ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение принадлежит к области химической фармацевтики и, согласно ему предложен ряд агонистов рецептора глюкагоноподобного пептида-1 (GLP-1R). Настоящее изобретение также относится к фармацевтическим композициям, содержащим данные соединения, и применению этих соединений в изготовлении лекарственного средства для лечения таких заболеваний, как сахарный диабет.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Сахарный диабет поражает миллионы людей во всем мире и считается одним из основных факторов риска смерти людей в 21ом столетии. Со временем неконтролируемый сахарный диабет может повредить системы организма, в том числе сердце, кровеносные сосуды, глаза, почки и нервы. Сахарный диабет является тяжелым социально-экономическим бременем во всем мире.
Существуют два основных типа сахарного диабета, обозначаемых диабетом 1 типа и 2 типа, при этом случаев сахарного диабета 2 типа (T2DM) насчитывается более 90% из числа всех пациентов с сахарным диабетом во всем мире. Сахарный диабет 1 типа характеризуется дефицитом инсулина, вызываемым главным образом аутоиммунно опосредуемым разрушением бета-клеток поджелудочной железы. Сахарный диабет 2 типа характеризуется аномальной секрецией инсулина и впоследствии толерантностью к инсулину. Чтобы предотвратить кетоацидоз, пациенты с сахарным диабетом 1 типа должны принимать экзогенный инсулин для выживания. Пациентам с сахарным диабетом 2 типа может потребоваться экзогенный инсулин для регуляции у них уровней глюкозы в крови, хотя они не так зависят от экзогенного инсулина, как пациенты с сахарным диабетом 1 типа.
Глюкагоно подобный пептид-1 (GLP-1), инкретин, секретируется L-клетками кишечного эпителия и оказывает физиологические эффекты посредством связывания со своим рецептором. Рецептор GLP-1 (GLP-1R) принадлежит к подсемейству G-белок-связанных рецепторов. Когда GLP-1 связывается с рецептором GLP-1, запускается ряд биологических эффектов. Показано, что GLP-1 способствует секреции инсулина глюкозозависимым образом, т.е., когда в организме концентрация глюкозы в крови повышается, GLP-1 стимулирует усиление секреции инсулина островковыми клетками, и тем самым снижается уровень глюкозы в крови. Агонист рецептора GLP-1 представляет собой гипогликемическое лекарственное средство нового типа, которое может эффективно регулировать уровень глюкозы в крови без вызывания гипогликемии и эффективно снижать массу тела с целью достижения регулирования массы посредством возрастания чувства сытости, замедления опорожнения желудка, подавления аппетита, уменьшения накопление жира и тому подобного.
В настоящее время пептидные лекарственные средства на основе агониста рецептора GLP-1, такие как лираглутид, эксенатид и семаглутид, нашли применение у пациентов с ожирением и сахарным диабетом 2 типа, а также в случае просто субъектов с ожирением или избыточным весом, и все они показали значительные эффекты по снижению массы тела. Однако, их прием часто сопровождается неблагоприятными эффектами со стороны желудочно-кишечного тракта, такими как тошнота и рвота. В научно-исследовательских институтах предпринимаются попытки использования пероральных непептидных лекарственных средств для лечения сахарного диабета 2 типа и регулирования массы, но разработка низкомолекулярных лекарственных средств для воздействия на рецептор глюкагоноподобного пептида-1 ограничена затруднением, связанным с имитацией взаимодействий между рецептором и пептидом в случае малых молекул.
В настоящее время, патентные заявки, раскрывающее агонисты рецептора GLP-1 непептидной природы, включают WO 2009/111700, WO 2010/114824, WO 2011/1142 71, WO 2013/090454, WO 2018/056453, WO 2018/109607, WO 2019239319, WO 2019239371, WO 2020103815 и т.д., при этом только ТТР-273 от vTv и PF-06882961 от Pfizer вступили в фазу II клинического исследования.
Таким образом, настоящее изобретение направлено на открытие новых непептидных агонистов рецептора GLP-1, особенно новых соединений, которые обладают хорошими биологическими свойствами и могут быть безопасно применены на людях. Согласно настоящему изобретению также предложен подход для предупреждения и/или лечения GLP-1-ассоциируемых заболеваний, особенно связанных с сахарным диабетом заболеваний.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложен ряд производных бензимидазола, способы их получения и их фармацевтическое применение.
В частности, согласно настоящему изобретению предложены соединение формулы (I) или его стереоизомер, таутомер либо фармацевтически приемлемая соль, при этом все переменные являются такими, как определено в данном описании.
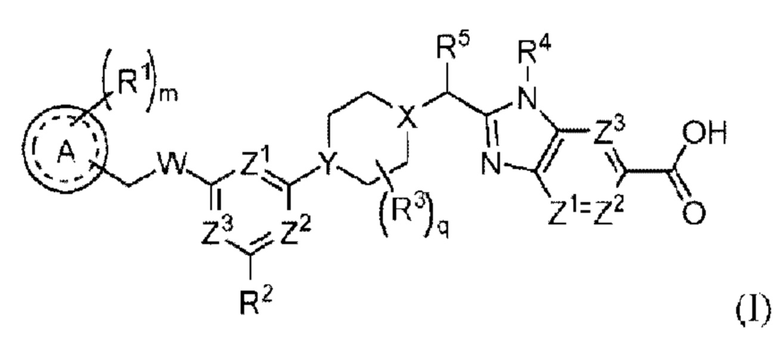
Эти соединения являются агонистами рецептора глюкагоноподобного пептида-1 (GLP-1R). Настоящее изобретение также относится к фармацевтическим композициям, содержащим данные соединения и к применению этих соединений в изготовлении лекарственного средства для лечения таких заболеваний, как сахарный диабет.
В частности, цель настоящего изобретения достигается следующими техническими решениями.
Соединение формулы (I):
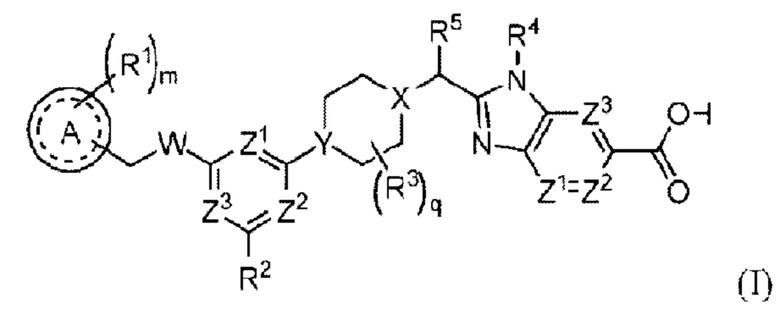
или его стереоизомер, таутомер либо фармацевтически приемлемая соль, где
А выбран из группы, состоящей из фенильного кольца, гетероароматического кольца и 8-10-членного конденсированного ароматического кольца;
R1 выбран из группы, состоящей из атома водорода, галогена, циано, алкила, алкокси, галогеналкила, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила, алкила, замещенного одним или несколькими R7, арила, замещенного одним или несколькими R8, и гетероарила, замещенного одним или несколькими R8;
m равно 0, 1, 2 или 3;
W представляет собой О или NH;
R2 выбран из группы, состоящей из атома водорода, галогена, циано, алкила, галогеналкила, алкокси и алкоксиалкила;
R3 выбран из группы, состоящей из атома фтора, гидроксила, циано, оксо (=O), С1-3залкила, ОС1-3алкила, С3-4циклоалкила и С3-4спироалкила, образованного путем циклизации двух R3 вместе, при этом С1-3алкил, ОС1-3алкил, С3-4циклоалкил и С3-4спироалкил могут быть замещены 0-3 атомами фтора или 0-1 гидроксилом, если позволяет валентность;
q равно 0, 1 или 2;
X представляет собой СН или N;
Υ представляет собой СН или Ν;
R5 выбран из группы, состоящей из галогена, гидроксила, алкила, галогеналкила, алкокси, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила, алкилциклоалкила и алкилгетероциклоалкила;
R4 представляет собой -С1-3алкил, -С0-3алкилен-С3-6циклоалкил, -С0-3алкилен-R9 или -С1-3алкилен-R6, где алкил может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из 0-3 атомов F, или 0-1 заместителем, выбранным из группы, состоящей из -С0-1алкилен-CN, -С0-1алкилен-ORO и -N(RN)2, если позволяет валентность, и где алкилен и циклоалкил могут быть независимо замещены 0-2 заместителями, независимо выбранными из группы, состоящей из 0-2 атомов F, или 0-1 заместителем, выбранным из группы, состоящей из - С0-1алкилен -CN, -С0-1алкилен-ORO и -N(RN)2, если позволяет валентность;
R9 представляет собой 4-6-членный гетероциклоалкил, при этом, если позволяет валентность, гетероциклоалкил может быть замещен 0-2 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-1 оксо (=O),
0-1 -CN,
0-2 атомов F и
0-2 заместителей, независимо выбранных из группы, состоящей из -С1-3алкила и -ОС1-3алкила, при этом, если позволяет валентность, алкил указанных -С1-3алкила и -ОС1-3алкила может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-3 атомов F,
0-1 -CN и
0-1 -ORO;
R6 представляет собой 5-6-членный гетероарил, при этом, если позволяет валентность, гетероарил может быть замещен 0-2 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-2 атомов галогена,
0-1 заместителя, выбранного из группы, состоящей -ORO и -N(RN)2, и
0-2 -С1-3алкилов, при этом, если позволяет валентность, алкил может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-3 атомов F и
0-1 -ORO;
каждый RO независимо представляет собой Η или -С1-3алкил, при этом С1-3алкил может быть замещен 0-3 атомами F;
каждый RN независимо представляет собой Η или-С1-3алкил;
Ζ1, Ζ2 и Ζ3 каждый независимо представляет собой -CRZ или N, а каждый RZ независимо представляет собой Н, F, Cl или -СН3;
R7 выбран из группы, состоящей из галогена, циано, гидроксила, циклоалкила, гетероциклоалкила и сульфонила; и
R8 выбран из группы, состоящей из галогена, циано, алкила, галогеналкила, алкокси, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила и сульфонила.
В предпочтительном воплощении настоящего изобретения соединение или его стереоизомер, таутомер либо фармацевтически приемлемая соль показаны формулой (III):
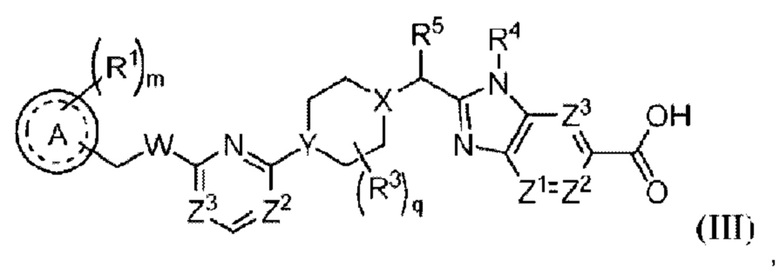 где
где
А выбран из группы, состоящей из фенильного кольца, гетероароматического кольца и 8-10-членного конденсированного ароматического кольца;
R1 выбран из группы, состоящей из атома водорода, галогена, циано, алкила, алкокси, галогеналкила, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила, алкила, замещенного одним или несколькими R7, арила, замещенного одним или несколькими R8, и гетероарила, замещенного одним или несколькими R8;
m равно 0, 1, 2 или 3;
W представляет собой О или NH;
R3 выбран из группы, состоящей из атома фтора, гидроксила, циано, оксо (=O), С1-3алкила, ОС1-3алкила, С3-4циклоалкила и С3-4спироалкила, образованного путем циклизации двух R3 вместе, при этом С1-3алкил, ОС1-3алкил, С3-4циклоалкил и С3-4спироалкил могут быть замещены 0-3 атомами фтора или 0-1 гидроксилом, если позволяет валентность;
q равно 0, 1 или 2;
X представляет собой СН или N;
Υ представляет собой СН или Ν;
R5 выбран из группы, состоящей из галогена, гидроксила, алкила, галогеналкила, алкокси, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила, алкилциклоалкила и алкилгетероциклоалкила;
R4 представляет собой -С1-3алкил, -С0-3алкилен-С3-6циклоалкил, -С0-3алкилен-R9 или -С1-3алкилен-R6, при этом алкил может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из 0-3 атомов F, или 0-1 заместителем, выбранным из группы, состоящей из -С0-1алкилен-CN, -С0-1алкилен-ORO и -N(RN)2, если позволяет валентность, и при этом алкилен и циклоалкил могут быть независимо замещены 0-2 заместителями, независимо выбранными из группы, состоящей из 0-2 атомов F, или 0-1 заместителем, выбранным из группы, состоящей из -С0-1алкилен-CN, -С0-1алкилен-ORO и -N(RN)2, если позволяет валентность;
R9 представляет собой 4-6-членный гетероциклоалкил, при этом, если позволяет валентность, гетероциклоалкил замещен 0-2 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-1 оксо (=O),
0-1 -CN,
0-2 атомов F и
0-2 заместителей, независимо выбранных из группы, состоящей из -С1-3алкила и -ОС1-3алкила, при этом, если позволяет валентность, алкил указанных -С1-3алкила и -ОС1-3алкила может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-3 атомов F,
0-1 -CN и
0-1 -ORO;
R6 представляет собой 5-6-членный гетероарил, при этом гетероарил, если позволяет валентность, может быть замещен 0-2 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-2 атомов галогена,
0-1 заместителя, выбранного из группы, состоящей из -ORO и -N(RN)2, и
0-2 - С1-3алкилов, при этом алкил, если позволяет валентность, может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-3 атомов F и
0-1 -ORO;
каждый RO независимо представляет собой Η или -С1-3алкил, при этом С1-3алкил может быть замещен 0-3 атомами F;
каждый RN независимо представляет собой Η или -С1-3алкил;
Ζ1, Ζ2 и Ζ3 каждый независимо представляет собой -CRZ или N, а каждый RZ независимо представляет собой Н, F, Cl или -СН3;
R7 выбран из группы, состоящей из галогена, циано, гидроксила, циклоалкила, гетероциклоалкила и сульфонила; и
R8 выбран из группы, состоящей из галогена, циано, алкила, галогеналкила, алкокси, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила и сульфонила.
В предпочтительном воплощении настоящего изобретения соединение или его стереоизомер, таутомер либо фармацевтически приемлемая соль показаны формулой (II):
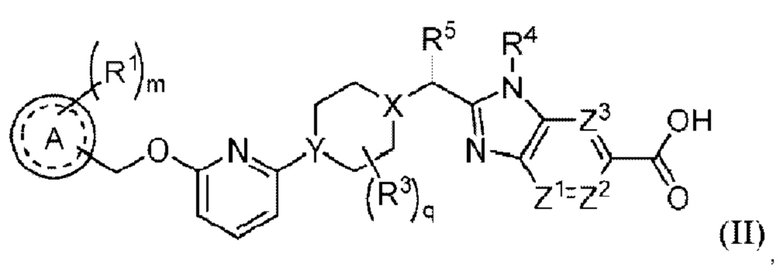 где
где
А выбран из группы, состоящей из фенильного кольца, гетероароматического кольца и 8-10-членного конденсированного ароматического кольца;
R1 выбран из группы, состоящей из атома водорода, галогена, циано, алкила, алкокси, галогеналкила, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила, алкила, замешенного одним или несколькими R7, арила, замещенного одним или несколькими R8, и гетероарила, замещенного одним или несколькими R8;
m равно 0, 1, 2 или 3;
R3 выбран из группы, состоящей из атома фтора, гидроксила, циано, С1-3алкила, ОС1-3алкила, С3-4циклоалкила и С3-4спироалкила, образованного путем циклизации двух R3 вместе, при этом С1-3алкил, ОС1-3алкил, С3-4циклоалкил и С3-4спироалкил могут быть замещены 0-3 атомами фтора или 0-1 гидроксилом, если позволяет валентность;
q равно 0, 1 или 2;
X представляет собой СН или N;
Υ представляет собой СН или Ν;
R5 выбран из группы, состоящей из галогена, гидроксила, алкила, галогеналкила, алкокси, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила, алкилциклоалкила и алкилгетероциклоалкила;
R4 представляет собой -С1-3алкил, -С0-3алкилен-С3-6циклоалкил, -С0-3алкилен-R9 или -С1-3алкилен-R6, при этом алкил может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из 0-3 атомов F, или 0-1 заместителем, выбранным из группы, состоящей из -С0-1алкилен-CN, -С0-1алкилен-ORO и -N(RN)2, если позволяет валентность, и при этом алкилен и циклоалкил могут быть независимо замещены 0-2 заместителями, независимо выбранными из группы, состоящей из 0-2 атомов F, или 0-1 заместителем, выбранным из группы, состоящей из -C0-1алкилен-CN, -С0-1алкилен-ORO и -N(RN)2, если позволяет валентность;
R9 представляет собой 4-6-членный гетероциклоалкил, при этом, если позволяет валентность, гетероциклоалкил может быть замещен 0-2 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-1 оксо (=O),
0-1 -CN,
0-2 атомов F и
0-2 заместителей, независимо выбранных из группы, состоящей из -С1-3алкила и -ОС1-3алкила, при этом алкил указанных -С1-3алкила и -ОС1-3алкила, если позволяет валентность, может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из группы ниже:
0-3 атомов F,
0-1 -CN и
0-1 -ORO;
R6 представляет собой 5-6-членный гетероарил, при этом гетероарил, если позволяет валентность, может быть замещен 0-2 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-2 атомов галогена,
0-1 заместителя, выбранного из группы, состоящей из -ORO и -N(RN)2, и
0-2 -С1-3алкилов, при этом алкил, если позволяет валентность, может быть замещен 0-3 заместителями, независимо выбранными из группы, состоящей из указанных ниже заместителей:
0-3 атомов F и
0-1 -ORO;
каждый RO независимо представляет собой Η или -С1-3алкил, при этом С1-3алкил может быть замещен 0-3 атомами F;
каждый RN независимо представляет собой Η или -С1-3алкил;
Ζ1, Ζ2 и Ζ3 каждый независимо представляет собой -CRZ или N, а каждый RZ независимо представляет собой Н, F, Cl или -СН3;
R7 выбран из группы, состоящей из галогена, циано, гидроксила, циклоалкила, гетероциклоалкила и сульфонила; и
R8 выбран из группы, состоящей из галогена, циано, алкила, галогеналкила, алкокси, галогеналкокси, алкоксиалкила, циклоалкила, гетероциклоалкила и сульфонила.
В предпочтительном воплощении настоящего изобретения алкил относится к линейной, разветвленной или циклической насыщенной углеводородной цепи, содержащей конкретное количество атомов углерода, предпочтительно к С1-6алкилу, выбранному из группы, состоящей из метила, этила, пропила, изопропила, н-бутила, изобутила, втор-бутила, трет-бутила, н-пентила, втор-пентила, 1-этилпропила, 2-метилбутила, трет-пентила, 1,2-диметилпропила, изопентила, неопентила, н-гексила, изогексила, втор-гексила, трет-гексила, неогексила, 2-метилпентила, 1,2-диметилбутила и 1-этилбутила.
В предпочтительном воплощении настоящего изобретения алкокси относится к радикалу простого алкилового эфира, предпочтительно С1-6алкокси, выбранному из группы, состоящей из метокси, этокси, пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентилокси, втор-пентилокси, 1-этилпропокси, 2-метилбутокси, трет-пентилокси, 1,2-диметилпропокси, изопентилокси, неопентилокси, н-гексилокси, изогексилокси, втор-гексилокси, трет-гексилокси, неогексилокси, 2-метилпентилокси, 1,2-диметилбутокси и 1-этилбутокси.
В предпочтительном воплощении настоящего изобретения алкоксиалкил относится к алкилу, у которого один или более атомов водорода замещены группой алкокси, предпочтительно к С1-4алкокси-С1-4алкилу, более предпочтительно метоксиметилу, метоксиэтилу, метоксипропилу, метоксибутилу, этоксиметилу, этоксиэтилу, этоксипропилу, этоксибутилу, пропоксиметилу, пропоксиэтилу, пропоксипропилу, пропоксибутилу, бутоксиметилу, бутоксиэтилу, бутоксипропилу, бутоксибутилу и тому подобному.
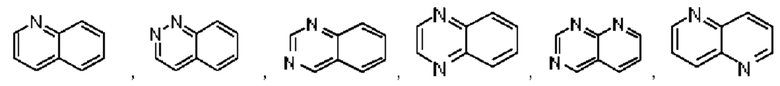
В предпочтительном воплощении настоящего изобретения галоген выбран из группы, состоящей из атома фтора, хлора, брома и йода; галогеналкил относится к алкилу, у которого один или более чем один атом водорода замещен галогеном; галогеналкокси относится к алкокси, у которого один или более чем один атом водорода замешен галогеном; гидроксиалкил относится к алкилу, у которого один или более чем один атом водорода замещен гидроксилом; гетероциклоалкил относится к алкилу, у которого один или более чем один атом водорода замещен гетероциклилом; и циклоалкилметилен относится к метилу, у которого один или более чем один атом водорода замещен циклоалкилом. В предпочтительном воплощении настоящего изобретения конденсированное ароматическое кольцо выбрано из группы, состоящей из нафталина и конденсированного гетероароматического кольца, при этом конденсированное гетероароматическое кольцо образуется путем конденсирования ароматического кольца или гетероароматического кольца с гетероароматическим кольцом, и гетероатом у гетероароматического кольца выбран из группы, состоящей из атомов азота, кислорода и серы, и могут присутствовать один или более гетероатомов.
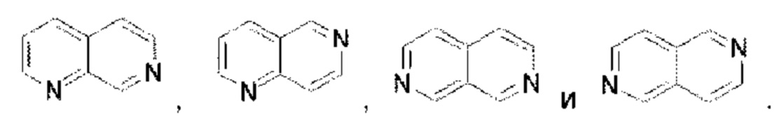
В предпочтительном воплощении настоящего изобретения конденсированное гетероароматическое кольцо выбрано из группы, состоящей из индазола, хинолина, изохинолина, хиноксалина, индола, изоиндола, циннолина, хиназолина, фгалазина, пурина, нафгиридина, птеридина, бензофурана, бензотиофена, бензоксазола, бензотиазола, бензизоксазола, бензизотиазола, бензоксадиазола, бензотиадиазола, бензотриазола, бензотриазина, бензимидазола, пиразолопиразина, пиразинопиримидина, пиразинопиридазина, пиразинотриазина, пиразолопиримидина, им идазолопирим идина, триазолопиримидина, пиримидотриазина, пиримидопиридазина, имидазолопиридазина, пиразолопиридазина, триазолопиридазина, пиридазинотриазина, имидазолотриазина, пиразолотриазина, триазолотриазина, имидазолопиридина, пиридопиридазина, пиразолопиридина, пиридопиримидина, пиридотриазина, оксазолопиридина, тиазолопиридина, изоксазолопиридина, изотиазолопиридина, оксадиазолопиридина, тиадиазолопиридина, фуранопиридина, пирролопиридина, имидазолопиразина, триазолопиразина, оксазолопиразина, тиазолопиразина, изоксазолопиразина, изотиазолопиразина, оксадиазолопиразина, тиадиазолопиразина, фуранопиразина, пирролопиразина, оксазолопиримидина, тиазолопиримидина, изоксазолопиримидина, изотиазолопиримидина, оксадиазолопиримидина, тиадиазолопиримидина, фуранопиримидина, пирролопиримидина, оксазолопиридазина, тиазолопиридазина, изоксазолопиридазина, изотиазолопиридазина, оксадиазолопиридазина, тиадиазолопиридазина, фуранопиридазина, пиррол опиридазина, оксазолотриазина, тиазолотриазина, изоксазолотриазина, изотиазолотриазина, оксадиазолотриазина, тиадиазолотриазина, фуранотриазин и пирролотриазина. Нафтиридин выбран из группы, состоящей из 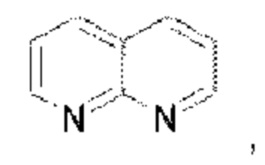
 Имидазолопиридин выбран из группы, состоящей из
Имидазолопиридин выбран из группы, состоящей из 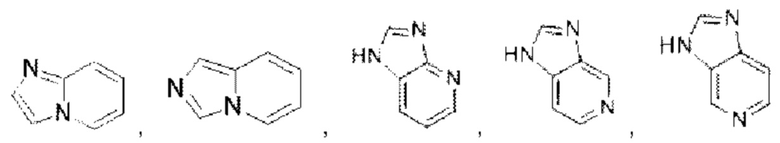 и
и 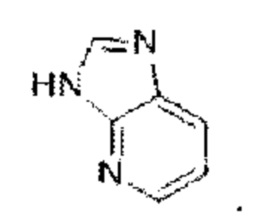 Имидазолопиразин выбран из группы, состоящей из
Имидазолопиразин выбран из группы, состоящей из 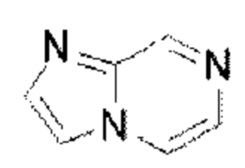 и
и 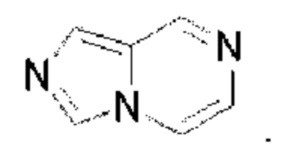 Триазолопиразин выбран из группы, состоящей из
Триазолопиразин выбран из группы, состоящей из 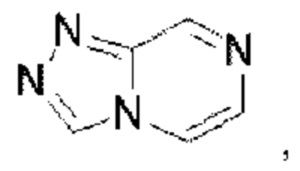
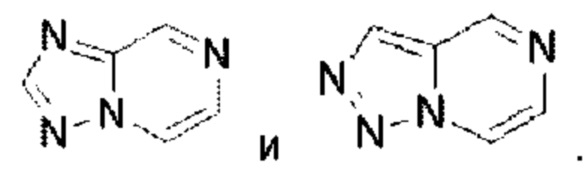 Пиразолопиримидин выбран из группы, состоящей из
Пиразолопиримидин выбран из группы, состоящей из 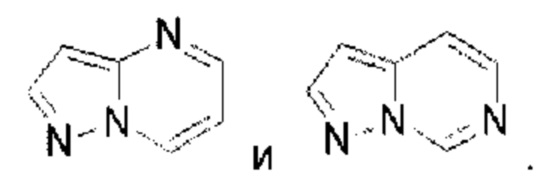 Имидазолопиримидин выбран из группы, состоящей из
Имидазолопиримидин выбран из группы, состоящей из 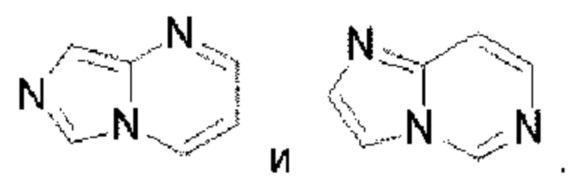 Триазолопиримидин выбран из группы, состоящей из
Триазолопиримидин выбран из группы, состоящей из 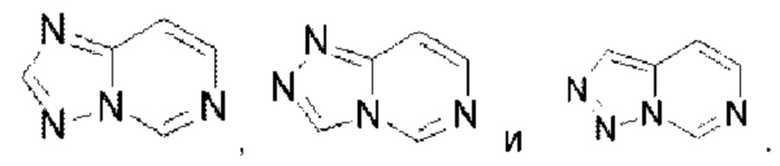 Имидазолопиридазин выбран из группы, состоящей из
Имидазолопиридазин выбран из группы, состоящей из 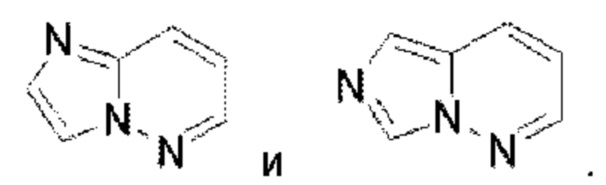 Тиазолопиридазин выбран из группы, состоящей из
Тиазолопиридазин выбран из группы, состоящей из 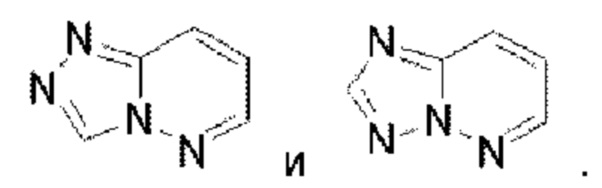 Имидазолотриазин выбран из группы, состоящей из
Имидазолотриазин выбран из группы, состоящей из 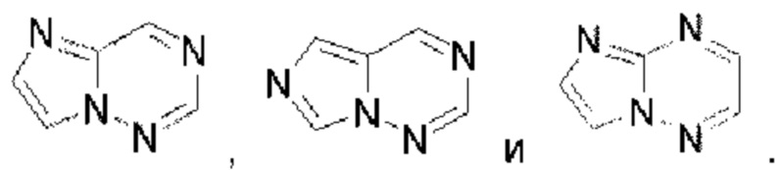 Пиридопиридазин выбран из группы, состоящей из
Пиридопиридазин выбран из группы, состоящей из 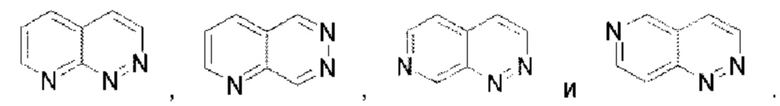 Пиразолопиридин выбран из группы, состоящей из
Пиразолопиридин выбран из группы, состоящей из 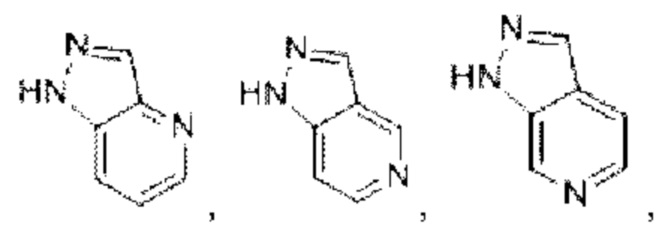
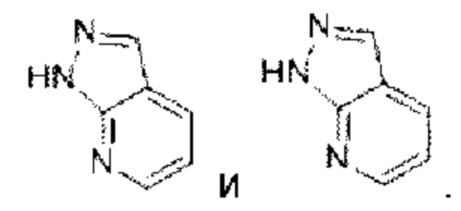 Пиридопиримидин выбран из группы, состоящей из
Пиридопиримидин выбран из группы, состоящей из 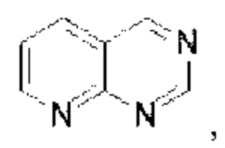
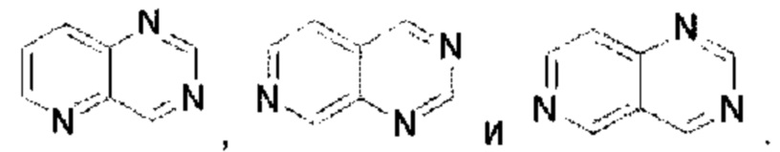 Пиридотриазин выбран из группы, состоящей из
Пиридотриазин выбран из группы, состоящей из 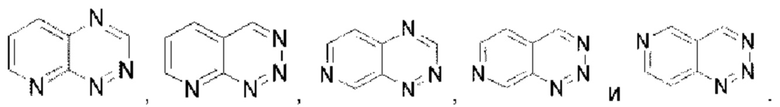 Пиримидотриазин выбран из группы, состоящей из
Пиримидотриазин выбран из группы, состоящей из 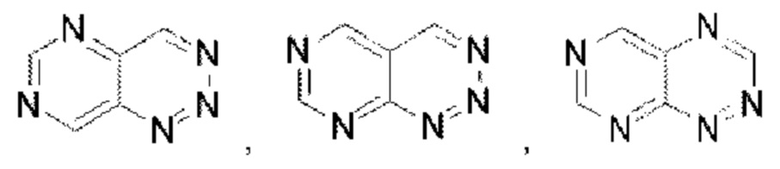 и
и 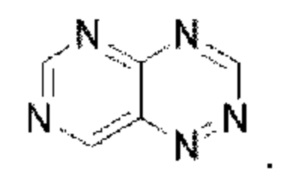
В предпочтительном воплощении настоящего изобретения гетероцикл в гетероциклоалкиле выбран из 4-10-членного гетероцикла, при этом 4-10-членный гетероцикл может быть выбран из группы, состоящей из 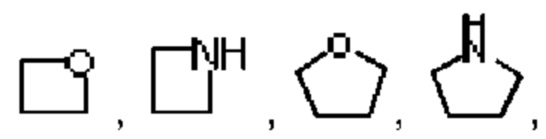
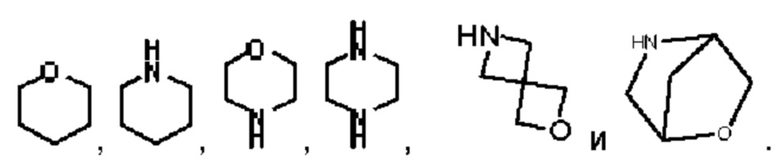 Арил выбран из фенила. Гетероарил выбран из 5-12-членного гетероарила, при этом 5-12-членный гетероарил может быть выбран из группы, состоящей из
Арил выбран из фенила. Гетероарил выбран из 5-12-членного гетероарила, при этом 5-12-членный гетероарил может быть выбран из группы, состоящей из 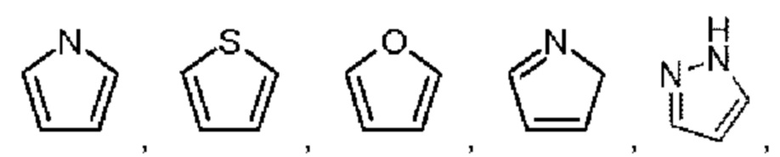
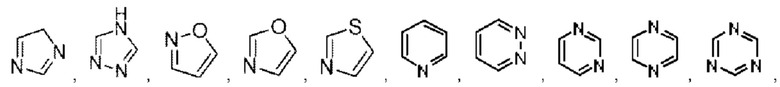
 и
и 
В предпочтительном воплощении настоящего изобретения "алкилен" относится к алкилу, который может быть независимо замещен 0-2 заместителями, если позволяет валентность; "циано" относится к -CN, "гидроксил" относится к -ОН, и "сульсронил" относится к -SO2.
В предпочтительном воплощении настоящего изобретения "гетероцикл" относится к насыщенному или ненасыщенному гетероциклу, содержащему один или более гетероатомов (азота, кислорода и серы). "Ароматическое кольцо" относится к фэнильному кольцу. "Гетероароматическое кольцо" относится к гетероциклу с 4n+2 тт-электронами в замкнутой сопряженной системе, который может представлять собой, например, фуран, тиофен, тетрагидропиррол, дигидропиразол, имидазол, тиазол, изотиазол, тиадиазол, оксазол, оксадиазол, изоксазол, пиперидин, пиперазин, пиридазин, пиразин, триазин, пиримидин, морсролин или пиридин. Выражение "более" (компонентов) относится к двум, трем или четырем компонентам.
В предпочтительном воплощении настоящего изобретения циклоалкил выбран из С3_6 циклоалкила, при этом С3-6циклоалкил может быть выбран из группы, состоящей из циклопропила, циклобутила, циклопентила и циклогексила.
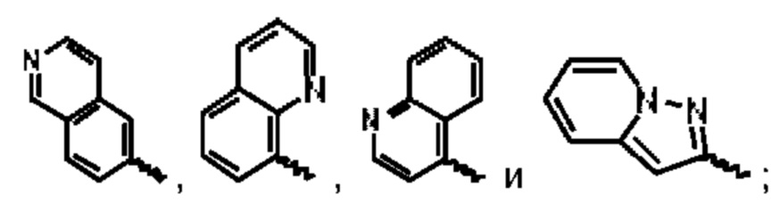
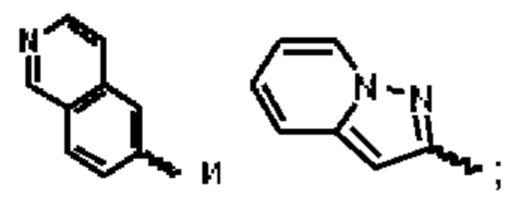
В предпочтительном воплощении настоящего изобретения А выбран из группы, состоящей из фенильного кольца, 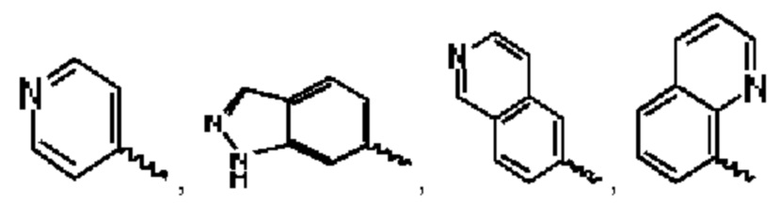 и
и 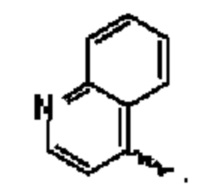
В предпочтительном воплощении настоящего изобретения соединение или его стереоизомер, таутомер, фармацевтически приемлемая соль представлены формулой (I), где:
А выбран из группы, состоящей из фенильного кольца, 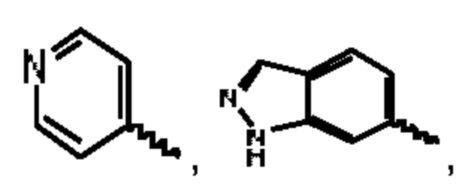
R1 выбран из группы, состоящей из атома водорода, фтора, хлора, брома, циано, дифторэтила и трифторэтила;
m равно 0, 1 или 2;
представляет собой О или NH;
R2 выбран из группы, состоящей из атома хлора, циано, трифторметила, метокси и метоксиметила;
q равно 0 или 1;
R3 представляет собой атом фтора, метил, оксо (=O), гидроксил, фторметил и метоксиэтил;
R4 представляет собой оксетан-2-илметил;
X представляет собой СН или N;
Υ представляет собой СН или Ν;
R5 выбран из группы, состоящей из метила, этила, циклопропилметила, фторэтила и метоксиэтила;
Ζ1 представляет собой СН;
Ζ2 представляет собой СН или CF; и
Ζ3 представляет собой СН, N или CF.
В предпочтительном воплощении настоящего изобретения соединение или его стереоизомер, таутомер, фармацевтически приемлемая соль представлены формулой (III), где:
А выбран из группы, состоящей из фенильного кольца, 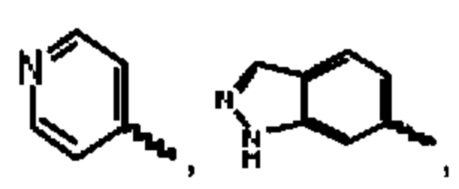
R1 выбран из группы, состоящей из атома водорода, фтора, хлора, брома, циано, дифторэтила и трифторэтила;
m равно 0, 1 или 2;
W представляет собой О или NH;
q равно 0 или 1;
R3 представляет собой атом фтора, метил, оксо (=O), гидроксил, фторметил или метоксиэтил;
R4 представляет собой оксетан-2-илметил;
X представляет собой СН или N;
Υ представляет собой СН или Ν;
R5 представляет собой метил, этил, циклопропилметил, фторэтил или метоксиэтил;
Ζ1 представляет собой СН;
Ζ2 представляет собой СН или CF; и
Ζ3 представляет собой СН, N или CF.
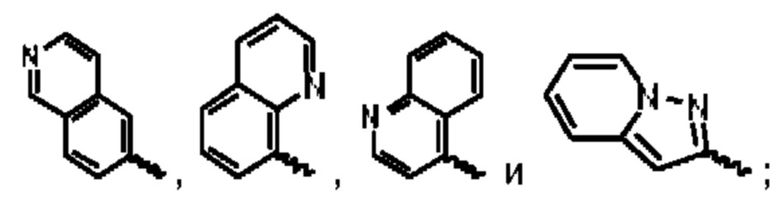
В предпочтительном воплощении настоящего изобретения соединение или его стереоизомер, таутомер, фармацевтически приемлемая соль представлены формулой (II), где:
А выбран из группы, состоящей из фенильного кольца, 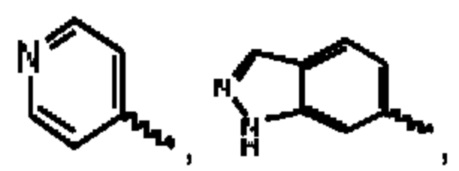
R1 выбран из группы, состоящей из атома водорода, фтора, хлора, брома, циано, дифторэтила и трифторэтила;
m равно 0, 1 или 2;
q равно 0;
R4 представляет собой оксетан-2-илметил;
X представляет собой СН или N;
Υ представляет собой СН или Ν;
R5 выбран из группы, состоящей из метила, этила, циклопропилметила, фторэтила и метоксиэтила;
Ζ1 представляет собой СН;
Ζ2 представляет собой СН или CF; и
Ζ3 представляет собой СН, N или CF.
В предпочтительном воплощении настоящего изобретения, в соединении или его стереоизомере, таутомере либо фармацевтически приемлемой соли, когда атом углерода, соединенный с R5, представляет собой хиральный атом углерода, этот хиральный атом углерода находится в S-конфигурации и/или R-конфигурации, предпочтительно в S-конфигурации.
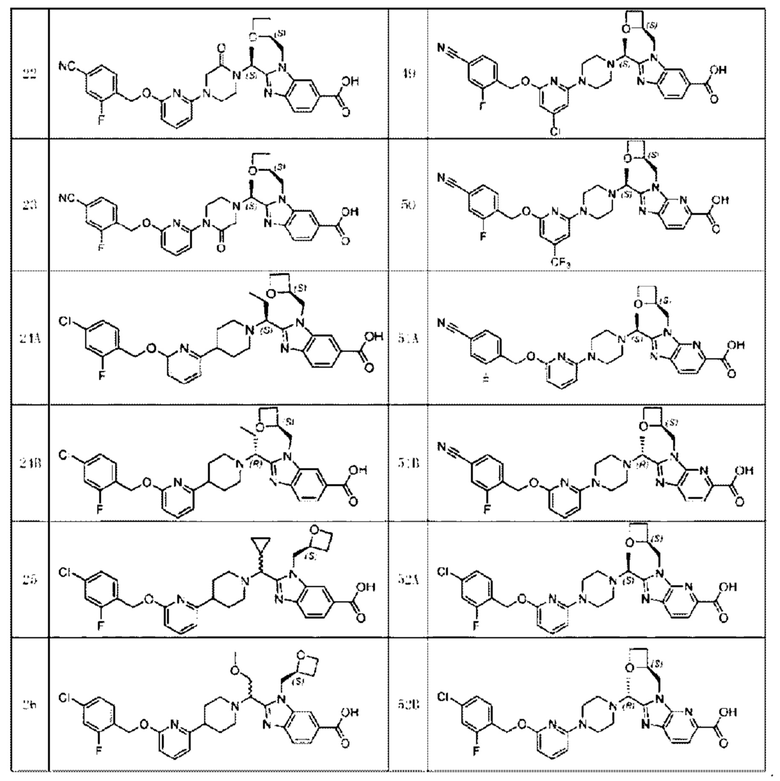
В предпочтительном воплощении настоящего изобретения соединение или его фармацевтически приемлемая соль выбраны из группы, состоящей из:
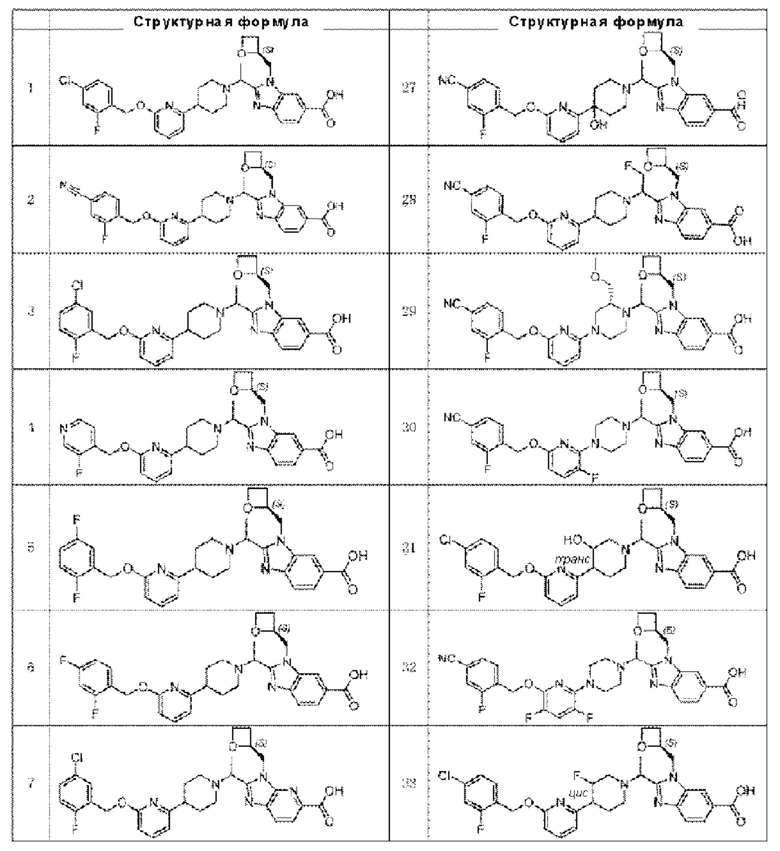
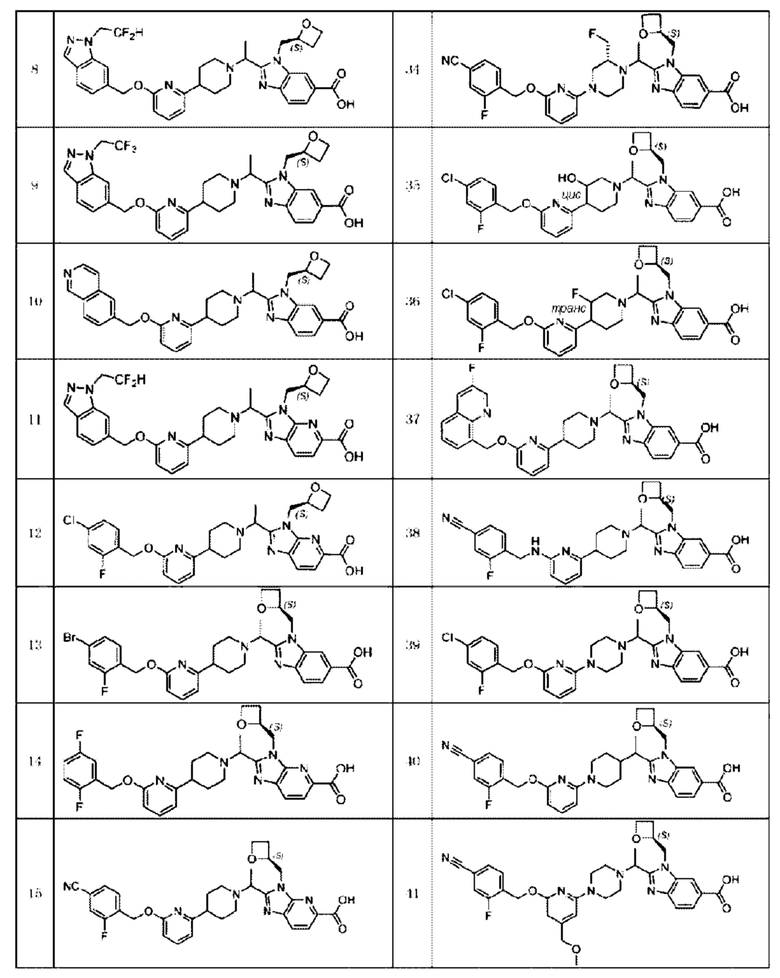
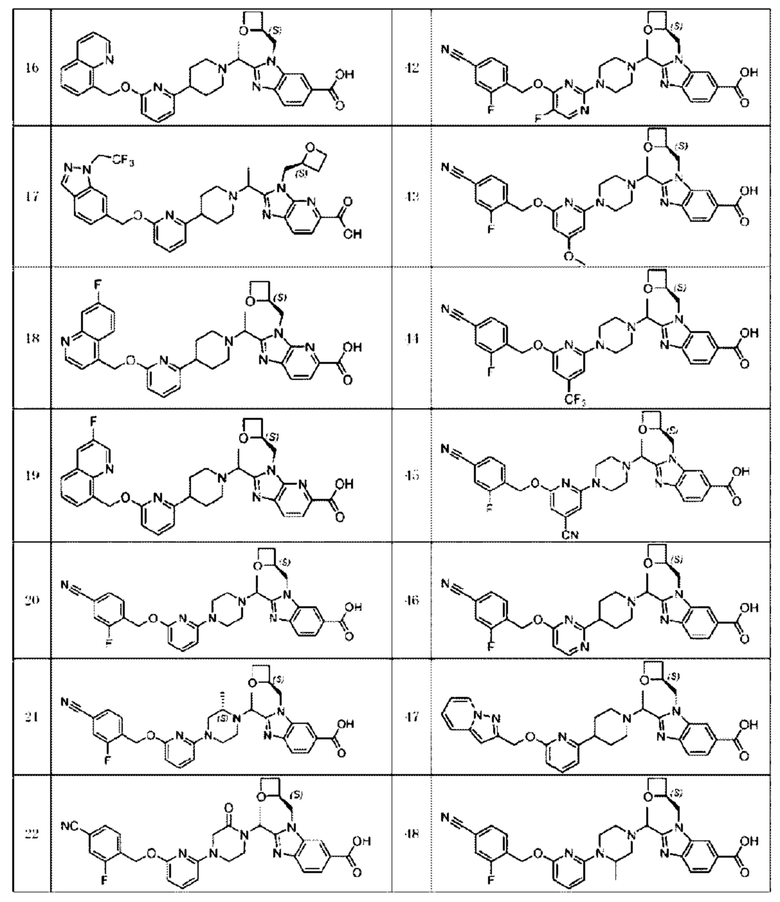
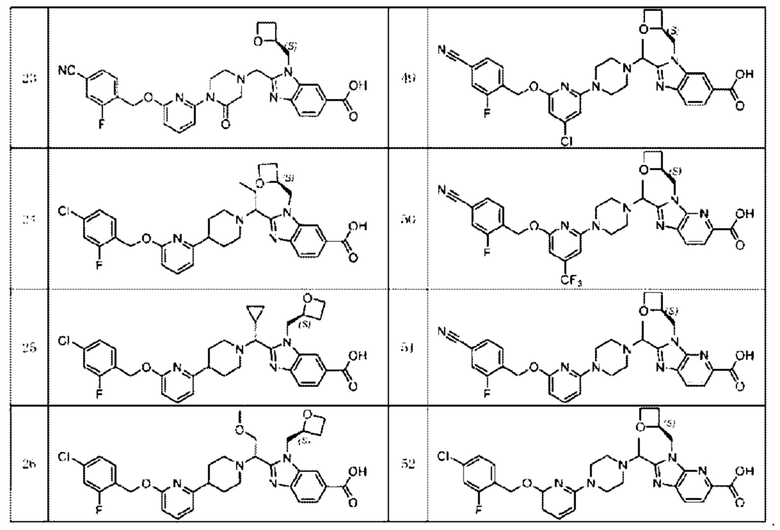
В предпочтительном воплощении настоящего изобретения соединение или его фармацевтически приемлемая соль выбраны из группы, состоящей из:
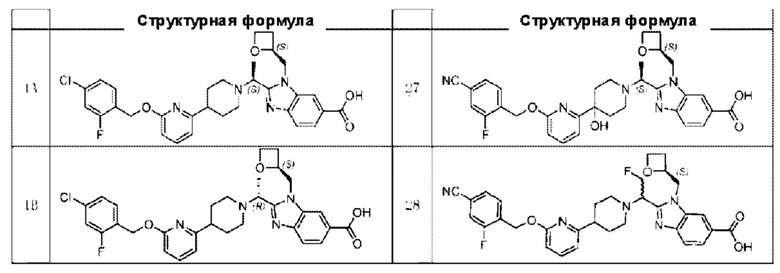
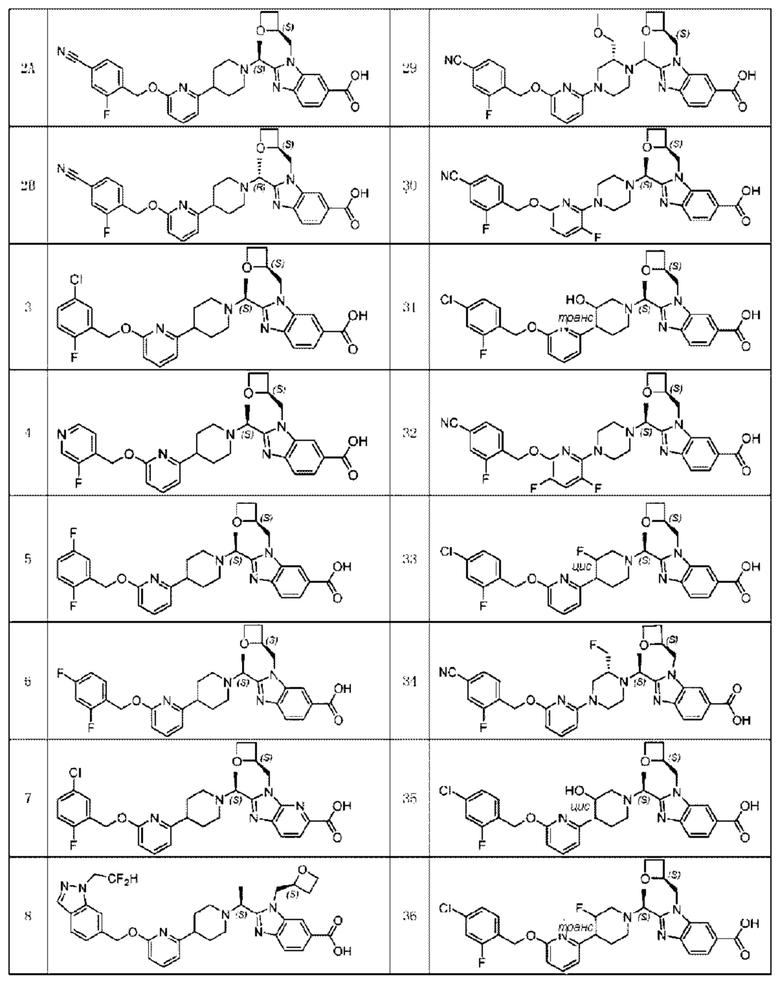
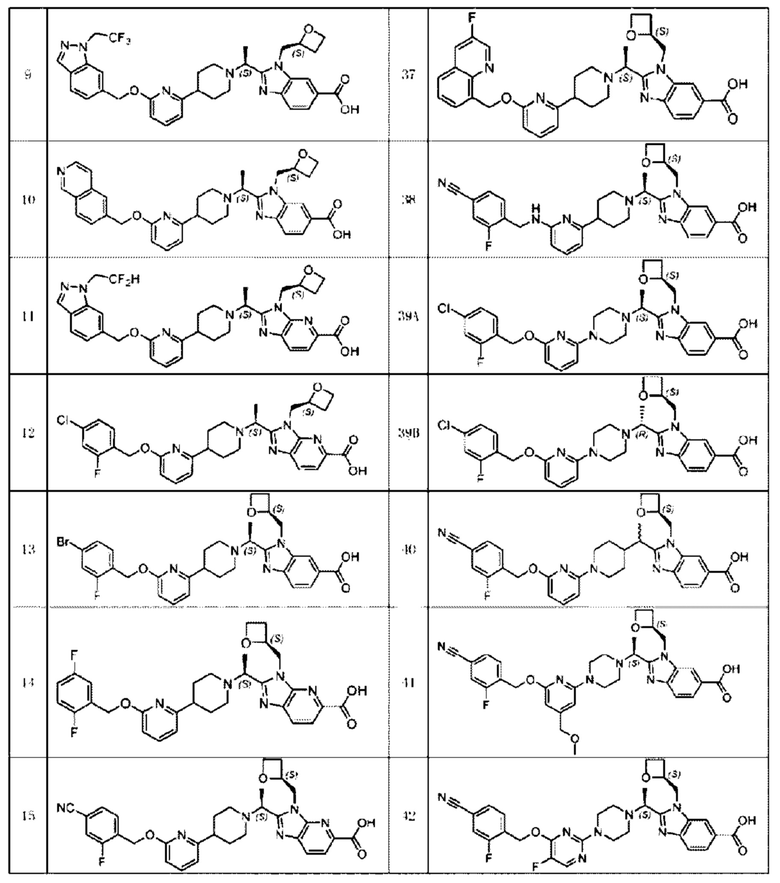
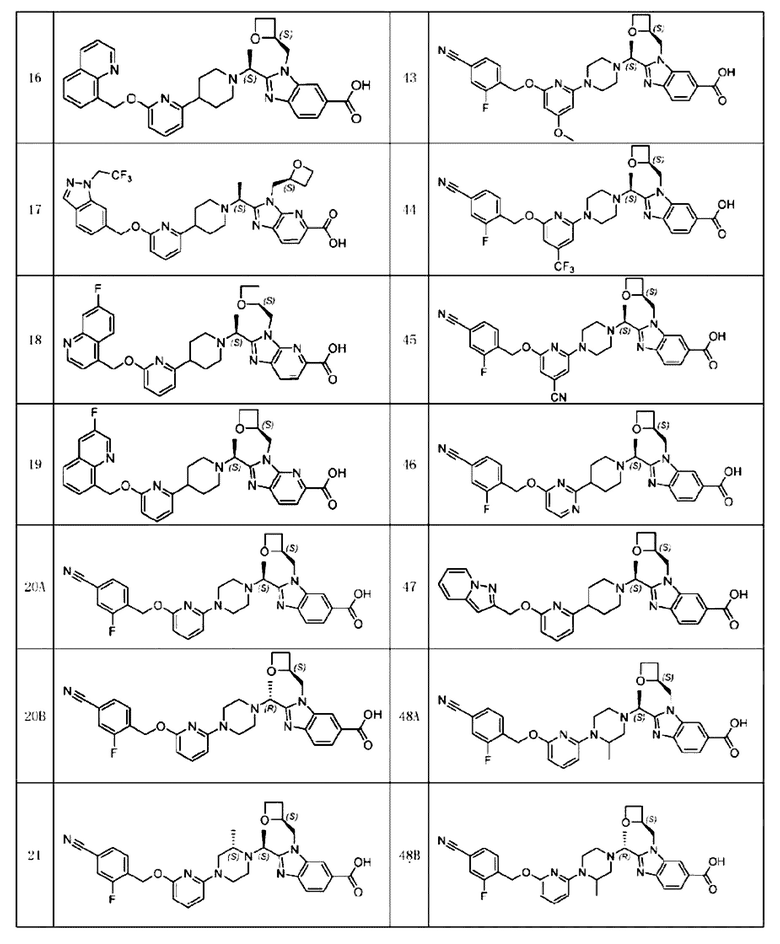
В предпочтительном воплощении настоящего изобретения фармацевтически приемлемая соль относится к соли, которую получают из соединения и фармацевтически приемлемой(ого) кислоты или основания.
В предпочтительном воплощении настоящего изобретения один или более атомов водорода в соединении заменены на изотоп дейтерий.
Другая цель настоящего изобретения заключается в разработке фармацевтической композиции, содержащей вышеупомянутое соединение формулы (I) или его стереоизомер, таутомер либо фармацевтически приемлемую соль и один или более чем один фармацевтически приемлемый носитель.
Другая цель настоящего изобретения заключается в предложении применения соединения формулы (I) или его стереоизомера, таутомера или фармацевтически приемлемой соли для изготовления лекарственного средства для лечения GLP-1-ассоциируемых заболеваний, особенно, связанных с сахарным диабетом заболеваний.
Если не указано иное, то подразумевается, что приведенные ниже термины и фразы, использованные в данном описании, имеют следующие значения. Конкретный термин или конкретную фразу не следует считать неопределенными или неясными в отсутствие специального определения, а следует понимать в соответствии с их обычным значением. Когда в данном описании появляется торговое название, то подразумевается, что оно относится к соответствующему ему продукту или его активному ингредиенту. Термин "фармацевтически приемлемый", использованный в данном описании, относится к таким соединениям, веществам, композициям и/или лекарственным формам, которые с медицинской точки зрения подходят для применения в контакте с тканями человека и животных без чрезмерных токсичности, раздражения, аллергической реакции или других проблем или осложнений, и соизмеримы с разумным соотношением польза/риск.
Термин "фармацевтически приемлемая соль" относится к соли соединений по настоящему изобретению, полученной исходя из соединений по настоящему изобретению, имеющих конкретные заместители, и фармацевтически приемлемых кислот или оснований.
Помимо солевой формы соединения, предложенные согласно настоящему изобретению, также имеют форму пролекарства. Пролекарства соединений, описанных в данной заявке, могут легко подвергаться химическим изменениям в физиологических условиях, в результате чего происходит их превращение в соединения по настоящему изобретению. Кроме того, пролекарства могут преобразовываться в соединения по настоящему изобретению химическими или биохимическими способами in vivo.
Некоторые соединения по настоящему изобретению могут присутствовать в несольватированной форме или в сольватированной форме, в том числе в гидратировэнной форме. Как правило, в объем настоящего изобретения включены и сольватированная форма, и несольватированная форма.
Соединения по настоящему изобретению могут существовать в конкретных геометрических или стереоизомерных формах. Подразумевается, что все такие соединения по настоящему изобретению, в том числе цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереоизомеры, (D)-изомеры, (L)-изомеры и рацемические смеси и другие их смеси, такие как энантиомерно или диастереомерно обогащенные смеси, находятся в пределах объема настоящего изобретения. В заместителе, таком как алкильная группа, может/могут присутствовать дополнительный(ые) асимметричный(ые) атом(ы) углерода. Все эти изомеры, а также их смеси, включены в объем настоящего изобретения.
Оптически активные (R)- и (S)-изомеры, а также D- и L-изомеры, могут быть получены хиральным синтезом или с использованием хиральных реагентов либо другими традиционными методами. Если желательно получить энантиомер соединения по настоящему изобретению, то его можно получить путем асимметрического синтеза или дериватизации с использованием хиральных вспомогательных веществ при этом полученную диастереомерную смесь разделяют и группу вспомогательного вещества отщепляют с получением желаемого чистого энантиомера. Альтернативно, если соединение содержит основную функциональную группу (например, амино) или кислотную функциональную группу (например, карбоксильную) в молекуле, то чистый энантиомер можно получить путем образования соли диастереоизомера с использованием молекулы с подходящей(им) оптически активной кислотой или основанием, затем отделения диастереоизомера традиционными методами, хорошо известными в данной области техники, и проведения последующей рециркуляции. Кроме того, отделение энантиомеров от диастереоизомеров обычно осуществляют посредством хроматографии, при этом для проведения указанной хроматографии применяют хиральную неподвижную фазу, и возможно в сочетании с химической дериватизацией (например, с образованием карбамата из амина).
Атомы в молекулах соединений по настоящему изобретению могут быть изотопами, и получение производных с изотопами обычно может приводить к увеличению периода полу выведения, снижению скорости выведения, улучшению метаболической стабильности, повышению активности in vivo и тому подобному. Кроме того, настоящее изобретение дополнительно включает воплощение, в котором по меньшей мере один атом заменен на атом, имеющий тот же атомный номер (число протонов) и разные массовые числа (сумма числа протонов и числа нейтронов). Примеры изотопов, инкорпорированных в соединения по настоящему изобретению, включают изотопы атома водорода, атома углерода, атома азота, атома кислорода, атома фосфора, атома серы, атома фтора, атома хлора, которые представляют собой 2Н, 3Н, 13С, 14С, 15N, 17O, 18O, 31Р, 32Р, 35S, 18F и 36Cl, соответственно. В частности, радиоактивные изотопы, испускающие излучение при своем распаде, например, 3Н или 14С, можно использовать в относящемся к топографической анатомии исследовании распределения фармацевтических композиций или соединений in vivo. Стабильные изотопы не распадаются или не изменяются в количественном отношении, так и не являются радиоактивными, поэтому их можно использовать без вреда для здоровья. Когда атомами, составляющими молекулы соединений по настоящему изобретению, должны быть изотопы, изотопное превращение можно проводить в соответствии с обычными методами путем замены реагента, используемого в синтезе, на реагент, содержащий соответствующие изотопы.
