ПРИТЯЗАНИЕ НА ПРИОРИТЕТ
По настоящей заявке испрашивается приоритет по временной заявке США № 62/989041, поданной 13 марта 2020 года, содержание которое включено, таким образом, в настоящее описание в качестве ссылки в полном объеме.
ЗАЯВЛЕНИЕ О ПОДДЕРЖКЕ ПО ФЕДЕРАЛЬНОМУ ГРАНТУ
Настоящее изобретение создано при поддержке государства по гранту № 2P50CA070907-21A1, выданному Национальным институтом онкологии США. Государство обладает некоторыми правами на изобретение.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к области медицины, фармакологии, молекулярной биологии и онкологии. Более конкретно, настоящее изобретение относится к способам и композициям для лечения рака с использованием последовательной терапии 6-тио-dG, ингибитором контрольных точек и/или лучевой терапии.
УРОВЕНЬ ТЕХНИКИ
Иммунотерапия произвела революцию в лечении многих видов рака в области иммуноонкологии (Brahmer et al., 2012; Hodi et al., 2010; Ribas and Wolchok, 2018; Topalian et al., 2012). Наиболее общеупотребительными способами иммунотерапии являются блокады контрольных точек PD-L1/PD-1, одобренные FDA для рака на поздних стадиях, таких как меланома, немелкоклеточный рак легких, рак молочной железы, рак шейки матки, рак толстого кишечника, рак головы и шеи, лимфома Ходжкина, рак печени, рак легких, почечно-клеточный рак, рак желудка, рак прямой кишки и любая солидная опухоль, неспособная к репарации ошибок в своей ДНК, возникающих во время репликации (Garon et al., 2015; Ribas et al., 2016; Rizvi et al., 2015b; Socinski et al., 2018; National Cancer Institute). Несмотря на успех иммунотерапии, многие пациенты не отвечают хорошо на эти способы терапии по причине иммуносупрессорного микроокружения опухоли, иммуногенности опухоли и возникновения первичной и адаптивной резистентности (Chen and Han, 2015; Gide et al., 2018). Хотя недавние исследования показали, что большое количество опухолевых мутаций и неоантигенов частично обуславливают ответы пациентов с раком на блокаду контрольных точек, существует значительное количество пациентов с высоким количеством мутаций и неоантигенов, не отвечающих хорошо (Le et al., 2017; Mandal et al., 2019; Rizvi et al., 2015a), что позволяет предполагать, что неоантигенов недостаточно для провоцирования противоопухолевых иммунных ответов. Таким образом, существует острая потребность в идентификации других факторов для лучших иммунных ответов и разработке новых подходов для улучшения общей выживаемости пациентов.
Возникновение эффективных противоопухолевых адаптивных иммунных ответов требует презентирования опухолевого антигена антигенпрезентирующими клетками, активация которых в значительной степени основывается на адекватном врожденном распознавании. Врожденное распознавание зачастую основывается на сигналах опасности, таких как белок 1 высокомобильной группы, внеклеточный АТФ и опухолевая ДНК, высвобождаемая подвергаемыми стрессу опухолевыми клетками (Kroemer et al., 2013; Pitt et al., 2017). Недавние исследования показали важность распознавания цитозольной ДНК при лучевой и повреждающей ДНК терапии (Deng et al., 2014; Sen et al., 2019). Присутствие ДНК в цитоплазме, например, в форме микроядер (небольших ДНК-содержащих органелл), теряющих внешние мембраны, может запускать иммунные ответы. Микроядра являются продуктами повреждения хромосом в результате генотоксического стресса и неправильного расщепления хромосом во время деления клеток (Fenech et al., 2011). Цитозольный сенсор ДНК cGAS распознает микроядра и превращает ГТФ (гуанозинтрифосфат) и АТФ (аденозинтрифосфат) во вторичный мессенджер cGAMP (циклический ГМФ-АМФ) (Wu et al., 2013). Затем адаптерный белок Стимулятор генов интерферонов (STING) связывается с cGAMP (Ablasser et al., 2013; Diner et al., 2013; Gao et al., 2013; Zhang et al., 2013). Этот сложный процесс приводит к активации TANK-связывающей киназы 1 (TBK1) и ИФН-регуляторного фактора 3 (IRF3) (Liu et al., 2015; Tanaka and Chen, 2012) и дальнейшей активации последующей транскрипции ИФН типа I и других цитокинов (обзор в (Li and Chen, 2018)), которые в конечном итоге повышают врожденное распознавание.
Эукариотические линейные хромосомы кэпированы специальными структурами, названными теломерами (TTAGGG), необходимыми для поддержания хромосомной стабильности (обзор в (Blackburn, 1991)). Теломеры составляют конечные ~10 т.п.н. всех хромосом человека и конечные 12-80 т.п.н. всех хромосом мыши (Lansdorp et al., 1996; Zijlmans et al., 1997). Во всех соматических клетках человека теломеры укорачиваются с каждым делением клетки из-за проблемы репликации концов и отсутствия механизма поддержания теломер (обзор в (Greider, 1996)). Однако, одноклеточные эукариоты, клетки зародышевой линии и бессмертные раковые клетки сохраняют свои теломеры с постоянной длиной почти всегда, активируя фермент теломеразу (Greider and Blackburn, 1985; McEachern and Blackburn, 1996; Morin, 1989; Nakamura et al., 1997; Singer and Gottschling, 1994; Yu et al., 1990). Теломераза представляет собой фермент обратную транскриптазу, удлиняющую теломеры, добавляя повторы TTAGGG на концы хромосом, и экспрессирующуюся в ~90% опухолей человека, но не в большинстве нормальных клеток (Shay and Bacchetti, 1997). Таким образом, теломераза является привлекательной мишенью для разработки противоопухолевых терапевтических средств.
Аналог нуклеозида, 6-тио-2’-дезоксигуанозин (6-тио-dG), представляет собой новый и эффективный терапевтический подход в области онкологии. Известно, что его встраивание в синтезируемые de novo теломеры с помощью теломеразы индуцирует повреждение теломерной ДНК (Mender et al., 2015a). Это приводит к быстрому уменьшению опухоли или прекращению роста во многих моделях ксенотрансплантатов, полученных из опухолей, с минимальными побочными эффектами (Mender et al., 2018; Sengupta et al., 2018; Zhang et al., 2018). Наиболее важным преимуществом этой направленной на теломеры терапии по сравнению с прямыми ингибиторами теломеразы является то, что 6-тио-dG не имеет длительного лаг-периода в отношении эффектов уничтожения опухоли. Кроме того, он не ингибирует напрямую теломеразу, но, главным образом, распознается теломеразой относительно других полимераз и встраивается в теломеры, что приводит незамедлительной терминации цепи ДНК. Важно, что его эффект не зависит от исходной длины теломеры благодаря захвату опухолей теломеразы, что приводит к нестабильным теломерам (Mender et al., 2015b).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Таким образом, в одном из аспектов настоящее изобретение относится к способам лечения рака у индивидуума, включающим введение указанному индивидууму эффективного количества 6-тио-2’-дезоксигуанозина (6-тио-dG) с последующим лечением ингибитором иммунных контрольных точек на цикл лечения. В некоторых вариантах осуществления рак выбран из одного или более из рака поджелудочной железы, рака легких, мезотелиомы, рака желудка, рака пищевода, рака печени, рака желчевыводящих протоков, рака мочевого пузыря, рака головы и шеи, рака роковой полости, рака носоглотки, рака головного мозга у взрослых, рака толстого кишечника, рака прямой кишки, колоректального рака, рака предстательной железы, рака яичников, рака шейки матки, рака матки, рака яичка, лимфомы, лейкоза, рака кожи, рака молочной железы, рака почки, нейробластомы, карциномы из клеток Меркеля, миелодиспластического синдрома, миелофиброза и множественной миеломы.
В некоторых вариантах осуществления ингибитор иммунных контрольных точек является ингибитором PD-1, ингибитором PD-L1 или ингибитором CTLA-4. В одном из вариантов осуществления ингибитор иммунных контрольных точек является комбинацией одного или более ингибиторов CTLA-4, одного или более ингибиторов PD-1 или одного или более ингибиторов PD-L1.
В некоторых вариантах осуществления ингибитор PD-1 выбран из одного или более из пембролизумаба, ниволумаба, цемиплимаба, JTx-4014, сасанлимаба, будигалимаба, BI 754091, спартализумаба, камрелизумаба, синтилимаба, тислелизумаба, зимберелимаба, торипалимаба, достарлимаба, INCMGA00012, AMP-224, REGN2810, BMS-936558, SHR1210, IBI308, PDR001, BGB-A317, BCD-100, JS001 и AMP-515.
В некоторых вариантах осуществления ингибитор PD-L1 выбран из одного или более из атезолизумаба, авелумаба, косибелимаба, бинтрафуспа альфа, дурвалумаба, MGD013, KNO35, KN046, AUNP12, CA-170 и BMS-9986189.
В некоторых вариантах осуществления ингибитор CTLA-4 выбран из одного или более из ипилимумаба и тремелимумаба.
В некоторых вариантах осуществления способов, представленных в настоящем описании, 6-тио-dG вводят в течение от приблизительно 1 до приблизительно 5 дней за терапевтический цикл. В некоторых вариантах осуществления ингибитор контрольных точек вводят в течение от приблизительно 1 до приблизительно 3 дней за терапевтический цикл.
В рамках изобретения термин "терапевтический цикл" означает от приблизительно 1 до приблизительно 12 недель между введениями терапевтических средств.
В одном из вариантов осуществления способов, представленных в настоящем описании, 6-тио-dG и ингибитор контрольных точек вводят в комбинации с химиотерапевтическим средством, гормональной терапией, терапией токсинами или хирургическим вмешательством.
В другом варианте осуществления настоящее изобретение относится к способам лечения рака у индивидуума, нуждающегося в лечении, включающим введение указанному индивидууму 6-тио-dG с последующим лечением цемиплимабом (Libtayo®), где рак выбран из одного или более из группы, состоящей из рака поджелудочной железы, рака легких, мезотелиомы, рака желудка, рака пищевода, рака печени, рака желчевыводящих протоков, рака мочевого пузыря, рака головы и шеи, рака роковой полости, рака носоглотки, рака головного мозга у взрослых, рака толстого кишечника, рака прямой кишки, колоректального рака, рака предстательной железы, рака яичников, рака шейки матки, рака матки, рака яичка, лимфомы, лейкоза, рака кожи, рака молочной железы, рака почки, нейробластомы, карциномы из клеток Меркеля, миелодиспластического синдрома, миелофиброза и множественной миеломы. В некоторых вариантах осуществления этого способа 6-тио-dG вводят в течение от приблизительно 1 до приблизительно 5 дней за терапевтический цикл. В некоторых вариантах осуществления способа цемиплимаб вводят в течение от приблизительно 1 до приблизительно 3 дней за терапевтический цикл. В одном из вариантов осуществления способа 6-тио-dG и цемиплимаб вводят в комбинации с химиотерапевтическим средством, гормональной терапией, терапией токсинами или хирургическим вмешательством.
В одном из вариантов осуществления настоящее изобретение относится к способам лечения рака у индивидуума, включающим введение указанному индивидууму 6-тио-dG с последующим лечением атезолизумабом, где рак выбран из одного или более из группы, состоящей из рака поджелудочной железы, рака легких, мезотелиомы, рака желудка, рака пищевода, рака печени, рака желчевыводящих протоков, рака мочевого пузыря, рака головы и шеи, рака роковой полости, рака носоглотки, рака головного мозга у взрослых, рака толстого кишечника, рака прямой кишки, колоректального рака, рака предстательной железы, рака яичников, рака шейки матки, рака матки, рака яичка, лимфомы, лейкоза, рака кожи, рака молочной железы, рака почки, нейробластомы, карциномы из клеток Меркеля, миелодиспластического синдрома, миелофиброза и множественной миеломы. В некоторых вариантах осуществления этого способа 6-тио-dG вводят в течение от приблизительно 1 до приблизительно 5 дней за терапевтический цикл. В некоторых вариантах осуществления способа атезолизумаб вводят в течение от приблизительно 1 до приблизительно 3 дней за терапевтический цикл. В одном из вариантов осуществления способа 6-тио-dG и атезолизумаб вводят в комбинации с химиотерапевтическим средством, гормональной терапией, терапией токсинами или хирургическим вмешательством.
В другом настоящее изобретение относится к способам лечения рака у индивидуума, включающим введение указанному индивидууму 6-тио-dG с последующим лечением ингибитором иммунных контрольных точек, вводимым в комбинации с лучевой терапией. В некоторых вариантах осуществления ингибитор контрольных точек является ингибитором PD-L1, PD-1 или CTAL-4. В некоторых вариантах осуществления ингибитор PD-L1 выбран из одного или более из атезолизумаба, авелумаба, косибелимаба, бинтрафуспа альфа, дурвалумаба, MGD013, KNO35, KN046, AUNP12, CA-170 и BMS-9986189. В некоторых вариантах осуществления ингибитор PD-L1 является атезолизумабом. В некоторых вариантах осуществления ингибитор PD-1 выбран из одного или более из пембролизумаба, ниволумаба, цемиплимаба, JTx-4014, сасанлимаба, будигалимаба, BI 754091, спартализумаба, камрелизумаба, синтилимаба, тислелизумаба, зимберелимаба, торипалимаба, достарлимаба, INCMGA00012, AMP-224, REGN2810, BMS-936558, SHR1210, IBI308, PDR001, BGB-A317, BCD-100, JS001 и AMP-515. В некоторых вариантах осуществления ингибитор PD-1 является цемиплимабом. В некоторых вариантах осуществления ингибитор CTLA-4 является ипилимумабом или тремелимумабом. В некоторых вариантах осуществления подвергаемый лечению рак выбран из одного или более из группы, состоящей из рака поджелудочной железы, рака легких, мезотелиомы, рака желудка, рака пищевода, рака печени, рака желчевыводящих протоков, рака мочевого пузыря, рака головы и шеи, рака роковой полости, рака носоглотки, рака головного мозга у взрослых, рака толстого кишечника, рака прямой кишки, колоректального рака, рака предстательной железы, рака яичников, рака шейки матки, рака матки, рака яичка, лимфомы, лейкоза, рака кожи, рака молочной железы, рака почки, нейробластомы, карциномы из клеток Меркеля, миелодиспластического синдрома, миелофиброза и множественной миеломы. В некоторых вариантах осуществления подвергаемый лечению рак является раком поджелудочной железы, раком легких, раком желудка, раком печени, раком мочевого пузыря, раком головы и шеи, раком ротовой полости, раком носоглотки, раком головного мозга, раком толстого кишечника, раком предстательной железы, раком яичников, раком шейки матки, раком яичка, лимфомой, лейкозом, раком кожи или раком молочной железы. В некоторых вариантах осуществления рак головного мозга является раком головного мозга взрослых. В некоторых вариантах осуществления сначала проводят лучевую терапию с последующим лечением одним или более ингибиторами контрольных точек. В некоторых вариантах осуществления лучевую терапию проводят после введения одного или более ингибиторов контрольных точек.
В некоторых вариантах осуществления способов по изобретению подвергаемый лечению рак является раком легких, колоректальным раком, раком печени, меланомой, раком поджелудочной железы, раком яичников или раком головного мозга (взрослых).
В некоторых вариантах осуществления способов по изобретению, подвергаемый лечению рак является раком поджелудочной железы, раком легких, раком желудка, раком печени, раком мочевого пузыря, раком головы и шеи, раком ротовой полости, раком носоглотки, раком головного мозга, раком толстого кишечника, раком предстательной железы, раком яичников, раком шейки матки, раком яичка, лимфомой, лейкозом, раком кожи или раком молочной железы
В других вариантах осуществления способов по изобретению общая доза 6-тио-dG, вводимого за приблизительно 1-5 дней терапии, составляет приблизительно 10-2000 мг, или приблизительно 15-2000 мг, или приблизительно 20-2000 мг, или приблизительно 10-4800 мг за терапевтический цикл.
В одном из вариантов осуществления способов по изобретению подвергаемый лечению рак является метастатическим.
В некоторых вариантах осуществления способов по изобретению подвергаемый лечению рак является рецидивирующим.
В некоторых вариантах осуществления способов по изобретению, подвергаемый лечению рак является резистентным к терапии. В одном из вариантов осуществления резистентный к терапии рак является резистентным к терапии ингибиторами контрольных точек. В другом варианте осуществления резистентный к терапии рак является резистентным к одному или более из ингибиторов PD-1, PD-L1 и/или CTLA-4. В некоторых вариантах осуществления рак является резистентным к ингибитору тирозинкиназы, таким как, в качестве неограничивающих примеров, эрлотиниб.
В некоторых вариантах осуществления способов, представленных в настоящем описании, подвергаемого лечению индивидуума ранее лечили ингибитором контрольных точек. В одном из вариантов осуществления индивидуума ранее лечили одним или более из ингибиторов PD-1, PD-L1 или CTLA-4. В другом варианте осуществления индивидуума ранее лечили ингибитором тирозинкиназы.
В некоторых вариантах осуществления способов, представленных в настоящем описании, введение 6-тио-dG с последующим лечением ингибитором контрольных точек повторяют по меньшей мере однократно.
В некоторых вариантах осуществления способов, представленных в настоящем описании, 6-тио-dG и ингибитор контрольных точек вводят системно. В других вариантах осуществления 6-тио-dG и ингибитор контрольных точек вводят локально или регионарно в очаг опухоли. В одном из вариантов осуществления 6-тио-dG вводят локально или регионарно в очаг опухоли и ингибитор контрольных точек вводят системно.
В некоторых вариантах осуществления способов, представленных в настоящем описании, введение 6-тио-dG и ингибитора контрольных точек приводит к ингибированию роста опухоли.
В некоторых вариантах осуществления способов, представленных в настоящем описании, введение 6-тио-dG и ингибитора контрольных точек приводит к ремиссии подвергаемого лечению рака.
В некоторых вариантах осуществления способов, представленных в настоящем описании, введение 6-тио-dG и одного или более ингибиторов контрольных точек приводит к снижению опухолевой нагрузки.
В некоторых вариантах осуществления способов, представленных в настоящем описании, введение 6-тио-dG и одного или более ингибиторов контрольных точек приводит к ингибированию метастазирования раковых клеток.
В некоторых вариантах осуществления способов, представленных в настоящем описании, введение 6-тио-dG и одного или более ингибиторов контрольных точек приводит к эрадикации опухоли.
В другом аспекте настоящее изобретение относится к способам лечения рака у индивидуума, включающим введение указанному индивидууму терапевтически эффективной дозы 6-тио-dG с последующим лечением с помощью лучевой терапии. В некоторых вариантах осуществления рак выбран из группы, состоящей из рака поджелудочной железы, рака легких, мезотелиомы, рака желудка, рака пищевода, рака печени, рака желчевыводящих путей, рака мочевого пузыря, рака головы и шеи, рака ротовой полости, рака носоглотки, рака головного мозга взрослых, рака толстого кишечника, рака прямой кишки, колоректального рака, рака предстательной железы, рака яичников, рака шейки матки, рака матки, рака яичка, лимфомы, лейкоза, рака кожи, рака молочной железы, рака почки, нейробластомы, карциномы из клеток Меркеля, миелодиспластического синдрома, миелофиброза и множественной миеломы. В некоторых вариантах осуществления подвергаемый лечению рак является раком поджелудочной железы, раком легких, раком желудка, раком печени, раком мочевого пузыря, раком головы и шеи, раком ротовой полости, раком носоглотки, раком головного мозга, раком толстого кишечника, раком предстательной железы, раком яичников, раком шейки матки, раком яичка, лимфомой, лейкозом, раком кожи или раком молочной железы. В некоторых вариантах осуществления рак головного мозга является раком головного мозга взрослых.
В другом аспекте настоящее изобретение относится к способам лечения рака у индивидуума, включающим введение указанному индивидууму терапевтически эффективной дозы 6-тио-dG с предшествующим лечением с помощью лучевой терапии. В некоторых вариантах осуществления рак выбран из группы, состоящей из рака поджелудочной железы, рака легких, мезотелиомы, рака желудка, рака пищевода, рака печени, рака желчевыводящих путей, рака мочевого пузыря, рака головы и шеи, рака ротовой полости, рака носоглотки, рака головного мозга (взрослых), рака толстого кишечника, рака прямой кишки, колоректального рака, рака предстательной железы, рака яичников, рака шейки матки, рака матки, рака яичка, лимфомы, лейкоза, рака кожи, рака молочной железы, рака почки, нейробластомы, карциномы из клеток Меркеля, миелодиспластического синдрома, миелофиброза и множественной миеломы. В некоторых вариантах осуществления подвергаемый лечению рак является раком поджелудочной железы, раком легких, раком желудка, раком печени, раком мочевого пузыря, раком головы и шеи, раком ротовой полости, раком носоглотки, раком головного мозга, раком толстого кишечника, раком предстательной железы, раком яичников, раком шейки матки, раком яичка, лимфомы, лейкозом, раком кожи или раком молочной железы. В некоторых вариантах осуществления рак является раком головного мозга взрослых.
В одном из вариантов осуществления способов, представленных в настоящем описании, введение 6-тио-dG и лучевую терапию повторяют по меньшей мере однократно.
Рак может проявлять теломеразную активность. 6-тио-dG и ингибитор PD-1, PD-L1 и CTLA-4, такой как атезолизумаб, авелумаб, косибелимаб, бинтрафусп альфа, дурвалумаб, MGD013, KNO35, KN046, AUNP12, CA-170, BMS-9986189, пембролизумаб, ниволумаб, цемиплимаб, JTx-4014, сасанлимаб, будигалимаб, BI 754091, спартализумаб, камрелизумаб, синтилимаб, тислелизумаб, зимберелимаб, торипалимаб, достарлимаб, INCMGA00012, AMP-224, REGN2810, BMS-936558, SHR1210, IBI308, PDR001, BGB-A317, BCD-100, JS001 AMP-515, ипилимумаб и тремелимумаб, можно вводить в комбинации с химиотерапевтическим средством, лучевой терапией, гормональной терапией, терапией токсинами или хирургическим вмешательством. Суточная доза вводимого 6-тио-dG может составлять от приблизительно 0,15 мг/кг до приблизительно 70 мг/кг. Интервал между введением 6-тио-dG и введением ингибитора PD-L1, PD-1 и/или CTLA-4 может составлять приблизительно 1-14 дней, например, приблизительно 1-4 дня, или приблизительно 2-4 дня, или приблизительно 2-5 дней, или приблизительно 2-6 дней, или приблизительно 2-7 дней, или приблизительно 2-8 дней, или приблизительно 2-9 дней, или приблизительно 2-10 дней, или приблизительно 2-11 дней, или приблизительно 2-12 дней, или приблизительно 2-13 дней. Способ может дополнительно включать стадию оценки теломеразной активности в раковой клетке головного мозга взрослого от указанного индивидуума. Введение 6-тио-dG и ингибитора PD-1, PD-L1 и/или CTLA-4 может приводить к ингибированию роста опухоли, ремиссии указанного рака, снижению опухолевой нагрузки, ингибированию метастазирования раковых клеток или эрадикации опухоли.
Рак может являться раком поджелудочной железы, раком легких, раком желудка, раком печени, раком мочевого пузыря, раком головы и шеи, раком ротовой полости, раком носоглотки, раком головного мозга, раком толстого кишечника, раком предстательной железы, раком яичников, раком шейки матки, раком яичка, лимфомой, лейкозом или раком кожи. Рак может являться метастатическим, и/или рецидивирующим, и/или резистентным к терапии. Резистентный к терапии рак может являться резистентным к терапии ингибитором контрольных точек, например, резистентным к ингибиторам PD-L1, PD-1 и/или CTLA-4. Индивидуума могли ранее подвергать терапии ингибитором контрольных точек, такой как терапия ингибиторами PD-L1, PD-1 и/или CTLA-4. Введение 6-тио-dG с последующим лечением ингибитором PD-1, PD-L1 и/или CTLA4 повторяют по меньшей мере однократно. 6-тио-dG и ингибитор PD-1, PD-L1 и/или CTLA4 можно вводить системно или вводить локально или регионарно в очаг опухоли. 6-тио-dG можно вводить тем же или иным путем, чем ингибитор PD-1, PD-L1 и/или CTLA4.
Другие цели, признаки и преимущества настоящего изобретения будут очевидны из следующего подробного описания. Однако следует понимать, что подробное описание и конкретные примеры при указании конкретных вариантов осуществления настоящего изобретения приведены исключительно в иллюстративных целях, т.к. различные изменения и модификации в пределах сущности и объема настоящего изобретения будут очевидны специалистам в этой области из настоящего подробного описания.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Следующие чертежи представляют собой часть настоящего описания и включены для дополнительной демонстрации некоторых аспектов настоящего изобретения. Настоящее изобретение может быть более понятным со ссылкой на один или более из этих чертежей в комбинации с подробным описанием конкретных вариантов осуществления, представленных в настоящем описании.
Фиг. 1A-G. Терапевтический эффект 6-тио-dG зависит от CD8+ T-клеток. (фиг. 1A) Жизнеспособность клеток (IC50) в случае 6-тио-dG в клетках MC38. Клетки обрабатывали 6-тио-dG в течение 5 дней. (фиг. 1B и фиг. 1C) Анализ образования колоний в случае 6-тио-dG в клетках MC38 в указанных дозах в течение 13 дней. Клетки обрабатывали 6-тио-dG каждые 3 дня, затем фиксировали и окрашивали кристалл-виолетом. Типичное изображение трех биологических параллелей показано на фиг. 1B, и данные количественного анализа показаны на фиг. 1C. (фиг. 1D и фиг. 1E) Мышам (n=5) C57BL/6 WT (фиг. 1D) или Rag1-/- (фиг. 1E) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). (фиг. 1F и фиг. 1G) Мышам (n=5) C57BL/6 инокулировали 5×105 опухолевых клеток MC38 и вводили им 6-тио-dG (3 мг/кг, дни 7, 8, 9). 200 мкг антитела против CD4 (фиг. 1F) или антитела против CD8 (фиг. 1G) вводили за день до начала лечения, а затем дважды в неделю в течение 3 недель. Рост опухоли измеряли каждые 3 дня. Данные приведены как среднее значение ± SEM для 2-3 независимых экспериментов. Значение P определяли с помощью двухстороннего непарного t-критерия Стьюдента (фиг. 1C) или двухстороннего ANOVA (фиг. 1D-G). Также см. фиг. 9A-D.
Фиг. 2A-F. Введение 6-тио-dG повышаешь ответ опухолеспецифических T-клеток. (фиг. 2A и фиг. 2B) Мышам C57BL/6 (n=4-5) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Через шесть дней после последнего введения инфильтрирующие опухоль T-клетки анализировали по доле всех T-клеток (фиг. 2A) и Ki67+CD8+ T-клеток (фиг. 2B). (фиг. 2C) Мышам C57BL/6 (n=5), несущим опухоль MC38-OVA, вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Через три дня после последнего введения инфильтрирующие опухоль T-клетки анализировали на OVA-специфические CD8+ T-клетки с помощью тетрамера H-2Kb-OVA257-264. (фиг. 2D и фиг. 2E) По той же схеме эксперимента, что и на (A), спленоциты собирали и повторно стимулировали облученными опухолевыми клетками MC38 в течение 48 ч. ИФНγ-продуцирующие клетки определяли с помощью анализа ELISPOT. Типичные пятна показаны на фиг. 2D, и данные количественного анализа (n=5) показаны на фиг. 2E. (фиг. 2F) ИФНγ-репортерным мышам (n=3) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Через одиннадцать дней после последнего введения, опухоли измельчали и расщепляли для проточного цитометрического анализа YFP+ T-клеток. Значение p определяли с помощью двухстороннего непарного t-критерия Стьюдента (фиг. 2A-C, фиг. 2E и фиг. 2F). Также см. фиг. 10A-F.
Фиг. 3A-F. Введение 6-тио-dG повышает способность дендритных клеток к перекрестному примированию. (фиг. 3A) Мышам C57BL/6 (n=5) инокулировали 5×105 MC38 опухолевых клеток и вводили им 6-тио-dG (3 мг/кг, дни 7, 8, 9). 200 мкг антитела против CSF1R вводили за день до начала лечения, а затем дважды в неделю в течение 3 недель. (фиг. 3B) Мышам Batf3-/- (n=5) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Рост опухоли измеряли каждые 3 дня. (фиг. 3C) Процент мышей без опухолей среди мышей WT и Batf3-/- (n=5) после введения 6-тио-dG. (фиг. 3D) BMDC культивировали с опухолевыми клетками MC38, предварительно обработанными 200 нМ 6-тио-dG или носителя в течение ночи, а затем очищали DC и сокультивировали с наивными T-клетками OT-1. Через 48 ч. супернатант собирали и тестировали на продукцию ИФНγ с помощью цитометрического анализа на бусах (CBA). (фиг. 3E) BMDC культивировали с опухолевыми клетками MC38, предварительно обработанными 200 нМ 6-тио-dG или носителем в течение 18 ч., собирали супернатант для ELISA на ИФНβ. (F) Мышам Ifnar1-/- (n=5) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Рост опухоли измеряли каждые 3 дня. Данные приведены как среднее значение ± SEM для 2-3 независимых экспериментов. Значение p определяли с помощью двухстороннего ANOVA (фиг. 3A, фиг. 3B и фиг. 3F) или двухстороннего непарного t-критерия Стьюдента (фиг. 3C-E).
Фиг. 4A-G. Для индуцируемого 6-тио-dG врожденного распознавания необходима передача сигнала STING в организме-хозяине. (фиг. 4A и фиг. 4B) Мышам Myd88-/- (фиг. 4A) или Tmem173-/- (фиг. 4B) (n=5) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Рост опухоли измеряли каждые 3 дня. (фиг. 4C и ФИГ. 4D) Мышам C57BL/6 (n=5) инокулировали 5×105 опухолевых клеток MC38 Tmem173 KO (фиг. 4C) или Mb21d1 KO (фиг. 4D) и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Рост опухоли измеряли каждые 3 дня. (фиг. 4E и фиг. 4F) Опухолевые клетки MC38 обрабатывали 1 мкМ 6-тио-dG в течение 24 ч. С помощью анализа TIF (анализа фокусов индуцированных дисфункцией теломер) подтверждали индукцию TIF при обработке 6-тио-dG клеток MC38. n=100 (контроль), n=100 (6-тио-dG). (фиг. 4G) BMDC культивировали с клетками рака толстого кишечника человека HCT116, предварительно обработанными 500 нМ 6-тио-dG или носителем в течение 4 ч., затем очищали DC и выделяли цитозольную ДНК. Относительное содержание MT-CO1 и 18S человека в цитозоле DC определяли посредством qPCR. Данные приведены как среднее значение ± SEM для 2-3 независимых экспериментов. Значение p определяли с помощью двухстороннего ANOVA (A-D) или двухстороннего непарного t-критерия Стьюдента (фиг. 4F и фиг. 4G). Также см. фиг. 11A-H.
Фиг. 5A-F. 6-тио-dG преодолевает резистентность к блокаде PD-L1 в моделях опухолей на поздних стадиях. (фиг. 5A) Мышам C57BL/6, несущим опухоль MC38 (n=4-5), вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Через 7 дней после первого введения тестировали долю PD-1+CD8+ T-клеток (слева) и MFI PD-1 (справа). (фиг. 5B и фиг. 5C) Мышам C57BL/6 (n=5) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дней 10, 11). 50 мкг антитела против PD-L1 вводили в дни 13 и 17. Показаны рост опухоли (фиг. 5B) и выживаемость (фиг. 5C). (фиг. 5D) Мышам C57BL/6 (n=5), несущим опухоль MC38, вводили 6-тио-dG (3 мг/кг, дней 10, 11), или антитело против PD-L1 (2,5 мг/кг, день 10), или комбинированное лечение из них двоих. Через 7 дней после первого введения собирали дренирующий лимфоузел и стимулировали облученными опухолевыми клетками MC38 или опухолевыми клетками LLC для ELISPOT ИФНγ. (фиг. 5E и фиг. 5F) Мышам C57BL/6 (n=5) инокулировали 1×106 опухолевых клеток легких мыши LLC и вводили 6-тио-dG (3 мг/кг, дней 4, 5, 6 и 10, 11). 200 мкг антитела против PD-L1 вводили в день 8 и день 13. Рост опухоли измеряли каждые 3-4 дня (фиг. 5E). Через шесть недель мышей без опухолей (n=4) в группе последовательного лечения и контрольных мышей повторно стимулировали с помощью 5×106 опухолевых клеток LLC (правый бок) и 5×106 опухолевых клеток MC38 (левый бок). Рост опухоли измеряли каждые 3-4 дня (фиг. 5F). Данные приведены как среднее значение ± SEM для двух независимых экспериментов. Значение p определяли с помощью двухстороннего непарного t-критерия Стьюдента (фиг. 5A, фиг. 5D), или двухстороннего ANOVA (фиг. 5B, фиг. 5E и фиг. 5F), или логарифмического рангового критерия (фиг. 5C). Также см. фиг. 12.
