Настоящая заявка относится к новым соединениям и их применению в качестве антагонистов гистаминовых рецепторов H4. Соединения, описанные в настоящем документе, могут быть полезны для лечения или профилактики заболеваний, в которые вовлечены рецепторы Н4. Заявка также относится к фармацевтическим композициям, включающим эти соединения, и к получению и применению этих соединений и композиций для профилактики или лечения таких заболеваний, в которые вовлечены рецепторы Н4.
Уровень техники изобретения
Гистамин представляет собой биогенный амин короткого действия, образующийся в тучных клетках, где он хранится в цитозольных гранулах и высвобождается в ответ на различные иммунологические и неиммунологические стимулы. Высвобождение гистамина из тучных клеток традиционно связывают с признаками и симптомами от легкой до тяжелой степени, характерными для реакций гиперчувствительности, включая эритему, крапивницу, зуд, тахикардию, гипотензию, фибрилляцию желудочков, бронхоспазм, остановка дыхания и сердечной деятельности. На сегодняшний день идентифицированы многочисленные дополнительные источники, включая базофилы, нейроны и раковые клетки. В дополнение к модуляции широкого спектра физиологических процессов гистамин участвует в патологических состояниях, включая аллергию и анафилаксию, астму и хроническое воспаление, аутоиммунные, сердечно-сосудистые, нервно-психиатрические и эндокринные расстройства, а также рак.
Гистамин проявляет свое плейотропное действие в основном за счет связывания с четырьмя типами рецепторов, связанных с G-белком (GPCR), обозначенных как H1-H4, которые по-разному экспрессируются в различных типах клеток и демонстрируют значительные различия между видами. Рецептор H2 отвечает за секрецию желудочного сока; рецептор H3 контролирует высвобождение гистамина и других нейромодуляторов в ЦНС и рецептор H1 связан с нарушением сна и воспалительной реакцией.
Идентифицированный в 2000 году высокоаффинный Н4-рецептор проявляет конститутивную активность и экспрессируется в основном, но не исключительно, на клетках иммунной системы, включая тучные клетки, моноциты, дендритные клетки, эозинофилы, базофилы, нейтрофилы и Т-клетки. Это открытие привело к привлекательной перспективе новой мишени лекарственного средства с терапевтическим потенциалом при остром и хроническом воспалении, аутоиммунных заболеваниях, иммунной защите организма и невропатической боли.
H4R имеет только 40% гомологию со своим ближайшим соседом H3R, и ни антагонисты H2, ни H1 не ингибируют индуцированный гистамином хемотаксис эозинофилов. Было показано, что гистамин ингибирует форсколин-индуцированные ответы сАМР чувствительным к коклюшному токсину (PTx) образом, предполагая, что H4R передает сигналы через гетеротримерные белки Gαi/o. Транзиентная экспрессия H4R в системах гетерологичных клеток (например, клетках HEK293) является широко используемым способом измерения передачи сигналов и связывания лиганда H4 для получения оценок функциональной активности и аффинности рецептора, соответственно.
Открытие антагонистов H4R с использованием этих методов и их изучение на моделях животных различных заболеваний, включая астму, хронический зуд, дерматит, ревматоидный артрит, ульцерогенез желудка и колит, подтвердило, что антагонизм H4R приводит к глубокому противовоспалительному эффекту и подтвердил терапевтический эффект воздействия на этот рецептор. Уже проведено первое клиническое исследование фазы 2а антагониста H4R у пациентов, страдающих атопическим дерматитом от умеренной до тяжелой степени, что еще раз подтвердило, что H4 является поддающийся воздействию лекарственных средств мишенью у пациентов.
Несмотря на ряд опубликованных лигандов H4R, остается необходимость в разработке новых антагонистов H4R с хорошим качеством потенциального лекарственного препарата. Эти антагонисты должны демонстрировать превосходную активность и аффинность при низких нМ при полной селективности в отношении рецепторов H1-H3. Они не должны проявлять агонистическую активность из-за рисков, связанных с индукцией провоспалительных реакций, и в идеале должны иметь сходный фармакологический профиль у разных видов, чтобы поддерживать ФК/ФД в различных животных моделях заболевания. Они должны быть метаболически стабильными, с отличной ФК, нетоксичными и демонстрировать отличную специфичность к H4 в широком профиле панели безопасности.
Ген специфических калиевых каналов сердца человека (hERG) кодирует порообразующую субъединицу быстро активирующегося калиевого канала замедленного выпрямления (IKr), который играет важную роль в реполяризации желудочков и в определении интервала QT на электрокардиограмме, причем интервал QT является временем, необходимым для деполяризации и реполяризации желудочков. Общепризнано, что hERG очень чувствителен к ингибированию широким спектром структурно разнообразных соединений. Когда способность каналов проводить электрический ток через клеточную мембрану ингибируется или нарушается применением лекарств, это может привести к потенциально смертельному расстройству, называемому синдромом QT. Ряд клинически эффективных препаратов на рынке имеют тенденцию ингибировать hERG и создавать сопутствующий риск внезапной смерти в качестве побочного эффекта, что делает ингибирование hERG важной анти-мишенью, которую необходимо избегать при разработке лекарственных средств.
Соединения по изобретению являются антагонистами рецептора Н4. Некоторые соединения обладают низким ингибированием hERG, что делает их особенно полезными.
ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к соединениям, обладающим активностью антагонистов рецептора Н4. Более конкретно, изобретение относится к соединениям, которые сочетают антагонизм в отношении Н4-рецепторов с низкой активностью hERG.
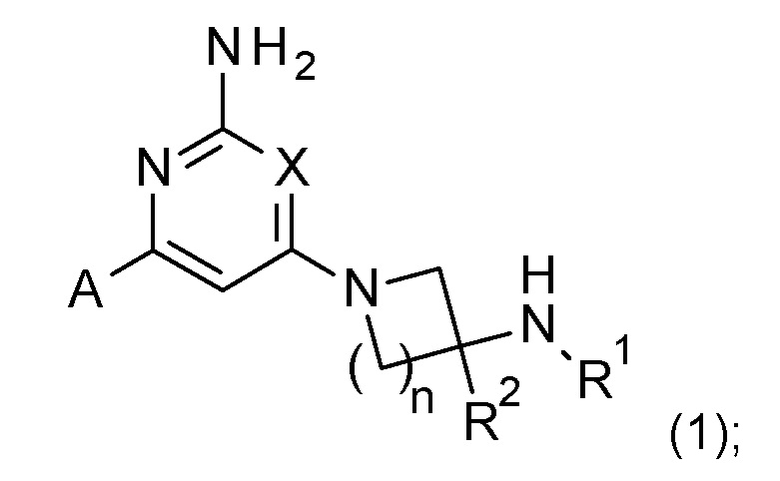
Соответственно, изобретение предоставляет соединение формулы (1):
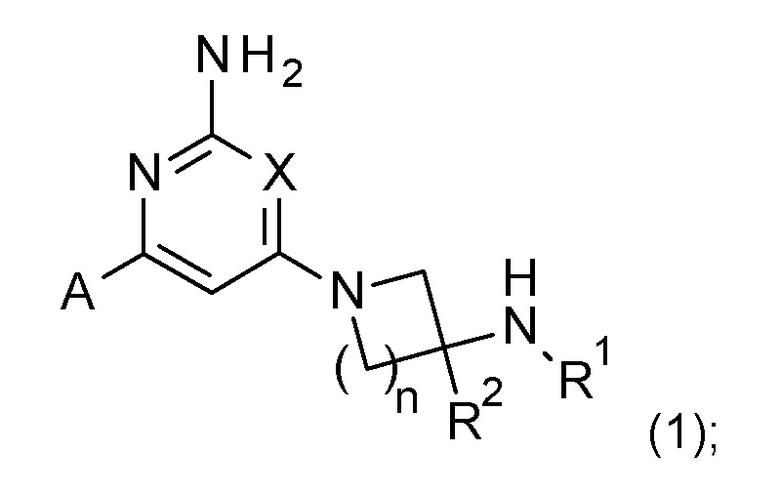
или его соль,
где
X представляет собой CH или N;
n имеет значение 1 или 2;
R1 выбран из H или C1-3 алкила;
R2 представляет собой H; и
A представляет собой необязательно замещенное пиразольное кольцо;
где соединение выбрано из группы, состоящей из:
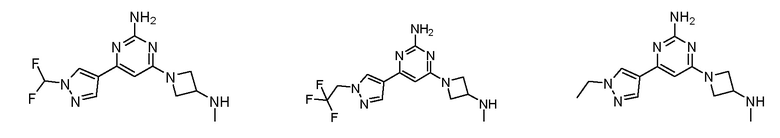
или его соль.
Соединения могут быть использованы в качестве антагонистов рецептора H4. Соединения могут быть использованы в получении лекарственных средств. Соединения или лекарственные средства могут быть использованы для лечения, профилактики, облегчения, контроля или снижения риска воспалительных заболеваний, включая астму, хронический зуд, дерматит, ревматоидный артрит, ульцерогенез желудка и колит.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Изобретение относится к новым соединениям. Изобретение также относится к применению новых соединений в качестве антагонистов рецептора Н4. Изобретение также относится к применению новых соединений в получении лекарственных средств для применения в качестве антагонистов рецептора Н4 или для лечения дисфункции системы Н4. Изобретение также относится к соединениям, композициям и лекарственным средствам, которые являются селективными антагонистами Н4-рецепторов.
Изобретение также относится к соединениям, композициям и лекарственным средствам, применимым для лечения острого и хронического воспаления, аутоиммунного заболевания, нарушений иммунной защиты организма и невропатической боли. Изобретение также относится к соединениям, композициям и лекарственным средствам, применимым для лечения воспалительных заболеваний, включая астму, хронический зуд, дерматит, ревматоидный артрит, ульцерогенез желудка и колит.
Соединения по изобретению включают соединения формулы (1):
или их соль,
где
X представляет собой CH или N;
n имеет значение 1 или 2;
R1 выбран из H или C1-3 алкила;
R2 представляет собой H; и
A представляет собой необязательно замещенное пиразольное кольцо;
где соединение выбрано из группы, состоящей из:
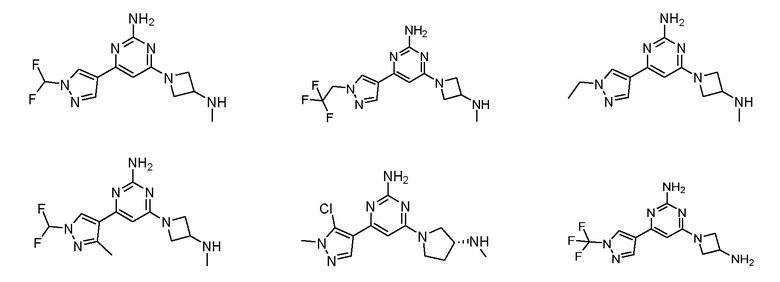
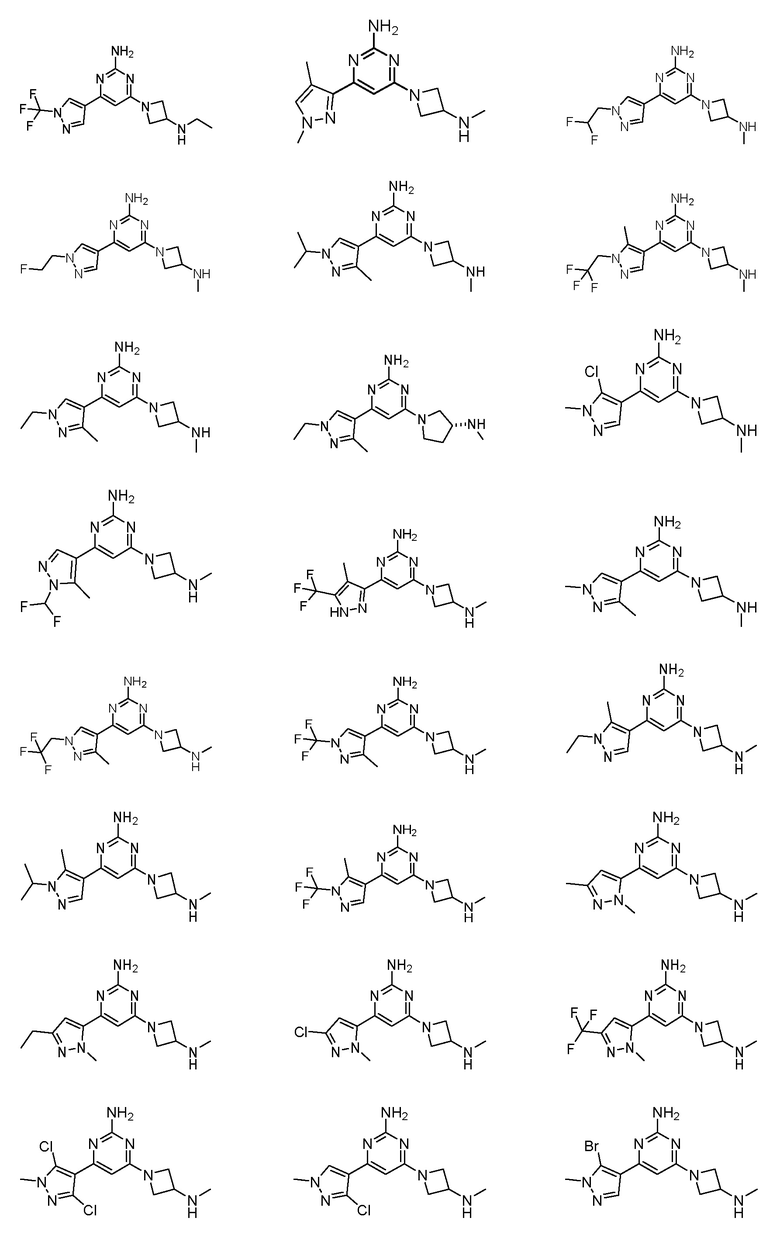
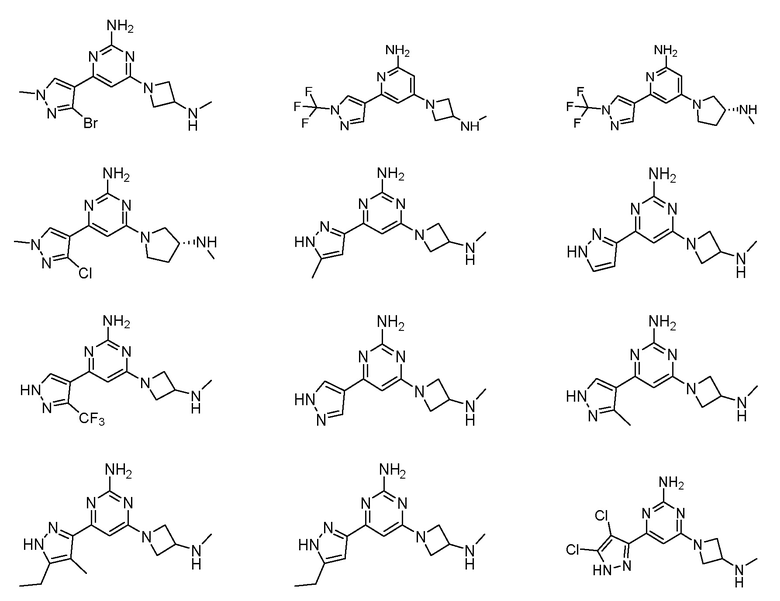
или его соль.