Соединения по настоящему изобретению могут содержать изотопы атомов в несуществующих в природе пропорциях в случае одного или более атомов, составляющих соединение. Например, соединения могут быть помечены такими радиоактивными изотопами, как дейтерий (2Н), йод-125 (125I) или С-14 (14С). Все соединения по настоящему изобретению с преобразованным изотопным составом, независимо от того, радиоактивны они или нет, включены в объем настоящего изобретения.
Кроме того, один или более атомов водорода в соединениях по настоящему изобретению может быть заменен изотопом дейтерием (2Н), и дейтерий-замещенные соединения по настоящему изобретению имеют преимущества, относящиеся к увеличению периода полувыведения, снижению скорости выведения, улучшению метаболической стабильности, повышению активности in vivo и тому подобному.
Способ получения изотопных производных обычно включает метод катализа с фазовым переносом. Например, в предпочтительном методе дейтерирования используют катализаторы межфазного переноса (например, соли тетраалкиламмония, NBu4HSO4). Более высокого содержания дейтерия можно добиться с использованием катализатора межфазного переноса для обмена протонов метилена в соединениях дифенилметана, чем с использованием дейтерированного силана (такого как дейтерированный триэтилсилан) в присутствии кислоты (такой как метансульфоновая кислота) или с использованием дейтерированного бората натрия с кислотой Льюиса, такой как трихлорид алюминия, для восстановления.
Термин "фармацевтически приемлемый носитель" относится к любому(ой) применяемому(ой) при приготовлении композиций носителю или среде, которые можно использовать для доставки эффективного количества активного вещества по изобретению, которые не оказывают влияния на биологическую активность активного вещества и не вызывают никаких токсических побочных эффектов у реципиента или пациента. Репрезентативные носители включают воду, масло, вещества растительного происхождения и минеральные вещества, основу для крема, основу для лосьона, мазевую основу и так далее. Эти основы включают суспендирующий агент, средство для повышения клейкости, усилитель проницаемости и так далее. Технологии их изготовления хорошо известны специалистам в области создания лекарственных средств для косметического или местного применения. В качестве дополнительной информации по носителям можно упомянуть работу Remington: The Science and Practice of Pharmacy, 21st Ed., Lippincott, Williams & Wilkins (2005), содержание которой включено в данное описание посредством ссылки.
Термин "эксципиент" обычно относится к носителю, разбавителю и/ил и среде, необходимым для приготовления эффективной фармацевтической композиции.
Что касается лекарственного средства или фармакологически активного агента, то термин "эффективное количество" или "терапевтически эффективное количество" относится к достаточному количеству лекарственного средства или агента, которое не оказывает токсического действия, но способствует достижению желаемого эффекта. Что касается пероральной лекарственной формы по настоящему изобретению, то термин "эффективное количество" активного вещества в композиции относится к количеству, необходимому для достижения желаемого эффекта в случае использования в комбинации с другим активным веществом композиции. Определение эффективного количества варьирует от человека к человеку и зависит от возраста и общего состояния здоровья субъекта, а также от конкретного активного вещества. Соответствующее эффективное количество в отдельных случаях может быть определено специалистами в данной области техники на основе обычных экспериментов.
Термины "активный ингредиент", "терапевтический агент", "активное вещество" или "активный агент" относятся к химическому соединению, которым можно эффективно лечить расстройства, заболевания или состояния субъекта.
Термин "возможный" или "возможно" относится к ситуации, когда описываемое далее событие или обстоятельство может иметь место, но происходит не обязательно, и данное описание включает ситуации, когда указанное событие или обстоятельство происходит, а также ситуации, в которых указанное событие или обстоятельство не происходит.
 показывает связь.
показывает связь.
Соединения по настоящему изобретению могут быть получены разнообразными способами синтеза, известными специалистам в данной области техники, включая конкретные воплощения, приведенные ниже, воплощения, являющиеся результатом их сочетания с другими методами химического синтеза, и эквиваленты, известные специалистам в данной области техники. Предпочтительные воплощения включают, но не ограничиваются этим, воплощения настоящего изобретения.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1 представляет собой схематическую диаграмму монокристаллической структуры соединения 39А из примера 39.
ПОДРОБНОЕ ОПИСАНИЕ
Данное изобретение описано ниже более подробно вместе с воплощениями, но воплощения по изобретению этим не ограничиваются.
Структуру соединений определяют с использованием ядерного магнитного резонанса (ЯМР) или масс-спектрометрии (MS). Сдвиги ЯМР (δ) приведены в 10-6 (млн-1). ЯМР-спектр снимают, используя ЯМР-спектрометр AVANCE-III от Bruker с дейтерированным диметилсульфоксидом (DMSO-d6) и дейтерированным хлороформом (CDCl3) в качестве растворителя для определения и тетраметилсилан (TMS) в качестве внутреннего стандарта.
MS-спектр снимают, используя масс-спектрометр ISQ™ ЕС (производитель: Thermo, модель: ISQ™ ЕС).
Анализ с применением высокоэффективной жидкостной хроматографии (HPLC) выполняют, используя хроматограф для высокоэффективной жидкостной хроматографии серии U3000 HPLC с DAD (диодный матричный детектор) HPLC от Thermo и хроматограф для высокоэффективной жидкостной хроматографии Agilent 1260.
Прибор для быстрой подготовки CombiFlash используется с CombiFlash Rf+LUMEN™ (TELEDYNE ISCO).
В качестве платин с силикагелем для тонкослойной хроматографии используют пластины с силикагелем HSGF254 или GF254 от Yantai Yinlong. По техническому регламенту толщина слоя силикагеля на пластине, используемой для тонкослойной хроматографии (TLC), составляет 0,17-0,23 мм, а толщина слоя силикагеля на пластине, используемой для тонкослойной хроматографии с целью разделения и очистки продуктов, составляет 0,4-0,5 мм.
В случае проведения хроматографии на колонке с силикагелем обычно в качестве носителя используют силикагель 100-200 меш от Rushan Shangbang silica gel.
Агенты: LDA, диизопропиламид лития; THF, тетрагидрофуран; TEA, триэтиламин; 1,4-диоксан; ВН3, боран; EDCl, 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорид; НОВТ, 1-гидроксибензотриазол; DIPEA, N,N-диизопропилэтиламин; DMF, N,N-диметилформамид; K2CO3, карбонат калия; DMSO, диметилсульфоксид; EtOH, этанол; NaH, гидрид натрия; толуол; к.т., комнатная температура; PdCl2(dppf), 1,1'-бис(дифенилфосфино) ферроцен-палладия дихлорид; Pd(OAc)2, ацетат палладия; PhMe, толуол; HCl/диоксан, соляная кислота в диоксане; Na2CO3, карбонат натрия; МеОН, метанол; DCE, дихлорэтан; NCS, N-х лорсукцинимид; TfOH, трифторметансульфоновая кислота; TFA, трифторуксусная кислота; АсОН, уксусная кислота; (CH2O)n, полиформальдегид; CDI, N,N'-карбонилдиимидазол; LAH, алюмогидрид лития; РРА, полифосфорная кислота; TsOH, п-толуолсульфоновая кислота; NBS, N-бромсукцинимид; ВРО, бензоилпероксид; HATU, 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; KOH.H2O, гидроксид калия моногидрат; NaOH, гидроксид натрия; Cs2CO3, карбонат цезия; AIBN, азодиизобутиронитрил; MeCN, ацетонитрил; DEAD, диэтилазодикарбоксилат; DAST, трифторид диэтиламиносеры; PPh3, трифенилфосфин; DCM, дихлорметан; DMAP, 4-диметиламинопиридин; TBD, 1,5,7-триазидобицикло[4.4.0]декан-5-ен; DIEA, N,N-диизопропилэтиламин; BINAP 2,2'-бис(дифенилфосфино)-1,1'-бинафталин; NMP, N-метилпирролидон.
Пример 1. 2-((S)-1-(4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (1А) и 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (1В)
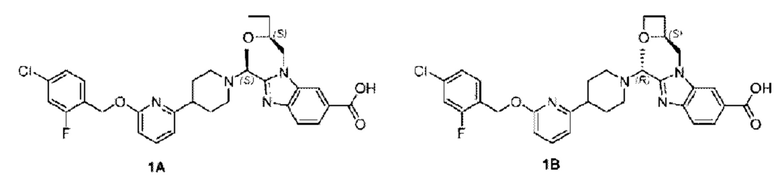
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 2-хлор-6-((4-хлор-2-фторбензил)окси)пиридина
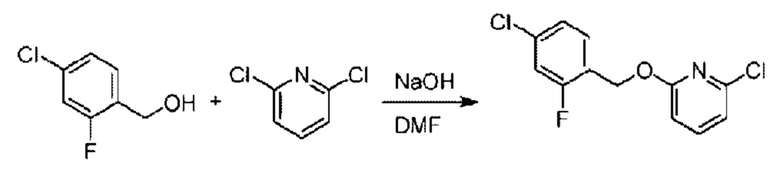
(4-Хлор-2-фгорфенил)метанол (1,6 г; 10,0 ммоль), 2,6-дихлорпиридин (1,627 г; 11,0 ммоль) и гидроксид натрия (1,2 г; 30,0 ммоль) растворяли в Ν,Ν-диметилформамиде (30,0 мл) и перемешивали при 120°С в течение 12 часов.
По завершении реакции реакционный раствор выливали в 100 мл ледяной воды. Раствор этой смеси экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, сушили и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: н-гексан/этилацетат = 10/1), получая 1,2 г 2-х лор-6-((4-хлор-2-фторбензил)окси)пиридина в виде белого твердого вещества (выход: 44,2%). LC-MS (жидкостная хроматография-масс-спектрометрия): RT (время удерживания) = 2,29 мин, [М+Н]+=272,04.
Стадия В. Синтез трет-бутил-6-((4-хлор-2-фторбензил)окси)-3',6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилата
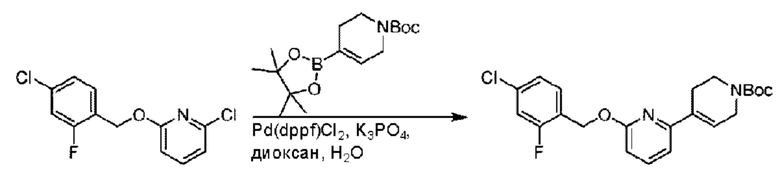
2-Хлор-6-((4-хлор-2-фторбензил)окси)пиридин (1,2 г; 4,4 ммоль), трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборинан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (1,3 г; 4,4 ммоль), [1,1-бис(дифенилфосфин)ферроцен]палладия дихлорид (323,0 мг), фосфат калия (1,8 г; 8,8 ммоль) растворяли в смеси диоксана (20 мл) и воды (4 мл). Температуру поднимали до 75°С и раствор этой смеси перемешивали в течение 8 часов.
По завершении реакции реакционный раствор выливали в 20 мл воды. Раствор этой смеси экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли, сушили и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией (элюент: н-гексан/этилацетат = 10/1), получая 1,2 г трет-бутил-6-((4-хлор-2-фторбензил)окси)-3',6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилата в виде светло-желтого твердого вещества (выход: 65,2%). LC-MS: RT=2,43 мин, [М+Н]+=419,24.
Стадия С. Синтез трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата
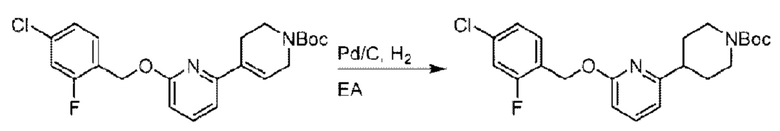
трет-Бутил-6-((4-хлор-2-фторбензил)окси)-3',6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилат (1,1 г; 2,6 ммоль) растворяли в этилацетате (55,0 мл) и к раствору этой смеси добавляли палладий-на-угле (275,0 мг). Взаимодействие проводили при комнатной температуре в атмосфере водорода (при стандартном атмосферном давлении) в течение 6 часов.
По завершении реакции реакционный раствор фильтровали и твердое вещество упаривали на роторном испарителе, получая 1,2 г трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата в виде бесцветного маслянистого продукта (выход: 80,0%). LC-MS: RT=2,43 мин, [М+Н-56]+=365,20.
Стадия D. Синтез 2-((4-хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридина
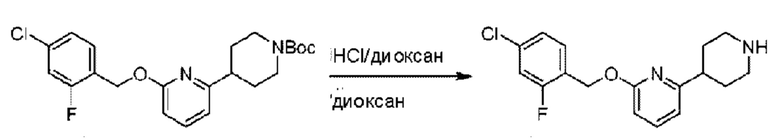
трет-Бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилат (1,1 г; 4,5 ммоль) растворяли в диоксане (5,0 мл) и к раствору этой смеси добавляли соляную кислоту в диоксане (35,0 мл) в ледяной бане. Взаимодействие проводили при комнатной температуре в течение 2 часов.
По завершении реакции твердое вещество осаждали, реакционный раствор фильтровали и твердое вещество упаривали на роторном испарителе, получая 500 мг 2-((4-хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридина в виде светло-желтого твердого вещества.
Стадия Е. Синтез этил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионата
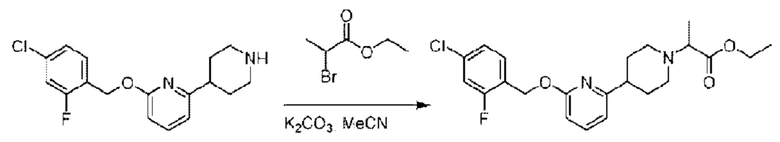
2-((4-Хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридин (500,0 мг; 1,6 ммоль), этил-2-бромпропионат (309,3 мг; 1,7 ммоль) и карбонат калия (441,0 мг; 3,2 ммоль) растворяли в ацетонитриле (8,0 мл) при комнатной температуре и раствор этой смеси перемешивали при комнатной температуре в течение ночи.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, сушили и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/3), получая 353,0 мг этил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионата в виде белого твердого вещества (выход: 52,5%). LC-MS: RT=1,82 мин, [М+Н]+=421,25.
Стадия F. Синтез 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионовой кислоты
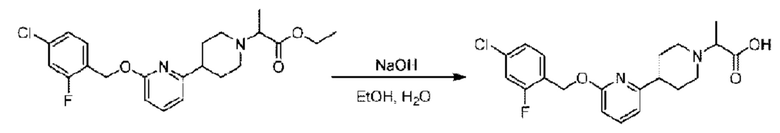
Этил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионат (323 мг; 0,7 ммоль) растворяли в смеси растворителей (3,6 мл; этанол/вода = 5/1) при комнатной температуре и к раствору этой смеси добавляли гидроксид натрия (121,6 мг; 3,0 ммоль), затем перемешивали при комнатной температуре в течение 12 часов.
По завершении реакции в реакционный раствор добавляли разбавленную 0,5 н. соляную кислоту и рН подводили до 2 для осаждения белого твердого вещества. Реакционный раствор фильтровали и твердое вещество упаривали на роторном испарителе, получая 302,0 мг 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионовой кислоты в виде белого твердого вещества. LC-MS: RT=1,84 мин, [М+Н]+=393,21.
Стадия G. Синтез метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионамидо)-3-((((5)-оксетан-2-ил)метил)амино)бензоата
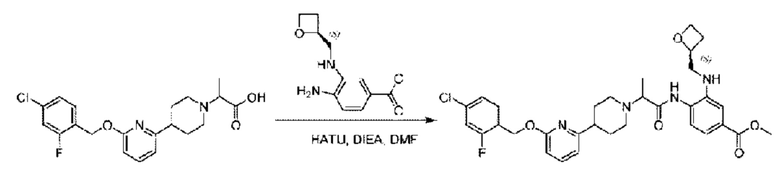
2-(4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионовую кислоту (262,0 мг; 0,7 ммоль) растворяли в Ν,Ν-диметилформамиде (2,5 мл), затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат (380,0 мг; 1,0 ммоль) и Ν,Ν-диизопропилэтиламин (0,5 мл) при комнатной температуре. Раствор этой смеси перемешивали в течение 0,5 часа, добавляли метил-(3)-4-амино-3-(((оксетан-2-ил)метил)амино)бензоат (158,0 мг; 0,6 ммоль) и далее перемешивали при комнатной температуре в течение ночи.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/1), получая 395,0 мг метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионамидо)-3-((((3)-оксетан-2-ил)метил)амино)бензоата в виде светло-желтого твердого вещества (выход: 92,5%). LC-MS: RT=1,82 мин, [М+Н]+=611,31.
Стадия Н. Синтез метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
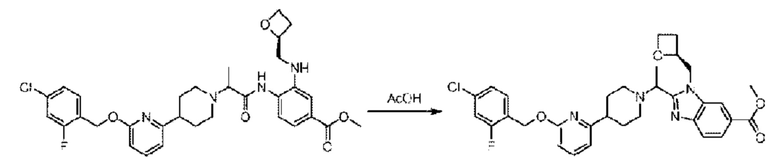
Метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоат (355,0 мг; 0,6 ммоль) растворяли в уксусной кислоте (4,0 мл). Температуру поднимали до 60°С и реакцию проводили в течение 5 часов.
По завершении реакции реакционный раствор выливали в 50,0 мл насыщенного водного раствора бикарбоната натрия и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который разделяли препаративной TLC (подвижная фаза: дихлорметан/метанол = 13/1), получая 118,0 мг метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде светло-желтого твердого вещества (выход: 33,0%). LC-MS: RT=1,84 мин, [М+Н]+=593,31.
Стадия I. Синтез 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (1А) и 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (1В)
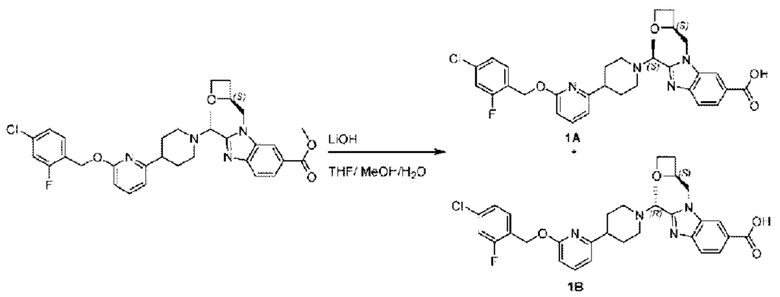
Метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (70,0 мг; 0,2 ммоль) растворяли в смеси растворителей (2,5 мл; тетрагидрофуран/метанол/вода = 1/3/1) при комнатной температуре, затем добавляли гидроксид лития (48,0 мг; 1,1 ммоль) и взаимодействие проводили при комнатной температуре в течение 24 часов.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида аммония и реакционный раствор экстрагировали смесью растворителей (15 мл × 3 раза; дихлорметан/метанол = 15/1). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который разделяли обращенно-фазовой HPLC в виде элюата, лиофилизацией) в следующих условиях разделения: хроматографическая колонка: препаративная 5-С18 от Agilent, 100 мм × 30 мм, 5 мкм; подвижная фаза: (для элюирования) вода (содержащая 0,1% аммиака)и ацетонитрил; скорость потока: 20 мл/мин; градиент: соединения 1Аи 1В элюировались 27%-ным ацетонитрилом в моменты времени 9,07 мин и 12,00 мин, соответственно; длина волны детектирования: 254 нм.
Последовательно получали 8,0 мг 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (1А) (выход: 7,0%) и 4,0 мг 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (1 В) (выход: 3,5%) в виде белого твердого вещества.
Соединение 1А: Η PLC: RT=9,07 мин.
LC-MS: RT=1,82 мин, [M+H]+=579,29. 1Н ЯМР (400 МГц, DMSO) δ 8.20 (brs, 1Н), 7.80 (dd, J=8,4; 1,3 Гц, 1Н), 7.63-7.53 (m, 3Н), 7.45 (dd, J=10,0; 1,9 Гц, 1Н), 7.28 (dd, J=8,2; 2,0 Гц, 1 Η), 6.84 (d, J=7,3 Гц, 1 Η), 6.66 (d, J=8,2 Гц, 1 Η), 5.36 (s, 2H), 5.22-5.1 6 (m, 1 Η), 4.87-4.78 (m, 1H), 4.62 (dd, J=15,2; 5,2 Гц, 1Η), 4.49-4.41 (m, 2H), 4.18 (dt, J=9,4; 5,9 Гц, 1Η), 2.98-2.96 (m, 1Η), 2.65-2.60 (m, 2H), 2.59-2.49 (m, 2H), 2.38-2.21 (m, 2H), 1.86-1.68 (m, 3Н), 1.58-1.48 (m, 1Η), 1.47 (d, J=6,7 Гц, 3Н).
Соединение 1В: ΗPLC: RT=12,00 мин.
LC-MS: RT=1,84 мин, [M+H]+=579,31. 1H ЯМР (400 МГц, DMSO) δ 8.38 (s, 1H), 7.90 (d, J=7,8 Гц, 1 Η), 7.79 (d, J=8,1 Гц, 1Η), 7.70-7,57 (m, 2H), 7.47 (d, J=9,8 Гц, 1H), 7.36-7,26 (m, 1Η), 6.91-6,89 (m, 1Η), 6.74-6,72 (m, 1Η), 5.38 (s, 2H), 5.14-4.91 (m, 2H), 4.71-4.46 (m, 2H), 3.60-3.15 (m, 3Н), 2.90-2.75 (m, 1Η), 2.50-2.39 (m, 1H), 2.14-1.92 (m, 4H), 1.79-1.62 (m, 3Н), 1.28-1.22 (m, 4H).
Пример 2. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (2А) и 2-((R)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (2В)
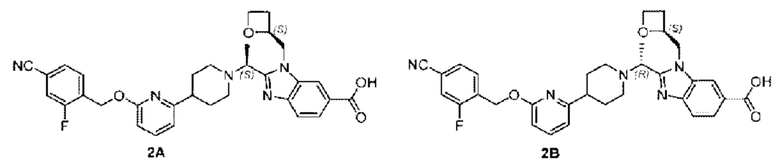
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата
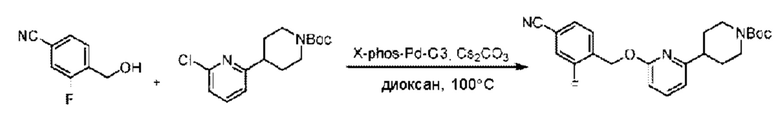
3-Фтор-4-(гидроксиметил)бензонитрил (3,45 г; 22,88 ммоль), трет-бутил-4-(6-хлорпиридин-2-ил)пиперидин-1-карбоксилат (4,50 г; 15,25 ммоль), палладиевый катализатор (644 мг) и карбонат цезия (10 г; 30,50 ммоль) растворяли в диоксане (80 мл). Температуру поднимали до 100°С и раствор этой смеси перемешивали в течение 8 часов.
По завершении реакции реакционный раствор фильтровали под вакуумом через диатомовую землю и промывали дихлорметаном капельным методом. Фильтрат концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией (элюент: этилацетат/н-гексан = 10/1), получая 5 г трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата в виде светло-желтой маслянистой жидкости (выход: 80%). LC-MS: RT=2,27 мин, [М-55]+=356,21.
Стадия В. Синтез 3-фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрила
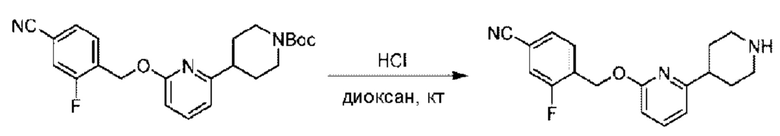
трет-Бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилат (5,00 г) растворяли в соляной кислоте в диоксане (40,0 мл) и раствор этой смеси перемешивали при комнатной температуре.
По завершении реакции реакционный раствор упаривали на роторном испарителе при пониженном давлении, получая 4,5 г 3-фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрила в виде светло-желтого твердого вещества. LC-MS: RT=1,65 мин, [М+Н]+=312,21.
Стадия С. Синтез этил-2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионата
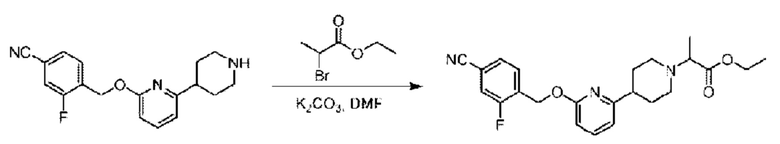
3-Фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрил (4,50 г; 14,46 ммоль) и карбонат калия (4,00 г; 28,92 ммоль) растворяли в Ν,Ν-диметилформамиде (50 мл) при комнатной температуре, затем добавляли этил-2-бромпропионат (3,16 мг; 17,35 ммоль) и раствор этой смеси перемешивали при комнатной температуре.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, сушили и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/3), получая 3,7 г этил-2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионата в виде белого твердого вещества (выход: 62%). LC-MS: RT=1,70 мин, [М+Н]+=412,25.
Стадия D. Синтез 2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионовой кислоты
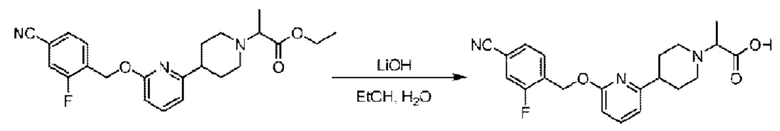
Этил-2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионат (1,00 г; 2,43 ммоль) растворяли в смеси растворителей (22 мл; этанол/вода = 5/1) при комнатной температуре и к раствору этой смеси добавляли гидроксид лития (185 мг; 4,87 ммоль), все это перемешивали при комнатной температуре в течение 48 ч асов.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/1), получая 200 мг 2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионовой кислоты в виде белого твердого вещества (выход: 21%). LC-MS: RT=1,73 мин, [М+Н]+=384,27.
Стадия Е. Синтез метил-4-(2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата
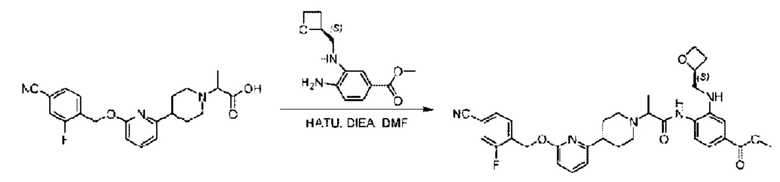
2-(4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионовую кислоту (200 мг; 0,51 ммоль) растворяли в Ν,Ν-диметилформамиде (10 мл) при комнатной температуре, затем добавляли HATU (388 мг; 1,02 ммоль) и DIEA(2 мл). Раствор этой смеси перемешивали в течение 0,5 часа, добавляли метил-(3)-4-амино-3-(((оксетан-2-ил)метил)амино)бензоат (144 мг; 0,61 ммоль) и затем перемешивали при комнатной температуре.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/1), получая 183 мг метил-4-(2-(4-(6-((4-циано-2-фторбензил)окси)пи ридин-2-ил)пиперидин-1-ил)пропионамидо)-3-((((3)-оксетан-2-ил)метил)амино)бензоата в виде светло-желтого твердого вещества (выход: 60%). LC-MS: RT=1,77 мин, [М+Н]+=602,32.
Стадия F. Синтез метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
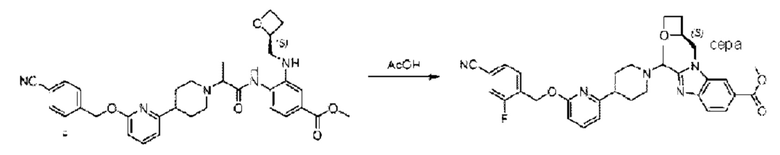
Метил-4-(2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоат (183 мг; 0,30 ммоль) растворяли в уксусной кислоте (2 мл) и взаимодействие проводили при температуре 60°С.
По завершении реакции получали неочищенный продукт незамедлительно после концентрирования и очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 7/1), получая 150 мг метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде коричневато-желтого твердого вещества (выход: 86%). LC-MS: RT=1,79 мин, [М+Н]+=584,32.
Стадия G Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (2А) и 2-((R)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (2В)
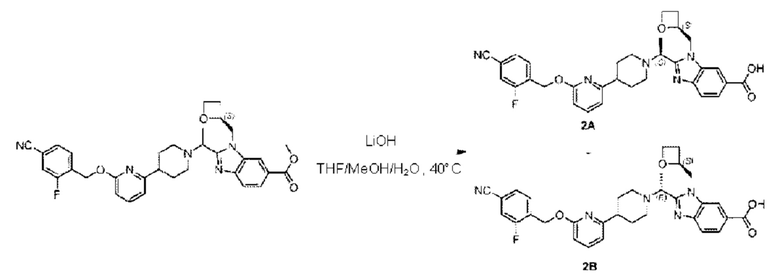
Метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((3)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (150 мг; 0,26 ммоль) растворяли в смеси растворителей (5 мл; тетрагидрофуран/метанол/вода=2,5/2,5/1) при комнатной температуре, затем добавляли гидроксид лития (40 мг; 1,05 ммоль) и взаимодействие проводили при температуре 40°С.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида аммония и реакционный раствор последовательно экстрагировали этилацетатом (10 мл × 2 раза) и смесью растворителей (10 мл × 2 раза; дихлорметан/метанол = 30/1). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который разделяли обращенно-фазовой HPLC в виде элюата, лиофилизацией) в следующих условиях разделения: хроматографическая колонка: препаративная 5-С18 от Agilent, 100 мм × 30 мм, 5 мкм; подвижная фаза: (для элюирования) вода (содержащая 0,1% аммиака) и ацетонитрил; скорость потока: 20 мл/мин; градиент: соединения 2А и 2В элюировались 23%-ным ацетонитрилом, в моменты времени 13,77 мин и 15,7 мин; соответственно; длина волны детектирования: 254 нм. Последовательно получали 18,93 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (2А) в виде белого твердого вещества (выход: 13%) и 31,15 мг 2-((R)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (2В) в виде белого твердого вещества (выход: 21%).
Соединение 2А: Η PLC: RT=13,77 мин.
LC-MS: RT=1,77 мин, [M+H]+=570,31. 1Н ЯМР (400 МГц, DMSO) δ 12.77 (brs, 1Н), 8.29-8.28 (m, 1Н), 7.87 (d, J=10,4 Гц, 1Н), 7.80 (dd, J=8,4; 1,6 Гц, 1Н), 7.70-7.66 (m, 3Н), 7.62 (dd, J=8,2; 7,4 Гц, 1 Η), 6.84 (d, J=7,1 Гц, 1 Η), 6.70 (d, J=7,7 Гц, 1H), 5.44 (brs, 2H), 5.19-5.14 (m, 1 Η), 4.83 (dd, J=15,5; 3,1 Гц, 1Η), 4.68 (dd, J=15,4; 5,6 Гц, 1Η), 4.50-4.39 (m, 2H), 4.18 (dt, J=9,1; 5,7 Гц, 1H), 2.98-2.94 (m, 1H), 2.67-2.45 (m, 4H), 2.37-2.19 (m, 2H), 1.84-1.61 (m, 3Н), 1.46 (d, J=6,8 Гц, 3Н), 1.54-1.43 (m, 1H).
Соединение 2В: Η PLC: RT=15,74 мин.
LC-MS: RT=1,77 мин, [M+H]+=570,35. 1Η ЯМР (400 МГц, DMSO-d6) δ 8.24 (s, 1Η), 7.87 (d, J=10,1 Гц, 1Η), 7.78 (dd, J=8,4; 1,4 Гц, 1Η), 7.72-7.67 (m, 2Η), 7.63 (dd, J=8,0; 6,2 Гц, 2H), 6.86 (d, J=7,3 Гц, 1H), 6.70 (d, J=8,1 Гц, 1Η), 5.46 (s, 2Η), 5.12-5.00 (m, 1Н), 4.89 (dd, J=15,1; 8,6 Гц, 1H), 4.63-4.40 (m, 4Н), 3.04 (d, J=10,8 Гц, 1H), 2.78-2.69 (m, 1Η), 2.61 (t, J=1 0,4 Гц, 1H), 2.55 (s, 2Н), 2.26 (t, J=1 0,7 Гц, 3Н), 1.96-1.59 (m, 4Н), 1.55-1.39 (m, 4Н).
Пример 3. 2-((S)-1-(4-(6-((5-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
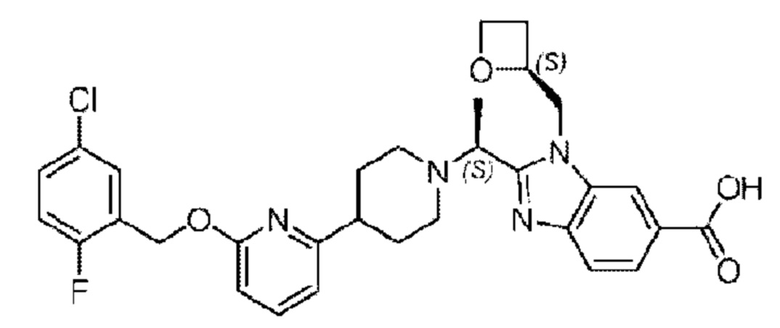
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 5-хлор-2-фторбензилметанола
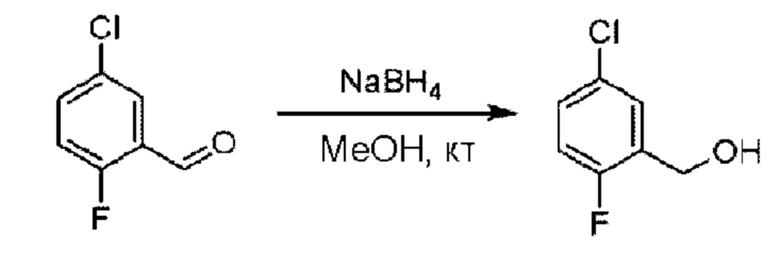
5-Хлор-2-фгорбензальдегид (500 мг; 3,16 ммоль) растворяли в метаноле (15 мл) при комнатной температуре и раствор этой смеси охлаждали до 0°С в ледяной бане, затем добавляли боргидрид натрия (234 мг; 6,33 ммоль). Раствор перемешивали при комнатной температуре.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия. Реакционный раствор экстрагировали дихлорметаном (30 мл), дважды промывали водой, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/3), получая 410 мг 5-хлор-2-фторбензилметанола в виде бесцветной маслянистой жидкости (выход: 81%). LC-MS: RT=1,78 мин.
Стадия В. Синтез трет-бутил-4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата
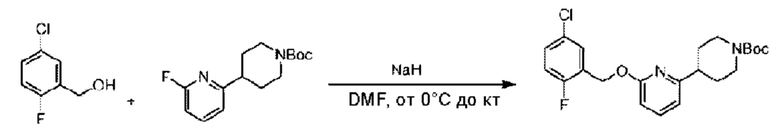
5-Хлор-2-фторбензилметанол (410 мг; 2,56 ммоль) растворяли в Ν,Ν-диметилформамиде (10 мл) и раствор этой смеси охлаждали до 0°С в ледяной бане, затем добавляли гидрид натрия (270 мг; 5,12 ммоль). Смесь перемешивали в течение 0,5 часа при комнатной температуре, добавляли трет-бутил-4-(6-фторпиридин-2-ил)пиперидин-1-карбоксилат (718 мг; 2,56 ммоль) и взаимодействие проводили при комнатной температуре.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия, реакционный раствор экстрагировали этилацетатом (30 мл), дважды промывали водой, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2), получая 550 мг трет-бутил-4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата в виде бесцветной маслянистой жидкости (выход: 51%). LC-MS: RT=2,41 мин, [М-55]+=365,17.
Стадия С. Синтез 2-((5-хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридина
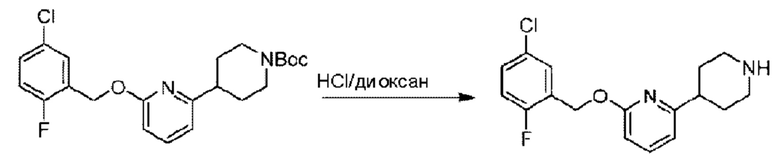
трет-Бутил-4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилат (550 мг; 1,31 ммоль) растворяли в соляной кислоте в диоксане (10 мл) и раствор этой смеси перемешивали при комнатной температуре. По завершении реакции после концентрирования при пониженном давлении получали 520 мг 2-((5-хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридина в виде белого твердого вещества. LC-MS: RT=1,72 мин, [М+Н]+=321,18.
Стадия D. Синтез метил-2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
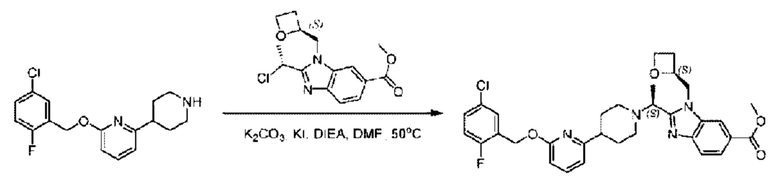
2-((5-Хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридин (150 мг; 0,47 ммоль), DIEA(1 мл), иодид калия (156 мг; 0,94 ммоль) и карбонат калия (130 г; 0,94 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре, затем добавляли метил-2-((R)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (144 мг; 0,47 ммоль) и раствор этой смеси нагревали до 50°С.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (20 мл × 2 раза). Органические фазы объединяли, сушили и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/8), получая 90 мг соединения метил-2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого твердого вещества (выход: 32%). LC-MS: RT=1,86 мин, [М+Н]+=593,31.
Стадия Е. Синтез 2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
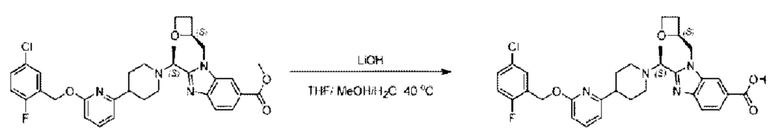
Соединение метил- 2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (90,0 мг; 0,15 ммоль) растворяли в смеси растворителей (5 мл; тетрагидрофуран/метанол/вода = 2,5/2,5/1) при комнатной температуре, затем добавляли гидроксид лития (23,0 мг; 0,61 ммоль) и взаимодействие проводили при температуре 40°С.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида аммония, реакционный раствор последовательно экстрагировали этилацетатом (10 мл × 2 раза) и смесью растворителей (10 мл × 2 раза, дихлорметан/метанол = 30/1). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20/1), получая 31,88 мг 2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 21%).
LC-MS: RT=1,81 мин, [М+Н]+=579,30. 1Н ЯМР (400 МГц, DMSO-d6) δ 12.72 (s, 1Η), 8.28 (s, 1Η), 7.79 (s, 1Η), 7.64 (dd, J=30,9; 7,9 Гц, 3Н), 7.41 (d, J=3,6 Гц, 1H), 7.26 (t, J=9,2 Гц, 1Η), 6.83 (d, J=7,4 Гц, 1Η), 6.67 (d, J=8,0 Гц, 1Η), 5.36 (d, J=9,1 Гц, 2H), 5.1 6 (s, 1H), 4.84 (d, J=1 3,6 Гц, 1Η), 4.69 (s, 1Η), 4.54-4.31 (m, 2H), 4.17 (dt, J=11,4; 5,7 Гц, 1Η), 2.97 (s, 1H), 2.62 (d, J=6,0 Гц, 2H), 2.24 (d, J=54,1 Гц, 3Н), 1.73 (dd, J=28,8; 21,5 Гц, 4H), 1.59-1.35 (m, 4H).
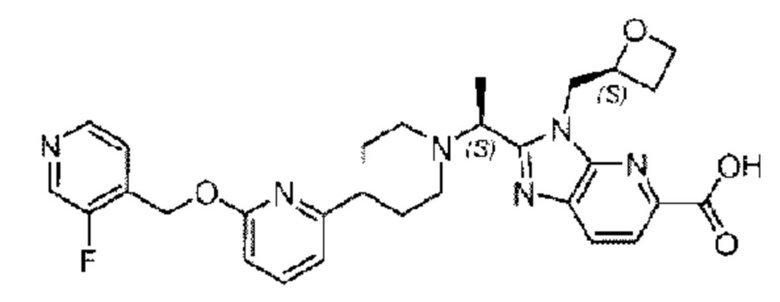
Пример 4. 2-((S)-1-(4-(6-((3-Фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновая кислота
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-формиата
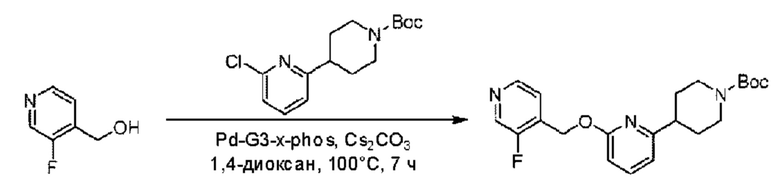
(3-Фторпиридин-4-ил)метанол (86 мг; 0,68 ммоль) и трет-бутил-4-(6-хлорпиридин-2-ил)пиперидин-1-формиат (201 мг; 0,68 ммоль) растворяли в 4 мл безводного диоксана, затем добавляли карбонат цезия (443 мг; 1,36 ммоль) и метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (58 мг; 0,068 ммоль). Три раза осуществляли заполнение азотом и взаимодействие проводили при 100°С в течение 7 часов.
По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (50 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта, который разделяли колоночной хроматографией (н-гексан/этилацетат = 4/1), получая 101 мг трет-бутил-4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-формиата в виде светло-желтого твердого вещества (выход: 38,3%).
Стадия В. Синтез 4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидина
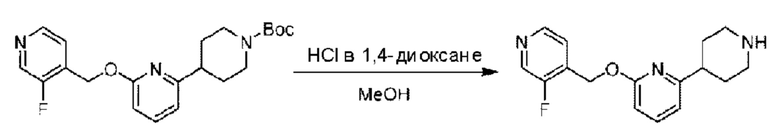
трет-Бутил-4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-формиат (101 мг; 0,26 ммоль) растворяли в 3 мл метанола и добавляли 4 Μ раствор соляной кислоты в диоксане (3 мл) при 0°С. Раствор этой смеси нагревали до комнатной температуры и реакцию проводили в течение 40 мин.
По завершении реакции реакционный раствор незамедлительно упаривали на роторном испарителе, получая 98 мг 4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидина в виде белого твердого вещества, которое использовали непосредственно на следующей стадии реакции. LC-MS: RT=1,56 мин, [М+Н]+=288,22.
Стадия С. Синтез метил-4-((S)-2-хлорпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата
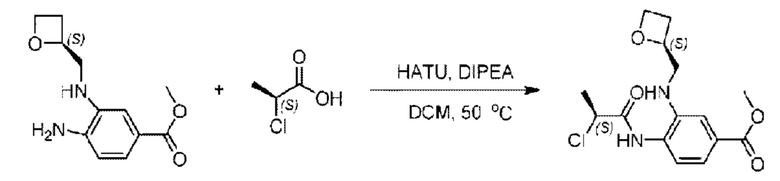
Метил-(3)-4-амино-3-(((оксетан-2-ил)метил)амино)бензоат (500,0 мг; 2,25 ммоль), (S)-2-хлорпропионовую кислоту (243,0 мг; 2,25 ммоль) растворяли в дихлорметане (10,0 мл) при комнатной температуре, затем добавляли 2-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфэсфат (1,28 г; 3,38 ммоль) и N,N-диизопропилэтиламин (3 мл). Взаимодействие проводили при 50°С в течение 2,0 часов в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: н-гексан/этилацетат = 2/1), получая 580,0 мг метил-4-((S)-2-хлорпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата в виде светло-желтого твердого вещества (выход: 79,1%). LC-MS: RT=1,82 мин, [М+Н]+=327,18.
Стадия D. Синтез метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
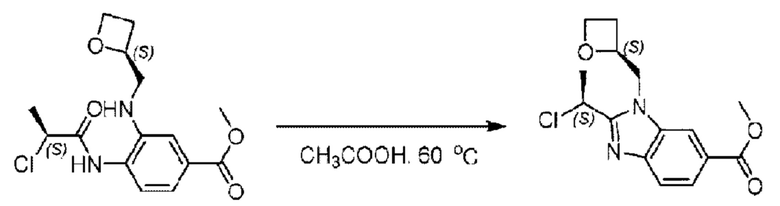
Метил-4-((S)-2-хлорпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)-бензоат (580,0 мг; 1,78 ммоль) растворяли в уксусной кислоте (10,0 мл) при комнатной температуре и взаимодействие проводили при 60°С в течение 12,0 часа в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и рН реакционного раствора подводили до 6 раствором бикарбоната натрия (0,5 моль/л). Реакционный раствор экстрагировали дихлорметаном (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: н-гексан:этилацетат = 4:1), получая 520,0 мг метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде светло-красного маслянистого вещества (выход: 94,8%). LC-MS: RT=1,86 мин, [М+Н]+=309,23.
Стадия Е. Синтез метил-2-((S)-1-(4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилата
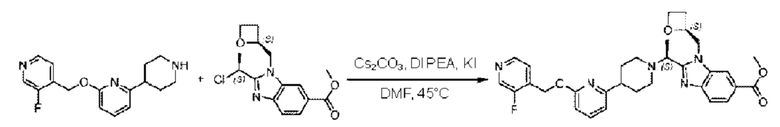
Метил-4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин (55,0 мг; 0,19 ммоль), метил-2-((3)-1-хлорэтил)-1-(((3)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (58,5 мг; 0,19 ммоль), иодид калия (31,5 мг; 0,19 ммоль) и карбонат цезия (123,1 мг; 0,38 ммоль) добавляли к Ν,Ν-диметилформамиду (10,0 мл) при комнатной температуре, затем добавляли Ν,Ν-диизопропилэтиламин (1,0 мл). Взаимодействие проводили при 45°С в течение 3,0 часа в атмосфере Ν2.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20/1), получая 65,0 мг метил-2-((S)-1-(4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилата в виде светло-желтого твердого вещества (выход: 58,6%). LC-MS: RT=1,73 мин, [М+Н]+=560,34.
Стадия F. Синтез 2-((S)-1-(4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновой кислоты
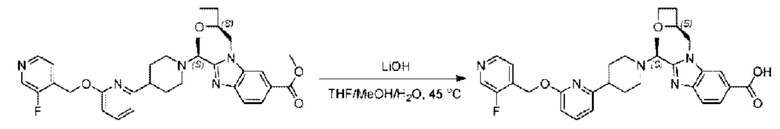
Гидроксид лития (20,0 мг; 0,48 ммоль) в воде (1,0 мл) по каплям добавляли к тетрагидрофурану (3,0 мл), содержащему метил-2-((S)-1-(4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилат (65,0 мг; 0,11 ммоль), при комнатной температуре, затем добавляли 1,0 мл метанола. Взаимодействие проводили при 45°С в течение 3,0 часа.
Реакцию гасили водой и рН подводили до 6 водным раствором хлорида аммония (0,5 моль/л). Реакционный раствор экстрагировали дихлорметаном (30 мл × 3 раза) и органические фазы объединяли, промывали водным раствором хлорида аммония (30 мл × 2 раза), сушили с использованием безводного сульфата натрия с получением неочищенного продукта, который очищали разделением на колонке (элюент: дихлорметан/метанол = 15/1), получая 32 мг 2-((S)-1-(4-(6-((3-фторпиридин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновой кислоты в виде светло-желтого твердого вещества (выход: 51,1%). LC-MS: RT=1,68 мин, [М+Н]+=546,31.
Пример 5. 2-((S)-1-(4-(6-((2,5-Дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
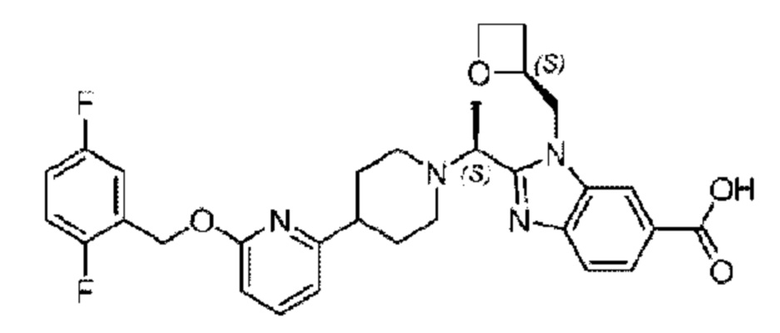
Конкретный путь синтеза приведен ниже.
Стадия А: Синтез (2,5-ди фторфенил)метанола
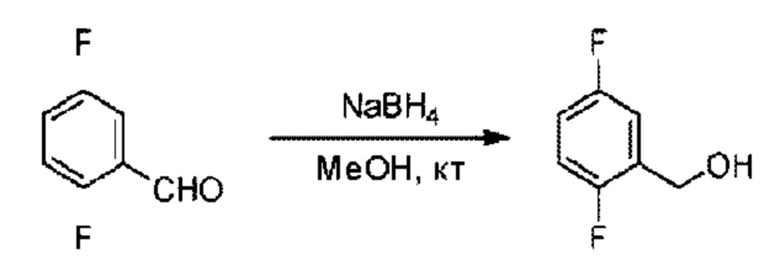
2,5-Дифторбензальдегид (500,0 мг; 3,52 ммоль) добавляли к метанолу (30,0 мл) в ледяной бане, затем порциями добавляли боргидрид натрия (200,6 мг; 5,28 ммоль). Реакцию проводили с перемешиванием при комнатной температуре в течение 1,0 часа в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении, получая 402,0 мг (2,5-дифторфенил)метанола в виде бесцветного маслянистого вещества (выход: 80,4%).
Стадия В. Синтез метил-4-((R)-2-хлорпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата
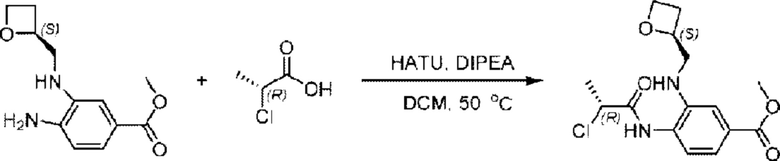
Метил-(S)-4-амино-3-(((оксетан-2-ил)метил)амино)бензоат (500,0 мг; 2,25 ммоль), (Р)-2-хлорпропионовую кислоту (243,0 мг; 2,25 ммоль) растворяли в дихлорметане (10,0 мл) при комнатной температуре, затем добавляли 2-(7-азабензотриазол-1-ил)-N;N,N',N'-тетраметилурония гексафторфосфат (1,28 г; 3,38 ммоль) и N,N-диизопропилэтиламин (3 мл). Взаимодействие проводили при 50°С в течение 2,0 часа в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: н-гексан/этилацетат = 2:1), получая 586,0 мг метил-4-((R)-2-хлорпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата в виде светло-желтого твердого вещества (выход: 79,8%). LC-MS: RT = 1,82 мин, [M+Н]+ = 327,17.
Стадия С.Синтез метил-2-((R)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
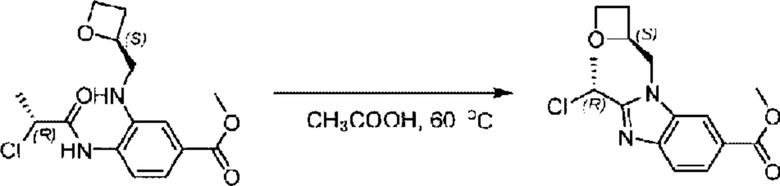
Метил-4-((R)-2-хлорпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоат (586,0 мг; 1,80 ммоль) растворяли в уксусной кислоте (10,0 мл) при комнатной температуре и взаимодействие проводили при 60°С в течение 12,0 часа в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и рН реакционного раствора подводили до 6 раствором бикарбоната натрия (0,5 моль/л). Реакционный раствор экстрагировали дихлорметаном (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: н-гексан:этилацетат = 4:1), получая 408,0 мг метил-2-((R)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде светло-красного маслянистого вещества (выход: 73,5%). LC-MS: RT = 1,84 мин, [М+Н]+ = 309,23.
Стадия D. Синтез трет-бутил-4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-формиата
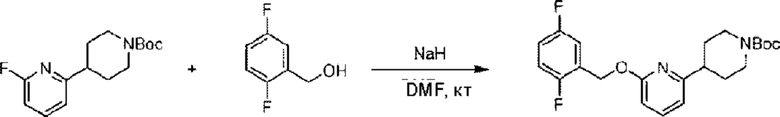
(2,5-Дифторфенил)метанол (50,0 мг; 0,35 ммоль) добавляли к N,N-
диметилформамиду (4,0 мл) в ледяной бане, затем порциями добавляли 60 масс. % гидрида натрия (28,0 мг; 0,70 ммоль). После перемешивания в течение 0,5 часа при комнатной температуре в атмосфере N2 добавляли трет-бутил-4-(6-фторпиридин-2-ил)пиперидин-1-формиат (98,0 мг; 0,35 ммоль) и реакцию проводили в течение 1,0 часа при комнатной температуре.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 50/1), получая 88,0 мг трет-бутил-4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-формиата в виде белого твердого вещества (выход: 62,2%). LC-MS: RT = 2,33 мин, [М+Н-56]+ = 349,33.
Стадия Е. Синтез 2-((2,5-фторбензил)окси)-6-(пиперидин-4-ил)пиридина
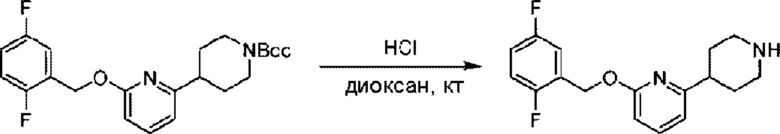
трет-Бутил-4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-формиат (88,0 мг; 0,22 ммоль) добавляли к раствору соляной кислоты в диоксане (1,00 ммоль/мл, 3 мл) при комнатной температуре и взаимодействие проводили при комнатной температуре в течение 1 часа в атмосфере N2.
По завершении реакции реакционный раствор концентрировали при пониженном давлении. Получали 58,0 мг 2-((2,5-фторбензил)окси)-6-(пиперидин-4-ил)пиридина в виде молочно-белого твердого вещества (выход: 87,4%), которое использовали непосредственно на следующей стадии реакции. LC-MS: RT = 1,68 мин, [М+Н]+ = 305,22.
Стадия F. Синтез метил-2-(S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
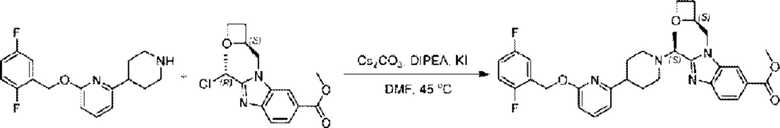
2-((2,5-Фторбензил)окси)-6-(пиперидин-4-ил)пиридин (58,0 мг; 0,19 ммоль), метил-2-((R)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (58,5 мг; 0,19 ммоль), иодид калия (31,5 мг; 0,19 ммоль) и карбонат цезия (123,1 мг; 0,38 ммоль) добавляли к N,N-диметилформамиду (10,0 мл) при комнатной температуре, затем добавляли N,N-диизопропилэтиламин (1,0 мл). Взаимодействие проводили при 45°С в течение 3,0 часа в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20/1), получая 68,0 мг метил- 2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде светло-желтого твердого вещества (выход: 62,1%). LC-MS: RT = 1,85 мин, [М+Н]+ = 577,39.
Стадия G. Синтез 2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
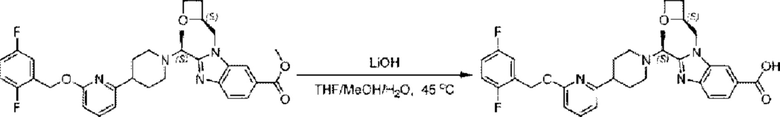
Гидроксид лития (20,0 мг; 0,48 ммоль) в воде (1,0 мл) по каплям добавляли к тетрагидрофурану (3,0 мл), содержащему метил-2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (68,0 мг; 0,12 ммоль), при комнатной температуре, затем добавляли 1,0 мл метанола. Взаимодействие проводили при 45°С в течение 3,0 часа.
По завершении реакции реакционную смесь гасили водой и рН подводили до 6 водным раствором хлорида аммония (0,5 моль/л). Реакционный раствор экстрагировали дихлорметаном (30 мл × 3 раза), органические фазы объединяли, промывали водным раствором хлорида аммония (30 мл × 2 раза) и сушили с использованием безводного сульфата натрия, получая неочищенный продукт, который очищали препаративной высокоэффективной жидкостной хроматографией. Условия очистки приведены ниже: колонка: препаративная 5-С18 от Agilent, 100 мм×30 мм, 5 мкм; подвижная фаза: вода (содержащая 0,05% аммиака) и ацетонитрил; скорость потока: 20 мл/мин; градиент: продукт элюировался 28%-ным ацетонитрилом в момент времени 8,00 мин; длина волны детектирования: 254 нм. После очистки и лиофилизации получали 24,0 мг соединения 2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в 30 виде белого твердого вещества (выход: 35,8%).
LC-MS: RT = 1,79 мин, [М+Н]+ = 563,32. 1Н ЯМР (400 МГц, DMSO) δ 12.70 (brs, 1Н), 8.30-8.29 (m, 1Н), 7.83-7.79 (m, 1Н), 7.69 (d, J = 8,5 Гц, 1Н), 7.65-7.60 (m, 1Н), 7.38-7.34 (m, 1Н), 7.31-7.19 (m, 2Н), 6.85 (d, J = 7,2 Гц, 1 H), 6.69 (d, J = 8,2 Гц, 1H), 5.36 (brs, 2Н), 5.20-5.15 (m, 1Н), 4.89-4.82 (m, 1Н), 4.68 (dd, J = 15,1; 5,2 Гц, 1H), 4.50-4.42 (m, 2Н), 4.19 (dt, J = 9,0; 5,7 Гц, 1 H), 2.99-2.96 (m, 1Н), 2.70-2.49 (m, 4Н), 2.38-2.21 (m, 2Н), 1.86-1.68 (m, 3Н), 1.59-1.45 (m, 1Н), 1.48 (d, J = 6,6 Гц, 3Н).
Пример 6. 2-((S)-1-(4-(6-((2,4-Дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этал)-1-(((S)-оксетан-2-ил)метал)-1Н-бензо[d]имидазол-6-карбоновая кислота
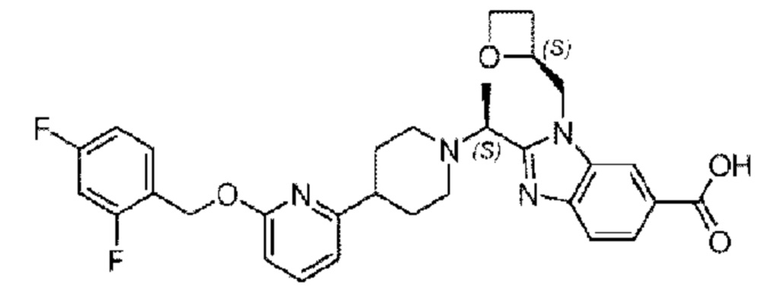
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-((2,4-дифторбензил)окси)пиридин-2-ил)пиперидин-1-формиата
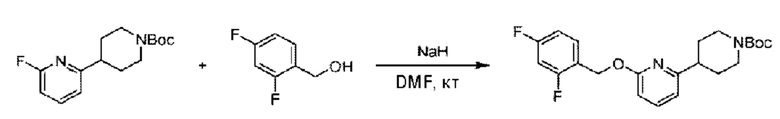
(2,4-Дифторфенил)метанол (50,0 мг; 0,35 ммоль) добавляли к N,N-диметилформамиду (4,0 мл) в ледяной бане, затем порциями добавляли 60 масс. %-ный гидрид натрия (28,0 мг; 0,70 ммоль). После перемешивания в течение 0,5 часа при комнатной температуре в атмосфере N2 добавляли трет-бутил-4-(6-фторпиридин-2-ил)пиперидин-1-формиат (98,0 мг; 0,35 ммоль) и реакцию проводили в течение 1,0 ч аса при комнатной температуре.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 50/1), получая 76,0 мг трет-бутил-4-(6-((2,4-дифторбензил)окси)пиридин-2-ил)пиперидин-1-формиата в виде белого твердого вещества (выход: 54,2%). LC-MS: RT = 2,33 мин, [М+Н-56]+ = 349,33.
Стадия В. Синтез 2-((2,4-дифторбензил)окси)-6-(пиперидин-4- ил)пиридина
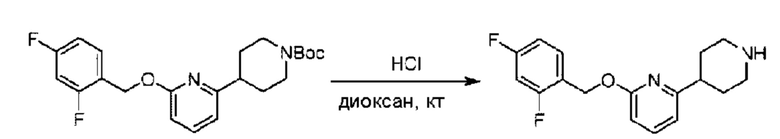
трет-Бутил-4-(6-((2,4-дифторбензил)окси)пиридин-2-ил)пиперидин-1-формиат (76,0 мг; 0,19 ммоль) добавляли к раствору соляной кислоты в диоксане (1,00 ммоль/мл; 3 мл) при комнатной температуре и взаимодействие проводили при комнатной температуре в течение 1 часа в атмосфере N2.
По завершении реакции реакционный раствор концентрировали при пониженном давлении. Получали 43,0 мг 2-((2,4-дифторбензил)окси)-6-(пиперидин-4-ил)пиридина в виде молочно-белого твердого вещества (выход: 74,4%), которое использовали непосредственно на следующей стадии реакции. LC-MS: RT = 1,68 мин, [М+Н]+ = 305,22.
Стадия С.Синтез метил-2-((S)-1-(4-(6-((2,4-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
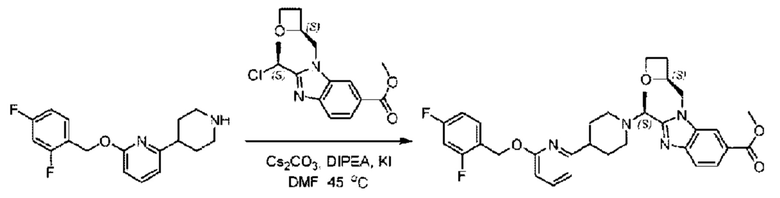
2-((2,4-Дифторбензил)окси)-6-(пиперидин-4-ил)пиридин (43,0 мг; 0,14 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (43,5 мг; 0,14 ммоль), иодид калия (23,2 мг; 0,14 ммоль) и карбонат цезия (91,3 мг; 0,28 ммоль) добавляли к N,N-диметилформ амиду (10,0 мл) при комнатной температуре, затем добавляли N,N-диизопропилэтиламин (1,0 мл). Взаимодействие проводили при 45°С в течение 3,0 часа в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20/1), получая 59,0 мг метил-2-((S)-1-(4-(6-((2,4-дифторбензил)окси)пиридин-2-ил)пипери дин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде светло-желтого твердого вещества (выход: 73,2%). LC-MS: RT = 1,85 мин, [М+Н]+ = 577,39.
