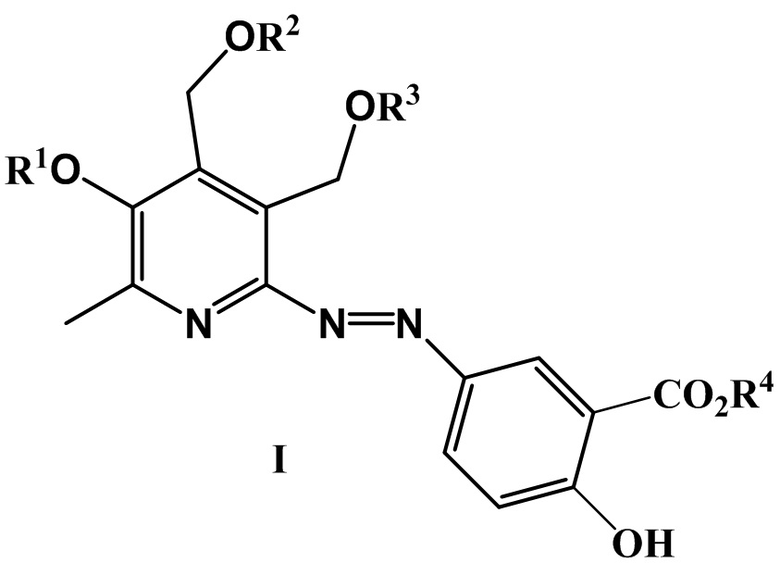
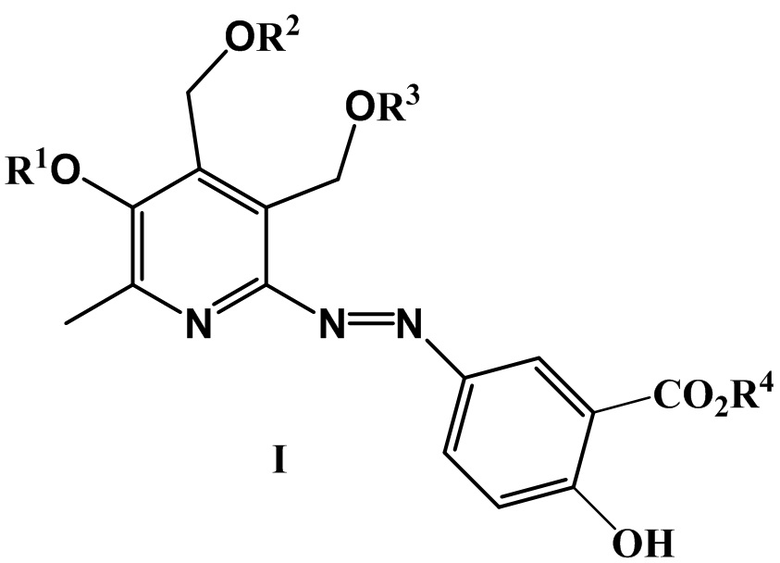
Изобретение относится к химии органических гетероциклических соединений, а именно, к новым азосоединениям на основе 5-аминосалициловой кислоты общей формулы (I), обладающим высокой антигликирующей активностью (блокируют образование конечных продуктов гликирования).
 ,
,
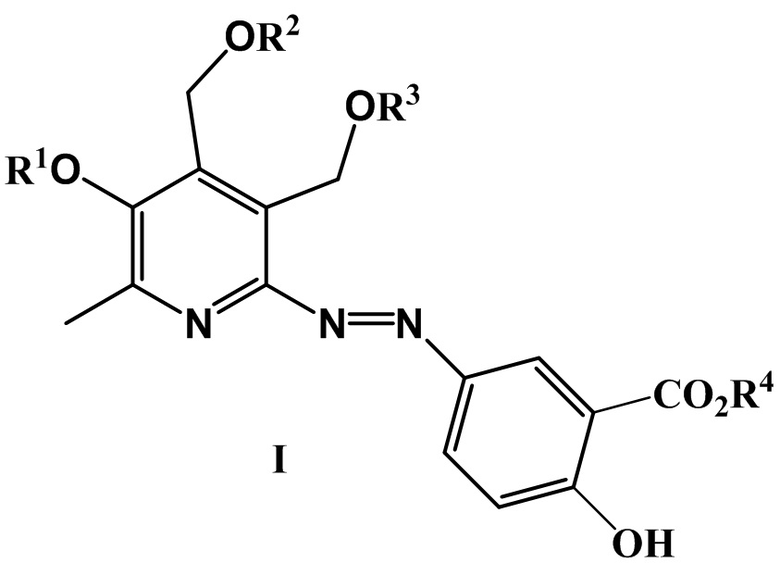
где R1 = CH3, H и другие фармацевтически приемлемые катионы, такие как Na, K, а R2 = R3 = H, или R2 и R3 вместе образуют заместитель вида СR5R6, где R5 = H, CH3 и R6 = H, CH3 и другие линейные алкильные заместители, такие как C2H5, н-C3H7, н-C7H15, н-C8H17;
R4 = Et3NH, H и другие фармацевтически приемлемые катионы органической и неорганической природы, такие как K, NH4.
Соединения формулы (I) являются эффективными ингибиторами образования конечных продуктов гликирования (далее КПГ) и могут найти широкое применение в медицине в области лечения широкого спектра социально значимых заболеваний, а именно -- осложнений сахарного диабета, атеросклероза, ревматоидного артрита, остеоартрита, нейродегенеративных заболеваний, включая болезни Альцгеймера и Паркинсона, катаракты, заболеваний, связанных со старением, и других болезней, причиной которых является образование КПГ.
КПГ - это широкий круг органических соединений, образующихся в результате взаимодействия карбонильных соединений с биологическими макромолекулами. Классический путь формирования КПГ включает в себя реакцию Майяра (взаимодействие восстанавливающих углеводов со свободными аминогруппами белков), перегруппировку получившегося основания Шиффа в продукт Амадори и последующие перегруппировки и/или фрагментацию с образованием КПГ (например, карбоксиметиллизин, глюкозепан, пентосидин и др., при этом количество выявленных КПГ постоянно растёт ). Другим важным механизмом образования КПГ является перекисное окисление сахаров, липидов и жирных кислот, приводящее к многочисленным диальдегидам (малоновый диальдегид, глиоксаль и др.). Обладающие высокой реакционной способностью дикарбонильные соединения быстро реагируют со свободными аминогруппами, приводя к формированию КПГ [Brings S., Fleming T., Freichel M., Muckenthaler M.U., Herzig S., Nawroth P.P. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention / Int. J. Mol. Sci. - 2017. - V. 18, N. 5. - P. 984].
Образование КПГ приводит к ковалентным кросс-сшивкам макромолекул и, как следствие, нарушению их структуры, что сказывается на выполнении их биологических функций. Под действием КПГ изменяется состав внеклеточного матрикса с повышенной экспрессией фибронектина, коллагена типа III, IV, VI и ламинина. Процесс гликирования также вызывает посттрансляционное сшивание эластина, снижая эластичность кожи и сосудов. Помимо непосредственного нарушения работы макромолекул накопление КПГ приводит к активации специфических рецепторов КПГ (РКПГ), что приводит к окислительному стрессу. Всё перечисленное в совокупности приводит к указанным ниже заболеваниям.
На дату подачи настоящей заявки внутри- и внеклеточное накопление КПГ считают важным фактором патогенеза таких заболеваний, как атеросклероз, сердечная недостаточность, воспаление, ревматоидный артрит и остеоартрит, нейродегенеративные заболевания, включая болезни Альцгеймера и Паркинсона. Особенно интенсивно данный процесс протекает при сахарном диабете, приводя к тяжелым последствиям сахарного диабета, таким как диабетические атеросклероз, нефро-, нейро-, ретино-, кардио-, ангиопатии, которые являются причиной высокого риска инвалидизации и смертности среди пациентов с сахарным диабетом [Munch G., Westcott B., Menini T., Gugliucci A. Advanced glycation endproducts and their pathogenic roles in neurological disorders / Amino Acids. - 2012. - V. 42. - P. - 1221-1236; Zeng C., Li Yu., Ma J., Niu L., Tay F.R. Clinical/Translational Aspects of Advanced Glycation End-Products / Trends in Endocrinology & Metabolism. - 2019. - V. 30, N. 12. - P. - 959-973].
Таким образом, применение соединений, обладающих высокой антигликирующей активностью, позволит уменьшить образование КПГ в организме, тем самым улучшая качество жизни пациентов. Все вышеперечисленное обусловливает повышенный интерес в мире к поиску ингибиторов образования конечных продуктов гликирования (КПГ), поскольку препаратов, специфически угнетающих образование КПГ и разрешенных для клинического применения, на дату представления настоящей заявки в мире не заявителем не выявлено.
Первым и наиболее изученным веществом, ингибирующим гликирование белков, является аминогуанидин (АГ) [A. Cerami, P.C. Ulrich, M. Brownlee, Pat US4758583A, опубл. 19.07.1988]. Краткой сущностью данного изобретения является то, что АГ предотвращает формирование КПГ и глюкозо-производных поперечно-сшитых молекул коллагена. Механизм антигликирующего действия аминогуанидина связывают с его способностью захватывать реактивные дикарбонильные интермедиаты. Однако клинические испытания данного лекарственного средства были остановлены из-за серьезных побочных эффектов (желудочно-кишечные расстройства, гриппоподобные симптомы, нарушение функции печени, анемию и васкулит).