Соединения по изобретению включают соединения формулы (1a):
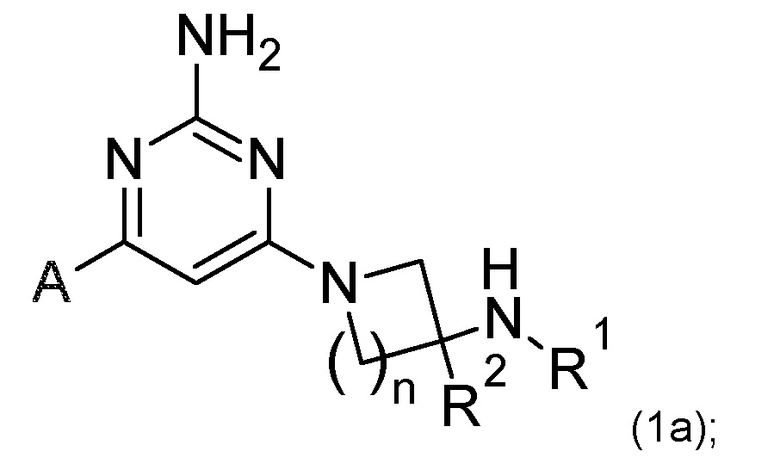
или его соль,
где
n имеет значение 1 или 2;
R1 выбран из H или C1-3 алкила;
R2 представляет собой H; и
A представляет собой необязательно замещенное пиразольное кольцо;
где соединение выбрано из группы, состоящей из:
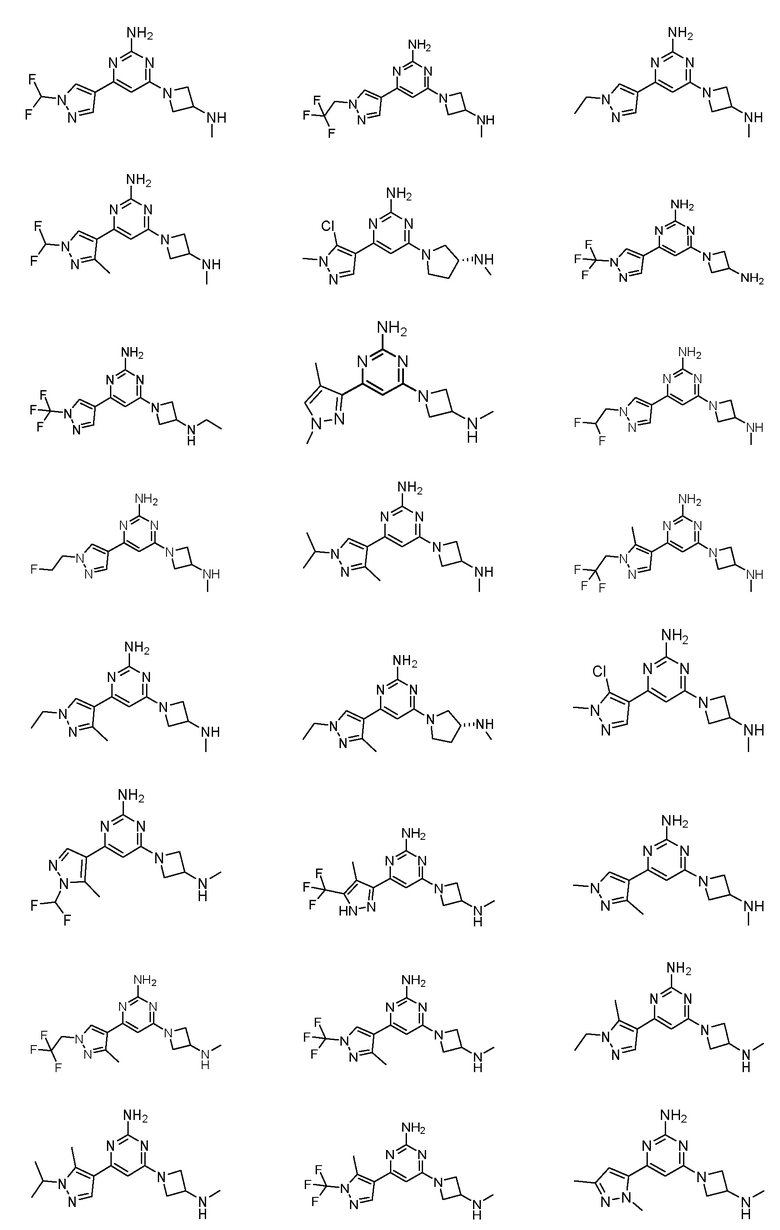
или его соль.
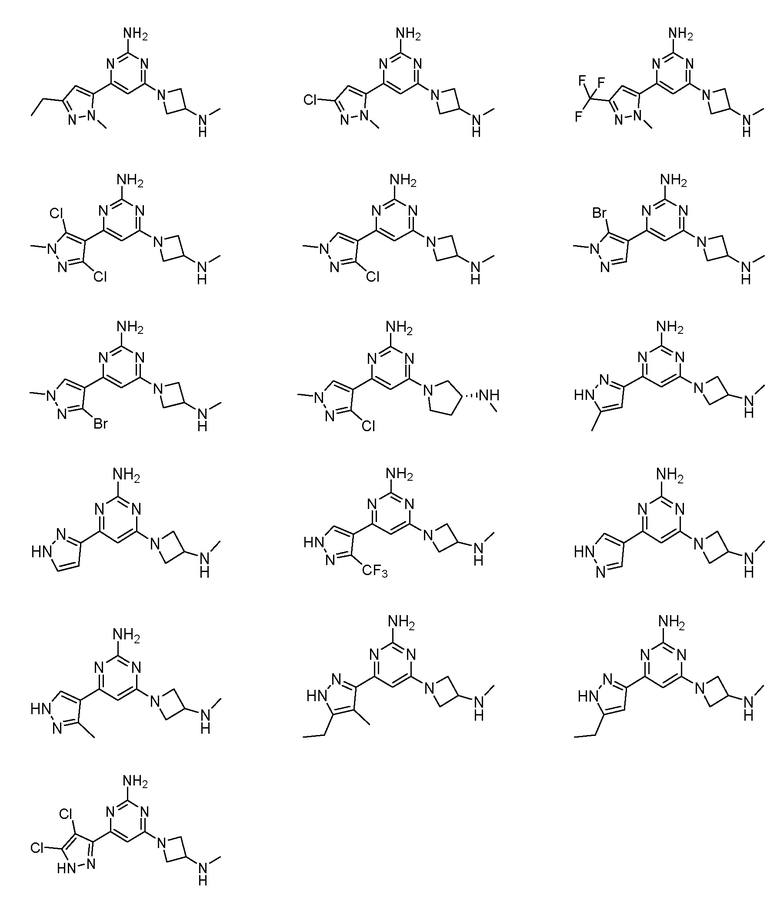
Соединение может быть выбрано из группы, состоящей из:
или его соль.
Соединение может быть соединением, выбранным из группы, состоящей из:
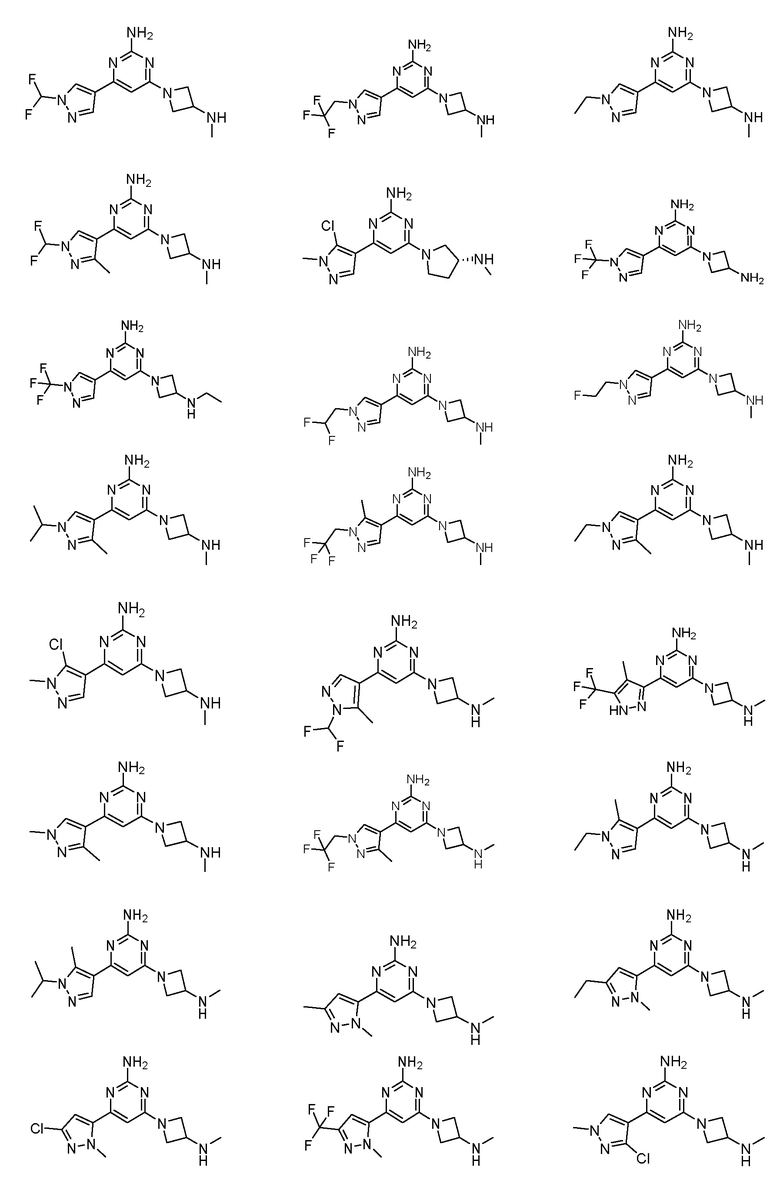
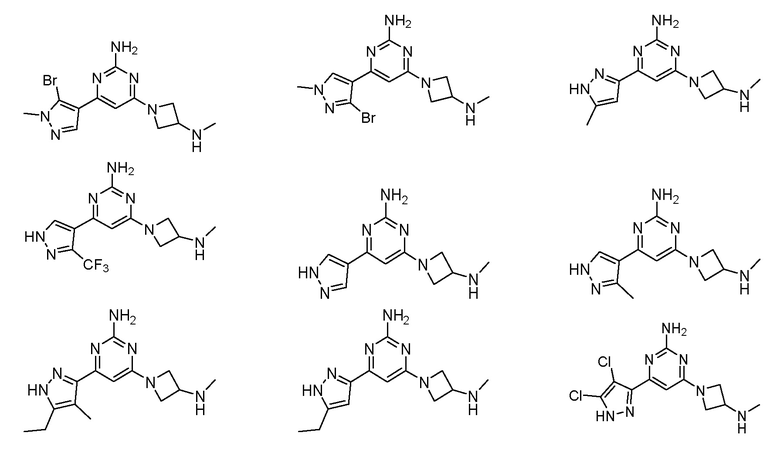
или его солью.
Соединение может быть соединением, выбранным из группы, состоящей из:
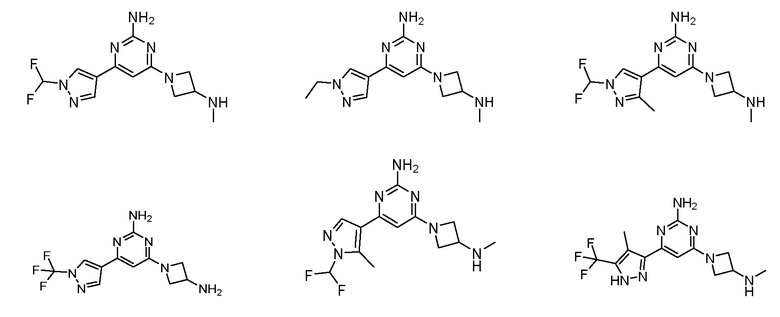
или его солью.
Конкретные примеры соединений включают соединения с низкой hERG-активностью. Соединения по изобретению проявляют низкую hERG-активность, что особенно полезно по причинам, изложенным выше в разделе «Уровень техники». Соединения, демонстрирующие низкую hERG-активность, в частности, представляют собой соединения с pIC50 hERG 4,5 и ниже.
Определения
В настоящей заявке применяются следующие определения, если не указано иное.
Термин «лечение» в отношении применения любого из соединений, описанных в настоящем документе, используется для описания любой формы вмешательства, когда соединение вводят субъекту, страдающему или подверженному риску заболевания или расстройства, или потенциально подверженному риску заболевания или расстройства, о котором идет речь. Таким образом, термин «лечение» охватывает как превентивное (профилактическое) лечение, так и лечение, при котором проявляются измеримые или обнаруживаемые симптомы заболевания или расстройства.
Термин «эффективное терапевтическое количество», используемый в настоящем документе (например, в отношении способов лечения заболевания или состояния), относится к количеству соединения, которое является эффективным для получения желаемого терапевтического эффекта. Например, если состояние представляет собой боль, то эффективное терапевтическое количество представляет собой количество, достаточное для обеспечения желаемого уровня облегчения боли. Желаемым уровнем облегчения боли может быть, например, полное устранение боли или снижение ее интенсивности.
В той степени, в какой любое из описанных соединений имеет хиральные центры, настоящее изобретение распространяется на все оптические изомеры таких соединений, будь то в форме рацематов или разделенных энантиомеров. Изобретение, описанное в настоящем документе, относится ко всем кристаллическим формам, сольватам и гидратам любого из раскрытых соединений, независимо от того, получены ли они таким образом. В той степени, в которой любое из раскрытых в настоящем документе соединений имеет кислотные или основные центры, такие как карбоксилаты или аминогруппы, все формы солей указанных соединений включены в настоящее описание. В случае фармацевтического применения соль следует рассматривать как фармацевтически приемлемую соль.
Соли или фармацевтически приемлемые соли, которые могут быть упомянуты, включают соли присоединения кислот и соли присоединения оснований. Такие соли могут быть образованы обычными способами, например, путем реакции свободной кислоты или свободного основания соединения с одним или несколькими эквивалентами соответствующей кислоты или основания, необязательно в растворителе или в среде, в которой соль нерастворима, с последующим удалением указанного растворителя или указанной среды с помощью стандартных способов (например, в вакууме, сушки вымораживанием или фильтрацией). Соли также могут быть получены путем обмена противоиона соединения в форме соли на другой противоион, например, с использованием подходящей ионообменной смолы.
Примеры фармацевтически приемлемых солей включают соли присоединения кислоты, полученные из минеральных кислот и органических кислот, и соли, полученные из металлов, таких как натрий, магний, калий и кальций.
Примеры соли присоединения кислоты включают соли присоединения кислоты, образованные с уксусной, 2,2-дихлоруксусной, адипиновой, альгиновой, арилсульфоновой кислотами (например, бензолсульфоновой, нафталин-2-сульфоновой, нафталин-1,5-дисульфоновой и п-толуолсульфоновой), аскорбиновой (например, L-аскорбиновой), L-аспарагиновой, бензойной, 4-ацетамидобензойной, бутановой, (+) камфорной, камфор-сульфоновой, (+)-(1S)-камфор-10-сульфоновой, каприновой, капроновой, каприловой, коричной, лимонной, цикламовой, додецилсерной, этан-1,2-дисульфоновой, этансульфоновой, 2-гидроксиэтансульфоновой, муравьиной, фумаровой, галактаровой, гентизиновой, глюкогептоновой, глюконовой (например, D-глюконовой), глюкуроновой (например, D-глюкуроновой), глутаминовой (например, L-глутаминовой), α-оксоглутаровой, гликолевой, гиппуровой, бромистоводородной, хлористоводородной, йодистоводородной, изетионовой, молочной (например, (+)-L-молочной и (±)-DL-молочной), лактобионовой, малеиновой, яблочной (например, (-)-L-яблочной), малоновой, (±)-DL-миндальной, метафосфорной, метансульфоновой, 1-гидрокси-2-нафтойной, никотиновой, азотной, олеиновой, оротовой, щавелевой, пальмитиновой, памовой, фосфорной, пропионовой, L-пироглутаминовой, салициловой, 4-аминосалициловой, себациновой, стеариновой, янтарной, серной, дубильной, винной (например, (+)-L-винной), тиоциановой, ундециленовой и валериановой кислотами.
Также охватываются любые сольваты соединений и их соли. Предпочтительными сольватами являются сольваты, образованные путем включения в структуру твердого состояния (например, кристаллическую структуру) соединений по изобретению молекул нетоксичного фармацевтически приемлемого растворителя (называемого ниже сольватирующим растворителем). Примеры таких растворителей включают воду, спирты (такие как этанол, изопропанол и бутанол) и диметилсульфоксид. Сольваты могут быть получены путем рекристаллизации соединений изобретения с использованием растворителя или смеси растворителей, содержащих сольватирующий растворитель. Образовался ли сольват в любом конкретном случае, можно определить, подвергая кристаллы соединения анализу с использованием хорошо известных и стандартных методов, таких как термогравиметрический анализ (TGA), дифференциальная сканирующая калориметрия (DSC) и рентгеновская кристаллография.
Сольваты могут быть стехиометрическими или нестехиометрическими сольватами. Конкретные сольваты могут быть гидратами, и примеры гидратов включают полугидраты, моногидраты и дигидраты. Для более подробного обсуждения сольватов и способов, используемых для их получения и характеристики, см. Bryn et al., Solid-State Chemistry of Drugs, Second Edition, опубликовано SSCI, Inc of West Lafayette, IN, USA, 1999, ISBN 0-967-06710-3.
Термин «фармацевтическая композиция» в контексте настоящего изобретения означает композицию, включающую активный агент и дополнительно включающую один или несколько фармацевтически приемлемых носителей. Композиция может дополнительно содержать ингредиенты, выбранные из, например, разбавителей, адъювантов, эксципиентов, носителей, консервантов, наполнителей, средств для улучшения распадаемости, смачивающих агентов, эмульгирующих агентов, суспендирующих агентов, подсластителей, вкусовых агентов, ароматизаторов, антибактериальных агентов, противогрибковых агентов, смазывающих агентов и диспергирующих агентов в зависимости от характера способа введения и лекарственных форм. Композиции могут иметь форму, например, таблеток, драже, порошков, эликсиров, сиропов, жидких препаратов, включая суспензии, спреи, средства, применяемые при ингаляциях, таблетки, пастилки, эмульсии, растворы, облатки, гранулы, капсулы и суппозитории, а также жидкие препараты для инъекций, включая липосомальные препараты.
