ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
[0001]
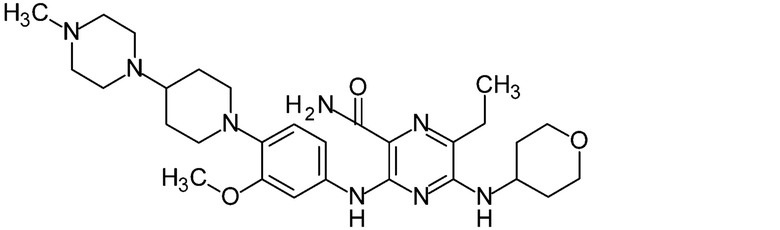
Настоящее изобретение относится к стабильной фармацевтической композиции для перорального введения, содержащей 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид, или его фармацевтически приемлемую соль.
УРОВЕНЬ ТЕХНИКИ
[0002]
6-Этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид (в дальнейшем в данном документе называемый соединением A в некоторых случаях) представляет собой соединение, представленное следующей химической структурной формулой. Имеются сообщения, что соединение A или его фармацевтически приемлемая соль обладает, например, ингибирующей активностью в отношении киназной активности белка слияния EML4 (ассоциированный с микротрубочками белок иглокожих, тип 4)-ALK (киназа анапластической лимфомы), и является полезным(-ой) в качестве активного ингредиента фармацевтической композиции для лечения рака (Документ Патентной литературы 1).
[0003]
[Химическое соединение 1]
[0004]
С точки зрения безопасности для пациентов, желательно, чтобы во время хранения составов вырабатывание сопутствующих веществ ингибировалось. Например, Министерство здравоохранения, труда и социального обеспечения в Японии опубликовало спецификацию для лекарственных продуктов, а именно, группу сопутствующих веществ (примесей) в лекарственных продуктах, отмечаемую в ходе проведения испытаний стабильности (Бюро по безопасности фармацевтических препаратов и пищевых продуктов, Регистрация отделения по оценке и лицензированию № 0624001 "Revision of the Guideline on the Impurities in the Medicinal Products with New Active Ingredients"). В соответствии с пересмотренным руководством, например, в том случае, когда количество лекарственного вещества, которое должно быть введено за сутки, составляет от 10 мг до 100 мг, порог для сопутствующих веществ, требующий квалификации по безопасности, в лекарственном продукте составляет менее либо 0,5% с учетом процентного содержания сопутствующих веществ, содержащихся в лекарственном средстве, либо 200 мкг с учетом общего суточного потребления сопутствующих веществ. Таким образом, полезно предоставить стабильный состав, содержащий Соединение A или его фармацевтически приемлемую соль, в котором ингибируется вырабатывание сопутствующих веществ во время хранения.
СПИСОК ПРОТИВОПОСТАВЛЕННЫХ МАТЕРИАЛОВ
ПАТЕНТНАЯ ЛИТЕРАТУРА
[0005]
[Документ Патентной литературы 1] WO 2010/128659
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
ТЕХНИЧЕСКАЯ ЗАДАЧА
[0006]
Задачей настоящего изобретения является предоставление стабильной фармацевтической композиции для перорального введения, содержащей Соединение А или его фармацевтически приемлемую соль, где ингибируется вырабатывание сопутствующих веществ во время хранения.
РЕШЕНИЕ ЗАДАЧИ
[0007]
Гемифумарат Соединения A как таковой является стабильным в теплой и влажной среде, и увеличение сопутствующих веществ не отмечается при некоторых условиях хранения лекарственных препаратов, как например, в условиях испытания по определению порогового значения уровня серьезности, или тому подобных условиях. Однако, при получении фармацевтической композиции Сравнительного Примера 1, описываемой ниже, в соответствии с некоторым вариантом осуществления способа влажной грануляции, способа грануляции с большим усилием сдвига, путем грануляции гемифумарата Соединения A вместе с микрокристаллической целлюлозой и тому подобным, которая(-ое) не обуславливает несовместимость с гемифумаратом Соединения A, использования воды, и сушки гранулированного продукта с изготовлением состава, обнаружено, что неожиданно увеличивается количество сопутствующих веществ. Для того, чтобы ингибировать вырабатывание сопутствующих веществ Соединения A во время хранения, авторы изобретения провели глубокое изучение, и в итоге обнаружили, что вырабатывание сопутствующих веществ Соединения А может быть ингибировано путем ингибирования снижения доли кристаллов гемифумарата Соединения A во время стадии приготовления состава, и завершили тем самым настоящее изобретение.
[0008]
Настоящее изобретение предоставляет:
[1] стабильную фармацевтическую композицию для перорального введения, содержащую 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид или его фармацевтически приемлемую соль, где доля кристаллов 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида или его фармацевтически приемлемой соли составляет 60% или более относительно общего количества 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида или его фармацевтически приемлемой соли;
[2] фармацевтическую композицию для перорального введения по пункту [1], где процентное содержание сопутствующего вещества 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида составляет 0,20% или менее, после хранения фармацевтической композиции для перорального введения в открытом режиме при 40°C и относительной влажности 75% в течение 1 месяца;
[3] фармацевтическую композицию для перорального введения по пункту [1] или [2], дополнительно содержащую фармацевтическую добавку, способную регулировать содержание воды в составе;
[4] фармацевтическую композицию для перорального введения по пункту [3], где фармацевтическая добавка, способная регулировать содержание воды в составе, представляет собой сахара и/или сахарные спирты;
[5] фармацевтическую композицию для перорального введения по пункту [4], где сахара и/или сахарные спирты представляют собой лактозу и/или D-маннит;
[6] фармацевтическую композицию для перорального введения по любому из пунктов [3]-[5], где содержание фармацевтической добавки, способной регулировать содержание воды в составе, составляет от 20% по массе до 90% по массе относительно общей массы фармацевтической композиции для перорального введения;
[7] способ изготовления стабильной фармацевтической композиции для перорального введения, где упомянутый способ включает в себя:
(1) смешение 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида или его фармацевтически приемлемой соли с фармацевтической добавкой, способной регулировать содержание воды в составе,
(2) гранулирование смеси с тем, чтобы доля кристаллов 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида или его фармацевтически приемлемой соли составляла 60% или более относительно общего количества 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида или его фармацевтически приемлемой соли, и
(3) формование компрессионным прессованием гранулированного продукта;
[8] способ изготовления фармацевтической композиции для перорального введения по пункту [7], где грануляцию проводят при содержании воды в гранулированном продукте, составляющем 30% или менее;
[9] способ изготовления фармацевтической композиции для перорального введения по пункту [7] или [8], где фармацевтическая добавка, способная регулировать содержание воды в составе, представляет собой сахара и/или сахарные спирты;
[10] способ стабилизирования 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида или его фармацевтически приемлемой соли, в стабильной фармацевтической композиции для перорального введения, содержащей 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид или его фармацевтически приемлемую соль, путем доведения доли кристаллов 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида или его фармацевтически приемлемой соли относительно общего количества 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида или его фармацевтически приемлемой соли до 60% или более, и/или путем добавления фармацевтической добавки, способной регулировать содержание воды в составе;
[11] применение фармацевтической добавки, способной регулировать содержание воды в составе, в изготовлении стабильной фармацевтической композиции для перорального введения, содержащей 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид или его фармацевтически приемлемую соль;
[12] стабильную фармацевтическую композицию для перорального введения, содержащую 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид или его фармацевтически приемлемую соль, и лактозу и/или D-маннит; и
[13] стабильную фармацевтическую композицию для перорального введения, содержащую 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид или его фармацевтически приемлемую соль, и D-маннит.
