Область техники
Настоящее изобретение относится к катализатору селективного гидрирования и способу его получения, и также к способу селективного гидрирования.
Уровень техники
Этилен является одним из важнейших основных сырьевых материалов для нефтехимической промышленности. В качестве мономера для синтеза различных полимеров этилен в подавляющем большинстве случаев получают путем парового крекинга нефтяных углеводородов (таких как этан, пропан, бутан, нафта, легкое дизельное топливо и т.д.). Фракция С2 с преобладанием этилена, полученная указанным способом, содержит от 0,5% до 2,3% по массе ацетилена. При применении в полимеризации ацетилен в этилене усложняет способ полимеризации этилена и ухудшает свойства полимера. Кроме того, он также снижает активность катализатора полимеризации и влияет на физические свойства полимера, и, следовательно, должен быть удален.
Селективное гидрирование в настоящее время широко применяют в промышленности для удаления ацетилена из этилена с применением в основном катализатора на основе благородных металлов, таких как Pd, Pt и Au. Концевое гидрирование С2 и начальное гидрирование С2 зависят от положения реактора гидрирования ацетилена относительно колонны удаления метана. То есть, в случае начального гидрирования С2 реактор гидрирования расположен перед колонной удаления метана, и в случае концевого гидрирования реактор гидрирования расположен после колонны удаления метана.
Преимущества способа пост-гидрирования заключаются в наличии множества средств управления способом гидрирования, возможности легко избежать неуправляемого нагрева и простоте эксплуатации. Однако этот способ довольно сложен и требует отдельной подачи водорода. В способе концевого гидрирования С2 из-за низкого содержания водорода в гидрирующих материалах происходит гидрирование и димеризация ацетилена с образованием фракций С4, которые дополнительно полимеризуются с образованием олигомеров с широким диапазоном молекулярной массы, обычно известных как «зеленое масло». Зеленое масло адсорбируется на поверхности катализатора с последующим образованием кокса, который блокирует каналы катализатора, предотвращая диффузию реагентов на поверхность активного центра катализатора и, таким образом, приводит к снижению активности катализатора.
Катализаторы на основе драгоценных металлов более активны, но при применении они, как правило, образуют зеленое масло, что вызывает закоксовывание и деактивацию катализатора и влияет на стабильность катализатора и срок службы. В патенте CN 200810119385.8 раскрыт катализатор селективного гидрирования на основе недрагоценного металла, содержащий носитель и первичный активный компонент, и также вторичный активный компонент, нанесенный на носитель, и способ его получения и применения, где первичный активный компонент представляет собой Ni, и вторичный активный компонент выбран из по меньшей мере одного из Mo, La, Ag, Bi, Cu, Nd, Cs, Се, Zn и Zr; как первичный, так и вторичный активные компоненты находятся в аморфных формах со средним диаметром частиц <10 нм; носитель представляет собой неокислительный пористый материал; и катализатор получают способом микроэмульгирования.
В способе концевого гидрирования С2 обычно применяют трехступенчатый реактор гидрирования: общая конверсия ацетилена в реакторе первой ступени составляет от 50 до 80% с отношением водорода к алкину от 1,0 до 1,4; общая конверсия ацетилена в реакторе второй ступени составляет от 40 до 20% с отношением водорода к алкину от 1,4 до 2,0; и остаточный ацетилен полностью превращается в реакторе третьей ступени с отношением водорода к алкину от 2,5 до 4,0, и содержание ацетилена на выходе из реактора третьей ступени обычно составляет 1 ч/млн или менее. Во время реакций концевого гидрирования С2 происходит димеризация ацетилена водородом, что приводит к образованию ряда олигомеров с различными молекулярными массами. Эти олигомеры не могут двигаться с газофазным материалом или двигаются с очень низкой скоростью, и, следовательно, прилипают к поверхности катализатора или поступают в каналы в течение длительного времени, вызывая засорение пор катализатора. Из-за медленного движения они постепенно агломерируются, при этом указанные олигомеры содержат большое количество ненасыщенных связей, которые могут дополнительно полимеризоваться и в конечном итоге образовывать кокс, что приводит к значительному снижению активности и селективности катализатора.
Количество продукта димеризации при гидрировании тесно связано с условиями гидрирования. Когда отношение водорода к алкину низкое, реакция димеризации при гидрировании ацетилена очень интенсивна из-за недостаточного количества водорода; скорость закоксовывания катализатора будет очень быстрой.
Во время реакций реактор первой ступени также генерирует наибольшее количество зеленого масла из-за большой нагрузки удаления ацетилена в реакторе первой ступени. Реакция гидродимеризации протекает наиболее интенсивно на входе в реактор первой ступени, и часть зеленого масла полимеризуется на входе в реактор первой ступени, что приводит к быстрому снижению активности катализатора на этом участке. Другим местом является выход из реактора первой ступени, поскольку по мере протекания реакции гидрирования отношение водорода к алкину становится все ниже и ниже, из-за чего снова повышается скорость реакции гидродимеризации, в то время как повышение температуры, в свою очередь, ускоряет полимеризацию зеленого масла.
В некоторых из установок концевого гидрирования С2 применяют двухстадийный способ гидрирования. Часть зеленого масла, образующегося в реакторе первой ступени, поступает в реактор второй ступени и накапливается на входе в реактор второй ступени, резко ухудшая эффект гидрирования реактора второй ступени, и содержание ацетилена на выходе из реактора быстро повышается до 1 ч/млн или выше. Чистота этилена низкая, что влияет на полимеризацию олефинов, и, таким образом, катализатор нуждается в регенерации.
В некоторых установках применяют изотермический реактор, в котором температура в реакторе постепенно падает по направлению массового потока, и зеленое масло, образующееся в верхней части реактора, не накапливается в нижней части реактора, при этом постепенно образуется кокс, вызывающий быстрое снижение селективности.
Некоторые установки по производству этилена, где фракции С2 гидрируют в одной секции, предъявляют более высокие требования к стабильности катализатора. Как правило, селективность катализатора значительно снижается после 3 месяцев работы из-за влияния зеленого масла.
В некоторых установках для регулировки нагрузки гидрирования каждого реактора иногда количество распределяемого водорода уменьшают вручную таким образом, чтобы входное отношение водорода к ал кину в реакторе было даже ниже 1, что значительно ускоряет закоксовывание катализатора. Хотя нагрузка каждого реактора регулируется, это приводит к значительному сокращению цикла работы катализатора.
Как только количество кокса достигает 10% или более от массы самого катализатора гидрирования С2, производительность значительно ухудшается. Образование зеленого масла оказывает такое значительное влияние на производительность катализаторов концевого гидрирования С2, и все же димеризация при гидрировании неизбежна. Таким образом, сокращение образования зеленого масла и замедление закоксовывания является одной из неизменных задач в разработке катализаторов.
В US 5856262 описан способ получения низкокислотных палладиевых катализаторов с применением модифицированного гидроксидом калия (или гидроксидом бария, стронция, рубидия и т.д.) кремнезема в качестве носителя, с молярной долей ацетилена на выходе менее 1×10-7 и селективностью по этилену до 56% при скорости воздуха 3000 ч-1, температуре на входе 35°С, молярной доле ацетилена на входе 0,71% и молярном соотношении водорода к ацетилену 1,43.
В US 4404124 катализатор селективного гидрирования с распределением активных компонентов в оболочке получают способом ступенчатой пропитки. Его можно применять для селективного гидрирования фракции С2 для удаления ацетилена из этилена. В US 5587348 катализатор гидрирования С2 с превосходными характеристиками получают с применением оксида алюминия в качестве носителя, добавлением серебра в качестве сокатализатора для взаимодействия с палладием и добавлением фтора, который химически связан со щелочными металлами. Катализатор обладает характеристиками снижения образования зеленого масла, улучшения селективности по этилену и снижения количества образующихся окисленных соединений.
CN 1736589 A описывает катализатор селективного гидрирования Pd/γ-Al2O3, полученный способом полной адсорбционной пропитки, и катализатор демонстрирует более высокое образование зеленого масла при применении. CN 200810114744.0 раскрывает катализатор селективного гидрирования ненасыщенных углеводородов и способ его получения. В катализаторе в качестве носителя применяют оксид алюминия, и в качестве активного компонента - палладий, и улучшают стойкость катализатора к примеси и закоксовыванию за счет добавления редкоземельных и щелочноземельных металлов и фтора. Однако его каталитическая селективность не является удовлетворительной
Все катализаторы, полученные вышеуказанными способами имеют размеры пор с одиночным распределением. Во время реакции с неподвижным слоем селективность катализатора низкая из-за влияния внутренней диффузии. ZL 971187339 раскрывает катализатор гидрирования, в котором носитель представляет собой носитель сотового типа с большим размером пор и эффективно улучшает селективность катализатора. CN 1129606 A раскрывает катализатор конверсии углеводородов, в котором катализатор-носитель содержит оксид алюминия, оксид никеля, оксид железа и т.п. Катализатор содержит два типа пор, один для улучшения поверхности каталитической реакции, и другой для облегчения диффузии. CN 101433842 A раскрывает катализатор гидрирования, характеризующийся бимодальным распределением пор по размерам, в котором малая часть пор имеет наиболее заполняемый радиус 2-50 нм, и большая часть пор имеет наиболее заполняемый радиус 100-500 нм. Катализатор обладает хорошей гидрирующей активностью наряду с хорошей селективностью и высоким приращением этилена благодаря бимодальному распределению пор по размерам.
В реакции гидрирования С2 образование зеленого масла и закоксовывание катализатора являются важными факторами, влияющими на срок службы катализатора. Активность, селективность и срок службы катализатора вносят вклад в общую производительность катализатора. Вышеупомянутые способы, перечисленные выше, либо предлагают лучший способ улучшения активности и селективности катализатора, но не решают проблему склонности катализаторов к закоксовыванию; либо они решают проблему склонности к образованию зеленого масла и закоксовыванию катализаторов, но не решают проблему селективности. Хотя носитель с крупнопористой структурой может улучшить селективность, более крупные молекулы, образующиеся в результате реакции полимеризации и роста цепи, также имеют тенденцию накапливаться в больших порах носителя. Это приводит к закоксовыванию и деактивации катализатора, что влияет на срок службы катализатора.
CN 201310114070.5 раскрывает способ отбора фракции С2 с применением катализатора, в котором активные компоненты Pd и Ag обеспечивают пропиткой водным раствором, и Ni обеспечивают пропиткой микроэмульсией типа вода-в-масле. CN 201310114077.7 раскрывает катализатор гидрирования, в котором носитель катализатора имеет бимодальное распределение пор по размерам. Активными компонентами в этом катализаторе являются Pd, Ag и Ni, причем Pd и Ag расположены в мелких порах, и Ni расположен в больших порах. CN 201310114079.6 раскрывает способ получения катализатора, в котором носитель применяемого катализатора имеет бимодальное распределение пор по размерам. При получении микроэмульсии типа вода-в-масле с диаметром частиц, большим, чем диаметр мелких пор носителя, и содержащей металлическую соль никеля, поскольку кинетический объем микроэмульсии больше, чем размер мелких пор, частицы микроэмульсии могут проникать только в более крупные поры носителя. Pd и Ag обеспечивают с помощью способа в растворе, и поскольку мелкие поры имеют более сильный эффект сифонирования, большая часть Pd и Ag попадает в большие поры носителя, таким образом, получают Ni, главным образом, расположенный в больших порах, и Pd и Ag, главным образом, расположенные в мелких порах. Благодаря применению указанного способа, Pd/Ag и Ni расположены в каналах с различными размерами пор, и зеленое масло, образующееся в результате реакции, насыщается и гидрируется в больших порах, уменьшая количество закоксовывания катализатора. Кроме того, катализатор должен быть восстановлен перед подачей катализатора для работы. Как правило, температура восстановления катализаторов из драгоценных металлов низкая, но температура восстановления Ni часто достигает примерно 500°С, при которой атомы Pd в восстановленном состоянии очень склонны к агрегации, что приводит к значительному снижению активности катализатора на 30% или более. Это требует значительного увеличения эквивалентного количества активного компонента для компенсации потери активности, что в свою очередь вызывает снижение селективности.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является обеспечение катализатора для селективного гидрирования алкина и способа его получения, в частности, катализатора для селективного гидрирования алкина, обладающего лучшей устойчивостью к закоксовыванию и способного снижать температуру восстановления катализатора, и способа его получения.
Для достижения вышеуказанного в настоящем изобретении предложен катализатор для селективного гидрирования алкинов, где носитель катализатора представляет собой оксид алюминия или по существу оксид алюминия и имеет бимодальное распределение пор по размерам, где размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu; в расчете на 100% по массе носителя содержание Pd составляет 0,03-0,1%, содержание Ni составляет 0,5-5%, и массовое отношение Cu к Ni составляет 0,1-1,0: 1,0; где Ni, Cu и часть Pd получены из микроэмульсии, и диаметр частиц микроэмульсии не меньше максимального размера мелких пор и не больше максимального размера крупных пор.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно носитель катализатора, предложенного в настоящем изобретении, представляет собой оксид алюминия или по существу оксид алюминия и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu; в расчете на 100% по массе носителя содержание Pd составляет 0,035-0,08% (более предпочтительно 0,035-0,07%), содержание Ni составляет 0,5-5%, и массовое отношение Cu к Ni составляет 0,1-1,0: 1; где Ni, Cu и часть Pd получены из микроэмульсии, и диаметр частиц микроэмульсии не меньше максимального размера мелких пор и не больше максимального размера крупных пор (предпочтительно больше максимального размера мелких пор и меньше максимального размера крупных пор); количество Pd, полученного из микроэмульсии, составляет 1/100-1/200 от общего содержания Ni и Cu, и остальную часть Pd получают из раствора.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно носитель катализатора, предложенный в настоящем изобретении, представляет собой оксид алюминия и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu в качестве активных компонентов; в расчете на 100% по массе носителя содержание Pd составляет 0,03-0,09% (более предпочтительно 0,035-0,075%), содержание Ni составляет 0,5-5%, и массовое отношение Cu к Ni составляет 0,1-1,0: 1,0; при этом Ni, Cu и часть Pd получены из микроэмульсии. Кроме того, предпочтительно количество Pd, полученного из микроэмульсии, составляет 1/100-1/200 от общего содержания Ni+Cu, и остальную часть Pd получают из раствора. Катализатор предпочтительно подходит для реакции гидрирования фракции С2.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно носитель катализатора, предложенного в настоящем изобретении, представляет собой оксид алюминия или по существу оксид алюминия и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu в качестве активных компонентов; в расчете на 100% по массе носителя содержание Pd составляет 0,06-0,1% (более предпочтительно 0,07-0,1%), содержание Ni составляет 0,5-5%, и массовое отношение Cu к Ni составляет 0,1-1,0:1,0. Кроме того, предпочтительно количество Pd, полученного из микроэмульсии, составляет 1/100-1/200 от общего содержания Ni+Cu, и остальную часть Pd получают из раствора. Катализатор предпочтительно подходит для способа селективного гидрирования в способе концевого гидрирования С2 с применением неочищенного водорода в качестве источника водорода.
В соответствии с конкретным вариантом реализации настоящего изобретения для конкретного носителя катализатора размер мелких пор и размер крупных пор представляют собой диапазон размеров, соответственно. Диаметр частиц микроэмульсии не ниже (больше) максимального размера мелких пор и не выше (меньше) максимального размера крупных пор, что означает, что диаметр частиц микроэмульсии, полученной во время нанесения, не меньше (больше) верхнего предела диапазона размеров мелких пор и не больше (меньше) верхнего предела диапазона размеров крупных пор конкретного носителя катализатора. Согласно конкретному варианту реализации настоящего изобретения диаметр частиц микроэмульсии может составлять 50-500 нм или более 50 нм и менее 500 нм.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно носитель катализатора, предложенного в настоящем изобретении, представляет собой оксид алюминия или по существу оксид алюминия и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu; в расчете на 100% по массе носителя содержание Pd составляет 0,06-0,08%, содержание Ni составляет 0,5-5%, и массовое отношение Cu к Ni составляет 0,1-1,0:1,0; при этом Ni, Cu и часть Pd получены из микроэмульсии; диаметр частиц микроэмульсии не меньше, чем максимальный размер мелких пор, и не больше, чем максимальный размер крупных пор; количество Pd, полученного из микроэмульсии, составляет 1/100-1/200 от общего содержания Ni+Cu, и остальную часть Pd получают из раствора. Катализатор подходит как для реакции гидрирования фракции С2, так и для способа селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно катализатор согласно настоящему изобретению дополнительно содержит Ag, получаемый из раствора, в количестве 0,03-0,5%, более предпочтительно в количестве 0,08-0,21%. Ag действует с образованием сплава с Pd и уменьшает количество образующегося зеленого масла для улучшения селективности гидрирования ацетилена.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно носитель катализатора, предложенного в настоящем изобретении, представляет собой оксид алюминия или по существу оксид алюминия и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ag, Ni и Cu; в расчете на 100% по массе носителя содержание Pd составляет 0,035-0,07%, содержание Ag составляет 0,08-0,21%, содержание Ni составляет 0,5-5%, и массовое отношение Cu к Ni составляет 0,1-1,0:1,0; при этом Ni, Cu и часть Pd получены из микроэмульсиии в основном распределены в крупных порах носителя. Количество Pd, полученного из микроэмульсии, составляет 1/100-1/200 от общего содержания Ni+Cu, и Ag и остальную часть Pd получают способом раствора.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно носитель катализатора, предложенного в настоящем изобретении, представляет собой оксид алюминия или по существу оксид алюминия и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ag, Ni и Cu; из расчета на 100% по массе носителя содержание Pd составляет 0,07-0,1%, содержание Ag составляет 0,03-0,5%, содержание Ni составляет 0,5-5% и содержание Cu составляет 0,5-5%, и общее содержание Ni+Cu составляет 1-5,5%. Катализатор также предпочтительно подходит для способа селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, катализатор согласно настоящему изобретению имеет удельную площадь поверхности 20-50 м2/г.
Обеспечение способом микроэмульсии, описанное в настоящем изобретении, означает обеспечение способом пропитки, в котором пропиточный раствор представляет собой микроэмульсию; то есть предшественники активных компонентов (например, Pd, Ni, Cu и т.д.) обеспечивают путем создания микроэмульсии для пропитки вместо создания раствора для пропитки. Обеспечение способом раствора, описанное в настоящем изобретении, означает обеспечение способом пропитки, в котором пропиточный раствор представляет собой раствор. В катализаторе согласно настоящему изобретению часть Pd получают способом микроэмульсии, в то время как остальную часть Pd получают способом пропитки. Кроме того, количество Pd, полученного из микроэмульсии, составляет 1/100-1/200, более предпочтительно 1/110-1/200 от общего содержания Ni+Cu.
Согласно конкретному варианту реализации согласно настоящему изобретению предпочтительно носитель представляет собой оксид алюминия или по существу оксид алюминия, и оксид алюминия находится в кристаллической форме θ, кристаллической форме α или их смешанной форме. Когда носитель катализатора представляет собой по существу оксид алюминия, оксид алюминия в носителе составляет 80% или более. Носитель с бимодальным распределением пор по размерам, применяемый в настоящем изобретении, обеспечивает высокую активность катализатора, в то время как наличие больших пор может снизить эффект внутренней диффузии, тем самым улучшая селективность катализатора.
В соответствии с конкретным вариантом реализации настоящего изобретения, предпочтительно, обеспечение способом микроэмульсии включает растворение соли-предшественника в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества и тщательное перемешивание с образованием микроэмульсии, при этом масляная фаза представляет собой алкан или циклоалкан, поверхностно-активное вещество представляет собой ионное поверхностно-активное вещество и/или неионогенное поверхностно-активное вещество, и дополнительное поверхностно-активное вещество представляет собой органический спирт.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно обеспечение способом микроэмульсии удовлетворяет следующим требованиям: массовое отношение водной фазы к масляной фазе составляет 2-3, массовое отношение поверхностно-активного вещества к масляной фазе составляет 0,15-0,6, и массовое отношение поверхностно-активного вещества к дополнительному поверхностно-активному веществу составляет 1-1,2, и диаметр частиц микроэмульсии составляет более 50 нм и менее 500 нм.
В соответствии с конкретным вариантом реализации настоящего изобретения, предпочтительно, в обеспечении способом микроэмульсии, масляная фаза представляет собой С6-С8 насыщенный алкан или циклоалкан, предпочтительно циклогексан или н-гексан; поверхностно-активное вещество представляет собой ионное поверхностно-активное вещество и/или неионогенное поверхностно-активное вещество, предпочтительно неионогенное поверхностно-активное вещество, более предпочтительно полиэтиленгликоль-октилфениловый эфир или бромид цетилтриметиламмония; и дополнительное поверхностно-активное вещество представляет собой С4-С6 спирт, предпочтительно н-бутанол и/или н-пентиловый спирт.
В соответствии с конкретным вариантом реализации настоящего изобретения, предпочтительно, во время получения катализатора порядок обеспечения раствора Pd, и обеспечения микроэмульсии Ni и Cu, не ограничен; предпочтительно стадию обеспечения микроэмульсии Pd проводят после стадии обеспечения микроэмульсии Ni и Cu. Предпочтительно стадию обеспечения раствора Ag осуществляют после стадии обеспечения раствора Pd.
В катализаторе согласно настоящему изобретению селективное гидрирование ацетилена происходит в основных активных центрах, состоящих из Pd, и Ni и Cu, обеспечиваемых в виде микроэмульсии в больших порах носителя, причем зеленое масло, образующееся в реакции, подвергается недеструктивному гидрированию в активных центрах, состоящих из Cu и Ni.
Для реакции гидрирования обычно необходимо сначала провести восстановление катализатора гидрирования перед применением катализатора для обеспечения присутствия активных компонентов в металлическом состоянии, чтобы катализатор обладал активностью гидрирования. Это связано с тем, что активация представляет собой высокотемпературный способ прокаливания во время получения катализатора, и в этом способе соли металлов обычно разлагаются на оксиды металлов, которые образуют кластеры, обычно нанометрового размера. Различные оксиды необходимо восстанавливать при разных температурах из-за их различных химических свойств. Однако для наноразмерных металлов температура примерно 200°С является важной критической температурой, и металлические частицы агломерируются значительно выше такой температуры. Следовательно, снижение температуры восстановления активных компонентов до 200°С или менее имеет большое значение для катализатора гидрирования.
Решения для закоксовывания катализатора согласно настоящему изобретению рассматриваются следующим образом.
Реакция селективного гидрирования ацетилена происходит в основных активных центрах, состоящих из Pd (или основных активных центрах, состоящих из Pd и Ag, когда Ag присутствует), и макромолекулы, такие как зеленое масло, образующиеся в реакции, имеют тенденцию проникать в большие поры катализатора. В больших порах катализатора обеспечивают компоненты Ni/Cu, где Ni имеет функцию недеструктивного гидрирования, и компонент зеленого масла может подвергаться реакции недеструктивного гидрирования в активных центрах, состоящих из Ni/Cu. Поскольку двойные связи насыщаются при гидрировании, компонент зеленого масла больше не может подвергаться полимеризации или скорость реакции полимеризации значительно снижается. Цепная реакция роста прекращается или замедляется, и не могут образовываться высокомолекулярные густые кольцевые соединения и компонент зеленого масла можно легко выводить из реактора с основным материалом. В результате степень закоксовывания на поверхности катализатора значительно снижается, и срок эксплуатации катализатора значительно продлевается.
Носитель согласно настоящему изобретению должен иметь бимодальную структуру распределения пор по размерам, в частности, иметь размер больших пор 80-500 нм и размер малых пор 15-50 нм.
Локализацию сплава Ni/Cu в больших порах катализатора контролируют способом согласно настоящему изобретению, в котором Ni/Cu загружают в виде микроэмульсии, имеющей диаметр частиц, больший, чем размер малых пор носителя, и меньший, чем максимальный размер больших пор. Соли металлов Ni и Cu содержатся в микроэмульсии, и им трудно проникать в поры носителя с меньшими размерами из-за стерического затруднения и в основном они попадают в большие поры носителя. Следовательно, в больших порах и малых порах катализатора образуются активные центры с различными функциями гидрирования. Большие поры включают активные центры, состоящие из Ni/Cu и Pd, которые имеют хорошую функцию недеструктивного гидрирования для молекул зеленого масла, что позволяет молекулам зеленого масла, входящим в большие поры, больше не полимеризоваться и, следовательно, постепенно выходить из реактора, и вероятность закоксовывания снижается.
Для реакции гидрирования обычно необходимо сначала провести восстановление катализатора гидрирования перед применением катализатора для обеспечения присутствия активных компонентов в металлическом состоянии, чтобы катализатор обладал активностью гидрирования. Это связано с тем, что активация представляет собой высокотемпературный способ прокаливания во время получения катализатора, и в этом способе соли металлов обычно разлагаются на оксиды металлов, которые образуют кластеры, обычно нанометрового размера. Различные оксиды необходимо восстанавливать при разных температурах из-за их различных химических свойств. Однако для наноразмерных металлов температура примерно 200°С является важной критической температурой, и металлические частицы агломерируются значительно выше такой температуры. Следовательно, снижение температуры восстановления активных компонентов имеет большое значение для катализатора гидрирования.
Авторы настоящего изобретения обнаружили, что температура восстановления Ni может быть резко снижена после обеспечения Cu вместе с Ni. Это связано с тем, что температура восстановления NiO обычно достигает 450°С или более, при этой температуре Pd имеет тенденцию к агломерации, тогда как после добавления Cu образуется сплав Cu/Ni, н его температура восстановления может быть снижена на 100°С или более до 350°С по сравнению с температурой чистого Ni, тем самым ослабляя агломерацию Pd.
Авторы настоящего изобретения также обнаружили, что если часть Pd дополнительно обеспечивают на поверхности сплава Ni/Cu, температура восстановления сплава Ni/Cu может быть еще более значительно снижена до температуры 200°С или ниже или даже до 150°С, что позволяет избежать агломерации активных центров Pd во время восстановления NiO при высокой температуре.
Таким образом, оптимальный катализатор имеет Pd, в основном присутствующий в небольших порах катализатора, и Ni/Cu, расположенный в больших порах катализатора, причем часть Pd также присутствует в больших порах вместе с Ni/Cu, в частности, на его поверхности.