Фиг. 6A-E. 6-тио-dG снижает нагрузку рака толстого кишечника человека в модели на гуманизированных мышах. (фиг. 6A) Общая выживаемость у пациентов с колоректальной аденокарциномой с высокой и низкой экспрессией TERT (теломеразной обратной транскриптазы, каталитической субъединицы теломеразы) из баз данных TCGA. (фиг. 6B) Жизнеспособность клеток (IC50) в случае 6-тио-dG в клетках рака толстого кишечника человека HCT116. Клетки обрабатывали 6-тио-dG в течение 5 дней. (фиг. 6C) Схема для модели опухоли на гуманизированных мышах. (фиг. 6D и фиг. 6E) Мышам NSG-SGM3 (n=5) (фиг. 6D) или гуманизированным мышам NSG-SGM3 (n=4) (фиг. 6E) инокулировали 1×106 опухолевых клетках HCT116 и вводили 6-тио-dG (3 мг/кг, дней 8, 9, 10). Рост опухоли измеряли каждые 3 дня. Данные приведены как среднее значение ± SEM для двух независимых экспериментов. Значение p определяли посредством логарифмического рангового критерия (фиг. 6A) или двухстороннего ANOVA (фиг. 6D и фиг. 6E). Также см. фиг. 13A-F.
Фиг. 7. Схема индукции 6-тио-dG c-GAS/STING/ИФН.
Фиг. 8A-B. Доказательство того, что 6-тио-dG с последующим введением ингибитора PD-L1 приводят к полной ремиссии опухоли и иммунной памяти.
Фиг. 9A-D (относится к фиг. 1A-G).(фиг. 9A) Жизнеспособность клеток (IC50) в случае 6-тио-dG в раковых клетках легких мыши LLC. Клетки обрабатывали 6-тио-dG в течение 4 дней. (фиг. 9B) Мышам C57BL/6 (n=5) инокулировали 1×106 опухолевых клеток LLC и вводили 6-тио-dG (3 мг/кг, дней 4, 5, 6). Рост опухоли измеряли каждые 3 дня. (фиг. 9C) IC50 6-тио-dG в клетках рака толстого кишечника мыши CT26. (фиг. 9D) Мышам BALB/C (n=5) инокулировали 5×105 опухолевых клеток CT26 и вводили 6-тио-dG (3 мг/кг, дней 5, 6, 7). Рост опухоли измеряли каждые 3 дня. Данные приведены как среднее значение ± SEM для двух независимых экспериментов. Значение p определяли посредством двухстороннего ANOVA.
Фиг. 10A-F (относится к фиг. 2A-G).(фиг. 10A-D) Мышам C57BL/6 (n=4-5) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Через 7 дней после первого введения опухоли анализировали на CD8+ T-клетки среди CD45+ клеток (фиг. 10A) и среди всех опухолевых клеток (фиг. 10B), инфильтрирующие опухоль T-клетки анализировали по доле CD4+Foxp3+ Treg-клеток (фиг. 10C) и NK-клеток (фиг. 11D). (фиг. 10E) Мышам C57BL/6 (n=5) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). 200 мкг антитела против NK1.1 вводили за день до начала лечения, а затем дважды в неделю в течение 3 недель. (фиг. 10F) ИФНγ-репортерным мышам (n=3) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дни 7, 8, 9). Через одиннадцать дней после последнего введения, опухоли измельчали и расщепляли для проточной цитометрической детекции YFP+ T-клеток. Показано типичное гейтирование проточной цитометрии. Данные приведены как среднее значение ± SEM для двух независимых экспериментов. Значение p определяли с помощью двухстороннего непарного t-критерия Стьюдента (фиг. 10A-D) или двухстороннего ANOVA (фиг. 10E).
Фиг. 11A-H (относится к фиг. 4A-G).(фиг. 11A) BMDC культивировали с опухолевыми клетками MC38, предварительно обработанными 0,2 мкМ или 1 мкМ 6-тио-dG в течение 6 ч., а затем DC очищали с помощью магнитных бус и подвергали вестерн-блоттингу. (фиг. 11B) BMDC из мышей дикого типа (WT) или Tmem173KO культивировали с опухолевыми клетками MC38, предварительно обработанными 200 нМ 6-тио-dG в течение ночи, и затем DC очищали с помощью магнитных бус и осуществляли qPCR для тестирования относительного содержания ИФНβ. (фиг. 11C и 11D) Мышам C57BL/6 (n=3) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дней 10, 11 и 12). Через 3 дня после последней инъекции мышей умерщвляли; опухоли собирали и фиксировали для окрашивания TIF (фокусов, индуцированных дисфункцией теломер). Изображения получали с помощью флуоресцентного микроскопа (100x). Красными точками указано повреждение ДНК (γ-H2AX), зелеными точками показаны теломеры и желтыми точками показаны TIF (повреждение ДНК на теломерах). Масштабные линейки, 10 мкМ. (фиг. 11E и 11F) Введение 6-тио-dG индуцировало микроядра в клетках MC38. (фиг. 11E) Типичное изображение двух дочерних клеток в поздней телофазе содержит сигналы теломер и покрытые и непокрытые микроядра в клетках MC38. Зелеными точками показаны теломерные сигналы и красным показан ламин A/C (биомаркер ядерной оболочки). (фиг. 11F) Количественный анализ индуцированных введением 1 мкМ 6-тио-dG микроядер через 48 ч. (фиг. 11G и 11H) 100000 клеток MC38 высевали в 6-луночный планшет и метили клетки 25 мкМ EdU. Через 2 дня клетки вымывали и инкубировали с 1 мкМ 6-тио-dG в свежей среде O/N. Затем клетки вымывали и сокультивировали с DC O/N. На следующий день DC очищали с помощью магнитных бус. Затем очищенные DC фиксировали и цитоцентрифугировали для иммуно-FISH. Теломерный зонд: зеленый, EdU: красный, DAPI: голубой. Изображения получали при увеличении 63X с помощью Axio Imager Z2, оборудованного автоматической системой захвата, и анализировали с помощью программного обеспечения ISIS (камера: CoolCube 1 от MetaSystems). Показаны типичное изображение (фиг. 11G) и данные количественного анализа (фиг. 11H), n=100. Данные приведены как среднее значение ± SEM для 2-3 независимых экспериментов. Значение p определяли с помощью двухстороннего непарного t-критерия Стьюдента (B, F и H).
ФИГ. 12 (относится к фиг. 5A-G). Мышам C57BL/6 (n=5) инокулировали 5×105 опухолевых клеток MC38 и вводили 6-тио-dG (3 мг/кг, дней 10, 11). 50 мкг антитела против PD-L1 вводили в день 13 и 17. Измеряли массу тела мышей. Данные приведены как среднее значение ± SEM.
Фиг. 13A-F (относится к фиг. 6A-E). (фиг. 13A-C) Через 12 недель после реконституции гуманизированной мыши CD45+ клетки и CD3+ T-клетки человека в периферической крови мыши тестировали посредством проточной цитометрии. Типичный график проточной цитометрии показан на фиг. 13A. Доли CD45 и CD3 в контрольной группе и группе 6-тио-dG перед введением показаны на фиг. 13B и 13C, n=5. (фиг. 13D) Жизнеспособность клеток (IC50) в случае 6-тио-dG в раковых клетках меланомы человека A375. Клетки обрабатывали 6-тио-dG в течение 4 дней. (фиг. 13E) Мышам NSG-SGM3 (n=5) инокулировали 2×106 опухолевых клеток A375 и вводили 6-тио-dG (3 мг/кг, день 7 и день 8), или антитело против PD-L1 с антителом против CTLA-4 (200 мкг i.p., день 10 и день 13), или комбинацию 6-тио-dG с антителом против PD-L1 и антителом против CTLA-4. Рост опухоли измеряли каждые 3 дня. (фиг. 13F) Гуманизированным мышам NSG-SGM3 (n=5-7) инокулировали 2×106 опухолевых клеток A375 и вводили 6-тио-dG (3 мг/кг, день 13 и день 14), или антитело против PD-L1 с антителом против CTLA-4 (200 мкг i.p., день 16 и день 19), или комбинацию 6-тио-dG с антителом против PD-L1 и антителом против CTLA-4. Рост опухоли измеряли каждые 3 дня. Данные приведены как среднее значение ± SEM. Значение p определяли с помощью двухстороннего непарного t-критерия Стьюдента (фиг. 13B и 13C, n.s. p>0,05) или двухстороннего ANOVA (фиг. 13F).
На фиг. 14 показаны эффекты 6-тио-dG и средства против PD-1 цемиплимаба (Libtayo®) в отношении объема опухоли у мышей, несущих полученные из клеток LLC опухоли (NSCLC). Дозы составляли 3 мг/кг 6-тио-dG (i.p) и 10 мг/кг цемиплимаб а(i.p). Введение разным группам показано в таблице ниже. День 1 (12/31/2020): 1000K клеток LLC инокулировали 35 мышам B6. День 11-13: Начало эксперимента. В этом исследовании использовали 3 мг/кг 6-тио-dG и 10 мг/кг Libtayo.
2021
2021
2021
2021
2021
2021
2021
2021
2021
2021
2021
На фиг. 15 показаны эффекты 6-тио-dG со средством против PD-1 цемиплимабом (Libtayo®) в отношении объема опухоли у мышей, несущих полученные из клеток LLC опухоли (NSCLC). Дозы составляли 3 мг/кг 6-тио-dG (i.p) и 10 мг/кг цемиплимаба (i.p). Введение разным группам показано в таблице выше. День 1 (12/31/2020): 1000K клеток LLC инокулировали 35 мышам B6. День 11-13: Начало эксперимента. В этом исследовании использовали 3 мг/кг 6-тио-dG и 10 мг/кг Libtayo.
На фиг. 16 показан эффект 6-тио-dG в комбинации со средством против PD-1 пембролизумабом в модели мелкоклеточного рака легких (SCLC) на гуманизированных мышах.
На фиг. 17 показан 6-тио-dG в комбинации с ингибитором PD-L1 и лучевой терапией в модели HCC на мышах.
На фиг. 18A-18D показан 6-тио-dG в комбинации с ингибитором PD-L1 и радиацией в модели HCC на мышах. Фиг. 18A, схема введения. Фиг. 18B, клетки рака печени HCC53N (нокаут p53 и NRAS), обработанные in vivo исходно фокальным IR с последующими 3 дозами 6-тио-dG с последующими 2 обработками антителом против PD-L1, что приводило к полной ремиссии опухолей. Фиг. 18C, при повторной стимуляции с использованием в 10 раз большего количества клеток HCC53N опухоли не вырастали снова, что позволяет предположить наличие иммунологической памяти; и фиг. 18D, при тестировании наивных мышей опухоли быстро росли.
ПОДРОБНОЕ ОПИСАНИЕ НАСТОЯЩЕГО ИЗОБРТЕНИЯ
Теломераза почти повсеместно экспрессируется в опухолевых клетках. Теломераза-опосредованное, нацеленное на теломеры лекарственное средство, 6-тио-dG, снижает лаг-период между исходным введением и ответом на терапию, напрямую индуцируя повреждение теломер в теломераза-положительных раковых клетках, но не в нормальных клетках с "молчащей" теломеразой. В этом исследовании авторы настоящего изобретения были нацелены на изучение того, может ли 6-тио-dG, индуцирующий теломерный стресс в теломераза-положительных раковых клетках, инициировать быстрое повреждение ДНК для врожденного распознавания. Они использовали сингенных мышей дикого типа и мышей с генетическим дефектом для оценки того, как 6-тио-dG запускает врожденное распознавание и участвует в противоопухолевом иммунитете организма-хозяина. Важно, что они демонстрируют, что 6-тио-dG преодолевает резистентность к блокаде PD-L1 в опухолях на поздней стадии. Неожиданно, 6-тио-dG индуцирует ДНК-опосредованное врожденное распознавание и активацию иммунных ответов в организме-хозяине STING-зависимым образом, что приводит к улучшенной противоопухолевой эффективности. Кроме того, 6-тио-dG с последовательной терапией средством против PD-L1 может приводить к полной элиминации опухолей на поздних стадиях. Таким образом, 6-тио-dG является нацеленным на опухоль и иммуностимулирующим лекарственным средством, которое может принести пользу пациентам с теломераза-положительными и PD-L1-резистентными раковыми заболеваниями в клинических условиях.
Эти и другие аспекты настоящего изобретения подробно описаны ниже.
I. Теломеры, теломераза и дисфункция теломер
Во время митоза клетки делают копии своего генетического материала. Половина генетического материала переходит в каждую новую дочернюю клетку. Для обеспечения того, что информация успешно передастся от одного поколения следующему, каждая хромосома имеет специальный защитный кэп под названием "теломера", находящийся на концах ее "плеч". Теломеры контролируются наличием фермента теломеразы.
Теломера является повторяющейся последовательностью ДНК (например, TTAGGG) на концах хромосом организма. Теломера может достигать длины 15000 пар оснований. Теломеры функционируют, предотвращая потерю хромосомами последовательностей пар оснований на их концах. Они также предотвращают слияние хромосом друг с другом. Однако, каждый раз, когда клетка делится, некоторые из теломер утрачиваются (как правило, 25-200 пар оснований на деление). Когда теломера становится слишком короткой, хромосома достигает "критической длины" и больше не может реплицироваться. Это означает, что клетка становится старой и погибает в ходе процесса под названием "апоптоз" или подвергается сенесценции. Активность теломер контролируется двумя механизмами: эрозией и добавлением. Эрозия, как указано, происходит при каждом делении клетки из-за неспособности к завершению синтеза отстающей цепи ДНК до самого конца. Добавление определяют по активности теломеразы.
Теломераза, также названная терминальной трансферазой теломер, является ферментом, состоящим из белковых и РНК-субъединиц, элонгирующих хромосомы посредством добавления последовательностей TTAGGG на концы существующих хромосом. Теломераза обнаружена в тканях плода, половых клетках взрослых, а также опухолевых клетках. Теломеразная активность регулируется во время развития и имеет очень низкую, почти недектируемую активность в соматических клетках (организма). Поскольку эти соматические клетки не используют теломеразу регулярно, они стареют. Результатом старения клеток является старение организма. Если теломераза активирована в клетке, клетка будет продолжать расти и делиться. Эта теория "бессмертной клетки" важна в двух исследовательских областях: старении и онкологии.
Клеточное старение, или сенесценция, является процессом, посредством которого клетка становится старой и прекращает расти или умирает. Это происходит из-за укорочения хромосомных теломер до такой степени, что хромосома достигает критической длины. Клеточное старение аналогично часовому механизму. Если часы остаются заведенными, клетка становится бессмертной и постоянно продуцирует новые клетки. Если часы замедляют ход, клетка прекращает продуцировать клетки и подвергается тому, что называется репликативной сенесценцией или погибает. Клетки постоянно стареют. Возможность сделать клетки способными пролонгировать свою способность к репликации, разумеется, создает некоторые захватывающие возможности, особенно в случае заболевания, ассоциированного с генетическим наследованием коротких теломер (что называют теломеропатиями или нарушениями теломерного спектра). Таким образом, исследование теломеразы может привести к важным открытиям, связанным с процессом старения.
Раковые клетки избегают явления нормального старения из-за коротких теломер и становятся злокачественными клетками. Злокачественные клетки делятся, пока не образуют опухоль, которая растет неконтролируемо и распространяется в отдаленные ткани по всему организму человека. Теломеразу определяют почти во всех раковых клетках человека. Это обеспечивает преимущество избирательного роста для многих типов опухолей. Если теломеразная активность должна быть отключена, то теломеры в раковых клетках будут все больше укорачиваться, так же, как и в нормальных клетках организма. Это предотвращало бы неконтролируемое деление раковых клеток на ранних стадиях их развития. В случае, когда опухоль уже полностью развилась, ее можно удалить и проводить противотеломеразную терапию для профилактики рецидива. По существу, предотвращение выполнения теломеразой своей функции сделает раковые клетки из бессмертных в смертные. Однако прямым ингибиторам теломеразы необходим лаг-период от начала лечения до уменьшения опухоли, и не продвинулись в клинической разработке из-за повышенной токсичности. Таким образом, настоящее изобретение относится к способам снижения лаг-периода, но для него теломеразная активность должна быть эффективной и потенциально снижать побочные эффекты.
II. Лечение рака
A. Терапевтические средства для последовательной терапии
1. В некоторых вариантах осуществления ингибитор PD-L1 выбран из одного или более из атезолизумаба, авелумаба, косибелимаба, бинтрафуспа альфа, дурвалумаба, MGD013, KNO35, KN046, AUNP12, CA-170 и BMS-9986189. В некоторых вариантах осуществления ингибитор PD-L1 является атезолизумабом. Атезолизумаб (торговое название Tecentriq®) является полностью гуманизированным, сконструированным моноклональным антителом изотипа IgG1 против лиганда белка программируемой гибели клеток-1 (PD-L1). В 2015 году он проходил клинические испытания в качестве иммунотерапии нескольких типов солидных опухолей. В мае 2016 года он был одобрен FDA для лечения рака мочевого пузыря, но в мае 2017 года он потерпел неудачу в исследовании фазы III в качестве лечения второй линии рака мочевого пузыря. В октябре 2016 года FDA одобрило атезолизумаб для уротелиальной карциномы и лечения пациентов с метастатическим немелкоклеточным раком легких (NSCLC), у которых заболевание прогрессировало во время или после химиотерапии соединениями платины. Пациенты с геномными опухолевыми аберрациями EGFR или ALK должен иметь прогрессирование заболевания во время одобренной FDA терапии по причине этих аберраций перед введением атезолизумаба. В сентябре 2018 года было анонсировано, что, согласно результатам исследования, представленным на 19-ой Всемирной конференции по раку легких (WCLC) в Торонто, Канада, атезолизумаб пролонгирует выживаемость при лечении мелкоклеточного рака легких на поздней стадии. В октябре 2018 года завершилось комбинированное клиническое испытание лекарственного средства с наб-паклитакселом на пациентах с трижды негативным раком молочной железы на поздней стадии. В марте 2019 года он был одобрен в США в комбинации со связанным с белком паклитакселом для взрослых пациентов с нерезектабельным местнораспространенным или метастатическим трижды негативным раком молочной железы (TNBC), у которых опухоли экспрессируют PD-L1 (окрашенные на PD-L1 инфильтрирующие опухоль иммунные клетки с любой интенсивностью, охватывающие ≥1% площади опухоли), что определяют с помощью одобренного FDA теста. В марте 2019 года он был одобрен в США в комбинации с карбоплатином и этопозидом для лечения первой линии взрослых пациентов с мелкоклеточным раком легких на поздней стадии (ES-SCLC). Наиболее распространенными нежелательными явлениями в исследованиях являлись утомляемость, снижение аппетита, тошнота и инфекции. Инфекция мочевыводящих путей являлась наиболее распространенным тяжелым нежелательным явлением.
Атезолизумаб блокирует взаимодействие PD-L1 с белком программируемой гибели клеток 1 (PD-1) и рецепторами CD80 (B7-1R). PD-L1 может высоко экспрессироваться на некоторых опухолях, что, как считают, приводит к сниженной активации иммунных клеток (цитотоксических T-клеток в частности), которые в ином случае распознают и атакуют рак. Ингибирование PD-L1 атезолизумабом может устранять этот ингибиторный эффект и, таким образом, вызывать противоопухолевый ответ. Это один из нескольких путей блокирования ингибиторных сигналов, относящихся к активации T-клеток, более общей стратегии, известной как ингибирование иммунных контрольных точек. В случае некоторых рак (в частности, мочевого пузыря) вероятность получения пользы связана с экспрессией PD-L1, но большинство раковых заболеваний с экспрессией PD-L1 все равно не отвечают, а некоторые (приблизительно 15%) без экспрессии PD-L1 - отвечают.
Авелумаб (Bavencio®) является полностью человеческим антителом IgG1, разработанным Merck Serono и Pfizer. Авелумаб одобрен FDA для лечения метастатической карциномы из клеток Меркеля. Он потерпел неудачу в клинических испытаниях фазы III для рака желудка.
Дурвалумаб (Imfinzi®) является полностью человеческим антителом IgG1, разработанным AstraZeneca. Дурвалумаб одобрен FDA для лечения уротелиальной карциномы и нерезектабельного немелкоклеточного рака легких после химиолучевой терапии.
KN035 является единственным антителом против PD-L1 с подкожным составом, в настоящее время находящимся на стадии клинической оценки в США, Китае и Японии.
AUNP12 является 29-мерным пептидом в качестве первого пептидного ингибитора PD-1/PD-L1, разработанного Aurigene и Laboratoires Pierre Fabre, оцениваемым в рамках клинических испытаний для лечения рака.
CA-170, разработанный Aurigene/Curis в качестве антагониста PD-L1 и VISTA, в настоящее время проходит клиническое испытание фазы I для лечения мезотелиомы.
2. Ингибиторы PD-1, такие как цемиплимаб, пембролизумаб, ниволумаб, JTx-4014, сасанлимаб, будигалимаб, BI 754091, спартализумаб, камрелизумаб, синтилимаб, тислелизумаб, зимберелимаб, торипалимаб, достарлимаб, INCMGA00012, AMP-224, REGN2810, BMS-936558, SHR1210, IBI308, PDR001, BGB-A317, BCD-100, JS001 и AMP-515. В некоторых вариантах осуществления ингибитор PD-1 является цемиплимабом или пембролизумабом.
Цемиплимаб, продаваемый под торговым названием Libtayo®, является лекарственным средством на основе моноклонального антитела для лечения плоскоклеточного рака кожи, базально-клеточной карциномы кожи и немелкоклеточного рака легких. Цемиплимаб принадлежит к классу лекарственных средств, связывающихся с рецептором программируемой гибели клеток-1 (PD-1), блокирующих путь PD-1/PD-L1. В сентябре 2018 года он был одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) для лечения людей с метастатической плоскоклеточной карциномой кожи (CSCC) или местнораспространенной CSCC, не являющихся кандидатами для радикального хирургического вмешательства или радикальной лучевой терапии. Цемиплимаб исследуют в отношении лечения меланомы, рака шейки матки, рака головного мозга, рака головы и шеи, почечноклеточной карциномы и лимфомы Ходжкина.
Пембролизумаб (ранее известный как ламбролизумаб, продаваемый под торговым названием Keytruda®) является гуманизированным антителом, используемым в иммунотерапии рака. Пембролизумаб одобрен для медицинского использования в США в 2014 году. В 2017 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило его для любой нерезектабельной или метастатической солидной опухоли с некоторыми генетическими отклонениями (дефицитом репарации ошибочно спаренных нуклеотидов или микросателлитной недостаточностью). Одобренные показания для Keytruda® в настоящее время включают, помимо прочего, метастатическую меланому, NSCLC, рак головы и шеи, лимфому Ходжкина, и метастатическую плоскоклеточную карциному пищевода. Пембролизумаб вводят посредством медленной инъекции в вену.
3. Тиопурины, такие как 6-тиогуанин и 6-меркаптопурин, в настоящее время используют в клинической практике в качестве противовоспалительных, противолейкозных и иммуносупрессорных средств. В реакциях активации 6-тиогуанин превращается в 6-тиогуанозинмонофосфат под действием фермента гипоксантингуанинфосфорибозилтрансферазы (HPRT). Затем 6-тиогуанозинмонофосфат дополнительно под действием киназ и РНК-редуктаз метаболизируется в 6-тио-2′-дезоксигуанозин-5′-трифосфат, который в конечном итоге может встраиваться в цепи ДНК во время репликации ДНК. Встроенный в ДНК 6-тиогуанин также может приводить к образованию активных форм кислорода, которые могут вызывать дополнительное повреждение ДНК, белков и других клеточных макромолекул и, таким образом, блокировать клеточную репликацию. Хотя тиопурины используют в клинических условиях для лечения некоторых типов лейкоза, их пригодность для лечения солидных опухолей ограничена, частично, из-за повышенной токсичности и разработки других способов терапии.
Одним конкретным тиопурином является 6-тио-dG. Это соединение является аналогом нуклеозида, и доказано, что он является теломераза-опосредовано разрушающим теломеры соединением. В связи с этим, раковые клетки очень чувствительны к 6-тио-dG с наблюдаемыми значениями IC50 в диапазоне 0,7-2,9 мкМ в зависимости от типа клеток, даже включая резистентные к терапии раковые заболевания (Mender et al., 2018). Структура приведена ниже:
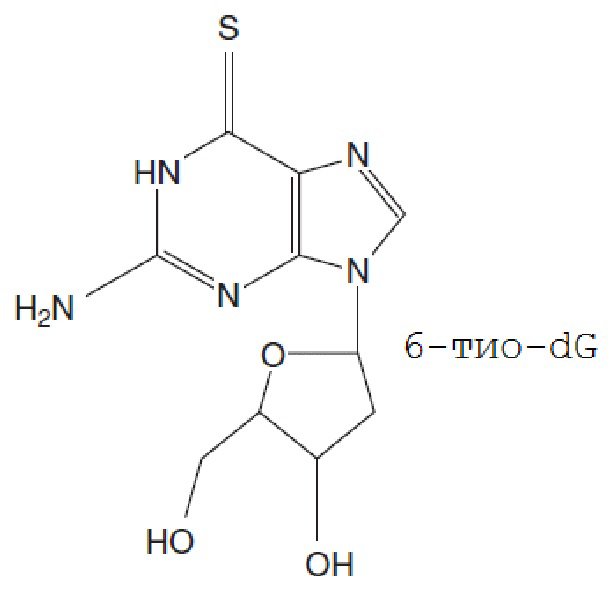
B. Схемы лечения
Настоящее изобретение относится к последовательному лечению рака с использованием лечения 6-тио-dG с последующей терапией ингибиторами PD-L1, PD-1 и/или CTLA-4. Периоды каждого лечения могут варьироваться, и предполагают, что короткий интервал между лечениями будет благоприятным. Например, лечение 6-тио-dG может занимать всего 2 дня, но может занимать 3, 4 или более дней, включая 2-4 дня. Интервал перед лечением ингибиторами PD-L1, PD-1 и/или CTLA-4 должен составлять по меньшей мере один день и может составлять до 14 дней, например, 2-4 дня. Следует избегать пересечения между 6-тио-dG и ингибиторами PD-L1, PD-1 и/или CTLA-4 из-за потенциально неблагоприятных эффектов 6-тио-dG в отношении активированных эффекторных T-клеток.
Суточная доза 6-тио-dG будет составлять от 0,5 мг/кг до 10 мг/кг, предпочтительно, внутривенно или перорально. Доза ингибиторов PD-L1, PD-1 и/или CTLA-4 будет соответствовать текущим одобренным схемам введения.
C. Теломераза-положительные раковые заболевания
Теломераза-положительные раковые заболевания гораздо более восприимчивы к способам по изобретению, чем теломераза-отрицательные раковые заболевания. Таким образом, тестирование биоптата для определения того, является ли рак теломераза-положительным, очень полезно, хоть и не является необходимым.
Наиболее распространенными способами детекции теломеразной активности являются способы амплификации теломерных повторов (TRAP), позволяющие осуществлять полуколичественные и количественные анализы с использованием некоторых из их модификаций (названных ddTRAP, что означает капельный цифровой TRAP). Эти модификации включают сцинтилляционный анализ сближения, анализ защиты гибридизации, анализ амплификации транскрипции и анализ выделения на магнитных бусах.
Способ амплификация теломерных повторов можно разделить на три основные стадии: элонгацию праймера, амплификацию теломераза-синтезированной ДНК и в конечном итоге ее детекцию. На стадии элонгации теломерные повторы добавляют на имитирующий теломеру олигонуклеотид с помощью теломеразы, присутствующей в клеточном экстракте. ПЦР-амплификацию теломераза-синтезированный ДНК осуществляют с использованием имитирующих теломеры и обратных праймеров. В теломераза-синтезированную ДНК можно встраивать разные метки. После этой стадии следует детекция (например, электрофоретическое разделение и визуализация продуктов ПЦР).
Другие способы включают количественное выделение теломеразы и последующее измерение общей активности теломеразы из указанного количества клеток, которую можно сравнивать с подходящими стандартами. После выделения и тестирования теломеразы in vitro можно использовать широкий спектр способов мечения и детекции.
D. Раковые заболевания, резистентные к лекарственным средствам
Термин "антионеопластическая резистентность", зачастую используемый взаимозаменяемо с термином "резистентность к химиотерапии", означает резистентность неопластических (раковых) клеток или способность раковых клеток выживать и расти, несмотря на противоопухолевую терапию. В некоторых случаях раковые заболевания могут приобретать резистентность к множеству лекарственных средств, что называют множественной лекарственной устойчивостью.
Существует две основные причины неудачи антинеопластической терапии: врожденные генетические характеристики, дающие раковым клеткам их резистентность, и приобретенная резистентность после воздействия лекарственного средства, основанная на концепции гетерогенности раковых клеток. Характеристики резистентных клеток включают измененный мембранный транспорт, повышенную репарацию ДНК, дефекты апоптотических путей, изменение целевых молекул, белков и механизмов путей, таких как ферментативная деактивация. Поскольку раковые заболевания являются генетическими заболеваниями, в основе приобретенной лекарственной резистентности лежат два геномных явления: геномные изменения (например, амплификация и делеция гена) и эпигенетические модификации. Раковые клетки постоянно используют различные инструменты, включающие гены, белки и измененные пути, для обеспечения своего выживания в условиях антинеопластических лекарственных средств.
Термин "антинеопластическая резистентность", синонимичный термину "резистентность к химиотерапии", означает способность раковых клеток выживать и расти, несмотря на разную противоопухолевую терапию, т.е. их множественную лекарственную устойчивость. Существует две основные причины неудачи антинеопластической терапии: (i) врожденная резистентность, такая как генетические характеристики, дающие раковым клеткам их резистентность с самого начала, что основано на концепции гетерогенности раковых клеток; и (ii) приобретенная резистентность после воздействия лекарственного средства.
Поскольку раковые заболевания являются генетическими заболеваниями, в основе приобретенной лекарственной резистентности лежат два геномных явления: геномные изменения (например, амплификация и делеция гена) и эпигенетические модификации.
Хромосомная перестройка из-за геномной нестабильности может вызвать амплификацию и делецию гена. Амплификация гена представляет собой повышение количества копий области хромосомы, что часто происходит в солидных опухолях и может вносить вклад в эволюцию опухоли в результате измененной экспрессии гена.
Исследование на клетках хомяка в 1993 году показало, что эти амплификации в гене DHFR, участвующем в синтезе ДНК, начинаются с поломки хромосомы ниже гена, и последующие циклы разрыв-слияние-мостик приводят к крупным внутрихромосомным повторам. Гиперамплификация онкогенов может происходить в ответ на химиотерапию, что, как считают, может являться основополагающим механизмом для нескольких классов резистентности. Например, амплификация DHFR происходит в ответ на метотрексат, амплификация TYMS (участвующего в синтезе ДНК) происходит в ответ на 5-фторурацил, и амплификация BCR-ABL происходит в ответ на иматиниба мезилат. Определение областей амплификации гена в клетках пациентов с раком имеет огромное клиническое значение. Делеция гена противоположна амплификации гена, при ней область хромосомы утрачивается, и лекарственная резистентность возникает в результате утраты генов опухолевых супрессоров, таких как TP53.
Геномная нестабильность может происходить, когда репликационная вилка нарушена или заблокирована в своей миграции. Это может происходить при участии барьеров репликационной вилки, белков, таких как PTIP, CHD4 и PARP1, которые в норме устраняются с помощью сенсоров повреждения ДНК клетки, нуклеаз Surveyor и респондеров BRCA1 и BRCA2.
Эпигенетические модификации при резистентности к антинеопластическим лекарственным средствам играют основную роль в развитии рака и лекарственной резистентности, т.к. они участвуют в регуляции экспрессии генов. Два основных типа эпигенетического контроля представляют собой метилирование ДНК и метилирование/ацетилирование гистонов. Метилирование ДНК представляет собой процесс добавления метильных групп к ДНК, как правило, в вышележащие промоторные области, что останавливает транскрипцию ДНК в области и эффективно приводит к сайленсингу отдельных генов. Модификации гистонов, такие как деацетилирование, изменяют образование хроматина и приводят к сайленсингу крупных хромосомных областей. В раковых клетках, где нормальная регуляция экспрессии генов нарушается, онкогены активируются посредством гипометилирования, а опухолевые супрессоры подвергаются сайленсингу посредством гиперметилирования. Аналогично, предполагают, что при развитии лекарственной резистентности эпигенетические модификации могут приводить к активации и гиперэкспрессии способствующих лекарственной резистентности генов.
