Изобретение относится к области медицины и может быть использовано в фармацевтической промышленности, а именно для производства лекарственного средства для терапии спинальной мышечной атрофии. Изобретение представляет из себя способ получения высокоочищенного антисмыслового олигонуклеотида нусинерсена, в котором применяется комбинация высокоэффективного твердофазного синтеза и очистки.
Терапевтические олигонуклеотиды – довольно новый вид терапевтических препаратов. Первые работы по химическому синтезу были опубликованы в 1955 году, с тех пор на рынок выведено 18 лекарственных препаратов, которые можно условно разделить на четыре больших класса: антисмысловые олигонуклеотиды (ASO); олигонуклеотиды с переключением сплайсинга (SSO); малые интерферирующие РНК (siRNA); РНК-аптамеры. Среди одних из самых известных препаратов на основе синтетических олигонуклеотидов является препарат Спинраза (мнн: нусинерсен). Данный препарат относится к классу SSO. Нусинерсен – антисмысловой олигонуклеотид, воздействующий на ген выживаемости мотонейронов (SMN2), применяется для терапии спинальной мышечной терапии (СМА).
Существует два основных подхода к синтезу антисмысловых терапевтических нуклеотидов – твердофазный и жидкофазный. В индустрии широко используется твердофазная стратегия с применением фосфорамидитной химии. Фосфорамидитный метод является хорошо известным в данной области органического синтеза. Общая стратегия твердофазного синтеза олигонуклеотидов хорошо описана в различных источниках (например, Beaucage и Caruthers 1981, Beaucage и Iyer 1991, WO 1994002499 A1, WO 2023149564 A1). Синтез антисмысловых олигонуклеотидов широко представлен в литературе (например, US 6007995A).
Основным методом производства синтетических олигонуклеотидов является способ синтеза на твердом сорбенте удлинением цепи до достижения необходимой последовательности. Получаемая таким образом цепочка олигонуклеотидов элиминируется с твердой подложки, с сохранением 5' гидроксильной группы или без нее, и отправляется на очистку.
В результате синтеза олигонуклеотидов могут образоваться различные примеси. Некоторые авторы предлагают способ очистки синтетических олигонуклеотидов путем тонкослойной хроматографии, электрофореза в полиакриламидном геле. Однако данные способы являются сугубо лабораторными и применяются в исследовательских целях.
В настоящее время известно несколько способов очистки синтетических олигонуклеотидов. Основной целью очистки является удаление близкородственных примесей, образующихся в результате синтеза, примесей, сопутствующих исходному сырью, органических растворителей и перевод в буфер готовой лекарственной формы.
Патент RU 2738093 раскрывает способ твердофазного синтеза нусинерсена и очистки методом анионообменной хроматографии. Авторы используют автоматизированный твердофазный синтез по фосфорамидитной схеме. В качестве твердого носителя используется стекло с контролируемым размером пор (500Å). Масштаб синтеза составил 90–100 мкмоль. В качестве детритилирующего агента использовался 3% раствор дихлоруксусной кислоты в дихлорметане. Активатором стадии присоединения выступает 5-этилтиотетразол (ETT). Для стадии кэпирования использовалась смесь пропионового ангидрида и N-метилимидазола. Для тиолирования использовался 3-[(диметиламинометилен)амино]-3H-1,2,4-дитиазол-5-тион (DDTT). Синтез вели с удалением последней DMT группы с 5' конца цепи. Реакцию аммонолиза вели 3 суток при комнатной температуре. Продукт фильтровали, упаривали, снова растворяли в воде и высаживали этанолом для получения технического нусинерсена. Недостатком данного решения является малый масштаб синтеза, который не позволяет получать терапевтический олигонуклеотид в достаточном количестве. Использование в качестве носителя стекла с контролируемым размером пор и нагрузкой 45 мкмоль/г уменьшает потенциальную возможность масштабировать процесс. Продолжительное время аммонолиза при комнатной температуре затратно и приводит к большому количеству побочных продуктов. В процессе выделения авторы используют осаждение продукта, что понижает выход продукта. Кроме того, в качестве детритилирующего агента используется раствор 3% дихлоруксусной кислоты в дихлорметане, который является токсичным галогенкалканом, трудным для утилизации и переработки.
В заявке WO 215/061246 A1 рассматривается оптимизированный подход к стадии удаления тритильной защиты с 5' конца, в том числе на примере нусинерсена, рассматривается влияние различных условий детритилирования на количество примесей с удаленными основаниями (депуринизация). Рассматривается влияние температуры, pH и концентрации на реакцию удаления DMT с 5' конца олигонуклеотида. В качестве лучших условий предлагается использовать муравьиную кислоту вместо уксусной, а реакцию детритилирования проводить при pH 2,5 и температуре 10°С. Остановку реакции проводили подщелачиванием до pH 5-7. Недостатком данного патента является необходимость охлаждения реакционной массы, что может потребовать специального технологического оборудования, а также малый масштаб описываемых реакции. Кроме того, авторы приводят данные только по проценту депуриниизации, без указания чистоты исходных и конечных олигонуклеотидов.
Из уровня техники известны и иные решения получения антисмысловых олигонуклеотидов. В заявке US 20060035224 A1 рассматривается возможность очистки на полиэтилениминовой фазе; в публикации «Automated determination of early eluting oligonucleotide impurities using ion-pair reversed-phase liquid chromatography high resolution-mass spectrometry» Stilianos G. Roussis*, Isaiah Cedillo, Claus Rentel Ionis Pharmaceuticals, Carlsbad, CA, 92010, USA, раскрыт способ очистки олигонуклеотидов, состоящий из двух стадий: анионообменной хроматографии и обращенно-фазовой хроматографии; в патенте US 9243023 B2 раскрыт способ очистки олигонуклеотидов методом реакции полимеризации; патент EP 3870594 B1 раскрывает очистку олигонуклеотидов методом обращенно-фазовой хроматографии; в публикации «Evaluating and Isolating Synthetic Oligonucleotides The Complete Guide» Dr. Alex Andrus Printed in the USA, 04/2002 Part Number 4304603B раскрыт способ очистки олигонуклеотидов при помощи специфичных картриджей; в заявке WO 2017/218454 A1 раскрыт способ очистки олигонуклеотидов методом гидрофобной хроматографии.
Публикации, раскрывающие полностью способ, позволяющий провести полный цикл синтеза и очистки нусинерсена, пригодный для производства лекарственного средства, в уровне техники не представлены.
Раскрытый в патенте RU 2738093 подход имеет ряд недостатков по сравнению с предложенным в заявленном изобретении решением, а именно слишком малый масштаб синтеза, который не позволяет получать количества, пригодные для промышленной наработки. Выделение и очистка нусинерсена осуществляется методом осаждения, что приводит к большим потерям продукта и невозможностью контролировать параметры процесса. Описанный способ очистки синтетических олигонуклеотидов методом анионообменной и обращенно-фазовой жидкостной хроматографии имеет ряд недостатков. Во-первых, используется аналитический анионообменный сорбент, который имеет размер гранул 13 нм, что не позволяет его упаковать в промышленные колонны. В решении представлен аналитический масштаб, который сложно масштабировать до промышленных объемов. Во-вторых, авторы приводят данные по чистоте олигонуклеотида, не уточняя содержание конкретных примесей.
Способ очистки синтетического олигонуклеотида на полиэтилеиминовой фазе US 20060035224 A1 включает нетехнологичные стадии, которые сложно масштабируемы и удорожают производство. Недостатком также является низкая чистота полученного продукта (около 90%).
Недостатком способа, раскрытого в публикации Stilianos G. Roussis, является отсутствие информации о качестве и соотношении близкородственных примесей в конечном продукте, что может привести к получению продукта низкого качества. Помимо этого, явным технологическим недостатком является применение в качестве основной стадии очистки ВЭЖХ, что требует от производителя наличия дорогостоящего оборудования, оборот большого количества токсичных растворителей, от которых впоследствии необходимо избавляться.
Основным недостатком способа по патенту US 9243023 B2 является удаление одной конкретной примеси P=O. Кроме того, данный способ не описывает возможность образования побочных продуктов реакции полимеризации.
Недостатком способа очистки обращенно-фазовой ВЭЖХ по патенту EP 3870594 B1 помимо вышеперечисленных, относящихся к ОФ ВЭЖХ, является низкий выход олигонуклеотида (44%) и низкая чистота конечного продукта (88%).
Способ выделения олигонуклеотидов при помощи OPC картриджей, связывающих олигонуклеотид по аффинному механизму, раскрытый в публикации Dr. Alex Andrus, не позволяет оценить наличие примесей в полученном продукте.
Условия проведения очистки по заявке WO 2017/218454 A1 позволяют снизить содержание примесей, однако одна из основных примесей n+1 остается на довольно высоком уровне.
Основным и общепринятым способом является очистка олигонуклеотидов методом жидкостной хроматографии. В случае очистки олигонуклеотидов с защитной группой на 5’ конце, представляющей из себя 5’-диметокситритил, наиболее часто используемым способом является обращено-фазовая высокоэффективная жидкостная хроматография.
В документе RU 2738093 указан способ производства и, в частности, выделения и очистки антисмыслового олигонуклеотида методом анионообменной хроматографии на сорбенте Tosoh Tskgel superQ-5pw. Подвижная фаза при данном процессе состоит из 20 мм триса, 10% ацетонитрила, и перхлората, хлорида или бромида натрия. Исходя из представленных данных, чистота олигонуклеотида после проведения стадии составляет 91-93%. В качестве второй стадии очистки используется обращенно-фазовая хроматография на сорбенте Waters Xbdrige Peptide ВЕН C18 180А 4.6×250. Полученный раствор олигонуклеотида упаривают под вакуумом, затем осаждают последовательно ацетоном и этиловым спиртом, для удаления ацетата аммония или триэтиламмония. После осаждения олигонуклеотид обессоливают методом гель-фильтрации на сорбенте Sephadex G-25 (колонна HiPrep 26/10 Desalting). Пик фракционировали, выбирали фракции с поглощением более 200 mAU, электропроводностью больше 30 мСм/см. Затем фракции анализируют методом масс-спектрометрии. Фракции с чистотой более 95% объединяют для дальнейшей лиофилизиации.
Предложенный способ производства обладает ключевым недостатком, которое заключается в применении аналитического анионообменного сорбента Tosoh Tskgel superQ-5pw. Данный сорбент имеет размер гранул 13 нм, что не позволяет применять его в реальном промышленном процессе как из-за конструктивных ограничений хроматографических колонн (сорбент будет просачиваться через верхний и нижний фритт колонны), так и из-за невозможности создавать производственным оборудованием необходимых условий для протока подвижной фазы как при упаковке колонны, так и при непосредственном проведении технологической стадии. Аналитический масштаб предлагаемого способа очистки подтверждает использование авторами сугубо аналитической системы ВЭЖХ Dionex Ultimate 3000. Последняя стадия очисти методом гель-фильтрации вызывает вопросы, авторы решения заявляют объем нанесения 15 мл на колонну объемом 53 мл или 28%, что является слишком большой нагрузкой для данного типа сорбента с учетом типа сорбента и целевой молекулы. На низкую эффективность удаления низкомолекулярных примесей указывает слишком высокое значением электропроводности в целевых фракциях (более 30 мСм/см), при этом в качестве подвижной фазы используется вода очищенная (электропроводность стремится к 0 мСм/см). Такое несоответствие электропроводности целевых фракций и подвижной фазы указывает на то, что в процессе очистки, возможно, происходит неполное удаление низкомолекулярных примесей и остаточных растворителей, которые могут в итоге попасть в конечный продукт.
