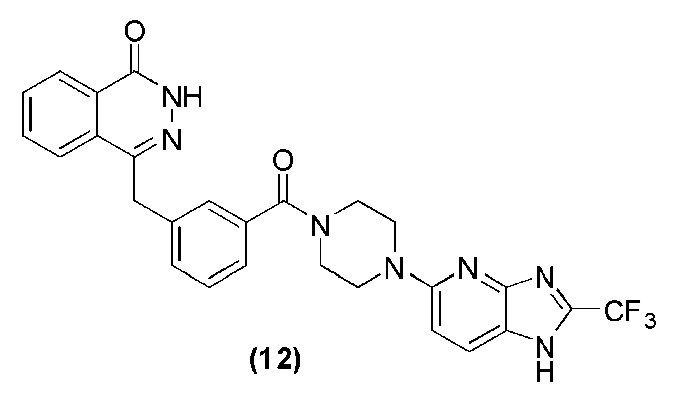
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением c и соединением j получают соединение (12): 4-(3-(4-(2-трифторметил)-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)бензил)фталазин-1(дигидро)-кетон (36 мг, коэффициент выработки 42%). MS (ESI) m/z: 534 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,56 (br, 1H), 8,22 (d, 1H, J=8,1 Hz), 7,95-7,87 (m, 3H), 7,83-7,76 (m, 3H), 7,42-7,36 (m, 2H), 7,22-7,17 (m, 1H ), 6,91 (d, 1H, J=9,0 Hz), 4,30 (s, 2H), 3,72 (br, 2H), 3,61 (br, 2H), 3,42 (br, 4H).
Пример реализации 13
Соединение (13): получение 4-(3-(4-(lH-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
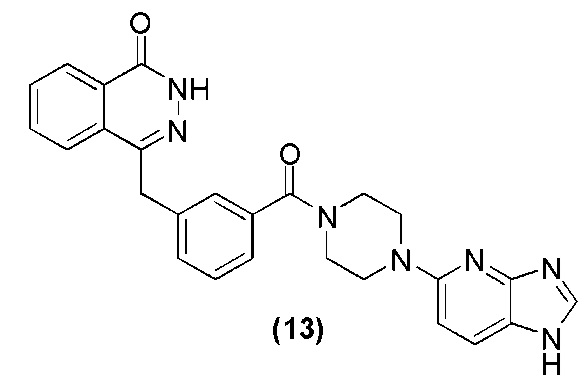
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением c и соединением l получают соединение (13): 4-(3-(4-(lH-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)фталазин-1(дигидро)-кетон (48 мг, коэффициент выработки 58%). MS (ESI) m/z: 466 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,57 (br, 1H), 12,50 (br, 1H), 8,23 (d, 1H, J=7,6 Hz), 8,00 (s, 1H), 7,96-7,93 (m, 1H), 7,88-7,72 (m, 3H), 7,40-7,34 (m, 3H), 7,25-7,24 (m, 1H), 6,79-6,73 (m, 1H), 4,33 (s, 2H ), 3,68-3,38 (m, 8H).
Пример реализации 14
Соединение (14): получение 4-(3-(4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-метоксилбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:

Стадия 1: получение 3-бром-4-метоксилметилбензоата.
В водный раствор (10 мл), содержащий 4-метоксилметилбензоат (1,5 г, 9 моль), при комнатной температуре медленно добавляют бромат калия (251 мг, 1,5 ммоль) и жидкий бром (722 мг, 4,5 ммоль). В течение 2,5 часов производят перемешивание при температуре реакционной системы не более 30°C. В описанную выше реакционную систему добавляют метилтретбутиловый эфир (25 мл), после экстракции органическую фазу промывают насыщенным солевым раствором. Вещество, оставшееся после дегидратации и выпаривания, подвергают флэш-хроматографии (соотношение петролейного эфира к этилацетату составляет 10:1) и получают твердое соединение d′ белого цвета: 3-бром-4-метоксилметилбензоат (2,1 г, коэффициент выработки95%).
Стадия 2: получение 3-циано-4-метоксилметилбензоата.
В раствор диметилформамида (10 мл), содержащий соединение d′ (1,1 г, 4,4 моль), добавляют цианистую медь (1,2 г, 13,33 ммоль), нагревают до 140°C и перемешивают в течение 6 часов. Описанную выше реакционную систему охлаждают и добавляют этилацетат (25 мл), после экстракции органическую фазу промывают насыщенным солевым раствором. Вещество, оставшееся после дегидратации и выпаривания, подвергают флэш-хроматографии (соотношение петролейного эфира к этилацетату составляет 10:1) и получают твердое соединение e′ белого цвета: 3-циано-4-метоксилметилбензоат (662 мг, коэффициент выработки 79%).
Стадия 3: получение 5-(метилол)-2-метоксиланилина.
В раствор тетрагидрофурана (25 мл), содержащий соединение e′ (1 г, 5,2 моль), добавляют боргидрил лития (0,45 г, 20,7 ммоль). При комнатной температуре производят перемешивание в течение ночи. Описанную выше реакционную систему подвергают дегидратации и выпариванию. Остаточное вещество подвергают флэш-хроматографии (соотношение петролейного эфира к этилацетату составляет 2:1) и получают твердое соединение f′ белого цвета: 5-(метилол)-2-метоксиланилин (845 мг, коэффициент выработки 100%).
Стадия 4: получение 5-формил-2-метоксилметилбензоата.
В раствор дихлорметана (50 мл), содержащий соединение f (845 мг, 5,2 моль), добавляют (1,1,1-триацетил)-1,1-дигидро-1,2-бензолйодацил-3(1H)-кетон (2,6 г, 6,2 ммоль). В течение 2 часов производят перемешивание при комнатной температуре. Описанную выше реакционную систему подвергают дегидратации и выпариванию. Остаточное вещество подвергают флэш-хроматографии (соотношение петролейного эфира к этилацетату составляет 3:1) и получают твердое соединение g′ белого цвета: 5-формил-2-метоксилметилбензоат (845 мг, коэффициент выработки 100%).
Стадия 5: получение 2-метоксил-5-((3-оксоизобензофуран-1(3H)-иден)метил)фенилциана.
Используется метод, аналогичный методу получения соединения e на стадии 5 в примере реализации 1, путем реакции между соединением g′ и 3-оксо-1,3-дигидроизобензофуран-1-ил-диметилортофосфатом получают соединение h′: 2-метоксилоксоизобензофуран-1(3H)-иден)метил)анилин (795 мг, коэффициент выработки 67%).
Стадия 6: получение 2-метоксил-5-((4-оксо-3,4-дигидрофталазил-1-ил)метилбензойной кислоты.
Используется метод, аналогичный методу получения соединения f на стадии 6 в примере реализации 1, путем реакции гидролиза из соединения h′ получают соединение i′: 2-метоксил-5-((4-оксо-3,4-дигидрофталазил-1-ил)-метил-бензойная кислота (318 мг, коэффициент выработки 63%). MS (ESI) m/z: 311 [M+l]+.
Стадия 7: получение 4-(3-(4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-метоксилбензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 1 в примере реализации 1: путем реакции конденсации между соединением r и соединением l получают соединение (14): 4-(3-(4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-метоксилбензил)фталазин-1(дигидро)-кетон (77 мг, коэффициент выработки 49%) MS (ESI) m/z: 496 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,55 (br, 1H), 8,23 (d, 1H, J=7,6 Hz), 8,19 (s, 1H), 7,95 (d, 1H, J=8,4 Hz), 7,88-7,80 (m, 3H), 7,39-7,31 (m, 1H), 7,16-7,15 (m, 1H), 7,01 (d, 1H, J=8,4 Hz), 6,80 (d, 1H, J=9,2 Hz), 4,24 (s, 2H), 3,73 (s, 3H), 3,70-3,69 (m, 2H), 3,56-3,54 (m, 2H), 3,37-3,36 (m, 2H), 3,18-3,16 (m, 2H).