Исследования на линиях раковых клеток показали, что гипометилирование (утрата метилирования) промотора гена MDR1 вызывала гиперэкспрессию и множественную лекарственную устойчивость.
В резистентных к метотрексату линиях клеток рака молочной железы без захвата лекарственного средства и экспрессии носителя фолата, введение DAC, ингибитора метилирования ДНК, улучшало захват лекарственного средства и экспрессию носителя фолата.
При приобретенной резистентности к алкилирующему лекарственному средству фотемустину в клетках меланомы наблюдали высокую активность MGMT, связанную с гиперметилированием экзонов гена MGMT.
Показано, что в резистентных к иматинибу (Gleevec®) линиях клеток сайленсинг гена SOCS-3 в результате метилирования вызывает активацию белка STAT3, что вызывает неконтролируемую пролиферацию.
Раковые клетки могут становиться резистентными к множеству лекарственных средств в результате измененного мембранного транспорта, повышенной репарации ДНК, дефектов путей апоптоза, изменения целевых молекул, белков и механизмов путей, таких как ферментативная деактивация.
Многие классы антинеопластических лекарственных средств действуют на внутриклеточные компоненты и пути, подобные ДНК, ядерным компонентам, что означает, что им необходимо проникать в раковые клетки. P-гликопротеин (P-gp), или белок множественной лекарственной устойчивости, является фосфорилированным и гликозилированным мембранным транспортером, который может транспортировать лекарственные средства из клетки, таким образом, снижая или устраняя эффективность лекарственного средства. Этот транспортный белок кодируется геном MDR1 и также называется белком АТФ-связывающей кассеты (ABC). MDR1 обладает смешанной субстратной специфичностью, что позволяет ему транспортировать многие структурно отличающиеся соединения через мембрану клетки, в основном, гидрофобные соединения. В исследованиях обнаружено, что ген MDR1 может активироваться и гиперэкспрессироваться в ответ на фармацевтические лекарственные средства, таким образом, формируя основу для резистентности ко многим лекарственным средствам. Гиперэкспрессия гена MDR1 в раковых клетках используется для поддержания внутриклеточных уровней антинеопластических лекарственных средств ниже уровней уничтожения клеток.
Например, обнаружено, что антибиотик рифампицин индуцирует экспрессию MDR1. Эксперименты на линиях клеток, резистентных к разным лекарственным средствам, и ДНК пациента, позволили выявить перестановки генов, инициировавшие активацию или гиперэкспрессию MDR1. Полиморфизм C3435T в экзоне 226 MDR1 также сильно коррелировал с активностью p-гликопротеина.
MDR1 активируется через NF-κB, белковый комплекс, действующий как фактор транскрипции. У крыс участок связывания NF-κB является смежным с геном mdr1b, NF-κB может быть активным в опухолевых клетках из-за мутантного гена NF-κB или того, что его ингибиторный ген IκB мутировал в условиях химиотерапии. В клетках колоректального рака ингибирование NF-κB или MDR1 вызывало повышенный апоптоз в ответ на химиотерапевтическое средство.
Повышенная репарация ДНК играет важную роль в способности раковых клеток преодолевать индуцированные лекарственным средством повреждения ДНК.
Химиотерапевтические средства на основе платины, такие как цисплатин, направленно воздействуют на опухолевые клетки посредством перекрестной сшивки цепей ДНК, вызывая мутацию и повреждение. Такое повреждение будет запускать программируемую гибель клеток (например, апоптоз) в раковых клетках. Резистентность к цисплатину возникает, когда раковые клетки развивают повышенную способность реверсировать такое повреждение, удаляя цисплатин из ДНК и репарируя любое произошедшее повреждение. Резистентные к цисплатину клетки положительно регулируют экспрессию кросс-комплементирующего гена эксцизионной репарации (ERCC1) и его белка.
Некоторыми химиотерапевтическими средствами являются алкилирующие средства, что означает, что они присоединяют алкильную группу к ДНК, предотвращая ее считывание. O-6-метилгуанин-ДНК-метилтрансфераза (MGMT) является ферментом репарации ДНК, удаляющим алкильные группы из ДНК. Экспрессия MGMT подвергается положительной регуляции во многих раковых клетках, что защищает их от алкилирующих средств. Повышенная экспрессия MGMT обнаружена при раке толстого кишечника, раке легких, неходжкинской лимфоме, раке молочной железы, глиомах, миеломе и раке поджелудочной железы.
TP53 является геном опухолевого супрессора, кодирующим белок p53, отвечающий на повреждение ДНК репарацией ДНК, арестом клеточного цикла или апоптозом. Утрата TP53 в результате делеции гена может позволить клеткам непрерывно реплицироваться, несмотря на повреждение ДНК. Толерантность к повреждению ДНК может обеспечивать раковые клетки способом резистентности к этим лекарственным средствам, в норме индуцирующим апоптоз посредством повреждения ДНК.
Другие гены, участвующие в апоптотическом пути, связанные с лекарственной резистентностью, включают h-ras и bcl-2/bax. Обнаружено, что онкогенный h-ras повышает экспрессию ERCC1, что приводит к повышенной репарации ДНК (см. выше). Обнаружено, что ингибирование h-ras повышает чувствительность к цисплатину в клетках глиобластомы. Подвергнутая положительной регуляции экспрессия Bcl-2 в лейкозных клетках (неходжкинской лимфомы) приводила к сниженным уровням апоптоза в ответ на химиотерапевтические средства, т.к. Bcl-2 является способствующим выживанию онкогеном.
Во время таргетированной терапии зачастую мишень модифицирует саму себя и снижает свою экспрессию до такой степени, что терапии более не эффективна. Одним из примеров этого является утрата рецептора эстрогена (ER) и рецептора прогестерона (PR) после антиэстрогенового лечения рака молочной железы. Опухоли с утратой ER и PR больше не отвечают на тамоксифен или другие антиэстрогеновые способы лечения, и, хотя раковые клетки остаются в некоторой степени отвечающими на ингибиторы синтеза эстрогена, они в конечном итоге становятся неотвечающими на эндокринное воздействие и больше не зависят от эстрогена в отношении роста.
Другой линий терапевтических средств, используемых для лечения рака молочной железы, является таргетинг киназа-подобного рецептора эпидермального фактора роста 2 человека (HER2) из семейства EGFR. Мутации зачастую возникают в гене HER2 после лечения ингибитором, при этом обнаружено, что приблизительно 50% пациентов с раком легких имеют мутацию-гейткипер EGFR-T790M.
Лечение хронического миелолейкоза (CML) включает ингибитор тирозинкиназы, нацеленный на слитый ген BCR/ABL, под названием "иматиниб". У некоторых людей, резистентных к иматинибу, ген BCR/ABL реактивирован или амплифицирован, или в гене возникает отдельная точечная мутация. Эти точечные мутации повышают аутофосфорилирование белка BCR-ABL, что приводит к стабилизации АТФ-связывающего участка в его активную форму, которая не может связываться иматинибом для правильной активации лекарственного средства.
Топоизомераза является привлекательной мишенью для терапии рака благодаря своей критической роли в качестве фермента в репликации ДНК, и получено множество ингибиторов топоизомеразы. Резистентность может возникать, когда уровни топоизомеразы снижаются, или когда разные изоформы топоизомеразы по-разному распределяются внутри клетки. Также обнаружены мутантные ферменты в лейкозных клетках пациента, а также мутации в других раковых заболеваниях, придающие резистентность к ингибиторам топоизомеразы.
Одним из механизмов антинеопластической резистентности является гиперэкспрессия метаболизирующих лекарственные средства ферментов или молекул-носителей. При повышении экспрессии метаболических ферментов, лекарственные средства быстрее превращаются в конъюгаты лекарственных средств или неактивные формы, которые затем могут экскретироваться. Например, повышенная экспрессия глутатиона способствует лекарственной резистентности, т.к. электрофильные свойства глутатиона позволяют ему реагировать с цитотоксическими средствами, инактивируя их. В некоторых случаях сниженная экспрессия или утрата экспрессии метаболизирующих лекарственное средство ферментов придает резистентность, т.к. ферменты необходимы для процессирования лекарственного средства из неактивной формы в активную форму. Арабинозид, общеупотребительное химиотерапевтическое средство для лейкоза и лимфом, превращается в цитозинарабинозидтрифосфат под действием дезоксицитидинкиназы. Мутация дезоксицитидинкиназы или утрата экспрессии приводит к резистентности к арабинозиду. Это является формой ферментативной деактивации.
Уровни экспрессии факторов роста также могут способствовать резистентности антинеопластическим способам терапии. При раке молочной железы обнаружено, что резистентные к лекарственному средству клетки экспрессируют высокие уровни ИЛ-6, в то время как чувствительные клетки не экспрессировали значимые уровни фактора роста. ИЛ-6 активируется энхансер CCAAT-связывающие белковые факторы транскрипции, активирующие экспрессию гена MDR1.
Другим типом антинеопластической резистентности является резистентность к ингибиторам контрольных точек. Первичная резистентность к блокаде иммунных контрольных точек возникает у приблизительно от 40% до 65% пациентов с меланомой, которых лечили с помощью терапии, направленной против PD-1. Эта клиническая проблема возникает при невозможности индуцировать эффективный противоопухолевый иммунный ответ на любой из трех стадий иммунного цикла рака. К настоящему времени факторы, ассоциированные с первичной резистентностью, включают повышенные исходные уровни ЛДГ в сыворотке, повышенную исходную опухолевую нагрузку, отсутствие экспрессии PD-L1 в образцах ткани меланомы на исходном уровне, отсутствие инфильтрации T-клетками, отсутствие T-клеток с PD-1 и макрофагов с PD-L1 в биоптатах меланомы, полученных на ранних стадиях лечения, недостаточность неоантигенов и низкая мутационная нагрузка, наличие врожденной сигнатуры резистентности к средствам против PD-1 (IPRES), транскрипционная сигнатура или отсутствие сигнатуры интерферона.
Приобретенная резистентность к иммунотерапии может развиться, если происходит селекция субпопуляций опухолевых клеток с генетическими и эпигенетическими признаками, позволяющим им избегать иммунного надзора. Примером является то, что утрата экспрессии B2M была обнаружена в линиях клеток меланомы от пациентов, которых лечили с помощью иммунотерапии и цитокиновой генотерапия. Это приводило к утрате экспрессии MHC класса I и, таким образом, последующему снижению распознавания CD8+ T-клетками. Недавно мутации JAK1/2 также идентифицированы как генетические маркеры приобретенной резистентности к иммунотерапии при меланоме. Эти мутации в опухолевых клетках приводят к сниженной чувствительности к ИФНγ, в конечном итоге предотвращая индуцируемый ИФНγ арест роста клеток. Мутации с утратой функции в генах кодирующий JAK1 или JAK2 обнаружены в рецидивирующих опухолях после полноэкзомного секвенирования биоптатов на исходном уровне и после прогрессирования; все пациенты имели объективный ответ на лечение пембролизумабом, а затем прогрессировали. Кроме того, приобретенная резистентность также может возникать на уровне отдельных клеток, если опухолевые клетки изменяют экспрессию своих генов в ответ на молекулы иммунной системы в микроокружении опухоли. Например, PD-L1 может подвергаться положительной регуляции опухолевыми клетками в ответ на цитокины, такие как ИФНγ, высвобождаемый T-клетками, таким образом, ограничивая функцию T-клеток, и это может происходить и при первичной, и при приобретенной резистентности.
III. Фармацевтические составы и пути введения
Если предусмотрено клиническое использование, фармацевтические композиции будут получать в форме, подходящей для предполагаемого использования. Как правило, это будет включать получение композиций, по существу, несодержащих пирогены, а также другие примеси, которые могут причинять вред людям или животным.
Как правило, желательным будет использование подходящих солей и буферов, чтобы сделать лекарственные средства стабильными и сделать возможным захват клетками-мишенями. Водные композиции по изобретению содержат эффективное количество лекарственного средства, растворенного или диспергированного в фармацевтически приемлемом носителе или водной среде. Фраза "фармацевтически или фармакологически приемлемый" относится к молекулярным соединениям и композициям, невызывающим побочные, аллергические или другие нежелательные реакции при введении животному или человеку. В рамках изобретения термин "фармацевтически приемлемый носитель" включает растворители, буферы, растворы, дисперсионные среды, покрытия, антибактериальные и противогрибковые средства, изотонические и замедляющие абсорбцию средства и т.п., приемлемые для использования в составлении фармацевтических средств, таких как фармацевтические средства, подходящие для введения людям. Использование таких сред и средств для фармацевтически активных веществ хорошо известно в этой области. За исключением случаев, когда любая общепринятая среда или средство несовместимо с активными ингредиентами по изобретению, предусмотрено его использование в терапевтических композициях. Дополнительные активные ингредиенты также можно включать в композиции, при условии, что они не инактивируют средства в композициях.
Активные композиции по изобретению могут включать классические фармацевтические препараты. Введение этих композиций в соответствии с настоящим изобретением можно осуществлять любым общепринятым путем при условии, что целевая ткань доступна при использовании этого пути, но, как правило, это включает системное введение. Это включает пероральный, назальный или буккальный путь. Альтернативно, введение можно осуществлять посредством внутрикожной, подкожной, внутримышечной, интраперитонеальной или внутривенной инъекции, или внутриопухолево или регионарно в опухоль, например, в сосудистое русло опухоль. Такие композиции, как правило, будут вводить в виде фармацевтически приемлемых композиций, как описано выше.
Активные соединения также можно вводить парентерально или интраперитонеально. В качестве иллюстрации, растворы активных соединений в виде свободного основания или фармакологически приемлемых солей можно получать воде, соответствующим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также можно получать в глицерине, жидких полиэтиленгликолях и их смесях и в маслах. В общепринятых условиях хранения и использования эти препараты, как правило, содержат консервант для предотвращения роста микроорганизмов.
Фармацевтические формы, подходящие для инъекционного использования, включают, например, стерильные водные растворы или дисперсии и стерильные порошки для экстемпорального получения стерильных инъекционных растворов или дисперсий. Как правило, эти препараты являются стерильными и жидкими до такой степени, что их легко вводить с помощью инъекции. Препараты должны быть стабильными в условиях производства и хранения и должны быть защищены от контаминирующего действия микроорганизмов, таких как бактерии и грибы. Подходящие растворители или дисперсионные среды могут содержать, например, воду, этанол, полиол (например, глицерин, пропиленгликоль, жидкий полиэтиленгликоль и т.п.), их подходящие смеси и растительные масла. Правильную текучесть можно поддерживать, например, с использованием покрытия, такого как лецитин, посредством поддержания необходимого размера частиц в случае дисперсии и с использованием поверхностно-активных веществ. Предотвращения действия микроорганизмов можно достигать посредством различных антибактериальных и противогрибковых средств, например, парабенов, хлорбутанола, фенола, сорбиновой кислоты, тиомерсала и т.п. Во многих случаях предпочтительно включать изотонические средства, например, сахара или хлорид натрия. Пролонгированной абсорбции инъецированных композиций можно достигать с использованием композиций средств, замедляющих абсорбцию, например, моностеарата алюминия и желатина.
Стерильные инъецируемые растворы можно получать посредством включения активных соединений в соответствующем количестве в растворитель вместе с любыми другими ингредиентами (например, перечисленными выше), при желании, с последующей стерилизацией фильтрацией. Как правило, дисперсии получают посредством включения различных стерилизованных активных ингредиентов в стерильный носитель, содержащий основную дисперсионную среду и желаемые другие ингредиенты, например, перечисленные выше. В случае стерильных порошков для получения стерильных инъецируемых растворов, предпочтительные способы получения включают вакуумную сушку и способы лиофилизации, которыми получают порошок активных ингредиентов с любым дополнительным желаемым ингредиентом из его ранее стерилизованного фильтрацией раствора.
Композиции по изобретению, как правило, можно составлять в нейтральной форме или форме соли. Фармацевтически приемлемые соли включают, например, кислые соли присоединения (образованные со свободными аминогруппами белка), полученные из неорганических кислот (например, соляной или фосфорной кислот) или органических кислот (например, уксусной, щавелевой, винной, миндальной и т.п.). Соли, образованные со свободными карбоксильными группами белка также можно получать из неорганических оснований (например, гидроксидов натрия, калия, аммония, кальция или железа) или органических оснований (например, изопропиламина, триметиламина, гистидина, прокаина и т.п.).
После составления растворы предпочтительно вводят способом, совместимым с лекарственным составом, и в количестве, являющемся терапевтически эффективным. Составы легко можно вводить в различных лекарственных формах, таких как инъецируемые растворы, капсулы для высвобождения лекарственного средства и т.п. Для парентерального введения в водном растворе, например, раствор, как правило, соответствующим образом забуферивают и жидкий дилюент сначала делают изотоническим, например, с помощью достаточного количества физиологического раствора или глюкозы. Такие водные растворы можно использовать, например, для внутривенного, внутримышечного, подкожного и интраперитонеального введения. Предпочтительно, стерильные водные среды используют так, как известно специалистам в этой области, в частности, в свете настоящего изобретения. В качестве иллюстрации, однократную дозу можно растворять в 1 мл изотонического раствора NaCl и добавлять к 1000 мл жидкости для гиподермоклизиса или инъецировать в запланированное место инфузии (см., например, "Remington's Pharmaceutical Sciences" 15-ое издание, стр. 1035-1038 and 1570-1580). Обязательно будут осуществлять некоторое изменение в зависимости от состояния индивидуума, подвергаемого лечению. Специалист, отвечающий за введение, в любом случае будет определять подходящую дозу для отдельного индивидуума. Кроме того, в случае введения человеку, препараты должны соответствовать стандартам стерильности, пирогенности, общей безопасности и чистоты, как этого требует Office of Biologics standards FDA.
IV. Комбинированная терапия
В контексте настоящего изобретения также предусмотрено, что 6-тио-dG/средство против PD-L1, такое как атезолизумаб, или 6-тио-dG/средство против PD-1, такое как Libtayo®, или средство против CTAL-4 можно использовать в комбинации с химиотерапией, или лучевой терапией, или другим лечением. Эффективным также может оказаться, в частности, комбинирование 6-тио-dG/средства против PD-L1, средства против PD-1 или средства против CTLA-4 с другими способами терапии, направленными на разные аспекты функционирования раковых клеток.
Для уничтожения клеток, ингибирования роста клеток, ингибирования метастазирования, ингибирования ангиогенеза или иного реверсирования или уменьшения злокачественного фенотипа опухолевых клеток, с использованием способов и композиций по изобретению, как правило, можно приводить "целевую" клетку в контакт с 6-тио-dG и по меньшей мере одним другим средством. Эти композиции будут вводить в последовательном или комбинированном количестве, эффективном для уничтожения или ингибирования пролиферации клетки. Это может включать приведение клеток в контакт с 6-тио-dG/средством против PD-L1, средством против PD-1 или средством против CTLA-4 и, в то же время, другими средствами или факторами. Этого можно достигать посредством приведения клетки в контакт с отдельной композицией или фармакологическим составом, включающим оба средства, или посредством приведения клетки в контакт с двумя отдельными композициями или составами одновременно, где одна композиция включает пролекарства интерферона по изобретению, а другая включает другое средство.
Альтернативно, терапия 6-тио-dG/средством против PD-L1, средством против PD-1 или средством против CTLA-4 может предшествовать или следовать за лечением другим средством с интервалами в диапазоне от минут до недель. В вариантах осуществления, в которых другое средство и пролекарства интерферона используют в отношении клетки раздельно, как правило, будут избегать значительного периода времени между каждой доставкой таким образом, что средство и экспрессирующая конструкция все еще смогут иметь благоприятный комбинированный эффект в отношении клетки. В таких случаях, предполагают, что можно приводить клетку в контакт с обоими вариантами лечения в пределах приблизительно 12-24 часов друг относительно друга, и более предпочтительно - в пределах приблизительно 6-12 часов друг относительно друга, при этом время задержки всего лишь приблизительно 12 часов является наиболее предпочтительным. В некоторых случаях желательным может являться значительное увеличение периода лечения, однако, когда между соответствующими введениями проходит от нескольких дней (2, 3, 4, 5, 6 или 7) до нескольких недель (1, 2, 3, 4, 5, 6, 7 или 8).
Также возможно, что желательным будет более одного введения пролекарств интерферона или другого средства. Можно использовать различные комбинации, где терапия 6-тио-dG/средством против PD-L1, средством против PD-1 или средством против CTLA-4 обозначена как "A", а другая терапия обозначена как "B", примеры чего приведены ниже:
A/B/A, B/A/B, B/B/A, A/A/B, B/A/A, A/B/B, B/B/B/A, B/B/A/B, A/A/B/B, A/B/A/B, A/B/B/A, B/B/A/A, B/A/B/A, B/A/A/B, B/B/B/A, A/A/A/B, B/A/A/A, A/B/A/A, A/A/B/A, A/B/B/B, B/A/B/B, B/B/A/B
Предусмотрены другие комбинации. И снова, для достижения уничтожения клеток оба средства доставляют к клетке в комбинированном количестве, эффективном для уничтожения клетки.
Средства или факторы, подходящие для противоопухолевой терапии, включают любое химическое соединение или способ лечения, индуцирующий повреждение ДНК при использовании в отношении клетки. Такие средства и факторы включают радиацию и излучение, индуцирующее повреждение ДНК, такое как радиационное облучение, микроволны, испускание электронов и т.п. Можно использовать различные химические соединения, также описанные как "химиотерапевтические" или "генотоксические средства". Этого можно достигать посредством облучения локализованного очага опухоли; альтернативно, опухолевые клетки можно приводить в контакт со средством посредством введения индивидууму терапевтически эффективного количества фармацевтической композиции.
Для использования в настоящем изобретении предусмотрены различные классы химиотерапевтических средств. Ниже описан иметелстат. Другие химиотерапевтические средства включают селективные антагонисты эстрогеновых рецепторов ("SERM"), такие как тамоксифен, 4-гидрокситамоксифен (афимоксфен), фалсодекс, ралоксифен, базедоксифен, кломифен, фемарель, лазоксифен, ормелоксифен и торемифен. Средства камптотецин, актиномицин-D и митомицин C являются общеупотребительными химиотерапевтическими лекарственными средствами. Настоящее изобретение также включает использование комбинации одного или более ДНК-повреждающих средств, на основе радиации или конкретных соединений, таких как использование рентгеновского излучения с цисплатином или использование цисплатина с этопозидом. Средство можно получать и использовать в качестве комбинированной терапевтической композиции.
Белок теплового шока 90 является регуляторным белком, обнаруживаемым во многих эукариотических клеток. Показано, что ингибиторы HSP90 можно использовать в лечении рака. Такие ингибиторы включают гелданамицин, 17-(аллиламино)-17-деметоксигелданамицин, PU-H71 и рифабутин.
Также предусмотрены средства, напрямую перекрестно сшивающие ДНК или образующие аддукты. Можно использовать средства, такие как цисплатин, и другие ДНК-алкилирующие средства. Цисплатин широко используют для лечения рака, при этом эффективные дозы, используемые в клинических условиях, составляют 20 мг/м2 в течение 5 дней каждые три недели всего в трех курсах. Цисплатин не абсорбируется при пероральном введении, и, таким образом, его необходимо вводить посредством инъекции внутривенно, подкожно, внутриопухолево или интраперитонеально.
Средства, повреждающие ДНК, также включают соединения, противодействующие репликации ДНК, митозу и хромосомной сегрегации. Такие химиотерапевтические соединения включают адриамицин, также известный как доксорубицин, этопозид, верапамил, подофиллотоксин и т.п. Широко используемые в клинических условиях для лечения неоплазий, эти соединения вводят посредством болюсных инъекций внутривенно в дозах в диапазоне 25-75 мг/м2 с 21-дневными интервалами в случае доксорубицин, 35-50 мг/м2 в случае этопозида внутривенно или двойной внутривенной дозе перорально. Также предусмотрены ингибиторы микротрубочек, такие как таксаны. Эти молекулы являются дитерпенами, продуцируемыми растениями рода Taxus и включают паклитаксел и доцетаксел.
Ингибиторы рецептора эпидермального фактора роста, такие как иресса, mTOR, мишень рапамицина в клетках млекопитающих (также известный как FK506-связывающий белок 12-рапамицин-ассоциированный белок 1 (FRAP1)), является серин/треониновой протеинкиназой, регулирующей рост клеток, пролиферацию клеток, подвижность клеток, выживание клеток, синтез белка и транскрипцию. Рапамицин и его аналоги ("рапалоги"), таким образом, предусмотрены для использования в противоопухолевой терапии по настоящему изобретению. Другим ингибитором EGFR, который можно использовать в настоящем изобретении, является гефитиниб.
Другая возможная терапия представляет собой ФНОα (фактор некроза опухоли-альфа), цитокин, участвующий в системном воспалении, и член группы цитокинов, стимулирующих реакцию острой фазы. Основной ролью ФНО является регуляция иммунных клеток. ФНО также может индуцировать апоптотическую гибель клеток, индуцировать воспаление и ингибировать образование опухоли и репликацию вируса.
Средства, нарушающие синтез и точность предшественников нуклеиновых кислот и субъединиц также приводит к повреждению ДНК. В связи с этим, разработан ряд предшественников нуклеиновых кислот. Особенно подходят средства, подвергшиеся обширному тестированию и легкодоступные. В связи с этим, средства, такие как 5-фторурацил (5-FU), преференциально используются неопластической тканью, что делает это средство особенно пригодным для таргетинга неопластических клеток. Хотя он и достаточно токсичен, 5-FU можно использовать в широком спектре носителей, включая топические, однако внутривенное введение в дозах в диапазоне от 3 до 15 мг/кг/сутки является общеупотребительным.
Другие факторы, вызывающие повреждение ДНК и широко используемые, включают то, что общеизвестно как γ-излучение, рентгеновское излучение и/или направленная доставка радиоактивных изотопов в опухолевые клетки. Также предусмотрены другие формы ДНК-повреждающих факторов, такие как микроволны и УФ-излучение. Скорее всего, все эти факторы вызывают широкий спектр повреждения ДНК в отношении предшественников ДНК, репликации и репарации ДНК и сборки и поддержания хромосом. Диапазоны доз в случае рентгеновского излучения составляют от суточных доз от 50 до 200 рентген в течение длительных периодов времени (от 3 до 4 недель) до однократных доз от 2000 до 6000 рентген. Диапазоны доз в случае радиоактивных изотопов широко варьируются и зависят от времени полужизни изотопа, силы и типа испускаемого излучения и захвата неопластическими клетками.
Кроме того, также предполагают, что можно использовать различную иммунотерапию, гормональную терапию, терапию токсинами и/или хирургическое вмешательство.
Специалистам в этой области могут использовать "Remington’s Pharmaceutical Sciences", 15-ое издание, глава 33, в частности, стр. 624-652. Обязательно будут осуществлять некоторые вариации дозы в зависимости от состояния индивидуума, подвергаемого лечению. Специалист, отвечающий за введение, в любом случае будет определять подходящую дозу для отдельного индивидуума. Кроме того, в случае введения человеку препараты должны соответствовать стандартам стерильности, пирогенности, общей безопасности и чистоты, как этого требует Office of Biologics standards FDA.
V. Примеры
В следующем разделе "Примеры" приведены дополнительные подробности, касающиеся примеров различных вариантов осуществления. Специалистам в этой области следует понимать, что способы, описанные в следующих примерах, представляют собой способы и/или композиции, которые, как обнаружили авторы настоящего изобретения, хорошо работают. Однако в свете настоящего описания специалисты в этой области должны понимать, что можно осуществлять множество изменений в конкретных описанных вариантах осуществления и все равно получать подобный или схожий результат без отклонения от сущности и объема настоящего изобретения. С помощью этих примеров иллюстрируют способы и системы, представленные в настоящем описании, и они не предназначены для ограничения объема настоящего изобретения. Их неограничивающие примеры включают представленное ниже.
Пример 1 - Материалы и способы
Мыши. Самок мышей C57BL/6J, BALB/c, Myd88-/-, Tmem173-/-, Batf3-/- и OT-1, трансгенных по CD8+ T-клеточному рецептору, с фоном C57BL/6J и мышей NSG-SMG3 приобретали в The Jackson Laboratory. Rag1-/- мышей и ИФН-репортерных мышей (Ifngtm3.1Lky/J) с фоном C57BL/6 приобретали в виварии Юго-западного медицинского центра Техасского университета. Ifnαr1-/- мышей предоставляла Dr. Anita Chong из Университета Чикаго. Всех мышей держали в специальных беспатогенных условиях. Содержание и эксперименты на животных осуществляли по протоколу и руководствам организации и Национальных институтов здравоохранения США. Это исследование одобрено Институциональным комитетом по содержанию и использованию животных Юго-западного медицинского центра Техасского университета.
Линии клеток и реагента. Клетки MC38, CT26, LLC A375 и HCT116 приобретали в ATCC. Клетки MC38-OVA получали посредством лентивирусной трансдукции гена OVA. Все линии клеток общепринятым образом тестировали с использованием набора для определения заражения микоплазмой (R&D) и культивировали в модифицированной по способу Дульбекко среде Игла, дополненной 10% термически инактивированной эмбриональной телячьей сывороткой, 100 ед./мл пенициллина и 100 ед./мл стрептомицина, при 5% CO2 и 37°C.
mAb против CD4 (GK1.5), против NK1.1 (PK136), против CD8 (53-5.8) и против CSF1R (AFS98) приобретали в BioXCell. Антитела против PD-L1 (атезолизумаб) и против CTLA-4 (ипилимумаб) были любезно предоставлены Аптечной службой Онкологического центра Симмонса Юго-западного медицинского центра Техасского университета. 6-тио-dG приобретали в Metkinen Oy. Для исследований in vitro 6-тио-dG растворяли в DMSO/воде (1:1) для получения 10 мМ стоковых растворов. Для исследований in vivo подготавливали 3 мг/кг 6-тио-dG в 5% DMSO (в 1-ркатном PBS) для интраперитонеальной инъекции. Лекарственные средства хранили замороженными при -20°C до использования.
Анализ жизнеспособности клеток. Для определения IC50 с использованием анализов пролиферации клеток, линии раковых клеток мыши и человека подвергали скринингу с использованием 6-тио-dG с серией 2-кратных разведений в 8 разных точках в 96-луночных планшетах. Клетки высевали за 24 ч. до добавления лекарственного средства, инкубировали в течение 4-5 дней и анализировали с использованием анализа пролиферации клеток CellTiter 96® Aqueous One Solution по инструкциям производителя (Promega). Количество клеток на лунку находилось в диапазоне от 1000 до 10000 клеток на лунку, являясь обратно пропорциональным времени удвоения. Получали кривые доза-эффект и вычисляли IC50s с использованием Graphpad Prism. Все образцы анализировали в трех параллелях и получали стандартные отклонения для 2-3 независимых экспериментов.
Анализ колониеобразования. Клетки MC38 высевали в трех разных концентрациях в 6-луночные планшеты (1000-4000 клеток/лунку) и обрабатывали различными концентрациями лекарственных средств каждые 3-4 дня. После обработки в течение 13 дней клетки фиксировали и окрашивали раствором 6% глутаральдегида (Fisher Scientific) с 0,5% кристалл-виолет (Sigma). После промывки проточной водой клетки сушили на воздухе и получали изображения с использованием G-BOX (Syngene, модель: G-BOX F3).
Анализ фокусов, индуцированных дисфункцией теломер (TIF) и анализы микроядер. Анализ TIF основан на детекции колокализации повреждения ДНК с помощью антитела против факторов ответа на повреждение ДНК, таких как гамма-H2AX, 53BP1, и антитела против теломерных белков или теломер с использованием специфического в отношении последовательности теломер зонда пептид-нуклеиновой кислоты (ПНК) (Mender and Shay, 2015). В кратком изложении, клетки высевали в 4-луночные слайд-камеры. На следующий день клетки обрабатывали 1 мкМ 6-тио-dG в течение 24 ч. (в случае анализа TIF) или 1-3 мкМ 6-тио-dG в течение 48 ч. (в случае анализа микроядер). Затем слайд-камеры дважды промывали PBS и фиксировали в 4% формальдегиде (Thermo Fisher) в PBS в течение 10 мин. Затем клетки дважды промывали PBS и пермеабилизовали в 0,5% Triton X-100 в PBS в течение 10 мин. После пермеабилизации клетки промывали три раза PBS. Клетки блокировали 10% сывороткой козы в 0,1% PBST (TritonX-100) в течение 1 ч. Гамма-H2AX (анализ TIF, мышь, 1:1000) (Millipore) или ламин A/C (анализ микроядер, мышь, 1:500) (Santa Cruz) разводили в блокирующем растворе и инкубировали на клетках в течение 2 ч. После трех промывок PBST (1-кратный PBS в 0,1% Triton) и 3 промывок PBS клетки инкубировали с конъюгированным с Alexa Fluor 568 антителом козы против мыши (1:500) (Invitrogen) в течение 40 мин, затем промывали пять раз 0,1% PBST. Клетки фиксировали в 4% формальдегиде в PBS в течение 20 мин при RT. Препараты последовательно дегидратировали с помощью 70%, 90%, 100% этанола с последующей денатурацией с помощью гибридизационного буфера, содержащего FAM-конъюгированный, специфический в отношении последовательности теломеры (C-богатой) зонд ПНК, 70% формамида, 30% 2-кратного SSC, 10% (масс./об.) MgCl2·6H2O (Fisher Sci), 0,25% (масс./об.) блокирующего реагента для гибридизации и детекции нуклеиновых кислот (Roche) в течение 7 мин при 80°C на термоблоке с последующей инкубацией в течение ночи при RT. Препараты промывали последовательно 70% формамидом (Ambion)/0,6-кратным SSC (Invitrogen) (2×1 ч.), 2-кратным SSC (1×15 мин), PBS (1×5 мин) и последовательно дегидратировали 70%, 90%, 100% этанолом, затем заключали в среду для заливки Vectashield с DAPI (Vector Laboratories). Изображения получали с помощью флуоресцентного микроскопа с использованием объектива 100X. TIF количественно анализировали с использованием Image J.