Стадия D. Синтез 2-((S)-1-(4-(6-((2,4-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
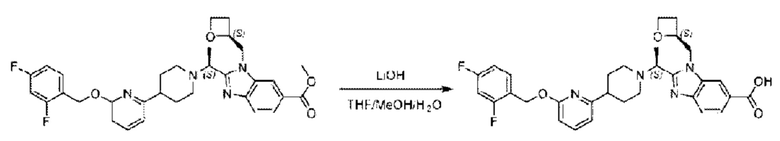
Гидроксид лития (16,8 мг; 0,40 ммоль) в воде (1,0 мл) по каплям при комнатной температуре добавляли к тетрагидрофурану (3,0 мл), содержащему метил-2-((S)-1-(4-(6-((2,4-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (59,0 мг; 0,10 ммоль), затем добавляли 1,0 мл метанола. Взаимодействие проводили при 45°С в течение 3,0 часа.
Реакцию гасили водой и рН подводили до 6 водным раствором хлорида аммония (0,5 моль/л). Реакционный раствор экстрагировали дихлорметаном (30 мл × 3 раза) и органические фазы объединяли, промывали водным раствором хлорида аммония (30 мл × 2 раза), сушили с использованием безводного сульфата натрия, получая неочищенный продукт, который очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 10/1), получая 17,3 мг 2-((S)-1-(4-(6-((2,4-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде светло-желтого твердого вещества (выход: 32,0%). LC-MS: RT = 1,79 мин, [М+Н]+ = 563,32.
1Н ЯМР (500 МГц, DMSO) δ 8.29 (s, 1Н), 8.04 (s, 1Н), 7.81 (dd, J = 8,4; 1,6 Гц, 1Н), 7.69 (d, J = 8,5 Гц, 1Н), 7.30-7.23 (m, 1Н), 7.11-7.04 (m, 1Н), 6.84 (d, J = 7,2 Гц, 1Н), 6.65 (d, J = 7,7 Гц, 1Н), 6.15 (t, J = 6,7 Гц, 1Н), 5.35 (s, 2Н), 5.27 (d, J = 4,2 Гц, 1Н), 5.04 (t, J = 5,2 Гц, 1Н), 4.85 (dd, J = 15,5; 3,1 Гц, 1Н), 4.69 (dd, J = 15,8; 5,8 Гц, 1Н), 4.49-4.42 (m, 3Н), 4.07 (d, J = 2,1 Гц, 3Н), 3.80-3.77(m, 1Н), 2.11 (dd, J = 8,4; 6,8 Гц, 3Н), 1.72 (dd, J = 16,0; 7,2 Гц, 3Н), 1.48 (d, J = 6,7 Гц, 3Н).
Пример 7. 2-((S)-1-(4-(6-((5-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновая кислота
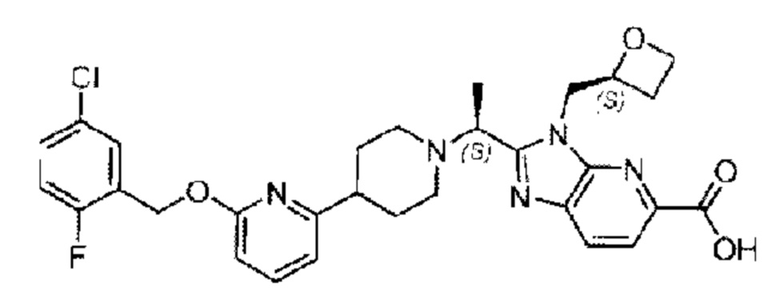
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-5-((S)-2-хлорпропионамидо)-6-((((S)-оксетан-2-ил)метил)амино)пиколината
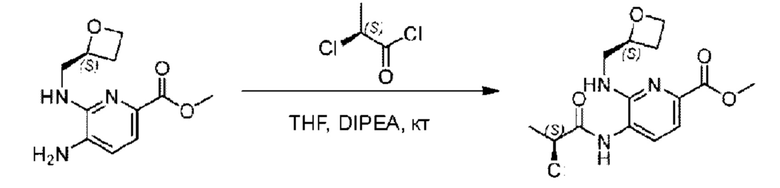
Метил-(S)-5-амино-6-(((оксетан-2-ил)метил)амино)пиколинат (0,8 г; 2,Зммоль) растворяли в тетрагидроф/ране (30,0 мл), затем добавляли триэтиламин (1,0 мл; 8,0 ммоль). Раствор этой смеси перемешивали при 0°С в течение 15 мин, затем медленно по каплям добавляли раствор (S)-2-хлорпропионилхлорида (350,0 мг; 2,76 ммоль) в тетрагидроф/ране (10,0 мл). Раствор перемешивали при комнатной температуре в течение 1 часа и за ходом реакции до ее завершения следили с использованием LC-MS.
К реакционному раствору добавляли воду (20,0 мл), затем его экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (25,0 мл × 3 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении с получением остатка, который очищали хроматографией на колонке с силикагелем, получая 600 мг метил-5-((S)-2-хлорпропионамидо)-6-((((S)-оксетан-2-ил)метил)амино)пиколината в виде светло-желтого твердого вещества (выход: 62,2%). LC-MS: RT = 1,73 мин, [М+Н]+ = 328,21.
Стадия В. Синтез метил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилата
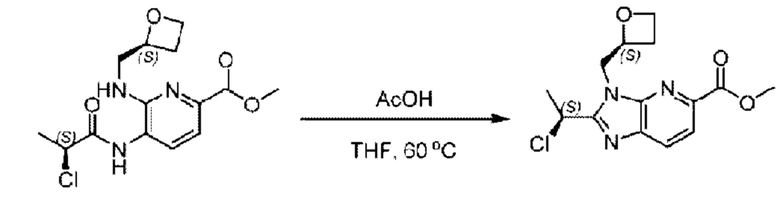
Метил-5-((S)-2-хлорпропионамидо)-6-((((S)-оксетан-2-ил)метил)амино)-пиколинат (0,4 г; 1,23 ммоль) растворяли в тетрагидроф/ране (10,0 мл), затем добавляли уксусную кислоту (1,0 мл). Раствор этой смеси перемешивали при 60°С в течение 2 часов и заходом реакции до ее завершения следили с использованием LC-MS.
К реакционному раствору добавляли воду (20,0 мл), затем экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (25,0 мл × 3 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении с получением остатка, который очищали хроматографией на колонке с силикагелем, получая 300 мг метил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)- имидазоло[4,5-b]пиридин-5-карбоксилата в виде желтого твердого вещества (выход: 78,3%). LC-MS: RT = 1,79 мин, [М+Н]+ = 310,11.
Стадия С. Синтез метил-2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилата
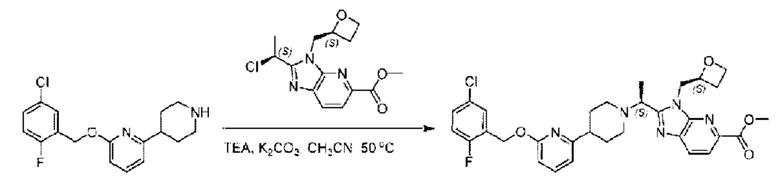
2-((5-Хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридин (100,0 мг; 0,28 ммоль) растворяли в ацетонитриле (10,0 мл) и триэтиламине (2,0 мл), затем добавляли карбонат калия (77,0 мг; 0,56 ммоль), иодид калия (93,0 мг; 0,56 ммоль) и метил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилат (82,0 мг; 0,28 ммоль). Раствор этой смеси перемешивали при 50°С в течение 2 часов и за ходом реакции до ее завершения следили с использованием LC-MS.
К реакционному раствору добавляли воду (30,0 мл), затем экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (25,0 мл × 3 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 120 мг метил-2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилата в виде желтого твердого вещества (выход: 72,3%). LC-MS: RT = 1,81 мин, [М+Н]+ = 594,21.
Стадия D. Синтез 2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновой кислоты
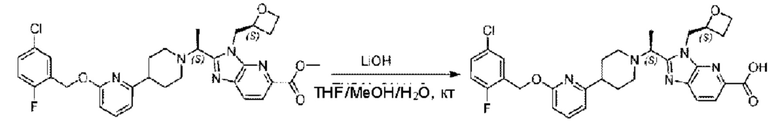
Метил-2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилат (120,0 мг; 0,73 ммоль) растворяли в тетрагидрофуране (30,0 мл) и метаноле (10,0 мл), затем добавляли раствор гидроксида лития (84,0 мг; 2,0 ммоль) в воде (10,0 мл) и раствор этой смеси перемешивали при комнатной температуре в течение 2 часов.
По завершении реакции по данным LC-MS реакционный раствор экстрагировали смесью дихлорметан/метанол (9/1; 20,0 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (15,0 мл × 3 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем, получая 46 мг 2-((S)-1-(4-(6-((5-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновой кислоты в виде белого твердого вещества (выход: 63,8%).
LS-MS: RT = 1,77 мин, [М+Н]+ = 580,31. 1Н ЯМР (400 МГц, DMSO) δ 8.17 (d, J = 8,2 Гц, 1Н), 7.99 (d, J = 8,2 Гц, 1Н), 7.61 (dd, J = 10,8; 4,9 Гц, 2Н), 7.45-7.38 (m, 1Н), 7.27 (d, J = 9,3 Гц, 1Н), 6.84 (d, J = 7,3 Гц, 1Н), 6.67 (d, J = 8,2 Гц, 1Н), 5.36 (s, 2Н), 5.31 (s, 1Н), 4.85 (dd, J = 15,0; 4,3 Гц, 1Н), 4.79-4.68 (m, 1Н), 4.61 (d, J = 6,6 Гц, 1Н), 4.41 (dd, J = 13,7; 7,7 Гц, 1Н), 4.13-4.02 (m, 1Н), 3.00 (d, J = 11,4 Гц, 3Н), 2.60 (dd, J = 22,6; 10,7 Гц, 4Н), 2.36 (s, 1Н), 2.30-2.21 (m, 1Н), 1.80 (d, J = 23,1; 2Н), 1.73-1.63 (m, 1Н), 1.56 (d, J = 8,6 Гц, 1Н), 1.48 (d, J = 6,6 Гц, 3Н).
Пример 8. 2-((S)-1-(4-(6-((1-(2,2-Дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
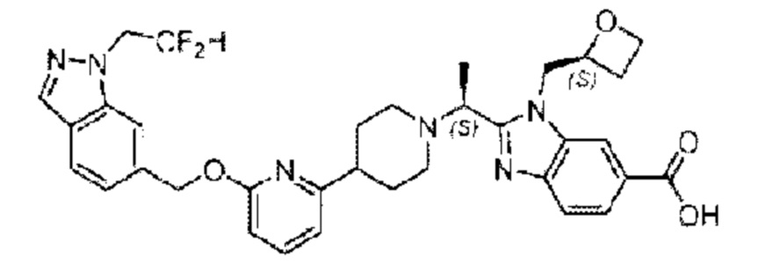
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этал)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
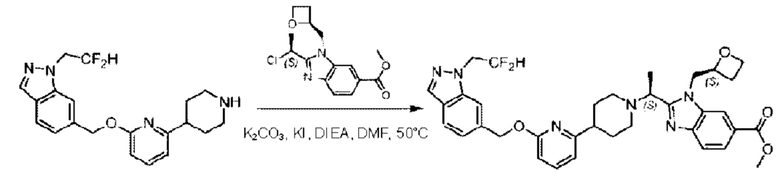
1-(2,2-Дифторэтил)-6-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)-1Н-индазол (150 мг; 0,40 ммоль), DIEA (1 мл), иодид калия (133 мг; 0,80 ммоль) и карбонат калия (110 мг; 0,80 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре, затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (124 мг; 0,40 ммоль) и взаимодействие проводили при температуре 50°С.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (20 мл × 2 раза). Органические фазы объединяли, сушили и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/8), получая 69,0 мг метил-2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого твердого вещества (выход: 27%). LC-MS: RT = 1,79 мин, [М+Н]+ = 645,42.
Стадия а Синтез 2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этал)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
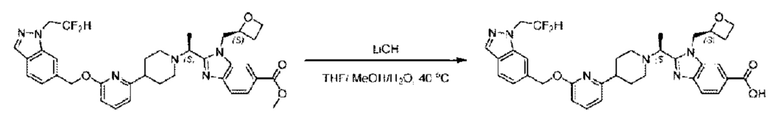
Метил-2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (69,0 мг; 0,11 ммоль) растворяли в смеси растворителей (5 мл; тетрагидрофуран/метанол/вода = 2,5/2,5/1) при комнатной температуре, затем добавляли гидроксид лития (16,0 мг; 0,44 ммоль) и взаимодействие проводили при температуре 40°С.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида аммония, реакционный раствор последовательно экстрагировали этилацетатом (10 мл × 2 раза) и смесью растворителей (дихлорметан/метанол = 30/1; 10 мл × 2 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20/1), получая 19,73 мг 2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 29%).
LS-MS: RT = 1,75 мин, [М+Н]+ = 631,40. 1Н ЯМР (400 МГц, DMSO-d6) δ 12.73 (s, 1Н), 8.27 (s, 1Н), 8.1 2 (s, 1Н), 7.77 (dd, J = 1 8,7; 7,8 Гц, 3Н), 7.70-7.56 (m, 2Н), 7.26 (d, J = 8,3 Гц, 1Н), 6.82 (d, J = 7,4 Гц, 1Н), 6.67 (d, J = 8,1 Гц, 1Н), 6.52 (s, 0.31Н), 6.39 (t, J = 3,4 Гц, 0.47Н), 6.25 (s, 0.29Н), 5.45 (s, 2Н), 5.16 (s, 1Н), 4.90 (d, J = 12,1 Гц, 1H), 4.66 (d, J = 10,9 Гц, 1H), 4.52-4.37 (m, 2Н), 4.22-4.11 (m, 1Н), 2.96 (s, 1Н), 2.62 (s, 2Н), 2.39-2.11 (m, 3Н), 1.77 (dd, J = 33,1; 20,9 Гц, 4Н), 1.51 (dd, J = 35,0; 7,7 Гц, 4Н).
Пример 9. 1-(((S)-Оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоновая кислота
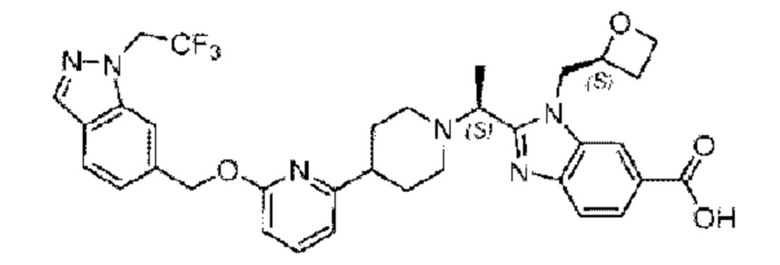
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилата
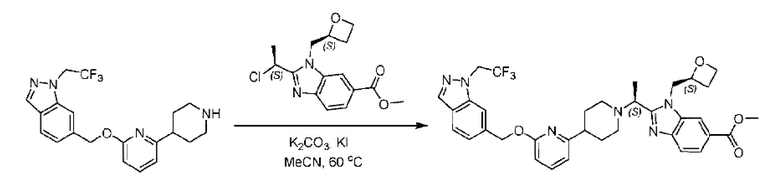
6-(((6-(Пиперидин-4-ил)пиридин-2-ил)окси)метил)-1-(2,2,2-трифторэтил)-1Н-индазол (75 мг; 0,176 ммоль), карбонат калия (48,6 мг; 0,352 ммоль) и иодид калия (58 мг; 0,352 ммоль) растворяли в 5 мл безводного ацетонитрила, затем добавляли метил-2-(S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (54 мг; 0,176 ммоль) и взаимодействие проводили при 60°С в течение 2 часов.
По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении, получая 96 мг метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилата в виде светло-желтого твердого вещества. LC-MS: RT = 1,86 мин, [М+Н]+ = 663,38.
Стадия В. Синтез 1-(((S)-оксетан-2-ил)мети n)-2-((S)-1-(4-(6-((1 -(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
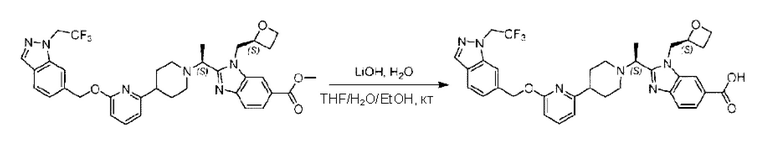
Метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1H-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилат (96 мг; 0,145 ммоль) растворяли в 4 мл тетрагидрофурана, затем добавляли гидроксида лития моногидрат (31 мг; 0,723 ммоль), 1 мл воды и 1 мл этанола. Взаимодействие проводили при комнатной температуре в течение 50 мин.
По завершении реакции реакционный раствор нейтрализовали, добавляя 20 мл насыщенного раствора хлорида аммония, и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией, получая 42 мг 1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде светло-желтого твердого вещества.
1Н ЯМР (500 МГц, DMSO) δ 12.65 (s, 1Н), 8.29 (s, 1Н), 8.19 (d, J = 0,9 Гц, 1Н), 7.88 (s, 1Н), 7.77-7.83 (m, 2Н), 7.69 (d, J = 8,4; 1Н), 7.59-7.65 (m, 1Н), 7.31 (dd, J = 8,3; 1,2 Гц, 1Н), 6.84 (d, J = 7,3 Гц, 1Н), 6.69 (d, J = 8,2 Гц, 1Н), 5.48 (s, 2Н), 5.42 (q, J = 9,2 Гц, 2Н), 5.12-5,18 (m, 1Н), 4.80-4.84 (m, 1Н), 4.66-4.70 (m, 1Н), 4.40-4.49 (m, 2Н), 4.16-4.20 (m, 1Н), 2.95-3.02 (m, 1Н), 2.54-2.69 (m, 4Н), 2.22-2.35 (m, 2Н), 1.85-1.90 (m, 1Н), 1.72-1.78 (m, 2Н), 1.53-1.60 (m, 1Н), 1.48 (d, J = 6,5 Гц, 3Н). LS-MS: RT = 1,77 мин, [М+Н]+ = 649,37.
Пример 10. 2-((S)-1-(4-(6-(Изохинолин-6-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
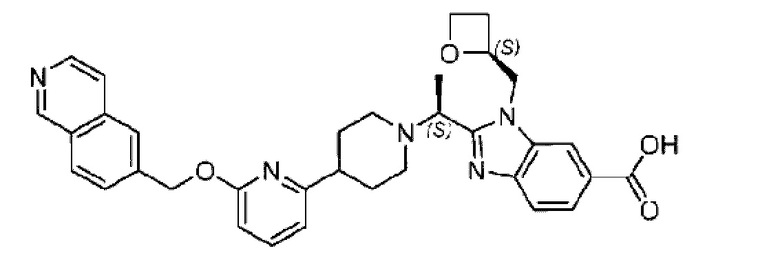
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-((S)-1-(4-(6-(изохинолин-6-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
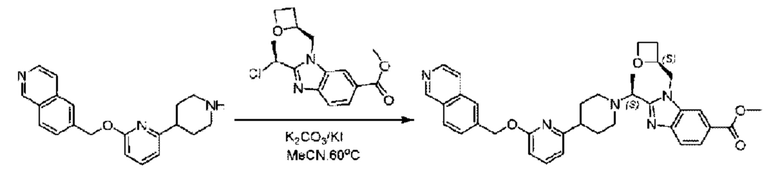
6-(((6-(Пиперидин-4-ил)пиридин-2-ил)окси)метил)изохинолин (100 мг; 0,281 ммоль), карбонат калия (73,1 мг; 0,563 ммоль) и иодид калия (92 мг; 0,563 ммоль) растворяли в 5 мл безводного ацетонитрила, затем добавляли метил-2-(S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (86 мг; 0,281 ммоль) и взаимодействие проводили при 60°С в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении, получая 78 мг метил-2-((S)-1-(4-(6-(изохинолин-6-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде светло-желтого твердого вещества. LC-MS: RT = 1,71 мин, [М+Н]+ = 592,28.
Стадия В. Синтез 2-((S)-1-(4-(6-(изохинолин-6-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
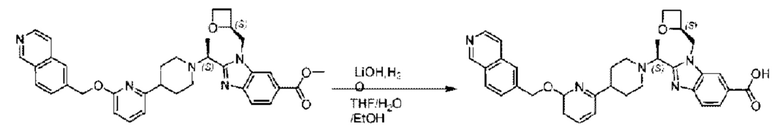
Метил-2-((S)-1-(4-(6-(изохинолин-6-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (78 мг; 0,131 ммоль) растворяли в 4 мл тетрагидрофурана, затем добавляли гидроксида лития моногидрат (28 мг; 0,655 ммоль), 1 мл воды и 1 мл этанола. Взаимодействие проводили при комнатной температуре в течение 50 мин.
По завершении реакции реакционный раствор нейтрализовали, используя 20 мл насыщенного раствора хлорида аммония, и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении с получением неочищенного продукта, который очищали колоночной хроматографией, получая 38 мг 2-((S)-1-(4-(6-(изохинолин-6-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде светло-желтого твердого вещества.
1Н ЯМР (400 МГц, DMSO) δ 12.20 (s, 1Н), 9.27 (s, 1Н), 8.40 (d, J = 5,5 Гц, 1Н), 8,28-8,33 (m, 1Н); 8.10 (d, J = 8,5 Гц, 1Н), 7.89-8.04 (s, 1Н), 7.80-7.85 (m, 1Н), 7.68-7.78 (m, 3Н), 7.60-7.67 (m, 1Н), 6.84 (d, J = 7,1 Гц, 1Н), 6.73 (d, J = 8,0 Гц, 1Н), 5.56 (s, 2Н), 5.15-5,20 (m, 1Н), 4.81-4.86 (m, 1Н), 4.64-4.74 (m, 1Н), 4.38-4.51 (m, 2Н), 4.15-4.20 (m, 1Н), 2.94-3.00 (m, 1Н), 2.57-2.66 (m, 2Н), 2.1 4-2.35 (m, 4Н), 1.77-1.85 (m, 2Н), 1.66-1.75 (m, 2Н), 1.45-1.52 (m, 3Н). LS-MS: RT = 1,60 мин, [М+Н]+ = 578,39.
Пример 11. 2-((S)-1-(4-(6-((1-(2,2-Дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота
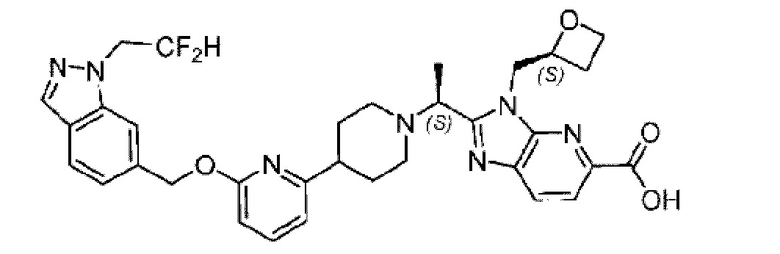
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата
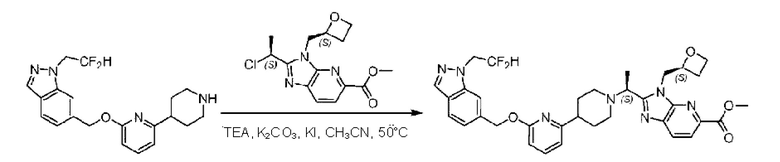
1-(2,2-Дифторэтил)-6-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)-1Н-индазол (150,0 мг; 0,37 ммоль) растворяли в ацетонитриле (10,0 мл) и триэтиламине (2,0 мл), затем добавляли карбонат калия (102,0 мг; 0,74 ммоль), иодид калия (123,0 мг; 0,74 ммоль) и метил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилат (114,0 мг; 0,37 ммоль). Раствор этой смеси перемешивали при 50°С в течение 2 часов.
После завершение реакции по данным TLC реакционный раствор разбавляли водой (30,0 мл) и экстрагировали этилацетатом (40 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (25,0 мл × 3 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 180 мг метил-2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбокс плата в виде желтого твердого вещества. LC-MS: RT = 1,78 мин, [М+Н]+ = 646,34.
Стадия В. Синтез 2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты
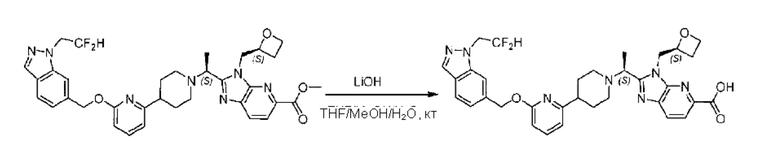
Метил-2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (180,0 мг; 0,28 ммоль) растворяли в тетрагидрофуране (6,0 мл) и метаноле (2,0 мл), затем добавляли гидроксид лития (21,0 мг; 0,5 ммоль) в воде (2,0 мл) и раствор этой смеси перемешивали при комнатной температуре в течение 1 часа.
По завершении реакции по данным LC-MS реакционный раствор экстрагировали смесью дихлорметан/метанол (9/1; 20,0 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (15,0 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем, получая 64 мг 2-((S)-1-(4-(6-((1-(2,2-дифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты в виде белого твердого вещества (выход 36,2%).
LS-MS: RT = 1,73 мин, [М+Н]+ = 632,35. 1Н ЯМР (400 МГц, DMSO) δ 8.14 (s, 1Н), 8.06 (d, J = 8,1 Гц, 1Н), 7.95 (d, J = 8,1 Гц, 1Н), 7.81 (s, 1Н), 7.75 (d, J = 8,3 Гц, 1Н), 7.61 (t, J = 7,8 Гц, 1Н), 7.26 (d, J = 8,2 Гц, 1Н), 6.82 (d, J = 7,3 Гц, 1Н), 6.67 (d, J = 8,2 Гц, 1Н), 6.54 (t, J = 3,8 Гц, 0,27Н), 6.40 (m, 0,49Н), 6.27 (m, 0,28Н), 5.45 (d, J = 3,2, Гц, 2Н), 5.20 (s, 1Н), 4.89 (td, J = 15,4; 3,5 Гц, 2Н), 4.76 (s, 2Н), 4.65-4.57 (m, 1Н), 4.34 (dd, J = 13,5; 7,8 Гц, 1Н), 4.06-4.01 (m, 1Н), 4.00-3,93 (m, 1Н), 2.98 (d, J = 10,5; 1Н), 2.61 (d, J = 11,0; 2Н), 2.23 (t, J = 10,8; 1 H), 2.10 (d, J = 8,7; 1 H), 1.99 (s, 1H), 1.92-1.67 (m, 3H), 1.62-1.50 (m, 1H), 1.47 (d, J = 6,7; 3H), 1.17 (t, J = 7,1; 1H), 1.17 (t, J = 7,1; 1H).
Пример 12. 2-((S)-1-(4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновая кислота
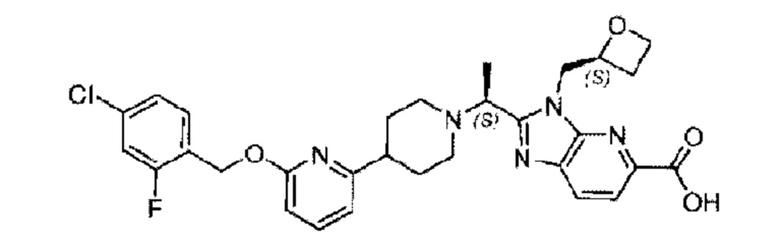
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилата
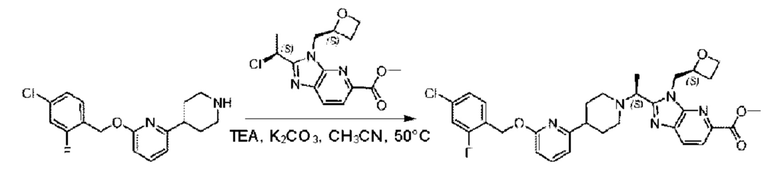
2-((4-Хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридин (100 мг; 0,28 ммоль) растворяли в ацетонитриле (10,0 мл) и триэтиламине (2,0 мл), затем добавляли карбонат калия (77,0 мг; 0,56 ммоль), иодид калия (93,0 мг; 0,56 ммоль) и метил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилат (82,0 мг; 0,28 ммоль). Раствор этой смеси перемешивали при 50°С в течение 2 часов.
По завершении реакции по данным TLC реакционный раствор разбавляли водой (30,0 мл) и экстрагировали этилацетатом (40,0 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (25,0 мл × 3 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 120 мг метил- 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилата в виде желтого твердого вещества (выход: 72,3%). LC-MS: RT = 1,83 мин, [М+Н]+ = 594,21.
Стадия В. Синтез 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновой кислоты
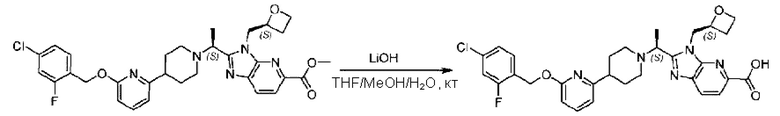
Метил-2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоксилат (120 мг; 0,73 ммоль) растворяли втетрагидрофуране (30,0 мл) и метаноле (10 мл), затем добавляли гидроксид лития (84 мг; 2,0 ммоль) в воде (10,0 мл) и раствор этой смеси перемешивали при комнатной температуре в течение 5 часов.
По завершении реакции по данным LC-MS реакционный раствор экстрагировали смесью дихлорметан/метанол (9/1; 20,0 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (15,0 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем, получая 16 мг 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазоло[4,5-b]пиридин-5-карбоновой кислоты в виде белого твердого вещества (выход: 23,8%). LC-MS: RT = 1,81 мин, [М+Н]+ = 580,31.
Пример 13. 2-((5)-1-(4-(6-((4-Бром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
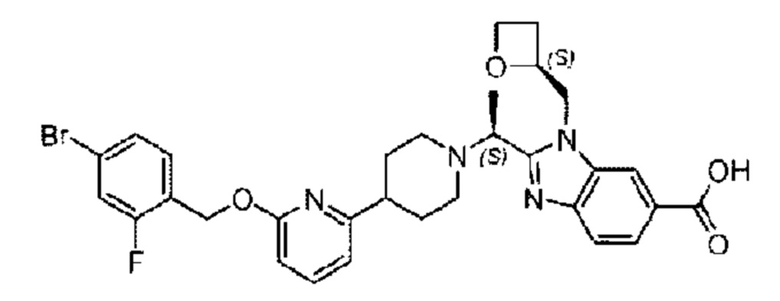
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез (2,5-дифторфенил)метанола
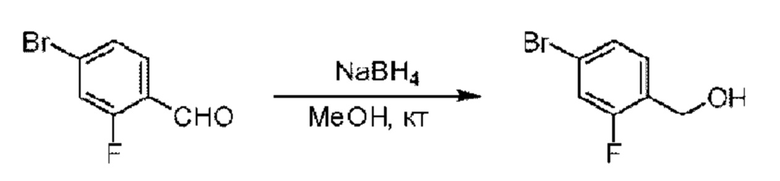
2,5-Дифторбензальдегид (500,0 мг; 2,47 ммоль) добавляли к метанолу (25,0 мл) в ледяной бане, затем порциями добавляли боргидрид натрия (140,6 мг; 3,70 ммоль). Взаимодействие проводили с перемешиванием при комнатной температуре в течение 1,0 часа в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении, получая 380,3 мг (2,5-дифторфенил)метанола в виде желтого маслянистого вещества (выход: 76,0%).
Стадия В. Синтез трет-бутил-4-(6-((4-бром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата
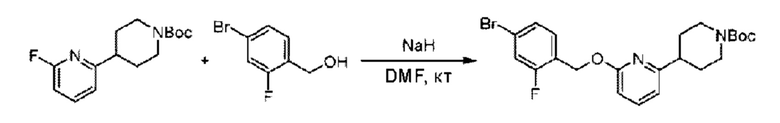
(4-Бром-2-фторфенил)метанол (60,0 мг; 0,29 ммоль) добавляли к N,N-диметилформамиду (3,0 мл) в ледяной бане, затем порциями добавляли 60 масс. %-ный гидрид натрия (23,2 мг; 0,58 ммоль). После перемешивания в течение 0,5 часа при комнатной температуре в атмосфере N2 добавляли трет-бутил-4-(6-фторпиридин-2-ил)пиперидин-1-формиат (81,2 мг; 0,29 ммоль) и реакцию проводили в течение 1,0 часа при комнатной температуре.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 50/1), получая 82,0 мг трет-бутил-4-(6-((4-бром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата в виде белого твердого вещества (выход: 61,0%). LC-MS: RT = 2,45 мин, [М+Н-56]+ = 409,17.
Стадия С.Синтез 2-((4-бром-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридина
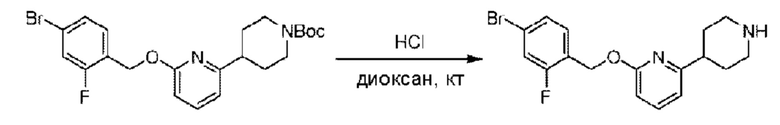
трет-Бутил-4-(6-((4-бром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилат (82,0 мг; 0,18 ммоль) добавляли к соляной кислоте в диоксане (1,00 ммоль/мл; 3 мл) при комнатной температуре и взаимодействие проводили при комнатной температуре в течение 1 часа в атмосфере N2.
По завершении реакции реакционный раствор концентрировали при пониженном давлении. Получали 50,2 мг 2-((4-бром-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридина в виде молочно-белого твердого вещества (выход: 76,4%о), которое использовали непосредственно на следующей стадии реакции. LC-MS: RT = 1,77 мин, [М+Н]+ = 365,16.
Стадия D. Синтез метил-2-((S)-1-(4-(6-((4-бром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
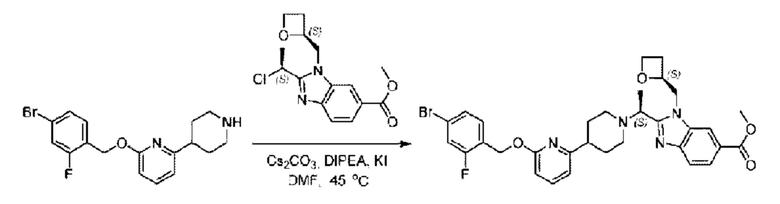
2-((4-Бром-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридин (50,2 мг; 0,14 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (43,5 мг; 0,14 ммоль), иодид калия (23,2 мг; 0,14 ммоль) и карбонат цезия (91,3 мг; 0,28 ммоль) добавляли к N,N-диметилформ амиду (10,0 мл) при комнатной температуре, затем добавляли N,N-диизопропилэтиламин (1,0 мл). Взаимодействие проводили при 45°С в течение 3,0 часа в атмосфере N2.
По завершении реакции реакционную смесь гасили водой и реакционный раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20/1), получая 66,3 мг метил-2-((S)-1-(4-(6-((4-6ром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбокс плата в виде светло-желтого твердого вещества (выход: 74,4%). LC-MS: RT = 1,88 мин, [М+Н]+ = 637,26.
Стадия Е. Синтез 2-((S)-1-(4-(6-((4-бром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
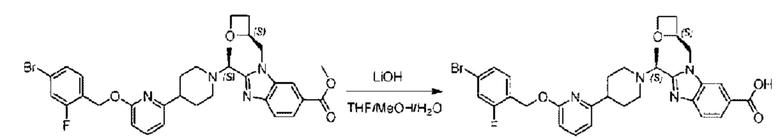
Гидроксид лития (16,8 мг; 0,40 ммоль) в воде (1,0 мл) по каплям при комнатной температуре добавляли к тетрагидрофурану (3,0 мл), содержащему метил-2-((S)-1-(4-(6-((4-6ром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (66,3 мг; 0,10 ммоль), затем добавляли 1,0 мл метанола. Взаимодействие проводили при 45°С в течение 3,0 часа.
По завершении реакции реакционную смесь гасили водой и рН подводили до 6 водным раствором хлорида аммония (0,5 моль/л). Реакционный раствор экстрагировали дихлорметаном (30 мл × 3 раза), органические фазы объединяли, промывали водным раствором хлорида аммония (30 мл × 2 раза) и сушили с использованием безводного сульфата натрия, получая неочищенный продукт, который очищали препаративной высокоэффективной жидкостной хроматографией. Условия очистки приведены ниже: колонка: препаративная 5-С18 от Agilent, 100 мм×30 мм, 5 мкм; подвижная фаза: вода (содержащая 0,05% аммиака) и ацетонитрил; скорость потока: 20 мл/мин; градиент: продукт элюировался 25%-ным ацетонитрилом в момент времени 7,50 мин; длина волны детектирования: 254 нм. После очистки и лиофилизации получали 19,3 мг 2-((S)-1-(4-(6-((4-бром-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 30,0%). LC-MS: RT = 1,84 мин, [М+Н]+ = 623,28.
Пример 14. 2-((S)-1-(4-(6-((2,5-Дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
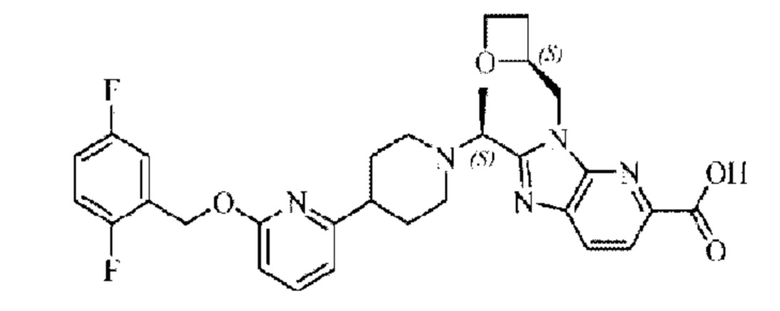
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез (2,5-дифторфенил)метанола
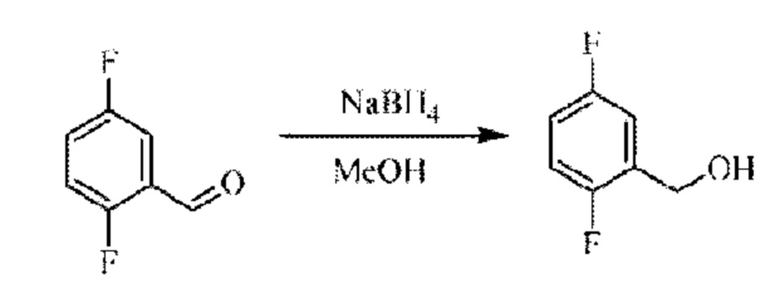
2,5-Дифторбензальдегид (300 мг; 2,11 ммоль) растворяли в метаноле (15 мл) при комнатной температуре, затем добавляли боргидрид натрия (156 мг; 4,23 ммоль) при 0°С. Взаимодействие проводили при комнатной температуре с перемешиванием. По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия. Реакционный раствор экстрагировали дихлорметаном (30 мл) и дважды промывали водой. Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/3), получая 240 мг (2,5-дифторфенил)метанола в виде бесцветной маслянистой жидкости (выход: 80%). LC-MS: RT = 1,78 мин.
Стадия В. Синтез трет-бутил-4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата
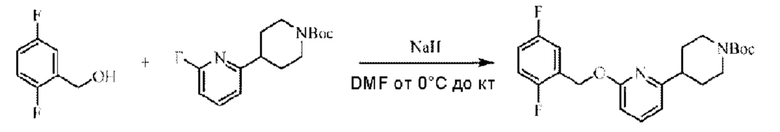
(2,5-Дифторфенил)метанол (240 мг; 1,67 ммоль) растворяли в N,N-диметилформ амиде (10 мл). Раствор этой смеси охлаждали до 0°С и добавляли гидрид натрия (80 мг; 3,34 ммоль). Реакционный раствор перемешивали при комнатной температуре в течение 0,5 часа и добавляли трет-бутил-4-(6-фторпиридин-2-ил)пиперидин-1-карбоксилат (476 мг; 1,67 ммоль). Взаимодействие проводили при комнатной температуре. По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия. Реакционный раствор экстрагировали этилацетатом (30 мл) и дважды промывали водой. Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2), получая 280 мг трет-бутил-4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата в виде бесцветной маслянистой жидкости (выход: 48%). LC-MS: RT = 2,32 мин, [М-55]+ = 349,25.
Стадия С.Синтез 2-((2,5-дифторбензил)окси)-6-(пиперидин-4-ил)пиридина
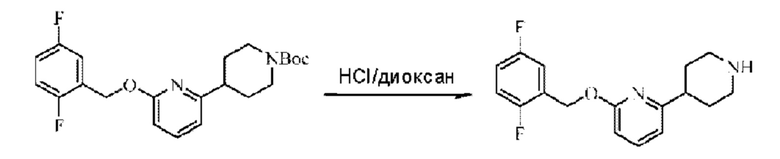
трет-Бутил-4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилат (280 мг; 0,69 ммоль) растворяли в соляной кислоте в диокеане (5 мл) и раствор этой смеси перемешивали при комнатной температуре. По завершении реакции после концентрирования при пониженном давлении получали 300 мг неочищенного продукта, который представлял собой 2-((2,5-дифторбензил)окси)-6-(пиперидин-4-ил)пиридин в виде белого твердого вещества. LC-MS: RT = 1,68 мин, [М+Н]+ = 305,25.
Стадия D. Синтез изопропил-2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
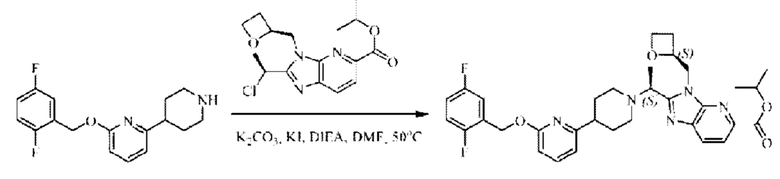
2-((2,5-Дифторбензил)окси)-6-(пиперидин-4-ил)пиридин (60 мг; 0,20 ммоль), DIEA (0,5 мл), иодид калия (65 мг; 0,9 ммоль) и карбонат калия (54 мг; 0,39 ммоль) растворяли в N,N-диметилформамиде (5 мл) при комнатной температуре, затем добавляли изопропил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (67 мг; 0,20 ммоль) и взаимодействие проводили при температуре 50°С.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (20 мл × 2 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/8), получая 65 мг изопропил-2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого твердого вещества (выход: 32%). LC-MS: RT = 1,93 мин, [М+Н]+ = 606,38.
Стадия Е Синтез 2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
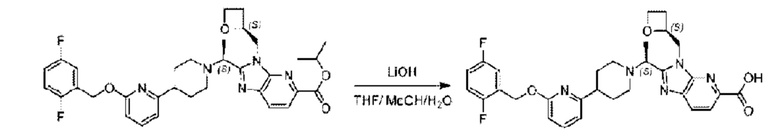
Изопропил-2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (65 мг; 0,11 ммоль) растворяли в смеси растворителей (6 мл; тетрагидрофуран/метанол/вода = 2,5/2,5/1) при комнатной температуре, затем добавляли гидроксид лития (16 мг; 0,44 ммоль) и взаимодействие проводили при комнатной температуре.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида аммония, реакционный раствор последовательно экстрагировали этилацетатом (10 мл × 2 раза) и смесью растворителей (10 мл × 1 раз; дихлорметан/метанол = 30/1). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта, который очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 10/1), получая 30,75 мг 2-((S)-1-(4-(6-((2,5-дифторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белоготвердоговещества (выход: 21%).
LS-MS: RT = 1,77 мин, [М+Н]+ = 564,31. 1Н ЯМР (400 МГц, DMSO-d6) δ 8.15 (d, J = 8,2 Гц, 1Н), 7.98 (d, J = 8,2 Гц, 1Н), 7.64-7.58 (m, 1Н), 7.39-7.1 4 (m, 4Н), 6.83 (d, J = 7,3 Гц, 1Н), 6.67 (dd, J = 8,1; 4,4 Гц, 1Н), 5.32 (dd, J = 1 9,5; 6,1 Гц, 2Н), 4.84 (dd, J = 15,0; 4,2 Гц, 1Н), 4.72 (dd, J = 15,1; 3,6 Гц, 1Н), 4.60 (d, J = 6,8 Гц, 1Н), 4.39 (dd, J = 13,6; 7,7 Гц, 1Н), 4.07 (dd, J = 15,0; 6,0 Гц, 1Н), 2.98 (d, J = 11,0; 1Н), 2.57 (dd, J = 11,7; 8,4 Гц, 3Н), 2.26 (dd, J = 22,2; 11,6 Гц, 2H), 1.84-1.63 (m,4H), 1.54 (dd, J = 12,0; 3,7 Гц, 1H), 1.46 (t, J = 6,5 Гц, 3Н).
Пример 15. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота
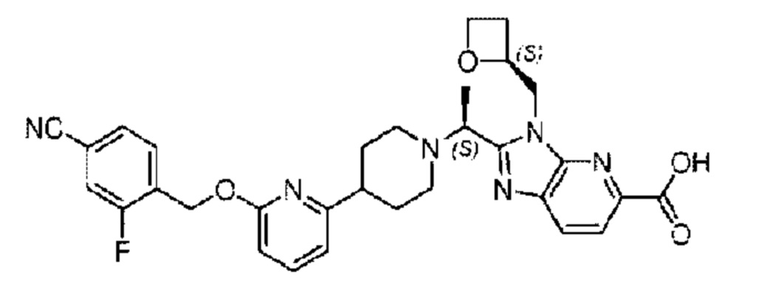
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата
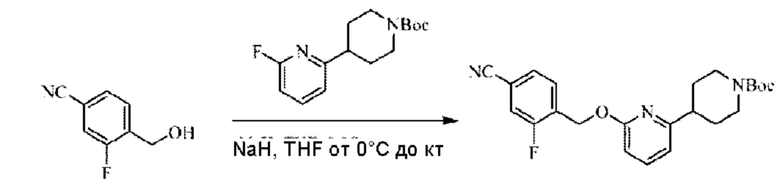
При 0°С гидрид натрия (357 мг; 60%-ную дисперсию в минеральном масле; 8,94 ммоль) добавляли к тетрагидрофурану (5,0 мл), содержащему 3-фтор-4-(гидроксиметил)бензонитрил (648 мг; 4,29 ммоль), и перемешивали в течение 20 мин, затем добавляли трет-бутил-4-(6-фторпиридин-2-ил)пиперидин-1-формиат (1,0 г; 3,57 ммоль) и взаимодействие проводили при комнатной температуре в течение 4 часов.
По завершении реакции реакционную смесь гасили водой и раствор экстрагировали этилацетатом (50 мл × 3 раза), промывали насыщенным солевым раствором (50 мл) и сушили с использованием безводного сульфата натрия. Фильтрат концентрировали при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/10). Получали 850 мг трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилата в виде белого твердого вещества (выход: 57,9%). LC-MS: RT = 2,28 мин, [М-Н]- = 410,32.
Стадия В. Синтез 3-фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрила
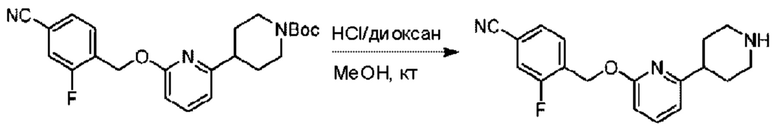
При 0°С соляную кислоту в 1,4-диоксане (2 мл; 4 М раствор) добавляли к метанолу (4 мл), содержащему трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-карбоксилат (850 мг; 2,07 ммоль), и взаимодействие проводили при комнатной температуре в течение 40 мин.
По завершении реакции реакционный раствор незамедлительно концентрировали, получая 720 мг 3-фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрила в виде белого твердого вещества, которое использовали непосредственно на следующей стадии. LC-MS: RT = 1,65 мин, [М+Н]+ = 312,26.
Стадия С.Синтез изопропил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата
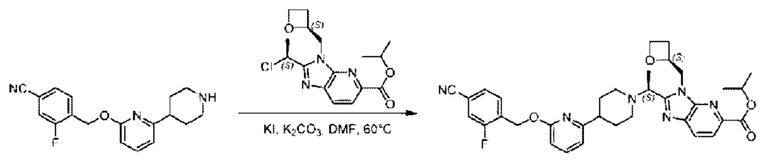
При комнатной температуре добавляли изопропил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (98 мг; 0,29 ммоль), иодид калия (96 мг; 0,58 ммоль) и карбонат калия (80 мг; 0,58 ммоль) к N,N-диметилформамиду (4,0 мл), содержащему 3-фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрил (120 мг; 0,35 ммоль), и взаимодействие проводили при 60°С в течение 4 часов.
По завершении реакции реакционную смесь гасили водой, экстрагировали этилацетатом (50 мл × 3 раза), промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/5). Получали 60 мг соединения, изопропил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата, в виде желтого твердого вещества (выход: 33,7%). LC-MS: RT = 1,84 мин, [М+Н]+ = 613,38.
Стадия D. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты
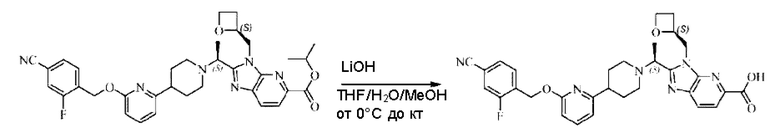
При 0°С по каплям добавляли водный раствор (1 мл) гидроксида лития (17 мг; 0,40 ммоль) к тетрагидрофурану (3 мл) и метанолу (1 мл), содержащему изопропил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (60 мг; 0,10 ммоль), и взаимодействие проводили при комнатной температуре в течение 30 мин.
По завершении реакции реакционную смесь гасили водой, экстрагировали этилацетатом (50 мл × 3 раза), промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 1/10). Получали 43 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты в виде белого твердого вещества (выход: 75,3%). LCMS: RT = 1,75 мин, [М+Н]+ = 571,36.
1Н ЯМР (400 МГц, DMSO) δ 8.22-8.08 (m, 1Н), 7.99 (dd, J = 8,2; 5,2 Гц, 1Н), 7.86 (d, J = 10,3 Гц, 1Н), 7.68 (d, J = 3,9 Гц, 2Н), 7.66-7.60 (m, 1Н), 6.85 (t, J = 7,5 Гц, 1Н), 6.70 (dt, J = 13,3; 6,6 Гц, 1Н), 5.45 (s, 2Н), 5,36-5,02 (m, 3Н), 4.86-4.46 (m, 4Н), 4.16-4.08 (m, 1Н), 4.07-3.98 (m, 1Н), 3.13-2,91 (m, 2Н), 2.67-2,54 (m, 2Н), 2.31-2,14 (m, 2Н), 2.02-1,90 (m, 1Н), 1.23 (d, J = 3,7 Гц, 3Н).
Пример 16. 1-(((S)-Оксетан-2-ил)метил)-2-((S)-1-(4-(6-(хинолин-8-ил-метокси)пиридин-2-ил)пиперидин-1 -ил)этил)-1Н-бензо[d]имидазол-6-карбоновая кислота
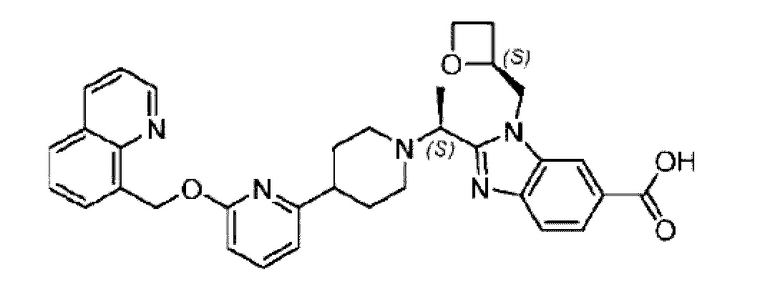
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(хинолин-8-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилата
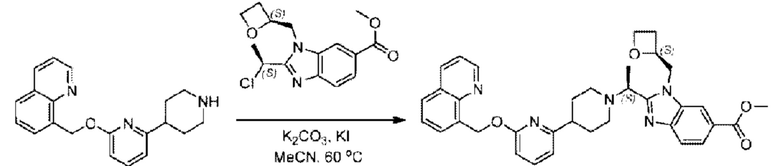
8-(((6-(Пиперидин-4-ил)пиридин-2-ил)окси)метил)хинолин (60 мг; 0,169 ммоль), карбонат калия (47,0 мг; 0,338 ммоль) и иодид калия (40,5 мг; 0,253 ммоль) растворяли в 5 мл безводного ацетонитрила, затем добавляли метил-2-((S)-1-хлорэтил)-1-((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (52,1 мг; 0,169 ммоль) и взаимодействие проводили при 60°С в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой, экстрагировали этилацетатом (30 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией, получая 76 мг метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(хинолин-8-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества. LOMS: RT = 1,82 мин, [М+Н]+ = 592,30.
Стадия В. Синтез 1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(хинолин-8-ил-метокси)пиридин-2-ил)пиперидин-1 -ил)этил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
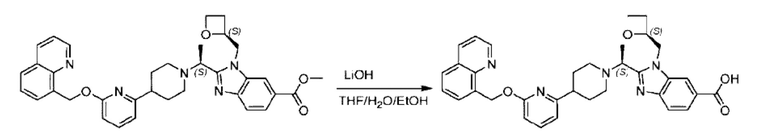
Метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(хинолин-8-ил-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилат (76 мг; 0,128 ммоль) растворяли в 4 мл тетрагидрофурана, затем добавляли гидроксида лития моногидрат (27 мг; 0,643 ммоль), 1 мл воды и 1 мл этанола и взаимодействие проводили при комнатной температуре в течение 50 мин. По завершении реакции реакционный раствор нейтрализовали, добавляя 20 мл насыщенного раствора хлорида аммония, и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 24 мг 1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(хинолин-8-илметокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде бледно-желтого твердого вещества.
LCMS: RT = 1,75 мин, [М-Н]- = 578,37. 1Н ЯМР (400 МГц, DMSO) δ 12.71 (s, 1Н), 8.96 (dd, J = 4,2; 1,8 Гц, 1Н), 8.41 (dd, J = 8,3; 1,7 Гц, 1Н), 8.28 (s, 1Н), 7.93-7.96 (m, 1Н), 7.79-7.86 (m, 2Н), 7.56-7.71 (m, 4Н), 6.84 (d, J = 7,2 Гц, 1Н), 6.73 (d, J = 8,3 Гц, 1Н), 6.00 (s, 2Н), 5,11-5,1 6 (m, 1Н), 4.75-4.81 (m, 1Н), 4.56-4.64 (m, 1Н), 4.36-4.46 (m, 2Н), 4.10-4,17 (m, 1Н), 2.90-2,98 (m, 1Н), 2.54-2,65 (m, 4Н), 2.18-2,33 (m, 2Н), 1.67-1.87 (m, 4H), 1.43-1.48 (m, 3Н).
Пример 17. 3-(((S)-Оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота
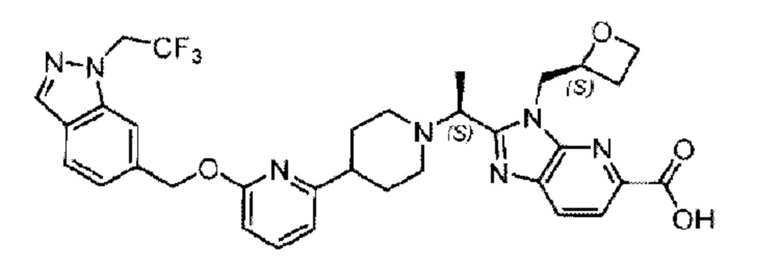
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез изопропил-3-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата
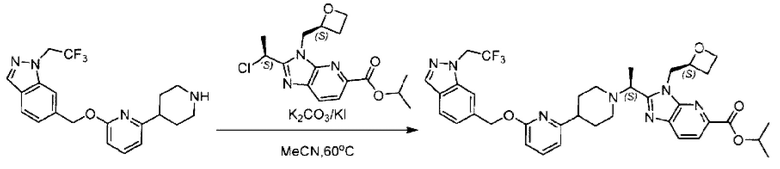
6-(((6-(Пиперидин-4-ил)пиридин-2-ил)окси)метил)-1-(2,2,2-трифторэтил)-1Н-индазол (78 мг; 0,183 ммоль), карбонат калия (50,5 мг; 0,366 ммоль) и иодид калия (43,8 мг; 0,274 ммоль) растворяли в 5 мл безводного ацетонитрила, затем добавляли изопропил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (61,6 мг; 0,183 ммоль) и взаимодействие проводили при 60°С в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 92 мг изопропил-3-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата в виде бледно-желтого твердого вещества. LC-MS: RT = 1,81 мин, [М+Н]+ = 692,27.
Стадия В. Синтез 3-(((S)-оксетан-2-ил)мети n)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты
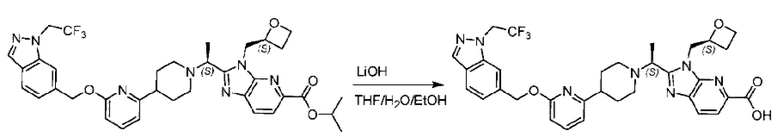
Изопропил-3-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (92 мг; 0,133 ммоль) растворяли в 4 мл тетрагидрофурана, затем добавляли гидроксида лития моногидрат (28 мг; 0,664 ммоль), 1 мл воды и 1 мл этанола и взаимодействие проводили при комнатной температуре в течение 50 мин. По завершении реакции реакционный раствор нейтрализовали, используя 20 мл насыщенного раствора хлорида аммония, и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 22 мг 3-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-((1-(2,2,2-трифторэтил)-1Н-индазол-6-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты в виде бледно-желтого твердого вещества.
LCMS: RT = 1,75 мин, [М+Н]+ = 650,39. 1Н ЯМР (500 МГц, DMSO) δ 8.19 (s, 1Н), 8.15 (d, J = 8,2 Гц, 1Н), 7.99 (d, J = 8,3 Гц, 1Н), 7.88 (s, 1Н), 7.78 (d, J = 8,2 Гц, 1Н), 7.62 (t, J = 7,8 Гц, 1Н), 7.31 (d, J = 8,2 Гц, 1Н), 6.84 (d, J = 7,4 Гц, 1Н), 6.69 (d, J = 8,1 Гц, 1Н), 5.48 (d, J = 1,7 Гц, 2Н), 5.42 (dd, J = 18,6; 9,4 Гц, 2Н), 5.30-5.26 (m, 1Н), 4.84-4.71 (m, 2Н), 4.41-4.37 (m, 1Н), 4.08-4,03 (m, 2Н), 3.03-2,99 (m, 1Н), 2.68-2,52 (m, 4Н), 2.22-2,30 (m, 2Н), 1.70-1.90 (m, 4Н), 1.49 (d, J = 6,7 Гц, 3Н).
Пример 18. 2-((S)-1-(4-(6-((7-Фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота
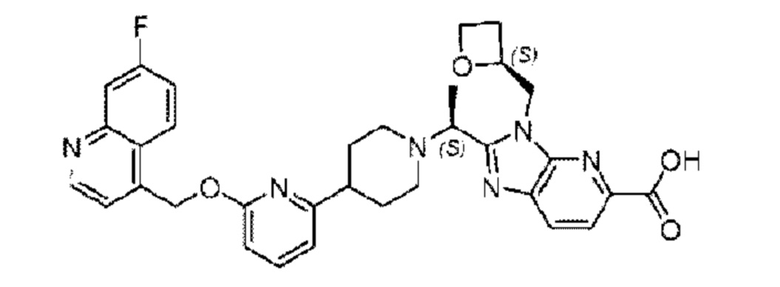
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 7-фтор-4-метилхинолина
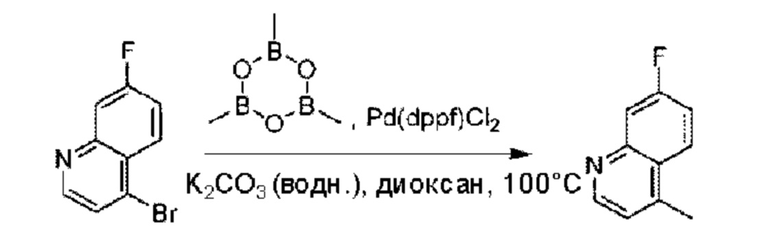
4-Бром-7-фторхинолин (1,00 г; 4,42 ммоль) растворяли в 1,4-диоксане (15,0 мл), затем добавляли триметилбороксин (1,3 мл; 4,42 ммоль), [1,1-бис(дифенилфосфино)ферроцен]дихлорпалладий (323,4 мг; 0,44 ммоль) и водный раствор карбоната калия (3,3 мл; 6,63 ммоль). Взаимодействие проводили при 100°С в течение 4 часов при перемешивании в атмосфере азота. Твердое вещество удаляли фильтрованием, фильтрат упаривали на роторном испарителе и очищали колоночной хроматографией (этилацетат/н-гексан = 1/5), получая 530,0 мг 7-фтор-4-метилхинолина в виде бесцветной жидкости (выход: 74,3%). LC-MS: RT = 0,84 мин, [М+Н]+ = 162,14.
Стадия В. Синтез4-(бромметил)-7-фторхинолина
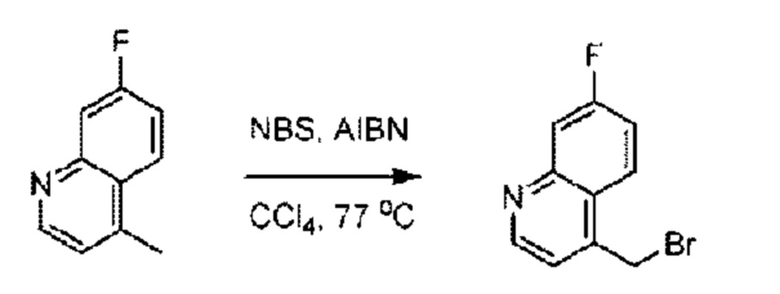
7-Фтор-4-метилхинолин (530,0 мг; 3,29 ммоль) растворяли в тетрахлорметане (10,0 мл), затем добавляли N-бромсукцинимид (703,0 мг; 3,95 ммоль) и азодиизобутиронитрил (54,2 мг; 0,33 ммоль). Реакционный раствор нагревали до 77°С и реакцию проводили в течение 2 часов при перемешивании. Твердое вещество удаляли фильтрованием и фильтрат упаривали на роторном испарителе, получая 780,0 мг 4-(бромметил)-7-фторхинолина в виде бежевого твердого вещества (выход: 98,8%). LC-MS: RT = 1,88 мин, [М+Н]+ = 240,08.
Стадия С. Синтез метил-(7-фторхинолин-4-ил)ацетата
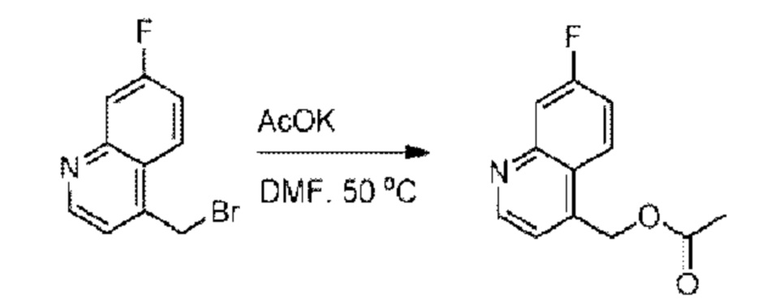
4-(Бромметил)-7-фторхинолин (775,0 мг; 3,23 ммоль) растворяли в N,N-диметилформамиде (6,0 мл), затем добавляли ацетат калия (634,0 мг; 6,46 ммоль). Реакционный раствор нагревали до 50°С и реакцию проводили в течение 1 часа при перемешивании. Реакционный раствор разбавляли этилацетатом (50 мл), промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, упаривали на роторном испарителе для удаления растворителя и разделяли колоночной хроматографией (этилацетат:н-гексан = 1:2), получая 400,0 мг (7-фторхинолин-4-ил)метилацетата в виде бежевого твердого вещества (выход: 56,5%). LC-MS: RT = 1,73 мин, [М+Н]+ = 220,15.
