ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к синергической комбинации ингибитора активности CXCL8 (интерлейкина-8) и кортикостероида и ее применению в терапии. Более конкретно, настоящее изобретение относится к применению синергической комбинации ингибитора активности CXCL8, такого как ладариксин или DF2755, и кортикостероида, такого как дексаметазон, для лечения астмы, резистентной к кортикостероидам.
УРОВЕНЬ ТЕХНИКИ
Астма представляет собой хроническое воспалительное аллергическое заболевание дыхательных путей, которое вызывает переменную обструкцию дыхательных путей в сочетании с гиперреактивностью дыхательных путей (AHR) и перепроизводством слизи (Fanta CH. Asthma. N Engl J Med 2009; 360:1002-14). Астмой страдают до 300 миллионов человек во всем мире, и распространенность астмы продолжает расти, особенно в странах Африки, Латинской Америки, Восточной Европы и Азии.
Большинство пациентов с астмой имеют заболевание легкой и средней степени тяжести, которое можно хорошо контролировать с помощью стандартных методов лечения, включая регулярное применение ингаляционных кортикостероидов (ICS), часто в сочетании с ингаляционными β2-агонистами короткого действия (SABA) или ингаляционными β2-агонистами длительного действия. (LABA).
Однако у части пациентов с астмой наблюдается полное отсутствие или очень ограниченный ответ на традиционную терапию кортикостероидами. Этот тип астмы называется астмой, нечувствительной к кортикостероидам, или астмой, резистентной к кортикостероидам. Этим пациентам часто назначают высокие дозы кортикостероидов без фармакологических доказательств их полезности и с усиливающимися побочными эффектами. У многих из этих пациентов астма остается неконтролируемой и связана с особенно высоким риском обострений, госпитализаций и смерти, а также с серьезным ухудшением качества жизни (Chanez P et al, J Allergy Clin Immunol 2007; 119:1337-48; Dockrell M et al, Allergy 2007; 62:134-41).
Для определения эффективных методов лечения этой группы пациентов, страдающих астмой, были предприняты многочисленные усилия.
Например, клиническое исследование, проведенное с анти-IL-5 антителами (меполизумаб), показало снижение частоты обострений и улучшение качества жизни пациентов согласно AQLQ (опроснику качества жизни при астме). В дополнение к действию на астму, это исследование также показало терапевтический эффект против полипозной риносинусопатии (Haldar P et al., Mepolizumab and exacerbations of refractory eosinophilic asthma (N Engl J Med. 2009 Mar 5;360(10):973-84).
В настоящее время анти-IgE агент омализумаб, анти-IL-5 агенты меполизумаб и реслизумаб, а также анти-IL-5α-рецептор бенрализумаб являются биологическими препаратами, одобренными в качестве дополнительной терапии тяжелой астмы с эозинофильным фенотипом (Ther Adv Respir Dis 2018, Vol. 12: 1-12DOI: 10.1177/ 1753466618808490).
В US 10420792 описан способ лечения астмы, нечувствительной к кортикостероидам, у пациента с иммунным профилем Th1, который включает снижение активности фактора регуляции интерферона 5 (IRF5), например, с использованием антисмысловых или РНК-интерференционных реагентов (таких как miRNA или siRNA) или антитела или фрагмент антитела.
В US 9078894 описан способ лечения тяжелой персистирующей астмы, резистентной к кортикостероидам, путем введения ингибитора тирозинкиназы или ингибитора тучных клеток, такого как маситиниб или его соль.
Несмотря на усилия исследователей, проведенные до настоящего времени, по-прежнему существует острая потребность в новом и более эффективном лечении, позволяющем адекватно контролировать астму, резистентную к кортикостероидам.
Хемокины представляют собой семейство структурно родственных пептидов, которые на поверхности лейкоцитов регулируют воспаление посредством связанных с G-белком на поверхности клетки рецепторов. Они опосредуют разнообразную биологическую и биохимическую активность, например, эндотелиальную адгезию, направленную миграцию и активацию цитотоксической активности, такой как респираторный взрыв и экзоцитоз (Baggiolini, M. et al, (1997) Annu. Rev. Immunol. 15, 675-705).
Хемокины были разделены на четыре семейства (C, CC, CXC и CX3C) в зависимости от количества и положения консервативных остатков цистеина на N-конце. Большинство хемокинов связывают и активируют более одного хемокинового рецептора, и многие хемокиновые рецепторы активируются несколькими хемокинами (Murphy, P. M. et al (2000) Pharmacol. Rev. 52, 145-176).
Интерлейкин-8 (IL-8; CXCL8) считается основным медиатором рекрутирования PMN (полиморфноядерных нейтрофилов) и участвует в ряде патологий, включая псориаз, ревматоидный артрит, хроническую обструктивную болезнь легких и реперфузионное повреждение трансплантированного органа.
Биологическая активность CXCL8 опосредована взаимодействием с двумя рецепторами, CXCR1 и CXCR2, которые экспрессируются на поверхности PMN человека. CXCR1 селективен в отношении CXCL8, тогда как CXCR2 является более разнородным рецептором, связывающим ряд различных цитокинов и хемокинов, таких как CXCL1, CXCL2, CXCL3, CXCL5, CXCL6 и CXCL7 (Baggiolini, M., (2000) Immunol. Rev. 177, 5-7).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Заявитель неожиданно обнаружил, что в модели животных с астмой, резистентной к кортикостероидам, сочетание ингибитора активности CXCL8, такого как ладариксин или DF2755, с традиционным лечением кортикостероидом, таким как дексаметазон, способно восстанавливать противовоспалительный эффект кортикостероидов посредством синергетического механизма, предотвращая нейтрофильное воспаление дыхательных путей и утечку белка, ремоделирование тканей, гиперреактивность бронхов и улучшение легочной механики.
Далее, в первом аспекте, настоящее изобретение относится к комбинации ингибитора активности CXCL8 и кортикостероида, а также к указанной комбинации для применения при лечении астмы, резистентной к кортикостероидам.
Во втором аспекте настоящее изобретение относится к фармацевтической композиции, содержащей комбинацию согласно первому объекту изобретения и по меньшей мере один инертный фармацевтически приемлемый эксципиент, для применения при лечении астмы, резистентной к кортикостероидам.
В третьем аспекте настоящее изобретение относится к способу лечения астмы, нечувствительной к кортикостероидам, у субъекта, нуждающегося в этом, включающему введение субъекту фармацевтической композиции, содержащей комбинацию ингибитора активности CXCL8 и кортикостероида и по меньшей мере одного инертного фармацевтически приемлемого эксципиента.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Вариант осуществления настоящего изобретения описан далее в качестве примера со ссылкой на прилагаемые чертежи, на которых:
На фигуре 1 представлены данные, полученные при оценке просачивания крови в дыхательные пути: общих лейкоцитов (панель A), макрофагов (панель B), нейтрофилов (панель C), эозинофилов (панель D), лимфоцитов (панель E) и общего белка (панель F), определенные, как описано в примере 1a), на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем (Veh), дексаметазоном (Dex), ладариксином (Ldx) или дексаметазоном и ладариксином (Dex+Ldx);
На фигуре 2 показаны уровни TNF-α (панель A) и IL-17A (панель B) в паренхиме легкого, определенные, как описано в примере 1b) на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем (Veh), дексаметазоном (Dex), ладариксином (Ldx) или дексаметазоном и ладариксином (Dex+Ldx);
На фигуре 3 показаны гистологические срезы легкого (краситель H&E), полученные, как описано в примере 1d, (увеличение в 400 раз) на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем (Veh), дексаметазоном (Dex), ладариксином (Ldx) или дексаметазоном и ладариксином (Dex+Ldx), где стрелки указывают на перибронхиальное воспаление;
На фигуре 4 показана количественная оценка активности MPO в легких (панель A) и оценка воспаления легких с точки зрения оценки пространства дыхательных путей (панель B), паренхиматозного пространства (панель C) и оценки воспаления (панель D), определенные, как описано в примерах 1c) и 1d), на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем (Veh), дексаметазоном (Dex), ладариксином (Ldx) или дексаметазоном и ладариксином (Dex+Ldx);
На фигуре 5 показана динамическая растяжимость (Cdyn) и резистентность легких, вызванная метахолином (ΔRl), определенные, как описано в примере 1e), на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем (Veh), дексаметазоном (Dex), ладариксином (Ldx) или дексаметазоном и ладариксином (Dex+Ldx).