Детекция ДНК в полученных из костного мозга дендритных клетках. Клетки метили EdU, как описано ранее (Min et al., 2019). В кратком изложении, 100000 клеток MC38 высевали в 6-луночный планшет и метили с помощью 25 мкМ EdU. Через два дня клетки вымывали и обрабатывали 1 мкМ 6-тио-dG в течение 24 ч. Клетки снова вымывали и сокультивировали с BMDC в течение ночи. На следующий день DC отсортировывали с помощью магнитных бус, промывали, фиксировали и цитоцентрифугировали. Затем препараты окрашивали флуоресцентным азидом 6-карбокситетраметилродамином (Invitrogen) в свежем, самостоятельно подготовленном окрашивающем растворе EdU (PBS, содержащем 1 мМ CuSO4, 2 мМ аскорбиновую кислоту) в течение 30 мин. Затем препараты интенсивно промывали PBS в течение по меньшей мере 1 ч., а затем следовали стадиям FISH теломер с использованием зонда FAM-TelG, как описано в частях способа в разделе "Анализ фокусов, индуцированных дисфункцией теломер (TIF) и анализ микроядер". Изображения получали при увеличении 63X с помощью Axio Imager Z2, оборудованном автоматической системой для захвата метафазы (камерой Coolcube1) и анализировали с помощью программного обеспечения ISIS (Metasystems).
Иммуно-FISH. В кратком изложении тканевые срезы толщиной 5 мкм депарафинизировали с помощью ксилола (2×5 мин), 100% этанола (2×2 мин), 95% этанола (1×2 мин), 75% этанола (1×2 мин) и 50% этанола (1×2 мин), а затем промывали проточной водой (2×3 мин). Депарафинизированные тканевые срезы инкубировали в буфере цитрата натрия (10 мМ цитрата Na, 0,05% Tween 20, pH=6,0) под воздействием микроволн в течение 20 мин для демаскирования антигенов. После охлаждения тканевых срезов, их промывали 1-кратным PBS в течение 5 мин, а затем дегидратировали в 95% этаноле в течение 3 мин. Денатурацию осуществляли с помощью гибридизационного буфера (70% формамида, 30% 2-кратного SSC, 10% (масс./об.) MgCl2·6H2O (Fisher Sci), 0,25% (масс./об.) блокирующий реагент (Roche)), содержащего FITC-конъюгированный, специфический в отношении последовательности теломер (TTAGGG)3 зонд ПНК, в течение 7 мин при 80°C на термоблоке. Препараты промывали последовательно 70% формамидом/0,6-кратным SSC (3×15 мин), 2-кратным SSC (1×15 мин), PBS (1×5 мин), PBST (PBS+0,1% Tween 20; 1×5 мин) и инкубировали с блокирующим буфером (4% BSA в PBST) в течение 30 мин. Срезы инкубировали с антителом против фосфо-гистона H2AX (1:500) (Cell Signaling) в блокирующем буфере при RT в течение 1 ч. После промывок PBST 2×5 мин тканевые срезы инкубировали с конъюгированным с Alexa Fluor 568 антителом козы против кролика в блокирующем буфере при RT в течение 1 ч. Срезы промывали последовательно PBST (3×5 мин) и PBS (1×5 мин). Препараты заключали в среду для заливки Vectashield с DAPI. Изображения получали с помощью флуоресцентного микроскопа с использованием объектива 100X. TIF количественно анализировали с использованием Image J.
Рост опухоли и введение. Всего инокулировали 5×105 клеток MC38, 5×105 клеток CT26 или 1×106 клеток LLC подкожно дорзально в правый бок мышей в 100 мкл фосфатно-солевого буфера (PBS). Несущих опухолей мыши случайным образом группировали в исследуемые группы, когда опухоли достигали приблизительно 100 мм3. В случае однократного введения 6-тио-dG, 3 мг/кг 6-тио-dG вводили интраперитонеально в дни 7, 8 и 9 в случае опухоли MC38 и опухоли LLC и в дни 5, 6, 7 в случае опухоли CT26. Для истощения CSF1R, NK1.1, CD4+ и CD8+ T-клеток 200 мкг антител инъецировали интраперитонеально за 1 день до начала лечения, а затем дважды в недели в течение 2 недель. Для PD-L1-блокирующего комбинированного лечения в модели MC38 6-тио-dG вводили в день 10 и день 11, 50 мкг антитела к PD-L1 инъецировали интраперитонеально в день 13 и 17. Для PD-L1-блокирующего комбинированного лечения в модели LLC, 6-тио-dG вводили в день 4, 5, 6, 10 и 11, 200 мкг антитела к PD-L1 инъецировали интраперитонеально в день 8 и 13. Объемы опухолей измеряли по длине (a), ширине (b) и высоте (h) и вычисляли как объем опухоли=abh/2.
Модели опухолей на гуманизированных мышах. Получение гуманизированных мышей описано ранее (Qiao et al., 2019). В кратком изложении, самок мышей NSG-SGM3 возрастом четыре недели облучали в дозе 100 сГр (рентгеновское излучение, облучатель X-RAD 320) за один день до переноса CD34+ клеток человека. Пуповинную кровь получали из Мемориального госпиталя Паркленд Юго-западного Техасского университета. CD34+ клетки человека очищали из пуповинной крови посредством центрифугирования в градиенте плотности (Ficoll® Paque Plus, GE Healthcare) с последующей положительной иммуномагнитной селекцией с помощью микрочастиц с антителом против CD34 человека (Stemcell). 1×105 CD34+ клеток инъецировали внутривенно каждой мыши-реципиенту. Через 12 недель после пересадки гуманизированным мышам, имеющим более чем 50% реконституции CD45+ клеток человека и совпадающим по возрасту и полу с негуманизированными мышами, подкожно инокулировали 1×106 опухолевых клеток HCT116 в правый бок. 3 мг/кг 6-тио-dG вводили интраперитонеально в дни 7, 8 и 9. Объемы опухолей измеряли дважды в неделю. Эксперименты осуществляли в соответствии с протоколом Комитета по исследованиям на людях UTSW и Институционального комитета по содержанию и использованию животных UTSW.
Линия клеток MC38 с нокаутом Tmem173 и Mb21d1. Гены Tmem173 и Mb21d1 в клетках MC38 подвергали нокауту с помощью технологии CRISPR/Cas9. Гидовую последовательность 5’-CACCTAGCCTCGCACGAACT-3’ (SEQ ID NO: 1) для Tmem173 и 5’-CGCAAAGGGGGGCTCGATCG-3’ (SEQ ID NO: 2) для Mb21d1 клонировали в плазмиды px458 (неинтегрирующаяся плазмида с селективным маркером GFP), а затем с помощью них транзиторно трансфицировали опухолевые клетки с использованием липофектамина 2000 (Thermo Fisher). Через 24 часа GFP-положительные клетки сортировали и культивировали еще в течение недели. Затем сортированные клетки высевали в 96-луночные планшеты. Еще через неделю GFP-отрицательные клоны пассировали в 12-луночный планшет и осуществляли вестерн-блоттинг для идентификации нокаутных клонов. И наконец, все нокаутные клоны объединяли для экспериментов.
Способ иммуноферментных пятен (ELISPOT) для анализа ИФНγ. Опухоли MC38 инъецировали подкожно в правый бок мышей C57BL/6. В случае однократного введения 6-тио-dG 3 мг/кг 6-тио-dG вводили интраперитонеально в дни 7, 8 и 9; для PD-L1-блокирующего комбинированного лечения в модели MC38 3 мг/кг 6-тио-dG вводили в день 10 и день 11, 50 мкг антитела к PD-L1 инъецировали интраперитонеально в день 11. Через 7 дней после последнего введения собирали дренирующий опухоль лимфоузел и селезенку несущих опухоли мышей и получали суспензию отдельных клеток. Облученные опухолевые клетки MC38 и контрольные опухолевые клетки LLC использовали для повторной стимуляции опухолеспецифических T-клеток. 1,5×105 клеток дренирующих лимфоузлов или спленоцитов и 7,5×104 облученных опухолевых клеток сокультивировали в течение 48 ч. и осуществляли анализ ELISPOT с использованием набора для ELISPOT ИФНγ (BD Bioscience) по инструкциям производителя. Пятна ИФНγ подсчитывали с помощью анализатора CTL-ImmunoSpot® S6 (Cellular Technology Limited).
Сокультивирование in vitro дендритных клеток костного мозга (BMDC) и T-клеток. Суспензии отдельных клеток костного мозга (BM) получали из большеберцовых костей и бедренных костей мышей C57BL/6. Клетки BM помещали в чашку 10 см и культивировали с полной средой RPMI 1640, содержащей 20 нг/мл рекомбинантного ГМ-КСФ мыши (BioLegend). Свежую среду добавляли в культуру в день 3 и день 6. BMDC собирали в день 7. CD8+ T-клетки выделяли из лимфоузлов и селезенки трансгенных мышей OT-1 с помощью набора для выделения CD8+ T-клеток посредством отрицательной селекции (Stemcell). Клетки MC38-OVA предварительно обрабатывали 200 нМ 6-тио-dG в течение 4 ч. Затем вымывали лекарственное средство, опухолевые клетки продолжали культивировать в течение 72 ч. и собирали в тот же день, что и BMDC. Затем клетки MC38-OVA сокультивировали с BMDC в течение ночи. Супернатант собирали для анализа ELISA ИФНβ (PBL). BMDC сортировали с помощью набора для положительной селекции по CD11c+ (Stemcell) и сокультивировали с OT-1 CD8+ T-клетками в течение 48 ч. Собирали супернатанты и измеряли ИФНγ с помощью цитометрического анализа на бусах (BD Biosciences).
Выделение цитозольной ДНК и количественная ПЦР в реальном времени. Клетки HCT116 предварительно обрабатывали 500 нМ 6-тио-dG в течение 4 ч. Затем лекарственное средство вымывали, опухолевые клетки продолжали культивировать в течение 72 ч. и собирали в тот же день, что и BMDC. Затем клетки HCT116 смешивали 1:1 с 1×106 BMDC в течение 4 ч. BMDC очищали и разделяли на две равные аликвоты. Одну аликвоту подвергали выделению тотальной геномной ДНК с помощью набора для выделения геномной ДНК Purelink (Invitrogen), и она служила в случае нормализационного контроля. Другую аликвоту ресуспендировали в 100 мкл буфера для цитозольного выделения, содержащего 150 мМ NaCl, 50 мМ HEPES и 25 мг/мл дигитонина (Sigma) и инкубировали в течение 10 мин при RT для пермеабилизации плазматической мембраны (West et al., 2015). Затем клетки центрифугировали для осаждения интактных клеток. Цитозольные супернатанты собирали и центрифугировали при 12000×g в течение 10 мин для осаждения остального клеточного детрита. Затем цитозольную ДНК выделяли с помощью набора для выделения геномной ДНК Purelink (Invitrogen). Количественную ПЦР осуществляли и с помощью экстрактов целых клеток, и с помощью цитозольных фракций с использованием праймеров для ДНК человека и праймеров ДНК мыши (Xu et al., 2017).
Расщепление опухоли. Опухолевые ткани иссекали и расщепляли с помощью 1 мг/мл коллагеназы I (Sigma) и 0,5 мг/мл ДНКазы I (Roche) при 37°C в течение 30 мин, затем опухоль пропускали через клеточное сито 70 мкм для удаления больших фрагментов нерасщепленного опухолевого материала. Инфильтрирующие опухоль клетки дважды промывали PBS, содержащим 2 мМ ЭДТА.
Проточная цитометрия. Суспензии отдельных клеток инкубировали с антителом против рецептора FcγIII/II (клон 2.4G2) в течение 15 минут для блокирования неспецифического связывания перед окрашиванием с помощью конъюгированных антител, а затем инкубировали с указанным антителом в течение 30 мин при 4°C в темноте. Фиксируемый краситель для оценки жизнеспособности eFluor 506 или eFluor780 (eBioscience) использовали для исключения погибших клеток. Foxp3 и Ki67 окрашивали внутриклеточно с использованием набора буферов для факторов транскрипции True-Nuclear (BioLegend) по инструкциям производителя. Данные получали с помощью проточного цитометра CytoFLEX (Beckman Coulter, Inc) и анализировали с использованием программного обеспечения FlowJo (Tree Star Inc., Ashland, OR).
Количественная ПЦР в реальном времени. ПЦР в реальном времени осуществляли с использованием SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad) по инструкциям производителя с разными наборами праймеров (MT-CO1 человека, прямой праймер 5’-CGCCACACTCCACGGAAGCA-3’ (SEQ ID NO: 3), обратный праймер 5’-CGGGGCATTCCG GATAGGCC-3’ (SEQ ID NO: 4); рРНК 18s человека, прямой праймер 5’-ACCGATTGGATGGTTTAGTGAG-3’(SEQ ID NO: 5), обратный праймер 5’-CCTACGGAAACCTTGTTACGAC-3’ (SEQ ID NO: 6); ИФНβ мыши, прямой праймер 5’-ATGAGTGGTGGTTGCAGGC-3’ (SEQ ID NO: 7), обратный праймер 5’-TGACCTTTCAAATGCAGTAGATTCA-3’ (SEQ ID NO: 8); GAPDH мыши, прямой праймер 5’-CATCAAGAA GGTGGTGAAGC-3’ (SEQ ID NO: 9), обратный праймер 5’-CCTGTTGCTGTAGCCGTATT-3’ (SEQ ID NO: 10)), GAPDH мыши использовали в качестве внутреннего контроля. Способ 2−ΔΔCt использовали для вычисления относительных изменений экспрессии.
Иммуноблоттинг. Обработку BMDC и MC38 проводили так же, как описано в разделе "Сокультивирование in vitro дендритных клеток костного мозга". Через 6 ч. после сокультивирования DC выделяли с помощью набора для положительной селекции по CD11c+ (Stemcell). Получение образца белка и иммуноблоттинг осуществляли, как описано ранее (Liu et al., 2019). Белки определяли с помощью моноклональных антител кролика против pSTING (Cell signaling, 72971), STING (Cell signaling, 50494), pTBK1 (Cell signaling, 5483), TBK1 (Cell signaling, 3504). Белковую нагрузку определяли с помощью антител против циклофилина A (Cell signaling, 2175). Антитело против кролика (1:2000 в 5% BSA) использовали в качестве вторичного антитела (Cell signaling, 7074). Рентгеновскую пленку (GeneMate, F-9024-8X10) использовали для проявления мембран. Субстрат Clarity Max Western ECL Substrate (Biorad, 1705062) или хемилюминесцентный субстрат Supersignal West PicoPlus Chemiluminescent Substrate (Thermoscientific, 34577) использовали для хемилюминесцентного вестерн-блоттинга.
Количественный анализ и статистический анализ. Анализ всех данных осуществляли с помощью статистического программного обеспечения GraphPad Prism и представляли в виде среднего значения ± SEM. Значение p определяли посредством двухстороннего ANOVA для роста опухоли, или посредством логарифмического рангового критерия для выживания, или непарного двухстороннего t-критерия Стьюдента для другого анализа. Значение p<0,05 считали статистически значимым.
Пример 2 - Результаты
Терапевтический эффект 6-тио-dG зависит от CD8+ T-клеток. Все предыдущие исследования с использованием моделей ксенотрансплантатов показали, что с помощью ежедневного интенсивного введения 6-тио-dG в течение 10 дней можно частично контролировать рост опухоли во многих моделях опухолей (Mender et al., 2015a; Mender et al., 2018; Zhang et al., 2018). Однако, потенциальная роль этого лекарственного средства во взаимодействии между опухолями и адаптивной иммунной системой неизвестна. Для исследования того, индуцирует ли 6-тио-dG распознавание ДНК на основе теломер в случае T-клеточных ответов, авторы настоящего изобретения сначала определяли ингибирование 6-тио-dG жизнеспособности клеток на теломераза-положительных клетках рака толстого кишечника мыши (MC38) в иммунокомпетентном хозяине. Опухолевые клетки MC38 чувствительны к 6-тио-dG с концентрацией IC50 370 нМ (фиг. 1A). Они также подтверждали чувствительность к 6-тио-dG в клетках MC38 посредством анализа образования отдельных колоний. Клетки MC38 обрабатывали 6-тио-dG каждые три дня в течение 13 дней, что приводило к тому, что менее 50% клеток образовывали колонии при обработке 0,5 мкМ 6-тио-dG (фиг. 1B и 1C). Для оценки того, снижает ли 6-тио-dG опухолевую нагрузку в моделях на сингенных мышах in vivo, авторы настоящего изобретения подкожно инокулировали клетки MC38 иммунокомпетентным мышам C57BL/6 дикого типа (WT). Через семь дней после инокуляции опухоли (когда объем опухоли достигал ~100 мм3) 3 мг/кг 6-тио-dG вводили ежедневно в течение всего лишь трех дней и значимо снижали рост опухоли (фиг. 1D) по сравнению с контрольной опухолью. Это не было уникальным ответом на модель опухоли MC38, т.к. авторы настоящего изобретения также наблюдали ингибирование жизнеспособности клеток in vitro и значительную задержку роста опухоли in vivo в моделях опухолей теломераза-положительной LLC (карциномы легких Льюиса мышей, полученной из мыши C57BL/6) и CT26 (карцинома толстого кишечника мыши, полученная из мыши BALB/C) при лечении всего лишь в течение трех дней (фиг. 9A-D).
Поскольку авторы настоящего изобретения использовали такую небольшую продолжительность лечения 6-тио-dG по сравнению со стратегией интенсивного введения в моделях ксенотрансплантатов (5 мг/кг, ежедневно в течение двух недель) и достигали лучшего противоопухолевого эффекта в моделях на сингенных мышах, они предполагали, что 6-тио-dG может выполнять иммуностимулирующую роль in vivo. Таким образом, они инокулировали опухоли Rag1-нокаутным мышам, которые не могут образовывать зрелые T- и B-клетки. Фактически, терапевтический эффект 6-тио-dG полностью уменьшался (фиг. 1E), что свидетельствует о том, что клетки адаптивного иммунитета, главным образом, необходимы для контроля опухоли in vivo. Для обнаружения того, какая субпопуляция T-клеток вносит вклад в 6-тио-dG-опосредованный противоопухолевый эффект, авторы настоящего изобретения истощали CD4+ или CD8+ T-клетки во время курса введения 6-тио-dG и наблюдали незначительное влияние истощения CD4+ T-клеток (фиг. 1F). Однако истощение CD8+ T-клеток полностью устраняло терапевтический эффект 6-тио-dG (фиг. 1G). В совокупности, данные можно интерпретировать так, чтобы предположить значительную роль CD8+ T-клеток в лечении 6-тио-dG.
Введение 6-тио-dG повышает ответ опухолеспецифических T-клеток. Поскольку терапевтический эффект 6-тио-dG зависит от T-клеток, авторы настоящего изобретения предположили, что введение 6-тио-dG может изменять экспансию иммунных клеток в микроокружении опухоли. Для проверки этого они анализировали количество инфильтрирующих опухоль лимфоцитов (TIL) через 6 дней после последней из трех суточных доз при введении 6-тио-dG. Авторы настоящего изобретения обнаружили повышение доли CD3+ T-клеток и CD8+ T-клеток среди TIL после введения 6-тио-dG (фиг. 2A, 10A и 10B). Они также наблюдали значительную положительную регуляцию пролиферации CD8+ T-клеток, о чем свидетельствует повышенная экспрессия Ki67 (фиг. 2B), но не наблюдали значимых изменений Treg-клеток (фиг. 10C). Хотя инфильтрирующие опухоль NK-клетки также повышались, авторы настоящего изобретения не наблюдали влияния истощения NK-клеток на терапевтический эффект 6-тио-dG (фиг. 10D и 10E). Вместе с экспериментами по истощению CD8, это позволяет предполагать, что NK-клетки не являются необходимыми, но ответы CD8+ T-клеток необходимы для 6-тио-dG-опосредованных противоопухолевых эффектов.
Авторы настоящего изобретения дополнительно тестировали ответ антиген-специфических T-клеток после введения 6-тио-dG с использованием модели опухоли MC38-OVA, позволяющей отслеживать антиген-специфические T-клетки в опухолевой ткани. Фактически, они наблюдали повышение опухолеспецифических CD8+ T-клеток в опухолях через 6 дней после введения 6-тио-dG (фиг. 2C). Они также наблюдали повышение ответов опухолеспецифических цитотоксических T-клеток в модели опухоли MC38 посредством измерения ИФНγ-продуцирующих T-клеток после введения 6-тио-dG (фиг. 2D и 2E). Для прямой оценки способности T-клеток продуцировать ИФНγ in vivo авторы настоящего изобретения использовали ИФНγ-репортерных мышей с YFP, позволяющих отслеживать ИФНγ-продуцирующие T-клетки с экспрессией YFP (Reinhardt et al., 2009). Введение 6-тио-dG значимо повышало YFP+ T-клетки в опухоли, что позволяет предполагать повышенную способность T-клеток продуцировать ИФНγ (фиг. 2F и 10F). Характерным признаком адаптивного иммунного ответа является образование памяти, инициирующей быстрый вторичный иммунный ответ при появлении того же антигена. Для определения того, индуцирует ли введение 6-тио-dG вторичный ответ, мышей с полностью регрессировавшими опухолями после введения 6-тио-dG тестировали в течение 5 недель и повторно стимулировали той же опухолью MC38, но с использованием в 10 раз большего количества опухолевых клеток в противоположный бок (левый бок), и опухолевые клетки LLC инокулировали в качестве контроля в правый бок. Когда наивным мышам (никогда не подвергавшимся воздействию клеток MC38 или 6-тио-dG) инъецировали то же количество клеток MC38, опухоли росли агрессивно. Примечательно, что все излеченные с помощью введения 6-тио-dG мыши спонтанно отторгали повторно подсаженные опухоли MC38.
Введение 6-тио-dG повышает способность дендритных клеток к перекрестному примированию. Перекрестное презентирование антигена антигенпрезентирующими клетками (APC), такими как DC или макрофаги, приводит к активации опухолеспецифических CD8+ T-клеток. Для исследования того, какая субпопуляция APC участвует в 6-тио-dG-индуцируемой активации T-клеток, авторы настоящего изобретения сначала использовали антитело против CSF1R для истощения макрофагов. Они обнаружили, что 6-тио-dG действовал даже лучше в группе с истощением макрофагов (фиг. 3A), что можно объяснить аддитивным эффектом удаления иммуносупрессорных опухолеассоциированных макрофагов. BATF3 (основной ATF-подобный фактор транскрипции с лейциновой молнией 3)-зависимые DC являются критическими для примирования антиген-специфических CD8+ T-клеток (Broz et al., 2014; Edelson et al., 2010). Введение 6-тио-dG мышам с дефицитом Batf3 частично замедляло рост опухоли, но было значимо менее эффективным по сравнению с мышами WT (фиг. 3B). Примечательно, что 60% мышей WT полностью не имели опухолей, но ни одна из Batf3-/- мышей не была свободна от опухолей (фиг. 3C), что позволяет предположить важную роль BATF3-зависимых DC в терапевтическом эффекте 6-тио-dG.
Чтобы прямо продемонстрировать, что введение 6-тио-dG повышает способность DC к перекрестному примированию, авторы настоящего изобретения сокультивировали предварительно обработанные 6-тио-dG опухолевые клетки MC38-OVA с полученными из костного мозга DC (BMDC) в течение ночи. Затем DC очищали и сокультивировали с наивными трансгенными CD8+ T-клетками OT-1, экспрессирующими TCR со специфичностью для распознавания эпитопа OVA257-264. Они наблюдали значимое повышение продукции ИФНγ CD8+ T-клетками в группе, которой вводили 6-тио-dG (фиг. 3D), что свидетельствовало о повышенной способности DC к перекрестному примированию после введения 6-тио-dG. Поскольку передача сигнала ИФН типа I способствует способности DC к перекрестному примированию (Diamond et al., 2011; Le Bon et al., 2003; Sanchez-Paulete et al., 2017), авторы настоящего изобретения анализировали продукцию ИФНβ DC после их сокультивирования с обработанными 6-тио-dG опухолевые клетки. Фактически, продукция ИФНβ значимо повышалась в группе, которой вводили 6-тио-dG, что свидетельствует о повышенном врожденном распознавании DC (фиг. 3E). Они дополнительно исследовали, является ли путь ИФН типа I необходимым для 6-тио-dG-опосредованного противоопухолевого эффекта. Используя Ifnar1-/- мышей, авторы настоящего изобретения показали, что утрата пути ИФН типа I у организма-хозяина устраняет противоопухолевый эффект 6-тио-dG (фиг. 3F), что свидетельствует о неотъемлемой роли передачи сигнала ИФН типа I в лечении 6-тио-dG.
Передача сигнала STING в организме-хозяине необходима для индуцируемого 6-тио-dG врожденного распознавания. Опухолевые клетки в стрессовых условиях могут высвобождать дистресс-ассоциированные молекулярные паттерны (DAMP) для вовлечения путей TLR/Myd88 в APC и инициации передачи сигала ИФН типа I. ДНК опухолевого происхождения также может запускать путь распознавания цитозольной ДНК cGAS/STING и активировать пути ИФН типа I (Deng et al., 2014; Li et al., 2019). Для дальнейшего выявления того, какой восходящий путь является важным в запускаемой 6-тио-dG активации передачи сигнала ИФН типа I в клетках-хозяевах, авторы настоящего изобретения инокулировали опухоли MC38 Myd88-/- и Tmem173-/- (Tmem173 кодирует STING) мышам. С помощью введения 6-тио-dG хорошо контролировали рост опухоли у Myd88-/- мышей, но он полностью терял эффективность у Tmem173-/- мышей (фиг. 4A и 4B), что позволяет предполагать важную роль передачи сигнала STING в организме-хозяине при запускаемом 6-тио-dG врожденном распознавании. Они дополнительно исследовали, активирует ли введение 6-тио-dG путь STING/ИФН типа I в организме-хозяине. Они наблюдали повышение фосфорилирования TBK1 в DC после сокультивирования с предварительно обработанными 6-тио-dG опухолевыми клетками, и фосфорилирование полностью исчезало в Tmem173 DC (фиг. 11A). Введение 6-тио-dG индуцировало продукцию ИФНβ в DC STING-зависимым образом (фиг. 11B). Поскольку в предыдущих исследования показано, что характерная для опухоли передача сигнала STING является критической в индуцирующих врожденное распознавание противоопухолевых способах терапии (Sen et al., 2019; Vanpouille-Box et al., 2017), авторы настоящего изобретения тестировали, вносит ли вклад характерная для опухоли передача сигнала STING в эффективность лечения 6-тио-dG. Они использовали CRISPR/Cas9 для нокаута Tmem173 и Mb21d1 (Mb21d1 кодирует cGAS) в опухолевых клетках MC38. В отличие от других исследований, характерная для опухоли передача сигнала STING играла несущественную роль, т.к. с помощью введения 6-тио-dG все равно контролировали рост опухоли у мышей, несущих опухолевые клетки Tmem173KO и Mb21d1KO (фиг. 4C и 4D).
Затем авторы настоящего изобретения пытались определить, как обработанные 6-тио-dG опухолевые клетки запускают врожденное распознавание в DC. т.к. 6-тио-dG является нацеленным на теломеры лекарственным средством, индуцируемый 6-тио-dG теломерный стресс может вносить вклад во врожденное распознавание DC посредством высвобождения ДНК. Таким образом, авторы настоящего изобретения сначала анализировали теломерный стресс посредством анализа TIF (анализа фокусов, индуцированных дисфункцией теломер) и показали, что 6-тио-dG индуцировал повреждение теломер в клетках MC38 (фиг. 4E и 4F). Поскольку теломеры представляют собой лишь небольшую фракцию геномной ДНК (~1/6000), важна любая колокализация теломер с повреждением ДНК. Они также наблюдали схожие повышения TIF в обработанных 6-тио-dG опухолевых тканях из мышей, несущих опухоль MC38 (фиг. 11C и 11D). 6-тио-dG также индуцировал образование "интерфазных мостиков" между двумя дочерними клетками во время телофазы и, т.к. многие из них содержали последовательности теломер, это может объяснить, почему многие микроядра содержат теломерные сигналы, когда клетки повторно входят в интерфазу после митоза (фиг. 11E). Эти цитозольные фрагменты образовывали микроядра с хрупкими ядерными оболочками (фиг. 11E и 11F), которые в конечном итоге могут распознаваться как дистресс-сигнал. Эти фрагменты ДНК высвобождаются из клеток и могут захватываться DC.
Для подтверждения этой гипотезы, авторы настоящего изобретения обрабатывали HCT116, линию клеток рака толстого кишечника человека, 6-тио-dG и сокультивировали их с BMDC мыши в течение 4 ч, а затем они выделяли DC и выделяли цитозольную ДНК. Кратковременное сокультивирование линии опухолевых клеток человека с BMDC мыши позволяло различать ДНК из разных источников. Они обнаружили повышение ДНК человека (MT-CO1 и 18S человека) в цитозоле DC мыши после обработки 6-тио-dG, что позволяет предполагать, что ДНК из опухолей проникает в DC организма-хозяина (фиг. 4G). Для определения того, повышает ли обработка 6-тио-dG захват DC уникальной теломерной ДНК, авторы настоящего изобретения метили опухолевые клетки EdU, а затем промывали клетки. Затем они обрабатывали 6-тио-dG, затем клетки снова промывали. И наконец, они сокультивировали опухолевые клетки с DC, а затем выделяли DC для анализа. Среди DC, имевших захват опухолевой ДНК (EdU+ DC) в цитозоль, авторы настоящего изобретения наблюдали повышение колокализации теломер с EdU после обработки 6-тио-dG, что позволяет предполагать значительный захват теломерной ДНК опухолевого происхождения (фиг. 11G и 11H). В целом, авторы настоящего изобретения показали, что 6-тио-dG запускает врожденное распознавание посредством активации пути распознавания STING/ИФН типа I цитозольной ДНК организма-хозяина.
6-тио-dG преодолевает резистентность к блокаде PD-L1 в опухолях на поздних стадиях. Хотя обработка 6-тио-dG активировала CD8+ T-клетки, она также приводила к положительной регуляции экспрессии PD-1 в части тотальных CD8+ T-клеток и в расчете на клетку (фиг. 5A). PD-1 является коингибиторной молекулой, ограничивающей активацию T-клеток. Повышенная экспрессия PD-1 в конечном итоге может ингибировать функционирование цитотоксических CD8+ T-клеток после обработки 6-тио-dG. Таким образом, авторы настоящего изобретения предполагали, что комбинация 6-тио-dG с блокадой PD-1/PD-L1 может усилить общий противоопухолевый иммунный ответ, особенно в условиях опухоли на поздней стадии, имеющих более иммуносупрессорное микроокружение, включающее множество механизмов резистентности, ограничивающих эффективность отдельного введения. Поскольку однократное введение 6-тио-dG являлось эффективным только при относительно небольших размерах опухоли ~100 мм3, в случае лечения опухолей на поздних стадиях авторы настоящего изобретения позволяли размерам опухоли достичь 150-200 мм3, а затем вводили 6-тио-dG и/или средство против PD-L1. При таких раковых заболеваниях на поздних стадиях, объем опухоли трудно контролировать с помощью двух ежедневных введений 6-тио-dG или двух введений средства против PD-L1 (фиг. 5B). Однако последовательное введение 6-тио-dG и средство против PD-L1 полностью ингибировало рост опухоли (фиг. 5B). Примечательно, что только мыши в группе комбинированного лечения достигали коэффициента выживания 100% (фиг. 5C), что свидетельствует о синергическом действии введения 6-тио-dG и блокады PD-L1. Кроме того, авторы настоящего изобретения не наблюдали какой-либо потери массы тела у мышей в группе комбинированного лечения (фиг. 12). Они дополнительно анализировали ответ опухолеспецифических T-клеток в дренирующих лимфоузлах (dLN) и обнаружили, что лечение средством против PD-L1 имело небольшой эффект в отношении активации T-клеток в опухолях на поздних стадиях. В отличие от этого, комбинированное лечение значимо повышало продукцию ИФНγ по сравнению с другими группами. Иммунный ответ являлся специфическим в отношении опухоли MC38, т.к. почти не было пятен ИФНγ в контрольной группе стимуляции опухоли LLC (фиг. 5D).