Соединения по изобретению могут содержать один или несколько изотопных замещений, и ссылка на конкретный элемент включает в себя все изотопы этого элемента. Например, ссылка на водород включает 1H, 2H (D) и 3H (T). Аналогично, ссылки на углерод и кислород включают в свой объем, соответственно, 12C, 13C и 14C и 16O и 18O. Аналогичным образом ссылка на конкретную функциональную группу также включает изотопные вариации, если в контексте не указано иное. Например, ссылка на алкильную группу, такую как этильная группа, или алкоксигруппу, такую как метоксигруппа, также охватывает варианты, в которых один или несколько атомов водорода в группе находятся в форме изотопа дейтерия или трития, например, в этильной группе, в которой все пять атомов водорода находятся в изотопной форме дейтерия (пердейтероэтильная группа), или в метоксигруппе, в которой все три атома водорода находятся в изотопной форме дейтерия (тридейтерометоксигруппа). Изотопы могут быть радиоактивными или нерадиоактивными.
Терапевтические дозы могут варьироваться в зависимости от потребностей пациента, тяжести состояния, подлежащего лечению, и используемого соединения. Определение надлежащей дозы для конкретной ситуации находится в компетенции специалиста в данной области. Как правило, лечение начинают с меньших доз, которые меньше оптимальной дозы соединения. После этого дозу увеличивают небольшими шагами повышения дозы, пока не будет достигнут оптимальный эффект при данных обстоятельствах. Для удобства общую суточную дозу можно разделить и вводить порциями в течение дня, если это необходимо.
Величина эффективной дозы соединения будет, конечно, варьироваться в зависимости от характера тяжести состояния, подлежащего лечению, а также от конкретного соединения и пути его введения. Выбор подходящих дозировок находится в пределах компетенции специалиста в данной области без чрезмерной нагрузки. Как правило диапазон суточной дозы может составлять от примерно 10 мкг до примерно 30 мг на кг массы тела человека и животного, предпочтительно от примерно 50 мкг до примерно 30 мг на кг массы тела человека и животного, например, от примерно 50 мкг до примерно 10 мг на кг массы тела человека и животного, например, от примерно 100 мкг до примерно 30 мг на кг массы тела человека и животного, например, от примерно 100 мкг до примерно 10 мг на кг массы тела человека и животного, и наиболее предпочтительно от примерно 100 мкг до примерно 1 мг на кг массы тела человека и животного.
Способы получения соединений по изобретению
Предложен способ получения соединения, как определено выше, включающий:
(A) взаимодействие соединения формулы (10):
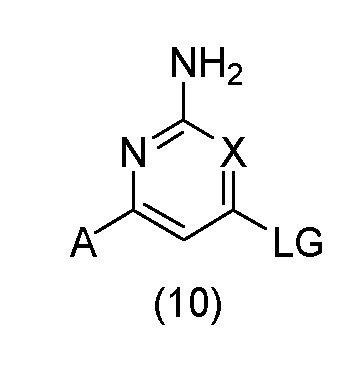
с соединением формулы (11):
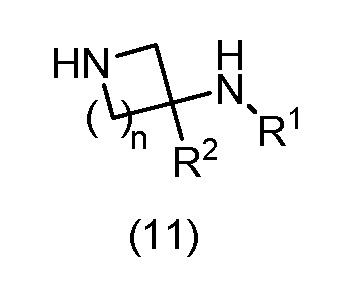
в условиях SNAr или в катализируемых переходным металлом условиях сочетания; где А представляет собой необязательно замещенное пиразольное кольцо; R1 представляет собой H, метил или этил; R2 представляет собой H; X представляет собой N или CH; n имеет значение 1 или 2; и LG представляет подходящую уходящую группу; или
(B) взаимодействие соединения формулы (12):
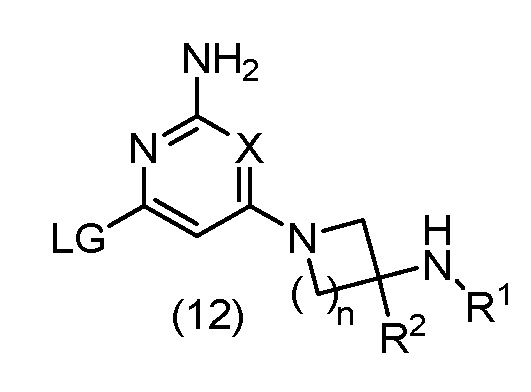
с соединением формулы (13):
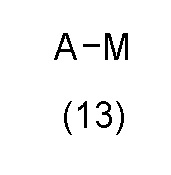
в катализируемых переходным металлом условиях сочетания или в условиях SNAr; где A, R1, R2, X и n определены выше, LG представляет подходящую уходящую группу и M, который может присутствовать или отсутствовать, представляет собой соответствующим образом замещенный металл или неметалл; или
В варианте способа (А) соединение формулы (10) может быть подвергнуто взаимодействию с соединением формулы (11) в условиях SNAr. Реакцию SNAr обычно проводят с использованием либо избытка соединения формулы (11), либо стехиометрического количества соединения формулы (11) в присутствии основания, которое может представлять собой основание третичного амина, такое как TEA или DIPEA, или неорганическое основание, такое как K2CO3, Cs2CO3 или NaHCO3, необязательно в подходящем растворителе, таком как H2O, MeCN, 1,4-диоксан, THF, MeOH, EtOH, IPA, BuOH, DMF, NMP или DMSO, или в комбинации подходящих растворителей, при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления, необязательно в присутствии добавки, такой как KF или соль серебра. Необязательно, соединение формулы (11) может присутствовать в реакции в виде кислой соли, такой как HCl, HBr или TFA соль, необязательно в присутствии третичного основания, такого как TEA или DIPEA. Уходящая группа LG в соединении формулы (10) может представлять собой галоген, такой как F, Cl или Br; алкоксигруппу, такую как OMe; арилоксигруппу, такую как пентафторфенокси; сульфенильную группу, такую как SMe, сульфинильную группу, такую как SOMe, сульфонильную группу, такую как SO2Me, сульфонилоксигруппу, такую как OTs, OMs, ONs или OTf; или уходящую группу, образованную реакцией гидроксильной группы с конденсирующим реагентом для образования пептидной связи, такую как BOP, PyBOP или HATU.
Альтернативно, в варианте способа (А) соединение формулы (10) может быть подвергнуто взаимодействию с соединением формулы (11) в катализируемых переходным металлом условиях сочетания. Реакцию сочетания, катализируемую переходным металлом, обычно проводят с использованием соединения формулы (11) в присутствии неорганического основания, такого как NaOtBu, KOtBu, K3PO4, K2CO3 или Cs2CO3, в подходящем растворителе, таком как 1,4-диоксан, THF, DME или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, Pd(dppf)Cl2, Pd(PPh3)2Cl2 или Pd(PPh3)4, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как PPh3, PBu3, PtBu3, XPhos, Xantphos или BINAP, при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления. Уходящая группа LG в соединении формулы (10) может представлять собой галоген, такой как Cl, Br или I, или сульфонилоксигруппу, такую как OTs, OMs, ONs или OTf.
Соединения формулы (10) могут быть получены по реакции, показанной на схеме 1 ниже:
Схема 1
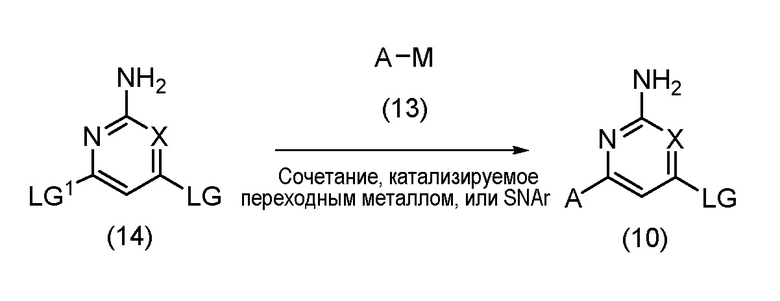
Таким образом, соединение формулы (14), где X имеет значение, определенное выше, и LG и LG1 могут быть одинаковыми или различными и представляют собой подходящие уходящие группы, может быть подвергнуто взаимодействию с соединением формулы (13), где A имеет значение, как определено выше, и М, который может присутствовать или отсутствовать, представляет собой соответствующим образом замещенный металл или неметалл, в катализируемых переходным металлом условиях сочетания или в условиях SNAr с образованием соединения формулы (10). Реакцию сочетания, катализируемую переходным металлом, или реакцию SNAr обычно проводят, как описано ниже, в варианте процесса (B), и соединения формулы (13) и формулы (14) могут быть коммерчески доступными или могут быть легко получены стандартными способами, описанными в опубликованной литературе из простых исходных веществ, известных специалисту в данной области. Иногда из-за их нестабильности может возникнуть необходимость получения соединений формулы (13), в которых присутствует М, in-situ при низких температурах, например, от примерно -78°C до комнатной температуры, и их дальнейшее взаимодействие в реакции сочетания, катализируемой переходным металлом, без их предварительного выделения. Подробности таких способов известны в опубликованной литературе, например, как описано Oberli and Buchwald in Org. Lett., 2012, Vol. 14, No. 17, p 4606.
Альтернативно, соединения формулы (10), где X представляет собой N, а LG представляет собой Cl, обычно можно получить с помощью последовательности реакций, показанной на схеме 2 ниже:
Схема 2
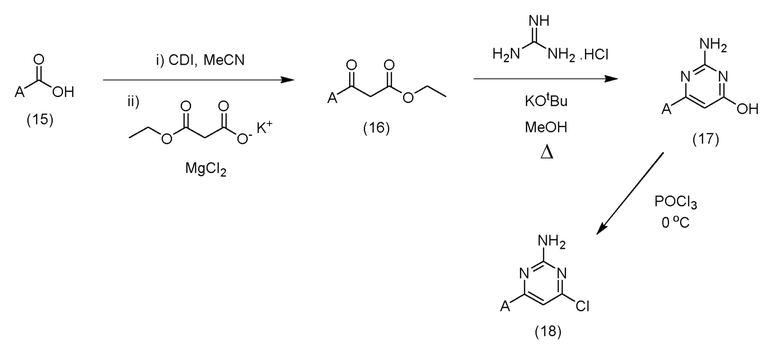
Таким образом, карбоновая кислота формулы (15) может быть гомологизирована до соответствующего бета-кетоэфира (16) путем ее предварительной активации с помощью ряда стандартных способов, известных специалисту в данной области, например, путем реакции с CDI в подходящем растворителе, таком как MeCN, и затем реакцией с производным малоновой кислоты, таким как калия 3-этокси-3-оксопропаноат, в присутствии кислоты Льюиса, такой как MgCl2. После образования бета-кетоэфир (16) может быть циклизован до аналога аминогидроксипиримидина (17) путем реакции с гуанидином или подходящей солью гуанидина в присутствии подходящего основания, такого как KOtBu, в подходящем растворителе, таком как МеОН. Полученный таким образом аминогидроксипиримидиновый аналог (17) можно затем подвергнуть взаимодействию с POCl3 в присутствии или в отсутствие подходящего растворителя с образованием соединения формулы (18). Соединения формулы (15) могут быть коммерчески доступны или могут быть легко получены стандартными способами, описанными в опубликованной литературе, из простых исходных веществ, известных специалисту в данной области.
Соединения формулы (11) могут быть коммерчески доступны или могут быть легко получены стандартными способами, описанными в опубликованной литературе, из простых исходных веществ, известных специалисту в данной области.
В варианте способа (В) соединение формулы (12) может быть подвергнуто взаимодействию с соединением формулы (13) в катализируемых переходным металлом условиях сочетания. Реакцию сочетания, катализируемую переходным металлом, обычно проводят с использованием соединения формулы (13), где М присутствует. Например, когда M представляет собой бороновую кислоту -B(OH)2, или бороновый эфир, такой как -B(OMe)2, -B(OiPr)2 или Bpin, или триалкилборат лития, такой как -B(OiPr)3Li, в таком случае обычно проводят реакцию сочетания, катализируемую переходным металлом, в присутствии неорганического основания, такого как NaHCO3, Na2CO3, K2CO3, Cs2CO3 или K3PO4, в подходящем растворителе, таком как H2O, MeCN, 1,4-диоксан, THF, Et2O, DME, EtOH, IPA, DMF, NMP или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, Pd(dppf)Cl2, Pd(PPh3)2Cl2, Pd(PPh3)4, или прекатализатора на основе переходного металла, такого как XPhos Pd G2, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как PPh3, PtBu3, PCy3 или XPhos, при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I, или сульфонилоксигруппу, такую как OTs, OMs или OTf.
В качестве альтернативы, когда M представляет собой соль трифторбората BF3-, в таком случае обычно проводят реакцию сочетания, катализируемую переходным металлом, в присутствии неорганического основания, такого как Na2CO3, K2CO3, Cs2CO3 или K3PO4, в подходящем растворителе, таком как H2O, MeCN, 1,4-диоксан, THF, MeOH или EtOH, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как PPh3, PCy3 или RuPhos при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I.
Альтернативно, когда M представляет собой группу триалкилолова, такую как SnMe3 или SnBu3, тогда реакцию сочетания, катализируемую переходным металлом, обычно проводят в подходящем растворителе, таком как 1,4-диоксан, THF, DMF, или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, Pd(PPh3)2Cl2 или Pd(PPh3)4, необязательно в присутствии неорганического основания, такого как K2CO3 или CsF, необязательно в присутствии добавки, такой как LiCl, CuI, Bu4NBr или Et4NCl, при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I.
Альтернативно, когда М отсутствует, в таком случае обычно проводят реакцию сочетания, катализируемую переходным металлом, в присутствии неорганического основания, такого как NaOtBu, KOtBu, K3PO4, K2CO3 или Cs2CO3, в подходящем растворителе, таком как 1,4-диоксан, THF, DME или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как Pd(OAc)2, Pd2(dba)3, Pd(dppf)Cl2, Pd(PPh3)2Cl2 или Pd(PPh3)4, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как PPh3, PBu3, PtBu3, XPhos, Xantphos или BINAP, при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I, или сульфонилоксигруппу, такую как OTs, OMs, ONs или OTf.
Альтернативно, когда М отсутствует, в таком случае обычно проводят реакцию сочетания, катализируемую переходным металлом, в присутствии неорганического основания, такого как K3PO4, K2CO3 или Cs2CO3, в подходящем растворителе, таком как 1,4-диоксан, DMF, DMSO или толуол, или комбинации подходящих растворителей, в присутствии субстехиометрического количества катализатора на основе переходного металла, такого как CuI, необязательно в присутствии субстехиометрического количества амина, такого как (S)-пролин или транс-N1,N2-диметилциклогексан-1,2-диамин, при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl, Br или I.
Альтернативно, когда М отсутствует, в таком случае обычно проводят реакцию сочетания, катализируемую переходным металлом, в присутствии органического основания, такого как nBu4OAc, в подходящем растворителе, таком как 1,4-диоксан, в присутствии субстехиометрического количества прекатализатора на основе переходного металла, такого как XPhos Pd G2, необязательно в присутствии субстехиометрического количества фосфинового лиганда, такого как XPhos, при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как Cl.
Альтернативно, в варианте способа (В) соединение формулы (12) может быть подвергнуто взаимодействию с соединением формулы (13) в условиях SNAr. Реакцию SNAr обычно проводят с использованием соединения формулы (13), где М отсутствует, в присутствии основания третичного амина, такого как TEA или DIPEA, или неорганического основания, такого как K2CO3, Cs2CO3, KOtBu или NaH, в подходящем растворителе, таком как THF, DMF, H2O, DMSO или NMP, или комбинации подходящих растворителей, при температуре от приблизительно комнатной температуры до приблизительно 200°C, с использованием обычного нагревания или необязательно путем нагревания с микроволновым излучением, в открытом сосуде или необязательно в герметичном сосуде, необязательно при давлении выше атмосферного давления. Уходящая группа LG в соединении формулы (12) может представлять собой галоген, такой как F, Cl или Br; алкоксигруппу, такую как OMe; арилоксигруппу, такую как пентафторфенокси; сульфенильную группу, такую как SMe, сульфинильную группу, такую как SOMe, сульфонильную группу, такую как SO2Me, или сульфонилоксигруппу, такую как OTs, OMs, ONs или OTf.
Соединение формулы (12) может быть получено последовательностью реакций, показанной на схеме 3 ниже:
Схема 3
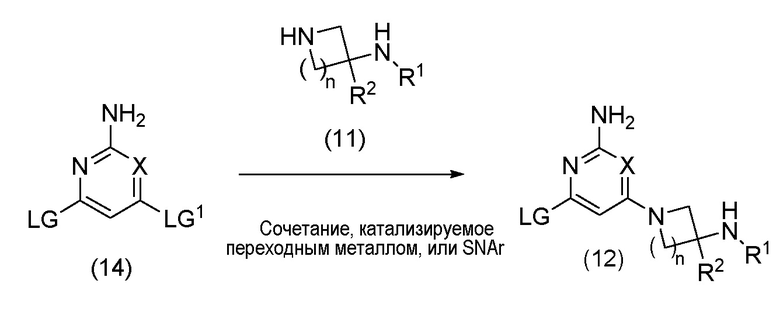
Таким образом, соединение формулы (14), где X имеет значение, определенное выше, и LG и LG1 могут быть одинаковыми или различными и представлять собой подходящие уходящие группы, может быть подвергнуто взаимодействию с соединением формулы (11), где R1, R2 и n определены выше, в условиях SNAr или в катализируемых переходным металлом условиях сочетания, с образованием соединения формулы (12). Реакцию SNAr или реакцию сочетания, катализируемую переходным металлом, обычно проводят, как описано выше в варианте способа (А).