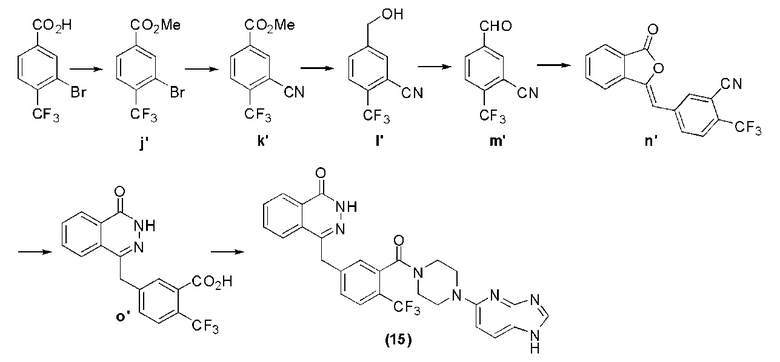
Пример реализации 15
Соединение (15): получение 4-(3-(4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
Стадия 1: получение 3-бром-4-трифторметилбензойной кислоты метиловый эфир.
В раствор (30 мл) метанола, содержащий 3-бром-4-трифторметил бензойную кислоту (4,1 г, 15,4 ммоль), при нормальной температуре медленно добавляют концентрированную серную кислоту (1 мл). Реакционную систему нагревают до 60°C и производят перемешивание в течение 6 часов. Охлаждают до нормальной температуры, в описанную выше реакционную систему добавляют этилацетат (25 мл), после экстракции органическую фазу промывают насыщенным солевым раствором. Вещество, оставшееся после дегидратации и выпаривания, подвергают флэш-хроматографии (соотношение петролейного эфира к этилацетату составляет 10:1) и получают твердое соединение j′ белого цвета: 3-бром-4-трифторметилбензойной кислоты метиловый эфир (4,2 г, коэффициент выработки 96%).
Стадия 2: получение 3-циано-4-трифторметилбензойной кислоты метиловый эфир.
Используется метод, аналогичный методу получения соединения e′ на стадии 2 в примере реализации 14: путем реакции цианирования из соединения f получают соединение k′: 3-циано-4-трифторметилбензойной кислоты метиловый эфир (1,6 г, коэффициент выработки 64%). MS (ESI) m/z: 230 [M+l]+.
Стадия 3: получение 5-(метилол)-2-трифторметиланилина.
Используется метод, аналогичный методу получения соединения f на стадии 3 в примере реализации 14, путем реакции восстановления из соединения k′ получают соединение l′: 5-(метилол)-2-трифторметиланилин (1,2 г, коэффициент выработки 87%). MS (ESI) m/z: 202 [M+l]+.
Стадия 4: получение 5-формил-2-трифторметиланилина.
Используется метод, аналогичный методу получения соединения g на стадии 4 в примере реализации 14, путем реакции восстановления из соединения k′ получают соединение m′: 5-формил-2-трифторметиланилин (1,3 г, коэффициент выработки 96%). MS (ESI) m/z: 200 [M+l]+.
Стадия 5: получение 2-трифторметил-5-((3-оксоизобензофуран-1(3H)-иден)метил)анилина.
Используется метод, аналогичный методу получения соединения e на стадии 5 в примере реализации 1, путем реакции между соединением m′ и 3-оксо-1,3-дибензоизофуран-1-ил-диметил ортофосфатом получают соединение n′: 2-трифторметил-5-((3-оксоизобензофуран-1(3H)-иден)метил)анилин (721 мг, коэффициент выработки 69%).
Стадия 6: получение 2-трифторметил-5-((4-оксо-3,4-дифталазил-1-ил)метил)бензойной кислоты.
Используется метод, аналогичный методу получения соединения f на стадии 6 в примере реализации 1, путем реакции гидролиза из соединения n′ получают соединение о′: получение 2-трифторметил-5-((4-оксо-3,4-дифталазил-1-ил)метил)бензойная кислота (678 мг, коэффициент выработки 86%). MS (ESI) m/z: 349 [M+l]+.
Стадия 7: получение 4-(3-(4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетона.
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением о′ и соединением l получают соединение (15): 4-(3-(4-(1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетон (65 мг, коэффициент выработки 53%). MS (ESI) m/z: 534 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,57 (s, 1H), 8,24 (d, 1H, J=0,8 Hz), 8,23 (s, 1H), 7,96-7,80 (m, 4H), 7,73 (d, 1H, J=8,0 Hz), 7,54 (d, 1H, J=8,0Hz), 7,50 (s, 1H), 6,77 (d, 1H, J=8,4Hz), 4,42 (s, 2H), 3,82-3,77 (m, 1H), 3,68-3,62 (m, 1H), 3,59-3,52 (m, 2H), 3,36-3,29 (m, 2H), 3,19-3,10 (m, 2H).
Пример реализации 16
Соединение (16): получение 4-(3-(4-(lH-[1,2,3]триазол[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
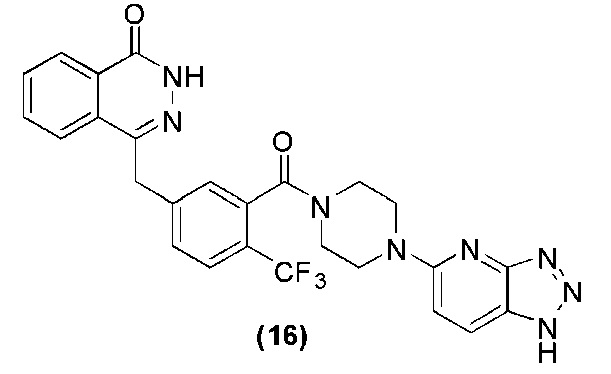
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением o′ и соединением d получают соединение (16): 4-(3-(4-(lH-[1,2,3]триазол[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетон (70 мг, коэффициент выработки 57%). MS (ESI) m/z: 535 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,57 (s, 1H), 8,24 (d, 1H, J=7,2Hz), 8,17 (d, 1H, J=8,8Hz), 7,95-7,81 (m, 3H), 7,74 (d, 1H, J=8,0Hz), 7,55 (d, 1H, J=8,0Hz), 7,51 (s, 1H), 6,98 (d, 1H, J=9,6Hz), 4,42 (s, 2H), 3,80-3,62 (m, 4H), 3,50-3,46 (m, 2H), 3,36-3,30 (m, 2H).
Пример реализации 17
Соединение (17): получение 4-(3-(4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
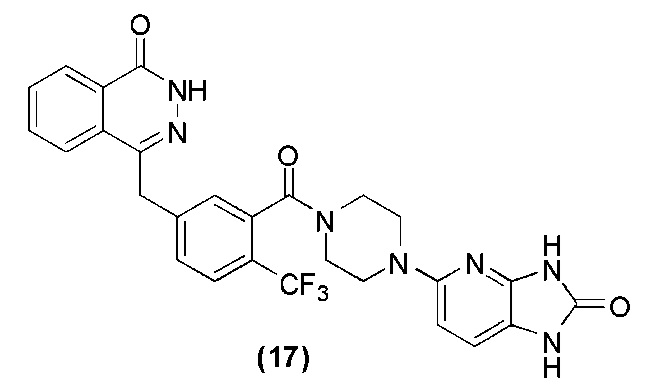
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением o′ и соединением n получают соединение (17): 4-(3-(4-(2-оксо-2,3-дигидро-1H-имидазо[4,5-b]пиридин-5-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетон (67 мг, коэффициент выработки 53%). MS (ESI) m/z: 550 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,57 (s, 1H), 10,95 (s, 1H), 10,37 (s, 1H), 8,23 (d, 1H, J=7,2Hz), 7,94-7,80 (m, 3H), 7,73 (d, 1H, J=8,0Hz), 7,54 (d, 1H, J=8,0Hz), 7,47 (s, 1H), 7,09 (d, 1H, J=8,0Hz), 6,33 (d, 1H, J=8,0Hz), 4,42 (s, 2H), 3,70-3,64 (m, 1H), 3,64-3,59 (m, 1H), 3,42-3,25 (m, 2H), 3,14-3,08 (m, 4H).