Стадия D. Синтез (7-фторхинолин-4-ил)метанола
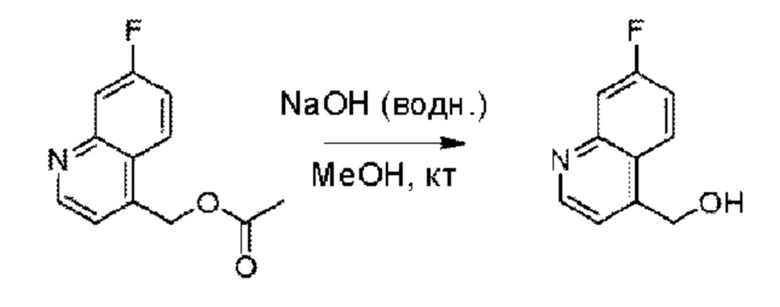
Метил-(7-фторхинолин-4-ил)ацетат (400,0 мг; 1,82 ммоль) растворяли в метаноле (3,0 мл), затем добавляли водный раствор гидроксида натрия (1,8 мл; 3,64 ммоль) и смесь перемешивали в течение 1 часа при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении для удаления метанола, разбавляли этилацетатом (50 мл), экстрагировали, промывали насыщенным солевым раствором, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении для удаления органического растворителя. Продукт концентрировали и разделяли колоночной хроматографией (этилацетат:н-гексан = 2:1), получая 120,0 мг (7-фторхинолин-4-ил)метанола в виде белого твердого вещества (выход: 37,1%). LC-MS: RT = 0,60 мин, [М+Н]+ = 178,13.
Стадия Е. Синтез трет-бутил-4-(6-((7-фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-карбоксилата
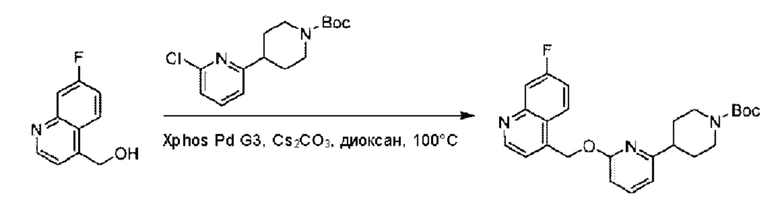
(7-Фторхинолин-4-ил)метанол (90,0 мг; 0,51 ммоль) растворяли в 1,4-диоксане (5 мл), затем добавляли трет-бутил-4-(6-хлорпиридин-2-ил)пиперидин-1-карбоксилат (151,4 мг; 0,51 ммоль), метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (42,3 мг; 0,05 ммоль) и карбонат цезия (249,3 мг; 0,77 ммоль). Реакционный раствор нагревали до 100°С и реакцию проводили в течение ночи при перемешивании в атмосфере азота. Твердое вещество удаляли фильтрованием и фильтрат концентрировали досуха, затем продукт разделяли колоночной хроматографией (этилацетат:н-гексан = 1:2), получая 190,0 мг трет-бутил-4-(6-((7-фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-карбоксилата в виде бежевого твердого вещества (выход: 85,5%). LC-MS: RT = 2,25 мин, [М+Н]+ = 438,32.
Стадия F. Синтез 7-фтор-4-((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)хинолина
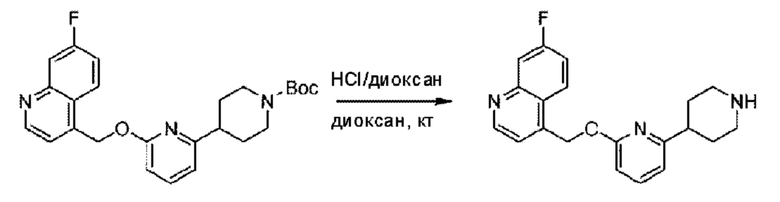
трет-Бутил-4-(6-((7-фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-карбоксилат (190,0 мг; 0,43 ммоль) растворяли в 1,4-диоксане (1,0 мл), затем добавляли соляную кислоту в 1,4-диоксане (1,0 мл; 4,0 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Растворитель удаляли путем концентрирования, получая 149,0 мг 7-фтор-4-((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)хинолина в виде белого твердого вещества (выход: 91,8%). LC-MS: RT = 1,63 мин, [М+Н]+ = 338,28.
Стадия G. Синтез изопропил-2-((S)-1-(4-(6-((7-фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата
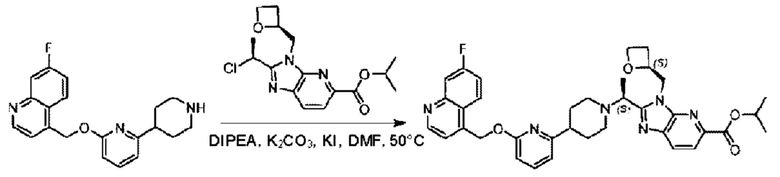
7-Фтор-4-((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)хинолин (70,0 мг; 0,19 ммоль) растворяли в N,N-диметилформамиде (3,0 мл), затем добавляли N,N-диизопропилэтиламин (132 мкл, 0,80 ммоль), и все это растворяли при перемешивании. Затем последовательно добавляли пропил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (64 мг; 0,19 ммоль), карбонат калия (39 мг; 0,28 ммоль) и иодид калия (46 мг; 0,28 ммоль). Реакционный раствор нагревали до 50°С и реакцию проводили в течение 2 часов при перемешивании. Раствор разбавляли этилацетатом (50 мл), экстрагировали, последовательно промывали водой и насыщенным солевым раствором, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали для удаления растворителя. Продукт разделяли колоночной хроматографией (этилацетат:н-гексан = 5:1), получая 80,0 мг изопропил-2-((S)-1-(4-(6-((7-фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата в виде бежевого твердого вещества (выход: 66,9%). LC-MS: RT = 1,81 мин, [М+Н]+ = 639,43.
Стадия Н. Синтез 2-((S)-1-(4-(6-((7-фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты
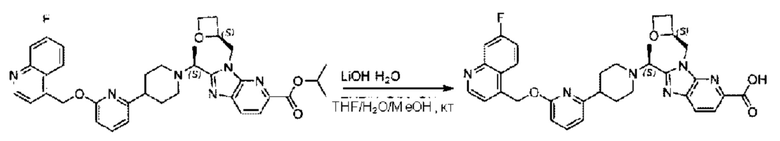
Изопропил-2-((S)-1-(4-(6-((7-фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (80,0 мг; 0,13 ммоль) растворяли в смеси растворителей тетрагидрофурана (0,3 мл), метанола (0,1 мл) и воды (0,1 мл), затем добавляли гидроксида лития моногидрат (26,3 мг; 0,63 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов. Растворитель удаляли путем концентрирования и продукт разделяли колоночной хроматографией (метанол:дихлорметан = 7:93), получая 51,3 мг 2-((S)-1-(4-(6-((7-фторхинолин-4-ил)метокси)пиридин-2-ил)пиперидин-1-ил)зтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты в виде белого твердого вещества (выход: 68,8%).
LCMS: RT = 1,70 мин, [М+Н]+ = 597,34. 1Н ЯМР (400 МГц, DMSO-d6) δ (млн1) 8.86 (d, J = 4,3 Гц, 1Н), 8.25 (dd, J = 9,1; 6,2 Гц, 1Н), 8.13 (d, J = 8,2 Гц, 1Н), 7.97 (d, J = 8,2 Гц, 1Н), 7.88 (dd, J = 10,3; 2,4 Гц, 1Н), 7.68-7.56 (m, 2Н), 7.53 (d, J = 4,3 Гц, 1Н), 6.84 (d, J = 7,4 Гц, 1Н), 6.74 (d, J = 8,2 Гц, 1Н), 5.87 (s, 2Н), 5.31-5.18 (m, 1Н), 4.83-4.73 (m, 1Н), 4.71-4.63 (m, 1Н), 4.61-4.52 (m, 1Н), 4.41-4.32 (m, 1Н), 4.09-4.00 (m, 1Н), 2.98-2.89 (m, 1Н), 2.63-2,51 (m, 3Н), 2.38-2.10 (m, 3Н), 1.84-1.57 (m, 4Н), 1.44 (d, J = 6,6 Гц, 3Н).
Пример 19. 2-((S)-1-(4-(6-((3-Фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота
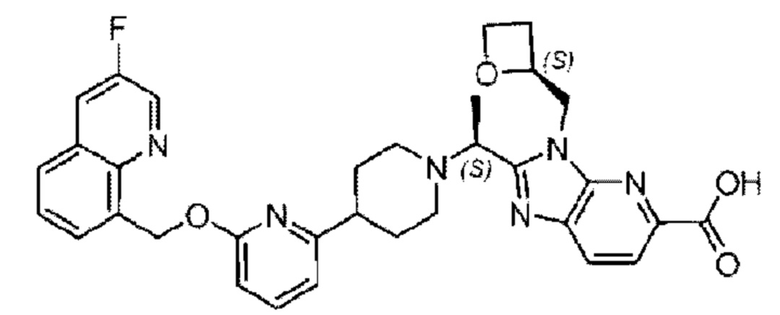
Стадия А. Синтез 3-фтор-8-метилхинолина
При комнатной температуре последовательно добавляли триметилбороксин (0,25 мл; 0,89 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (32 мг; 0,044 ммоль) и карбонат калия (244 мг; 1,77 ммоль) к раствору смеси 1,4-диоксан/вода (3,4 мл/1,0 мл), содержащему 8-бром-3-фторхинолин (200 мг; 0,89 ммоль). Затем реакционную систему продували азотом и взаимодействие проводили при 100°С в течение 3 часов. По завершении реакции раствор экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/10). Получали 128 мг 3-фтор-8-метилхинолина в виде белого твердого вещества (выход: 89,8%). LC-MS: RT = 2,01 мин, [М+Н]+ = 162,14.
Стадия В. Синтез 8-(бромметил)-3-фторхинолина
При комнатной температуре к тетрахлорметану (4,0 мл), содержащему 3-фтор-8-метилхинолин (128 мг; 0,80 ммоль), последовательно добавляли N-бромсукцинимид (156 мг; 0,88 ммоль) и азодиизобутиронитрил (5 мг; 0,032 ммоль), затем реакционный раствор нагревали до 80°С и реакцию проводили в течение 1 часа. По завершении реакции реакционный раствор разбавляли дихлорметаном (100 мл), экстрагировали, последовательно промывали насыщенным раствором бикарбоната натрия (50 мл) и солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/15). Получали 90 мг 8-(бромметил)-3-фторхинолина в виде белого твердого вещества (выход: 46,9%). LC-MS: RT = 2,05 мин, [М+Н]+ = 242,05.
Стадия С. Синтез (3-фторхинолин-8-ил)метанола
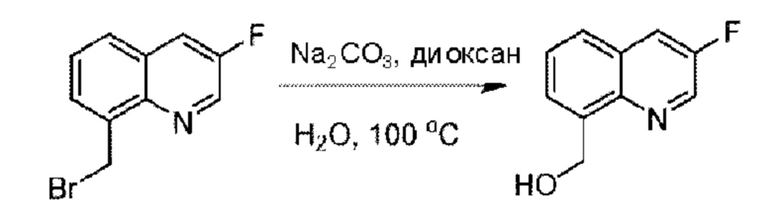
При комнатной температуре добавляли карбонат натрия (119 мг; 1,13 ммоль) к раствору смеси 1,4-диоксан/вода (1,5 мл/1,5 мл), содержащему 8-(бромметил)-3-фторхинолин (90 мг; 0,38 ммоль), реакционный раствор нагревали до 100°С и реакцию проводили в течение 3 часов.
По завершении реакции реакционный раствор охлаждали до комнатной температуры, экстрагировали этилацетатом (50 мл × 3 раза), промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Затем полученный остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/8). Получали 40 мг (3-фторхинолин-8-ил)метанола в виде белого твердого вещества (выход: 59,5%). LC-MS: RT = 1,72 мин, [М+Н]+ = 178,15.
Стадия D. Синтез трет-бутил-4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-формиата
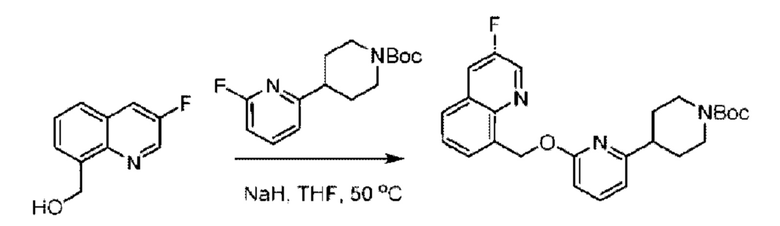
В бане с ледяной водой и в атмосфере азота добавляли гидрид натрия (180 мг; 60%-ную дисперсию в минеральном масле; 4,5 ммоль) к раствору тетрагидрофурана (5,0 мл), содержащему (3-фторхинолин-8-ил)метанол (266 мг; 1,50 ммоль). После добавления баню с ледяной водой удаляли и реакционный раствор перемешивали при комнатной температуре в течение 30 мин. Добавляли раствор трет-бутил-4-(6-фторпиридин-2-ил)пиперидин-1-формиата в тетрагидрофуране (2,0 мл), реакционный раствор нагревали до 50°С и реакцию проводили в течение 3 часов.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида аммония. Реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/8). Получали 450 мг трет-бутил-4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-формиата в виде бледно-желтого маслянистого продукта (выход: 68,5%). LC-MS: RT = 2,37 мин, [М+Н]+ = 438,24.
Стадия Е. Синтез 3-фтор-8-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)хинолина
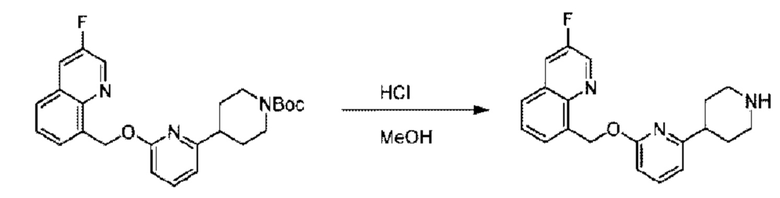
При комнатной температуре добавляли соляную кислоту в 1,4-диоксане (1,3 мл; 4,0 моль/л; 5,14 ммоль) к раствору трет-бутил-4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-формиата (450 мг; 1,03 ммоль) в метаноле (4,0 мл). После добавления смесь перемешивали при комнатной температуре в течение 2 часов. По завершении реакции растворитель выпаривали досуха при пониженном давлении, получая 420 мг 3-фтор-8-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)хинолина в виде белого твердого вещества. LC-MS: RT = 1,73 мин, [М+Н]+ = 338,27.
Стадия F. Синтез изопропил-2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата
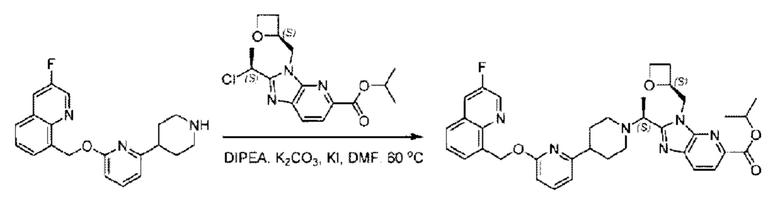
При комнатной температуре добавляли N,N-диизопропилэтиламин (132 мкл; 0,80 ммоль) к N,N-диметилформамиду (2,0 мл), содержащему 3-фтор-8-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)хинолин (153 мг; 0,342 ммоль), и растворяли при перемешивании. Последовательно добавляли изопропил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (90 мг; 0,27 ммоль), карбонат калия (74 мг; 0,53 ммоль) и иодид калия (89 мг; 0,53 ммоль). После добавления реакционный раствор нагревали до 60°С и перемешивали в течение 1 часа.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида аммония. Реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 8/92). Получали 90 мг продукта, изопропил-2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата, в виде бесцветного маслянистого вещества (выход: 52,8%). LC-MS: RT = 1,89 мин, [М+Н]+ = 639,37.
Стадия G Синтез 2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты
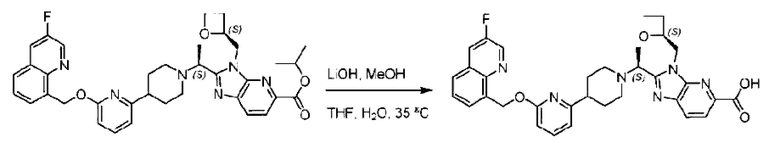
При комнатной температуре добавляли гидроксида лития моногидрат (45 мг; 1,07 ммоль) к раствору смеси тетрагидрофуран/метанол/вода (2,5 мл/2,5 мл/1,0 мл), содержащему изопропил-2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (90 мг; 0,14 ммоль), реакционный раствор нагревали до 35°С и реакцию проводили в течение 1 часа. По завершении реакции реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 8/92). Получали 34 мг 2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты в виде белого твердого вещества, (выход: 40,4%). LC-MS: RT = 1,80 мин, [М+Н]+ = 597,40.
1Н ЯМР (400 МГц, DMSO) δ 9.04-8.96 (m, 1Н), 8.32 (dd, J = 9,6; 2,9 Гц, 1Н), 8.09 (d, J = 8,2 Гц, 1Н), 7.96 (d, J = 8,2 Гц, 2Н), 7.82 (d, J = 6,7 Гц, 1Н), 7.63 (dd, J = 15,5; 7,6 Гц, 2Н), 6.83 (d, J = 7,2 Гц, 1Н), 6.71 (d, J = 8,1 Гц, 1Н), 5.97 (s, 2Н), 5.29-5.19 (m, 1Н), 4.72 (ddd, J = 38,7; 14,9; 4,0 Гц, 2Н), 4.55 (q, J = 6,6 Гц, 1Н), 4.41-4.32 (m, 1Н), 4.05 (dt, J = 9,2; 5,9 Гц, 2Н), 2.95 (d, J = 10,9 Гц, 2Н), 2.63-2.52 (m, 4Н), 2.35-2.17 (m, 2Н), 1.88-1.62 (m, 3Н), 1.57-1.48 (m, 1Н), 1.46 (d, J = 6,7 Гц, 3Н).
Пример 20. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (20А) и 2-((R)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (20В)
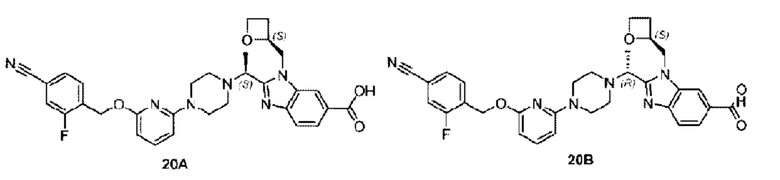
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-хлорпиридин-2-ил)пиперазин-1-карбоксилата
трет-Бутилпиперазин-1-карбоксилат (300,0 мг; 1,61 ммоль) растворяли в 1,4-диоксане (12,0 мл), затем добавляли 2,6-дихлорпиридин (262,0 мг; 1,77 ммоль), метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (135,4 мг; 0,16 ммоль) и карбонат цезия (788,5 мг; 2,42 ммоль). В атмосфере азота реакционный раствор нагревали до 100°С и реакцию проводили в течение 5 часов при перемешивании. Затем растворитель выпаривали на роторном испарителе и продукт разделяли колоночной хроматографией (этилацетат:н-гексан = 1:10), получая 150,0 мг трет-бутил-4-(6-хлорпиридин-2-ил)пиперазин-1-карбоксилата в виде бежевого твердого вещества (выход: 31,3%). LC-MS: RT = 1,82 мин, [M-1Bu+H]+ = 242,36.
Стадия В. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата
трет-Бутил-4-(6-хлорпиридин-2-ил)пиперазин-1-карбоксилат (150,0 мг; 0,51 ммоль) растворяли в 1,4-диоксане (5,0 мл), затем добавляли 3-фтор-4-(гидроксиметил)бензонитрил (76,3 мг; 0,51 ммоль), метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (43,2 мг; 0,05 ммоль) и карбонат цезия (246,8 мг; 0,76 ммоль). В атмосфере азота реакционный раствор нагревали до 100°С и реакцию проводили в течение 15 часов при перемешивании. Растворитель выпаривали на роторном испарителе и продукт разделяли колоночной хроматографией (этилацетат:н-гексан = 1:10), получая 102,0 мг трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата в виде бежевого твердого вещества (выход: 49,1%). LC-MS: RT = 2,23 мин, [M-1Bu+H]+ = 357,23.
Стадия С.Синтез 3-фтор-4-(((6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила
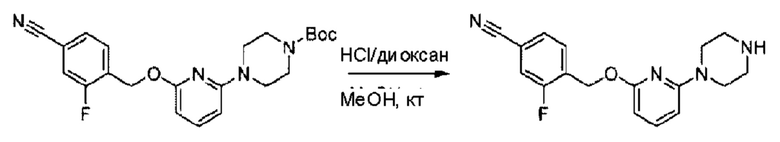
трет-Бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилат (102,0 мг; 0,25 ммоль) растворяли в метаноле (2,0 мл), затем добавляли соляную кислоту в 1,4-диоксане (0,5 мл; 2,0 ммоль). Взаимодействие проводили при комнатной температуре в течение 3 часов с перемешиванием. Растворитель выпаривали на роторном испарителе, получая 73,7 мг 3-фтор-4-(((6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила в виде белого твердого вещества (выход: 73,5%). LC-MS: RT = 1,68 мин, [М+Н]+ = 313,25.
Стадия D. Синтез метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
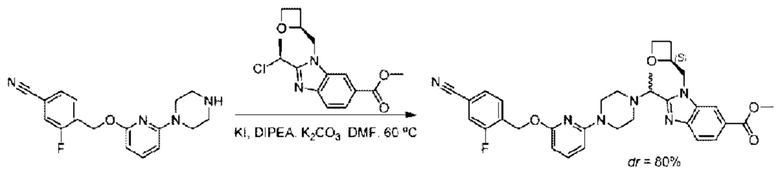
3-Фтор-4-(((6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрил (73,7 мг; 0,21 ммоль) растворяли в N,N-диметилформамиде (3,0 мл), затем добавляли N,N-диизопропилэтиламин (54,7 мг; 0,42 ммоль), карбонат калия (58,5 мг; 0,42 ммоль), иодид калия (52,8 мг; 0,32 ммоль) и метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (71,7 мг; 0,23 ммоль). Реакционный раствор нагревали до 60°С и реакцию проводили в течение 4 часов при перемешивании. По завершении реакции реакционный раствор разбавляли этилацетатом (50 мл), экстрагировали, промывали насыщенным солевым раствором и сушили с использованием безводного сульфата натрия. Затем растворитель выпаривали на роторном испарителе и продукт разделяли колоночной хроматографией (этилацетат:н-гексан = 4:1), получая 102,0 мг метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бежевого твердого вещества (выход: 82,6%, dr (степень высушивания) = 80%). LC-MS: RT = 1,86 мин, [М+Н]+ = 585,42.
Стадия Е. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (20А) и 2-((R)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (20В)
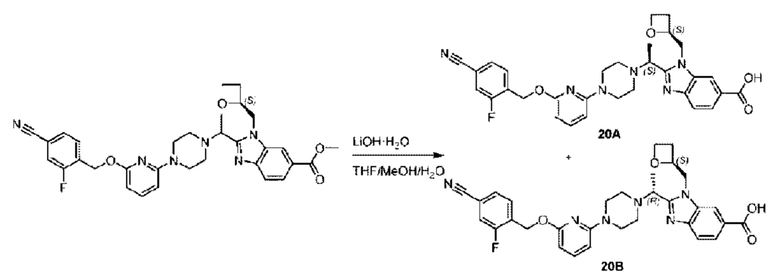
Метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (102,0 мг; 0,17 ммоль) растворяли в смеси растворителей тетрагидрофурана (3,0 мл), метанола (3,0 мл) и воды (1,0 мл), затем добавляли гидроксида лития моногидрат (36,6 мг; 0,87 ммоль). Взаимодействие проводили при комнатной температуре в течение 4 часов с перемешиванием. Растворитель выпаривали на роторном испарителе и продукт разделяли препаративной жидкостной хроматографией, получая 21,0 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (20А) в виде белого твердого вещества (выход: 20,6%) и 2,2 мг 2-(R)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (20 В) в виде белого твердого вещества (выход 2,3%). Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией. Условия разделения приведены ниже: колонка: препаративная 5-С18 от Agilent, 100 мм×30 мм, 5 мкм; подвижная фаза: вода (содержащая 0,1% аммиака) и ацетонитрил; скорость потока: 20 мл/мин; градиент: соединения 20А и 20В элюировались 23%-ным ацетонитрилом в моменты времени 7,67 мин и 8,54 мин; соответственно; длина волны детектирования: 254 нм.
Соединение 20А: HPLC: RT = 7,67 мин. LC-MS: RT = 1,79 мин. [М+Н]+ = 571,39. 1Н Я-1P (500 МГц, DMSO-d6) δ (млн-1) 8.06 (s, 1Н), 7.87 (dd, J = 9,9; 1,2 Гц, 1Н), 7.78 (d, J = 8,3 Гц, 1Н), 7.69 (dd, J = 7,9; 1,4 Гц, 1Н), 7.64 (d, J = 7,4 Гц, 1Н), 7.46 (s, 1Н), 7.44 (s, 1Н), 6.29 (d, J = 8,2 Гц, 1Н), 6.09 (d, J = 7,8 Гц, 1Н), 5.37 (s, 2Н), 5.20-5.13 (m, 1Н), 4.75 (dd, J = 1 5,5; 3,6 Гц, 1Н), 4.55-4.53 (щ 1Н), 4.48-4.42 (m, 2Н), 4.18-4.12 (m, 1Н), 3.40-3.30 (m, 4Н), 2.65-2.63 (m, 1Н), 2.58-2,50 (m, 4Н), 2.35-2.29 (m, 1Н), 1.42(d, J = 6,7 Гц, 3Н).
Соединение 20В: HPLC: RT = 8,54 мин. LC-MS: RT = 1,78 мин. [М+Н]+ = 571,33. 1Н ЯМР (500 МГц, DMSO-d6) δ (млн-1) 12.74 (s, 1Н), 8.27 (d, J = 1,2 Гц, 1Н), 7.87 (dd, J = 9,9; 1,4 Гц, 1Н), 7.79 (dd, J = 8,4; 1,5 Гц, 1Н), 7.69 (dd, J = 7,9; 1,5 Гц, 1Н), 7.68-7.61 (m, 2Н), 7.46 (t, J = 8,0 Гц, 1Н), 6.31 (d, J = 8,2 Гц, 1Н), 6.11 (d, J = 7,8 Гц, 1Н), 5.39 (s, 2Н), 5.08-5.03 (m, 1Н), 4.93-4.87 (m, 1Н), 4.59-4.56 (m, 1Н), 4.51-4.47 (m, 3Н), 3.42-3.39 (m, 4Н), 2.78-2.73 (m, 1Н), 2.70-2,58 (m, 2Н), 2.54-2.45 (m, 3Н), 1.41 (d, J = 6,7 Гц, 3Н).
Пример 21. 2-((S)-1-((S)-4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
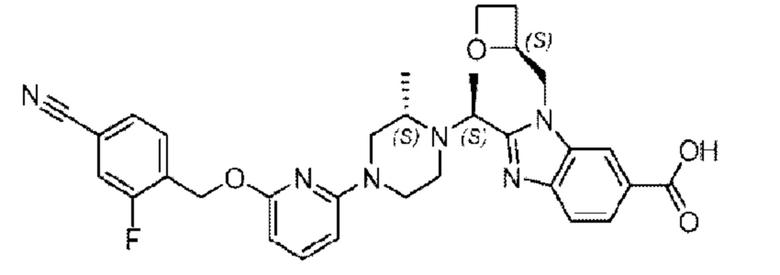
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 4-((6-хлорпиридин-2-ил)окси)метил)-3-фторбензонитрила
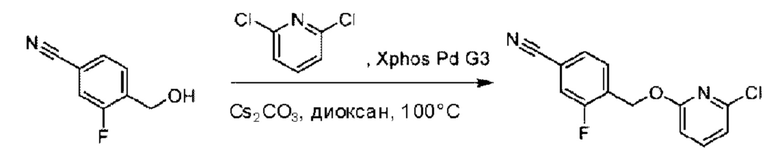
3-Фтор-4-(гидроксиметил)бензонитрил (1,00 г; 6,62 ммоль) растворяли в 1,4-диоксане (25,0 мл), затем добавляли 2,6-дихлорпиридин (1,18 г; 7,94 ммоль), метансульфонато(2-ди циклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (558,7 мг; 0,66 ммоль) и карбонат цезия (3,24 г; 9,93 ммоль). В атмосфере азота реакционный раствор нагревали до 100°С и реакцию проводили в течение ночи при перемешивании. Твердое вещество удаляли фильтрованием, фильтрат упаривали на роторном испарителе и продукт очищали колоночной хроматографией (этилацетат:н-гексан = 1:10), получая 330,0 мг 4-((6-хлорпиридин-2-ил)окси)метил)-3-фторбензонитрила в виде белого твердого вещества (выход: 19,0%). LC-MS: RT = 2,13 мин, [М+Н]+ = 263,09.
Стадия В. Синтез трет-бутил-(S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперазин-1-карбоксилата
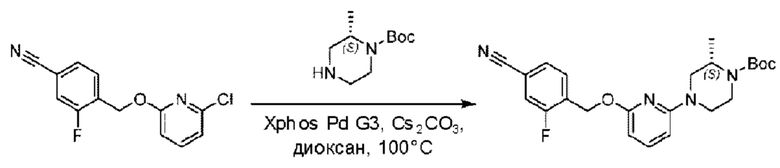
4-((6-Хлорпиридин-2-ил)окси)метил)-3-фторбензонитрил (330,0 мг; 1,26 ммоль) растворяли в 1,4-диокеане (15,0 мл), затем добавляли трет-бутил-(З)-2-метилпиперазин-1-карбоксилат (302,4 мг; 1,51 ммоль), метансульфонато(2-ди циклогексилфос фино-2',4',6'-триизоп ропил-1,1'-бифенил)(2'-амин о-1,1'-бифенил-2-ил)палладий(II) (110,0 мг; 0,13 ммоль) и карбонат цезия (615,8 мг; 1,89 ммоль). В атмосфере азота реакционный раствор нагревали до 100°С и реакцию проводили в течение 14 часов при перемешивании. Твердое вещество удаляли фильтрованием, фильтрат упаривали на роторном испарителе и продукт разделяли колоночной хроматографией (этилацетат:н-гексан = 1:5), получая 260,3 мг трет-бутил-(S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперазин-1-карбоксилата в виде белого твердого вещества (выход: 48,6%). LC-MS: RT = 2,27 мин, [М-1Bu+Н]+ = 371,21.
Стадия С.Синтез (S)-3-фтор-4-((6-(3-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила
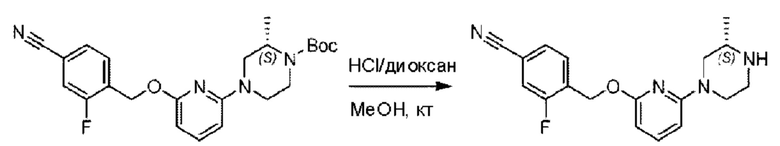
трет-Бутил-(S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперазин-1-карбоксилат (260,3 мг; 0,61 ммоль) растворяли в метаноле (3,0 мл), затем добавляли соляную кислоту в 1,4-диоксане (1 мл; 4,0 ммоль). Взаимодействие в реакционном растворе проводили при комнатной температуре в течение 2 часов с перемешиванием. Растворитель выпаривали на роторном испарителе, получая 220,1 мг (S)-3-фтор-4-((6-(3-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила в виде белого твердого вещества (выход: 99,4%). LC-MS: RT = 1,68 мин, [М+Н]+ = 327,25.
Стадия D. Синтез метил-2-((S)-1-((S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
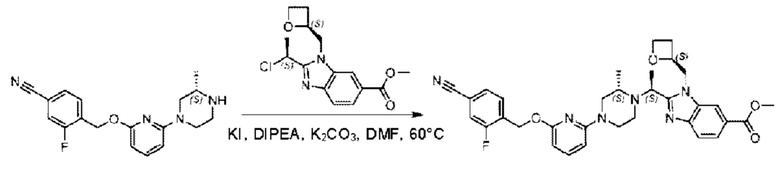
(S)-3-Фтор-4-((6-(3-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)-бензонитрил (110,5 мг; 0,30 ммоль) растворяли в N,N-диметилформамиде (5,0 мл), затем добавляли N,N-диизопропилэтиламин (78,4 мг; 0,61 ммоль), карбонат калия (83,9 мг; 0,61 ммоль), иодид калия (75,7 мг; 0,46 ммоль) и метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (102,9 мг; 0,33 ммоль). Реакционный раствор нагревали до 60°С и реакцию проводили в течение ночи при перемешивании. Реакционный раствор разбавляли этилацетатом (50 мл), экстрагировали, промывали насыщенным солевым раствором и сушили с использованием безводного сульфата натрия. Затем растворитель выпаривали на роторном испарителе и продукт очищали колоночной хроматографией (этилацетат:н-гексан = 4:1), получая 140,0 мг метил-2-((S)-1-((S)-4-(6-((4-циано-2-фторбензил)окси)пи ридин-2-ил)-2-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бежевого твердого вещества (выход: 76,8%). LC-MS: RT = 1,93 мин, [М+Н]+ = 599,35.
Стадия Е Синтез2-((S)-1-((S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
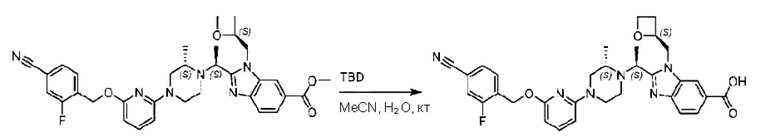
Метил-2-((S)-1-((S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперазин-1-ил)этил)-1-((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (140,0 мг; 0,23 ммоль) растворяли в смеси растворителей ацетонитрила (5,0 мл) и воды (1,0 мл), затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (65,1 мг; 0,47 ммоль). Взаимодействие проводили при комнатной температуре в течение ночи с перемешиванием. Значение рН системы подводили до 6 путем добавления водного раствора лимонной кислоты (2 моль/л) и полученный раствор экстрагировали дихлорметаном (30 мл × 3 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия и упаривали на роторном испарителе для удаления растворителя. Продукт разделяли колоночной хроматографией (метанол:дихлорметан = 8:92), получая 45,0 мг 2-((S)-1-((S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 32,1%).
LC-MS: RT = 1,84 мин. [М+Н]+ = 585,39. 1Н ЯМР (400 МГц, DMSO-d6) δ (млн-1) 8.26 (d, J = 1,0 Гц, 1Н), 7.87 (dd, J = 10,0; 1,4 Гц, 1Н), 7.81 (dd, J = 8,4; 1,6 Гц, 1Н), 7.71-7.66 (m, 2Н), 7.61 (t, J = 7,6 Гц, 1Н), 7.44 (t, J = 8,0 Гц, 1Н), 6.28 (d, J = 8,2 Гц, 1Н), 6.1 0 (d, J = 7,8 Гц, 1Н), 5.37 (s, 2Н), 5.25-5.1 7 (m, 1Н), 5.03 (dd, J = 15,6; 3,0 Гц, 1Н), 4.95 (q, J = 6,6 Гц, 1Н), 4.62 (dd, J = 15,4; 5,0 Гц, 1Н), 4.49-4.39 (m, 1Н), 4.19-4.11 (m, 1Н), 4.07-4.00 (m, 1Н), 3.88 (d, J = 12,6 Гц, 1Н), 2.67-2.54 (m, 2Н), 2.40-2.28 (m, 2Н), 2.27-2.18 (m, 1Н), 1.66-1.56 (m, 2Н), 1.36 (d, J = 6,6 Гц, 3Н), 1.17 (d, J = 6,2 Гц, 3Н).
Пример 22. 2-((5)-1-(4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)-2-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
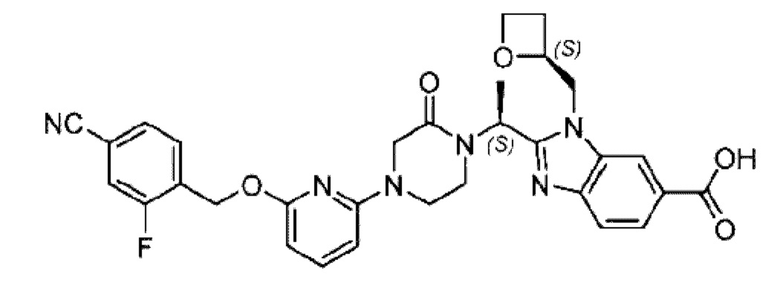
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-((S)-1-(4-(трет-бутоксикарбонил)-2-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
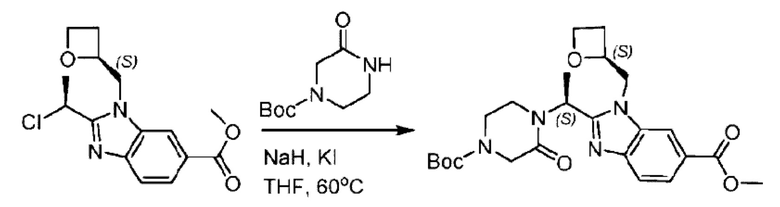
трет-Бутил-3-оксопиперазин-1-карбоксилат (380 мг; 1,90 ммоль), гидрид натрия (380 мг; 9,5 ммоль) и иодид калия (473 мг; 2,85 ммоль) растворяли в 15 мл безводного тетрагидрофурана, затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (600 мг; 1,94 ммоль) и взаимодействие проводили при 60°С в течение 20 мин. По завершении реакции реакционный раствор выливали в 200 мл раствора хлорида аммония и реакционный раствор экстрагировали этилацетатом (50 мл × 3 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (60 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 110 мг метил-2-((S)-1-(4-(трет-бутоксикарбонил)-2-оксопиперази н-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества. LC-MS: RT=2,12 мин, [М+Н]+=473,27.
Стадия В. Синтез метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(2-оксопиперазин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилата
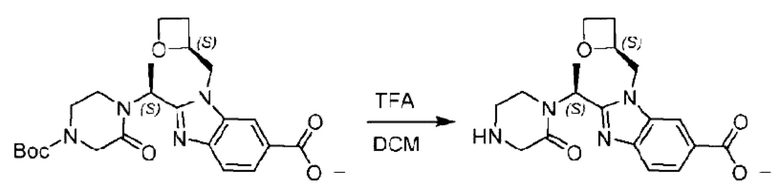
Метил-2-((S)-1-(4-(трет-бутоксикарбонил)-2-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (110 мг; 0,233 ммоль) растворяли в 4 мл безводного дихлорметана, затем добавляли 2 мл трифторуксусной кислоты. Взаимодействие проводили при комнатной температуре в течение 40 мин. По завершении реакции реакционный раствор выливали в 100 мл раствора бикарбоната натрия и экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, получая 90 мг неочищенного продукта метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(2-оксопиперазин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилата. LC-MS: RT=1,96 мин, [М+Н]+=373,27.
Стадия С. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
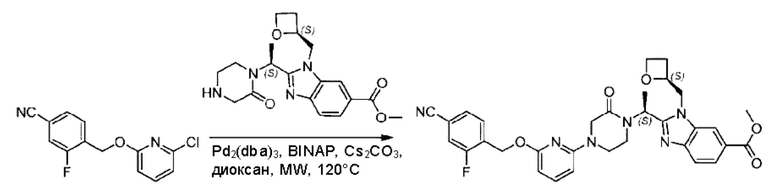
Метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(2-оксопиперазин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилат (90 мг; 0,241 ммоль), 4-(((6-хлорпиридин-2-ил)окси)метил)-3-фторбензонитрил (63,2 мг; 0,241 ммоль), карбонат цезия (157 мг; 0,482 ммоль) и 1,1'-бинафтил-2,2'-бис-дифенилфосфин (15,2 мг; 0,0241 ммоль) растворяли в 5 мл безводного 1,4-диоксана, затем добавляли трис(дибензилиденацетон)дипалладий (Rd2(dba)3; 22,1 мг; 0,0241 ммоль). Систему три раза продували азотом и реакционный раствор выдерживали при 120°С в условиях облучения микроволнами (MW) в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (30 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 72 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества. LC-MS: RT=2,08 мин, [М+Н]+=599,27.
Стадия D. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
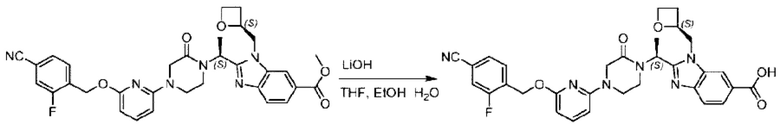
Метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-оксопиперазин-1-ил)этил)-1-((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (72 мг; 0,121 ммоль) растворяли в 4 мл тетрагидрофурана, затем добавляли гидроксида лития моногидрат (26 мг; 0,602 ммоль), 1 мл воды и 1 мл этанола и взаимодействие проводили при комнатной температуре в течение 50 мин. По завершении реакции реакционный раствор нейтрализовали путем добавления 20 мл насыщенного раствора хлорида аммония и экстрагировали этилацетатом (30 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 37 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пи ридин-2-ил)-2-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества.
LC-MS: RT=1,95 мин, [М+Н]+=585,38. 1Н ЯМР (500 МГц, DMSO) δ 12.67 (s, 1 Н), 8.30 (d, J=1,0 Гц, 1 Н), 7.89 (dd, J=9,7; 0,9 Гц, 1 Н), 7.82 (dd, J=8,4; 1,6 Гц, 1 Н), 7.75 (t, J=8,0 Гц, 1 Н), 7.72-7.70 (m, 3Н), 7.54-7.51 (m, 1 Н), 6.73 (m, 1 Н), 5.43 (s, 2Н), 5.15-5.09 (m, 1 Н), 4.75-4.70 (m, 2Н), 4.59-4.55 (m, 1 Н), 4.45-4.41 (m, 1 Н), 4.18-4.14 (m, 1 Н), 3.80-3.72 (m, 2Н), 3.43 (d, J=11,6 Гц, 2Н), 2.94-2.88 (m, 2Н), 2.65-2.57 (m, 1 Н), 2.34-2.24 (m, 1 Н), 1.55 (d, J=6,8 Гц, 3Н).
Пример 23. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-ил)этал)-1-(((S)-оксетан-2-ил)метал)-1Н-бензо[d]имидазол-6-карбоновая кислота
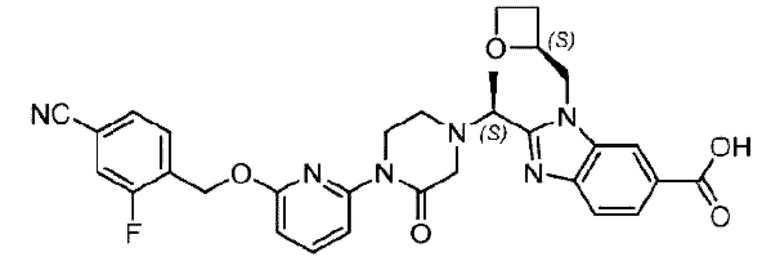
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-формиата
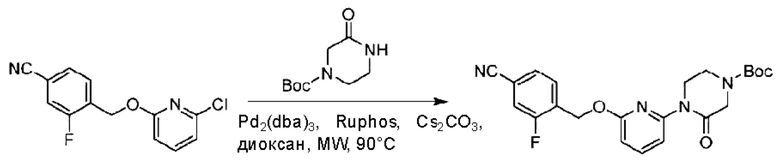
4-(((6-Хлорпиридин-2-ил)окси)метил)-3-фторбензонитрил (130 мг; 0,496 ммоль), трет-бутил-3-оксопиперазин-1-карбоксилат (99,2 мг; 0,496 ммоль), карбонат цезия (323 мг; 0,992 ммоль) и 2-дициклогексилфосфоний-2',6'-диизопропокси-1,1'-бифенил (Ruphos; 22,5 мг; 0,0496 ммоль) растворяли в 5 мл безводного 1,4-диоксана, затем добавляли трис(дибензилиденацетон)дипалладий (45,3 мг; 0,0496 ммоль). Систему трижды продували азотом и реакционный раствор выдерживали при 90°С в условиях облучения микроволнами в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой, экстрагировали этилацетатом (30 мл × 2 раза) и затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Затем полученный неочищенный продукт разделяли колоночной хроматографией, получая 151 мг трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-формиата в виде бледно-желтого твердого вещества. LC-MS: RT=2,08 мин, [М+Н]+=427,21.
Стадия В. Синтез 3-фтор-4-(((6-(2-оксопиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила
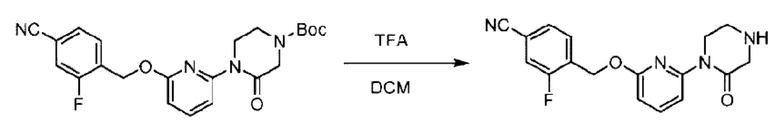
трет-Бутил-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-формиат (151 мг; 0,354 ммоль) растворяли в 4 мл безводного дихлорметана, затем добавляли 2 мл трифторуксусной кислоты и взаимодействие проводили при комнатной температуре в течение 40 мин. По завершении реакции реакционный раствор выливали в 60 мл раствора бикарбоната натрия и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении, получая 120 мг неочищенного продукта 3-фтор-4-(((6-(2-оксопиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила. LC-MS: RT=1,89 мин, [М+Н]+=327,27.
Стадия С.Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
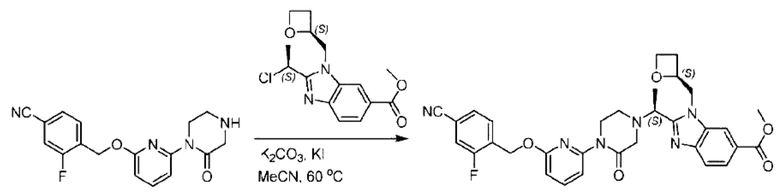
3-Фтор-4-(((6-(2-оксопиперазин-1-ил)пи ридин-2-ил)окси)метил)бензонитрил (120 мг; 0,367 ммоль), карбонат калия (101 мг; 0,733 ммоль) и иодид калия (91,4 мг; 0,551 ммоль) растворяли в 5 мл безводного ацетонитрила, затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (113 мг; 0,367 ммоль) и взаимодействие проводили при 60°С в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (30 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. В конце полученный неочищенный продукт разделяли колоночной хроматографией, получая 146 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества. LC-MS: RT=1,93 мин, [М+Н]+=599,27.
Стадия D. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
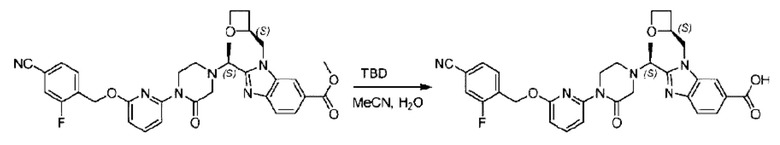
Метил-2-((S)-1-(4-(6-((4-циано-2-фгорбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-ил)этил)-1-((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (146 мг; 0,243 ммоль) растворяли в 4 мл ацетонитрила, затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (71 мг; 0,510 ммоль) и 1 мл воды и взаимодействие проводили при комнатной температуре в течение 7 часов. По завершении реакции реакционный раствор нейтрализовали путем добавления 20 мл насыщенного раствора хлорида аммония и экстрагировали дихлорметаном (30 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. В конце полученный неочищенный продукт разделяли колоночной хроматографией, получая 38 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-оксопиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества.
GC-MS (газовая хроматография-масс-спектрометрия): RT=1,87 мин, [М+Н]+=585,33. 1Н ЯМР (500 МГц, DM SO) δ 12.69 (s, 1 Н), 8.30 (d, J=1,0 Гц, 1 H), 7.89 (dd, J=9,7; 0,9 Гц, 1 H), 7.82 (dd, J=8,4; 1,6 Гц, 1 H), 7.75 (t, J=8,0 Гц, 1 H), 7.72-7.70 (m, 3Н), 7.54-7.51 (m, 1 Н), 6.73-6.70 (m, 1 Н), 5.43 (s, 2Н), 5.15-5.09 (m, 1 Н), 4.75-4.70 (m, 2Н), 4.59-4.55 (m, 1 Н), 4.45-4.41 (m, 1 Н), 4.17 (dt, 9,0; 5,8 Гц, 1 Н), 3.78-3.72 (m, 2Н), 3.43 (d, J=11,6 Гц, 2Н), 2.94-2.88 (m, 2Н), 2.65-2.57 (m, 1 Н), 2.34-2.24 (m, 1 Н), 1.51 (d, J=6,8 Гц, 3Н).
Пример 24. 2-((S)-1-(4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-муравьиная кислота (24А) и 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-муравьиная кислота (24В)
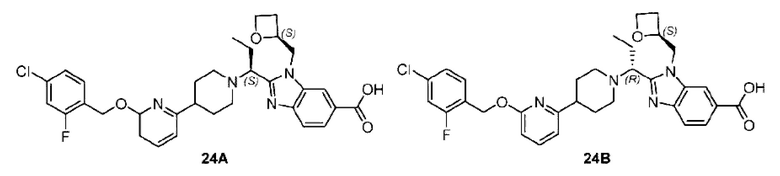
Конкретный путь синтеза соединения 24А приведен ниже.
Стадия А. Синтез метил-4-((R)-2-бромбутирамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата
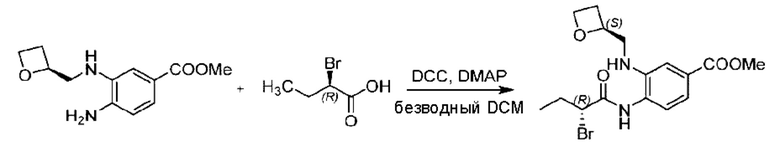
При комнатной температуре растворяли метил-4-амино-3-((((S)-оксетан-2-ил)метил)амино)бензоат (190,2 мг; 0,8 ммоль) и (R)-2-броммасляную кислоту (200,0 мг; 1,2 ммоль) в безводном дихлорметане (5 мл). К вышеупомянутому раствору последовательно добавляли 4-диметиламинопиридин (9,7 мг; 80,0 мкмоль) и дициклогексилкарбодиимид (494,4 мг; 2,4 ммоль). Взаимодействие проводили при комнатной температуре в течение ночи с перемешиванием и по завершении реакции реакционный раствор фильтровали при пониженном давлении с диатомовой землей и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 1/1), получая 300,3 мг метил-4-((R)-2-бромбутирамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата в виде бледно-желтого маслянистого продукта (выход: 97,1%). LC-MS: RT=2,45 мин, [М+Н]+=384,76; 387,02.
Стадия В. Синтез метил-2-((R)-1-бромпропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиата
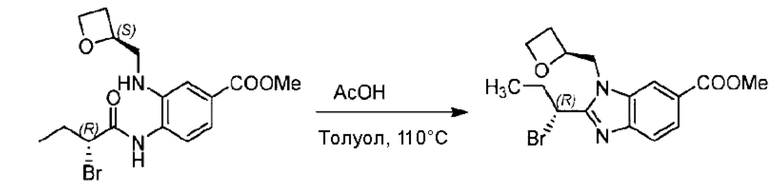
При комнатной температуре растворяли метил-4-((R)-2-бромбутирамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоат (300,3 мг; 0,8 ммоль) в толуоле (3 мл), затем добавляли уксусную кислоту (200 мкл). Взаимодействие проводили при 110°С в течение 3 часов с обратным холодильником и по завершении реакции реакционный раствор концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 3/1), получая 195,2 мг метил-2-((R)-1-бромпропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиата в виде желтого маслянистого продукта (выход: 68,3%). LC-MS: RT=2,34 мин, [М+Н]+=366,93; 369,12.
Стадия С.Синтез метил-2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиата
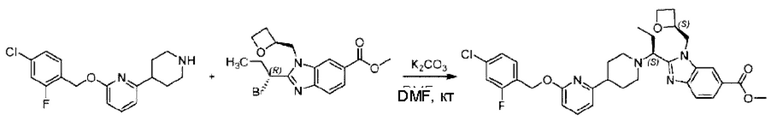
При комнатной температуре растворяли метил-2-((R)-1-бромпропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиат (195,2 мг; 0,5 ммоль) и 2-((4-хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридин (210,3 мг; 0,6 ммоль) в N,N-диметилформамиде (3 мл). К вышеупомянутому раствору добавляли безводный карбонат калия (208,5 мг; 1,5 ммоль) и взаимодействие проводили при комнатной температуре в течение ночи с перемешиванием. По завершении реакции реакционный раствор разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = от 1/1 до 15/1), получая 230,2 мг соединения, метил-2-((S)-1-(4-(6-((4-хлор- 2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[с!]имидазол-6-формиата, в виде белого твердого вещества (выход: 71,4%). LC-MS: RT=3,43 мин, [М+Н]+=607,07.
Стадия D. Синтез 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-муравьиной кислоты
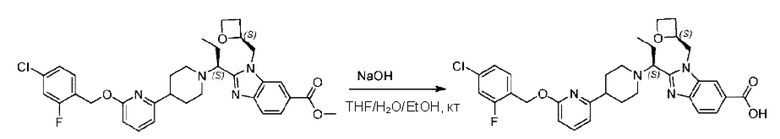
При комнатной температуре растворяли метил-2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пи ридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиат (230,2 мг; 0,4 ммоль) в смеси тетрагидрофурана (6 мл), воды (2 мл) и этанола (2 мл), затем добавляли гидроксид натрия (48,0 мг; 1,2 ммоль) и взаимодействие проводили при комнатной температуре в течение ночи с перемешиванием.
По завершении реакции по каплям добавляли водный раствор лимонной кислоты в концентрации 1 моль на литр до тех пор, пока значение рН раствора не достигало 5. Реакционный раствор разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (25 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали полупрепаративной HPLC (подвижная фаза: (вода, содержащая 0,1% аммиака)/ацетонитрил = от 90/10 до 40/60), получая 68,8 мг 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[с1]имидазол-6-муравьиной кислоты (выход: 30,7%).
LC-MS: RT=2,68 мин, [М+Н]+=593,21. 1Н ЯМР (400 МГц, CD3OD) δ 8.40 (s, 1 Н), 8.09 (dd, J=8,6; 1,5 Гц, 1 Н), 7.86 (d, J=8,5 Гц, 1 Н), 7.67 (t, J=7,8 Гц, 1 Н), 7.51 (t, J=8,0 Гц, 1 Н), 7.25 (ddd, J=10,7; 9,0; 2,0 Гц, 2Н), 6.91 (d, J=7,2 Гц, 1 Н), 6.75 (d, J=8,3 Гц, 1 Н), 5.43 (s, 2Н), 5.22 (q, J=7,9 Гц, 1 Н), 5.09 (dd, J=11,0; 3,9 Гц, 1 Н), 4.92 (s, 1 Н), 4.80 (d, J=1 5,3 Гц, 3Н), 4.04 (s, 1 Н), 3.64 (s, 2Н), 3.03 (s, 1 Н), 2.93 (р, J=7,4 Гц, 1 Н), 2.70 (р, J=8,3 Гц, 1 Н), 2.47 (d, J=36,2 Гц, 2Н), 2.20 (t, J=12,0 Гц, 3Н), 2.15-1.96 (m, 2Н), 0.98 (t, J=7,3 Гц, 3Н).
Конкретный путь синтеза соединения 24 В приведен ниже.
Стадия А. Синтез метил-4-((S)-2-бромбутирамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата
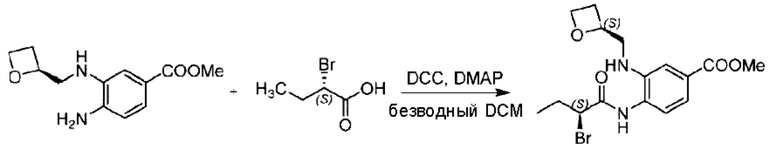
При комнатной температуре растворяли метил-4-амино-3-((((S)-оксетан-2-ил)метил)амино)бензоат (190,2 мг; 0,8 ммоль) и (S)-2-броммасляную кислоту (200,0 мг; 1,2 ммоль) в безводном дихлорметане (5 мл). К вышеупомянутому раствору последовательно добавляли 4-диметиламинопиридин (9,7 мг; 80,0 микромоль) и дициклогексилкарбодиимид (494,4 мг; 2,4 ммоль). Взаимодействие проводили при комнатной температуре в течение ночи с перемешиванием и по завершении реакции реакционный раствор фильтровали при пониженном давлении с использованием диатомовой земли и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 1/1), получая 300,3 мг метил-4-((S)-2-бромбутирамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата в виде бледно-желтого маслянистого продукта (выход: 97,1%). LC-MS: RT=2,45 мин, [М+Н]+=384,76; 387,02.
Стадия В. Синтез метил-2-((S)-1-бромпропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиата
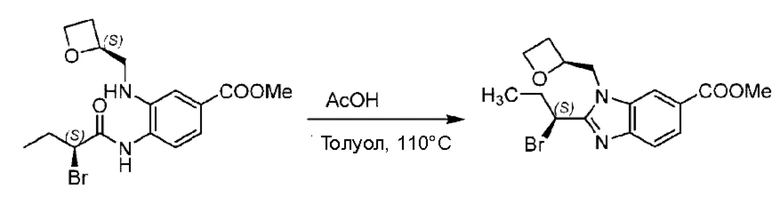
При комнатной температуре растворяли метил-4-((S)-2-бромбутирамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоат (300,3 мг; 0,8 ммоль) в толуоле (3 мл), затем добавляли уксусную кислоту (200 мкл). Взаимодействие проводили при 110°С в течение 3 часов с обратным холодильником и по завершении реакции реакционный раствор концентрировали при пониженном давлении. Затем полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 3/1), получая 195,2 мг метил-2-((S)-1-бромпропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиата в виде желтого маслянистого продукта (выход: 68,3%). LC-MS: RT=2,34 мин, [М+Н]+=366,93; 369,12.
Стадия С. Синтез метил-2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиата
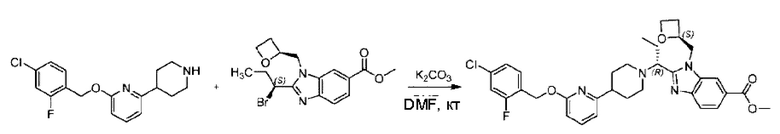
При комнатной температуре растворяли метил-2-((S)-1-бромпропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиат (195,2 мг; 0,5 ммоль) и 2-((4-хлор-2-фторбензил)окси)-6-(пиперидин-4-ил)пиридин (210,3 мг; 0,6 ммоль) в N,N-диметилформамиде (3 мл). К вышеупомянутому раствору добавляли безводный карбонат калия (208,5 мг; 1,5 ммоль) и взаимодействие проводили при комнатной температуре в течение ночи с перемешиванием. По завершении реакции реакционный раствор разбавляли водой (20 мл) и экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = от 1/1 до 15/1), получая 230,2 мг соединения, метил-2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиата, в виде белого твердого вещества (выход: 71,4%). LC-MS: RT=3,43 мин, [М+Н]+=607,07.
Стадия D. Синтез 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-муравьиной кислоты
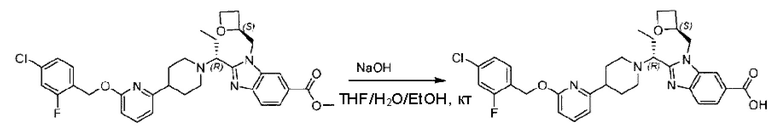
При комнатной температуре растворяли метил-2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пи ридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазо-6-формиат (230,2 мг; 0,4 ммоль) в смеси тетрагидрофурана (6 мл), воды (2 мл) и этанола (2 мл), затем добавляли гидроксид натрия (48,0 мг; 1,2 ммоль) и взаимодействие проводили при комнатной температуре в течение ночи с перемешиванием.
По завершении реакции по каплям добавляли водный раствор лимонной кислоты в концентрации 1 моль на литр до тех пор, пока значение рН раствора не достигало 5. Реакционный раствор разбавляли водой (20 мл), экстрагировали этилацетатом (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (25 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали полупрепаративной HPLC (подвижная фаза: (вода, содержащая 0,1% аммиака)/ацетонитрил = от 90/10 до 40/60), получая 68,8 мг 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)пропил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[с!]имидазол-6-муравьиной кислоты (выход: 30,7%).
LC-MS: RT=2,68 мин, [М+Н]+=593,21. 1Н ЯМР (400 МГц, CD3OD) δ 8.34 (d, J=1,5 Гц, 1 Н), 8.03 (dd, J=8,5; 1,5 Гц, 1 Н), 7.77 (d, J=8,5 Гц, 1 Н), 7.60 (dd, J=8,2; 7,3 Гц, 1 Н), 7.50 (t, J=8,3 Гц, 1 Н), 7.25-7.14 (m, 2Н), 6.83 (d, J=7,3 Гц, 1 Н), 6.66 (d, J=8,1 Гц, 1 Н), 5.41 (s, 2Н), 5.32 (qd, J=7,2; 2,5 Гц, 1 Н), 4.85 (dd, J=15,8; 2,7 Гц, 1 Н), 4.75 (dd, J=15,8; 5,9 Гц, 1 Н), 4.66 (td, J=8,0; 5,9 Гц, 1 Н), 4.51 (dd, J=10,6; 4,1 Гц, 1 Н), 4.40 (dt, J=9,3; 5,8 Гц, 1 Н), 3.22 (t, J=10,9 Гц, 2Н), 2.92 (td, J=10,8; 4,8 Гц, 1 Н), 2.80 (ddt, J=13,8; 8,1; 4,0 Гц, 1 Н), 2.68 (dq, J=10,9; 5,5; 4,8 Гц, 1 Н), 2.60 (td, J=12,1; 3,4 Гц, 1 Н), 2.56-2.46 (m, 1 Н), 2.35 (ddd, J=13,1; 10,6; 7,1 Гц, 1 Н), 2.19 (ddt, J=11,6; 7,3; 4,3 Гц, 1 Н), 1.96 (td, J=10,5; 9,5; 3,4 Гц, 2Н), 1.91-1.72 (m, 2Н), 0.99 (t, J=7,3 Гц, 3Н).
Пример 25. 2-((4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)(цикпопропил)метил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
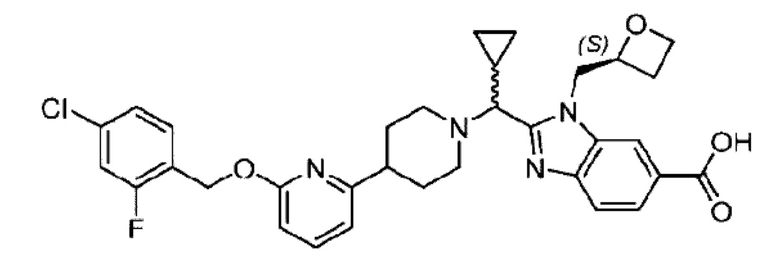
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез этил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилацетата
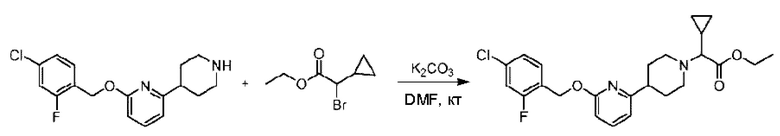
При комнатной температуре растворяли 4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин (155,0 мг; 483,2 мкмоль), этил-2-бром-2-циклопропил ацетат (200,0 мг; 966,4 мкмоль) и карбонат калия (267,3 мг; 1,9 ммоль) в N;N-диметилформамиде (5 мл) и взаимодействие проводили при комнатной температуре в течение 16 часов. По завершении реакции реакционный раствор разбавляли водой (80 мл), экстрагировали этилацетатом (50 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (50 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 5/1), получая 177,2 мг этил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилацетата (смеси S- и R-конфигурации) в виде бледно-желтого маслянистого продукта (выход: 82,5%). LC-MS: RT=3,00 мин, [М+Н]+=446,97.