В оптимальном способе получения катализатора после нанесения Ni и Cu небольшое количество Pd обеспечивают в больших порах с помощью способа микроэмульсии. Количество Pd, обеспечиваемое таким образом, составляет 1/100-1/200 от общего содержания Ni+Cu.
В настоящем изобретении также предложен способ получения вышеуказанного катализатора, включающий следующие стадии:
(1) растворение солей-предшественников Ni и Cu в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества, тщательное перемешивание с получением микроэмульсии и пропитку носителя полученной микроэмульсией в течение 0,5-4 часов, затем удаление остаточной жидкости фильтрованием и сушку и прокаливание при 400-600°С в течение 4 часов или более с получением полуфабриката катализатора А;
(2) растворение соли-предшественника Pd в воде, доведение рН до 1,5-2,5 и добавление полуфабриката катализатора А к раствору соли Pd для обеспечения пропитки и адсорбции в течение 0,5-4 часов, и затем сушку и прокаливание при 400-600°С в течение 4-6 часов с получением полуфабриката катализатора В;
(3) растворение соли-предшественника Pd в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества, тщательное перемешивание с получением микроэмульсии и добавление полуфабриката катализатора В к полученной микроэмульсии с обеспечением пропитки в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и сушку и прокаливание при 400-600°С в течение 4 ч или более с получением требуемого катализатора.
На вышеуказанных стадиях получения стадия (1) и стадия (2) являются взаимозаменяемыми, и стадия (3) следует за стадией (1).
Носитель на вышеуказанной стадии (1) представляет собой оксид алюминия или по существу оксид алюминия, и кристаллическая форма Al2O3 предпочтительно представляет собой θ, α или их смешанную форму. Носитель катализатора предпочтительно содержит оксид алюминия 80% или более, и носитель может также содержать другие оксиды металлов, такие как оксид магния, оксид титана и т.п.
Носитель на стадии (1) выше может иметь сферическую, цилиндрическую, клеверную форму, форму четырехлистного клевера и тому подобное.
Соли-предшественники Ni, Cu и Pd, описанные на стадиях (1) и (3) выше, представляют собой растворимые соли и могут представлять собой нитратные, хлоридные соли или другие их растворимые соли.
В соответствии с конкретным вариантом реализации настоящего изобретения предпочтительно массовое отношение Cu к Ni составляет 0,1-1:1, и количество Pd, обеспечиваемое эмульсионным способом, составляет 1/100-1/200 от содержания Ni+Cu.
В соответствии с конкретным вариантом реализации настоящего изобретения, когда катализатор дополнительно содержит Ag, Ag может образовывать сплав с Pd, обеспечиваемым на стадии (2), для улучшения селективности гидрирования ацетилена. Принцип заключается в том, что Ag образует сплав с Pd, и атомы Ag разделяют атомы Pd таким образом, что пространственное расстояние между адсорбированными молекулами ацетилена увеличивается, и, следовательно, промежуточные продукты реакции после гидрирования ацетилена имеют большее расстояние; взаимодействие промежуточных продуктов происходит с меньшей вероятностью, что уменьшает образование зеленого масла.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, когда катализатор содержит Ag, способ получения катализатора согласно настоящему изобретению может включать следующие способы I-III.
I. Способ получения катализатора включает следующие стадии:
(1) растворение солей-предшественников Ni и Cu в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества, тщательное перемешивание с получением микроэмульсии и пропитку носителя полученной микроэмульсией в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и сушку и прокаливание при 400-600°С в течение 4-6 ч с получением полуфабриката катализатора А;
(2) растворение соли-предшественника Pd в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества, тщательное перемешивание с получением микроэмульсии и добавление полуфабриката катализатора А к полученной микроэмульсии с обеспечением пропитки в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и сушку и прокаливание при 400-600°С в течение 4-6 ч с получением полуфабриката катализатора В;
(3) растворение соли-предшественника Pd в воде, доведение рН до 1,5-2,5 и добавление полуфабриката катализатора В к раствору соли Pd для обеспечения пропитки и адсорбции в течение 0,5-4 часов, и затем сушку и прокаливание при 400-600°С в течение 4-6 часов с получением полуфабриката катализатора С;
(4) растворение соли Ag в деионизированной воде, доведение рН до 1-5 и пропитку полуфабриката катализатора С полученным раствором до полного поглощения раствора, и затем сушку и прокаливание при 400-600°С в течение 4-6 часов с получением катализатора.
II. Способ получения катализатора включает следующие стадии:
(1) растворение солей-предшественников Ni и Cu в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества, тщательное перемешивание с получением микроэмульсии и пропитку носителя полученной микроэмульсией в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и сушку и прокаливание при 400-600°С в течение 4-6 ч с получением полуфабриката катализатора А;
(2) растворение соли-предшественника Pd в воде, доведение рН до 1,5-2,5 и добавление полуфабриката катализатора А к раствору соли Pd для обеспечения пропитки и адсорбции в течение 0,5-4 часов, и затем сушку и прокаливание при 400-600°С в течение 4-6 часов с получением полуфабриката катализатора В;
(3) обеспечение Ag путем насыщенной пропитки путем приготовления раствора соли Ag, который соответствует 80-110% поглощающей способности насыщения водой носителя, и доведения рН до 1-5 для обеспечения пропитки, и затем сушки и прокаливания при 400-600°С в течение 4-6 ч с получением полуфабриката катализатора С;
(4) растворение соли-предшественника Pd в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества, тщательное перемешивание с образованием микроэмульсии, добавление полуфабриката катализатора С к полученной микроэмульсии для обеспечения пропитки в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и сушку и прокаливание при 400-600°С в течение 4-6 ч с получением катализатора.
III. Способ получения катализатора включает следующие стадии:
(1) растворение солей-предшественников Ni и Cu в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества, тщательное перемешивание с получением микроэмульсии, пропитку носителя полученной микроэмульсией в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и сушку при 80-120°С в течение 1-6 ч и прокаливание при 300-600°С в течение 2-8 ч с получением полуфабриката катализатора А;
(2) обеспечение Ag путем насыщенной пропитки путем приготовления раствора соли Ag, который соответствует 80-110% поглощающей способности насыщения водой носителя, и доведения рН до 1-5 для обеспечения пропитки полуфабриката катализатора А, и затем сушки и прокаливания при 500-550°С в течение 4-6 ч с получением полуфабриката катализатора В;
(3) растворение соли-предшественника Pd в воде, доведение рН до 1,5-2,5 и добавление полуфабриката катализатора В к раствору соли Pd для обеспечения пропитки и адсорбции в течение 0,5-4 часов, с последующей сушкой в течение 1-4 часов и прокаливанием при 400-550°С в течение 2-6 часов с получением полуфабриката катализатора С;
(4) растворение соли-предшественника Pd в воде, добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества, тщательное перемешивание с получением микроэмульсии и добавление полуфабриката катализатора С к полученной микроэмульсии с обеспечением пропитки в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и сушку в течение 1-6 ч и прокаливание при 300-600°С в течение 2-8 ч с получением полуфабриката катализатора D;
(5) помещение полуфабриката катализатора D в реакционное устройство с неподвижным слоем для восстановления при 150-200°С в течение 4-8 часов с применением газовой смеси N2:H2=1:1 для получения готового катализатора Е.
Поверхностно-активное вещество на вышеуказанных стадиях (1) и (4) представляет собой ионное поверхностно-активное вещество или неионогенное поверхностно-активное вещество, предпочтительно неионогенное поверхностно-активное вещество, более предпочтительно октилфениловый эфир полиэтиленгликоля (Triton Х-100) или бромид цетилтриметиламмония (ЦТАБ).
Масляная фаза на вышеуказанных стадиях (1) и (4) представляет собой С6-С8 насыщенный алкан или циклоалкан, предпочтительно циклогексан или н-гексан.
Дополнительное поверхностно-активное вещество на вышеуказанных стадиях (1) и (4) представляет собой С4-С6 спирт, предпочтительно н-бутанол или н-пентанол.
Температура восстановления для катализатора согласно настоящему изобретению предпочтительно составляет 150-200°С.
Согласно конкретному варианту реализации настоящего изобретения во время получения катализатора предпочтительно стадию микроэмульсионного обеспечения Pd проводят после стадии микроэмульсионного обеспечения Ni и Cu.
Согласно конкретному варианту реализации настоящего изобретения во время получения катализатора предпочтительно порядок растворного обеспечения Pd и микроэмульсионного обеспечения Ni/Cu не ограничен.
Согласно конкретному варианту реализации настоящего изобретения во время получения катализатора, предпочтительно, стадию растворного обеспечения Ag проводят после стадии растворного обеспечения Pd.
Для предотвращения попадания Ni и Cu в мелкие поры и покрытия обеспечиваемого Pd предпочтительно растворное обеспечение Pd осуществляют после микроэмульсионного обеспечения Ni+Cu.
В настоящем изобретении предпочтительно способ микроэмульсионного обеспечения удовлетворяет следующим условиям: массовое отношение водной фазы к масляной фазе составляет 2-3, массовое отношение поверхностно-активного вещества к масляной фазе составляет 0,15-0,6, массовое отношение поверхностно-активного вещества к дополнительному поверхностно-активному веществу составляет 1-1,2, и диаметр частиц образовавшейся микроэмульсии составляет более 50 нм и менее 500 нм.
В настоящем изобретении также предложен способ селективного гидрирования алкинов во фракции С2, при этом сырье для гидрирования представляет собой фракцию С2 из верхней части предварительного деэтанизатора, подается в реактор с неподвижным слоем и подвергается гидрированию в газовой фазе для удаления ацетилена, при этом технологические условия для реакции селективного гидрирования представляют собой: температуру на входе в реактор 35-100°С, давление 1,5-3,0 МПа и объемную скорость газа 2000-11000 ч-1; и при этом катализатор представляет собой катализатор, предложенный в настоящем изобретении.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в катализаторе, применяемом в способе селективного гидрирования алкина во фракции С2, в качестве носителя применяют оксид алюминия, который имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu; из расчета 100% по массе носителя содержание Pd составляет 0,03-0,09%, содержание Ni составляет 0,5-5%, и массовое отношение Cu к Ni составляет 0,1-1,0:1,0; Ni, Cu и часть Pd обеспечивают микроэмульсией, и количество этой части Pd составляет 1/100-1/200 от общего содержания Ni+Cu; и диаметр частиц микроэмульсии составляет 50-500 нм. Предпочтительно катализатор дополнительно содержит 0,08-0,21% Ag, который обеспечивают раствором. Содержание Pd предпочтительно составляет 0,035-0,075%.
В предпочтительном варианте реализации катализатора согласно настоящему изобретению, обеспечение Pd осуществляют с помощью растворного способа с двумя способами обеспечения, при этом Pd, обеспечиваемый в каждом из двух способов обеспечения, распределен в разных областях. Более предпочтительно, обеспечение Pd осуществляют с помощью растворного способа с двумя способами обеспечения, где два способа обеспечения осуществляют с анионным предшественником и катионным предшественником, соответственно. Обеспечение анионным предшественником может легко образовывать оболочечное распределение Pd, которое в основном применяют для селективного гидрирования ацетилена. С катионным предшественником Pd может быть распределен по более широкой области и образовывать сплав с Ni/Cu, тем самым снижая температуру восстановления Ni/Cu, возможно, до 200°С или ниже.
В более предпочтительном варианте реализации настоящего изобретения обеспечение Pd осуществляют с помощью двух способов пропитки, один при помощи раствора, и другой при помощи микроэмульсии; Ni и Cu наносят с помощью микроэмульсионного способа; диаметр частиц микроэмульсии регулируют таким образом, чтобы он находился между максимальным размером мелких пор и максимальным размером крупных пор в катализаторе, так что Ni и Cu в основном распределены в крупных порах носителя. Количество Pd, обеспечиваемое в способе микроэмульсии, составляет 1/100-1/200 от общего содержания Ni и Cu, и микроэмульсионное обеспечение Pd осуществляют после микроэмульсионного обеспечения Ni и Cu. Пропитку большей части Pd осуществляют раствором. При обеспечении Pd раствором раствор, содержащий Pd, быстрее поступает в поры за счет сифонирующего эффекта мелких пор. Pd присутствует в форме хлорпалладат-ионов, и поскольку такие ионы могут образовывать химическую связь с гидроксильной группой на поверхности носителя, палладий быстро закрепляется, чем быстрее раствор поступает в поровые каналы, тем быстрее происходит обеспечение. Следовательно, в способе пропитки Pd пересыщенный пропиткой Pd легче загружается в мелкие поры.
В более предпочтительном варианте реализации настоящего изобретения активный компонент палладий сначала загружают в мелкие поры, и затем активные компоненты никель/медь вместе с малым количеством палладия загружают в большие поры катализатора; активный компонент палладий загружают в мелкие поры. Ацетилен или тому подобное подвергается селективной реакции гидрирования, главным образом в мелких порах, с получением этилена. Побочные продукты с большими молекулярными размерами, образующиеся в реакции, в основном фракции С4-С14, с большей вероятностью попадают в крупные поры и подвергаются реакции недеструктивного гидрирования в присутствии активных компонентов, таких как никель, в крупных порах. Поскольку эти молекулы насыщаются при гидрировании, их молекулярные цепи больше не растут и, таким образом, легко выводятся из реактора вместе с основными материалами.
Автором настоящего изобретения было обнаружено, что, когда Ni и Cu пропитывают одновременно, они образуют сплав, и из-за присутствия Cu температура восстановления Ni резко снижается до минимума 350°С, хотя эта температура все еще слишком высока для катализатора Pd. Автором настоящего изобретения также было обнаружено, что после нанесения небольшого количества Pd на катализатор Ni/Cu температура восстановления катализатора Ni/Cu значительно снижается, возможно, до 150°С, что является вполне приемлемой температурой для катализатора Pd, поскольку катализатор Pd, как правило, имеет температуру восстановления 100-150°С, и такой катализатор может работать при 120-130°С в течение длительного периода времени в некоторых случаях, что указывает на то, что агрегация активных компонентов может не происходить при 100-150°С.
Условия способа гидрирования согласно настоящему изобретению представляют собой: реактор с неподвижным слоем, который представляет собой адиабатический или изотермический реактор, с условиями реакции: температура на входе в реактор 35-100°С, реакционное давление 1,5-3,0 МПа и объемная скорость газа 2000-11000 ч-1.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в вышеуказанном способе селективного гидрирования алкинов во фракции С2 сырье для гидрирования представляет собой фракцию С2 с 65-93% (об./об.) этилена, 0,2-2,5% (об./об.) ацетилена и 0,01-0,8% (об./об.) фракции С3; более предпочтительно, 65-93% (об./об.) этилена, 5-34,69% (об./об.) этана, 0,3-2,5% (об./об.) ацетилена и 0,01-0,5% (об./об.) фракции С3. Указанное содержание рассчитывают из расчета 100% от общего объема сырья для гидрирования.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в вышеуказанном способе селективного гидрирования алкинов во фракции С2, когда для гидрирования применяют одноступенчатый реактор с неподвижным слоем, отношение водорода к алкину на входе в реактор составляет 1,3-2,2, предпочтительно 1,3-1,8. В настоящем изобретении отношение водорода к алкину относится к молярному отношению водорода к алкину.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в вышеуказанном способе селективного гидрирования алкинов во фракции С2, когда для гидрирования используют двухступенчатый реактор с неподвижным слоем, отношение водорода к алкину на входе в реактор первой ступени составляет 1,0-1,4, и отношение водорода к алкину на входе в реактор второй ступени составляет 1,5-2,5.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в вышеуказанном способе селективного гидрирования алкинов во фракции С2, когда для гидрирования применяют трехступенчатый реактор с неподвижным слоем, отношение водорода к алкину на входе в реактор первой ступени составляет 0,5-1,5, отношение водорода к алкину на входе в реактор второй ступени составляет 1,0-2,0, и отношение водорода к алкину на входе в реактор третьей ступени составляет 1,4-3,0; более предпочтительно отношение водорода к алкину на входе в реактор первой ступени составляет 0,8-1,5, отношение водорода к алкину на входе в реактор второй ступени составляет 1,2-1,6, и отношение водорода к алкину на входе в реактор третьей ступени составляет 1,5-2,5.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно в вышеуказанном способе селективного гидрирования алкинов во фракции С2 катализатор необходимо восстанавливать при температуре восстановления 150-200°С перед гидрированием.
Настоящее изобретение также относится к способу селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода, где материал для гидрирования представляет собой фракцию С2 из верхней части деэтанизатора, подается в реактор с неподвижным слоем и подвергается гидрированию в газовой фазе для удаления ацетилена; где водород, используемый в реакции гидрирования, представляет собой неочищенный водород с содержанием Н2 20-50 об.% и содержанием СО 0,1-1 об.%; где условия способа: температура на входе в реактор 55-130°С, давление 1,5-3,0 МПа и объемная скорость газа 1500-6000 ч-1; и где катализатор представляет собой катализатор по любому из пп. 1-19.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в способе селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода, гидрируемый материал представляет собой фракцию С2 из верхней части деэтанизатора и подается в реактор с неподвижным слоем и подвергается гидрированию в газовой фазе для удаления ацетилена; где водород, используемый в реакции гидрирования, представляет собой неочищенный водород с содержанием Н2 30-50 об.%, содержанием СО 0,1-1 об.% и остальное составляет метан; в следующих условиях способа: температура на входе в реактор 55-130°С, давление 1,5-3,0 МПа и объемная скорость газа 1500-4000 ч-1; и где катализатор представляет собой вышеуказанный катализатор, предложенный в настоящем изобретении.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно носитель, используемый в способе селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода, представляет собой оксид алюминия или по существу оксид алюминия и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм, и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu; в расчете на 100% по массе носителя содержание Pd составляет 0,06-0,1%, содержание Ni составляет 0,5-5%, и массовое отношение Cu к Ni составляет 0,1-1:1. Предпочтительно, катализатор имеет удельную площадь поверхности 20-50 м2/г. Предпочтительно катализатор также содержит 0,03-0,5% Ag, который обеспечивают раствором. В катализаторе содержание Pd предпочтительно составляет 0,07-0,1%, и общее содержание Ni и Cu предпочтительно составляет 1-5,5%.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в способе селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода, гидрируемый материал представляет собой материал из верхней части колонны деэтанизации в установке по производству этилена, который подают в реактор гидрирования С2 с неподвижным слоем вместе с дозированным неочищенным водородом для селективного гидрирования для удаления содержащегося в нем алкина.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в способе селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода, реактор представляет собой одноступенчатый реактор с неподвижным слоем. Отношение водорода к алкину в материале на входе в реактор можно регулировать так, чтобы оно составляло 2-3 (молярное отношение); температуру на входе в реактор можно регулировать так, чтобы она составляла 65-130°С (предпочтительно 65-125°С); и условия реакции можно регулировать при реакционном давлении 1,5-3,0 МПа и объемной скорости газа 1500-6000 ч-1.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в способе селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода, неочищенный водород имеет содержание СО 0,1-1 об./об.% и содержание водорода 20-50 об./об.%. Это содержание рассчитывают на основе 100% от общего объема неочищенного водорода.
Согласно конкретному варианту реализации настоящего изобретения, предпочтительно, в способе селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода, материал для каталитической реакции, который подается в одноступенчатый реактор с неподвижным слоем, содержит 60-93 об./об.% этилена, 0,2-1,0 об./об.% ацетилена и 1,0-7,0 об./об.% фракции С3. Указанное содержание рассчитывают на основе 100% от общего объема гидрируемого материала.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно в способе селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода катализатор необходимо восстанавливать при температуре восстановления 150-200°С перед гидрированием.
Согласно конкретному варианту реализации настоящего изобретения предпочтительно в способе селективного гидрирования фракции С2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода отношение водорода к алкину в материале на входе в одноступенчатый реактор составляет 2-3 (молярное отношение).
В способе постгидрирования фракции С2 с применением неочищенного водорода в качестве источника водорода содержание фракции С3 в общем материале может составлять до 7,0%. Из-за заметно более высокой устойчивости катализатора согласно настоящему изобретению к закоксовыванию это намного выше, чем содержание фракции С3, ограниченное обычными способами гидрирования, что позволяет блоку гидрирования работать нормально, даже когда есть большие колебания в блоке разделения и содержание тяжелой фракции в общем материале превышает предел.
Катализатор согласно настоящему изобретению имеет следующие особенности: в начале реакции гидрирования реакция селективного гидрирования ацетилена происходит главным образом в малых порах из-за высокой гидрирующей активности палладия и его распределения главным образом в малых порах. По мере увеличения времени работы катализатора на поверхности катализатора образуется часть побочных продуктов с большей молекулярной массой. Эти вещества из-за их большего молекулярного размера в основном попадают в большие поры и имеют более длительное время пребывания и подвергаются реакции гидрирования двойной связи в присутствии никелевого катализатора, в результате чего образуются насыщенные углеводороды или ароматические углеводороды, которые не содержат изолированную двойную связь, без получения веществ с еще большей молекулярной массой.
Автор настоящего изобретения обнаружил, что катализатор, полученный с применением этого способа, имеет значительно более высокую начальную активность и селективность по сравнению с катализатором с бимодальным распределением пор по размерам, который не содержит палладия.
Автор также обнаружил, что при применении этого катализатора не наблюдается тенденции к снижению активности и селективности катализатора, даже если реагенты включают более тяжелые фракции и образование зеленого масла на катализаторе значительно увеличивается.
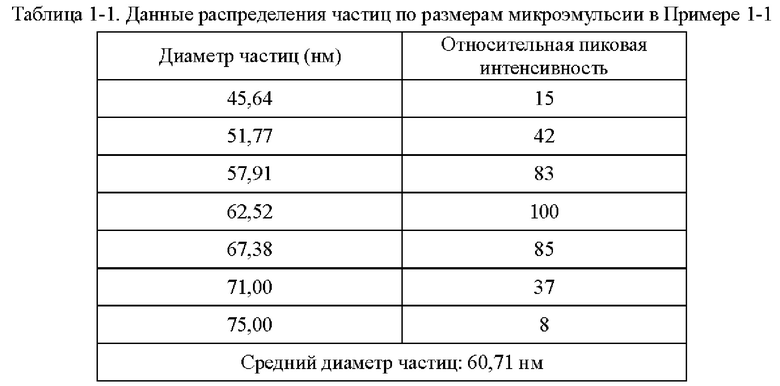
Краткое описание чертежей
На фиг. 1 показано распределение частиц по размеру микроэмульсии Ni и Cu, полученной в Примере 1-1.
На фиг. 2 показана диаграмма TPR катализатора согласно примеру 1-1.
Подробное описание изобретения
Для получения катализаторов согласно настоящему изобретению применяют следующие средства определения характеристик:
Анализатор динамического рассеяния света: анализатор динамического рассеяния света М286572 для анализа гранулометрического состава микроэмульсии сплава Ni/Cu;
Автоматический ртутный пьезометр: ртутный пьезометр типа 9510 (Mack, США), для анализа объема пор, удельной площади поверхности и распределения пор по размерам;
AA240FS атомно-абсорбционный спектрометр, для измерения содержания Pd, Ag, Ni и Cu в катализаторе;
Газовый хроматограф Agilent 7890А для измерения содержания водорода и ацетилена на выходе и входе реактора;
Электронные весы (0,1 мг), для измерения массы катализатора;
Первоначальная конверсия ацетилена: конверсия ацетилена, измеренная через 24 часа после начала подачи в реактор;
Конверсия ацетилена=[(содержание ацетилена на входе в реактор - содержание ацетилена на выходе из реактора) / содержание ацетилена на выходе из реактора]*100%;
Селективность по этилену={2-[(содержание водорода на входе в реактор - содержание водорода на выходе из реактора) / (содержание ацетилена на входе в реактор - содержание ацетилена на выходе из реактора)]}*100%;
Количество закоксовывания=[(масса катализатора после реакции - масса катализатора до реакции) / масса катализатора в реакторе]*100%.
Пример 1-1
Носитель катализатора: Использовали коммерчески доступный сферический носитель оксид алюминия с бимодальным распределением пор по размерам, диаметром 4 мм. После прокаливания при 1092°С в течение 4 ч бимодальное распределение пор по размерам находилось в диапазоне 15-38 нм и 80-350 нм, водопоглощение составляло 65%, и удельная площадь поверхности составляла 49,65 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 15,51 г безводного нитрата никеля и 1,05 г хлорида меди взвешивали и растворяли в 60 мл деионизированной воды, к которой добавляли 28,57 г циклогексана, 16,57 г Triton Х-100 и 16 г н-бутанола и тщательно перемешивали с получением микроэмульсии (данные гранулометрического состава микроэмульсии Ni и Cu приведены в таблице 1-1 и на фиг. 1). Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 30 мин, затем отфильтровывали остаточную жидкость, промывали деионизированной водой, сушили при 40°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А1.
(2) 0,0651 г нитрата палладия растворяли в 110 мл деионизированной воды и значение рН доводили до 1,5. Затем полуфабрикат катализатора А1 пропитывали в полученном солевом растворе Pd в течение 30 мин, сушили при 80°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В1.
(3) 0,046 г хлорида палладия растворяли в 60 мл деионизированной воды, к которой добавляли 28,57 г циклогексана, 16,57 г Triton Х-100 и 16 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В1, взвешенный в количестве 100 г, помещали в приготовленную микроэмульсию и встряхивали в течение 30 мин, затем отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 60,71 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 60,18 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и было определено, что в катализаторе, полученном в примере 1-1, содержание Pd составляло 0,0575%, содержание Ni составляло 5%, и содержание Cu составляло 0,5%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:Н2=1:1.
TPR этого катализатора показан на фиг. 2. Как показано на фиг. 2, температура восстановления сплава Ni/Cu составляет 350°С. Температура восстановления Ni/Cu при добавлении Pd составляет примерно 150°С.
Сравнительный Пример 1-1
Катализатор был приготовлен с применением того же носителя и условий, что и в примере 1-1, за исключением того, что Cu не обеспечивали.
