Область техники, к которой относится изобретение
[0001]
Настоящее изобретение относится к фармацевтической композиции, включающей 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид или его фармацевтически приемлемую соль.
Уровень техники
[0002]
6-Этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид, или 6-этил-3-({3-метокси-4-[4-(4-метоксипиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид (далее иногда указан по его международному непатентованному названию (INN) гилтеритиниб) представляет собой соединение, представленное химической структурной формулой, формулой (I). Сообщалось о том, что гилтеритиниб или его фармацевтически приемлемая соль обладает, например, активностью ингибитора киназной активности EML4 (белок, подобный белку иглокожих, ассоциированному с микротрубочками, тип 4)-ALK (киназа анапластической лимфомы) слитого белка и является полезным в качестве активного ингредиента фармацевтической композиции для лечения рака (Патентная литература 1: WО2010/128659, Патентная литература 2: WO2017/006855). Кроме того, например, препарат XOSPATA (зарегистрированная торговая марка) в форме таблеток 40мг, содержащих 40 мг гилтеритиниба фумарата, в расчете на гилтеритиниб, поступил в продажу в качестве терапевтического средства от острого миелоидного лейкоза (Непатентная литература 1: XOSPATA Таблетки 40 мг Инструкция по применению препарата (Япония), или PMDA, FDA или EMA веб-сайт). В этой связи, гилтеритиниб фумарат является непатентованным названием для соли соединения, включающей соотношение две молекулы гилтеритиниба к одной молекуле фумаровой кислоты, и также указан в настоящей заявке как гилтеритиниб гемифумарат.
[0003]
Формула (I)
[Химическая формула 1]
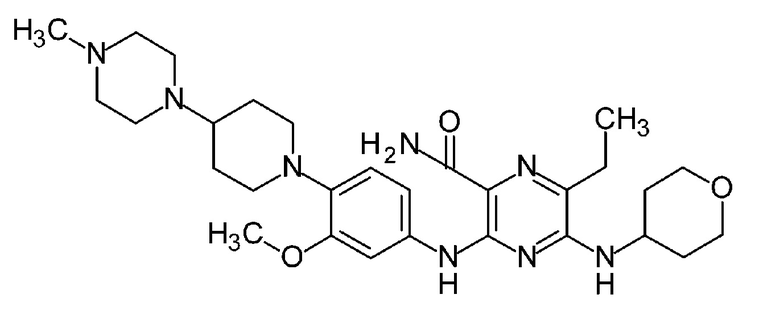
[0004]
Чтобы добиться лекарственного лечения рака, необходимо сформулировать лекарственное средство в такую композицию, чтобы оно проявляло надлежащую эффективность, и важным является получение композиции с хорошей комплаентностью. Если пациентом является младенец или ребенок, следует учитывать множество факторов, таких как лекарственная форма, масса, вкус, фармацевтические добавки и т.п. С точки зрения легкости проглатывания лекарственные формы, желательные для младенцев и детей, часто представляют собой жидкости, порошки, гранулы, сиропы и т.п. Однако трудно выбрать жидкость или сироп, поскольку многие лекарственные средства нестабильны в водном растворе. Кроме того, также требуется время для правильного отвешивания, чтобы отобрать нужное количество. В случае порошков или гранул, когда они упакованы в отдельный упаковочный лист, поскольку лекарственное средство извлекают, разрывая пакет, возникают проблемы, связанные с тем, что детям его трудно открыть, или с тем, что невозможно отобрать нужное количество, потому что часть может рассыпаться. Кроме того, в случае лекарственных средств с сильной горечью необходимо исследовать способы или фармацевтические добавки для уменьшения горечи при разработке жидкостей, порошков или гранул.
[0005]
В последние годы в качестве лекарственной формы, которая привлекает внимание, появились маленькие таблетки диаметром примерно от 1 до 4 мм (далее иногда называемые мини-таблетками или просто таблетками). Сообщается, что мини-таблетки можно принимать даже детям такого возраста, когда они не могут проглотить таблетки нормального размера (Непатентная литература 2: Takae et al., Pharmacology, Japanese Society of Pharmaceutical Sciences, 2015, vol. 75, p. 32-37). Кроме того, поскольку количество активного ингредиента на таблетку меньше, чем в таблетках нормального размера, можно легко скорректировать дозу, необходимую для педиатрических препаратов, регулируя количество таблеток.
[0006]
Что касается пациентов, которые не могут проглотить даже маленькие таблетки, существует метод растворения, диспергирования или суспендирования необходимой дозы таблеток в небольшом количестве подходящего растворителя. Однако, например, когда таблетки суспендируют в небольшом количестве воды, лекарственное средство растворяется, и эффект уменьшения горечи теряется, и, следовательно, есть возможности для дальнейшего улучшения.
Перечень цитируемых документов
Патентная литература
[0007]
[Патентная литература 1] WO2010/128659
[Патентная литература 2] WO2017/006855
Непатентная литература
[0008]
[Непатентная литература 1] XOSPATA Таблетки 40 мг Инструкция по применению препарата (Japan), или PMDA, FDA или EMA веб-сайт
[Непатентная литература 2] TAKAE, Seiji et al., Pharmacology, Japanese Society of Pharmaceutical Sciences, 2015, vol. 75, p. 32-37
Сущность изобретения
[0009]
Целью настоящего изобретения является обеспечение фармацевтической композиции, содержащей гилтеритиниб или его фармацевтически приемлемую соль, подсластитель для уменьшения горечи и добавку для придания связывающей способности, и обладающей отличной стабильностью растворения. Более конкретно, целью настоящего изобретения является обеспечение фармацевтической композиции, содержащей гилтеритиниб или его фармацевтически приемлемую соль, подсластитель для уменьшения горечи и добавку для придания связывающей способности, подавляющей снижение стабильности растворения гилтеритиниба из-за стресса, такого как высокая температура и/или влажность, с течением времени и обладающей отличной стабильностью растворения.
Решение задачи
[0010]
Для уменьшения горечи, наблюдаемой когда фармацевтическую композицию, содержащую гилтеритиниб или его фармацевтически приемлемую соль, суспендируют в небольшом количестве воды, авторы настоящего изобретения тщательно изучили фармацевтические добавки для уменьшения горечи гилтеритиниба или его фармацевтически приемлемой соли и в результате обнаружили подсластители, которые уменьшают специфическую горечь. Кроме того, авторы настоящего изобретения сфокусировались на стабильности растворения гилтеритиниба и провели всесторонние исследования, в результате которых было обнаружено, что фармацевтическую композицию, содержащую гилтеритиниб или его фармацевтически приемлемую соль и обладающую отличной стабильностью растворения, можно получить с использованием добавок для придания специфической связывающей способности, и было создано настоящее изобретение.
[0011]
Настоящее изобретение относится к следующим:
[1] Фармацевтическая композиция, включающая 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид или его фармацевтически приемлемую соль, подсластитель и два или более сахаров и/или сахарных спиртов.
[2] Фармацевтическая композиция по пункту [1], где подсластитель представляет собой одно, два или более соединений, выбранных из группы, состоящей из сахарина, ацесульфама калия, аспартама и сукралозы, и их смесь.
[3] Фармацевтическая композиция по пункту [1] и [2], где подсластитель представляет собой сукралозу.
[4] Фармацевтическая композиция по любому из пунктов [1] - [3], где сахара представляют собой дисахариды, а сахарные спирты содержат 6 или 12 атомов углерода.
[5] Фармацевтическая композиция по любому из пунктов [1] - [4], где два или более сахаров и/или сахарных спиртов выбраны из группы, состоящей из маннита, изомальта гидрата, мальтита, сорбита, лактозы, сахарозы и трегалозы и их смеси.
[6] Фармацевтическая композиция по любому из пунктов [1] - [5], где один из двух или более сахаров и/или сахарных спиртов представляет собой маннит.
[7] Фармацевтическая композиция по любому из пунктов [1] - [5], где один из двух или более сахаров и/или сахарных спиртов выбран из группы, состоящей из изомальта гидрата, мальтита, сорбита, сахарозы и трегалозы.
[8] Фармацевтическая композиция по любому из пунктов [1] - [5], где один из двух или более сахаров и/или сахарных спиртов представляет собой изомальт гидрат.
[9] Фармацевтическая композиция по пункту [7] или [8], где содержание изомальта гидрата, мальтита, сорбита, сахарозы или трегалозы, описанных в пункте [7] или [8], в расчете на массу фармацевтической композиции составляет от 1% масс. до 20% масс.
[10] Фармацевтическая композиция по пункту [7] или [8], где изомальт гидрат, мальтит, сорбит, сахарозу или трегалозу, описанные в пункте [7] или [8], используют в качестве связующего.
[11] Фармацевтическая композиция по любому из пунктов [1] - [10], где фармацевтически приемлемая соль представляет собой гемифумарат.
[12] Фармацевтическая композиция по любому из пунктов [1] - [11], где фармацевтическая композиция является твердой.
[13] Фармацевтическая композиция по любому из пунктов [1] - [12], дополнительно включающая разрыхлитель.
[14] Фармацевтическая композиция по пункту [12] или [13], где фармацевтическая композиция представляет собой таблетку.
[15] Фармацевтическая композиция по любому из пунктов [1] - [14],
где после хранения фармацевтической композиции по любому из пунктов [1] - [14] при 40°C и 75% относительной влажности в течение 1 месяца процент растворения 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамида через 15 минут после начала испытания на растворимость лопастным методом с использованием 900 мл 0,1 моль/л хлористоводородной кислоты, описанным в Японской фармакопее, семнадцатое издание, составляет 85% или больше; или
где после хранения фармацевтической композиции по любому из пунктов [1] - [14] при 40°C и 75% относительной влажности в течение 2 месяцев и/или 3 месяцев процент растворения 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамида через 15 минут после начала испытания на растворимость лопастным методом с использованием 900 мл 0,1 моль/л хлористоводородной кислоты, описанным в Японской фармакопее, семнадцатое издание, составляет 80% или больше.
[16] Фармацевтическая композиция по любому из пунктов [1] - [15], где фармацевтическая композиция растворена или диспергирована в подходящем растворителе и представляет собой раствор, суспензию, пасту или гель.
[17] Фармацевтическая композиция, включающая 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид гемифумарат, маннит, сукралозу и изомальт гидрат.
[18] Фармацевтическая композиция по любому из пунктов [1] - [17], полученная способом получения, включающим:
(1) получение связующей жидкости путем диспергирования или растворения по меньшей мере одного из сахаров и/или сахарных спиртов в растворителе;
(2) получение смеси путем смешивания 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамида или его фармацевтически приемлемой соли, подсластителя и по меньшей мере одного из сахаров и/или сахарных спиртов; и
(3) распыление или добавление связующей жидкости, полученной на стадии (1), к смеси, полученной на стадии (2), для образования гранул.