ПРЕИМУЩЕСТВЕННЫЕ ЭФФЕКТЫ ИЗОБРЕТЕНИЯ
[0009]
Согласно настоящему изобретению, может быть предоставлена стабильная фармацевтическая композиция для перорального введения, содержащая Соединение A или его фармацевтически приемлемую соль, где ингибируется вырабатывание сопутствующих веществ во время хранения.
ОПИСАНИЕ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
[0010]
Термин ʺстабильныйʺ в контексте данного документа означает наличие стабильности в условиях воздействия, например, нагрева, света, температуры, и/или влажности. Например, после выстаивания фармацевтической композиции для перорального введения в условиях, приведенных ниже, это определяют как вариант осуществления, в котором сопутствующие вещества Соединения A, содержащиеся в фармацевтической композиции для перорального введения, имеют заданное процентное содержание или менее. Например, после выстаивания фармацевтической композиции для перорального введения при 70°C в течение 9 дней, при 40°C и относительной влажности 75% (в дальнейшем в данном документе относительную влажность X% иногда приводят в сокращенном виде как отн.вл. X%) в течение 6 месяцев в некотором варианте осуществления, при 40°C и отн.вл. 75% в течение 3 месяцев в некотором варианте осуществления, при 40°C и отн.вл. 75% в течение 1 месяца в некотором варианте осуществления, при 25°C и отн.вл. 60% в течение 12 месяцев в некотором варианте осуществления, при 25°C и отн.вл. 60% в течение 6 месяцев в некотором варианте осуществления, при 25°C и отн.вл. 60% в течение 3 месяцев в некотором варианте осуществления, и при 25°C и отн.вл. 60% в течение 1 месяца в некотором варианте осуществления, это определяют как процентное содержание сопутствующих веществ Соединения A, содержащихся в фармацевтической композиции для перорального введения, измеряемое методом высокоэффективной жидкостной хроматографии (в дальнейшем в данном документе иногда сокращенно называемым как метод HPLC), составляющее, например, 0,50% или менее, 0,20% или менее в некотором варианте осуществления, и 0,10% или менее в некотором варианте осуществления. В некотором варианте осуществления, после выстаивания фармацевтической композиции для перорального введения в открытом режиме при 40°C и относительной влажности 75% в течение 1 месяца, 3 месяцев, или 6 месяцев, это определяют как процентное содержание сопутствующих веществ Соединения А, содержащихся в фармацевтической композиции для перорального введения, измеряемое методом HPLC, составляющее, например, 0,20% или менее, и 0,10% или менее в некотором варианте осуществления.
[0011]
Термин ʺсопутствующее вещество Соединения Aʺ определяется как, например, продукт окислительного разложения Соединения A, и в некотором варианте осуществления, как вещество, имеющее относительное время удерживания приблизительно 1,06 по отношению к пику, соответствующему Соединению A, которое измеряют методом HPLC, описываемым ниже. В связи с этим, сопутствующее вещество, имеющее относительное время удерживания, равное приблизительно 1,06, по отношению к пику Соединения А, как полагают, представляет собой продукт окислительного разложения Соединения A. Используемые численные значения интерпретируют как большие значения переменной, как правило, в пределах ошибки эксперимента (например, в пределах доверительного интервала при доверительной вероятности 95% для среднего значения), или в пределах ±10% относительно указанного значения, и все значения переменной.
[0012]
ʺДоля кристалловʺ Соединения A или его фармацевтически приемлемой соли определяется как доля кристаллов относительно общего количества Соединения A или его фармацевтически приемлемой соли, и может быть вычислена с применением спектроскопии в ближней инфракрасной области спектра (NIR), как описано ниже, или тому подобного.
[0013]
Термин ʺпотеря в массе при высушиванииʺ в контексте данного документа означает количество влаги, которое содержится в образце и теряется в результате высушивания. Потеря в массе при высушивании может быть вычислена, например, с помощью следующего уравнения:
Потеря в массе при высушивании (%)=(снижение веса (массы) в результате высушивания /вес (масса) образца в начале измерения потери в массе при высушивании) × 100
Конкретнее, потеря в массе при высушивании может быть вычислена с помощью следующего уравнения:
Потеря в массе при высушивании (%)=[(вес (масса) образца в начале измерения потери в массе при высушивании - вес (масса) образца в конце измерения потери в массе при высушивании)/(вес (масса) образца в начале измерения потери в массе при высушивании)] × 100
[0014]
Соединение A или его фармацевтически приемлемая соль, которое(-ую) используют в настоящем изобретении, является легко доступным(-ой) для приобретения, например, в результате применения способа, описанного в Документе Патентной литературы 1, или подобного тому способа.
[0015]
Соединение A может находиться в свободной форме, которая не образует соль, и может образовывать фармацевтически приемлемую соль с кислотой. Примеры такой соли включают соль присоединения кислоты, получаемую посредством неорганической кислоты, такой как соляная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, или тому подобное; и соль присоединения кислоты, получаемую посредством органической кислоты, такой как муравьиная кислота, уксусная кислота, пропионовая кислота, щавелевая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, гемифумаровая кислота, малеиновая кислота, молочная кислота, яблочная кислота, лимонная кислота, винная кислота, карбоновая кислота (углекислота), пикриновая кислота, метансульфоновая кислота, этансульфоновая кислота, глутаминовая кислота, или тому подобное. Эти соли могут быть получены обычно применяемыми способами. Гемифумарат может быть приведен в качестве примера в некотором одном варианте осуществления.
[0016]
Соединение A или его фармацевтически приемлемая соль проявляет, например, ингибирующую активность в отношении киназной активности белка слияния EML4-ALK, и является полезным(-ой) в качестве активного ингредиента фармацевтической композиции для лечения рака.
[0017]
Доза Соединения A или его фармацевтически приемлемой соли может быть надлежащим образом определена в зависимости от индивидуальных особенностей, принимаемых во внимание, например, симптомов, возраста пациента, пола, или тому подобного.
В случае обычного перорального введения, суточная дозировка для взрослого человека составляет подходящим образом от 0,001 мг/кг или более до 100 мг/кг или менее, предпочтительно от 0,005 мг/кг до 30 мг/кг, и более предпочтительно от 0,01 мг/кг до 10 мг/кг. Это вводят одной дозой, или делят на две - четыре дозы в день.
[0018]
Содержание Соединения A или его фармацевтически приемлемой соли составляет, например, относительно массы фармацевтической композиции для перорального введения, от 1% по массе или более до 70% массе или менее, от 5% по массе или более до 50% по массе или менее в некотором варианте осуществления, и от 10% по массе или более до 40% по массе или менее в некотором варианте осуществления. Количество, включающее в себя Соединение А или его фармацевтически приемлемую соль, составляет, в целом составе, от 1 мг или более до 200 мг или менее, от 5 мг или более до 150 мг или менее в некотором варианте осуществления, и от 40 мг или более до 50 мг или менее в некотором варианте осуществления.