Пример реализации 18
Соединение (18): получение 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-метоксилбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
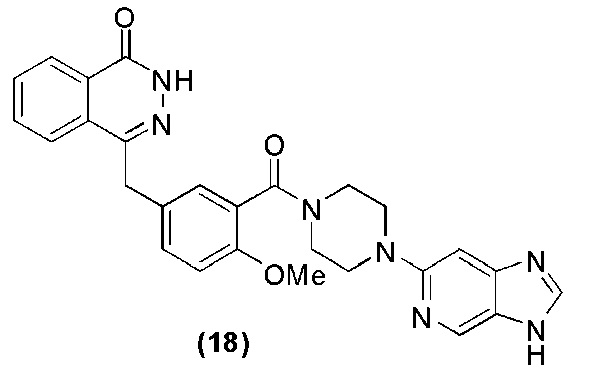
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1, путем реакции конденсации между соединением i′ и соединением r получают соединение (18): 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-метоксилбензил)фталазин-1(дигидро)-кетон (54 мг, коэффициент выработки 34%). MS (ESI) m/z: 496 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,55 (s, 1H), 8,56 (s, 1H), 8,23 (s, 1H), 8,22 (s, 1H), 7,93 (d, 1H, J=8,0Hz), 7,87-7,77 (m, 3H), 7,31 (d, 1H, J=8,0Hz), 7,15 (s, 1H), 7,00 (d, 1H, J=8,0Hz), 6,82 (s, 1H), 4,23 (s, 2H), 3,71 (br, 5H), 3,47-3,46 (m, 2H), 3,32-3,18 (m, 4H).
Пример реализации 19
Соединение (19): получение 4-(3-(4-(3H-имидазо[4,5-c]пиридин-6-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
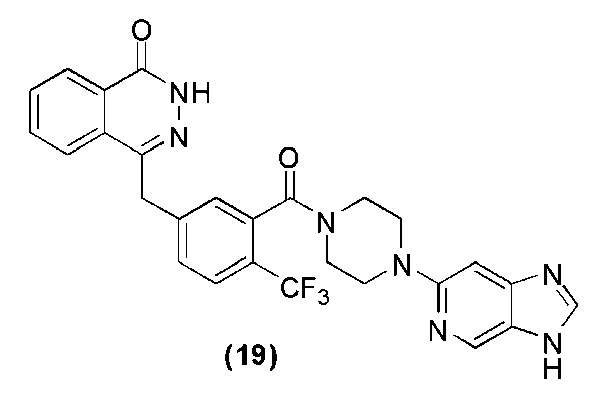
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением о′ и соединением r получают соединение (19): 4-(3-(4-(3H-имидазо[4,5-c]пиридин-6-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетон (46 мг, коэффициент выработки 46%). MS (ESI) m/z: 534 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,57 (s, 1H), 8,55 (s, 1H), 8,24 (d, 1H, J=8,0Hz), 8,17 (s, 1H), 7,95-7,72 (m, 5H), 7,55 (d, 1H, J=8,0Hz), 7,48 (s, 1H), 6,80 (s, 1H), 4,43 (s, 2H), 3,81-3,79 (m, 1H), 3,78-3,77 (m, 1H), 3,68-3,64 (m, 2H), 3,49-3,46 (m, 2H), 3,17-3,12 (m, 2H).
Пример реализации 20
Соединение (20): получение 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:

Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1: путем реакции конденсации между соединением о′ и соединением v получают соединение (20): 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-трифторметилбензил)фталазин-1(дигидро)-кетон (37 мг, коэффициент выработки 41%). MS (ESI) m/z: 534 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,57 (s, 1H), 8,47 (s, 1H), 8,24 (s, 1H), 8,22 (s, 1H), 7,95-7,73 (m, 5H), 7,55 (d, 1H, J=8,0 Hz), 7,50 (s, 1H), 7,49 (s, 1H), 4,43 (s, 2H), 3,85-3,82 (m, 1H), 3,73-3,70 (m, 1H), 3,20-3,19 (m, 4H), 2,98 (m, 2H).
Пример реализации 21
Соединение (21): получение 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-метоксилбензил)фталазин-1(дигидро)-кетона, ниже дано конкретное уравнение химической реакции:
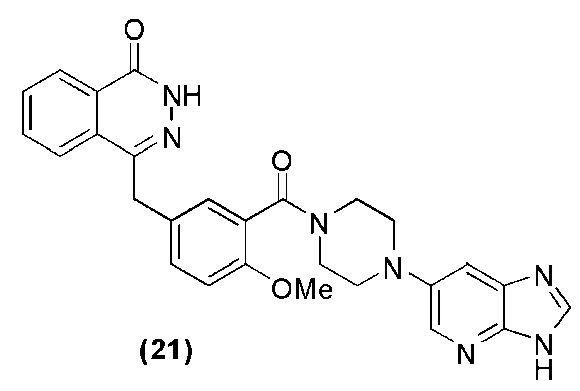
Используется метод, аналогичный методу получения соединения (1) на стадии 7 в примере реализации 1, путем реакции конденсации между соединением i′ и соединением v получают соединение (21): 4-(3-(4-(3H-имидазо[4,5-b]пиридин-6-ил)пиперазин-1-карбонил)-4-метоксилбензил)фталазин-1(дигидро)-кетон (66 мг, коэффициент выработки 42%). MS (ESI) m/z: 496 [M+l]+. 1H NMR (300 MHz, DMSO-d6): δ 12,55 (s, 1H), 8,37 (s, 1H), 8,23 (s, 1H), 8,21 (s, 1H), 7,94-7,76 (m, 3H), 7,49 (s, 1H), 7,31 (d, 1H, J=8,0 Hz), 7,15-7,12 (m, 1H), 7,00 (d, 1H, J=8,0 Hz), 6,93 (s, 1H), 4,23 (s, 2H), 3,77 (s, 3H), 3,76 (br, 2H), 3,23-3,13 (m, 4H), 3,05-2,97 (m, 2H).
Биологическая оценка
Пример 1. Испытание по оценке ферментативной активности PARP
Положения эксперимента
Образование нуклеопротеиновой поли-ADP-рибозы происходит после реакции на повреждение ДНК. Полное название PARP — поли(аденозиндифосфат рибоза)-полимераза. В присутствии NAD (никотинамидадениндинуклеотида) катализируют поли(АДФ-рибозил) на ближайший нуклеопротеин, тем самым вызывая механизм репарации ДНК, проходящей по путям эксцизионной репарации основных радикалов. HT Universal Chemiluminescent PARP Assay Kit, производимый компанией Trevigen, может измерить связанный уровень АДФ-рибозы и гистонов, маркированных биотином.
Реактивы и расходные материалы
1. HT Universal Chemiluminescent PARP Assay Kit with Histone-coated Strip Wells, производитель Trevigen, США, серийный номер: 4676-096-K.
2. Устройство считывания EnVision Multilabel Plate Reader, производитель Perkin Elmer, США.
Растворы и буферные растворы
Промывная жидкость: раствор PBS, содержащий 0,1% Triton X-100.
Буферный раствор PARP, 20X: Буферный раствор PARP, 20X, разбавляют деионизированной водой в 20 раз и получают 1X буферного раствора, который используется для разбавления рекомбинантных ферментов PARP, PARP Cocktail и измеряемого соединения.
PARP Cocktail, 10X: 1X PARP Cocktail готовится следующим образом: 10X PARP Cocktail 2,5 μl/well, 10X активируют DNA 2,5 μl/well, 1X PARP буферный раствор 20 μl/well.
PARP Enzyme: Перед использованием рекомбиназа осторожно разбавляется 1X буферного раствора PARP, разбавленный раствор ферментов необходимо как можно быстрее использовать, а неиспользованный уничтожить.
Strep-HRP: Перед использованием Strep-HRP 500 разбавляют 1X растворителя Strep в два раза и получают 1X раствора.
Хемолюминесцентный субстрат: Перед использованием растворы PeroxyGlow A и B аналогичного объема равномерно смешивают и получают субстрат пероксидаза хрена.
Техника эксперимента
Составление соединений
10 mM маточного раствора каждого измеряемого соединения, используя DMSO, разбавляют до 10 μM, 1 μM.
Перед началом эксперимента раствор градиентной концентрации каждого соединения, растворенного в DMSO, разбавляется 1X буферного раствора PARP 20 раз, получают 5X раствора соединения, который можно использовать в эксперименте. В лунках положительного (POSITIVE) и негативного (NEGATIVE) контроля находится 1X буферного раствора PARP (содержание DMSO 5%), в том числе контрольным соединением является соединение AZD2281 (Olaparib, производитель фармацевтическая компания AstraZeneca).