[19] Способ получения фармацевтической композиции, включающей 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид или его фармацевтически приемлемую соль и два или более сахаров и/или сахарных спиртов, при этом указанный способ включает:
(1) получение связующей жидкости путем диспергирования или растворения по меньшей мере одного из сахаров и/или сахарных спиртов в растворителе;
(2) получение смеси путем смешивания 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамида или его фармацевтически приемлемой соли, подсластителя и по меньшей мере одного из сахаров и/или сахарных спиртов; и
(3) распыление или добавление связующей жидкости, полученной на стадии (1), к смеси, полученной на стадии (2), для образования гранул.
Полезные эффекты изобретения
[0012]
В соответствии с настоящим изобретением можно обеспечить фармацевтическую композицию, включающую гилтеритиниб или его фармацевтически приемлемую соль, подсластитель, который уменьшает горечь, и два или более сахаров и/или сахарных спиртов и обладающую отличной стабильностью растворения, более конкретно фармацевтическую композицию, подавляющую снижение свойств растворимости гилтеритиниба из-за стресса, такого как высокая температура и/или влажность, и обладающую отличной стабильностью растворения.
В соответствии с настоящим изобретением можно обеспечить жидкую фармацевтическую композицию, такую как раствор, суспензия, паста или гель, полученную путем растворения или диспергирования фармацевтической композиции, включающей гилтеритиниб или его фармацевтически приемлемую соль, подсластитель, который уменьшает горечь, и два или более сахаров и/или сахарных спиртов, в подходящем растворителе.
В соответствии с настоящим изобретением, фармацевтическую композицию, включающую гилтеритиниб или его фармацевтически приемлемую соль, подсластитель, который уменьшает горечь, и два или более сахаров и/или сахарных спиртов, более конкретно фармацевтическую композицию, подавляющую снижение свойств растворимости гилтеритиниба из-за стресса, такого как высокая температура и/или влажность, и обладающую отличной стабильностью растворения, можно обеспечить способом, включающим стадию получения связующей жидкости путем диспергирования или растворения по меньшей мере одного из сахаров и/или сахарных спиртов в растворителе; стадию получения смеси путем смешивания гилтеритиниба или его фармацевтически приемлемой соли, подсластителя, который уменьшает горечь, и по меньшей мере одного из сахаров и/или сахарных спиртов; и стадию распыления или добавления связующей жидкости, полученной на вышеописанной стадии, к смеси, полученной на вышеописанной стадии, для образования гранул.
В соответствии с настоящим изобретением можно обеспечить фармацевтическую композицию, включающую гилтеритиниб или его фармацевтически приемлемую соль, маннит, сукралозу и изомальт гидрат, более конкретно фармацевтическую композицию, подавляющую снижение свойств растворимости гилтеритиниба из-за стресса, такого как высокая температура и/или влажность, и обладающую отличной стабильностью растворения.
В соответствии с настоящим изобретением можно обеспечить способ получения фармацевтической композиции, включающей гилтеритиниб или его фармацевтически приемлемую соль, подсластитель и два или более сахаров и/или сахарных спиртов, более конкретно способ, включающий стадию получения связующей жидкости путем диспергирования или растворения по меньшей мере одного из сахаров и/или сахарных спиртов в растворителе; стадию получения смеси путем смешивания гилтеритиниба или его фармацевтически приемлемой соли, подсластителя, который уменьшает горечь, и по меньшей мере одного из сахаров и/или сахарных спиртов; и стадию распыления или добавления связующей жидкости, полученной на вышеуказанной стадии к смеси, полученной на вышеуказанной стадии для образования гранул.
Описание вариантов осуществления
[0013]
Настоящее изобретение относится к фармацевтической композиции, включающей гилтеритиниб или его фармацевтически приемлемую соль, подсластитель и два или более сахаров и/или сахарных спиртов. Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей гилтеритиниб гемифумарат, маннит, сукралозу и изомальт гидрат.
[0014]
Термин “подсластитель” в контексте настоящей заявки означает добавку для подслащивания фармацевтической композиции. Примеры подсластителя включают сахарин или сахарин натрия, глицирризиновую кислоту, аспартам, стевию, тауматин, ацесульфам калия, натрия цикламат, адвантам, стевиол-гликозиды, неогесперидин дигидрохалкон, неотам, тауматин, другие сладкие белки, сапонины, такие как осладин или гициллидин, сукралозу, их смесь и т.п. “Для уменьшения горечи” в контексте настоящей заявки означает подавление горечи до такой степени, когда можно принимать гилтеритиниб или его фармацевтически приемлемую соль, которые по своей природе имеют горький вкус. Подсластителем, предпочтительным для уменьшения горечи, наблюдаемой когда гилтеритиниб или его фармацевтически приемлемую соль суспендируют или т.п. в небольшом количестве воды или т.п., является сахарин, ацесульфам калия, аспартам, сукралоза или их смесь, более предпочтительно аспартам или сукралоза, еще более предпочтительно сукралоза.
[0015]
Используемый в настоящей заявке термин “сахарин” также известен как о-сульфобензимид, о-сульфимид бензоат или 2-сульфобензоат имид и представляет собой подсластитель, обычно используемый в виде водорастворимой натриевой соли (сахаринат натрия).
Используемый в настоящей заявке “ацесульфам калия” представляет собой искусственный подсластитель.
Используемый в настоящей заявке “аспартам” представляет собой подсластитель, имеющий структуру, в которой аминогруппа метилового эфира фенилаланина, образованная дегидратационной конденсацией L-фенилаланина и метанола, и карбокси группа L-аспарагиновой кислоты дегидратированы и конденсированы с образованием пептидной связи.
Термин “сукралоза”, используемый в настоящей заявке, является зарегистрированной торговой маркой 4,1',6'-трихлоргалактосахарозы, и ее химическое название 1,6-дихлор-1,6-дидезокси-β-D-фруктофуранозил-4-хлор-4-дезокси-α-D-гарактопиранозид и представляет собой подсластитель, полученный путем селективной замены трех гидроксильных групп сахарозы атомами хлора.
[0016]
Доля подсластителя, необходимая для уменьшения горечи, наблюдаемой при суспендировании или т.п. гилтеритиниба или его фармацевтически приемлемой соли в небольшом количестве растворителя, такого как вода или т.п., может быть определена, например, методом in vitro с использованием устройства, обычно называемое датчиком вкуса, или методом in vivo, таким как сенсорный тест и т.п. с использованием тестовой панели. Примеры метода оценки сенсорного теста включают, но не ограничиваются этим, метод, в котором после того как испытываемый раствор, содержащий соответствующее количество подсластителя, в растворе, в котором соединение A или вещество, имеющее горечь, аналогичную горечи соединения A или его фармацевтически приемлемой соли, растворено в концентрации 10 мг/мл, вводится в рот и сразу же выплевывается, горечь оценивается сразу после выплевывания и до 30 минут после выплевывания. Подходящее содержание подсластителя различается в зависимости от типа подсластителя, но, в расчете на массу фармацевтической композиции, оно составляет типично от 0,001 до 70,0% масс., предпочтительно от 0,01 до 60,0% масс., более предпочтительно от 0,1 до 50,0% масс., еще более предпочтительно от 1,0 до 40,0% масс., еще более предпочтительно от 5,0 до 35,0% масс., еще более предпочтительно от 5,5 до 33,3% масс., еще более предпочтительно от 10,0 до 30,0% масс., еще более предпочтительно от 15,0 до 30,0% масс. и еще более предпочтительно от 20,0 до 25,0% масс. Каждый нижний предел и каждый верхний предел можно произвольно комбинировать по желанию.
[0017]
Фармацевтическая композиция по настоящему изобретению содержит два или более видов сахаров и/или сахарных спиртов. Следовательно, фармацевтическая композиция содержит по меньшей мере два сахара, по меньшей мере один сахар и по меньшей мере один сахарный спирт или по меньшей мере два сахарных спирта.
Примеры “сахаров” включают моносахариды, такие как глюкоза, галактоза и т.п., и дисахариды, такие как сахароза, лактоза, трегалоза, мальтоза и т.п., но не ограничиваются этим.
Термин “сахарные спирты” относится к типу сахара, полученному путем восстановления карбонильной группы альдозы или кетозы. Сахарные спирты представляют собой смесь соединений, полученных гидрированием сахаров и имеющих общую формулу HOCH2 (CHOH)nCH2OH, например маннит, ксилит, сорбит, инозит, мальтит, лактит и т.п. Примеры, в которых n равно 4 или 10, т.е. примеры, содержащие 6 или 12 атомов углерода, включают маннит, изомальт гидрат, мальтит, сорбит и т.п., но не ограничиваются этим.
[0018]
В “сахарах и/или сахарных спиртах”, используемых в настоящем изобретении, сахара могут быть выбраны предпочтительно из лактозы, сахарозы, трегалозы, мальтозы, глюкозы или фруктозы, более предпочтительно из лактозы, сахарозы или трегалозы. Сахарные спирты могут быть выбраны предпочтительно из маннита, изомальта гидрата, мальтита, сорбита, ксилита, лактита или эритрита, более предпочтительно из маннита, изомальта гидрата, мальтита или сорбита, и еще более предпочтительно из маннита или изомальта гидрата. Сахара и/или сахарные спирты могут быть выбраны предпочтительно из лактозы, маннита, изомальта гидрата, мальтита, сорбита, сахарозы или трегалозы, более предпочтительно из маннита, изомальта гидрата, мальтита или сорбита, еще более предпочтительно из маннита, изомальта гидрата или сорбита, и еще более предпочтительно из маннита или изомальта гидрата.
В качестве более предпочтительного варианта осуществления, один из двух или более сахаров и/или сахарных спиртов, содержащихся в фармацевтической композиции по настоящему изобретению, представляет собой маннит и может использоваться, например, в качестве наполнителя. В фармацевтической композиции, в которой один из двух или более сахаров и/или сахарных спиртов, содержащихся в фармацевтической композиции по настоящему изобретению, представляет собой маннит, один сахар и/или сахар спирт из остальных по меньшей мере одного сахара и/или сахарного спирта может использоваться, например, в качестве добавки для придания связывающей способности. Его можно выбрать предпочтительно из изомальта гидрата, мальтита, сорбита, сахарозы или трегалозы, более предпочтительно из изомальта гидрата, мальтита или сорбита, и еще более предпочтительно из изомальта гидрата или сорбита.