Предложенный способ производства нусинерсена не может быть масштабирован для удовлетворения потребности в лекарственном средстве для терапевтического использования.
В US 20060035224 A1 авторы описывают способ очистки олигонуклеотидов на ионообменном сорбенте с привитой полиэтилениминовой фазой. (Matrex Ion Exchange Silica PEI-300-15). В очистке используется карбонатный буферный раствор А, состоящий из 50 ммоль гидрокарбоната натрия pH=8,2; буферный раствор B, состоящий из 50 ммоль гидрокарбоната натрия pH=11,1 (pH доводят 0,1 М раствором NaOH), 0,1 М раствор натрия гидроксида (раствор С) и буферный раствор D, состоящий из 0,1М триса, 2 М натрия хлорида, pH=7,5. В растворе после аммонолиза доводят pH до 7,5 ортофосфорной кислотой. Полученный раствор наносят на колонну, упакованную ионообменным сорбентом. Колонну предварительно уравновешивают раствором А. После нанесения сорбент с сорбированным олигонуклеотидов промывают раствором А. Элюцию осуществляют градиентом раствора А в растворе В от 0 до 80% за 20 колоночных объемов, создавая тем самым градиент pH. Пик фракционируют, фракции с содержанием целевого олигонуклеотидов больше 90% объединяли, обессоливали методом ультрафильтрации и лиофильно высушивали. Недостатком данного метода очистки является низкая чистота целевого продукта (на уровне около 90%), а также использование низкотехнологичных стадий: диафильтрации и лиофилизации.
Также в данном решении указан способ очистки олигонуклеотидов на полиэтиленимин модифицированном силикагеле, отличающийся от предыдущего составом растворов для подвижной фазы. В качестве уравновешивающего буфера используют раствор 25 ммоль гидрокарбоната натрия, pH=7,5, буфер элюции имеет состав 100 ммоль гидроксида аммония, pH=11.1, в качестве раствора для промывки используют воду очищенную. Раствор олигонуклеотидов перед нанесением на колонну титруют 1,0 М уксусной кислотой до pH=7,5. Раствор олигонуклеотидов наносят на колонну, промывают водой очищенной и элюируют раствором с pH=11.1. Чистота полученного олигонуклеотида составила около 90%. Как и описанная выше методика очистки, рассматриваемый способ очистки не позволяет достичь чистоты целевого продукта более 90%.
Затем, согласно рассматриваемому решению, все полученные образцы обессоливают путем фильтрации в тангенциальном потоке. Как отмечалось выше, данный способ является нетехнологичным, т.к. для обессоливания нуклеотидов применяют кассеты с отсечкой менее 1кДа (Lajmi, A. R., Schwartz, L., & Sanghvi, Y. S. (2004). Membrane Purification of an Antisense Oligonucleotide. Organic Process Research & Development, 8(4), 651–657), что выражается в крайне высокой длительности данного технологического этапа.
Согласно публикации Stilianos G. Roussis анионообменную хроматографию проводят на сорбенте Q sepharose FF при температуре около 20°С. Уравновешивающим раствором является раствор гидроксида натрия с концентрацией 25 ммоль. Стартовый материал представляет собой раствор олигонуклеотида с тритильной защитой. Стартовый материал наносят на колонну, затем колонну промывают 20% градиентом раствора натрия гидроксида с концентрацией 25 ммоль, и натрия хлорида с концентрацией 2 моль. При проведении промывки элюируют молекулы олигонуклеотида без тритильной защиты, которые потенциально являются нежелательной примесью. Элюция целевого олигонуклеотида осуществляется линейным градиентом от 0% до 90% раствора, состоящего из 25 ммоль натрия гидроксида и 2 моль натрия хлорида против раствора, содержащего 25 ммоль натрия гидроксида. Так же этими авторами предложен способ очистки олигонуклеотида при помощи обращено-фазовой хроматографии. При обращено-фазовой хроматографии используют сорбент XTT30. Объем колонны составил 44 мл. Процесс проводят при длине волны 295 нм с длиной оптического пути 0,5 мм. Процесс проводят при скорости потока 153 см/ч. В стартовый материал добавляют 7,9% 3.0 М раствора ацетата натрия и 29,7% метанола по массе. Колонну уравновешивают 3.0 колоночными объемами смеси вода/3.0 М раствор ацетата натрия/ метанол в соотношении 62,4/7,9/29,7 по массе. После нанесения на колонну стартового материала проводят элюцию целевой молекулы с колонны. При этом олигонуклеотид без тритильной защитной группы десорбируется с колонны после промывки тремя колоночными объемами смеси вода:3.0 М раствор ацетата натрия: метанол в соотношении 50,5:9,7:39,8 (соотношение по массе). Олигонуклеотид с тритильной защитной группой элюируют с колонны промывкой 2,5 колоночных объемов смесью вода:3.0 М раствор ацетата натрия:метанол в соотношении 19,4:4,4:75,7 (соотношение по массе). Элюат, содержащий олигонуклеотид с тритильной защитной группой собирают, начиная с оптической плотности 250 мAu при длине волны 295 нм. Регенерируют колонну 1,5 колоночными объемами метанола. Олигонуклеотид с тритильной защитной группой осаждают добавлением метанола в соотношении 1:1,5 по объему. После отстаивания в течение суток, супернатант декантируют. Осадок растворяют в воде до концентрации 50 мг/г раствора. В полученном раствора доводят pH до значения 4,5 добавлением ледяной уксусной кислоты и инкубировали при температуре 40°C в течение 145 минут, останавливали реакцию добавлением 3.0М раствора ацетата натрия до pH=5,5. В публикации описаны только отдельные стадии производственного процесса, не в полной мере понятно, какой чистоты конечного препарата в итоге достигают авторы работы. Очевидным технологическим недостатком является использование стадии обращенно-фазовой хроматографии, требующей использования большого количества токсичных растворителей и последующие проблемы с их утилизацией. Дополнительным недостатком использования стадии ОФВЭЖХ является необходимость использования отдельного выделенного оборудования для проведения данной стадии. Авторы решения приводят информацию по чистоте только по раноэлюруемым примесям, при этом не приводят подтверждения очистки продукта от других типов примесей (коэлюируемые и поздно элюируемые примеси, элементные примеси, остаточные растворители).
Существует способ очистки нуклеотидов, описанный в US 9243023 B2, отражающий очистку полноразмерного олигонуклеотида при помощи реакции полимеризации с полиакриламидом. Полноразмерный олигонуклеотид предварительно очищают от олигонуклеотида с делециями методом обращенно-фазовой ВЭЖХ. Затем полноразмерный олигонуклеотид, содержащий примеси P=O, отделяют от целевого сульфированного олигонуклеотида при помощи реакции полимеризации. К смеси олигонуклеотидов добавляют полимеризующий агент (250 мкл N,N-диметилакриламида 1,69 М, N,N'-метиленбис(акриламид) 16,9 мМ). Колбу продувают азотом и при постоянно перемешивании добавиляют 5 мкл 10% раствора тиосульфата аммония и 5 мкл N,N,N',N'-тетраметилэтилендиамина. В реакцию полимеризации вступают олигонуклеотиды, имеющие связь P=O, при этом P=S остаются в растворе. Спустя 30 минут, полученный гель промыли водой, экстрагируя целевой олигонуклеотид в водный раствор. Очищенный раствор олигонуклеотида обессоливали при помощи гель-фильтрации с добавлением гидроксида аммония, для последующего высушивания. Авторы решения 5 описывают только отдельные стадии очистки олигонуклеотидов (метод очистки методом полимеризации и методом гель-фильтрации с последующей лиофилизацией). Недостатком данного решения является его направленность только на удаление примеси P=O, при этом указанный способ не позволяет удалять остальные примеси, значительное разнообразие которых образуется в процессе синтеза. Важным моментом, который не затрагивают авторы решения, является образование побочных продуктов реакции полимеризации, что несет дополнительные риски с точки зрения безопасности конечного препарата.
В EP 3870594 B1 указан способ очистки олигонуклеотида методом обращенно-фазовой ВЭЖХ. Раствор олигонуклеотида с тритильной защитной группой наносят на колонну с привитой фазой C18 (YMC Triart C18 preparative). Стартовый буфер представляет собой 50 ммоль/л раствор карбоната натрия; буфер элюции – чистый ацетонитрил. Затем очищенную фракцию разбавляют водой до концентрации 2 г/л, после чего материал концентрируют приблизительно в 20 раз, добавляют этанол до конечной концентрации 40% и проводят диафильтрацию против ацетатного буфера с концентрацией 100 ммоль/л, pH=3 для снятия тритильной защиты. Процесс проводят 4,8 часа. Для нейтрализации реакции добавляют 0,1 раствор натрия гидроксида и инкубируют в течение 6 минут. После чего продолжают диафильтрацию против смеси вода: этанол 4:1 в течение 2,4 часа. Общий выход с процесс составляет 44%, чистота по UV ВЭЖХ – 88%.
Предложенный способ очистки нуклеотидов содержит информацию только по отдельным стадиям очистки, при этом не описывая технологию комплексно. Недостатком данного способа является низкая чистота конечного продукта (88% по данным UV ВЭЖХ) и низкий выход в процессе (44%).
Согласно публикации Dr. Alex Andrus, используется способ выделения олигонуклеотидов при помощи OPC картриджей. Способ представляет собой сорбцию по аффинному механизму взаимодействия олигонуклеотидов с тритильной защитной группой на данный сорбент с последующей элюцией. Затем на сорбированном олигонуклеотиде проводят реакцию детритилирования путем понижения pH до значения 3 ед. Элюция проводится в растворе ацетонитрила. В дальнейшем обессоливание проводят методом гельфильтрации. Очевидным недостатком использования OPC картриджей является их крайне высокая цена и сугубо лабораторное применение. Как и в вышеуказанных решениях, авторы не раскрывают технологию целиком и не оценивают уровни примесей конечного продукта.