Также проводились клинические испытания пиридоксамина [R. Khalifah, B.G. Hudson, Pat US6716858B1, опубл. 06.04.2004], обладающего антигликирующими свойствами. Краткой сущностью данного изобретения является то, что пиридоксамин может быть использован для лечения артрита, рака, легочного респираторного дистресс-синдрома взрослых, инсультов, панкреатита и язв кишечника, заболеваний, вызванных воздействием ионизирующей радиации или химиотерапевтических средств. Результаты второй фазы клинических исследований этого препарата показали недостаточную эффективность. На данный момент клинические исследования пиридоксамина также были остановлены.
Известны ингибиторы образования КПГ на основе азопроизводных фенилсульфокислот, содержащих пиридоксиновый фрагмент [А.А. Спасов, Ю.Г. Штырлин, К.В. Балакин, В.А. Кузнецова, В.И. Петров, А.Д. Стрельник, Патент РФ №2634594 от 01.11.2017; А.А. Спасов, Ю.Г. Штырлин, К.В. Балакин, А.У. Зиганшин, В.А. Кузнецова, В.И. Петров, А.Д. Стрельник, Патент РФ №2628605 от 21.08.2017], однако они не совпадают с заявленным соединением по химической структуре.
При этом следует отметить, что все описанные выше лекарственные препараты, по мнению заявителя, не могут рассматриваться в качестве аналогов к заявленному изобретению вследствие того, что они не совпадают с заявленным соединением по химической структуре, хотя и обладают сходной в целом антигликирующей активностью (совпадают по назначению), сопоставимой с заявленным изобретением в большей или меньшей степени.
Технической проблемой, решаемой заявленным изобретением, и его техническим результатом является разработка средств для профилактики и лечения заболеваний, вызванных образованием и накоплением в организме конечных продуктов гликирования путем получения новых соединений общей формулы (I).
Сущностью заявленного технического решения являются азопроизводные 5-аминосалициловой кислоты общей формулы (I)
,
где R1 = CH3, Н и другие фармацевтически приемлемые катионы, такие как Na, K, а R2 = R3 = H, или R2 и R3 вместе образуют заместитель вида СR5R6, где R5 = H, CH3 и R6 = H, CH3 и другие линейные алкильные заместители, такие как C2H5, н-C3H7, н-C7H15, н-C8H17; R4 = Et3NH, H и другие фармацевтически приемлемые катионы органической и неорганической природы, такие как Na, K, NH4.
Азопроизводные 5-аминосалициловой кислоты по п.1, обладающие антигликирующей активностью.
Заявленное техническое решение иллюстрируется Фиг.1 - Фиг.4.
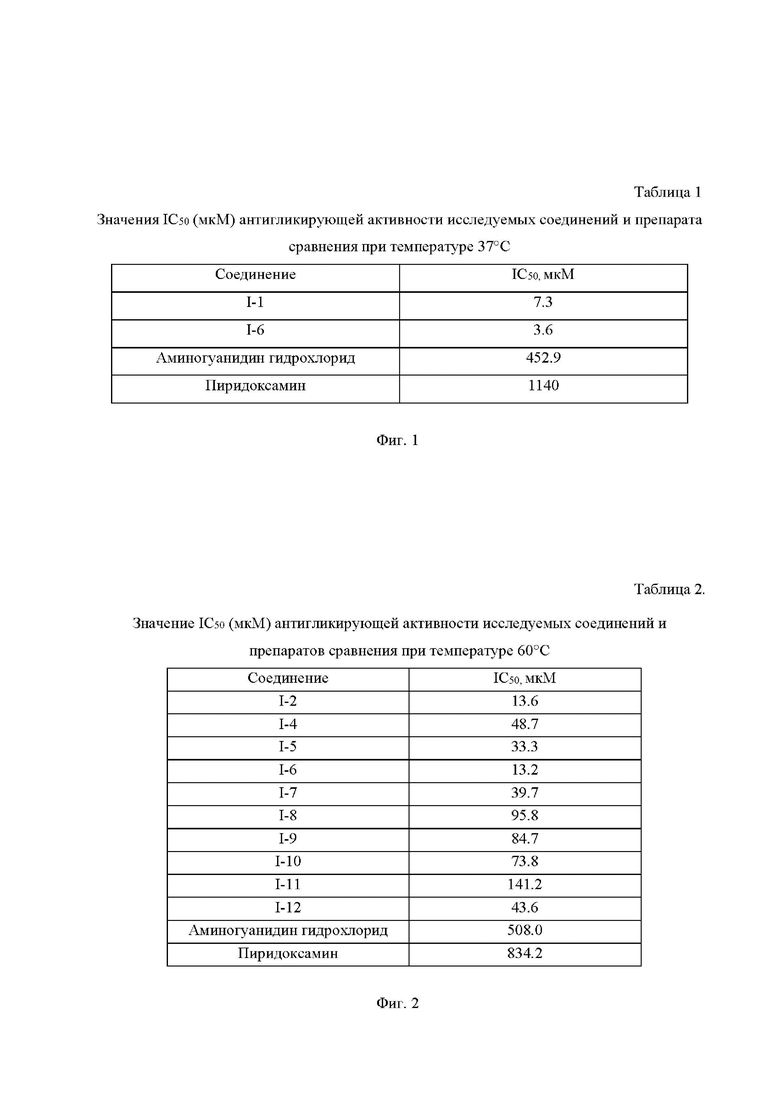
На Фиг.1 приведена Таблица 1, на которой представлены значения IC50 соединений формулы I и препарата сравнения (аминогуанидин гидрохлорид, пиридоксамин) в реакции образования КПГ при взаимодействии глюкозы с бычьим сывороточным альбумином при температуре 37°С.
На Фиг.2 приведена Таблица 2, на которой представлены значения IC50 соединений формулы I и препаратов сравнения (аминогуанидин гидрохлорид, пиридоксамин) в реакции образования КПГ при взаимодействии глюкозы с бычьим сывороточным альбумином при температуре 60°С.
Представленные на Фиг. 1, 2 результаты свидетельствуют о том, что заявляемые молекулы значительно превосходят препараты сравнения аминогуанидин гидрохлорид и пиридоксамин, так как для заявляемых соединений формулы I ингибирование образования гликированного бычьего сывороточного альбумина наблюдается в гораздо меньших концентрациях (в некоторых случаях разница была более чем на 2 порядка).
На Фиг.3 приведена Таблица 3, на которой представлены значения IC50 соединений формулы I и препаратов сравнения (аминогуанидин гидрохлорид, пиридоксамин, trolox) по данным теста перекисного окисления липидов.
На Фиг.4 приведена Таблица 4, на которой представлены значения IC50 соединений формулы I и препаратов сравнения (аминогуанидин гидрохлорид, пиридоксамин, кверцетин) по данным теста на антиоксидантную активность.
Представленные на Фиг. 3, 4 результаты свидетельствуют о том, что заявляемые молекулы способны ингибировать и второй путь образования КПГ, так как для заявляемых соединений формулы I ингибирование образования соответствующих реакций происходит в концентрациях, сопоставимых с таковыми для препаратов сравнения или даже меньших. В тесте перекисного окисления липидов (Фиг. 3) аминогуанидин и пиридоксамин оказались неактивны, IC50 препарата trolox составила 8.3 мкМ, тогда как IC50 наиболее активных заявляемых соединений - 4.3 - 6.7 мкМ. По данным теста на антиоксидантную активность IC50 препаратов сравнения аминогуанидин, пиридоксамин и кверцетин составила 16.5 мкМ, более 600 мкМ и 13.7 мкМ соответственно, значения IC50 заявляемых соединений находились в интервале 8.7-52.9 мкМ.
Далее заявителем приведено осуществление заявленного технического решения.
Заявленные соединения получают согласно нижеприведенным схемам 1-5, где заявляемые соединения обозначены номерами I-1 - I-12.
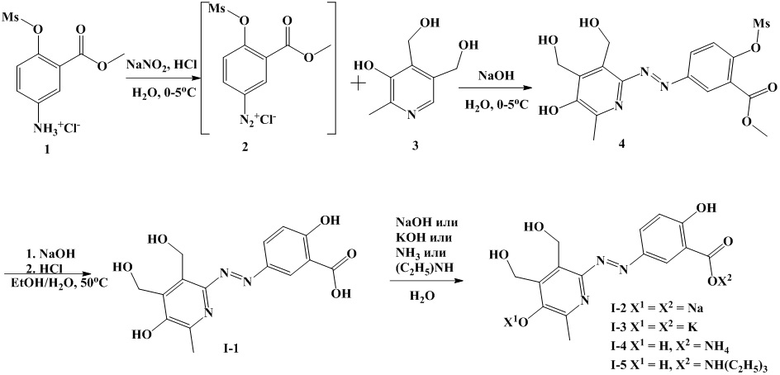
Схема 1.