В качестве еще более предпочтительного варианта осуществления, два или более сахаров и/или сахарных спиртов, содержащихся в фармацевтической композиции по настоящему изобретению, представляют собой маннит и изомальт гидрат. Изомальт гидрат представляет собой смесь 6-0-α-D-глюкопиранозил-D-сорбит и 1-0-α-D-глюкопиранозил-D-маннит, но он не ограничивается гидратированной формой.
[0019]
Содержание двух или более сахаров и/или сахарных спиртов, содержащихся в фармацевтической композиции по настоящему изобретению, в расчете на массу фармацевтической композиции, составляет типично от 1 до 90% масс., предпочтительно от 5 до 70% масс., более предпочтительно от 10 до 60% масс., еще более предпочтительно от 15 до 50% масс., еще более предпочтительно от 25 до 45% масс., еще более предпочтительно от 30 до 45% масс., еще более предпочтительно от 35 до 42% масс. и еще более предпочтительно от 36 до 42% масс.
В фармацевтической композиции, в которой один из двух или более сахаров и/или сахарных спиртов, содержащихся в фармацевтической композиции по настоящему изобретению, представляет собой маннит, содержание одного сахара и/или сахарного спирта из остальных по меньшей мере одного сахара и/или сахарного спирта, в расчете на массу фармацевтической композиции, составляет типично от 1 до 20% масс., предпочтительно от 3 до 15% масс., более предпочтительно от 5 до 15% масс., еще более предпочтительно от 7 до 12% и еще более предпочтительно от 9 до 11% масс. Содержание маннита в фармацевтической композиции по настоящему изобретению, содержащей маннит, составляет, в расчете на массу фармацевтической композиции, типично от 1 до 40% масс., предпочтительно от 10 до 40% масс., более предпочтительно от 20 до 40% масс., еще более предпочтительно от 25 до 35% масс. и еще более предпочтительно от 27 до 33% масс.
Каждый нижний предел и каждый верхний предел можно произвольно комбинировать по желанию.
[0020] Термин “стабильность растворения” в контексте настоящей заявки означает свойства растворимости гилтеритиниба после некоторого периода воздействия тепла и/или влажности. Фраза “отличная стабильность растворения” или “для подавления снижения стабильности растворения с течением времени” в контексте настоящей заявки означает, что после хранения фармацевтической композиции (например, таблетки в качестве лекарственной формы) типично при 70°C в течение 9 дней, предпочтительно при 40°C и 75% относительной влажности (далее X% относительной влажности иногда сокращенно указывается как X% RH) в течение 1 месяца, 2 месяцев, 3 месяцев или 6 месяцев процент растворения гилтеритиниба через 15 минут или 30 минут после начала испытания на растворимость лопастным методом с использованием 900 мл 0,1 моль/л хлористоводородной кислоты при скорости вращения лопасти 50 об/мин, описанным в Японской фармакопее, семнадцатое издание, является высоким. То, что процент растворения гилтеритиниба через 15 минут после начала испытания на растворимость является высоким, означает типично 80% или больше, предпочтительно 85% или больше. В другом варианте осуществления это означает, что процент растворения гилтеритиниба через 30 минут после начала испытания на растворимость составляет типично 90% или больше. В другом варианте осуществления это означает, что после хранения фармацевтической композиции при 40°C и 75% относительной влажности в течение 1 месяца 85% или более гилтеритиниба растворяется через 15 минут после начала испытания на растворимость лопастным методом с использованием 900 мл 0,1 моль/л хлористоводородной кислоты, описанным в Японской фармакопее, семнадцатое издание, или что после хранения фармацевтической композиции при 40°C и 75% относительной влажности в течение 2 месяцев и/или 3 месяцев 80% или более гилтеритиниба растворяется через 15 минут после начала испытания на растворимость лопастным методом с использованием 900 мл 0,1 моль/л хлористоводородной кислоты, описанным в Японской фармакопее, семнадцатое издание.
[0021]
Гилтеритиниб или его фармацевтически приемлемую соль, используемые в настоящем изобретении, легко можно получить, например, способом, описанным в Патентной литературе 1 (WO 2010/128659), или аналогичным способом.
[0022]
Гилтеритиниб может быть в свободной форме, которая не образует соли, и может образовывать фармацевтически приемлемую соль с кислотой. Примеры такой соли включают кислотно-аддитивную соль с неорганической кислотой, такую как гидрохлорид, гидробромид, гидроиодид, сульфат, нитрат, фосфат или т.п.; и кислотно-аддитивную соль с органической кислотой, такую как формиат, ацетат, пропионат, оксалат, малонат, сукцинат, фумарат, гемифумарат, малеат, лактат, малат, цитрат, тартрат, карбонат, пикрат, метансульфонат, этансульфонат, глутамат или т.п. Эти соли можно получить обычными методами. Гемифумарат является предпочтительным.
[0023]
Доля кристаллов гильтеритиниба или его фармацевтически приемлемой соли, которые используются в настоящем изобретении, конкретно не ограничивается, при условии, что она находится в диапазоне, при котором соединение стабильно во время хранения. Когда фармацевтическая композиция является твердой, доля кристаллов может быть рассчитана, например, методом дифференциальной сканирующей калориметрии (DSC анализ), методом порошковой рентгеновской дифракции, методом твердотельного ЯМР, методом ближней инфракрасной спектроскопии (NIR) и т.п. Например, в качестве метода расчета доли кристаллов гемифумарата гилтеритиниба в гемифумарате гилтеритиниба, например, спектр измеряют методом ближней инфракрасной спектроскопии с использованием спектрометра ближнего инфракрасного диапазона с преобразованием Фурье (MPA, Bruker Optics) (диапазон измерения: от 12500 см-1 до 5800 см-1, разрешение: 8 см-1, количество сканирований: 32), и полученный спектр вторично дифференцируют (метод свертки Савицкого-Голея), и его можно анализировать с использованием программного обеспечения для анализа ближнего инфракрасного спектра (например, OPUS, Bruker Optics). Фармацевтическую композицию измельчают с использованием ступки и пестика для измерения спектра. Перед измерением спектра фармацевтической композиции спектры препаратов, в которых кристаллы гемифумарата гилтеритиниба смешаны в различных пропорциях, подвергают регрессионному анализу методом частичных наименьших квадратов для создания калибровочной кривой, и каждый спектр, полученный из фармацевтической композиции, интерполируют в калибровочную кривую для расчета доли кристаллов гемифумарата гилтеритиниба.
[0024]
Доля кристаллов составляет, например, в расчете на общее количество гилтеритиниба или его фармацевтически приемлемой соли, типично 60% или больше, предпочтительно от 60% до 100%, более предпочтительно от 70% до 100%, еще более предпочтительно от 80% до 100% и еще более предпочтительно от 90% до 100%. Кроме того, доля кристаллов составляет, например, в расчете на общее количество гилтеритиниба или его фармацевтически приемлемой соли, предпочтительно от 60% до меньше чем 100%, более предпочтительно от 70% до меньше чем 100%, еще более предпочтительно от 80% до меньше чем 100% и еще более предпочтительно от 90% до меньше чем 100%. В связи с этим используемые числовые значения интерпретируются как большее значение переменной, как правило, в пределах экспериментальной ошибки (например, в пределах 95% доверительного интервала для среднего значения) или в пределах ± 10% от указанного значения, и все значения переменной.
[0025]
Дозу гилтеритиниба или его фармацевтически приемлемой соли можно соответствующим образом определить в зависимости от конкретных случаев, принимая во внимание симптомы, возраст, пол или т.п. пациента. Суточная доза для взрослого составляет типично от 5 до 300 мг, предпочтительно от 10 до 200 мг, более предпочтительно от 20 до 180 мг, еще более предпочтительно от 40 до 160 мг, еще более предпочтительно от 80 до 140 мг и еще более предпочтительно от 110 до 130 мг, в расчете на гилтеритиниб. Ее вводят в виде одной дозы или делят на две-четыре дозы в день. Каждый нижний предел и каждый верхний предел можно произвольно комбинировать по желанию.
[0026]
Содержание гилтеритиниба или его фармацевтически приемлемой соли составляет, например, в расчете на массу фармацевтической композиции, от 1 до 90% масс., предпочтительно от 5 до 50% масс., более предпочтительно от 10 до 40% масс. и еще более предпочтительно от 25 до 35% масс. Количество содержащегося гилтеритиниба или его фармацевтически приемлемой соли составляет, в композиции в целом, от 5 до 300 мг, предпочтительно от 10 до 200 мг, более предпочтительно от 10 до 50 мг и еще более предпочтительно от 10 до 40 мг. Каждый нижний предел и каждый верхний предел можно произвольно комбинировать по желанию.
[0027]
Фармацевтическая композиция по настоящему изобретению может представлять собой различные препараты, такие как таблетки, капсулы, порошки, гранулы, мелкие гранулы, сухие сиропы и т.п., но не ограничиваясь этим. Таблетки включают не содержащую покрытия таблетку без пленочного покрытия, таблетку с пленочным покрытием, таблетку, распадающуюся при пероральном введении, растворяющуюся таблетку и мини-таблетку, но не ограничиваются этим. Таблетки или капсулы являются предпочтительными, а таблетки являются более предпочтительными.
[0028]
Жидкий препарат по настоящему изобретению включает раствор, суспензию, сироп и т.п., но не ограничивается этим. Жидкий препарат по настоящему изобретению можно получить путем растворения, диспергирования, суспендирования фармацевтической композиции, содержащей гилтеритиниб или его фармацевтически приемлемую соль, подсластитель и два или более сахаров/сахарных спиртов в растворителе. Примеры растворителя включают воду, сок, молоко и т.п., но не ограничиваются этим.
[0029]
Масса фармацевтической композиции по настоящему изобретению конкретно не ограничивается при условии, что пациент может ее принимать. Масса фармацевтической композиции обычно составляет от 5 до 600 мг, предпочтительно от 270 до 600 мг, более предпочтительно от 10 до 500 мг, еще более предпочтительно от 15 до 300 мг, еще более предпочтительно от 30 до 270 мг, еще более предпочтительно от 35 до 180 мг, еще более предпочтительно от 100 до 140 мг и еще более предпочтительно от 30 до 50 мг. Каждый нижний предел и каждый верхний предел можно произвольно комбинировать по желанию.
[0030]
В фармацевтической композиции по настоящему изобретению можно подходящим образом использовать различные фармацевтические добавки, такие как связующие, корригенты, шипучие агенты, отдушки, буферы, антиоксиданты, поверхностно-активные вещества, суспензии, агенты пленочного покрытия, разрыхлители, смазывающие вещества и т.п., если желательно, с таким расчетом, чтобы могли достигаться эффекты настоящего изобретения. В настоящем изобретении эти фармацевтические добавки можно соответственно добавлять по отдельности или в виде комбинации двух или более в подходящих количествах.