Стадия В. Синтез 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилуксусной кислоты
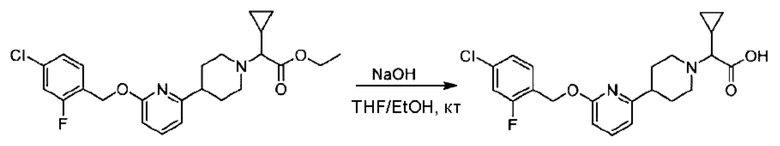
При комнатной температуре растворяли этил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилацетат (177,0 мг; 396,7 мкмоль) в смеси тетрагидрофурана (10 мл), этанола (3 мл) и воды (3 мл), затем добавляли гидроксид натрия (160,0 мг; 4,0 ммоль). Взаимодействие проводили при комнатной температуре в течение 48 часов. По завершении реакции по каплям добавляли водный раствор лимонной кислоты в концентрации 1 моль на литр до тех пор, пока значение рН раствора не достигало 5. Полученную смесь разбавляли водой (50 мл), экстрагировали этилацетатом (25 мл × 3 раза), промывали насыщенным солевым раствором (25 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = от 1/1 до 10/1), получая 132,0 мг 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилуксусной кислоты (смеси S- и R-конфигураций) в виде желтого маслянистого продукта (выход: 76,9%). LC-MS: RT=2,45 мин, [М+Н]+=419,05.
Стадия С. Синтез метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилацетамино)-3-(((S)-оксетан-2-ил)метил)бензоата
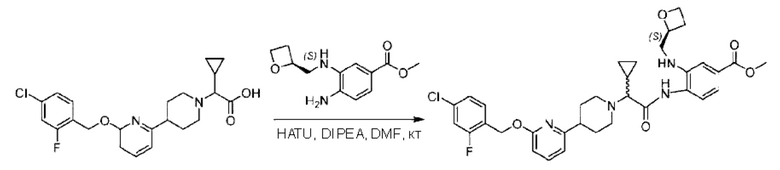
При комнатной температуре растворяли 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилуксусную кислоту (132,0 мг; 315,1 мкмоль), 2-(7-азабензотриазол)-N;N,N'N'-тетраметилурония гексафторфосфат (120,0 мг; 315,1 мкмоль) и N;N-диизопропилэтиламин (122,5 мг; 945,2 мкмоль) в N;N-диметилформамиде (5 мл). Взаимодействие проводили при комнатной температуре в течение получаса, затем добавляли метил-(S)-4-амино-3-((((S)-оксетан-2-ил)метил)амино)бензоат (74,3 мг; 315,1 мкмоль) и взаимодействие продолжали в течение 24 часов.
По завершении реакции реакционный раствор разбавляли этилацетатом (50 мл), экстрагировали, после этого промывали водой (30 мл × 3 раза), насыщенным физиологическим раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = от 5/1 до 1/1), получая 85,0 мг метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилацетамино)-3-(((S)-оксетан-2-ил)метил)бензоата (смеси S- и Реконфигураций) в виде желтого маслянистого продукта (выход: 42,2%). LC-MS: RT=3,38 мин, [М+Н]+=637,14.
Стадия D. Синтез метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)(цикпопропил)метил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
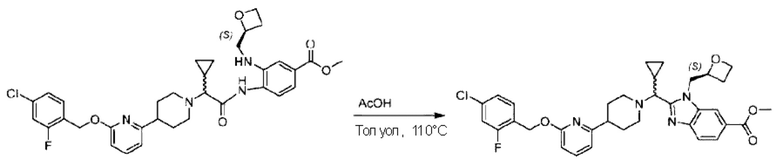
При комнатной температуре в толуол (5 мл) добавляли метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-циклопропилацетамино)-3-(((S)-оксетан-2-ил)метил)бензоат (85,0 мг; 137,2 мкмоль), затем добавляли уксусную кислоту (200 мкл). Реакционный раствор нагревали до 110°С и реакцию проводили в течение 4 часов. По завершении реакции реакционный раствор охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали этилацетатом (15 мл × 3 раза). Затем органические фазы объединяли, последовательно промывали насыщенным водным раствором карбоната натрия (15 мл × 3 раза) и насыщенным солевым раствором (15 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = от 1/1 до 10/1), получая 51,0 мг метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)(циклопропил)метил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата (смеси S- и R-конфигураций) в виде желтого маслянистого продукта (выход: 60,1%). LC-MS: RT=3,48 мин, [М+Н]+=619,29.
Стадия Е Синтез 2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)(циклопропил)метил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
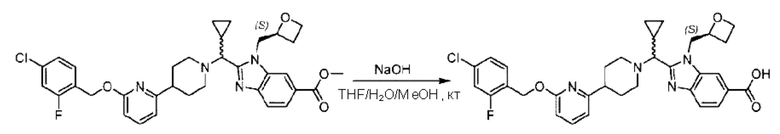
При комнатной температуре растворяли метил-2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)(циклопропил)метил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (51,0 мг; 82,4 мкмоль) в растворе тетрагидрофурана (6 мл), метанола (2 мл) и воды (2 мл), затем добавляли гидроксид натрия (32,9 мг; 823,7 мкмоль) и взаимодействие проводили при комнатной температуре в течение 24 часов. По завершении реакции по каплям добавляли водный раствор лимонной кислоты в концентрации 1 моль на литр до тех пор, пока значение рН раствора не достигало 5. Полученную смесь разбавляли водой (50 мл), экстрагировали этилацетатом (25 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (25 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали обращенно-фазовой HPLC (подвижная фаза: (вода, содержащая 0,1% аммиака)/ацетонитрил = от 90/10 до 40/60) и лиофилизировали, получая 9,2 мг 2-((4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)(ци клопропил)метил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (смеси S- и R-конфигураций) в виде белого твердого вещества (выход: 18,4%). LC-MS: RT=2,84 мин, [М+Н]+=605,22.
Пример 26. 2-(1-(4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-метоксиэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксипропионата
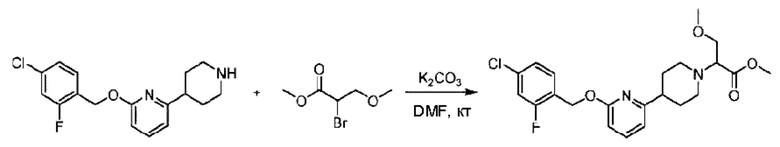
При комнатной температуре растворяли 4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин (155,0 мг; 483,2 мкмоль), метил-2-бром-3-метоксипропионат (200,0 мг; 966,4 мкмоль) и карбонат калия (267,3 мг; 1,9 ммоль) в N;N-диметилформамиде (5 мл) и взаимодействие проводили при комнатной температуре в течение 16 часов.
По завершении реакции реакционный раствор разбавляли водой (80 мл), экстрагировали этилацетатом (50 мл × 3 раза), промывали насыщенным солевым раствором (50 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 5/1), получая 177,2 мг метил- 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксипропионата (смеси S- и R-конфигурации) в виде бледно-желтого маслянистого продукта (выход: 82,5%). LC-MS: RT=2,63 мин, [М+Н]+=436,96.
Стадия В. Синтез 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксипропионовой кислоты
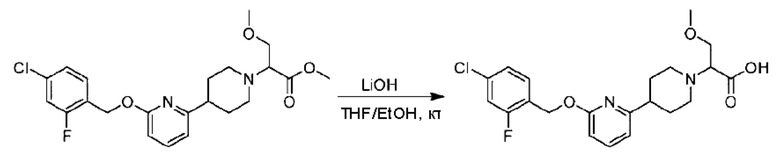
При комнатной температуре растворяли метил-2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксипропионат (177,0 мг; 396,7 мкмоль) в растворе тетрагидрофурана (10 мл), этанола (3 мл) и воды (3 мл), затем добавляли гидроксидлития (96,0 мг; 4,0 ммоль) и взаимодействие проводили при комнатной температуре в течение 6 часов. По завершении реакции по каплям добавляли водный раствор лимонной кислоты в концентрации 1 моль на литр до тех пор, пока значение рН раствора не достигало 5. Реакционный раствор разбавляли водой (50 мл), экстрагировали этилацетатом (25 мл × 3 раза), промывали насыщенным солевым раствором (25 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент : петролейный эфир/этилацетат = от 1/1 до 10/1), получая 139,0 мг 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксипропионовой кислоты (смеси S- и R-конфигураций) в виде желтого маслянистого продукта (выход: 69,2%). LC-MS: RT=2,37 мин, [М+Н]+=423,03.
Стадия С.Синтез метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксипропионамидо)-3-(((S)-оксетан-2-ил)метил)бензоата
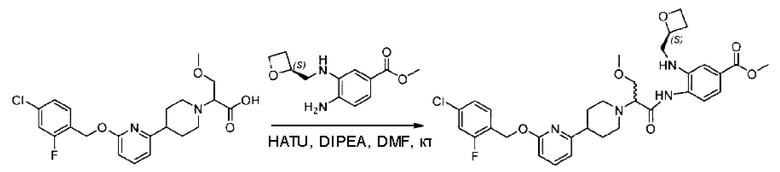
При комнатной температуре растворяли 2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксиуксусную кислоту (195,0 мг; 460,1 мкмоль), 2-(7-азабензотриазол)-N;N,N'N'-тетраметилурония гексафторфосфат (174,8 мг; 460,1 мкмоль) и N;N-диизопропилэтиламин (178,3 мг; 1,4 ммоль) в N;N-диметилформамиде (5 мл) и взаимодействие проводили при комнатной температуре в течение 0,5 часа. Затем добавляли метил-4-амино-3-((((S)-оксетан-2-ил)метил)амино)бензоат (109,0 мг; 460,1 мкмоль) и взаимодействие продолжали в течение 24 часов.
По завершении реакции реакционный раствор разбавляли этилацетатом (50 мл), экстрагировали, последовательно промывали водой (30 мл × 3 раза) и насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = от 2/1 до 1/1), получая 145,0 мг метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксипропионамидо)-3-(((S)-оксетан-2-ил)метил)бензоата (смеси S- и Реконфигураций) в виде желтого маслянистого продукта (выход: 49,0%). LC-MS: RT=3,12 мин, [М+Н]+=641,05.
Стадия D. Синтез метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-метоксиэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
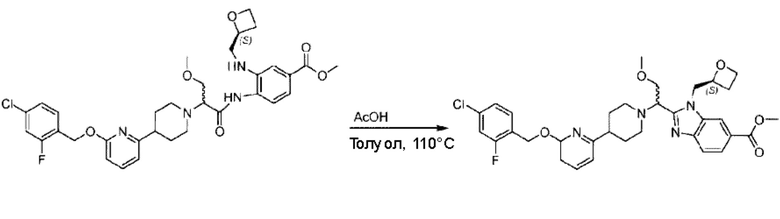
При комнатной температуре в толуол (5 мл) добавляли метил-4-(2-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-метоксипропионамидо)-3-(((S)-оксетан-2-ил)метил)бензоат (145,0 мг; 227,8 мкмоль), затем добавляли уксусную кислоту (100 мкл). Реакционный раствор нагревали до 110°С и реакцию проводили в течение 4 часов.
По завершении реакции реакционный раствор охлаждали до комнатной температуры, разбавляли водой (30 мл) и экстрагировали этилацетатом (15 мл × 3 раза). Затем органические фазы объединяли, последовательно промывали насыщенным водным раствором карбоната натрия (15 мл × 3 раза) и насыщенным солевым раствором (15 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = от 1/1 до 10/1), получая 98,5 мг метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-метоксиэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата (смеси S- и R-конфигураций) в виде желтого маслянистого продукта (выход: 68,0%). LC-MS: RT=3,48 мин, [М+Н]+=623,48.
Стадия Е. Синтез 2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-метоксиэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
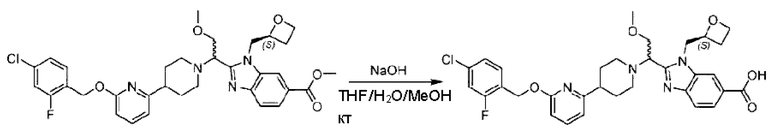
При комнатной температуре растворяли метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пи ридин-2-ил)пиперидин-1-ил)-2-метоксиэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (98,5 мг; 158,1 мкмоль) в растворе тетрагидрофурана (6 мл), метанола (2 мл) и воды (2 мл), затем добавляли гидроксид натрия (101,8 мг; 2,5 ммоль) и взаимодействие проводили при комнатной температуре в течение 24 часов. По завершении реакции по каплям добавляли водный раствор лимонной кислоты в концентрации 1 моль на литр дотех пор, пока значение рН раствора не достигало 5. Реакционный раствор разбавляли водой (50 мл), экстрагировали этилацетатом (25 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (25 мл × 3 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный остаток далее очищали обращенно-фазовой HPLC (подвижная фаза: (вода, содержащая 0,1% аммиака)/ацетонитрил = от 90/10 до 40/60) и лиофилизировали, получая 18,7 мг 2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пи ридин-2-ил)пиперидин-1-ил)-2-метоксиэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (смеси S- и R-конфигурации) в виде белого твердого вещества (выход: 20,5%). LC-MS: RT=2,82 мин, [М+Н]+=609,27.
Пример 27. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
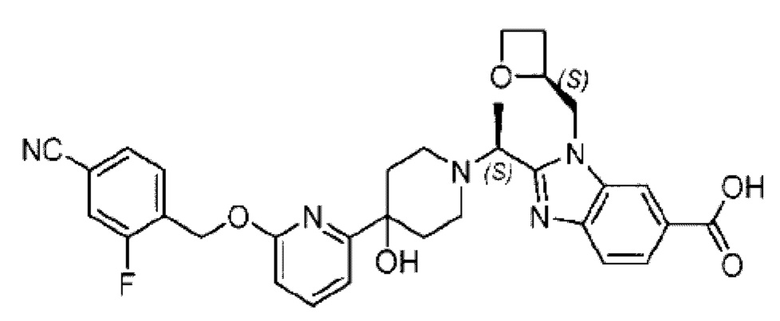
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-бромпиридин-2-ил)-4-гидроксипиперидин-1-формиата
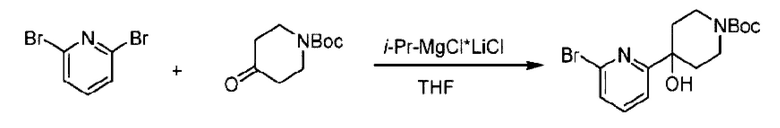
При комнатной температуре растворяли 2,6-дибромпиридин (5 г; 21,18 ммоль) в тетрагидрофуране (30 мл), затем добавляли хлорид изопропилмагния-хлорид лития (16,3 мл) в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 2 часов, затем добавляли трет-бутил-4-оксопиперидин-1-карбоксилат (4,23 г; 21,18 ммоль) и взаимодействие проводили при комнатной температуре в течение ночи.
По завершении реакции реакционный раствор гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 5/1), получая 2 г трет-бутил-4-(6-бромпиридин-2-ил)-4-гидроксипиперидин-1-формиата в виде бледно-желтой маслянистой жидкости (выход: 27%). LC-MS: RT=2,04 мин, [М-55]+=301,11.
Стадия В. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-карбоксилата
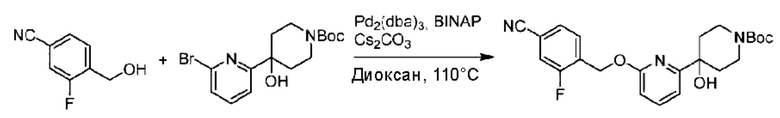
3-Фтор-4-(гидроксиметил)бензонитрил (425 мг; 2,81 ммоль), трет-бутил-4-(6-бромпиридин-2-ил)-4-гидроксипиперидин-1-формиат (1 г; 2,81 ммоль), палладиевый катализатор (128 мг), BINAP(175 мг; 0,28 ммоль) и карбонат цезия (1,83 г; 5,62 ммоль) растворяли в 1,4-диоксане (50 мл), реакционный раствор нагревали до 110°С и реакционный раствор перемешивали в течение 8 часов.
По завершении реакции реакционный раствор фильтровали, используя диатомовую землю, и элюировали дихлорметаном. Фильтрат концентрировали и полученный неочищенный продукт очищали колоночной хроматографией (элюент: этилацетат/н-гексан = 10/1), получая 820 мг трет-бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-карбоксилата в виде бледно-желтой маслянистой жидкости (выход: 68%). LC-MS: RT=2,12 мин, [М-55]+=372,33.
Стадия С.Синтез 3-фтор-4-(((6-(4-гидроксипиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрила
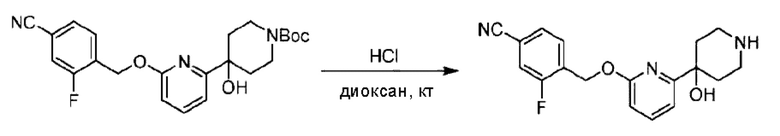
трет-Бутил-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-карбоксилат (820 мг; 1,92 ммоль) растворяли в соляной кислоте в 1,4-диоксане (15 мл) и смесь перемешивали при комнатной температуре.
По завершении реакции реакционный раствор упаривали на роторном испарителе при пониженном давлении, получая 410 мг в 3-фтор-4-(((6-(4-гидроксипиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрила в виде бледно-желтого твердого вещества (выход: 69%). LC-MS: RT=1,63 мин, [М+Н]+=328,22.
Стадия D. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
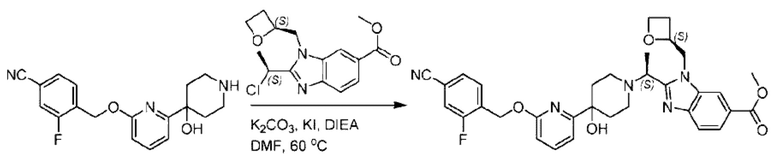
При комнатной температуре растворяли 3-фтор-4-(((6-(4-гидроксипиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрил (150 мг; 0,46 ммоль), иодид калия (153 мг; 0,92 ммоль) и карбонат калия (127 мг; 0,92 ммоль) в N;N-диметилформамиде (10 мл), затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (141 мг; 0,46 ммоль). Для осуществления взаимодействия реакционный раствор нагревали до 50°С.
По завершении реакции реакционный раствор гасили насыщенным раствором хлорида натрия и экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 10/1), получая 100 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого твердого вещества (выход: 36%). LC-MS: RT=1,73 мин, [М+Н]+=600,34.
Стадия Е. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
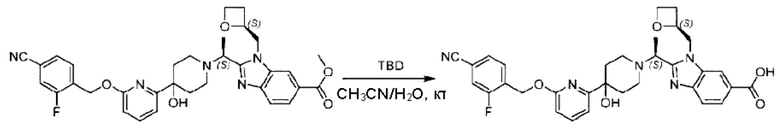
При комнатной температуре растворяли метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (100 мг; 0,17 ммоль) в смеси растворителей (6 мл; ацетонитрил/вода = 5/1), затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (47 мг; 0,34 ммоль). Взаимодействие проводили при комнатной температуре.
По завершении реакции реакционную смесь гасили раствором лимонной кислоты и экстрагировали этилацетатом (10 мл × 3 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Затем неочищенный продукт, полученный из концентрата, очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол=10/1), получая 58,26 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-4-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 58%).
LC-MS: RT=1,72 мин, [М+Н]+=585,36. 1Н ЯМР (400 МГц, DMSO-d6) δ 12.86 (s, 1 Н), 10.63 (s, 1 Н), 8.38 (s, 1 Н), 7.94-7.67 (m, 6Н), 7.24 (d, J=7,4 Гц, 1 Н), 6.78 (d, J=8,0 Гц, 1 Н), 5.70-5.40 (m, 3Н), 5.28-5,06 (m, 2Н), 4.90-4.72 (m, 2Н), 4.46 (d, J=6,2 Гц, 1 Н), 4.18 (dt, J=9,2; 5,9 Гц, 1 Н), 3.86-3.70 (m, 1 Н), 3.60-3.40 (m, 2Н), 3.24-3.10 (m, 2Н), 2.69-2.54 (m, 2Н), 2.30-2.22 (m, 1 Н), 1.82-1.64 (m, 4Н).
Пример 28. Синтез 2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-фторэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
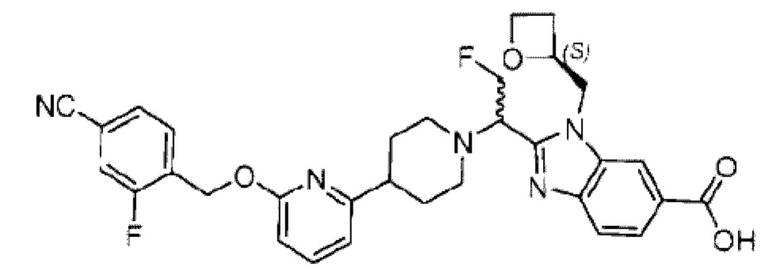
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-бром-3-гидроксипропионата
С использованием бани со смесью льда и соли в водный раствор (9,3 мл), содержащий DL-серин (1,2 г; 11,4 ммоль), последовательно добавляли бромистоводородную кислоту (2,8 мл; 48%-ный водный раствор), бромид калия (4,6 г; 38,8 ммоль) и карбонат калия (244 мг; 1,77 ммоль). Реакционный раствор перемешивали при этой температуре в течение 10 мин и взаимодействие проводили при комнатной температуре в течение 3 часов. По завершении реакции раствор экстрагировали этилацетатом (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении.
Полученный остаток растворяли в метаноле (8,8 мл), затем добавляли ацетилхлорид (924 мкл; 13,0 ммоль), далее нагревали с обратным холодильником и проводили взаимодействие в течение 3 часов. По завершении реакции растворитель удаляли выпариванием при пониженном давлении, полученный остаток разбавляли этилацетатом (100 мл), последовательно промывали насыщенным раствором бикарбоната натрия (50 мл) и насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт (1,5 г) непосредственно использовали на следующей стадии реакции.
Стадия В. Синтез метил-2-(4-(6-((4-циано-2-фторбензил)окси))пиридин-2-ил)пиперидин-1-ил)-3-гидроксипропионата
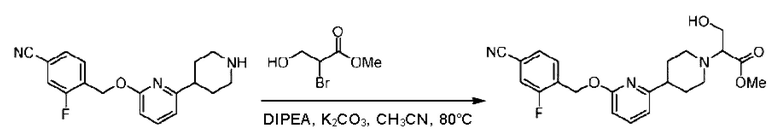
При комнатной температуре в ацетонитрил (4,0 мл), содержащий метил-2-бром-3-гидроксипропионат (1,5 г; 8,2 ммоль), последовательно добавляли 3-фтор-4-((((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)бензонитрил (950 мг; 2,73 ммоль), N;N-диизопропилэтиламин (1,0 мл; 6,06 ммоль) и карбонат калия (1,0 г; 7,25 ммоль). Реакционный раствор нагревали до 80°С и реакцию проводили в течение 2 часов. По завершении реакции реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 5/95), получая 800 мг метил-2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-)пиперидин-1-ил)-3-гидроксипрспионата (смеси S- и R-конфигурации) в виде желтого маслянистого продукта (выход: 71,3%). LC-MS: RT=1,70 мин, [М+Н]+=414,26.
Стадия С.Синтез метил-2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-фторпропионата
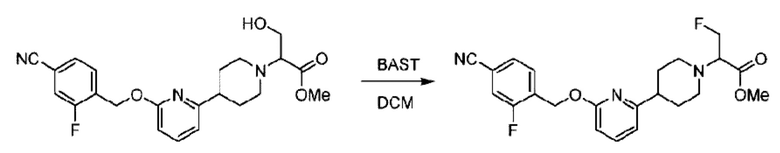
С использованием бани с ледяной водой к раствору метил-2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-гидроксипропионата (200 мг; 0,484 ммоль) в дихлорметане (5,0 мл) добавляли бис(2-метоксиэтил)аминотрифторсульфид (179 мкл; 0,969 ммоль), температура естественным образом поднималась до комнатной температуры, и реакцию проводили в течение 2 часов.
По завершении реакции полученную смесь охлаждали в бане с ледяной водой, гасили водой и экстрагировали дихлорметаном (80 мл). Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия (50 мл) и насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Затем полученный остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлор метан = 3/97), получая 160 мг метил-2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-фторпропионата (смеси S- и Реконфигураций) в виде бледно-желтого маслянистого продукта (выход: 79,7%). LC-MS: RT=1,73 мин, [М+Н]+=416,26.
Стадия D. Синтез метил-4-(2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-фторпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата
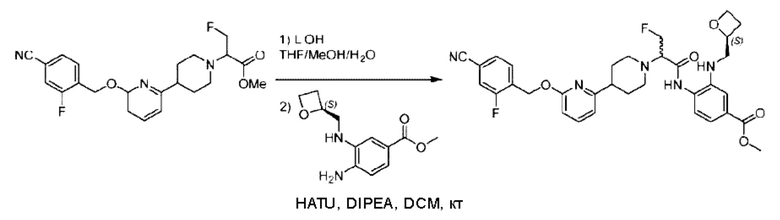
При комнатной температуре к смеси тетрагидрофуран/метанол/вода (3,0 мл/0,5 мл/0,5 мл), содержащей метил-2-(4-(6-((4-циано-2-фторбензил)окси)-пиридин-2-ил)пиперидин-1-ил)-3-фторпропионат (160 мг; 0,386 ммоль), добавляли гидроксида лития моногидрат (71 мг; 1,54 ммоль) и взаимодействие проводили при комнатной температуре в течение 0,5 часа. По завершении реакции реакционный раствор гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт использовали непосредственно на следующей стадии реакции. LC-MS: RT=1,72 мин, [М+Н]+=402,26.
Полученный выше неочищенный продукт растворяли в дихлорметане (4,0 мл), затем последовательно добавляли 2-(7-азабензотриазол)-N;N,N',N'-тетраметилурония гексафторфосфат (293 мг; 0,77 ммоль), N,N-диизопропилэтиламин (127 мкл, 0,77 ммоль) и метил-(S)-4-амино-3-(((оксетан-2-ил)метил)амино)бензоат (90 мг; 0,386 ммоль). Далее проводили взаимодействие при комнатной температуре в течение 40 мин. По завершении реакции реакционный раствор разбавляли дихлорметаном (100 мл), органическую фазу последовательно промывали водой (50 мл) и насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 5/95). Получали 200 мг метил-4-(2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-фторпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата (смесь S- и R-конфигураций) в виде бледно-желтого маслянистого продукта (выход: 83,6%). LC-MS: RT=1,79 мин, [М+Н]+=620,36.
Стадия Е. Синтез метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-фторэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
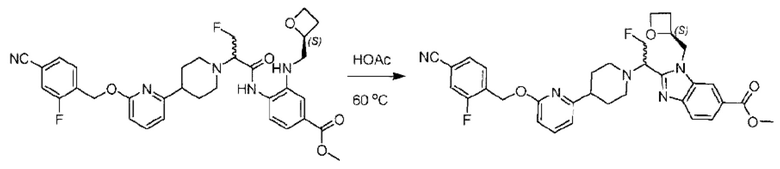
Метил-4-(2-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-3-фторпропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоат (200 мг; 0,323 ммоль) растворяли в уксусной кислоте (2,0 мл) и взаимодействие проводили при 60°С в течение 2 часов. По завершении реакции добавляли этилацетат (100 мл) и при перемешивании добавляли насыщенный раствор бикарбоната натрия до тех пор, пока не появлялись пузырьки. Затем органические фазы разделяли, последовательно промывали водой (50 мл) и насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 5/95). Получали 130 мг метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-фторэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата (смесь S- и R-конфигураций) в виде бледно-желтого масла (выход: 67,0%). LC-MS: RT=1,81 мин, [М+Н]+=602,30.
Стадия F. Синтез 2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-фторэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
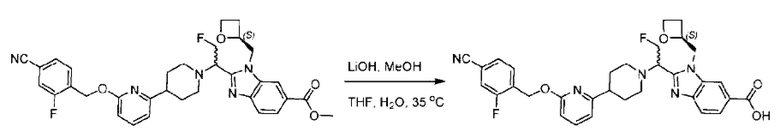
При комнатной температуре добавляли гидроксида лития моногидрат (36 мг; 0,86 ммоль) к раствору метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-фторэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата (130 мг; 0,216 ммоль) в смеси тетрагидрофуран/метанол/вода (2,5 мл/2,5 мл/1,0 мл), реакционный раствор нагревали до 35°С и реакцию проводили в течение 1 часа. По завершении реакции реакционный раствор гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 6/94). Получали 19 мг 2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)-2-фторэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (смесь S- и R-конфигураций) в виде белого твердого вещества, (выход: 15,0%). LC-MS: RT=1,77 мин, [М+Н]+=588,23.
Пример 29. 2-((S)-1-((R)-4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
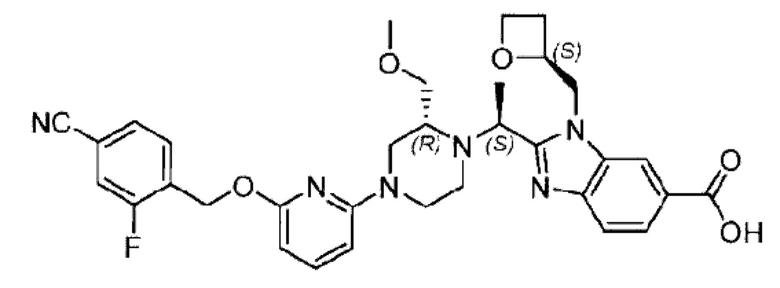
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 4-(((6-бромпиридин-2-ил)окси)метил)-3-фторбензонитрила
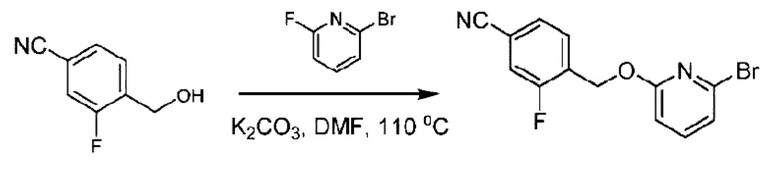
3-Фтор-4-(гидроксиметил)бензонитрил (5,00 г; 28,41 ммоль) растворяли в N;N-диметилформамиде (30 мл), затем добавляли карбонат калия (7,84 г; 56,82 ммоль) и 2-бром-6-фторпиридин (4,30 мг; 28,41 ммоль). Затем реакционный раствор нагревали до 110°С и проводили взаимодействие.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/10), получая 4,50 г 4-(((6-бромпиридин-2-ил)окси)метил)-3-фторбензонитрила в виде белого твердого вещества (выход: 52%). LC-MS: RT=2,14 мин, [М+Н]+=307,03.
Стадия В. Синтез трет-бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(гидроксиметил)пиперазин-1-формиата
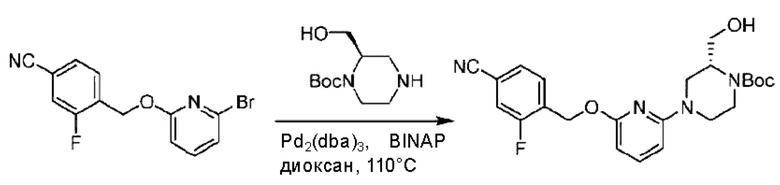
4-(((6-Бромпиридин-2-ил)окси)метил)-3-фторбензонитрил (1,00 г; 3,26 ммоль), трет-бутил-(R)-2-(гидроксиметил)пиперазин-1-карбоксилат (0,70 г; 3,26 ммоль), палладиевый катализатор (302 мг), BINAP (411 мг; 0,66 ммоль) и карбонат цезия (2 г; 6,52 ммоль) растворяли в 1,4-диоксане (20 мл), затем реакционный раствор нагревали до 100°С и реакционный раствор перемешивали в течение 8 часов.
По завершении реакции реакционный раствор фильтровали с отсасыванием через диатомовую землю и элюировали дихлорметаном. Фильтрат концентрировали и полученный неочищенный продукт очищали колоночной хроматографией (элюент: этилацетат/н-гексан = 1/1), получая 1,00 г трет-бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(гидроксиметил)пиперазин-1-формиата в виде бледно-желтой маслянистой жидкости (выход: 69%). LC-MS: RT=2,09 мин, [М-55]+=443,22.
Стадия С.Синтез трет-бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-формиата
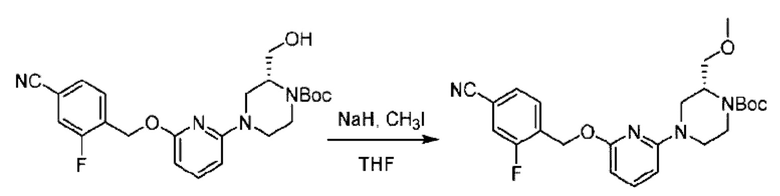
трет-Бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(гидроксиметил)пиперазин-1-формиат (100 мг; 0,23 ммоль) растворяли в тетрагидрофуране (5 мл). Раствор охлаждали до 0°С, затем добавляли гидрид натрия (11 мг; 0,46 ммоль). Смесь перемешивали при комнатной температуре в течение 0,5 часа, затем добавляли подметан (65 мг; 0,46 ммоль) и реакцию проводили в течение ночи при комнатной температуре.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия, реакционный раствор экстрагировали этилацетатом (30 мл) и дважды промывали водой. Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2), получая 280 мг трет-бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-формиата в виде бледно-желтого твердого вещества (выход: 48%). LC-MS: RT=2,25 мин, [М-55]+=457,27.
Стадия D. Синтез (R)-3-фтор-4-(((6-(3-(метоксиметил)пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила
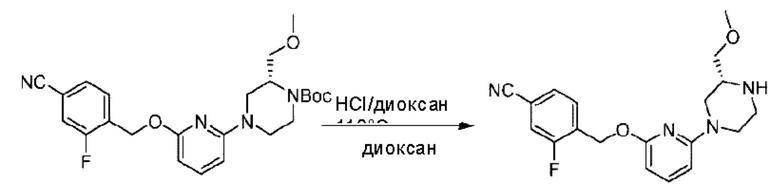
трет-Бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-формиат (50 мг; 0,11 ммоль) растворяли в соляной кислоте в 1,4-диоксане (5 мл) и перемешивали при комнатной температуре.
По завершении реакции раствор упаривали на роторном испарителе при пониженном давлении, получая 61 мг (R)-3-фтор-4-(((6-(3-(метоксиметил)пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила в виде неочищенного продукта. LC-MS: RT=1,70 мин, [М+Н]+=357,18.
Стадия Е Синтез метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
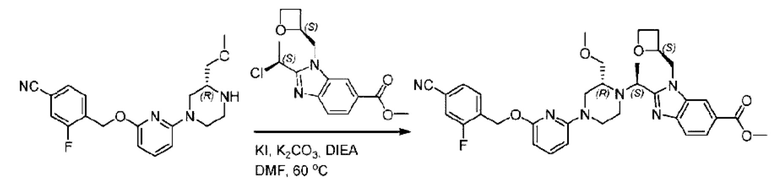
При комнатной температуре растворяли (R)-3-фтор-4-(((6-(3-(метоксиметил)пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрил (60 мг; 0,17 ммоль), DIEA (1 мл), иодид калия (56 мг; 0,34 ммоль) и карбонат калия (47 г; 0,34 ммоль) в N;N-диметилформамиде (5 мл), затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (52 мг; 0,17 ммоль) и взаимодействие проводили при повышенной до 60°С температуре.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (20 мл × 2 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 10/1), получая 20 мг метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества (выход: 19%). LC-MS: RT=1,97 мин, [М+Н]+=629,33.
Стадия F. Синтез 2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
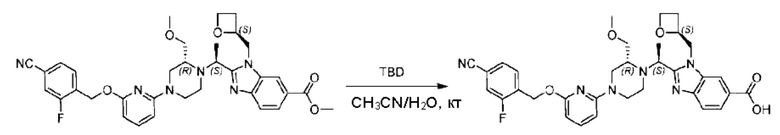
При комнатной температуре растворяли метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (20 мг; 0,03 ммоль) в смеси растворителей (3,5 мл; ацетонитрил/вода = 3/0,5), затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (9 мг; 0,06 ммоль) и взаимодействие проводили при комнатной температуре.
По завершении реакции реакционную смесь гасили лимонной кислотой и реакционный раствор экстрагировали этилацетатом (10 мл × 2 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 10/1), получая 12 мг 2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(метоксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества, (выход: 61%).
LC-MS: RT=1,86 мин, [М+Н]+=61 5,31. 1 Н ЯМР (400 МГц, DMSO-d6) δ 12.71 (s, 1 Н), 8.27 (s, 1 Н), 7.86 (dd, J=10,0; 1,3 Гц, 1 Н), 7.79 (dd, J=8,4; 1,5 Гц, 1 Н), 7.69-7.64 (m, 2Н), 7.61 (t, J=7,6 Гц, 1 Н), 7.44 (t, J=8,0 Гц, 1 Н), 6.26 (d, J=8,2 Гц, 1 Н), 6.10 (d, J=7,8 Гц, 1 Н), 5.37 (s, 2Н), 5.13 (d, J=13,0 Гц, 2Н), 4.87 (d, J=6,6 Гц, 1 Н), 4.61 (dd, J=15,8; 6,5 Гц, 1 Н), 4.44 (dd, J=13,4; 8,0 Гц, 1 Н), 4.20 (dt, J=9,0; 5,9 Гц, 1 Н), 4.08-3.96 (m, 1 Н), 3.82 (d, J=11,8 Гц, 1 Н), 3.65 (dd, J=10,5; 3,4 Гц, 1 Н), 3.53-3.41 (m, 1 Н), 3.33 (s, 3Н), 2.81 (t, J=11,3 Гц, 1 Н), 2.71-2.60 (m, 2Н), 2.35 (d, J=9,1 Гц, 2Н), 2.27-2.13 (m, 2Н), 1.39 (d, J=6,6 Гц, 3Н).
Пример 30. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
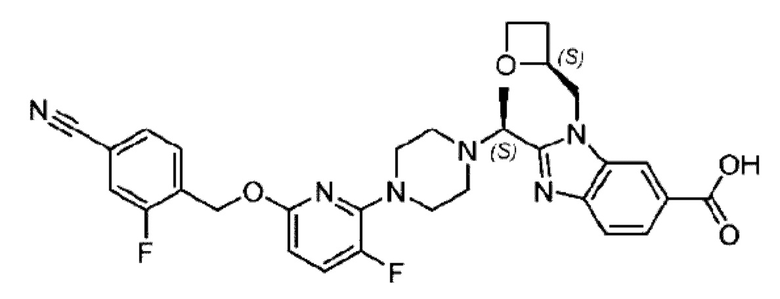
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-бром-3-фторпиридин-2-ил)пиперазин-1-формиата
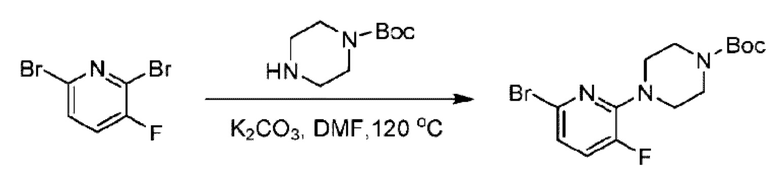
2,6-Дибром-3-фторпиридин (1,0 г; 3,93 ммоль), карбонат калия (1,08 г; 7,86 ммоль) и трет-бутилпиперазин-1-карбоксилат (731 мг; 3,93 ммоль) растворяли в 10 мл безводного N;N-диметилформамида и взаимодействие проводили при 120°С в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (50 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 1,18 г трет-бутил-4-(6-бром-3-фторпиридин-2-ил)пиперазин-1-формиата в виде белого твердого вещества. LC-MS: RT=1,98 мин, [М+Н]+=360,10.
Стадия В. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-формиата
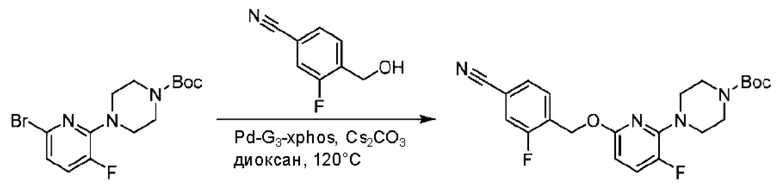
трет-Бутил-4-(6-бром-3-фторпиридин-2-ил)пиперазин-1-формиат (400 мг; 1,11 ммоль) и 3-фтор-4-(гидроксиметил)бензонитрил (168 мг; 1,11 ммоль) растворяли в 5 мл безводного 1,4-диоксана, затем добавляли карбонат цезия (723 мг; 2,22 ммоль) и метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий (94 мг; 0,111 ммоль) и систему три раза продували азотом. Взаимодействие проводили при 120°С в течение 5 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (50 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 367 мг трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-формиата в виде бледно-желтого твердого вещества. LC-MS: RT=1,95 мин, [М-Н]~=431,22.
Стадия С. Синтез 3-фтор-4-(((5-фтор-6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила
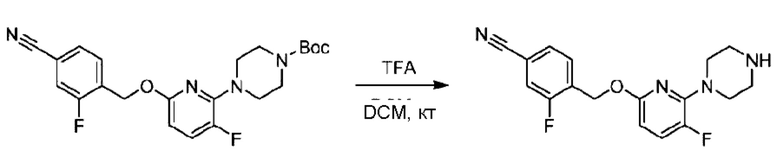
трет-Бутил-4-(6-((4-циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-формиат (367 мг; 0,85 ммоль) растворяли в 4 мл безводного дихлорметана, затем добавляли 2 мл трифторуксусной кислоты и взаимодействие проводили при комнатной температуре в течение 40 мин. По завершении реакции реакционный раствор выливали в 60 мл раствора бикарбоната натрия и экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, получая 260 мг 3-фтор-4-(((5-фтор-6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила. LC-MS: RT=1,81 мин, [М+Н]+=331,27.
Стадия D. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
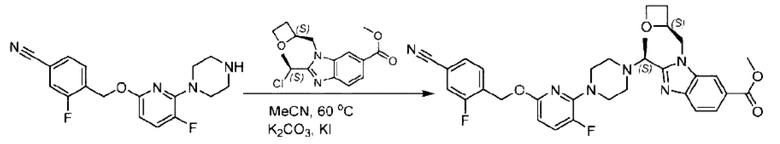
3-Фтор-4-(((5-фтор-6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрил (260 мг; 0,785 ммоль), карбонат калия (216 мг; 1,57 ммоль) и иодид калия (195 мг; 1,17 ммоль) растворяли в 5 мл безводного ацетонитрила, затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (241 мг; 0,785 ммоль) и взаимодействие проводили при 60°С в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (40 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 310 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества. LC-MS: RT=1,91 мин, [М+Н]+=603,27.
Стадия Е. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
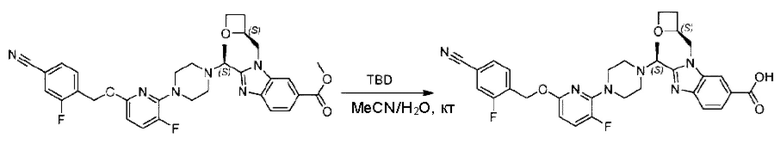
Метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (100 мг; 0,166 ммоль) растворяли в 4 мл ацетонитрила, затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (48,4 мг; 0,348 ммоль) и 1 мл воды и взаимодействие проводили при комнатной температуре в течение 7 часов. По завершении реакции реакционный раствор нейтрализовали, используя 20 мл насыщенного раствора хлорида аммония, и экстрагировали дихлорметаном (30 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 38 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3-фторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества.
LC-MS: RT=1,79 мин, [М+Н]+=589,27. 1Н ЯМР (400 МГц, DMSO) δ 12.78 (s, 1 Н), 8.29 (d, J=1,0 Гц, 1 Н), 7.88 (dd, J=10,0; 1,2 Гц, 1 Н), 7.81 (dd, J=8,4; 1,5 Гц, 1 Н), 7.72-7.64 (m, 3Н), 7.49-7.44 (m, 1 Н), 6.26 (dd, J=8,4; 1,6 Гц, 1 Н), 5.37 (s, 2Н), 5.19-5.11 (m, 1 Н), 4.83-4.78 (m, 1 Н), 4.69 (dd, J=15,6; 5,6 Гц, 1 Н), 4.49-4.43 (m, 2Н), 4.21-4.16 (m, 1 Н), 2.66-2.62 (m, 2Н), 2.60-2.55 (m, 6Н), 2.37-2.30 (m, 2Н), 1.44 (d, J=6,7 Гц, 3Н).
Пример 31. 2-((1S)-1-((3,4-транс)-4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
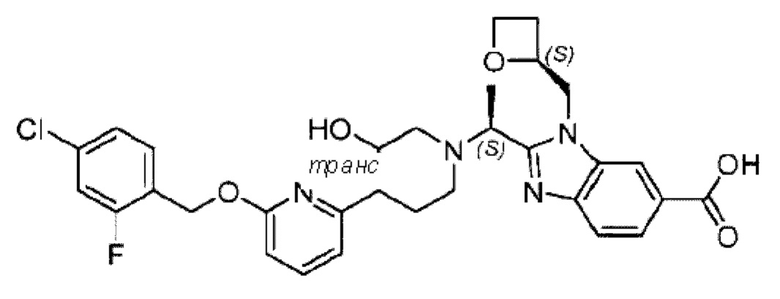
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез (3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-3-ола
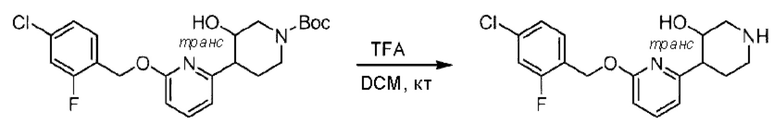
трет-Бутил-(3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-формиат (350 мг; 0,802 ммоль) растворяли в 5 мл безводного дихлорметана, затем добавляли 2 мл трифторуксусной кислоты и взаимодействие проводили при комнатной температуре в течение 40 мин. По завершении реакции реакционный раствор выливали в 60 мл раствора бикарбоната натрия и реакционный раствор экстрагировали этилацетатом (50 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, получая 270 мг (3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-3-ола. LC-MS: RT=1,81 мин, [М+Н]+=337,17.
Стадия В. Синтез метил-2-((1S)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
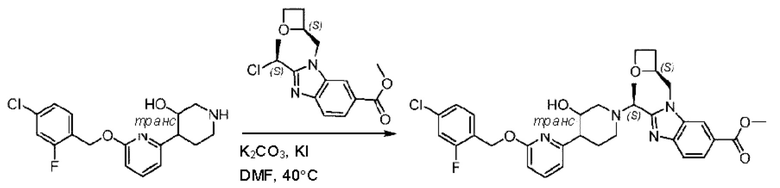
(3,4-транс)-4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-3-ол (270 мг; 0,801 ммоль), карбонат калия (222 мг; 1,61 ммоль) и иодид калия (199 мг; 1,21 ммоль) растворяли в 5 мл безводного ацетонитрила, затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (246 мг; 0,801 ммоль) и взаимодействие проводили при 60°С в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (40 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 320 мг метил-2-((13)-1-((3,4-транс)-4-(6-((4-хлор-2-фгорбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества. LC-MS: RT=1,86 мин, [М+Н]+=609,27.
Стадия С. Синтез 2-((1 S)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
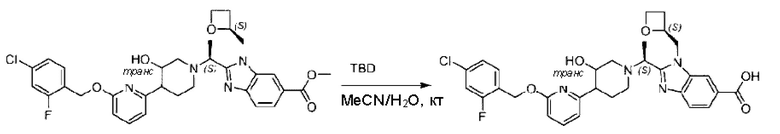
Метил-2-((13)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (150 мг; 0,246 ммоль) растворяли в 4 мл ацетонитрила, затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (71,8 мг; 0,517 ммоль) и 1 мл воды и взаимодействие проводили при комнатной температуре в течение 7 часов. По завершении реакции реакционный раствор нейтрализовали, используя 20 мл насыщенного раствора хлорида аммония, и экстрагировали дихлорметаном (30 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 41 мг 2-((13)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 8,6% затри стадии).
LC-MS: RT=1,78 мин, [М+Н]+=595,25. 1Н ЯМР (400 МГц, DMSO) δ 12.78 (s, 1 Н), 8.30 (dd, J=5,1; 1,0 Гц, 1 Н), 7.83-7.80 (m, 1 Н), 7.74-7.71 (m, 1 Н), 7.70-7.68 (m, 1 Н), 7.61-7.54 (m, 2Н), 7.48-7.42 (m, 1 Н), 7.28-7.24 (m, 1 Н), 6.84-6.81 (m, 1 Н), 6.55 (d, J=8,2 Гц, 1 Н), 5.42-5.32 (m, 2Н), 5.24-5.14 (m, 1 Н), 4.85-4.80 (m, 1 Н), 4.72-4.66 (m, 1Н), 4.55-4.41 (m, 3Н), 4.22-4.16 (m, 1 Н), 3.20-3.17 (m, 2Н), 2.70-2.60 (m, 2Н), 2.40-2.32 (m, 3Н), 1.88(t, J=5,9 Гц, 2H), 1.48 (dd, J=6,7; 2,7 Гц, 3Н).
Пример 32. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
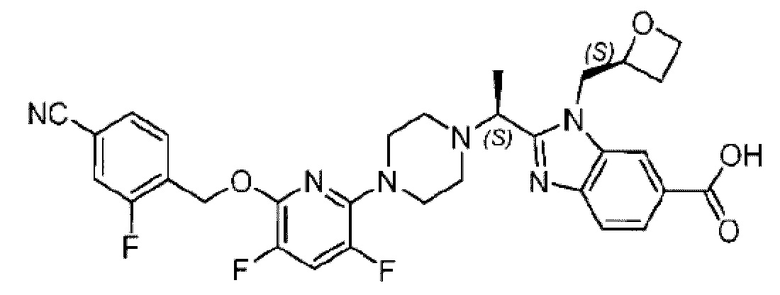
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 3-фтор-4-(((3,5,6-трифторпиридин-2-ил)окси)метил)бензонитрила
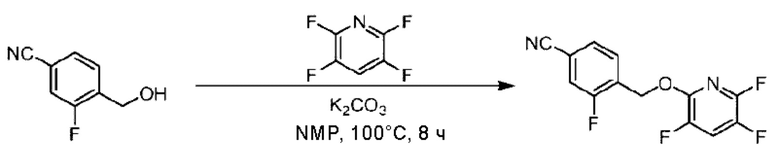
3-Фтор-4-(гидроксиметил)бензонитрил (151 мг; 1 ммоль) и 2,3,5,6-тетрафторпиридин (151 мг; 1 ммоль) растворяли в 2 мл N-метилпирролидона, затем добавляли карбонат калия (414 мг; 3 ммоль). Систему продували 3 раза азотом и взаимодействие проводили при 100°С в течение 8 часов.
По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (50 мл × 3 раза), органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией (н-гексан/этилацетат = 8/1), получая 212 мг 3-фтор-4-(((3,5,6-трифторпиридин-2-ил)окси)метил)бензонитрила (выход: 76,3%) в виде бледно-желтого твердого вещества. LC-MS: RT=2,09 мин, [М-Н]+=279,13.
Стадия В. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-формиата
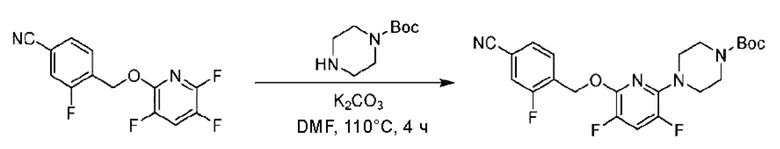
3-Фтор-4-(((3,5,6-трифторпиридин-2-ил)окси)метил)бензонитрил (212 мг; 0,76 ммоль) и трет-бутилпиперазин-1-карбоксилат (157 мг; 0,84 ммоль) растворяли в 2 мл N;N-диметилформамида, затем добавляли карбонат калия (210 мг; 1,52 ммоль). Систему 3 раза продували азотом и взаимодействие проводили при 110°С в течение 8 часов.
По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (50 мл × 3 раза), затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией (н-гексан/этилацетат = 8/1), получая 198 мг трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-формиата в виде бледно-желтого твердого вещества (выход: 58,0%).
Стадия С. Синтез 4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазина
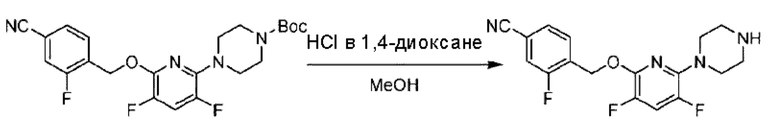
трет-Бутил-4-(6-((4-циано-2-фторбензи л)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-формиат (198 мг; 0,44 ммоль) растворяли в 3 мл метанола и при 0°С добавляли 4 М соляную кислоту в 1,4-диоксане (3 мл), реакционную смесь нагревали до комнатной температуры и взаимодействие осуществляли в течение 40 мин.
По завершении реакции реакционный раствор непосредственно упаривали на роторном испарителе, получая 215 мг 4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазина в виде белого твердого вещества, которое использовали непосредственно на следующей стадии реакции. LC-MS: RT=1,68 мин, [М+Н]+=349,16.
Стадия D. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
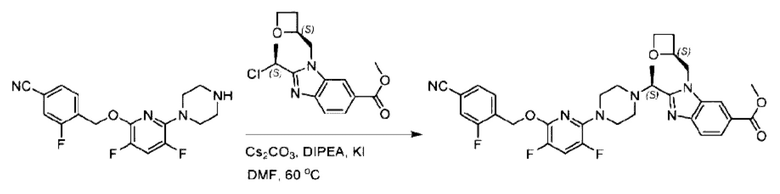
При комнатной температуре добавляли 4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин (215 мг; 0,44 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (135 мг; 0,44 ммоль), иодидкалия (73 мг; 0,44 ммоль) и карбонат цезия (287 мг; 0,88 ммоль) к N;N-диметилформамиду (10,0 мл), затем добавляли N;N-диизопропилэтиламин (1,0 мл). Взаимодействие проводили при 60°С в течение 3,0 часов в атмосфере азота.
По завершении реакции реакционную смесь гасили водой и раствор экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученную смесь очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20/1), получая 205 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества (выход: 75,1%). LC-MS: RT=1,89 мин, [М+Н]+=621,34.
Стадия Е. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
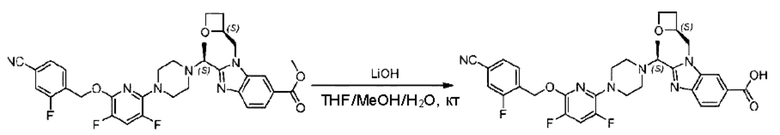
При комнатной температуре к тетрагидрофурану (3,0 мл), содержащему метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (205 мг; 0,33 ммоль), по каплям добавляли раствор гидроксида лития (16 мг; 0,66 ммоль) в воде (1,0 мл), затем добавляли 1,0 мл метанола. Взаимодействие проводили при 25°С в течение 3,0 часов.
По завершении реакции реакционную смесь гасили водой, значение рН подводили до 6 водным раствором хлорида аммония (0,5 моль/л) и полученную смесь экстрагировали дихлорметаном (30 мл × 3 раза). Затем органические фазы объединяли, промывали водным раствором хлорида аммония (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли и очищали колоночной хроматографией (элюент: дихлорметан/метанол 15/1), получая 132 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-3,5-дифторпиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде бледно-желтого твердого вещества (выход: 51,1%).
LC-MS: RT=1,79 мин, [М+Н]+=607,29. 1Н ЯМР (400 МГц, DMSO-d6) δ 12.74 (s, 1 Н), 8.27 (s, 1 Н), 7.92-7.86 (m, 1 Н), 7.83-7.75 (m, 2Н), 7.72-7.62 (m, 3Н), 5.45 (d, J=13,7 Гц, 2Н), 5.13 (d, J=2,9 Гц, 1 Н), 4.83-4.74 (m, 1 Н), 4.67 (dd, J=15,4; 5,5 Гц, 1 Н), 4.49-4.40 (m, 2Н), 4.17 (dt, J=9,1; 5,8 Гц, 1 Н), 3.29-3.18 (m, 3Н), 2.67-2.52 (m, 5H),2.29(dd, J=1 8,2; 7,3 Гц, 2Н), 1.42 (t, J=8,0 Гц, 3Н).
Пример 33. 2-((13)-1-((3,4-цис)-4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
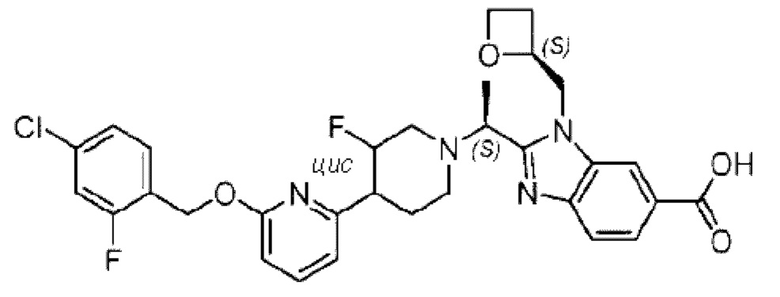
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-(3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-формиата
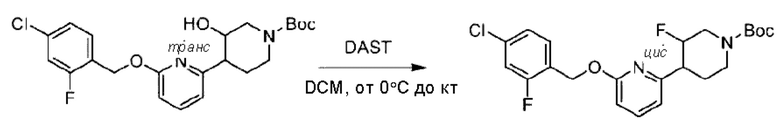
трет-Бутил-(3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-формиат (500 мг; 1,14 ммоль) растворяли в 5 мл безводного дихлорметана, при 0°С добавляли трифторид диэтиламиносеры (220 мг; 1,37 ммоль) и взаимодействие проводили при 0°С в течение 30 мин. По завершении реакции реакционный раствор выливали в 60 мл ледяной воды и экстрагировали этилацетатом (40 мл × 2 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении, получая 485 мг трет-бутил-(3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-формиата. LC-MS: RT=1,97 мин, [М+Н]+=439,22.
Стадия В. Синтез 2-((4-хлор-2-фторбензил)окси)-6-((3,4-цис)-3-фторпиперидин-4-ил)пиридина
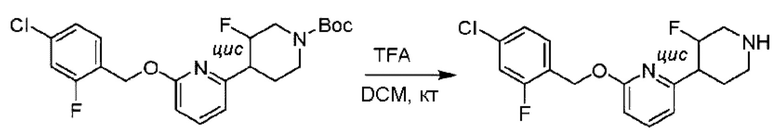
трет-Бутил-(3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-формиат (485 мг; 1,10 ммоль) растворяли в 5 мл безводного дихлорметана, затем добавляли 2 мл трифторуксусной кислоты и взаимодействие проводили при комнатной температуре в течение 40 мин. По завершении реакции реакционный раствор выливали в 60 мл раствора бикарбоната натрия и экстрагировали этилацетатом (50 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении, получая 330 мг 2-((4-хлор-2-фторбензил)окси)-6-((3,4-цис)-3-фторпиперидин-4-ил)пиридина. LC-MS: RT=1,92 мин, [М+Н]+=339,1 7.
Стадия С. Синтез метил-2-((13)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
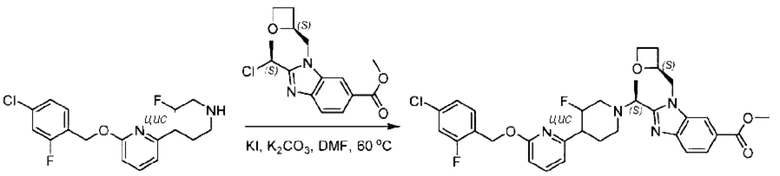
2-((4-Хлор-2-фторбензил)окси)-6-((3,4-цис)-3-фторпиперидин-4-ил)пиридин (330 мг; 0,973 ммоль), карбонат калия (268 мг; 1,94 ммоль) и иодид калия (242 мг; 1,46 ммоль) растворяли в 5 мл безводного ацетонитрила, затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (299 мг; 0,973 ммоль) и взаимодействие проводили при 60°С в течение 2 часов. По завершении реакции реакционный раствор разбавляли водой и экстрагировали этилацетатом (40 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия и окончательно концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 510 мг метил-2-((1S)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества. LC-MS: RT=2,08 мин, [М+Н]+=611,23.
Стадия D. Синтез 2-((1S)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
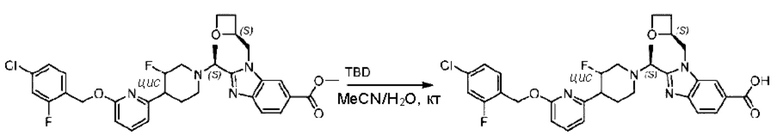
Метил-2-((1 3)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (200 мг; 0,327 ммоль) растворяли в 4 мл ацетонитрила, затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (95,5 мг; 0,687 ммоль) и 1 мл воды и взаимодействие проводили при комнатной температуре в течение 7 часов. По завершении реакции реакционный раствор нейтрализовали, используя 20 мл насыщенного раствора хлорида аммония, и экстрагировали дихлорметаном (40 мл × 2 раза). Затем органические фазы объединяли, промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 47 мг 2-((1S)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 6,9% за четыре стадии).
LC-MS: RT=1,96 мин, [М+Н]+=597,23. 1Н ЯМР (400 МГц, DMSO) δ 12.77 (s, 1 Н), 8.31 (s, 1 Н), 7.84-7.80 (m, 1 Н), 7.73-7.69 (m, 1 Н), 7.66-7.60 (m, 1 Н), 7.57-7.53 (m, 1 Н), 7.42 (ddd, J=10,0; 4,8; 2,0 Гц, 1 Н), 7.28-7.23 (m, 1 Н), 6.90 (d, J=7,4 Гц, 1 Н), 6.72 (d, J=8,2 Гц, 1 Н), 5.43-5.34 (m, 2Н), 5.20-5.14 (m, 1 Н), 5.02-4.82 (m, 1 Н), 4.78-4.68 (m, 2H), 4.62-4.53 (m, 2H), 4.43 (dd, J=13,5; 7,8 Гц, 1 H), 4.21-4.16 (m, 1 H), 3.03-2.91 (m, 1H), 2.77-2.57 (m, 3Н), 2.34-2.18 (m, 2H), 1.85-1.61 (m, 2H), 1.50 (dd, J=6,7; 2,8 Гц, 3Н).