Во многих реакциях, описанных выше, может быть необходимо защитить одну или несколько групп для предотвращения протекание реакции в нежелательном месте молекулы. Примеры защитных групп и способы защиты и удаления защиты функциональных групп можно найти в Greene's Protective Groups in Organic Synthesis, Fifth Edition, Editor: Peter G. M. Wuts, John Wiley, 2014, (ISBN: 9781118057483). В частности, полезная защитная группа для манипуляции соединениями формулы (10) или формулы (12) включает 2,5-диметил-1Н-пиррольную группу; полезные защитные группы для манипуляции соединениями формулы (11) или формулы (12) включают BOC и CBZ; и полезные защитные группы для манипуляции соединениями формулы (13) включают SEM и THP.
Соединения, полученные вышеуказанными способами, могут быть выделены и очищены любым из множества способов, хорошо известных специалистам в данной области, и примеры таких способов включают методы перекристаллизации и хроматографии, такие как колоночная хроматография (например, флэш-хроматография), ВЭЖХ и SFC.
Фармацевтические композиции
Хотя активное соединение можно вводить отдельно, предпочтительно представлять его в виде фармацевтической композиции (например, лекарственной формы).
Соответственно, изобретение предоставляет фармацевтическую композицию, содержащую по меньшей мере одно соединение по настоящему изобретению, как определено выше, вместе по меньшей мере с одним фармацевтически приемлемым эксципиентом.
Композиция может представлять собой таблетированную композицию.
Композиция может представлять собой композицию капсулы.
Фармацевтически приемлемый эксципиент(эксципиенты) можно выбрать, например, из носителей (например, твердый, жидкий или полутвердый носитель), адъювантов, разбавителей (например, твердых разбавителей, таких как наполнители или объемообразующие агенты, и жидких разбавителей, таких как растворители и сорастворители), гранулирующих агентов, связующих веществ, агентов для повышения текучести, покрывающих агентов, агентов, контролирующих высвобождение (например, полимеры или воски, замедляющие или задерживающие высвобождение), связующих веществ, разрыхлителей, буферных агентов, смазывающих веществ, консервантов, противогрибковых и антибактериальных агентов, антиоксидантов, буферных агентов, агентов, регулирующих тоничность, загустителей, ароматизаторов, подсластителей, пигментов, пластификаторов, агентов, маскирующих вкус, стабилизаторов или любых других эксципиентов, обычно используемых в фармацевтических композициях.
Термин «фармацевтически приемлемый» в контексте настоящего документа означает соединения, материалы, композиции и/или лекарственные формы, которые, в рамках здравого медицинского суждения, подходят для применения в контакте с тканями субъекта (например, субъекта-человека) без чрезмерной токсичности, раздражения, аллергической реакции или других проблемы или осложнений, соответствующих разумному соотношению польза/риск. Каждый эксципиент также должен быть «приемлемым» в смысле совместимости с другими ингредиентами композиции.
Фармацевтические композиции, содержащие соединения по изобретению, могут быть получены в соответствии с известными методами, см., например, Remington’s Pharmaceutical Sciences, Mack Publishing Company, Easton, PA, USA.
Фармацевтические композиции могут быть в любой форме, пригодной для перорального, парентерального, местного, интраназального, внутрибронхиального, сублингвального, офтальмологического, ушного, ректального, интравагинального или чрескожного введения.
Фармацевтические лекарственные формы, пригодные для перорального введения, включают таблетки (с покрытием или без покрытия), капсулы (с твердой или мягкой оболочкой), каплеты, пилюли, леденцы, сиропы, растворы, порошки, гранулы, эликсиры и суспензии, сублингвальные таблетки, облатки или пластыри, такие как буккальные пластыри.
Композиции таблеток могут содержать стандартную дозу активного соединения вместе с инертным разбавителем или носителем, таким как сахар или сахарный спирт, например; лактоза, сахароза, сорбит или маннит; и/или разбавителем, не содержащим сахара, таким как карбонат натрия, фосфат кальция, карбонат кальция или целлюлоза или ее производное, такое как микрокристаллическая целлюлоза (MCC), метилцеллюлоза, этилцеллюлоза, гидроксипропилметилцеллюлоза и крахмалы, такие как кукурузный крахмал. Таблетки также могут содержать такие стандартные ингредиенты, как связующие и гранулирующие агенты, такие как поливинилпирролидон, разрыхлители (например, поддающиеся разбуханию сшитые полимеры, такие как перекрестно-сшитая карбоксиметилцеллюлоза), смазывающие агенты (например, стеараты), консерванты (например, парабены), антиоксиданты (например, BHT), буферные агенты (например, например, фосфатный или цитратный буферы) и шипучие агенты, такие как смеси цитрата/бикарбоната. Такие эксципиенты хорошо известны и не нуждаются в подробном обсуждении в настоящем документе.
Таблетки могут быть предназначены для высвобождения лекарственного средства либо при контакте с желудочным соком (таблетки с немедленным высвобождением), либо для контролируемого высвобождения (таблетки с контролируемым высвобождением) в течение длительного периода времени или в определенной области желудочно-кишечного тракта.
Фармацевтические композиции обычно содержат от приблизительно 1% (масс./масс.) до приблизительно 95%, предпочтительно % (масс./масс.) активного ингредиента и от 99% (масс./масс.) до 5% (масс./масс.) фармацевтически приемлемого эксципиента (например, как определено выше) или комбинацию таких эксципиентов. Предпочтительно композиции содержат от приблизительно 20% (масс./масс.) до приблизительно 90% (масс./масс.) активного ингредиента и от 80% (масс./масс.) до 10% фармацевтического эксципиента или комбинации эксципиентов. Фармацевтические композиции содержат от приблизительно 1% до приблизительно 95%, предпочтительно от приблизительно 20% до приблизительно 90% активного ингредиента. Фармацевтические композиции по изобретению могут быть, например, в форме стандартных лекарственных форм, таких как ампулы, флаконы, суппозитории, предварительно заполненные шприцы, драже, порошки, таблетки или капсулы.
Таблетки и капсулы могут содержать, например, 0-20% разрыхлителей, 0-5% смазывающих веществ, 0-5% агентов для повышения текучести и/или 0-99% (масс./масс.) наполнителей/ или объемообразующих агентов (в зависимости от дозы лекарственного средства). Они также могут содержать 0-10% (масс./масс.) полимерных связующих, 0-5% (масс./масс.) антиоксидантов, 0-5% (масс./масс.) пигментов. Таблетки с замедленным высвобождением, кроме того, обычно содержат 0-99% (масс./масс.) полимеров, контролирующих высвобождение (например, задерживающих) (в зависимости от дозы). Пленочные покрытия таблетки или капсулы обычно содержат 0-10% (масс./масс.) полимеров, 0-3% (масс./масс.) пигментов и/или 0-2% (масс./масс.) пластификаторов.
Парентеральные композиции обычно содержат 0-20% (масс./масс.) буферов, 0-50% (масс./масс.) сорастворителей и/или 0-99% (масс./масс.) воды для инъекций (WFI) (в зависимости от дозы и в случае сублимационной сушки). Композиции для внутримышечных депо также могут содержать 0-99% (масс./масс.) масел.
Фармацевтические композиции могут быть представлены пациенту в «упаковках для пациентов», содержащих весь курс лечения в одной упаковке, обычно в блистерной упаковке.
Соединения по изобретению, как правило, представлены в виде стандартной лекарственной формы и, как таковые, обычно содержат достаточное количество соединения для обеспечения желаемого уровня биологической активности. Например, композиция может содержать от 1 нанограмма до 2 граммов активного ингредиента, например, от 1 нанограмма до 2 миллиграммов активного ингредиента. В пределах этих диапазонов конкретные поддиапазоны соединения составляют от 0,1 миллиграмма до 2 граммов активного ингредиента (чаще от 10 миллиграммов до 1 грамма, например, от 50 миллиграммов до 500 миллиграммов) или от 1 микрограмма до 20 миллиграммов (например, от 1 микрограмма до 10 миллиграммов, например, от 0,1 миллиграмма до 2 миллиграммов активного ингредиента).
Для пероральных композиций стандартная лекарственная форма может содержать от 1 миллиграмма до 2 граммов, более типично от 10 миллиграммов до 1 грамма, например, от 50 миллиграммов до 1 грамма, например, от 100 миллиграммов до 1 грамма активного соединения.
Активное соединение вводят пациенту, нуждающемуся в этом (например, пациенту-человеку или животному) в количестве, достаточном для достижения желаемого терапевтического эффекта (эффективное количество). Точные количества вводимого соединения могут быть определены лечащим врачом в соответствии со стандартными процедурами.
ПРИМЕРЫ
Теперь изобретение будет проиллюстрировано, но не ограничено, со ссылкой на следующие примеры.
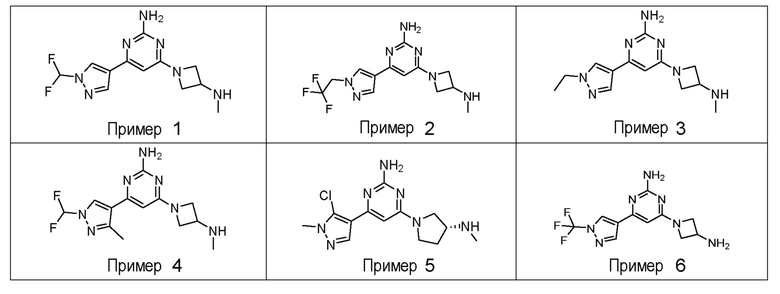
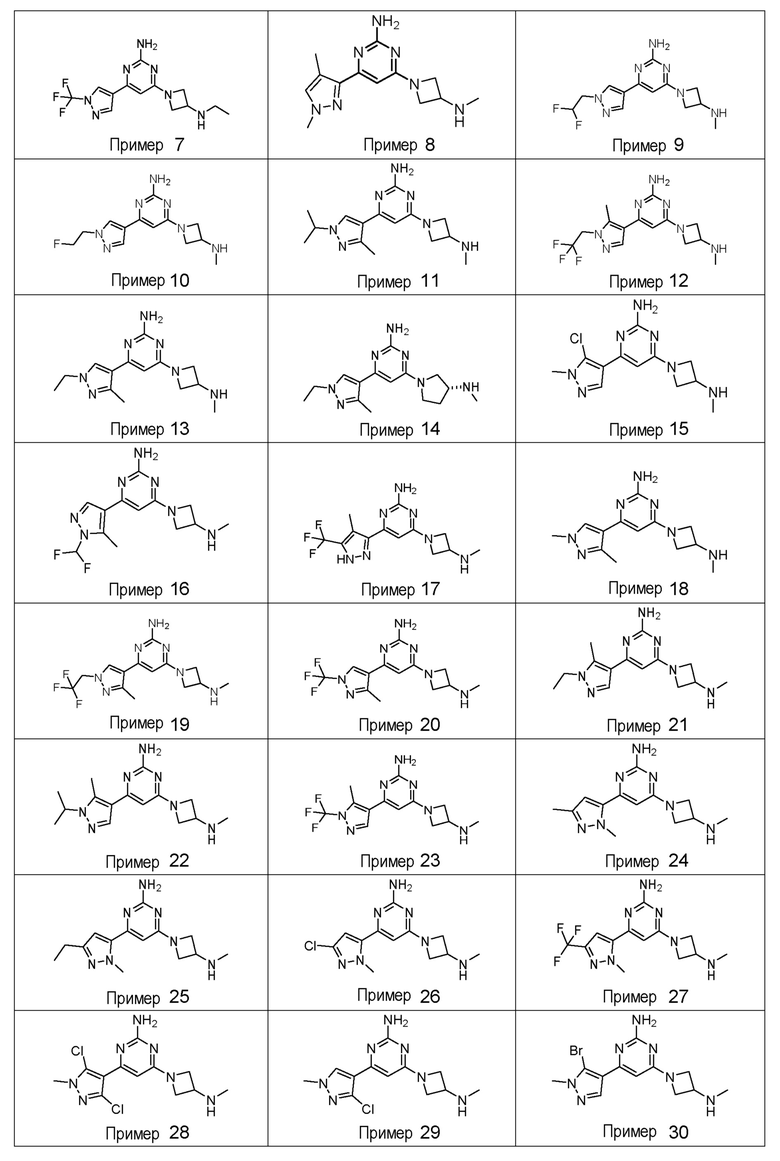
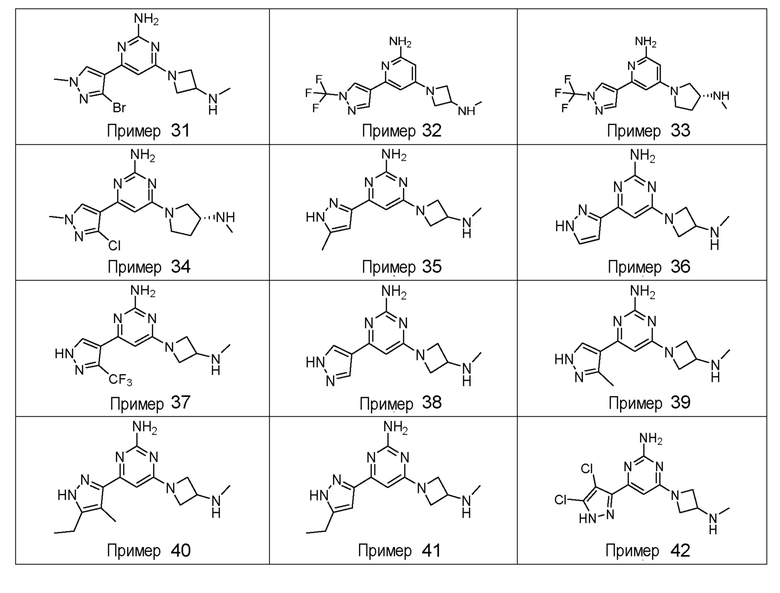
ПРИМЕРЫ 1-42
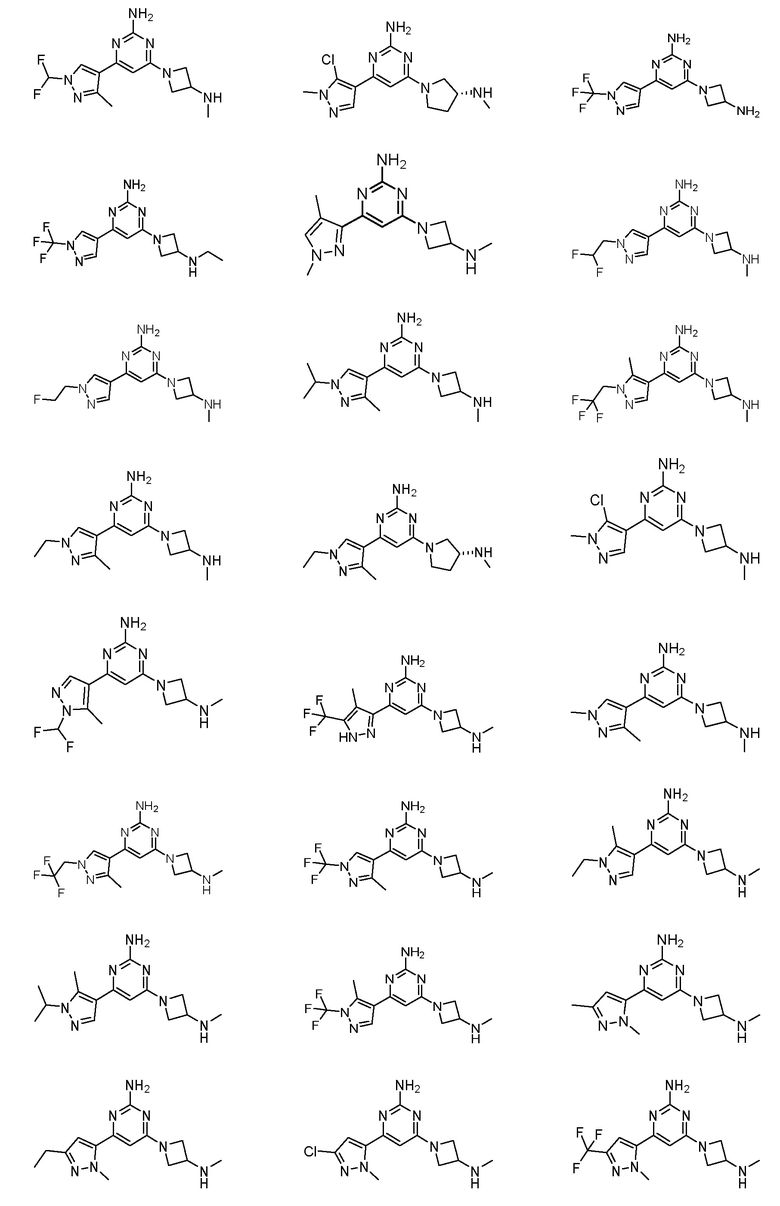
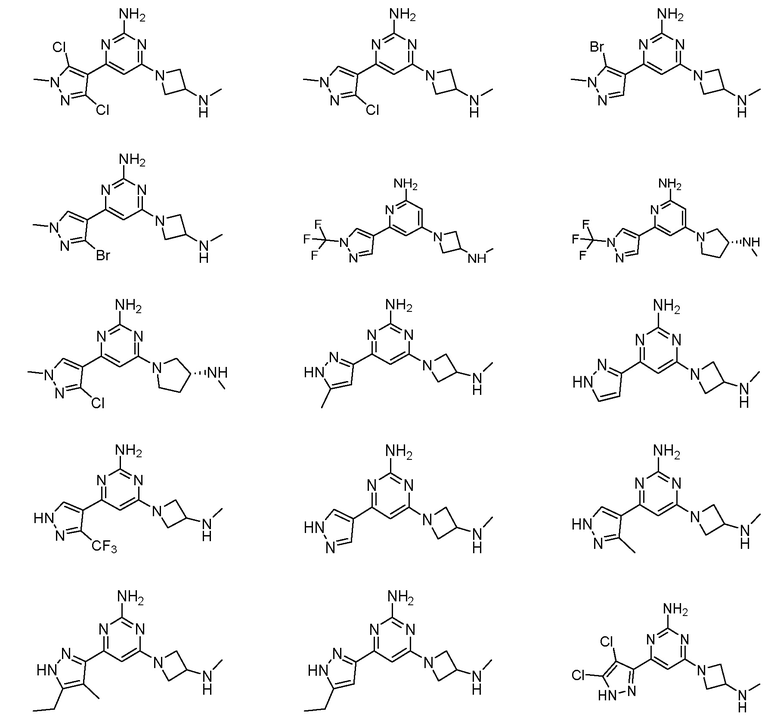
Соединения примеров 1-42, показанные в таблице 1 ниже, были получены. Их ЯМР и LCMS свойства, а также способы их получения приведены в таблице 3. Исходные вещества для каждого из примеров перечислены в таблице 2. Для примеров 20, 23, 32 и 33 показаны предлагаемые пути синтеза.