[0031]
“Связующее”, используемое в настоящем изобретении, означает вещество, используемое для взаимодействия между частицами и поддержания их в виде агрегата. Примеры связующего включают сахара, сахарные спирты, производные целлюлозы, такие как гидроксипропилцеллюлоза (HPC), и растворимые полимеры, но не ограничиваются этим. Предпочтительно, сахара и/или сахарные спирты, содержащиеся в фармацевтической композиции по настоящему изобретению, могут действовать как связующее. В этой связи, в настоящем изобретении при желании можно добавить дополнительное связующее с таким расчетом, чтобы могли достигаться эффекты настоящего изобретения, но фармацевтическая композиция по настоящему изобретению по существу не содержит HPC.
[0032]
Примеры корригентов включают лимонную кислоту, винную кислоту, яблочную кислоту и т.п., но не ограничиваются этим.
[0033]
Примеры шипучих агентов включают бикарбонат натрия и т.п., но не ограничиваются этим.
[0034]
Примеры отдушек включают лимонную, апельсиновую, вишневую, малиновую, ментоловую и т.п., но не ограничиваются этим.
[0035]
Примеры буферов включают лимонную кислоту, янтарную кислоту, фумаровую кислоту, винную кислоту, аскорбиновую кислоту и их соли; глутаминовую кислоту, глутамин, глицин, аспарагиновую кислоту, аланин, аргинин и их соли; оксид магния, оксид цинка, гидроксид магния, фосфорную кислоту, борную кислоту и их соли; и т.п., но не ограничиваются этим.
[0036]
Примеры антиоксидантов включают лимонную кислоту, нитрит натрия, аскорбиновую кислоту, эдетат натрия, лецитин соевых бобов, природный витамин E, пиросульфит натрия, дибутилгидрокситолуол и т.п., но не ограничиваются этим.
[0037]
Примеры поверхностно-активных веществ включают полисорбат 80, лаурилсульфат натрия, полиоксиэтилен-гидрогенизированное касторовое масло и т.п., но не ограничиваются этим.
[0038]
Примеры суспензий включают кристаллическую целлюлозу, кармеллозу натрия, ксантановую камедь, агар и т.п., но не ограничиваются этим.
[0039]
Фармацевтическая композиция по настоящему изобретению может дополнительно содержать вещество пленочного покрытия. Вещество пленочного покрытия конкретно не ограничивается при условии, что может быть получена фармацевтическая композиция, имеющая превосходную стабильность растворения. Примеры вещества пленочного покрытия включают фармацевтически приемлемые полимеры, такие как поливиниловый спирт, гидроксипропилметилцеллюлоза (HPMC), гидроксипропилцеллюлоза, этилцеллюлоза (EC), поливиниловый спирт (PVA), графт-сополимеры поливинилового спирта и полиэтиленгликоля и т.п.; фармацевтически приемлемые пластификаторы, такие как пропиленгликоль, полиэтиленгликоль (PEG), глицерин, триацетин (глицеринтриуксусная кислота), триэтилцитрат (TEC) и т.п.; масла, такие как минеральное масло, растительное масло и т.п.; фармацевтически приемлемые смазывающие вещества или осветлители, такие как тальк, воск, карнаубский воск и т.п.; фармацевтически приемлемые красители, такие как оксид титана, сесквиоксид железа и т.п.; подсластители; отдушки, такие как мята, ягодная отдушка, ваниль и т.п.; модификаторы вязкости, такие как полидекстроза, крахмал, аравийская камедь, ксантановая камедь и подобные; и т.п., но не ограничиваются этим. В качестве вещества пленочного покрытия предпочтительныя является коммерчески доступный покрывающий агент с быстрым высвобождением, такой как Opadry (зарегистрированная торговая марка) (изготовитель Colorcon Japan), содержащий PVA, HPMC или графт-сополимеры поливинилового спирта и полиэтиленгликоля в качестве полимера, PEG в качестве пластификатора, тальк в качестве смазывающего вещества и сесквиоксид железа в качестве красителя; и более предпочтительным является коммерчески доступный агент покрытия с быстрым высвобождением, такой как Opadry (зарегистрированная торговая марка) (изготовитель Colorcon Japan), содержащий HPMC в качестве полимера, PEG в качестве пластификатора, тальк в качестве смазывающего вещества и сесквиоксид железа в качестве красителя.
[0040]
Фармацевтическая композиция по настоящему изобретению дополнительно содержит разрыхлитель. Разрыхлитель конкретно не ограничивается при условии, что он может обеспечить фармацевтическую композицию, имеющую превосходную стабильность растворения. Примеры разрыхлителя включают кармеллозу, кармеллозу кальция, кроскармеллозу натрия, низкозамещенную гидроксипропилцеллюлозу, кукурузный крахмал, картофельный крахмал, рисовый крахмал, частично прежелатинизированный крахмал, прежелатинизированный крахмал и кросповидон, но не ограничиваются этим. Предпочтительной является низкозамещенная гидроксипропилцеллюлоза. Разрыхлитель может быть частью смеси разрыхлитель-наполнитель, например маннит/крахмал 8:2 (PEARLITOL FLASH, изготовитель Roquette). Разрыхлитель, содержащийся в фармацевтической композиции, используемой в настоящем изобретении, можно добавлять отдельно или в виде комбинации двух или более.
[0041]
Содержание разрыхлителя, используемого в настоящем изобретении, составляет, в расчете на массу фармацевтической композиции, типично от 1 до 20% масс., предпочтительно от 2 до 15% масс., более предпочтительно от 3 до 10% масс., и еще более предпочтительно от 4 до 6% масс. Каждый нижний предел и каждый верхний предел можно произвольно комбинировать по желанию.
[0042]
Фармацевтическая композиция по настоящему изобретению может дополнительно содержать смазывающее вещество. Смазывающее вещество конкретно не ограничивается при условии, что во время стадии формулирования препарата (особенно стадии формования) можно подавить адгезию фармацевтической композиции, например, к ступке, пестику или т.п., и чрезмерное увеличение или т.п. усилия для извлечения таблетки из ступки (далее иногда называется давлением выталкивания) в устройстве для таблетирования, и может быть получена фармацевтическая композиция, имеющая превосходную стабильность растворения. Примеры смазывающего вещества включают стеарат магния (далее иногда указан как Mg-St), стеарат кальция, стеарилфумарат натрия и тальк, но не ограничиваются этим. Смазывающее вещество, содержащееся в фармацевтической композиции, используемой в настоящем изобретении, может быть добавлено отдельно или в виде комбинации двух или более.
[0043]
Содержание смазывающего вещества, используемого в настоящем изобретении, составляет, в расчете на массу фармацевтической композиции, типично от 0,5 до 5% масс., предпочтительно от 1 до 3% масс. и более предпочтительно от 1,5 до 2% масс. Каждый нижний предел и каждый верхний предел можно произвольно комбинировать по желанию.
[0044]
Настоящее изобретение также относится к способу получения фармацевтической композиции, включающей гилтеритиниб или его фармацевтически приемлемую соль и два или более сахаров и/или сахарных спиртов, при этом указанный способ включает:
(1) получение связующей жидкости путем диспергирования или растворения по меньшей мере одного из сахаров и/или сахарных спиртов в растворителе;
(2) получение смеси путем смешивания гилтеритиниба или его фармацевтически приемлемой соли, подсластителя и по меньшей мере одного из сахаров и/или сахарных спиртов; и
(3) распыление или добавление связующей жидкости, полученной на стадии (1), к смеси, полученной на стадии (2), для образования гранул.
[0045]
«Связующее» в контексте настоящего изобретения означает агент, используемый для взаимодействия между частицами и поддержания их в виде агрегата. «Жидкое связующее» в контексте настоящего изобретения означает жидкость, полученную путем диспергирования или растворения связующего в растворителе. Примеры растворителя включают воду, ацетон, метанол и этанол, но не ограничиваются этим.
[0046]
Что касается терминов «подсластитель», «сахара и/или сахарные спирты» или т.п., которые используются в способе получения по настоящему изобретению, их объяснения, описанные для фармацевтической композиции по настоящему изобретению, могут быть непосредственно применимы.
Что касается содержания каждого компонента, способа смешивания или т.п. в способе получения по настоящему изобретению, их объяснения, описанные для фармацевтической композиции по настоящему изобретению, могут быть непосредственно применимы.
[0047]
Способ получения фармацевтической композиции по настоящему изобретению будет объяснен ниже, но включает известный способ, включающий, например, измельчение, смешивание, гранулирование, сушку, просеивание, сортировку по размерам, формование (таблетирование), нанесение пленочного покрытия, кристаллизацию и т.п.
[0048]
Стадия измельчения и стадия смешивания
На стадии измельчения ни устройство, ни средства конкретно не ограничиваются при условии, что это способ, в котором гилтеритиниб или его фармацевтически приемлемую соль и соответствующие фармацевтические добавки можно измельчать обычным используемым в фармацевтике способом. Примеры измельчающего устройства включают молотковую мельницу, шаровую мельницу, струйную мельницу, коллоидную мельницу и т.п., но не ограничиваются этим. Условия для измельчения можно выбрать соответствующим образом, и они конкретно не ограничиваются.
На стадии смешивания компонентов после стадии измельчения ни устройство, ни средства конкретно не ограничиваются при условии, что это способ, в котором компоненты можно однородно смешивать обычным используемым в фармацевтике способом.
[0049]
Стадия гранулирования
На стадии гранулирования ни устройство, ни средства конкретно не ограничиваются при условии, что это способ, в котором гилтеритиниб или его фармацевтически приемлемую соль и соответствующие фармацевтические добавки можно гранулировать обычным используемым в фармацевтике способом.
Примеры способа гранулирования и устройства для гранулирования, которые используют для влажной грануляции с использованием растворителя, такого как вода, или связующей жидкости, полученной путем диспергирования или растворения подходящего количества связующего в воде или т.п., включают метод гранулирования с высоким усилием сдвига, гранулирование методом перемалывания (измельчения), метод гранулирования в псевдоожиженном слое, гранулирование методом экструзии, метод гранулирования в барабанном грануляторе и метод гранулирования распылением; и устройства и т.п., которые используют в этих способах, но не ограничиваются этим.