Пример 34. 2-((S)-1-((R)-4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)-2-(фторметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
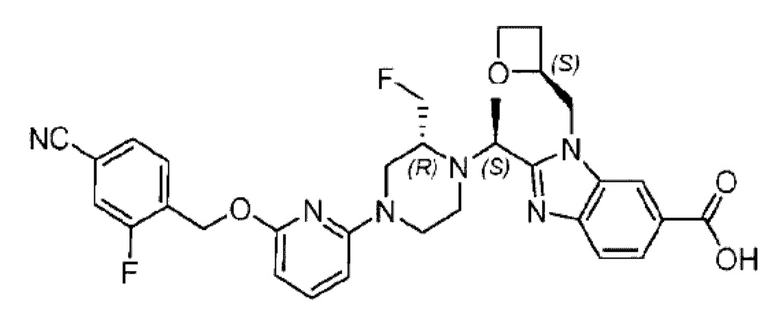
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(гидроксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
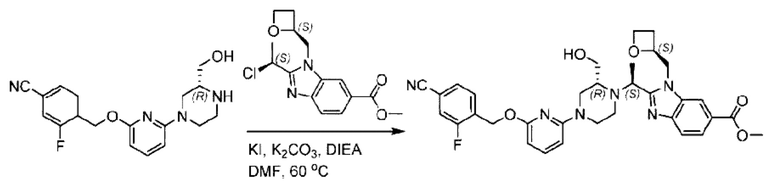
При комнатной температуре растворяли (R)-3-фтор-4-(((6-(3-(гидроксиметил)пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрил (160 мг; 0,47 ммоль), DI ЕА(1 мл), иодид калия (155 мг; 0,93 ммоль) и карбонат калия (129 г; 0,93 ммоль) в N;N-диметилформамиде (5 мл), затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (144 мг; 0,47 ммоль), реакционный раствор нагревали до 50°С и осуществляли взаимодействие.
По завершении реакции реакционный раствор гасили насыщенным раствором хлорида натрия и экстрагировали этилацетатом (20 мл × 2 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 10/1), получая 94 мг метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(гидроксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества (выход: 33%). LC-MS: RT=1,81 мин, [М+Н]+=615,30.
Стадия В. Синтез метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(фторметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
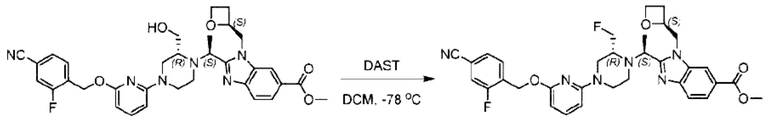
Метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(гидроксиметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (90 мг; 0,15 ммоль) растворяли в дихлорметане (2 мл). Затем раствор охлаждали до -78°С, после чего добавляли DAST (63 мг; 0,29 ммоль) и перемешивали при низкой температуре.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (10 мл × 2 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 10/1) и получали 80 мг метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(фторметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества (выход: 33%). LOMS: RT=2,09 мин, [М+Н]+=617,27.
Стадия С. Синтез 2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(фторметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
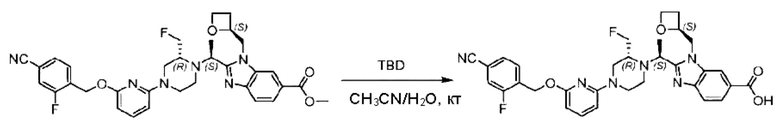
Метил-2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(фторметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (80 мг; 0,13 ммоль) растворяли в смеси растворителей (6 мл; ацетонитрил/вода = 5/1), затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (36 мг; 0,26 ммоль) и взаимодействие проводили при комнатной температуре.
По завершении реакции реакционную смесь гасили лимонной кислотой и реакционный раствор экстрагировали этилацетатом (10 мл × 2 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали и неочищенный продукт, полученный из концентрата, подвергали высокоэффективной жидкостной хроматографии, получая 9 мг 2-((S)-1-((R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-2-(фторметил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 12%). LC-MS: RT=1,97 мин, [М+Н]+=603,31.
Пример 35. 2-((1S)-1-((3,4-цис)-4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
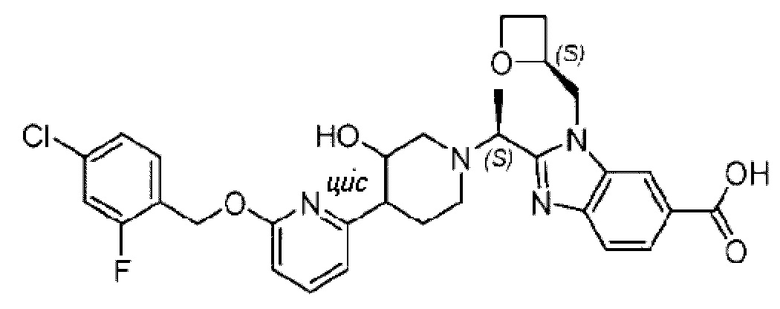
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-6-((4-хлор-2-фторбензил)окси)-3',6'-дигидро-[2,4'-дипиридил]-1'(2' Н)-карбоксилата
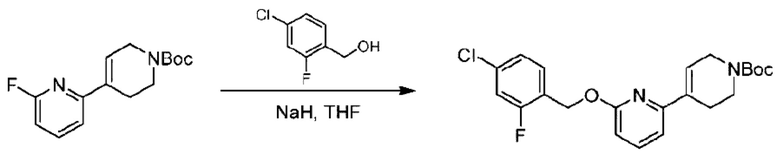
(4-Хлор-2-фторфенил)метанол (17 г; 106,5 ммоль) растворяли в тетрагидрофуране (300 мл), затем охлаждали до 0°С, после чего добавляли гидрид натрия (5,6 г; 142 ммоль). Смесь перемешивали при комнатной температуре в течение 0,5 часа, затем добавляли трет-бутил-6-фтор-3',6'-дигидро-[2,4'-дипиридил]-1'(2'Н)-карбоксилат (20 г; 71 ммоль) и перемешивали при комнатной температуре в течение ночи.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (30 мл), дважды промывали водой, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/10), получая 10 г трет-бутил-6-((4-хлор-2-фторбензил)окси)-3',6'-дигидро-[2,4'-дипиридил]-1'(2'Н)-карбоксилата в виде белого твердого вещества (выход: 34%). LC-MS: RT=2,15 мин, [М+Н]+=419,23.
Стадия В. Синтез трет-бутил-(3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-формиата
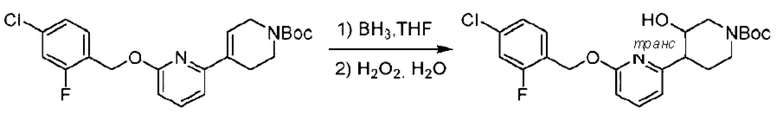
трет-Бутил-6-((4-хлор-2-фторбензил)окси)-3',6'-дигидро-[2,4'-дипиридин]-1'(2'Н)-карбоксилат (10 г; 24 ммоль) растворяли в тетрагидрофуране (150 мл) и раствор охлаждали до 0°С, затем медленно по каплям добавляли боргидрид в тетрагидрофуране (1 М раствор; 26 мл; 26 ммоль). Смесь перемешивали при комнатной температуре в течение 0,5 часа, затем добавляли гидроксид натрия (1 М раствор; 2,4 г) и перекись водорода (30%-ную, 10 мл) и перемешивали при комнатной температуре в течение ночи.
По завершении реакции реакционную смесь гасили насыщенным раствором бисульфита натрия и насыщенным раствором бикарбоната натрия и реакционный раствор экстрагировали этилацетатом (30 мл × 3 раза). Затем органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали и полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/5), получая 9 г трет-бутил-(3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2- ил)-3-гидроксипиперидин-1-формиата в виде белого твердого вещества (выход: 90%). LC-MS: RT=1,90 мин, [М+Н]+=437,37.
Стадия С. Синтез трет-бутил-(3,4-цис)-4-(6-(((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-((4-нитробензоил)окси)пиперидин-1-формиата
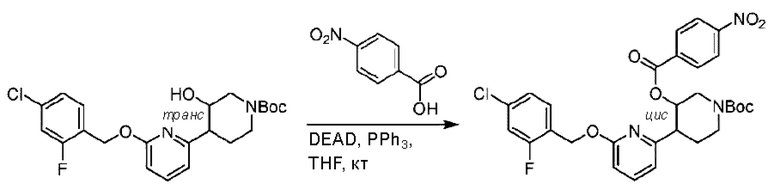
Трифенилфосфин растворяли в тетрагидрофуране и добавляли диэтилазодикарбоксилат (800 мг; 4,6 ммоль) вледяной бане. Смесь перемешивали в течение 0,5 часа, после чего добавляли трет-бутил-(3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-формиат (1 г; 2,3 ммоль) и перемешивали в течение 15 мин. Затем добавляли л-нитробензойную кислоту (768 мг; 4,6 ммоль) и смесь перемешивали при комнатной температуре в течение 16 часов.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и экстрагировали этилацетатом (100 мл × 3 раза). Затем органические фазы объединяли, дважды промывали водой, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/10), получая 800 мг трет-бутил-(3,4-цис)-4-(6-(((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-((4-нитробензоил)окси)пиперидин-1-формиата в виде белого твердого вещества (выход: 45%). LC-MS: RT=2,13 мин, [М+Н]+ = 586,19.
Стадия D. Синтез трет-бутил-(3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-формиата
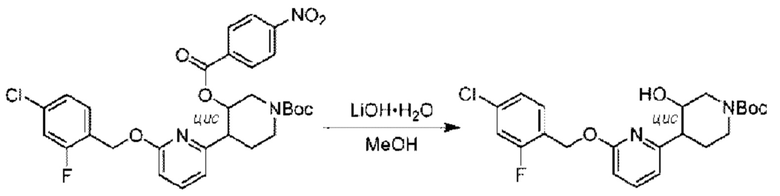
трет-Бутил-(3,4-цис)-4-(6-(((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-((4-нитробензоил)окси)пиперидин-1-формиат (500 мг; 0,85 ммоль) растворяли в метаноле (10 мл) и воде (2 мл), затем добавляли гидроксида лития моногидрат (107 мг; 2,55 ммоль) и взаимодействие проводили при комнатной температуре в течение 1 часа.
По завершении реакции реакционный раствор гасили, разбавляли водой и экстрагировали этилацетатом (20 мл × 3 раза). Затем органические фазы объединяли, дважды промывали насыщенным раствором бикарбоната натрия, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали, получая 400 мг трет-бутил-(3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-формиата в виде желтого твердого вещества (выход: 98%). LC-MS: RT=1,90 мин, [М+Н]+ = 437,25.
Стадия Е. Синтез (3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-3-ола
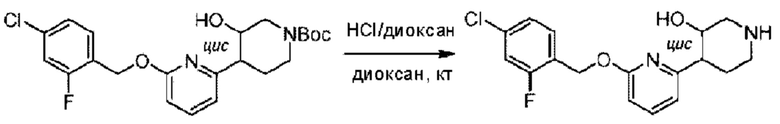
При комнатной температуре растворяли трет-бутил-(3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-формиат (150 мг; 0,34 ммоль) в 1,4-диоксане (2 мл) и добавляли соляную кислоту в 1,4-диоксане (2 мл) в бане с ледяной водой. Смесь перемешивали при комнатной температуре в течение 0,5 часа.
По завершении реакции после концентрирования получали 120 мг (3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-3-ола в виде белого твердого вещества. LC-MS: RT=1,65 мин, [М+Н]+ = 337,25.
Стадия F. Синтез метил-2-((1S)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
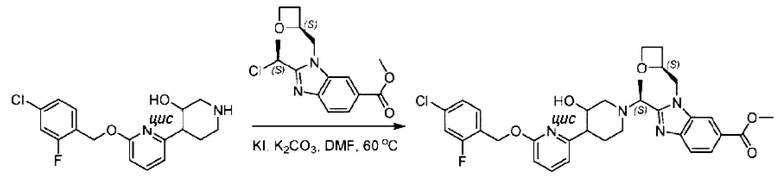
При комнатной температуре растворяли (3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-3-ол (120 мг; 0,34 ммоль) в N,N-диметилформамиде (2 мл), после этого добавляли метил-2-((S)-(1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (157 мг; 0,51 ммоль), иодид калия (113 мг; 0,68 ммоль) и карбонат калия (88 мг; 0,68 ммоль) и затем смесь перемешивали при 60°С в течение 12 часов.
По завершении реакции реакционную смесь гасили насыщенным раствором хлорида натрия и реакционный раствор экстрагировали этилацетатом (10 мл × 2 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали и полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/1), получая 60 мг метил-2-((1S)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бесцветной маслянистой жидкости (выход: 42%). LC-MS: RT=1,97 мин, [М+Н]+ = 609,35.
Стадия G. Синтез 2-((1S)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[сПимидазол-6-карбоновой кислоты
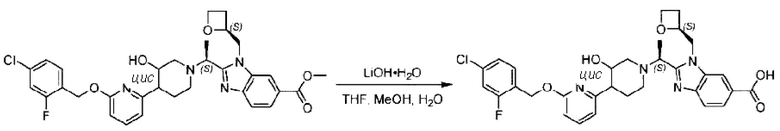
Метил-2-((1S)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (60 мг; 0,1 ммоль) растворяли в смеси растворителей, состоящей из тетрагидрофурана (3 мл), метанола (1 мл) и воды (1 мл), затем добавляли гидроксида лития моногидрат (12 мг; 0,3 ммоль) при комнатной температуре и реакцию проводили в течение 4 часов.
По завершении реакции реакционный раствор концентрировали и полученный неочищенный продукт очищали высокоэффективной жидкостной хроматографией, получая 18,22 мг 2-((1S)-1-((3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 30%). LC-MS: RT=1,81 мин, [М+Н]+ = 595,27.
Пример 36. 2-((1S)-1-((3,4-транс)-4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
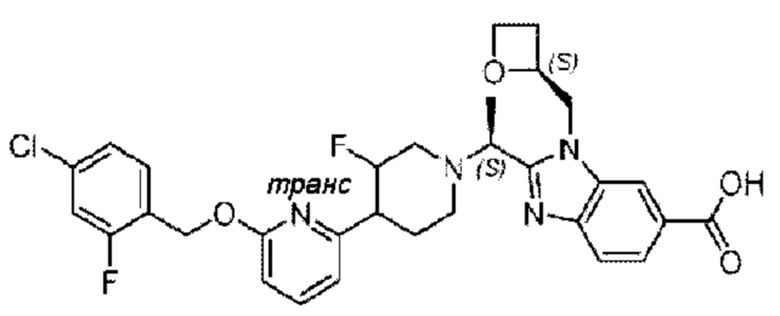
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-(3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-формиата
трет-Бутил-(3,4-цис)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-гидроксипиперидин-1-формиат (200 мг; 0,45 ммоль) растворяли в тетрагидрофуране, добавляли трифторид диэтиламиносеры (147 мг; 0,9 ммоль) в ледяной бане и раствор этой смеси перемешивали в течение 0,5 ч.
По завершении реакции реакционный раствор гасили путем добавления насыщенного раствора хлорида натрия, экстрагировали этилацетатом (50 мл × 3 раза), дважды промывали водой, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали. Затем неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/10), получая 150 мг трет-бутил-(3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-формиата в виде желтого твердого вещества (выход: 76%). LC-MS: RT=2,03 мин, [М+Н]+ = 439,1 9.
Стадия В. Синтез 2-((4-хлор-2-фторбензил)окси)-6-((3,4-транс)-3-фторпиперидин-4-ил)-пиридина
трет-Бутил-(3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-формиат (150 мг; 0,34 ммоль) растворяли в 1,4-диоксане (2 мл) при комнатной температуре и добавляли соляную кислоту в 1,4-диоксане (2 мл) в ледяной бане. Раствор этой смеси перемешивали при комнатной температуре в течение 0,5 ч.
По завершении реакции реакционный раствор концентрировали, получая 120 мг 2-((4-хлор-2-фторбензил)окси)-6-((3,4-транс)-3-фторпиперидин-4-ил)-пиридина в виде белого твердого вещества. LC-MS: RT=1,68 мин, [М+Н]+ = 339,29.
Стадия С. Синтез метил-2-((1S)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
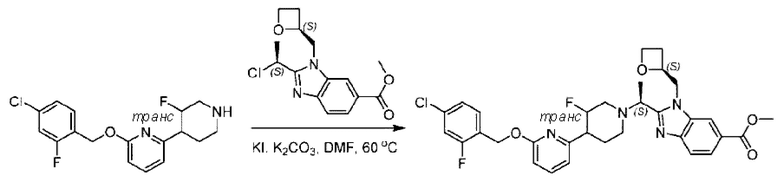
2-((4-Хлор-2-фторбензил)окси)-6-((3,4-транс)-3-фторпиперидин-4-ил)-пиридин (120 мг; 0,34 ммоль) растворяли в N,N-диметилформамиде (2 мл) при комнатной температуре, добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (157 мг; 0,51 ммоль), иодид калия (113 мг; 0,68 ммоль) и карбонат калия (88 мг; 0,68 ммоль) и раствор этой смеси перемешивали при 60°С в течение 12 ч.
По завершении реакции реакционный раствор гасили путем добавления насыщенного раствора хлорида натрия и экстрагировали этилацетатом (10 мл × 2 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали и полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/1), получая 60 мг метил-2-((1S)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бесцветной маслянистой жидкости (выход: 42%). LC-MS: RT=1,99 мин, [М+Н]+ = 611,38.
Стадия D. Синтез 2-((1S)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
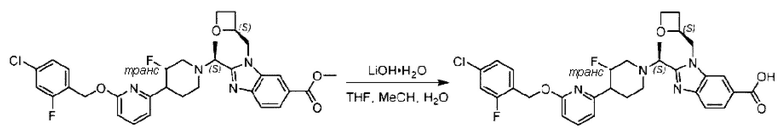
Метил-2-((1S)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (60 мг; 0,1 ммоль) растворяли в смеси растворителей тетрагидрофурана (3 мл), метанола (1 мл) и воды (1 мл), добавляли гидроксида лития моногидрат (12 мг; 0,3 ммоль) при комнатной температуре и реакцию проводили в течение 4 ч.
По завершении реакции реакционный раствор концентрировали и неочищенный продукт очищали высокоэффективной жидкостной хроматографией, получая 18,22 мг 2-((1S)-1-((3,4-транс)-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)-3-фторпиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 30%). LC-MS: RT=1,88 мин, [М+Н]+ = 597,23.
Пример 37. 2-((S)-1-(4-(6-((3-Фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
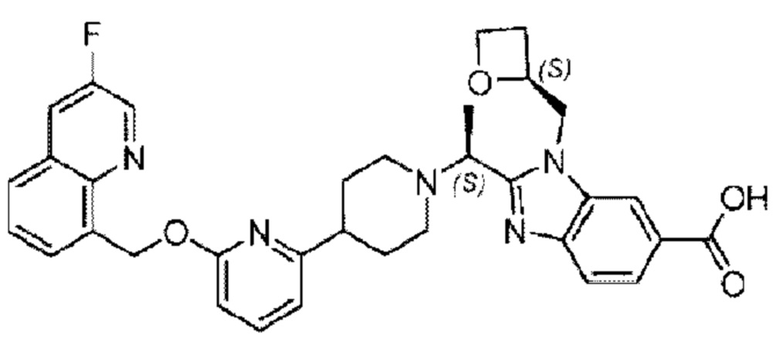
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1 Н-бензо[d]имидазол-6-карбоксилата
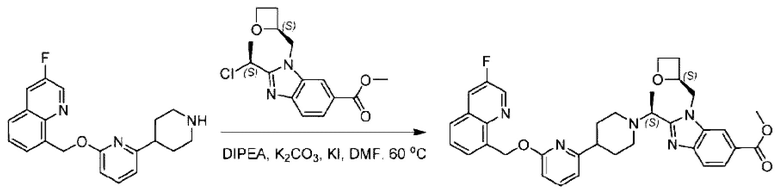
N,N-Диизопропилэтиламин (161 мкл; 0,975 ммоль) при комнатной температуре добавляли в раствор N,N-диметилметанамида (2,0 мл), содержащий 3-фтор-8-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)хинолин (132 мг; 0,325 ммоль), и растворяли при перемешивании. Последовательно добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (100 мг; 0,325 ммоль), карбонат калия (90 мг; 0,65 ммоль) и иодид калия (108 мг; 0,65 ммоль). После добавления раствор этой смеси нагревали до 60°С и перемешивали в течение 1 ч.
По завершении реакции реакционный раствор гасили путем добавления насыщенного раствора хлорида аммония и экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 6/94). Получали 111 мг метил-2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бесцветного маслянистого продукта (выход: 56,0%). LC-MS: RT=1,87 мин, [М+Н]+ = 610,33.
Стадия В. Синтез 2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[сПимидазол-6-карбоновой кислоты
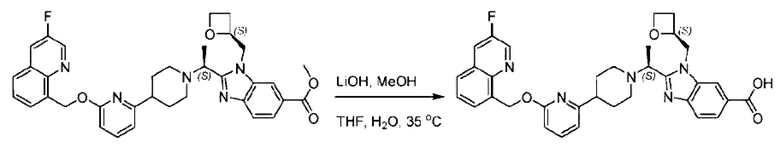
Гидроксида лития моногидрат (31 мг; 0,73 ммоль) добавляли при комнатной температуре в раствор смеси тетрагидрофуран/метанол/вода (2,5 мл/2,5 мл/1,0 мл), содержащий метил-2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (111 мг; 0,18 ммоль), смешанный раствор нагревали до 35°С и реакцию проводили в течение 1 часа. По завершении реакции реакционный раствор гасили путем добавления насыщенного раствора хлорида аммония и экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (30 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 8/92). Получали 27 мг 2-((S)-1-(4-(6-((3-фторхинолин-8-ил)метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 24,9%). LC-MS: RT=1,82 мин, [М+Н]+ = 596,32. 1Н ЯМР (400 МГц, DMSO) δ 12.75 (s, 1Н), 9.00 (d, J=2,9 Гц, 1Н), 8.30 (dd, J=9,6; 2,9 Гц, 1Н), 8.27 (d, J=0,8 Гц, 1Н), 7.95 (d, J=8,1 Гц, 1Н), 7.85-7.77 (m, 2Н), 7.70-7.58 (m, 3Н), 6.83 (d, J=7,3 Гц, 1Н), 6.71 (d, J=8,1 Гц, 1Н), 5.97 (s, 2Н), 5.18-5.10 (m, 1Н), 4.77 (dd, J=15,5; 2,7 Гц, 1Н), 4.62 (dd, J=15,5; 5,4 Гц, 1Н), 4.46-4.35 (m, 2Н), 4.14 (dt, J=9,1; 5,8 Гц, 1Н), 2.94 (d, J=10,6 Гц, 1H), 2.66-2.51 (m, 4Н), 2.34-2.17 (m, 2Н), 1.85-1.62 (m, 3Н), 1.55-1.47 (m, 1Н), 1.45 (d, J=6,7 Гц, 3Н).
Пример 38. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетанил-2-ил)метил)-1-Н-бензо[d]имидазол-6-карбоновая кислота
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-6-амино-3'6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилата
трет-Бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-3,6-дигидропиридин-1(2Н)-карбоксилат (500,4 мг; 1,6 ммоль), 2-амино-6-хлорпиридин (420,3 мг; 3,2 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (Pd(dppf)Cl2; 146,3 мг; 0,2 ммоль) и карбонат цезия (1,2 г; 3,5 ммоль) добавляли при комнатной температуре в смесь растворителей 1,4-диоксана (30 мл) и воды (6 мл). В атмосфере азота смешанный раствор нагревали до 90°С и кипятили с обратным холодильником в течение 24 ч.
По завершении реакции реакционный раствор фильтровали через диатомовую землю и фильтрат концентрировали при пониженном давлении. Концентрат разбавляли путем добавления воды (100 мл), экстрагировали этилацетатом (50 мл × 3 раза), затем промывали насыщенным солевым раствором (50 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 6/1), получая 310,7 мг трет-бутил-6-амино-3'6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилата в виде желтого маслянистого продукта (выход: 69,6%).
Стадия В. Синтез трет-бутил-4-(6-аминопиридин-2-ил)пиперидин-1-карбоксилата
трет-Бутил-6-амино-3'6'-дигидро-[2,4'-бипиридин]-1'(2'Н)-карбоксилат (310,2 мг; 1,1 ммоль) растворяли в метаноле (10 мл) при комнатной температуре и добавляли диоксид платины (12,5 мг; 55,0 ммоль). Три раза осуществляли заполнение водородом и реакцию проводили в течение 24 ч в атмосфере водорода. По завершении реакции реакционный раствор фильтровали через диатомовую землю и фильтрат концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 1/1), получая 103,5 мг трет-бутил-4-(6-аминопиридин-2-ил)пиперидин-1-карбоксилата в виде белого маслянистого продукта (выход: 33,0%). LC-MS: RT=3,25 мин, [М+Н]+ = 278,21.
Стадия С. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-карбоксилата
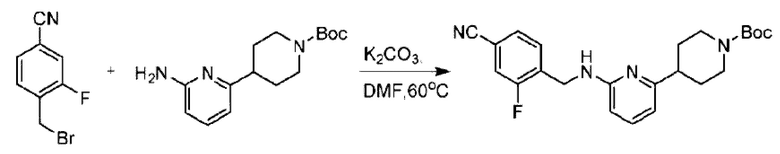
трет-Бутил-4-(6-хлорпиридин-2-ил)пиперидин-1-карбоксилат (103,2 мг; 371,4 ммоль), 4-(бромметил)-3-фторбензонитрил (159,02 мг; 742,8 мкмоль) и карбонат калия (151,8 мг; 1,1 ммоль) растворяли в N;N-диметилформамиде (3 мл) при комнатной температуре. Раствор этой смеси нагревали до 60°С и реакцию проводили в течение 6 ч.
По завершении реакции реакционный раствор разбавляли путем добавления воды (20 мл), экстрагировали этилацетатом (10 мл × 3 раза), затем промывали насыщенным солевым раствором (15 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = 5/1), получая 101,2 мг трет-бутил-4-(6-((4-циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-карбоксилата в виде желтого маслянистого продукта (выход: 66,4%). LC-MS: RT=4,07 мин, [М+Н]+ = 433,25.
Стадия D. Синтез 3-фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)амино)метил)бензонитрила
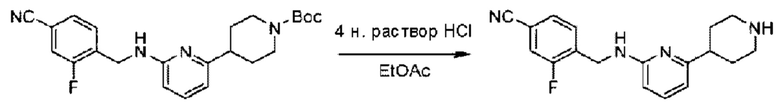
трет-Бутил-4-(6-((4-циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-карбоксилат (101,3 мг; 246,0 мкмоль) растворяли в растворе хлористого водорода в этилацетате (5 мл) с концентрацией 4 моль/л при комнатной температуре и раствор этой смеси перемешивали в течение 1 ч для постепенного осаждения белого твердого вещества. По завершении реакции реакционный раствор концентрировали при пониженном давлении, разбавляли путем добавления воды (20 мл), по каплям добавляли насыщенный водный раствор карбоната натрия, чтобы сделать раствор этой смеси щелочным. Водный слой экстрагировали этилацетатом (10 мл × 3 раза), затем промывали насыщенным солевым раствором (15 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Получали 76,4 мг 3-фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)амино)метил)бензонитрила в виде белого порошка, который использовали непосредственно в следующей реакции без очистки. LC-MS: RT=2,69 мин, [М+Н]+ = 311,04.
Стадия Е. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
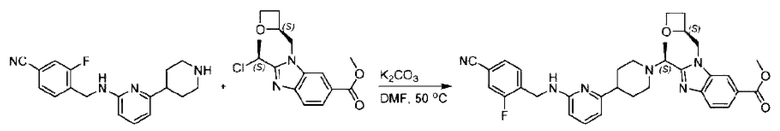
3-Фтор-4-(((6-(пиперидин-4-ил)пиридин-2-ил)амино)метил)бензонитрил (76,1 мг; 244,7 мкмоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-формиат (83,2 мг; 269,2 мкмоль) и карбонат калия (170,3 мг; 1,2 ммоль) растворяли в N;N-диметилформамиде (10 мл) при комнатной температуре и взаимодействие проводили при 50°С в течение 16 ч.
По завершении реакции реакционный раствор разбавляли путем добавления воды (20 мл), экстрагировали этилацетатом (50 мл × 3 раза), затем промывали насыщенным солевым раствором (20 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: петролейный эфир/этилацетат = от 1/1 до 0/1), получая 102,9 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата (выход: 72,1%). LC-MS: RT=1,97 мин, [М+Н]+ = 583,73.
Стадия F. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
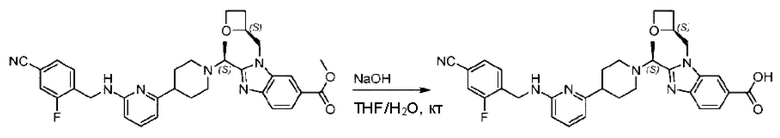
метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1-Н-бензо[d]имидазол-6-карбоксилат (102,9 мг; 171,8 мкмоль) растворяли в смеси тетрагидрофурана (10 мл), этанола (3 мл) и воды (3 мл) при комнатной температуре. Затем добавляли гидроксид натрия (76,0 мг; 1,9 ммоль) и взаимодействие проводили при комнатной температуре в течение 16 ч.
По завершении реакции по каплям добавляли водный раствор лимонной кислоты с концентрацией 1 моль/л до достижения значения рН, равного 5. Реакционный раствор разбавляли путем добавления воды (20 мл), экстрагировали этилацетатом (25 мл × 3 раза), пром ывали насыщенным солевым раствором (25 мл × 3 раза), сушили с использованием безводного сульфата натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали полупрепаративной HPLC (подвижная фаза: (вода, содержащая 0,1% аммиака)/ацетонитрил = от 90/10 до 40/60), получая 20,5 мг 2-((3)-1-(4-(6-((4-циано-2-фторбензил)амино)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1-Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 20,4%). LC-MS: RT=1,08 мин, [М+Н]+ = 569,60.
Пример 39. 2-((S)-1-(4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (39А) и 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (39В)
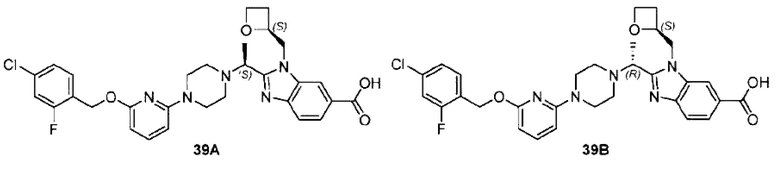
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 2-хлор-6-((4-хлор-2-фторбензил)окси)пиридина
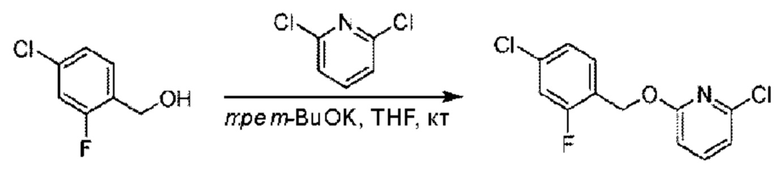
трет-Бутилат калия (4,88 г; 43,60 ммоль) добавляли в раствор тетрагидрофурана (50,0 мл), содержащий (4-хлор-2-фторфенил)метанол (5,0 г; 31,14 ммоль), при 0°С и раствор этой смеси перемешивали в течение 30 мин. Затем добавляли 2,6-дихлорпиридин (4,2 г; 28,38 ммоль) и реакцию проводили в течение ночи при 25°С.
По завершении реакции реакционный раствор гасили путем добавления воды (50 мл) и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл × 3 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 7,53 г 2-х лор-6-((4-хлор-2-фторбензил)окси)пиридина в виде желтого маслянистого продукта (выход: 88,9%). LC-MS: RT=2,27 мин, [М+Н]+ = 272,05.
Стадия В. Синтез трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1 -карбоксил ата
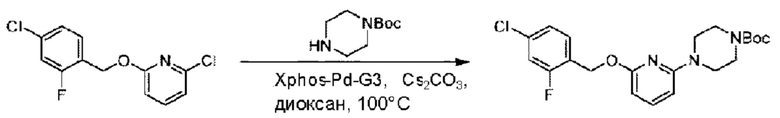
трет-Бутилпиперазин-1-карбоксилат (5,15 г; 27,68 ммоль), карбонат цезия (18,05 г; 55,36 ммоль) и метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (702,5 мг; 0,83 ммоль) добавляли в раствор 1,4-диоксана, содержащего 2-хлор-6-((4-хлор-2-фторбензил)окси)пиридин (7,53 г; 27,68 ммоль), при 25°С и взаимодействие проводили при 100°С в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 4,7 г трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата в виде белого твердого вещества (выход: 40,2%). LC-MS: RT=2,39 мин, [М+Н]+ = 422,22.
Стадия С. Синтез 4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазина
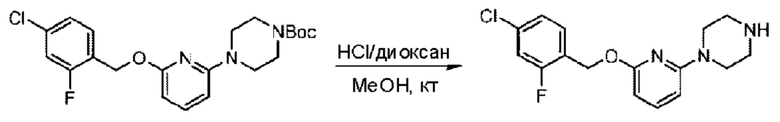
Соляную кислоту в 1,4-диоксане (20,0 мл; 4 моль/л) добавляли в раствор метанола (30 мл), содержащий трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилат (4,34 г; 10,28 ммоль), и взаимодействие проводили при комнатной температуре в течение 3 ч.
По завершении реакции реакционный раствор незамедлительно упаривали на роторном испарителе, получая 3,68 г 4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазина в виде белого твердого вещества, которое использовали непосредственно на следующей стадии. LC-MS: RT=1,65 мин, [М+Н]+ = 312,26.
Стадия D. Синтез метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
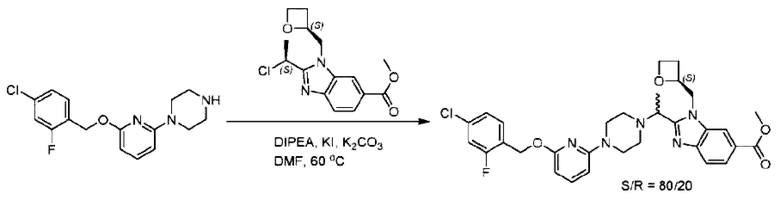
N,N-Диизопропилэтиламин (1,0 мл), иодид калия (420 мг; 3,13 ммоль), карбонат калия (577 мг; 4,18 ммоль), N;N-диметилформамид (10,0 мл) добавляли в раствор ацетонитрила (8,0 мл), содержащий 4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин (750 мг; 2,09 ммоль). Затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (650 мг; 2,09 ммоль) и взаимодействие проводили при 60°С в течение 3 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (30 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 2/1). Получали 770 мг метил- 2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде желтого твердого вещества (выход: 62,1%, dr = 60%). LC-MS: RT=1,92 мин, [М+Н]+ = 594,26.
Стадия Е. Синтез 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (39А) и 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (39В)
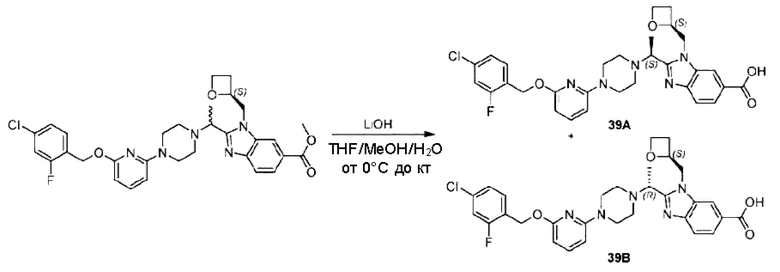
Гидроксид лития (307 мг; 7,30 ммоль) в воде (1 мл) при 0°С по каплям добавляли в раствор смеси тетрагидрофурана (3 мл) и метанола (1 мл), содержащий метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (770 мг; 1,30 ммоль), и взаимодействие проводили при 25°С в течение 30 мин.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 1/10). Получали 313 мг 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (39А) (выход: 41,6%) и 78 мг 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (39 В) (выход: 10,4%). Неочищенные продукты очищали препаративной высокоэффективной жидкостной хроматографией. Условия разделения приведены ниже: хроматографическая колонка: препаративная 5-С18 от Agilent, 100 мм×30 мм, 5 мкм; подвижная фаза: (для элюирования) вода (содержащая 0,1% аммиака) и ацетонитрил; скорость потока: 20 мл/мин; градиент: соединения 39А и 39В элюировались 25%-ным ацетонитрилом в моменты времени 15,23 мин и 17,06 мин; соответственно; длина волны детектирования: 254 нм.
Соединение 39А: HPLC: RT=15,23 мин. LC-MS: RT=1,88 мин, [М+Н]+ = 580,26. 1Н ЯМР(400 МГц, DMSO-d6) δ 8.26 (s, 1Н), 7.80 (dd, J=8,4; 1,5 Гц, 1Н), 7.66 (d, J=8,4 Гц, 1Н), 7.49 (t, J=8,2 Гц, 1Н), 7.44 (dt, J=16,0; 4,9 Гц, 2Н), 7.28 (dd, J=8,2; 1,8 Гц, 1Н), 6.28 (d, J=8,1 Гц, 1Н), 6.06 (d, J=7,8 Гц, 1Н), 5.29 (d, J=8,8 Гц, 2Н), 5.14 (d, J=3,0 Гц, 11-0, 4.80 (dd, J=15,5; 2,9 Гц, 1Н), 4.68 (dd, J=15,5; 5,5 Гц, 1H), 4.52-4.40 (m, 2Н), 4.18 (dt, J=9,1; 5,7 Гц, 1H), 3.40 (d, J=5,3 Гц, 4H), 2.68-2.59 (m, 11-0,2.54 (d, J=16,1 Гц, 4H), 2.38-2.26 (m, 1H), 1.45 (d, J=6,7 Гц, 3Н).
Монокристаллическая структура соединения 39А показана на Фиг. 1, а специфические параметры кристалла приведены ниже.
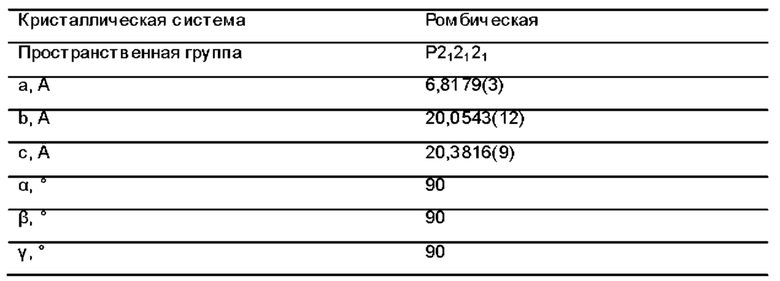
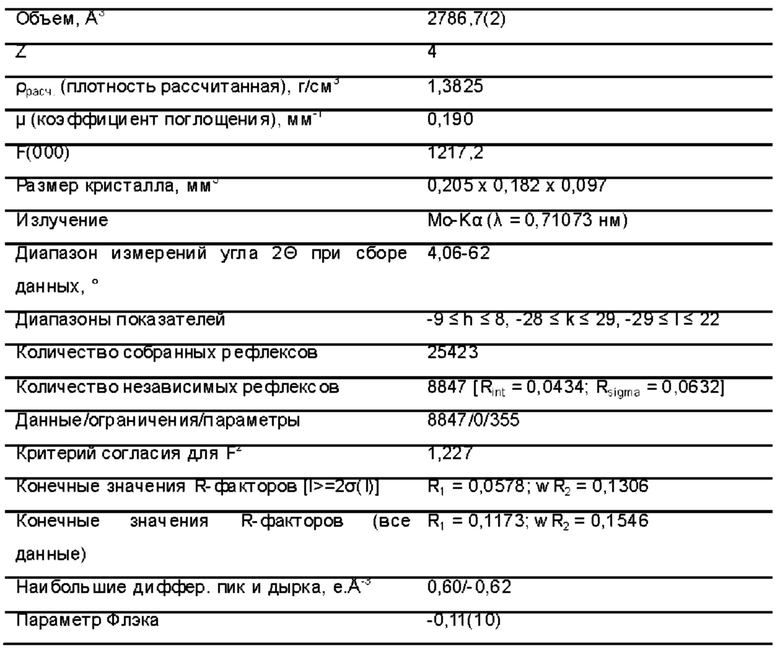
Соединение 39 В: HPLC: RT=17,06 мин. LC-MS: RT=1,88 мин, [М+Н]+ = 580,26. 1Н ЯМР (400 МГц, DMSO-d6) δ 12.74 (s, 1Н), 8.27-8.26 (m, 1Н), 7.79 (dd, J=9,9; 1,4 Гц, 1Н), 7.66 (dd, J=8,4; 1,5 Гц, 1Н), 7.50 (t, J=8,0 Гц, 1Н), 7.46-7.42 (m, 2Н), 7.29-7.27 (m, 1Н), 6.30 (d, J=8,2 Гц, 1Н), 6.07 (d, J=7,8 Гц, 1Н), 5.29 (s, 2Н), 4.93-4.87 (m, 1Н), 4.60-4.55 (m, 1Н), 4.52-4.47 (m, 3Н), 3.47-3.38 (m, 5Н), 2.79-2.72 (m, 1Н), 2.64-2.61 (m, 2Н), 2.56-2.47 (m, 2Н), 1.42 (d, J=6,7 Гц, 3Н).
Пример 40. 2-(1-(1-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
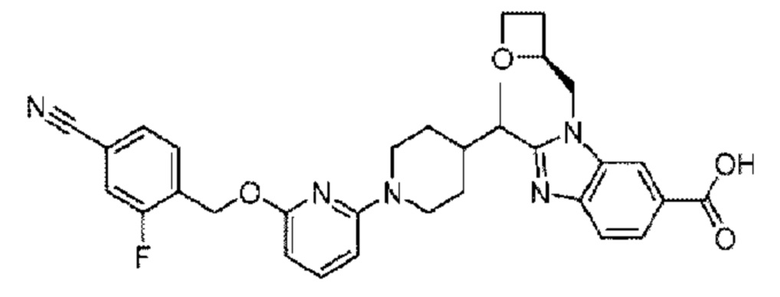
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез этил-2-(пиперидин-4-ил)пропионата
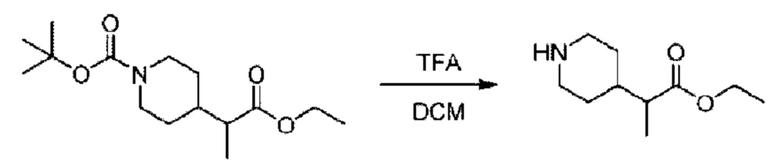
трет-Бутил-4-(1-этокси-1-оксипропан-2-ил)пиперидин-1-формиат (520 мг; 1,82 ммоль) растворяли в 4 мл безводного дихлорметана и добавляли 2 мл трифторуксусной кислоты. Взаимодействие проводили при комнатной температуре в течение 40 мин. По завершении реакции реакционный раствор выливали в 50 мл раствора бикарбоната натрия и раствор этой смеси экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 310 мг этил-2-(пиперидин-4-ил)пропи оната (выход: 92,1%).
Стадия В. Синтез этил-2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионата
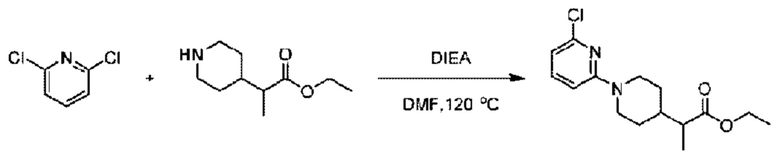
N,N-Диизопропилэтиламин (432 мг; 3,35 ммоль) добавляли в раствор N,N-диметил форм амида (10 мл), содержащий этил-2-(пиперидин-4-ил)пропионат (310 мл; 1,67 ммоль) и раствор этой смеси перемешивали в течение 2 мин. Затем добавляли 2,6-дихлорпиридин (245 мг; 1,67 ммоль) и взаимодействие проводили при 120°С в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды (50 мл) и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 422 мг этил-2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионата в виде бледно-желтого твердого вещества (выход: 85,1%). LC-MS: RT=2,21 мин, [М+Н]+ = 297,23.
Стадия С. Синтез 2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионовой кислоты
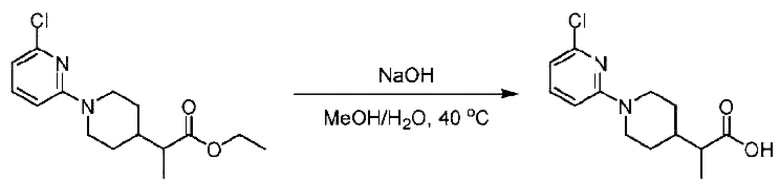
Этил-2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионат (422 мг; 1,42 ммоль) растворяли в 10 мл смеси растворителей метанола и воды и добавляли твердый гидроксид натрия (142 мг; 3,55 ммоль). Затем проводили взаимодействие при 40°С в течение 3 ч.
По завершении реакции реакционный раствор нейтрализовали разбавленной соляной кислотой до значения рН, равного примерно 6, и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 264 мг 2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионовой кислоты (выход: 69,1%). LC-MS: RT=1,94 мин, [М+Н]+ = 269,11.
Стадия D. Синтез метил-4-(2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоата
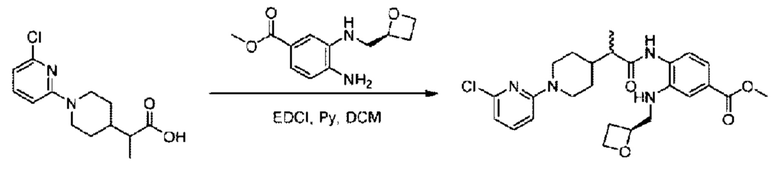
Пиридин (Ру; 141 мг; 1,78 ммоль) добавляли в раствор дихлорметана (10,0 мл), содержащий 2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионовую кислоту (240 мг; 0,892 ммоль) и 1-(3-диметиламинопропил)-3-этилкарбодиимид (255 мг; 1,34 ммоль), и раствор этой смеси перемешивали в течение 10 мин. Затем добавляли метил-(S)-4-амино-3-(((оксетан-2-ил)метил)амино)бензоат (211 мг; 0,892 ммоль) и взаимодействие проводили при комнатной температуре в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды (50 мл) и экстрагировали дихлорметаном (50 мл × 3 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/1). Получали 166 мг метил-4-(2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоатав виде бледно-желтого твердого вещества (выход: 38,2%). LC-MS: RT=1,98 мин, [М+Н]+ = 487,23.
Стадия Е Синтез метил-2-(1-(1-(6--хлорпиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
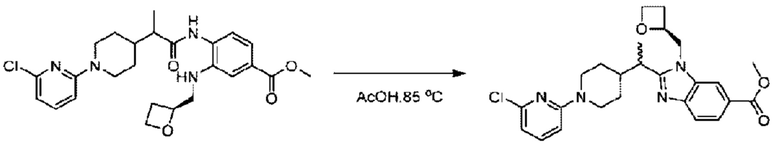
Метил-4-(2-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)пропионамидо)-3-((((S)-оксетан-2-ил)метил)амино)бензоат (166 мг; 0,341 ммоль) растворяли в 5 мл уксусной кислоты и взаимодействие проводили при 85°С в течение 12 ч.
По завершении реакции реакционный раствор нейтрализовали насыщенным раствором бикарбоната натрия до тех пор, пока значение рН не составляло примерно 7, и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 86 мг метил-2-(1-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата (выход: 53,7%). LC-MS: RT=2,24 мин, [М+Н]+ = 469,23.
Стадия F. Синтез метил-2-(1-(1-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[сПимидазол-6-карбоксилата
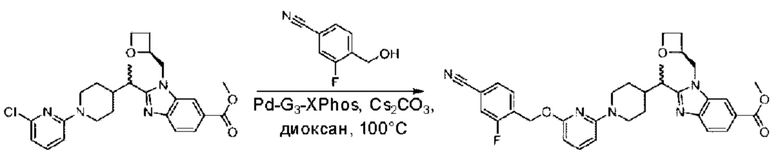
Карбонат цезия (119 мг; 0,366 ммоль) и метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (15,5 мг; 0,0183 ммоль) добавляли при комнатной температуре в раствор 1,4-диоксана (5 мл), содержащий метил-2-(1-(1-(6-хлорпиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (86,0 мг; 0,183 ммоль) и 3-фтор-4-(гидроксиметил)бензонитрил (33,2 мг; 0,221 ммоль), и взаимодействие проводили при 100°С в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (20 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 5/1). Получали 52 мг метил-2-(1-(1-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества (выход: 48,6%). LC-MS: RT=2,12 мин, [М+Н]+ = 584,27.
Стадия G. Синтез 2-(1-(1-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
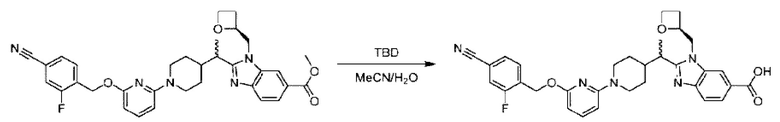
Метил-2-(1-(1-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (52,0 мг; 0,089 ммоль) растворяли в 4 мл ацетонитрила, затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (25 мг; 0,187 ммоль) и 1 мл воды и взаимодействие проводили при комнатной температуре.
По завершении реакции реакционный раствор нейтрализовали путем добавления 20 мл насыщенного раствора хлорида аммония и экстрагировали дихлорметаном (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли препаративной HPLC (аммиак), получая 22 мг 2-(1-(1-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперидин-4-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 43,4%). 1Н ЯМР (400 МГц, DMSO) δ 12.71 (s, 1Н),8.20 (dd, J=6,1; 1,3 Гц, 1Н), 7.87-7.90 (m, 1Н), 7.79 (dd, J=8,4; 1,6 Гц, 1Н), 7.68-7.71 (m, 1Н), 7.59-7.66 (m, 2Н), 7.41-7.45 (m, 1Н), 6.30 (dd, J=8,2; 4,8 Гц, 1Н), 6.05-6.08 4(m, 1Н), 5.38 (s, 2Н), 4.95-5.05 (m, 1Н), 4.44-4.55 (m, 2Н), 4.25-4.28 (m, 1Н), 4.06-4.13 (m, 1Н), 3.06-3.15 (m, 2Н), 2.58-2.77 (m, 5Н), 2.32-2.38 (m, 1Н),1.96-2.10 (m, 2Н), 1.80-1.95 (m, 2Н), 1.28 (t, J=6,7 Гц, 3Н). LC-MS: RT=1,97 мин, [М+Н]+ = 570,29.
Пример 41. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d|имидазол-6-карбоновая кислота
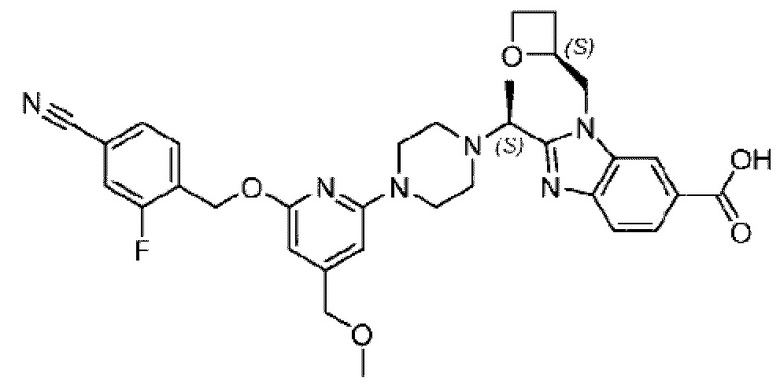
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 2,6-дихлор-4-(метоксиметил)пиридина
В раствор тетрагидрофурана (10,0 мл), содержащий (2,6-дихлорпиридин-4-ил)метанол (1,00 г; 5,68 ммоль), при 0°С добавляли 60% гидрида натрия (272 мг; 6,81 ммоль) и раствор этой смеси перемешивали в течение 5 мин. Затем добавляли подметан (873 мг; 6,24 ммоль) и взаимодействие проводили при комнатной температуре в течение 2 ч.
По завершении реакции реакционный раствор гасили путем добавления воды (50 мл) и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/5). Получали 670 мг 2,6-дихлор-4-(метоксиметил)пиридина в виде белого твердого вещества (выход: 61,4%). LC-MS: RT=2,24 мин, [М+Н]+ = 192,23.
Стадия В. Синтез трет-бутил-4-(6-хлор-4-(метоксиметил)пиридин-2-ил)пиперазин-1-формиата
Карбонат калия (960 мг; 6,96 ммоль) добавляли в раствор ацетонитрила (10,0 мл), содержащий 2,6-дихлор-4-(метоксиметил)пиридин (670 мг; 3,48 ммоль) и трет-бутилпиперазин-1-карбоксилат (647 мг; 3,48 ммоль), и взаимодействие проводили при 95°С в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды (50 мл) и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/3). Получали 746 мг трет-бутил-4-(6-хлор-4-(метоксиметил)пиридин-2-ил)пиперазин-1-формиата в виде бледно-желтого твердого вещества (выход: 62,6%). LC-MS: RT=2,18 мин, [М+Н]+ = 342,17.
Стадия С. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперидин-1-формиата
Карбонат цезия (477 мг; 1,46 ммоль) и метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (62,1 мг; 0,0733 ммоль) добавляли в раствор 1,4-диоксана (5 мл), содержащий трет-бутил-4-(6-хлор-4-(метоксиметил)пиридин-2-ил)пиперазин-1-формиат (250 мг; 0,733 ммоль) и 3-фтор-4-(гидроксиметил)бензонитрил (122 мг; 0,806 ммоль), при комнатной температуре и взаимодействие проводили при 100°С в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 320 мг трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперидин-1-формиата в виде бледно-желтого твердого вещества (выход: 69,5%). LC-MS: RT=1,98 мин, [М+Н]+ = 456,27.
Стадия D. Синтез 3-фтор-4-(((4-(метоксиметил)-6-пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила
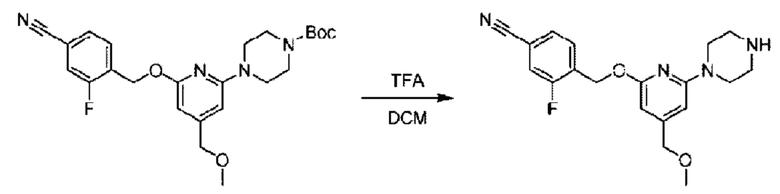
трет-Бутил-4-(6-((4-циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперидин-1-формиат (320 мг; 0,701 ммоль) растворяли в 4 мл безводного дихлорметана и добавляли 2 мл трифторуксусной кислоты. Взаимодействие проводили при комнатной температуре в течение 40 мин.
По завершении реакции реакционный раствор выливали в 50 мл раствора бикарбоната натрия и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 236 мг 3-фтор-4-(((4-(метоксиметил)-6-пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила (выход: 94,4%). LC-MS: RT=1,83 мин, [М+Н]+ = 356,27.
Стадия Е. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
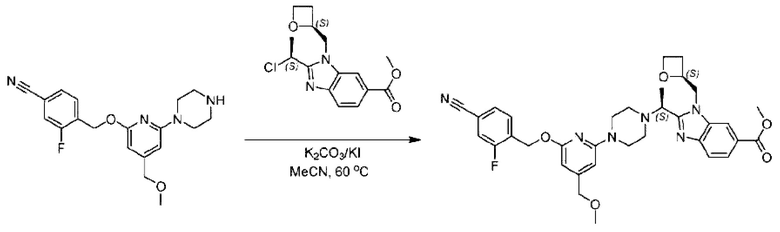
метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (204 мг; 0,662 ммоль), иодид калия (165 мг; 0,993 ммоль) и карбонат калия (183 мг; 1,32 ммоль) добавляли при комнатной температуре в раствор ацетонитрила (8,0 мл), содержащий 3-фтор-4-(((4-(метоксиметил)-6-пиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрил (236 мг; 0,662 ммоль), и взаимодействие в реакционном растворе проводили при 60°С в течение 5 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (30 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 5/1). Получали 296 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бледно-желтого твердого вещества (выход: 71,1%). LC-MS: RT=1,89 мин, [М+Н]+ = 629,27.
Стадия F. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
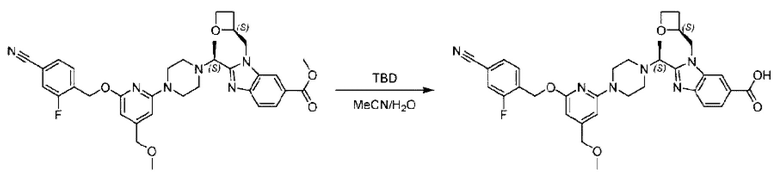
метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (120 мг; 0,191 ммоль) растворяли в 4 мл ацетонитрила, затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (55,7 мг; 0,401 ммоль) и 1 мл воды и взаимодействие проводили при комнатной температуре в течение 7 ч.
По завершении реакции реакционный раствор нейтрализовали путем добавления 20 мл насыщенного раствора хлорида аммония и экстрагировали дихлорметаном (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 36 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(метоксиметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 30,7%). 1Н ЯМР (400 МГц, DMSO) δ 12.77 (s, 1Н), 8.28 (d, J=0,9 Гц, 1Н), 7.85-7.91 (m, 1Н), 7.81 (dd, J=8,4; 1,5 Гц, 1Н), 7.67-7.72 (m, 2Н), 7.60-7.66 (m, 1Н), 6.23 (s, 1Н), 6.05 (s, 1Н), 5.39 (s, 2Н), 5.12-5.19 (m, 1Н), 4.80 (dd, J=16,2; 3,7 Гц, 1Н), 4.65-4.73 (m, 1Н), 4.42-4.52 (m, 2Н), 4.29 (s, 2Н), 4.14-4.23 (m, 1Н), 3.27 (s, 3Н), 3.14-3.23 (m, 2Н), 2.60-2.68 (m, 2Н), 2.52-2.56 (m, 3Н), 2.30-2.36(m, 1Н), 1.81-1.96 (m, 2Н), 1.44 (d, J=6,7 Гц, 3Н). LC-MS: RT=1,79 мин, [М+Н]+ = 615,38.
Пример 42. 2-((S)-1-(4-(4-((4-Циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
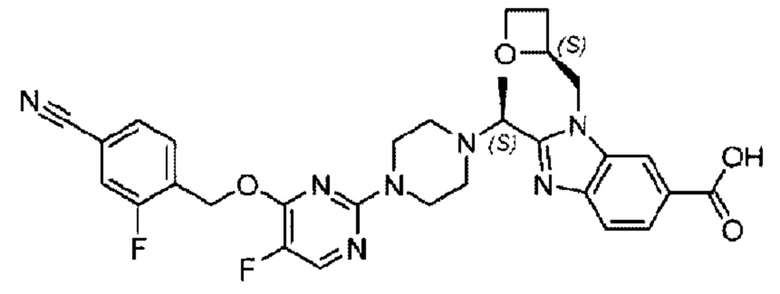
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 4-(((2-хлор-5-фторпиримидин-4-ил)окси)метил)-3-фторбензонитрила
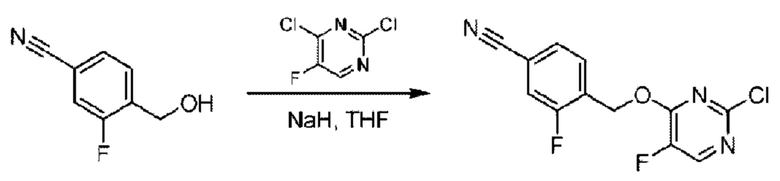
(4-Циано2-фторфенил)метанол (2 г; 13,2 ммоль) растворяли в тетрагидрофуране (100 мл). Раствор этой смеси охлаждали до 0°С и затем добавляли гидрид натрия (0,8 г; 19,5 ммоль). Раствор этой смеси перемешивали при комнатной температуре в течение 0,5 ч, затем добавляли 2,4-дихлор-5-фторпиримидин (3,3 г; 71 ммоль). Реакцию проводили в течение ночи при комнатной температуре.
По завершении реакции реакционный раствор гасили путем добавления насыщенного раствора хлорида натрия, экстрагировали этилацетатом (30 мл), дважды промывали водой, сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/10), получая 2 г 4-(((2-хлор-5-фторпиримидин-4-ил)окси)метил)-3-фторбензонитрила в виде белого твердого вещества (выход: 53%). LC-MS: RT=2,12 мин, [М+Н]+ = 282,23.
Стадия В. Синтез трет-бутил-4-(4-(((4-циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин)-1-формиата
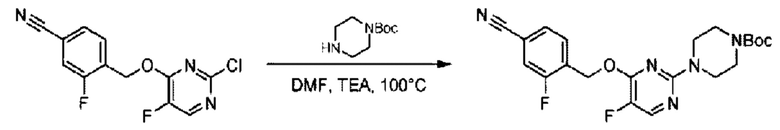
4-(((2-Циано-5-фторпиримидин-4-ил)окси)метил)-3-фторбензонитрил (1 г; 3,5 ммоль) растворяли в тетрагидрофуране (50 мл), затем добавляли трет-бутилпиперазин-1-карбоксилат (1 г; 5,3 ммоль) и реакцию проводили в течение ночи при 90°С.
По завершении реакции реакционный раствор гасили путем добавления насыщенного раствора хлорида натрия и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, сушили и концентрировали при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/5), получая 1 г трет-бутил-4-(4-(((4-циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин)-1-формиата в виде белого твердого вещества (выход: 44%). LC-MS: RT=1,89 мин, [М+Н]+ = 432,37.
Стадия С. Синтез 3-фтор-4-(((5-фтор-2-(пиперазин-1-ил)пиримидин-4-ил)окси)метил)бензонитрила
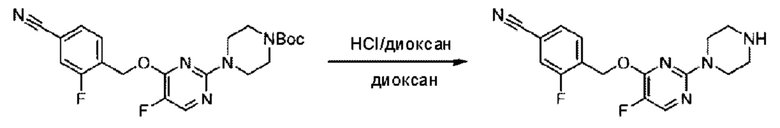
трет-Бутил-4-(4-(((4-циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин)-1-формиат (1 г; 2,3 ммоль) растворяли в диоксане (10 мл) при комнатной температуре и добавляли раствор хлористого водорода в диоксане (10 мл) с использованием ледяной бани. Раствор этой смеси перемешивали при комнатной температуре в течение 0,5 ч.
По завершении реакции реакционный раствор концентрировали, получая 120 мг 3-фтор-4-(((5-фтор-2-(пиперазин-1-ил)пиримидин-4-ил)окси)метил)-бензонитрила в виде белого твердого вещества. LC-MS: RT=1,62 мин, [М+Н]+ = 332,25.
Стадия D. Синтез метил-2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
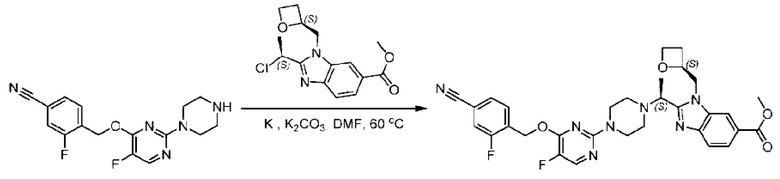
3-Фтор-4-(((5-фтор-2-(пиперазин-1-ил)пиримидин-4-ил)окси)метил)бензонитрил (200 мг; 0,6 ммоль) растворяли в N,N-диметилформамиде (10 мл) при комнатной температуре и затем добавляли метил-(S)-2-(хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (186 мг; 0,6 ммоль), иодид калия (200 мг; 1,2 ммоль) и карбонат калия (166 мг; 1,2 ммоль). Смесь перемешивали при 60°С в течение 12 ч.
По завершении реакции реакционный раствор гасили путем добавления насыщенного раствора хлорида натрия и экстрагировали этилацетатом (10 мл × 2 раза). Органические фазы объединяли, сушили с использованием безводного сульфата натрия, фильтровали и концентрировали и неочищенный продукт очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/1), получая 150 мг метил-2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде бесцветной маслянистой жидкости (выход: 41%). LC-MS: RT=1,95 мин, [М+Н]+ = 603,35.