Приготовление катализатора:
(1) 15,61 г безводного нитрата никеля взвешивали и растворяли в 60 мл деионизированной воды, к которой добавляли 28,57 г циклогексана, 16,57 г Triton Х-100 и 16 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 30 мин, затем отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А1-1.
(2) 0,0651 г нитрата палладия растворяли в 110 мл деионизированной воды и значение рН доводили до 1,5. Затем полуфабрикат катализатора А1-1 пропитывали в полученном солевом растворе Pd в течение 30 мин, сушили при 80°С в течение 6 ч и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В1-1.
(3) 0,046 г хлорида палладия растворяли в 60 мл деионизированной воды, к которой добавляли 28,57 г циклогексана, 16,57 г Triton Х-100 и 16 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В1-1, полученный на стадии (2), помещали в приготовленную микроэмульсию и встряхивали в течение 30 мин, затем отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 62,12 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 60,18 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и было определено, что в катализаторе, полученном в сравнительном примере 1-1, содержание Pd составляло 0,0575%, и содержание Ni составляло 5%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при 200°С с применением газовой смеси с молярным соотношением N2:Н2=1:1.
Пример 1-2
Носитель: Применяли коммерчески доступный сферический носитель оксид алюминия с бимодальным распределением пор по размерам, 3 мм в диаметре. После прокаливания при 1111°С в течение 4 часов бимодальное распределение пор по размерам находилось в диапазоне 20-40 нм и 120-400 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 39,71 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 1,098 г хлорида никеля и 1,47 г нитрата никеля взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 34,14 г н-гексана, 20 г ЦТАБ и 19 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 6 ч с получением полуфабриката катализатора А2.
(2) 0,068 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А2 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора В2.
(3) 0,0167 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 34,14 г н-гексана, 20 г ЦТАБ и 19 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В2, полученный на стадии (2), помещали в приготовленную микроэмульсию и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 55,48 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 54,40 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в Примере 1-2, содержание Pd составляло 0,05%, содержание Ni составляло 5%, и содержание Cu составляло 0,5%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при 150°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 1-2
Катализатор был приготовлен с применением того же носителя и условий, что и в примере 1-2, за исключением того, что Cu обеспечивали при помощи растворного способа.
Носитель: Применяли коммерчески доступный сферический носитель оксид алюминия с бимодальным распределением пор по размерам, 3 мм в диаметре. После прокаливания при 111°С в течение 4 часов бимодальное распределение пор по размерам находилось в диапазоне 20-40 нм и 120-400 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 39,71 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 1,098 г хлорида никеля взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 34,14 г н-гексана, 20 г ЦТАБ и 19 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 6 ч с получением полуфабриката катализатора А2.
(2) 0,068 г хлорида палладия и 1,47 г нитрата никеля растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А2 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора В2.
(3) 0,0167 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 34,14 г н-гексана, 20 г ЦТАБ и 19 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В2, полученный на стадии (2), помещали в приготовленную микроэмульсию и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 55,48 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 54,40 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в Примере 1-2, содержание Pd составляло 0,05%, содержание Ni составляло 5%, и содержание Cu составляло 0,5%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при 150°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Пример 1-3
Носитель: Использовали коммерчески доступный сферический носитель оксид алюминия с бимодальным распределением пор по размерам, диаметром 4 мм. После прокаливания при 1128°С в течение 4 часов бимодальное распределение пор по размерам находилось в диапазоне 25-50 нм и 95-500 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 20,19 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 6,203 г безводного нитрата никеля и 2,11 г хлорида меди взвешивали и растворяли в 76 мл деионизированной воды, к которой добавляли 26 г циклогексана, 4,9 г Triton Х-100 и 4,49 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 240 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора A3.
(2) 0,108 г нитрата палладия растворяли в 90 мл деионизированной воды и значение рН доводили до 2. Затем полуфабрикат катализатора A3 пропитывали в полученном солевом растворе Pd в течение 90 мин, сушили при 120°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора В3.
(3) 0,33 г нитрата серебра растворяли в 68,2 мл деионизированной воды и значение рН доводили до 4. Полуфабрикат катализатора В3, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 150°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора С3.
(4) 0,043 г нитрата палладия растворяли в 76 мл деионизированной воды, к которой добавляли 26 г циклогексана, 4,9 г Triton Х-100 и 4,49 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С3, полученный на стадии (3), растворяли в полученной микроэмульсии, встряхивали в течение 240 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 500°С в течение 5 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микро эмульсии, полученной на стадии (1), составлял 403,65 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 401,83 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-3, содержание Pd составляло 0,07%, содержание Ni составляло 2%, содержание Cu составляло 1%, и содержание Ag составляло 0,21%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 150°С с помощью чистого водорода.
Сравнительный Пример 1-3
Катализатор был приготовлен с применением того же носителя и условий, что и в Примере 1-3, за исключением того, что Pd не обеспечивали с помощью растворного способа. Приготовление катализатора:
(1) 6,203 г безводного нитрата никеля и 2,11 г хлорида меди взвешивали и растворяли в 76 мл деионизированной воды, к которой добавляли 26 г циклогексана, 4,9 г Triton Х-100 и 4,49 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 100 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора А3-1.
(2) 0,151 г нитрата палладия растворяли в 90 мл деионизированной воды и значение рН доводили до 2. Затем полуфабрикат катализатора А3-1 пропитывали в полученном солевом растворе Pd в течение 90 мин, сушили при 120°С в течение 4 ч и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора В3-1.
(3) 0,33 г нитрата серебра растворяли в 68,2 мл деионизированной воды и значение рН доводили до 4. Полуфабрикат катализатора В3-1, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 150°С в течение 2 часов и прокаливали при 500°С в течение 5 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 403,65 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-3, содержание Pd составляло 0,07%, содержание Ni составляло 2%, содержание Cu составляло 1%, и содержание Ag составляло 0,21%
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 150°С с применением чистого водорода.
Пример 1-4
Носитель: применяли коммерчески доступный сферический носитель оксид алюминия-диоксид титана с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и содержание диоксида титана 20%. После прокаливания при 1118°С в течение 4 ч бимодальное распределение пор по размерам составляло 23-47 нм и 90-450 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 30,28 м2/г.100 г носителя взвешивали.
Приготовление катализатора:
(1) 2,21 г хлорида никеля и 2,94 г нитрата меди взвешивали и растворяли в 80 мл деионизированной воды, к которой добавляли 40,00 г н-гексана, 24 г ЦТАБ и 23 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 180 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора А4.
(2) 0,033 г хлорида палладия растворяли в 80 мл деионизированной воды, к которой добавляли 40,00 г н-гексана, 24 г ЦТАБ и 23 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А4 пропитывали полученной микроэмульсией и встряхивали в течение 180 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора В4.
(3) 0,075 г хлорида палладия растворяли в 80 мл деионизированной воды и значение рН доводили до 2,5. Затем полуфабрикат катализатора В4 пропитывали в полученном солевом растворе Pd в течение 120 мин, сушили при 130°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора С4.
(4) 0,126 г нитрата серебра растворяли в 58 мл деионизированной воды и значение рН доводили до 5. Полуфабрикат катализатора С4, полученный на стадии (3), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 100°С и прокаливали при 550°С в течение 5 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 52,83 нм, и диаметр частиц микроэмульсии, полученной на стадии (2), составлял 52,61 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-4, содержание Pd составляло 0,064%, содержание Ni составляло 1%, содержание Cu составляло 1%, и содержание Ag составляло 0,08%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 1-4
Катализатор был приготовлен с применением того же носителя и условий, что и в примере 1-4, за исключением отсутствия Ni в этом сравнительном примере.
(1) 2,94 г нитрата меди взвешивали и растворяли в 80 мл деионизированной воды, к которой добавляли 40,00 г н-гексана, 24 г ЦТАБ и 23 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 180 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора А4-1.
(2) 0,033 г хлорида палладия растворяли в 80 мл деионизированной воды, к которой добавляли 40,00 г н-гексана, 24 г ЦТАБ и 23 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А4-1 пропитывали полученной микроэмульсией и встряхивали в течение 180 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора В4-1.
(3) 0,075 г хлорида палладия растворяли в 80 мл деионизированной воды и значение рН доводили до 2,5. Затем полуфабрикат катализатора В4-1 пропитывали в полученном солевом растворе Pd в течение 120 мин, сушили при 130°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора С4-1.
(4) 0,126 г нитрата серебра растворяли в 58 мл деионизированной воды и значение рН доводили до 5. Полуфабрикат катализатора С4-1, полученный на стадии (3), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 100°С и прокаливали при 550°С в течение 5 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 52,87 нм, и диаметр частиц микроэмульсии, полученной на стадии (2), составлял 52,65 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-4, содержание Pd составляло 0,064%, содержание Cu составляло 1%, и содержание Ag составляло 0,08%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Пример 1-5
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия и магния с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и содержание оксида магния 3%. После прокаливания при 999°С в течение 4 ч бимодальное распределение пор по размерам составляло 23-47 нм и 80-380 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 45,08 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 0,076 г хлорида палладия растворяли в 80 мл деионизированной воды и значение рН доводили до 2. Затем носитель пропитывали в полученном солевом растворе Pd в течение 120 мин, сушили при130°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А5.
(2) 0,158 г нитрата серебра растворяли в 58 мл деионизированной воды и значение рН доводили до 5. Полуфабрикат катализатора А5, полученный на стадии (1), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 100°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора В5.
(3) 3,295 г хлорида никеля и 1,45 г нитрата меди взвешивали и растворяли в 80 мл деионизированной воды, к которой добавляли 35,00 г н-гексана, 20,36 г Triton Х-100 и 19,39 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В5, полученный на стадии (2), пропитывали полученной микроэмульсией и встряхивали в течение 180 мин, после чего отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора С5.
(4) 0,017 г хлорида палладия растворяли в 80 мл деионизированной воды, к которой добавляли 35,00 г н-гексана и 19,39 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полученный полуфабрикат катализатора С5 пропитывали в полученной микроэмульсии и встряхивали в течение 180 мин, затем отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 66,38 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 65,22 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-5, содержание Pd составляло 0,055%, содержание Ni составляло 1,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,10%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 180°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 1-5
Катализатор получали с применением того же носителя и условий, что и в Примере 1-5, за исключением того, что количество добавленного Ni уменьшали до 0,3%. Приготовление катализатора:
(1) 0,076 г хлорида палладия растворяли в 80 мл деионизированной воды и значение рН доводили до 2. Затем носитель пропитывали в полученном солевом растворе Pd в течение 120 мин, сушили при130°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А5-1.
(2) 0,157 г нитрата серебра растворяли в 58 мл деионизированной воды и значение рН доводили до 5. Полуфабрикат катализатора А5-1, полученный на стадии (1), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 100°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора В5-1.
(3) 0,659 г хлорида никеля и 1,45 г нитрата меди взвешивали и растворяли в 80 мл деионизированной воды, к которой добавляли 35,00 г н-гексана, 20,36 г Triton Х-100 и 19,39 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В5-1, полученный на стадии (2), пропитывали полученной микроэмульсией и встряхивали в течение 180 мин, после чего отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч, с получением полуфабриката катализатора С5-1.
(4) 0,017 г хлорида палладия растворяли в 80 мл деионизированной воды, к которой добавляли 35,00 г н-гексана и 19,39 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полученный полуфабрикат катализатора С5-1 пропитывали полученной микроэмульсией и встряхивали в течение 180 мин, затем отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 66,32 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 65,24 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-5, содержание Pd составляло 0,055%, содержание Ni составляло 0,28%, содержание Cu составляло 0,5%, н содержание Ag составляло 0,10%
Пример 1-6
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия и магния с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и содержание оксида магния 10%. После прокаливания при 999°С в течение 4 ч бимодальное распределение пор по размерам составляло 23-47 нм и 80-380 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 45,08 м2/г.100 г носителя взвешивали.
Приготовление катализатора:
(1) 2,20 г хлорида никеля и 2,93 г нитрата меди взвешивали и растворяли в 80 мл деионизированной воды, к которой добавляли 36,00 г н-гексана, 20,36 г Triton Х-100 и 19,50 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 180 мин, затем отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А6.
(2) 0,076 г хлорида палладия растворяли в 80 мл деионизированной воды и значение рН доводили до 2,5. Полуфабрикат катализатора А6 пропитывали в полученном солевом растворе Pd в течение 120 мин, сушили при 130°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора В6.
(3) 0,157 г нитрата серебра растворяли в 58 мл деионизированной воды и значение рН доводили до 5. Полуфабрикат катализатора В6, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 100°С и прокаливали при 550°С в течение 6 ч с получением полуфабриката катализатора С6.
(4) 0,017 г хлорида палладия растворяли в 80 мл деионизированной воды, к которой добавляли 36,00 г н-гексана, 20,36 г Triton Х-100 и 19 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С6, полученный на стадии (3), растворяли в полученной микроэмульсии и встряхивали в течение 180 мин с последующим фильтрованием остаточной жидкости, сушкой при 70°С и прокаливанием при 400°С в течение 6 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 66,32 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 65,36 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-6, содержание Pd составляло 0,055%, содержание Ni составляло 1%, содержание Cu составляло 1%, и содержание Ag составляло 0,10%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 1-6
Катализатор получали с применением того же носителя и условий, что и в примере 1-6, за исключением того, что количество Pd, добавляемого во время получения микроэмульсии на стадии (4), уменьшено до 1/3 от количества, указанного в примере 1-6, и составляло менее 1/200 от содержания Ni+Cu.
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия и магния с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и содержание оксида магния 10%. После прокаливания при 1000°С в течение 4 ч бимодальное распределение пор по размерам составляло 23-47 нм и 80-380 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 45,08 м2/г.100 г носителя взвешивали.
Приготовление катализатора:
(1) 2,20 г хлорида никеля и 2,93 г нитрата меди взвешивали и растворяли в 80 мл деионизированной воды, к которой добавляли 36,00 г н-гексана, 20,36 г Triton Х-100 и 19,50 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 180 мин, затем отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А6-1.
(2) 0,076 г хлорида палладия растворяли в 80 мл деионизированной воды и значение рН доводили до 2,5. Полуфабрикат катализатора А6-1 пропитывали в полученном солевом растворе Pd в течение 120 мин, сушили при 130°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора В6-1.
(3) 0,157 г нитрата серебра растворяли в 58 мл деионизированной воды и значение рН доводили до 5. Полуфабрикат катализатора В6-1, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 100°С и прокаливали при 550°С в течение 6 ч с получением полуфабриката катализатора С6-1.
(4) 0,0057 г хлорида палладия растворяли в 80 мл деионизированной воды, к которой добавляли 36,00 г н-гексана, 20,36 г Triton Х-100 и 19 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С6-1, полученный на стадии (3), растворяли в полученной микроэмульсии и встряхивали в течение 180 мин с последующим фильтрованием остаточной жидкости, сушкой при 70°С и прокаливанием при 400°С в течение 6 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 66,32 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 65,36 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-6, содержание Pd составляло 0,0483%, содержание Ni составляло 1%, содержание Cu составляло 1%, и содержание Ag составляло 0,10%
Пример 1-7
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм, содержание оксида алюминия 97% и содержание диоксида титана 3%. После прокаливания при 978°С в течение 4 ч бимодальное распределение пор по размерам составляло 20-35 нм и 90-200 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 49,81 м2/г.100 г носителя взвешивали.
Приготовление катализатора:
(1) 0,05 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем носитель пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при100°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А7.
(2) 0,126 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора А7, полученный на стадии (1), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 600°С в течение 6 ч с получением полуфабриката катализатора В7.
(3) 1,56 г нитрата никеля и 1,06 г хлорида меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,0 г н-гексана, 13,75 г ЦТАБ и 12,73 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В7 пропитывали полученной микроэмульсией и встряхивали в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора С7.
(4) 0,009 г хлорида палладия взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,50 г н-гексана, 13,75 г ЦТАБ и 12,50 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С7 пропитывали полученной микроэмульсией и встряхивали в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 100,60 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 100,28 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-7, содержание Pd составляло 0,035%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,08%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 150°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 1-7
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм, содержание оксида алюминия 97% и содержание диоксида титана 3%. После прокаливания при 940°С бимодальное распределение пор по размерам составляло 10-20 нм и 30-97 нм, водопоглощение составляло 65%, и удельная площадь поверхности составляла 75,21 м2/г.100 г носителя взвешивали. Приготовление катализатора:
(1) 0,05 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем носитель пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при100°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А7-1.
(2) 0,126 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора А7-1, полученный на стадии (1), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 600°С в течение 6 ч с получением полуфабриката катализатора В7-1.
(3) 1,56 г хлорида никеля и 1,06 г нитрата меди взвешивали и растворяли в 71,5 г деионизированной воды, к которой добавляли 27,0 г н-гексана, 13,75 г ЦТАБ и 12,73 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В7-1 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора С7-1.
(4) 0,009 г хлорида палладия взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,50 г н-гексана, 13,75 г ЦТАБ и 12,50 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С7-1 пропитывали полученной микроэмульсией, встряхивали в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 600°С в течение 4 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 100,60 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 100,23 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-7, содержание Pd составляло 0,035%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,08%
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 150°С с применением газовой смеси с молярным соотношением N2:Н2=1:1.
Пример 1-8
Катализатор примера 1-8 получали с применением того же носителя и условий, что и в примере 1-7, за исключением того, что порядок стадий (1) и (2) был обратным.
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм, содержание оксида алюминия 97% и содержание диоксида титана 3%. После прокаливания при 978°С бимодальное распределение пор по размерам составляло 20-35 нм и 90-200 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 49,81 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 0,126 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Затем 100 г прокаленного носителя пропитывали полученным раствором нитрата серебра, содержащим серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А8.
(2) 0,05 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А8, полученный на стадии (1), пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 600°С в течение 6 ч с получением полуфабриката катализатора В8.
(3) 1,56 г нитрата никеля и 1,06 г хлорида меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,0 г н-гексана, 13,75 г ЦТАБ и 12,73 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В7, полученный на стадии (2), пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора С8.
(4) 0,009 г хлорида палладия взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г н-гексана, 13,75 г ЦТАБ и 12,45 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С8, полученный на стадии (3), пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 100,60 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 100,23 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-8, содержание Pd составляло 0,035%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, н содержание Ag составляло 0,08%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 150°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 1-8
Катализатор получали с применением того же носителя и условий, что и в примере 1-8, за исключением того, что диаметр частиц микроэмульсии, полученной на стадии (4), был больше максимального размера пор носителя.
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм, содержание оксида алюминия 97% и содержание диоксида титана 3%. После прокаливания при 980°С бимодальное распределение пор по размерам составляло 20-35 нм и 90-200 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 49,81 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 0,126 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Затем 100 г прокаленного носителя пропитывали полученным раствором нитрата серебра, содержащим серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А8-1.
(2) 0,05 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А8-1, полученный на стадии (1), пропитывали в полученном солевом растворе Pd в течение 60 мин, и затем сушили при 100°С и прокаливали при 600°С в течение 6 часов с получением полуфабриката катализатора В8-1.
(3) 1,56 г нитрата никеля и 1,06 г хлорида меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,10 г н-гексана, 13,75 г ЦТАБ и 12,73 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В8-1, полученный на стадии (2), пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч, с получением полуфабриката катализатора С8-1.
(4) 0,009 г хлорида палладия взвешивали и растворяли в 65 г воды, к которой добавляли 22,26 г циклогексана, 4,22 г Triton Х-100 и 3,70 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С8-1, полученный на стадии (3), пропитывали полученной микро эмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 100,60 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 398,76 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-8, содержание Pd составляло 0,031%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,08%
Пример 1-9
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм. После прокаливания при 1115°С в течение 4 ч бимодальное распределение пор по размерам составляло 28-48 нм и 102-499 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 20,73 м2/г. 100 г носителя взвешивали.
(1) 0,068 г хлорида палладия растворяли в 120 мл деионизированной воды и значение рН доводили до 2,5. Затем носитель пропитывали в полученном солевом растворе Pd в течение 1 ч, сушили при 120°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А9.
(2) 0,33 г нитрата серебра растворяли в 50 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора А9 пропитывали в полученном растворе и встряхивали в течение 15 мин, сушили при 120°С и прокаливали при 600°С в течение 4 ч, с получением полуфабриката катализатора В9.
(3) 15,58 г безводного нитрата никеля и 1,471 г нитрата меди взвешивали и растворяли в 65 г воды, к которой добавляли 22,40 г циклогексана, 4,25 г Triton Х-100 и 3,80 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В9, полученный на стадии (2), пропитывали полученной микроэмульсией в течение 4 ч, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора С9.
(4) 0,059 г нитрата палладия взвешивали и растворяли в 65 г воды, к которой добавляли 22,40 г циклогексана, 4,25 г Triton Х-100 и 3,80 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С9, полученный на стадии (3), пропитывали полученной микроэмульсией в течение 4 ч, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 60°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 398,56 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 398,75 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-9, содержание Pd составляло 0,0675%, содержание Ni составляло 5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,21%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 8 часов.
Сравнительный Пример 1-9
Приготовление катализатора:
Катализатор получали с применением того же носителя и стадий, что и в Примере 1-9, за исключением того, что диаметр частиц микроэмульсии составлял 622,38 нм при обеспечении Ni/Cu.
(1) 0,068 г хлорида палладия растворяли в 120 мл деионизированной воды и значение рН доводили до 2,5. Затем носитель пропитывали в полученном солевом растворе Pd в течение 70 мин, сушили при 120°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А9-1.
(2) 0,33 г нитрата серебра растворяли в 50 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора А9-1 пропитывали в полученном растворе и встряхивали в течение 15 мин, сушили при 120°С и прокаливали при 600°С в течение 4 ч, с получением полуфабриката катализатора В9-1.
(3) 15,58 г безводного нитрата никеля и 1,471 г нитрата меди взвешивали и растворяли в 65 г воды, к которой добавляли 22,40 г циклогексана, 2,75 г Triton Х-100 и 2,75 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В9-1, полученный на стадии (2), пропитывали в полученной микроэмульсии в течение 4 ч, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 600°С в течение 4 ч, с получением полуфабриката катализатора С9-1.
(4) 0,059 г нитрата палладия взвешивали и растворяли в 65 г воды, к которой добавляли 22,26 г циклогексана, 4,25 г Triton Х-100 и 3,80 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С9, полученный на стадии (3), пропитывали полученной микроэмульсией в течение 4 ч, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (3), составлял 621,67 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 399,62 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-9, содержание Pd составляло 0,0675%, содержание Ni составляло 1,34%, содержание Cu составляло 0,12%, и содержание Ag составляло 0,21%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 8 часов.
Пример 1-10
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм. После прокаливания при 1118°С в течение 4 ч бимодальное распределение пор по размерам составляло 30-43 нм и 100-498 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 20,35 м2/г.100 г носителя взвешивали.
(1) 5,52 г хлорида никеля и 7,38 г нитрата меди взвешивали и растворяли в 69 мл деионизированной воды, к которой добавляли 23,2 г н-пентана, 3,45 г ЦТАБ и 2,88 г н-октанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А10.
(2) 0,051 г хлорида палладия растворяли в 69 мл деионизированной воды, к которой добавляли 23,2 г н-пентана, 3,50 г ЦТАБ и 2,91 г н-октанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А10 пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В10.
(3) 0,084 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора В10 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора С10.
(4) 0,158 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора С10, полученный на стадии (3), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 500°С в течение 6 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 497,65 нм, и диаметр частиц микроэмульсии, полученной на стадии (2), составлял 495,32 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-10, содержание Pd составляло 0,08%, содержание Ni составляло 2,5%, содержание Cu составляло 2,5%, и содержание Ag составляло 0,10%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 200°С, выдержка в течение 4 часов.
Сравнительный Пример 1-10
Катализатор получали в тех же условиях, что и в примере 1-10, за исключением того, что удельная площадь поверхности составляла менее 20 м2/г.
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм. После прокаливания при 1155°С в течение 4 ч бимодальное распределение пор по размерам составляло 41-76 нм и 114-684 нм, водопоглощение составляло 60%, и удельная площадь поверхности составляла 14,29 м2/г.100 г носителя взвешивали.
(1) 5,52 г хлорида никеля и 7,38 г нитрата меди взвешивали и растворяли в 69 мл деионизированной воды, к которой добавляли 23,2 г н-пентана, 3,45 г ЦТАБ и 2,88 г н-октанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А10.
(2) 0,051 г хлорида палладия растворяли в 69 мл деионизированной воды, к которой добавляли 23 г н-пентана, 3,50 г ЦТАБ и 2,91 г н-октанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А10-1 пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В10-1.
(3) 0,084 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора В10 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора С10-1.
(4) 0,158 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора С10-1, полученный на стадии (3), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 500°С в течение 6 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 497,61 нм, и диаметр частиц микроэмульсии, полученной на стадии (2), составлял 495,33 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-10, содержание Pd составляло 0,08%, содержание Ni составляло 2,5%, содержание Cu составляло 2,5%, и содержание Ag составляло 0,10%
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 200°С, выдержка в течение 4 часов.
Пример 1-11
Носитель: применяли коммерчески доступный сферический носитель на основе оксида алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм. После прокаливания при 1100°С в течение 4 ч бимодальное распределение пор по размерам составляло 30-45 нм и 300-450 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 47 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 1,10 г хлорида никеля и 1,47 г нитрата меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 35 г н-гексана, 21 г ЦТАБ и 20 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора А11.
(2) 0,067 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора А11 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора В11.
(3) 0,126 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора В11, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката для катализатора С11.
(4) 0,018 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 3 5 г н-гексана, 21 г ЦТАБ и 20 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С11 пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 500°С в течение 5 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 51,61 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 50,39 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-11, содержание Pd составляло 0,05%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,08%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 1-11
Катализатор получали с применением того же носителя и условий, что и в примере 1-11, за исключением того, что диаметр частиц микроэмульсии был больше, чем максимальный размер мелких пор носителя.
Приготовление катализатора:
(1) 1,10 г хлорида никеля и 1,47 г нитрата меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 37 г н-гексана, 30 г ЦТАБ и 30 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора А11-1.
(2) 0,067 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора А11 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора В11-1.
(3) 0,126 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора В11-1, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 500°С в течение 5 ч с получением полуфабриката катализатора С11-1.