MC38 известны как модель иммуногенной опухоли. Для анализа того, может ли комбинированное лечение также преодолевать резистентность к блокаде PD-L1 в моделях менее иммуногенных опухолей, авторы настоящего изобретения использовали модель опухоли LLC на мышах, которая, как описано, резистентна к блокаде PD-L1 (Bullock et al., 2019; Li et al., 2017). В соответствии с предыдущими сообщениями, отдельное введение средства против PD-L1 не имело терапевтического эффекта (фиг. 5E). Примечательно, что комбинация 6-тио-dG со средством PD-L1 значимо снижала опухолевую нагрузку у мыши, и 40% мышей в конечном итоге полностью отторгали опухоли (фиг. 5E). Авторы настоящего изобретения повторно стимулировали неимеющих опухоли мышей через 6 недель после регрессирования опухоли для проверки вторичного ответа. Все мыши, которых подвергли комбинированному лечению, спонтанно отторгали опухоли LLC, но не опухоли MC38, что позволяет предполагать наличие долговременной опухолеспецифической иммунной памяти (фиг. 5F). Учитывая эти результаты, введение 6-тио-dG позволяет преодолеть резистентность к блокаде PD-L1 в опухолях на поздней стадии. Это потенциально принесет пользу резистентным к блокаде PD-1/PD-L1 пациентам в клинических условиях.
6-тио-dG снижает нагрузку рака толстого кишечника человека в модели на гуманизированных мышах. В предыдущих исследованиях показано, что пациенты с высокой экспрессией TERT (каталитической субъединицы теломераза) имеют плохие клинические исходы при различных раковых заболеваниях, таких как немелкоклеточный рак легких и B-клеточный хронический лимфоцитарный лейкоз (Terrin et al., 2007; Wang et al., 2002). Таким образом, авторы настоящего изобретения анализировали пациентов с колоректальной аденокарциномой из базы данных TCGA и обнаруживали, что пациенты с аномальной высокой экспрессией TERT, имели значимо более плохую общую выживаемость по сравнению с пациентами с раком толстого кишечника с низкой экспрессией TERT (фиг. 6A). Чтобы напрямую продемонстрировать, могут ли пациенты с раком получить пользу от индуцируемого 6-тио-dG теломерного стресса в более клинически значимой модели, авторы настоящего изобретения разрабатывали модель на гуманизированным мышах с использованием мыши NSG-SGM3, имеющей трансгенную экспрессию SCF-1, ГМ-КСФ и ИЛ-3 человека, поддерживающей лучшее развитие миелоидных клеток человека. Они воспроизводили иммунную систему человека в мышах NSG-SGM3 с помощью CD34+ гемапоэтических стволовых клеток (HSC) человека. Через 12 недель после переноса HSC гуманизированные мыши имели в среднем более 60% CD45+ клеток человека и более 20% T-клеток человека среди CD45+ клеток человека в кровотоке (фиг. 13A-13C). Затем авторы настоящего изобретения инокулировали HCT116, линию клеток рака толстого кишечника человека, чувствительную к обработке 6-тио-dG с IC50 0,73 мкМ (фиг. 6B), контрольным мышам NSG-SGM3 и гуманизированным мышам NSG-SGM3. Контрольная группа гуманизированных мышей имела состав иммунных клеток человека, схожий с группой, которой вводили 6-тио-dG перед началом лечения (фиг. 13B и 13C). После трех доз 6-тио-dG иммунокомпрометированные мыши не имели значимых различий по сравнению с контрольной группой (фиг. 6D). Примечательно, что у гуманизированных мышей значимо замедлялся рост опухоли при введении 6-тио-dG (фиг. 6C и 6E). Затем авторы настоящего изобретения тестировали линию клеток меланомы человека A375, чувствительную к 6-тио-dG in vitro (фиг. 13D). Они не наблюдали какого-либо эффекта у иммунокомпрометированных мышей NSG-SGM3, т.к. они осуществляли относительно кратковременное лечение 6-тио-dG (фиг. 13E). Примечательно, что они обнаружили, что лечение с помощью двух доз 6-тио-dG частично замедляло рост опухоли у гуманизированных мышей. Кроме того, комбинация с блокадой иммунных контрольных точек дополнительно снижала опухолевую нагрузку, что позволяет предполагать, что предварительное введение 6-тио-dG сенсибилизирует опухоли человека к блокаде иммунных контрольных точек (фиг. 13F). Учитывая, что гуманизированные мыши имеют лишь частично восстановленный иммунитет человека из-за отсутствия некоторых иммунных клеток и ограниченного количества T-клеток человека, неудивительно, что авторы настоящего изобретения не наблюдали полного регрессирования опухоли.
Авторы настоящего изобретения показали (фиг. 8A-B), что трехдневное лечение 6-тио-dG с последующими двумя днями введения средства против PD-L1 приводит к полной ремиссии опухолей при карциноме легких Льюиса. Это очень агрессивный тип опухоли, как показано на фиг. 8A. В пределах 20 дне подкожно инъецированная опухоль достигала более 1000 мм3. Они наблюдали такой же быстрый рост опухоли при лечении только средством против PD-L1. Однако, всего три введения 6-тио-dG ("тио") приводят к значительному контролю опухоли в течение 20 дней. Неожиданно, лечение с помощью "тио" с последующим средством против PDL-L1 (атезолизумабом) приводило к полному регрессированию опухоли. Авторы настоящего изобретения держали вылеченных мышей еще в течение 5 недель, повторно стимулировали тех же мышей в пять раз большим количество опухолевого материала LLC и не наблюдали роста опухоли. Однако если они инъецировали вылеченным от LLC мышам MC38 опухоли росли и средство против PD-L1 не имело эффекта (фиг. 8B). У контрольных мышей, которым никогда не вводили "тио", контрольные опухоли LLC росли аналогично MC38. Это позволяет предполагать наличие опухолеспецифической иммунной памяти.
В целом, эти данные можно интерпретировать как подтверждение того, что 6-тио-dG индуцирует теломераза-зависимое повреждение ДНК и повышает захват DC опухолевой ДНК. Повышение цитозольной ДНК запускает характерный для DC путь STING/ИФН типа I, что приводит к повышенной способности DC к перекрестному примированию и последующей активации опухолеспецифических T-клеток. Кроме того, 6-тио-dG позволяет преодолеть резистентность к блокаде PD-L1 в опухолях на поздней стадии. В этом исследовании 6-тио-dG определили как новое иммуностимулирующее лекарственное средство, которое потенциально принесет пользу широкой популяции пациентов с раком в клинических условиях.
Пример 3 - Обсуждение
Высокую экспрессию теломеразы в опухолевых клетках считают плохим прогностическим фактором при развитии рака (Zhang et al., 2018). Авторы настоящего изобретения описывают ранее не определенную роль теломераза-зависимой, нацеленной на теломеры терапии (6-тио-dG) в индуцировании противоопухолевых иммунных ответов в моделях рака толстого кишечника и рака легких на сингенных мышах и моделях рака на гуманизированных мышах. Этот эффект опосредован запуском пути STING/ИФН типа I распознавания цитозольной ДНК в DC, что в конечном итоге повышает способность DC к перекрестному примированию и последующую активацию опухолеспецифических T-клеток. Это является замечательным результатом, т.к. теломераза является универсальным опухолевым маркером, и ее потенциально можно использовать в случае многих других теломераза-положительных рака. Кроме того, последовательное введение 6-тио-dG и средства против PD-L1 позволяет преодолеть резистентность PD-L1 в резистентных к блокаде PD-L1 опухолях, что позволяет предполагать, что комбинированное лечение может принести пользу PD-L1-резистентным пациентам в клинических условиях.
Существующая догма заключается в том, что лечение 6-тио-dG приводит к уничтожению опухолевых клеток, в основном, посредством повреждения теломер и индуцирования повреждения ДНК. Это исследование свидетельствует о том, что это лекарственное средство также позволяет контролировать опухоли, главным образом, в зависимости от распознавания ДНК и T-клеточных ответов. В большинстве предыдущих исследований использовали модели ксенотрансплантатов без интактной иммунной системы. В этих моделях они могут исследовать только характерные для опухоли эффекты или часть врожденных иммунных ответов. Хотя они могут являться важными факторами, T-клетки необходимы для длительного контроля опухоли. Кроме того, в большинстве предыдущих исследований стремились использовать высокую дозу или стратегии интенсивного введения, напрямую приводящие к более эффективному уничтожению опухолевых клеток, но на самом деле угнетающих иммунные ответы из-за токсичности для иммунных клеток или неиммуногенной гибели опухолевых клеток (Galluzzi et al., 2017; Kroemer et al., 2013). Кроме того, эти стратегии интенсивного введения зачастую приводят к возникновению механизмов опухолевой резистентности. В настоящем исследовании авторы настоящего изобретения воспользовались преимуществом моделей на сингенных мышах с интактными иммунными системами и модели на гуманизированных мышах с большей клинической значимостью, чтобы полностью оценить влияние более низких доз и более коротких схем лечения с использованием 6-тио-dG на иммунные ответы организма-хозяина у несущих опухоли мышей. Это открытие, заключающееся в том, что 6-тио-dG является иммуностимулирующим лекарственным средством, может сделать возможным дизайн лучшего комбинированного лечения, включая иммунотерапию, для усиления начального иммунитета.
Накапливающиеся исследования показывают, что опосредованное опухолевой ДНК врожденное распознавание является критическим для индуцирования противоопухолевых иммунных ответов, и путь STING/ИФН типа I, главным образом, участвует в инициировании противоопухолевого иммунного ответа, но что является более необходимым - STING организма-хозяина или опухолевый автономный STING - зависит от разных схем лечения (Deng et al., 2014; Li et al., 2019; Qiao et al., 2017; Sen et al., 2019; Vanpouille-Box et al., 2017; Woo et al., 2014). Это расхождение, вероятно, можно объяснить относительной силой активации STING организма-хозяина по сравнению с опухолевыми клетками, например, опухолевые клетки могут иметь супрессию или низкую активность пути STING (Xia et al., 2016). Авторы настоящего изобретения показали, что запускаемое лечением 6-тио-dG врожденное распознавание зависит от передачи сигнала STING организма-хозяина, т.к. 6-тио-dG полностью теряет свою эффективность у мышей с дефицитом Tmem173, но не в опухолях с дефицитом Tmem173. Поскольку передача сигнала STING в опухолях MC38 является активной, одним из объяснений является то, что после введения 6-тио-dG собственный STING опухоли активируется, но большинство опухолевых клеток погибло, поэтому может продуцироваться мало ИФН типа I. Другой возможностью является то, что может существовать внутренний механизм, ограничивающий активацию STING в опухолевых клетках, все еще плохо определенный. Недавно показало, что передача сигнала STING также может вносить вклад в активацию аутофагии, что с меньшей вероятностью будет способствовать терапевтическому эффекту 6-тио-dG (Gui et al., 2019; Nassour et al., 2019), т.к. авторы настоящего изобретения не наблюдали активацию аутофагии в опухолевых клетках после введения 6-тио-dG (данные не представлены). Кроме того, 6-тио-dG теряет эффективность у мышей с дефицитом Ifnar1, что позволяет предполагать вовлечение передачи сигнала ИФН типа I. Однако активация аутофагии под действием STING не зависит от передачи сигнала ИФН типа I.
По сравнению с общими подходами индуцирования повреждения ДНК, например, лучевой терапией или химиотерапией, неселективно индуцирующих повреждение ДНК во всех пролиферирующих клетках, одним из уникальных признаков 6-тио-dG является специфическое индуцирование теломера-ассоциированного повреждения ДНК в теломераза-экспрессирующих клетках, главным образом, опухолевых клетках, незатрагивающие иммунные клетки и другие соматические клетки с молчащей теломеразой. Важно, что 6-тио-dG можно преимущественно встраивать в синтезированные de novo теломеры и вызывать уменьшение размеров опухоли. Однако прямые ингибиторы теломеразы действуют через ингибирование теломеразной активности и основаны на прогрессирующем укорочении теломер. В отличие от этого, 6-тио-dG быстро действует независимо от начальной длины теломер. Это критично для снижения токсичности по сравнению с прямым ингибитором теломеразы (Gryaznov et al., 2007; Mender et al., 2015b). Авторы настоящего изобретения показали, что индуцируемое 6-тио-dG повреждение ДНК в значительной степени колокализовано с теломерами, что свидетельствует об образовании фокусов, индуцированных дисфункцией теломер (TIF). Теломеры составляют только ~1/6000 геномной ДНК, таким образом, любой TIF очень значим. Кроме того, некоторые TIF захватываются DC и дополнительно запускают STING-зависимую передачу сигнала ИФН типа I.
Несмотря на впечатляющий успех блокады иммунных контрольных точек, особенно блокады PD-1/PD-L1, в клинических условиях, лишь небольшая часть пациентов хорошо отвечала. И первичная, и адаптивная резистентность ограничивает клиническую пользу терапии PD-1/PD-L1 (Chen and Han, 2015; Gide et al., 2018; Zaretsky et al., 2016; Zou et al., 2016). Авторы настоящего изобретения считают, что отсутствие правильного врожденного распознавания может ограничивать активацию T-клеток в микроокружении опухоли, таким образом, крайне необходимо комбинированное лечение, нацеленное на клетки врожденного и адаптивного иммунитета. Блокада PD-L1 приводит к возобновлению адаптивных иммунных ответов посредством "отпускания тормоза", в то время как индуцируемое 6-тио-dG врожденное распознавание - посредством "добавления топлива". Авторы настоящего изобретения предполагали, что комбинация 6-тио-dG с блокадой PD-L1, должна усиливать общие противоопухолевые иммунные ответы. Фактически, это исследование показало, что последовательное введение 6-тио-dG и средства против PD-L1 имеет синергическое действие в опухолях на поздней стадии и резистентных к блокаде PD-L1 опухолях. Необходимо провести дополнительные исследования, касающиеся оптимальных схем комбинированного лечения.
В целом, эти результаты показывают ранее неопределенную роль 6-тио-dG, теломераза-зависимого, нацеленного на теломеры низкомолекулярного лекарственного средства, в потенцировании противоопухолевых иммунных ответов. Механистически, 6-тио-dG индуцирует дисфункцию теломер и повышает высвобождение цитозольной ДНК. Важно, что эти фрагменты теломерной ДНК захватываются DC и активируют внутренний путь DC STING/ИФН, что приводит к повышенной способности DC к перекрестному примированию и последующей активации опухолеспецифических T-клеток. Кроме того, это исследование, показавшее значительную эффективность последовательного введения 6-тио-dG и средства против PD-L1 в опухолях на поздней стадии и резистентных к блокаде PD-L1 опухолей, обеспечивает сильное научное обоснование продвижения комбинированного лечения до клинических испытаний. Авторы настоящего изобретения ожидают, что эти результаты будут претворены в жизнь в ближайшем будущем и принесут пользу большему количеству пациентов в клинических условиях.
Все из способов, описанных и заявленных в настоящем описании, можно осуществлять без излишнего экспериментирования в свете настоящего описания. Хотя композиции и способы по настоящему изобретению описаны в терминах конкретных вариантов осуществления, специалистам в этой области очевидно, что можно осуществлять изменения в способах и стадиях или последовательности стадий способа, представленного в настоящем описании, без отклонения от концепции, сущности и объема изобретения. Более конкретно, очевидно, что некоторые средства, являющиеся химически и физиологически родственными, можно заменять средствами, представленными в настоящем описании, при этом будут достигать тех же или схожих результатов. Все такие схожие замены и модификации, очевидные специалистам в этой области, считают находящимся в пределах сущности, объема и концепции изобретения, определяемого формулой изобретения.
VI. Ссылки
Следующие ссылки, в той степени, в которой они представляют примерные процедурные или другие подробности, дополняющие изложенное в настоящем описании, конкретно включены в настоящее описание в качестве ссылки.
Ablasser, A., Goldeck, M., Cavlar, T., Deimling, T., Witte, G., Rohl, I., Hopfner, K. P., Ludwig, J., and Hornung, V. (2013). cGAS produces a 2'-5'-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380-384.
Blackburn, E. H. (1991). Structure and function of telomeres. Nature 350, 569-573.
Brahmer, J. R., Tykodi, S. S., Chow, L. Q., Hwu, W. J., Topalian, S. L., Hwu, P., Drake, C. G., Camacho, L. H., Kauh, J., Odunsi, K., et al. (2012). Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366, 2455-2465.
Broz, M. L., Binnewies, M., Boldajipour, B., Nelson, A. E., Pollack, J. L., Erle, D. J., Barczak, A., Rosenblum, M. D., Daud, A., Barber, D. L., et al. (2014). Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 26, 938.
Bullock, B. L., Kimball, A. K., Poczobutt, J. M., Neuwelt, A. J., Li, H. Y., Johnson, A. M., Kwak, J. W., Kleczko, E. K., Kaspar, R. E., Wagner, E. K., et al. (2019). Tumor-intrinsic response to IFNgamma shapes the tumor microenvironment and anti-PD-1 response in NSCLC. Life Sci Alliance 2.
Chen, L., and Han, X. (2015). Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 125, 3384-3391.
Deng, L., Liang, H., Xu, M., Yang, X., Burnette, B., Arina, A., Li, X. D., Mauceri, H., Beckett, M., Darga, T., et al.(2014). STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843-852.
Diamond, M. S., Kinder, M., Matsushita, H., Mashayekhi, M., Dunn, G. P., Archambault, J. M., Lee, H., Arthur, C. D., White, J. M., Kalinke, U., et al. (2011). Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med 208, 1989-2003.
Diner, E. J., Burdette, D. L., Wilson, S. C., Monroe, K. M., Kellenberger, C. A., Hyodo, M., Hayakawa, Y., Hammond, M. C., and Vance, R. E. (2013). The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell reports 3, 1355-1361.
Edelson, B. T., Kc, W., Juang, R., Kohyama, M., Benoit, L. A., Klekotka, P. A., Moon, C., Albring, J. C., Ise, W., Michael, D. G., et al. (2010). Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med 207, 823-836.
Fenech, M., Kirsch-Volders, M., Natarajan, A. T., Surralles, J., Crott, J. W., Parry, J., Norppa, H., Eastmond, D. A., Tucker, J. D., and Thomas, P. (2011). Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis 26, 125-132.
Galluzzi, L., Buque, A., Kepp, O., Zitvogel, L., and Kroemer, G. (2017). Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17, 97-111.
Gao, P., Ascano, M., Wu, Y., Barchet, W., Gaffney, B. L., Zillinger, T., Serganov, A. A., Liu, Y., Jones, R. A., Hartmann, G., et al. (2013). Cyclic [G(2',5')pA(3',5')p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094-1107.
Garon, E. B., Rizvi, N. A., Hui, R., Leighl, N., Balmanoukian, A. S., Eder, J. P., Patnaik, A., Aggarwal, C., Gubens, M., Horn, L., et al. (2015). Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372, 2018-2028.
Gide, T. N., Wilmott, J. S., Scolyer, R. A., and Long, G. V. (2018). Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin Cancer Res 24, 1260-1270.
Greider, C. W. (1996). Telomere length regulation. Annual review of biochemistry 65, 337-365.
Greider, C. W., and Blackburn, E. H. (1985). Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43, 405-413.
Gryaznov, S. M., Jackson, S., Dikmen, G., Harley, C., Herbert, B. S., Wright, W. E., and Shay, J. W. (2007). Oligonucleotide conjugate GRN163L targeting human telomerase as potential anticancer and antimetastatic agent. Nucleosides Nucleotides Nucleic Acids 26, 1577-1579.
Gui, X., Yang, H., Li, T., Tan, X., Shi, P., Li, M., Du, F., and Chen, Z. J. (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262-266.
Hodi, F. S., O'Day, S. J., McDermott, D. F., Weber, R. W., Sosman, J. A., Haanen, J. B., Gonzalez, R., Robert, C., Schadendorf, D., Hassel, J. C., et al. (2010). Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363, 711-723.
Kroemer, G., Galluzzi, L., Kepp, O., and Zitvogel, L. (2013). Immunogenic Cell Death in Cancer Therapy. Annual Review of Immunology 31, 51-72.
Lansdorp, P. M., Verwoerd, N. P., van de Rijke, F. M., Dragowska, V., Little, M. T., Dirks, R. W., Raap, A. K., and Tanke, H. J. (1996). Heterogeneity in telomere length of human chromosomes. Human molecular genetics 5, 685-691.
Le Bon, A., Etchart, N., Rossmann, C., Ashton, M., Hou, S., Gewert, D., Borrow, P., and Tough, D. F. (2003). Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol 4, 1009-1015.
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K., Lu, S., Kemberling, H., Wilt, C., Luber, B. S., et al. (2017). Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409-413.
Li, H. Y., McSharry, M., Bullock, B., Nguyen, T. T., Kwak, J., Poczobutt, J. M., Sippel, T. R., Heasley, L. E., Weiser-Evans, M. C., Clambey, E. T., and Nemenoff, R. A. (2017). The Tumor Microenvironment Regulates Sensitivity of Murine Lung Tumors to PD-1/PD-L1 Antibody Blockade. Cancer Immunol Res 5, 767-777.
Li, T., and Chen, Z. J. (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. The Journal of experimental medicine 215, 1287-1299.
Li, X., Liu, Z., Zhang, A., Han, C., Shen, A., Jiang, L., Boothman, D. A., Qiao, J., Wang, Y., Huang, X., and Fu, Y. X. (2019). NQO1 targeting prodrug triggers innate sensing to overcome checkpoint blockade resistance. Nat Commun 10, 3251.
Liu, S., Cai, X., Wu, J., Cong, Q., Chen, X., Li, T., Du, F., Ren, J., Wu, Y. T., Grishin, N. V., and Chen, Z. J. (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630.
Liu, Z., Han, C., Dong, C., Shen, A., Hsu, E., Ren, Z., Lu, C., Liu, L., Zhang, A., Timmerman, C., et al.(2019). Hypofractionated EGFR tyrosine kinase inhibitor limits tumor relapse through triggering innate and adaptive immunity. Sci Immunol 4.
Mandal, R., Samstein, R. M., Lee, K. W., Havel, J. J., Wang, H., Krishna, C., Sabio, E. Y., Makarov, V., Kuo, F., Blecua, P., et al. (2019). Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485-491.
McEachern, M. J., and Blackburn, E. H. (1996). Cap-prevented recombination between terminal telomeric repeat arrays (telomere CPR) maintains telomeres in Kluyveromyces lactis lacking telomerase. Genes & development 10, 1822-1834.
Mender, I., Gryaznov, S., Dikmen, Z. G., Wright, W. E., and Shay, J. W. (2015a). Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2'-deoxyguanosine. Cancer Discov 5, 82-95.
Mender, I., Gryaznov, S., and Shay, J. W. (2015b). A novel telomerase substrate precursor rapidly induces telomere dysfunction in telomerase positive cancer cells but not telomerase silent normal cells. Oncoscience 2, 693-695.
Mender, I., LaRanger, R., Luitel, K., Peyton, M., Girard, L., Lai, T. P., Batten, K., Cornelius, C., Dalvi, M. P., Ramirez, M., et al.(2018). Telomerase-Mediated Strategy for Overcoming Non-Small Cell Lung Cancer Targeted Therapy and Chemotherapy Resistance. Neoplasia 20, 826-837.
Mender, I., and Shay, J. W. (2015). Telomere Dysfunction Induced Foci (TIF) Analysis. Bio-protocol 5.
Min, J., Wright, W. E., and Shay, J. W. (2019). Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev 33, 814-827.
Morin, G. B. (1989). The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59, 521-529.
Nakamura, T. M., Morin, G. B., Chapman, K. B., Weinrich, S. L., Andrews, W. H., Lingner, J., Harley, C. B., and Cech, T. R. (1997). Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955-959.
Nassour, J., Radford, R., Correia, A., Fuste, J. M., Schoell, B., Jauch, A., Shaw, R. J., and Karlseder, J. (2019). Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565, 659-663.
Pitt, J. M., Kroemer, G., and Zitvogel, L. (2017). Immunogenic and Non-immunogenic Cell Death in the Tumor Microenvironment. Adv Exp Med Biol 1036, 65-79.
Qiao, J., Liu, Z., Dong, C., Luan, Y., Zhang, A., Moore, C., Fu, K., Peng, J., Wang, Y., Ren, Z., et al. (2019). Targeting Tumors with IL-10 Prevents Dendritic Cell-Mediated CD8(+) T Cell Apoptosis. Cancer Cell 35, 901-915 e904.
Qiao, J., Tang, H., and Fu, Y. X. (2017). DNA sensing and immune responses in cancer therapy. Curr Opin Immunol 45, 16-20.
Reinhardt, R. L., Liang, H. E., and Locksley, R. M. (2009). Cytokine-secreting follicular T cells shape the antibody repertoire. Nat Immunol 10, 385-393.
Ribas, A., Hamid, O., Daud, A., Hodi, F. S., Wolchok, J. D., Kefford, R., Joshua, A. M., Patnaik, A., Hwu, W. J., Weber, J. S., et al. (2016). Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 315, 1600-1609.
Ribas, A., and Wolchok, J. D. (2018). Cancer immunotherapy using checkpoint blockade. Science 359, 1350-1355.
Rizvi, N. A., Hellmann, M. D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J. J., Lee, W., Yuan, J., Wong, P., Ho, T. S., et al. (2015a). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124-128.
Rizvi, N. A., Mazieres, J., Planchard, D., Stinchcombe, T. E., Dy, G. K., Antonia, S. J., Horn, L., Lena, H., Minenza, E., Mennecier, B., et al. (2015b). Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol 16, 257-265.
Sanchez-Paulete, A. R., Teijeira, A., Cueto, F. J., Garasa, S., Perez-Gracia, J. L., Sanchez-Arraez, A., Sancho, D., and Melero, I. (2017). Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Ann Oncol 28, xii44-xii55.
Sen, T., Rodriguez, B. L., Chen, L., Corte, C. M. D., Morikawa, N., Fujimoto, J., Cristea, S., Nguyen, T., Diao, L., Li, L., et al.(2019). Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov 9, 646-661.
Sengupta, S., Sobo, M., Lee, K., Senthil Kumar, S., White, A. R., Mender, I., Fuller, C., Chow, L. M. L., Fouladi, M., Shay, J. W., and Drissi, R. (2018). Induced Telomere Damage to Treat Telomerase Expressing Therapy-Resistant Pediatric Brain Tumors. Molecular cancer therapeutics 17, 1504-1514.
Shay, J. W., and Bacchetti, S. (1997). A survey of telomerase activity in human cancer. European journal of cancer 33, 787-791.
Singer, M. S., and Gottschling, D. E. (1994). TLC1: template RNA component of Saccharomyces cerevisiae telomerase. Science 266, 404-409.
Socinski, M. A., Jotte, R. M., Cappuzzo, F., Orlandi, F., Stroyakovskiy, D., Nogami, N., Rodriguez-Abreu, D., Moro-Sibilot, D., Thomas, C. A., Barlesi, F., et al. (2018). Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N Engl J Med 378, 2288-2301.
Tanaka, Y., and Chen, Z. J. (2012). STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Science signaling 5, ra20.
Terrin, L., Trentin, L., Degan, M., Corradini, I., Bertorelle, R., Carli, P., Maschio, N., Bo, M. D., Noventa, F., Gattei, V., et al.(2007). Telomerase expression in B-cell chronic lymphocytic leukemia predicts survival and delineates subgroups of patients with the same igVH mutation status and different outcome. Leukemia 21, 965-972.
Topalian, S. L., Hodi, F. S., Brahmer, J. R., Gettinger, S. N., Smith, D. C., McDermott, D. F., Powderly, J. D., Carvajal, R. D., Sosman, J. A., Atkins, M. B., et al. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443-2454.
Vanpouille-Box, C., Alard, A., Aryankalayil, M. J., Sarfraz, Y., Diamond, J. M., Schneider, R. J., Inghirami, G., Coleman, C. N., Formenti, S. C., and Demaria, S. (2017). DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun 8, 15618.
Wang, L., Soria, J. C., Kemp, B. L., Liu, D. D., Mao, L., and Khuri, F. R. (2002). hTERT expression is a prognostic factor of survival in patients with stage I non-small cell lung cancer. Clin Cancer Res 8, 2883-2889.
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., Bestwick, M., Duguay, B. A., Raimundo, N., MacDuff, D. A., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553-557.
Woo, S. R., Fuertes, M. B., Corrales, L., Spranger, S., Furdyna, M. J., Leung, M. Y., Duggan, R., Wang, Y., Barber, G. N., Fitzgerald, K. A., et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830-842.
Wu, J., Sun, L., Chen, X., Du, F., Shi, H., Chen, C., and Chen, Z. J. (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826-830.
Xia, T., Konno, H., Ahn, J., and Barber, G. N. (2016). Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 14, 282-297.
Xu, M. M., Pu, Y., Han, D., Shi, Y., Cao, X., Liang, H., Chen, X., Li, X. D., Deng, L., Chen, Z. J., et al. (2017). Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein alpha Signaling. Immunity 47, 363-373 e365.
Yu, G. L., Bradley, J. D., Attardi, L. D., and Blackburn, E. H. (1990). In vivo alteration of telomere sequences and senescence caused by mutated Tetrahymena telomerase RNAs. Nature 344, 126-132.
Zaretsky, J. M., Garcia-Diaz, A., Shin, D. S., Escuin-Ordinas, H., Hugo, W., Hu-Lieskovan, S., Torrejon, D. Y., Abril-Rodriguez, G., Sandoval, S., Barthly, L., et al.(2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819-829.
Zhang, G., Wu, L. W., Mender, I., Barzily-Rokni, M., Hammond, M. R., Ope, O., Cheng, C., Vasilopoulos, T., Randell, S., Sadek, N., et al. (2018). Induction of Telomere Dysfunction Prolongs Disease Control of Therapy-Resistant Melanoma. Clin Cancer Res 24, 4771-4784.
Zhang, X., Shi, H., Wu, J., Zhang, X., Sun, L., Chen, C., and Chen, Z. J. (2013). Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Molecular cell 51, 226-235.
Zijlmans, J. M., Martens, U. M., Poon, S. S., Raap, A. K., Tanke, H. J., Ward, R. K., and Lansdorp, P. M. (1997). Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proceedings of the National Academy of Sciences of the United States of America 94, 7423-7428.
Zou, W., Wolchok, J. D., and Chen, L. (2016). PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8, 328rv324.