Таблица 1 - Примеры соединений
Общие процедуры
Если не указаны пути получения, то соответствующий промежуточный продукт является коммерчески доступным. Коммерческие реагенты использовали без дополнительной очистки. Комнатная температура (комн.темп.) означает приблизительно 20-27°C. 1H-ЯМР спектры регистрировали при 400 МГц на приборе Bruker или Jeol. Значения химического сдвига выражены в частях на миллион (ppm), т.е. (δ)-значения. Следующие сокращения используются для мультиплетности сигналов ЯМР: с=синглет, ушир.=уширенный, д=дублет, т=триплет, кв=квартет, квинт.=квинтет, тд=триплет дублетов, тт=триплет триплетов, qd=квартет дуплетов, ддд=двойной дублет дуплетов, ддт=двойной дублет триплетов, м=мультиплет. Постоянные спин-спинового взаимодействия указаны как значения J, измеренные в Гц. Результаты ЯМР и масс-спектроскопии были скорректированы для учета пиков фона. Хроматография относится к колоночной хроматографии, проводимой с использованием силикагеля 60-120 меш и проводимой в условиях давления азота (флеш-хроматография). Колоночная хроматография, выполненная с использованием «основного силикагеля», относится к использованию силикагеля Biotage® KP-NH. Колоночная хроматография, выполненная в условиях обращенной фазы с использованием ‘силикагеля C18’, относится к использованию силикагеля Biotage® KP-C18. ТСХ для мониторинга реакций относится к ТСХ с использованием указанной подвижной фазы и силикагеля F254 в качестве неподвижной фазы от Merck. Опосредованные микроволнами реакции проводили в микроволновых реакторах Biotage Initiator или CEM Discover.
LCMS-анализ
LCMS-анализ соединений проводили в условиях электрораспыления с использованием приборов и способов, указанных в таблицах ниже:
50×4,6 мм,
3,5 мкм или
эквивалент
50×4,6 мм,
3,5 мкм или эквивалент
Данные LCMS в экспериментальной части и таблицах 2 и 3 представлены в следующем формате: (система приборов, способ): масса ионов, время удерживания, длина волны УФ-детектирования.
Очистка соединения
Окончательную очистку соединений проводили препаративной ВЭЖХ с обращенной фазой с использованием прибора и способов, подробно описанных ниже, где данные представлены в следующем формате: Методика очистки: [фаза (описание колонки, длина колонки х внутренний диаметр, размер частиц), скорость потока растворителя, градиент - указан как % подвижной фазы B в подвижной фазе A (с течением времени), подвижной фазы (A), подвижной фазы (B)].
Очистка препаративной ВЭЖХ:
Бинарная система Shimadzu LC-20AP с с УФ-детектором SPD-20A
Система полупрепаративной ВЭЖХ Gilson Система полупрепаративной ВЭЖХ Gilson с насосом 321, жидкостным манипулятором GX-271 и Gilson 171 DAD, управляемая с помощью программного обеспечения Gilson Trilution
Способ очистки A
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 12 мл/мин, градиент 0% - 30% (более 17 мин), 100% (более 1 мин), 100%-0% (более 4 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки B
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 12 мл/мин, градиент 0% - 15% (более 24 мин), 15% - 15% (более 3 мин), 100% (более 2 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки C
Препаративная ВЭЖХ: [Обращенная фаза (YMC-Actus Triart C-18, 250 х 20 мм, 5 мкм), 16 мл/мин, градиент 5% - 15% (более 18 мин), 15% - 15% (более 2 мин), 100% (более 2 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки D
Препаративная ВЭЖХ: [Обращенная фаза (X-select CSH Phenyl Hexyl C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 3% - 3% (более 40 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки E
Препаративная ВЭЖХ: [Обращенная фаза (Gemini-NX C-18, 150 х 21,2 мм, 5 мкм), 16 мл/мин, градиент 5% - 30% (более 25 мин), 100% (более 3 мин), 100% - 5% (более 4 мин), подвижная фаза (A): 5 мМ бикарбонат аммония+0,1% аммония в воде, (B): 100% ацетонитрил].
Способ очистки F
Препаративная ВЭЖХ: [Обращенная фаза (YMC-Actus Triart C-18, 250 х 20 мм, 5 мкм), 13 мл/мин, градиент 5% - 20% (более 22 мин), 20% - 20% (более 3 мин), 100% (более 2 мин), 100% - 5% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки G
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 13 мл/мин, градиент 0% - 15% (более 24 мин), 15% - 15% (более 5 мин), 100% (более 2 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки H
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 30% (более 17 мин), 100% (более 1 мин), 100% - 0% (более 4 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки I
Препаративная ВЭЖХ: [Обращенная фаза (YMC-Actus Triart C-18, 250 х 20 мм, 5 мкм), 15 мл/мин, градиент 0% - 15% (более 25 мин), 15% - 15% (более 4 мин), 100% (более 2 мин), 100% - 5% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки J
Препаративная ВЭЖХ: [Обращенная фаза (YMC-Actus Triart C-18, 250 х 20 мм, 5 мкм), 15 мл/мин, градиент 5% - 12% (более 28 мин), 100% (более 2 мин), 100% - 5% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки K
Препаративная ВЭЖХ: [Обращенная фаза (YMC-Actus Triart C-18, 250 х 20 мм, 5 мкм), 15 мл/мин, градиент 0% - 15% (более 18 мин), 15% - 15% (более 5 мин), 100% (более 2 мин), 100% - 0% (более 3 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки L
Препаративная ВЭЖХ: [Обращенная фаза (X-select CSH Phenyl Hexyl C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 20% (более 23 мин), 100% (более 2 мин), 100% - 0% (более 3 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки M
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 18 мм, 5 мкм), 14 мл/мин, градиент 0% - 20% (более 22 мин), 100% (более 2 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки N
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 18 мм, 5 мкм), 15 мл/мин, градиент 0% - 20% (более 22 мин), 100% (более 2 мин), 100% - 0% (более 2 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки O
Препаративная ВЭЖХ: [Обращенная фаза (X-select CSH Phenyl Hexyl C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 5% (более 22 мин), 5% - 5% (более 2 мин), 100% (более 2 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки P
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 13 мл/мин, градиент 10% - 15% (более 24 мин), 100% (более 2 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки Q
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 20 мм, 5 мкм), 15 мл/мин, градиент 0% - 30% (более 17 мин), 100% (более 1 мин), 100% - 0% (более 4 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки R
Препаративная ВЭЖХ: [Обращенная фаза (X-Bridge C-18, 250 х 19 мм, 5 мкм), 14 мл/мин, градиент 10% - 30% (более 20 мин), 30% - 30% (более 2 мин), 100% (более 2 мин), 100% - 10% (более 6 мин), подвижная фаза (A): 5 мМ бикарбонат аммония+0,1% аммония в воде, (B): 100% ацетонитрил].
Способ очистки S
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 20% (более 20 мин), 100% (более 2 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки T
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 5% - 20% (более 20 мин), 100% (более 2 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки U
Препаративная ВЭЖХ: [Обращенная фаза (YMC-Actus Triart C-18, 250 х 20 мм, 5 мкм), 12 мл/мин, градиент 0% - 20% (более 25 мин), 100% (более 2 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки V
Препаративная ВЭЖХ: [Обращенная фаза (Gemini NX C-18, 150 х 21,2 мм, 5 мкм), 15 мл/мин, градиент 0% - 15% (более 18 мин), 100% (более 2 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки W
Препаративная ВЭЖХ: [Обращенная фаза (Gemini NX C-18, 150 х 21,2 мм, 5 мкм), 16 мл/мин, градиент 0% - 8% (более 18 мин), 100% (более 2 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки X
Препаративная ВЭЖХ: [Обращенная фаза (X-select CSH Phenyl Hexyl C-18, 250 х 19 мм, 5 мкм), 14 мл/мин, градиент 0% - 20% (более 20 мин), 100% (более 3 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки Y
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 14 мл/мин, градиент 0% - 15% (более 20 мин), 15% - 15% (более 2 мин), 100% (более 2 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки Z
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 14 мл/мин, градиент 0% - 15% (более 20 мин), 100% (более 2 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AA
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 10% (более 25 мин), 10% - 10% (более 2 мин), 100% (более 3 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AB
Препаративная ВЭЖХ: [Обращенная фаза (X-Bridge C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 5% - 27% (более 26 мин), 100% (более 3 мин), 100% - 5% (более 5 мин), подвижная фаза (A): 5 мМ бикарбонат аммония+0,1% аммония в воде, (B): 100% ацетонитрил].
Способ очистки AC
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 20 мм, 5 мкм), 15 мл/мин, градиент 0% - 30% (более 17 мин), 100% (более 1 мин), 100% - 0% (более 4 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AD
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 10% (более 18 мин), 10% - 10% (более 2 мин), 100% (более 2 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AE
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 150 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 10% (более 12 мин), 100% (более 2 мин), 100% - 0% (более 2 мин), подвижная фаза (A): 0,1% муравьиной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AF
Препаративная ВЭЖХ: [Обращенная фаза (X-Bridge C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 12% (более 25 мин), 100% (более 2 мин), 100% - 0% (более 8 мин), подвижная фаза (A): 5 мМ бикарбонат аммония+0,1% аммония в воде, (B): 100% ацетонитрил].
Способ очистки AG
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 15% (более 18 мин), 100% (более 3 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AH
Препаративная ВЭЖХ: [Обращенная фаза (X-Bridge C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 12% (более 20 мин), 100% (более 2 мин), 100% - 0% (более 5 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AI
Препаративная ВЭЖХ: [Обращенная фаза (Sunfire C-18, 250 х 19 мм, 5 мкм), 17 мл/мин, градиент 0% - 20% (более 17 мин), 100% (более 2 мин), 100% - 0% (более 4 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AJ
Препаративная ВЭЖХ: [Обращенная фаза (X-Bridge C-18, 250 х 19 мм, 5 мкм), 15 мл/мин, градиент 0% - 10% (более 28 мин), 10% - 10% (более 6 мин), 100% (более 2 мин), 100% - 0% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Способ очистки AK
Препаративная ВЭЖХ: [Обращенная фаза (YMC-Actus Triart C-18, 250 х 20 мм, 5 мкм), 16 мл/мин, градиент 5% - 20% (более 24 мин), 100% (более 2 мин), 100% - 5% (более 6 мин), подвижная фаза (A): 0,1% трифторуксусной кислоты в воде, (B): 100% ацетонитрил].
Аббревиатуры
CDI=карбонилдиимидазол
DAST=трифторид диэтиламиносеры
DCM=дихлорметан
DIPEA=N,N-диизопропилэтиламин
ESI=ионизация электрораспылением
EtOAc=этилацетат
ч=час(часы)
H2O=вода
HCl=хлороводород, хлористоводородная кислота
ВЭЖХ=высокоэффективная жидкостная хроматография
IPA=пропан-2-ол
LC=жидкостная хроматография
MeCN=ацетонитрил
MeOH=метанол
мин=минута(минуты)
MS=масс-спектрометрия
нм=нанометр(ы)
ЯМР=ядерный магнитный резонанс
POCl3=оксихлорид фосфора
Комн.темп.=комнатная температура
насыщ.=насыщенный
SFC=сверхкритическая флюидная хроматография
TEA=триэтиламин
TFA=трифторуксусная кислота
THF=тетрагидрофуран
TLC=тонкослойная хроматография.
Синтез промежуточных соединений:
Путь 1
Типичная процедура получения пиримидинов на примере получения промежуточного соединения 3, трет-бутил (1-(2-амино-6-хлорпиримидин-4-ил)азетидин-3-ил)(метил)карбамата
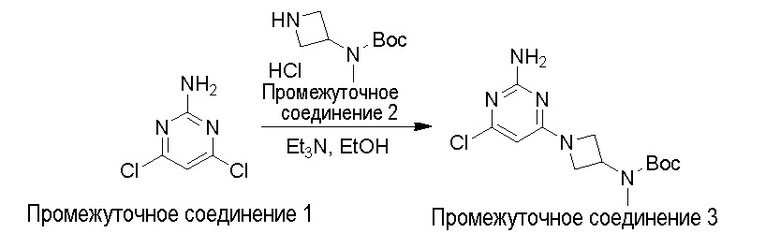
4,6-Дихлорпиримидин-2-амин промежуточное соединение 1 (2 г, 12,27 ммоль) порциями добавляли к перемешиваемому раствору трет-бутил N-(азетидин-3-ил)-N-метилкарбамат гидрохлорида промежуточное соединение 2 (3,0 г, 12,9 ммоль) в EtOH (50 мл), затем Et3N (8 мл, 30,6 ммоль) при комнатной температуре. Полученную суспензию нагревали до температуры кипения с обратным холодильником и поддерживали в течение 2 ч. Смесь охлаждали и по каплям добавляли воду (30 мл). Полученное твердое вещество выделяли, промывали водой и сушили с получением трет-бутил (1-(2-амино-6-хлорпиримидин-4-ил)азетидин-3-ил)(метил)карбамата промежуточное соединение 3 (3 г, 79%) в виде белого твердого вещества. Данные для промежуточного соединения 3 приведены в таблице 2.
Общие процедуры синтеза:
Путь A
Типичная процедура получения пиримидинов на примере получения примера 1, 4-(1-(дифторметил)-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амин
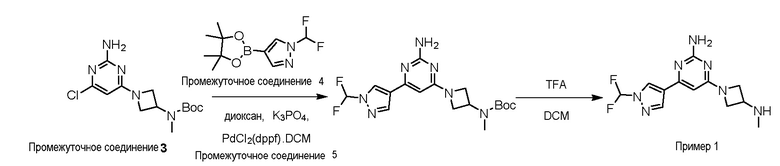
трет-Бутил (1-(2-амино-6-хлорпиримидин-4-ил)азетидин-3-ил)(метил)карбамат, промежуточное соединение 3, (0,350 г, 1,11 ммоль), 1-(дифторметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол промежуточное соединение 4 (0,300 г, 1,23 ммоль), K3PO4 (0,711 г, 3,36 ммоль) растворяли в 1,4-диоксане (4 мл) и воде (1 мл) и сырьевой материал дегазировали в течение 15 мин. Добавляли Pd(dppf)Cl2.DCM промежуточное соединение 5 (0,090 г, 0,11 ммоль) и сырьевой материал перемешивали при 70oC в течение 16 ч. Реакционную массу разбавляли водой (10 мл), экстрагировали этилацетатом (3×20 мл), объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали с получением неочищенного остатка, который очищали колоночной хроматографией (нейтральный оксид алюминия, 0-35% EtOAc: гексан) с получением трет-бутил (1-(2-амино-6-(1-(дифторметил)-1H-пиразол-4-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамата (0,4 г, 90,7%) в виде не совсем белого твердого вещества.