В заявке WO 2017/218454 A1 описывается способ очистки синтетических олигонуклеотидов методом гидрофобной хроматографии. Авторы изучают влияние нагрузки на сорбент в диапазоне 8,8-100% от максимальной, влияние различных неорганических солей. В качестве примера используют сульфат аммония. В стартовый материал, содержащий олигонуклеотид с тритильной защитой добавили аммония сульфат до концентрации 765 мМ, трис 50 мМ, pH=8,5. Хроматографию проводили на сорбенте Phenyl Sepharose FF HS. Сорбент уравновешивают буферным раствором, состоящим из 850 мМ сульфата аммония, 50 мМ триса в течение 4 колоночных объемов. Процесс проводят при скорости потока 200 см/ч. После нанесения стартового материала колонну промывают 7 колоночными объемами раствора, состоящего из 440 мМ сульфата аммония и 50 мМ триса. Целевой олигонуклеотид элюируют раствором, состоящим из 40 мМ аммония сульфата и 50 мМ триса. В результате при наибольшей нагрузке удалось снизить количество примесей: примесь (n-1) с 3,2% до 2,6%, P=O с 2,6% до 2,1%, примесь (n+1) с 1,25% до 0,2%, CNet с 0,31% до 0,20%. Однако не указано, какой чистоты достигают авторы для конечного продукта. Содержание всех примесей, за исключением n+1, остается на высоком уровне после проведения последней стадии очистки.
На данный момент в литературе не представлено полного комплексного описания технологии производства антисмысловых олигонуклеотидов, рассматриваемые решения содержат информацию только по отдельным стадиям производства. Почти во всех решениях предлагается использование стадии очистки методом ОФВЭЖХ и диафильтрации, которые имеют ряд существенных недостатков. Все рассмотренные решения относятся к области лабораторного применения, не могут быть масштабированы и использованы для промышленного производства лекарственных средств на основе нусинерсена.
Наиболее близким аналогом к заявленному изобретению является способ, раскрытый в патенте РФ №2738093. Данное решение описывает технологию синтеза и очистки нусинерсена, однако, авторы описывают синтез лабораторного масштаба, а процесс очистки в аналитическом масштабе с применением соответствующих аналитических хроматографических сорбентов и оборудования. Таким образом, предлагаемая технология не может быть масштабирована и применяться для выпуска коммерческой продукции. Дополнительным недостатком предлагаемой в патенте РФ №2738093 технологии является использование стадии обращенно-фазовой хроматографии, которая несет в себе ряд недостатков, описанных выше. Авторы работы не указывают информацию по итоговому выходу финального продукта.
Проблема существующих способов получения нусинерсена методом твердофазного синтеза заключается в неспособности достижения достаточного количества финального препарата высокой чистоты при сохранении высоких выходов по целевому продукту. Решение данной проблемы позволит повысить доступность данного препарата при сохранении высоких требований к его качеству.
Задача настоящего изобретения – разработка способа синтеза и очистки нусинерсена, позволяющего получать высокоочищенный продукт с высоким выходом в количествах, достаточных для удовлетворения терапевтических потребностей пациентов.
Технический результат – повышение эффективности очистки тиофосфатных олигонуклеотидов при сохранении высоких выходов целевого продукта.
Поставленная задача решается, а технический результат достигается путем создания способа получения модифицированного тиофосфатного олигонуклеотида, содержащего 2'-O-(2-метоксиэтил)рибонуклеотиды, где каждый нуклеозид содержит 2'-O-(2-метоксиэтил) модифицированный остаток сахара,
причем олигонуклеотид синтезируют на твердофазном носителе с использованием амидофосфитного метода,
причем в качества активатора используется 5-(этилтио)-1H-тетразол (ETT),
при этом способ получения содержит следующие этапы:
a) для получения тиофосфатных групп используют гидрид ксантана (5-амино-1,2,4-дитиазол-3-тион) в 20% растворе пиридина в ацетонитриле, аммонолиз проводят в 23-35% растворе гидроксида аммония в течение от 16 до 24 часов, доводят значение удельной электропроводности (УЭП) до 145,0–170,0 мСм/см,
b) проводят очистку на гидрофобном сорбенте с размером гранул от 45 до 165 мкм для удаления основного количества низкомолекулярных компонентов и родственных примесей, проводят финальное детритилирование промежуточного полупродукта,
c) затем проводят хроматографическую очистку в присутствии полярных апротонных растворителей на анионообменном сорбенте для эффективного удаления родственных примесей нусинерсена
d) и далее проводят очистку гель-фильтрационной хроматографией для удаления остаточных органических растворителей и элементных примесей.
Согласно предпочтительным вариантам реализации указанный технический результат также достигается тем, что:
- гидрофобный сорбент выбран из группы, включающей Butyl Sepharose FF, Phenyl Sepharose FF, Phenyl-650M, Capto Phenyl;
- анионобменный сорбент выбран из группы, включающей Q Sepharose FF, 30 Q Source, GigaCap Q650M, BioPro IEX SmartSep Q30 (YMC), WorkBeads 40Q.
- хроматографическая очистка на анионобменном сорбенте проводится для удаления полноразмерных олигонуклеотидов, имеющих связь P=O, примесей (n-1) и (n-2), (n+1), примесей с удаленным азотистым основанием.
- этап очистки гель-фильтрационной хроматографией проводится на сорбенте, выбранном из группы, включающей Sephadex G-25, Superdex 75, HW-50F.
- очистку на гидрофобном сорбенте проводят для удаления основного количества низкомолекулярных компонентов и родственных примесей и отделения последовательности с делециями без групп DMT и разветвленных соединений, содержащих две или более группы DMT, от неочищенного продукта с одной группой DMT.
- после этапа очистки гель-фильтрационной хроматографией продукт концентрируют и проводят его фильтрацию.
- фильтрацию продукта проводят через фильтр 0,2 мкм для снижения микробной нагрузки.
- модифицированный тиофосфатный олигонуклеотид имеет последовательность UCACUUUCAUAAUGCUGG.
- модифицированный тиофосфатный олигонуклеотид представляет собой нусинерсен.
Краткое описание чертежей
Изобретение поясняется следующими чертежами.
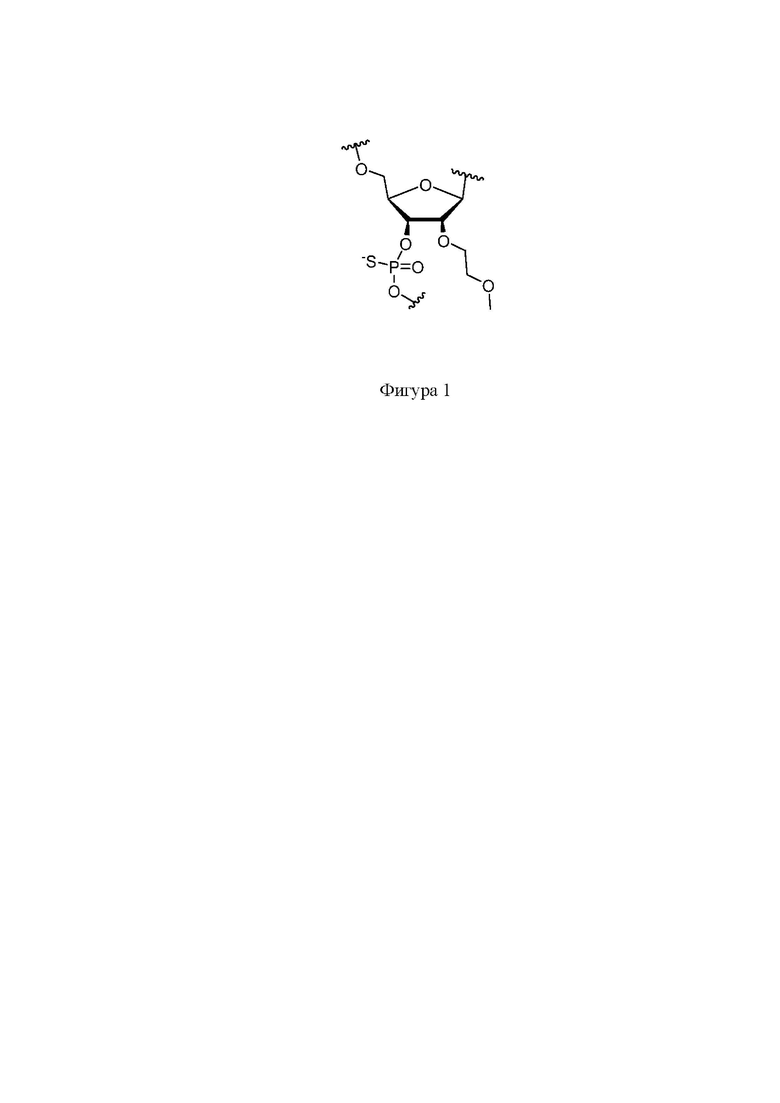
На фигуре 1 представлена структура фосфоротиоатного линкера.
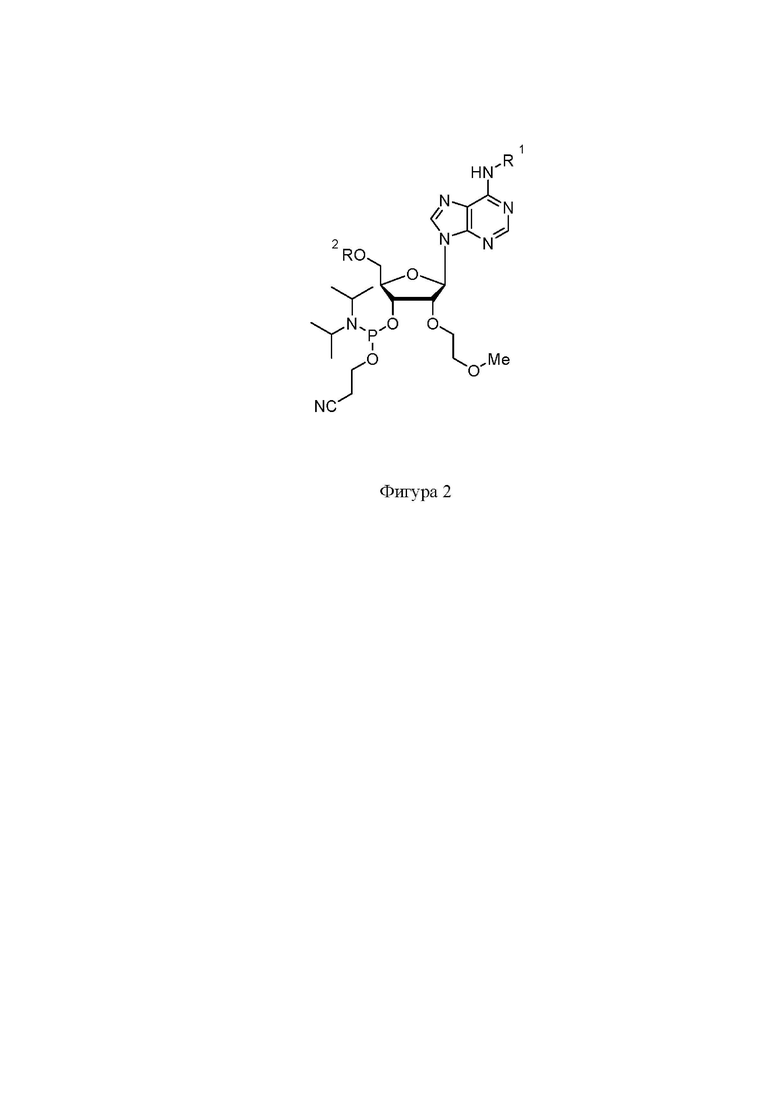
На фигуре 2 представлена структура защищенного A-2’-MOE фосфорамидита.