Стадия Е. Синтез 2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
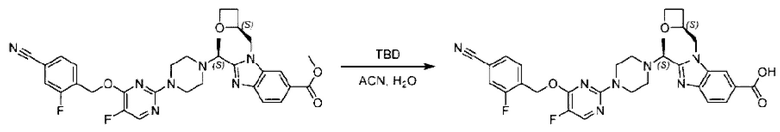
метил-2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (150 мг; 0,25 ммоль) растворяли в ацетонитриле (10 мл) и воде (2 мл), затем добавляли 1,3,4,6,7,8-гексагидро-2Н-пиримидин[1,2-а]пиримидин (139 мг; 1,0 ммоль) и взаимодействие проводили при комнатной температуре в течение 4 ч.
По завершении реакции реакционный раствор концентрировали. Полученный неочищенный продукт очищали высокоэффективной жидкостной хроматографией, получая 55,11 мг 2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)-5-фторпиримидин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 37%). LC-MS: RT=1,80 мин, [М+Н]+ = 590,27.
Пример 43. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
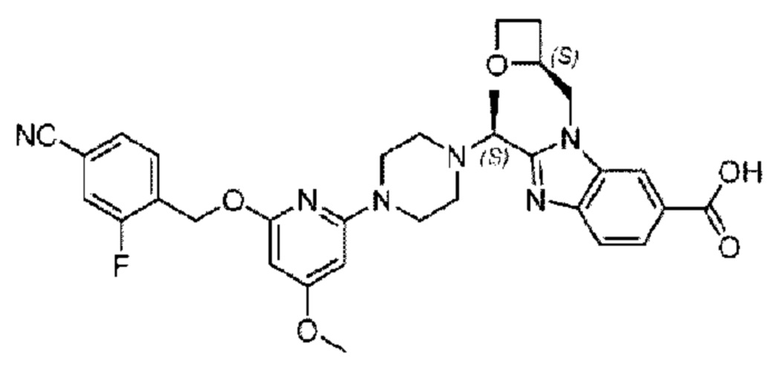
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 2-хлор-6-((4-циано-2-фторбензил)окси)-4-метоксипиридина
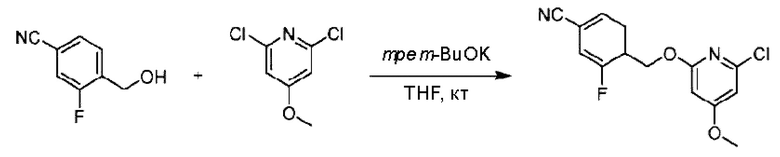
трет-Бутилат калия (0,98 г; 8,72 ммоль) добавляли в раствор тетрагидрофурана (10,0 мл), содержащий (4-циано-2-фторфенил)метанол (1,0 г; 6,23 ммоль), при 0°С и раствор этой смеси перемешивали в течение 30 мин. Затем добавляли 2,6-дихлор-4-метоксипиридин (1,1 г; 6,23 ммоль) и реакцию проводили в течение ночи при 25°С.
По завершении реакции реакционный раствор гасили путем добавления воды (50 мл) и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл × 3 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 0,98 г 2-хлор-6-((4-циано-2-фторбензил)окси)-4-метоксипиридина в виде желтого маслянистого продукта (выход: 53,9%). LC-MS: RT=2,15 мин, [М+Н]+ = 293,05.
Стадия В. Синтез трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-карбоксилата
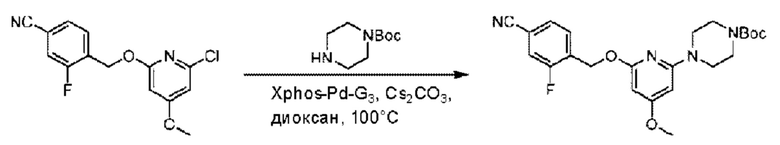
трет-Бутилпиперазин-1-карбоксилат (0,62 г; 3,36 ммоль), карбонат цезия (2,2 г; 6,72 ммоль) и метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (288 мг; 0,34 ммоль) добавляли в раствор 1,4-диоксана, содержащий 2-хлор-6-((4-циано-2-фторбензил)окси)-4-метоксипиридин (0,98 г; 3,36 ммоль), при 25°С и взаимодействие проводили при 100°С в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 1,2 г трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-карбоксилата в виде белого твердого вещества (выход: 80,8%). LC-MS: RT=2,23 мин, [М+Н]+ = 443,29.
Стадия С. Синтез 4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазина
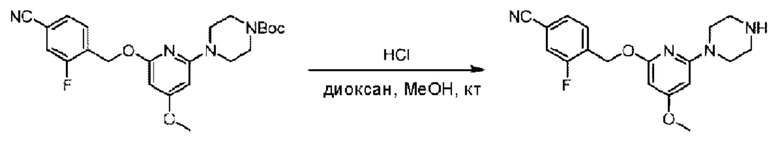
Соляную кислоту в 1,4-диоксане (10,0 мл; 4 моль/л) добавляли в раствор метанола (20 мл), содержащий трет-бутил-4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-карбоксилат (1,2 г; 2,71 ммоль), при 25°С и взаимодействие проводили при 25°С в течение 3 ч.
По завершении реакции реакционный раствор незамедлительно упаривали на роторном испарителе, получая 1,33 г 4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазина в виде белого твердого вещества, которое использовали непосредственно на следующей стадии. LC-MS: RT=1,72 мин, [М+Н]+ = 343,18.
Стадия D. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
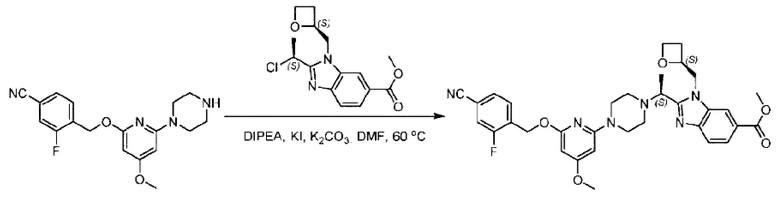
При 25°С добавляли N,N-диизопропилэтиламин (1,0 мл), иодид калия (676 мг; 4,07 ммоль), карбонат калия (748 мг; 5,42 ммоль) и N,N-диметилформамид (10,0 мл) в раствор ацетонитрила (8,0 мл), содержащий 4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазина гидрохлорид (1,33 мг; 2,71 ммоль), и затем добавляли метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (837 мг; 2,71 ммоль). Взаимодействие проводили при 60°С в течение 3 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (30 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 2/1). Получали 900 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде желтого твердого вещества (выход: 54,0%). LC-MS: RT=1,92 мин, [М+Н]+ = 615,31.
Стадия Е. Синтез 2-((3)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
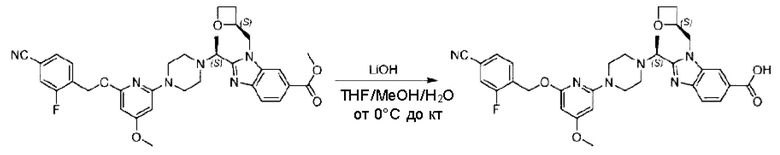
Гидроксид лития (307 мг; 7,30 ммоль) в воде (1 мл) по каплям добавляли в раствор смеси тетрагидрофурана (3 мл) и метанола (1 мл), содержащий метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (900 мг; 1,46 ммоль), при 0°С и взаимодействие проводили при 25°С в течение 30 мин.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 1/10). Получали 430 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-метоксипиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 49,0%). LC-MS: RT=1,79 мин, [М+Н]+ = 601,39. 1Н ЯМР (400 МГц, DMSO-d6) δ 12.75 (s, 1Н), 8.27 (t, J=2,4 Гц, 1Н), 7.86 (dd, J=10,0; 1,3 Гц, 1Н), 7.79 (ddd, J=5,6; 4,1; 1,5 Гц, 1Н), 7.71-7.65 (m, 2Н), 7.62 (t, J=7,6 Гц, 1Н), 5.83 (t, J=4,1 Гц, 1Н), 5.76-5.71 (m, 1Н), 5.35 (s, 2Н), 5.12 (d, J=3,0 Гц, 1Н), 4.78 (dd, J=15,5; 2,9 Гц, 1Н), 4.67 (dd, J=15,5; 5,5 Гц, 1Н), 4.52-4.41 (m, 2Н), 4.20-4.11 (m, 1Н), 3.71 (d, J=3,0 Гц, 3Н), 3.33 (s, 4Н), 2.67-2.59 (m, 1Н), 2.50 (d, J=1,2 Гц, 4Н), 2.31 (dt, J=18,4, 7,4 Гц, 1Н), 1.48-1.39 (m, 3Н).
Пример 44. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d|имидазол-6-карбоновая кислота
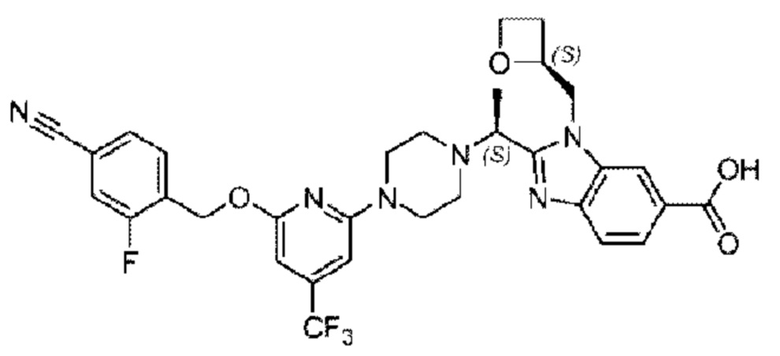
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-хлор-4-(трифторметил)пиридин-2-ил)пиперазин-1-формиата
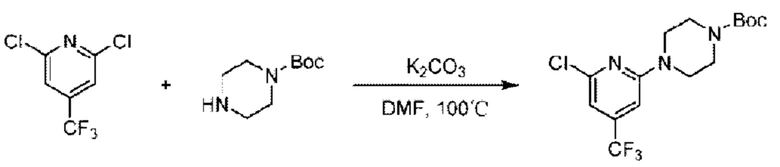
2,6-Дихлор-4-(трифторметил)пиридин (500,0 мг; 2,32 ммоль), трет-бутилпиперазин-1-карбоксилат (431,5 мг; 2,32 ммоль) и карбонат калия (641,3 мг; 4,64 ммоль) добавляли к N,N-диметилформамиду (20,0 мл) при комнатной температуре. Взаимодействие проводили при 100°С в течение 6 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 5/1), получая 677,0 мг трет-бутил-4-(6-хлор-4-(трифторметил)пиридин-2-ил)пиперазин-1-формиата в виде бледно-желтого твердого вещества (выход: 79,6%). LC-MS: RT=1,93 мин, [М+Н]+-56 = 310,11.
Стадия В. Синтез трет-бутил-4-(6-(((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин)-1-формиата
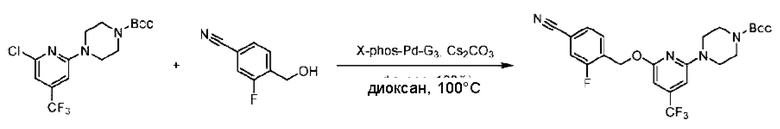
трет-Бутил-4-(6-хлор-4-(трифторметил)пиридин-2-ил)пиперазин-1-формиат (300,0 мг; 0,82 ммоль), 3-фтор-4-(гидроксиметил)бензонитрил (148,6 мг; 0,98 ммоль) и карбонат цезия (534,6 мг; 1,64 ммоль) добавляли к диоксану (15,0 мл) при комнатной температуре, затем добавляли метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (35,0 мг; 0,04 ммоль). Взаимодействие проводили при 100°С в течение 0,5 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали дихлорметаном (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), затем сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: н-гексан:этилацетат = 10:1), получая 240,0 мг трет-бутил-4-(6-(((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин)-1-формиата в виде бледно-желтого маслянистого продукта (выход: 60,1%). LC-MS: RT=1,95 мин, [М-56+Н]+ = 425,26.
Стадия С. Синтез 3-фтор-4-((((6-(пиперазин-1-ил)-4-(трифторметил)пиридин-2-ил)окси)метил)бензонитрила
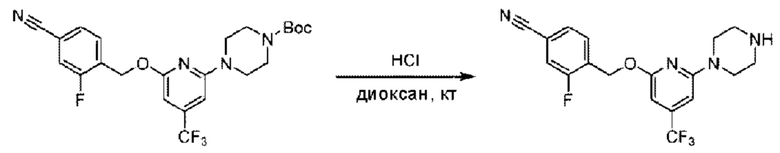
трет-Бутил-4-(6-(((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин)-1-формиат (240,0 мг; 0,50 ммоль) добавляли к раствору соляной кислоты в 1,4-диоксане (1,00 ммоль/мл; 5 мл) при комнатной температуре. Взаимодействие проводили при комнатной температуре в течение 1 ч в атмосфере N2.
По завершении реакции реакционный раствор концентрировали при пониженном давлении. Получали 195,0 мг 3-фтор-4-((((6-(пиперазин-1-ил)-4-(трифторметил)пиридин-2-ил)окси)метил)бензонитрила в виде бледно-желтого твердого вещества (выход: 100,0%), которое использовали непосредственно в следующей реакции. LC-MS: RT=1,72 мин, [М+Н]+ = 381,17.
Стадия D. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[сПимидазол-6-карбоксилата
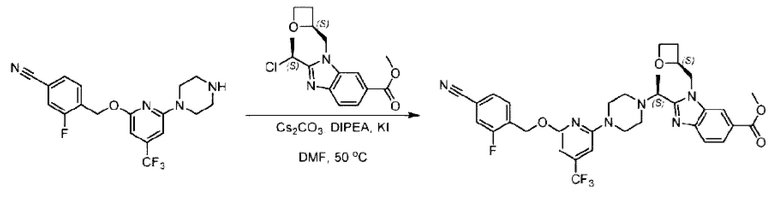
3-Фтор-4-((((6-(пиперазин-1-ил)-4-(трифторметил)пиридин-2-ил)окси)метил)бензонитрил (50,0 мг; 0,13 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (40,0 мг; 0,13 ммоль), карбонат цезия (40,4 мг; 0,13 ммоль) и иодид калия (22,0 мг; 0,13 ммоль) добавляли к N,N-диметилформамиду (5,0 мл), затем добавляли N,N-диизопропилэтиламин (0,50 мл) при комнатной температуре. Взаимодействие проводили при 50°С в течение 4,0 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), затем сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20:1), получая 50,2 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого твердого вещества (выход: 59,1%). LC-MS: RT=1,83 мин, [М+Н]+ = 653,27.
Стадия Е. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
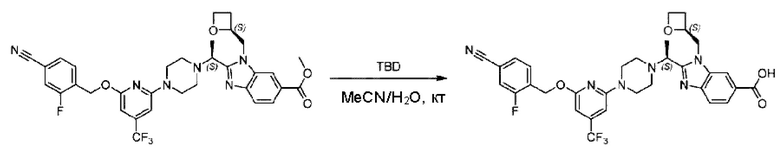
Раствор 1,5,7-триазабицикло[4.4.0]дец-5-ена (33,4 мг; 0,24 ммоль) в воде (3,0 мл) по каплям при комнатной температуре добавляли в раствор ацетонитрила (12,0 мл), содержащий метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (50,2 мг; 0,08 ммоль). Взаимодействие проводили при комнатной температуре в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и значение рН подводили до 6 водным раствором лимонной кислоты (0,5 моль/л). Раствор этой смеси экстрагировали дихлорметаном (30 мл × 3 раза). Органические фазы объединяли, промывали водным раствором лимонной кислоты (30 мл × 2 раза) и сушили с использованием безводного сульфата натрия. Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией. Условия разделения приведены ниже: хроматографическая колонка: препаративная 5-С18 от Agilent, 100 мм×30 мм, 5 мкм; подвижная фаза: вода (содержащая 0,1% аммиака) и ацетонитрил; скорость потока: 20 мл/мин; градиент: продукт элюировался 32,3%-ным ацетонитрилом в момент времени 8,12 мин; длина волны детектирования: 254 нм. После очистки и лиофилизации получали 22,0 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 44,1%). LC-MS: RT=1,75 мин, [М+Н]+ = 639,33. 1Н ЯМР (400 МГц, DMSO) δ 8.26 (d, J=0,9 Гц, 1Н), 7.88 (dd, J=10,0; 1,2 Гц, 1Н), 7.79 (dd, J=8,4; 1,5 Гц, 1Н), 7.68 (qd, J=7,8; 3,6 Гц, 3Н), 6.55 (s, 1Н), 6.33 (s, 1Н), 5.43 (s, 2Н), 5.13 (dd, J=13,9; 8,9 Гц, 1Н), 4.70 (ddd, J=25,6; 10,8; 4,3 Гц, 2Н), 4.46 (dd, J=17,0; 4,8 Гц, 2Н), 4.20-4.12 (m, 1Н), 3.48 (d, J=5,4 Гц, 4Н), 2.67-2.51 (m, 5Н), 2.38-2.24 (m, 1Н), 1.43 (d, J=6,7 Гц, 3Н).
Пример 45. 2-((S)-1-(4-(4-Циано-6-((4-циано-2-фторбензил)оксо)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[сПимидазол-6-карбоновая кислота
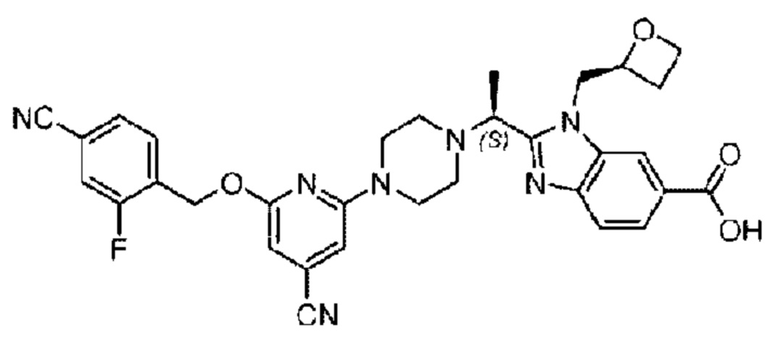
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез трет-бутил-4-(6-хлор-4-цианопиридин-2-ил)пиперазин-1-карбоксилата
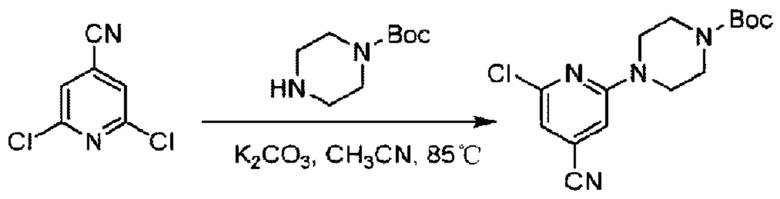
2,6-Дихлор-4-цианопиридин (1,0 г; 5,78 ммоль), N-Вос-пиперазин (Вое означает трет-бутилоксикарбонил; 1,29 г; 6,94 ммоль) и карбонат калия (1,6 г; 11,56 ммоль) добавляли к 10 мл безводного ацетонитрила при комнатной температуре и взаимодействие проводили при 85°С в течение 4 ч.
По завершении реакции реакционный раствор охлаждали до комнатной температуры и выливали в 40 мл ледяной воды. Раствор этой смеси перемешивали в течение 30 мин и фильтровали с отсасыванием. Осадок на фильтре промывали водой и сушили, получая 1,7 г трет-бутил-4-(6-хлор-4-цианопиридин-2-ил)пиперазин-1-карбоксилата в виде белого твердого вещества (выход: 91,4%). LC-MS: RT=2,18 мин, [М+Н]+ = 223,10.
Стадия В. Синтез трет-бутил-4-(4-циано-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата
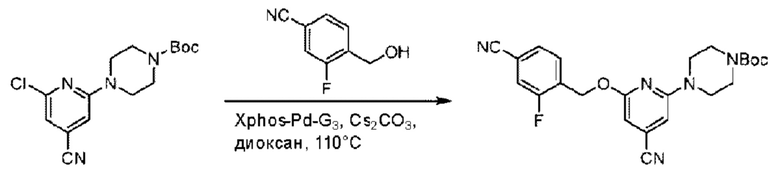
Промежуточный трет-бутил-4-(6-хлор-4-цианопиридин-2-ил)пиперазин-1-карбоксилат (500 мг; 1,55 ммоль), 3-фтор-4-гидроксиметилбензонитрил (258 мг; 1,70 ммоль), метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (65,7 мг; 0,08 ммоль) и карбонат цезия (1012 мг; 3,1 ммоль) добавляли к 10 мл раствора безводного диоксана при комнатной температуре. Взаимодействие проводили при 110°С в течение 2 ч в атмосфере N2.
По завершении реакции реакционный раствор упаривали, экстрагировали этилацетатом (20 мл), сушили с использованием безводного сульфата натрия и упаривали, получая бледно-желтое твердое вещество, которое очищали хроматографией на колонке с силикагелем (н-гексан/этилацетат = 5/2) с получением 383 мг трет-бутил-4-(4-циано-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата в виде бледно-желтого твердого вещества (выход: 56,1%). LC-MS: RT=2,21 мин, [М-56]+ = 379,25.
Стадия С. Синтез 2-((4-циано-2-фторбензил)окси)-6-(пиперазин-1-ил)изоникотинонитрила
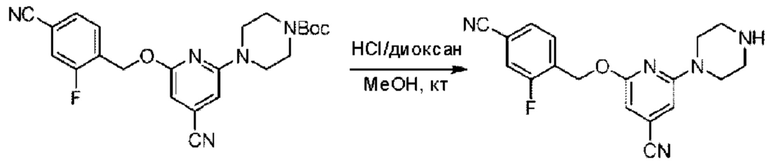
трет-Бутил-4-(4-циано-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилат (380 мг; 0,87 ммоль) растворяли в 10 мл метанола при комнатной температуре и затем в ледяной бане добавляли 10 мл раствора хлористого водорода в диоксане с концентрацией 4 моль/л. Взаимодействие проводили при комнатной температуре в течение 2 ч.
По завершении реакции реакционный раствор упаривали, получая 340 мг (2-((4-циано-2-фторбензил)окси)-6-(пиперазин-1-ил)изоникотинонитрила в виде белого твердого вещества (выход: 116,0%). LC-MS: RT=1,70 мин, [М+Н]+ = 338,18.
Стадия D. Синтез метил-2-((S)-1-(4-(4-циано-6-((4-циано-2-фторбензил)оксо)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[сПимидазол-6-карбоксилата
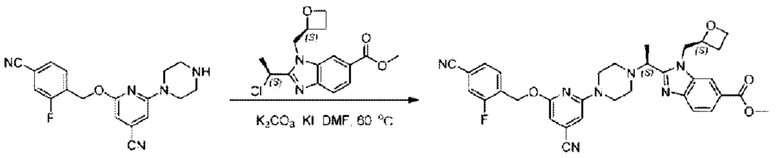
Промежуточный 2-((4-циано-2-фторбензил)окси)-6-(пиперазин-1-ил)изоникотинонитрил (380 мг; 0,87 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (267 мг; 0,87 ммоль), карбонат калия (240 мг; 1,74 ммоль) и иодид калия (217 мг; 1,3 ммоль) добавляли к 10 мл DMF при комнатной температуре и взаимодействие проводили при 60°С в течение 10 ч.
По завершении реакции реакционный раствор выливали в ледяную воду и твердое вещество осаждали. Затем смесь фильтровали с отсасыванием и осадок на фильтре промывали водой, получая 300 мг метил-2-((S)-1-(4-(4-циано-6-((4-циано-2-фторбензил)оксо)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого продукта (выход: 56,6%). LC-MS: RT=1,93 мин, [М+Н]+ = 610,34.
Стадия Е. Синтез 2-((S)-1-(4-(4-циано-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
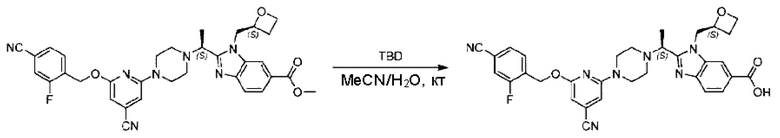
Промежуточный метил-2-((S)-1-(4-(4-циано-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (100 мг; 0,16 ммоль) растворяли в 5 мл раствора смеси ацетонитрил/вода (4:1) при комнатной температуре, затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (48 мг; 0,34 ммоль) при комнатной температуре и взаимодействие проводили при комнатной температуре в течение 4 ч.
По завершении реакции реакционный раствор выливали в 15%-ный водный раствор лимонной кислоты, и значение рН устанавливалось в диапазоне 3-4. Реакционный раствор экстрагировали дихлорметаном (20 мл), 3 раза промывали водой, сушили с использованием безводного сульфата натрия и упаривали, получая бледно-желтое твердое вещество. После очистки хроматографией на колонке с силикагелем (дихлорметан/метанол = 10/1) получали 60 мг 2-((S)-1-(4-(4-циано-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 61,4%). LC-MS: RT=1,82 мин, [М+Н]+ = 596,29. 1Н ЯМР (400 МГц, DMSO) δ 8.25 (s, 1Н), 7.88 (dd, J=10,0; 1,3 Гц, 1Н), 7.79 (dd, J=8,4; 1,4 Гц, 1Н), 7.71-7.60 (m, 3Н), 6.76 (s, 1Н), 6.46 (s, 1Н), 5.42 (d, J=13,5 Гц, 2Н), 5.18-5.06 (m, 1Н), 4.81-4.72 (m, 1Н), 4.66 (dd, J=15,4; 5,4 Гц, 1Н), 4.52-4.36 (m, 2Н), 4.16 (dt, J=9,1; 5,8 Гц, 1Н), 3.50-3.42 (m, 4Н), 2.67-2.56 (m, 1Н), 2.56-2.50 (m, 4Н), 2.37-2.21 (m, 1Н), 1.42 (d, J=6,7 Гц, 3Н).
Пример 46. 2-((S)-1-(4-(4-((4-Циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
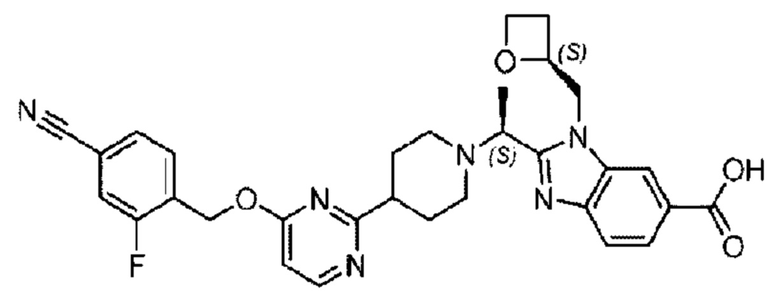
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 4-(((2-хлорпиримидин-4-ил)окси)метил)-3-фторбензонитрила
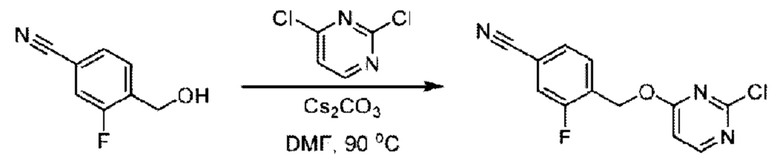
Карбонат цезия (860 мг; 2,64 ммоль) добавляли в раствор N,N-диметилформамида (10 мл), содержащий (4-циано-2-фторфенил)метанол (2,0 г; 1,32 ммоль), и раствор этой смеси перемешивали в течение 2 мин. Затем добавляли 2,4-дихлорпиримидин (1,97 г; 1,32 ммоль) и взаимодействие проводили при 90°С в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды (50 мл) и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 2,3 г 4-(((2-фторпиримидин-4-ил)окси)метил)-3-фторбензонитрила в виде желтого маслянистого продукта (выход: 67,6%). LC-MS: RT=2,36 мин, [М+Н]+ = 264,17.
Стадия В. Синтез трет-бутил-4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)-3,6-дигидропиридин-1(2Н)-формиата
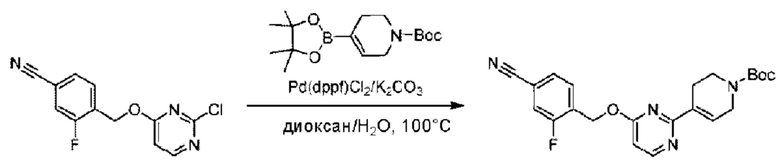
Карбонат калия (731 мг; 5,30 ммоль) и 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий(II) (193 мг; 0,265 ммоль) добавляли в раствор смеси 1,4-диоксана (5 мл) и воды, содержащий 4-(((2-фторпиримидин-4-ил)окси)метил)-3-фторбензонитрил (700 мг; 2,65 ммоль) и трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-ди оксаборолан-2-ил)-3,6-ди гидропиридин-1(2Н)-карбоксилат (820 мг; 2,65 ммоль), при комнатной температуре. Систему три раза продували азотом и взаимодействие проводили при 100°Св течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 570 мг трет-бутил-4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)-3,6-дигидропиридин-1(2Н)-формиата в виде бледно-желтого твердого вещества (выход: 52,3%). LC-MS: RT=2,22 мин, [М+Н]+ = 411,22.
Стадия С. Синтез трет-бутил-4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-формиата
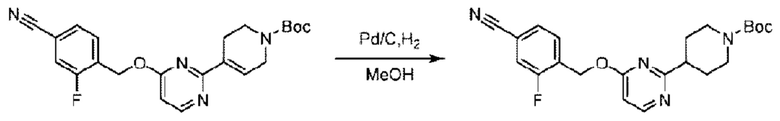
трет-Бутил-4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)-3,6-дигидропиридин-1(2Н)-формиат (300 мг; 0,729 ммоль) растворяли в 10 мл метанола и добавляли 30 мг влажного палладия-на-угле (10%). Взаимодействие проводили при комнатной температуре в течение 3 ч в атмосфере водорода.
По завершении реакции реакционный раствор фильтровали, затем фильтрат сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 186 мг трет-бутил-4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-формиата (выход: 61,8%). LC-MS: RT=2,03 мин, [М+Н]+ = 413,23.
Стадия D. Синтез 3-фтор-4-(((2-(пиперидин-4-ил)пиримидин-4-ил)окси)метил)бензонитрила
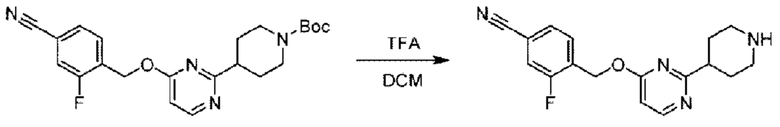
трет-Бутил-4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-формиат (186 мг; 0,451 ммоль) растворяли в 4 мл безводного дихлорметана и добавляли 2 мл трифторуксусной кислоты. Затем проводили взаимодействие при комнатной температуре в течение 40 мин.
По завершении реакции реакционный раствор выливали в 50 мл раствора бикарбоната натрия и раствор этой смеси экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении, получая 110 мг 3-фтор-4-(((2-(пиперидин-4-ил)пиримидин-4-ил)окси)метил)бензонитрила (выход: 78,1%). LC-MS: RT=1,86 мин, [М+Н]+ = 313,27.
Стадия Е. Синтез метил-2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
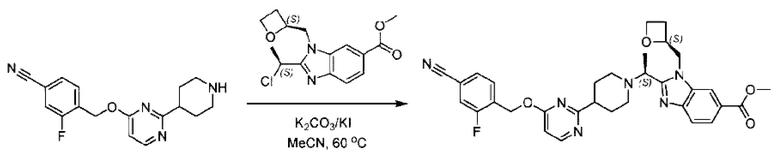
метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (109 мг; 0,352 ммоль), иодид калия (234 мг; 1,41 ммоль) и карбонат калия (242 мг; 1,76 ммоль) добавляли в раствор ацетонитрила (8,0 мл), содержащий 3-фтор-4-(((2-пиперидин-4-ил)пиримидин-4-ил)окси)метил)бензонитрил (110 мг; 0,352 ммоль), при комнатной температуре и взаимодействие в реакционном растворе проводили при 60°Св течение 5 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (30 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 5/1). Получали 84 мг метил-2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде желтого твердого вещества (выход: 70,4%). LC-MS: RT=1,97 мин, [М+Н]+ = 585,31.
Стадия F. Синтез 2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
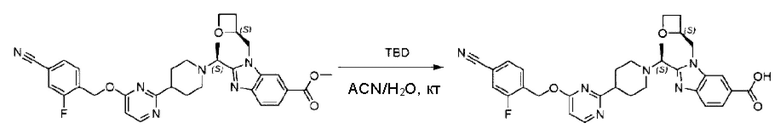
метил-2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (82,0 мг; 0,140 ммоль) растворяли в 4 мл ацетонитрила и затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (41,1 мг; 0,294 ммоль) и 1 мл воды. Взаимодействие проводили при комнатной температуре в течение 7 ч.
По завершении реакции реакционный раствор нейтрализовали путем добавления 20 мл насыщенного раствора хлорида аммония и экстрагировали дихлорметаном (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Полученный неочищенный продукт разделяли колоночной хроматографией, получая 26 мг 2-((S)-1-(4-(4-((4-циано-2-фторбензил)окси)пиримидин-2-ил)пиперидин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 32,5%). 1Н ЯМР (400 МГц, DMSO) δ 12.75 (s, 1Н), 8.49 (d, J=5,1 Гц, 1Н), 8.27 (d, J=1,1 Гц, 1Н), 7.91 (dd, J=10,2; 1,1 Гц, 1Н), 7.80 (dd, J=8,4; 1,5 Гц, 1Н), 7.74-7.76 (m, 1Н), 7.71-7.73 (m, 1Н), 7.67 (d, J=8,7 Гц, 1Н), 7.07 (d, J=5,1 Гц, 1Н), 5.48 (s, 2Н), 5.13-5.19 (m, 1Н), 4.83 (dd, J=16,2; 3,7 Гц, 1Н), 4.68 (dd, J=15,5; 5,7 Гц, 1Н), 4.41-4.50 (m, 2Н), 4.16-4.21 (m, 1Н), 2.96-2.98 (m, 1Н), 2.54-2.67 (m, 4Н), 2.21-2.35 (m, 2Н), 1.83-1.86 (m, 2Н), 1.67-1.76 (m, 2Н), 1.47(d, J=6,8 Гц, 3Н). LC-MS: RT=1,70 мин, [М+Н]+ = 571,31.
Пример 47. 1-(((S)-Оксетан-2-ил)метил)-2-((S)-1-(4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоновая кислота
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 2-(((6-хлорпиридин-2-ил)окси)метил)пиразоло[1,5-а]пиридина
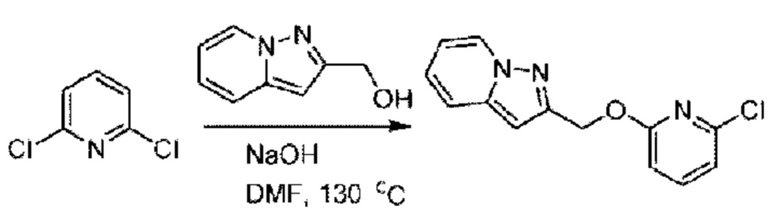
2,6-Дихлорпиридин (375 мг; 2,55 ммоль), пиразоло[1,5-а]пиридин-2-метанол (453 мг; 3,06 ммоль) и гидроксид натрия (510 мг; 12,75 ммоль) добавляли к 10 мл безводного DMF и взаимодействие проводили при повышенной до 130°С температуре в течение 2 ч.
По завершении реакции реакционный раствор охлаждали до комнатной температуры и выливали в 20 мл ледяной воды. Раствор этой смеси перемешивали в течение 30 мин и затем фильтровали с отсасыванием. Осадок на фильтре промывали водой и сушили, получая 480 мг 2-(((6-хлорпиридин-2-ил)окси)метил)пиразоло[1,5-а]пиридина в виде белого твердого вещества (выход: 72,7%). LC-MS: RT=2,04 мин, [М+Н]+ = 260,10.
Стадия В. Синтез трет-бутил-6-(пиразоло[1,5-а]пиридин-2-метокси)-3',6'-дигидро-[2,4'-бипиридин]1'(2'Н)-карбоксилата
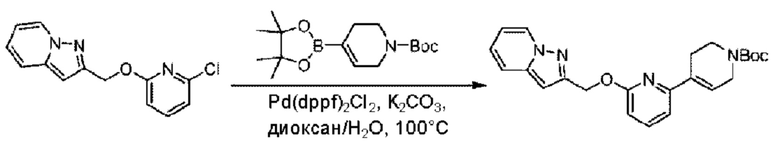
Промежуточный 2-(((6-хлорпиридин-2-ил)окси)метил)пиразоло[1,5-а]пиридин (300 мг; 1,16 ммоль), пинаколовый эфир н-бутилоксикарбонил-1,2,5,6-тетрагидропиридин-4-борной кислоты (430 мг; 1,39 ммоль), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (127,3 мг; 0,17 ммоль) и карбонат калия (320 мг; 2,32 ммоль) добавляли при комнатной температуре в смесь диоксан/вода (4/1, 10 мл). Взаимодействие проводили при 100°С в течение 2 ч в атмосфере N2.
Реакционный раствор упаривали по завершении реакции и затем экстрагировали этилацетатом, сушили с использованием безводного сульфата натрия и упаривали, получая бледно-желтое твердое вещество. После очистки хроматографией на колонке с силикагелем (н-гексан/этилацетат = 5/1) получали 266 мг трет-бутил-6-(пиразоло[1,5-а]пиридин-2-метокси)-3',6'-дигидро-[2,4'-бипиридин]1'(2'Н)-карбоксилата (выход: 56,5%). LC-MS: RT=2,21 мин, [М+Н]+ = 407,21.
Стадия С. Синтез трет-бутил-4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиперидин-1-карбоксилата
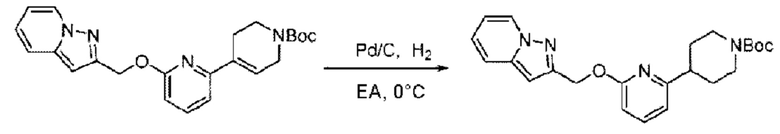
трет-Бутил-6-(пиразоло[1,5-а]пиридин-2-метокси)-3',6'-дигидро-[2,4'-бипиридин]1'(2'Н)-карбоксилат (240 мг; 0,59 ммоль) растворяли в 5 мл метанола в ледяной бане и добавляли 10% палладия-на-угле (24 мг; 0,0059 ммоль) в ледяной бане. Взаимодействие проводили при 0°С в течение 12 ч в атмосфере Н2.
По завершении реакции реакционный раствор фильтровали с отсасыванием и фильтрат упаривали, получая 200 мг трет-бутил-4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиперидин-1-карбоксилата в виде белого маслянистого продукта (выход: 83,0%). LC-MS: RT=2,21 мин, [М+Н]+ = 409,29.
Стадия D. Синтез 2-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)пиразоло[1,5-а]пиридина
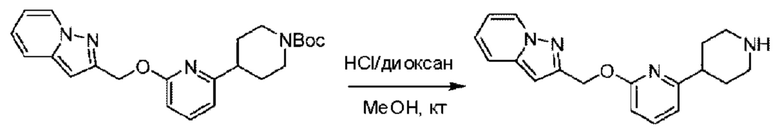
трет-Бутил-4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиридин-1-карбоксилат (200 мг; 0,49 ммоль) растворяли в 10 мл метанола при комнатной температуре и добавляли 5 мл раствора хлористого водорода в диоксане с концентрацией 4 M в ледяной бане. Взаимодействие проводили при комнатной температуре в течение 2 ч.
По завершении реакции реакционный раствор упаривали, получая 200 мг 2-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)пиразоло[1,5-а]пиридина в виде белого твердого вещества (выход: 132,5%). LC-MS: RT=1,65 мин, [М+Н]+ = 309,22.
Стадия Е. Синтез метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилата
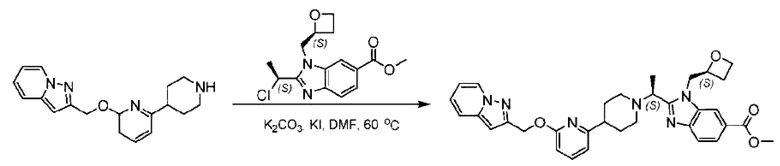
Промежуточный 2-(((6-(пиперидин-4-ил)пиридин-2-ил)окси)метил)пиразоло[1,5-а]пиридин (200 мг; 0,49 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (150 мг; 0,49 ммоль), карбонат калия (136,2 мг; 0,98 ммоль) и иодид калия (122 мг; 0,98 ммоль) добавляли к 10 мл DMF при комнатной температуре и взаимодействие проводили при температуре 60°С в течение 10 ч.
По завершении реакции реакционный раствор выливали в ледяную воду, и твердое вещество выпадало в осадок. Смесь фильтровали с отсасыванием и осадок на фильтре промывали водой, получая 200 мг метил-1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого продукта (выход: 70,4%). LC-MS: RT=1,79 мин, [М+Н]+ = 581,38.
Стадия F. Синтез 1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d|имидазол-6-карбоновой кислоты
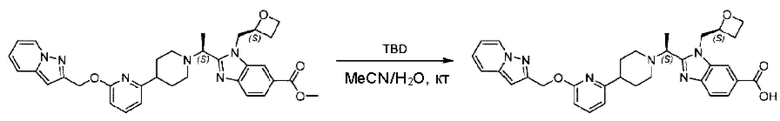
Промежуточный метил-1-(((S)-оксетан-2-ил)метил)-2-((3)-1-(4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоксилат (200 мг; 0,34 ммоль) растворяли в 5 мл смеси растворителей ацетон итрил/вода (4:1) при комнатной температуре и затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (101 мг; 0,71 ммоль) при комнатной температуре. Взаимодействие проводили при комнатной температуре в течение 7 ч.
По завершении реакции реакционный раствор выливали в 15%-ный водный раствор лимонной кислоты, и значение рН устанавливалось в диапазоне 3-4. Реакционный раствор экстрагировали дихлорметаном (20 мл), 3 раза промывали водой, сушили с использованием безводного сульфата натрия и упаривали, получая бледно-желтое твердое вещество. После очистки колоночной хроматографией (дихлорметан/метанол = 10/1) получали 130 мг 1-(((S)-оксетан-2-ил)метил)-2-((S)-1-(4-(6-(пиразоло[1,5-а]пиридин-2-метокси)пиридин-2-ил)пиперидин-1-ил)этил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 67,7%). LC-MS: RT=1,73 мин, [М+Н]+ = 567,35.
Пример 48. 2-((S)-1-((R)-4-(6-(((4-Циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (48А) и 2-((S)-1-((S)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота (48В)
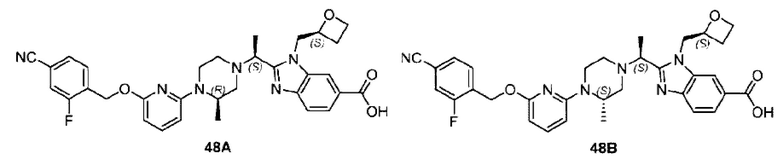
Конкретный путь синтеза соединения 48А приведен ниже.
Стадия А. Синтез трет-бутил-(R)-4-(6-хлорпиридин-2-ил)-3-метилпиперазин-1-карбоксилата
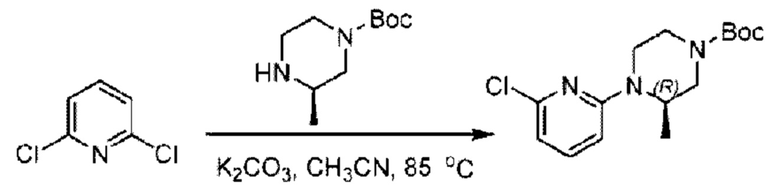
2,6-Дихлорпиридин (1,5 г; 10 ммоль), трет-бутил-(R)-3-метилпиперазин-1-карбоксилат (2,64 г; 13 ммоль) и карбонат калия (2,76 г; 20 ммоль) добавляли к 20 мл безводного ацетонитрила и взаимодействие проводили при температуре 85°С в течение 15 ч.
По завершении реакции реакционный раствор охлаждали до комнатной температуры и выливали в 40 мл ледяной воды. Раствор этой смеси перемешивали в течение 30 мин, затем фильтровали с отсасыванием. Осадок на фильтре промывали водой, сушили и очищали колоночной хроматографией (н-гексан/этилацетат = 5/1). Получали 350 мг трет-бутил-(R)-4-(6-хлорпиридин-2-ил)-3-метилпиперазин-1-карбоксилата в виде белого твердого вещества (выход: 11%). LC-MS: RT=2,20 мин, [М-56]+ = 256,16.
Стадия В. Синтез трет-бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-карбоксилата
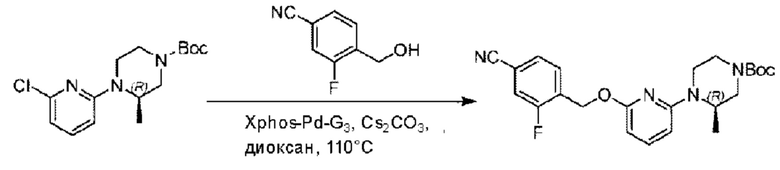
Промежуточный трет-бутил-(R)-4-(6-хлорпиридин-2-ил)-3-метилпиперазин-1-карбоксилат (150 мг; 0,48 ммоль) и 3-фгор-4-гидроксиметилбензонитрил (80 мг; 0,53 ммоль), метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (20,4 мг; 0,024 ммоль) и карбонат цезия (314 мг; 0,96 ммоль) добавляли к 10 мл безводного диоксана при комнатной температуре. Взаимодействие проводили при 110°С в течение 2 ч в атмосфере N2.
По завершении реакции реакционный раствор упаривали, затем экстрагировали этилацетатом (20 мл), сушили с использованием безводного сульфата натрия и упаривали, получая бледно-желтое твердое вещество. После очистки колоночной хроматографией (н-гексан/этилацетат = 5/2) получали 150 мг трет-бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-карбоксилата в виде бледно-желтого твердого вещества (выход: 73,5%). LC-MS: RT = 2,27 мин, [М-56]+=371,19.
Стадия С. Синтез (R)-3-фтор-4-(((6-(2-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила
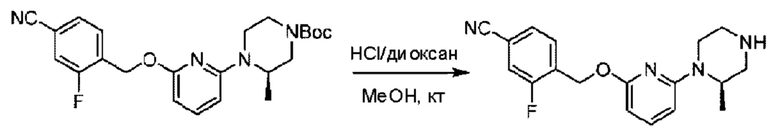
трет-Бутил-(R)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-карбоксилат (150 мг; 0,35 ммоль) растворяли в 10 мл метанола при комнатной температуре и добавляли 5 мл раствора хлористого водорода в диоксане с концентрацией 4 M в ледяной бане. Взаимодействие проводили при комнатной температуре в течение 2 ч.
По завершении реакции реакционный раствор упаривали, получая 118 мг (R)-3-фтор-4-(((6-(2-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила в виде белого твердого вещества (выход: 103,5%). LC-MS: RT=1,70 мин, [М+Н]+=327,21.
Стадия D. Синтез метил-2-((S)-1-((R)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
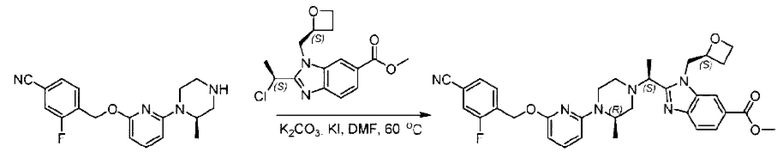
Промежуточный (R)-3-фтор-4-(((6-(2-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрил (150 мг; 0,35 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (108 мг; 0,35 ммоль), карбонат калия (97,2 мг; 0,70 ммоль) и иодид калия (87,7 мг; 0,53 ммоль) добавляли к 10 мл DMF при комнатной температуре и взаимодействие проводили при температуре 60°С в течение 10 ч.
По завершении реакции реакционный раствор выливали в ледяную воду, и белые твердые вещества выпадали в осадок. Затем смесь фильтровали с отсасыванием и осадок на фильтре промывали водой, получая 140 мг метил-2-((S)-1-((R)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбокс плата в виде белого продукта (выход: 67,0%). LC-MS: RT=1,97 мин, [М+Н]+=599,34.
Стадия Е. Синтез 2-((S)-1-((R)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (48А)
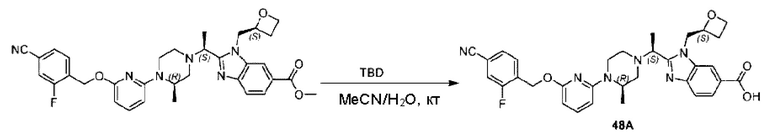
Промежуточный метил-2-((S)-1-((R)-4-(6-(((4-циано-2-фторбензил)окси)пи ридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (100 мг; 0,17 ммоль) растворяли в 5 мл раствора смеси ацетонитрил/вода (4:1) при комнатной температуре и затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (58 мг; 0,42 ммоль) при комнатной температуре. Взаимодействие проводили при комнатной температуре в течение 4 ч.
По завершении реакции реакционный раствор выливали в 15%-ный водный раствор лимонной кислоты, и значение рН устанавливалось в диапазоне 3-4. Реакционный раствор экстрагировали дихлорметаном (20 мл), 3 раза промывали водой, сушили с использованием безводного сульфата натрия и упаривали, получая бледно-желтое твердое вещество. После очистки колоночной хроматографией (дихлорметан/метанол = 10/1) получали 38 мг 2-((S)-1-((R)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (48А) в виде белого твердого вещества (выход: 38,2%). LC-MS: RT=1,86 мин, [М+Н]+=585,31.
1Н ЯМР (400 МГц, DMSO) δ 8.26 (s, 1Н), 7.87 (dd, J=10,0; 1,3 Гц, 1Н), 7.80 (dd, J=8,4; 1,4 Гц, 1Н), 7.67 (td, J=5,8; 3,0 Гц, 2Н), 7.60 (t, J=7,6 Гц, 1Н), 7.42 (t, J=8,0 Гц, 1Н), 6.20 (d, J=8,2 Гц, 1Н), 6.07 (d, J=7,8 Гц, 1Н), 5.41 (d, J=1 3,7 Гц, 1Н), 5.32 (d, J=13,7 Гц, 1Н), 5.20-5.07 (m, 1Н), 4.86-4.76 (m, 1Н), 4.71 (dd, J=1 5,5; 5,6 Гц, 1Н), 4.45 (dt, J=13,4; 7,3 Гц, 3Н), 4.17 (dt, J=9,1; 5,8 Гц, 1Н), 3.73 (d, J=11,7 Гц, 1Н), 2.82-2.56 (m, 4Н), 2.52 (d, J=9,6 Гц, 1Н), 2.32-2.22 (m, 1Н), 2.17 (dd, J=11,5; 8,3 Гц, 1Н), 1.43 (d, J=6,7 Гц, 3Н), 1.01 (d, J=6,5 Гц, 3Н).
Конкретный путь синтеза соединения 48В приведен ниже.
Стадия А. Синтез трет-бутил-(S)-4-(6-хлорпиридин-2-ил)-3-метилпиперазин-1-карбоксилата
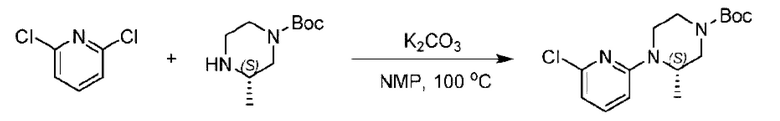
2,6-Дихлорпиридин (1,5 г; 10,00 ммоль), трет-бутил-(S)-3-метилпиперазин-1-карбоксилат (2,4 г; 12,00 ммоль) и карбонат калия (2,7 г; 20,00 ммоль) добавляли к N,N-диметилформамиду (50,0 мл) при комнатной температуре. Взаимодействие проводили при 100°С в течение 16,0 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 5/1), получая 108,0 мг трет-бутил-(S)-4-(6-хлорпиридин-2-ил)-3-метилпиперазин-1-карбоксилата в виде бледно-желтого твердого вещества (выход: 3,5%). LC-MS: RT=2,21 мин, [М-56+Н]+=256,11.
Стадия В. Синтез трет-бутил-(S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-карбоксилата
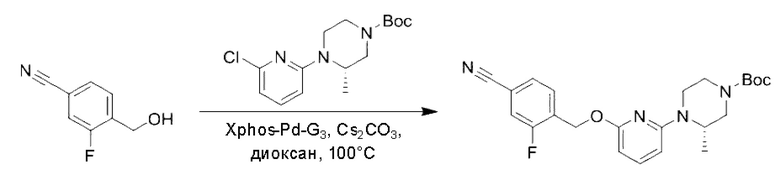
трет-Бутил-(S)-4-(6-хл орпиридин-2-ил)-3-метилпиперазин-1-карбоксилат (108,0 мг; 0,35 ммоль), 3-фтор-4-(гидроксиметил)бензонитрил (63,4 мг; 0,42 ммоль) и карбонат цезия (228,2 мг; 0,70 ммоль) добавляли к диоксану (10,0 мл) при комнатной температуре, затем добавляли метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (12,0 мг; 0,01 ммоль). Взаимодействие проводили при 100°С в течение 5,0 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали дихлорметаном (30 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), затем сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: н-гексан/этилацетат = 10:1), получая 76,5 мг трет-бутил-(S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-карбоксилата в виде коричневого маслянистого твердого вещества (выход: 51,3%). LC-MS: RT=2,28 мин, [М+Н-56]+=371,26.
Стадия С. Синтез (S)-3-фтор-4-(((6-(2-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила
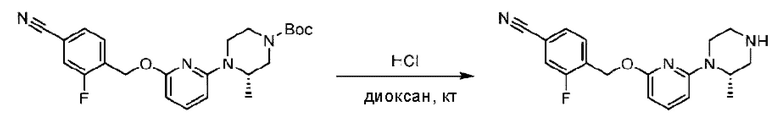
трет-Бутил-(S)-4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-карбоксилат (76,5 мг; 0,18 ммоль) добавляли к соляной кислоте в 1,4-диоксане (1,00 ммоль/мл; 5 мл) при комнатной температуре. Взаимодействие проводили при комнатной температуре в течение 1 ч в атмосфере N2.
По завершении реакции реакционный раствор концентрировали при пониженном давлении. Получали 60,0 мг (S)-3-фтор-4-(((6-(2-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрила в виде бледно-желтого твердого вещества (выход: 100,0%), которое использовали непосредственно в следующей реакции. LC-MS: RT=1,74 мин, [М+Н]+=327,14.
Стадия D. Синтез метил-2-((S)-1-((S)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-металпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата

(S)-3-Фтор-4-(((6-(2-метилпиперазин-1-ил)пиридин-2-ил)окси)метил)бензонитрил (60,0 мг; 0,18 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (55,4 мг; 0,18 ммоль), карбонат цезия (117,4 мг; 0,36 ммоль) и иодид калия (30,0 мг; 0,18 ммоль) добавляли к N;N-диметил формам иду (5,0 мл) при комнатной температуре и в конце добавляли N;N-диизопропилэтиламин (0,50 мл). Взаимодействие проводили при 50°С в течение 4,0 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), затем сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20:1), получая 43,0 мг метил-2-((S)-1-((S)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого твердого вещества (выход: 40,1%). LC-MS: RT=2,00 мин, [М+Н]+=599,37.
Стадия Е. Синтез 2-((S)-1-((S)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты (48В)
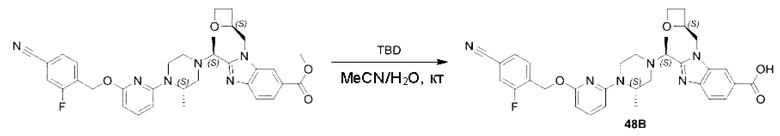
Раствор 1,5,7-триазабицикло[4.4.0]дец-5-ена (77,8 мг; 0,56 ммоль) в воде (3,0 мл) по каплям при комнатной температуре добавляли в раствор ацетонитрила (12,0 мл), содержащий метил-2-((S)-1-((S)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (43,0 мг; 0,07 ммоль). Взаимодействие проводили при комнатной температуре в течение 2,0 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и рН подводили до 6 водным раствором лимонной кислоты (0,5 моль/л). Затем экстрагировали дихлорметаном (30 мл × 3 раза), органические фазы объединяли, промывали водным раствором лимонной кислоты (30 мл × 2 раза) и сушили с использованием безводного сульфата натрия. Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией. Условия разделения приведены ниже: хроматографическая колонка: препаративная 5-С18 от Agilent, 100 мм × 30 мм, 5 мкм; подвижная фаза: вода (содержащая 0,1% аммиака) и ацетонитрил; скорость потока: 20 мл/мин; градиент: продукт элюировался 36,3%-ным ацетонитрилом в момент времени 8,23 мин; длина волны детектирования: 254 нм. После очистки и лиофилизации получали 9,0 мг 2-((S)-1-((S)-4-(6-(((4-циано-2-фторбензил)окси)пиридин-2-ил)-3-метилпиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (48В) (выход: 22,1%). LC-MS: RT=1,89 мин, [М+Н]+=585,37.
1Н ЯМР (500 МГц, DMSO) δ 8.23 (s, 1Н), 7.85 (dd, J=10,0; 1,3 Гц, 1Н), 7.79 (dd, J=8,4; 1,5 Гц, 1Н), 7.69-7.63 (m, 2Н), 7.60 (t, J=7,6 Гц, 1Н), 7.43 (t, J=8,0 Гц, 1Н), 6.21 (d, J=8,2 Гц, 1Н), 6.07 (d, J=7,8 Гц, 1Н), 5.41 (d, J=13,7 Гц, 1Н), 5.32 (d, J=13,7 Гц, 1Н), 5.1 7-5.09 (m, 1Н), 4.89 (dd, J=15,5; 2,6 Гц, 1Н), 4.71 (dd, J=15,5; 5,7 Гц, 1Н), 4.55-4.39 (m, 2Н), 4.27 (s, 1Н), 4.19 (d, J=9,1 Гц, 1Н), 3.88 (d, J=12,1 Гц, 1Н), 3.20 (s, 1Н), 3.01-2.84 (m, 2Н), 2.66-2.57 (m, 1Н), 2.32 (dd, J=31,7; 17,9 Гц, 3Н), 1.43 (d, J=6,7 Гц, 3Н), 0.77 (d, J=6,5 Гц, 3Н).
Пример 49. 2-((S)-1-(4-(4-Хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновая кислота
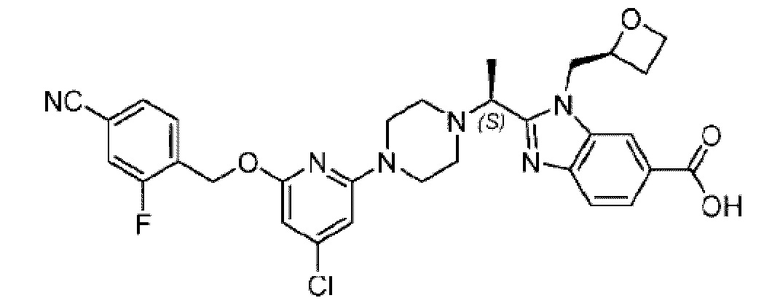
Стадия А. Синтез трет-бутил-4-(6-бром-4-хлорпиридин-2-ил)пиперазин-1-карбоксилата
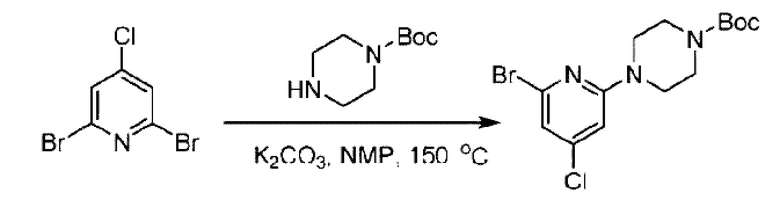
2,6-Дихлор-4-бромпиридин (200 мг; 1,1 ммоль), N-Boc-пиперазин (214 мг; 1,15 ммоль) и карбонат калия (302 мг; 2,2 ммоль) добавляли к 10 мл безводного NMP и взаимодействие проводили при температуре 150°С в течение 1 ч.
По завершении реакции реакционный раствор охлаждали до комнатной температуры и выливали в 20 мл ледяной воды. Раствор этой смеси перемешивали в течение 30 мин и затем фильтровали с отсасыванием. Осадок на фильтре промывали водой и сушили, получая 208 мг трет-бутил-4-(6-бром-4-хлорпиридин-2-ил)пиперазин-1-карбоксилата в виде белого твердого вещества (выход: 50,5%). LC-MS: RT=2,30 мин, [М-55]+=320,1 0.
Стадия В. Синтез трет-бутил-4-(4-хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата
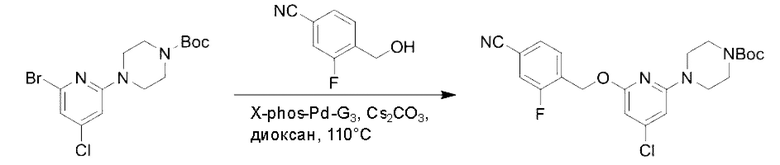
Промежуточный трет-бутил-4-(6-бром-4-хлорпиридин-2-ил)пиперазин-1-карбоксилат (168 мг; 0,45 ммоль) и 3-фтор-4-гидроксиметилбензонитрил (74,4 мг; 0,49 ммоль), метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (19 мг; 0,02 ммоль) и карбонат цезия (293,4 мг; 0,90 ммоль) добавляли к 10 мл безводного диоксана при комнатной температуре. Взаимодействие проводили при 110°С в течение 2 ч в атмосфере N2.
По завершении реакции реакционный раствор упаривали, затем экстрагировали, используя 20 мл этилацетата. Этилацетатный слой отделяли, сушили с использованием безводного сульфата натрия и упаривали, получая бледно-желтое твердое вещество. После очистки колоночной хроматографией (н-гексан/этилацетат = 5/2) получали 82 мг трет-бутил-4-(4-хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата в виде бледно-желтого твердого вещества (выход: 40,8%). LC-MS: RT=2,29 мин, [М-56+]+=391,18.
Стадия С. Синтез 4-(((4-хлор-6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)-3-фторбензонитрила
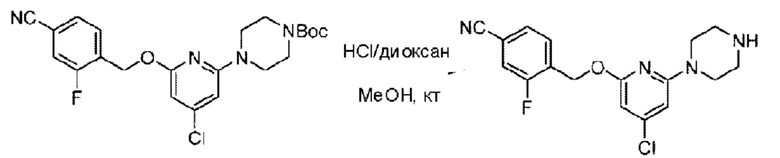
трет-Бутил-4-(4-хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилат (82 мг; 0,18 ммоль) растворяли в 10 мл метанола при комнатной температуре и добавляли 10 мл раствора хлористого водорода в диоксане с концентрацией с концентрацией 4Mb ледяной бане. Взаимодействие проводили при комнатной температуре в течение 2 ч.
По завершении реакции реакционный раствор упаривали, получая 85 мг 4-(((4-хлор-6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)-3-фторбензонитрила в виде белого твердого вещества (выход: 140,5%). LC-MS: RT=1,77 мин, [М+Н]+=347,19.