Последовательность операций
В каждую лунку добавляют гистоны, пропитанные 50 μl 1X PARP буферным раствором, при комнатной температуре культивируют в течение 30 минут на культуральном планшете, затем из лунок удаляют 1X буферного раствора PARP, а остаточная жидкость стряхивается на бумажную салфетку.
В соответствии с порядком соединений с (1) по (21) и контрольного соединения AZD2281, 5X разбавленного раствора соединения добавляют в соответствующие лунки по 10 μl в каждую, лунки положительного (POSITIVE) и отрицательного (NEGATIVE) контроля предназначены для 1X буферного раствора PARP (содержание DMSO 5%).
Используя 1X буферного раствора PARP, фермент PARP разбавляют так, чтобы каждые 15 μl раствора содержали 0,5 Unit. Затем во все лунки, кроме лунки негативного контроля, добавляют по 15 μl раствора фермента, а в лунку негативного контроля добавляют 1X буферного раствора PARP и культивируют при комнатной температуре в течение 10 минут.
Далее в каждую лунку добавляют по 25 μl 1X буферного раствора PARP Cocktail.
Культивируют на планшете в течение 60 минут при температуре 27°C.
По окончании культивирования реакционный раствор экстрагируется, а остаточная жидкость стряхивается на бумажную салфетку. Далее раствором PBS, содержащим 0,1% Triton X-100, 4 раза промывают планшет, каждый раз для каждой лунки используется 200 μl, а остаточная жидкость стряхивается на бумажную салфетку.
Далее в каждую лунку добавляют IX разбавленного раствора Strep-HRP раствор, затем культивируют на планшете в течение 60 минут при температуре 27°C.
По окончании культивирования реакционный раствор экстрагируется, а остаточная жидкость стряхивается на бумажную салфетку. Далее раствором PBS, содержащим 0,1% Triton X-100, 4 раза промывают планшет, каждый раз для каждой лунки используется 200 μl, а остаточная жидкость стряхивается на бумажную салфетку.
После промывки планшета равномерно смешивают растворы PeroxyGlow A и B одинакового объема, добавляют в каждую лунку по 100 μl, немедленно ставят планшет в считывающее устройство для записи хемолюминесцентных сигналов.
Обработка данных
Показания каждой лунки должны быть сконвертированы в коэффициент торможения. Коэффициент торможения для соединений можно рассчитать по следующей формуле:

Примечание: Показания лунки положительного контроля — это показания лунки positive, и означают 100%-ную активность ферментов; показания лунки негативного контроля — это показания лунки negative и означают 0%-ную активность ферментов. Активность X — это показания для уровня концентрации каждого образца.
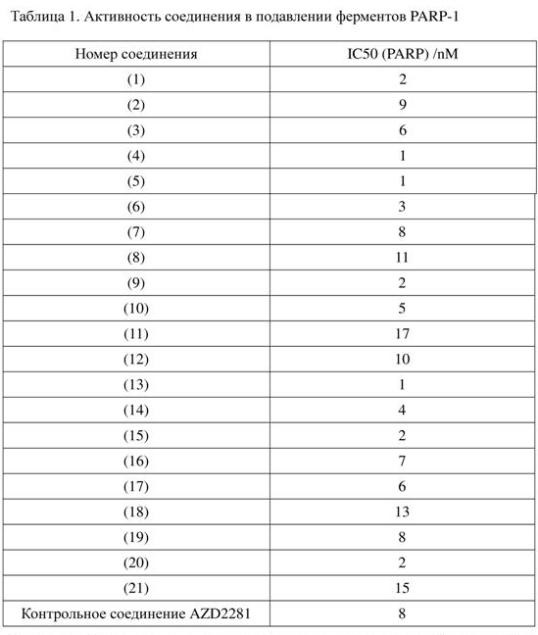
Вывод: наиболее предпочтительное соединение настоящего изобретения обладает очевидным эффектом в подавлении ферментов PARP-1.
Пример 2. Испытания по оценке цитостаза
Следующий эксперимент проводился в искусственных условиях для оценки эффекта цитостаза, оказываемого соединениями, указанными в настоящем изобретении, на штамм MDA-MB-436 клеток рака молочной железы тройного негативного фенотипа.
Реактивы и расходные материалы
1 штамм прошедших испытания на микоплазму опухолевых клеток, MDA-MB-436, предоставленный компанией HD Biosciences Co., Ltd (Шанхай).
Питательный раствор L15, производитель Invitrogen США, серийный номер: 11415-064.
Эмбриональная телячья сыворотка, производитель Hyclone, США, серийный номер: CH30160.03.
Раствор пенициллина-стрептомицина, производитель Invitrogen, США, серийный номер: 15140-122.
Диметилсульфоксид (ДМСО), производитель Sigma, США, серийный номер: D4540.
96-луночный культуральный планшет, производитель Corning, США, серийный номер: 3610.
CellTiter-Glo Luminescent Cell Viability Assay, производитель Promega, США, серийный номер: G7571.
Считывающее устройство EnVision Multilabel Plate Reader, производитель Perkin Elmer, США,
Питательный раствор
1. Готовый питательный раствор L15: раствор L15, содержащий 10% эмбриональной телячьей сыворотки, 100U пенициллина и 100 мг/мл стрептомицина.
Техника эксперимента
Составление соединений
Используя DMSO, из каждого анализируемого соединения подготавливают 60 мл маточного раствора, который хранится порционно при температуре -80°C. Используя DMSO, маточный раствор для каждого анализируемого соединения разбавляют до нескольких растворов с градиентной концентрацией 6 mM, 2 mM, 0,6 mM, 0,2 mM, 60 μM, 20 μM.
Перед началом испытаний в стерильных условиях готовой клеточной питательной средой разбавляют подготовленный раствор соединения с градиентной концентрацией 100 раз. В это время градиентная концентрация соединения составляет 60 μM, 20 μM, 6 μM, 2 μM, 0,6 μM, 0,2 μM. Эти растворы представляют собой 2x растворы соединений, которые можно использовать для обработки клеток.
Используя DMSO, маточные растворы упомянутых выше соединений с (1) по (21), а также контрольного соединения AZD2281, разбавляют до нескольких растворов с градиентной концентрацией 20 μM, 2 μM, 0,2 μM, 0,02 μM, 0,002 μM, 0,0002 μM. Перед началом испытаний в стерильных условиях готовой клеточной питательной средой разбавляют подготовленный раствор соединения с градиентной концентрацией 100 раз. В это время градиентная концентрация положительных соединений составляет 200 μM, 20 μM, 2 μM, 0.2 μM, 0,02 μM, 0,002 μM. Эти растворы представляют собой 2x растворы соединений, которые можно использовать для обработки клеток.
Последовательность операций
За день до анализа соединения производят посев клеток на питательную среду на 96-луночный культуральный планшет. Плотность посева микроорганизмов на питательную среду: 8000 клеток / 50 μl / лунка.
На следующий день уже готовый 2х раствор соединения в соответствии с диаграммой Парето вносится на культуральный планшет по 50 μl в каждую лунку.
Планшет осторожно встряхивают, инкубируют при температуре 37°C в течение 120 часов.
По окончании культивирования в соответствии с инструкцией на культуральный планшет добавляют реактив CellTiter Glo, после тщательного перемешивания культивируют в течение 10 минут при комнатной температуре в защищенном от света месте.
Клеточная пластинка вводится в считывающее устройство для анализа, устанавливается режим чтения хемилюминесценции и записи данных.
Обработка данных
Данные по каждой лунке должны быть сконвертированы в коэффициент клеточной выживаемости. Коэффициент клеточной выживаемости можно вычислить, используя следующую формулу:

После обработки данных, используя аналитическое программное обеспечение GraphPad Prism 5, выполняют нелинейный регрессионный анализ и получают кривую зависимости «доза-эфффект», а также вычисляют концентрацию измеряемого соединения, необходимую для поражения половины клеток штамма MDA-MB-436.

Вывод: наиболее предпочтительные соединения настоящего изобретения обладают очевидным эффектом в подавлении размножения клеток MDA-MB-436.