[0019]
Доля кристаллов Соединения А или его фармацевтически приемлемой соли, которая используется в настоящем изобретении, особым образом не ограничивается, если она находится в пределах диапазона, где Соединение А или его фармацевтически приемлемая соль является стабильным(-ой) во время хранения. Доля кристаллов может быть вычислена, например, с использованием анализа методом дифференциальной сканирующей калориметрии (анализ методом DSC), методом порошковой рентгеновской дифракции, методом ЯМР твердого тела, методом спектроскопии в ближней инфракрасной области спектра (NIR), или тому подобного.
[0020]
В качестве способа вычисления доли кристаллов гемифумарата Соединения A в гемифумарате Соединения A, например, измеряют спектр, в случае измерения методом спектроскопии в ближней инфракрасной области спектра, с помощью Фурье-спектрометра ближнего инфракрасного диапазона (MPA, Bruker Optics K.K.)(диапазон измерений; от 12500 см-1 до 5800 см-1, разрешение; 8 см-1, число сканирований; 32), и полученный спектр преобразуют с получением второй производной (метод свертки Савицкого-Голея), и анализируют с помощью программного обеспечения для анализа спектра в ближней инфракрасной области (например, OPUS, Bruker Optics K.K.). Фармацевтическую композицию для перорального введения превращают в порошок с помощью ступки и пестика для измерения спектра. Перед спектральным измерением фармацевтической композиции для перорального введения, спектры препаратов, в которых кристаллы гемифумарата Соединения А смешаны в различных долях, подвергают регрессионному анализу методом дробных наименьших квадратов с получением калибровочной кривой, и каждый спектр, полученный для фармацевтической композиции для перорального введения, интерполируют в калибровочную кривую с вычислением доли кристаллов гемифумарата Соединения A.
[0021]
Доля кристаллов относительно общего количества Соединения А или его фармацевтически приемлемой соли составляет, например, 60% или более, от 60% или более до 100% или менее в некотором варианте осуществления, от 70% или более до 100% или менее в некотором варианте осуществления, от 80% или более до 100% или менее в некотором варианте осуществления, от 90% или более до 100% или менее в некотором варианте осуществления, от 60% или более до менее 100% в некотором варианте осуществления, от 70% или более до менее 100% в некотором варианте осуществления, от 80% или более до менее 100% в некотором варианте осуществления, и от 90% или более до менее 100% в некотором варианте осуществления. В соответствии с этим, используемые численные значения определяют как большее значение переменной, как правило, в пределах экспериментальной ошибки (например, в пределах доверительного интервала при доверительной вероятности 95% для среднего значения), или в пределах ±10% относительно указанного значения, и все значения переменной.
[0022]
Фармацевтическая композиция для перорального введения по настоящему изобретению может дополнительно содержать фармацевтическую добавку, способную регулировать содержание воды во время стадии приготовления состава и/или его хранения (в дальнейшем в данном документе иногда называемую как фармацевтическая добавка, способная регулировать содержание воды в составе). Фармацевтическая добавка, способная регулировать содержание воды в составе, особым образом не ограничивается, если добавка сама по себе проявляет потерю в массе при высушивании, способную поддерживать композицию, содержащую Соединение А или его фармацевтически приемлемую соль, в стабильном состоянии; или стабильная фармацевтическая композиция для перорального введения, содержащая Соединение А или его фармацевтически приемлемую соль, может быть предоставлена путем удерживания содержания воды в композиции, содержащей Соединение А или его фармацевтически приемлемую соль, во время стадии приготовления состава (в частности, стадии грануляции) на низком уровне, или путем дополнительного снижения содержания воды в составе и поддерживания содержания воды. Примеры добавки включают сахара и/или сахарные спирты, и добавка представляет собой D-маннит, мальтозу, мальтит, эритрит, ксилит, лактозу (гидрат лактозы), сахарозу, глюкозу, сорбит, трегалозу, лактит, фруктозу, арабинозу, или трегалозу в некотором варианте осуществления, лактозу (гидрат лактозы) или D-маннит в некотором варианте осуществления, и D-маннит в некотором варианте осуществления.
[0023]
Потеря в массе при высушивании фармацевтической добавки, способной регулировать содержание воды в составе, может быть измерена, например, аналогично измерению в Испытании по оцениванию потери в массе при высушивании, которое определено в the General Tests of The Japanese Pharmacopoeia, Sixteenth Edition. В некотором варианте осуществления, потеря в массе при высушивании может быть измерена в результате обеспечения выстаивания фармацевтической добавки в заранее заданных условиях температуры и влажности с тем, чтобы увлажнить ее до достижения постоянного значения веса (массы), и затем высушивания ее в условиях заранее заданных температуры и влажности до тех пор, пока вес (масса) не достигнет постоянного своего значения. В некотором варианте осуществления, потеря в массе при высушивании фармацевтической добавки, измеряемая путем размещения фармацевтической добавки в бутылке, обеспечения выстаивания бутылки в открытом режиме при 40°C и отн.вл. 75% в течение 1 недели, и измерения потери в массе при высушивании после хранения в соответствии с испытанием по оцениванию потери в массе при высушивании (например, галогенный анализатор влажности HR73 (изготовленный в METTLER TOLEDO) используют в качестве прибора, и измерение выполняют до тех пор, пока масса образца не достигнет постоянного значения при 80°C.), составляет, например, 20% или менее, 1,0% или менее в некотором варианте осуществления, 0,5% или менее в некотором варианте осуществления, и 0,4% или менее в некотором варианте осуществления.
[0024]
Потеря в массе при высушивании фармацевтической композиции (например, таблетки) может быть измерена, например, аналогично измерениям в соответствии с Испытанием по оцениванию потери в массе при высушивании, которое определено в the General Tests of The Japanese Pharmacopoeia, Sixteenth Edition. В некотором варианте осуществления, потеря в массе при высушивании может быть измерена в результате обеспечения выстаивания фармацевтической композиции (например, таблетки) в заранее заданных условиях температуры и влажности с тем, чтобы увлажнить ее до достижения постоянного значения веса (массы), и затем высушивания ее в условиях заранее заданных температуры и влажности до тех пор, пока вес (масса) не достигнет постоянного своего значения. В некотором варианте осуществления, потеря в массе при высушивании фармацевтической композиции (например, таблетки), измеряемая путем размещения фармацевтической композиции (например, таблетки) в бутылке, обеспечения выстаивания бутылки в открытом режиме при 40°C и отн.вл. 75% в течение 1 недели, и измерения потери в массе при высушивании после хранения в соответствии с испытанием по оцениванию потери в массе при высушивании (например, галогенный анализатор влажности HR73 (изготовленный в METTLER TOLEDO) используют в качестве прибора, и измерение выполняют до тех пор, пока масса образца не достигнет постоянного значения при 80°C.), составляет, например, 4,0% или менее, 3,0% или менее в некотором варианте осуществления, и 2,0% или менее в некотором варианте осуществления.
[0025] Фармацевтическая добавка, способная регулировать содержание воды в составе может быть надлежащим образом добавлена как таковая, или в виде комбинации двух или более добавок, в соответственных количествах.