На фигуре 6 показаны данные, полученные путем оценки общего количества лейкоцитов (панель A) в дыхательных путях, подчеркивая рекрутирование нейтрофилов (панель B), эозинофилов (панель C), макрофагов (панель D) и лимфоцитов (панель E) определенных, как описано в примере 2a), на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем, дексаметазоном (Dexa), DF2755A или дексаметазоном и DF2755A (Dexa+DF2755A). Накопление нейтрофилов (панель F) и эозинофилов (панель G) в легких оценивали по активности MPO и EPO, соответственно, как описано в примере 2c), на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем, дексаметазоном (Dexa), DF2755A или дексаметазоном и DF2755A (Dexa+DF2755A). В каждой экспериментальной группе было по 8 животных. * Р<0,05, ** Р<0,01, *** Р<0,001.
На фигуре 7 показана оценка уровней хемокинов CXCL1 (панель A), CCL2 (панель B), CCL24 (панель C) и цитокинов IFN-γ (панель D), IL-17A (панель E) и TNF-α (панель F) в легких, определенных, как описано в примере 2b) на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем, дексаметазоном (Dexa), DF2755A или дексаметазоном и DF2755A (Dexa+DF2755A). В каждой экспериментальной группе было по 8 животных. * Р<0,05, ** Р<0,01, *** Р<0,001.
На фигуре 8 показана оценка функциональных параметров легких, проведенная через 24 ч после последней провокации; функциональные параметры легких представлены в виде объемов легких (панель A) и резистентности (панель B), определенных, как описано в примере 2d) на мышиной модели астмы, резистентной к кортикостероидам, после лечения носителем, дексаметазоном (Dexa), DF2755A или дексаметазоном и DF2755A (Dexa+DF2755A). В каждой экспериментальной группе было по 8 животных. * Р<0,05, ** Р<0,01, *** Р<0,001.
На фигуре 9 показаны гистологические срезы легкого (окрашенные H&E), полученные как описано в примере 2e) на мышиной модели астмы, резистентной к кортикостероидам, от контрольной мыши (панель A), зараженной OVA (панель B), обработанной DF2755A (панель C), обработанной дексаметазоном (панель D) и обработанной как DF2755A, так и дексаметазоном (панель E). Панель F представляет собой оценку воспаления. Масштабная линейка: 50 мкм. В каждой экспериментальной группе было по 8 животных. * Р<0,05, ** Р<0,01, *** Р<0,001.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Первым объектом настоящего изобретения является комбинация ингибитора активности CXCL8 и кортикостероида.
Предпочтительно, вышеуказанная комбинация предназначена для лечения астмы, резистентной к кортикостероидам.
Предпочтительно, указанная астма, резистентная к кортикостероидам, представляет собой астму, характеризующуюся менее чем 15% улучшением исходного FEV1 (объем форсированного выдоха за 1 секунду) после 14-дневного курса перорального приема преднизолона (40 мг/день) у пациентов, у которых наблюдается более чем 15% улучшение FEV1 после лечения ингаляционным агонистом β2.
Термин «ингибитор активности CXCL8» в соответствии с настоящей заявкой относится к соединению, способному частично или полностью ингибировать биологическую активность CXCL8. Такое соединение может действовать путем снижения экспрессии CXCL8 или его рецептора(ов) или путем ингибирования запуска внутриклеточной передачи сигналов, активируемой рецептором(ами) CXCL8. Предпочтительно, чтобы указанный ингибитор активности CXCL8 был способен ингибировать по меньшей мере 60%, предпочтительно, по меньшей мере 70%, более предпочтительно, по меньшей мере 80%, еще более предпочтительно, по меньшей мере 90% хемотаксиса PMN, индуцированного оптимальной концентрацией CXCL8 (1 нМ) при концентрации равной или ниже 500 нМ, предпочтительно, ниже 100 нМ.
В соответствии с предпочтительным вариантом осуществления ингибитор активности CXCL8 по настоящему изобретению ингибирует активность CXCL8, опосредованную рецептором CXCR1 или опосредованную как рецепторами CXCR1, так и рецепторами CXCR2. Предпочтительно, в соответствии с этим вариантом осуществления, указанный ингибитор активности CXCL8 представляет собой ингибитор рецептора(ов) CXCL8, который ингибирует связывание CXCL8 с его рецептором(ами) или внутриклеточную передачу сигналов, активируемую связыванием CXCL8 с его рецептором(ами). Предпочтительно, указанный ингибитор рецептора CXCL8 представляет собой либо аллостерический ингибитор, либо ортостерический антагонист рецептора CXCR1 или как рецептор CXCR1, так и рецептор CXCR2.
Более предпочтительно, ингибитор рецептора(ов) CXCL8 в соответствии с изобретением имеет значение IC50 по отношению к рецептору CXCR1 в низком наномолярном диапазоне, предпочтительно, в диапазоне 0,02-5 наномолей.
В соответствии с предпочтительным вариантом осуществления, также в сочетании с любым из предыдущих вариантов осуществления, указанный ингибитор активности CXCL8 выбран из соединений с низкой молекулярной массой, пептидов и антител, более предпочтительно, представляет собой соединение с низкой молекулярной массой.
Ингибиторы активности CXCL8 в соответствии с приведенным выше определением, способные ингибировать активность CXCL8, опосредованную рецептором CXCR1 или опосредованную как рецепторами CXCR1, так и рецепторами CXCR2, хорошо известны в данной области.
На сегодняшний день описано несколько ингибиторов активности CXCL8, таких как небольшие молекулы, пептиды и антитела, ряд из которых в настоящее время проходит клинические испытания или используются в терапии (Jie Jack, Expert Opinion Ther. Patents, 2001, 11(12), Chao J. et al., Bioorganic & Medicinal Chemistry Letters 17, 2007, p. 3778-3783, Busch-Petersen J. Current Topics in Medicinal Chemistry, 2006, 6, p. 1345-135, Allegretti et al, Immunology Letters 2012, Vol. 145, p. 68-78).
Предпочтительными ингибиторами активности CXCL8 в соответствии с настоящим изобретением являются ингибиторы рецепторов CXCR1 и CXCR2, описанные в WO2000/024710A1 и WO2005/090295, где также описан способ их синтеза, их активность в качестве ингибиторов рецептора CXCL8, а также их применение в качестве ингибиторов хемотаксиса нейтрофилов и дегрануляции, индуцированной CXCL8, и при лечении CXCL8-зависимых патологий. Эти ингибиторы активности CXCL8 являются ингибиторами рецептора(ов) CXCL8.
Из них предпочтительны ингибиторы активности CXCL8, особенно ингибиторы следующей формулы (I):
 (I),
(I),
где
R1 выбран из линейного или разветвленного C1-C6 алкила, бензоила, фенокси и трифторметансульфонилокси;
R2 выбран из атома водорода и линейного или разветвленного C1-C6 алкила; и
R3 представляет собой линейный или разветвленный C1-C6 алкил или трифторметил,
и их фармацевтически приемлемые соли.
Предпочтительно, R1 выбран из бензоила, изобутила и трифторметансульфонилокси. Более конкретно, R1 предпочтительно связан с фенильным кольцом в 3-м или 4-м положении. Согласно наиболее предпочтительному варианту осуществления R1 представляет собой 3-бензоил, 4-изобутил или 4-трифторметансульфонилокси.
Предпочтительно, R2 выбран из атома водорода или метила.