LCMS (Система 1, способ 1): m/z 395 (M+H)+ (ESI +ve), около 2,88 мин, 220 нм
трет-Бутил (1-(2-амино-6-(1-(дифторметил)-1H-пиразол-4-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамат (0,400 г, 0,10 ммоль) растворяли в DCM (4 мл), добавляли по каплям TFA (2 мл) при 0oC и сырьевой материал перемешивали при комнатной температуре в течение 3 ч. Растворитель выпаривали в вакууме и подвергали азеотропной перегонке с толуолом (3×10 мл) с получением неочищенного остатка, который очищали по способу очистки A с получением ди TFA соли 4-(1-(дифторметил)-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амина пример 1 (245 мг, 82,0%) в виде белого твердого вещества. Данные для примера 1 приведены в таблице 3.
Путь B
Типичная процедура получения пиримидинов на примере получения примера 4, 4-(1-(дифторметил)-3-метил-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амин, и примера 16, 4-(1-(дифторметил)-5-метил-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амин
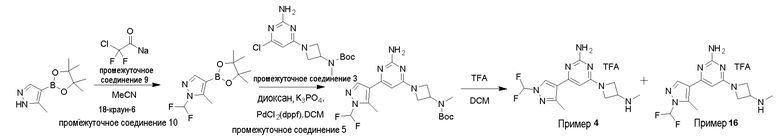
5-Метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол промежуточное соединение 8 (1,0 г, 4,81 ммоль) растворяли в ацетонитриле (10 мл), добавляли 18-краун-6 промежуточное соединение 10 (254 мг, 0,96 ммоль) и хлордифторацетат натрия промежуточное соединение 9 (879 мг, 5,77 ммоль) и реакционную смесь нагревали при 80oC в течение 24 ч. После охлаждения осадок удаляли фильтрованием и фильтрат концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией (нейтральный оксид алюминия, 0-35% этилацетат в гексане) с получением смеси 1-(дифторметил)-5-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол и 1-(дифторметил)-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (вместе 1 г, 80,6%) в виде не совсем белого твердого вещества.
LCMS (система 2, способ 2): Изомер 1, только УФ около 3,52 мин, 202 нм и изомер 2, только УФ около 3,63 мин, 202 нм.
1-(Дифторметил)-5-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол и 1-(дифторметил)-3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (в целом 1 г, 3,87 ммоль) и трет-бутил (1-(2-амино-6-хлорпиримидин-4-ил)азетидин-3-ил)(метил)карбамат промежуточное соединение 3 (1,1 г, 3,50 ммоль) растворяли в 1,4-диоксане (10 мл) при комнатной температуре. Добавляли воду (2 мл), K3PO4 (2,23 г, 10,51 ммоль) и смесь дегазировали в течение 15 мин. Добавляли PdCl2(dppf).DCM промежуточное соединение 5 (280 мг, 0,35 ммоль) и смесь перемешивали при 70oC в течение 16 ч. Реакционную смесь разделяли между H2O (50 мл) и этилацетатом (35 мл), водный слой дополнительно экстрагировали этилацетатом (2 х 35 мл), объединенные органические слои сушили (Na2SO4), фильтровали и растворитель концентрировали с получением неочищенного остатка, который очищали колоночной хроматографией (нейтральный оксид алюминия, 0-2% метанола в DCM) с получением смеси трет-бутил (1-(2-амино-6-(1-(дифторметил)-5-метил-1H-пиразол-4-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамата и трет-бутил (1-(2-амино-6-(1-(дифторметил)-3-метил-1H-пиразол-4-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамата (в целом 630 мг, 39,7%) в виде желтой смолы.
LCMS (система 2, способ 2): Изомер 1, m/z 410,2 (M+H)+ (ES+), около 3,35 мин, 202 нм и изомер 2, m/z 410,2 (M+H)+ (ES+), около 3,40 мин, 202 нм.
трет-Бутил (1-(2-амино-6-(1-(дифторметил)-5-метил-1H-пиразол-4-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамат и трет-бутил (1-(2-амино-6-(1-(дифторметил)-3-метил-1H-пиразол-4-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамат (в целом 800 мг, 1,95 ммоль) растворяли в DCM (8 мл) при 0oC, TFA (4 мл) добавляли по каплям и смесь перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь концентрировали и неочищенный остаток подвергали азеотропной перегонке с толуолом (3×5 мл) с получением неочищенного продукта, который очищали способом очистки D с получением 4-(1-(дифторметил)-3-метил-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амина TFA соли пример 4 (362 мг, 43,8%) в виде белого твердого вещества и 4-(1-(дифторметил)-5-метил-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амина TFA соли пример 16 (150 мг, 18,1%) в виде белого твердого вещества. Данные для примера 4 и примера 16 приведены в таблице 3.
Путь C
Типичная процедура получения пиримидинов на примере получения примера 5, (R)-4-(5-хлор-1-метил-1H-пиразол-4-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амин
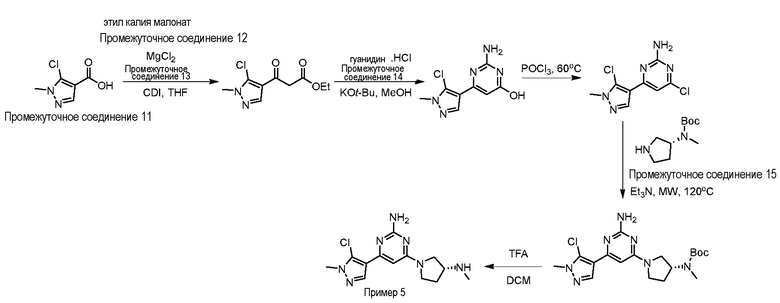
5-Хлор-1-метил-1H-пиразол-4-карбоновую кислоту промежуточное соединение 11 (1,0 г, 6,25 ммоль) растворяли в THF (50 мл) и охлаждали до 0°C. Добавляли карбонилдиимидазол (1,51 г, 9,37 моль) при интенсивном перемешивании и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали до 0°C, добавляли этил калия малонат промежуточное соединение 12 (1,59 г, 9,37 моль) и хлорид магния промежуточное соединение 13 (0,89 г, 9,37 моль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Растворитель выпаривали при пониженном давлении, реакционную смесь разбавляли H2O (50 мл), подкисляли добавлением 1М раствора HCl (10 мл), экстрагировали EtOAc (3×100 мл), объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4 и концентрировали с получением этил 3-(5-хлор-1-метил-1H-пиразол-4-ил)-3-оксопропаноата (1,01 г, 70%) в виде не совсем белого твердого вещества.
LCMS (система 2, способ 2): m/z 231,1 (M+H)+ (ES+), at 2,63 мин, 254 нм.
Этил 3-(5-хлор-1-метил-1H-пиразол-4-ил)-3-оксопропаноат (0,9 г, 3,9 ммоль) растворяли в MeOH (15,0 мл) при 0oC, добавляли трет-бутоксид калия (1,31 г, 11,7 ммоль) и гуанидин гидрохлорид промежуточное соединение 14 (0,743 г, 7,82 ммоль) и реакционную смесь нагревали при 70°C в течение 16 ч. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении с получением желтого твердого вещества, которое суспендировали в воде (50 мл), подкисляли добавлением 1М раствора HCl (10 мл), экстрагировали DCM (3×50 мл), объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении с получением 2-амино-6-(5-хлор-1-метил-1H-пиразол-4-ил)пиримидин-4-ола (0,8 г, 91%) в виде не совсем белого твердого вещества.
LCMS (система 2, способ 2): m/z 226,1 (M+H)+ (ES+), около 1,81 мин, 230 нм.
В сосуд для микроволновой обработки, содержащий 2-амино-6-(5-хлор-1-метил-1H-пиразол-4-ил)пиримидин-4-ол (0,8 г, 3,5 ммоль) добавляли оксихлорид фосфора (3,0 мл) при 0°C и полученный раствор нагревали при 70°C в течение 16 ч. Реакционную смесь охлаждали до комнатной температуры, выливали в ледяную воду (20 мл), водный слой нейтрализовали добавлением твердого NaHCO3 и экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (100 мл), сушили над Na2SO4 и концентрировали с получением 4-хлор-6-(5-хлор-1-метил-1H-пиразол-4-ил)пиримидин-2-амина (0,38 г, 44%) в виде белого твердого вещества.
LCMS (система 2, способ 1): m/z 244,1 (M+H)+ (ES+), около 2,54 мин, 240 нм.
4-Хлор-6-(5-(трифторметил)-1H-пиразол-3-ил)пиримидин-2-амин (0,18 г, 0,74 ммоль) и трет-бутил (R)-метил(пирролидин-3-ил)карбамат промежуточное соединение 15 (0,296 г. 1,48 моль) растворяли в Et3N (5,0 мл) в 35 мл сосуде для микроволновой обработки и полученную смесь нагревали при 120°C в течение 16 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры, добавляли DCM (100 мл), органический слой промывали H2O (50 мл) и насыщенным солевым раствором (50 мл) и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель 60-120, 0-5% MeOH в DCM) с получением трет-бутил (R)-(1-(2-амино-6-(5-хлор-1-метил-1H-пиразол-4-ил)пиримидин-4-ил)пирролидин-3-ил)(метил)карбамата (0,230 г, 76%) в виде коричневой липкой смолы.
LCMS (система 2, способ 1): m/z 408,2 (M+H)+ (ES+), около 3,19 мин, 254 нм.
трет-Бутил (R)-(1-(2-амино-6-(5-хлор-1-метил-1H-пиразол-4-ил) пиримидин-4-ил)пирролидин-3-ил)(метил)карбамат (0,23 г, 0,57 ммоль) растворяли в DCM (1,0 мл), добавляли TFA (1,0 мл) при 0oC и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали при пониженном давлении и остаток очищали по способу очистки E с получением (R)-4-(5-хлор-1-метил-1H-пиразол-4-ил)-6-(3-(метиламино)пирролидин-1-ил)пиримидин-2-амина пример 5 (35 мг, 20%) в виде белого твердого вещества. Данные для примера 5 приведены в таблице 3.
Путь D
Типичная процедура получения пиримидинов на примере получения примера 19, 4-(3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амин
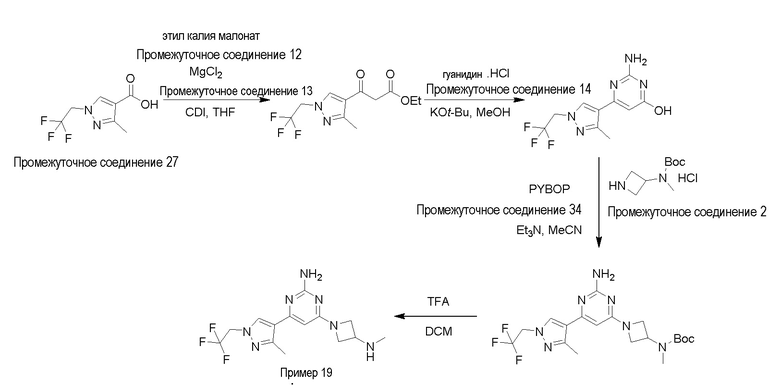
3-Метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-карбоновую кислоту промежуточное соединение 27 (1,0 г, 4,8 ммоль) растворяли в сухом THF (100 мл) в атмосфере газа N2 и раствор охлаждали до 0°C. Добавляли карбонилдиимидазол (1,6 г, 9,6 ммоль) и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали до 0°C, добавляли этил калия малонат промежуточное соединение 12 (1,63 г, 9,6 ммоль) и хлорид магния промежуточное соединение 13 (0,9 г, 9,6 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 16 ч. По прошествии этого времени растворитель выпаривали, остаток разбавляли H2O (50 мл), водный слой подкисляли добавлением 1 н. раствора HCl (20 мл) и экстрагировали EtOAc (3×100 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель; 60-120 меш; 0-40% EtOAc в гексане) с получением этил 3-(3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-3-оксопропаноата (0,52 г, 40%) в виде не совсем белого твердого вещества.
LCMS (система 2, способ 2): m/z 277,0 (M+H)+ (ES+), около 2,84 мин, 244 нм
Этил 3-(3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-3-оксопропаноат (0,5 г, 1,79 моль) растворяли в MeOH (15 мл) при 0°C, добавляли трет-бутоксид калия (0,6 г, 0,00539 моль) и гуанидин гидрохлорид промежуточное соединение 14 (0,345 г, 3,59 ммоль) и реакционную смесь нагревали при 70°C в течение 16. По прошествии этого времени растворитель выпаривали с получением желтого твердого вещества, которое суспендировали в воде (50 мл), подкисляли добавлением 1М раствора HCl (10 мл), экстрагировали DCM (3×50 мл), объединенные органические слои сушили над Na2SO4 и концентрировали при пониженном давлении с получением 2-амино-6-(3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)пиримидин-4-ола (0,38 г, 77%) в виде не совсем белого твердого вещества.
LCMS (система 2, способ 2): m/z 274,2 (M+H)+ (ES+), около 1,85 мин, 240 нм
2-Амино-6-(3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)пиримидин-4-ол (0,38 г, 1,39 ммоль) и трет-бутил азетидин-3-ил(метил)карбамат промежуточное соединение 2 (0,463 г. 2,08 ммоль) растворяли в Et3N (5,0 мл) и MeCN (8,0 мл) при 0°C. Добавляли PYBOP промежуточное соединение 34 (1,08 г, 2,08 ммоль) при 0oC и реакционную смесь нагревали при 80°C в течение 16 ч. По прошествии этого времени реакционную смесь охлаждали до комнатной температуры, добавляли DCM (100 мл), органический слой промывали H2O (50 мл) и насыщенным солевым раствором (50 мл) и растворители концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель 60-120 меш, 0-5% MeOH в DCM) с получением трет-бутил (1-(2-амино-6-(3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамата (0,410 г, 66%) в виде коричневой липкой смолы.
LCMS (система 2, способ 1): m/z 442,3 (M+H)+ (ES+), около 3,03 мин, 214 нм
трет-Бутил (1-(2-амино-6-(3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамат (0,41 г, 0,929 ммоль) растворяли в DCM (2,0 мл) при 0°C, добавляли TFA (2,0 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали при пониженном давлении и остаток очищали по способу очистки R с получением 4-(3-метил-1-(2,2,2-трифторэтил)-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амина пример 19 (27 мг, 33%) в виде белого твердого вещества. Данные для примера 19 приведены в таблице 3.
Путь E
Типичная процедура получения пиримидинов на примере получения примера 36, 4-(3-(метиламино)азетидин-1-ил)-6-(1H-пиразол-3-ил)пиримидин-2-амин
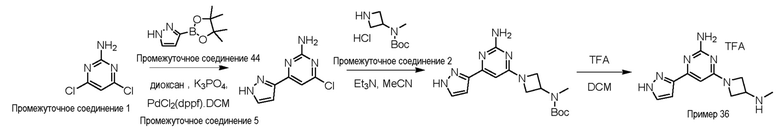
4,6-Дихлорпиримидин-2-амин промежуточное соединение 1 (1 г, 12,0 ммоль), 3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол промежуточное соединение 44 (1,40 г, 7,20 ммоль) и K3PO4 (3,88 г, 18,9 ммоль) растворяли в 1,4-диоксане (20 мл) и воде (4 мл) в атмосфере азота и дегазировали в течение 20 мин. Добавляли Pd(dppf)Cl2.DCM промежуточное соединение 5 (497 мг, 0,60 ммоль) и реакционную смесь перемешивали при 80°C в течение 16 ч. После охлаждения, реакционную смесь разбавляли водой (50 мл), экстрагировали этилацетатом (3×50 мл), объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали с получением неочищенного остатка, который очищали колоночной хроматографией (силикагель; 60-120 меш, 0-28% EtOAc в гексане) с получением 4-хлор-6-(1H-пиразол-3-ил)пиримидин-2-амина (230 мг, 19,5%) в виде не совсем белого твердого вещества.
LCMS (система 1, способ 1): m/z 196 (M+H)+ (ES+), около 2,62 мин, 254 нм
4-Хлор-6-(1H-пиразол-3-ил)пиримидин-2-амин (210 мг, 1,0 ммоль) и трет-бутил азетидин-3-ил(метил)карбамат гидрохлорид промежуточное соединение 2 (401 мг, 2,1 ммоль) растворяли в MeCN (10 мл), добавляли Et3N (4 мл) и реакционную смесь перемешивали при 90°C в течение 6 ч. Реакционную смесь охлаждали, разбавляли водой (30 мл), экстрагировали этилацетатом (3×30 мл), объединенные органические слои сушили (Na2SO4), фильтровали и концентрировали с получением неочищенного трет-бутил (1-(2-амино-6-(1H-пиразол-3-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамата (предполагается 100%) в виде не совсем белого твердого вещества, который использовали в неочищенном виде без очистки.