Содержание составляет, относительно общей массы фармацевтической композиции для перорального введения, например, от 20% по массе или более до 90% по массе или менее, от 30% по массе или более до 80% по массе или менее в некотором варианте осуществления, от 40% по массе или более до 70% по массе или менее в некотором варианте осуществления, от 50% по массе или более до 70% по массе или менее в некотором варианте осуществления, и от 50% по массе или более до 60% по массе или менее в некотором варианте осуществления.
[0026]
Фармацевтическая композиция для перорального введения по настоящему изобретению может иметь различные составы, такие как таблетки, капсулы, порошки, гранулы, мелкодисперсные гранулы, сухие сиропы, или тому подобное. Она представляет собой таблетку или капсулу в одном варианте осуществления, и таблетку в некотором другом варианте осуществления.
В фармацевтической композиции для перорального введения по настоящему изобретению, различные фармацевтические добавки, такие как связующие, агенты для улучшения распадаемости таблеток, корригирующие вещества, шипучие агенты, подсластители, ароматизирующие вещества, скользящие вещества, буферные растворы, антиоксиданты, стабилизаторы, поверхностно-активные вещества, агенты для нанесения пленочной оболочки, и тому подобное, могут быть надлежащим образом использованы, при желании, в той степени, чтобы могли быть достигнуты эффекты настоящего изобретения.
[0027]
Примеры связующих включают аравийскую камедь, гипромеллозу, гидроксипропилцеллюлозу, гидроксиэтилцеллюлозу, и тому подобное.
[0028]
Примеры агентов для улучшения распадаемости таблеток включают кукурузный крахмал, картофельный крахмал, кальциевую соль кармеллозы, натриевую соль кармеллозы, гидроксипропилцеллюлозу с низкой степенью замещения, и тому подобное.
[0029]
Примеры корригирующих веществ включают лимонную кислоту, винную кислоту, яблочную кислоту, и тому подобное.
[0030]
Примеры шипучих агентов включают бикарбонат натрия, и тому подобное.
[0031]
Примеры подсластителей включают сахарин натрия, глицирризиновую кислоту, аспартам, экстракт листьев медовой травы стевии, тауматин, и тому подобное.
[0032]
Примеры ароматизирующих веществ включают сок лимона, сок лимона и лайма, сок апельсина, ментол, и тому подобное.
[0033]
Примеры скользящих веществ включают стеарат магния, стеарат кальция, и тому подобное.
[0034]
Примеры буферных растворов включают лимонную кислоту янтарную кислоту, фумаровую кислоту винную кислоту, аскорбиновую кислоту, и их соли; глутаминовую кислоту, глутамин, глицин, аспарагиновую кислоту, аланин, аргинин, и их соли; оксид магния, оксид цинка, гидроксид магния, фосфорную кислоту, борную кислоту, и их соли; и тому подобное.
[0035]
Примеры антиоксидантов включают лимонную кислоту, нитрит натрия, аскорбиновую кислоту, сложный эфир L-аскорбиновй кислоты и стеариновой кислоты, гидронитрит натрия, сульфит натрия, α-тиоглицерин, эдетат натрия, эриторбиновую кислоту, гидрохлорид цистеина, обезвоженный сульфит натрия, дихлоризоцианурат калия, соевый лецитин, тиогликолят натрия, тиомалат натрия, натуральный витамин E, токоферол, d-δ-токоферол, сложный эфир токоферола и уксусной кислоты, смешанный концентрат токоферолов, сложный эфир аскорбиновой кислоты и пальмитиновой кислоты, пиросульфит натрия, бутилгидроксианизол, 1,3-бутиленгликоль, бензотриазол, пентаэритритил-тетракис[3-(3,5-ди-трет-бутил-4-гидроксифенил)пропионат], 2-меркаптобензимидазол, пропилгаллат, дибутилгидрокситолуол, и тому подобное.
[0036]
Антиоксиданты также действуют в качестве стабилизаторов. Примеры стабилизаторов включают лимонную кислоту; и гидрат лимонной кислоты, цитрат кальция, гидрат цитрата натрия, дигидроцитрат натрия, цитрат динатрия, и тому подобное в некотором варианте осуществления.
[0037]
Примеры поверхностно-активных веществ включают полисорбат 80, лаурилсульфат натрия, полиоксиэтиленовое гидрогенизированное касторовое масло, и тому подобное.
[0038]
Примеры агентов для нанесения пленочной оболочки включают гипромеллозу, поливиниловый спирт, и тому подобное.
[0039]
Эти фармацевтические добавки могут быть должным образом добавлены как таковые, или в виде комбинации двух или более добавок, в соответственных количествах. Что касается содержаний фармацевтических добавок, то каждая фармацевтическая добавка может быть использована в количестве, при котором могут быть достигнуты желательные эффекты настоящего изобретения.
[0040]
Фармацевтическая композиция для перорального введения по настоящему изобретению может быть получена известными способами, включающими в себя стадии, например, пульверизации, смешения, грануляции, сушки, формования (таблетирования), нанесения пленочного покрытия, кристаллизации, и тому подобного. Способ изготовления фармацевтической композиции для перорального введения по настоящему изобретению будет разъяснен ниже.
[0041]
Стадия пульверизации и стадия смешения
На стадии пульверизации, как установка, так и средство особым образом не ограничиваются, если они обеспечивают способ, в котором Соединение A или его фармацевтически приемлемая соль и соответственные фармацевтические добавки могут быть подвергнуты пульверизации обычным для фармацевтики образом. Примеры пульверизатора включают молотковую дробилку, шаровую мельницу, струйную мельницу, коллоидную мельницу, и тому подобное. Условия для пульверизации могут быть соответственно подобраны и особым образом не ограничены.
На стадии смешения компонентов, следующей за стадией пульверизации, как установка, так и средство особым образом не ограничиваются, если они обеспечивают способ, в котором компоненты могут быть равномерно смешаны обычным для фармацевтики способом.
[0042]
Стадия грануляции
На стадии грануляции, как установка, так и средство особым образом не ограничиваются, если они обеспечивают способ, в котором Соединение A или его фармацевтически приемлемая соль и соответственные фармацевтические добавки могут быть гранулированы обычным для фармацевтики образом.
Примеры способа грануляции и установки для грануляции, которые применяют во влажной грануляции с использованием растворителя, такого как вода, включают в себя способ грануляции с большим усилием сдвига, способ грануляции измельчением (пульверизацией), способ грануляции в псевдоожиженном слое, способ грануляции экструзией, способ грануляции обработкой в барабане, и способ грануляции распылением; и установки и тому подобное, которые используются в этих способах. Способ грануляции в псевдоожиженном слое и гранулятор с псевдоожиженным слоем являются предпочтительными, и способ сушки особым образом не ограничивается, если сушка может быть осуществлена обычным для фармацевтики образом.
[0043]
Во время грануляции, предпочтительно, чтобы содержание воды являлось низким, с тем, чтобы ингибировать снижение доли кристаллов Соединения А или его фармацевтически приемлемой соли. Содержание воды во время грануляции составляет, например, 30% или менее, 5% или менее в одном варианте осуществления, 3% или менее в некотором варианте осуществления, 2% или менее в одном варианте осуществления, и 1% или менее в одном варианте осуществления. Способ грануляции особым образом не ограничивается, если содержание воды может быть отрегулировано в рамках диапазона. Примеры такого способа грануляции включают способ грануляции измельчением (пульверизацией), способ грануляции в псевдоожиженном слое, способ грануляции обработкой в барабане, и способ грануляции распылением; и способ грануляции в псевдоожиженном слое в одном варианте осуществления.