(4) 0,018 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 37 г н-гексана, 30 г ЦТАБ и 30 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С11 пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 500°С в течение 5 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 30,87 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 30,24 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-11, содержание Pd составляло 0,05%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,08%
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:Н2=1:1.
Пример 1-12
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1088°С в течение 4 ч бимодальное распределение пор по размерам составляло 20-46 нм и 85-350 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 39,29 м2/г.100 г носителя взвешивали.
(1) 0,08 г хлорида палладия растворяли в 140 мл деионизированной воды и значение рН доводили до 2. Затем полученный носитель пропитывали в солевом растворе Pd в течение 50 мин, сушили при 100°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора А12.
(2) 4,93 г нитрата никеля и 1,47 г нитрата меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г н-гексана, 13,80 г ЦТАБ и 12,73 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А12 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора В12.
(3) 0,021 г нитрата палладия взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г н-гексана, 13,80 г ЦТАБ и 12,73 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В12 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 450°С в течение 6 ч с получением полуфабриката катализатора С12.
(4) 0,291 г нитрата серебра полностью растворяли в 49,5 мл деионизированной воды и значение рН доводили до 2. Затем полуфабрикат катализатора С12 пропитывали в полученном растворе и встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 550°С в течение 6 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (2), составлял 98,78 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 99,31 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-12, содержание Pd составляло 0,057%, содержание Ni составляло 1,57%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,18%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 4 часов.
Сравнительный Пример 1-12
Катализатор получали с применением того же носителя и условий, что и в примере 1-12, за исключением того, что количество носителя Cu было ниже 1/10 от количества Ni.
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1090°С в течение 4 ч бимодальное распределение пор по размерам составляло 20-46 нм и 85-350 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 39,29 м2/г. 100 г носителя взвешивали.
(1) 0,08 г хлорида палладия растворяли в 140 мл деионизированной воды и значение рН доводили до 2. Затем полученный носитель пропитывали в солевом растворе Pd в течение 50 мин, сушили при 110°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатор а А12-1.
(2) 4,93 г нитрата никеля и 0,294 г нитрата меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г н-гексана, 13,80 г ЦТАБ и 12,73 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А12-1 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора В12-1.
(3) 0,021 г нитрата палладия взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г н-гексана, 13,80 г ЦТАБ и 12,73 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В12-1 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 450°С в течение 6 ч с получением полуфабриката катализатора С12-1.
(4) 0,291 г нитрата серебра полностью растворяли в 49,5 мл деионизированной воды и значение рН доводили до 2. Затем полуфабрикат катализатора С12-1 пропитывали в полученном растворе и встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 550°С в течение 6 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (2), составлял 98,78 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 99,31 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-12, содержание Pd составляло 0,057%, содержание Ni составляло 1,57%, содержание Cu составляло 0,1%, и содержание Ag составляло 0,18%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 4 часов.
Пример 1-13
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1112°С в течение 4 ч бимодальное распределение пор по размерам составляло 26-47 нм и 95-450 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 25,45 м2/г 100 г носителя взвешивали.
(1) 0,075 г нитрата палладия растворяли в 44 мл деионизированной воды и значение рН доводили до 2. Затем полученный раствор распыляли на полученный носитель. После полной абсорбции раствора носитель сушили при 100°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А13.
(2) 12,46 г безводного нитрата никеля и 2,94 г нитрата меди взвешивали и растворяли в 70,00 г воды, к которой добавляли 35,00 г циклогексана, 21,00 г Triton Х-100 и 20,80 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А13 пропитывали в полученной микроэмульсии в течение 3 ч, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора В13.
(3) 0,0416 г нитрата палладия взвешивали и растворяли в 70,00 г воды, к которой добавляли 35,00 г циклогексана, 21,00 г Triton Х-100 и 20,80 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В13 пропитывали в полученной микроэмульсии в течение 3 ч, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С13.
(4) 0,21 г нитрата серебра растворяли в 60 мл деионизированной воды и значение рН доводили до 4. Полученный раствор распыляли на полуфабрикат катализатора С13. После полной абсорбции раствора катализатор сушили при 110°С и прокаливали при 550°С в течение 4 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (2), составлял 50,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 50,32 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-13, содержание Pd составляло 0,054%, содержание Ni составляло 4%, содержание Cu составляло 1%, и содержание Ag составляло 0,13%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 200°С, выдержка в течение 8 часов.
Сравнительный Пример 1-13
Катализатор был приготовлен с применением того же носителя и количества активных компонентов, что и в Примере 1-13, за исключением того, что Pd обеспечивали с помощью растворного способа.
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1112°С в течение 4 ч бимодальное распределение пор по размерам составляло 26-47 нм и 95-450 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 25,45 м2/г. 100 г носителя взвешивали.
(1) 12,46 г безводного нитрата никеля и 2,94 г нитрата меди взвешивали и растворяли в 70,00 г воды, к которой добавляли 35,00 г циклогексана, 21,00 г Triton Х-100 и 20,80 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А13 пропитывали в полученной микроэмульсии в течение 3 ч, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч, с получением полуфабриката катализатора А13-1.
(2) 0,1166 г нитрата палладия взвешивали и растворяли в 70,00 г воды, к которой добавляли 35,00 г циклогексана, 21,00 г Triton Х-100 и 20,80 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А13-1 пропитывали в полученной микроэмульсии в течение 3 ч, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора В13-1.
(3) 0,21 г нитрата серебра полностью растворяли в 60 мл деионизированной воды и значение рН доводили до 4. Затем полученный раствор распыляли на полуфабрикат катализатора С13-1. После полной абсорбции раствора катализатор сушили при 110°С и прокаливали при 550°С в течение 4 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микро эмульсии, полученной на стадии (1), составлял 50,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (2), составлял 50,34 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-13, содержание Pd составляло 0,054%, содержание Ni составляло 4%, содержание Cu составляло 1%, и содержание Ag составляло 0,13%
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 200°С, выдержка в течение 8 часов.
Пример 1-14
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм. После прокаливания при 1088°С в течение 4 ч бимодальное распределение пор по размерам составляло 20-46 нм и 85-350 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 40,23 м2/г.100 г носителя взвешивали.
(1) 4,93 г безводного нитрата никеля и 1,47 г нитрата меди взвешивали и растворяли в 70 мл воды, к которой добавляли 26,92 г н-гексана, 16,16 г ЦТАБ и 16,00 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полученный носитель пропитывали в полученной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А14.
(2) 0,07 г нитрата палладия растворяли в 140 мл деионизированной воды и значение рН доводили до 2. Затем полученный полуфабрикат катализатора А14 пропитывали в солевом растворе Pd в течение 50 мин, сушили при 110°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора В14.
(3) 0,023 г нитрата палладия взвешивали и растворяли в 70 мл воды, к которой добавляли 26,92 г н-гексана, 16,16 г ЦТАБ и 16,00 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полученный полуфабрикат катализатора В14 пропитывали в полученной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С14.
(4) 0,291 г нитрата серебра растворяли в 49,5 мл деионизированной воды и значение рН доводили до 2. Полуфабрикат катализатора С14 пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 550°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 80,28 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 80,56 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-14, содержание Pd составляло 0,043%, содержание Ni составляло 1,57%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,18%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 4 часов.
Сравнительный Пример 1-14
Приготовление катализатора:
Катализатор был приготовлен с применением того же носителя и условий, что и в Примере 1-14, за исключением того, что медь и никель обеспечивали в растворном способе.
(1) 4,93 г безводного нитрата никеля и 1,47 г нитрата меди взвешивали и растворяли в 71,5 г воды и тщательно перемешивали. Полученный носитель пропитывали в полученном растворе в течение 80 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А14-1.
(2) 0,07 г нитрата палладия растворяли в 140 мл деионизированной воды и значение рН доводили до 2. Затем полученный полуфабрикат катализатора А14-1 пропитывали в солевом растворе Pd в течение 50 мин, сушили при 110°С и прокаливали при 550°С в течение 4 ч с получением требуемого полуфабриката катализатора В14-1.
(3) 0,023 г нитрата палладия взвешивали и растворяли в 70 мл воды, к которой добавляли 26,92 г н-гексана, 16,16 г ЦТАБ и 16,00 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В14-1 пропитывали в полученной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С14-1.
(4) 0,291 г нитрата серебра растворяли в 49,5 мл деионизированной воды и значение рН доводили до 2. Полуфабрикат катализатора С14 пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 550°С в течение 4 ч с получением требуемого катализатора.
Диаметр частиц микроэмульсии, полученной на стадии (3), как определено динамическим рассеянием света, составлял 80,56 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-14, содержание Pd составляло 0,043%, содержание Ni составляло 1,57%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,18%
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 4 часов.
Пример 1-15
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1092°С в течение 4 ч бимодальное распределение пор по размерам составляло 20-45 нм и 85-350 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 39,47 м2/г. 100 г носителя взвешивали.
(1) 4,93 г безводного нитрата никеля и 1,47 г нитрата меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г изо-пентана, 13,75 г ЦТАБ и 12,60 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полученный носитель пропитывали в полученной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А15.
(2) 0,064 г нитрата палладия растворяли в 140 мл деионизированной воды и значение рН доводили до 2. Затем полученный полуфабрикат катализатора А15 пропитывали в солевом растворе Pd в течение 50 мин, сушили при 110°С и прокаливали при 550°С в течение 6 ч с получением полуфабриката катализатора В15.
(3) 0,022 г нитрата палладия взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г изо-пентана, 13,75 г ЦТАБ и 12,60 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В15 пропитывали в полученной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С15.
(4) 0,291 г нитрата серебра растворяли в 49,5 мл деионизированной воды и значение рН доводили до 2. Полуфабрикат катализатора С15 пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 500°С в течение 6 часов с получением требуемого катализатора.
Диаметр частиц микроэмульсии, полученной на стадиях (1) и (3), как определено динамическим рассеянием света, составлял 101,39 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 1-15, содержание Pd составляло 0,04%, содержание Ni составляло 1,57%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,18%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 4 часов.
Сравнительный Пример 1-15
Применяли носитель с унимодальным распределением пор по размерам.
Получение катализатора: применяли коммерчески доступный сферический носитель оксид алюминия с унимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1092°С в течение 4 ч распределение по размерам пор составляло 20-45 нм, то есть представляло собой унимодальное распределение по размерам пор, водопоглощение составляло 55%, и удельная площадь поверхности составляла 39,47 м2/г. 100 г носителя взвешивали.
(1) 4,93 г безводного нитрата никеля и 1,47 г нитрата меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г изо-пентана, 13,75 г ЦТАБ и 12,60 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полученный носитель пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А15-1.
(2) 0,064 г нитрата палладия растворяли в 140 мл деионизированной воды и значение рН доводили до 2. Затем полученный полуфабрикат катализатора А15-1 пропитывали в солевом растворе Pd в течение 50 мин, сушили при 110°С и прокаливали при 550°С в течение 6 ч с получением полуфабриката катализатора В15-1.
(3) 0,022 г нитрата палладия взвешивали и растворяли в 71,5 г воды, к которой добавляли 27,5 г изо-пентана, 13,75 г ЦТАБ и 12,60 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В15-1 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 70°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С15-1.
(4) 0,291 г нитрата серебра растворяли в 49,5 мл деионизированной воды и значение рН доводили до 2. Полуфабрикат катализатора С15-1 пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 500°С в течение 6 часов с получением требуемого катализатора.
Диаметр частиц микроэмульсии, полученной на стадиях (1) и (3), как определено динамическим рассеянием света, составлял 101,39 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 1-15, содержание Pd составляло 0,034%, содержание Ni составляло 0,32%, содержание Cu составляло 0,11%, и содержание Ag составляло 0,18%
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 4 часов.
Характеристики катализатора, применяемого в реакции концевого гидрирования С2
Способ оценки:
Катализатор загружали в одноступенчатый реактор с неподвижным слоем в количестве 100 мл (записанная масса) с количеством наполнителя: 50 мл, газовой скоростью реакционной массы: 5000/ч; рабочим давлением: 2,0 МПа; отношением водорода к алкину: 1,15; температурой на входе в реактор: 50°С. Результаты оценки рассчитывали, как показано в Таблице 1-2.


Начальная селективность представляет собой селективность, измеренную через 24 часа после начала подачи в реактор.
Начальная активность представляет собой активность, измеренную через 24 часа после начала подачи в реактор (конверсия ацетилена).
Состав реакционной массы показан в Таблице 1-3.

Результаты оценки катализатора приведены в Таблице 1-4.
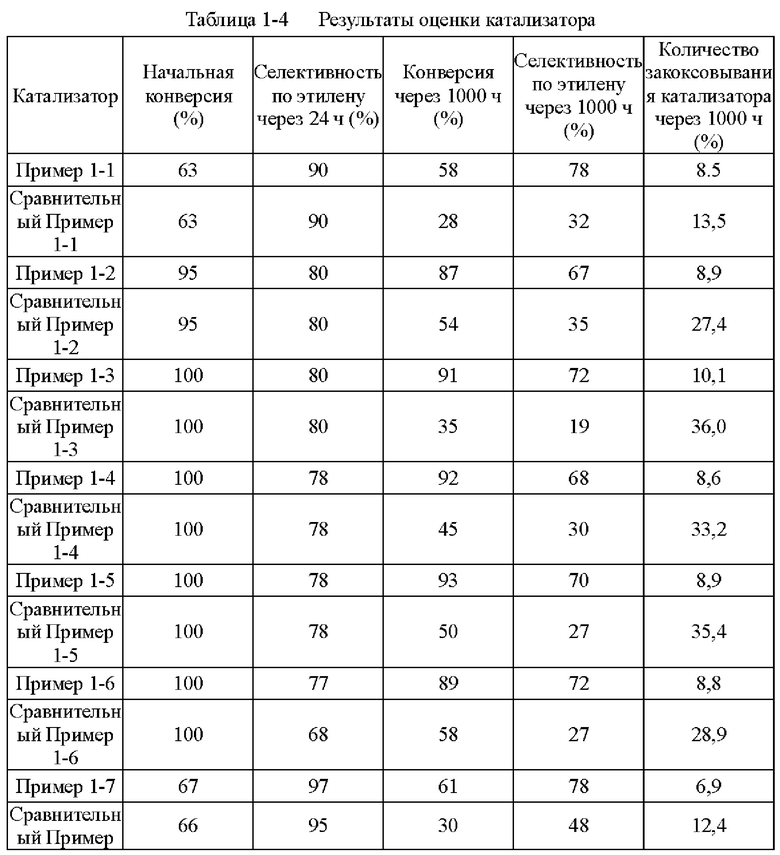
Путем сравнения результатов оценки катализатора из таблиц 1-2 и 1-3:
В сравнительном примере 1-1, в котором Cu не обеспечивали, и температура реакции составляла 200°С, начальная конверсия ацетилена и селективность были по существу такими же, как в примере 1, но были значительно ниже через 1000 ч по сравнению с примером 1. Это указывает на то, что обеспечение Cu или температура восстановления катализатора важны для улучшения характеристик против закоксовывания. Другая возможность заключается в том, что активный центр с функцией недеструктивного гидрирования не функционирует так, как ожидалось, при температуре восстановления 200°С.
В сравнительном примере 1-2, в котором Cu обеспечивали растворным способом, Cu была очень равномерно диспергирована в носителе и не действовала для эффективного снижения температуры восстановления Ni по сравнению с примером 1-2. По мере протекания реакции количество закоксовывания катализатора значительно увеличивалось, и отличие от катализатора примера 1-2 становилось более очевидным.
В сравнительном примере 1-3, в котором Pd обеспечивали растворным способом, Pd входил в мелкие поры и обладал очень высокой активностью по сравнению с примером 1-3. Начальная конверсия ацетилена в сравнительном примере 1-3 достигала 100%. Учитывая, что Pd не присутствовал в больших порах в сравнительном примере 1-3, Ni-Cu не восстанавливали при 150°С, наблюдалось большое количество закоксовывания через 1000 ч, и производительность катализатора существенно снизилась.
В сравнительном примере 1-4, в котором не было обеспечения Ni, из-за снижения недеструктивного гидрирования зеленого масла количество закоксовывания катализатора через 1000 часов было большим, и производительность катализатора существенно снизилась по сравнению с примером 1-4.
В сравнительном примере 1-5 содержание Ni было снижено до 0,3%. При этом содержании и условиях реакции активность гидрирования активного центра Ni-Cu стала недостаточной для обеспечения недеструктивного гидрирования выбранных побочных продуктов гидрирования. По сравнению с примером 1-5, производительность катализатора значительно снизилась через 1000 ч.
В сравнительном примере 1-6, в котором катализатор получали при тех же условиях, что и в примере 1-6, за исключением того, что количество Pd, обеспечиваемое способом микроэмульсии, было значительно уменьшено, из-за снижения содержания Pd, не получилось значительно снизить температуру восстановления Ni, и, следовательно, Ni недостаточно функционировал при недеструктивном гидрировании побочных продуктов, также с заметной разницей в производительности через 1000 часов.
По сравнению с примером 1-7, в сравнительном примере 1-7 удельная площадь поверхности была слишком большой, и размер активного центра Pd на катализаторе был слишком малым. Первоначальная активность была явно недостаточной, несмотря на хорошую начальную селективность. Кроме того, из-за чрезмерно большой удельной площади поверхности активность в активном центре Ni-Cu была недостаточной, и количество закоксовывания через 1000 часов было значительно выше, чем в примере 1-7.
В примерах 1-8 катализатор был получен сначала путем нанесения Ag, и Ag образовал сплав с Pd. Более высокое содержание Ag в этой структуре сплава снижало активность селективного гидрирования, поэтому его начальная активность была хуже, чем в примере 1-7.
В сравнительном примере 1-8, когда Pd обеспечивали микроэмульсией, диаметр частиц микроэмульсии был больше, чем максимальный размер пор носителя, так что Pd не мог проникать в поры носителя, и только его часть обеспечивали на внешней поверхности катализатора, в то время как другая часть терялась вместе с раствором. Несмотря на хорошую начальную селективностью, Pd не мог эффективно образовывать сплав с Ni-Cu, и, таким образом, не мог эффективно снижать температуру восстановления Ni-Cu. Кроме того, недеструктивное гидрирование молекул зеленого масла протекало неэффективно. Следовательно, через 1000 часов производительность катализатора в сравнительном примере 1-8 была значительно хуже, чем в примере 1-8.
В сравнительном примере 1-9 микроэмульсия для обеспечения Pd имела диаметр частиц 398 нм, и микроэмульсия для обеспечения Ni-Cu имела диаметр частиц 621 нм, что было больше, чем максимальный размер пор носителя. Микроэмульсия, содержащая Ni-Cu, не могла попасть в поры носителя, но только ее часть адсорбировалась на поверхности носителя, и другая часть терялась вместе с микроэмульсией. Поскольку адсорбированная часть не могла образовывать сплав с Ni-Cu, часть Ni-Cu не могла быть эффективно восстановлена при температуре восстановления 150°С, и недеструктивное гидрирование молекул зеленого масла проходило неэффективно. Следовательно, через 1000 часов результаты реакции катализатора в сравнительном примере 1-9 были хуже, чем в примере.
В сравнительном примере 1-10 удельная площадь поверхности катализатора была небольшой, и размер активного центра Pd был слишком большим для того же обеспеченного количества, что приводило к высокой активности, но плохой селективности. Кроме того, активный центр Ni-Cu имел еще большую размерность и функционировал не только при недеструктивном гидрировании побочных продуктов, но и при гидрировании этилена, что приводило к плохой селективности при гидрировании этилена. Что касается количества закоксовывания, хотя в сравнительном примере 1-10 через 1000 часов закоксовывания было меньше, селективность была значительно ниже, чем в примере.
В сравнительном примере 1-11, когда компоненты соответственно обеспечивали микроэмульсией, размер пор эмульсии был меньше, чем максимальный размер мелких пор носителя, так что каждый компонент обеспечивали в мелких порах. Поскольку все эти компоненты, обладающие активностью гидрирования, были собраны в мелких порах, активный центр гидрирования в мелких порах имел очень высокую активность, но плохую начальную селективность, и было получено еще больше побочных продуктов. Из-за неподходящего распределения положения активных центров Ni-Cu полученные побочные продукты не могли быть недеструктивным гидрированием, закоксовывание через 1000 часов было больше, и активность и селективность по этилену были хуже, чем в примере 1-11.
В сравнительном примере 1-12 было обеспечено очень небольшое количество Cu. Хотя Pd, Ni и Cu образуют сплав в больших порах, Cu может способствовать восстановлению Ni, поскольку его восстановление может быть более легким, чем восстановление Ni. Однако содержание Cu в этом сравнительном примере было слишком низким, чтобы способствовать восстановлению Ni, и закоксовывание катализатора через 1000 часов также было более интенсивным.
В сравнительном примере 1-13 весь Pd обеспечивали способом микроэмульсии, так что Pd не обеспечивали в оптимальном размере пор 20-30 нм. Кроме того, для всего сплава, образованного из Pd вместе с Ni и Cu, его активные центры были расположены в больших порах, и, несмотря на хорошую активность одного активного центра, собственная селективность была плохой, и под влиянием диффузии активные центры в больших порах не могли легко контактировать с молекулами ацетилена, что приводило к плохой начальной активности и плохой селективности.
В сравнительном примере 1-14 Ni и Cu были обеспечены раствором, и они были равномерно распределены в носителе, и было трудно сформировать каталитический реакционный центр с хорошей активностью. Часть Pd, обеспечиваемого растворным способом, образовывала активные центры на наружных слоях носителя, но присутствие Ni и Cu было неблагоприятным для улучшения селективности активных центров Pd и даже оказывало отрицательное влияние. Таким образом, катализатор обладал достаточной начальной активностью, но плохой начальной селективностью. Без активных центров недеструктивного гидрирования побочных продуктов гидрирования наблюдалось большое количество закоксовывания и серьезное ухудшение производительности через 1000 часов.
В сравнительном примере 1-15 применяли носитель с унимодальным распределением пор по размерам, и диаметр частиц полученной микроэмульсии был больше, чем максимальный размер пор носителя, что не позволяло эмульсии поступать в носитель. Некоторые из активных компонентов могли быть распределены только в самом внешнем слое носителя, и некоторые из них не могли быть эффективно обеспечены и были потеряны. Хотя они могли функционировать при недеструктивном гидрировании до определенной степени на наружной поверхности, каталитический эффект был еще хуже, чем в примере 1-15.
Пример 2-1
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1058°С в течение 4 ч бимодальное распределение пор по размерам составляло 15-32 нм и 80-204 им, водопоглощение составляло 60%, и удельная площадь поверхности составляла 47,28 м2/г. 100 г носителя взвешивали.
(1) 15,94 г безводного нитрата никеля и 1,47 г нитрата меди взвешивали и растворяли в 72 мл деионизированной воды, к которой добавляли 36 г н-гексана, 21,6 г ЦТАБ и 21,50 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель пропитывали в полученной микроэмульсии в течение 30 мин, отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А.
(2) 0,079 г хлорида палладия растворяли в 120 мл деионизированной воды и значение рН доводили до 1,5. Полуфабрикат катализатора А пропитывали в полученном солевом растворе Pd в течение 30 мин, сушили при 100°С и прокаливали при 400°С в течение 6 ч, с получением полуфабриката катализатора В.
(3) 0,071 г хлорида палладия растворяли в 72 мл деионизированной воды, к которой добавляли 36 г н-гексана, 21,6 г ЦТАБ и 21,50 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В пропитывали в полученной микроэмульсии в течение 30 мин, отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 50,29 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 51,45 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в примере 2-1, содержание Pd составляло 0,08%, содержание Ni составляло 5,0%, и содержание Cu составляло 0,5%.
Восстановление катализатора: восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 8 часов.
Сравнительный пример 2-1А:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1058°С в течение 4 ч бимодальное распределение пор по размерам составляло 15-32 нм и 80-204 нм, водопоглощение составляло 60%, и удельная площадь поверхности составляла 47,28 м2/г. 100 г носителя взвешивали.
(1) 15,94 г безводного нитрата никеля взвешивали и растворяли в 72 мл деионизированной воды, к которой добавляли 36 г н-гексана, 21,6 г ЦТАБ и 21,50 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель пропитывали в полученной микроэмульсии в течение 30 мин, отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А1-1.
(2) 0,079 г хлорида палладия растворяли в 120 мл деионизированной воды и значение рН доводили до 1,5. Полуфабрикат катализатора А-1 пропитывали в полученном солевом растворе Pd в течение 30 мин, сушили при 100°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В1-1.
(3) 0,071 г хлорида палладия растворяли в 72 мл деионизированной воды, к которой добавляли 36 г н-гексана, 21,6 г ЦТАБ и 21,50 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В1-1 пропитывали в полученной микроэмульсии в течение 30 мин, отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 50,31 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 51,38 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и было определено, что в катализаторе, полученном в сравнительном примере 2-1 А, содержание Pd составляло 0,08%, и содержание Ni составляло 5,2%.
Восстановление катализатора: восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 8 часов.
Сравнительный Пример 2-1В
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1058°С в течение 4 ч бимодальное распределение пор по размерам составляло 15-32 нм и 80-204 нм, водопоглощение составляло 60%, и удельная площадь поверхности составляла 47,28 м2/г. 100 г носителя взвешивали.
(1) 1,47 г нитрата меди взвешивали и растворяли в 72 мл деионизированной воды, к которой добавляли 36 г н-гексана, 21,6 г ЦТАБ и 21,50 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель пропитывали в полученной микроэмульсии в течение 30 мин, отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора А1-2.
(2) 0,079 г хлорида палладия растворяли в 120 мл деионизированной воды и значение рН доводили до 1,5. Полуфабрикат катализатора А1-2 пропитывали в полученном солевом растворе Pd в течение 30 мин, сушили при 100°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В1-2.
(3) 0,071 г нитрата палладия растворяли в 72 мл деионизированной воды, к которой добавляли 36 г н-гексана, 21,6 г ЦТАБ и 21,50 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В1-2 пропитывали в полученной микроэмульсии в течение 30 мин, отфильтровывали остаточную жидкость, сушили при 40°С и прокаливали при 400°С в течение 6 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 50,31 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 51,49 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и было определено, что в катализаторе, полученном в сравнительном примере 2-1В, содержание Pd составляло 0,08%, и содержание Cu составляло 0,52%.