Пример 3. Испытания по оценке соединений настоящего изобретения в подавлении опухоли у мышей
Испытания ниже проводились на живых организмах для оценки и сравнения эффективности соединений, упомянутых в настоящем изобретении, при лечении перевиваемых опухолей штамма MDA-MB-436 человеческого рака молочной железы, перевиваемых опухолей штамма MX-1 человеческого рака молочной железы или перевиваемых опухолей штамма CAPAN-1 человеческого рака поджелудочной железы.
Испытываемое соединение
Соединение (4) из примера реализации
Лабораторные животные
Голые мыши BALB/cA-nude, 6-7 недель, закупленные у Shanghai Slac Laboratory Animal Co., LTD. Сертификат качества: SCXK (Шанхай) 2012-0002. Условия выращивания: СПФ-статус.
Ход эксперимента
Под кожу голых мышей инокулируются клетки человеческого рака молочной железы штамма MDA-MB-436 или MX-1, или клетки человеческого рака поджелудочной железы штамма CAPAN-1. По достижении опухолью размеров 100-200 мм3, животные случайным образом распределялись по группам (D0). Доза и схема приема лекарственного средства показаны в таблице 1. Каждую неделю 2-3 раза измерялся объем опухоли, мыши взвешивались, все данные записывались. Макроскопический объём опухоли (V) рассчитывается по следующей формуле:
V=l/2xaxb2,
где a, b означают длину, ширину соответственно.
T/C (%) = (T-T0)/(C-Co) X 100,
где T, C — это макроскопический объём опухоли по окончании испытаний; T0, C0 — это макроскопический объём опухоли на начало испытаний.

Вывод: некоторые наиболее предпочтительные соединения настоящего изобретения в самостоятельном виде обладают выраженной противораковой активностью по отношению к перевиваемой опухоли MDA-MB-436 человеческого рака молочной железы.
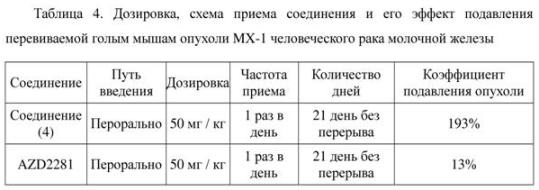
Вывод: некоторые наиболее предпочтительные соединения настоящего изобретения в отдельном виде обладают выраженной противораковой активностью по отношению к перевиваемой опухоли MX-1 человеческого рака молочной железы.

Вывод: некоторые наиболее предпочтительные соединения настоящего изобретения в отдельном виде обладают выраженной противораковой активностью по отношению к перевиваемой опухоли CAPAN-1 человеческого рака поджелудочной железы.
Основные признаки настоящего изобретения перечислены в следующих пунктах.
1. Гетероциклическое имидазольное соединение общей формулы (I) или его фармацевтически приемлемая соль:
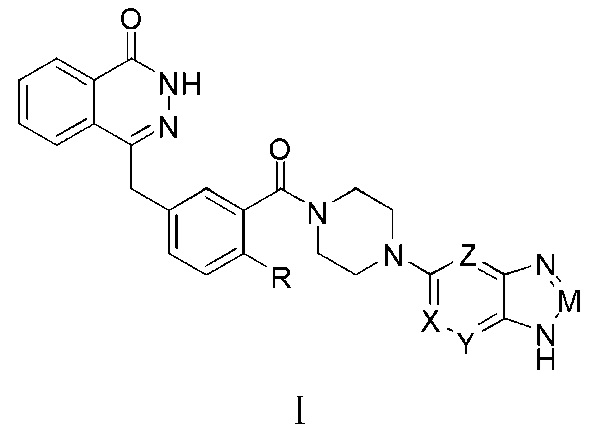 ,
,
где в общей формуле (I)
R представляет собой водород, галоген, C1-C6алкил или C1-C6галогеналкил;
один из X, Y и Z представляет собой азот, а другие представляют собой CH; или один из X, Y и Z представляет собой CH, а другие представляют собой азот; и
M представляет собой азот или CR1, в котором
R1 представляет собой водород, кислород, алкил, алкокси или галогеналкил.
2. Соединение общей формулы I по пункту 1, где
R представляет собой водород, галоген, C1-C3алкокси или C1-C3галогеналкил.
3. Соединение общей формулы I по любому из пунктов 1-2, где
R представляет собой водород, кислород, метокси или трифторметил.
4. Соединение общей формулы I по любому из пунктов 1-3, где
X представляет собой азот, а Y и Z представляют собой CH; или Z представляет собой азот, а X и Y представляют собой CH; или Y представляет собой азот, а X и Z представляют собой CH; или X и Z представляют собой азот, а Y представляет собой CH.
5. Соединение общей формулы I по любому из пунктов 1-4, где
R1 представляет собой водород, кислород C1-C6алкил или C1-C6галогеналкил.
6. Соединение общей формулы I по любому из пунктов 1-5, где
R1 представляет собой водород, кислород C1-C3алкил или C1-C3галогеналкил.
7. Соединение общей формулы I по любому из пунктов 1-6, где
R1 представляет собой водород, кислород, метил или трифторметил.
8. Соединение общей формулы I по любому из пунктов 1-7, которое выбрано из группы, состоящей из соединений 1-21, или его фармацевтически приемлемая соль:

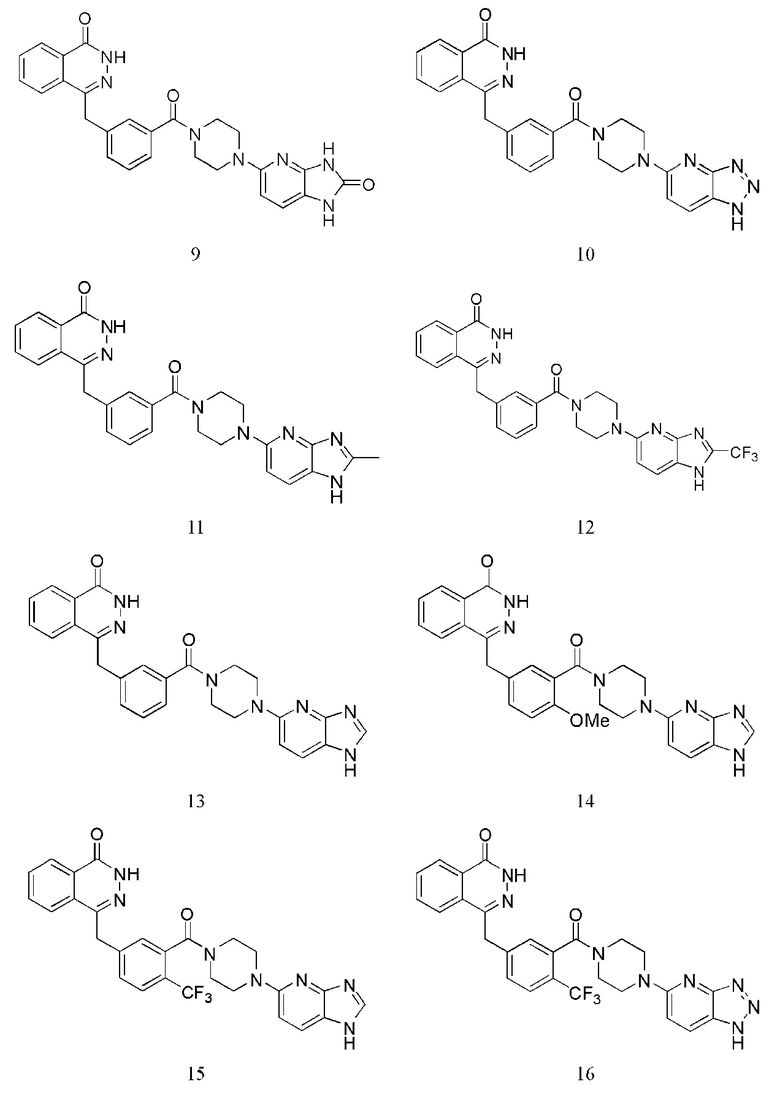
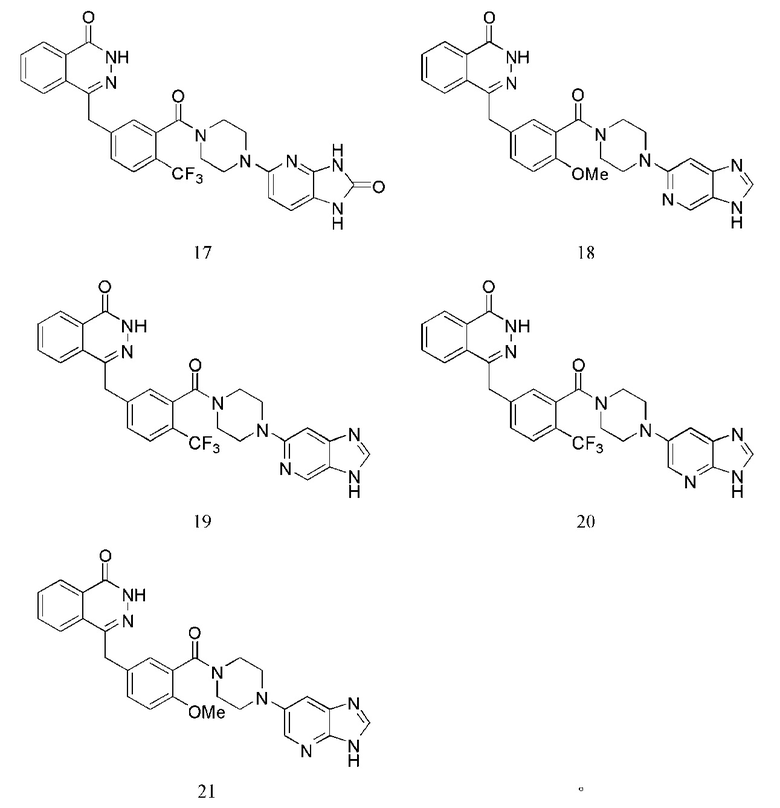
.