В качестве способа гранулирования, например, смесь, содержащую гилтеритиниб или его фармацевтически приемлемую соль и подсластитель, можно гранулировать путем распыления или добавления связующей жидкости, полученной путем диспергирования или растворения изомальта гидрата в растворителе, таком как вода, с получением гранулированного продукта. В качестве способа, не использующего воду в процессе гранулирования, можно выбрать способ влажного гранулирования с использованием неводного растворителя или способ сухого гранулирования без использования растворителя.
[0050]
Стадия сушки
На стадии сушки ни устройство, ни средства конкретно не ограничиваются при условии, что это способ, в котором гранулированный продукт можно высушить обычным используемым в фармацевтике способом. Примеры устройства включают сушилку с принудительной подачей воздуха, сушилку, работающую при пониженном давлении, вакуумную сушилку, грануляционную сушилку с псевдоожиженным слоем и т.п., но не ограничиваются этим.
[0051]
Стадия просеивания и сортировки по размерам
На стадии просеивания и сортировки по размерам ни устройство, ни средства конкретно не ограничиваются при условии, что это способ, в котором высушенный продукт может быть проесеян или отсортирован обычным используемым в фармацевтике способом. Примеры устройства включают сито, конусную мельницу, силовую мельницу и т.п., но не ограничиваются этим.
[0052]
Стадия формования (таблетирования)
На стадии формования ни устройство, ни средства конкретно не ограничиваются при условии, что это способ формования фармацевтической композиции по настоящему изобретению. Примеры способа включают способ, в котором, без стадии гранулирования и сушки, гилтеритиниб или его фармацевтически приемлемую соль и соответствующие фармацевтические добавки смешивают и сразу же осуществляют формование прессованием с получением фармацевтической композиции; способ, в котором гилтеритиниб или его фармацевтически приемлемую соль и соответствующие фармацевтические добавки гранулируют и сушат и сразу же осуществляют формование прессованием с получением фармацевтической композиции; способ, в котором гилтеритиниб или его фармацевтически приемлемую соль и соответствующие фармацевтические добавки гранулируют и затем смешивают со смазывающим веществом и смесь подвергают формованию прессованием с получением фармацевтической композиции (например, таблетки без покрытия); и т.п., но не ограничиваясь этим.
[0053]
Примеры таблетировочной машины включают ротационную таблетировочную машину, гидравлический пресс и т.п. Условия для таблетировани, такие как давление таблетирования, конкретно не ограничиваются при условии, что это давление таблетирования, приемлемое для формования прессованием. Например, давление таблетирования (основное давление), когда формование прессованием осуществляют с использованием ротационной таблетировочной машины (EX-10, изготовитель HATA TEKKOSHO), может составлять от 0,5 до 20,0 кН, предпочтительно от 1,0 до 10,0 кН и более предпочтительно от 1,5 до 4,5 кН. Каждый нижний предел и каждый верхний предел можно произвольно комбинировать по желанию.
[0054]
Прочность таблетки без покрытия конкретно не ограничивается при условии, что таблетка от процесса ее изготовления до распределения не повреждается. Например, прочность, когда прочность таблетированного продукта измеряют с использованием ротационной таблетировочной машины (HT-CVX-TYPEIII20, изготовитель HATA TEKKOSHO), составляет 30 Н или больше, предпочтительно 42 Н или больше, более предпочтительно 64 Н или больше и еще более предпочтительно 80 Н или больше. Верхний предел прочности составляет 400 Н или меньше. Когда, например, ротационную таблетировочную машину, гидравлический пресс или т.п. используют в качестве таблетировочной машины, поскольку гилтеритиниб или его фармацевтически приемлемая соль обладает свойством сильной адгезии к металлу, непрерывное таблетирование вызывает прилипание к пестику, трудности, связанные с удалением сформованных прессованием таблеток из ступки, и повышение давления выталкивания. Поскольку в случае прилипания или повышения давления выталкивания не только ухудшается внешний вид таблетки, это также вызывает нагрузку на ступку и пестик и таблетировочную машину, что следует исправить. Примеры способа усовершенствования включают обработку ступки и пестика твердым хромом или обработку нитридом хрома и т.п., но не ограничиваются этим.
[0055]
Стадия нанесения пленочного покрытия
После таблетирования поверхность фармацевтической композиции (например, таблеток без покрытия) может быть покрыта пленкой. Способ нанесения пленочного покрытия конкретно не ограничивается при условии, что покрытие можно наносить обычным используемым в фармацевтике способом. Примеры нанесения покрытия включают способ с использованием установки для нанесения оболочки на таблетки, нанесение покрытия струйным обливом, покрытие погружением и т.п., но не ограничиваются этим.
[0056]
Стадия кристаллизации
Когда доля кристаллов гилтеритиниба или его фармацевтически приемлемой соли снижается, можно использовать стадию промотирования кристаллизации. Примеры стадии включают обработку путем микроволнового облучения, обработку ультразвуком, обработку путем низкочастотного облучения, обработку путем термоэлектронного облучения и т.п., но не ограничиваются этим.
[0057]
В качестве микроволновой обработки, например, можно использовать длину волны от 10 МГц до 25 ГГц, но не ограничиваясь этим. Хотя время обработки зависит от уровня начальной доли кристаллов или фармацевтических добавочных компонентов, оно может составлять, например, от 10 секунд до 60 минут. Облучение можно осуществлять непрерывно или периодически и в любое время.
[0058]
В качестве ультразвуковой обработки, например, можно использовать звуковые волны с частотой от 10 кГц до 600 кГц, но не ограничиваясь этим. Хотя время обработки зависит от уровня доли кристаллов или фармацевтических добавочных компонентов, оно может составлять, например, от 10 секунд до 24 часов. Облучение можно осуществлять непрерывно или периодически и в любое время.
ПРИМЕРЫ
[0059]
Гилтеритиниб гемифумарат, который использовали в Примерах или т.п., был получен в соответствии со способом, описанным в Патентной литературе 1 (WO 2010/128659), или аналогичным способом.
[0060]
В качестве соединений или веществ, описанных в Примерах, или т.п. использовали следующие:
PEARLITOL (зарегистрированная торговая марка) 50C (изготовитель ROQUETTE), который представлял собой маннит;
сукралоза (зарегистрированная торговая марка) (P) (изготовитель San-Ei Gen F.F.I.), которая представляла собой сукралозу;
Ajinomoto KK аспартам (изготовитель Ajinomoto), который представлял собой аспартам;
HPC L (изготовитель Nippon Soda), которая представляла собой гидроксипропилцеллюлозу (далее иногда указана как HPC);
galenIQ 721 (изготовитель BENEO-PALATINIT), который представлял собой изомальт гидрат;
Sweet Peral (зарегистрированная торговая марка) P200 (изготовитель ROQUETTE), который представлял собой мальтит;
NEOSORB (зарегистрированная торговая марка) XTAB 290 (изготовитель ROQUETTE), который представлял собой сорбит;
сахароза (UE-E) (изготовитель KANTO CHEMICAL), которая представляла собой сахарозу;
трегалоза (P) (изготовитель Asahi Kasei), которая представляла собой трегалозу;
L-HPC (зарегистрированная торговая марка) LH-21 (изготовитель Shin-Etsu Chemical), которая представляла собой низкозамещенную гидроксипропилцеллюлозу;
Parteck (зарегистрированная торговая марка) LUB MST (изготовитель Merck KGaA), который представлял собой Mg-St;
Opadry (зарегистрированная торговая марка) (изготовитель Colorcon Japan) или Opadry (зарегистрированная торговая марка) QX (изготовитель Colorcon Japan), которые представляли собой вещества для пленочного покрытия;
[0061]
<<Экспериментальн Пример>>
К раствору, полученному путем растворения гилтеритиниба гемифумарата в воде при концентрации 11,05 мг/мл, несколько видов подсластителей добавляли с получением анализируемого раствора. Сенсорное испытание полученного анализируемого раствора показало, что, в частности, сукралоза уменьшала горечь гилтеритиниба гемифумарата.
[0062]
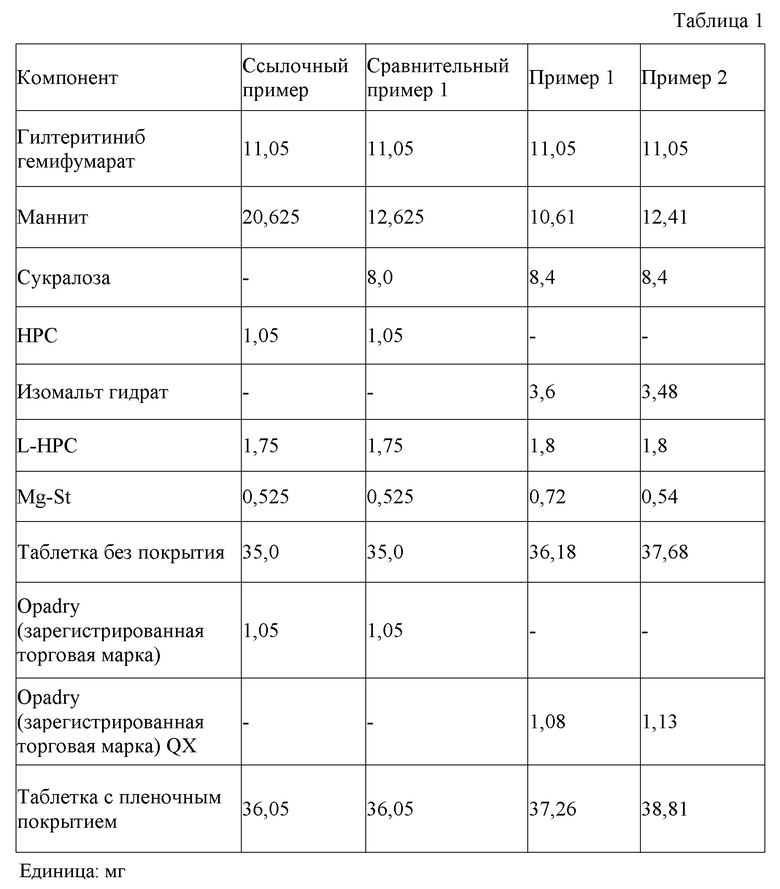
<<Композиции Ссылочного примера, Сравнительного примера 1, Примера 1 и Примера 2>>
Таблица 1 показывает композицию Ссылочного примера, которая представляет собой композицию, не содержащую два или более сахаров и/или сахарных спиртов, композицию Сравнительного примера 1, которая представляет собой композицию, в которой часть маннита в Ссылочном примере заменяли сукралозой в качестве подсластителя, композицию Примера 1, в которой HPC в Сравнительном примерае 1 заменяли изомальтом гидратом, и композицию Примера 2, в которой соотношение компонентов несколько отличалось от композиции Примера 1. “Ссылочный Пример” в контексте настоящей заявки означает пример по существу не содержащий подсластителя, “Пример” означает пример, содержащий подсластитель и обладающий отличной стабильностью растворения, и “Сравнительный пример” означает пример, содержащий подсластитель и имеющий низкую стабильность растворения.