Восстановление катализатора: восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 8 часов.
Эффект внедрения
Условия реакции гидрирования:
Применяли двухступенчатый реактор с массовой скоростью воздуха 4000/ч, рабочим давлением 3,0 МПа и объемом загрузки катализатора 200 мл. Условия реактора приведены в Таблице 2-1. Результаты реакции приведены в Таблице 2-2.
Массовый состав: вход в реактор первой ступени, ацетилен: 1,8% (об./об.), этилен 80% (об./об.).
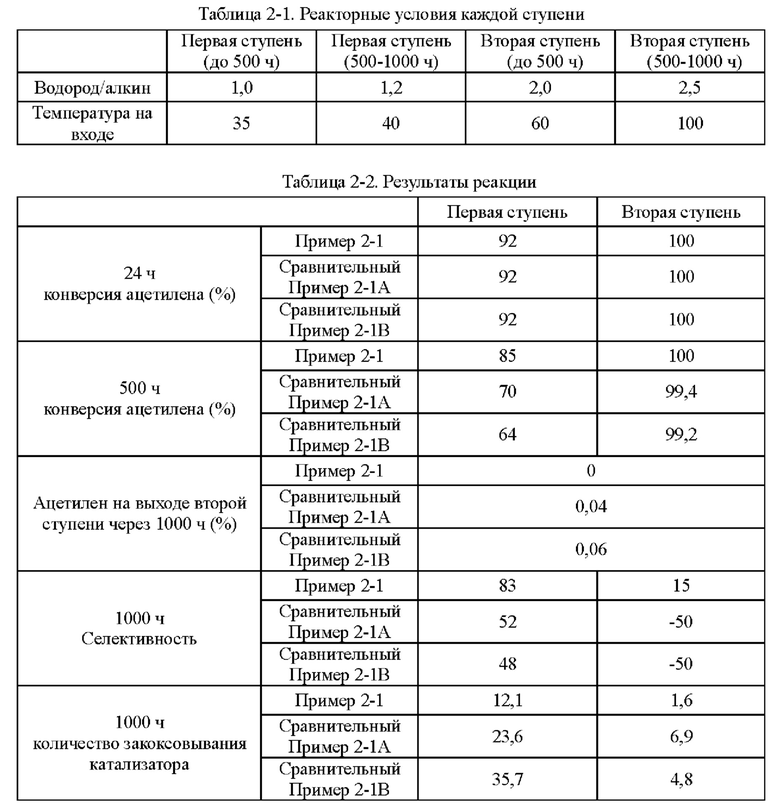
Из результатов оценки, приведенных в Таблице 2-2, активность реакции и селективность в Сравнительных примерах 2-1А и 1В были такими же, как в Примере 2-1, в течение 24 часов. Однако через 500 и 1000 часов реакционная активность и селективность в сравнительных примерах 2-1А и 1В становились все более низкими, чем в примере 2-1. В результате отсутствия Ni и Cu гидрирующая активность насыщения для побочных продуктов гидрирования была недостаточной, что приводило к сильному закоксовыванию катализатора, и, следовательно, активность и селективность снижались. Ацетилен на выходе из реактора превысил требуемое значение, и этилен был недостаточно качественным.
Пример 2-2
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1115°С в течение 4 ч бимодальное распределение пор по размерам составляло 30-43 нм и 112-496 нм, водопоглощение составляло 50%, и удельная площадь поверхности составляла 21,74 м2/г. 100 г носителя взвешивали.
(1) 3,19 г безводного нитрата никеля и 2,95 г нитрата меди взвешивали и растворяли в 72 мл деионизированной воды, к которой добавляли 26,67 г циклогексана, 4,0 г Triton Х-100 и 3,50 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Носитель пропитывали полученной микроэмульсией в течение 4 ч, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А2.
(2) 0,043 г нитрата палладия растворяли в 80 г деионизированной воды, к которой добавляли 26,67 г циклогексана, 4,0 г Triton Х-100 и 3,50 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А2 пропитывали в полученной микроэмульсии в течение 4 ч, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора В2.
(3) 0,067 г хлорида палладия растворяли в 120 мл деионизированной воды и значение рН доводили до 2,5. Полуфабрикат катализатора В2 пропитывали в полученном солевом растворе Pd в течение 1 ч, сушили при 120°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света (лазерным рассеянием света), диаметр частиц микроэмульсии, полученной на стадии (1), составлял 487,63 нм, и диаметр частиц микроэмульсии, полученной на стадии (2), составлял 486,19 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и было определено, что в катализаторе, полученном в примере 2-2, содержание Pd составляло 0,065%, содержание Ni составляло 1,1%, и содержание Cu составляло 1,1%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 200°С, выдержка в течение 8 часов.
Сравнительный Пример 2-2
Приготовление катализатора:
Применяли коммерчески доступный унимодальный сферический носитель оксид алюминия, имеющий диаметр 4 мм. После прокаливания при 1115°С в течение 4 ч бимодальное распределение пор по размерам составляло 30-43 нм и 32-88 нм, водопоглощение составляло 50%, и удельная площадь поверхности составляла 21,74 м2/г. 100 г носителя взвешивали.
(1) 3,19 г безводного нитрата никеля и 2,95 г нитрата меди взвешивали и растворяли в 72 мл деионизированной воды, к которой добавляли 26,67 г циклогексана, 4,0 г Triton Х-100 и 3,50 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Носитель пропитывали полученной микроэмульсией в течение 4 ч, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А2-1.
(2) 0,043 г нитрата палладия растворяли в 80 г деионизированной воды, к которой добавляли 26,67 г циклогексана, 4,0 г Triton Х-100 и 3,50 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А2-1 на стадии (1) пропитывали в приготовленной микроэмульсии в течение 4 ч, отфильтровывали остаточную жидкость, промывали деионзированной водой, сушили при 80°С и прокаливали при 600°С в течение 4 ч, с получением полуфабриката катализатора В2-1.
(3) 0,067 г хлорида палладия растворяли в 120 мл деионизированной воды и значение рН доводили до 2,5. Полуфабрикат катализатора В2 пропитывали в полученном солевом растворе Pd в течение 1 ч, сушили при 120°С и прокаливали при 600°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 487,65 им, и диаметр частиц микроэмульсии, полученной на стадии (2), составлял 486,22 им.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и было определено, что в катализаторе, полученном в сравнительном примере 2-2, содержание Pd составляло 0,06%, содержание Ni составляло 1,0%, и содержание Cu составляло 1,1%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 200°С, выдержка в течение 8 часов.
Эффект внедрения
Условия реакции гидрирования 1
Применяли способ с трехступенчатым реактором с массовой скоростью воздуха 11000/ч, рабочим давлением 1,5 МПа и объемом загрузки катализатора 300 мл. Реакторные условия приведены в Таблице 2-3. Результаты реакции приведены в Таблице 2-4.
Массовый состав: вход в реактор первой ступени, ацетилен: 2,5% (об./об.), этилен 93% (об./об.).
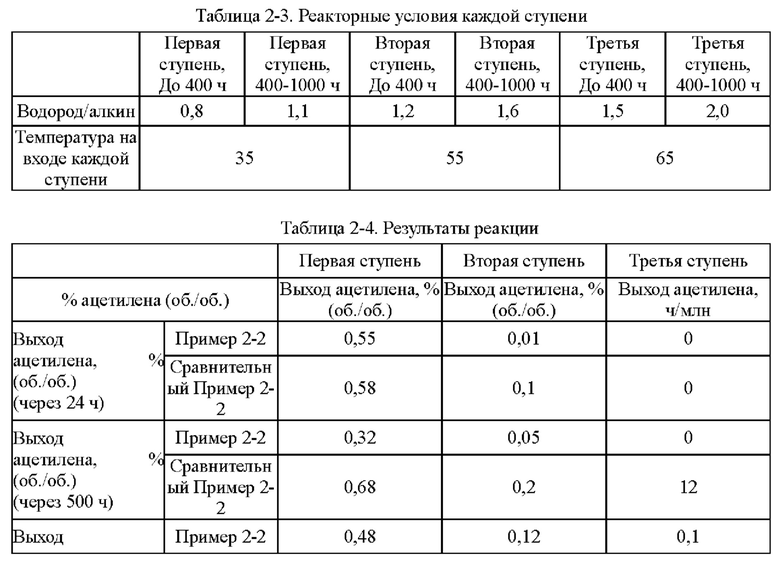
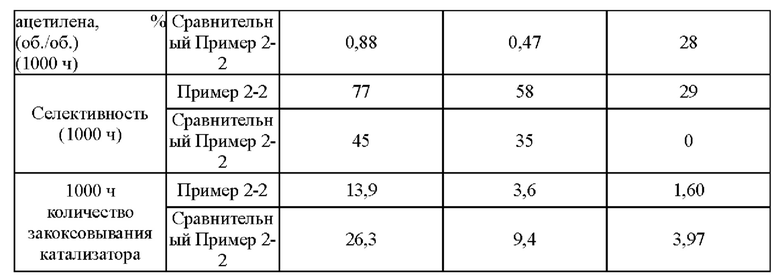
Условия реакции гидрирования 2
Применяли способ с одноступенчатым реактором с массовой скоростью воздуха 4000/ч, рабочим давлением 1,6 МПа и объемом загрузки катализатора 200 мл.
Массовый состав на входе в реактор: ацетилен 0,2% (об./об.), этилен 65% (об./об.), С3 0,8% (об./об.). Реакторные условия приведены в Таблице 2-5.
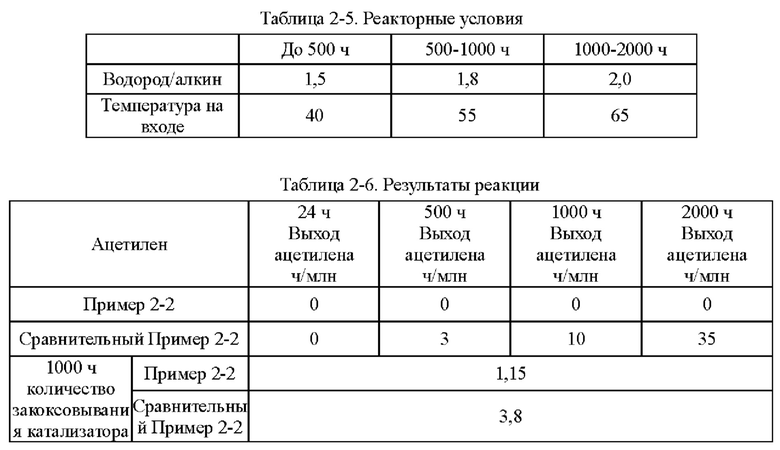
Из результатов, приведенных в таблице 2-4, сравнительный пример 2-2 и пример 2-2 отличались в начале реакции тем, что носитель в сравнительном примере 2-2 имел унимодальное распределение пор по размерам без больших пор, и, следовательно, активные компоненты в микроэмульсии не могли проникать в поры носителя или полуфабриката катализатора, полностью присутствуя на внешней поверхности носителя и закупоривая поровые каналы носителя, что приводило к снижению активности катализатора.
По мере протекания реакции образующиеся побочные продукты накапливались в слое оболочки катализатора и образовывали закоксовывание. Характеристики катализатора в Сравнительном примере 2-2 постепенно снижались. Активный компонент, расположенный на поверхности катализатора, до некоторой степени функционировал при недеструктивном гидрировании побочных продуктов, но допускал только часть побочных продуктов для недеструктивного гидрирования. Засорение пор постепенно ухудшалось, и производительность катализатора значительно снижалась.
В таблице 2-6 показаны результаты одноступенчатого реактора. Хотя содержание ацетилена на входе в реактор было низким, высокое содержание С3 также способствовало образованию большего количества побочных продуктов в реакции гидрирования. До 2000 часов содержание ацетилена на выходе из реактора последовательно составляло 0 в Примере, но в Сравнительном примере, поскольку только небольшое количество Ni-Cu адсорбировалось на внешней поверхности катализатора, и недеструктивное гидрирование большинства побочных продуктов гидрирования было неудачным, побочные продукты накапливались с образованием закоксовывания, в то время как селективность катализатора снижалась, и удаление ацетилена было неэффективным. Через 1000 часов результаты реакции были недостаточно хорошими.
Пример 2-3
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1092°С в течение 4 ч бимодальное распределение пор по размерам составляло 20-46 нм и 85-350 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 39,58 м2/г. 100 г носителя взвешивали.
(1) 0,051 г хлорида палладия растворяли в 140 мл деионизированной воды и значение рН доводили до 2. Носитель пропитывали в полученном солевом растворе Pd в течение 50 мин, сушили при 110°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора A3.
(2) 1,59 г безводного нитрата никеля и 1,47 г нитрата меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 28,80 г н-гексана, 17,16 г ЦТАБ и 15,6 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора A3 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора В3.
(3) 0,0107 г нитрата палладия взвешивали и растворяли в 71,5 г воды, к которой добавляли 28,80 г н-гексана, 17,16 г ЦТАБ и 15,6 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В3 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора С3.
(4) 0,126 г нитрата серебра полностью растворяли в 49,5 мл деионизированной воды и значение рН доводили до 2. Затем полуфабрикат катализатора С3 пропитывали в полученном растворе и встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 500°С в течение 5 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (2), составлял 78,38 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 77,64 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и было определено, что в катализаторе, полученном в Примере 2-3, содержание Pd составляло 0,035%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,08%.
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 4 часов.
Сравнительный Пример 2-3
Приготовление катализатора:
Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1092°С в течение 4 ч бимодальное распределение пор по размерам составляло 20-46 нм и 85-350 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 39,58 м2/г. 100 г носителя взвешивали.
(1) 0,059 г хлорида палладия растворяли в 140 мл деионизированной воды и значение рН доводили до 2. Носитель пропитывали в полученном солевом растворе Pd в течение 50 мин, сушили при 110°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора А3-1.
(2) 1,59 г безводного нитрата никеля и 1,47 г нитрата меди взвешивали и растворяли в 71,5 г воды, к которой добавляли 28,80 г н-гексана, 17,16 г ЦТАБ и 15,6 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора А3-1 пропитывали в приготовленной микроэмульсии в течение 80 мин, отфильтровывали остаточную жидкость, промывали деионизированной водой до нейтрального состояния, сушили при 80°С и прокаливали при 550°С в течение 5 ч с получением полуфабриката катализатора В3-1.
(3) 0,126 г нитрата серебра полностью растворяли в 49,5 мл деионизированной воды и значение рН доводили до 2. Затем полуфабрикат катализатора В3-1 пропитывали в полученном растворе и встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 500°С в течение 5 ч с получением требуемого катализатора.
Диаметр частиц микроэмульсии, полученной на стадии (2), определяемый динамическим рассеянием света, составлял 78,45 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 2-3А, содержание Pd составляло 0,035%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,08%
Восстановление катализатора:
Восстановительный газ: водород, скорость восстановительного газа: 100 ч-1, температура 150°С, выдержка в течение 4 часов.
Условия реакции гидрирования:
Применяли способ с одноступенчатым реактором с массовой скоростью воздуха 2000/ч, рабочим давлением 3,0 МПа и объемом загрузки катализатора 200 мл. Реакторные условия приведены в Таблице 2-7. Результаты реакции приведены в Таблице 2-8.
Массовый состав:
Материал на входе в реактор: ацетилен 0,5% (об./об.), этилен 70%(об./об.), С3 0,8%(об./об.).

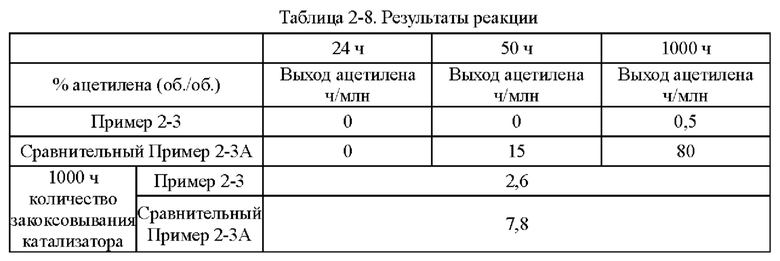
Из результатов, приведенных в Таблице 2-8, в Сравнительном примере 2-3А, ацетилен на выходе из реактора находился в крайнем избытке через 1000 часов, что указывает на то, что производительность катализатора была полностью недостаточной. Это было главным образом связано с тем, что палладий не обеспечивали эмульсионным способом и что оксиды никеля и меди не восстанавливались до их металлического состояния при 150°С и, таким образом, не могли воздействовать на недеструктивное гидрирование тяжелых фракций побочных продуктов, что приводило к более быстрому закоксовыванию катализатора.
Пример 2-4
Носитель: Применяли коммерчески доступный сферический носитель оксид алюминия с бимодальным распределением пор по размерам, 3 мм в диаметре. После прокаливания при 1078°С в течение 4 ч бимодальное распределение пор по размерам находилось в диапазоне 20-35 нм и 90-250 нм, водопоглощение составляло 57%, и удельная площадь поверхности составляла 44,82 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 3,18 г безводного нитрата никеля и 0,74 г нитрата меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 35 г н-гексана, 21 г ЦТАБ и 20 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора А4.
(2) 0,069 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А4 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора В4.
(3) 0,236 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора В4, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 550°С в течение 6 ч с получением полуфабриката катализатора С4.
(4) 0,016 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 35 г н-гексана, 21 г ЦТАБ и 20 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С4 растворяли в полученной микроэмульсии, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 6 часов с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 51,19 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 50,85 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в примере 2-4 содержание Pd составляло 0,05%, содержание Ni составляло 1%, содержание Cu составляло 0,25%, и содержание Ag составляло 0,14%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 190°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 2-4А
Приготовление катализатора:
Носитель: Применяли коммерчески доступный сферический носитель оксид алюминия с бимодальным распределением пор по размерам, 3 мм в диаметре. После прокаливания при 1078°С в течение 4 ч бимодальное распределение пор по размерам находилось в диапазоне 20-35 нм и 90-250 нм, водопоглощение составляло 62%, и удельная площадь поверхности составляла 44,82 м2/г. 100 г носителя взвешивали.
(1) 3,18 г безводного нитрата никеля и 0,74 г нитрата меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 35 г н-гексана, 21 г ЦТАБ и 20 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора А4.
(2) 0,069 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А4-1 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора В4-1.
(3) 0,236 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора В4-1, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 550°С в течение 6 ч с получением полуфабриката катализатора С4-1.
(4) 0,016 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 38 г н-гексана, 24 г ЦТАБ и 24 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С4-1 растворяли в полученной микроэмульсии, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 6 ч с получением требуемого катализатора.
Диаметр частиц микроэмульсии, полученной на стадии (1), как определено динамическим рассеянием света, составлял 52,18 нм.
Диаметр частиц микроэмульсии, полученной на стадии (4), определяемый динамическим рассеянием света, составлял 26,58 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 2-4А, содержание Pd составляло 0,05%, содержание Ni составляло 1%, содержание Cu составляло 0,25%, и содержание Ag составляло 0,15%
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 190°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Сравнительный Пример 2-4В
Носитель: Применяли коммерчески доступный сферический носитель оксид алюминия с бимодальным распределением пор по размерам, 3 мм в диаметре. После прокаливания при 1078°С в течение 4 ч бимодальное распределение пор по размерам находилось в диапазоне 20-35 нм и 90-250 нм, водопоглощение составляло 57%, и удельная площадь поверхности составляла 44,82 м2/г. 100 г носителя взвешивали.
(1) 3,18 г безводного нитрата никеля и 0,74 г нитрата меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 35 г н-гексана, 21 г ЦТАБ и 20 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Носитель после высокотемпературного прокаливания, взвешенный в количестве 100 г, пропитывали в приготовленной микроэмульсии и встряхивали в течение 90 мин, затем отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора А4-2.
(2) 0,069 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А4-2 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 100°С и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора В4-2.
(3) 0,236 г нитрата серебра растворяли в 57 мл деионизированной воды и значение рН доводили до 3. Полуфабрикат катализатора В4-2, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 140°С и прокаливали при 550°С в течение 6 ч с получением полуфабриката катализатора С4-2.
(4) 0,016 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 22,15 г н-гексана, 2,79 г ЦТАБ и 2,75 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С4-2 растворяли в полученной микроэмульсии, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 6 ч с получением требуемого катализатора.
Диаметр частиц микроэмульсии, полученной на стадии (1), как определено динамическим рассеянием света, составлял 52,13 нм.
Диаметр частиц микроэмульсии, полученной на стадии (4), как определено динамическим рассеянием света, составлял 595,09 нм.
Элементное содержание полученных катализаторов определяли с помощью атомно-абсорбционной спектроскопии. В катализаторе, полученном в сравнительном примере 2-4В, содержание Pd составляло 0,05%, содержание Ni составляло 1,0%, содержание Cu составляло 0,25%, и содержание Ag составляло 0,15%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 190°С с применением газовой смеси с молярным соотношением N2:H2=1:1.
Эффект внедрения
Условия реакции гидрирования 1
Применяли способ с двухступенчатым реактором с массовой скоростью воздуха 5000/ч, рабочим давлением 2,2 МПа и объемом загрузки катализатора 200 мл. Реакторные условия приведены в Таблице 2-9. Результаты реакции приведены в Таблице 2-10.
Массовый состав:
материал на входе в реактор первой ступени: ацетилен 1,4% (об./об.), этилен 85% (об./об.), С3 0,5%(об./об.).
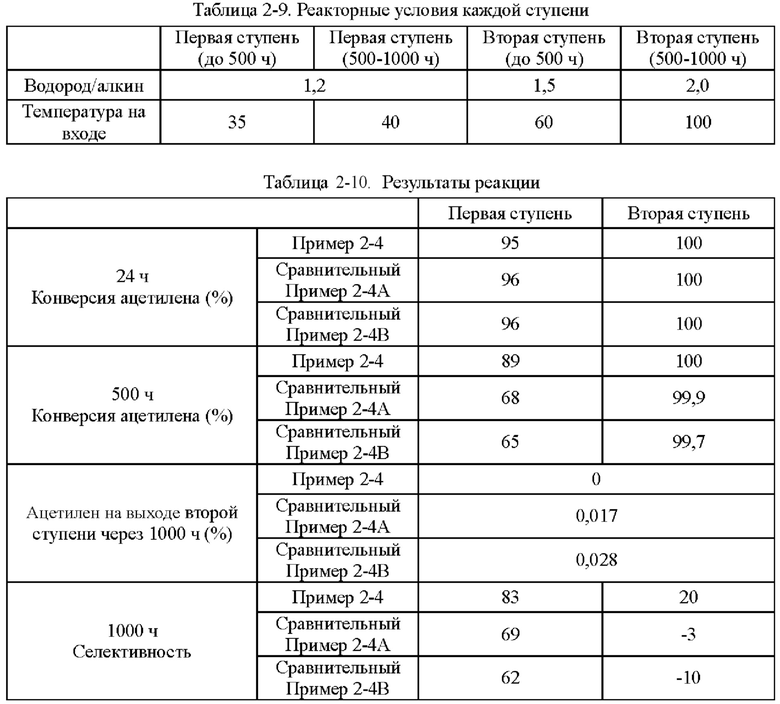
Условия реакции гидрирования 2
Применяли способ с трехступенчатым реактором, в котором первая и вторая ступени находились в адиабатических реакторах с массовой скоростью воздуха 5000/ч, и третья ступень находилась в изотермическом реакторе с массовой скоростью воздуха 11000/ч. Рабочее давление составляло 2,5 МПа, и объем загрузки катализатора на первой и второй ступенях составлял 1000 мл. Реакторные условия приведены в Таблице 2-11. Результаты реакции приведены в Таблице 2-12.
Массовый состав: ацетилен на входе в реактор первой ступени: 1,5% (об./об.), этилен 93% (об./об.).
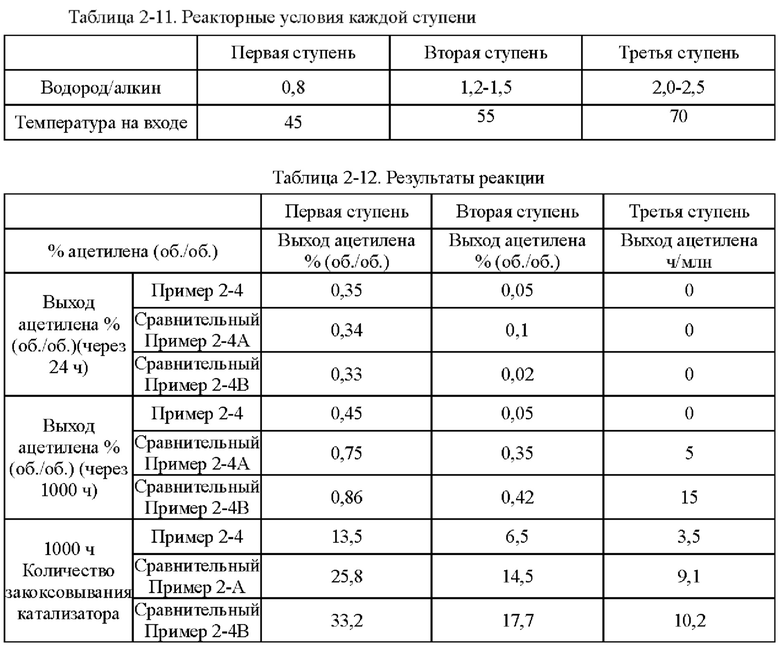
Из результатов, приведенных в таблицах 2-10 и 2-12, активность и селективность сравнительных примеров 2-4А и 4В были немного выше, чем в примере 2-4 в начале реакции, что указывает на то, что независимо присутствующий палладий обладает активностью гидрирования и может реагировать с ацетиленом с образованием этилена и тому подобное. Однако по мере протекания реакции селективность активности катализаторов сравнительных примеров 2-4А и 4В становилась все более уступающей селективности катализатора примера 2-4. К 1000 часам содержание ацетилена на выходе из реактора было несоответствующим. Что касается количеств закоксовывания в таблицах 2-12, закоксовывание катализатора в реакторе первой ступени было значительным, что указывает на то, что низкое содержание водорода/ацетилена с большей вероятностью вызывало закоксовывание катализатора. Дополнительно продемонстрировано, что настоящий катализатор обладает превосходными антикоксовыми свойствами.