9. Способ получения соединения общей формулы I по любому из пунктов 1-8 или его фармацевтически приемлемой соли, где реакционная схема является следующей:
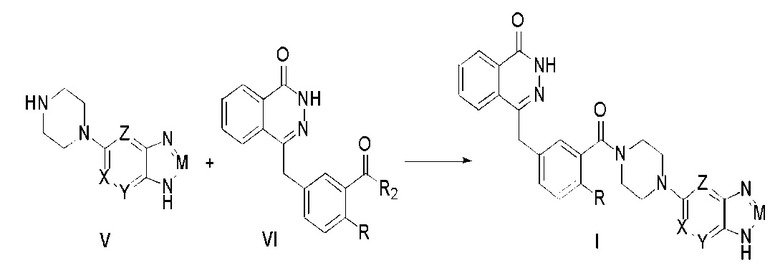 ,
,
где R, X, Y, Z и M определены в п. 1, а R2 представляет собой гидроксил, галоген или диимидазол-1-ил; при этом способ, в частности, предусматривает:
конденсацию промежуточного соединения V с помощью производного фталазинкарбоновой кислоты VI с получением соединения общей формулы I.
10. Способ получения соединения общей формулы I по п. 9, где производное фталазинкарбоновой кислоты VI представляет собой соединение, приведенное ниже:
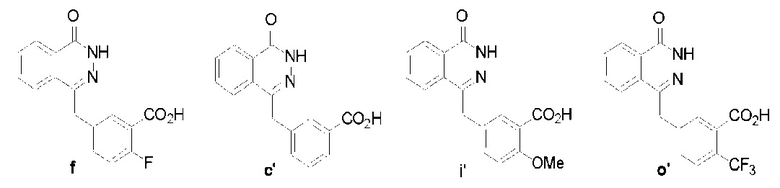 .
.
11. Способ получения соединения формулы I по любому из пунктов 9-10, где конденсирующее средство, применяемое в реакции конденсации выбрано из группы, состоящей из 1,1-карбонилиимидазола, 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорида, 2-(7-азабензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата и бензотриазол-N,N,N',N'-тетраметилурония гексафторфосфата.
12. Промежуточное соединение для получения гетероциклического имидазольного соединения общей формулы I по любому из пунктов 1-8 или его фармацевтически приемлемой соли, при этом промежуточное соединение имеет структуру, представленную ниже:
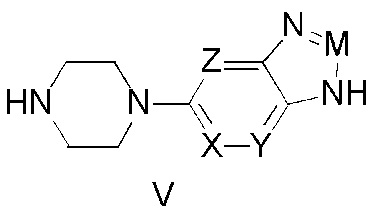
где в промежуточном соединении V
один из X, Y и Z представляет собой азот, а другие представляют собой CH; или один из X, Y и Z представляет собой CH д, а другие представляют собой азот; и
M представляет собой азот или CR1, в котором
R1 представляет собой водород, кислород, алкил, алкокси или галогеналкил.
13. Промежуточное соединение по пункту 12, где в промежуточном соединении (V)
X и Z представляют собой азот, а Y представляет собой CH; или X представляет собой азот, а Y и Z представляют собой CH; или Z представляет собой азот, а X и Y представляют собой CH; или Y представляет собой азот, а X и Z представляют собой CH; и
R1 представляет собой водород, кислород, метил или трифторметил.
14. Промежуточное соединение по пункту 12 или пункту 13, имеющее структуру, как показано при помощи соединений, представленных ниже:
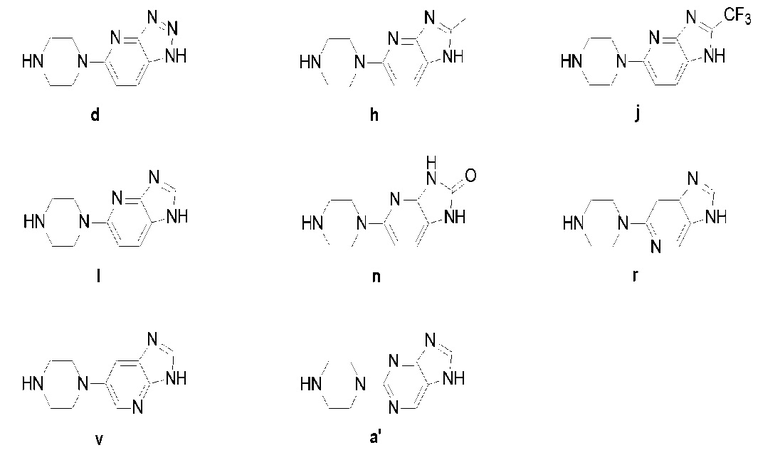 .
.
15. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения общей формулы I по любому из пунктов 1-8 или его фармацевтически приемлемой соли, а также один или более фармацевтически приемлемых носителей или вспомогательных веществ.
16. Фармацевтическая композиция по пункту 15, которая составлена в форме таблеток, капсул, водной суспензии, масляной суспензии, диспергируемого порошка, гранул, таблеток для рассасывания, эмульсии, сиропа, крема, суппозиториев или растворов для инъекций.
17. Фармацевтическая композиция по пункту 15, где соединение общей формулы (I) существует в свободной форме.
18. Применение соединения общей формулы (I) по любому из пунктов 1-8 или его фармацевтически приемлемой соли или фармацевтической композиции по любому из пунктов 15-17 для получения лекарственных средств для лечения заболеваний, уменьшение выраженности которых достигается посредством ингибирования активности ПАРП.
19. Применение по пункту 18, при котором заболевания, уменьшение выраженности которых достигается посредством ингибирования активности ПАРП, включают заболевания сосудов, септический шок, ишемические повреждения, нейротоксические симптомы, геморрагический шок, воспалительное заболевание, рассеянный склероз, нейродегенеративные заболевания или диабет.
20. Применение соединения общей формулы I по любому из пунктов 1-8 или его фармацевтически приемлемой соли или фармацевтической композиции по любому из пунктов 15-17 в получении вспомогательных лекарственных средств для лечения форм рака, в качестве химиотерапевтических средств для лечения форм рака.
21. Применение соединения общей формулы (I) по любому из пунктов 1-8 или его фармацевтически приемлемой соли или фармацевтической композиции для получения химиотерапевтических препаратов для лечения форм рака или лекарственных средств для усиления лучевой терапии рака.
22. Применение по любому из пунктов 12-21, где рак вызван дефектом путей репарации двухнитевого разрыва ДНК, зависимой от гомологичной рекомбинации.