[0063]
[0064]
<<Получение таблеток Ссылочного примера>>
В соответствии с композицией, описанной в Таблице 1, 2223,0 г гилтеритиниба гемифумарата и 4112,0 г маннита смешивали с использованием гранулятора с псевдоожиженным слоем (GPCG-PRO-5, изготовитель Powrex) и смесь гранулировали путем распыления 3008 г водного раствора HPC (содержание твердого вещества: 7% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов к полученному гранулированному продукту добавляли 326,8 г L-HPC и 98,05 г Mg-St и смешивали с использованием смесителя (Container Mixer PM200 (60-л контейнер), изготовитель HIROSHIMA METAL & MACHINERY) с получением смешанного продукта. Полученный смешанный продукт формовали в таблетки с использованием ротационной таблетировочной машины (HT-CVX-TYPEIII20, изготовитель HATA TEKKOSHO) с получением таблеток без покрытия. Полученные таблетки без покрытия (5158,8 г) помещали в машину для нанесения пленочного покрытия (PRC-20/60 (20-л контейнер), изготовитель Powrex) и пленочное покрытие наносили с использованием жидкости, полученной путем диспергирования или растворения Opadry (зарегистрированная торговая марка) в очищенной воде, с получением таблеток с пленочным покрытием Ссылочного примера.
[0065]
<<Получение таблеток Сравнительного примера 1>>
В соответствии с композицией, описанной в Таблице 1, 165,8 г гилтеритиниба гемифумарата, 189,4 г маннита и 120,0 г сукралозы смешивали с использованием гранулятора с псевдоожиженным слоем (FLO-1, изготовитель Freund Corporation) и смесь гранулировали путем распыления 225 г водного раствора HPC (содержание твердого вещества: 7% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 196,3 г из 473,4 г полученного гранулированного продукта и добавляли к ним 10,5 г L-HPC и 3,15 г Mg-St и смешивали вручную с использованием полиэтиленового пакета с получением смешанного продукта. Полученный смешанный продукт формовали в таблетки с использованием ротационной таблетировочной машины (EX-10, изготовитель HATA TEKKOSHO) с получением таблеток без покрытия. Полученные таблетки без покрытия (35,0 г) помещали в машину для нанесения пленочного покрытия (Flow Coater mini, изготовитель Powrex) и пленочное покрытие наносили с использованием жидкости, полученной путем диспергирования или растворения Opadry (зарегистрированная торговая марка) в очищенной воде, с получением таблеток с пленочным покрытием Сравнительного примера 1.
[0066]
<<Получение таблеток Примера 1>>
В соответствии с композицией, описанной в Таблице 1, 2,21 кг гилтеритиниба гемифумарата, 2122,0 г маннита и 1680,0 г сукралозы смешивали с использованием гранулятора с псевдоожиженным слоем (GPCG-PRO-5, изготовитель Powrex) и смесь гранулировали путем распыления 3602 г водного раствора изомальта гидрата (содержание твердого вещества: 20% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 5385,6 г из 6377,5 г полученного гранулированного продукта и добавляли к ним 288,0 г L-HPC и 115,2 г Mg-St и смешивали с использованием смесителя (Container Mixer PM200 (60-л контейнер), изготовитель HIROSHIMA METAL & MACHINERY) с получением смешанного продукта. Полученный смешанный продукт формовали в таблетки с использованием ротационной таблетировочной машины (HT-CVX-TYPEIII20, изготовитель HATA TEKKOSHO) с получением таблеток без покрытия. Полученные таблетки без покрытия (900,6 г) помещали в машину для нанесения пленочного покрытия (HCT-30, изготовитель Freund Corporation) и пленочное покрытие наносили с использованием жидкости, полученной путем диспергирования или растворения Opadry (зарегистрированная торговая марка) QX в очищенной воде, с получением таблеток с пленочным покрытием Примера 1.
[0067]
<<Получение таблеток Примера 2>>
В соответствии с композицией, описанной в Таблице 1, 165,75 г гилтеритиниба гемифумарата, 186,15 г маннита и 126,0 г сукралозы смешивали с использованием гранулятора с псевдоожиженным слоем (GPCG-1, изготовитель Powrex) (далее указан как GPCG-1) и смесь гранулировали путем распыления 475 г водного раствора изомальта гидрата (содержание твердого вещества: 10% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 212,08 г из 401,9 г полученного гранулированного продукта и добавляли к ним 10,84 г L-HPC и 3,23 г Mg-St и смешивали вручную с использованием полиэтиленового пакета с получением смешанного продукта. Полученный смешанный продукт формовали в таблетки с использованием ротационной таблетировочной машины (EX-10, изготовитель HATA TEKKOSHO) с получением таблеток без покрытия. Полученные таблетки без покрытия (37,69 г) помещали в машину для нанесения пленочного покрытия (MINI COATER/DRIER-2, изготовитель CALEVA) и пленочное покрытие наносили с использованием жидкости, полученной путем диспергирования или растворения Opadry (зарегистрированная торговая марка) QX в очищенной воде, с получением таблеток с пленочным покрытием Примера 2.
[0068]
<<Экспериментальный Пример 2>>
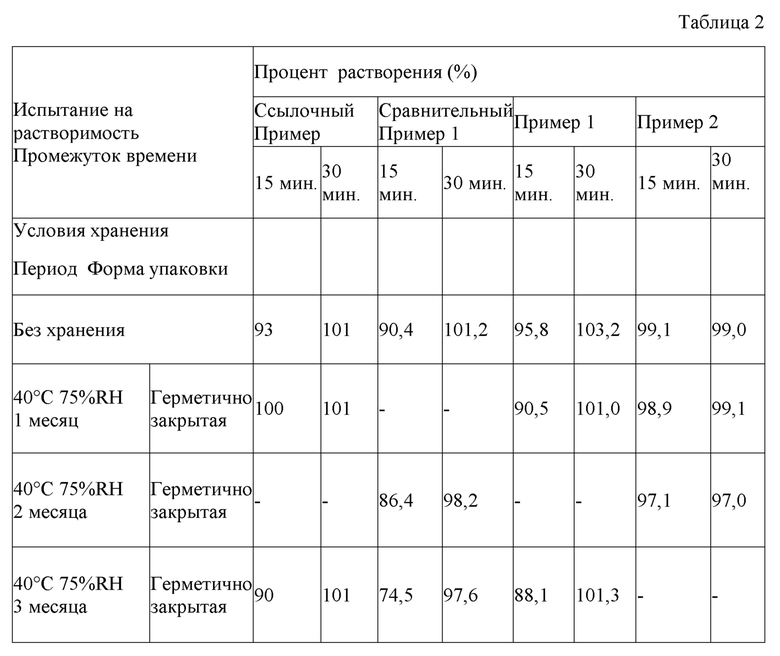
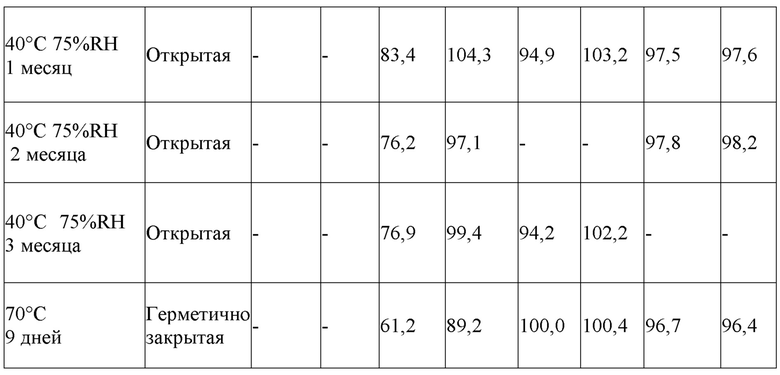
Каждую таблетку с пленочным покрытием, полученную в Ссылочном примере, Сравнительном примере 1, Примере 1 и Примере 2, помещали в отдельную бутыль из полиэтилена высокой плотности и выдерживали при 40°C и 75% RH в течение 1 месяца, 2 месяцев или 3 месяцев или при 70°C в течение 9 дней с получением образца в каждых условиях хранения. В этой связи, условия укупоривания были такими, что горлышко бутыли уплотняли индукционно запаянной прокладкой и крышку закрывали, а открытые условия были такими, что бутыль не закрывали. Что касается образцов, полученных в каждых условиях хранения, испытание на растворимость осуществляли с использованием тестера растворения (серии NTR-6100, серии NTR-6200 или серии NTR-6400, изготовитель TOYAMA SANGYO), в соответствии с испытанием на растворимость лопастным методом Японской фармакопеи, семнадцатое издание, с использованием 900 мл 0,1 моль/л хлористоводородной кислоты в качестве жидкости для испытания на растворимость, при скорости вращения лопасти 50 об/мин. Через 15 минут и 30 минут после начала испытания площадь пика гилтеритиниба в жидкости для испытания на растворимость измеряли с использованием спектрофотометрического метода в ультрафиолетовой и видимой областях спектра (УФ метод) или метода высокоэффективной жидкостной хроматографии (ВЭЖХ метод) и концентрацию гилтеритиниба рассчитывали из полученной площади пика для расчета степени растворения. В УФ методе измерение осуществляли с использованием спектрофотометра для измерений в ультрафиолетовой и видимой областях спектра (UV-1800, изготовитель Shimadzu Corporation) (длина волны: 313 нм). В ВЭЖХ методе измерение осуществляли с использованием системы Alliance HPLC (зарегистрированная торговая марка) (изготовитель Nihon Waters) (длина волны: 314 нм). В методе ВЭЖХ использовали колонку CAPCELLPAK C18 AQ (внутренний диаметр: 4,6 мм, длина: 150 мм, размер частиц: 3 мкм, изготовитель OSAKA SODA) или ее эквивалент. Колонку использовали, поддерживая ее при 40°C, и в качестве подвижной фазы использовали смешанную жидкость раствор перхлорной кислоты (pH 2,2)/ацетонитрил=65/35. Процент растворения гилтеритиниба в каждом образце без хранения и в каждом образце после хранения показан в Таблице 2.