Содержание воды может быть измерено, например, способом оценивания потери в массе при высушивании, или тому подобным способом. В качестве прибора, может быть использован, например, галогенный анализатор влажности (METTLER TOLEDO).
Способ грануляции с большим усилием сдвига может быть выбран в том случае, когда используют условия, способные снизить содержание воды в гранулах во время грануляции.
В качестве способа без использования воды во время грануляции, также может быть применен способ влажной грануляции с использованием неводного растворителя, или способ сухой грануляции.
[0044]
Стадия сушки
На стадии сушки как установка, так и средство особым образом не ограничиваются, если имеет место способ, в котором гранулированный продукт может быть подвергнут сушке обычным для фармацевтики образом. Примеры установки включают сушильную камеру с принудительной подачей воздуха, сушильную камеру, работающую под сниженным давлением, вакуумный сушильный шкаф, грануляционную сушильную установку с псевдоожиженным слоем, и тому подобное.
После сушки высушенный продукт может быть просеян через сито и отсортирован по размеру с помощью сита, конусной мельницы, или тому подобного, если желательно.
[0045]
Стадия формования
На стадии формования, как установка, так и средство особым образом не ограничиваются, если обеспечивается способ формования фармацевтической композиции для перорального введения по настоящему изобретению. Примеры способа включают способ, в котором, в отсутствии стадии грануляции и сушки, Соединение A или его фармацевтически приемлемую соль и подходящие фармацевтические добавки смешивают, и напрямую формуют компрессионным прессованием с получением фармацевтической композиции для перорального введения; способ, в котором Соединение A или его фармацевтически приемлемую соль и подходящие фармацевтические добавки подвергают грануляции и сушке, и формуют компрессионным прессованием с получением фармацевтической композиции для перорального введения; способ, в котором Соединение A или его фармацевтически приемлемую соль и надлежащие фармацевтические добавки подвергают грануляции, и дополнительно смешивают со скользящим веществом, и эту смесь формуют компрессионным прессованием с получением фармацевтической композиции для перорального введения; и тому подобное.
Примеры таблеточной машины включают роторную таблеточную машину, масляный пресс, и тому подобное.
Условия для таблетирования, такие как давление таблетирования, особым образом не ограничиваются, если оно соответствует давлению таблетирования, которое может обеспечивать формование компрессионным прессованием.
Твердость таблетированного продукта особым образом не ограничивается, если он не претерпевает повреждения во время процесса изготовления, процесса распределения, и тому подобного. Твердость может иметь значение, например, от 40 до 200 Н.
[0046]
Стадия нанесения пленочной оболочки
После таблетирования, поверхность фармацевтической композиции для перорального введения может быть покрыта пленочной оболочкой.
Способ нанесения пленочной оболочки особым образом не ограничивается, если она может быть нанесена обычным для фармацевтики образом. Примеры нанесения оболочки на таблетку включают дражирование (нанесение оболочки на таблетку в дражировочном котле), нанесение оболочки на таблетку погружением, и тому подобное.
Агент для нанесения пленочной оболочки может быть подходящим образом добавлен как таковой, или в виде комбинации двух или более агентов, в соответственных количествах.
Норма нанесения оболочки на таблетку особым образом не ограничивается, если может быть образована пленка. Норма нанесения оболочки на таблетку составляет, например, относительно общей массы фармацевтической композиции для перорального введения, от 1% по массе до 10% по массе, или тому подобное.
Во время нанесения пленочной оболочки или после нанесения пленочной оболочки, продукт с нанесенной оболочкой может быть подвергнут сушке. Способ сушки особым образом не ограничивается, если сушка может быть осуществлена обычным для фармацевтики образом. Условия для сушки особым образом не ограничиваются, если они надлежащим образом подбираются с учетом, например, стабильности фармацевтической композиции для перорального введения.
[0047]
Стадия кристаллизации
При снижении доли кристаллов Соединения A или его фармацевтически приемлемой соли, может быть введена стадия промотирования кристаллизации. Примеры этой стадии включают обработку микроволновым (сверхвысокочастотным) излучением, обработку ультразвуковым излучением, обработку низкочастотным излучением, обработку излучением тепловых электронов, и тому подобное.
В качестве обработки микроволновым излучением может быть использовано излучение, например, с длиной волны в диапазоне от 10 МГц до 25 ГГц. Хотя продолжительность обработки зависит от величины исходной доли кристаллов, или компонентов фармацевтической добавки, ее проводят, например, в течение 10 секунд - 60 минут. Облучение может быть выполнено в непрерывном или прерывистом режиме, и в произвольный момент времени.
В качестве обработки ультразвуковым излучением может быть использовано излучение, например, звуковых волн с частотой от 10 кГц до 600 кГц. Хотя продолжительность обработки зависит от величины доли кристаллов, или компонентов фармацевтической добавки, ее проводят, например, в течение 10 секунд - 24 часов. Облучение может быть выполнено в непрерывном или прерывистом режиме, и в произвольный момент времени.
[0048]
Настоящее изобретение включает в себя способ стабилизирования Соединения А или его фармацевтически приемлемой соли путем обеспечения конкретной доли кристаллов Соединения А или его фармацевтически приемлемой соли, и/или обеспечения фармацевтической добавки, способной регулировать содержание воды в составе.
Настоящее изобретение включает в себя применение фармацевтической добавки, способной регулировать содержание воды в составе, в изготовлении стабильной фармацевтической композиции для перорального введения, содержащей Соединение A или его фармацевтически приемлемую соль.
В отношении ʺкристаллов Соединения A или его фармацевтически приемлемой солиʺ, ʺфармацевтической добавки, способной регулировать содержание воды в составеʺ и ʺСоединения A или его фармацевтически приемлемой солиʺ, которые используются в способе стабилизирования по настоящему изобретению, и в применении фармацевтической добавки, способной регулировать содержание воды в составе по настоящему изобретению, могут быть напрямую применены разъяснения этого, описанные в отношении фармацевтической композиции для перорального введения по настоящему изобретению.
В отношении содержания каждого компонента, способа их смешивания, и тому подобного в способе стабилизирования по настоящему изобретению, и в применении фармацевтической добавки, способной регулировать содержание воды в составе по настоящему изобретению, могут быть напрямую применены разъяснения этого, описанные в отношении фармацевтической композиции для перорального введения по настоящему изобретению и способа получения таковой.
ПРИМЕРЫ
[0049]
Ниже настоящее изобретение будет дополнительно проиллюстрировано с помощью следующих Сравнительных Примеров, Примеров, и Экспериментальных Примеров, но ни в какой мере не будет ограничено этим.
[0050]
<<Сравнительный Пример 1 и Примеры 1-3>>
Составы Сравнительного Примера 1 и Примеров 1-3 показаны в Таблицах 1 и 2. Гемифумарат Соединения A, который используют ниже, получен согласно способу, описанному в международной публикации WO 2010/128659, или аналогично тому.