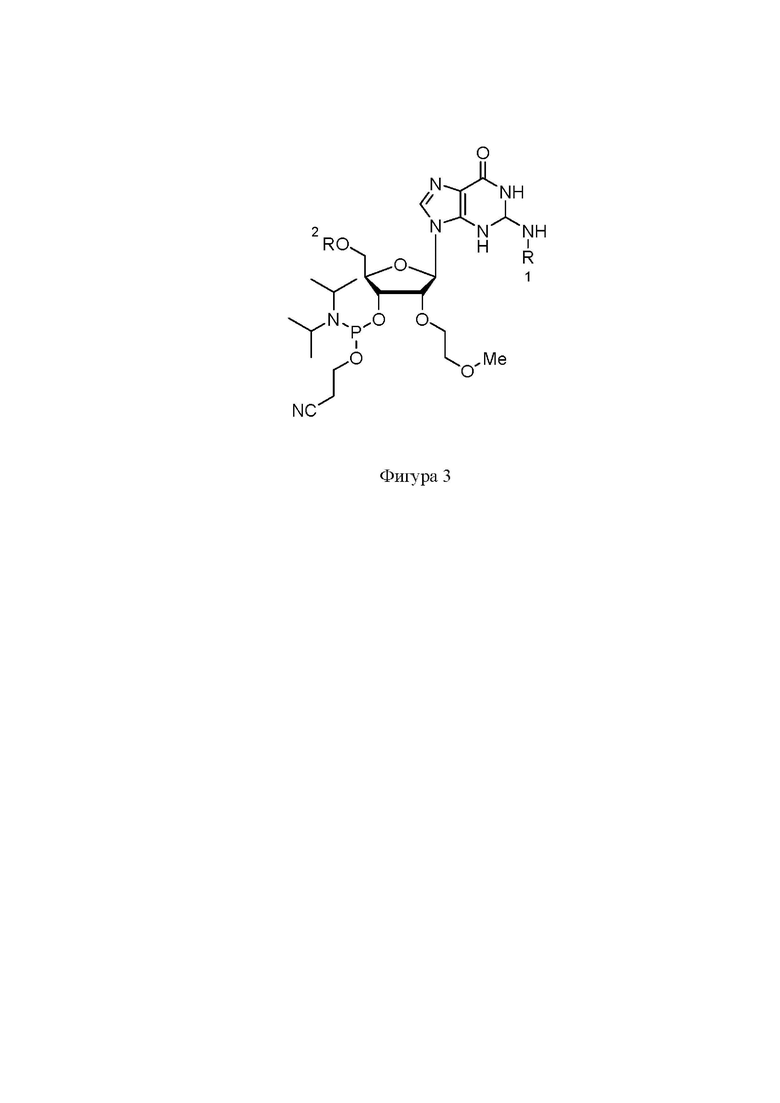
На фигуре 3 представлена структура защищенного G-2’-MOE фосфорамидита.
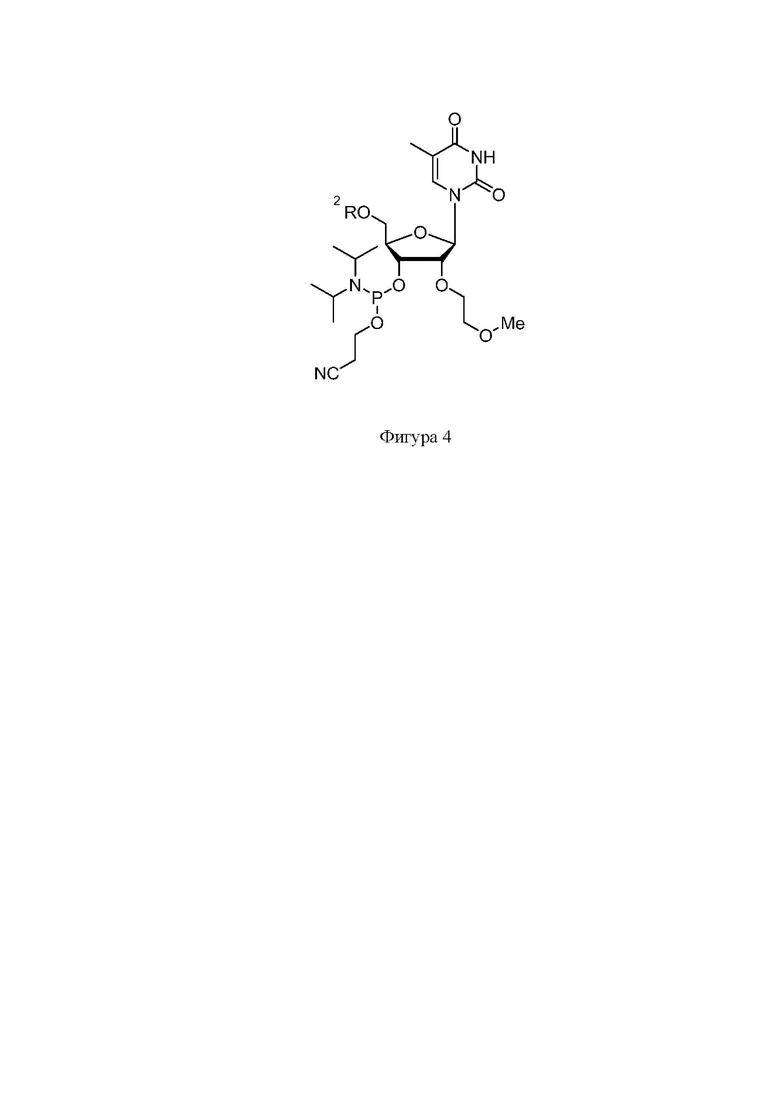
На фигуре 4 представлена структура защищенного T-2’-MOE фосфорамидита
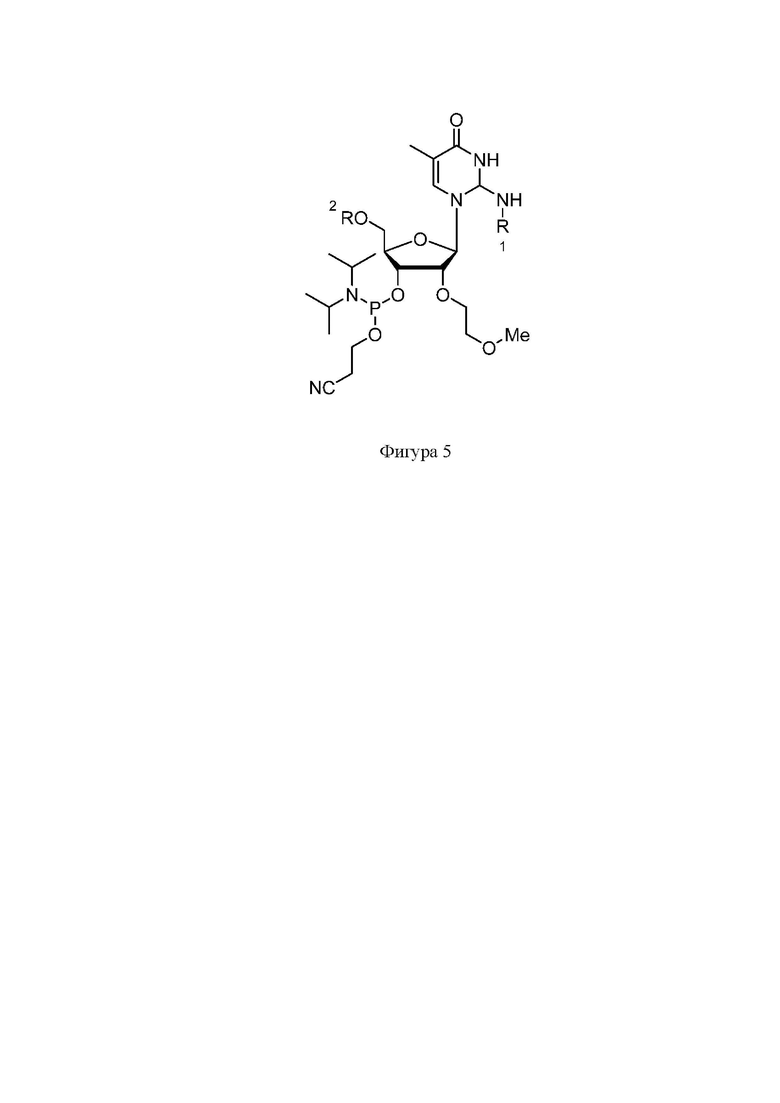
На фигуре 5 представлена структура защищенного C-2’-MOE фосфорамидита.
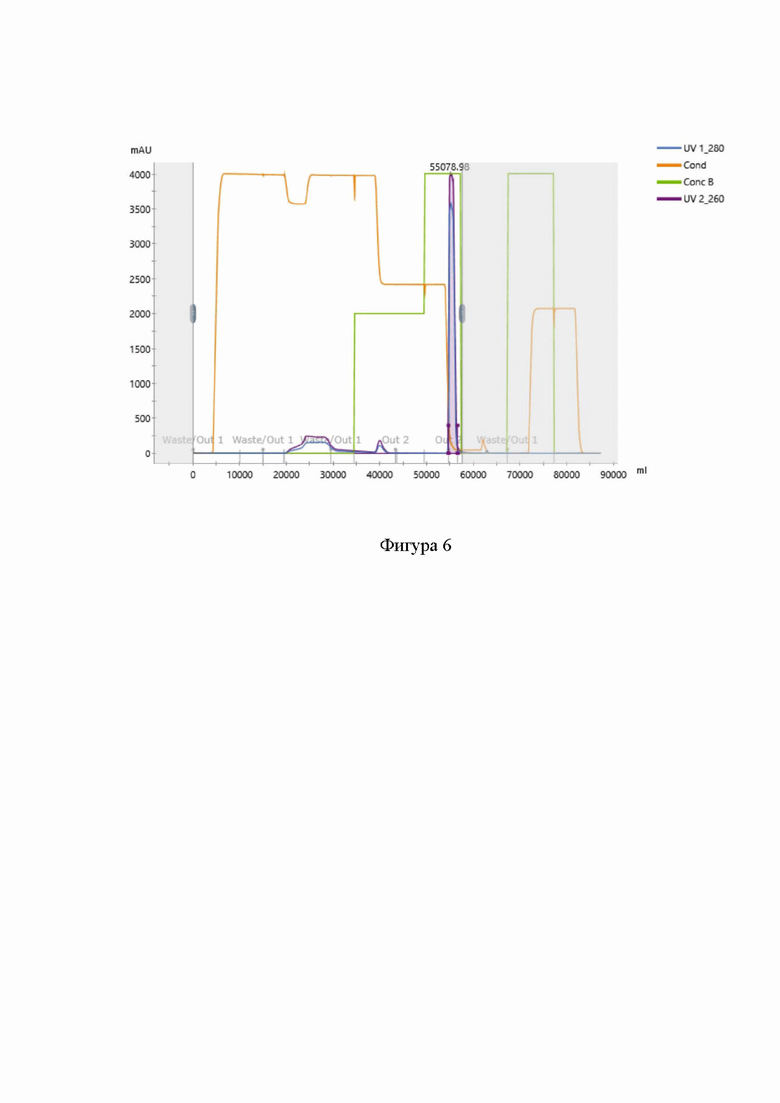
На фигуре 6 представлена хроматограмма очистки нусинерсена методом гидрофобной хроматографии. При этом:
“UV 1_280” представляет собой хроматограмму выхода раствора целевого продукта с детекцией УФ поглощения раствора на длине волны 280 нм,
“UV 2_260” представляет собой хроматограмму выхода раствора целевого продукта с детекцией УФ поглощения раствора на длине волны 260 нм,
“Cond” представляет собой график проводимости раствора,
“Conс B” представляет собой график доли буфера B в смеси буферов A и B.