[0069]
[0070]
В образцах, в которых таблетка Ссылочного примера хранилась при 40°C и 75% RH в условиях укупоривания в течение 1 месяца и 3 месяцев, процент растворения гилтеритиниба через 15 минут после начала испытания составлял 85% или больше. С другой стороны, в образцах, в которых таблетка Сравнительного примера 1 хранилась при 40°C и 75% RH в условиях укупоривания в течение 3 месяцев и при 70°C в условиях укупоривания в течение 9 дней, процент растворения гилтеритиниба через 15 минут после начала испытания составлял 75% или меньше. Однако в Примерах 1 и 2, в которых связующее представляло собой изомальт гидрат, при всех условиях хранения процент растворения гилтеритиниба через 15 минут после начала испытания составлял 85% или больше.
[0071]
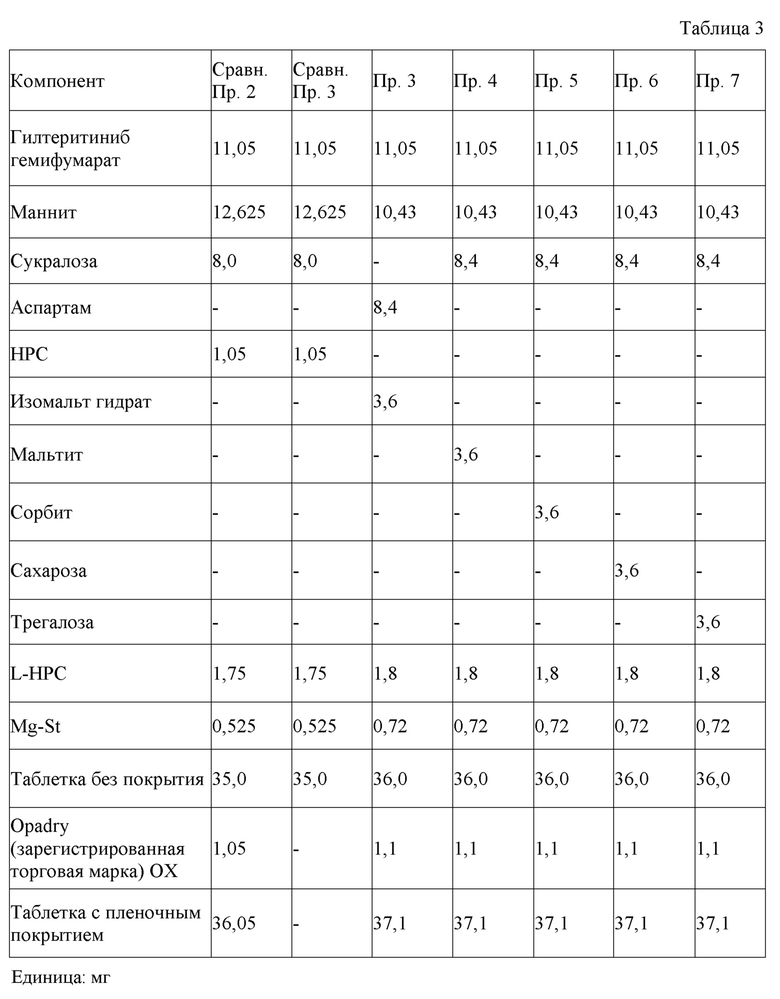
<<Композиции Сравнительных примеров 2-3 и Примеров 3-7>>
Композиция Сравнительного примера 2, в которой тип вещества для пленочного покрытия Сравнительного примера 1 был изменен, и композиция Сравнительного примера 3 без пленочного покрытия показаны в Таблице 3. Кроме того, композиция Примера 3, в которой подсластитель Примера 1 был заменен на аспартам, и композиции Примеров 4-7, в которых изомальт гидрат Примера 1 был заменен на мальтит, сорбит, сахарозу или трегалозу, показаны в Таблице 3.
[0072]
[0073]
<<Получение таблеток Сравнительного примера 2 и Сравнительного примера 3>>
В соответствии с композициями, описанными в Таблице 3, 165,76 г гилтеритиниба гемифумарата, 189,26 г маннита и 119,98 г сукралозы смешивали с использованием GPCG-1 и смесь гранулировали путем распыления 225,0 г водного раствора HPC (содержание твердого вещества: 7% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 196,35 г из 296,83 г полученного гранулированного продукта и к ним добавляли 10,51 г L-HPC и 3,15 г Mg-St и смешивали и формовали в таблетки таким же способом, как в Примере 2, с получением таблеток без покрытия Сравнительного примера 3. На полученные таблетки без покрытия (10,5152 г) наносили пленочное покрытие таким же способом, как в Примере 2, с получением таблеток с пленочным покрытием Сравнительного примера 2.
[0074]
<<Получение таблеток Примера 3>>
В соответствии с композицией, описанной в Таблице 3, 165,77 г гилтеритиниба гемифумарата, 156,47 г маннита и 126,02 г аспартама смешивали с использованием GPCG-1 и смесь гранулировали путем распыления 270,0 г водного раствора изомальта гидрата (содержание твердого вещества: 20% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 267,85 г из 442,52 г полученного гранулированного продукта и к ним добавляли 14,43 г L-HPC и 5,76 г Mg-St и смешивали и формовали в таблетки таким же способом, как в Примере 2, с получением таблеток без покрытия. На полученные таблетки без покрытия (10,8147 г) наносили пленочное покрытие таким же способом, как в Примере 2, с получением таблеток с пленочным покрытием Примера 3.
[0075]
<<Получение таблеток Примера 4>>
В соответствии с композицией, описанной в Таблице 3, 165,76 г гилтеритиниба гемифумарата, 156,45 г маннита и 126,02 г сукралозы смешивали с использованием GPCG-1 и смесь гранулировали путем распыления 270,0 г водного раствора мальтита (содержание твердого вещества: 20% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 267,85 г из 459,84 г полученного гранулированного продукта и к ним добавляли 14,40 г L-HPC и 5,76 г Mg-St и смешивали и формовали в таблетки таким же способом, как в Примере 2, с получением таблеток без покрытия. На полученные таблетки без покрытия (10,7941 г) наносили пленочное покрытие таким же способом, как в Примере 2, с получением таблеток с пленочным покрытием Примера 4.
[0076]
<<Получение таблеток Примера 5>>
В соответствии с композицией, описанной в Таблице 3, 165,75 г гилтеритиниба гемифумарата, 156,44 г маннита и 126,02 г сукралозы смешивали с использованием GPCG-1 и смесь гранулировали путем распыления 270,0 г водного раствора сорбита (содержание твердого вещества: 20% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 267,84 г из 425,03 г полученного гранулированного продукта и к ним добавляли 14,41 г L-HPC и 5,76 г Mg-St и смешивали таким же способом, как в Примере 2, и формовали в таблетки с использованием ручной настольной машины для формования таблеток (HANDTAB-200, изготовитель Ichihashi Seiki) с получением таблеток без покрытия. На полученные таблетки без покрытия (870,8 мг) наносили пленочное покрытие таким же способом, как в Примере 2, с получением таблеток с пленочным покрытием Примера 5.
[0077]
<<Получение таблеток Примера 6>>
В соответствии с композицией, описанной в Таблице 3, 165,74 г гилтеритиниба гемифумарата, 156,45 г маннита и 126,00 г сукралозы смешивали с использованием GPCG-1 и смесь гранулировали путем распыления 270,2 г водного раствора сахарозы (содержание твердого вещества: 20% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 267,84 г из 458,3 г полученного гранулированного продукта и к ним добавляли 14,41 г L-HPC и 5,76 г Mg-St и смешивали и формовали в таблетки таким же способом, как в Примере 2, с получением таблеток без покрытия. На полученные таблетки без покрытия (10,8116 г) наносили пленочное покрытие таким же способом, как в Примере 2, с получением таблеток с пленочным покрытием Примера 6.
[0078]
<<Получение таблеток Примера 7>>
В соответствии с композицией, описанной в Таблице 3, 165,76 г гилтеритиниба гемифумарата, 156,45 г маннита и 126,03 г сукралозы смешивали с использованием GPCG-1 и смесь гранулировали путем распыления 270,0 г водного раствора трегалозы (содержание твердого вещества: 20% масс.) в качестве связующего и сушили с получением гранулированного продукта. После просеивания через сито для удаления агрегатов отвешивали 267,84 г из 469,39 г полученного гранулированного продукта и к ним добавляли 14,42 г L-HPC и 5,76 г Mg-St и смешивали и формовали в таблетки таким же способом, как в Примере 2, с получением таблеток без покрытия. На полученные таблетки без покрытия (10,8207 г) наносили пленочное покрытие таким же способом, как в Примере 2, с получением таблеток с пленочным покрытием Примера 7.
[0079]
<<Экспериментальный Пример 3>>
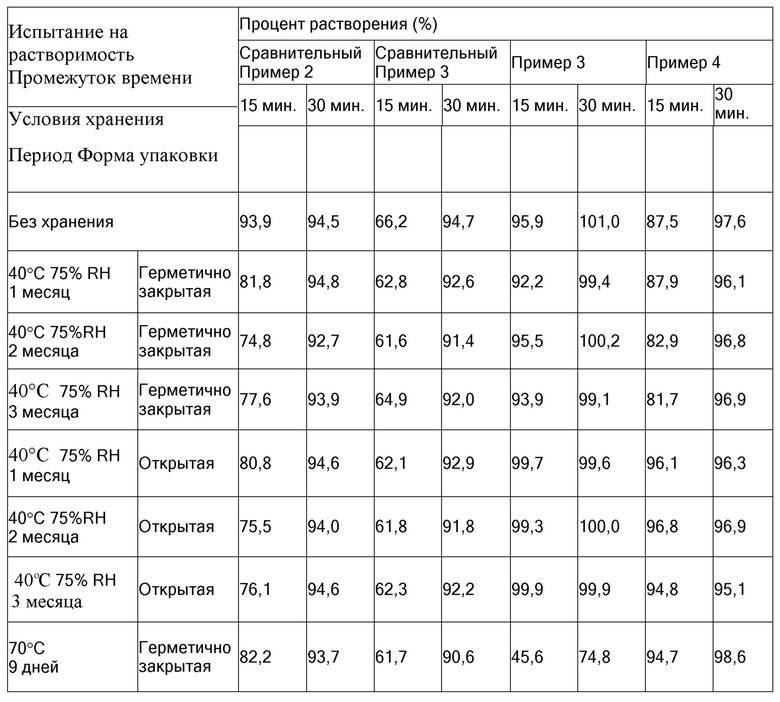
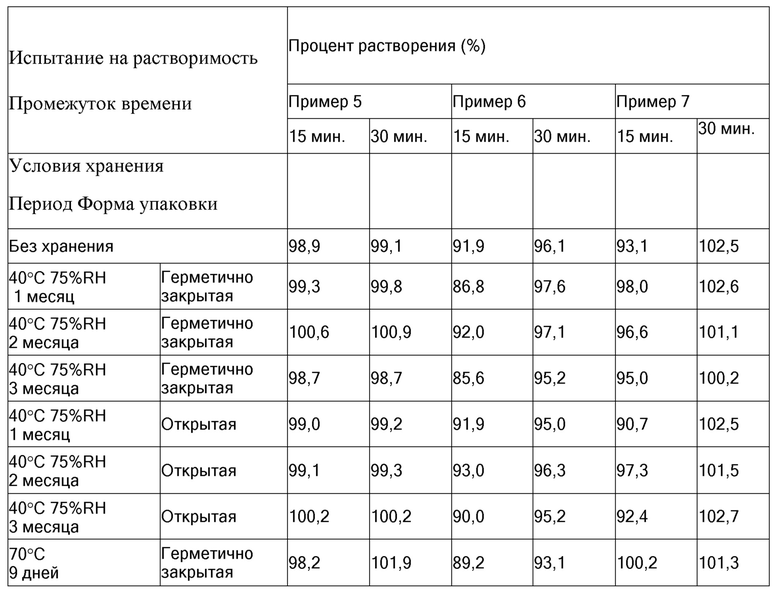
Каждую таблетку с пленочным покрытием или таблетку без покрытия, полученную в Сравнительных примерах 2-3 и Примерах 3-7, помещали в отдельную бутыль из полиэтилена высокой плотности и выдерживали при 40°C и 75% RH в течение 1 месяца, 2 месяцев или 3 месяцев или при 70°C в течение 9 дней с получением образца в каждых условиях хранения. Что касается образцов, полученных в каждых условиях хранения, испытание на растворимость осуществляли в таких же условиях, как в Экспериментальном примере 2, и рассчитывали процент растворения. Процент растворения гилтеритиниба в каждом образце без хранения и образце после хранения в каждых условиях показан в Таблице 4.