[0051]
[Таблица 1]
Единица измерения: мг
[0052]
[Таблица 2]
Единица измерения: мг
[0053]
Pharmatose 200M (название продукта, изготовленного в FrieslandCampina DMV BV) используют в качестве гидрата лактозы, HPC-L (название продукта, изготовленного в Nippon Soda Co., Ltd.) используют в качестве гидроксипропилцеллюлозы, Parteck LUB MST (название продукта, изготовленного в Merck KGaA) используют в качестве стеарата магния, и PEARLITOL 5°C (название продукта, изготовленного в ROQUETTE) используют в качестве D-маннита.
[0054]
<<Сравнительный Пример 1>>
В соответствии с составом, описанным в Таблице 1, 110,5 г гемифумарата Соединения А, 537,5 г гидрата лактозы, 45 г микрокристаллической целлюлозы (название продукта: Ceolus PH-101, изготовленного в Asahi Kasei Chemicals Corporation), 90 г гидроксипропилцеллюлозы с низкой степенью замещения (название продукта: L-HPC LH-21, изготовленного в Shin-Etsu Chemical Co., Ltd.), и 18 г гидроксипропилцеллюлозы смешивают с помощью гранулятора с большим усилием сдвига (название изделия: VG-05, изготовленного в Powrex Corporation), и к этому дополнительно добавляют 300 г очищенной воды, и эту смесь подвергают грануляции. Содержание воды в гранулированном продукте во время грануляции составляет 27%. Проводят две дополнительные серии грануляции, и в результате сушки в течение 15 часов с помощью вакуумного сушильного шкафа получают гранулированный продукт (название продукта: DB-30, изготовленного в ULVAC, Inc.). После просеивания 2403 г полученного гранулированного продукта, к тому добавляют 135 г микрокристаллической целлюлозы (название продукта: Ceolus PH-102, изготовленного в Asahi Kasei Chemicals Corporation), 135 г гидроксипропилцеллюлозы с низкой степенью замещения (название продукта: L-HPC LH-11, изготовленного в Shin-Etsu Chemical Co., Ltd.), и 27 г стеарата магния, и смешивают с помощью смесителя (название изделия: Контейнерный Смеситель LM20, изготовленный в Kotobuki Industries Co., Ltd.) с получением смешанного продукта (гранулы для компрессионного прессования для получения таблеток). Полученному смешанному продукту придают форму таблеток с помощью роторной таблеточной машины (название изделия: HT-X20, изготовленного в HATA TEKKOSHO Co., Ltd.) с получением таблеток (таблеток без оболочки). На полученные таблетки без оболочки (1350 г) наносят пленочную оболочку с помощью машины для нанесения пленочного покрытия (название изделия: HCT-30, изготовленного в Freund Corporation) с использованием жидкости, приготовленной в результате растворения/диспергирования OPADRY 03F42203 (название продукта, изготовленного в Colorcon) в очищенной воде, так чтобы в итоге концентрация OPADRY 03F42203 составляла 10% по массе (концентрация твердых компонентов). Проводят дополнительную серию нанесения пленочной оболочки, с получением таблеток (таблетки с пленочной оболочкой) Сравнительного Примера 1.
[0055]
<<Пример 1>>
В соответствии с составом, описанным в Таблице 1, 442 г гемифумарата Соединения А, 2150 г гидрата лактозы, 180 г микрокристаллической целлюлозы (название продукта: Ceolus PH-101, изготовлен в Asahi Kasei Chemicals Corporation), 360 г гидроксипропилцеллюлозы с низкой степенью замещения (название продукта: L-HPC LH-21, изготовлен в Shin-Etsu Chemical Co., Ltd.), и 72 г гидроксипропилцеллюлозы смешивают с помощью гранулятора с большим усилием сдвига (название изделия: VG-25, изготовлено в Powrex Corporation), и эту смесь подвергают грануляции при добавлении 1170 г очищенной воды. Содержание воды в гранулированном продукте во время грануляции составляет 27%. Проводят девять дополнительных серий грануляции, и в результате сушки в течение 1 часа с помощью грануляционной сушильной установки с псевдоожиженным слоем получают гранулированный продукт (название продукта: GPCG-PRO-5, изготовлен в Powrex Corporation). После просеивания 32040 г полученного гранулированного продукта, к тому добавляют 1800 г микрокристаллической целлюлозы (название продукта: Ceolus PH-102, изготовлен в Asahi Kasei Chemicals Corporation), 1800 г гидроксипропилцеллюлозы с низкой степенью замещения (название продукта: L-HPC LH-11, изготовлен в Shin-Etsu Chemical Co., Ltd.), и 360 г стеарата магния, и смешивают с помощью смесителя (название изделия: Контейнерный Смеситель PM200, изготовлен в Kotobuki Industries Co., Ltd.) с получением смешанного продукта (гранулы для компрессионного прессования для получения таблеток). Полученному смешанному продукту придают форму таблеток с помощью роторной таблеточной машины (название изделия: HT-CVX-TYPEIII20, изготовлено в HATA TEKKOSHO Co., Ltd.) с получением таблеток (таблеток без оболочки). На полученные таблетки без оболочки (36000 г) наносят пленочную оболочку с помощью машины для нанесения пленочного покрытия (название изделия: PRC-20/60, изготовлено в Powrex Corporation) с использованием жидкости, приготовленной в результате растворения/диспергирования OPADRY 03F42203 (название продукта, изготовленного в Colorcon) в очищенной воде, так чтобы в итоге концентрация OPADRY 03F42203 составляла 10% по массе (концентрация твердых компонентов), с получением таблеток (таблетки с пленочной оболочкой) Примера 1.
[0056]
<<Example 2>>
В соответствии с составом, описанным в Таблице 2, 6630 г гемифумарата Соединения A и 12375 г D-маннита смешивают с помощью грануляционной сушильной установки с псевдоожиженным слоем (название изделия: GPCG-PRO-15, изготовлено в Powrex Corporation). После смешения, эту смесь подвергают грануляции при распылении 9000 г водного раствора гидроксипропилцеллюлозы (содержание твердого вещества: 7% по массе) в качестве связующего, и сушат с получением гранулированного продукта. Содержание воды в гранулированном продукте во время грануляции составляет максимально 0,43%. После просеивания 19635 г полученного гранулированного продукта, к тому добавляют 1050 г гидроксипропилцеллюлозы с низкой степенью замещения (название продукта: L-HPC LH-21, изготовлен в Shin-Etsu Chemical Co., Ltd.) и 315 г стеарата магния, и смешивают с помощью смесителя (название изделия: Контейнерный Смеситель PM200, изготовлен в Kotobuki Industries Co., Ltd.) с получением смешанного продукта (гранулы для компрессионного прессования для получения таблеток). Полученному смешанному продукту придают форму таблеток с помощью роторной таблеточной машины (название изделия: HT-CVX-TYPEIII20, изготовлено в HATA TEKKOSHO Co., Ltd.) с получением таблеток (таблеток без оболочки). На полученные таблетки без оболочки (21000 г) наносят пленочную оболочку с помощью машины для нанесения пленочного покрытия (название изделия: PRC-20/60, изготовлено в Powrex Corporation) с использованием жидкости, приготовленной в результате растворения/диспергирования OPADRY 03F42203 (название продукта, изготовленного в Colorcon) в очищенной воде, так чтобы в итоге концентрация OPADRY 03F42203 составляла 10% по массе (концентрация твердых компонентов), с получением таблеток (таблетки с пленочной оболочкой) Примера 2.