Пример 2-5
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия и магния с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и содержание оксида магния 3%. После прокаливания при 1065°С в течение 4 ч бимодальное распределение пор по размерам составляло 23-47 нм и 80-380 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 42,38 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 0,051 г хлорида палладия растворяли в 80 мл деионизированной воды и значение рН доводили до 3. Затем носитель пропитывали в полученном солевом растворе Pd в течение 120 мин, сушили при 130°C и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А5.
(2) 0,33 г нитрата серебра растворяли в 58 мл деионизированной воды и значение рН доводили до 5. Полуфабрикат катализатора А5, полученный на стадии (1), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 100°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора В5.
(3) 0,017 г хлорида палладия взвешивали и растворяли в 80 мл деионизированной воды, к которой добавляли 36,00 г н-гексана, 20,88 г Triton Х-100 и 19,4 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В5 пропитывали полученной микроэмульсией, встряхивали в течение 180 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора С5.
(4) 1,648 г хлорида палладия и 2,175 г нитрата меди растворяли в 80 мл деионизированной воды, к которой добавляли 36,00 г н-гексана, 20,88 г Triton Х-100 и 19,60 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полученный полуфабрикат катализатора С5 на стадии (2) пропитывали полученной микроэмульсией, встряхивали в течение 180 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 65,0 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 65,24 нм.
Элементное содержание полученных катализаторов определяли с помощью атомно-абсорбционной спектроскопии. В катализаторе, полученном в Примере 2-5, содержание Pd составляло 0,04%, содержание Ni составляло 0,75%, содержание Cu составляло 0,75%, и содержание Ag составляло 0,21%.
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 180°С с применением газовой смеси с молярным соотношением N2:Н2=1:1.
Сравнительный Пример 2-5
Катализатор был приготовлен с применением того же носителя и условий, что и в Примере 2-5, за исключением того, что Pd не обеспечивали растворным способом.
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия и магния с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и содержание оксида магния 3%. После прокаливания при 1000°С в течение 4 ч бимодальное распределение пор по размерам составляло 23-47 нм и 80-380 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 40 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 0,069 г хлорида палладия растворяли в 80 мл деионизированной воды и значение рН доводили до 3. Затем носитель пропитывали в полученном солевом растворе Pd в течение 120 мин, сушили при 130°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А5-1.
(2) 0,33 г нитрата серебра растворяли в 58 мл деионизированной воды и значение рН доводили до 5. Полуфабрикат катализатора А5-1, полученный на стадии (1), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 100°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора В5-1.
(3) 1,648 г хлорида никеля и 2,175 г нитрата меди взвешивали и растворяли в 80 мл деионизированной воды, к которой добавляли 36,00 г н-гексана, 20,88 г Triton Х-100 и 19,4 г н-гексанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В5-1, полученный на стадии (2), пропитывали полученной микроэмульсией, встряхивали в течение 180 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 500°С в течение 4 ч с получением требуемого катализатора.
Диаметр частиц микроэмульсии, полученной на стадии (4), определяемый динамическим рассеянием света, составлял 65,20 нм.
Полученный катализатор исследовали с помощью атомно-абсорбционной спектрометрии, и в катализаторе, полученном в сравнительном примере 2-5, содержание Pd составляло 0,04%, содержание Ni составляло 0,75%, содержание Cu составляло 0,75%, b содержание Ag составляло 0,21%
Восстановление катализатора:
Катализатор помещали в реакционное устройство с неподвижным слоем перед применением и восстанавливали в течение 8 часов при температуре 180°С с применением газовой смеси с молярным соотношением N2:Н2=1:1.
Эффект внедрения
Условия реакции гидрирования
Применяли способ с трехступенчатым реактором, в котором каждая ступень представляла собой адиабатический реактор с массовой скоростью воздуха 11000/ч, рабочим давлением 2,3 МПа и объемом загрузки катализатора 1000 мл. Реакторные условия приведены в Таблице 2-13. Результаты реакции приведены в Таблице 2-14.
Массовый состав: ацетилен на входе в реактор первой ступени: 1,5% (об./об.), этилен 70% (об./об.).
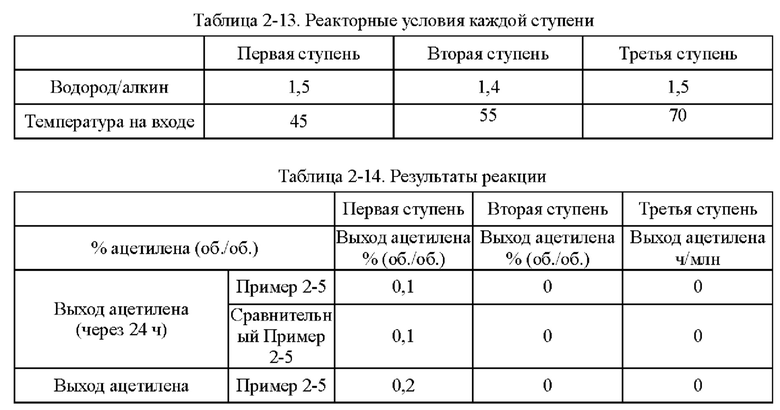
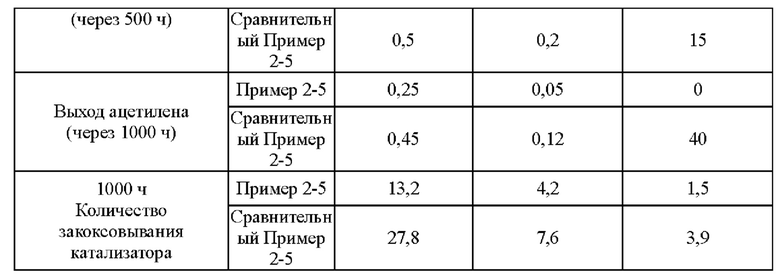
В таблице 2-14 для катализатора сравнительного примера 2-5 содержание ацетилена на выходе из реактора достигло 40 частей на миллион через 1000 часов, несмотря на то, что содержание ацетилена на входе в реактор третьей ступени составляло всего 0,12%, что указывало на то, что низкий уровень водорода/ацетилена на третьей ступени отрицательно сказывался на удалении ацетилена. Однако катализатор примера 2-5 все еще способен полностью превращать ацетилен, что указывает на то, что катализатор согласно настоящему изобретению имеет минимальное ухудшение производительности после 1000 часов работы. Напротив, катализатор сравнительного примера 2-5 был намного хуже, поскольку Pd не обеспечивали эмульсионным способом, что не позволяло восстанавливать никель и медь при 180°С.
Пример 3-1
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1005°С в течение 4 ч бимодальное распределение пор по размерам составляло 15-35 нм и 80-203 нм, водопоглощение составляло 63%, и удельная площадь поверхности составляла 48,45 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 15,6 г безводного нитрата никеля и 1,48 г нитрата меди взвешивали и растворяли в 63 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 400°С в течение 8 ч с получением полуфабриката катализатора А1.
(2) 0,151 г нитрата палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 1,3. Затем полуфабрикат катализатора А1 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В1.
(3) Полуфабрикат катализатора С, полученный на стадии (2), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 5 ч при температуре 190°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе примера 3-1 содержание Pd составляло 0,07%, содержание Ni составляло 5%, и содержание Cu составляло 0,50%.
Сравнительный Пример 3-1
Катализатор был приготовлен с применением того же носителя, условий и содержания других компонентов, что и в Примере 3-1, за исключением того, что не обеспечивали Cu.
(1) 1,59 г безводного нитрата никеля взвешивали и растворяли в 63 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 400°С в течение 8 ч с получением полуфабриката катализатора А1-1.
(2) 0,151 г нитрата палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 1,3. Затем полуфабрикат катализатора А1-1 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В1-1.
(3) Полуфабрикат катализатора В1-1, полученный на стадии (2), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 5 ч при температуре 190°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе сравнительного примера 3-1 содержание Pd составляло 0,07%, и содержание Ni составляло 5%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя) с массовой скоростью воздуха 6000 ч-1, рабочим давлением 1,5 МПа, отношением водорода к алкину 2,0 и температурой на входе в реактор 70°С. Состав реакционной массы представлен в Таблице 3-1, и результаты работы представлены в Таблице 3-2. Состав неочищенного водорода: Н2 20%, содержание СО 1%.
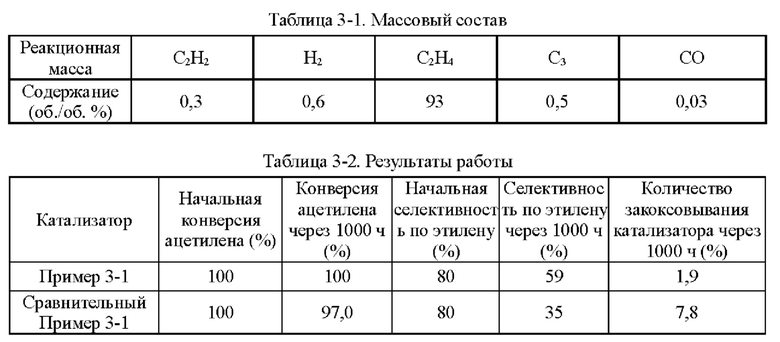
По сравнению с примером 3-1, в сравнительном примере 3-1 не обеспечивают Cu.
В примере 3-1 из-за большого количества обеспеченных Ni и Cu наблюдалась тенденция к равномерному распределению в носителе, и количество Pd, обеспечиваемого позднее, было намного ниже, чем в первом случае, и на его распределение влияла кислотность раствора. Поэтому часть Pd образовывала отдельные активные центры, и часть образовывала сплав с Ni и Cu. Температура восстановления сплава Ni-Cu может быть снижена до 200°С или ниже в присутствии Pd, что позволяет проводить недеструктивное гидрирование побочных продуктов селективного гидрирования и обеспечивает лучшую производительность катализатора.
В сравнительном примере 3-1 из-за отсутствия Cu одного Pd было недостаточно для снижения температуры восстановления Ni до 200°С или ниже. Следовательно, катализатор не функционировал при недеструктивном гидрировании побочных продуктов селективного гидрирования, и закоксовывание катализатора происходило быстрее. Селективность и активность через 1000 ч были значительно ниже, чем в Примере 3-1.
Пример 3-2
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1005°С в течение 4 ч бимодальное распределение пор по размерам составляло 15-35 нм и 80-203 нм, водопоглощение составляло 63%, и удельная площадь поверхности составляла 48,45 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 15,6 г безводного нитрата никеля и 1,48 г нитрата меди взвешивали и растворяли в 63 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 400°С в течение 8 ч с получением полуфабриката катализатора А2.
(2) 0,140 г нитрата палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 2,5. Затем полуфабрикат катализатора А2 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В2.
(3) 0,011 г нитрата палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 1,0. Затем полуфабрикат катализатора В2 пропитывали в полученном растворе нитрата Pd в течение 20 мин, оставляли стоять в течение 4 ч или более, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора С2.
(4) Полуфабрикат катализатора С2, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 5 ч при температуре 190°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе примера 3-2 содержание Pd составляло 0,07%, содержание Ni составляло 5%, и содержание Cu составляло 0,50%.
Сравнительный Пример 3-2
Катализатор был приготовлен с применением того же носителя, условий и содержания других компонентов, что и в Примере 3-2, за исключением того, что отсутствовало обеспечение Cu.
Приготовление катализатора:
(1) 15,6 г безводного нитрата никеля взвешивали и растворяли в 63 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 400°С в течение 8 ч с получением полуфабриката катализатора А2-1.
(2) 0,140 г нитрата палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 2,5. Затем полуфабрикат катализатора А2-1 пропитывали в полученном растворе нитрата Pd в течение 20 мин, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В2-1.
(3) 0,011 г нитрата палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 1,0. Затем полуфабрикат катализатора В2 пропитывали в приготовленном растворе нитрата Pd в течение 20 мин, оставляли стоять в течение 4 ч или более, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора С2-1.
(4) Полуфабрикат катализатора С2-1, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 5 ч при температуре 190°С с применением газовой смеси с молярным соотношением N2:Н2=1:1 с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе сравнительного примера 3-2 содержание Pd составляло 0,07%, и содержание Ni составляло 5%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя) с массовой скоростью воздуха 4000 ч-1, рабочим давлением 1,5 МПа, отношением водорода к алкину 2,0 и температурой на входе в реактор 65°С. Состав реакционной массы представлен в Таблице 3-3, и результаты работы представлены в Таблице 3-4. Состав неочищенного водорода: H2 20%, содержание СО 1%.

По сравнению с примером 3-2, в сравнительном примере 3-2 не обеспечивают Cu.
В примере 3-2 для первого обеспечения Pd количество кислоты было низким из-за равномерного распределения Ni и Cu, и эта часть Pd была распределена в более внешних слоях оболочки и образовывала отдельные активные центры. Для второго обеспечения Pd кислотность раствора была высокой, и эта часть Pd образовывала сплав с Ni-Cu, так что температура восстановления сплава Ni-Cu была снижена до 200°С или ниже, что обеспечивало возможность недеструктивного гидрирования побочных продуктов селективного гидрирования и обеспечивало лучшую производительность катализатора.
В сравнительном примере 3-2 из-за отсутствия Cu одного Pd было недостаточно для снижения температуры восстановления Ni до 200°С или ниже. Следовательно, катализатор не функционировал при недеструктивном гидрировании побочных продуктов селективного гидрирования, и закоксовывание катализатора происходило быстрее. Селективность и активность через 1000 ч были значительно ниже, чем в Примере 3-2.
В примере 3-2, поскольку Pd был нанесен дважды, область, в которой был нанесен Pd, могла бы лучше контролироваться с образованием большего количества сплава Ni-Cu-Pd. Это было выгодно для недеструктивного гидрирования выбранных побочных продуктов гидрированием, и селективность после 1000 часов в примере 3-2 была лучше, чем в примере 3-1.
Пример 3-3
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1005°С в течение 4 ч бимодальное распределение пор по размерам составляло 15-35 нм и 80-203 нм, водопоглощение составляло 63%, и удельная площадь поверхности составляла 48,45 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 15,6 г безводного нитрата никеля и 1,48 г нитрата меди взвешивали и растворяли в 63 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 400°С в течение 8 ч с получением полуфабриката катализатора A3.
(2) 0,140 г хлорида палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 2,5. Затем полуфабрикат катализатора A3 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В3.
(3) 0,011 г нитрата палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 1,0. Затем полуфабрикат катализатора В3 пропитывали в полученном растворе нитрата Pd в течение 20 мин, оставляли стоять в течение 4 ч или более, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора С3.
(4) Полуфабрикат катализатора С3, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 5 ч при температуре 190°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе примера 3-3 содержание Pd составляло 0,07%, содержание Ni составляло 5%, и содержание Cu составляло 0,50%.
Сравнительный Пример 3-3
Катализатор был приготовлен с применением того же носителя и условий, что и в Примере 3-3, за исключением того, что не обеспечивали Cu.
Приготовление катализатора:
(1) 15,6 г безводного нитрата никеля взвешивали и растворяли в 63 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 10 мин, сушили при 100°С и прокаливали при 400°С в течение 8 ч с получением полуфабриката катализатора А3-1.
(2) 0,140 г хлорида палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 2,0. Затем полуфабрикат катализатора А3-1 пропитывали в полученном растворе нитрата Pd в течение 20 мин, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора В31.
(3) 0,011 г нитрата палладия растворяли в 70 мл деионизированной воды и значение рН доводили до 1,0. Затем полуфабрикат катализатора В3-1 пропитывали в полученном растворе нитрата Pd в течение 20 мин, оставляли стоять в течение 4 ч или более, сушили при 120°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора С3-1.
(4) Полуфабрикат катализатора С3-1, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 5 ч при температуре 190°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе сравнительного примера 3-3 содержание Pd составляло 0,07%, и содержание Ni составляло 5%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя) с массовой скоростью воздуха 5000 ч-1, рабочим давлением 1,5 МПа, отношением водорода к алкину 2,0 и температурой на входе в реактор 85°С. Состав реакционной массы представлен в Таблице 3-5, и результаты работы представлены в Таблице 3-6. Состав неочищенного водорода: Н2 20%, содержание СО 1%.
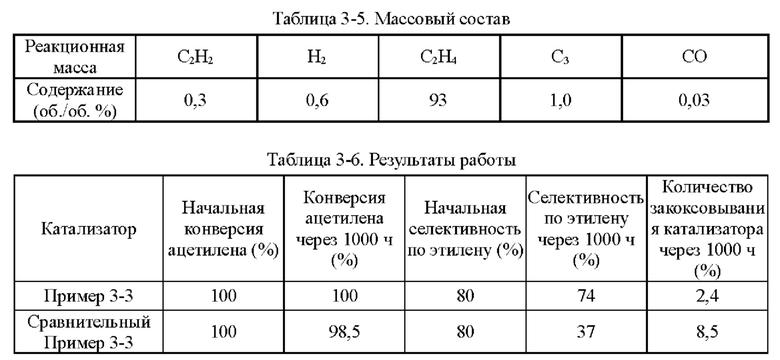
По сравнению с примером 3-3, в сравнительном примере 3-3 не обеспечивают Cu.
В примере 3-3 для первого обеспечения Pd применяли анионный предшественник, и Pd легче распределялся во внешнем слое оболочки, образуя отдельные активные центры, которые превратились в активный центр селективного гидрирования ацетилена. Вторую часть Pd обеспечивали при высокой кислотности раствора и длительной продолжительности обеспечения и имела тенденцию к более легкому образованию сплава с Ni и Cu. Температуру восстановления сплава Ni-Cu снижали до 200°С или ниже. Эта часть активных центров функционировала при недеструктивном гидрировании побочных продуктов селективного гидрирования и обеспечивала превосходные рабочие характеристики катализатора.
В сравнительном примере 3-3 из-за отсутствия Cu одного Pd было недостаточно для снижения температуры восстановления Ni до 200°С или ниже. Следовательно, катализатор не функционировал при недеструктивном гидрировании побочных продуктов селективного гидрирования, и закоксовывание катализатора происходило быстрее. Селективность и активность через 1000 ч были значительно ниже, чем в Примере 3-3.
Благодаря обеспечению с применением анионного предшественника он легко связывался с гидроксильной группой носителя и имел более узкое распределение, и, следовательно, селективность гидрирования была выше. Таким образом, как исходная селективность, так и селективность через 1000 ч в примере 3-3 были лучше, чем в примере 3-2.
Пример 3-4
Носитель: Применяли имеющийся в продаже цилиндрический носитель оксида алюминия с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и длину 3 мм. После прокаливания при 1018°С в течение 4 ч бимодальное распределение пор по размерам составляло 18-38 нм и 85-270 нм, водопоглощение составляло 61%, и удельная площадь поверхности составляла 43,62 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 4,41 г хлорида никеля и 2,12 г хлорида меди взвешивали и растворяли в 61 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 20 мин, сушили при 120°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А4.
(2) 0,093 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А4 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 100°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора В2.
(3) 0,0108 г нитрата палладия растворяли в 61 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора В2, полученный на стадии (2), пропитывали в полученном растворе и встряхивали в течение 10 мин, оставляли стоять в течение 4 ч или более, сушили при 100°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора С2.
(4) Полуфабрикат катализатора С2, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 7 часов при температуре 150°С с применением водорода с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе примера 3-4 содержание Pd составляло 0,060%, содержание Ni составляло 2,3%, и содержание Cu составляло 1,1%.
Сравнительный Пример 3-4
Катализатор был приготовлен с применением того же носителя и условий, что и в Примере 3-4, за исключением того, что не обеспечивали Ni.
Приготовление катализатора:
(1) 2,12 г хлорида меди взвешивали и растворяли в 61 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 20 мин, сушили при 120°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А4-1.
(2) 0,093 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А4 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 100°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора В4-1.
(3) 0,0108 г нитрата палладия растворяли в 61 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора В4-1, полученный на стадии (2), пропитывали в полученном растворе и встряхивали в течение 10 мин, оставляли стоять в течение 4 ч или более, сушили при 100°С и прокаливали при 400°С в течение 6 ч с получением полуфабриката катализатора С4-1.
(4) Полуфабрикат катализатора С4-1, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 7 часов при температуре 150°С с применением водорода с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе сравнительного примера 3-4 содержание Pd составляло 0,060%, и содержание Cu составляло 1,1%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя) с массовой скоростью воздуха 1500 ч-1, рабочим давлением 1,7 МПа, отношением водорода к алкину 2,5 и температурой на входе в реактор 55°С. Состав реакционной массы представлен в Таблице 3-7, и результаты работы представлены в Таблице 3-8. Состав неочищенного водорода: H2 50%, содержание СО 0,5%.
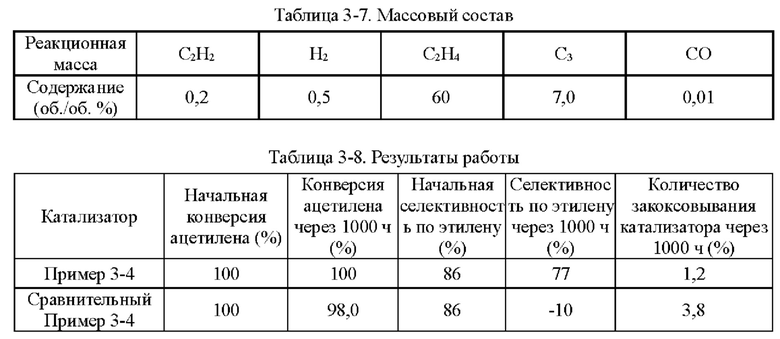
По сравнению с примером 3-4, в сравнительном примере 3-4 не обеспечивают Ni.
В сравнительном примере 3-4 из-за отсутствия никеля, хотя Cu и Pd образуют сплав, который легче восстанавливается, чем Ni, гидрирующая активность образованного активного центра была недостаточной для эффективного недеструктивного гидрирования побочных продуктов селективного гидрирования. Это привело к увеличению закоксовывания катализатора, недостаточной конверсии ацетилена в течение 1000 часов и плохой селективности.
Пример 3-5
Носитель: Использовали коммерчески доступный носитель оксида алюминия в форме клеверного листа с бимодальным распределением пор по размерам, имеющий диаметр 2 мм и длину 4 мм. После прокаливания при 1118°С в течение 4 ч бимодальное распределение пор по размерам составляло 30-49 нм и 187-499 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 20,80 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 8,29 г безводного нитрата никеля и 5,08 г хлорида меди взвешивали и растворяли в 55 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 20 мин, сушили при 80°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А5.
(2) 0,124 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А5 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 110°C и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора В5.
(3) 0,057 г нитрата палладия растворяли в 55 мл деионизированной воды и значение рН доводили до 1,3. Затем полуфабрикат катализатора В5, полученный на стадии (2), пропитывали в полученном растворе и встряхивали в течение 30 мин, оставляли стоять в течение 4 ч или более, сушили при 80°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора С5.
(4) Полуфабрикат катализатора С4, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 6 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:Н2=1:1 с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе примера 3-5 содержание Pd составляло 0,1%, содержание Ni составляло 2,60%, и содержание Cu составляло 2,40%.
Сравнительный Пример 3-5
Носитель: Использовали коммерчески доступный носитель оксида алюминия в форме клевера с унимодальным распределением пор по размерам, имеющий диаметр 2 мм и длину 4 мм. После прокаливания при 1118°С в течение 4 ч унимодальное распределение пор по размерам составляло 30-49 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 20,80 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 8,29 г безводного нитрата никеля и 5,08 г хлорида меди взвешивали и растворяли в 55 мл деионизированной воды. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 20 мин, сушили при 80°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А5-1.
(2) 0,124 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А5-1 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 110°C и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора В5-1.
(3) 0,057 г нитрата палладия растворяли в 55 мл деионизированной воды и значение рН доводили до 1,3. Затем полуфабрикат катализатора В5-1, полученный на стадии (2), пропитывали в полученном растворе и встряхивали в течение 30 мин, оставляли стоять в течение 4 ч или более, сушили при 80°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора С5-1.
(4) Полуфабрикат катализатора С5-1, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 6 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:Н2=1:1 с получением требуемого катализатора.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе сравнительного примера 3-5 содержание Pd составляло 0,79%, содержание Ni составляло 2,60%, и содержание Cu составляло 2,40%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 100 мл (50 мл наполнителя) с массовой скоростью воздуха 4000/ч, рабочим давлением 2,0 МПа, отношением водорода к алкину 2 и температурой на входе в реактор 75°С. Состав реакционной массы представлен в Таблице 3-9, и результаты работы представлены в Таблице 3-10. Состав неочищенного водорода: Н2 30%, содержание СО 0,8%.

В Сравнительном примере 3-5 применяли носитель с унимодальным распределением пор по размерам.
Во время селективного гидрирования фракции С2 поры в диапазоне 50 нм или менее благоприятны для того, чтобы молекулы ацетилена контактировали и реагировали с активным центром из-за распределения основного активного компонента в слое внешней оболочки. В этом сравнительном примере из-за применения носителя с унимодальным распределением размеров пор подавляющее большинство пор имело размер 50 нм или менее, для сплава, образованного Ni-Cu-Pd, из-за высокого содержания активных компонентов он все еще обладал активностью гидрирования для ацетилена и этилена в этом диапазоне размеров пор. Однако собственная селективность Ni-Cu была низкой, что также приводило к гидрированию части этилена, и, таким образом, начальная селективность сравнительного примера 3-5 была ниже, чем у примера 3-5. В сравнительном примере активный центр имел большой размер, что позволяло адсорбировать несколько молекул одновременно, и легко происходили гидрирование и димеризация ацетилена, что приводило к образованию большего количества зеленого масла, чем в примере 3-5, и как активность, так и селективность через 1000 часов были ниже, чем в примере 3-5.