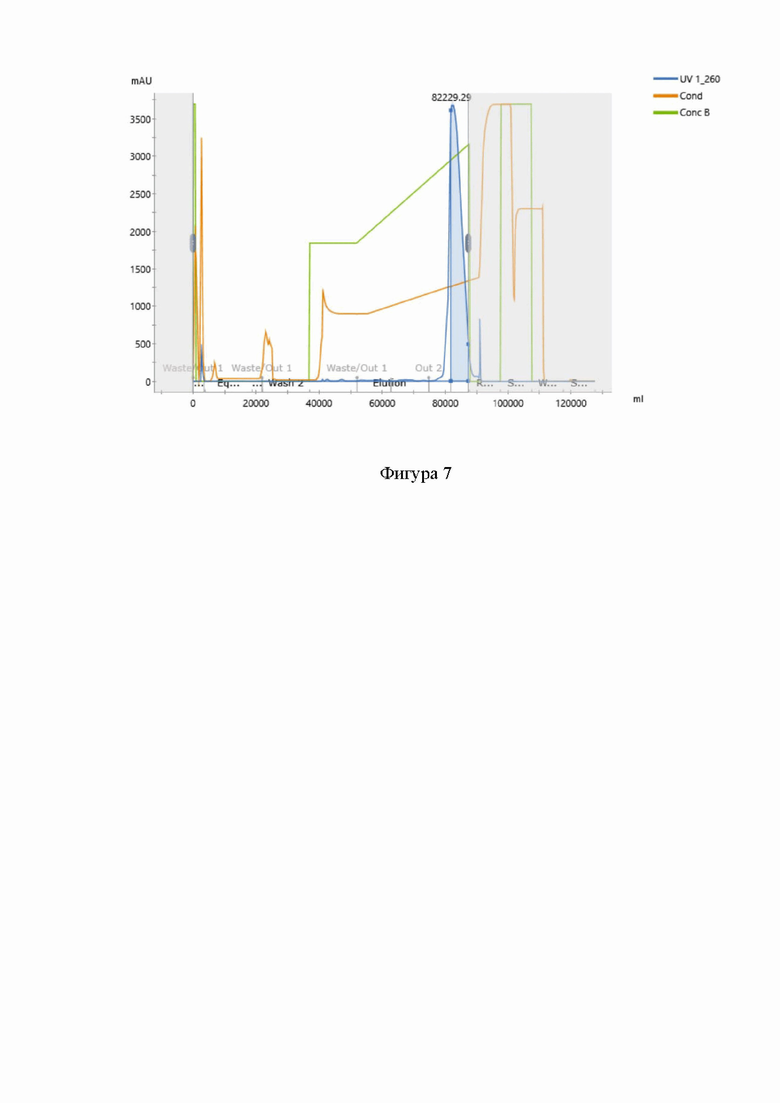
На фигуре 7 представлена типичная хроматограмма очистки нусинерсена методом анионообменной хроматографии. При этом:
“UV 1_260” представляет собой хроматограмму выхода раствора целевого продукта с детекцией УФ поглощения раствора на длине волны 260 нм,
“Cond” представляет собой график проводимости раствора,
“Conс B” представляет собой график доли буфера B в смеси буферов A и B.
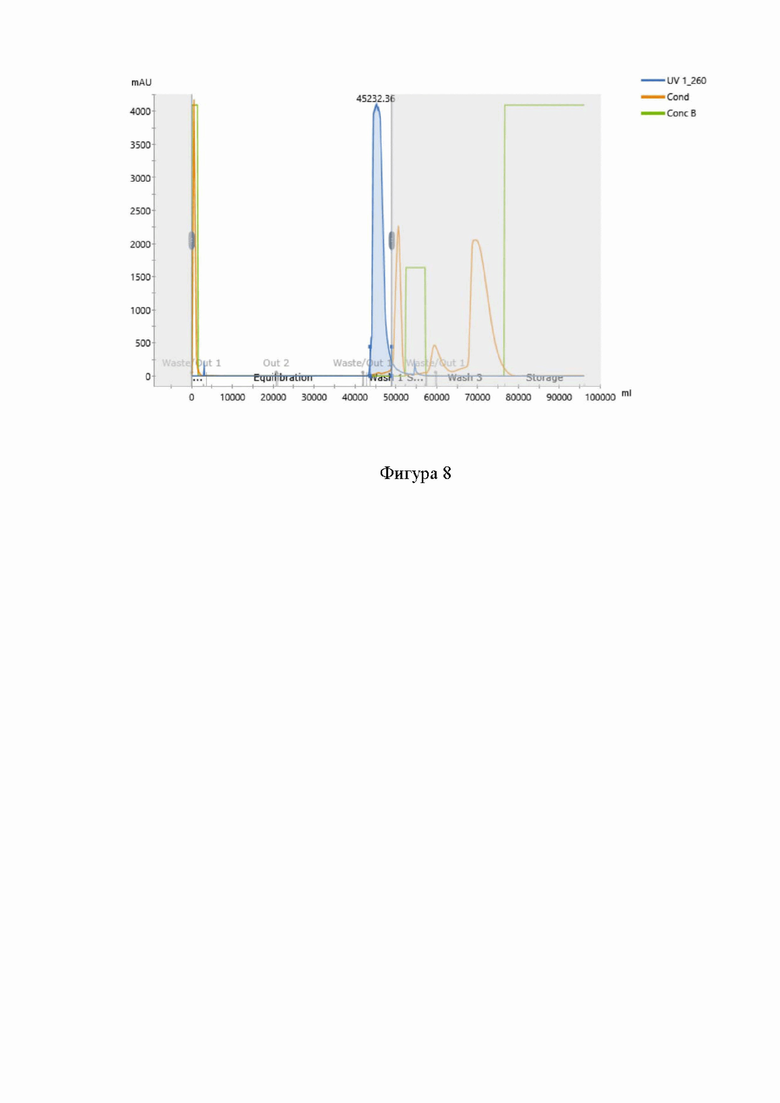
На фигуре 8 представлена типичная хроматограмма очистки нусинерсена методом гель-фильтрационной хроматографии. При этом:
“UV 1_260” представляет собой хроматограмму выхода раствора целевого продукта с детекцией УФ поглощения раствора на длине волны 260 нм,
“Cond” представляет собой график проводимости раствора,
“Conс B” представляет собой график доли буфера B в смеси буферов A и B.
Преимуществами предлагаемого согласно настоящему изобретению способа являются:
- высокоэффективный твердофазный синтез с низким расходом ключевого дорогостоящего сырья;
- оптимизированная схема синтеза, позволяющая получать полупродукт с чистотой по основной форме не менее 90 % (метод ВЭЖХ МС);
- применение стадии высокоэффективной гидрофобной хроматографии, позволяющей селективно удалять низкомолекулярные примеси и продуты синтеза, не содержащие синтетический олигонуклеотид с защитой группой на 5’ конце, что позволяет получать целевой продукт с выходом не менее 85 % и чистотой не менее 91-92%;
- применение высокоэффективной двухстадийной анионообменной стадии очистки, позволяющей получать олигонуклеотид с чистотой не менее 96 % (метод ВЭЖХ МС) и выходом не менее 60%;
- применение стадии гель-фильтрационной хроматографии для удаления остаточных растворителей, позволяющей избежать трудозатратой и дорогой комбинации стадий диафильтрации и последующей лиофилизации, при этом выход со стадии составляет не менее 90% и чистота целевого продукта составляет не менее 96% (метод ВЭЖХ МС);
- комбинация описанных способов твердофазного синтеза и хроматографической очистки, позволяющая получать финальный продукт с чистотой не менее 96% по основной форме и уровнем примесей: не более 1% для примеси (n-1); не более 1,5% для примеси Р=О; не более 0,5% для примеси (n+1); не более 0,3% для примесей Loss A, Аbasic и суммарным выходом на уровне не менее 50%.
В ходе разработки процесса синтеза нусинерсена была повышена эффективность выхода основного продукта за счет изменения процедуры аммонолиза, а также достигнута хорошая масштабируемость хроматографической очистки за счет применения сорбентов с более крупным размером гранул.
Нусинерсен представляет собой 18-членный фосфоротиоатный олигонуклеотид, несущий в своей структуре 2-метилоксиэтильные группы во втором положении рибозы (Фигура 1), а также метилированный по 5 положению цитидин.
Синтез нусинерсена осуществлялся фосфорамидитным методом на твердом носителе. Защищенные фосфорамидиты были приобретены в коммерческих источниках (Hongene Biotech, WuXi AppTec, Chemgenes и др.). Структуры фосфорамидитов представлены на Фигурах 2-5, где R1: Бензоил (Bz), изобутирил (iBu), ацетил (Ac); R2 – 4,4'-диметокситритильная группа (DMT).
Олигонуклеотид синтезировался на синтезаторе ÄKTA oligopilot 100, на реакционных колоннах для синтеза объемом от 20 до 60 мл (КО). В качестве твердого носителя для синтеза использовался коммерчески доступный полистирольный сорбент NittoPhase HL (Kinovate) с емкостью от 300 до 400 мкмоль/г. Масштаб синтеза составил от 180 до 1800 ммоль. Дорогостоящие фосфорамидиты вносят самый большой вклад в себестоимость олигонуклеотида. Оптимизация процесса синтеза позволила снизить количество используемых на каждой стадии фосфорамидитов до 1,3-1,5 мольных эквивалентов, что существенно снизило себестоимость продукта. Дорогостоящие безводные растворители также вносят значительный вклад в себестоимость. Оптимизация процесса позволила значительно сократить количество используемого ацетонитрила.
Твердый носитель и присоединенные мономеры детритилируют деблокирующим раствором, который состоит из 3-5% раствора дихоруксусной кислоты в толуоле или дихлорметане, предпочтительным является 5% раствор дихлоруксусной кислоты в толуоле.
Для активации стадии присоединения используются 4,5-дицианоимидазол (DCI), 1H-тетразол, предпочтительным является 5-этилтиотетразол (ETT).