--->
СПИСОК ПОСЛЕДОВАТЕЛЬНОСТЕЙ
<110> The Board of Regents of the University of Texas System
<120> ПОСЛЕДОВАТЕЛЬНОЕ ЛЕЧЕНИЕ РАКА С ИСПОЛЬЗОВАНИЕМ 6-тио-dG, ИНГИБИТОРОВ
КОНТРОЛЬНЫХ ТОЧЕК И ЛУЧЕВОЙ ТЕРАПИИ
<130> UTFD.P3648WO
<140> Неизвестно
<141> 2021-03-12
<150> 62/989,041
<151> 2020-03-13
<160> 10
<170> PatentIn version 3.5
<210> 1
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 1
cacctagcct cgcacgaact 20
<210> 2
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 2
cgcaaagggg ggctcgatcg 20
<210> 3
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 3
cgccacactc cacggaagca 20
<210> 4
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 4
cggggcattc cggataggcc 20
<210> 5
<211> 22
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 5
accgattgga tggtttagtg ag 22
<210> 6
<211> 22
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 6
cctacggaaa ccttgttacg ac 22
<210> 7
<211> 19
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 7
atgagtggtg gttgcaggc 19
<210> 8
<211> 25
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 8
tgacctttca aatgcagtag attca 25
<210> 9
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 9
catcaagaag gtggtgaagc 20
<210> 10
<211> 20
<212> ДНК
<213> Искусственная последовательность
<220>
<223> Синтетический олигонуклеотид
<400> 10
cctgttgctg tagccgtatt 20
<---
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ЛЕЧЕНИЯ СУБЪЕКТА С РАКОМ ТОЛСТОГО КИШЕЧНИКА ИЛИ РАКОМ ЛЕГКИХ | 2014 |
|
RU2713555C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ TUSC2-ИММУНОТЕРАПИИ | 2017 |
|
RU2755903C2 |
| МОДИФИЦИРОВАННЫЕ МОНОЦИТЫ/МАКРОФАГИ, ЭКСПРЕССИРУЮЩИЕ ХИМЕРНЫЕ АНТИГЕННЫЕ РЕЦЕПТОРЫ, И ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2766690C2 |
| ЛЕЧЕНИЕ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛИ С ИСПОЛЬЗОВАНИЕМ ХИМЕРНОГО РЕЦЕПТОРА АНТИГЕНА ПРОТИВ CD19 | 2015 |
|
RU2718542C2 |
| СПОСОБЫ EX VIVO ЭКСПАНСИИ ЕСТЕСТВЕННЫХ КЛЕТОК-КИЛЛЕРОВ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2814083C2 |
| РЕКОМБИНАНТНЫЙ АДЕНОВИРУС, КОТОРЫЙ СОДЕРЖИТ РИБОЗИМ, ОПОСРЕДУЮЩИЙ ТРАНС-СПЛАЙСИНГ, И ПРОТИВОРАКОВЫЙ ТЕРАПЕВТИЧЕСКИЙ ГЕН, И ЕГО ПРИМЕНЕНИЕ | 2012 |
|
RU2575620C2 |
| СРЕДСТВО, ПОВЫШАЮЩЕЕ ИММУНИТЕТ, ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОГО НОВООБРАЗОВАНИЯ С ПРИМЕНЕНИЕМ АНТАГОНИСТА АЛЛЕРГИНА-1 | 2016 |
|
RU2734777C2 |
| ЛЕЧЕНИЕ РАКА С ПОМОЩЬЮ ХИМЕРНОГО АНТИГЕННОГО РЕЦЕПТОРА К CD33 | 2015 |