[0080]
Таблица 4
[0081]
В образцах, в которых таблетки Сравнительных примеров 2-3 хранились при 40°C и 75% RH в условиях укупоривания или в открытых условиях в течение 2 месяцев и 3 месяцев, процент растворения гилтеритиниба через 15 минут после начала испытания составлял 80% или меньше. Однако в Примерах 4-7, в которых в качестве связующего использовали сахара и/или сахарные спирты, при всех условиях хранения процент растворения гилтеритиниба через 15 минут после начала испытания составлял 80% или больше. Кроме того, в образцах, в которых таблетка Примера 3, в которой в качестве связующего использовали сахара и/или сахарные спирты, хранилась при 40°C и 75% RH в условиях укупоривания или в открытых условиях в течение 2 месяцев и 3 месяцев, процент растворения гилтеритиниба через 15 минут после начала испытания составлял 80% или больше.
[0082]
Из представленных выше результатов можно видеть, что за счет содержания двух или более видов сахаров и/или сахарных спиртов можно обеспечить фармацевтическую композицию, включающую гилтеритиниб или его фармацевтически приемлемую соль и подсластитель, который уменьшает горечь, и обладающую отличной стабильностью растворения.
Промышленная применимость
[0083]
В соответствии с настоящим изобретением можно обеспечить фармацевтическую композицию, подавляющую снижение стабильности растворения с течением времени, включающую гилтеритиниб или его фармацевтически приемлемую соль и подсластитель, который уменьшает горечь, и демонстрирующую отличную стабильность растворения.
[0084]
Хотя настоящее изобретение было описано со ссылкой на конкретные варианты осуществления, возможны различные изменения и модификации, очевидные для специалистов в данной области техники, без отступления от объема прилагаемой формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ТВЕРДАЯ НИКОТИНСОДЕРЖАЩАЯ ДОЗИРОВАННАЯ ФОРМА СО СНИЖЕННЫМ НЕПРИЯТНЫМ ОРГАНОЛЕПТИЧЕСКИМ ВОЗДЕЙСТВИЕМ | 2013 |
|
RU2623018C2 |
| СТАБИЛЬНАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПЕРОРАЛЬНОГО ВВЕДЕНИЯ | 2016 |
|
RU2764750C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ОСЕЛЬТАМИВИР | 2020 |
|
RU2811475C2 |
| ВЫСОКОДОЗНАЯ КОМПОЗИЦИЯ ТРАНЕКСАМОВОЙ КИСЛОТЫ И СПОСОБЫ ЕЕ ПОЛУЧЕНИЯ | 2023 |
|
RU2814336C1 |
| КОМПОЗИЦИИ ПОДСЛАСТИТЕЛЕЙ, СОДЕРЖАЩИЕ РЕБАУДИОЗИД-D | 2016 |
|
RU2727640C2 |
| НОСИТЕЛЬ ДЛЯ ПЕРОРАЛЬНОЙ ДОСТАВКИ | 2016 |
|
RU2738205C1 |
| ТАБЛЕТКА, СОДЕРЖАЩАЯ ОТДЕЛЬНОЕ СВЯЗЫВАЮЩЕЕ ВЕЩЕСТВО И ЭРИТРИТ | 2017 |
|
RU2736072C1 |
| ТАБЛЕТКА ПРЕССОВАННОЙ ЖЕВАТЕЛЬНОЙ РЕЗИНКИ И СПОСОБ ЕЕ ПРОИЗВОДСТВА | 2003 |
|
RU2305948C2 |
| ПАСТИЛКА ИЗ БЕЗДЫМНОГО ТАБАКА И СПОСОБ ФОРМОВАНИЯ ДЛЯ ПОЛУЧЕНИЯ ПРОДУКТОВ ИЗ БЕЗДЫМНОГО ТАБАКА | 2011 |
|
RU2612626C2 |
| КОНДИТЕРСКИЙ ПРОДУКТ, ОСВЕЖАЮЩИЙ ПОЛОСТЬ РТА (ВАРИАНТЫ) | 2010 |
|
RU2488277C2 |
Группа изобретений относится к химии и фармацевтике, а именно к вариантам фармацевтической композиции в форме таблетки и к способу ее получения. В одном из вариантов предложенная фармацевтическая композиция включает 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид (гилтеритиниб) или его фармацевтически приемлемую соль, сукралозу и два или более сахаров и/или сахарных спиртов, при этом два или более сахаров и/или сахарных спиртов выбраны из изомальта гидрата, мальтита, сорбита, сахарозы или трегалозы и содержатся в количестве 3-15 мас.% и по меньшей мере один из двух или более сахаров и/или сахарных спиртов представляет собой манит. В другом варианте композиция включает гилтеритиниба гемифумарат, манит, сукралозу и изомальт гидрат, который содержится в количестве 3-15 мас.%. Группа изобретений обеспечивает стабильность растворения и уменьшение горечи гилтеритиниба или его фармацевтически приемлемой соли. 3 н. и 6 з.п. ф-лы, 4 табл., 14 пр.
1. Фармацевтическая композиция, включающая 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид или его фармацевтически приемлемую соль, сукралозу и два или более сахаров и/или сахарных спиртов,
где два или более сахаров и/или сахарных спиртов выбраны из группы, состоящей из изомальта гидрата, мальтита, сорбита, сахарозы и трегалозы, и содержатся в количестве от 3% масс. до 15% масс. и по меньшей мере один из двух или более сахаров и/или сахарных спиртов представляет собой маннит;
где фармацевтическая композиция представляет собой таблетку.
2. Фармацевтическая композиция по п.1, где один из двух или более сахаров и/или сахарных спиртов представляет собой изомальт гидрат.
3. Фармацевтическая композиция по п.1 или 2, где изомальт гидрат, мальтит, сорбит, сахарозу или трегалозу, описанные в п.1 или 2, используют в качестве связующего.
4. Фармацевтическая композиция по любому из пп.1-3, где фармацевтически приемлемая соль представляет собой гемифумарат.
5. Фармацевтическая композиция по любому из пп.1-4, дополнительно включающая разрыхлитель.
6. Фармацевтическая композиция по любому из пп.1-5, где фармацевтическая композиция растворена или диспергирована в подходящем растворителе и представляет собой раствор, суспензию, пасту или гель.
7. Фармацевтическая композиция, включающая 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид гемифумарат, маннит, сукралозу и изомальт гидрат,
где изомальт гидрат содержится в количестве от 3% масс. до 15% масс.;
где фармацевтическая композиция представляет собой таблетку.
8. Фармацевтическая композиция по любому из пп.1-7, полученная способом получения, включающим:
(1) получение связующей жидкости путем диспергирования или растворения по меньшей мере одного из сахаров и/или сахарных спиртов в растворителе;
(2) получение смеси путем смешивания 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамида или его фармацевтически приемлемой соли, сукралозы и по меньшей мере одного из сахаров и/или сахарных спиртов; и
(3) распыление или добавление связующей жидкости, полученной на стадии (1), к смеси, полученной на стадии (2), для образования гранул.
9. Способ получения фармацевтической композиции, включающей 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамид или его фармацевтически приемлемую соль, сукралозу и два или более сахаров и/или сахарных спиртов,
где два или более сахаров и/или сахарных спиртов выбраны из группы, состоящей из изомальта гидрата, мальтита, сорбита, сахарозы и трегалозы, и содержатся в количестве от 3% масс. до 15% масс. и по меньшей мере один из двух или более сахаров и/или сахарных спиртов представляет собой маннит,
при этом указанный способ включает:
(1) получение связующей жидкости путем диспергирования или растворения по меньшей мере одного из сахаров и/или сахарных спиртов в растворителе;
(2) получение смеси путем смешивания 6-этил-3-{3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]анилино}-5-[(оксан-4-ил)амино]пиразин-2-карбоксамида или его фармацевтически приемлемой соли, сукралозы и по меньшей мере одного из сахаров и/или сахарных спиртов; и
(3) распыление или добавление связующей жидкости, полученной на стадии (1), к смеси, полученной на стадии (2), для образования гранул;
где фармацевтическая композиция представляет собой таблетку.
| US 20180185359 A1, 05.07.2018 | |||
| Способ уничтожения насекомых | 1933 |
|
SU31697A1 |
| US 20180110763 A1, 26.04.2018 | |||
| US 20150141517 A1, 21.05.2015 | |||
| Gerad K Bolhuis et al | |||
| Polyols as filler-binders for disintegrating tablets prepared by direct compaction / Journal Drug Development and Industrial Pharmacy, 2009, V | |||
| Скоропечатный станок для печатания со стеклянных пластинок | 1922 |
|
SU35A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Электрический терморегулятор для термостатов | 1924 |
|
SU671A1 |
| US 20170105988 A1, 20.04.2017. | |||