23. Применение по любому из пунктов 12-21, где рак включает один или несколько видов раковых клеток, у которых, в отличие от нормальных клеток, понижена или утрачена способность к репарации двухнитевого разрыва ДНК, зависимой от гомологичной рекомбинации.
24. Применение по любому из пунктов 12-21, где рак представляет собой рак, имеющий дефективный или мутантный фенотип BRCA1 или BRCA2.
25. Применение по любому из пунктов 12-21, где рак представляет собой дефектный или мутантный рак BRCA1 и/или BRCA2.
26. Применение по любому из пунктов 12-21, где рак представляет собой рак молочной железы, рак яичников, рак поджелудочной железы, рак предстательной железы, рак прямой кишки, рак ободочной кишки или рак печени.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДИГИДРОПИРИДОФТАЛАЗИНОНОВЫЕ ИНГИБИТОРЫ ПОЛИ(АДФ-РИБОЗА)ПОЛИМЕРАЗЫ | 2009 |
|
RU2514937C2 |
| ПРОИЗВОДНЫЕ ИМИДАЗОПИРИДИН-2-ОНА, ОБЛАДАЮЩИЕ mTOR ИНГИБИРУЮЩЕЙ АКТИВНОСТЬЮ | 2009 |
|
RU2478636C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| БИЦИКЛИЧЕСКИЕ НИТРОИМИДАЗОЛЫ, КОВАЛЕНТНО СОЕДИНЕННЫЕ С ЗАМЕЩЕННЫМИ ФЕНИЛОКСАЗОЛИДИНОНАМИ | 2009 |
|
RU2504547C2 |
| ИСПОЛЬЗОВАНИЕ ЛИГАНДОВ РЕЦЕПТОРА ЕР4 ДЛЯ ЛЕЧЕНИЯ ОПОСРЕДОВАННЫХ IL-6 ЗАБОЛЕВАНИЙ | 2003 |
|
RU2285527C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PRMT5 | 2018 |
|
RU2797822C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ ДЛЯ УСИЛЕНИЯ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ СНИЖЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2822216C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2018 |
|
RU2768629C2 |
| АЗАБЕНЗИМИДАЗОЛЬНЫЕ СОЕДИНЕНИЯ И ФАРМКОМПОЗИЦИЯ | 2019 |
|
RU2804485C2 |
Изобретение относится к гетероциклическому имидазольному соединению общей формулы (I), в которой R представляет собой водород, галоген, C1-С6алкокси или C1-С6галогеналкил; один из X, Y и Z представляет собой азот, а другие представляют собой СН или один из X, Y и Z представляет собой СН, а другие представляют собой азот; М представляет собой азот или CR1, R1 представляет собой водород, кислород, C1-С6алкил или C1-С6галогеналкил. Изобретение также относится к способу получения соединения общей формулы (I), к применению соединения. Технический результат: получены новые гетероциклические имидазольные соединения общей формулы (I), обладающие свойствами ингибитора поли(АДФ-рибоза)-полимеразы (ПАРП). 3 н. и 11 з.п. ф-лы, 5 табл., 24 пр.
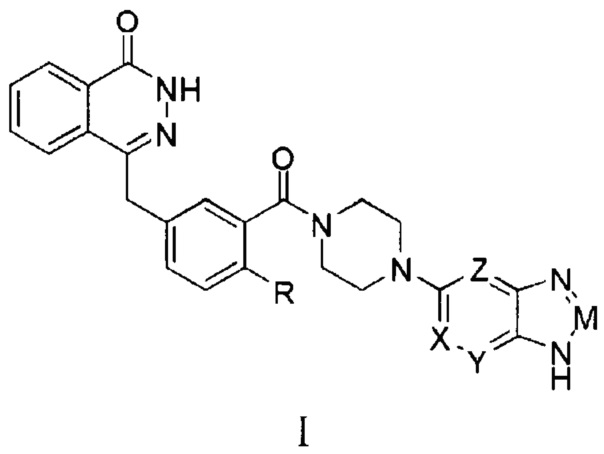
1. Гетероциклическое имидазольное соединение общей формулы (I):

где в общей формуле (I)
R представляет собой водород, галоген, C1-С6алкокси или C1-С6галогеналкил;
один из X, Y и Z представляет собой азот, а другие представляют собой СН или один из X, Y и Z представляет собой СН, а другие представляют собой азот и
М представляет собой азот или CR1, в котором
R1 представляет собой водород, кислород, C1-С6алкил или C1-С6галогеналкил.
2. Соединение общей формулы I по п. 1, где
R представляет собой водород, галоген, С1-С3алкокси или C1-С3галогеналкил.
3. Соединение общей формулы I по п. 1, где
R представляет собой водород, фтор, метокси или трифторметил.
4. Соединение общей формулы I по п. 1, где