Пример 3-6
Использовали коммерчески доступный носитель оксида алюминия в форме клеверного листа с бимодальным распределением пор по размерам, имеющий диаметр 2 мм и длину 4 мм. После прокаливания при 1054°С в течение 4 ч бимодальное распределение пор по размерам составляло 20-42 нм и 90-360 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 37,32 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 9,64 г безводного нитрата никеля и 5,08 г хлорида меди взвешивали и растворяли в 65 мл деионизированной воды, к которой добавляли 25 г н-гексана, 13,60 г ЦТАБ и 12,5 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 70 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А6.
(2) 0,126 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А6 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 110°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора В6.
(3) 0,055 г нитрата палладия растворяли в 65 мл деионизированной воды, к которой добавляли 25 г н-гексана, 13,60 г ЦТАБ и 12,5 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Затем полуфабрикат катализатора В6, полученный на стадии (2), пропитывали в полученном растворе и встряхивали в течение 70 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора С6.
(4) Полуфабрикат катализатора С6, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 6 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:Н2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 82,36 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 81,76 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в Примере 3-6, содержание Pd составляло 0,1%, содержание Ni составляло 3,1%, содержание Ag составляло 0,5%, и содержание Cu составляло 2,4%.
Сравнительный Пример 3-6
Катализатор получали с применением того же носителя и условий, что и в Примере 3-6, за исключением того, что не добавляли Cu.
Приготовление катализатора:
(1) 9,64 г безводного нитрата никеля взвешивали и растворяли в 65 мл деионизированной воды, к которой добавляли 25 г н-гексана, 13,60 г ЦТАБ и 12,5 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали в полученном растворе, встряхивали в течение 70 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А6-1.
(2) 0,126 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Затем полуфабрикат катализатора А6-1 пропитывали в полученном растворе нитрата Pd в течение 60 мин, сушили при 110°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора В6-1.
(3) 0,055 г нитрата палладия растворяли в 65 мл деионизированной воды, к которой добавляли 25 г н-гексана, 13,60 г ЦТАБ и 12,5 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Затем полуфабрикат катализатора В6-1, полученный на стадии (2), пропитывали в полученном растворе и встряхивали в течение 70 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора С6-1.
(4) Полуфабрикат катализатора С6-1, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 6 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 82,36 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 81,76 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в сравнительном примере 3-6, содержание Pd составляло 0,1%, содержание Ni составляло 3,1%, и содержание Ag составляло 0,5%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя) с массовой скоростью воздуха 4000 ч-1, рабочим давлением 3,0 МПа, отношением водорода к алкину 2,5 и температурой на входе в реактор 55°С. Состав реакционного материала представлен в Таблице 3-11, и результаты работы представлены в Таблице 3-12. Состав неочищенного водорода: Н2 50%, содержание СО 0,1%.
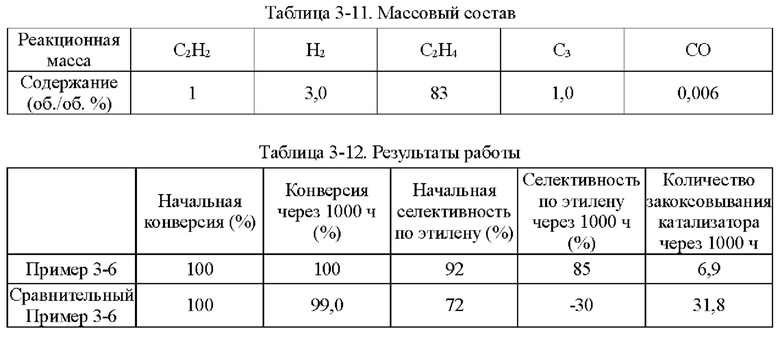
В примере 3-6 растворный способ применяли для обеспечения части Pd, которую в основном обеспечивали в меньших порах носителя. Микроэмульсионный предшественник применяли для обеспечения Cu, Ni и части Pd в мелких порах носителя с образованием сплава Cu-Ni-Pd. В этой структуре сплава Ni был основным активным компонентом, который обладал функцией высокого недеструктивного гидрирования для обеспечения недеструктивного гидрирования зеленого масла, образовавшегося в ходе селективного гидрирования. Хотя Ni и Pd также в определенной степени обладают активностью гидрирования, из-за низкого содержания Pd эти два компонента в основном способствовали снижению температуры восстановления Ni-содержащего сплава до 200°С или ниже. В Сравнительном примере 3-6 отсутствовала Cu. Следовательно, активный центр в крупных порах не участвовал во всем способе гидрирования. Закоксовывание в Сравнительном примере 3-6 было сильным из-за высокого содержания ацетилена и высокого образования зеленого масла.
Первоначальная селективность настоящего примера была выше, чем у катализатора, полученного в растворном способе, поскольку Cu и Ni находились только в больших порах и не участвовали в гидрировании этилена по сравнению с катализатором, обеспечиваемым в растворном способе.
Пример 3-7
Носитель: Использовали имеющийся в продаже носитель оксида алюминия в форме четырехлистного клевера с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и длину 3 мм. После прокаливания при 1092°С в течение 4 ч бимодальное распределение пор по размерам составляло 22-45 нм и 70-420 нм, водопоглощение составляло 56%, и удельная площадь поверхности составляла 29,67 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 1,11 г хлорида никеля и 1,49 г нитрата меди взвешивали и растворяли в 60 мл деионизированной воды, к которой добавляли 27 г н-гексана, 15,00 г Triton Х-100 и 14,4 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией и встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А7.
(2) 0,11 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора А7 растворяли в полученном солевом растворе Pd в течение 60 мин, сушили при 90°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора В7.
(3) 0,084 г хлорида палладия взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 31,5 г н-гексана, 17,50 г Triton Х-100 и 16,8 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В7, полученный на стадии (2), пропитывали полученной микроэмульсией и встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора С7.
(4) Полуфабрикат катализатора С7, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 8 часов при температуре 150°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 66,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 67,49 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в Примере 3-7, содержание Pd составляло 0,07%, содержание Ni составляло 3,5%, и содержание Cu составляло 0,5%.
Сравнительный Пример 3-7
Катализатор был приготовлен с применением того же носителя и условий, что и в Примере 3-7, за исключением того, что обеспечение Ni отсутствовало. Приготовление катализатора:
(1) 1,49 г нитрата меди взвешивали и растворяли в 60 мл деионизированной воды, к которой добавляли 27 г н-гексана, 15,00 г Triton Х-100 и 14,4 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией и встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А7-1.
(2) 0,11 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора А7-1 растворяли в полученном солевом растворе Pd в течение 60 мин, сушили при 90°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора В7-1.
(3) 0,084 г хлорида палладия взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 31,5 г н-гексана, 17,50 г Triton Х-100 и 16,8 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В7-1, полученный на стадии (2), пропитывали полученной микроэмульсией и встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч, с получением полуфабриката катализатора С7-1.
(4) Полуфабрикат катализатора С7-1, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 8 часов при температуре 150°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 66,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 67,49 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в сравнительном примере 3-7, содержание Pd составляло 0,07%, и содержание Cu составляло 0,5%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя) с массовой скоростью воздуха 3000 ч-1, рабочим давлением 3,0 МПа, отношением водорода к алкину 3 и температурой на входе в реактор 135°С. Состав реакционной массы представлен в Таблице 3-13, и результаты работы представлены в Таблице 3-14. Состав неочищенного водорода: Н2 30%, содержание СО 1%.
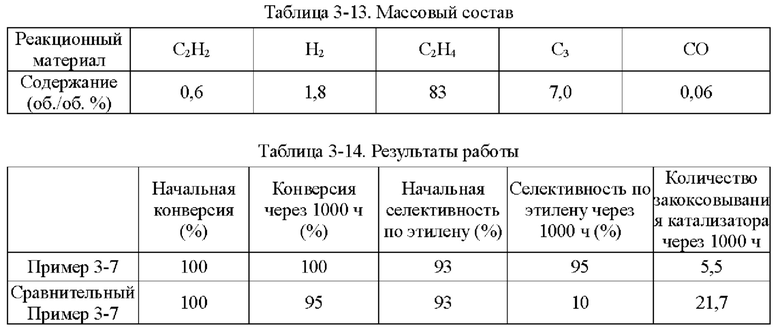
Поскольку в Сравнительном примере 3-7 не был обеспечен Ni, и было обеспечено низкое содержание Cu, активность эффективного гидрирования побочных продуктов реакции не была обеспечена, несмотря на высокую температуру реакции. Через 1000 ч селективность гидрирования снизилась до 10%. Тем не менее, исходные селективности примера 3-7 и сравнительного примера 3-7 были выше, чем у катализатора, полученного растворным способом.
Пример 3-8
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1128°С в течение 4 ч бимодальное распределение пор по размерам составляло 25-47 нм и 300-498 нм, водопоглощение составляло 50%, и удельная площадь поверхности составляла 21,46 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 9,34 г безводного нитрата никеля и 1,06 г хлорида меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 23,3 г н-гексана, 3,5 г ЦТАБ и 2,95 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией, встряхивали в течение 100 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора А8.
(2) 0,093 г хлорида палладия растворяли в 50 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора В8, полученный на стадии (1), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 90°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора В8.
(3) 0,0107 г нитрата палладия растворяли в 70 мл деионизированной воды, к которой добавляли 23,3 г н-гексана, 3,5 г ЦТАБ и 2,95 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В8, полученный на стадии (2), пропитывали полученной микроэмульсией, встряхивали в течение 100 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора С8.
(4) Полуфабрикат катализатора С8, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 4 ч при температуре 180°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 496,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 497,23 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в примере 3-8, содержание Pd составляло 0,6%, содержание Ni составляло 3,00%, и содержание Cu составляло 0,5%.
Сравнительный Пример 3-8
Катализатор был приготовлен с применением того же носителя, что и в Примере 3-8, за исключением того, что палладий обеспечивали в количестве менее 0,06%.
Приготовление катализатора:
(1) 9,34 г безводного нитрата никеля и 1,06 г хлорида меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 23,3 г н-гексана, 3,5 г ЦТАБ и 2,95 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией, встряхивали в течение 100 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора А8.
(2) 0,0465 г хлорида палладия растворяли в 50 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора В8, полученный на стадии (1), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 90°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора В8.
(3) 0,0054 г нитрата палладия растворяли в 70 мл деионизированной воды, к которой добавляли 23,3 г н-гексана, 3,5 г ЦТАБ и 2,95 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В8, полученный на стадии (2), пропитывали полученной микроэмульсией, встряхивали в течение 100 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора С8.
(4) Полуфабрикат катализатора С8, полученный на стадии (3), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 4 ч при температуре 180°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 496,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 497,23 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в сравнительном примере 3-8, содержание Pd составляло 0,3%, содержание Ni составляло 3,00%, и содержание Cu составляло 0,5%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый изотермический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя). Состав неочищенного водорода: Н-2 40%, содержание СО 0,3%. Скорость воздуха реакционной массы: 3000 ч-1, рабочее давление: 3,0 МПа, отношение водорода к алкину: 2, температура на входе в реактор: 85°С. Состав реакционной массы представлен в Таблице 3-15, и результаты работы представлены в Таблице 3-16.
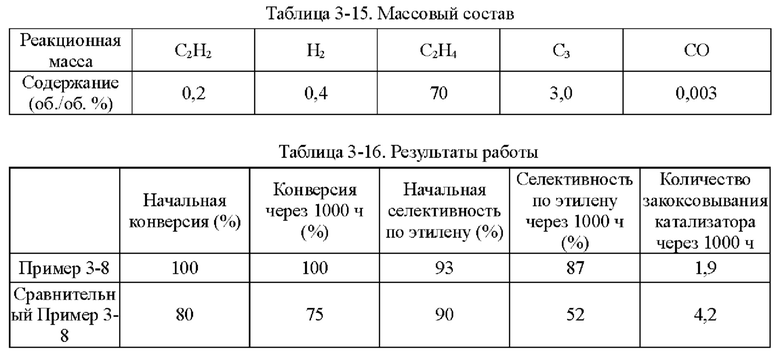
В сравнительном примере 3-8 из-за недостаточного содержания палладия активность гидрирования была низкой и не могла соответствовать требованиям удаления алкина.
Пример 3-9
Носитель: Использовали коммерчески доступный носитель оксида алюминия в форме клеверного листа с бимодальным распределением пор по размерам, имеющий диаметр 2 мм и длину 4 мм. После прокаливания при 1055°С в течение 4 ч бимодальное распределение пор по размерам составляло 25-42 нм и 90-360 нм, водопоглощение составляло 58%, и удельная площадь поверхности составляла 37,32 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 9,64 г безводного нитрата никеля и 5,08 г хлорида меди взвешивали и растворяли в 65 мл деионизированной воды, к которой добавляли 25 г н-гексана, 13,50 г ЦТАБ и 12,5 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией, встряхивали в течение 70 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А9.
(2) 0,79 г нитрата серебра взвешивали и растворяли в 64 г деионизированной воды, и значение рН доводили до 1,0 с помощью раствора азотной кислоты. Полуфабрикат катализатора А6 пропитывали в полученном растворе в течение 30 мин и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора В8.
(3) 0,126 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора А9 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 110°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора С9.
(4) 0,055 г нитрата палладия взвешивали и растворяли в 65 мл деионизированной воды, к которой добавляли 25 г н-гексана, 13,50 г ЦТАБ и 12,5 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С9, полученный на стадии (3), пропитывали полученной микроэмульсией, встряхивали в течение 70 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч, с получением полуфабриката катализатора D9.
(5) Полуфабрикат катализатора D9, полученный на стадии (4), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 6 часов при температуре 200°С с применением газовой смеси с молярным соотношением N2:Н2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 82,36 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 81,76 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в Примере 3-9, содержание Pd составляло 0,1%, содержание Ni составляло 3,1%, содержание Ag составляло 0,5%, и содержание Cu составляло 2,4%.
Сравнительный Пример 3-9
Катализатор получали в тех же условиях, что и в примере 3-9, за исключением того, что применяли унимодальный пористый носитель.
Применяли коммерчески доступный носитель оксида алюминия в форме клевера с унимодальным распределением пор по размерам, имеющий диаметр 2 мм и длину 4 мм. После прокаливания при 1060°С в течение 4 ч распределение пор по размерам составляло 59-113 нм, водопоглощение составляло 55%, и удельная площадь поверхности составляла 37,32 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 9,64 г безводного нитрата никеля и 5,08 г хлорида меди взвешивали и растворяли в 65 мл деионизированной воды, к которой добавляли 25 г н-гексана, 13,50 г ЦТАБ и 12,5 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией, встряхивали в течение 70 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора А9-1.
(2) 0,126 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора А9-1 пропитывали в полученном солевом растворе Pd в течение 60, сушили при 110°С и прокаливали при 500°С в течение 4 ч с получением полуфабриката катализатора В9-1.
(3) 0,055 г нитрата палладия взвешивали и растворяли в 65 мл деионизированной воды, к которой добавляли 25 г н-гексана, 13,50 г ЦТАБ и 12,5 г н-пентанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора В9-1, полученный на стадии (3), пропитывали полученной микроэмульсией, встряхивали в течение 70 мин, отфильтровывали остаточную жидкость, сушили при 60°С и прокаливали при 500°С в течение 4 ч, с получением полуфабриката катализатора С9-1.
(4) Полуфабрикат катализатора С9-1, полученный на стадии (4), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 6 ч при температуре 200°С с применением газовой смеси с молярным соотношением N2:Н2=1:1, с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 82,36 нм, и диаметр частиц микроэмульсии, полученной на стадии (3), составлял 81,76 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в сравнительном примере 3-9, содержание Pd составляло 0,10%, содержание Ni составляло 3,1%, и содержание Cu составляло 2,4%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя) с массовой скоростью воздуха 1500 ч-1, рабочим давлением 3,0 МПа, отношением водорода к алкину 2,5 и температурой на входе в реактор 55°С. Состав реакционной массы представлен в Таблице 3-17, и результаты работы представлены в Таблице 3-18. Состав неочищенного водорода: Н2 50%, СО 0,1%.
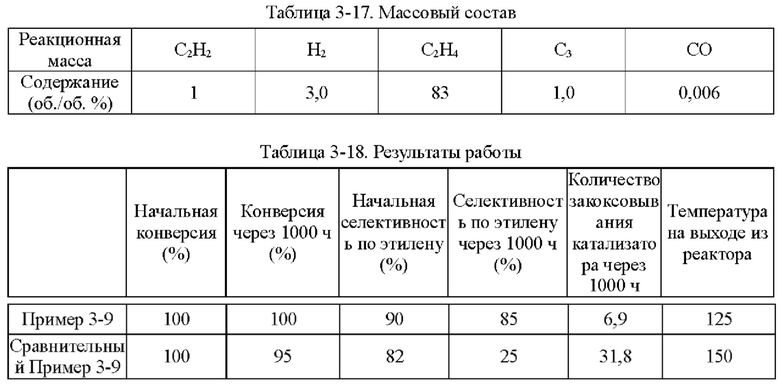
Пример 3-10
Носитель: Применяли имеющийся в продаже сферический носитель оксид алюминия с бимодальным распределением пор по размерам, имеющий диаметр 4 мм. После прокаливания при 1125°С в течение 4 ч бимодальное распределение пор по размерам составляло 25-47 нм и 300-498 нм, водопоглощение составляло 50%, и удельная площадь поверхности составляла 21,46 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 9,34 г безводного нитрата никеля и 1,06 г хлорида меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 23,3 г н-гексана, 3,5 г ЦТАБ и 3,0 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией, встряхивали в течение 100 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора А10.
(2) 0,095 г нитрата серебра взвешивали и растворяли в 55 мл деионизированной воды, и рН доводили до 3. Полуфабрикат катализатора А10 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 80°С и прокаливали при 530°С в течение 5 ч с получением полуфабриката катализатора В10.
(3) 0,129 г нитрата палладия растворяли в 50 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора В10, полученный на стадии (2), растворяли в полученном растворе нитрата серебра, содержащем серебро. После встряхивания и полной абсорбции раствора полученный продукт сушили при 90°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С10.
(4) 0,0645 г нитрата палладия растворяли в 70 мл деионизированной воды, к которой добавляли 23,3 г н-гексана, 3,5 г ЦТАБ и 3,0 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С10, полученный на стадии (3), пропитывали полученной микроэмульсией, встряхивали в течение 100 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора D10.
(5) Полуфабрикат катализатора D10, полученный на стадии (4), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 4 ч при температуре 180°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 496,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 497,14 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в примере 3-10, содержание Pd составляло 0,10%, содержание Ni составляло 3,00%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,06%.
Сравнительный Пример 3-10
Катализатор получали с применением того же носителя и условий, что и в примере 3-10, за исключением того, что количество обеспечиваемого Ni было снижено до 1/10 от количества в примере 3-10.
Приготовление катализатора:
(1) 0,934 г безводного нитрата никеля и 1,06 г хлорида меди взвешивали и растворяли в 70 мл деионизированной воды, к которой добавляли 23,3 г н-гексана, 3,5 г ЦТАБ и 3,0 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией, встряхивали в течение 100 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора А10-1.
(2) 0,095 г нитрата серебра взвешивали и растворяли в 55 мл деионизированной воды, и рН доводили до 3. Полуфабрикат катализатора А10, полученный на стадии (1), пропитывали полученным раствором нитрата серебра, содержащим серебро. После встряхивания и полной абсорбции раствора полученный продукт пропитывали в течение 60 мин, сушили при 80°С и прокаливали при 530°С в течение 5 ч с получением полуфабриката катализатора В10-1.
(3) 0,129 г нитрата палладия растворяли в деионизированной воде и значение рН доводили до 1,8. Полуфабрикат катализатора В10-1 растворяли в полученном растворе нитрата серебра, содержащем серебро, и встряхивали, сушили при 90°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С10-1.
(4) 0,0645 г нитрата палладия растворяли в 70 мл деионизированной воды, к которой добавляли 23,3 г н-гексана, 3,5 г ЦТАБ и 3,0 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С10-1, полученный на стадии (3), пропитывали полученной микроэмульсией, встряхивали в течение 100 мин, отфильтровывали остаточную жидкость, сушили при 70°С и прокаливали при 450°С в течение 5 ч с получением полуфабриката катализатора D10.
(5) Полуфабрикат катализатора D10, полученный на стадии (4), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 4 ч при температуре 180°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 496,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 497,14 нм. Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в сравнительном примере 3-10, содержание Pd составляло 0,9%, содержание Ni составляло 0,3%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,06%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый изотермический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя). Состав неочищенного водорода: водород 40%, СО 0,3%. Массовая скорость воздуха: 3000 ч-1, рабочее давление: 3,0 МПа, отношение водорода к алкину: 2, температура на входе в реактор: 65°С. Состав реакционного материала представлен в Таблице 3-19, и результаты работы представлены в Таблице 3-20.
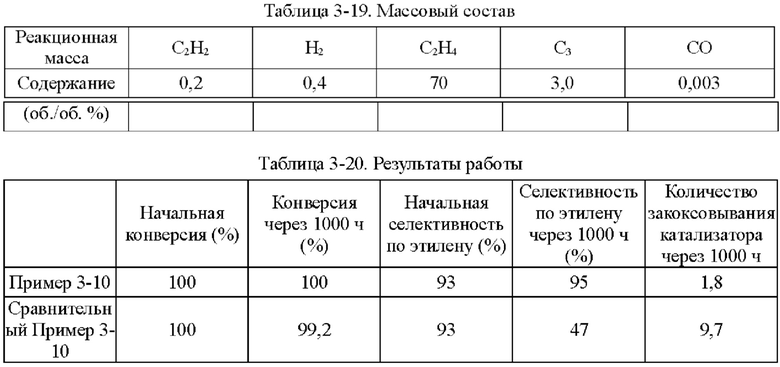
В сравнительном примере 3-10 содержание обеспечиваемого Ni было уменьшено таким образом, что активность активного центра Ni и Cu больше не обеспечивала недеструктивное гидрирование побочных продуктов гидрирования. Таким образом, хотя начальная активность реактора и селективность в реакторе были хорошими, по мере протекания реакции накапливались побочные продукты гидрирования, и образовавшееся закоксовывание приводило к постоянному снижению производительности катализатора в этом сравнительном примере, так что конверсия ацетилена через 1000 ч снижалась до 95%, и селективность снижалась до половины начального значения.
Пример 3-11
Носитель: Использовали имеющийся в продаже носитель оксида алюминия в форме четырехлистного клевера с бимодальным распределением пор по размерам, имеющий диаметр 3 мм и длину 3 мм. После прокаливания при 1093°С в течение 4 ч бимодальное распределение пор по размерам составляло 22-45 нм и 70-420 нм, водопоглощение составляло 56%, и удельная площадь поверхности составляла 29,67 м2/г. 100 г носителя взвешивали.
Приготовление катализатора:
(1) 1,11 г хлорида никеля и 1,49 г нитрата меди взвешивали и растворяли в 60 мл деионизированной воды, к которой добавляли 27 г н-гексана, 15,00 г Triton Х-100 и 14,4 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора A11.
(2) 0,26 г нитрата серебра взвешивали и растворяли в 45 г деионизированной воды, и значение рН доводили до 5,0 с помощью раствора азотной кислоты. Полуфабрикат катализатора А11 пропитывали в полученном растворе Ag в течение 60 мин, сушили при 120°С и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора В11.
(3) 0,11 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора В11 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 90°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С11.
(4) 0,084 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 31,5 г н-гексана, 17,50 г Triton Х-100 и 16,8 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С11, полученный на стадии (3), пропитывали полученной микро эмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора D11.
(5) Полуфабрикат катализатора D11, полученный на стадии (4), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 8 часов при температуре 150°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 66,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 67,49 нм.
Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в Примере 3-11, содержание Pd составляло 0,07%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,16%.
Сравнительный Пример 3-11
Катализатор был приготовлен с применением того же носителя и условий, что и в Примере 3-11, за исключением того, что содержание Pd было низким.
Приготовление катализатора:
(1) 1,11 г хлорида никеля и 1,49 г нитрата меди взвешивали и растворяли в 60 мл деионизированной воды, к которой добавляли 27 г н-гексана, 15,00 г Triton Х-100 и 14,4 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Взвешенный носитель пропитывали полученной микроэмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч с получением полуфабриката катализатора А11-1.
(2) 0,26 г нитрата серебра взвешивали и растворяли в 45 г деионизированной воды, и значение рН доводили до 5,0 с помощью раствора азотной кислоты. Полуфабрикат катализатора А11-1 пропитывали в полученном растворе Ag в течение 60 мин, сушили при 120°С и прокаливали при 500°С в течение 6 ч с получением полуфабриката катализатора В11-1.
(3) 0,084 г хлорида палладия растворяли в 100 мл деионизированной воды и значение рН доводили до 1,8. Полуфабрикат катализатора В11-1 пропитывали в полученном солевом растворе Pd в течение 60 мин, сушили при 90°С и прокаливали при 550°С в течение 4 ч с получением полуфабриката катализатора С11-1.
(4) 0,0085 г хлорида палладия растворяли в 70 мл деионизированной воды, к которой добавляли 31,5 г н-гексана, 17,50 г Triton Х-100 и 16,8 г н-бутанола и тщательно перемешивали с получением микроэмульсии. Полуфабрикат катализатора С11-1, полученный на стадии (3), пропитывали полученной микро эмульсией, встряхивали в течение 90 мин, отфильтровывали остаточную жидкость, сушили при 80°С и прокаливали при 600°С в течение 4 ч, с получением полуфабриката катализатора D11-1.
(5) Полуфабрикат катализатора D11-1, полученный на стадии (4), помещали в реакционное устройство с неподвижным слоем и восстанавливали в течение 8 часов при температуре 150°С с применением газовой смеси с молярным соотношением N2:H2=1:1 с получением требуемого катализатора.
Как определено динамическим рассеянием света, диаметр частиц микроэмульсии, полученной на стадии (1), составлял 66,68 нм, и диаметр частиц микроэмульсии, полученной на стадии (4), составлял 67,49 нм. Содержание активных компонентов полученных катализаторов определяли методом атомно-абсорбционной спектроскопии. В катализаторе, полученном в Примере 3-11, содержание Pd составляло 0,055%, содержание Ni составляло 0,5%, содержание Cu составляло 0,5%, и содержание Ag составляло 0,16%.