LCMS (система 1, способ 1): m/z 346 (M+H)+ (ES+), около 2,98 мин, 240 нм
трет-Бутил (1-(2-амино-6-(1H-пиразол-3-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамат (570 мг, 1,6 ммоль) растворяли в DCM (3 мл) при 0°C, TFA (1,5 мл) добавляли по каплям и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Растворитель концентрировали и остаток подвергали азеотропной перегонке с толуолом (3×3 мл) с получением неочищенного продукта, который очищали способом очистки AI с получением 4-(3-(метиламино)азетидин-1-ил)-6-(1H-пиразол-3-ил)пиримидин-2-амина пример 36 (151 мг, 37,3%) в виде белого твердого вещества. Данные для примера 36 приведены в таблице 3.
Путь F
Процедура получения примера 20, 4-(3-метил-1-(трифторметил)-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амин, и примера 23, 4-(5-метил-1-(трифторметил)-1H-пиразол-4-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амин
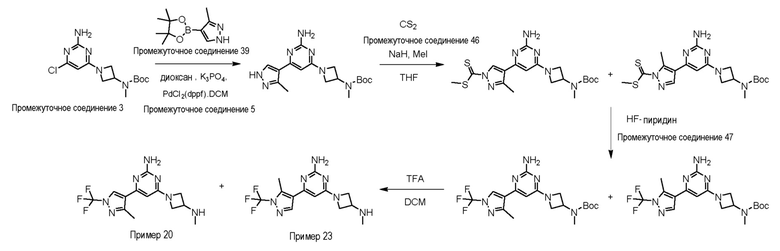
Путь G
Процедура получения примера 32, 4-(3-(метиламино)азетидин-1-ил)-6-(1-(трифторметил)-1H-пиразол-4-ил)пиридин-2-амин, и примера 33, (R)-4-(3-(метиламино)пирролидин-1-ил)-6-(1-(трифторметил)-1H-пиразол-4-ил)пиридин-2-амин
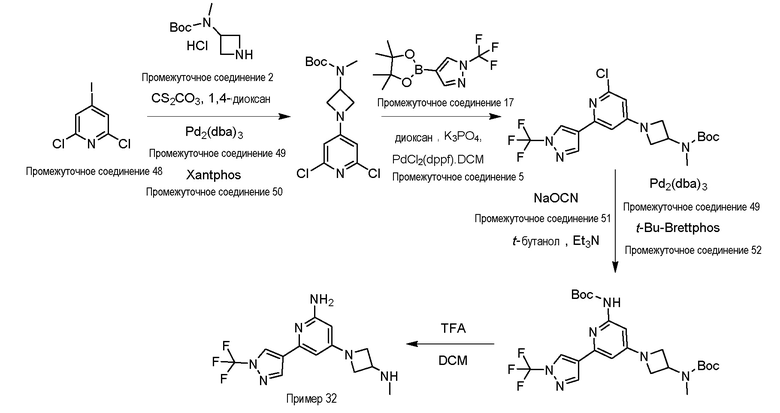
Для синтеза примера 33 замещают промежуточное соединение 2 промежуточным соединением 17.
Путь H
Процедура получения примера 40, 4-(5-этил-4-метил-1H-пиразол-3-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амин
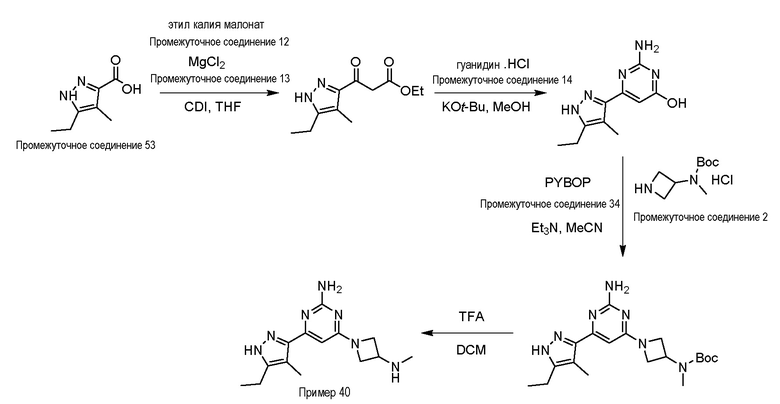
5-Этил-4-метил-1H-пиразол-3-карбоновую кислоту промежуточное соединение 53 (0,75 г, 4,87 ммоль) растворяли в сухом THF (100 мл) и раствор охлаждали до 0°C. Добавляли карбонилдиимидазол (1,57 г, 9,74 ммоль) при интенсивном перемешивании и смесь перемешивали при комнатной температуре в течение 1 ч. Реакционную смесь охлаждали до 0°C, затем добавляли этил калия малонат промежуточное соединение 12 (1,62 г, 9,74 ммоль) и хлорид магния промежуточное соединение 13 (0,935 г, 9,74 моль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции растворитель выпаривали при пониженном давлении, реакционную смесь разбавляли H2O (50 мл), водный слой подкисляли добавлением 1 н. раствора HCl (30 мл), экстрагировали EtOAc (3×100 мл) и объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель; 60-120 меш; 0-40% EtOAc/гексан) с получением этил 3-(5-этил-4-метил-1H-пиразол-3-ил)-3-оксопропаноата (0,71 г, 65%) в виде не совсем белого твердого вещества.
LCMS (система 1, способ 2): m/z 225,3 (M+H)+ (ES+), около 2,80 мин, 240 нм
Этил 3-(5-этил-4-метил-1H-пиразол-3-ил)-3-оксопропаноат (0,7 г, 3,12 ммоль) растворяли в MeOH (15 мл), добавляли трет-бутоксид калия (1,05 г, 9,37 ммоль) и гуанидин гидрохлорид промежуточное соединение 14 (0,6 г, 6,25 ммоль) при 0°C, реакционную смесь нагревали до комнатной температуры и затем нагревали при 70°С в течение ночи. После завершения реакции реакционную смесь охлаждали до комнатной температуры, растворитель выпаривали при пониженном давлении с получением желтого твердого вещества, которое подкисляли добавлением по каплям 1 н. HCl (5 мл) и водный слой экстрагировали EtOAc (3×50 мл). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4 и концентрировали при пониженном давлении с получением 2-амино-6-(5-этил-4-метил-1H-пиразол-3-ил)пиримидин-4-ола (0,68 г, 99%) в виде не совсем белого твердого вещества.
LCMS (система 1, способ 2): m/z 220,3 (M+H)+ (ES+), около 1,81 мин, 202 нм
POCl3 (2,0 мл) добавляли к 2-амино-6-(5-этил-4-метил-1H-пиразол-3-ил)пиримидин-4-олу (0,68 г, 3,1 ммоль) при 0°C и реакционную смесь нагревали при 70°C в течение ночи. После завершения реакции, реакционную смесь выливали на баню со льдом, нейтрализовали добавлением твердого NaHCO3, водный слой экстрагировали EtOAc (3×50 мл) и объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над Na2SO4, фильтровали и концентрировали с получением неочищенного остатка, который очищали колоночной хроматографией (силикагель; 60-120 меш; 0-40% EtOAc/гексан) с получением 4-хлор-6-(5-этил-4-метил-1H-пиразол-3-ил)пиримидин-2-амина (401 мг, 54%) в виде желтого твердого вещества.
LCMS (система 2, способ 2): m/z 238,3 (M+H)+ (ES+), около 2,75 мин, 214 нм
4-Хлор-6-(5-этил-4-метил-1H-пиразол-3-ил)пиримидин-2-амин (189 мг, 7,97 ммоль) и трет-бутил азетидин-3-ил(метил)карбамат промежуточное соединение 2 (0,354 г. 1,59 ммоль) растворяли в триэтиламине (5,0 мл) и реакционную смесь нагревали при 120°C в течение ночи. После завершения реакции, реакционную смесь охлаждали до комнатной температуры, добавляли DCM (100 мл), органический слой промывали H2O (50 мл), насыщенным солевым раствором (50 мл), и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией (силикагель 60-120 меш, 0-5% MeOH: DCM) с получением трет-бутил (1-(2-амино-6-(5-этил-4-метил-1H-пиразол-3-ил)пиримидин-4-ил)азетидин-3-ил)(метил)карбамата (230 мг, 74%) в виде коричневой липкой смолы.
LCMS (система 2, способ 1): m/z 273,3 (M+H)+ (ES+), около 3,03 мин, 202 нм
трет-Бутил (1-(2-амино-6-(5-этил-4-метил-1H-пиразол-3-ил) пиримидин-4-ил)азетидин-3-ил)(метил)карбамат (230 мг, 5,94 ммоль) растворяли в DCM (2,0 мл), добавляли TFA (2,0 мл) при 0°C и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали при пониженном давлении и полученный неочищенный продукт очищали препаративной ВЭЖХ с получением дитрифторацетатной соли 4-(5-этил-4-метил-1H-пиразол-3-ил)-6-(3-(метиламино)азетидин-1-ил)пиримидин-2-амина пример 40 (33 мг, 19%) в виде белого твердого вещества. Данные для примера 40 приведены в таблице 3.
Таблица 2 - Промежуточные соединения
CAS: 56-05-3
CAS: 943060-59-1
CAS: 1206640-82-5
CAS: 95464-05-4
CAS: 1049730-42-8
CAS: 847818-70-6
CAS: 936250-20-3
CAS: 1895-39-2
CAS: 17455-13-9
CAS: 54367-66-7
CAS: 6148-64-7
CAS: 7786-30-3
CAS: 50-01-1
CAS: 392338-15-7
CAS: 217806-26-3
CAS: 1046831-98-4
CAS: 929716-69-8
CAS: 2223043-80-7
CAS: 1049730-40-6
CAS: 1049730-39-3
CAS: 2068065-34-7
CAS: 288251-53-6
CAS: 2019997-43-2
CAS: 1623156-88-6
CAS: 1046832-21-6
CAS: 113100-55-3
CAS: 887408-72-2
CAS: 1007541-94-7
CAS: 847818-79-5
CAS: 26308-42-9
CAS: 173841-02-6
CAS: 344591-91-9
CAS: 128625-52-5
CAS: 134589-53-0
CAS: 137343-52-3
CAS: 54367-67-8
CAS: 1218790-40-9
CAS: 936250-20-3
CAS: 115964-19-7
CAS: 1399653-86-1
CAS: 269410-08-4
CAS: 402-61-9
CAS: 844501-71-9
CAS: 4027-59-2
CAS: 75-15-0
CAS: 62778-11-4
CAS: 98027-84-0
CAS: 51364-51-3
CAS: 161265-03-8
CAS: 917-61-3
CAS: 1160861-53-9
CAS: 957129-38-3
Таблица 3 - Примеры соединений
No.
система и способ
3, 4 и 5
Способ A
Способ 1
3, 6 и 5
Способ B
Способ 2
3, 7 и 5
Способ C
Способ 2
8, 9, 10, 3 и 5
Способ D
Способ 2
11, 12, 13, 14 и 15
Способ E
Способ 1
1, 16, 17 и 5
Способ F
Способ 2
1, 18, 17 и 5
Способ G
Способ 2
3, 19 и 5
Способ H
Способ 1
3, 20 и 5
Способ I
Способ 2
3, 21 и 5
Способ J
Способ 1
3, 22 и 5
Способ K
Способ 2
23, 12, 13, 14 и 2
Способ L
Способ 1
3, 24 и 5
Способ M
Способ 2
1, 15, 24 и 5
Способ N
Способ 2
11, 12, 13, 14 и 2
Способ O
Способ 1
8, 9, 10, 3 и 5
Способ D
Способ 2
25, 12, 13, 14 и 2
Способ P
Способ 1
3, 26 и 5
Способ Q
Способ 1
27, 12, 13, 14, 2 и 34
Способ R
Способ 1
28, 12, 13, 14 и 2
Способ S
Способ 2
29, 12, 13, 14 и 2
Способ T
Способ 2
3, 30 и 5
Способ U
Способ 2
31, 12, 13, 14 и 2
Способ V
Способ 2
32, 12, 13, 14 и 2
Способ W
Способ 1
3, 33 и 5
Способ X
Способ 2
35, 12, 13, 14, 2 и 34
Способ Y
Способ 1
36, 12, 13, 14 и 2
Способ Z
Способ 1
37, 12, 13, 14 и 2
Способ AA
Способ 1
41, 12, 13, 14, 2 и 34
Способ AF
Способ 2
36, 12, 13, 14 и 15
Способ AB
Способ 1
43, 12, 13, 14, 2 и 34
Способ AH
Способ 2
1, 44, 5, и 2
Способ AI
Способ 2
3, 38 и 5
Способ AC
Способ 1
3, 42 и 5
Способ AG
Способ 2
3, 39 и 5
Способ AD
Способ 2
53, 12, 13, 14, 2 и 34
Способ AK
Способ 1
45, 12, 13, 14, 2 и 34
Способ AJ
Способ 2
40, 12, 13, 14 и 2
Способ AE
Способ 1
Биологическая активность
ПРИМЕР А
Анализ функционального cAMP Gi антагониста H4
Клетки HEKf инфицировали в течение ночи бакуловирусом, экспрессирующим человеческий рецептор H4, затем центрифугировали при 1200 об/мин в течение 5 мин, замораживали в среде для замораживания клеток (Sigma) и хранили при -150°C. В день анализа клетки размораживали и ресуспендировали в HBSS с 500 нМ IBMX до достижения плотности 1500 клеток/лунку. Лиганды H4 получали в DMSO и вносили с помощью акустического дозатора LabCyte ECHO по 25 нл в планшеты малого объема. Клетки по 10 мкл/лунку высевали в присутствии 1 мкМ форсколина, центрифугировали при 1200 об/мин в течение 1 мин и инкубировали в течение 30 мин перед добавлением реагентов для обнаружения Cisbio cAMP до общего объема 20 мкл/лунку. Для анализа антагонистов клетки предварительно инкубировали с лигандами-антагонистами H4 в течение 30 минут, затем добавляли гистамин в концентрации EC80 и инкубировали еще 30 минут. После добавления реагента для детекции и встряхивания при комнатной температуре в течение 60 мин, измеряли накопление cAMP с использованием HTRF на планшет-ридере PheraStar. Значения EC50 были получены с использованием 4-параметрического логистического эмпирического уравнения для количественной оценки активности агонистов. Значения функциональной аффинности антагониста получали с использованием уравнения Ченга-Прусоффа для расчета значения pKb с использованием данных анализа антагониста.
Анализ функционального динамического перераспределения массы антагониста H4
Клетки HEKf инфицировали бакуловирусом, экспрессирующим человеческий рецептор H4, высевали на покрытые фибронектином планшеты EPIC с плотностью 10000 клеток/лунку и инкубировали в течение ночи при 37°C. Среду на клетках заменяли на 30 мкл HBSS с 20 мМ HEPES на лунку и добавляли 30 нл DMSO на лунку с помощью акустического дозатора LabCyte ECHO. После 2 ч уравновешивания при комнатной температуре 30 нл лигандов H4, полученных в DMSO, вносили с помощью акустического дозатора LabCyte ECHO в планшеты EPIC с посевом и отслеживали динамическое перераспределение клеточной массы с помощью планшет-ридера Corning EPIC. После 45 мин измерения добавляли 30 нл/лунку гистамина EC80 и контролировали для получения данных анализа антагониста. Для построения кривых концентрация-эффект использовали максимальные скорректированные по базовой линии ответы в pm. Значения EC50 были получены с использованием 4-параметрического логистического эмпирического уравнения для количественной оценки активности агонистов. Значения функциональной аффинности антагониста получали с использованием уравнения Ченга-Прусоффа для расчета значения pKb с использованием данных анализа антагониста.
Анализ hERG
Данные анализа hERG были определены компанией Metrion Biosciences, Cambridge, UK, с использованием экспериментальных протоколов, подробно описанных ниже:
Линия клеток яичника китайского хомячка (CHO), стабильно экспрессирующая ген, родственный ether-a-go-go человека, выращивали и пассировали в стандартных условиях культивирования. Клетки получали для анализов с использованием протоколов диссоциации, разработанных для оптимизации нормального состояния клеток, выхода и запечатывания и качества анализа. Образцы для исследований были предоставлены в виде 10 мМ исходных растворов в 100% DMSO. Все манипуляции с образцами и серийные разведения проводили с использованием стеклянных контейнеров и эмалированных планшетов. Максимальную рабочую концентрацию 30 мкМ получали из исходного раствора образца 10 мМ с использованием 1:333-кратного разбавления во внешнем регистрирующем растворе (0,3% DMSO об./об.). В анализе с одной концентрацией исследуемые образцы подвергали скринингу при 30 мкМ против как минимум трех отдельных клеток. В анализе pIC50 исследуемые образцы подвергали скринингу при 1, 3, 10 и 30 мкМ против как минимум трех отдельных клеток. Каждая четырехточечная кривая концентрация-эффект была построена с использованием кумулятивных двойных добавок каждой концентрации в одну и ту же клетку.
Все эксперименты проводили на автоматизированной платформе для патч-клампа QPatch. Композиция внешнего и внутреннего растворов для записи экспериментов QPatch показан в таблице A ниже. Все растворы фильтровали (0,2 мкм) перед каждым экспериментом.
Таблица A: Композиция растворов для наружного и внутреннего применения (в мМ), использованных в исследовании hERG.
(мM)
(мM)
Все записи были сделаны в стандартной конфигурации целой клетки и выполнены при температуре окружающей среды (~21°C) с использованием стандартных чипов с одним каналом (Rchip 1,5-4 МОм MΩ). Последовательное сопротивление (4-15 МОм) было компенсировано > 80%. Токи вызывали от исходного потенциала -90 мВ с использованием стандартного промышленного протокола напряжения «+40/-40», как показано на Фигуре 1; это применялось при частоте стимула 0,1 Гц.
Протокол напряжения QPatch, используемый для анализа hERG.
При достижении конфигурации целой клетки носитель (0,3% DMSO об./об. во внешнем регистрирующем растворе) наносили на каждую клетку двумя болюсными добавлениями с двухминутным периодом записи между каждым добавлением для обеспечения стабильных записей. После периода введения носителя либо:
i) Для анализа с одной концентрацией - применяли одну концентрацию исследуемого образца в концентрации 30 мкМ в виде пяти болюсных добавок на каждую исследуемую концентрацию с двухминутными интервалами; или
ii) Для анализа pIC50 - применяли четыре концентрации исследуемого образца от 1 мкМ до 30 мкМ в виде двух болюсных добавок на каждую исследуемую концентрацию с двухминутными интервалами;
и затем в течение четырехминутного периода записи измеряли эффект на амплитуду следового тока hERG. Для каждого диапазона протокола напряжения мембранный ток и пассивные свойства отдельных клеток регистрировали с помощью программного обеспечения для анализа QPatch. Пиковая амплитуда выходного следового тока, вызванная во время тестового импульса до -40 мВ, была измерена относительно мгновенного тока утечки, измеренного во время начального предварительного импульса до -40 мВ. Для целей контроля качества минимальная амплитуда тока для анализа составляет > 200 пА пикового исходящего тока, измеренного в конце периода использования носителя. Программное обеспечение для анализа QPatch вычисляет средний пиковый ток для последних трех разверток в конце каждого периода применения концентрации, и данные экспортируются в Excel и обрабатываются с помощью пакета биоинформатики, разработанного в Pipeline Pilot (Biovia, USA). Матрица рассчитывает процент ингибирование для каждого периода применения исследуемой концентрации как снижение среднего пикового тока или заряда по сравнению со значением, измеренным в конце периода контроля (т.е. носителя). Значения процента ингибирования для каждой клетки используются для построения кривых концентрация-реакция с использованием четырехпараметрической логистической аппроксимации с уровнями ингибирования 0 и 100%, зафиксированными при очень низких и очень высоких концентрациях, соответственно, и свободным угловым коэффициентом Хилла. Затем определяют IC50 (50% ингибирующая концентрация) и коэффициент Хилла, но включают только данные для клеток с угловыми коэффициентами Хилла в пределах 0,5>nH<2,0. Приведенные ниже данные IC50 представляют собой среднее значение по меньшей мере для трех отдельных клеток (N≥3). Обычно исследуемый образец, который не смог достичь >40% блокировки при максимальной концентрации, даст неоднозначное значение IC50 из-за плохой или неограниченной подгонки. В этом случае возвращается произвольное значение IC50, которое на 0,5 log единицы выше самой высокой испытанной концентрации. Например, если образец не демонстрирует среднее ингибирование блока > 40% при максимальной концентрации 30 мкМ, то сообщается значение IC50, равное 100 мкМ, т.е. pIC50 ≤ 4,0.
pIC50
2 Jennifer D. Venable et al, J. Med. Chem., 48, (2005), 8289-8298.
3 Brad M. Savall et al, J. Med. Chem., 57, (2014), 2429−2439.
4 Robin L Thurmond et al, Ann Pharmacol Pharm., 2, (2017), 1-11.
5 Charles E. Mowbray et al, Bioorg. Med. Chem. Lett., 21, (2011), 6596-6602.
6 Rogier A. Smits et al, Bioorg. Med. Chem. Lett., 23, (2013), 2663-2670.
7 Chan-Hee Park et al, J. Med. Chem., 61, (2018), 2949−2961.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОКСАДИАЗОЛЫ В КАЧЕСТВЕ АГОНИСТОВ МУСКАРИНОВОГО РЕЦЕПТОРА M1 И/ИЛИ M4 | 2019 |
|
RU2817018C2 |
| 6, 6-БИЦИКЛИЧЕСКИЕ КОЛЬЦЕВЫЕ ЗАМЕЩЕННЫЕ ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗ | 2005 |
|
RU2379308C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| Бициклические азотсодержащие соединения как агонисты М1 мускариновых рецепторов | 2019 |
|
RU2811601C1 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2018 |
|
RU2767857C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
| ПРОИЗВОДНЫЕ 1,2,4-ТРИАЗИН-3-АМИНА | 2011 |
|
RU2771819C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ ДЕЛЬТА ЧЕЛОВЕКА | 2013 |
|
RU2661896C2 |
| ГЕТЕРОАРИЛ-БИФЕНИЛ АМИНЫ ДЛЯ ЛЕЧЕНИЯ PD-L1 ЗАБОЛЕВАНИЙ | 2020 |
|
RU2837843C1 |
| 3,5-ДИЗАМЕЩЕННОЕ АЛКИНИЛБЕНЗОЛЬНОЕ СОЕДИНЕНИЕ И ЕГО СОЛЬ | 2013 |
|
RU2576384C1 |
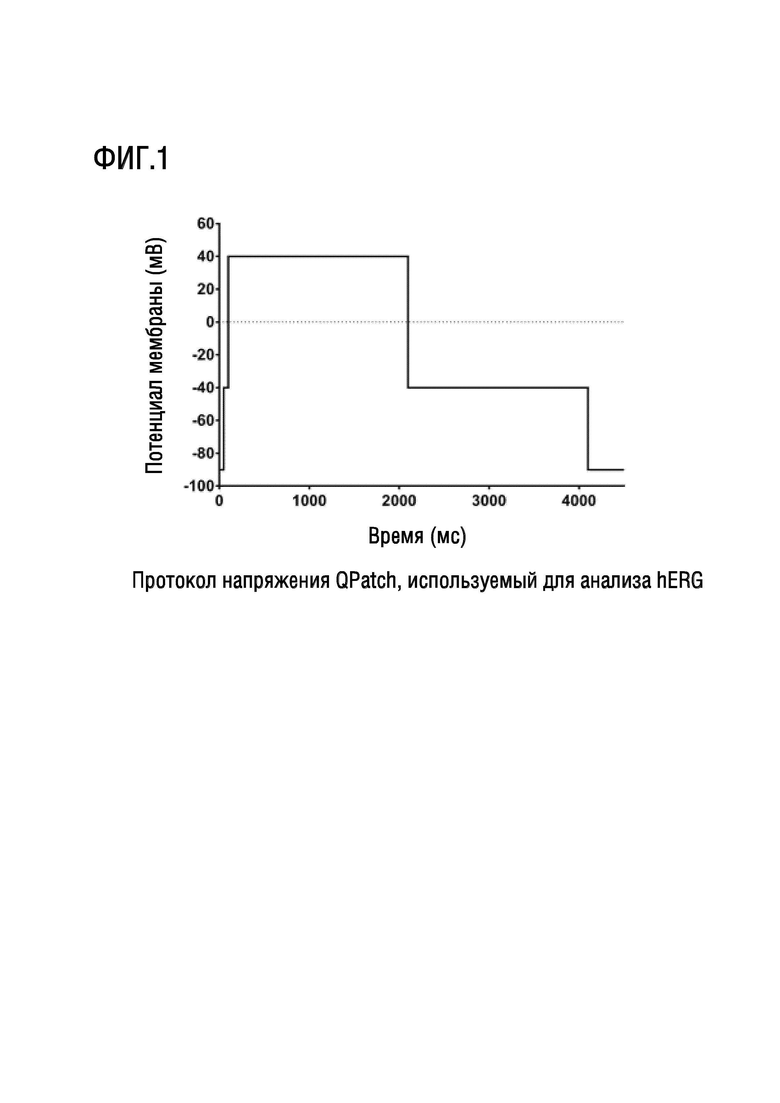
Изобретение относится к новым соединениям, обладающим активностью антагонистов гистаминового рецептора Н4. Предложены соединение, выбранное из группы соединений, указанных ниже, или его фармацевтически приемлемая соль, а также фармацевтическая композиция, содержащая такое соединение. Предложенные соединения сочетают антагонизм в отношении гистаминовых Н4-рецепторов с низкой активностью hERG и могут применяться для лечения воспалительных заболеваний, связанных с H4-рецепторами. 2 н. и 7 з.п. ф-лы, 1 ил., 4 табл., 43 пр.
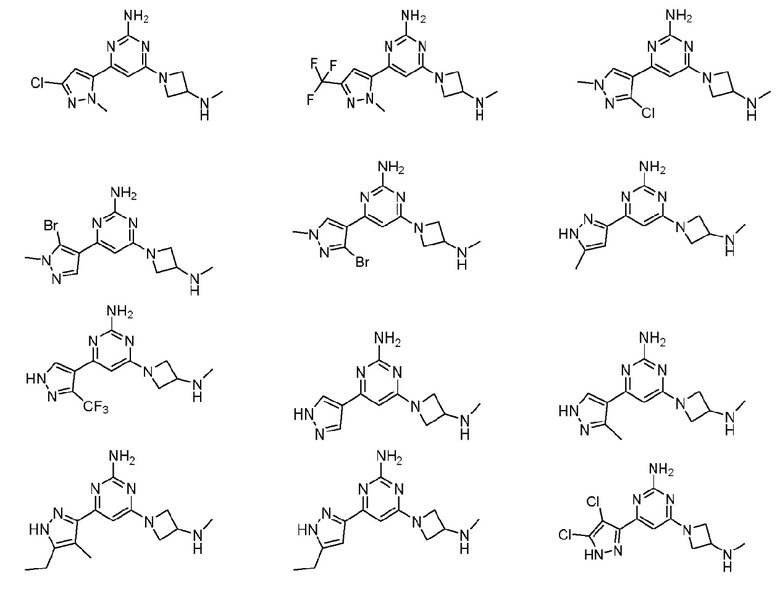
1. Соединение, выбранное из группы, состоящей из:
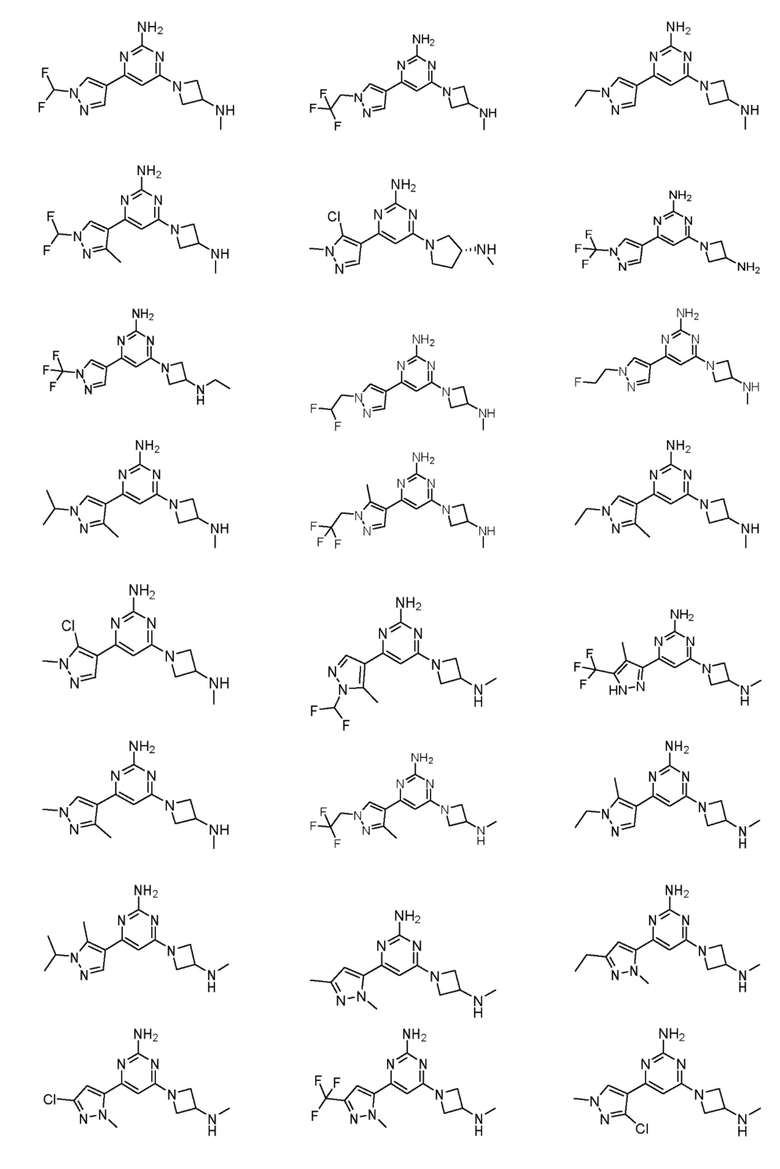
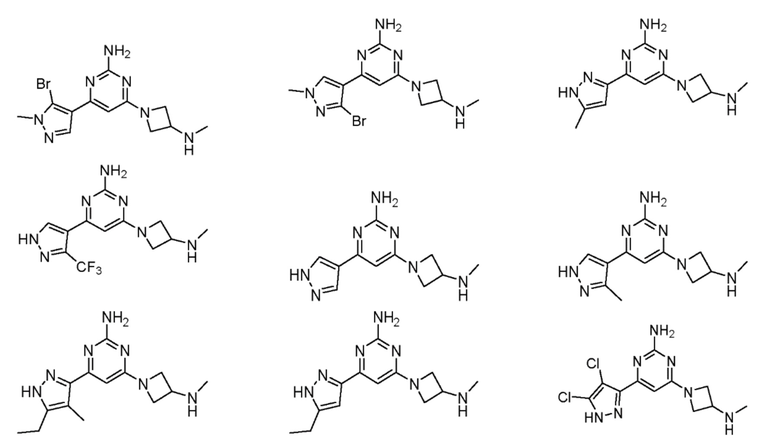
или его фармацевтически приемлемая соль.
2. Соединение по п.1, выбранное из группы, состоящей из:
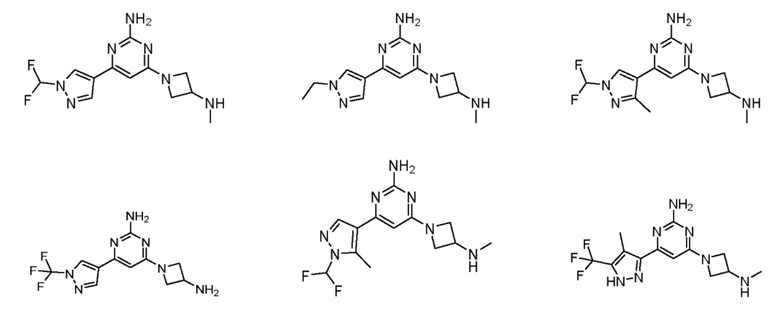
или его фармацевтически приемлемая соль.
3. Фармацевтическая композиция, обладающая активностью рецептора Н4, содержащая соединение или его фармацевтически приемлемую соль по любому из пп. 1-2 и фармацевтически приемлемый эксципиент.
4. Соединение по любому из пп. 1-2 или его фармацевтически приемлемая соль для применения в медицине в качестве лекарственного средства, обладающего активностью рецептора Н4.
5. Фармацевтическая композиция по п. 3 для применения в медицине в качестве лекарственного средства, обладающего активностью рецептора Н4.
6. Соединение по п.1 или его фармацевтически приемлемая соль, обладающее активностью рецептора Н4.
7. Соединение по п. 6 или его фармацевтически приемлемая соль, проявляющее низкую активность в отношении hERG.
8. Соединение или его фармацевтически приемлемая соль по любому одному из пп. 1-2 для применения при лечении воспалительных заболеваний, в которые вовлечены рецепторы Н4, включая астму, хронический зуд, дерматит, ревматоидный артрит, ульцерогенез желудка и колит.
9. Фармацевтическая композиция по п. 3 для применения при лечении воспалительных заболеваний, в которые вовлечены рецепторы Н4, включая астму, хронический зуд, дерматит, ревматоидный артрит, ульцерогенез желудка и колит.
| WO 2005054239 A1, 16.06.2005 | |||
| WO 2007072163 A2, 28.06.2007 | |||
| WO 2009077608 A1, 25.06.2009 | |||
| WO 2009068512 A1, 04.06.2009 | |||
| WO 2008100565 A1, 21.08.2008 | |||
| WO 2008031556 A2, 20.03.2008 | |||
| ПРОИЗВОДНЫЕ 4-АМИНОПИРИМИДИНА | 2008 |
|
RU2489430C2 |
| ПРОИЗВОДНЫЕ АМИНОАЛКИЛПИРИМИДИНА В КАЧЕСТВЕ АНТАГОНИСТОВ H РЕЦЕПТОРА ГИСТАМИНА | 2010 |
|
RU2573828C2 |