[0057]
<<Пример 3>>
Таблетки (таблетки с пленочной оболочкой) Примера 3 изготавливают аналогично способу Примера 2, в соответствии с составом, описанным в Таблице 2.
[0058]
<<Экспериментальный Пример 1: Вычисление доли кристаллов>>
В отношении таблеток (таблеток с пленочной оболочкой), получаемых в Сравнительном Примере 1, Примере 1, Примере 2, и Примере 3, вычисляют долю кристаллов гемифумарата Соединения А после получения с применением спектроскопии в ближней инфракрасной области спектра.
Конкретнее, измеряют спектр с помощью Фурье-спектрометра ближнего инфракрасного диапазона (название изделия: MPA, Bruker Optics K.K.)(диапазон измерений; от 12500 см-1 до 5800 см-1, разрешение; 8 см-1, число сканирований; 32), и полученный спектр преобразуют с получением второй производной (метод свертки Савицкого-Голея), и анализируют с помощью программного обеспечения для анализа спектра в ближней инфракрасной области (название продукта: OPUS, Bruker Optics K.K.). Таблетки превращают в порошок с помощью ступки и пестика, и спектры измеряют. Перед спектральным измерением таблеток, спектры препаратов, в которых кристаллы гемифумарата Соединения А смешаны в различных долях, подвергают регрессионному анализу методом дробных наименьших квадратов с получением калибровочной кривой, и каждый спектр, полученный для таблеток, интерполируют в калибровочную кривую с вычислением доли кристаллов гемифумарата Соединения A. Результаты показаны в Таблице 4.
[0059]
<<Экспериментальный Пример 2: Измерение сопутствующих веществ>>
Таблетки (таблетки с пленочной оболочкой), получаемые в Сравнительном Примере 1, Примере 1, Примере 2, и Примере 3, помещают в бутылки, и оставляют выстаиваться в открытом режиме при 40°C и отн.вл. 75% в течение 1 месяца и 3 месяцев. Сопутствующие вещества после хранения измеряют методом HPLC. Измерение сопутствующих веществ проводят в следующих условиях:
В качестве колонки для HPLC, используют колонку Kinetex XB-C18, размер частиц: 2,6 мкм, 4,6 мм (внутренний диаметр) × 75 мм (изготовленную в Phenomenex Inc.), или ее эквивалент, и поддерживают при 40°C.
В качестве подвижной фазы A, используют раствор перхлората (pH 2,2), и в качестве подвижной фазы B, используют ацетонитрил.
В качестве растворов образцов, используют образцы, разбавленные смесью раствор перхлората (pH 2,2)/ацетонитрил (=4/1) так, чтобы концентрация соединения A составляла 0,8 мг/мл.
В качестве эталонного раствора, используют эталон, разбавленный смесью раствор перхлората (pH 2,2)/ацетонитрил (=4/1) так, чтобы концентрация соединения A составляла 0,008 мг/мл.
Измерение сопутствующих веществ проводят с помощью спектрофотометра ультрафиолетового поглощения (длина волны: 220 нм), в соответствии с программой градиента, приведенной в Таблице 3 ниже, и процентное содержание каждого сопутствующего вещества вычисляют на основе отношения площади пика для каждого сопутствующего вещества к площади пика для эталонного раствора.
Результаты измерений для сопутствующего вещества, имеющего относительное время удерживания, равное приблизительно 1,06, относительно пика, соответствующего Соединению A, приведены в Таблице 4.
[0060]
[Таблица 3]
[0061]
[Таблица 4]
Открытый режим при 40°С и отн.вл. 75%
LOQ: Предел количественного обнаружения; N.T.: Не испытано
[0062]
Образец Сравнительного Примера 1 изготавливают способом грануляции с большим усилием сдвига, аналогично способу грануляции образца Примера 1, но они отличаются друг от друга величиной доли кристаллов. Полагают, что это имеет место в силу различия продолжительности сушки, обусловленной различными способами сушки.
В таблетке Примера 1, в которой доля кристаллов гемифумарата Соединения А составляет 64%, процентное содержание сопутствующего вещества составляет 0,11% после хранения в открытом режиме при 40°C и отн.вл. 75% в течение 1 месяца, и процентное содержание сопутствующего вещества составляет 0,26% после хранения в течение 3 месяцев. В таблетках Примеров 2 и 3, процентное содержание сопутствующего вещества после хранения в течение 1 месяца имеет меньшую величину, чем предел количественного обнаружения (LOQ). Таблетки этих Примеров содержат небольшое количество сопутствующего вещества, в сравнении с таблеткой Сравнительного Примера, и являются стабильными. Для справочной информации, значение LOQ составляет 0,05%.
Как описано выше, получено подтверждение тому, что при увеличении доли кристаллов гемифумарата Соединения A, вырабатывание сопутствующих веществ может быть ингибировано.
[0063]
<<Примеры 4-15>>
После добавления воды к кристаллам гемифумарата Соединения А, проводят сушку с получением гемифумарата Соединения А, доля кристаллов которого составляет 62%. Различные фармацевтические добавки, приведенные в Таблице 5, физически смешивают с полученным гемифумаратом Соединения А в массовом соотношении 1:1, и полученные фармацевтические композиции помещают в бутылки и оставляют выстаиваться в открытом режиме при 40°C и отн.вл. 75% в течение 1 месяца и 3 месяцев.
[0064]
<<Экспериментальный Пример 3: Измерение сопутствующих веществ>>
Сопутствующие вещества, содержащиеся в фармацевтических композициях Примеров, измеряют аналогично способу измерения Экспериментального Примера 2. Результаты измерений для сопутствующего вещества, имеющего относительное время удерживания, равное приблизительно 1,06, относительно пика, соответствующего Соединению A, приведены в Таблице 5.
[0065]
<<Экспериментальный Пример 4: Измерение потери в массе при высушивании фармацевтических добавок>>
Измерение потери в массе при высушивании для различных фармацевтических добавок, приведенных в Таблице 5, проводят аналогично измерениям в испытании в отношении вышеупомянутой потери в массе при высушивании. Используемые фармацевтические добавки представляют собой гидрат лактозы (название продукта: Pharmatose 200M, изготовлен в FrieslandCampina DMV BV), гидроксипропилцеллюлозу (название продукта: HPC-L, изготовлен в Nippon Soda Co., Ltd.), стеарат магния (название продукта: Parteck LUB MST, изготовлен в Merck KGaA), D-маннит (название продукта: PEARLITOL 50C, изготовлен в ROQUETTE), микрокристаллическую целлюлозу (название продукта: Ceolus PH-101, изготовлен в Asahi Kasei Chemicals Corporation), безводный двухосновный фосфат кальция (название продукта: GS, изготовлен в Kyowa Chemical Industry Co., Ltd.), гипромеллозу (название продукта: TC-5E, Shin-Etsu Chemical Co., Ltd.), кукурузный крахмал (название продукта: кукурузный крахмал, изготовлен в Nihon Shokuhin Kako Co., Ltd.), гидроксипропилцеллюлозу с низкой степенью замещения (название продукта: L-HPC LH-21, гидроксипропилцеллюлоза с низкой степенью замещения), кроскармеллозу натрия (название продукта: KICCOLATE ND-2HS, изготовлен в Nichirin Chemical Industries, Ltd.), стеарат кальция (название продукта: Parteck LUB CST, изготовлен в Merck KGaA), и тальк (название продукта: Hi-filler, изготовлен в Matsumura Sangyo Co., Ltd.).