Предпочтительно, R3 выбран из линейного или разветвленного C1-C6 алкила, более предпочтительно, из линейного или разветвленного C1-C3 алкила. Согласно наиболее предпочтительному варианту осуществления R3 представляет собой метил.
Предпочтительно, хиральный углерод соединений формулы (I) имеет RS- или R-конфигурацию, более предпочтительно, он имеет R-конфигурацию.
Следующие соединения являются особенно предпочтительными из указанных ингибиторов активности CXCL8 формулы (I):
- 2-(4-изобутилфенил)пропионилметансульфонамид, предпочтительно, R-(-)-2-(4-изобутилфенил)пропионилметансульфонамид и его фармацевтически приемлемые соли, предпочтительно, его лизиновая соль (также известная как репариксин), и
- 2-(4-трифторметансульфонилокси)фенил]-N-метансульфонил пропионамид, предпочтительно, R(-)-2-(4-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамид, и его фармацевтически приемлемые соли, в частности, его натриевая соль (также известная как ладариксин или DF2156A).
Кроме того, другими предпочтительными ингибиторами активности CXCL8 в соответствии с настоящим изобретением являются ингибиторы, описанные в WO2010/031835, где также описан способ их синтеза, их активность в качестве ингибиторов рецептора CXCL8, а также их применение в качестве ингибиторов хемотаксиса и дегрануляции нейтрофилов, индуцированных CXCL8, и при лечении CXCL8-зависимых патологий. Эти соединения являются ингибиторами рецептора(ов) CXCL8.
Из них предпочтительны ингибиторы активности CXCL8, особенно ингибиторы следующей формулы (II):
 (II)
(II)
и их фармацевтически приемлемые соли, где:
R1 представляет собой водород;
X представляет собой OH;
R2 представляет собой водород или линейный C1-C4 алкил,
Y представляет собой гетероатом, выбранный из S, O и N,
Z выбран из линейного или разветвленного C1-C4 алкила, линейного или разветвленного C1-C4 алкокси, галогено C1-C3 алкила и галогено C1-C3 алкокси.
Предпочтительно, хиральный атом углерода соединений формулы (II) имеет RS- или S-конфигурацию, более предпочтительно, S-конфигурацию.
Из указанных ингибиторов активности CXCL8 формулы (II) особенно предпочтительным является 2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропионовая кислота, предпочтительно, (2S)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)-пропионовая кислота (DF2755Y), или ее натриевая соль (также известная как DF2755A).
Наиболее предпочтительными ингибиторами активности CXCL8 согласно настоящему изобретению являются R(-)-2-(4-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамид или (2S)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)-пропионовая кислота, предпочтительно, в виде натриевых солей.
Любой из ингибиторов активности CXCL8, описанных выше, может быть в виде соли с фармацевтически приемлемой органической или неорганической кислотой или основанием, или быть в виде пролекарства, или быть частично или полностью дейтерированным.
Термин «кортикостероид» в соответствии с настоящей заявкой относится к классу природных стероидных гормонов и к их синтетическим аналогам, которые включают глюкокортикоиды и минералкортикоиды.
В соответствии с предпочтительным вариантом осуществления настоящего изобретения кортикостероиды в комбинации по настоящему изобретению представляют собой глюкокортикоиды. Глюкокортикоиды хорошо известны в данной области.
Предпочтительными глюкокортикоидами, подходящими для использования в комбинации по настоящему изобретению, являются глюкокортикоиды, используемые для лечения астмы, предпочтительно, амцинонид, беклометазона дипропионат, бетаметазон, бетаметазона ацетат, бетаметазона бензоат, бетаметазона дипропионат, бетаметазона натрия фосфат, бетаметазона валерат, будесонид, карбеноксолон натрия, клокортолона ацетат, клокортолона пивалат, клопреднол, кортикотропин (инъекция), кортикотропин (репозиторий), гидроксид кортикотропина цинка, кортизона ацетат, кортивазол, десцинолона ацетонид, дексаметазон, дексаметазона натрия фосфат, дифлукортолон, дифлукортолона пивалат, флуклоронид, флуметазон, флуметазона пивалат, флунизолид, флуоцинолона ацетонид, флуоциномид, флуокортолон, флуокортолона капроат, флуорометолон, флуперолона ацетат, флупреднизолон, флупреднизолона валерат, флурандренолид, формокортал, флутиказон, гидрокортизон, гидрокортизона ацетат, бутепрат гидрокортизона, гидрокортизона бутират, гидрокортизона ципионат, гидрокортизона натрия фосфат, гидрокортизона натрия сукцинат, гидрокортизона валерат, медризон, метилпреднизолон, метилпреднизолона ацетат, метилпреднизолона натрия фосфат, метилпреднизолона натрия сукцинат, нивазол, параметазона ацетат, предникарбат, преднизолон, преднизолона ацетат, преднизолона гемисукцинат, преднизолона натрия фосфат, преднизолона натрия сукцинат, преднизолона тебутат, преднизолон, преднивал, тикабезона пропионат, тралонид, триамцинолон, триамцинолона ацетонид, триамцинолона ацетонид фосфат натрия, триамцинолона диацетат триамцинолона и гексацетонид триамцинолона.
Предпочтительно, в комбинации согласно настоящему изобретению глюкокортикоиды выбран из беклометазона дипропионата, бетаметазона, бетаметазона ацетата, бетаметазона бензоата, бетаметазона дипропионата, бетаметазона натрия фосфата, бетаметазона валерата, будесонида, кортизона ацетата, дексаметазона, флуметазона, флуметазона пивалата, гидрокортизона, гидрокортизона ацетата, гидрокортизона бутепрата, гидрокортизона бутирата, гидрокортизона ципионата, гидрокортизона натрия фосфата, флутиказона, гидрокортизона натрия сукцината, гидрокортизона валерата, метилпреднизолона, метилпреднизолона ацетата, метилпреднизолона натрия фосфата, метилпреднизолона натрия сукцината, параметазона ацетата, преднизолона, преднизолона ацетата, преднизолона гемисукцината, преднизолона натрия фосфата, преднизолона натрия сукцината, преднизолона тебутата, преднизона и триамцинолона.
Более предпочтительно, в комбинации согласно настоящему изобретению глюкокортикоиды выбраны из беклометазона, флуметазона, триамцинолона, бетаметазона, будесонида, дексаметазона, флутиказона, преднизолона, метилпреднизолона и преднизона. Из них особенно предпочтительными являются дексаметазон, преднизолон, метилпреднизолон и преднизон. Наиболее предпочтительным глюкокортикоидом по изобретению является дексаметазон.
В соответствии с особенно предпочтительным вариантом осуществления комбинация согласно первому объекту изобретения представляет собой комбинацию ингибитора активности CXCL8 R(-)-2-(4-трифторметансульфонилокси)фенил]-N-метансульфонил пропионамида, предпочтительно, в виде натриевой соли (также известной как ладариксин или DF2156A) или (2S)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропионовой кислоты, предпочтительно, в виде натриевой соли (также известной как DF2755A) и дексаметазона.
Ингибитор активности CXCL8 и кортикостероидные соединения по настоящему изобретению могут образовывать стабильные фармацевтически приемлемые соли кислот или оснований с фармацевтически приемлемыми органическими или неорганическими кислотами или основаниями, и в таких случаях может быть подходящим введение соединения в виде соли.
Примеры кислотно-аддитивных солей включают ацетат, адипат, аскорбат, бензоат, бензолсульфонат, бикарбонат, бисульфат, бутират, камфорат, камфорсульфонат, холин, цитрат, циклогексилсульфамат, диэтилендиамин, этансульфонат, фумарат, глутамат, гликолят, гемисульфат, 2-гидроксиэтилсульфонат, гептаноат, гексаноат, гидрохлорид, гидробромид, гидройодид, гидроксималеат, лактат, малат, малеат, метансульфонат, меглумин, 2-нафталинсульфонат, нитрат, оксалат, памоат, персульфат, фенилацетат, фосфат, дифосфат, пикрат, пивалат, пропионат, хинат, салицилат, стеарат, сукцинат, сульфамат, сульфанилат, сульфат, тартрат, тозилат (п-толуолсульфонат), трифторацетат и ундеканоат.
Примеры солей присоединения оснований включают соли аммония; соли щелочных металлов, такие как соли натрия, лития и калия; соли щелочноземельных металлов, такие как соли алюминия, кальция и магния; соли с органическими основаниями, такие как соли дициклогексиламина и N-метил-D-глюкамина; и соли с аминокислотами, такими как аргинин, лизин, орнитин и т.д. Кроме того, основные азотсодержащие группы можно кватернизовать такими агентами, как: низшие алкилгалогениды, такие как метил, этил, пропил и бутилгалогениды; диалкилсульфаты, такие как диметил, диэтил, дибутил; диамилсульфаты; галогениды с длинной цепью, такие как децил, лаурил, миристил и стеарилгалогениды; арилалкилгалогениды, такие как бензилбромид и другие. Предпочтительны нетоксичные физиологически приемлемые соли, хотя могут быть использованы и другие соли, например, при выделении или очистке продукта.
Соли могут быть образованы обычными способами, такими как взаимодействие свободной формы продукта с одним или несколькими эквивалентами соответствующей кислоты или основания в растворителе или среде, в которой соль нерастворима, такой как, например, вода или этанол, которые удаляют в вакууме или сушкой вымораживанием.
Настоящее изобретение также включает пролекарства, стереоизомеры и энантиомеры ингибитора рецептора CXCL8 и кортикостероидного соединения, описанные выше.
Как используется в настоящем документе, термин «пролекарство» относится к агенту, который преобразуется в исходное лекарственное средство in vivo посредством какого-либо физиологического химического процесса (например, пролекарство при доведении до физиологического pH преобразуется в желаемую лекарственную форму). Пролекарства часто используются, поскольку в некоторых ситуациях их легче вводить, чем исходное лекарственное средство. Они могут, например, быть биодоступными при пероральном введении, в то время как исходное лекарственное средство нет. Пролекарство также может иметь улучшенную растворимость в фармакологических композициях по сравнению с исходным лекарственным средством. Примером, без ограничений, пролекарства может быть соединение по настоящему изобретению, когда его вводят в виде сложного эфира («пролекарство») для облегчения прохождения через клеточную мембрану, где растворимость в воде не является благоприятной, но затем оно метаболически гидролизуется внутри клетки, где растворимость в воде является благоприятной.
Пролекарства обладают многими полезными свойствами. Например, пролекарство может быть более растворимым в воде, чем конечное лекарственное средство, что облегчает внутривенное введение лекарственного средства. Пролекарство также может иметь более высокий уровень пероральной биодоступности, чем конечное лекарственное средство. После введения пролекарство ферментативно или химически расщепляется для доставки конечного лекарственного средства в кровь или ткань.
В частности, рассматриваются сложноэфирные пролекарства ингибитора активности CXCL8 и кортикостероидные соединения, описанные в настоящем документе. Без ограничения, сложный эфир может представлять собой алкиловый эфир, ариловый эфир или гетероариловый эфир. Термин «алкил» имеет значение, обычно понятное специалистам в данной области, и он относится к неразветвленным, разветвленным или циклическим алкильным группам. Особенно используются C1-6 алкиловые эфиры, где алкильная часть сложного эфира имеет от 1 до 6 атомов углерода и включает, но этим не ограничивается, метил, этил, пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, изомеры пентила, изомеры гексила, циклопропил, циклобутил, циклопентил, циклогексил и их комбинации, имеющие от 1 до 6 атомов углерода.
Некоторые ингибиторы активности CXCL8 и кортикостероидные соединения могут существовать в таутомерных формах, и данное изобретение включает все такие таутомерные формы этих соединений, если не указано иное.
Если не указано иное, структуры, изображенные в настоящем документе, также включают все стереохимические формы структуры; т. е. конфигурации R и S для каждого асимметричного центра. Таким образом, в объем изобретения входят отдельные стереохимические изомеры, а также энантиомерные и диастереомерные смеси настоящих соединений. Таким образом, данное изобретение охватывает каждый диастереомер или энантиомер, по существу свободный от других изомеров (>90% и, предпочтительно, >95%, свободный от других стереоизомеров в молярном отношении), а также смесь таких изомеров.
Конкретные оптические изомеры могут быть получены разделением рацемических смесей в соответствии с обычными способами, например, образованием диастереомерных солей, обработкой оптически активной кислотой или основанием. Примерами подходящих кислот являются винная, диацетилвинная, дибензоилвинная, дитолуоилвинная и камфорсульфоновая кислоты, а затем разделение смеси диастереомеров путем кристаллизации с последующим высвобождением оптически активных оснований из этих солей. Другой способ разделения оптических изомеров включает использование хиральной хроматографической колонки, оптимально выбранной для максимального разделения энантиомеров. Еще один способ включает синтез ковалентных диастереомеров путем взаимодействия соединений по изобретению с оптически чистой кислотой в активированной форме или с оптически чистым изоцианатом. Синтезированные диастереомеры можно разделить обычными способами, такими как хроматография, перегонка, кристаллизация или возгонка, а затем гидролизовать с получением энантиомерно чистого соединения. Оптически активные соединения по изобретению могут быть получены с использованием активных исходных материалов. Эти изомеры могут быть в виде свободной кислоты, свободного основания, сложного эфира или соли.
Согласно одному варианту осуществления ингибитор активности CXCL8 и кортикостероид по изобретению вводят одновременно или последовательно в раздельных фармацевтических композициях.
Согласно альтернативному варианту осуществления ингибитор активности CXCL8 и кортикостероид вводят вместе в одной и той же фармацевтической композиции.
Соответственно, вторым объектом настоящего изобретения является фармацевтическая композиция, содержащая комбинацию ингибитора активности CXCL8 и кортикостероида в соответствии с первым объектом изобретения и по меньшей мере один инертный фармацевтически приемлемый эксципиент.
Предпочтительно, указанная фармацевтическая композиция предназначена для лечения астмы, нечувствительной к кортикостероидам.
Предпочтительно, фармацевтическую композицию по настоящему изобретению готовят в виде подходящих лекарственных форм, содержащих эффективное количество комбинации ингибитора активности CXCL8 и кортикостероида, его соли с фармацевтически приемлемой органической или неорганической кислотой или основанием или его пролекарства и по меньшей мере одного инертного вещества.
Путь введения фармацевтической композиции по настоящему изобретению соответствует известным способам, например, ингаляция, инъекция или инфузия внутривенно, парентерально, местно, ректально, назально, трансбуккально, вагинально или через имплантированный резервуар или с помощью систем с замедленным высвобождением. Как используется в настоящем документе, термин парентеральный включает внутривенную, внутрибрюшинную, внутримозговую, подоболочечную, внутричерепную, внутримышечную, внутрисуставную, интрасиновиальную, интрастернальную, внутриглазную, внутриартериальную, подкожную, внутрикожную или внутриочаговую инъекцию или инфузию.
В одном предпочтительном воплощении изобретение относится к лечению астмы, нечувствительной к кортикостероидам, путем прямого введения фармацевтической композиции по настоящему изобретению в дыхательные пути. Фармацевтическая композиция по настоящему изобретению может использоваться в самых разных устройствах, предназначенных для прямой доставки фармацевтических композиций и терапевтических составов в дыхательные пути. В одном аспекте настоящего изобретения фармацевтическую композицию по настоящему изобретению вводят в виде аэрозоля или ингаляции. Фармацевтическую композицию по настоящему изобретению можно вводить в аэрозольном составе в виде сухого порошка или в растворе или суспензии с разбавителем.
В настоящем описании и в последующей формуле изобретения формулировка «эффективное количество» означает количество соединения или композиции, достаточное для значительного и положительного изменения симптомов и/или состояний, подлежащих лечению (например, обеспечить положительный клинический ответ). Эффективное количество активного ингредиента для применения в фармацевтической композиции будет варьироваться в зависимости от конкретного состояния, подвергаемого лечению, тяжести состояния, продолжительности лечения, характера сопутствующей терапии, конкретного используемого активного ингредиента (ингредиентов), конкретного фармацевтически приемлемого эксципиента(ов)/носителя(ей) и подобных факторов в пределах знаний и опыта лечащего врача.
Как описано в настоящем документе, фармацевтическая композиция по настоящему изобретению содержит комбинацию ингибитора активности CXCL8 и кортикостероида вместе с фармацевтически приемлемым эксципиентом, который, как используется в настоящем документе, включает любые и все растворители, разбавители или другие носители, добавки для дисперсии или суспензии, поверхностно-активные вещества, изотонические вещества, загустители или эмульгаторы, консерванты, твердые связующие вещества, смазывающие вещества и тому подобное, подходящие для конкретной желаемой лекарственной формы.
Некоторые примеры веществ, которые могут служить в качестве фармацевтически приемлемых эксципиентов, включают, но этим не ограничиваются, сахара, такие как лактоза, глюкоза и сахароза; крахмалы, такие как кукурузный крахмал и картофельный крахмал; целлюлоза и ее производные, такие как натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; порошкообразный трагакант; солод; желатин; тальк; наполнители, такие как масло какао и воск для суппозиториев; масла, такие как арахисовое масло, хлопковое масло; сафлоровое масло; кунжутное масло; оливковое масло; кукурузное масло и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные агенты, такие как гидроксид магния и гидроксид алюминия; альгиновую кислоту; апирогенную воду; изотонический раствор; стерилизованную воду; раствор Рингера; забуференный солевой раствор; раствор декстрозы; раствор мальтодекстрина; этиловый спирт; и фосфатно-буферные растворы.
Кроме того, композиция по настоящему изобретению может быть изготовлена в виде лекарственных форм для ингаляции или инъекций, таких как растворы, суспензии и эмульсии, путем дополнительного добавления разбавителей, диспергаторов и поверхностно-активных веществ.
Кроме того, композиция по настоящему изобретению может быть соответствующим образом составлена с использованием соответствующих способов, известных в данной области, или с помощью метода, описанного в Remingtonʼs Pharmaceutical Science (последнее издание), Mack Publishing Company, Easton Pa.
Термины «фармацевтически приемлемый» и «физиологически приемлемый» предназначены для определения, без каких-либо конкретных ограничений, любого вещества, пригодного для изготовления фармацевтической композиции для введения живому существу.
Лекарственные формы также могут содержать другие традиционные ингредиенты, такие как: консерванты, стабилизаторы, поверхностно-активные вещества, буферы, соли для регулирования осмотического давления, эмульгаторы, подсластители, красители, ароматизаторы и тому подобное.
Количество указанного ингибитора активности CXCL8 и кортикостероидных соединений в фармацевтической композиции по настоящему изобретению представляет собой количество, обычно используемое при их индивидуальном применении.
Количество может варьироваться в широком диапазоне в зависимости от известных факторов, например, используемого соединения, тяжести заболевания, веса тела пациента, лекарственной формы, выбранного пути введения и количества введений в день. Однако специалист в данной области техники может определить оптимальное количество простым и рутинным способом.
Кроме того, конкретные дозы и схемы лечения для каждого конкретного пациента будут зависеть от множества факторов, включая, например, активность и период полувыведения конкретного используемого соединения, возраст, вес тела, общее состояние здоровья, пол, режим питания, тяжесть и течение заболевания.
Лекарственные формы фармацевтической композиции по настоящему изобретению могут быть изготовлены методами, известными химику-фармацевту, и включают смешивание, грануляцию, прессование, растворение, стерилизацию и тому подобное.
Третий аспект настоящего изобретения относится к способу лечения астмы, нечувствительной к кортикостероидам, у субъекта, нуждающегося в этом, включающему введение субъекту фармацевтической композиции, содержащей комбинацию ингибитора активности CXCL8 и кортикостероида в соответствии со вторым аспектом настоящего изобретения.
Далее изобретение будет описано в следующих примерах, которые не ограничивают объем изобретения, описанный в формуле изобретения.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
На фигурах все результаты выражены как среднее значение±стандартная ошибка среднего. Нормализованные данные анализировали с помощью однофакторного дисперсионного анализа с посттестом Тьюки с использованием программного обеспечения GraphPad Prism 7.0. Различия считались значимыми при P<0,05. n=8; * Р<0,05; ** Р<0,01; *** р<0,001.
Пример 1
Исследование проводилось с использованием ранее описанной мышиной модели астмы, нечувствительной к глюкокортикоидам Th17 (Campa CC et al., Nat Commun (2018), v. 9, p. 5232, и Dejager Let al., Mucosal Immunol (2015), v. 8, p 1212-25).
Подробно, в день 0 мыши BALB/c получали системную иммунизацию путем подкожной инъекции 20 мкг овальбумина куриного яйца (OVA класс V, чистота >98%; Sigma, St Louis, MO) в физиологическом растворе, эмульгированном в 75 мкл CFA (Sigma-Aldrich).
С 28-го дня после первой иммунизации до 31-го дня мышей обрабатывали PBS (носитель), ладариксином, дексаметазоном или ладариксином/дексаметазоном, как описано ниже, и через 60 минут подвергали воздействию аэрозольного овальбумина (1% в физиологическом растворе) в течение 20 минут в день. Ладариксин вводили перорально через желудочный зонд (10 мг/кг в физиологическом растворе 0,9%), как описано ранее (Bertini et al., Br J Pharmacol (2012), v.165, p. 436-54), один раз в день. Дексаметазон вводили подкожно (5 мг/кг в физиологическом растворе), один раз в день.
a) Просачивание лейкоцитов в дыхательные пути
Оценку общего содержания лейкоцитов в дыхательных путях проводили путем проведения бронхоальвеолярного лаважа (BALF), как описано ранее (Russo et al Am J Respir Cell Mol Biol (2009), v. 40, p. 410-21; Campa CC et al., Nat Commun (2018), v. 9, p. 5232). Вкратце, в трахее делали разрез, куда вставляли полиэтиленовый катетер с наружным диаметром 1,7 мм. Затем дыхательные пути промывали 2 раза 1 мл фосфатно-солевого буфера (PBS). Образцы центрифугировали при 300×g в течение 10 минут при температуре 4ºC, осадок собирали, ресуспендировали в 100 мкл 3% BSA в PBS, и образец из этого раствора использовали для оценки общего количества лейкоцитов в дыхательных путях. Оставшуюся часть центрифугировали при 300×g в течение 10 минут при температуре 4ºC на предметном стекле для гистологии для оценки дифференциального подсчета макрофагов, лимфоцитов, эозинофилов и нейтрофилов с помощью оптической микроскопии. Общий белок количественно определяли в супернатанте BALF с использованием реагента для анализа белка Bio-Rad (Bio-Rad, США) для измерения возможной утечки белка, как описано ранее (Campa CC et al., Nat Commun (2018), v. 9, p. 5232).
Результаты проиллюстрированы на фигуре 1.
Как видно из полученных данных, у мышей, получавших носитель, наблюдается значительный приток лейкоцитов в дыхательные пути с преобладанием нейтрофилов и макрофагов и менее повышенное количество эозинофилов и лимфоцитов с повышенной утечкой белка в дыхательные пути.
Лечение ладариксином снижает общий приток лейкоцитов в альвеолярное пространство за счет заметного снижения количества макрофагов и нейтрофилов, в то время как дексаметазон не снижает рекрутирование этих клеток. Однако неожиданно комбинированное лечение ладариксином и дексаметазоном дает синергетический эффект со значительно большим снижением общего притока лейкоцитов и количества инфильтрированных макрофагов и нейтрофилов по сравнению с ладариксином в отдельности.
Также наблюдается синергетический эффект в отношении рекрутирования эозинофилов в дыхательные пути, который несколько снижается при лечении дексаметазоном и усиливается при введении ладариксина. Удивительно, но одновременное лечение дексаметазоном и ладариксином приводит к более значительному снижению рекрутирования этих клеток, чем при применении только дексаметазона.
b) Количественное определение цитокинов в легочной ткани
Уровни IL17A и TNF-α также измеряли в различных экспериментальных группах мышей. Вкратце, после BALF, правое легкое удаляли и образец 100 мг гомогенизировали в 1 мл PBS, содержащего антипротеазы (0,1 мМ фенилметилсульфонилфторида, 0,1 мМ хлорида бензетония, 10 мМ ЭДТА и 20 KI апротинина (А)). Гомогенат центрифугировали при 8000×g в течение 10 минут при температуре 4ºC. Супернатант замораживали для дальнейшего анализа ELISA. Уровни IL-17A и TNF-α измеряли методом ELISA с использованием коммерческих наборов DuoSet от R&D Systems в соответствии с инструкциями производителя. Результаты, показанные на фигуре 2, выражены в пг цитокинов на мл лизата легких (пг/мл).
Как показано на фигуре 2, цитокины Th17 IL-17A и TNF-α значительно снижались при лечении только ладариксином или ладариксином в комбинации с дексаметазоном.
c) Активность пероксидазы нейтрофилов (MPO)
Анализ MPO проводили на легочной ткани различных экспериментальных групп мышей.
Подробно анализ MPO проводили в 96-луночном микропланшете путем добавления в каждую лунку 25 мкл 3,3ʼ-5,5ʼ-тетраметилбензидина (TMB Sigma), разведенного в диметилсульфоксиде (ДМСО; Merck, Darmstadt, Germany) при конечной концентрации 1,6 мМ; 100 мкл H2O2 0,02% об./об. в PBS (pH 5,4), содержащем HTAB и 25 мкл супернатанта обработанной ткани. Реакция начиналась при температуре 37ºС добавлением раствора TMB и супернатанта в микропланшет, а через 5 мин добавляли H2O2 с последующей новой инкубацией при температуре 37ºC в течение 5 мин. Реакцию останавливали добавлением 100 мкл 1М H2SO4 и количественно определяли при длине волны 450 нм на спектрофотометре (VERSA max, Molecular Devices, CA).
Полуколичественный гистопатологический анализ проводили, как описано (Campa CC et al., Nat Commun (2018), v. 9, p. 5232). Вкратце, десять случайных изображений на одно легкое были получены с 200-кратным увеличением, и оценку воспаления легких регистрировали как шестибалльную, где «0» соответствует менее 1% площади легочной ткани, «1» до 9% площади легкого поражено воспалением, «2» от 10% до 29% площади легочной ткани, «3», если поражено от 30% до 49% площади легочной ткани, «4», если поражено от 50% до 69% площади легочной ткани, и «5», если воспалением поражено более 70% площади легочной ткани.
Как показано на фигуре 4, при оценке инфильтрации лейкоцитов в легочную ткань с помощью MPO наблюдался синергетический эффект между обработкой дексаметазоном и ладариксином. Ладариксин, но не только дексаметазон, способен предотвращать накопление нейтрофилов в паренхиме легких после воздействия OVA. Однако добавление дексаметазона к лечению ладариксином дополнительно снижает инфильтрацию нейтрофилов.
d) Гистологический анализ
После BALF удаляли левое легкое и фиксировали в 4% формалине в PBS (pH 7,4), как описано ранее. Ткань постепенно обезвоживали в этаноле, заливали в парафин, разрезали на срезы толщиной 4 мкм и окрашивали H&E, перйодной кислотой Шиффа (PAS) или трихромом Гомори. Микрофотографии получали при 400-кратном увеличении и анализировали с помощью программы Image Pro Plus (Image-Pro Plus 4.1, Media Cybernetics, USA) в условиях световой микроскопии. От девяти до двенадцати областей бронхов на легкое количественно определяли содержание PAS или трихрома Гомори, и результаты выражали в виде положительной площади (пиксели). Фиброз определяли с помощью срезов, окрашенных трихромом по Гомори. Полуколичественный гистопатологический анализ проводили, как описано Campa CC et al., Nat Commun (2018), v. 9, p. 5232 et al, 2018. Вкратце, десять случайных изображений на одно легкое были получены с 200-кратным увеличением, и оценку воспаления легких регистрировали в соответствии с шестибальной шкалой, где «0» соответствует менее 1% площади легочной ткани, «1» до 9% площади легкого поражено воспалением, «2» от 10% до 29% площади легочной ткани, «3» при поражении от 30 до 49% площади легочной ткани, «4» при поражении от 50 до 69% площади легочной ткани, «5» при поражении более 70% площади легочной ткани.
Оценивая гистологию легкого и показатель патологии, можно было наблюдать, что у контрольных мышей воспаления нет, но у мышей-носителей показатель воспаления очень высок. Лечение ладариксином, но не дексаметазоном, было эффективным для уменьшения воспаления (см. фигуру 4, панель D), но дальнейшее снижение оценки воспаления наблюдалось, когда дексаметазон связан с ладариксином, что свидетельствовало о синергическом эффекте, когда два соединения связаны. Эти данные также подтверждаются гистологическими изображениями на фигуре 3, где стрелки указывают на перибронхиальное воспаление.
e) Оценка легочной механики
Мышей из разных экспериментальных групп анестезировали подкожной инъекцией кетамина и ксилазина (130 мг/кг кетамина и 8,5 мг/кг ксилазина) для поддержания спонтанного дыхания под наркозом. Мышей подвергали трахеостомии и помещали в плетизмограф всего тела, подключенный к управляемому компьютером вентилятору (Forced Pulmonary Maneuver System®, Buxco Research Systems©, Wilmington, North Carolina USA). При механическом дыхании определяли динамическую податливость (Cdyn) и легочное сопротивление (Rl) с помощью теста сопротивления и податливости RC.
Мыши под наркозом были инструментами для непосредственного измерения дыхательного потока и давления в дыхательных путях. Из этих прямых измерений рассчитывали расход воздуха (RI) и давление (Cdyn).
Полученные результаты, показанные на фигуре 5, показывают, что воспаление дыхательных путей Th17 приводит к дисфункции тканей, такой как снижение потока в дыхательных путях, и к эластичности легких, которые не поддаются лечению дексаметазоном. Однако совместное лечение ладариксином и дексаметазоном сохраняло динамическую комплаентность (фигура 5, панель A). Что наиболее важно, ладариксин или дексаметазон в сочетании с ладариксином были способны снижать гиперреактивность дыхательных путей, вызванную метахолином (фигура 5, панель B).
В совокупности эти результаты продемонстрировали, что при нечувствительном к кортикостероидам Th17-нейтрофильном воспалении дыхательных путей и гиперреактивности, вызванных распылением OVA у мышей, ладариксин способен восстанавливать противовоспалительный эффект дексаметазона посредством синергетического механизма.
Действительно, антагонизм CXCR1 и CXCR2 при пероральном введении ладариксина плюс дексаметазон предотвращали нейтрофильное воспаление дыхательных путей и утечку белка, ремоделирование тканей, гиперреактивность бронхов и улучшали легочную механику в модели астмы, нечувствительной к GC.
Пример 2
Исследование проводили на той же мышиной модели, что и в примере 1.
Подробно, в день 0 мыши BALB/c получали системную иммунизацию путем подкожной инъекции 20 мкг овальбумина куриного яйца (OVA класс V, чистота >98%; Sigma, St Louis, MO) в физиологическом растворе, эмульгированном в 75 мкл CFA (Sigma-Aldrich).
С 28-го дня после первой иммунизации до 31-го дня мышей обрабатывали PBS (носитель), DF2755A, дексаметазоном или DF2755A/дексаметазоном, как описано ниже, и через 60 минут подвергали воздействию аэрозольного овальбумина (1% в физиологическом растворе), в течение 20 минут в день. DF2755A вводили перорально через желудочный зонд два раза в день, так что одно из введений всегда было за 60 минут до заражения OVA. DF2755A разводили в 0,9% солевом растворе только во время применения при комнатной температуре и применяли в дозе 10 мг/кг перорально. Дексаметазон разводили в 0,9% физиологическом растворе только во время использования при комнатной температуре и давали в дозе 5 мг/кг.
a) Оценка содержания лейкоцитов в дыхательных путях
Для оценки лейкоцитарной инфильтрации в дыхательных путях проводили бронхоальвеолярный лаваж (BALF). На 32-й день после иммунизации делали разрез в трахее, куда вставляли полиэтиленовый катетер с наружным диаметром 1,7 мм. Затем дыхательные пути промывали 2 раза 1 мл фосфатно-солевого буфера (PBS). Образцы центрифугировали при 300×g в течение 10 минут при температуре 4ºC и к осадку добавляли 100 мкл 3% BSA в PBS. Образец этого раствора использовали для оценки общего количества лейкоцитов в дыхательных путях. Оставшийся раствор центрифугировали при 300×g в течение 10 минут при температуре 4ºC и помещали на предметное стекло для гистологии для проведения дифференциального подсчета макрофагов, лимфоцитов, эозинофилов и нейтрофилов с помощью оптической микроскопии. Супернатант использовали для количественного определения количества общего белка и уровней хемокинов и цитокинов. Анализ общего белка определяли с использованием красителя для анализа белка Bio-Rad (Bio-Rad, США) для измерения возможной утечки белка.
Как показано на фигуре 6, в дыхательных путях мышей, получавших носитель, было обнаружено массивное пополнение лейкоцитов (фигура 6A) с преобладанием нейтрофилов (фигура 6B) и эозинофилов (фигура 6C) и, в меньшей степени, повышенное количество макрофагов (фигура 6D) и лимфоцитов (фигура 6E). Как и ожидалось, лечение DF2755A было способно уменьшить приток нейтрофилов, эозинофилов и лимфоцитов в дыхательные пути (фигуры 6A, B и E), в то время как лечение только дексаметазоном не уменьшало приток нейтрофилов и лимфоцитов в дыхательные пути. Комбинация DF2755A с дексаметазоном дает синергетический эффект со значительно большим снижением рекрутирования большинства проанализированных подмножеств лейкоцитов, за заметным исключением макрофагов (фигура 6).
b) Анализ хемокинов и цитокинов
После BALF удаляли правое легкое и образец 100 мг гомогенизировали в 1 мл PBS (0,4 M NaCl и 10 мМ NaPO4), содержащего антипротеазы (0,1 мМ фенилметилсульфонилфторида, 0,1 мМ хлорида бензетония, 10 мМ EDTA и 20 KI апротинина А). Гомогенат центрифугировали при 8000×g в течение 10 минут при температуре 4ºC. Супернатант замораживали для дальнейшего анализа ELISA, а осадок трижды замораживали/оттаивали с использованием жидкого азота для оценки активности миелопероксидазы нейтрофилов (MPO) и пероксидазы эозинофилов (EPO).
Уровни хемокинов CXCL1/KC, CCL2/MCP-1 и CCL24/Eotaxin2 и цитокинов TNF-α, IFN-γ и IL-17A в супернатанте легочной ткани измеряли с помощью ELISA. Результаты выражали в виде уровней цитокинов (пг/мл или нг/мл) на 0,1 г влажной ткани/мл экстракционного буфера. В частности, повышенные уровни IFN-γ, IL-17 и TNF-α являются отличительными признаками астмы, нечувствительной к Th17 GC.
Как показано на фигуре 7, лечение DF2755A значительно снижало уровни CXCL1 (фигура 7A), CCL2 (фигура 7B), CCL24 (фигура 7C), IL-17 (фигура 7E) и TNF-α (фигура 7F), в то время как снижение уровня IFN-γ было незначительным по сравнению с мышами, получавшими носитель (фигура 7D). Примерно такой же эффект наблюдался после лечения дексаметазоном, за исключением IL-17 (фигура 7E) и TNF-α (фигура 7F), которые показали меньшее снижение его уровней по сравнению с мышами, получавшими DF2755A. Опять же, комбинированное лечение DF2755A и дексаметазоном было более эффективным по сравнению с однократным лечением в снижении уровня большинства анализируемых цитокинов и хемокинов (фигура 7D-F).
Хотя лечение DF2755A, как правило, уменьшало воспаление как дыхательных путей, так и легких, его введение в сочетании с дексаметазоном было неожиданно более эффективным для предотвращения рекрутирования лейкоцитов и продукции хемокинов и цитокинов в легких.
c) Анализ активности миелопероксидазы нейтрофилов и пероксидазы эозинофилов
Активность миелопероксидазы нейтрофилов (MPO) и пероксидазы эозинофилов (EPO) измеряли следующим образом. Анализ MPO проводили в 96-луночном микропланшете, добавляя в каждую лунку 25 мкл 3,3ʼ-5,5ʼ-тетраметилбензидина (TMB Sigma), разведенного в диметилсульфоксиде (ДМСО; Merck, Darmstadt, Germany) при конечной концентрации 1,6 мМ; 100 мкл H2O2 0,02% об./об. в PBS (pH 5,4), содержащем HTAB, и 25 мкл супернатанта обработанной ткани. Реакция начиналась при температуре 37ºС добавлением раствора TMB и супернатанта в микропланшет, а через 5 мин добавляли H2O2 с последующей новой инкубацией при температуре 37ºC в течение 5 мин. Реакцию останавливали добавлением 100 мкл 1М H2SO4 и количественно определяли при длине волны 450 нм на спектрофотометре (VERSAmax, Molecular Devices, CA). Для анализа EPO супернатант обработанных образцов распределяли в 96-луночном микропланшете с последующим добавлением 75 мкл OPD (1,5 мМ в трис-HCl 0,05 М, рН 8,0, с добавлением 6,6 мМ H2O2). После 30-минутной инкубации при комнатной температуре реакцию останавливали добавлением 50 мкл 1М H2SO4 и определяли количество при 492 нм на спектрофотометре.
Как показано на фигуре 6, панели F и G, в то время как дексаметазон не смог уменьшить накопление нейтрофилов в легких, DF2755A предотвратил инфильтрацию как нейтрофилов (фигура 6F) и эозинофилов (фигура 6G) в ткань. Кроме того, комбинированное лечение DF2755A и дексаметазоном улучшало предотвращение рекрутмента по сравнению с двумя видами лечения по отдельности.
d) Оценка легочной механики
Мышей анестезировали подкожной инъекцией кетамина и ксилазина (130 мг/кг кетамина и 8,5 мг/кг ксилазина) для поддержания спонтанного дыхания под анестезией. Мышей подвергали трахеостомии, помещали в бодиплетизмограф и подключали к аппарату искусственной вентиляции легких с компьютерным управлением (Forced Pulmonary Maneuver System®, Buxco Research Systems©, Wilmington, North Carolina USA). Эта лабораторная установка, специально разработанная для использования на мышах, обеспечивает три различных маневра: закон Бойля FRC, маневры квазистатического давления-объема и быстрого потока-объема. Сначала анестезированному животному навязывали среднюю частоту дыхания 160 вдохов/мин с помощью искусственной вентиляции легких до достижения регулярного дыхания и полного выдоха при каждом дыхательном цикле, учитывая, что Rinx (индекс отторжения)=0. При механическом дыхании определяли динамическую податливость (Cdyn) и легочное сопротивление (Rl) с помощью теста сопротивления и податливости RC. Для измерения общей емкости легких (TLC) и остаточного объема (RV) были выполнены маневры давление-объем, при которых легкие раздуваются до стандартного давления +30 см, а затем происходит медленный выдох, пока не будет достигнуто отрицательное давление -30 см H2O. Для измерения объема форсированного выдоха (FEV) и кривой «поток-объем» были выполнены маневры «быстрый поток-объем». При этом маневре легкие сначала раздувались до +30 см H2O (TLC) и сразу же применяли отрицательное давление (-30 см H2O), чтобы усилить выдох. Для оценки AHR те же мыши, которые использовались в предыдущих экспериментах (исходное состояние), получали метахолин, 1 мг/кг (хлорид ацетил-β-метилхолина, A-2251, Sigma-Aldrich St. Louis, MO, USA) внутривенно, и через 20 секунд был проведен новый набор маневров для оценки изменений Rl. Субоптимальные маневры были отклонены, и для каждого теста на каждой отдельной мыши было проведено менее чем по меньшей мере три приемлемых маневра, чтобы получить надежное среднее значение для всех числовых параметров.