Стадия D. Синтез метил-2-((S)-1-(4-(4-хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата
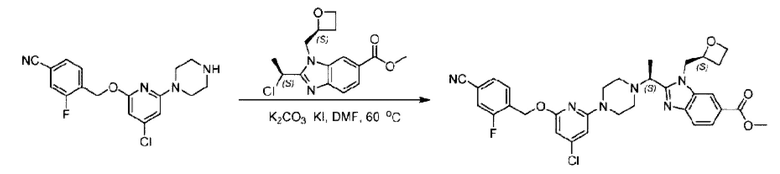
Промежуточный 4-(((4-хлор-6-(пиперазин-1-ил)пиридин-2-ил)окси)метил)-3-фторбензонитрил (85 мг; 0,19 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (59 мг; 0,19 ммоль), карбонат калия (52,4 мг; 0,38 ммоль) и иодид калия (63,0 мг; 0,38 ммоль) добавляли к 1 0 мл DMF при комнатной температуре и взаимодействие проводили при температуре 60°Св течение 10 ч.
По завершении реакции реакционный раствор выливали в ледяную воду, и белое твердое вещество выпадало в осадок. Затем смесь фильтровали с отсасыванием и осадок на фильтре промывали водой, получая 63 мг метил-2-((S)-1-(4-(4-хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилата в виде белого продукта (выход: 53,8%). LC-MS: RT=1,98 мин, [М+Н]+=619,30.
Стадия Е. Синтез 2-((S)-1-(4-(4-хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты
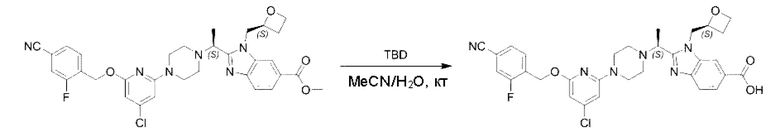
Промежуточный метил-2-((S)-1-(4-(4-хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (63 мг; 0,10 ммоль) растворяли в 5 мл раствора смеси ацетонитрил/вода (4:1) при комнатной температуре и затем добавляли 1,5,7-триазабицикло[4.4.0]дец-5-ен (34,8 мг; 0,25 ммоль) при комнатной температуре. Взаимодействие проводили при комнатной температуре в течение 4 ч.
По завершении реакции реакционный раствор выливали в 15%-ный водный раствор лимонной кислоты, и значение рН устанавливалось в диапазоне 3-4. Реакционный раствор экстрагировали дихлорметаном (20 мл), 3 раза промывали водой, сушили с использованием безводного сульфата натрия и упаривали, получая бледно-желтое твердое вещество. После очистки колоночной хроматографией (дихлорметан/метанол = 10/1) получали 30 мг 2-((S)-1-(4-(4-хлор-6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоновой кислоты в виде белого твердого вещества (выход: 49,8%).
LC-MS: RT = 1,88 мин, [М+Н]+=605,37. 1Н ЯМР (400 МГц, DMSO) δ 8.24 (s, 1Н), 7.87 (d, J=10,1 Гц, 1Н), 7.79 (dd, J=8,4; 1,3 Гц, 1Н), 7.66 (ddd, J=15,1; 9,5; 7,9 Гц, 3Н), 6.39 (s, 1Н), 6.18 (d, J=0,9 Гц, 1Н), 5.38 (s, 2Н), 5.13 (s, 1Н), 4.77 (d, J=12,8 Гц, 1Н), 4.65 (dd, J=15,5; 5,5 Гц, 1Н), 4.45 (td, J=13,4; 7,3 Гц, 2Н), 4.16 (dt, J=9,0; 5.7 Гц, 1Н), 3.41 (m, 4Н), 2.61 (dd, J=16,7; 8,7 Гц, 1Н), 2.50 (m, 4Н), 2.29 (dd, J=18,0; 7.8 Гц, 1Н), 1.42 (d, J=6,7 Гц, 3Н).
Пример 50. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота
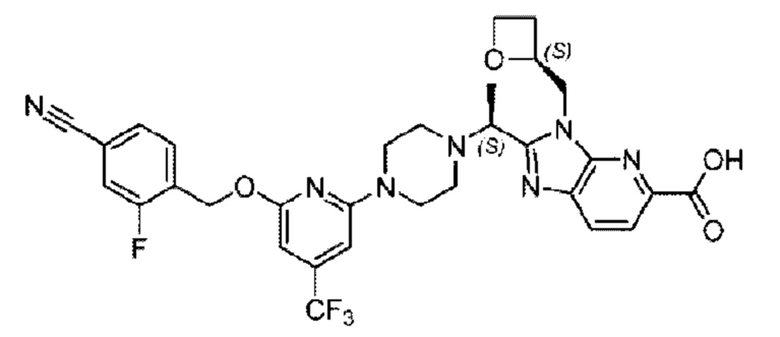
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата
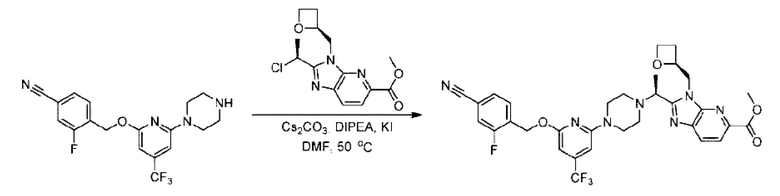
3-Фтор-4-((((6-(пиперазин-1-ил)-4-(трифторметил)пиридин-2-ил)окси)метил)бензонитрил (50,0 мг; 0,13 ммоль), метил-2-((S)-1-хлорэтил)-1-(((S)-оксетан-2-ил)метил)-1Н-бензо[d]имидазол-6-карбоксилат (44,0 мг; 0,13 ммоль), карбонат цезия (40,4 мг; 0,13 ммоль) и иодид калия (22,0 мг; 0,13 ммоль) добавляли к N;N-диметил формам иду (5,0 мл) при комнатной температуре и в конце добавляли N;N-диизопропилэтиламин (0,50 мл). Взаимодействие проводили при 50°С в течение 4,0 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, промывали насыщенным солевым раствором (30 мл × 3 раза), затем сушили с использованием безводного сульфата натрия и в конце очищали хроматографией на колонке с силикагелем (элюент: дихлорметан/метанол = 20:1), получая 37,2 мг метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата в виде желтого твердого вещества (выход: 43,7%). LC-MS: RT=1,81 мин, [М+Н]+=654,33.
Стадия В. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты
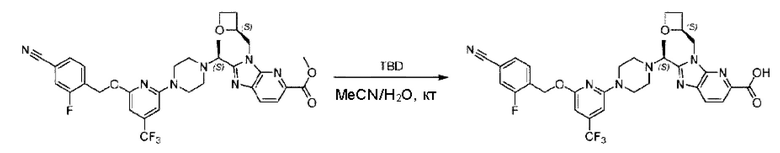
Раствор 1,5,7-триазабицикло[4.4.0]дец-5-ена (66,7 мг; 0,48 ммоль) в воде (3,0 мл) по каплям при комнатной температуре добавляли в раствор ацетонитрила (12,0 мл), содержащий метил-2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (37,2 мг; 0,06 ммоль). Взаимодействие проводили при комнатной температуре в течение 3,0 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и рН подводили до 6 водным раствором лимонной кислоты (0,5 моль/л). Затем смесь экстрагировали дихлорметаном (30 мл × 3 раза), органические фазы объединяли, промывали водным раствором лимонной кислоты (30 мл × 2 раза) и сушили с использованием безводного сульфата натрия. Неочищенный продукт очищали препаративной высокоэффективной жидкостной хроматографией. Условия разделения приведены ниже: хроматографическая колонка: препаративная 5-С18 от Agilent, 100 мм × 30 мм, 5 мкм; подвижная фаза: вода (содержащая 0,1% аммиака) и ацетонитрил; скорость потока: 20 мл/мин; градиент: продукт элюировался 33,7%-ным ацетонитрилом в момент времени 7,52 мин; длина волны детектирования: 254 нм. После очистки и лиофилизации получали 10,0 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)-4-(трифторметил)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты в виде белого твердого вещества (выход: 26,1%). LC-MS: RT=1,69 мин, [М+Н]+=640,33.
Пример 51. 2-((S)-1-(4-(6-((4-Циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота (51 А) и 2-((R)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота (51В)
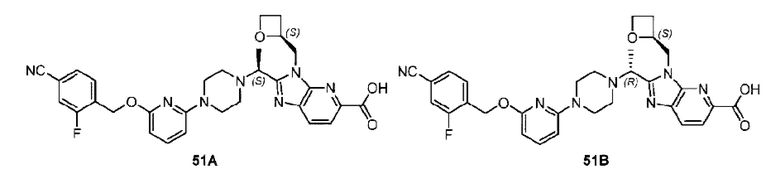
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата
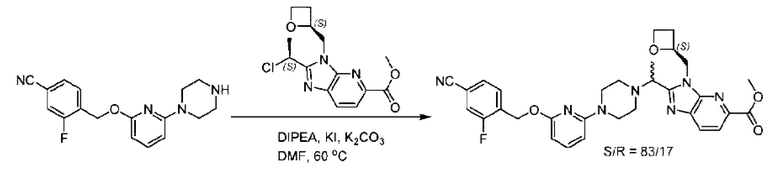
При 25°С добавляли N;N-диизопропилэтиламин (1,0 мл), иодид калия (420 мг; 3,13 ммоль) и карбонат калия (577 мг; 4,18 ммоль) в раствор ацетонитрила (8,0 мл), содержащий 4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазина гидрохлорид (652 мг; 2,09 ммоль), и затем добавляли N,N-диметилформамид (10 мл). В конце добавляли метил-2-((S)-1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (650 мг; 2,09 ммоль). Взаимодействие проводили при 60°С в течение 3,0 ч в атмосфере N2.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (30 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 2/1). Получали 690 мг метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата в виде желтого твердого вещества (выход: 56,4%, dr = 66%). LC-MS: RT=1,93 мин, [М+Н]+=586,26.
Стадия В. Синтез 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты (51А) и 2-(R)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты (51В)
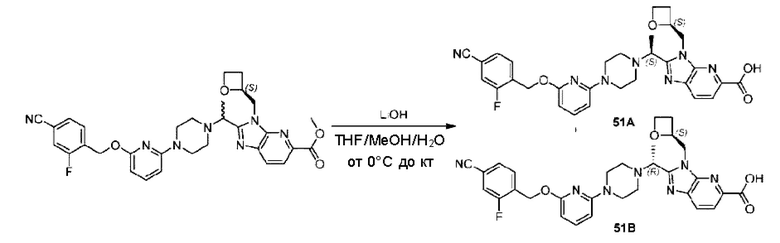
Раствор гидроксида лития (198 мг; 4,72 ммоль) в воде (1 мл) по каплям при 0°С добавляли в раствор смеси тетрагидрофурана (3 мл) и метанола (1 мл), содержащий метил-2-(1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (690 мг; 1,18 ммоль), и взаимодействие проводили при 25°С в течение 30 мин.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 1/10). Получали 290 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты (51А) в виде белого твердого вещества (выход: 43,0%) и 59 мг 2-((S)-1-(4-(6-((4-циано-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты (51В) в виде белого твердого вещества (выход: 8,7%). Неочищенные продукты очищали препаративной высокоэффективной жидкостной хроматографией. Условия разделения приведены ниже: хроматографическая колонка: препаративная 5-С18 от Agilent, 100 мм × 30 мм, 5 мкм; подвижная фаза: элюировали водой (содержащей 0,1% аммиака) и ацетонитрилом; скорость потока: 20 мл/мин; градиент: соединения 51А и 51В элюировались 21%-ным ацетонитрилом в моменты времени 10,12 мин и 12,17 мин; соответственно; длина волны детектирования: 254 нм.
Соединение 51А: HPLC: RT = 10,12 мин. LC-MS: RT = 1,79 мин, [М+Н]+=572,30. 1Н ЯМР(400 МГц, DMSO-d6) δ 8.14 (d, J=8,2 Гц, 1Н), 7.97 (d, J=8,2 Гц, 1Н), 7.87 (dd, J=10,0; 1,5 Гц, 1Н), 7.69 (dd, J=7,9; 1,4 Гц, 1Н), 7.64 (t, J=7,6 Гц, 1Н), 7.45 (t, J=8,0 Гц, 1Н), 6.30 (d, J=8,1 Гц, 1Н), 6.10 (d, J=7,8 Гц, 1Н), 5.38 (s, 2Н), 5.30-5.24 (m, 1 H), 4.82-4.70 (m, 2H), 4.63-4.58 (m, 1 H), 4.44-4.39 (m, 1 H), 4.11-4.06 (m, 1H), 3.43-3.33 (m, 4H), 2.64-2.58 (m, 1 H), 2.56-2.52 (m, 4H), 2.39-2.30 (m, 1H), 1.45 (d, J=6,7 Гц, 3Н).
Соединение 51В: HPLC: RT = 12,17 мин. LC-MS: RT = 1,79 мин, [М+Н]+=572,30. 1Н ЯМР (400 МГц, DMSO) δ 8.09 (d, J=8,2 Гц, 1Н), 7.95 (d, J=8,2 Гц, 1Н), 7.87 (dd, J=10,0; 1,4 Гц, 1Н), 7.69 (dd, J=7,9; 1,5 Гц, 1Н), 7.67-7.61 (m, 1Н), 7.46 (t, J=8,0 Гц, 1Н), 6.31 (d, J=8,1 Гц, 1Н), 6.11 (d, J=7,8 Гц, 1Н), 5.39 (s, 2Н), 5.07-5.00 (m, 2Н), 4.69-4.49 (m, 4Н), 3.47-3.37 (m, 4Н), 2.79-2.70 (m, 1Н), 2.66-2.52 (m, 5Н), 1.42 (d, J=6,8 Гц, 3Н).
Пример 52. 2-((S)-1-(4-(6-((4-Хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота (52А) и 2-((R)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновая кислота (52В)
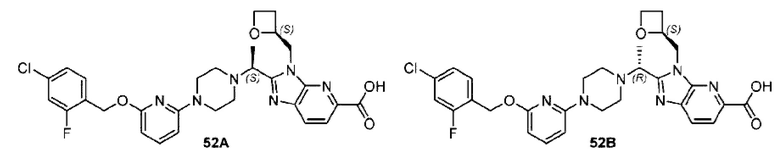
Конкретный путь синтеза приведен ниже.
Стадия А. Синтез 2-хлор-6-((4-хлор-2-фторбензил)окси)пиридина
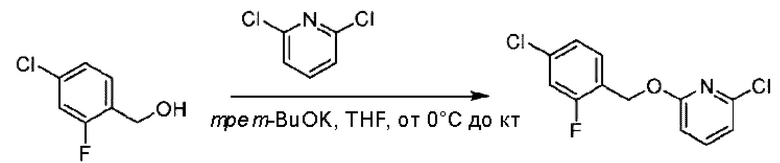
трет-Бутилат калия (4,88 г; 43,60 ммоль) добавляли при 0°С в раствор тетрагидрофурана (50,0 мл), содержащий (4-хлор-2-фторфенил)метанол (5,0 г; 31,14 ммоль), и раствор этой смеси перемешивали в течение 30 мин. Затем добавляли 2,6-дихлорпиридин (4,2 г; 28,38 ммоль) и реакцию проводили в течение ночи при комнатной температуре.
По завершении реакции реакционный раствор гасили путем добавления воды (50 мл) и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 7,53 г 2-хлор-6-((4-хлор-2-фторбензил)окси)пиридина в виде желтого маслянистого продукта (выход: 88,9%). LC-MS: RT=2,27 мин, [М+Н]+=272,05.
Стадия В. Синтез трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата
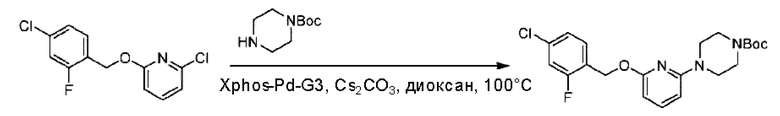
трет-Бутилпиперазин-1-карбоксилат (5,15 г; 27,68 ммоль), карбонат цезия (18,05 г; 55,36 ммоль) и метансульфонато(2-дициклогексилфосфино-2',4',6'-триизопропил-1,1'-бифенил)(2'-амино-1,1'-бифенил-2-ил)палладий(II) (702,5 мг; 0,83 ммоль) добавляли при комнатной температуре в раствор 1,4-диоксана, содержащий 2-хлор-6-((4-хлор-2-фторбензил)окси)пиридин (7,53 г; 27,68 ммоль), и взаимодействие проводили при 100°С в течение 3 ч.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (50 мл × 3 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (50 мл × 2 раза), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 1/2). Получали 4,7 г трет-бутил-4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилата в виде белого твердого вещества (выход: 40,2%). LC-MS: RT=2,39 мин, [М+Н]+=422,22.
Стадия С. Синтез 4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазина
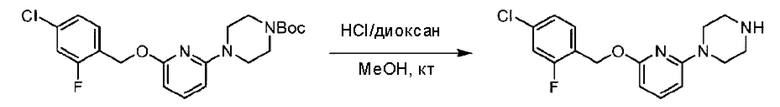
Соляную кислоту в диоксане (20,0 мл; 4 моль/л) добавляли при комнатной температуре в раствор метанола (30 мл), содержащий трет-бутил-4-(6-(((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-карбоксилат (4,34 г; 10,28 ммоль), и взаимодействие проводили при комнатной температуре в течение 3 ч.
По завершении реакции реакционный раствор незамедлительно упаривали на роторном испарителе, получая 3,68 г 4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазина гидрохлорида в виде белого твердого вещества, которое использовали непосредственно на следующей стадии. LC-MS: RT=1,73 мин, [М+Н]+=322,15.
Стадия D. Синтез метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата
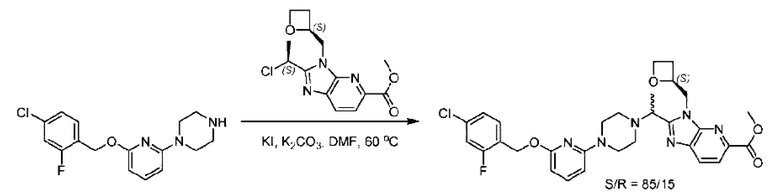
Метил-2-(1-хлорэтил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (650 мг; 2,09 ммоль), иодид калия (521 мг; 3,14 ммоль) и карбонат калия (577 мг; 4,18 ммоль) добавляли при комнатной температуре в раствор ацетонитрила (8,0 мл), содержащий 4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазина гидрохлорид (750 мг; 2,09 ммоль), и реакцию проводили в течение ночи при 60°С.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (30 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: этилацетат/н-гексан = 2/1). Получали 870 мг метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилата в виде желтого твердого вещества (выход: 70,0%). LC-MS: RT=1,93 мин, [М+Н]+=595,25.
Стадия Е. Синтез 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты (52А) и 2-((R)-1-(4-(6-((4-xnop-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты (52В)
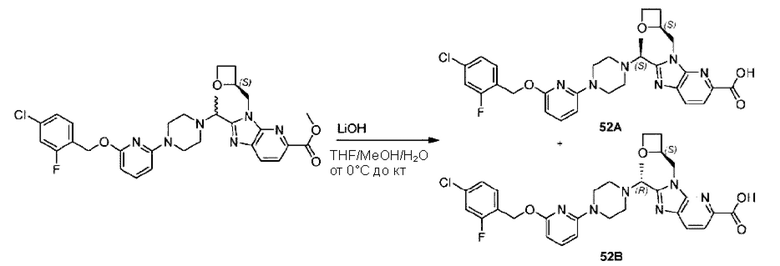
Раствор гидроксида лития (307 мг; 7,30 ммоль) в воде (1 мл) по каплям при 0°С добавляли в раствор смеси тетрагидрофурана (3 мл) и метанола (1 мл), содержащий метил-2-(1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоксилат (870 мг; 1,46 ммоль), и взаимодействие проводили при комнатной температуре в течение 30 мин.
По завершении реакции реакционный раствор гасили путем добавления воды и экстрагировали этилацетатом (30 мл × 2 раза). Органические фазы объединяли, затем промывали насыщенным солевым раствором (20 мл), сушили с использованием безводного сульфата натрия и концентрировали при пониженном давлении. Остаток очищали хроматографией на колонке с силикагелем (элюент: метанол/дихлорметан = 1/10). Получали 300 мг 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты (52А) в виде белого твердого вещества (выход: 35,4%) и 53 мг 2-((S)-1-(4-(6-((4-хлор-2-фторбензил)окси)пиридин-2-ил)пиперазин-1-ил)этил)-3-(((S)-оксетан-2-ил)метил)-3Н-имидазо[4,5-b]пиридин-5-карбоновой кислоты (52В) в виде белого твердого вещества (выход: 6,2%). Неочищенные продукты очищали препаративной высокоэффективной жидкостной хроматографией. Условия разделения приведены ниже: хроматографическая колонка: препаративная 5-С18 от Agilent, 100 мм × 30 мм, 5 мкм; подвижная фаза: элюировали водой (содержащей 0,1% аммиака) и ацетонитрилом; скорость потока: 20 мл/мин; градиент: соединения 52А и 52В элюировались 25%-ным ацетонитрилом в моменты времени 14,09 мин и 15,49 мин, соответственно; длина волны детектирования: 254 нм.
Соединение 52А: HPLC: RT = 14,09 мин. LC-MS: RT = 1,91 мин, [М+Н]+=581,25. 1Н ЯМР (400 МГц, DMSO) δ 8.14 (d, J=8,2 Гц, 1Н), 7.97 (d, J=8,2 Гц, 1Н), 7.49 (t, J=8,2 Гц, 1Н), 7.46-7.41 (m, 2Н), 7.28 (dd, J=8,2; 2,0 Гц, 1Н), 6.29 (d, J=8,1 Гц, 1Н),6.07 (d, J=7,8 Гц, 1Н), 5.28 (s, 2Н), 4.83-4.70 (m,2H), 4.62 (q, J=6,5 Гц, 1H), 4.44-4.39 (m, 1Н), 4.11-4.06 (m, 1Н), 3.42-3.19 (m, 5Н), 2.64-2.55 (m, 5Н), 2.39-2.32 (m, 1 И), 1.47 (d, J=6,8 Гц,3Н).
Соединение 52В: HPLC: RT=15,49 мин. LC-MS: RT=1,91 мин, [М+Н]+=581,25. 1Н ЯМР (400 МГц, DM SO) 5 8.03 (d, J=8,1 Гц, 1H), 7.92 (d, J=8,2 Гц, 1 H), 7.51 (d, J=8,0 Гц, 1H), 7.49-7.41 (m,2H), 7.28 (dd, J=8,2; 2,1 Гц, 1H), 6.31 (d, J=8,1 Гц, 1H), 6.07 (d, J=7,8 Гц, 1H), 5.29 (s, 2H), 5.04-5.00 (m, 2H), 4.69-4.50 (m, 3Н), 3.49-3.42 (m, 3Н), 3.29-3.19 (m, 2H), 2.79-2.71 (m, 1H), 2.67-2.55 (m, 4Н), 1.92-1.86 (m, 1H), 1.42 (d, J=6,6 Гц, 3Н).
Пример 53. Измерение in vitro уровня циклоаденозинмонофосфата (цАМФ) в качестве сигнала активации соединениями в клетках человека, экспрессирующих GLP1R
1. Культура клеток
В этом эксперименте использовали линию клеток, стабильно экспрессирующих GLP1R человека (hGLP1 R-U2OS). Клетки культивировали в ростовой среде (полной среде, DMEM (модифицированная Дульбекко среда Игла) + 10% фетальной телячьей сыворотки (FBS) + 500 мкг 3418/мл (Promega)) в инкубаторе при 37°С, 5% CO2 и насыщенной влагой атмосфере.
2. Определение цАМФ
Клетками hGLP1R-U2OS инокулировали лунки 384-луночного планшета в количестве 1,0×104 клеток/лунка (5 мкл) (рабочий раствор для клеток: DM ЕМ + 0,1% бычьего сывороточного альбумина (BSA) + 1 мМ IBMX (3-изобутил-1-метил-ксантин; поставляемый Bide Pharmatech Ltd.)), планшет помещали в инкубатор при 37°С и выдерживали в течение 30 мин. Предназначенные для тестирования соединения поэтапно разбавляли буфером 1 для стимуляции (1Х) из набора и разбавленные соединения добавляли в планшет с культурой в количестве 5 мкл/лунка. Инкубирование проводили в инкубаторе при 37°С в течение 30 мин. Затем последовательно добавляли раствор цАМФ-d2, приготовленный в буфере для лизиса и детектирования, в количестве 5 мкл/лунка и рабочий раствор антитела к цАМФ, меченного криптатом европия, в количестве 5 мкл/лунка и проводили инкубирование при комнатной температуре в течение 60 мин, после чего выполняли детектирование.
3. Обработка данных
Отношение сигнала, испускаемого донором, к сигналу, испускаемому акцептором, в каждой лунке рассчитывали так, как приведено ниже: соотношение сигналов гомогенной флуоресценции с разрешением по времени (HTRF) = (сигнал при 665 нм/сигнал при 620 нм) × 104, и затем HTRF соотношение преобразовывали в степень прохождения реакции (%). Кривую зависимости ответа от концентрации для каждого соединения из примеров строили, используя 4-параметрическое уравнение в виде Y = Bottom + (Тор - Bottom)/(1+10∧((LogIC50 - X)*Hill slope)), и рассчитывали полумаксимальную (50%) эффективную концентрацию (ЕС50). Результаты показаны в Таблице 1.
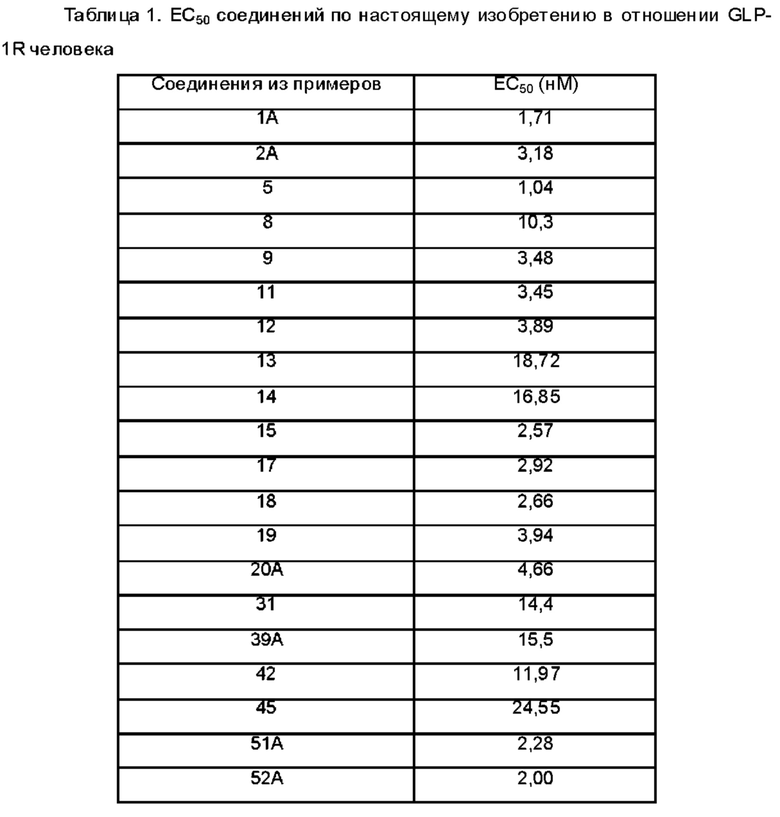
Заключение: соединения по настоящему изобретению обладают значительной активирующей активностью в отношении GLP-1R человека.
Пример 54. Фармакокинетическое изучение соединений по настоящему изобретению на крысах
1. Экспериментальные материалы
Крысы Sprague Daw ley (SD): самцы, 180-250 г; приобретены у Beijing Vital River Laboratory Animal Technology Co., Ltd.
Реагенты: DMSO (диметилсульфоксид), ПЭГ-400 (полиэтиленгликоль 400), физиологический раствор, гепарин, ацетонитрил, муравьиная кислота и пропранолол (внутренний стандарт), и все они имеются в продаже.
Приборы: Thermo Fisher Scientific LC-MS (U300 UPLC (сверхэффективная жидкостная хроматография), тройной квадрупольный масс-спектрометр TSQ QUANTUMN ULTRA).
2. Методика эксперимента
Отвешивали навеску соединения и растворяли в системе DMSO-n3l~400-физиологический раствор (5:60:35, об./об./об.). После этого крысам вводили соединение внутривенно (в.в.) или через желудочный зонд, отбирали по 200 мкл образцов венозной крови в моменты времени 15 мин, 30 мин, 1 ч, 2 ч, 5 ч, 7 ч и 24 ч (для группы с в.в. введением выполняли дополнительный отбор в момент времени 5 мин) в пробирки с гепарином типа эппендорф (ЕР), соответственно, которые центрифугировали при 12000 об./мин в течение 2 мин, плазму крови замораживали при -80°С для дальнейшего тестирования. Отвешивали точную навеску определенного количества тестируемого вещества и растворяли в DMSO, получая концентрированный раствор с концентрацией 2 мг/мл. Соответствующее количество концентрированного раствора соединения аккуратно отбирали пипеткой и разбавляли ацетонитрилом для приготовления стандартного серийного раствора. По 20 мкл каждого из указанных выше стандартных серийных растворов аккуратно отбирали пипеткой, добавляли по 180 мкл плазмы крови в качестве холостой пробы и перемешивали с использованием вортекса для приготовления образцов плазмы крови, соответствующих концентрациям в плазме крови, равным 0,3, 1, 3, 10, 30, 100, 300, 1000, 3000 нг/мл. Для построения стандартной кривой для каждой концентрации проводили анализ двух образцов. Отбирали по 30 мкл плазмы крови (для образцов, которые получали в моменты времени 5 мин, 15 мин, 30 мин и 1 ч после внутривенного введения, плазму крови разбавляли в 10 раз), добавляли по 200 мкл раствора пропранолола в ацетонитриле (50 нг/мл) в качестве внутреннего стандарта, перемешивали с использованием вортекса. Затем к смеси добавляли 100 мкл чистой воды и перемешивали с использованием вортекса и еще раз перемешивали. Смесь центрифугировали при 4000 об./мин в течение 5 мин и отбирали супернатант для проведения анализа посредством LC-MS. Условия детектирования с использованием LC-MS приведены ниже.
Хроматографическая колонка: колонка для UPLC HYPERSIL GOLD С-18, 100 мм × 2,1 мм, 1,7 мкм, от Thermo Scientific.
Подвижная фаза: вода (с 0,1% муравьиной кислот и)-ацетонитрил; градиентное элюирование проводили согласно приведенной ниже таблице.
3. Обработка данных
После определения концентраций лекарственных средств в крови с использованием LC-MS для расчета фармакокинетических параметров применяли программное обеспечение WinNonlin 6.1, основанное на некомпартнентной модели. Результаты показаны в Таблице 2.
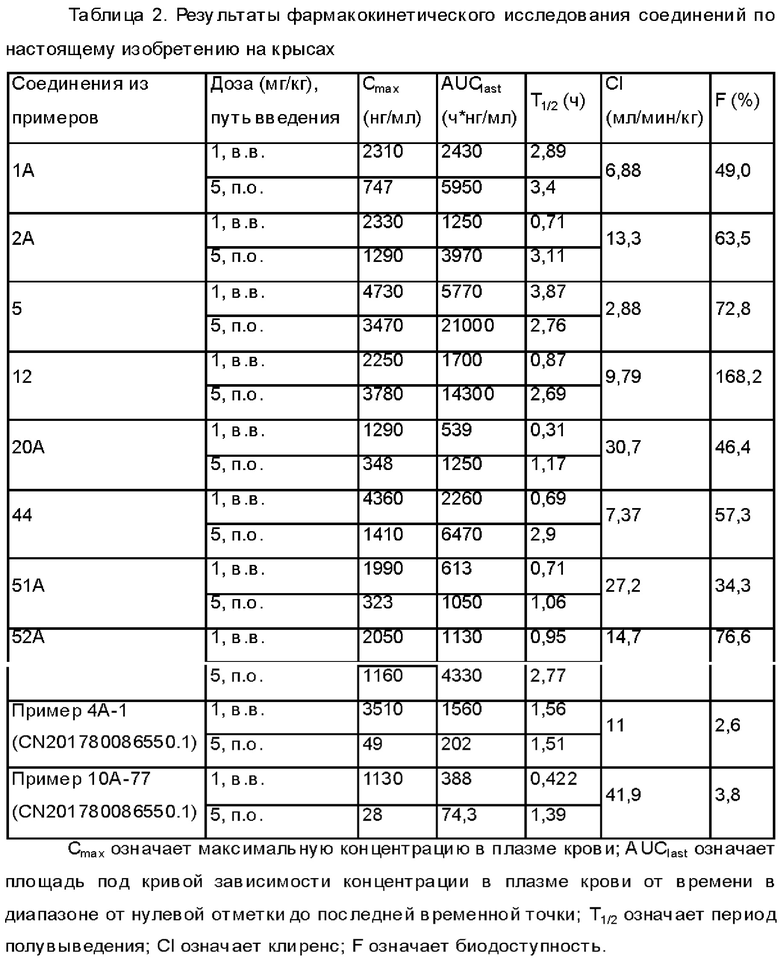
Заключение: на основании данных Таблицы 2 можно видеть, что соединения по настоящему изобретению характеризуются улучшенным пероральным (п.о.) всасыванием у крыс и обладают улучшенными экспозицией и биодоступностью.
Пример 55. Исследование влияния соединений по настоящему изобретению на калиевый ионный канал - калиевый канал человека (hERG)
1. Культура клеток
В этом эксперименте использовали линию клеток CHO-hERG из яичника китайского хомячка (СНО), которые стабильно экспрессируют калиевый канал hERG. Клетки культивировали в полной среде F12, содержащей 10% FBS, 1% антибиотиков (G41 8) и гигромицин в концентрации 89 мкг/мл. В качестве среды для восстановления использовали среду F12, содержащую 10% FBS. Культивирование проводили при 37°С и 5% CO2.
2. Определение hERG
Ингибирующую активность соединений по настоящему изобретению в сериях изменяющихся концентраций в отношении калиевого канала в CHO-hERG определяли с использованием ручного варианта системы пэтч-кламп.
3. Обработка данных
Текущее значение для hERG импортируется в EXCEL для анализа данных, и измеренные значения IC50 показаны в Таблице 3.

Заключение: на основании данных Таблицы 3 можно видеть, что соединения по настоящему изобретению оказывают слабое ингибирующее действие на hERG. Кроме того, значения IC50 для некоторых соединений, протестированных согласно приведенному выше методу эксперимента, даже превышают 100 мкМ, что указывает на то, что связанные с сердечно-сосудистой системой побочные эффекты, вызываемые действием hERG, могут быть значительно снижены.
Вышеупомянутые примеры представляют собой предпочтительные примеры настоящего изобретения. Однако, воплощения настоящего изобретения не ограничиваются вышеупомянутыми примерами, и любые другие изменения, модификации, замены, комбинации и упрощения, которые не отклоняются от сущности и принципа настоящего изобретения, следует рассматривать как эквивалентные замены, и все они включены в объем правовой охраны настоящего изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПИПЕРИДИНИЛПИРАЗОЛОПИРИМИДИНОНЫ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2713937C2 |
| АГОНИСТ РЕЦЕПТОРА GLP-1, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ЕГО, И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 2021 |
|
RU2800290C1 |
| АЗОТСОДЕРЖАЩИЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ В ЛЕКАРСТВЕННЫХ ПРЕПАРАТАХ | 2014 |
|
RU2694254C1 |
| МОДУЛЯТОРЫ ЯДЕРНЫХ РЕЦЕПТОРОВ | 2016 |
|
RU2715897C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2015 |
|
RU2667498C2 |
| АГОНИСТЫ РЕЦЕПТОРА GLP-1 И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2740135C1 |
| ПРОИЗВОДНЫЕ ЦИКЛИЧЕСКИХ АМИНОВ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА ЕР4 | 2011 |
|
RU2565596C2 |
| АГОНИСТЫ GLP-1 РЕЦЕПТОРА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2765721C1 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| БИЦИКЛИЧЕСКИЕ АРИЛЬНЫЕ МОНОБАКТАМОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2017 |
|
RU2733402C2 |
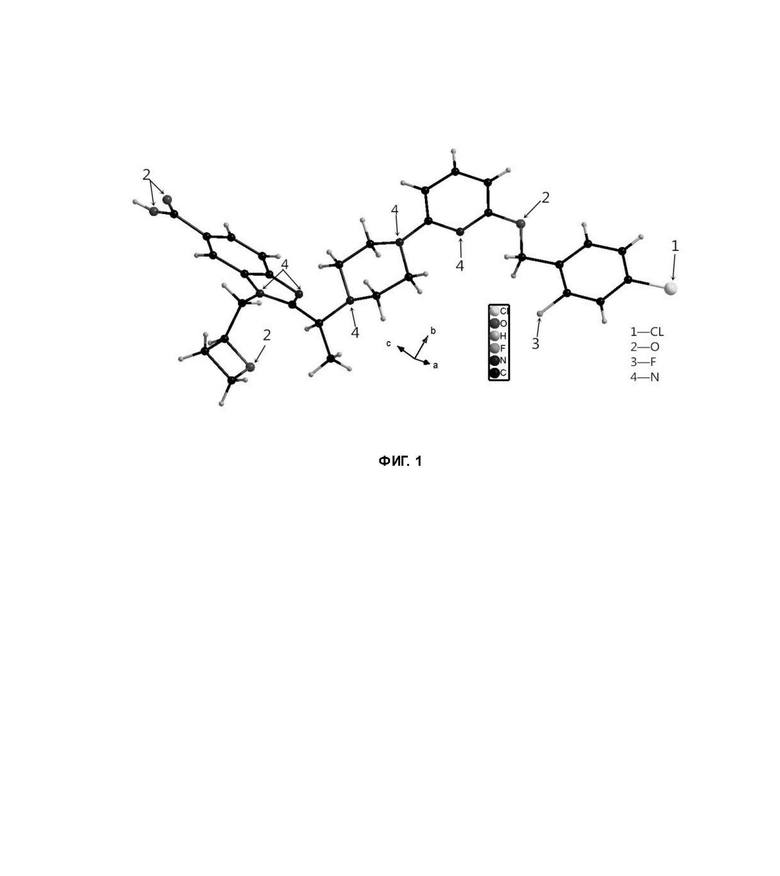
Изобретение относится к соединению формулы (I) или его стереоизомеру:
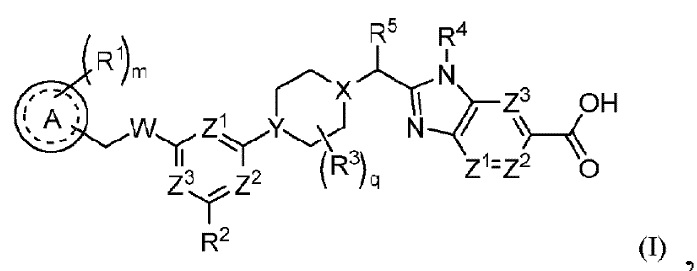
где A выбран из группы, состоящей из фенильного кольца,  ,
,  ,
,
 ,
,  ,
, и
и  ; R1 выбран из группы, состоящей из атома водорода, фтора, хлора, брома, циано, дифторэтила и трифторэтила; m равно 0, 1 или 2; W представляет собой O; R2 выбран из группы, состоящей из атома водорода, хлора, циано, трифторметила, метокси и метоксиметила; R3 выбран из группы, состоящей из атома фтора, гидроксила, оксо (=O), метила, фторметила и метоксиметила; q равно 0 или 1; X представляет собой CH или N; Y представляет собой CH или N; R5 представляет собой метил; R4 представляет собой оксетан-2-илметил; в
; R1 выбран из группы, состоящей из атома водорода, фтора, хлора, брома, циано, дифторэтила и трифторэтила; m равно 0, 1 или 2; W представляет собой O; R2 выбран из группы, состоящей из атома водорода, хлора, циано, трифторметила, метокси и метоксиметила; R3 выбран из группы, состоящей из атома фтора, гидроксила, оксо (=O), метила, фторметила и метоксиметила; q равно 0 или 1; X представляет собой CH или N; Y представляет собой CH или N; R5 представляет собой метил; R4 представляет собой оксетан-2-илметил; в 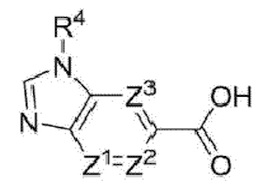 Z1 представляет собой CH; Z2 представляет собой CH; Z3 представляет собой CH или N; и в
Z1 представляет собой CH; Z2 представляет собой CH; Z3 представляет собой CH или N; и в 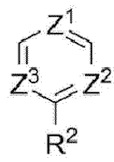 Z1 представляет собой N, Z2 представляет собой CH, или N, или CF, а Z3 представляет собой CH или CF, а также к фармацевтической композиции и применению соединения формулы (I) для изготовления лекарственного средства для лечения заболеваний, связанных с GLP-1. Технический результат: получены новые соединения, которые могут быть применимы для изготовления лекарственного средства для лечения GLP-1-ассоциируемых заболеваний, особенно связанных с сахарным диабетом. 3 н. и 6 з.п. ф-лы, 3 табл., 55 пр., 1 ил.
Z1 представляет собой N, Z2 представляет собой CH, или N, или CF, а Z3 представляет собой CH или CF, а также к фармацевтической композиции и применению соединения формулы (I) для изготовления лекарственного средства для лечения заболеваний, связанных с GLP-1. Технический результат: получены новые соединения, которые могут быть применимы для изготовления лекарственного средства для лечения GLP-1-ассоциируемых заболеваний, особенно связанных с сахарным диабетом. 3 н. и 6 з.п. ф-лы, 3 табл., 55 пр., 1 ил.
1. Соединение формулы (I)
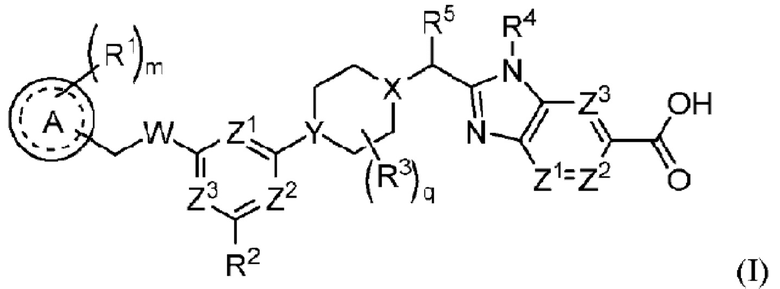
или его стереоизомер,
где А выбран из группы, состоящей из фенильного кольца,  ,
, 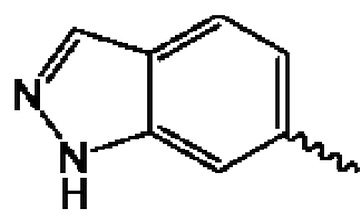 ,
, 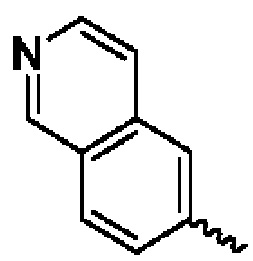 ,
, 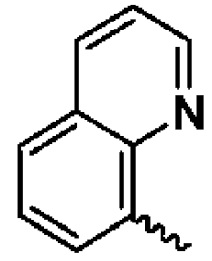 ,
, 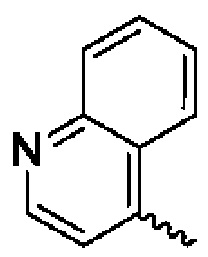 и
и  ;
;
R1 выбран из группы, состоящей из атома водорода, фтора, хлора, брома, циано, дифторэтила и трифторэтила;
m равно 0, 1 или 2;
W представляет собой О;
R2 выбран из группы, состоящей из атома водорода, хлора, циано, трифторметила, метокси и метоксиметила;
R3 выбран из группы, состоящей из атома фтора, гидроксила, оксо (=O), метила, фторметила и метоксиметила;
q равно 0 или 1;
X представляет собой СН или N;
Y представляет собой СН или N;
R5 представляет собой метил;
R4 представляет собой оксетан-2-илметил;
в 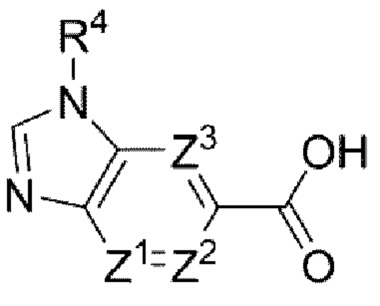 Z1 представляет собой СН; Z2 представляет собой СН; Z3 представляет собой СН или N; и
Z1 представляет собой СН; Z2 представляет собой СН; Z3 представляет собой СН или N; и
в 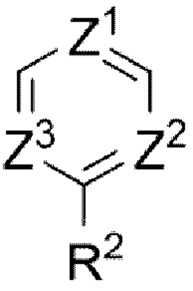 Z1 представляет собой N, Z2 представляет собой СН, или N, или CF, a Z3 представляет собой СН или CF.
Z1 представляет собой N, Z2 представляет собой СН, или N, или CF, a Z3 представляет собой СН или CF.
2. Соединение по п. 1 или его стереоизомер, где, когда атом углерода, соединенный с R5, представляет собой хиральный атом углерода, данный хиральный атом углерода находится в S-конфигурации и/или R-конфигурации.
3. Соединение по п. 2 или его стереоизомер, где, когда атом углерода, соединенный с R5, представляет собой хиральный атом углерода, данный хиральный атом углерода находится в S-конфигурации.
4. Соединение по п. 1 или его стереоизомер, где указанное соединение выбрано из группы, состоящей из:
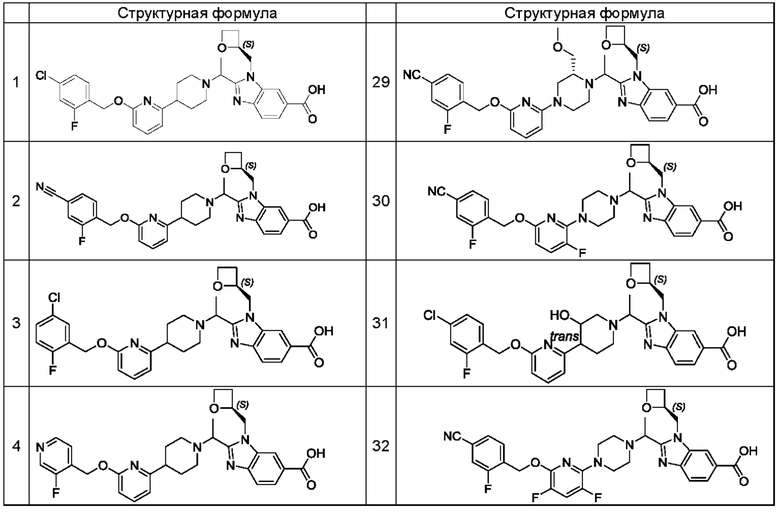
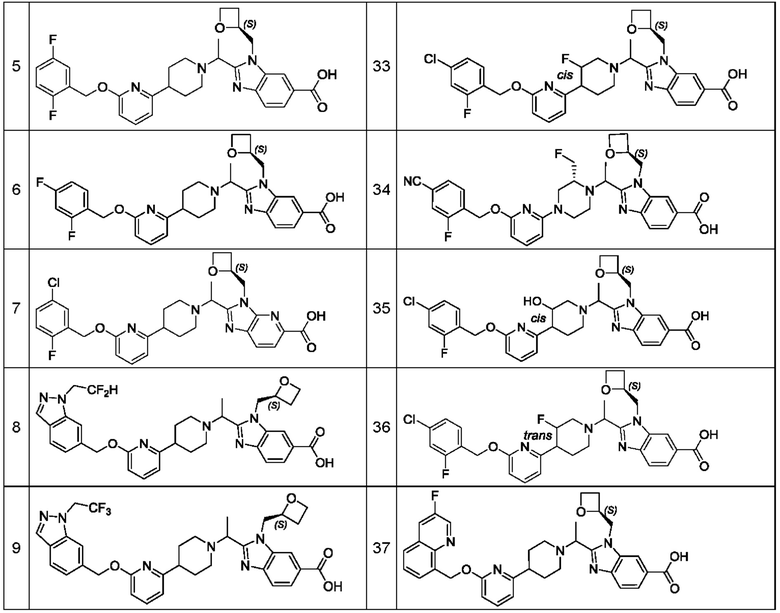
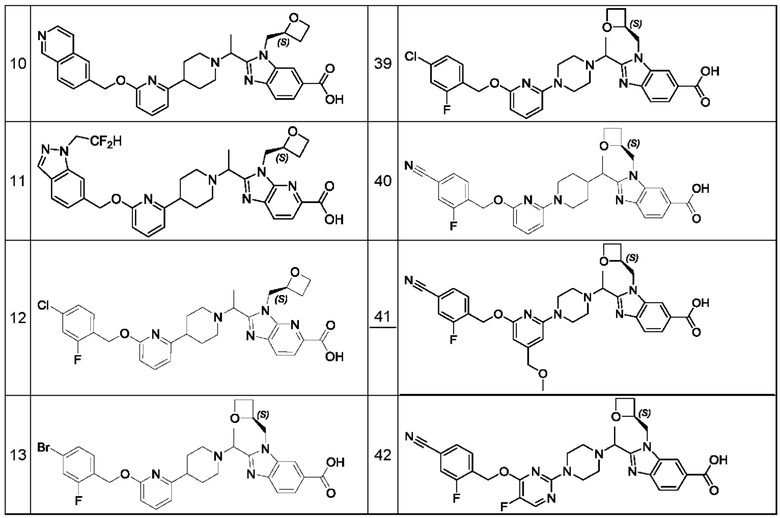
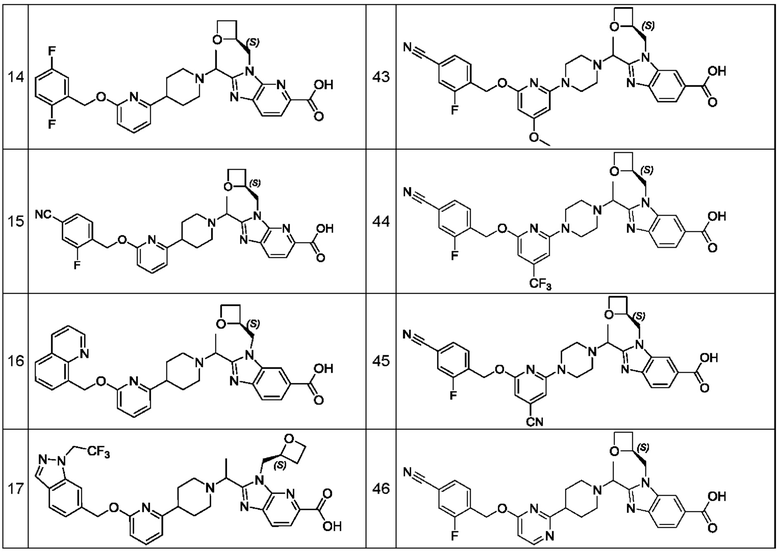
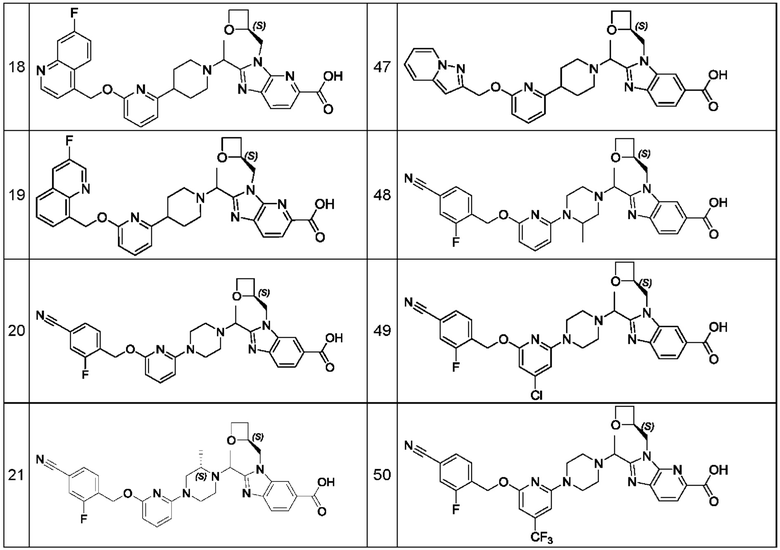
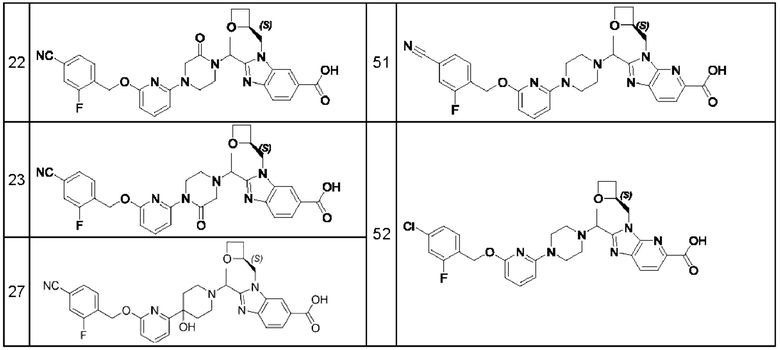
5. Соединение по п. 1 или его стереоизомер, где указанное соединение выбрано из группы, состоящей из:
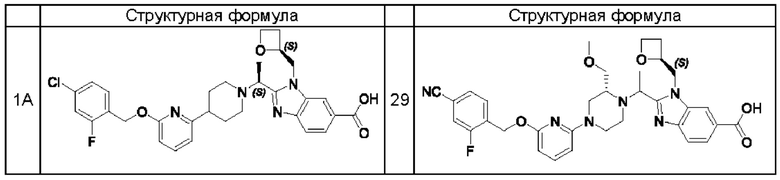
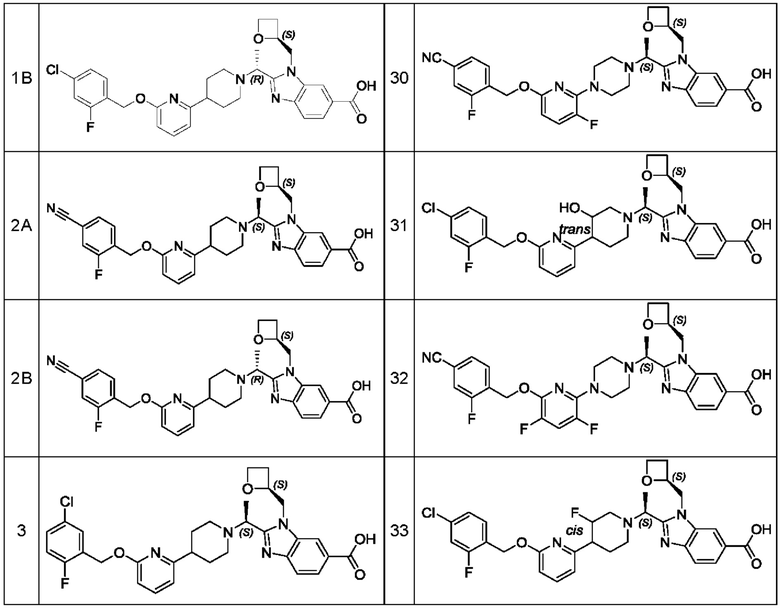
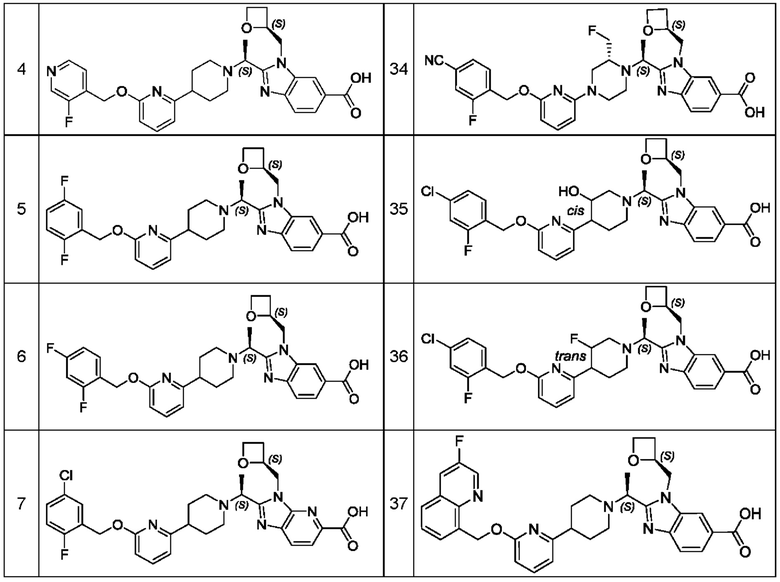
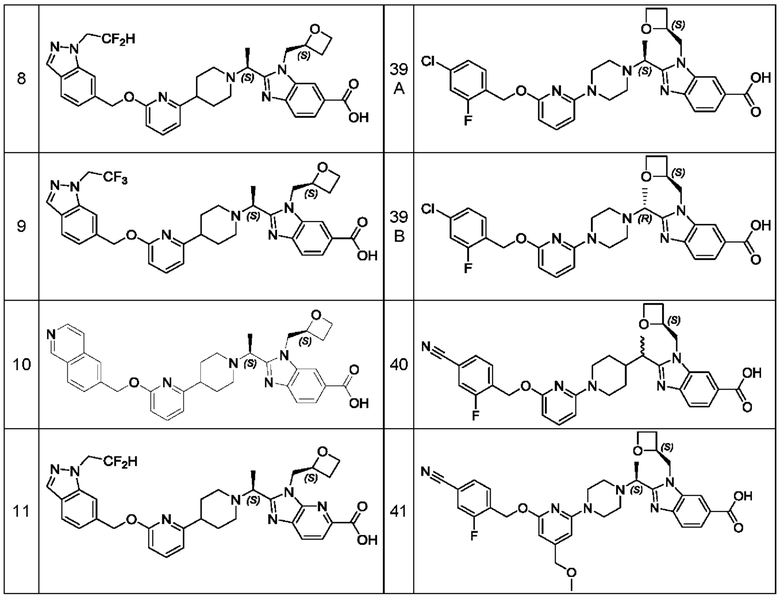
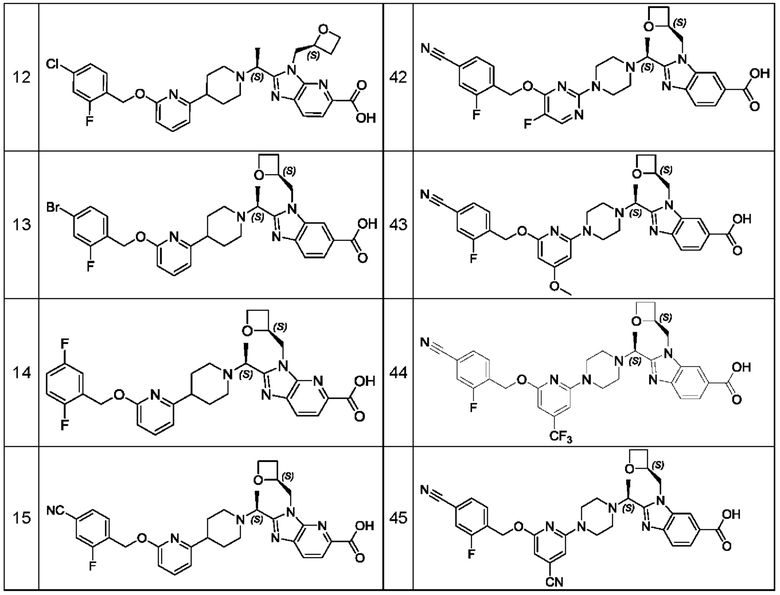
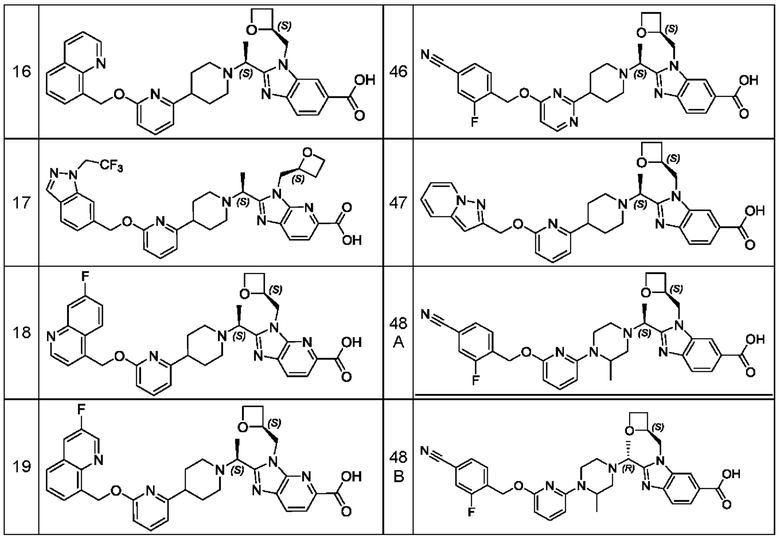
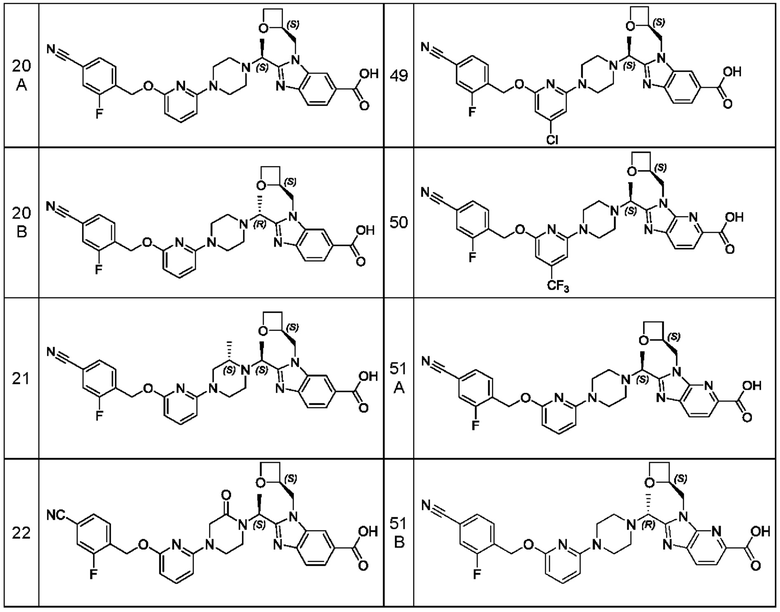
6. Соединение по любому из пп. 1-3 или его стереоизомер, где фармацевтически приемлемая соль относится к соли, которая получена из указанного соединения и фармацевтически приемлемых кислоты или основания.
7. Фармацевтическая композиция для лечения заболеваний, связанных с глюкагоноподобным пептидом-1 (GLP-1), содержащая эффективное количество соединения по любому из пп. 1-6 или его стереоизомера и один или более чем один фармацевтически приемлемый носитель.
8. Применение соединения по любому из пп. 1-6 или его стереоизомера для изготовления лекарственного средства для лечения заболеваний человека, связанных с GLP-1.
9. Применение по п. 8, где заболевания, связанные с GLP-1, представляют собой заболевания, связанные с сахарным диабетом.
| WO 2020103815 A1, 28.05.2020 | |||
| WO 2020207474 A1, 15.10.2020 | |||
| WO 2019239371 A1, 19.12.2019 | |||
| CN 110325530 A, 11.10.2019 | |||
| EA 201990518 A1, 30.12.2019. |