На стадии тиолирования оксиление проводят 3-(диметиламино-метилиденамино)-3H-1,2,4-дитиазол-3-тионом (DDTT), 3H-1,2-бензодитиол-3-он 1,1-диоксидом (реагентом Бокажа), предпочтительным является использование гидрида ксантана. В качестве кэпирующего агента используют смесь пропионового ангидрида, 2,6-лутидина, N-метилимидазола и ацетонитрила. Аммонолиз проводят в 23-35% растворе гидроксида аммония.
Синтетический процесс данного изобретения включает в себя следующие стадии:
1. детритилирование твердого носителя;
2. активация и присоединение следующего в последовательности фосфорамидита;
3. окислительное тиолирование эфира фосфита;
4. кэпирование непрореагировавших сайтов;
5. детритилирование присоединенного мономера для активации 5` гидроксила;
6. повторение шагов 2-5 до достижения необходимой длинны олигонуклеотида;
7. деблокирование тиофосфатных групп при помощи диэтиламина;
8. аммонолиз.
Последнее детритилирование в синтетической процедуре данного изобретения не проводится для облегчения дальнейшей очистки.
Предпочтительные варианты реализации изобретения
Основный технический результат, достигнутый в ходе осуществления настоящего изобретения – повышение эффективности очистки тиофосфатных олигонуклеотидов при сохранении высоких выходов целевого продукта – выразился в следующем: получен продукт с содержанием основного вещества не менее 96%; содержание специфицированных примесей олигонуклеотида составляло:
Способ может быть масштабируем от лабораторных до промышленных объемов. Основные стадии очистки предложенного способа:
1. Очистка промежуточного полупродукта на одном из вариантов гидрофобного сорбента Butyl Sepharose FF (Cytiva), Phenyl Sepharose FF (Cytiva), Phenyl-650M (Tosoh), Capto Phenyl (Cytiva) размером гранул от 45 до 165 мкм или иного гидрофобного сорбента с аналогичным принципом действия и размером гранул. На данной стадии происходит первичный захват продукта и эффективное удаление основного количества низкомолекулярных компонентов, а также родственных примесей. Промежуточный полупродукт содержит гидрофобную группу DMT, которая используется в ходе хроматографии в качестве средства, которое позволяет удалять примеси, такие как последовательности с делециями без групп DMT и разветвленные или другие соединения, содержащие две или более группы DMT, от неочищенного продукта с одной группой DMT;
2. После очистки промежуточного полупродукта от вариантов с делециями без групп DMT и содержащих несколько групп DMT проводят финальное детритилирование промежуточного полупродукта, после чего получают нусинерсен;
3. Стадия очистки в присутствии полярных апротонных растворителей на анионообменном сорбенте Q Sepharose FF (Cytiva), 30 Q Source (Cytiva), GigaCap Q650M (Tosoh), BioPro IEX SmartSep Q30 (YMC), WorkBeads 40Q (Bio-Works) размером гранул от 30 до 90 мкм или на ином сорбенте с аналогичным принципом действия и размером гранул является ключевой, поскольку на данном сорбенте эффективно удаляются родственные примеси нусинерсена, такие как полноразмерные олигонуклеотиды (P=O), примеси (n-1) и (n-2), (n+1), примеси с удаленным азотистым основанием (Loss A, Аbasic);
4. Стадия гель-фильтрационной хроматографии на сорбенте Sephadex G-25 (Cytiva), Superdex 75 (Cytiva), HW-50F (Tosoh) размером гранул от 30 до 90 мкм удаляет остаточные органические растворители и элементные примеси;
5. На стадии концентрирования получают субстанцию-концентрат нусинерсена с требуемой концентрацией нусинерсена;
6. На стадии упаковки проводят фильтрацию раствора нусинерсена через фильтр 0,2 мкм что обеспечивает снижение микробной нагрузки.
На первой стадии очистки был использован классический подход для разделения олигонуклеотидов, основанный на присутствии в составе полупродукта сильно гидрофобной группы DMT.
Промежуточный полупродукт после синтеза и аммонолиза содержит сильно гидрофобную группу DMT, которая используется в ходе хроматографии для очистки неочищенного продукта с одной группой DMT от примесей, таких как последовательности с делециями без групп DMT и разветвленные или другие соединения, содержащие две или более группы DMT. Для разделения этих соединений на основе их гидрофобных свойств может быть использована обратно-фазовая хроматография (RPC) или гидрофобная хроматография (HIC), не требующая использования органических растворителей и хроматографов и колонн высокого давления.
При использовании сильно гидрофобной группы DMT с помощью хроматографии гидрофобного взаимодействия на сорбенте Capto Phenyl (Cytiva) происходит полное разделение примесей без группы DMT; полупродукта, содержащего одну группу DMT; примесей, содержащих несколько групп DMT. Благодаря лиотропному эффекту в водной подвижной фазе с высоким содержанием солей гидрофобная молекула притягиваются к гидрофобным лигандам HIC. Связанные молекулы элюируются при снижении концентрации соли. Поскольку HIC-разделение проводится в мягких условиях элюирования, оно не требует использования органических растворителей и высокого давления, что является значительным преимуществом перед обратно-фазовой хроматографией.
В раствор нусинерсена добавляют хлорид натрия, доводя значение УЭП до проводимости 145,0-170,0 мСм/см, затем наносят на сорбент, промывают и элюируют подвижной фазой с низкой УЭП. Профиль элюирования непрерывно контролируется методом абсорбционной УФ спектроскопии. Примеси без группы DMT выходят в проскоке и промывках, затем элюируют продукт с группой DMT. Сбор элюата начинают при достижении оптической плотности 100 mAu и заканчивают при достижении 100 mAu в правом «плече» пика (длина волны 260 нм), собирая продукт с одной группой DMT. Полученный полупродукт, представляет собой нусинерсен с одной 5’-DMT защитной группой:
DMT – MeUMeCA MeCMeUMeU MeUMeCA MeUAA MeUGMeC MeUGG
Вторая стадия очистки:
Для дальнейшей тонкой очистки нусинерсена необходимо удалить 5’-DMT защитную группу. Механизм детритилирования аналогичен, используемому в синтезе нисунерсена на шаге 1. Детритилирование проводят при рН 3.5 ± 0.5 при перемешивании при комнатной температуре в течение 8–20 часов. В этих условиях происходит полное детритилирование полупродукта, в результате чего получают нусинерсен:
MeUMeCA MeCMeUMeU MeUMeCA MeUAA MeUGMeC MeUGG.
Третья стадия очистки:
Для тонкой очистки нусинерсена от родственных примесей, таких как (n-1) и (n-2), (n+1), примеси с удаленным азотистым основанием (Loss A, Аbasic), полноразмерные олигонуклеотиды (P=O), выбран метод анионообменной хроматографии. При этом примесь (n-1) – это примесь целевого олигонуклеотида, образующаяся во время синтеза, которая имеет в своей структуре на один нуклеотид меньше, чем целевая молекула (пропуск звена может быть расположен в любом месте цепочки); примесь (n-2) – это примесь целевого олигонуклеотида, образующаяся во время синтеза, которая имеет в своей структуре на два нуклеотида меньше чем целевая молекула (пропуски звеньев могут быть расположены в любом месте цепочки и образовываться независимо друг от друга); примесь (n+1) – это примесь целевого олигонуклеотида, образующаяся во время синтеза, которая имеет в своей структуре на один нуклеотид больше чем целевая молекула (присоединение лишнего нуклеотида может произойти в любое место цепочки).
Разделение нусинерсена и родственных примесей основано на различии в заряде между ними. Отрицательный заряд олигонуклеотида, содержащего фосфоротиоатную связь с серой, более поляризован, что приводит к более прочному связыванию со анионо-обменным сорбентом по сравнению с полноразмерным олигонуклеотидом (P=O). Нусинерсен имеет больший отрицательный заряд, чем примеси (n-1) и (n-2) и также более прочно связывается с анионообменным сорбентом по сравнению с ними. Это различие и позволяет провести разделение и удаление примесей из нусинерсена. Синтетические олигонуклеотиды, содержащие фосфоротиоатную связь с серой, могут на основе гидрофобных взаимодействий образовывать множество комплексов, таких как вторичные структуры (шпильки) или мультимерные комплексы; высокая концентрация олиго нуклеотидов также может повышать гидрофобность и способность к комплексообразованию. Образование гидрофобных комплексов и неспецифические взаимодействия являются факторами, усложняющими хроматографическое разделение с использованием анионообменной хроматографии. Для улучшения разделения используют хаотропы, которые обладают комплексоразрушающим эффектом:
– органические растворители в подвижной фазе (ацетонитрил, этанол, изопропанол), которые повышают растворимость фосфоротиоатных олигонуклеотидов и ингибируют неспецифические гидрофобные взаимодействия и тем самым усиливают ионное разделение;
– высокий рН;
– соли (NaCl, NaBr и NaClO4);
– повышенная температура.
Тестировали сорбенты BioPro IEX SmartSep Q30 (YMC), SOURCE 30Q (Cityva), WorkBeads 40Q (Bio-Works). Показано, что сорбент BioPro IEX SmartSep Q30 (YMC) обладает наилучшей способностью для разделения примесей нусинерсена. Также подобраны условия для повышения степени разделения: в подвижную фазу введен ацетонитрил и NaCl, определены критерии сбора целевой фракции, позволяющие отобрать продукт надлежащей чистоты:
• содержание основного вещества (не менее 96%);
• содержанию специфицированных примесей олигонуклеотида:
С целью удаления органических растворителей из полученного раствора нусинерсена использовали повторную очистку методом анионообменной хроматографии на сорбенте BioPro IEX SmartSep Q30 в режиме посадки-съем с использованием подвижных фаз, не содержащих органических растворителей. Данный подход позволил практически полностью удалить органические растворители из раствора нусинерсена.
Четвертая стадия очистки:
Из продукта необходимо удалить остаточные органические растворители, элементные примеси, соли и другие возможные низкомолекулярные примеси. Как правило, для выполнения этих задач используются две стадии – диафильтрация (удаление низкомолекулярных примесей) и лиофилизация (удаление остаточных органических растворителей). Стадия гель-фильтрации позволяет решить данные задачи за один этап. Хроматография на сорбенте Sephadex G-25 (Cytiva) удаляет остаточные органические растворители и элементные примеси до безопасного уровня. Результаты по содержанию остаточных растворителей и элементных примесей представлены в таблицах 1 и 2.
Таблица 1. Содержание остаточных органических растворителей в субстанции нусинерсена
Таблица 2. Содержание элементных примесей в субстанции нусинерсена
Пятая стадия очистки:
После стадии гель-фильтрации получают водный раствор нусинерсена, который необходимо сконцентрировать до концентрации 4±0,2 мг/мл, чтобы использовать в дальнейшем для производства лекарственного препарата. Концентрация 4±0,2 мг/мл позволяет при производстве препарата провести добавление компонентов БГФ (буфера готовой формы) в виде концентрата и получить лекарственный препарат с надлежащей концентрацией нусинерсена и вспомогательных веществ.