Эффект внедрения
Условия реакции гидрирования:
Катализатор загружали в одноступенчатый адиабатический реактор с неподвижным слоем в количестве 50 мл (50 мл наполнителя) с воздушной скоростью материала 4000 ч-1, рабочим давлением 3,0 МПа, отношением водорода к алкину 3 и температурой на входе в реактор 135°С. Состав реакционной массы представлен в Таблице 3-21, и результаты работы представлены в Таблице 3-22. Состав неочищенного водорода: Н2 30%, содержание СО 1%.
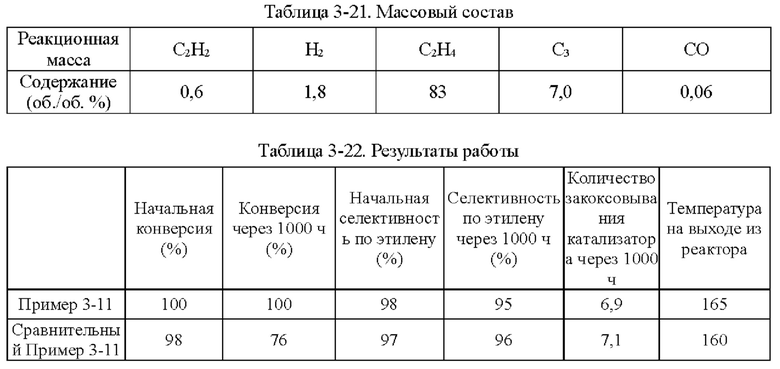
В этих условиях реакции, даже несмотря на то, что содержание ацетилена в материале не было самым высоким, содержание СО в массе составляло до 600 частей на миллион, что оказывало значительное ингибирующее действие на активность катализатора. Когда содержание Pd, обеспечиваемое растворным способом в сравнительном примере 3-11, было ниже предельного значения, ацетилен не мог быть эффективно удален только путем повышения температуры на входе в реактор. Следовательно, ацетилен не мог быть полностью преобразован даже после того, как температура на входе в реактор была повышена до 135°С.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ И КАТАЛИЗАТОР | 2005 |
|
RU2355670C2 |
| КАТАЛИЗАТОР СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ АЛКИНОВ С ЕГО ИСПОЛЬЗОВАНИЕМ | 2001 |
|
RU2259877C2 |
| КАТАЛИЗАТОР СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ ДИЕНОВ | 2023 |
|
RU2811194C1 |
| СПОСОБ СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ АЦЕТИЛЕНОВ | 2003 |
|
RU2310639C2 |
| КАТАЛИЗАТОР ДЛЯ СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ НЕНАСЫЩЕННЫХ УГЛЕВОДОРОДОВ И СПОСОБ ЕГО ПРИГОТОВЛЕНИЯ | 1995 |
|
RU2077945C1 |
| КАТАЛИЗАТОР СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ АЦЕТИЛЕНОВЫХ И ДИЕНОВЫХ УГЛЕВОДОРОДОВ В С-С-УГЛЕВОДОРОДНЫХ ФРАКЦИЯХ | 2014 |
|
RU2547258C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ ФУРФУРОЛА | 2018 |
|
RU2689417C1 |
| КАТАЛИЗАТОР СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ НЕНАСЫЩЕННЫХ ОЛЕФИНОВ, СПОСОБ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ КАТАЛИЗАТОРА | 2001 |
|
RU2278731C2 |
| Биметаллический катализатор для жидкофазного селективного гидрирования ацетиленовых углеводородов и способ его получения | 2022 |
|
RU2786218C1 |
| КАТАЛИЗАТОР СЕЛЕКТИВНОГО ГИДРИРОВАНИЯ ФУРФУРОЛА | 2018 |
|
RU2689418C1 |
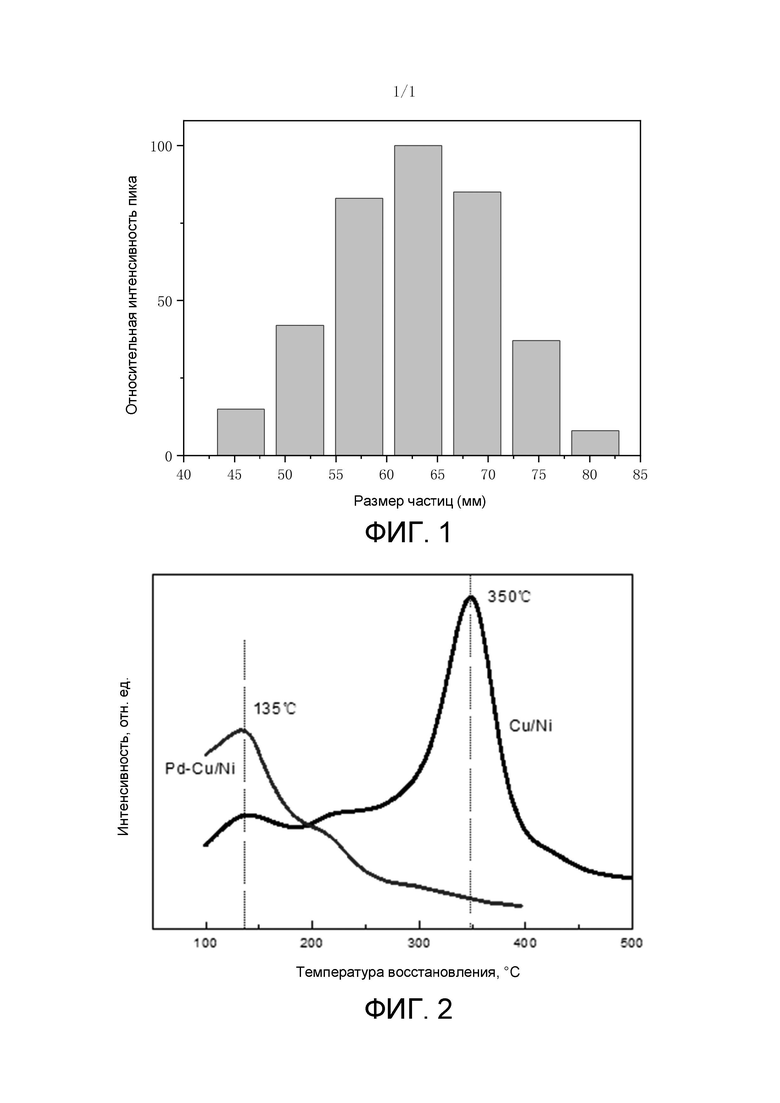
Изобретение может быть использовано в химической промышленности. Предложен катализатор селективного гидрирования алкинов, в котором носитель катализатора представляет собой оксид алюминия или оксид алюминия составляет 80% или более от носителя и имеет бимодальное распределение пор по размерам. Размер мелких пор составляет 15-50 нм, а размер крупных пор составляет 80-500 нм. Катализатор содержит по меньшей мере Pd, Ni и Cu, причем в расчете на 100 % по массе носителя содержание Pd составляет 0,03-0,1 %, содержание Ni составляет 0,5-5 %, массовое отношение Cu к Ni составляет 0,1-1,0:1,0. При этом Ni, Cu и часть Pd получены из микроэмульсии. Диаметр частиц микроэмульсии не меньше, чем максимальный размер мелких пор, и не больше, чем максимальный размер крупных пор. Предложены также способ получения катализатора, способ селективного гидрирования алкинов во фракции С2 и способ селективного гидрирования фракции С2. Группа изобретений позволяет получить катализатор с хорошими антикоксовыми характеристиками, который может поддерживать хорошую гидрирующую активность и превосходную селективность в течение относительно длительного времени. 4 н. и 30 з.п. ф-лы, 2 ил., 40 табл., 3 пр.
1. Катализатор селективного гидрирования алкинов, в котором носитель катализатора представляет собой оксид алюминия или оксид алюминия составляет 80 % или более от носителя и имеет бимодальное распределение пор по размерам,
где размер мелких пор составляет 15-50 нм и размер крупных пор составляет 80-500 нм;
где катализатор содержит по меньшей мере Pd, Ni и Cu;
где в расчете на 100 % по массе носителя содержание Pd составляет 0,03-0,1 %, содержание Ni составляет 0,5-5 % и массовое отношение Cu к Ni составляет 0,1-1,0:1,0;
где Ni, Cu и часть Pd получены из микроэмульсии и диаметр частиц микроэмульсии не меньше, чем максимальный размер мелких пор, и не больше, чем максимальный размер крупных пор.
2. Катализатор селективного гидрирования алкинов по п. 1, в котором носитель катализатора представляет собой оксид алюминия или оксид алюминия составляет 80 % или более от носителя и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu; в расчете на 100 % по массе носителя содержание Pd составляет 0,035-0,08 %, содержание Ni составляет 0,5-5 % и массовое отношение Cu к Ni составляет 0,1-1,0:1,0; при этом Ni, Cu и часть Pd получены из микроэмульсии и диаметр частиц микроэмульсии не меньше, чем максимальный размер мелких пор, и не больше, чем максимальный размер крупных пор; количество Pd, полученного из микроэмульсии, составляет 1/100-1/200 от общего содержания Ni и Cu и остальную часть Pd получают из раствора.
3. Катализатор селективного гидрирования алкинов по п. 1, в котором носитель катализатора представляет собой оксид алюминия и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu в качестве активных компонентов; в расчете на 100 % по массе носителя содержание Pd составляет 0,03-0,09 %, содержание Ni составляет 0,5-5 % и массовое отношение Cu к Ni составляет 0,1-1,0:1,0; при этом Ni, Cu и часть Pd получают из микроэмульсии.
4. Катализатор селективного гидрирования алкинов по п. 1, в котором носителем катализатора является оксид алюминия или оксид алюминия составляет 80% или более от носителя, при этом оксид алюминия имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu в качестве активных компонентов; в расчете на 100 % по массе носителя содержание Pd составляет 0,06-0,1 %, содержание Ni составляет 0,5-5 % и массовое отношение Cu к Ni составляет 0,1-1,0:1,0.
5. Катализатор по п. 1, в котором носитель катализатора представляет собой оксид алюминия или оксид алюминия составляет 80% или более от носителя и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ni и Cu; в расчете на 100 % по массе носителя содержание Pd составляет 0,06-0,08 %, содержание Ni составляет 0,5-5 % и массовое отношение Cu к Ni составляет 0,1-1,0:1,0; при этом Ni, Cu и часть Pd получены из микроэмульсии; и количество Pd, получаемое способом микроэмульсии, составляет 1/100-1/200 от общего содержания Ni и Cu.
6. Катализатор по любому из пп. 1-5, в котором катализатор дополнительно содержит Ag, который получен из раствора, и содержание Ag составляет 0,03-0,5 %, предпочтительно 0,08-0,21 %.
7. Катализатор по п. 6, в котором носитель катализатора представляет собой оксид алюминия или оксид алюминия составляет 80 % или более от носителя и имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм и размер крупных пор составляет 80-500 нм; катализатор содержит по меньшей мере Pd, Ag, Ni и Cu; в расчете на 100% по массе носителя содержание Pd составляет 0,035-0,07 %, содержание Ag составляет 0,08-0,21 %, содержание Ni составляет 0,5-5 % и массовое отношение Cu к Ni составляет 0,1-1,0:1,0; при этом Ni, Cu и часть Pd получены из микроэмульсии; количество Pd, получаемое способом микроэмульсии, составляет 1/100-1/200 от общего содержания Ni и Cu; и Ag и остальную часть Pd получают способом раствора.
8. Катализатор по п. 1, в котором носитель представляет собой оксид алюминия или оксид алюминия составляет 80 % или более от носителя и оксид алюминия находится в кристаллической форме θ или кристаллической форме α или их смеси.
9. Катализатор селективного гидрирования алкинов по п. 3, в котором содержание Pd составляет 0,035-0,075 % и количество части Pd, полученной из микроэмульсии, составляет 1/110-1/200 от общего содержания Ni и Cu.
10. Катализатор селективного гидрирования алкинов по п. 1, в котором носитель катализатора представляет собой оксид алюминия или оксид алюминия составляет 80 % или более от носителя, при этом оксид алюминия имеет бимодальное распределение пор по размерам, при этом размер мелких пор составляет 15-50 нм и размер крупных пор составляет 80-500 нм; катализатор содержит в качестве активных компонентов по меньшей мере Pd, Ag, Ni и Cu; в расчете на 100 % по массе носителя содержание Pd составляет 0,07-0,1 %, содержание Ag составляет 0,03-0,5 %, содержание Ni составляет 0,5-5 %, содержание Cu составляет 0,5-5 % и общее содержание Ni и Cu составляет 1-5,5 %.
11. Катализатор селективного гидрирования алкинов по любому из пп. 1-10, в котором указанный катализатор имеет удельную площадь поверхности 20-50 м2/г.
12. Катализатор селективного гидрирования алкинов по п. 1, в котором количество Pd, полученного из микроэмульсии, составляет 1/100-1/200 от общего содержания Ni и Cu.
13. Катализатор по п. 1, в котором способ получения микроэмульсии включает:
растворение соли-предшественника в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества и
тщательное перемешивание с получением микроэмульсии,
при этом масляная фаза представляет собой алкан или циклоалкан, поверхностно-активное вещество представляет собой ионное поверхностно-активное вещество и/или неионогенное поверхностно-активное вещество и дополнительное поверхностно-активное вещество представляет собой органический спирт.
14. Катализатор по п. 13, в котором микроэмульсия удовлетворяет следующим условиям: массовое отношение водной фазы к масляной фазе составляет 2-3, массовое отношение поверхностно-активного вещества к масляной фазе составляет 0,15-0,6, массовое отношение поверхностно-активного вещества к дополнительному поверхностно-активному веществу составляет 1-1,2 и диаметр частиц микроэмульсии составляет более 50 нм и менее 500 нм.
15. Катализатор по п. 13 или 14, в котором в указанной микроэмульсии масляная фаза представляет собой C6-C8 насыщенный алкан или циклоалкан, поверхностно-активное вещество представляет собой ионное поверхностно-активное вещество и/или неионогенное поверхностно-активное вещество и дополнительное поверхностно-активное вещество представляет собой C4-C6 спирт.
16. Катализатор по любому из пп. 13-15, в котором масляная фаза представляет собой циклогексан или н-гексан.
17. Катализатор по любому из пп. 13-15, в котором поверхностно-активное вещество представляет собой октилфениловый эфир полиэтиленгликоля или бромид цетилтриметиламмония.
18. Катализатор по любому из пп. 13-15, в котором дополнительное поверхностно-активное вещество представляет собой н-бутанол и/или н-пентанол.
19. Способ получения катализатора по любому из пп. 1-18, включающий стадии:
(1) растворение солей-предшественников Ni и Cu в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества,
тщательное перемешивание с образованием микроэмульсии,
пропитку носителя в полученной микроэмульсии в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и
сушку и прокаливание при 400-600 °С в течение 4 ч или более с получением полуфабриката катализатора А;
(2) растворение соли-предшественника Pd в воде,
доведение рН до 1,5-2,5,
добавление полуфабриката катализатора А в солевой раствор Pd для обеспечения пропитки и адсорбции в течение 0,5-4 ч и затем
сушку и прокаливание при 400-600 °С в течение 4-6 ч с получением полуфабриката катализатора В;
(3) растворение соли-предшественника Pd в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества,
тщательное перемешивание с образованием микроэмульсии, и
добавление полуфабриката катализатора B к полученной микроэмульсии для обеспечения пропитки в течение 0,5-4 ч, затем удаление остаточной жидкости путем фильтрации, и
сушку и прокаливание при 400-600 °С в течение 4 ч или более с получением катализатора.
20. Способ получения катализатора по п. 19, в котором, когда катализатор содержит Ag, способ включает стадии:
(1) растворение солей-предшественников Ni и Cu в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества,
тщательное перемешивание с образованием микроэмульсии, и
пропитку носителя в полученной микроэмульсии в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием, и
сушку и прокаливание при 400-600 °С в течение 4-6 ч с получением полуфабриката катализатора А;
(2) растворение соли-предшественника Pd в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества,
тщательное перемешивание с образованием микроэмульсии, и
добавление полуфабриката катализатора А к полученной микроэмульсии для обеспечения пропитки в течение 0,5-4 ч, затем удаление остаточной жидкости путем фильтрации, и
сушку и прокаливание при 400-600 °С в течение 4-6 ч с получением полуфабриката катализатора В;
(3) растворение соли-предшественника Pd в воде,
доведение рН до 1,5-2,5,
добавление полуфабриката катализатора В в солевой раствор Pd для обеспечения пропитки и адсорбции в течение 0,5-4 ч и затем
сушку и прокаливание при 400-600 °С в течение 4-6 ч с получением полуфабриката катализатора С;
(4) растворение соли Ag в деионизованной воде,
доведение рН до 1-5,
пропитку полуфабриката катализатора С в полученном растворе до полной абсорбции раствора и затем
сушку и прокаливание при 400-600 °С в течение 4-6 ч с получением катализатора;
предпочтительно способ включает стадии:
(1) растворение солей-предшественников Ni и Cu в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества,
тщательное перемешивание с образованием микроэмульсии, и
пропитку носителя в полученной микроэмульсии в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием, и
сушку и прокаливание при 400-600 °С в течение 4-6 ч с получением полуфабриката катализатора А;
(2) растворение соли-предшественника Pd в воде,
доведение рН до 1,5-2,5,
добавление полуфабриката катализатора А в солевой раствор Pd для обеспечения пропитки и адсорбции в течение 0,5-4 ч и затем
сушку и прокаливание при 400-600 °С в течение 4-6 ч с получением полуфабриката катализатора В;
(3) обеспечение Ag путем насыщенной пропитки путем приготовления раствора соли Ag, который соответствует 80-110 % поглощающей способности насыщения водой носителя, и доведение pH до 1-5 для обеспечения пропитки, и затем сушки и прокаливания при 400-600 °С в течение 4-6 ч с получением полуфабриката катализатора C;
(4) растворение соли-предшественника Pd в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества,
тщательное перемешивание с образованием микроэмульсии,
добавление полуфабриката катализатора С к полученной микроэмульсии для обеспечения пропитки в течение 0,5-4 ч, затем удаление остаточной жидкости путем фильтрации и
сушку и прокаливание при 400-600 °С в течение 4-6 ч с получением катализатора;
в качестве альтернативы способ включает стадии:
(1) растворение солей-предшественников Ni и Cu в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества,
тщательное перемешивание с образованием микроэмульсии,
пропитку носителя в полученной микроэмульсии в течение 0,5-4 ч, затем удаление остаточной жидкости фильтрованием и
сушку при 80-120 °С в течение 1-6 ч и прокаливание при 400-600 °С в течение 4-8 ч с получением полуфабриката катализатора А;
(2) нанесение Ag путем насыщенной пропитки путем приготовления раствора соли Ag, который соответствует 80-110 % поглощающей способности насыщения водой носителя, и доведения рН до 1-5, нанесение Ag на полуфабрикат катализатора A с последующим прокаливанием при 500-550 °С в течение 4-6 часов с получением полуфабриката катализатора B;
(3) растворение соли-предшественника Pd в воде,
доведение рН до 1,5-2,5,
добавление полуфабриката катализатора B к солевому раствору Pd для обеспечения пропитки и адсорбции в течение 0,5-4 ч и затем
сушку в течение 1-4 ч и прокаливание при 400-550 °С в течение 2-6 ч с получением полуфабриката катализатора С;
(4) растворение соли-предшественника Pd в воде,
добавление масляной фазы, поверхностно-активного вещества и дополнительного поверхностно-активного вещества,
тщательное перемешивание с образованием микроэмульсии, и
добавление полуфабриката катализатора С к полученной микроэмульсии для обеспечения пропитки в течение 0,5-4 ч, затем удаление остаточной жидкости путем фильтрации, и
сушку в течение 1-6 ч и прокаливание при 400-600 °С в течение 4-8 ч с получением полуфабриката катализатора D;
(5) помещение полуфабриката катализатора D в реактор с неподвижным слоем для обеспечения восстановления при 150-200 °С в течение 4-8 часов в среде газовой смеси N2:H2=1:1, с получением готового катализатора E.
21. Способ по п. 19 или 20, в котором микроэмульсия удовлетворяет следующим условиям: массовое отношение водной фазы к масляной фазе составляет 2-3, массовое отношение поверхностно-активного вещества к масляной фазе составляет 0,15-0,6, массовое отношение поверхностно-активного вещества к дополнительному поверхностно-активному веществу составляет 1-1,2 и диаметр частиц микроэмульсии составляет более 50 нм и менее 500 нм.
22. Способ селективного гидрирования алкинов во фракции C2, в котором сырье для гидрирования представляет собой фракцию C2 из верхней части предварительного деэтанизатора и его подают в реактор с неподвижным слоем и подвергают гидрированию в газовой фазе для удаления ацетилена, при этом технологические условия для реакции селективного гидрирования представляют собой: температура на входе в реактор 35-100 °С, давление 1,5-3,0 МПа и объемная скорость газа 2000-11000 ч-1; и при этом катализатор представляет собой катализатор по любому из пп. 1-18.
23. Способ по п. 22, в котором сырье для каталитического гидрирования содержит 65-93 % (об./об.) этилена и 0,2-2,5 % (об./об.) ацетилена.
24. Способ по п. 22, в котором сырье для каталитического гидрирования содержит 65-93 % (об./об.) этилена, 5-34,69 % (об./об.) этана, 0,3-2,5 % (об./об.) ацетилена и 0,01-0,8 % (об./об.) фракции C3.
25. Способ по любому из пп. 22-24, в котором при применении одноступенчатого реактора с неподвижным слоем для гидрирования отношение водорода к алкину на входе в реактор составляет 1,3-2,2, предпочтительно 1,3-1,8.
26. Способ по любому из пп. 22-24, в котором при применении двухступенчатого реактора с неподвижным слоем для гидрирования отношение водорода к алкину на входе в реактор первой ступени составляет 1,0-1,4 и отношение водорода к алкину на входе в реактор второй ступени составляет 1,5-2,5.
27. Способ по любому из пп. 22-24, в котором при применении трехступенчатого реактора с неподвижным слоем для гидрирования отношение водорода к алкину на входе в реактор первой ступени составляет 0,5-1,5, отношение водорода к алкину на входе в реактор второй ступени составляет 1,0-2,0 и отношение водорода к алкину на входе в реактор третьей ступени составляет 1,4-3,0; предпочтительно отношение водорода к алкину на входе в реактор первой ступени составляет 0,8-1,5, отношение водорода к алкину на входе в реактор второй ступени составляет 1,2-1,6 и отношение водорода к алкину на входе в реактор третьей ступени составляет 1,5-2,5.
28. Способ по п. 22, в котором катализатор восстанавливают при температуре 150-200 °С перед гидрированием.
29. Способ селективного гидрирования фракции C2 путем постгидрирования с применением неочищенного водорода в качестве источника водорода, в котором сырье для гидрирования представляет собой фракцию C2 из верхней части деэтанизатора и его подают в реактор с неподвижным слоем и подвергают гидрированию в газовой фазе для удаления ацетилена; при этом газообразный водород, который применяют в реакции гидрирования, представляет собой неочищенный водород с содержанием H2 20-50 об. % и содержанием CO 0,1-1 об. %; при этом условия способа: температура на входе в реактор 55-130 °С, давление 1,5-3,0 МПа и объемная скорость газа 1500-6000 ч-1; и при этом катализатор представляет собой катализатор по любому из пп. 1-18.
30. Способ по п. 29, в котором сырье для гидрирования представляет собой фракцию C2 из верхней части деэтанизатора и его подают в реактор с неподвижным слоем и подвергают гидрированию в газовой фазе для удаления ацетилена; при этом газообразный водород, который применяют в реакции гидрирования, представляет собой неочищенный водород с содержанием H2 30-50 об./об. %, содержанием CO 0,1-1 об./об. % и остальное составляет метан; при этом условия способа представляют собой: температура на входе в реактор 55-130 °С, давление 1,5-3,0 МПа и объемная скорость газа 1500-4000 ч-1; и при этом катализатор представляет собой катализатор по п. 4.
31. Способ по п. 29, в котором реактор представляет собой одноступенчатый реактор с неподвижным слоем.
32. Способ по п. 31, в котором сырье для каталитической реакции, подаваемое в одноступенчатый реактор с неподвижным слоем, содержит 60-93 об./об. % этилена, 0,2-1,0 об./об. % ацетилена и 1,0-7,0 об./об. % фракции C3.
33. Способ по п. 29, в котором катализатор восстанавливают при температуре 150-200 °С перед гидрированием.
34. Способ по п. 31, в котором отношение водорода к алкину в подаваемом материале на входе в одноступенчатый реактор с неподвижным слоем составляет 2-3.
| CN 104098426 A, 15.10.2014 | |||
| МАКРО- И МЕЗОПОРИСТЫЙ КАТАЛИЗАТОР С ОДНОРОДНО РАСПРЕДЕЛЕННОЙ АКТИВНОЙ НИКЕЛЕВОЙ ФАЗОЙ И СРЕДНИМ ДИАМЕТРОМ МАКРОПОР ОТ 50 ДО 300 нм И ЕГО ПРИМЕНЕНИЕ В ГИДРИРОВАНИИ УГЛЕВОДОРОДОВ | 2015 |
|
RU2683777C2 |
| НИКЕЛЕВЫЙ КАТАЛИЗАТОР ГИДРИРОВАНИЯ НА НОСИТЕЛЕ И СПОСОБ ПРИГОТОВЛЕНИЯ МОДИФИЦИРОВАННОГО НИКЕЛЕВОГО КАТАЛИЗАТОРА ГИДРИРОВАНИЯ НА НОСИТЕЛЕ | 1995 |
|
RU2095136C1 |
| CN 101433842 A, 20.05.2009 | |||
| WO 2015034521 A1, 12.03.2015 | |||
| Способ получения дифференциального спектра | 1978 |
|
SU764467A1 |
| СПОСОБ ОЦЕНКИ АДЕКВАТНОЙ КИНЕЗОТЕРАПЕВТИЧЕСКОЙ НАГРУЗКИ НА МЫШЕЧНО-СУСТАВНОЙ АППАРАТ | 1996 |
|
RU2134536C1 |