|
RU2747384C2 |
| СПОСОБЫ ЛЕЧЕНИЯ РАКА, ИМЕЮЩЕГО ГЕМИЗИГОТНУЮ ПОТЕРЮ ТР53 | 2016 |
|
RU2721953C2 |
| ПРОЛЕКАРСТВО ИНТЕРФЕРОНА ДЛЯ ЛЕЧЕНИЯ РАКА | 2018 |
|
RU2788736C2 |


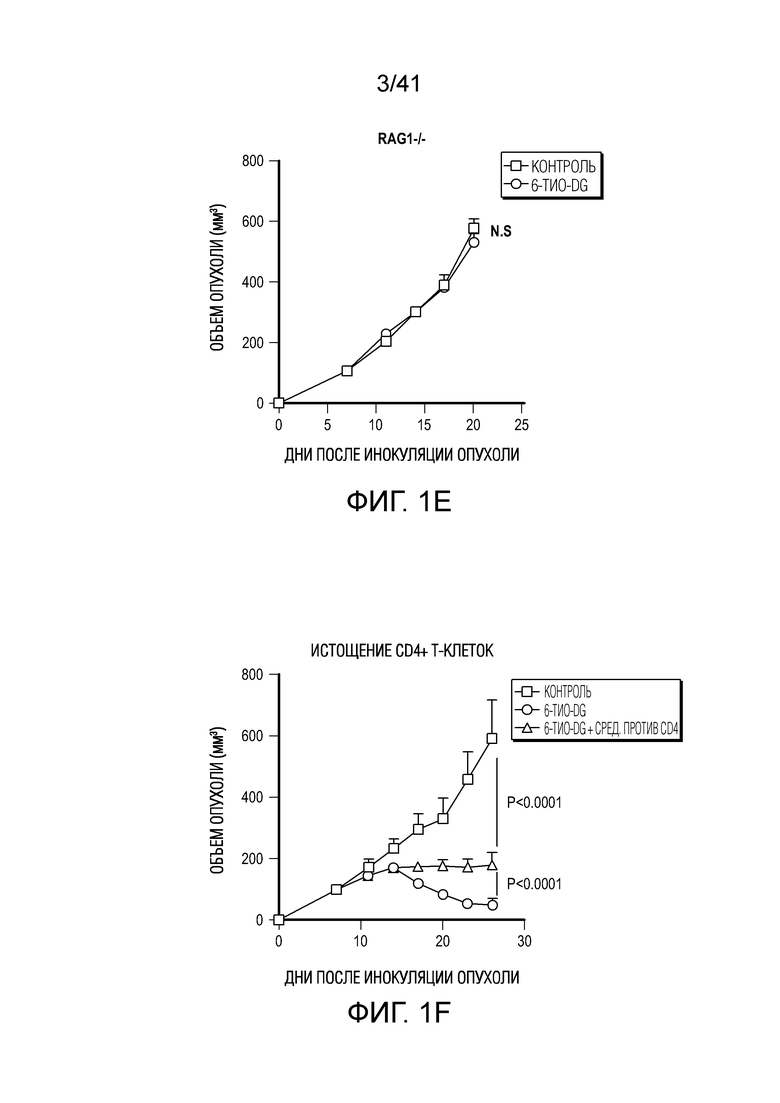





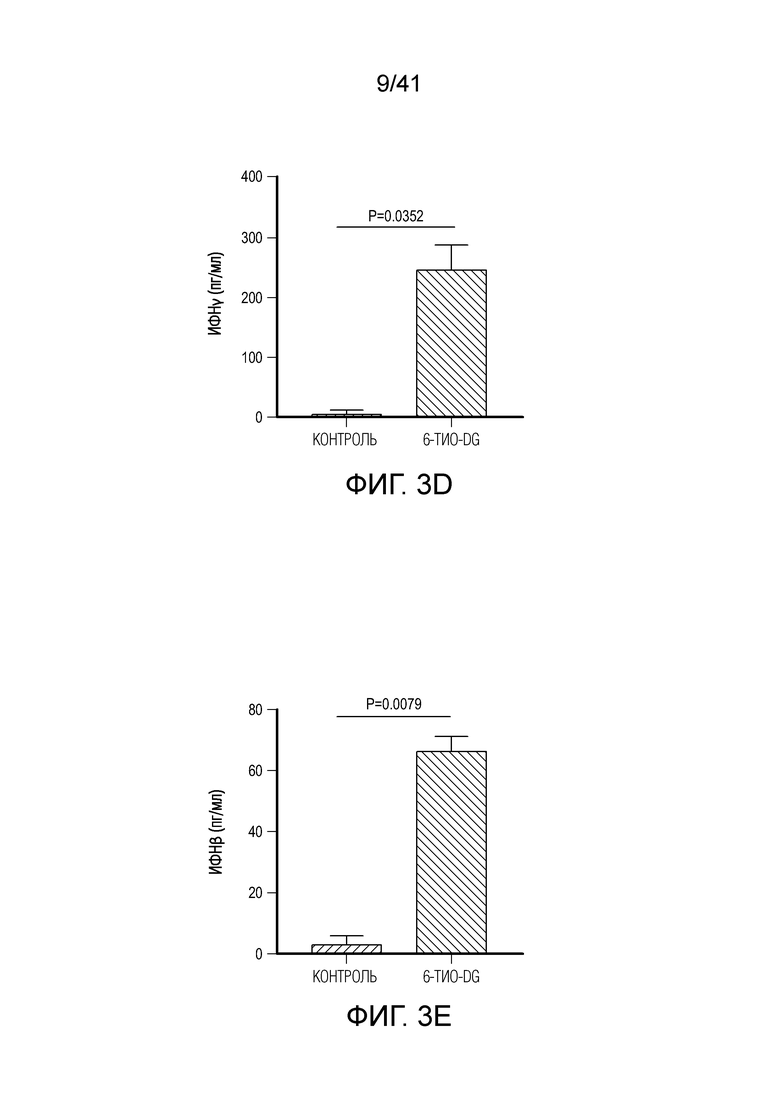
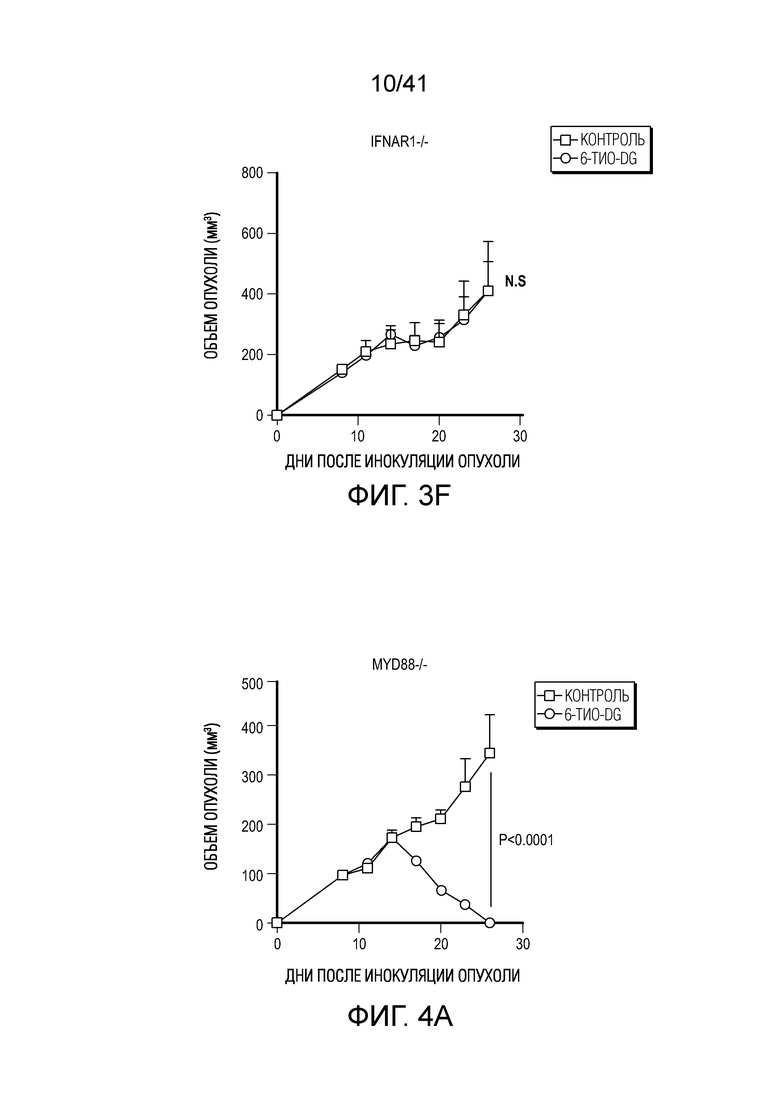
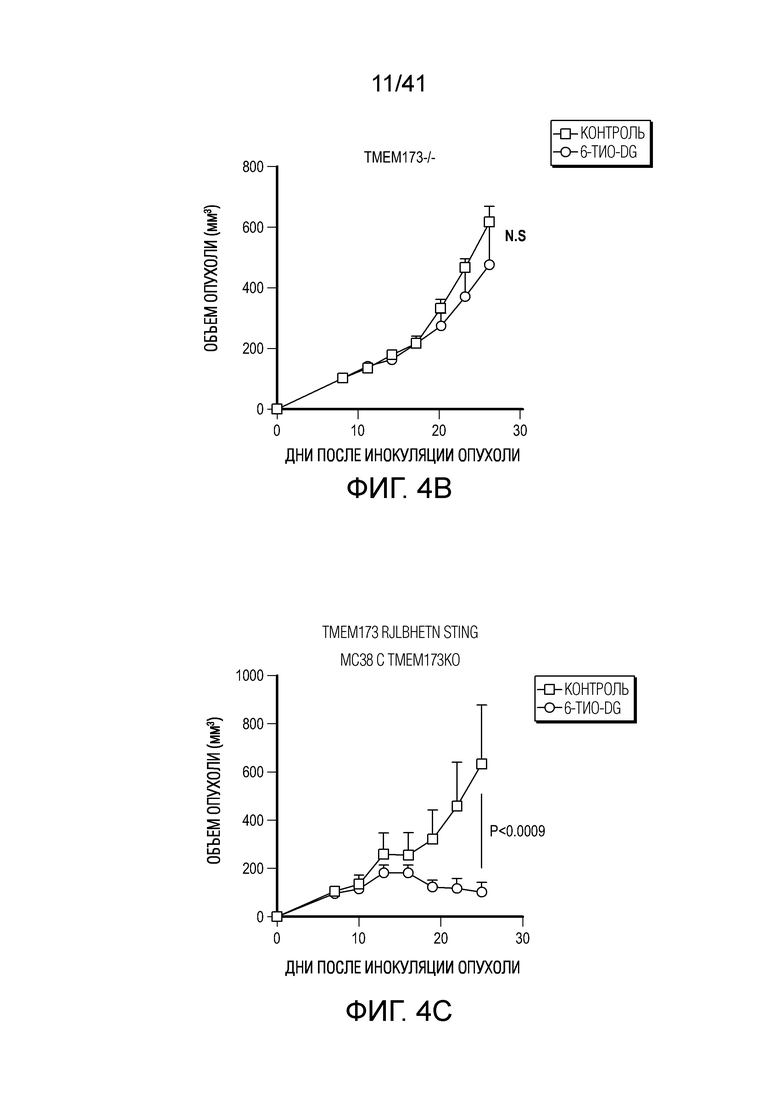
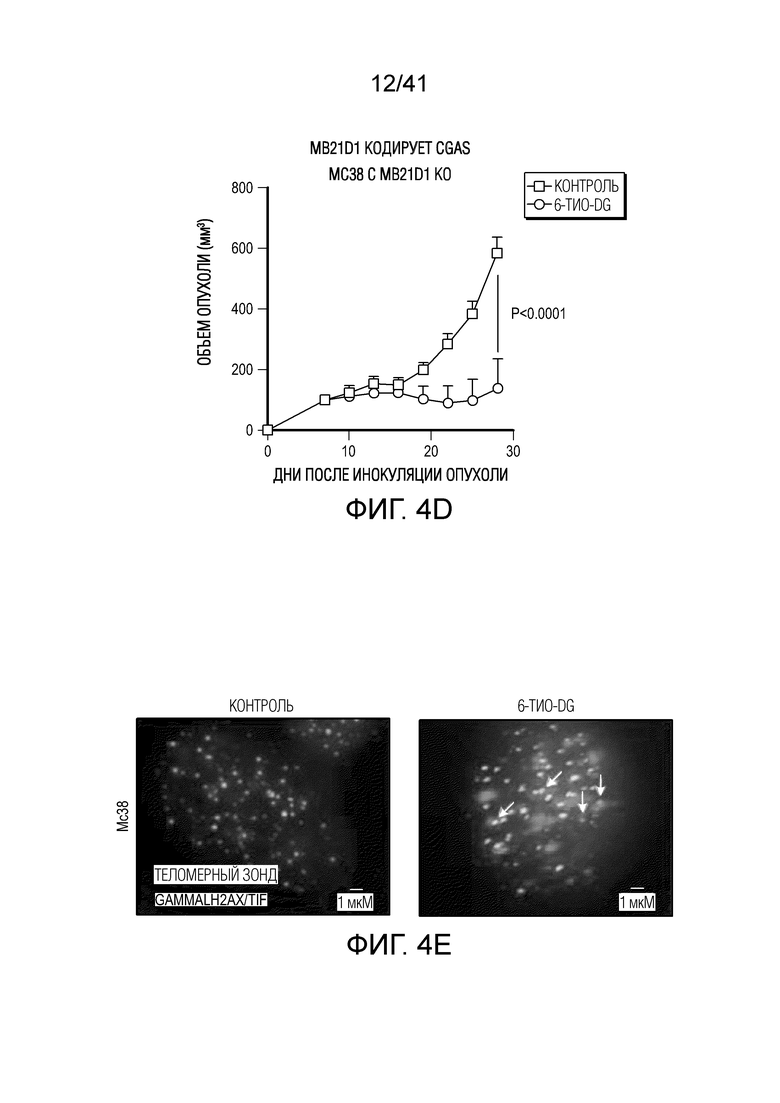
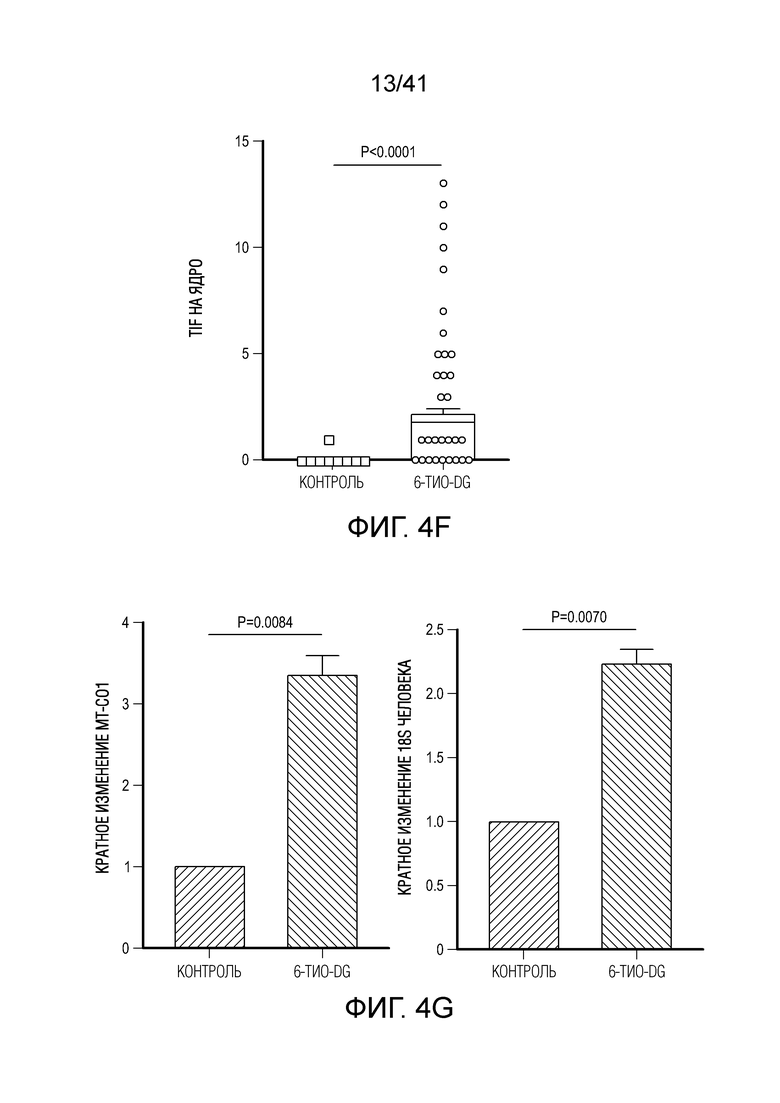
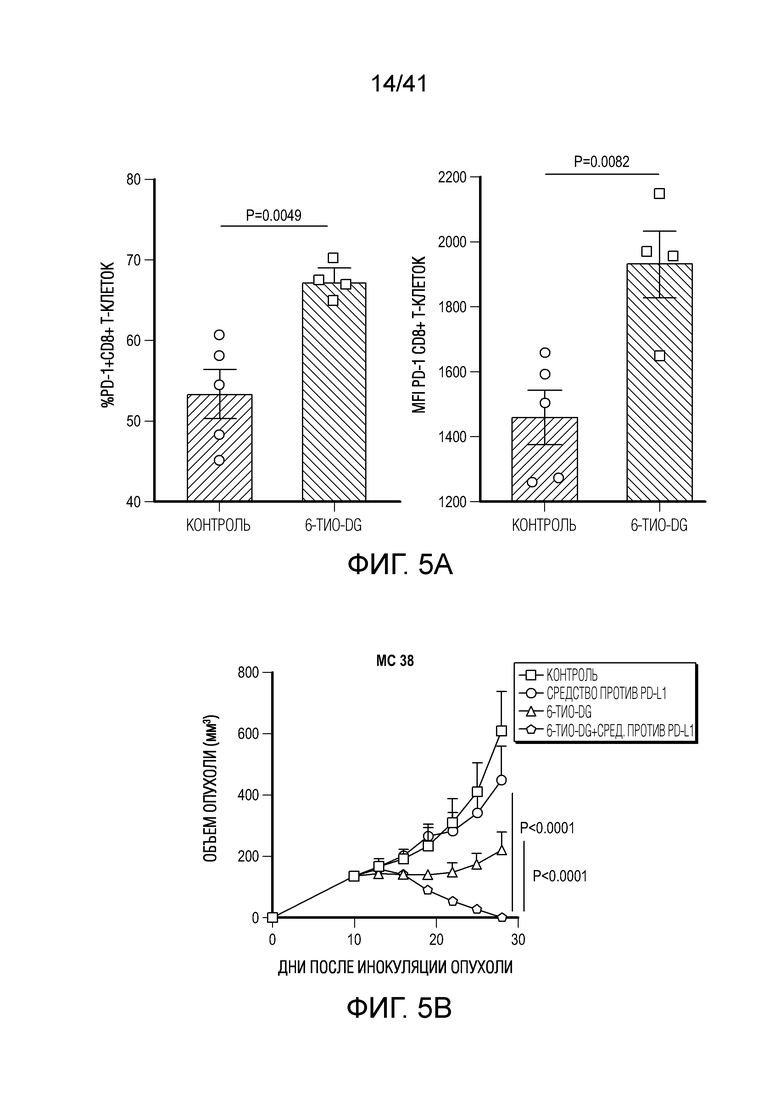
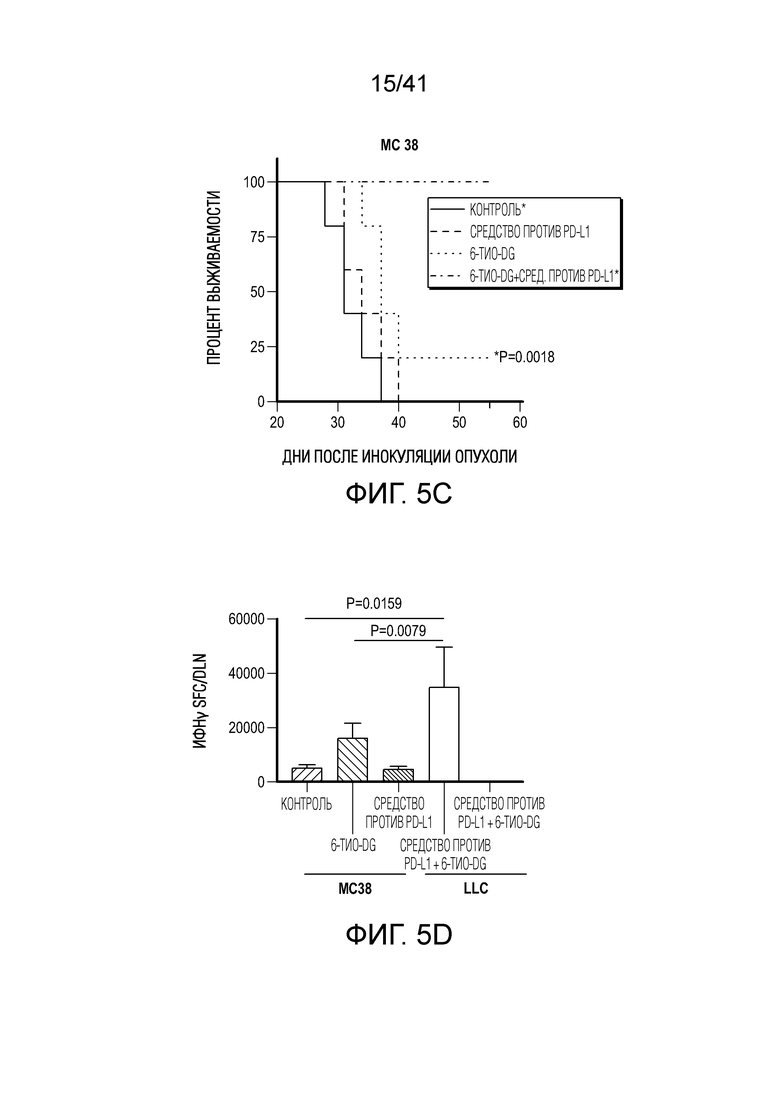
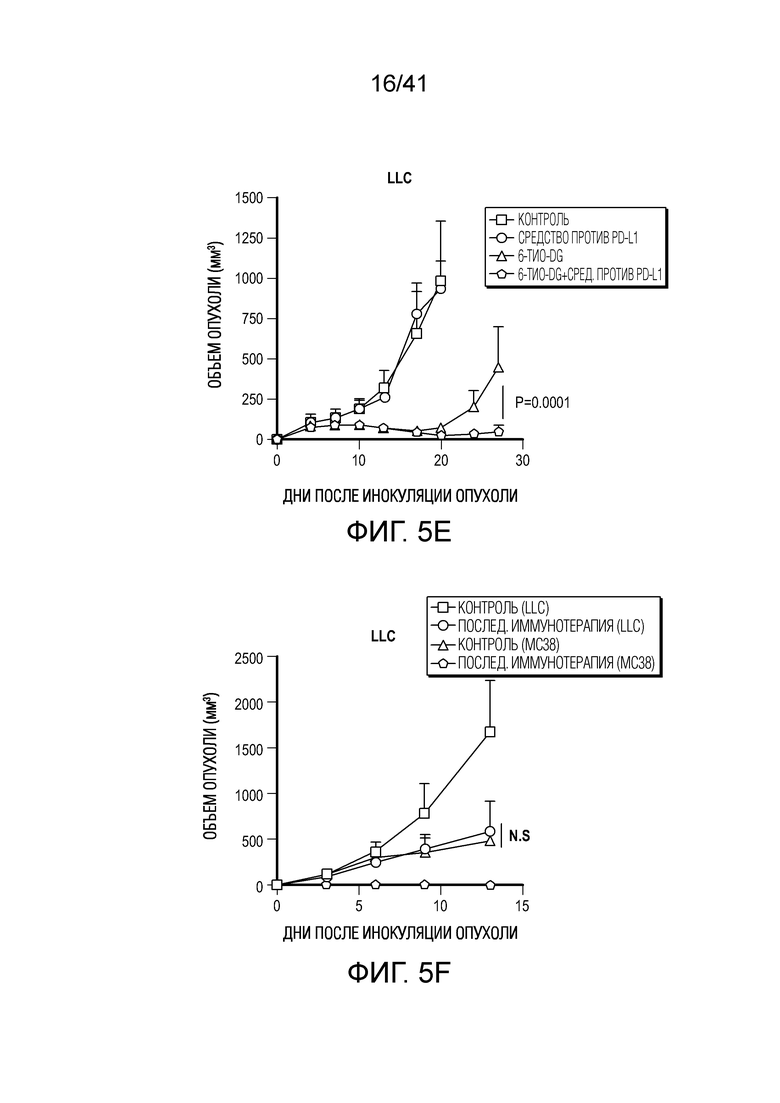
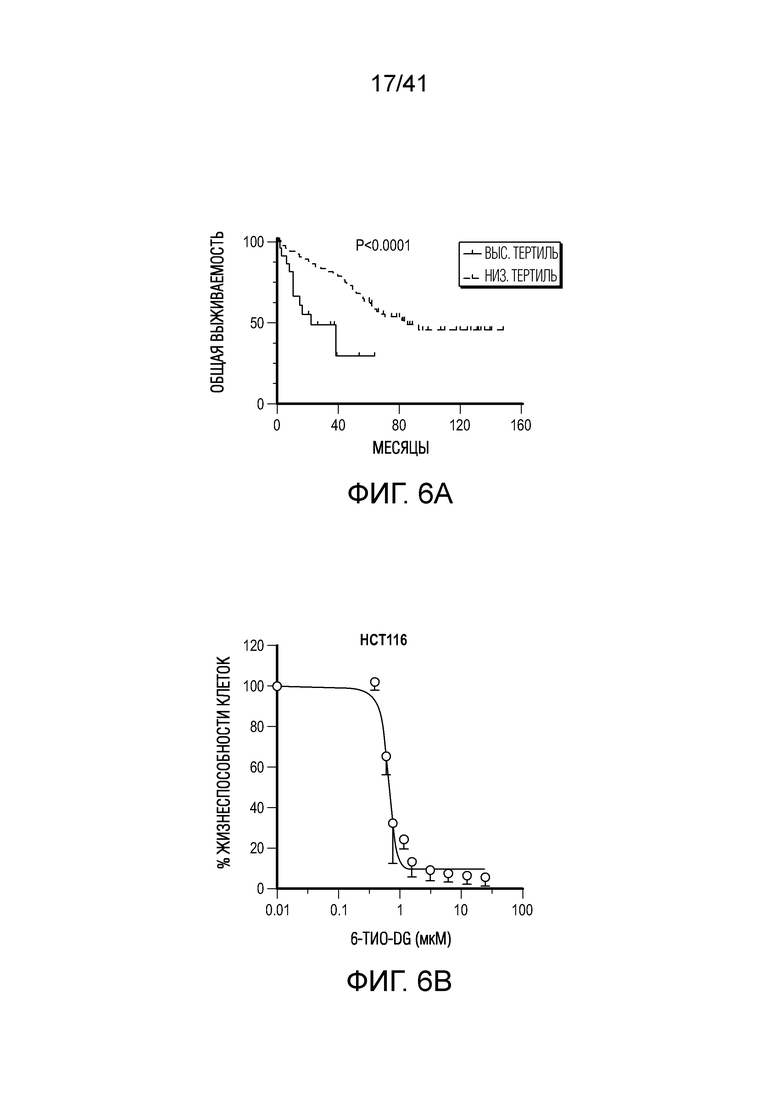
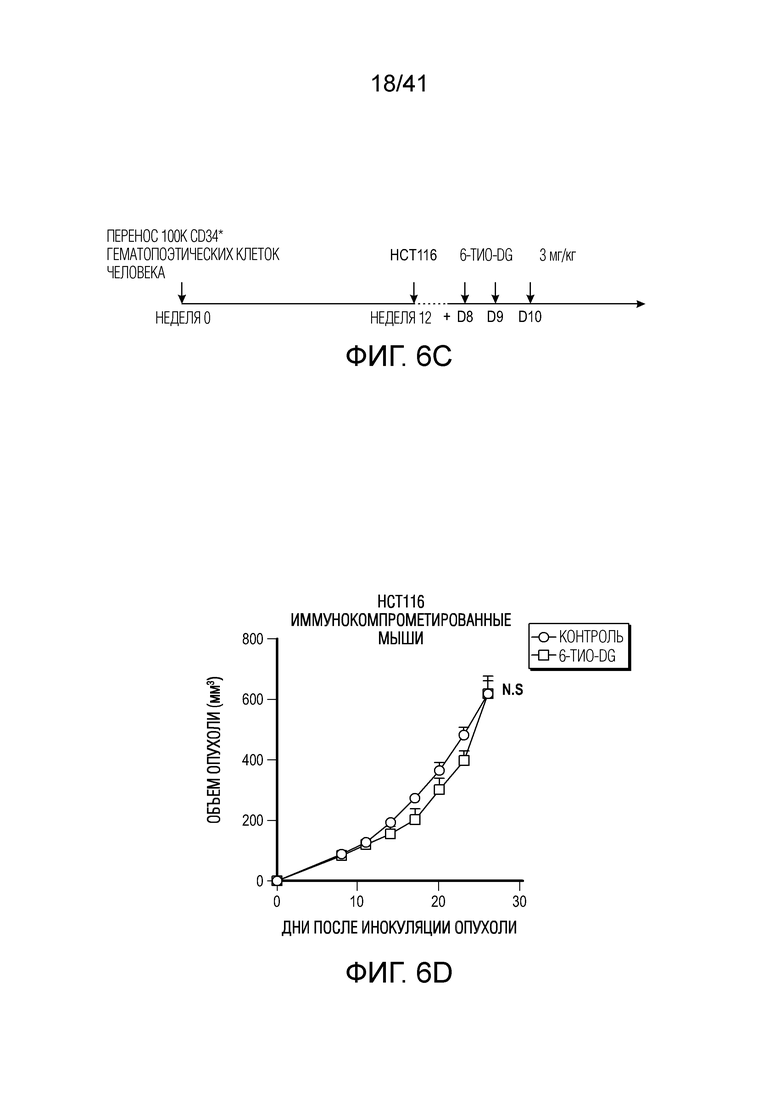
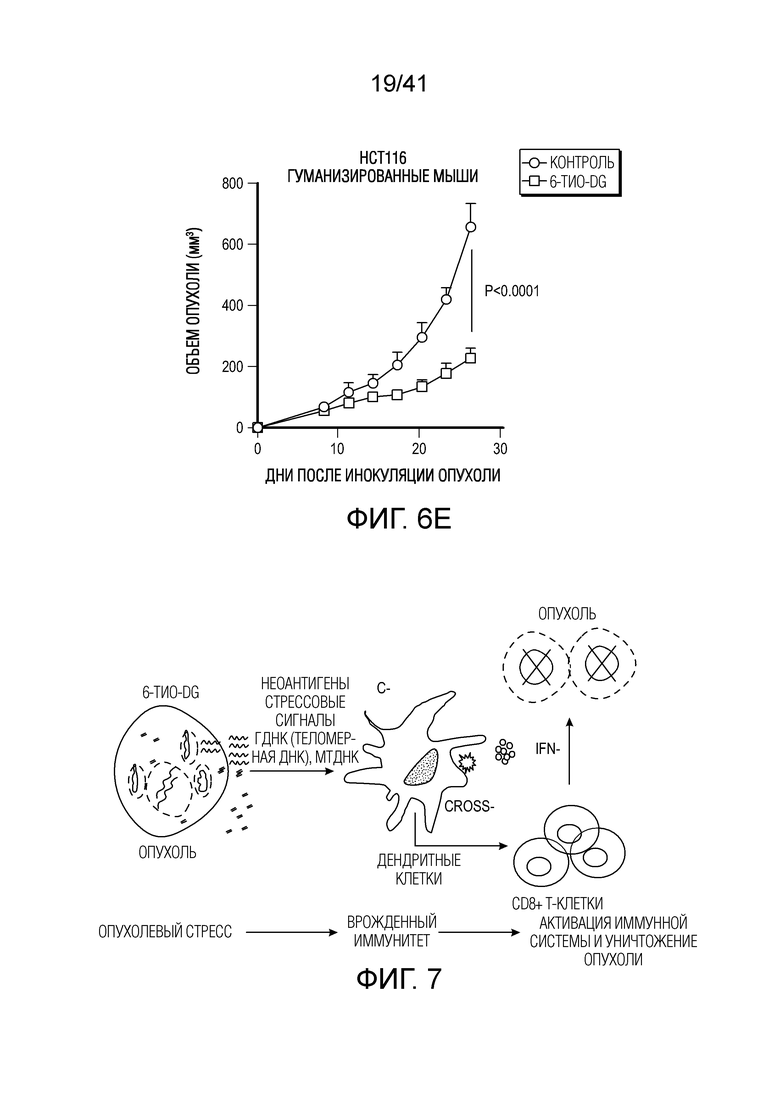
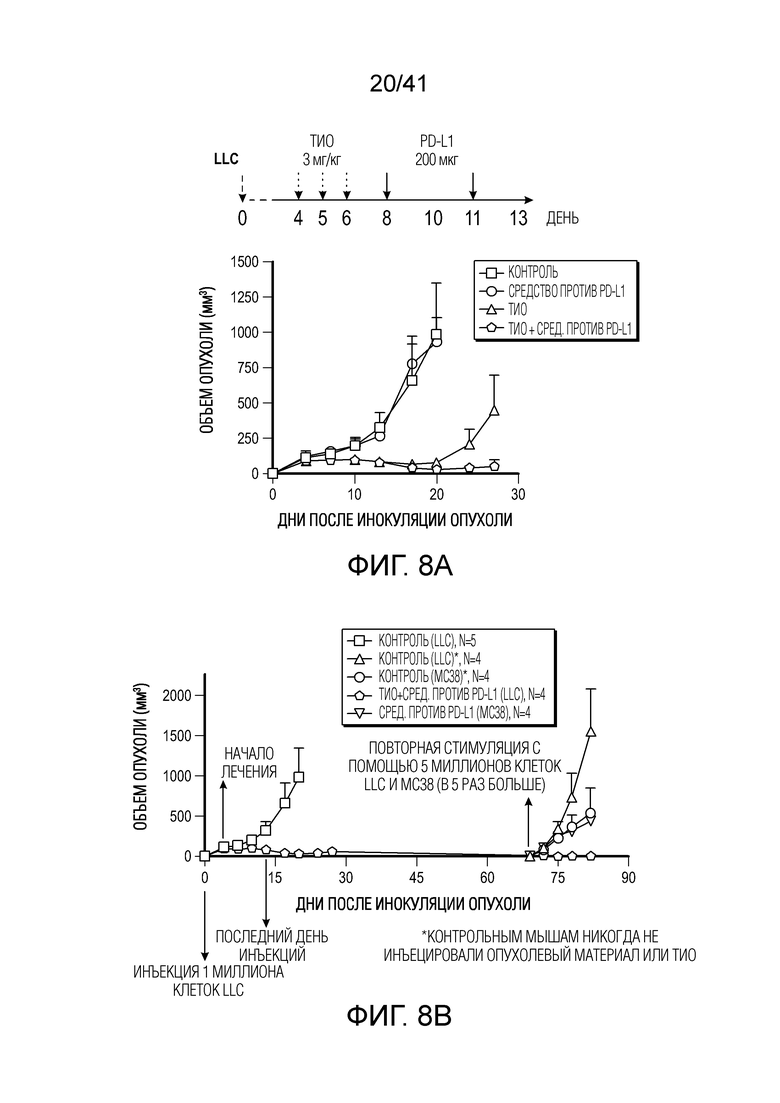
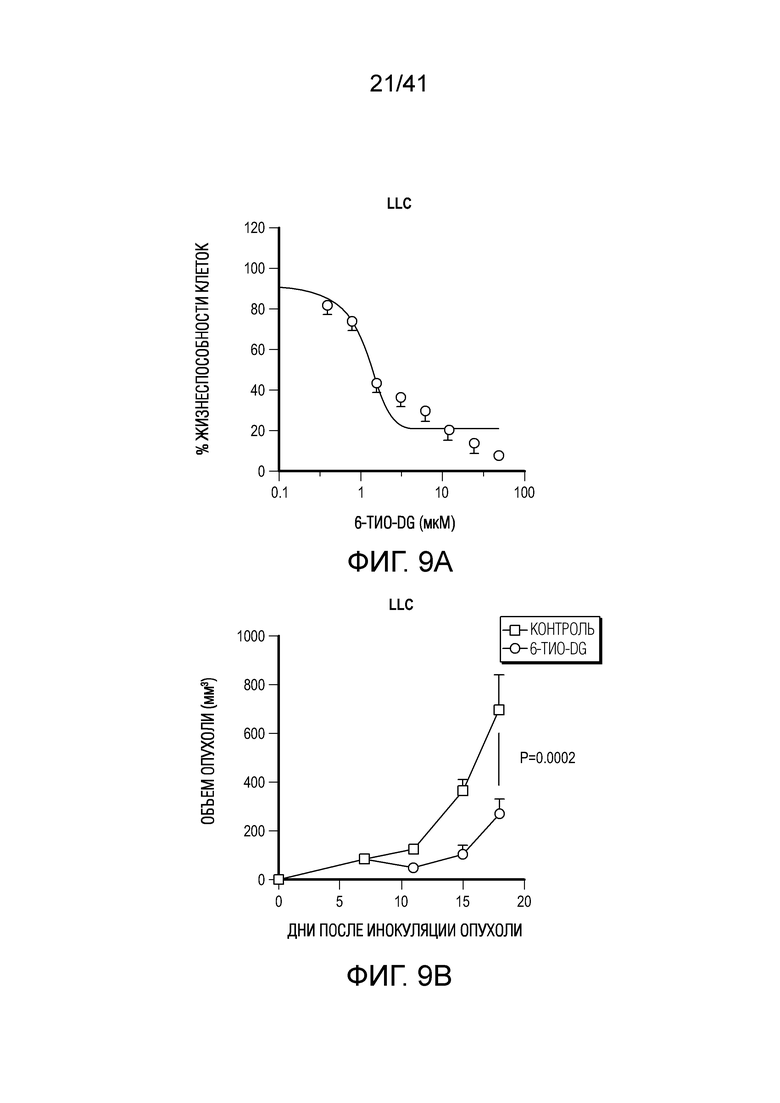
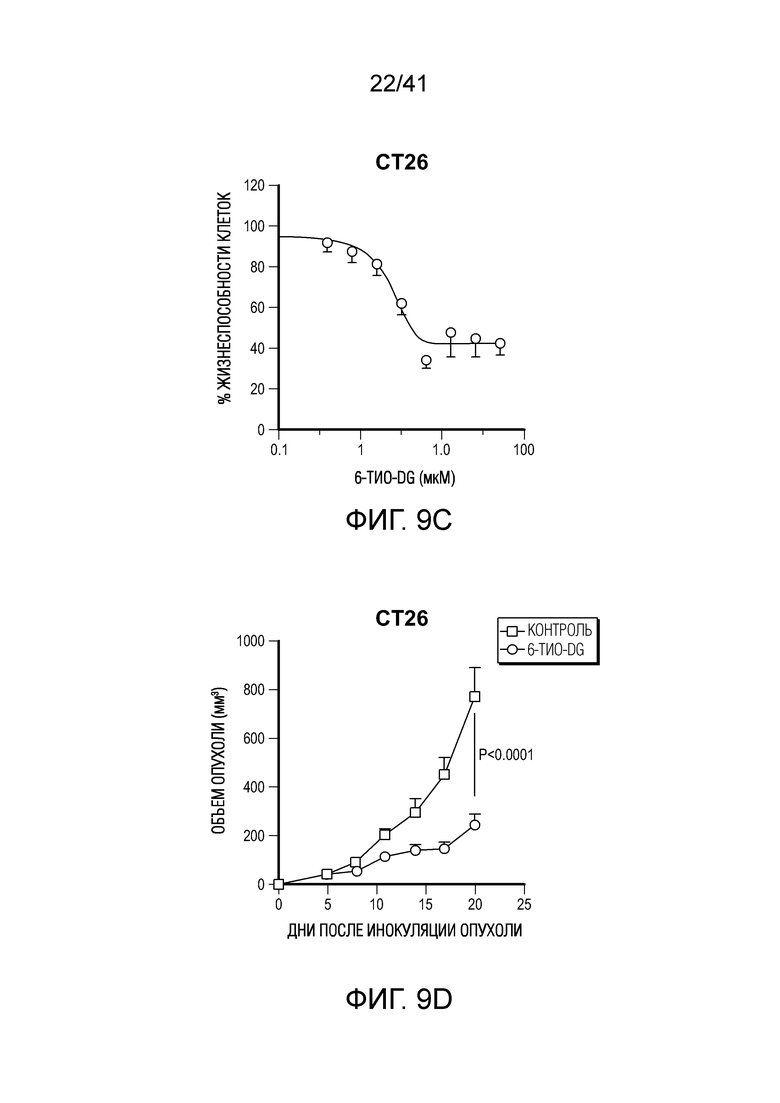
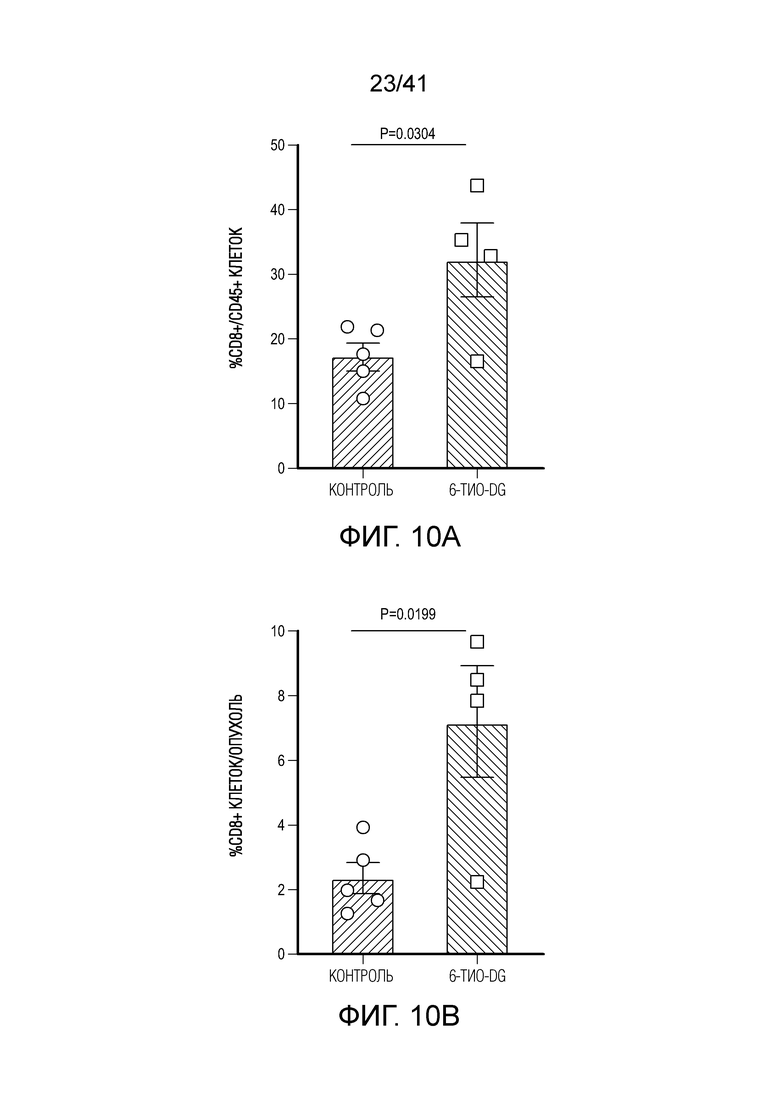
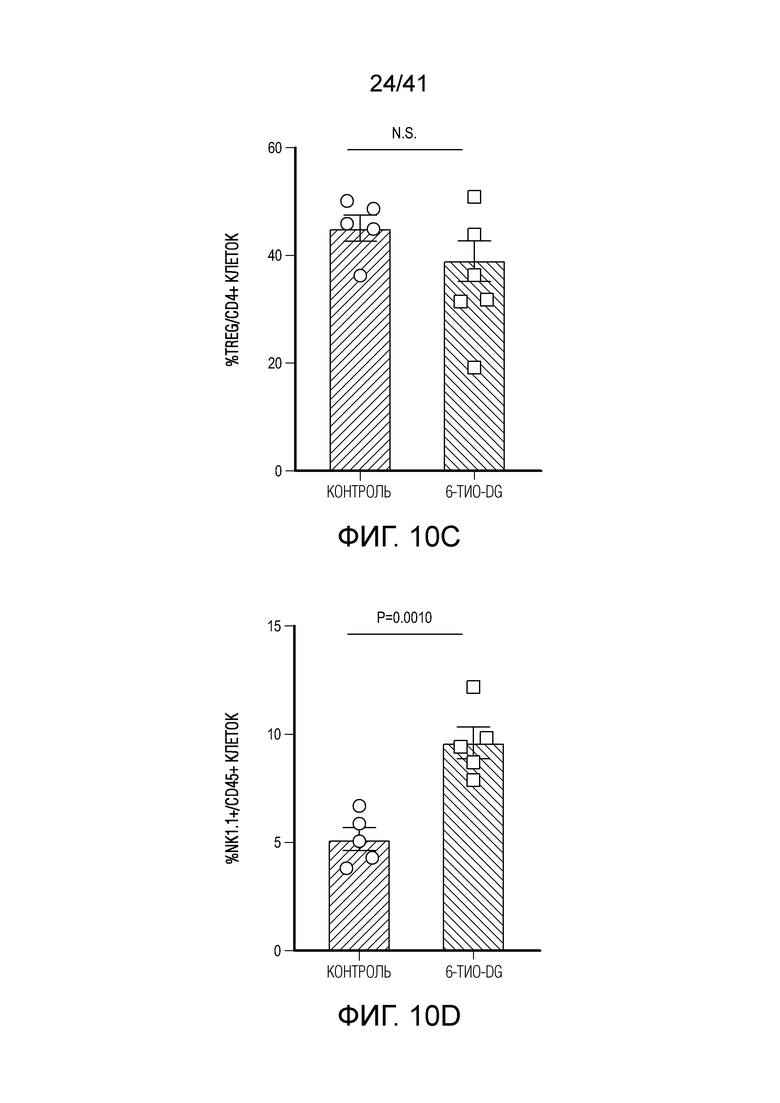
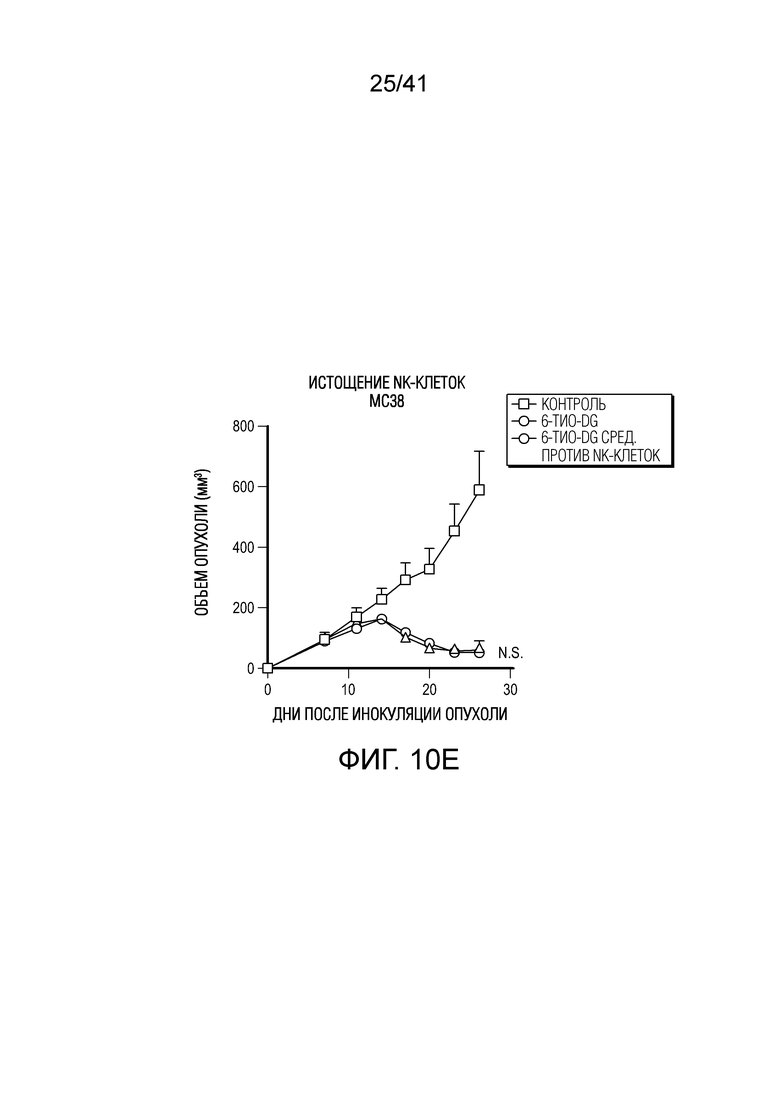
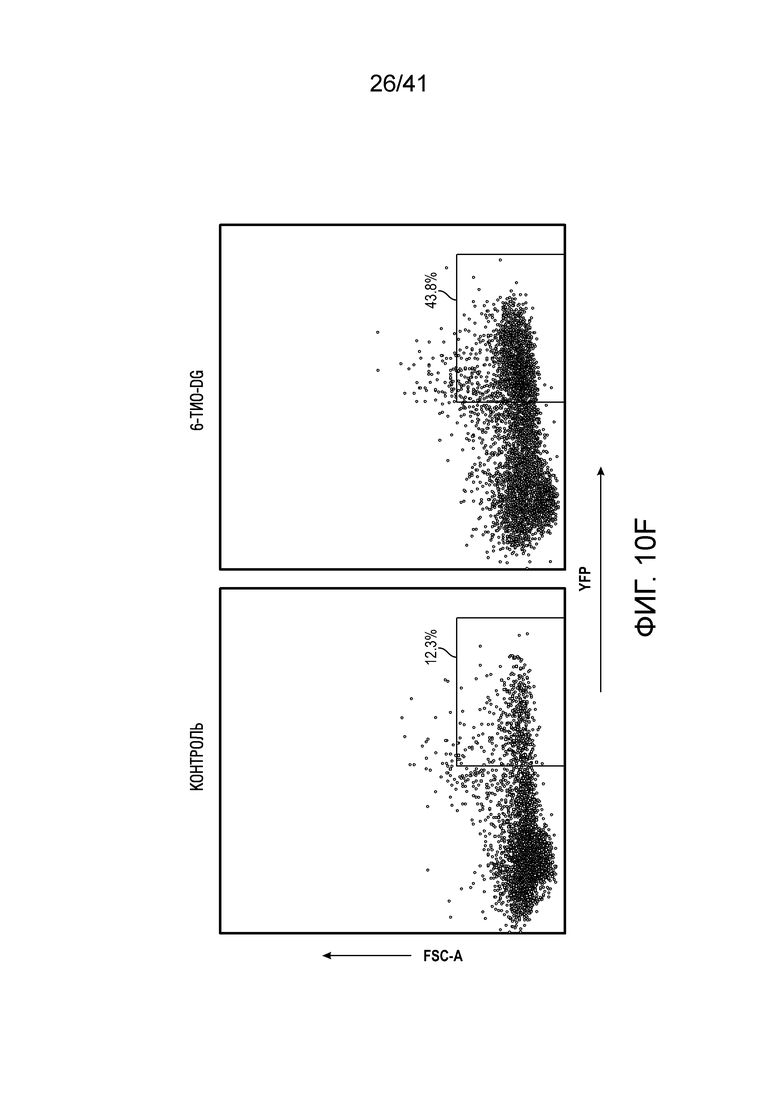
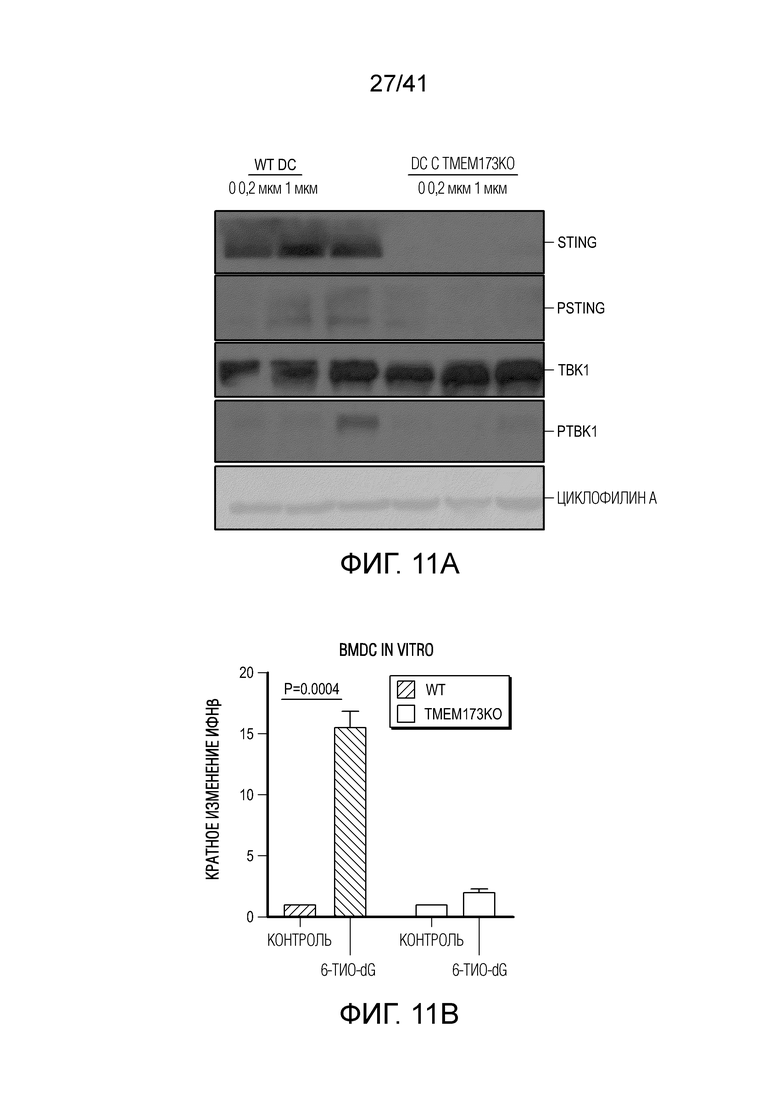
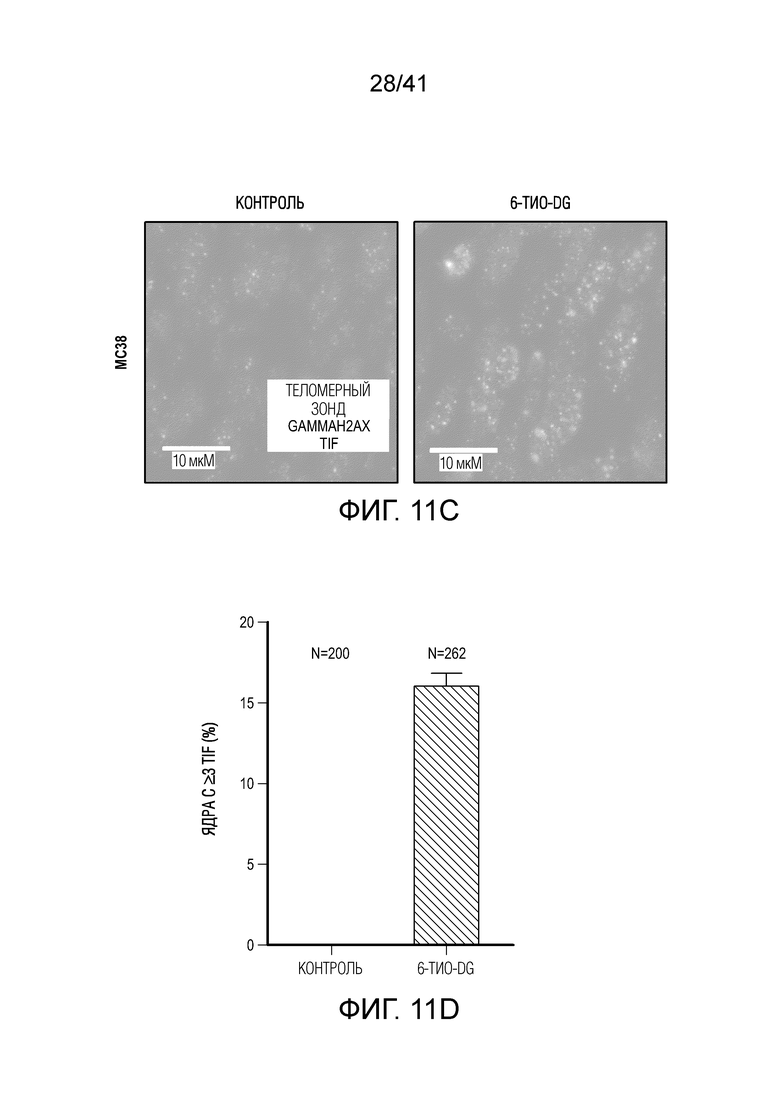
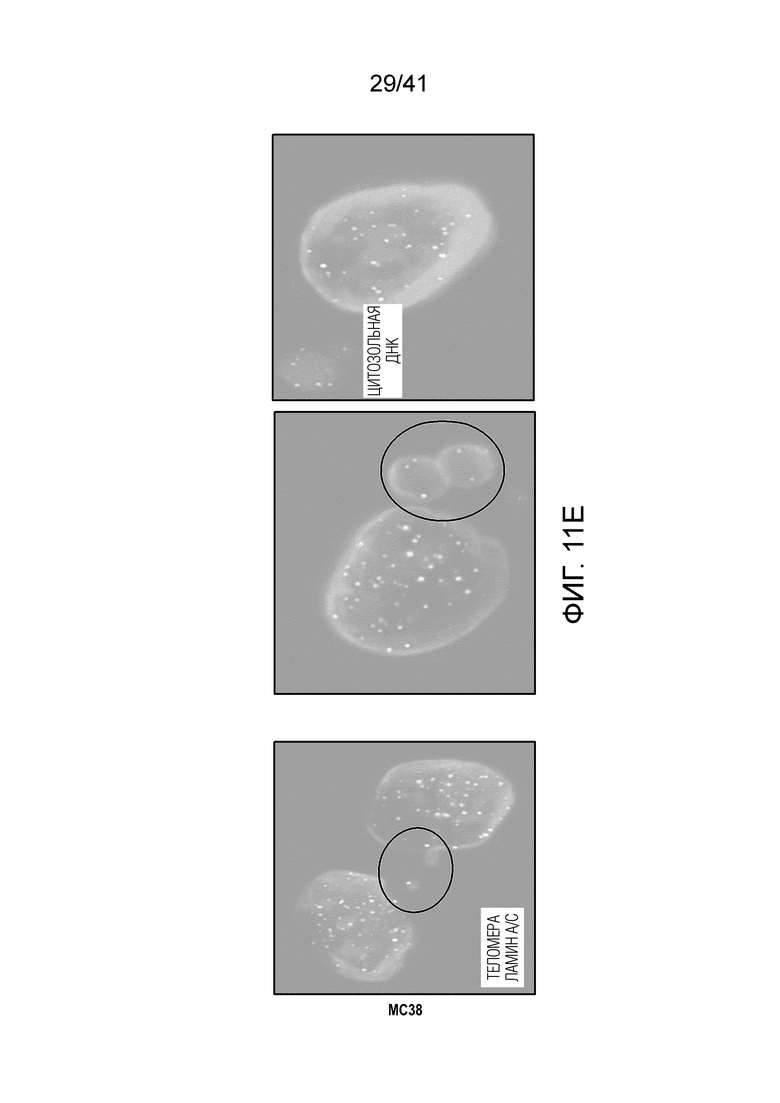
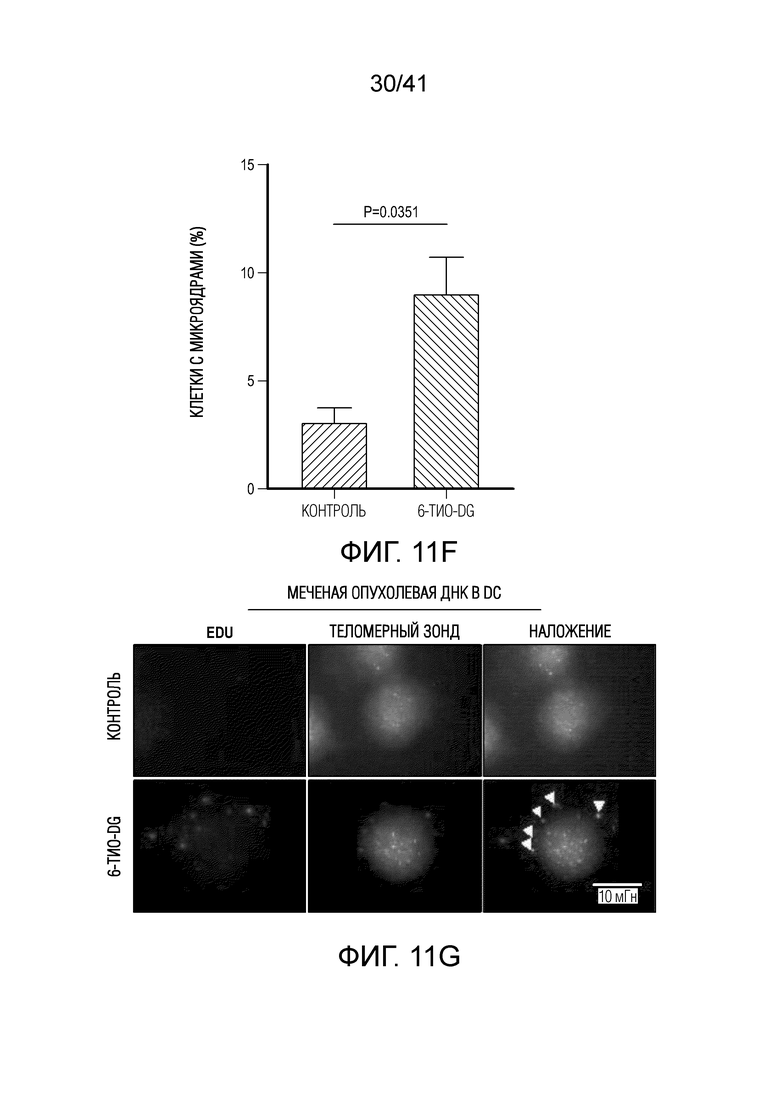
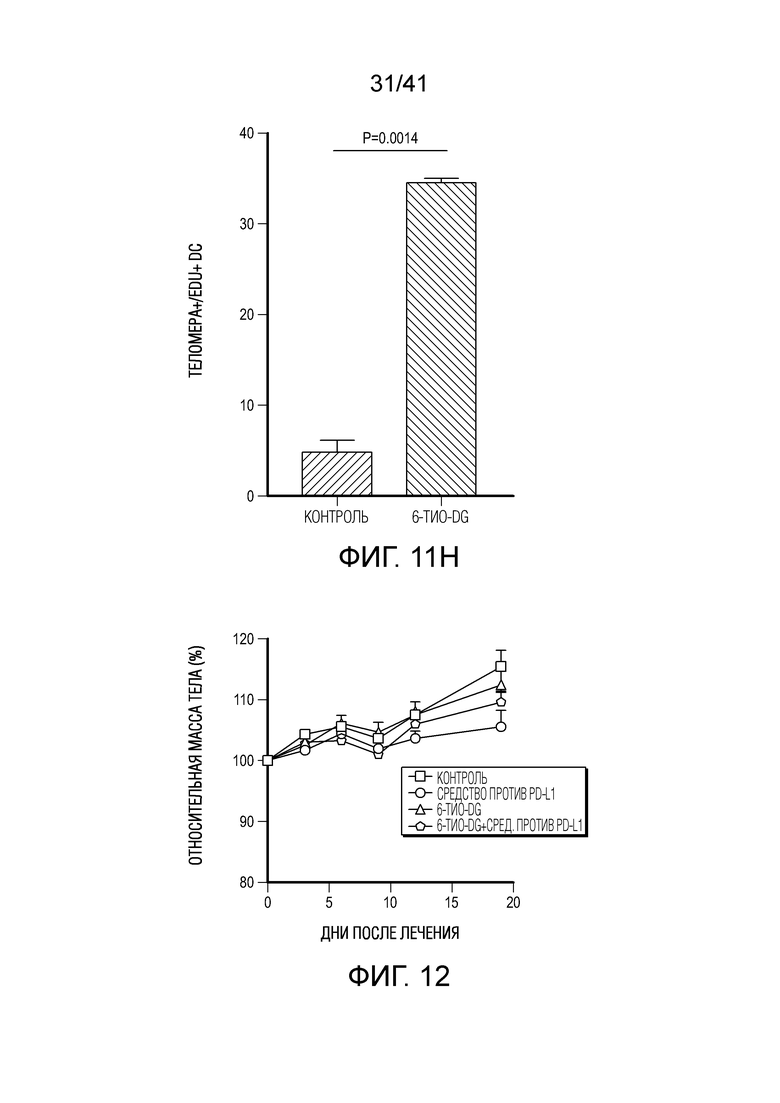
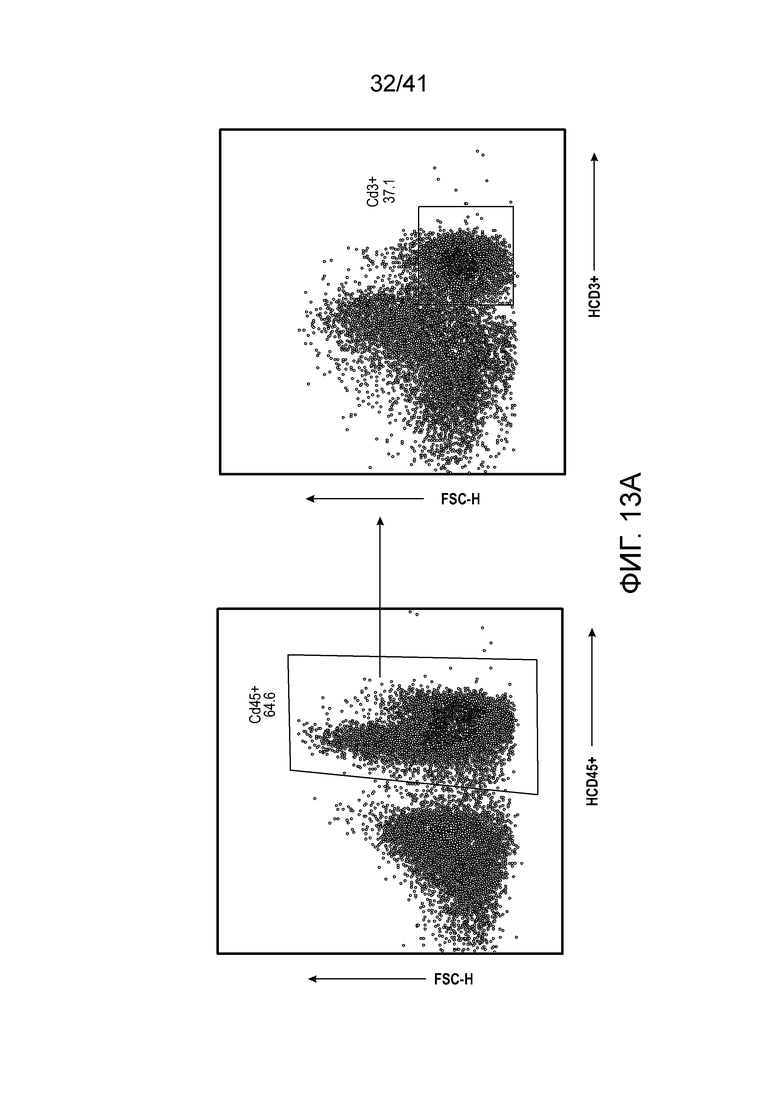
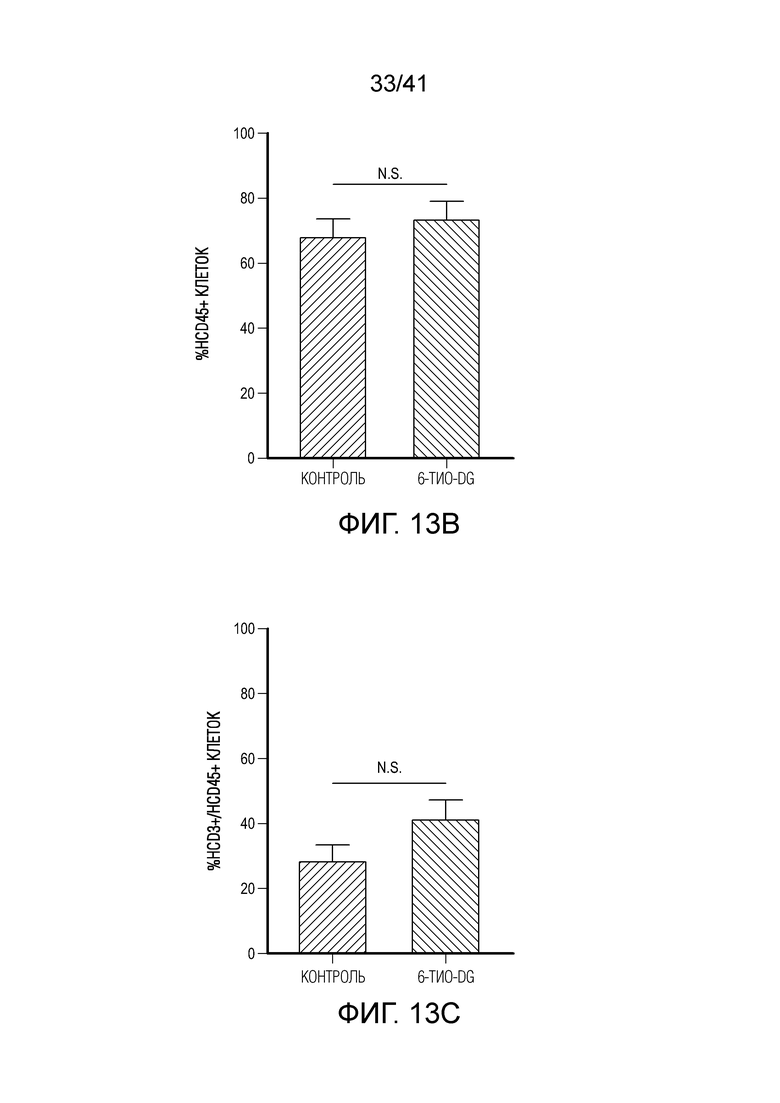
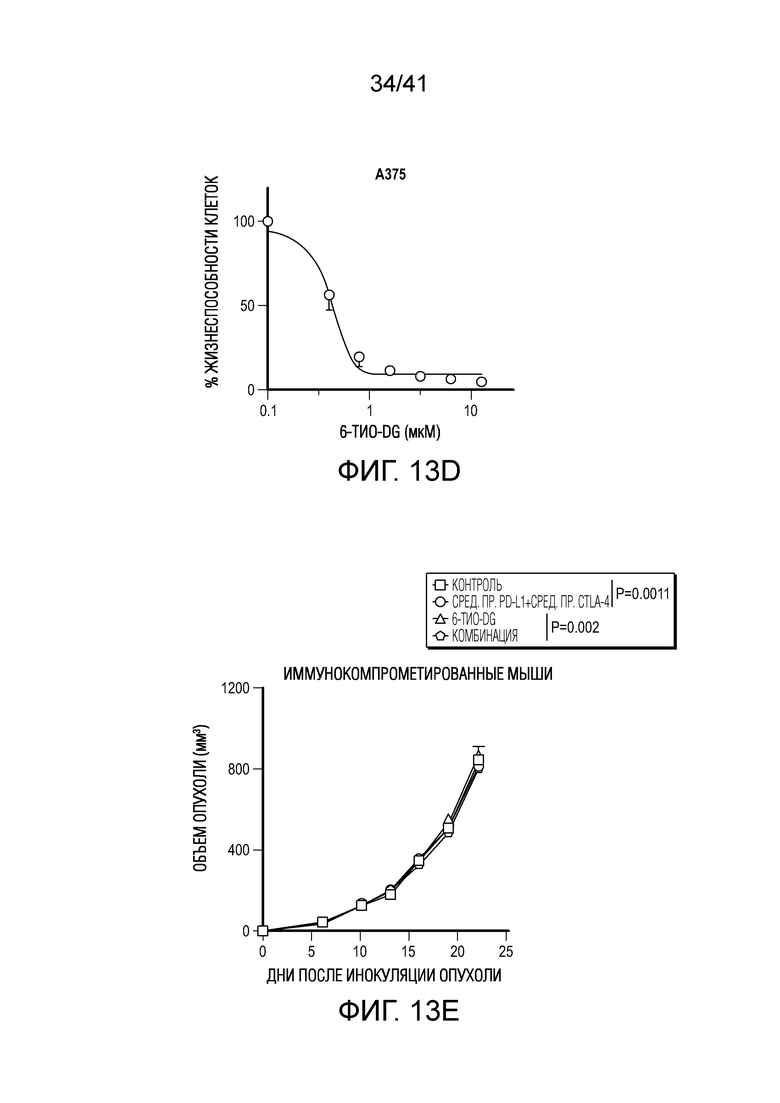
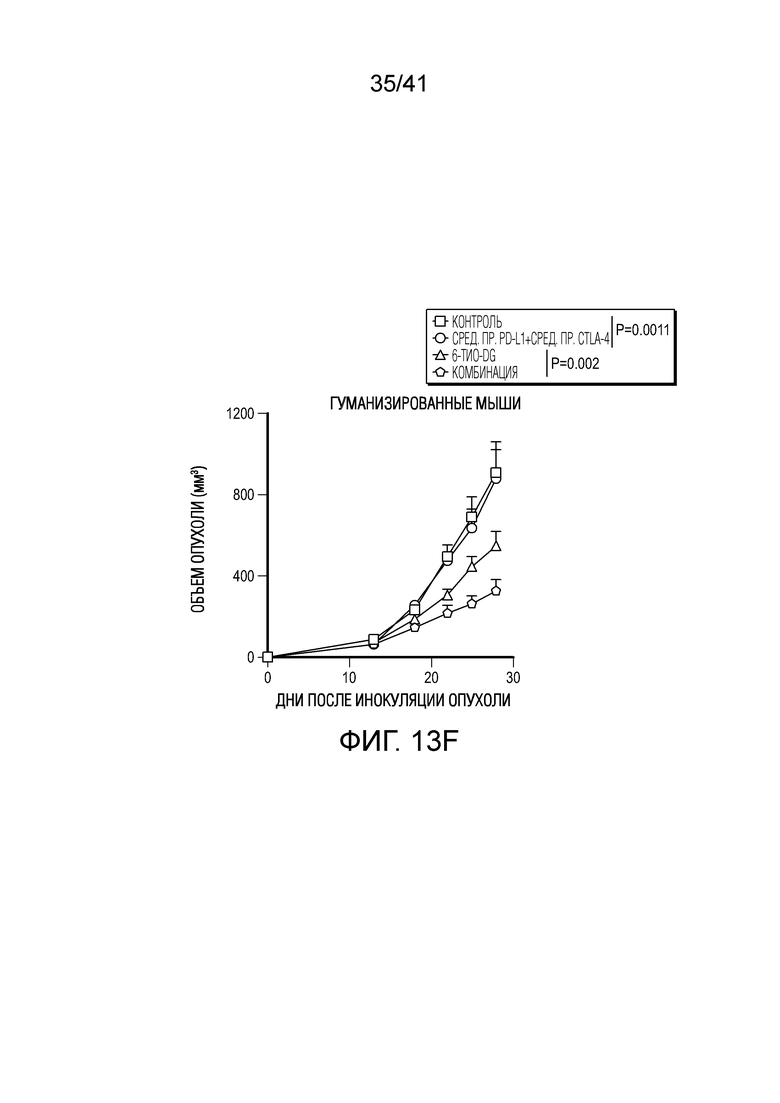
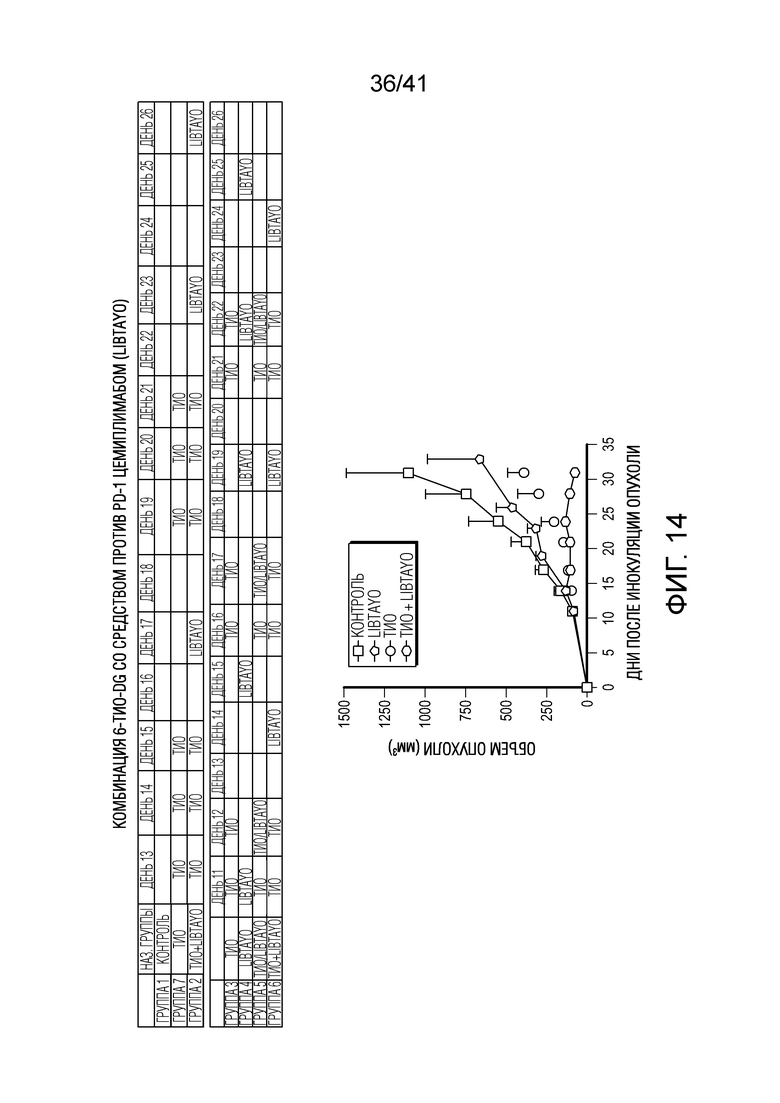
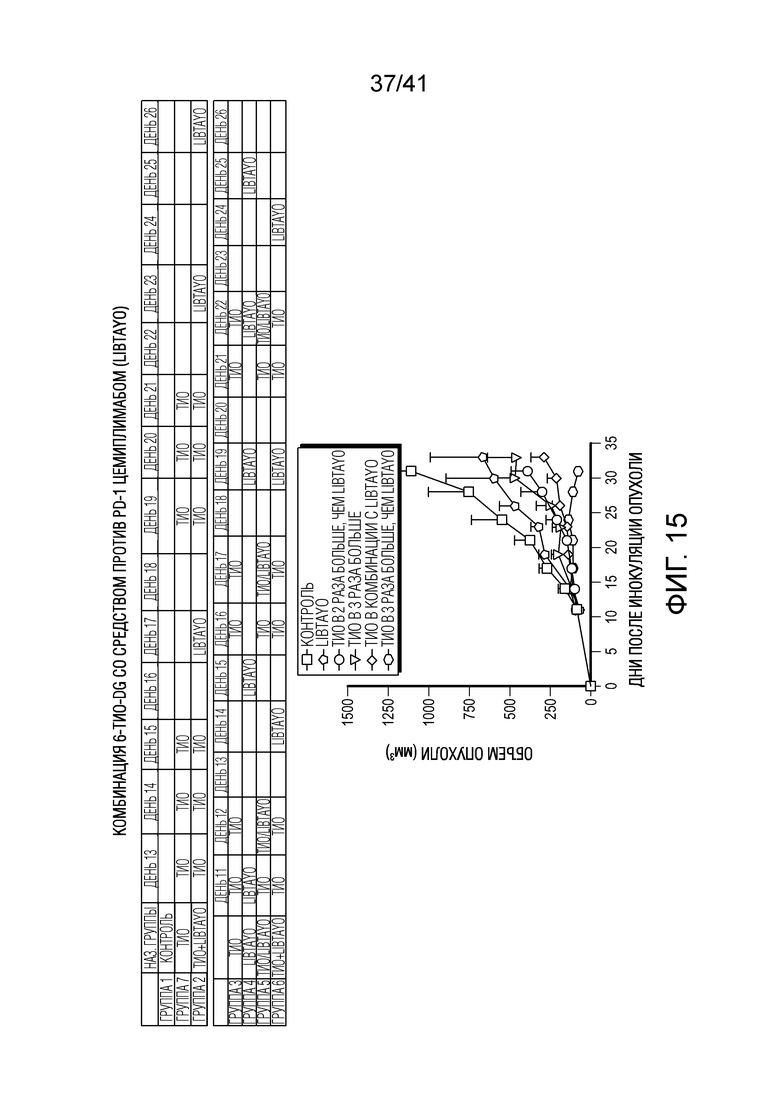
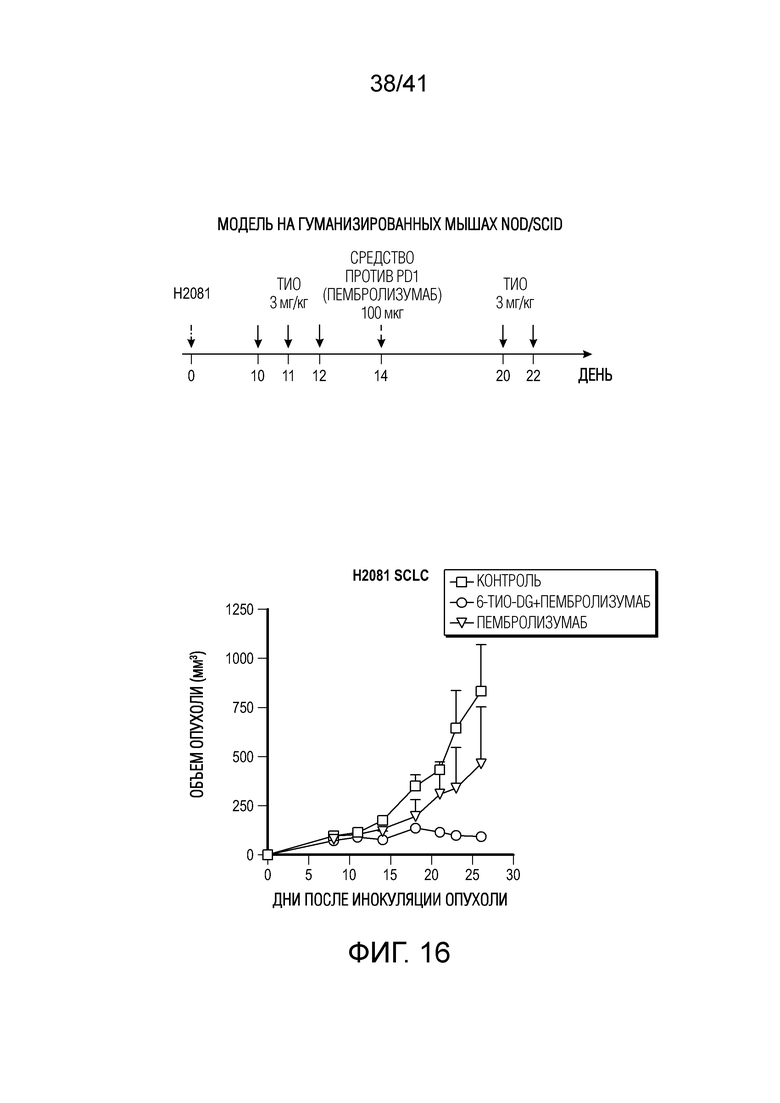
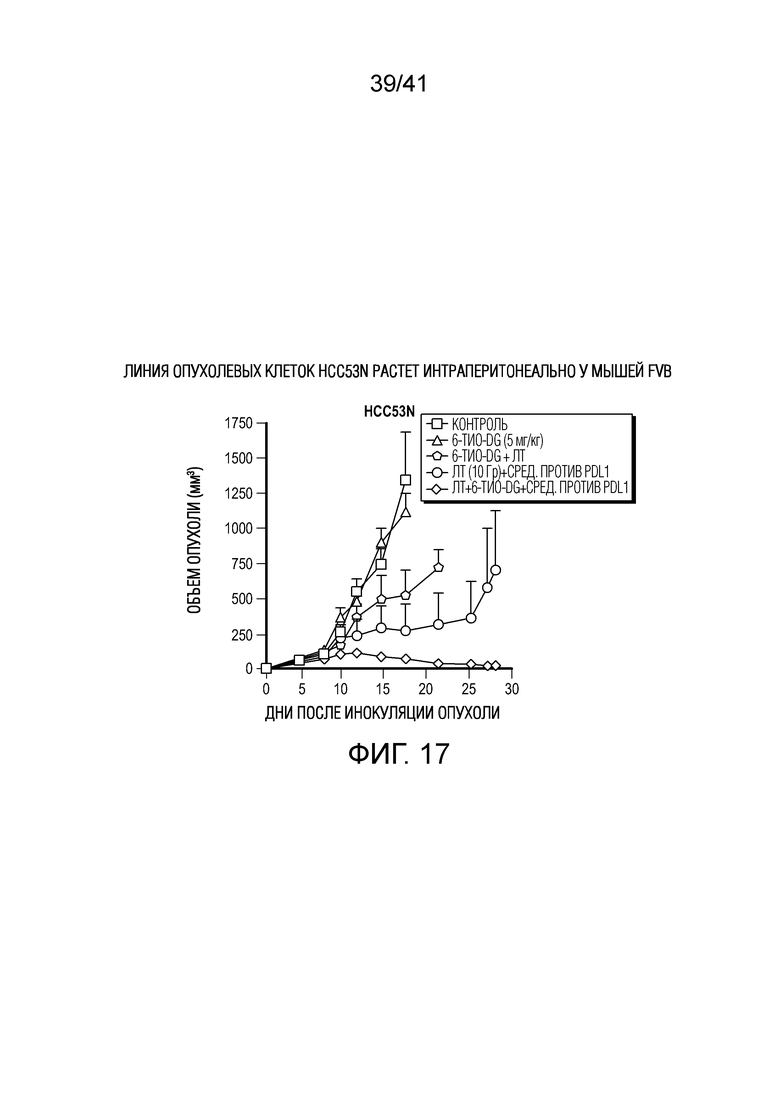
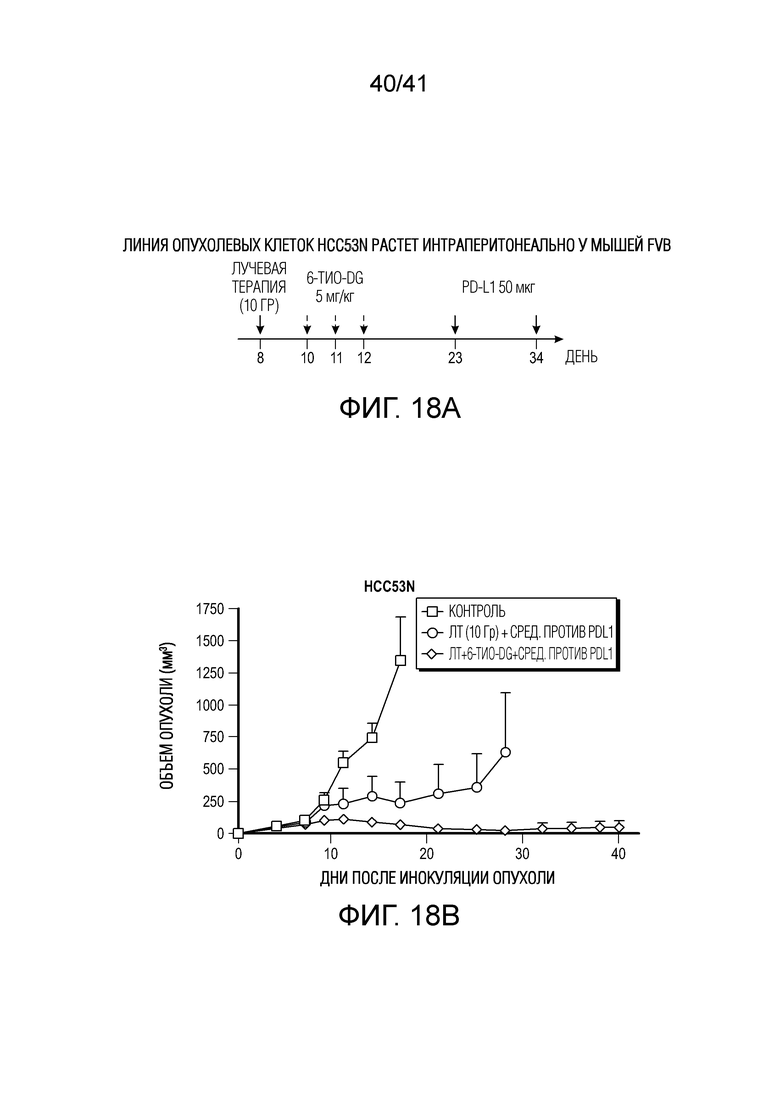
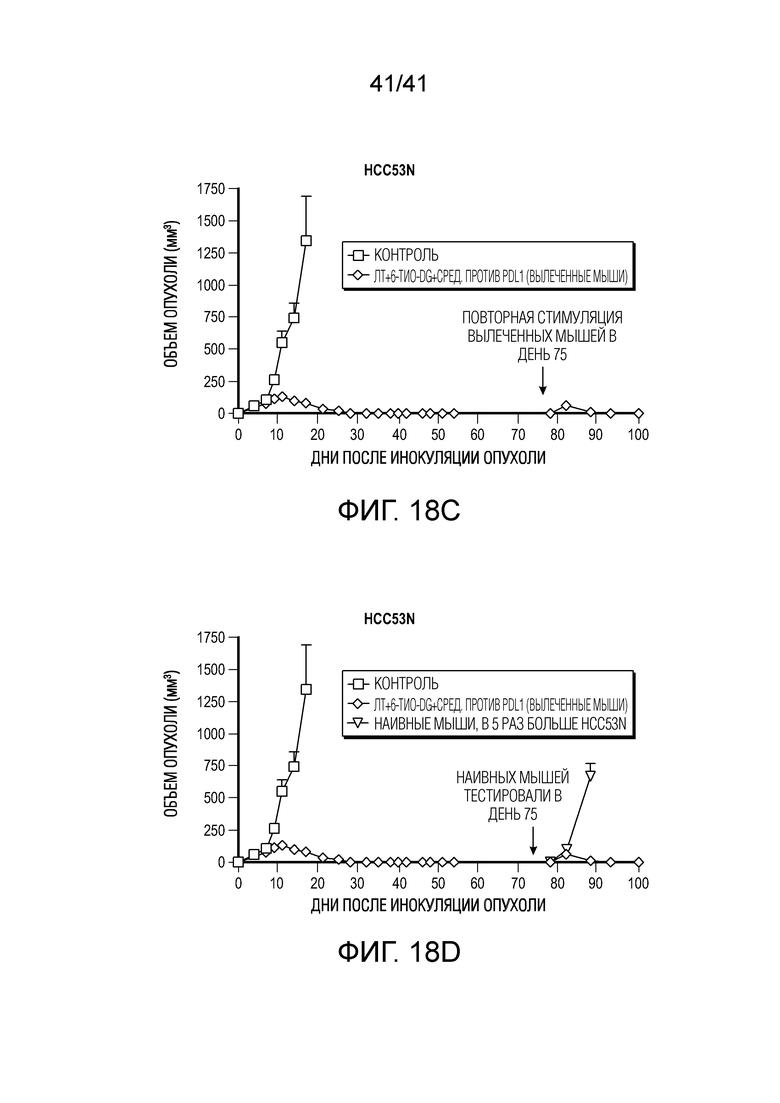
Изобретение относится к способам лечения рака. Предложен способ лечения устойчивого к ингибиторам контрольных точек рака у индивидуума, включающий введение указанному индивидууму 6-тио-2’-дезоксигуанозина (6-тио-dG) с последующим лечением ингибитором контрольных точек, выбранным из атезолизумаба (Tecentriq®) и цемиплимаба (Libtayo®), при этом рак выбран из группы, состоящей из рака легких, рака печени и колоректального рака. Предложенный способ обеспечивает регрессирование опухоли и позволяет преодолеть резистентность к блокаде PD-L1 в опухолях на поздней стадии. 16 з.п. ф-лы, 18 ил., 1 табл., 3 пр.