Для концентрирования выбран метод упаривания под вакуумом с использованием ротационного испарителя. Процесс ведут при давлении не более 15,0 мбар при температуре 25–35°С, при перемешивании колбы со скоростью 100-200 об/мин. Перед началом концентрирования определяют концентрацию нусинерсена в растворе; рассчитывают до какого объема нужно сконцентрировать раствор, чтобы получить концентрацию 4±0,2 мг/мл; после окончания концентрирования проверяют, что нужная концентрация достигнута.
Шестая стадия:
Для снижения потенциальной микробной нагрузки на стадии фасовки введена фильтрация раствора нусинерсена через стерилизующий фильтр 0,2 мкм в одноразовый стерильный пластиковый мешок, в котором хранится субстанция-концентрат нусинерсена.
При осуществлении изобретения достигаются следующие технические результаты: Получен олигонуклеотид нусинерсен с содержанием целевой формы на уровне не менее 96%, содержанием примеси P=O не более 1,5%, примеси (n-1) – не более 1%; примеси (n+1) – не более 0,5% , как определено методом ВЭЖХ МС (см. табл.3).
Выход по целевому продукту не менее 50 % от теоретического.
Далее приводятся примеры осуществления изобретения, которые, однако, не охватывают все возможные варианты осуществления изобретения и не ограничивают заявленное изобретение.
Примеры осуществления изобретения
Пример 1. Проведение этапа синтеза
Синтез нусинерсена
Олигонуклеотид нусинерсен (mU-mC-A-mC-mU-mU-mU-mC-A-mU-A-A-mU-G-mC-mU-G-G) синтезировали на твердом носителе NittoPhase 300 при помощи олигонуклеотидного синтезатора ÄKTA oligopilot 100, на колонне переменного объема (Adjustable Oligo column) объемом 60 мл. Масса загруженного сорбента составила 6,0 г, емкость сорбента составляла 302 ммоль/г).
Присоединение фосфорамидита G
Реакционную колонну с сорбентом промывают 3-5 КО сухого ацетонитрила. После чего реактор промывают 7 КО детритилирующего раствора на скорости 75 см/мин. После детритилирования реакционная колонна промывается 3 КО сухого ацетонитрила на скорости до 75 мл/мин. В колонну подается 2 экв. 0.15 М раствора фосфорамидита G в ацетонитриле в смеси с 60% (V/V) 0,3 M раствора ETT. Смесь рециркулируют через колонну 3 минуты на линейной скорости не ниже 75 см/мин. После окончания присоединения реакционную колонну промывают 3 КО сухого ацетонитрила. В колонну подается 2 экв. 0.15 М раствора фосфорамидита G в ацетонитриле в смеси с 60% (V/V) 0,3 M раствора ETT. Смесь рециркулируют через колонну 3 минуты на линейной скорости не ниже 75 см/мин. После окончания присоединения реакционную колонну промывают 3 КО сухого ацетонитрила. В реакционную колонну подается 3 экв. раствора гидрида ксантана 0,04 М в смеси 20% (V/V) пиридина с ацетонитрилом. Раствор тиолятора рециркулируют через колонну реактора 6 минут на линейной скорости не ниже 75 см/мин. После окончания цикла окислительного тиолирования реакционную колонну промывают 3 КО ацетонитрила. В реакционную колонну подается смесь 2 КО 50/50% (V/V) CapA (20% N-метилимидазол в ацетонитриле) и CapB (30/42/28 (V/V/V) пропионовый ангидрид, 2,6-лутидин, ацетонитрил). Колонну промывают 3 КО сухого ацетонитрила. В реакционную колонну подается смесь 2 КО 50/50% (V/V) CapA (20% N-метилимидазол в ацетонитриле) и CapB (30/42/28 (V/V/V) пропионовый ангидрид, 2,6-лутидин, ацетонитрил). Колонну промывают 3 КО сухого ацетонитрила.
Присоединение фосфорамидита G
Колоночный реактор промывают детритилирующим раствором на скорости 75 см/мин до падения пика поглощения ниже 50 mAU на длине волны 436 нм. После детритилирования реакционная колонна промывается 3 КО сухого ацетонитрила на скорости до 75 мл/мин. В колонну подается 2 экв. 0.15 М раствора фосфорамидита G в ацетонитриле в смеси с 60% (V/V) 0,3 M раствора ETT. Смесь рециркулируют через колонну 3 минуты на линейной скорости не ниже 75 см/мин. После окончания присоединения реакционную колонну промывают 3 КО сухого ацетонитрила. В реакционную колонну подается 3 экв. раствора гидрида ксантана 0,04 М в смеси 20% (V/V) пиридина с ацетонитрилом. Раствор тиолятора рециркулируют через колонну реактора 6 минут на линейной скорости не ниже 75 см/мин. После окончания цикла окислительного тиолирования, реакционную колонну промывают 3 КО ацетонитрила. В реакционную колонну подается смесь 2 КО 50/50% (V/V) CapA (20% N-метилимидазол в ацетонитриле) и CapB (30/42/28 (V/V/V) пропионовый ангидрид, 2,6-лутидин, ацетонитрил). Колонну промывают 3 КО сухого ацетонитрила.
Присоединение фосфорамидита mU
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mU.
Присоединение фосфорамидита mC
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mC.
Присоединение фосфорамидита G
Цикл проводят аналогично циклу 2 с использованием фосфорамидита G.
Присоединение фосфорамидита mU
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mU.
Присоединение фосфорамидита A
Цикл проводят аналогично циклу 2 с использованием фосфорамидита A.
Присоединение фосфорамидита A
Цикл проводят аналогично циклу 2 с использованием фосфорамидита A.
Присоединение фосфорамидита mU
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mU.
Присоединение фосфорамидита A
Цикл проводят аналогично циклу 2 с использованием фосфорамидита A.
Присоединение фосфорамидита mC
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mC.
Присоединение фосфорамидита mU
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mU.
Присоединение фосфорамидита mU
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mU.
Присоединение фосфорамидита mU
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mU.
Присоединение фосфорамидита mC
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mC.
Присоединение фосфорамидита A
Цикл проводят аналогично циклу 2 с использованием фосфорамидита A.
Присоединение фосфорамидита mC
Цикл проводят аналогично циклу 2 с использованием фосфорамидита mC.
Присоединение фосфорамидита mU
Колоночный реактор промывают детритилирующим раствором на скорости 75 см/мин до падения пика поглощения до 50 mAU на длине волны 436 нм. После детритилирования реакционная колонна промывается 3 КО сухого ацетонитрила на скорости до 75 мл/мин. В колонну подается 2 экв. 0.15 М раствора фосфорамидита mU в ацетонитриле в смеси с 60% (V/V) 0,3 M раствора ETT. Смесь рециркулируют через колонну 3 минуты на линейной скорости не ниже 75 см/мин. После окончания присоединения реакционную колонну промывают 3 КО сухого ацетонитрила. В реакционную колонну подается 3 экв. раствора гидрида ксантана 0,04 М в смеси 20% (V/V) пиридина с ацетонитрилом. Раствор тиолятора рециркулируют через колонну реактора 6 минут на линейной скорости не ниже 75 см/мин. После окончания цикло окислительного тиолирования реакционную колонну промывают 3 КО ацетонитрила. В реакционную колонну подается смесь 2 КО 50/50% (V/V) CapA (20% N-метилимидазол в ацетонитриле) и CapB (30/42/28 (V/V/V) пропионовый ангидрид, 2,6-лутидин, ацетонитрил). Колонну промывают 3 КО сухого ацетонитрила. Реакционную колонну промывают 2 КО 20% раствор N,N-диэтиламина в ацетонитриле. Колонну промывают 5 КО ацетонитрила.
Аммонолиз
Сорбент после синтеза сушат в токе инертного газа, загружают в 2 л реактор высокого давления и добавляют 900 мл раствора гидроксида аммония. Реакционную массу нагревают до температуры 55°С и выдерживают при этой температуре 16 ч. После окончания реакции реакционную массу охлаждают и фильтруют от смолы. Смолу на фильтре промывают 1 л 50% этанола. Объединенный фильтрат концентрируют в вакууме роторного испарителя для удаления аммиака. Полученный раствор DMTon полупродукта передают на стадию очистки. По результатам аммонолиза получают промежуточный раствор нусинерсена с содержанием основного вещества 12 г и чистотой на уровне 91,5%.
Пример 2. Проведение хроматографической очистки
Гидрофобная хроматография
Раствор нусинерсена с DMT-защитой на 5’-конце после стадии аммонолиза разбавляли буфером до УЭП 145–170 мСм/см и наносили на колонну с сорбентом Capto Phenyl HS (объем колонки (CV) 5,0 л), предварительно уравновешенную раствором 10 мМ NaOH, 2 M NaCl. После нанесения сорбент промывали 3 CV того же раствора. Связанный продукт элюировали путем постепенного снижения содержания хлорида натрия в подвижной фазе.
В результате реализации указанной стадии очистки было получено две ключевые фракции: проскок нанесения; целевой пик. Типичная хроматограмма данной стадии очистки представлена на фигуре 6.
Данная стадия позволяет высокоэффективно удалять близкородственные примеси целевого нуклеотида, не содержание DMT-защитную группу и низкомолекулярные примеси.
В результате проведенной стадии очистки чистота DMT-защищенного олигонуклеотида увеличивается с 91,5% до 92,3% при выходе 10,2 г.
Детритилирование
Раствор нуклеотида подвергают обработке низким значением рН для снятия DMT – защиты. Детритилирование осуществляли при рН 3.5 в течение 18 часов.
Анионообменная хроматография
В растворе олигонуклеотида со стадии детритилирования довели значение электропроводности до 26 мС/см водой для инъекций. Колонну, содержащую сорбент YMC Q30 объемом около 1 л, уравновесили 3 колоночными объемами раствора A2, состоящего из 0,4 г натрия гидроксида 200 г ацетонитрила в воде для инъекций на 1 кг раствора. Подготовленный раствор олигонуклеотида после детритилирования нанесли на колонну. Затем промыли 3 колоночными объемами раствора A2 и 3 колоночными объемами смеси раствора А2 и раствора B2, состоящего из 0,4 г натрия гидроксида, 200 г ацетонитрила и 58,5 г натрия хлорида в воде для инъекций на 1 кг раствора, в соотношении 1:1. Олигонуклеотид, сорбированный на колонне, элюировали градиентом раствора B2 против раствора А2 от 50 до 100% за 10 колоночных объемов. Сбор целевой фракции олигонуклеотида начинали при достижении оптической плотности значения максимума пика, заканчивали при значении оптической плотности 500 mAU.
Провели регенерацию и санацию сорбента последовательным пропусканием следующих растворов:
Сорбент уравновесили 3 КО раствора B1, состоящего из 0,4 г натрия гидроксида на 1 кг раствора в воде для инъекций. В растворе промежуточного продукта скорректировали значение электропроводности водой для инъекций до 27 мСм/см. Полученный раствор нанесли на колонну, заполненную сорбентом YMC Q30 объемом около 5 л, и промыли 3 КО раствора В1.