X представляет собой азот, a Y и Z представляют собой СН; или Z представляет собой азот, а X и Y представляют собой СН; или Y представляет собой азот, а X и Z представляют собой СН; или X и Z представляют собой азот, a Y представляет собой СН.
5. Соединение общей формулы I по п. 1, где
R1 представляет собой водород, кислород, С1-С3алкил или С1-С3галогеналкил.
6. Соединение общей формулы I по п. 1, где
R1 представляет собой водород, кислород, метил или трифторметил.
7. Соединение общей формулы I по п. 1, которое выбрано из группы, состоящей из соединений 1-21:
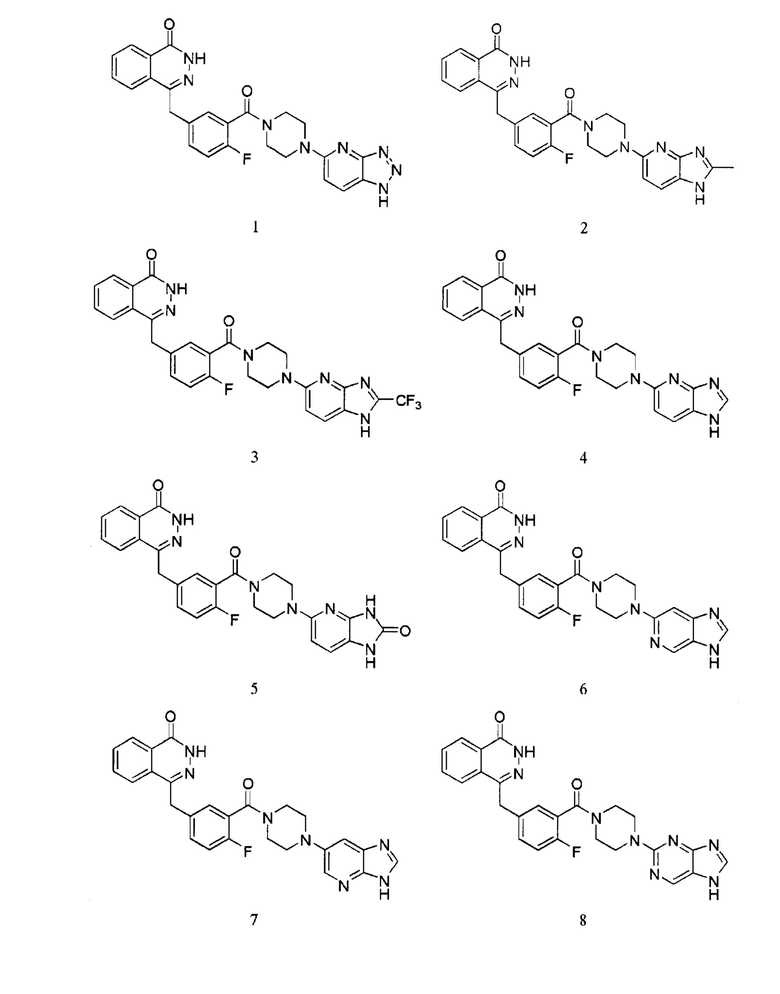
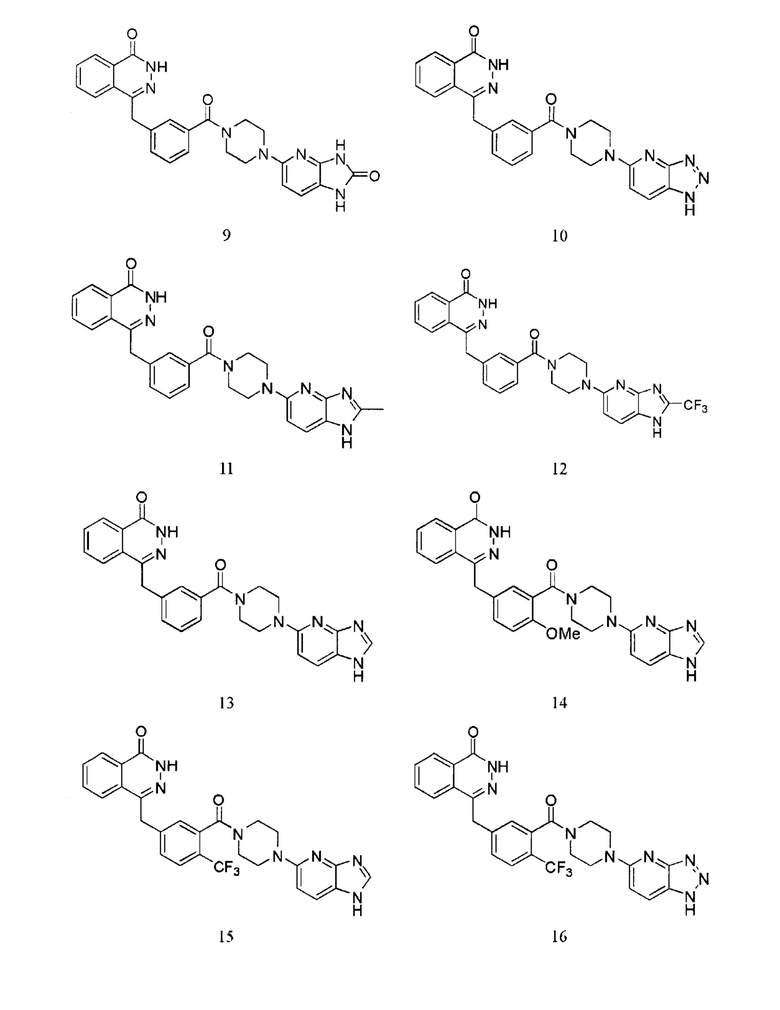
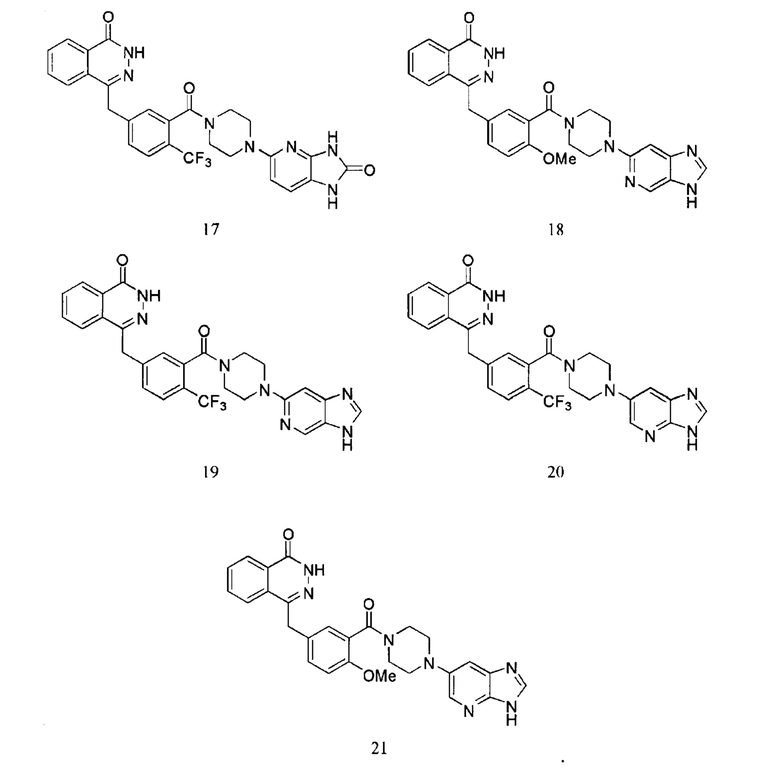
8. Способ получения соединения общей формулы I по любому из пп. 1-7, где реакционная схема является следующей:
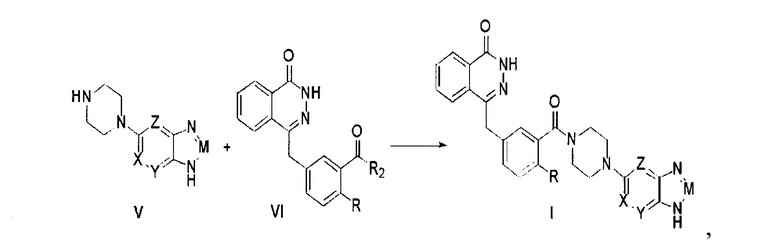
где R, X, Y, Z и М определены в п. 1 и R2 представляет собой гидроксил, галоген или диимидазол-1-ил; при этом способ, в частности, предусматривает:
конденсацию промежуточного соединения V с помощью производного фталазинкарбоновой кислоты VI в присутствии конденсирующего средства с получением соединения общей формулы I,
где конденсирующее средство, применяемое в реакции конденсации, выбрано из группы, состоящей из 1,1-карбонилдиимидазола,
1-этил-(3-диметиламинопропил)карбодиимида гидрохлорида, 2-(7-азабензотриазолил)-N,N,N',N'-тетраметилурония гексафторфосфата и бензотриазол-N,N,N',N'-тетраметилурония гексафторфосфата.
9. Способ получения соединения общей формулы I по п. 8, где производное фталазинкарбоновой кислоты VI представляет собой соединение, приведенное ниже:
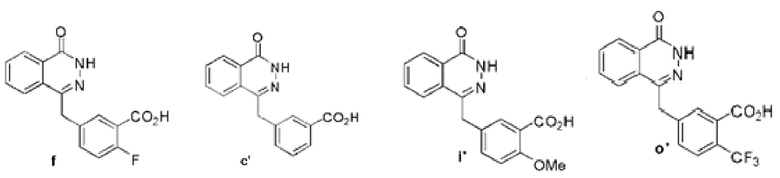 .
.
10. Применение соединения общей формулы I по любому из пп. 1-7 в качестве ингибитора поли(АДФ-рибоза)-полимеразы (ПАРП) во вспомогательных лекарственных средствах для лечения форм рака, в качестве химиотерапевтических средств для лечения форм рака или в качестве лекарственных средств для усиления лучевой терапии рака.
11. Применение по п. 10, где рак вызван дефектом путей репарации двухнитевого разрыва ДНК, зависимой от гомологичной рекомбинации.
12. Применение по п. 10, где рак включает один или несколько видов раковых клеток, у которых, в отличие от нормальных клеток, понижена или утрачена способность к репарации двухнитевого разрыва ДНК, зависимой от гомологичной рекомбинации.
13. Применение по п. 10, где рак представляет собой рак, имеющий дефективный или мутантный фенотип BRCA1 или BRCA2, и где рак представляет собой рак молочной железы, рак яичника или рак поджелудочной железы.
14. Применение по п. 10, где рак представляет собой дефектный или мутантный рак BRCA1 и/или BRCA2 и где рак представляет собой рак молочной железы, рак яичника или рак поджелудочной железы.
| CN 104003940 A, 27.08.2014 | |||
| ПРИСПОСОБЛЕНИЕ ДЛЯ ТОЧНОЙ УСТАНОВКИ В ФОРМАХ МЕТАЛЛИЧЕСКИХ ЧАСТЕЙ ПРИ ЗАЛИВКЕ ИХ В ОТЛИВАЕМЫЕ ИЗДЕЛИЯ | 1926 |
|
SU6300A1 |
| ПОЛИМОРФНАЯ ФОРМА 4-[3-(4-ЦИКЛОПРОПАНКАРБОНИЛПИПЕРАЗИН-1-КАРБОНИЛ)-4-ФТОРБЕНЗИЛ]-2Н-ФТАЛАЗИН-1-ОНА | 2007 |
|
RU2465270C2 |
| WO 2006021801 A1, 02.03.2006 | |||
| US 20080269234 A1, 30.10.2008 | |||
| CN 102898378 A, 30.01.2013. | |||