1. Способ лечения рака у индивидуума, где рак, подлежащий лечению, устойчив к ингибиторам контрольных точек, включающий введение указанному индивидууму 6-тио-2’-дезоксигуанозина (6-тио-dG) с последующим лечением ингибитором контрольных точек, выбранным из атезолизумаба (Tecentriq®) и цемиплимаба (Libtayo®), где рак выбран из группы, состоящей из рака легких, рака печени и колоректального рака.
2. Способ по п. 1, где 6-тио-dG вводят в течение от приблизительно 1 до приблизительно 5 дней за терапевтический цикл.
3. Способ по п. 1, где ингибитор контрольных точек вводят в течение от приблизительно 1 до приблизительно 3 дней за терапевтический цикл.
4. Способ по любому из пп. 1-3, где ингибитор контрольных точек представляет собой атезолизумаб.
5. Способ по любому из пп. 1-4, где указанный ингибитор контрольных точек представляет собой цемиплимаб (Libtayo®).
6. Способ по любому из пп. 1-5, где общая доза 6-тио-dG, вводимая за приблизительно 1-5 дней терапии, составляет приблизительно 20-2000 мг.
7. Cпособ по любому из пп. 1-6, где рак является резистентным к химиотерапии.
8. Способ по любому из пп. 1-7, где указанного индивидуума ранее лечили посредством терапии ингибитором контрольных точек.
9. Способ по п. 8, где индивидуума ранее лечили посредством одного или более из терапии ингибиторами контрольных точек PD-1 или PD-L1.
10. Способ по любому из пп. 1-9, где указанный 6-тио-dG и ингибитор контрольных точек вводят системно.
11. Способ по любому из пп. 1-9, где указанный 6-тио-dG и ингибитор контрольных точек вводят локально или регионарно в очаг опухоли.
12. Способ по любому из пп. 1-9, где указанный 6-тио-dG вводят локально или регионарно в очаг опухоли и ингибитор контрольных точек вводят системно.
13. Способ по любому из пп. 1-9, где введение 6-тио-dG и ингибитора контрольных точек приводит к ингибированию роста опухоли.
14. Способ по любому из пп. 1-9, где введение 6-тио-dG и ингибитора контрольных точек приводит к ремиссии указанного рака.
15. Способ по любому из пп. 1-9, где введение 6-тио-dG и ингибитора контрольных точек приводит к снижению опухолевой нагрузки.
16. Способ по любому из пп. 1-9, где введение 6-тио-dG и ингибитора контрольных точек приводит к ингибированию метастазирования раковых клеток.
17. Способ по любому из пп. 1-9, где введение 6-тио-dG и ингибитора контрольных точек приводит к эрадикации опухоли.
| WO 2017205756 A1, 30.11.2017 | |||
| WO 2017165675 A1, 28.09.2017 | |||
| WO 2019183482 A1, 26.09.2019 | |||
| WO 2018156494 A1, 30.08.2018 | |||
| US 20200138804 A1, 07.05.2020. |