[0066]
[Таблица 5]
1 месяц
3 месяца
LOQ: Предел количественного обнаружения
[0067]
Подтверждено, что фармацевтические композиции Примеров 4-15 являются стабильными по истечении 1 месяца и 3 месяцев хранения в открытом режиме при 40°C и отн.вл. 75%, и, что при увеличении доли кристаллов гемифумарата Соединения А, вырабатывание сопутствующих веществ может быть ингибировано. Полагают, что, в частности, гидрат лактозы (Пример 5) и D-маннит (Пример 6) подходят с точки зрения ингибирования вырабатывания сопутствующих веществ.
[0068]
<<Экспериментальный Пример 5: Измерение потери в массе при высушивании таблеток Примеров 2 и 3>>
Потерю в массе при высушивании для таблеток Примеров 2 и 3 после хранения в открытом режиме при 40°C и отн.вл. 75% в течение 1 недели измеряют аналогично способу измерения в Экспериментальном Примере 4. Результаты показаны в Таблице 6. Потеря в массе при высушивании для таблеток, которые содержат D-маннит в составах, является низкой.
[0069]
[Таблица 6]
[0070]
На основании вышеприведенных результатов, стабильный состав, содержащий Соединение А или его фармацевтически приемлемую соль, может быть обеспечен путем регулирования доли кристаллов Соединения А или его фармацевтически приемлемой соли, и/или путем использования фармацевтической добавки, способной регулировать содержание воды в составе.
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
[0071]
Согласно настоящему изобретению, предоставляется стабильная фармацевтическая композиция для перорального введения, содержащая Соединение А или его фармацевтически приемлемую соль, где ингибируется вырабатывание сопутствующих веществ во время хранения.
Хотя настоящее изобретение описано со ссылкой на конкретные варианты осуществления, различные изменения и модификации, очевидные для специалистов в данной области, могут быть выполнены в пределах объема прилагаемых пунктов формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 2020 |
|
RU2831451C2 |
| ДИАМИНОГЕТЕРОЦИКЛИЧЕСКОЕ КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2010 |
|
RU2526253C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[3, 4-b]ПИРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), ПРИМЕНЕНИЕ (ВАРИАНТЫ), КОМПОЗИЦИЯ (ВАРИАНТЫ) | 2003 |
|
RU2357967C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ КАТЕХОЛА ИЛИ ИХ СОЛЬ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 2019 |
|
RU2795227C2 |
| ПРОИЗВОДНЫЕ АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА В КАЧЕСТВЕ МЕДИКАМЕНТА | 2012 |
|
RU2600976C2 |
| СОЕДИНЕНИЯ 8-МЕТИЛ-1-ФЕНИЛИМИДАЗО[1, 5-а]ПИРАЗИНА | 2011 |
|
RU2560162C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
| ЗАМЕЩЕННЫЕ ПИРИДОПИРАЗИНЫ КАК НОВЫЕ ИНГИБИТОРЫ Syk | 2012 |
|
RU2569635C9 |
| ПИРАЗОЛО[3,4-b]ПИРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОДИЭСТЕРАЗ | 2003 |
|
RU2348633C2 |
| ПОЛИМОРФНАЯ ФОРМА 4-{ [4-({ [4-(2,2,2-ТРИФТОРЭТОКСИ)-1,2-БЕНЗИЗОКСАЗОЛ-3-ИЛ]ОКСИ} МЕТИЛ)ПИПЕРИДИН-1-ИЛ]МЕТИЛ} -ТЕТРАГИДРО-2Н-ПИРАН-4-КАРБОНОВОЙ КИСЛОТЫ | 2012 |
|
RU2616978C2 |
Предоставляют стабильную фармацевтическую композицию для перорального введения, содержащую 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамид (в дальнейшем в данном документе называемый как Соединение А) или его фармацевтически приемлемую соль, где ингибируется вырабатывание сопутствующих веществ во время хранения. В стабильной фармацевтической композиции для перорального введения доля кристаллов Соединения A или его фармацевтически приемлемой соли составляет 60% или более относительно общего количества Соединения А или его фармацевтически приемлемой соли. 3 н. и 3 з.п. ф-лы, 6 табл., 5 пр.
1. Стабильная фармацевтическая композиция для перорального введения, состоящая из гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида и фармацевтических добавок, представляющих собой D-маннит, гидроксипропилцеллюлозу, гидроксипропилцеллюлозу с низкой степенью замещения, стеарат магния и агент для нанесения пленочной оболочки, при этом доля кристаллов гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида составляет 60% или более относительно общего количества гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида.
2. Фармацевтическая композиция для перорального введения по п. 1, где процентное содержание сопутствующего вещества 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида составляет 0,20% или менее, после хранения фармацевтической композиции для перорального введения в открытом режиме при 40°C и относительной влажности 75% в течение 1 месяца.
3. Фармацевтическая композиция для перорального введения по п. 1 или 2, где содержание D-маннита в составе составляет от 20% по массе до 90% по массе относительно общей массы фармацевтической композиции для перорального введения.
4. Способ изготовления стабильной фармацевтической композиции для перорального введения, включающий:
(1) смешение гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида с фармацевтическими добавками, представляющими собой D-маннит, гидроксипропилцеллюлозу, гидроксипропилцеллюлозу с низкой степенью замещения и стеарат магния,
(2) гранулирование смеси с тем, чтобы доля кристаллов гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида составляла 60% или более относительно общего количества гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида,
(3) формование компрессионным прессованием гранулированного продукта и
(4) нанесение пленочной оболочки.
5. Способ изготовления фармацевтической композиции для перорального введения по п. 4, где грануляцию проводят при содержании воды в гранулированном продукте, составляющем 30% или менее.
6. Способ стабилизирования гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида в стабильной фармацевтической композиции для перорального введения, включающий доведение доли кристаллов гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида относительно общего количества гемифумарата 6-этил-3-({3-метокси-4-[4-(4-метилпиперазин-1-ил)пиперидин-1-ил]фенил}амино)-5-(тетрагидро-2H-пиран-4-иламино)пиразин-2-карбоксамида до 60% или более и путем добавления фармацевтических добавок, представляющих собой D-маннит, гидроксипропилцеллюлозу, гидроксипропилцеллюлозу с низкой степенью замещения и стеарат магния, и нанесения пленочной оболочки.
| ДИАМИНОГЕТЕРОЦИКЛИЧЕСКОЕ КАРБОКСАМИДНОЕ СОЕДИНЕНИЕ | 2010 |
|
RU2526253C2 |
| US 2013273161 A, 2013-10-17 | |||
| Raymond C Rowe, Handbook of Pharmaceutical Excipients FIFTH EDITION, Pharmaceutical Press and American Pharmacists Association 2006, стр | |||
| Способ получения нерастворимых лаков основных красителей в субстанции и на волокнах | 1923 |
|
SU132A1 |
| Gohel MC et al., A review of co-processed directly compressible excipients, J Pharm Pharmceut Sci | |||
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |

