Изобретение относится к способу получения десфлурана (1,2,2,2-тетрафторэтилдифторметилового эфира, HCF2OCFHCF3), представляющего класс простых фторированных эфиров. Являясь фторорганическим ингаляционным анестетиком последнего поколения, десфлуран нашел широкое применение в хирургической практике, вследствие высокой эффективности и низкой растворимости в крови, что способствует более быстрому его выведению и минимизации негативных процессов для организма.
УРОВЕНЬ ТЕХНИКИ
Известно несколько способов получения десфлурана реакцией фторирования изофлурана фтористым водородом в присутствии галогенидов сурьмы (WO 2006076342 A2, ЕР 2167453 А2).
В частности, в заявке ЕА 012298 В1 описан способ получения десфлурана взаимодействием изофлурана HCF2OCHClCF3 с фтористым водородом, взятым в недостатке, в присутствии каталитических количеств пентафторида сурьмы (1,0-2,5 масс. %). Реакция протекает при температуре 15-25°С в течение 1-1,5 часов с выходом продукта 60-70%. Патент US 6800786 B1 также раскрывает способ получения десфлурана взаимодействием изофлурана с фтористым водородом (1,3-2,2 эквивалента) и пентахлоридом сурьмы (0,7-1,2 мольн. %). Реакция протекает при температуре 9-18°С в течение 6-7 часов и выходом продукта 70-85%.
Причины, препятствующие получению в известном способе требуемого технического результата, заключаются в использовании в качестве фторирующего реагента фтористого водорода, обладающего высокой токсичностью и опасностью при обращении, а также использовании в качестве исходного субстрата изофлурана, являющегося достаточно дорогим коммерческим продуктом.
Известен способ получения десфлурана (GB 2219292 A) реакцией фтордехлорирования изофлурана фторидом щелочного металла, например, фторидом калия в полярном апротонном растворителе, например, сульфолане. Реакцию проводят в автоклавном оборудовании в присутствии катализаторов межфазного переноса, например, тетраметиламмоний хлорид, тетра-н-бутиламмоний бромид, эфир 18-краун-6, при температуре 170-220°С в течение 4 часов с конверсией 35-70% и селективностью 94-98,5%.
Причины, препятствующие получению в известном способе требуемого технического результата, заключаются в достижении невысокой конверсии исходного изофлурана и проведении процесса в автоклавном оборудовании при высокой температуре и давлении.
Известен способ получения десфлурана (US 6054626) фторированием 2-дифторметокси-1,1,1-трифторэтана CF3CH2OCHF2 трифторидом кобальта. Реакция протекает при температуре 220°С с конверсией 67% и селективностью 57%.
Недостатком данного способа является необходимость проведения непрерывной регенерации трифторида кобальта путем пропускания через реакционную массу элементного фтора, являющегося чрезвычайно агрессивным и токсичным реагентом.
Известен способ получения десфлурана (US 5205914) из 1,2,2,2-тетрафторэтилметилового эфира CF3CFHOCH3. На первой стадии данного способа обработкой CF3CFHOCH3 хлором в присутствии радикального инициатора и под воздействием ультрафиолетового излучения получают смесь из монохлор-, дихлор- и трихлорпроизводных соединений с последующим выделением из смеси целевого продукта - 1,2,2,2-тетрафторэтил-дихлорметилового эфира CF3CFHOCHCl2. На второй стадии газофазное фторирование CF3CFHOCHCl2 фторирующим агентом, например, безводным фтористым водородом приводит к образованию 1,2,2,2-тетрафторэтилдифторметилового эфира HCF2OCFHCF3. Фторирование проводят при температуре 180°С в присутствии катализатора пятихлористой сурьмы или четыреххлористого олова.
Причины, препятствующие получению в известном способе требуемого технического результата, заключаются в использовании в качестве исходного субстрата 1,2,2,2-тетрафторэтилметилового эфира, не являющегося коммерческим продуктом. Описано несколько способов получения CF3CFHOCH3: из трихлорацетальдегида (US 10683252), из фторальметил гемиацеталя (US 4972040). Все известные способы получения 1,2,2,2-тетрафторэтилметилового эфира являются не пригодными для его промышленного получения, ввиду использования дорогостоящего сырья и достижения невысоких выходов продукта.
Кроме того, существенными недостатками известного способа получения десфлурана являются низкая селективность образования целевого продукта хлорирования CF3CFHOCHCl2, а также использование на стадии фторирования высокотоксичного реагента - фтористого водорода.
Таким образом, существует необходимость разработки способа получения 1,2,2,2-тетрафторэтилдифторметилового эфира (десфлурана), лишенного недостатков, присущих известным способам.
РАСКРЫТИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Задача, на решение которой направлено изобретение, заключается в разработке способа получения десфлурана без использования дорогого или коммерчески недоступного сырья, с применением простых химико-технологических процессов синтеза, сопровождающихся получением продуктов с высоким выходом и чистотой.
В соответствии с заявляемым изобретением решение данной задачи достигается тем, что разработан способ получения десфлурана, заключающийся во взаимодействии фторангидридных соединений общей формулы R-COF, где R=CCl3, CF2J, COF, с окисью гексафторпропилена, в присутствии фторида щелочного металла и полярного апротонного растворителя, с образованием продуктов конденсации общей формулы R-CF2OCF(CF3)COF, где R имеют вышеупомянутые значения; обработке продуктов конденсации общей формулы R-CF2OCF(CF3)COF, где R=CCl3, CF2J, олеумом; взаимодействии дигалогенангидридов общей формулы R'-CF2OCF(CF3)COF, где R'=COCl, COF, со спиртами; омылении образующегося эфира щелочью и термическом декарбоксилировании полученной соли с одновременной отгонкой образующегося десфлурана.
Таким образом, предложенный способ получения десфлурана заключается в проведении следующих химических стадий:
а) каталитическая конденсация ацилфторидов (Ia-с) с окисью гексафторпропилена с получением продуктов конденсации (IIa-с):
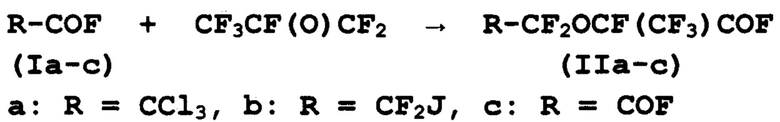
б) гидролиз продуктов конденсации (IIa, b) в олеуме с получением дигалогенангидридов (IIc, d):
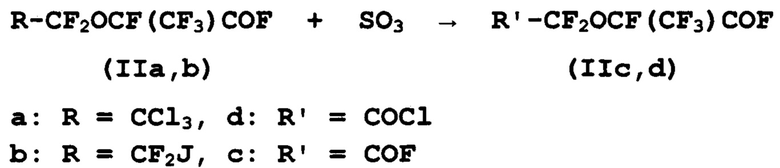
в) взаимодействие дигалогенангидридов (IIc, d) с метанолом с получением диметил-перфтор-2-метил-3-оксапентандиоата (III):
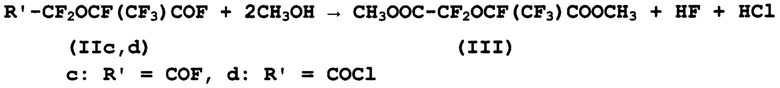
г) получение перфтор-2-метил-3-оксапентандиоата натрия (IV) и его термическое декарбоксилирование с получением целевого продукта - 1,2,2,2-тетрафторэтилдифторметилового эфира - десфлурана (V):
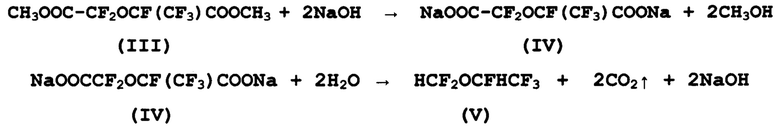
В соответствии с настоящим изобретением, конденсацию ацилфторидов (Ia-с) с окисью гексафторпропилена проводят при температуре 15-30°С. Конденсацию проводят в присутствии фторида щелочного металла и полярного апротонного растворителя. В предпочтительном варианте изобретения в качестве фторида щелочного металла используют фторид калия, в качестве полярного апротонного растворителя - сульфолан, смесь сульфолана и ацетонитрила. Допускается многократное использование каталитической системы без потери ее каталитической активности. В одном из вариантов настоящего изобретения подачу окиси гексафторпропилена проводят по сифону в зону реакции в непрерывном режиме.
В способе по настоящему изобретению реагенты используются в мольном отношении ацилфторйд (Ia-с): окись гексафторпропилена, равном 1:(0,36-0,60). Использование заявленного соотношения реагентов является существенным признаком настоящего изобретения и создает условия, препятствующие протеканию дальнейшей олигомеризации ацил фторидов (Ia-с), поэтому в качестве основных продуктов получают продукты конденсации (IIa-с) с селективностью 98% и выходом 95-97%.
В соответствии с настоящим изобретением, гидролиз продуктов конденсации (IIa, b) проводят в олеуме с содержанием серного ангидрида 40-65%, предпочтительно 60-65%, при температуре 60-70°С в течение 24 часов. Выделение дигалогенангидридов (IIс, d), образующихся в результате гидролиза продуктов (IIa, b) проводят отмывкой 100%-ной серной кислотой и фракционированием.
В способе по настоящему изобретению процесс гидролиза (IIa) проводят в присутствии катализаторов, выбранных из группы, включающей соли ртути (I), соли ртути (II) или их смеси, в частности сульфаты, нитраты, хлориды, бромиды; галогены, в частности, бром, хлор, йод. В предпочтительном варианте изобретения используют ртуть (II) сернокислую. Гидролиз (IIb) предпочтительно проводят без использования катализаторов.
В соответствии с настоящим изобретением, взаимодействие дигалогенангидридов (IIc, d) с низкомолекулярными спиртами, предпочтительно метанолом, проводят при температуре 5-30°С с образованием диметил-перфтор-2-метил-3-оксапентандиоата (III).
В соответствии с настоящим изобретением, омыление диметил-перфтор-2-метил-3-оксапентандиоата (III) проводят обработкой водным раствором гидроксида натрия или калия в высокотемпературном растворителе, выбранном из группы, включающей этиленгликоль, сульфолан, диглим, этилцеллозольв, с образованием перфтор-2-метил-3-оксапентандиоата натрия (IV). Суспензия полученной соли (IV) подвергается термическому разложению с выделением углекислого газа, образованием десфлурана (V) и его выделением по мере накопления из зоны реакции известными методами.
В способе по настоящему изобретению взаимодействие диметил-перфтор-2-метил-3-оксапентандиоата (III) с водным раствором гидроксида натрия проводят при температуре 25-55°С, предпочтительно 35-40°С. Реагенты расходуются в мольном отношении диметил-перфтор-2-метил-3-оксапентандиоат (III): вода: гидроокись натрия, равном 1,0:2,2:2,0. Дальнейшее термическое разложение (пиролиз) перфтор-2-метил-3-оксапентандиоата натрия (IV) проводят, нагревая суспензию соли (IV) в растворителе до температуры 135-185°С с одновременной отгонкой десфлурана (V) из зоны реакции.
В соответствии с настоящим изобретением, стадии омыления диметил-перфтор-2-метил-3-оксапентадионата (III) термическое декарбоксилирование перфтор-2-метил-3- оксапентандиоата натрия (IV) проводят в одном реакторе.
Сведения, подтверждающие возможность осуществления изобретения, но не ограничивающие его, иллюстрируются следующими примерами.
ПРИМЕРЫ
Пример №1.
Синтез 2-(трифторметил)-3-окса-2,4,4-трифтор-5,5,5-трихлорпентаноил-фторида (IIa).
В стеклянный реактор объемом 2 дм3, снабженный рубашкой, термопарой, перемешивающим устройством, линией подачи газообразных реагентов, обратным холодильником, при перемешивании загружают 400 г сульфолана и 17 г фторида калия, предварительно прокаленного при температуре 350°С.
При температуре в реакторе не менее 25°С проводят загрузку 1400 г (8,5 моль) трихлорацетилфторида (Ia) и выдержку при перемешивании в течение 30 минут. По окончании выдержки начинают дозировку окиси гексафторпропилена со скоростью 29-30 г/час. Дозировку 700 г (4,25 моль) окиси гексафторпропилена проводят при температуре 25-30°С в течение 24 часов.
По окончании синтеза реакционную массу расслаивают и отделяют 1880 г нижнего фторорганического слоя в виде бесцветной подвижной жидкости. Содержимое реактора нагревают при перемешивании до 135-140°С и отгоняют дополнительную порцию фторорганических продуктов. Суммарное количество фторорганических продуктов составило 2050 г, состав по данным анализа ГЖХ (мольн. %): трихлорацетилфторид - 46,3%; 2-(трифторметил)-3-окса-2,4,4-трифтор-5,5,5-трихлорпентаноилфторид - 52,1%; 2,5-ди(трифторметил)-3,6-диокса-2,4,4,5,7,7-гексафтор-8,8,8-трихлороктаноилфторид - 1,6%. Суспензию фторида калия в растворителе используют в рецикле без дополнительной очистки.
Фторорганические продукты синтеза ректифицируют, фракцию исходного трихлорацетилфторида (675 г) направляют в рецикл. В качестве основной фракции получают 1359 г 2-(трифторметил)-3-окса-2,4,4-трифтор-5,5,5-трихлорпентаноилфторида (IIa) с содержанием основного вещества 98,0%. Конверсия - 50%, выход продукта IIa от теоретического - 96,2%.
Пример №2.
Синтез 2-(трифторметил)-3-окса-2,4,4,5,5-пентафтор-5-йодпентаноилфторида (IIb).
В стеклянный реактор объемом 2 дм3, снабженный рубашкой, термопарой, перемешивающим устройством, линией подачи газообразных реагентов, обратным холодильником, при перемешивании загружают 430 г сульфолана и 22 г фторида калия, предварительно прокаленного при температуре 350°С.
При температуре в реакторе не менее 25°С проводят загрузку 1785 г (8,0 моль) дифторйодацетилфторида (Ib) и выдержку при перемешивании в течение 30 минут. По окончании выдержки начинают дозировку окиси гексафторпропилена со скоростью 28-29 г/час. Дозировку 800 г (4,8 моль) окиси гексафторпропилена проводят при температуре 25-30°С в течение 28 часов.
По окончании синтеза реакционную массу расслаивают и отделяют 2310 г нижнего фторорганического слоя в виде бесцветной подвижной жидкости. Содержимое реактора нагревают при перемешивании до 115-120°С и отгоняют дополнительную порцию фторорганических продуктов. Суммарное количество фторорганических продуктов составило 2515 г, состав по данным анализа ГЖХ (мольн. %): дифторйодацетилфторид - 40,7%; 2-(трифторметил)-3-окса-2,4,4,5,5-пентафтор-5-йодпентаноилфторид - 57,8%; 2,5-ди(трифторметил)-3,6-диокса-2,4,4,5,7,7,8,8-октафтор-8-йодоктаноилфторид - 1,5%. Суспензию фторида калия в растворителе используют в рецикле без дополнительной очистки.
Фторорганические продукты синтеза ректифицируют, фракцию исходного дифторйодацетилфторида (690 г) направляют в рецикл. В качестве основной фракции получают 1798 г 2-(трифторметил)-3-окса-2,4,4,5,5-пентафтор-5-йодпентаноилфторида (IIb) с содержанием основного вещества 98,4%. Конверсия - 60%, выход продукта IIb от теоретического - 97,3%.
Пример №3.
Синтез перфтор-2-метил-3-оксапентан-1,5-диоилфторида (IIc).
В стеклянный реактор объемом 2 дм3, снабженный рубашкой, термопарой, перемешивающим устройством, линией подачи газообразных реагентов, обратным холодильником, при перемешивании загружают 350 г смеси растворителей, состоящей из 70 г ацетонитрила и 280 г сульфолана, и 20 г фторида калия, предварительно прокаленного при температуре 350°С.
При температуре в реакторе не более 15°С проводят загрузку 940 г (10,0 моль) оксалилфторида (Ic) и проводят выдержку при перемешивании в течение 30 минут. По окончании выдержки начинают дозировку окиси гексафторпропилена со скоростью 30 г/час. Дозировку 600 г (3,6 моль) окиси гексафторпропилена проводят в течение 20 часов.
По окончании синтеза реакционную массу расслаивают и отделяют 1390 г нижнего фторорганического слоя в виде бесцветной подвижной жидкости. Содержимое реактора нагревают при перемешивании до 75°С и отгоняют дополнительную порцию фторорганических продуктов. Суммарное количество фторорганических продуктов составило 1510 г, состав по данным анализа ГЖХ (мольн. %): оксалилфторид - 51,2%, перфтор-2-метил-3-оксапентан-1,5-диоилфторид - 47,0%, перфтор-2,5-метил-3,6-диоксаоктан-1,8-диоилфторид - 1,8%. Суспензию фторида калия в смеси растворителей используют в рецикле без дополнительной очистки.
Фторорганические продукты синтеза ректифицируют, фракцию исходного оксалилфторида (579 г) направляют в рецикл. В качестве основной фракции получают 910 г перфтор-2-метил-3-оксапентан-1,5-диоилфторида (IIc) с содержанием основного вещества 98,4%. Конверсия - 37%, выход продукта IIc от теоретического - 95,7%.
Пример №4.
Синтез перфтор-2-метил-3-оксапентандиоил-1-фторид-5-хлорида (IId).
В стеклянный реактор объемом 2 дм3, снабженный рубашкой, термопарой, мешалкой и обратным холодильником, при перемешивании загружают 1365 г 60-65% олеума, 20 г ртути (II) сернокислой и 1359 г (4,1 моль) 2-(трифторметил)-3-окса-2,4,4-трифтор-5,5,5-трихлорпентаноилфторида (IIa). По окончании загрузки содержимое реактора выдерживают при перемешивании и температуре 60-65°С в течение 24 часов.
По окончании выдержки фторорганические продукты гидролиза отгоняют из реакционной массы до температуры куба 130°С. В результате получают 1480 г бесцветной дымящей во влажном воздухе подвижной жидкости. Полученный отгон отмывают 300 г 100%) серной кислоты при перемешивании в течение 10 минут. Верхний фторорганический слой отделяют и фракционируют с получением 665 г (96,5%) перфтор-2-метил-3-оксапентандиоил-1-фторид-5-хлорида (IId).
Выход продукта IId от теоретического - 78,7%.
Пример №5.
Синтез перфтор-2-метил-3-оксапентан-1,5-диоилфторида (IIc).
В стеклянный реактор объемом 2 дм, снабженный рубашкой, термопарой, мешалкой и обратным холодильником, при перемешивании загружают 1425 г 60-65% олеума и 1798 г 2-(трифторметил)-3-окса-2,4,4,5,5-пентафтор-5-йодпентаноилфторида (IIb). По окончании загрузки содержимое реактора выдерживают при перемешивании и температуре 65-70°С в течение 24 часов.
По окончании выдержки фторорганические продукты гидролиза отгоняют из реакционной массы до температуры куба 130°С. В результате получают 1277 г бесцветной дымящей во влажном воздухе подвижной жидкости. Полученный отгон отмывают 250 г 100%) серной кислоты при перемешивании в течение 10 минут. Верхний фторорганический слой отделяют и фракционируют с получением 951 г (97,6%) перфтор-2-метил-3-оксапентан-1,5-диоилфторида (IIc).
Выход продукта IIc от теоретического - 88,5%.
Пример №6.
Синтез диметил-перфтор-2-метил-3-оксапентандиоата (III).
В стеклянный реактор объемом 2 дм3, снабженный рубашкой, термопарой, обратным холодильником, мешалкой, донным сливом, капельной воронкой, при перемешивании загружают 500 мл абсолютного метилового спирта и 336 г (8,0 моль) фторида натрия, предварительно прокаленного при температуре 300°С.
Полученную суспензию охлаждают до температуры 5°С и проводят дозировку 910 г (3,5 моль) перфтор-2-метил-3-оксапентан-1,5-диоилфторида (IIc) в течение 4,5 часов. По окончании дозировки реакционную массу перемешивают 30 минут и отфильтровывают от бифторида натрия с промывкой осадка на фильтре 300 мл метилового спирта. Из объединенных фильтратов отгоняют метанол при пониженном давлении и получают в остатке 924 г диметил-перфтор-2-метил-3-оксапентандиоата (III), с содержанием основного вещества 97,3%. Выход от теоретического 92,9%.
Пример №7.
Синтез диметил-перфтор-2-метил-3-оксапентандиоата (III).
К охлажденному до температуры 5°С абсолютному метиловому спирту (300 мл) при перемешивании по каплям прибавляют 665 г перфтор-2-метил-3-оксапентандиоил-1-фторид-5-хлорида (IId). Реакционную массу кипятят в течение 1 часа, и размывают ледяной водой, органический слой промывают холодной водой, сушат хлористым кальцием. Получают 555 г диметил-перфтор-2-метил-3-оксапентандиоата (III), с содержанием основного вещества 93,4%. Выход от теоретического 80,1%.
Пример №8.
Синтез 1,2,2,2-тетрафторэтилдифторметилового эфира - десфлурана (V).
В стеклянный реактор объемом 5 дм, снабженный рубашкой, термопарой, прямым холодильником, мешалкой, донным сливом, капельной воронкой, охлаждаемой поглотительной емкостью объемом 4 л, низкотемпературной ловушкой объемом 0,5 л, загружают 1600 мл этиленгликоля, 100 мл дистиллированной воды и 260 г (6,5 моль) гидроокиси натрия.
Содержимое реактора перемешивают в течение 30 минут, охлаждают до температуры 10°С и прикапывают 924 г (3,25 моль) диметил-перфтор-2-метил-3-оксапентандиоата (III) в течение 3 часов, не допуская подъема температуры выше 30°С. По окончании дозировки реакционную массу выдерживают при перемешивании в течение 30 минут при комнатной температуре и получают суспензию перфтор-2-метил-3-оксапентандиоата натрия (IV) в этиленгликоле.
После чего реакционную массу нагревают до температуры 155°С, при которой начинается процесс декарбоксилирования полученной динатриевой соли (IV) и отгонка сырца десфлурана. Проходя через охлаждаемый до температуры +5°С адсорбер углекислого газа, заполненный 2,6 кг 25% водного раствора гидроокиси калия, сырец десфлурана конденсировался в низкотемпературной ловушке, охлаждаемой до температуры минус 35°С. В течение 5 часов температуру реакционной массы поднимали до 175°С и выдерживали до прекращения газовыделения еще в течение 3 часов.
Получают 481 г сырца десфлурана с содержанием основного вещества 98,9% (ГЖХ). Выход от теоретического 88%.
Цитируемая литература
1. Патент ЕА 012298 В1, «Способ получения 1,2,2,2-тетрафторэтилдифторметилового эфира», Росс К. Таррелл, Джошуа А. Левинсон, опубликовано 28.08.2009.
2. Патент US 6800786 B1, «Preparation of desfTurane», Leonid A. Rozov, Ralph A. Lessor, опубликовано 05.10.2004.
3. Патент WO 2006076342 A2, «Synthesis of fluorinated ethers», Swinson Joel, Jones Barry, and other, опубликовано 20.07.2006.
4. Патент GB 2219292 A, «Process of preparing of 1,2,2,2-tetrafluoroethyl-difluoromethyl ester», Kawai Toshikazu, опубликовано 06.12.1989.
5. Патент US 6054626, «Synthesis of fluorinated ethers», Owen Ross Chambers, Roderic Nigel Fraser Simpson, опубликовано 25.04.2000.
6. Патент US 5205914, «Synthesis of desflurane», Leonid A. Rozov, Chialang Huang, Gerald G. Vernice, опубликовано 27.04.1993.
7. Патент US 10683252, «Production method for 1,2,2,2-tetrafluoroethyldifluoromethyl ester (desflurane)», Kenji Hosoi et al., опубликовано 16.06.2020.
8. Патент US 4972040, «Process for the preparation of CHF2OCHFCF3», Mark L. Robin, Donald F. Halpern, опубликовано 20.11.1990.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения перфтор-4-(фторсульфонил)бутилвинилового эфира | 2022 |
|
RU2800857C1 |
| ПЕРФТОР(2-ФТОРСУЛЬФАТЭТОКСИ)ПРОПИОНИЛ ФТОРИД | 2010 |
|
RU2443685C1 |
| ПЕРФТОР[(2-ФТОРСУЛЬФАТ)ЭТИЛАЛЛИЛОВЫЙ] ЭФИР | 2010 |
|
RU2430914C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРАНГИДРИДА ТРИФТОРАКРИЛОВОЙ КИСЛОТЫ | 1992 |
|
RU2035449C1 |
| СПОСОБ ОЧИСТКИ ГЕКСАФТОРПРОПИЛЕНА | 1997 |
|
RU2150457C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОРИРОВАННЫХ ПРОСТЫХ ЭФИРОВ С КОНЦЕВЫМИ ФУНКЦИОНАЛЬНЫМИ ГРУППАМИ | 2000 |
|
RU2179548C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСАПЕРФТОРАЛКАНСУЛЬФОКИСЛОТ И ИХ СОЛЕЙ | 2012 |
|
RU2503659C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕКСАФТОРЭТАНА | 1997 |
|
RU2124493C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПЕРФТОР(2,2-ДИМЕТИЛ-1,3-ДИОКСОЛА) | 2016 |
|
RU2633352C1 |
| ФТОРСОДЕРЖАЩИЙ ПАРАФИН В КАЧЕСТВЕ СМАЗКИ ДЛЯ ОБРАБОТКИ ПОВЕРХНОСТИ ПЛАСТИКОВЫХ ЛЫЖ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2011 |
|
RU2473532C1 |
Изобретение относится к способу получения десфлурана - фторорганического ингаляционного анестетика последнего поколения, который нашел широкое применение в хирургической практике, вследствие высокой эффективности и низкой растворимости в крови, что способствует более быстрому его выведению и минимизации негативных процессов для организма. Предлагаемый способ включает стадию конденсации фторангидридных соединений общей формулы R-COF, где R=CCl3, CF2J, COF, с окисью гексафторпропилена в мольном отношении, равном 1:(0,36-0,60), в присутствии фторида щелочного металла и полярного апротонного растворителя с образованием продуктов конденсации общей формулы R-CF2OCF(CF3)COF, где R имеет вышеупомянутые значения. Далее осуществляют обработку продуктов конденсации общей формулы R-CF2OCF(CF3)COF, где R=CCl3, CF2J, олеумом при температуре 60-70°С в течение 24 часов и выделение образующихся дигалогенаногидридов отмывкой 100%-ной серной кислотой и фракционированием. Затем полученные дигалогенангидриды общей формулы R'-CF2OCF(CF3)COF, где R'=COCl, COF, подвергают взаимодействию с низкомолекулярными спиртами при температуре 5-30°С. Образующийся диалкил-перфтор-2-метил-3-оксапентандиоат омыляют щелочами в высокотемпературном растворителе с получением перфтор-2-метил-3-оксапентандиоата, который подвергают термическому декарбоксилированию при температуре 135-185°С, с одновременной отгонкой образующегося десфлурана. Технический результат – создание более технологичного в промышленном масштабе способа, исключающего использование фтористого водорода и нестандартного технологического оборудования при проведении всех стадий синтеза и очистки с высоким выходом и селективностью. 9 з.п. ф-лы, 8 пр.
1. Способ получения десфлурана, включающий следующие стадии:
(а) конденсацию фторангидридных соединений общей формулы R-COF, где R=CCl3, CF2J, COF, с окисью гексафторпропилена в мольном отношении, равном 1:(0,36-0,60), в присутствии фторида щелочного металла и полярного апротонного растворителя с образованием продуктов конденсации общей формулы R-CF2OCF(CF3)COF, где R имеет вышеупомянутые значения; (б) обработку продуктов конденсации общей формулы R-CF2OCF(CF3)COF, где R=CCl3, CF2J олеумом при температуре 60-70°С в течение 24 часов и выделением образующихся дигалогенаногидридов отмывкой 100%-ной серной кислотой и фракционированием; (в) взаимодействие дигалогенангидридов общей формулы R'-CF2OCF(CF3)COF, где R'=COCl, COF, с низкомолекулярными спиртами при температуре 5-30°С; (г) омыление образующегося диалкил-перфтор-2-метил-3-оксапентандиоата щелочами в высокотемпературном растворителе с получением перфтор-2-метил-3-оксапентандиоата и термическое декарбоксилирование полученной соли при температуре 135-185°С с одновременной отгонкой образующегося десфлурана.
2. Способ по п. 1, характеризующийся тем, что стадию (а) проводят в присутствии фторида калия и сульфолана.
3. Способ по п. 1, характеризующийся тем, что стадию (а) проводят в присутствии фторида калия, сульфолана и ацетонитрила.
4. Способ по пп. 1-3, характеризующийся тем, что на стадии (а) подачу окиси гексафторпропилена проводят по сифону в зону реакции в непрерывном режиме.
5. Способ по п. 1, характеризующийся тем, что стадию (б) проводят в присутствии катализаторов, выбранных из группы, включающей соли ртути (I), соли ртути (II) или их смеси, в частности сульфаты, нитраты, хлориды, бромиды; галогены, в частности бром, хлор, йод.
6. Способ по п. 1, характеризующийся тем, что стадию (б) проводят без использования катализаторов.
7. Способ по пп. 5, 6, характеризующийся тем, что стадию (б) проводят с использованием олеума с содержанием серного ангидрида 60-65%.
8. Способ по п. 1, характеризующийся тем, что стадию (в) проводят с использованием метанола.
9. Способ по п. 1, характеризующийся тем, что стадию (г) проводят с использованием водного раствора гидроксида натрия или калия в высокотемпературном растворителе, выбранном из группы, включающей этиленгликоль, сульфолан, диглим, этилцеллозольв.
10. Способ по п. 9, характеризующийся тем, что на стадии (г) омыление диметил-перфтор-2-метил-3-оксапентандиоата и термическое декарбоксилирование перфтор-2-метил-3-оксапентандиоата натрия проводят в одном реакторе.
| US 5205914 A1, 27.04.1993 | |||
| Прибор, сигнализирующий о повышении или понижении температуры | 1927 |
|
SU12298A1 |
| Hariharan Sivaramakrishnan et al | |||
| The Preparation of Desflurane by the Vapor-Phase Fluorination of Isoflurane | |||
| Organic Process Research & Development, 2011, 15(3), 585-592 | |||
| ИНФОРМАЦИОННАЯ СИСТЕМА | 1998 |
|
RU2167453C2 |
| US 9150480 B2, 06.10.2015. | |||