Олигонуклеотид, сорбированный на колонне, элюировали раствором В3, состоящим из 0,4 г натрия гидроксида и 58,5 г натрия хлорида на 1 кг раствора в воде для инъекций. Сбор элюата начали при достижении оптической плотности 200 mAu и закончили при 200 mAu в правом «плече» пика (длина волны 260 нм).
Провели регенерацию и санацию сорбента последовательным пропусканием следующих растворов:
В таблице 3 ниже приведены сравнительные параметры качества синтетического олигонуклеотида до и после проведения стадии анионообменной хроматографии.
Таблица 3. Сравнительные параметры качества нусинерсена до и после очистки методом анионообменной хроматографии
Типичная хроматограмма данной стадии очистки представлена на Фигуре 7.
Гель-фильтрационная хроматография
Раствор нусинерсена с после стадии анионообменной хроматографии наносили на колонну с сорбентом Sephadex G-25 (объем колонки (CV) 1,0 л), предварительно уравновешенную водой для инъекций. Объемная нагрузка на сорбент составляла не более 10 % от объема колонны. Целевой продукт собирали в несорбируемой фракции. После проведения стадии получали раствор нусинерсена в воде для инъекций с УЭП не более 20 мСм/см. Хроматограмма очистки нусинерсена методом гель-фильтрационной хроматографии приведена на Фигуре 8. Результаты анализа продукта после проведения стадии гель-фильтрации представлены в таблицах 4 и 5.
Данная стадия позволяет высокоэффективно удалять неорганические примеси и остаточные растворители.
В результате проведенной стадии очистки чистота целевого продукта составила 97,41% при выходе не менее 90% относительно предыдущей стадии.
Таблица 4. Содержание остаточных органических растворителей в растворе олигонуклеотида после проведения стадии гель-фильтрационной хроматографии
Таблица 5. Содержание элементных примесей в растворе олигонуклеотида после проведения стадии гель-фильтрационной хроматографии
Получение стерильной субстанции-раствора нусинерсена
После проведения стадии гель-фильтрации концентрировали полученный раствор нусинерсена до 4,0 мг/мл упариванием под вакуумом и подвергали фильтрации через фильтр 0,2 мкм.
Таким образом, предложенный в настоящем изобретении способ очистки тиофосфатных олигонуклеотидов позволяет снизить содержание остаточных растворителей и элементных примесей до уровня ниже чувствительности аналитической методики (см. таблицу 1 и 2).
| название | год | авторы | номер документа |
|---|---|---|---|
| АНТИКОАГУЛЯНТ ПРЯМОГО ДЕЙСТВИЯ НА ОСНОВЕ МОДУЛЬНОГО БИВАЛЕНТНОГО ДНК АПТАМЕРА, СЕЛЕКТИВНОГО К ТРОМБИНУ | 2016 |
|
RU2631829C1 |
| СПОСОБ СИНТЕЗА 2'-О-(2-МЕТОКСИЭТИЛ) ТИОФОСФАТНОГО ОЛИГОНУКЛЕОТИДА | 2019 |
|
RU2738093C1 |
| НОВЫЕ АНАЛОГИ 2`, 5`-ОЛИГОАДЕНИЛАТА ИЛИ ИХ ФАРМАКОЛОГИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ИХ ПРИМЕНЕНИЕ | 2003 |
|
RU2311422C2 |
| КАТИОННЫЕ ОЛИГОНУКЛЕОТИДЫ, АВТОМАТИЗИРОВАННЫЕ СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2006 |
|
RU2451022C2 |
| Способ получения олигонуклеотидной вакцины против вируса SARS-Cov-2 | 2023 |
|
RU2824405C1 |
| Лекарственная форма ДНК-аптамера | 2019 |
|
RU2730000C1 |
| СПОСОБ ИММОБИЛИЗАЦИИ МОДИФИЦИРОВАННЫХ НЕПРЕДЕЛЬНЫМИ ФРАГМЕНТАМИ ОЛИГОНУКЛЕОТИДОВ ПУТЕМ СОПОЛИМЕРИЗАЦИИ | 1999 |
|
RU2157377C1 |
| СПОСОБ ОЧИСТКИ ИНСУЛИНА-СЫРЦА, ПОЛУЧАЕМОГО ИЗ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ СВИНЕЙ | 1996 |
|
RU2126690C1 |
| СПОСОБ ПОЛУЧЕНИЯ ВЫСОКООЧИЩЕННОГО РЕКОМБИНАНТНОГО ИНГИБИТОРА С1-ЭСТЕРАЗЫ ЧЕЛОВЕКА ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ | 2020 |
|
RU2769201C2 |
| СПОСОБ ПОЛУЧЕНИЯ ПЭГИЛИРОВАННЫХ ОЛИГОНУКЛЕОТИДОВ | 2012 |
|
RU2564855C2 |
Изобретение относится к области медицины и может быть использовано в фармацевтической промышленности, а именно для производства лекарственного средства для терапии спинальной мышечной атрофии. Изобретение раскрывает способ получения высокоочищенного антисмыслового олигонуклеотида нусинерсена, в котором применяется комбинация высокоэффективного твердофазного синтеза и очистки. А именно раскрывается способ получения модифицированного тиофосфатного олигонуклеотида, содержащего 2'-О-(2-метоксиэтил)рибонуклеотиды, структуры UCACUUUCAUAAUGCUGG, где каждый нуклеозид содержит 2'-O-(2-метоксиэтил) модифицированный остаток сахара, причем олигонуклеотид синтезируют на твердофазном носителе с использованием амидофосфитного метода, причем в качестве активатора используется 5-(этилтио)-1H-тетразол (ЕТТ), при этом способ получения содержит следующие последовательные операции: для получения тиофосфатных групп используют гидрид ксантана (5-амино-1,2,4-дитиазол-3-тион) в 20% растворе пиридина в ацетонитриле, аммонолиз проводят в 23-35% растворе гидроксида аммония в течение от 16 до 24 часов, доводят значение удельной электропроводности (УЭП) до 145,0-170,0 мСм/см, затем проводят очистку на гидрофобном сорбенте, выбранном из группы, включающей Butyl Sepharose FF, Phenyl Sepharose FF, Phenyl-650M, Capto Phenyl с размером гранул от 45 до 165 мкм для удаления основного количества низкомолекулярных компонентов и родственных примесей, проводят финальное детритилирование промежуточного полупродукта, затем проводят хроматографическую очистку в присутствии полярных апротонных растворителей на анионообменном сорбенте, выбранном из группы, включающей Q Sepharose FF, 30 Q Source, GigaCap Q650M, BioPro IEX SmartSep Q30 (YMC), WorkBeads 40Q, при этом в подвижную фазу введен ацетонитрил и NaCl для эффективного удаления родственных примесей модифицированного тиофосфатного олигонуклеотида, и далее проводят очистку гель-фильтрационной хроматографией на сорбенте, выбранном из группы, включающей Sephadex G-25, Superdex 75, HW-50F для удаления остаточных органических растворителей и элементных примесей с получением модифицированного тиофосфатного олигонуклеотида, чистота которого составляет не менее 96%, при определении методом ВЭЖХ МС. Технический результат – повышение эффективности очистки тиофосфатных олигонуклеотидов при сохранении высоких выходов целевого продукта. 5 з.п. ф-лы, 8 ил., 5 табл., 2 пр.
1. Способ получения модифицированного тиофосфатного олигонуклеотида, содержащего 2'-О-(2-метоксиэтил)рибонуклеотиды, структуры UCACUUUCAUAAUGCUGG, где каждый нуклеозид содержит 2'-O-(2-метоксиэтил) модифицированный остаток сахара,
причем олигонуклеотид синтезируют на твердофазном носителе с использованием амидофосфитного метода,
причем в качестве активатора используется 5-(этилтио)-1H-тетразол (ЕТТ),
при этом способ получения содержит следующие последовательные операции:
для получения тиофосфатных групп используют гидрид ксантана (5-амино-1,2,4-дитиазол-3-тион) в 20% растворе пиридина в ацетонитриле, аммонолиз проводят в 23-35% растворе гидроксида аммония в течение от 16 до 24 часов, доводят значение удельной электропроводности (УЭП) до 145,0-170,0 мСм/см,
затем проводят очистку на гидрофобном сорбенте, выбранном из группы, включающей Butyl Sepharose FF, Phenyl Sepharose FF, Phenyl-650M, Capto Phenyl с размером гранул от 45 до 165 мкм для удаления основного количества низкомолекулярных компонентов и родственных примесей,
проводят финальное детритилирование промежуточного полупродукта,
затем проводят хроматографическую очистку в присутствии полярных апротонных растворителей на анионообменном сорбенте, выбранном из группы, включающей Q Sepharose FF, 30 Q Source, GigaCap Q650M, BioPro IEX SmartSep Q30 (YMC), WorkBeads 40Q, при этом в подвижную фазу введен ацетонитрил и NaCl для эффективного удаления родственных примесей модифицированного тиофосфатного олигонуклеотида,
и далее проводят очистку гель-фильтрационной хроматографией на сорбенте, выбранном из группы, включающей Sephadex G-25, Superdex 75, HW-50F для удаления остаточных органических растворителей и элементных примесей,
с получением модифицированного тиофосфатного олигонуклеотида, чистота которого составляет не менее 96%, при определении методом ВЭЖХ МС.
2. Способ по п. 1, где хроматографическая очистка на анионобменном сорбенте проводится для удаления полноразмерных олигонуклеотидов, имеющих связь Р=О, примесей (n-1) и (n-2), (n+1), примесей с удаленным азотистым основанием.
3. Способ по п. 1, где очистку на гидрофобном сорбенте проводят для удаления основного количества низкомолекулярных компонентов и родственных примесей и отделения последовательности с делециями без групп DMT и разветвленных соединений, содержащих две или более группы DMT, от неочищенного продукта с одной группой DMT.
4. Способ по п. 1, где после этапа очистки гель-фильтрационной хроматографией продукт концентрируют и проводят его фильтрацию.
5. Способ по п. 4, где фильтрацию продукта проводят через фильтр 0,2 мкм для снижения микробной нагрузки.
6. Способ по п. 1, где модифицированный тиофосфатный олигонуклеотид представляет собой нусинерсен.
| СПОСОБ СИНТЕЗА 2'-О-(2-МЕТОКСИЭТИЛ) ТИОФОСФАТНОГО ОЛИГОНУКЛЕОТИДА | 2019 |
|
RU2738093C1 |
| WO 2021045141 А1, 11.03.2021 | |||
| WO 2017223258 А1, 28.12.2017 | |||
| WO 2017218454 A1, 21.12.2017 | |||
| YANG, JIMIN et al | |||
| Solid-Phase Synthesis of Phosphorothioate Oligonucleotides Using Sulfurization Byproducts for in Situ Capping | |||
| The Journal of organic chemistry, 2018, v | |||
| Пуговица | 0 |
|
SU83A1 |
| Двухтактный двигатель внутреннего горения | 1928 |
|
SU11577A1 |