Как показано на фигуре 8, провокация OVA вызывала потерю объема легких (фигура 8A) и увеличение сопротивления легких и дыхательных путей (фигура 8B). Хотя DF2755A сам по себе не вызывал значительного улучшения легочной функции, комбинированное лечение с дексаметазоном полностью восстанавливало легочную механику (фигура 8). Что наиболее важно, комбинированное лечение DF2755A и дексаметазоном предотвращало гиперреактивность дыхательных путей в ответ на инъекцию метахолина (фигура 8B).
Эти результаты показывают, что лечение DF2755A, по-видимому, восстанавливает чувствительность GC, поскольку его введение в сочетании с дексаметазоном улучшает легочную механику и предотвращает развитие гиперреактивности дыхательных путей.
e) Гистология легких
После BALF удаляли левое легкое и фиксировали в 4% формалине в PBS (pH 7.4). Ткань постепенно обезвоживали в этаноле, заливали в парафин, разрезали на срезы толщиной 4 мкм и окрашивали гематоксилином и эозином. Микрофотографии получали при 400-кратном увеличении и анализировали с помощью программы Image Pro Plus (Image-Pro Plus 4.1, Media Cybernetics, USA).
Как показано на фигуре 9, в то время как у контрольных мышей были обнаружены нормальные гистологические данные (фигура 9A), у мышей, которые получали только носитель, наблюдалось значительное накопление лейкоцитов в паренхиме легких (фигуры 9B и F), с накоплением лейкоцитов, в основном обнаруживаемым в перибронхиальной и периваскулярной областях (фигура 9F). Лечение только дексаметазоном не смогло предотвратить воспаление легких (фигура 9D). Напротив, как DF2755A отдельно (фигуры 9C, 9F) в комбинации с дексаметазоном (фигуры 9E, 9F) были эффективны в снижении накопления лейкоцитов в ткани.
В совокупности эти результаты продемонстрировали, что лечение DF2755A отдельно или в комбинации с дексаметазоном оказывает благотворное влияние на мышиную модель астмы, нечувствительной к GC.
По этим причинам антагонизм CXCR1 и CXCR2 представляется безопасной, выгодной и новой потенциальной терапевтической стратегией для лечения астмы, нечувствительной к GC.









Изобретение относится к комбинации (i) ингибитора активности CXCL8, представляющего собой соединение формулы (I) или соединение формулы (II) или их фармацевтически приемлемую соль, и (ii) кортикостероида, представляющего собой дексаметазон, для применения при лечении астмы, нечувствительной к кортикостероидам, а также к фармацевтической композиции для лечения астмы, нечувствительной к кортикостероидам, содержащей эффективное количество такой комбинации и по меньшей мере один инертный фармацевтически приемлемый эксципиент. В общей формуле (I) R1 представляет собой рифторметансульфонилокси; R2 представляет собой атома водорода; и R3 представляет собой линейный или разветвленный C1-C6 алкил или трифторметил, в общей формуле (II) R1 представляет собой водород; X представляет собой OH; R2 представляет собой водород, Y представляет собой гетероатом, выбранный из S, Z представляет собой галогено C1-C3 алкила и галогено C1-C3 алкокси. Использование изобретения позволяет эффективно лечить астму, нечувствительную к кортикостероидам. 2 н. и 6 з.п. ф-лы, 9 ил., 2 пр.
 (I)
(I)  (II)
(II)
1. Комбинация (i) ингибитора активности CXCL8 и (ii) кортикостероида для применения при лечении астмы, нечувствительной к кортикостероидам, где указанный ингибитор активности CXCL8 представляет собой соединение формулы (I):
 (I),
(I),
где
R1 представляет собой трифторметансульфонилокси;
R2 представляет собой атома водорода; и
R3 представляет собой линейный или разветвленный C1-C6 алкил или трифторметил,
или его фармацевтически приемлемую соль; или
соединение формулы (II):
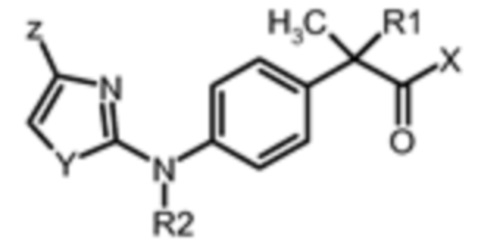 (II),
(II),
где:
R1 представляет собой водород;
X представляет собой OH;
R2 представляет собой водород,
Y представляет собой гетероатом, выбранный из S,
Z представляет собой галогено C1-C3 алкила и галогено C1-C3 алкокси,
или его фармацевтически приемлемую соль, и
где указанный кортикостероид представляет собой дексаметазон.
2. Комбинация по п.1, где указанный ингибитор активности CXCL8 ингибирует активность CXCL8, опосредованную рецептором CXCR1 или опосредованную как рецепторами CXCR1, так и рецепторами CXCR2.
3. Комбинация по пп.1, 2, где указанный ингибитор активности CXCL8 выбран из:
- 2-(4-изобутилфенил)пропионилметансульфонамида, предпочтительно R-(-)-2-(4-изобутилфенил)пропионилметан сульфонамида, и его фармацевтически приемлемых солей, предпочтительно его лизиновой соли, и
- 2-(4-трифторметансульфонилокси)фенил]-N-метансульфонил пропионамида, предпочтительно R(-)-2-(4-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамида, и его фармацевтически приемлемых солей, в частности его натриевой соли.
4. Комбинация по любому из пп.1-3, где указанный ингибитор активности CXCL8 представляет собой 2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропионовую кислоту, предпочтительно (2S)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)пропионовую кислоту, или его натриевую соль.
5. Комбинация по любому из пп.1-4, где указанный ингибитор активности CXCL8 представляет собой R(-)-2-(4-трифторметансульфонилокси)фенил]-N-метансульфонилпропионамид или (2S)-2-(4-{[4-(трифторметил)-1,3-тиазол-2-ил]амино}фенил)-пропионовую кислоту или натриевую соль, предпочтительно, в виде натриевой соли.
6. Комбинация по любому из пп.1-5, где указанная астма представляет собой астму, характеризующуюся менее чем 15% улучшением исходного FEV1 после 14-дневного курса перорального приема преднизолона (40 мг/день) у пациентов, у которых наблюдается более чем 15% улучшение FEV1 после лечения ингаляционным агонистом β2.
7. Фармацевтическая композиция для лечения астмы, нечувствительной к кортикостероидам, содержащая эффективное количество комбинации по пп.1-5 и по меньшей мере один инертный фармацевтически приемлемый эксципиент.
8. Фармацевтическая композиция по п.7, где указанная астма представляет собой астму, характеризующуюся менее чем 15% улучшением исходного FEV1 после 14-дневного курса перорального приема преднизолона (40 мг/день) у пациентов, у которых наблюдается более чем 15% улучшение FEV1 после лечения ингаляционным агонистом β2.
| WO 2009085880 A2, 09.07.2009 | |||
| WO 2003080053 A1, 02.10.2003 | |||
| US 20070249672 A1, 25.10.2007 | |||
| WO 2011154738 A1, 15.12.2011 | |||
| WO 2014131852 A1, 04.09.2014 | |||
| PURAKKATTLE BIJU et al., "Steroidal C-21 heteroaryl thioethers | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |