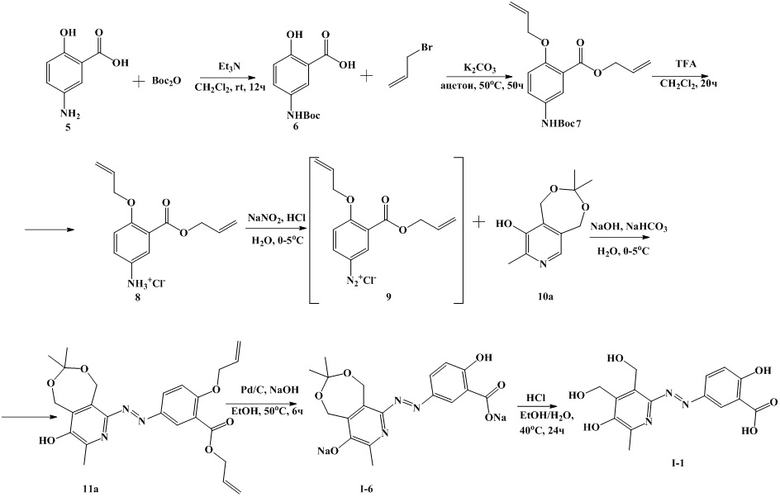
Схема 2
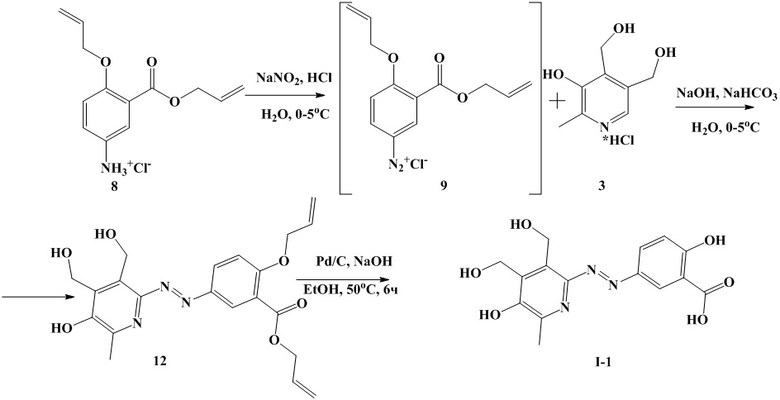
Схема 3
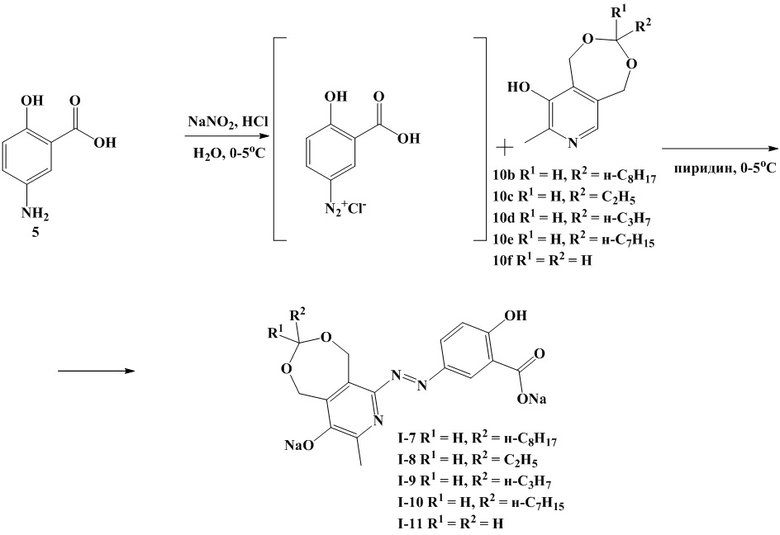
Схема 4
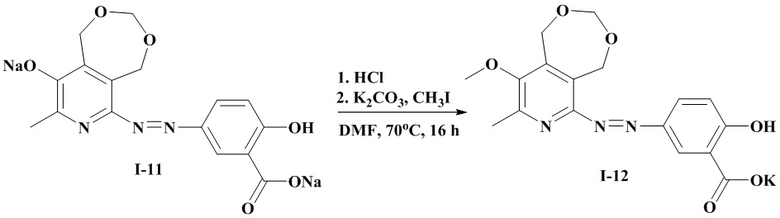
Схема 5
Характеристики новых соединений приведены далее в примерах конкретного выполнения. Структуры полученных соединений подтверждены методами масс-спектрометрии, 1Н и 13С ЯМР-спектроскопии. Спектры ЯМР регистрировали на приборе Bruker AVANCE-400. Химический сдвиг определялся относительно сигналов остаточных протонов дейтерированных растворителей (1H и 13С). Температуры плавления определялись с помощью прибора Stanford Research Systems MPA-100 OptiMelt. Контроль за ходом реакций и чистотой соединений проводили методом ТСХ на пластинах Sorbfil Plates.
HRMS-эксперимент был проведен с использованием масс-спектрометра высокого разрешения «TripleTOF 5600» AB Sciex (Германия) из раствора в метаноле методом турбо-ионной ионизации (TIS, от англ. turbo-ionic spray) при энергии столкновения с молекулами азота 10 эВ. Образцы с концентрацией вещества 5 мкмоль/л готовили путем растворения испытуемых соединений в метаноле (HPLC-UV Grade, LabScan).
Масс-спектры MS были получены на тройном квадрупольном масс-спектрометре c линейной ионной ловушкой AB Sciex QTrap 6500 (Сингапур) с использованием детектора высокой энергии IonDriveTM и источником ионизации IonDrive Turbo V (газообразный азот распылителя, положительная полярность ионизации, напряжение иглы 5200 В). Запись спектров проводилась в режиме “Q1” с энергией столкновения 10 эВ, потенциалом декластеризации 90 эВ.
Примеры конкретного выполнения заявленного технического решения
Пример 1. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-оксипиридин-2-ил)диазенил)-2-гидроксибензойной кислоты (I-1) по схеме 1, состоит из двух стадий.
Стадия 1. Синтез метил 5-((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)диазенил)-2-((метилсульфонил)окси)бензоата (4).
К навеске 0.37 г (1.3 ммоль) аммониевой соли 1 добавляют 75 мл воды и 0.44 г (3.9 ммоль) концентрированной соляной кислоты (33%) и перемешивают до растворения. Навеску 0.09 г (1.3 ммоль) нитрита натрия растворяют в 10 мл воды. К навеске 0.27 г (1.3 ммоль) пиридоксин гидрохлорида добавляют 25 мл воды и 0.24 г (6.0 ммоль) гидроксида натрия и перемешивают до растворения. Полученные растворы охлаждают до температуры 0-5°С.
Далее к охлажденному раствору аммониевой соли 1, добавляют раствор нитрита натрия, реакционную массу перемешивают 5 минут на ледяной бане, после чего ее добавляют к раствору пиридоксин гидрохлорида, охлажденному до 5°С.
Далее реакционную массу перемешивают 30 мин при комнатной температуре, после чего подкисляют концентрированной соляной кислотой до рН 5.5, выпавший осадок соединения 4 отфильтровывают, промывают 10 мл воды и высушивают.
Получают 0.42 г (76%) соединения (4).
ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д.: 2.41 (с, СH3, 3H); 3.48 (с, СH3, 3H); 3.90 (с, СH3, 3H); 4.91 (уш.с, СН2, 2Н); 5.02 (с, СН2, 2Н); 5.18 (уш.с, OH, 1Н); 7,69 (д, 3Jнн = 8.6 Гц, СH, 1 H); 8.20 (уш.д, 3Jнн = 8.6 Гц, СH, 1H); 8.30 (уш.с, СH, 1H).
Стадия 2. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-оксипиридин-2-ил)диазенил)-2-гидроксибензойной кислоты (I-1)
К навеске 0.20 г (0.47 ммоль) соединения 4 добавляют 7.5 мл этанола, 7.5 мл воды и 0.056 г гидроксида натрия (1.4 ммоль). Полученный раствор перемешивают при температуре 50 °С в течение 48 часов. Далее реакционную массу охлаждают до 25 °С и подкисляют концентрированной соляной кислотой до рН 1.5. Выпавший осадок отфильтровывают, промывают 10 мл воды и высушивают в вакууме при температуре 75°С. Получают 0.14 г (79%) соединения I-1.
Т. пл. 138 - 140°С (с разложением). Спектр ЯМР 1H (400 МГц, ДМСО-d6) δ, м.д.: 2.42 (с, 3H, CH3), 4.94 (с, 2H, CH2), 5.01 (с, 2H, CH2), 7.16 (д , 1H, 3JHH = 8.9 Гц, CHAr), 8.10 (д.д, 1H, 3JHH = 8.9 Гц, 4JHH = 2.4 Гц, CHAr), 8.33 (д, 1H, 4JHH = 2.4 Гц, CHAr).
Спектр ЯМР 13C (100 МГц, ДМСО-d6) δ, м.д.: 19.06 (с, СH3), 54.61 (с, CH2), 56.90 (с, CH2), 113.69 (с, CAr), 118.38 (с, CAr), 125.75 (с, CAr), 129.23 (с, CAr), 132.83 (с, CAr), 134.49 (с, CAr), 145.25 (с, CAr), 145.94 (с, CAr), 150.20 (с, CAr), 152.69 (с, CAr), 163.57 (с, CAr), 171.48 (с, C(O)OH).
Масс-спектр (HRMS-ESI): Найдено 334.1034 [М+H]+. Вычислено для [C15H16N3O6]+ - 334.1034.
Пример 2. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-оксипиридин-2-ил)диазенил)-2-гидроксибензоата натрия (I-2) по схеме 1.
К навеске 20.80 г (62.5 ммоль) соединения I-1 добавляют 300 мл воды и подщелачивают концентрированным (49%) раствором гидроксида натрия до рН 9.3 до полного растворения осадка. Получившийся раствор высушивают в вакууме при 75 °С до постоянной массы. Получают 22.0 г (94.6%) соединения I-2.
Т. пл. 238°С (с разложением). ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д.: 2.22 (с, СH3, 3H); 4.75 (c, СН2, 2Н); 4.93 (c, СН2, 2Н); 6.72 (д, 3Jнн = 8.7 Гц, СH, 1H); 7.69 (д.д, 3Jнн = 8.7 Гц, 4Jнн = 2.2 Гц, СH, 1H); 8.35 (д, 4Jнн = 2.2 Гц, СH, 1H).
ЯМР 13С {H} (100 МГц, ДМСО-d6), δ, м.д.: 19.70 (с, СH3); 55.31 (с, СH2); 59.31 (с, СH2); 116.58 (с, CAr); 119.77 (с, CAr); 124.49 (с, CAr); 125.62 (с, CAr); 131.19 (с, CAr); 133.28 (с, CAr); 144.33 (с, CAr); 144.39 (с, CAr); 147.65 (с, CAr); 165.28 (с, CAr); 165.63 (с, CAr); 171.43 (с, CО2.). Масс-спектр (HRМS-ESI): найдено 334.1034 [М-2Na+3Н]+, Вычислено для [C15H16N3O6]+ - 334.1034.
Пример 3. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-оксипиридин-2-ил)диазенил)-2-гидроксибензоата калия (I-3) по схеме 1.
Синтезируют и разрабатывают аналогично соединению I-2, используя гидроксид калия вместо гидроксида натрия. Выход 99%.
Т. разложения 205°С. ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д.: 2.20 (с, СH3, 3H); 4.69 (c, СН2, 2Н); 4.95 (c, СН2, 2Н); 6.81 (д, 3Jнн = 8.7 Гц, СH, 1H); 7.75 (д.д, 3Jнн = 8.7 Гц, 4Jнн = 2.5 Гц, СH, 1H); 8.19 (д, 4Jнн = 2.5 Гц, СH, 1H).
ЯМР 13С {H} (100 МГц, ДМСО-d6), δ, м.д.: 20.20 (с, СH3); 56.00 (с, СH2); 59.16 (с, СH2); 117.1 (с, CAr); 120.33 (с, CAr); 125.48 (с, CAr); 126.20 (с, CAr); 131.79 (с, CAr); 134.36 (с, CAr); 144.12 (с, CAr); 145.75 (с, CAr); 149.75 (с, CAr); 163.46 (с, CAr); 168.37 (с, CAr); 172.63 (с, CО2.).
Пример 4. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-гидроксипиридин-2-ил)диазенил)-2-гидроксибензоата аммония (I-4) по схеме 1.
Синтезируют и разрабатывают аналогично соединению I-2, используя 7М раствор аммиака в метаноле вместо гидроксида натрия. Выход 73.4%.
Т. разложения 121°С. ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д.: 2.39 (с, СH3, 3H); 4.93 (c, СН2, 2Н); 4.98 (c, СН2, 2Н); 5.10 (уш.c, ОН, 1Н); 6.04 (уш.c, ОН, 1Н); 6.81 (д, 3Jнн = 8.8 Гц, СH, 1H); 7.22 (уш.c, NH4, 4Н); 7.69 (д, 3Jнн = 8.8 Гц, СH, 1H); 8.25 (c, СH, 1H); 9.85 (уш.c, ОН, 1Н).
ЯМР 13С {H} (100 МГц, ДМСО-d6), δ, м.д.: 19.37 (с, СH3); 54.76 (с, СH2); 57.07 (с, СH2); 117.46 (с, CAr); 118.72 (с, CAr); 125.14 (с, CAr); 127.68 (с, CAr); 131.46 (с, CAr); 134.01 (с, CAr); 143.88 (с, CAr); 145.72 (с, CAr); 151.27 (с, CAr); 151.83 (с, CAr); 167.22 (с, CAr); 171.27 (с, CО2.).
Пример 5. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-гидроксипиридин-2-ил)диазенил)-2-гидроксибензоата триэтиламмония (I-5) по схеме 1.
Синтезируют и разрабатывают аналогично соединению I-2, используя триэтиламин вместо гидроксида натрия. Выход 90.5%. Гигроскопичные маслянистые кристаллы.
ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д.: 1.00 (т, 3Jнн = 7.3 Гц, СH3, 3H); 2.39 (с, СH3, 3H); 3.06 (кв, 3Jнн = 7.3 Гц, СH2, 2H); 4.93 (c, СН2, 2Н); 4.98 (c, СН2, 2Н); 5.10 (уш.c, ОН, 1Н); 6.78 (д, 3Jнн = 8.8 Гц, СH, 1H); 7.81 (д.д, 3Jнн = 8.8 Гц, 4Jнн = 2.5 Гц, СH, 1H); 8.24 (д, 4Jнн = 2.5 Гц, СH, 1H); 8.50 (уш.c, ОН, 1Н); 17.53 (уш.c, ОН, 1Н).
ЯМР 13С {H} (100 МГц, ДМСО-d6), δ, м.д.: 8.68 (с, СH3); 19.39 (с, СH3); 45.57 (с, СH2); 54.76 (с, СH2); 57.07 (с, СH2); 117.29 (с, CAr); 119.37 (с, CAr); 125.03 (с, CAr); 127.46 (с, CAr); 131.20 (с, CAr); 133.94 (с, CAr); 143.71 (с, CAr); 145.65 (с, CAr); 151.42 (с, CAr); 151.70 (с, CAr); 167.57 (с, CAr); 171.32 (с, CО2.).
Пример 6. Синтез 2-гидрокси-5-((3,3,8-триметил-9-окси-1,5-дигидро-[1,3]диксипино[5,6-c]пиридин-6-ил)диазенил)бензоата натрия (I-6) по схеме 2, состоит из 5 стадий.
Стадия 1. Синтез 5-((трет-бутоксикарбонил)амино)салициловой кислоты ( 6 )
В круглодонную колбу загружают 28.0 г (0.18 моль) 5-аминосалициловой кислоты (5), 350 мл хлористого метилена, 37.0 г (0.36 моль) триэтиламина и включают перемешивание. Затем добавляют раствор 43.9 г (0.20 моль) ди-трет-бутилдикарбоната в 50 мл хлористого метилена. Реакционную массу перемешивают 15 часов при температуре 20-25°С. Затем к реакционной массе в течение 5 мин добавляют при перемешивании 36.2 мл (0.36 моль) 32.5 % соляной кислоты, разбавленной водой до 200 мл. Затем полученную суспензию перемешивают еще 20 минут. Осадок отфильтровывают, дважды промывают водой порциями по 100 мл и высушивают в вакууме. Получают 43.9 г (94.5%) соединения 6.
Стадия 2. Синтез Аллил 2-(аллилокси)-5-((трет-бутоксикарбонил)амино) бензоата (7)
В круглодонную колбу загружают 42.9 г (0.17 моль) 5-((трет-бутоксикарбонил)амино)салициловой кислоты, 70.2 г (0.51 моль) карбоната калия, 600 мл ацетона 44 мл (0.51 моль) аллилбромида. Реакционную массу кипятят при перемешивании 37-80 часов, контроль за ходом реакции осуществляют методом ТСХ, используя в качестве элюента хлороформ. Окончание реакции подтверждается отсутствием синей окраски через 40 минут после обработки пластинки для ТСХ (Sorbfil ПТСХ-АФ-В-УФ) раствором хлорида железа (III).
Осадок из реакционной массы отфильтровывают под вакуумом на фильтре Шотта и промывают 100 мл ацетона. Фильтрат высушивают на роторном испарителе при температуре 55°С, сухой осадок измельчают и высушивают до постоянной массы. Получают 55.9 г (98.9 %) соединения 7.
Стадия 3. Синтез Аллил 2-(аллилокси)-5-аминобензоата (8)
В круглодонную колбу загружают 55.6 г (0.167 моль) аллил 2-(аллилокси)-5-((трет-бутоксикарбонил)амино)бензоата (7), 250 мл хлористого метилена, 68 мл (0.912 моль) трифторуксусной кислоты и перемешивают 20 часов при комнатной температуре.
Реакционную массу охлаждают до 5°С на бане со льдом. Затем к реакционной массе при перемешивании и охлаждении в течение 20 минут добавляют охлажденный раствор 36.5 г (0.912 моль) гидроксида натрия в 278 мл воды. Органический слой отделяют на делительной воронке и два раза промывают водой порциями по 250 мл.
Органическую фракцию отделяют и высушивают на роторном испарителе до постоянной массы. К полученному маслообразному аллил 2-(аллилокси)-5-аминобензоату добавляют 200 мл этанола и перемешивают до растворения, затем к раствору при перемешивании небольшими порциями в течение 10 мин добавляют 17.3 мл концентрированной соляной кислоты. Растворитель отгоняют в вакууме, продукт высушивают досуха при температуре 50°С и давлении 20 мбар до постоянной массы. Получают 43.0 г (98.3 %) соединения 8.
Стадия 4. Синтез Аллил 2-(аллилокси)-5-((9-гидрокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-c]пиридин-6-ил)диазенил)бензоата (11а)
К навеске 34.5 г (0.128 моль) гидрохлорида аллил 2-(аллилокси)-5-аминобензоата добавляют 830 мл воды и 33.45 мл (0.384 моль) концентрированной (34.57%) соляной кислоты, смесь перемешивают до полного растворения гидрохлорида аллил 2-(аллилокси)-5-аминобензоата. Навеску 9.0 г (0.130 моль) нитрита натрия растворяют в 50 мл воды. К навеске 26.8 г (0.128 моль) соединения 10а добавляют 400 мл воды и 27.4 г (0.320 моль) водного раствора гидроксида натрия (46.7%), после растворения соединения 10а к раствору добавляют 21.5 г (0.256 моль) гидрокарбоната натрия. Полученные растворы охлаждают до температуры 0-5°С.
К охлажденному раствору гидрохлорида аллил 2-(аллилокси)-5-аминобензоата при интенсивном перемешивании добавляют охлажденный раствор нитрита натрия. Реакционную массу перемешивают 5 мин и добавляют при интенсивном перемешивании к охлажденному раствору соединения 10а. При этом значения рН поддерживают в интервале 8.2-8.7 добавлением водного раствора гидроксида натрия (46.7%). После добавления раствора диазониевой соли реакционную массу перемешивают еще 30 мин до достижения стабильных значений pH.
Затем к реакционной массе при интенсивном перемешивании и охлаждении на бане со льдом добавляют раствор 15.9 г концентрированной соляной кислоты в 50.6 мл воды. Выпавший осадок отфильтровывают, промывают 200 мл воды и высушивают в вакууме при температуре 60°С до постоянной массы. Получают 57.3 г (98.8 %) соединения 11а.
Стадия 5. Синтез 2-гидрокси-5-((3,3,8-триметил-9-окси-1,5-дигидро-[1,3]диксипино[5,6-c]пиридин-6-ил)диазенил)бензоата натрия (I-6)
В колбу загружают 57.0 г соединения 11, 5.7 г палладия на угле (10%) и 570 г раствора гидроксида натрия в этиловом спирте (10%). Реакционную массу перемешивают 6 часов при температуре 50°С. К реакционной массе добавляют 310 мл воды и отфильтровывают от палладия на угле под вакуумом на фильтре Шотта. Фильтрат упаривают в 2-2.5 раза на роторном испарителе. Затем реакционную массу подкисляют соляной кислотой (6%) до достижения значения рН 9.10-9.15 и высушивают в вакууме до постоянной массы. Получившуюся смесь промывают 250 мл ацетона, затем к осадку добавили 455 мл этанола и 195 мл ацетона. Суспензию перемешивают при нагревании до 50°С, затем отфильтровывают от нерастворившегося осадка. Фильтрат высушивают в вакууме, получившийся продукт перекристаллизовывают из смеси изо-пропанол - вода 19:1. Получают 47.4 г (90%) соединения I-6.
Т разложения 222°С. Спектр ЯМР 1H (400 МГц, d6) δ, м.д.: 1.42 (с, 6H, 2СH3), 2.24 (с, 3H, CH3), 4.79 (с, 2H, CH2), 5.21 (с, 2H, CH2), 6.71 (д, 1H, 3JHH = 8.7 Гц, CHAr), 7.68 (д.д, 1H, 3JHH = 8.7 Гц, 4JHH = 2.6 Гц, CHAr), 8.17 (д, 1H, 4JHH = 2.6 Гц, CHAr), 17.06 (уш.с, 1H, OH).
Спектр ЯМР 13C {H} (100 МГц, ДМСО- d6) δ, м.д.: 19.69 (с, СH3), 23.92 (с, 2CH3), 58.79 (с, CH2), 60.11 (с, CH2), 101.53 (с, C(CH3)2), 116.66 (с, CAr), 119.72 (с, CAr), 124.45 (с, CAr), 125.72 (с, CAr), 131.62 (с, CAr), 133.57 (с, CAr), 143.32 (с, CAr), 144.36 (с, CAr), 146.35 (с, CAr), 165.34 (с, CAr), 171.50 (с, C(O)ONa).
Масс-спектр (HRMS-ESI): Найдено 374.1352 [М-2Na+3H]+. Вычислено для [C18H20N3O6]+ - 374.1347.
Пример 7. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-оксипиридин-2-ил)диазенил)-2-гидроксибензойной кислоты (I-1) по схеме 2.
К навеске 47.0 г (0.112 моль) соединения I-6 добавляют 700 мл этанола, 300 мл воды и 15 мл концентрированной соляной кислоты. Полученную суспензию перемешивают 6 часов при температуре 50°С, затем 12 часов при комнатной температуре. Осадок отфильтровывают, промывают 550 мл воды и высушивают в вакууме. Получают 35.8 г (95.5 %) соединения I-1.
Пример 8. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-оксипиридин-2-ил)диазенил)-2-гидроксибензойной кислоты (I-1) по схеме 3, состоит из 2 стадий.
Стадия 1. Синтез Аллил 2-(аллилокси)-5-((3,4-бис(гидроксиметил)-6-метил-5-оксипиридин-2-ил)диазенил)бензоата (12)
К навеске 10.0 г (37 ммоль) гидрохлорида аллил 2-(аллилокси)-5-аминобензоата 8 добавляют 500 мл воды и 12.3 г (111 ммоль) концентрированной (34.6%) соляной кислоты, смесь перемешивают до растворения гидрохлорида аллил 2-(аллилокси)-5-аминобензоата 8. К навеске 2.8 г (40 ммоль) нитрита натрия добавляют 50 мл воды. К навеске 7.6 г (37 ммоль) соединения 3 добавляют 100 мл воды, 18.5 г (203 ммоль) водного раствора гидроксида натрия (44.0%). Полученные растворы охлаждают до температуры 0-5°С.
К охлажденному раствору гидрохлорида аллил 2-(аллилокси)-5-аминобензоата 8 при интенсивном перемешивании добавляют охлажденный раствор нитрита натрия. Реакционную массу перемешивают 5 мин и добавляют при интенсивном перемешивании к охлажденному раствору соединения 3. При этом значение рН поддерживают в интервале 8.2-8.7 добавлением водного раствора гидроксида натрия (44.0 %). После добавления раствора диазониевой соли реакционную массу перемешивают еще 30 мин до достижения постоянного значения pH.
Затем реакционную массу при интенсивном перемешивании и охлаждении на бане со льдом подкисляют концентрированной соляной кислотой до рН 1. Выпавший осадок отфильтровывают, промывают 100 мл воды и высушивают в вакууме при температуре 60 °С до постоянной массы. Получают 11.5 г (66.8 %) соединения 12.
Стадия 2. Синтез 5-((3,4-бис(гидроксиметил)-6-метил-5-оксипиридин-2-ил)диазенил)-2-гидроксибензойной кислоты (I-1)
Навеску 10 г соединения 12 добавляют к 100 г раствора гидроксида натрия в этаноле (10%), затем в реакционную массу вносят 0.5 г палладия на угле (10%). Реакционную массу перемешивают 6 часов при температуре 50°С. К реакционной массе добавляют 70 мл воды и отфильтровывают от палладия на угле под вакуумом на фильтре Шотта. Фильтрат упаривают в 2 раза на роторном испарителе. Затем реакционную массу подкисляют концентрированной соляной кислотой до рН 1. Осадок отфильтроввывают, 2 раза промывают 50 мл воды и высушивают в вакууме. Получают 7.9 г (55.3 %) соединения I-1.
Пример 9. Синтез 2-гидрокси-5-((3-октил-8-метил-9-окси-1,5-дигидро-[1,3]диксипино[5,6-c]пиридин-6-ил)диазенил)бензоат натрия (I-7) по схеме 4.
В колбу на 100 мл добавляют 0.38 г (2.5 ммоль) 5-аминосалициловой кислоты, 20 мл воды и 0.10 г (2.5 ммоль) NaOH. После растворения 5-аминосалициловой кислоты к раствору добавляют 0.18 г (2.6 ммоль) нитрита натрия. Полученный раствор охлаждают до 5°С и добавляют к раствору 1.10 г концентрированной (33%) соляной кислоты в 5 мл воды, предварительно охлажденному до 5°С. Реакционную массу перемешивают 5 минут, после чего осадок диазониевой соли отфильтровывают на воронке Шотта. Осадок без дополнительной очистки добавляют к раствору 0.73 г (2.5 ммоль) ацеталя 10б в 50 мл пиридина, предварительно охлажденному до 5°С. Реакционную массу перемешивают 72 часа при температуре 0-5 °С. Реакционную массу фильтруют через слой силикагеля (1см), после чего растворитель отгоняют в вакууме. К получившейся смеси добавляют 7 мл воды, после чего добавляют концентрированный (48%) раствор NaOH в воде до растворения осадка. Продукт выделяют двукратным хроматографированием на колонке PuriFlash С18, в качестве элюента используют систему вода/этанол в соотношении от 1 к 9 до 1 к 1. Получают 58 мг (4.5%) соединения I-7.
Т. разложения 190 °С. ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д.: 0.86 (т, 3Jнн = 6.6 Гц, СH3, 3H); 1.17 - 1.40 (м, 6СH2, 12H); 1.53- 1.61 (м, СH2, 2H); 2.14 (с, СH3, 3H); 4.49, 5.08 (АВ, 2Jнн = - 14.6 Гц, СН2, 2Н); 4.85 (т,3Jнн = 5.6 Гц, СН, 1Н); 4.94, 5.56 (АВ, 2Jнн = - 14.8 Гц, СН2, 2Н); 6.66 (д, 3Jнн = 8.7 Гц, СH, 1H); 7.59 (дд, 3Jнн = 8.7 Гц, 4Jнн = 2.7 Гц, СH, 1H); 8.06 (д, 4Jнн = 2.7 Гц, СH, 1H); 17,05 (с, ОH, 1H).
ЯМР 13С {H} (100 МГц, ДМСО-d6), δ, м.д.: 14.06 (с, СH3); 20.10 (с, СH3); 22.19 (с, СH2); 24.43 (с, СH2); 28.74 (с, СH2); 28.83 (с, СH2); 28.99 (с, СH2); 31.37 (с, СH2); 33.54 (с, СH2); 63.39 (с, СH2); 64.13 (с, СH2); 105.75 (с, С); 116.38 (с, Car.); 120.03 (с, Car.); 123.41 (с, Car.); 125.52 (с, Car.); 129.90 (с, Car.); 135.07 (с, Car.); 144.72 (с, Car.); 148.08 (с, Car.); 164.50 (с, Car.); 171.34 (с, CО2). Масс-спектр (HRМS-ESI): найдено [М-2Na+Н]- 456.2135, C24H30N3O6-, вычислено [М-2Na+Н]- 456.2140.
Пример 10. Синтез 2-гидрокси-5-((3-этил-8-метил-9-окси-1,5-дигидро-[1,3]диксипино[5,6-c]пиридин-6-ил)диазенил)бензоат натрия (I-8) по схеме 4.
Синтезируют и разрабатывают аналогично соединению I-7, используя ацеталь 10c вместо ацеталя 10b. Выход 5.2 %.
Т. разложения 165°С. ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д.: 0.91 (т, 3Jнн = 7.5 Гц, СH3, 3H); 1.66 (дт, 3Jнн = 7.5 Гц, 3Jнн = 5.8 Гц, СH2, 2H); 2.43 (с, СH3, 3H); 4.80, 5.13 (АВ, 2Jнн = - 15.6 Гц, СН2, 2Н); 4.93 (т,3Jнн = 5.8 Гц, СН, 1Н); 5.10, 5.48 (АВ, 2Jнн = - 15.2 Гц, СН2, 2Н); 7.12 (д, 3Jнн = 8.8 Гц, СH, 1H); 8,08 (дд, 3Jнн = 8.7 Гц, 4Jнн = 2.3 Гц, СH, 1H); 8.31 (д, 4Jнн = 2.3 Гц, СH, 1H).
ЯМР 13С {H} (100 МГц, ДМСО-d6), δ, м.д.: 9.02 (с, СH3); 19.58 (с, СH3); 25.81 (с, СH2); 61.92 (с, СH2); 62.34 (с, СH2); 106.31 (с, С); 113.85 (с, Car.); 118.40 (с, Car.); 124.65 (с, Car.); 126.13 (с, Car.); 128.78 (с, Car.); 136.06 (с, Car.); 145.06 (с, Car.); 145.59 (с, Car..); 149.55 (с, Car.); 150.01 (с, Car.); 163.69 (с, Car.); 171.39 (с, CО2). Масс-спектр (МS-ESI): найдено [М-2Na+Н]- 374.3, C18H20N3O6+, вычислено [М-2Na+3Н]+ 374.134.
Пример 11. Синтез 2-гидрокси-5-((3-пропил-8-метил-9-окси-1,5-дигидро-[1,3]диксипино[5,6-c]пиридин-6-ил)диазенил)бензоата натрия (I-9) по схеме 4.
Синтезируют и разрабатывают аналогично соединению I-7, используя ацеталь 10d вместо ацеталя 10b. Выход 5.5%.
Т. разложения 156°С. ЯМР 1H (400 МГц, ДМСО-d6), δ, м.д.: 0.92 (т, 3Jнн = 7.5 Гц, СH3, 3H); 1.34-1.43 (м, СH2, 2H); 1.62-1.67 (м, СH2, 2H); 2.47 (с, СH3, 3H); 4.82, 5.14 (АВ, 2Jнн = - 15.6 Гц, СН2, 2Н); 5.02 (т,3Jнн = 5.7 Гц, СН, 1Н); 5.12, 5.48 (АВ, 2Jнн = - 15.4 Гц, СН2, 2Н); 7.15 (д, 3Jнн = 8.9 Гц, СH, 1H); 8,11 (д, 3Jнн = 8.9 Гц, СH, 1H); 8,34 (с, СH, 1H).
ЯМР 13С {H} (100 МГц, CD3OD), δ, м.д.: 14.26 (с, СH3); 17.64 (с, СH3); 19.08 (с, СH2); 36.77 (с, СH2); 62.67 (с, СH2); 63.43 (с, СH2); 106.62 (с, С); 114.59 (с, Car.); 119.53 (с, Car.); 126.42 (с, Car.); 129.20 (с, Car.); 129.51 (с, Car.); 135.73 (с, Car.); 140.71 (с Car.); 145.59 (с, Car.); 146.37 (с, Car.); 158.52 (с, Car.); 166.64 (с, Car.); 173.05 (с, CО2). Масс-спектр (МS-ESI): найдено [М-2Na+3Н]+ 388.4, C19H22N3O6+, вычислено [М-2Na+3Н]+ 388.150.
Пример 12. Синтез 2-гидрокси-5-((3-гептил-8-метил-9-окси-1,5-дигидро-[1,3]диксипино[5,6-c]пиридин-6-ил)диазенил)бензоата натрия (I-10) по схеме 4.
Синтезируют и разрабатывают аналогично соединению I-7, используя ацеталь 10e вместо ацеталя 10b. Выход 4.1%.
Т. разложения 141°С. ЯМР 1H (400 МГц, CD3OD), δ, м.д.: 0.86 (т, 3Jнн = 6.5 Гц, СH3, 3H); 1.15 - 1.46 (м, 5СH2, 10H); 1.64-1.75 (м, СH2, 2H); 2.57 (с, СH3, 3H); 4.83, 5.16 (АВ, 2Jнн = - 15.6 Гц, СН2, 2Н); 4.96 (т,3Jнн = 5.6 Гц, СН, 1Н); 5.16, 5.48 (АВ, 2Jнн = - 15.6 Гц, СН2, 2Н); 7.03 (д, 3Jнн = 8.8 Гц, СH, 1H); 8,04 (д, 3Jнн = 8.8 Гц, СH, 1H); 8,48 (с, СH, 1H).
ЯМР 13С {H} (100 МГц, CD3OD), δ, м.д.: 14.47 (с, СH3); 16.60 (с, СH3); 23.74 (с, СH2); 25.82 (с, СH2); 30.44 (с, СH2); 30.47 (с, СH2); 32.99 (с, СH2); 33.50 (с, СH2); 62.26 (с, СH2); 63.46 (с, СH2); 106.72 (с, С); 114.65 (с, Car.); 119.81 (с, Car.); 129.35 (с, Car.); 130.36 (с, Car.); 136.32 (с, Car.); 143.41 (с, Car.); 144.77 (с, Car.); 146.28 (с, Car.); 146.97 (с, Car.); 153.06 (с, Car.); 167.28 (с, Car.); 172.90 (с, CО2). Масс-спектр (МS-ESI): найдено [М-2Na+3Н]+ 444.5, C23H30N3O6+, вычислено [М-2Na+3Н]+ 444.213.
Пример 13. Синтез 2-гидрокси-5-((8-метил-9-окси-1,5-дигидро-[1,3]диксипино[5,6-c]пиридин-6-ил)диазенил)бензоат натрия (I-11) по схеме 4.
Синтезируют и разрабатывают аналогично соединению I-7, используя ацеталь 10f вместо ацеталя 10b. Выход 3%.
Т. разложения 150°С. ЯМР 1H (400 МГц, CD3OD), δ, м.д.: 2.70 (с, СH3, 3H); 5.02 (с, СН2, 2Н); 5.10 (с, СН2, 2Н); 5.43 (с, СН2, 2Н); 7,12 (д, 3Jнн = 8.8 Гц, СH, 1H); 8,16 (дд, 3Jнн = 8.8 Гц, 4Jнн = 2.2 Гц, СH, 1H); 8,61 (д, 4Jнн = 2.3 Гц, СH, 1H);
ЯМР 13С {H} (100 МГц, CD3OD), δ, м.д.: 15.11 (с, СH3); 64.33 (с, СH2); 65.71 (с, СH2); 98.04 (с, СH2); 114.82 (с, Car.); 120.16 (с, Car.); 129.69 (с, Car.); 131.34 (с, Car.); 137.27 (с, Car.); 143.67 (с, Car.); 144.86 (с, Car.); 146.19 (с, Car.); 147.43 (с, Car.); 153.36 (с, Car.); 168.18 (с, Car.); 172.73 (с, CО2.). Масс-спектр (МS-ESI): найдено [М-2Na+3Н]+ 346.3, C16H16N3O6+, вычислено [М-2Na+3Н]+ 346.103.
Пример 14. Синтез 2-гидрокси-5-((8-метил-9-метокси-1,5-дигидро-[1,3]диксипино[5,6-c]пиридин-6-ил)диазенил)бензоата калия (I-12) по схеме 5.
К раствору 0.22 г (0.58 ммоль) соединения I-11 в 15 мл воды добавляют концентрированную соляную кислоту до рН около 1. Выпавший осадок отфильтровавают на фильтре Шотта под вакуумом и высушивают в вакууме. Высушенный осадок растворяют в 25 мл ДМФА, к раствору добавляют 0.20 г (1.45 ммоль) карбоната калия и 0.82 г (0.58 ммоль) йодистого метила. Реакционную массу перемешивают 8 часов при температуре 70°C, после чего оставляют на 12 часов при комнатной температуре. Затем к реакционной массе добавляют еще 0.82 г (0.58 ммоль) йодистого метила и перемешивают 8 часов при температуре 70°C. После окончания реакции ДМФА отгоняют в вакууме, остаток растворяют в 5 мл смеси ацетон:этанол=10:1, продукт очищают колоночной хроматографией на селикагеле, в качестве элюента используют последовательно ацетон; смесь ацетон:этанол=10:1; смесь ацетон:этанол=6:1; смесь ацетон:этанол=4:1. Получают 0.045 г (20 %) соединения I-12; оранжевое кристаллическое вещество.
Т. разложения 160°С. Спектр 1H ЯМР (ДМСО-d6) δ, м.д.: 2.45 (с, 3H, CH3), 3.74 (с, 3H, CH3), 4.99 (с, 2H, CH2), 5.01 (с, 2H, CH2), 5.21 (с, 2H, CH2), 6.91 (д, 1H, 3JHН =8.8 Гц, CHAr), 7.93 (дд, 1Н, 3JHH =8.8 Гц, 4JHH = 2.5 Гц, СНAr), 8.30 (д, 1H, 3JHН =2.5 Гц, CHAr).
Спектр 13C{1H} ЯМР (ДМСО-d6) δ, м.д.: 18.82 (c, CH3), 61.16 (c, CH3), 63.71 (c, CH2), 64.33 (c, CH2), 96.88 (c, CH2), 117.13 (c, CAr), 118.17 (c, CAr), 126.65 (c, CAr), 127.71 (c, CAr), 130.72 (c, CAr), 142.90 (c, CAr), 143.69 (c, CAr), 150.16 (c, CAr), 151.76 (c, CAr), 153.46 (c, CAr), 167.86 (c, CAr), 170.85 (c, COOH). Масс-спектр (МS-ESI): найдено [М-K+2Н]+ 360.1, C17H17N3O6+, вычислено [М-K+2Н]+ 360.119.
Пример 15. Определение антигликирующей активности соединений при температуре 37°С.
Реакцию гликирования воспроизводят по взаимодействию глюкозы с бычьим сывороточным альбумином. Реакцию проводят в фосфатном буферном растворе 0.05 М рН 7.4. Состав реакционной среды: 0.5М раствор глюкозы, 1 мг/мл БСА, 0.02% натрия азид.
Исследуемые и контрольное соединения растворяют в диметилсульфоксиде (ДМСО), с конечной концентрацией 75 мМ. В 48-ми луночном планшете делают серию разведений с шагом два в ДМСО (всего 14 точек). После чего переносят разведения (50 мкл) в планшет с рабочим буфером (1200 мкл). Общий объем реакционной смеси составляет 1250 мкл.
Концентрация соединений и препарата сравнения для определения IC50 после их добавления в реакционную среду составляет 1500-0.73 мкМ (14 точек разведения).
Контрольные пробы содержат эквивалентный объем растворителя. Дополнительно делают аналогичную серию с буфером, не содержащим глюкозу и БСА, для определения фонового сигнала флюоресценции.
Образцы инкубируют в течение 9 дней при температуре 37°С.
Спектральные характеристики КПГ определяют на длинах волн возбуждения/испускания 370/440 нм.
Первичная математическая обработка включает определение коэффициента флуоресценции КПГ по формуле
Fly=10(log10(Exp)-log10(blank)), где
Fly - относительный показатель флуоресценции;
log10(Exp) и log10(blank) - фактические уровни флуоресценции образцов, содержащих индуктор гликирования, и соответствующих им проб, не содержащих таковой.
Процедура проведения теста.
Конечный объем реакционной смеси составляет 1250 мкл. Реакционная смесь содержит бычий сывороточный альбумин (1 мг/мл) и глюкозу (500 мМ) в фосфатном буфере (pH 7.4) и 0.02 % азид натрия. Для оценки адекватности воспроизведения метода используют аминогуанидин, раствор веществ добавляют в экспериментальные образцы в различных концентрациях в объеме 50 мкл. В контрольные пробы добавляют ДМСО в аналогичном объеме. Для предупреждения бактериального роста все манипуляции проводят в асептических условиях (с использованием ламинарного шкафа), с этой же целью в буферный раствор вносят азид натрия в конечной концентрации 0.02 %. Исследования выполняют в трипликатах.
Все экспериментальные образцы инкубируют в течение 9 суток при 37°С. Через каждые 24 часа отбирают по 30 мкл и проводят определение специфической флуоресценции гликированного бычьего сывороточного альбумина на спектрофотометре Tecan Infinite 200 PRO (TECAN, Швейцария) при длине волны возбуждения 370 нм и испускания 440 нм.
Эксперимент в одинаковых условиях повторяют три раза.
Проведенные исследования показывают, что исследуемые и контрольные вещества не обладают высоким фоновым сигналом флюоресценции и не вносят дополнительный вклад, который мог бы повлиять на эксперимент.
Кроме того, изучение изменения сигнала флюоресценции показывает, что максимальные значения наблюдаются на 7-е сутки, после чего сигнал перестает нарастать и наблюдается спад. В связи с этим для расчета используют данные, полученные на 7-е сутки инкубации, по которым и рассчитывали значения IC50, представленные на Фиг. 1 и Таблице 1.
Статистическую обработку результатов проводят с использованием редактора Microsoft Excel 2007, для расчета показателя IС50 используют программное обеспечение OriginPro 8, в котором строят графики доза-ответ (х - концентрация, мкМ; у - ингибирование, в процентах, вычисленное исходя из значений флуоресценции контроля и исследуемых образцов).
Полученные результаты свидетельствуют о том, что заявляемые соединения значительно превосходят препарат сравнения аминогуанидин, так как для I-1, I-6 ингибирование образования гликированного бычьего сывороточного альбумина наблюдается в гораздо меньших концентрациях.
Пример 16. Определение антигликирующей активности соединений при температуре 60°С.
Реакцию гликирования проводят в фосфатном буферном растворе 0.05М рН 7.4. Состав реакционной среды: 0.4 М раствор глюкозы и 1 мг/мл БСА (фракция V). Исследуемые соединения растворяют в 99% ДМСО, с конечной концентрацией в реакционной среде ~3%. Конечная концентрация веществ после их добавления в реакционную среду составляет 1000-30 мкМ (10000-1000 мкМ для аминогуанидина и пиридоксамина), с промежуточным логарифмическим шагом 0.5, с оценкой IC50. Контрольные пробы содержат эквивалентный объем растворителя. Образцы инкубируют 24 ч при 60 °С. В качестве вещества сравнения используют гидрохлорид аминогуанидина и пиридоксамин.
Регистрацию данных проводят спектро-флуориметрическим методом на длинах волн возбуждения/испускания 370/440 нм.
Первичная математическая обработка включает определение коэффициента флуоресценции КПГ по формуле 1
Формула 1: Fly=10(log10(Exp)-log10(blank))
, где Fly - относительный показатель флуоресценции, log10(Exp) и log10(blank) - фактические уровни флуоресценции образцов, содержащих индуктор гликирования, и соответствующих им проб, не содержащих таковой.
Оценка активности соединений в широком спектральном диапазоне и применение логарифмов в анализе первичных данных призваны минимизировать риск ложноположительного результата, связанного с 1) собственными оптическими/флуоресцентными свойствами исследуемых соединений, 2) интерференцией флуоресцентных свойств КПГ с собственными свойствами соединений.
Статистическую обработку данных проводят при помощи программного обеспечения MicrosoftExcel 2007, а также программы GraphPad Prism 7.0 с использованием однофакторного вариационного анализа и пост-теста Туке (p<0.05).
Рассчитанные значения IC50 заявляемых соединений и препарата сравнения (аминогуанидина гидрохлорида) представлены на Фиг. 2, Таблица 2.
Полученные результаты свидетельствуют о том, что заявляемые соединения значительно превосходят препараты сравнения аминогуанидина гидрохлорид и пиридоксамин, так как для I-2, I-4 - I-12 ингибирование образования гликированного бычьего сывороточного альбумина наблюдается в гораздо меньших концентрациях.
Пример 17. Методика определения активности соединений в перекисном окислении липидов.
Для приготовления лецитиновых липосом берут навеску яичного лецитина массой 100 мг, растворяют в 40 мл хлороформа, переносят в круглодонную колбу. Хлороформ упаривают на роторном испарителе при комнатной температуре с получением тонкой лецитиновой пленки. Далее в колбу приливают 40 мл трис-HCl-буфера (pH 7.4), суспензию подвергают ультразвуковому диспергированию в течение 90 секунд при средней мощности. Полученную гомогенную суспензию липосом доводят до конечного объема 50 мл трис-HCl-буфером, с получением конечной концентрации липосом 2 мг/мл.
Приготовление реагента ТВА (с тиобарбитуровой кислотой): раствор TBA - трихлоруксусная кислота - HCl получают растворением 0.416 мг тиобарбитуровой кислоты в 10 мл трихлоруксусной кислоты (16.8% w/v разведенной в 0.125 N HCl). Для лучшего растворения соединений смесь нагревают до 40°С в течение 15 минут.
Для анализа перекисного окисления липидов 10 мкл исследуемых соединений с различными концентрациями добавляют к 700 мкл липосом и выдерживают 5 минут при комнатной температуре. Тролокс используют в качестве положительного контроля в серийных разведениях (50; 25; 12.5; 6.25 мкМ). Далее в реакционную смесь добавляют 125 мкл раствора аскорбиновой кислоты, получая конечную концентрацию 0.2 мМ. Реакцию перекисного окисления инициируют добавлением 75 мкл свежеприготовленного раствора сульфата железа двухвалентного с получением конечной концентрации 10 мкМ. Реакционную смесь инкубируют в открытой пробирке Эппендорф с доступом кислорода воздуха в течение 60 минут при 37 °C (водяная баня) с умеренным покачиванием. Через 60 мин реакционную смесь в объеме 0.5 мл отбирают и переносят в другую пробирку, реакцию останавливают добавлением 1.5 мл ледяного раствора ТВА - трихлоруксусная кислота - HCl. Смесь нагревают 15 мин при 85°C, а затем центрифугируют 10 мин при 15000 об / мин, при 4 °C. Поглощение надосадка определяют при 535 нм против 600 нм.
После этого рассчитывают процент ингибирования I (%) по формуле (4):
 (4),
(4),
где A0 - поглощение контроля, в который не были внесены соединения (100% окисление липидов);
A1- поглощение образца с исследуемым соединением.
Далее строят график зависимости антиоксидантной активности I (%) от концентрации исследуемых соединений. График используют для определения концентраций соединений, ингибирующих перекисное окисление липидов на 50%. Значения IC50 (мкМ) исследуемых соединений и препаратов сравнения представлены на Фиг. 3 и Таблица 3.
Пример 18. Методика исследования антиоксидантной активности соединений.
Антиоксидантную активность определяют по нейтрализации радикала ABTS (2,2'-азино-бис(3-этилбензтиазолин-6-сульфоновой кислоты) исследуемыми соединениями, проводится расчет IC50 (мкM).
Реагент АВТS растворяют в воде до концентрации 7 мM. Радикальный катион ABTS (АВТS+) получают путем взаимодействия исходного раствора АВТS с 2.45 мМ персульфатом аммония 1:1 и выдерживания смеси в темноте при комнатной температуре в течение 12-16 часов перед использованием. Раствор ABTS+ разбавляют в смеси метанол/вода (1:1) до концентрации 0.7 мM и достижения оптической плотности 0.7 единиц при 738 нм. Различные концентрации соединений (1000, 500, 250, 125, 62.5, 31.2, 15.6, 7.8 μМ), а так же кверцетин в различных концентрациях (250, 125, 62.5, 31.2, 15.6, 7.8, 3.9, 2.0 μМ) в объеме 130 μL в смеси метанол/вода (1:1) смешивают с 130 μL катиона радикала ABTS (0.7 мM) и выдерживают при 37°С на водяной бане в течение 15 минут. Оптическую плотность измеряют при 734 нм на спектрофотометре Tecan Infinite 200 PRO (TECAN, Швейцария). Рассчитывают процент ингибирования поглощения относительно контроля и строят график зависимости от концентрации антиоксидантов. По графику определяют концентрацию потенциального антиоксиданта, ингибирующую оптическую плотность радикала ABTS на 50%. Значения IC50 (мкМ) антиоксидантной активности исследуемых соединений и препаратов сравнения представлены на Фиг. 4 и Таблица 4.
Таким образом, из описанного выше можно сделать вывод, что заявителем решена выявленная техническая проблема и достигнут заявленный технический результат, а именно - разработаны средства для профилактики и лечения заболеваний, вызванных образованием и накоплением в организме конечных продуктов гликирования, общей формулы (I), обладающих значительно большей в сравнении с аминогуанидином и пиридоксамином, известными ингибиторами образования КПГ, антигликирующей активностью.
Заявленное техническое решение соответствует критерию «новизна», предъявляемому к изобретениям, так как из исследованного уровня техники не выявлены технические решения, обладающие заявленной совокупностью отличительных признаков, обеспечивающих достижение заявленных результатов.
Заявленное техническое решение соответствует критерию «изобретательский уровень», предъявляемому к изобретениям, так как не является очевидным для специалиста в данной области науки и техники.
Заявленное техническое решение соответствует критерию «промышленная применимость», так как может быть реализовано на любом специализированном предприятии с использованием стандартного оборудования, известных отечественных материалов и технологий.
| название | год | авторы | номер документа |
|---|---|---|---|
| Азопроизводные аминофенолов, обладающие способностью ингибировать образование конечных продуктов гликирования | 2024 |
|
RU2839138C1 |
| Ингибиторы образования конечных продуктов гликирования на основе азопроизводных фенилсульфокислот | 2016 |
|
RU2634594C1 |
| Соединения фторхинолонового ряда на основе производных пиридоксина, обладающие антибактериальными свойствами | 2019 |
|
RU2713932C1 |
| Бис-аммониевые соединения на основе пиридоксина, обладающие антибактериальными и антимикотическими свойствами | 2020 |
|
RU2731999C1 |
| ПРИМЕНЕНИЕ ГЕТЕРОЦИКЛИЧЕСКИХ ГИДРАЗОНОВ В КАЧЕСТВЕ СРЕДСТВ, ОБЛАДАЮЩИХ АНТИГЛИКИРУЮЩЕЙ АКТИВНОСТЬЮ | 2016 |
|
RU2658819C2 |
| Четвертичные аммониевые соединения на основе 3-гидроксипиридина, обладающие антибактериальной активностью | 2021 |
|
RU2778507C1 |
| Применение азопроизводных фенилсульфокислот в качестве ингибиторов образования конечных продуктов гликирования | 2016 |
|
RU2628605C1 |
| Оксазолидиноны на основе производных пиридоксина, обладающие антибактериальной активностью | 2024 |
|
RU2836570C1 |
| БИС[1,2,4]ТРИАЗОЛО[4,3-b:3',4'-f][1,2,4,5]ТЕТРАЗИНЫ, ОБЛАДАЮЩИЕ АНТИГЛИКИРУЮЩЕЙ, АНТИГЛИКОКСИДАЦИОННОЙ И АНТИАГРЕГАНТНОЙ АКТИВНОСТЬЮ | 2020 |
|
RU2755897C1 |
| Применение сульфасалазина в качестве ингибитора образования конечных продуктов гликирования | 2017 |
|
RU2680844C1 |


Изобретение относится к новым азосоединениям на основе 5-аминосалициловой кислоты общей формулы (I), обладающим высокой антигликирующей активностью (способностью блокировать образование конечных продуктов гликирования).
 ,
,
где R1 = CH3, H и другие фармацевтически приемлемые катионы, такие как Na, K, а R2 = R3 = H, или R2 и R3 вместе образуют заместитель вида СR5R6, где R5 = H, CH3 и R6 = H, CH3 и другие линейные алкильные заместители, такие как C2H5, н-C3H7, н-C7H15, н-C8H17; R4 = Et3NH, H и другие фармацевтически приемлемые катионы органической и неорганической природы, такие как K, NH4. Технический результат - получены новые соединения, которые являются эффективными ингибиторами образования конечных продуктов гликирования (далее КПГ) и могут найти широкое применение в медицине в области лечения осложнений сахарного диабета, атеросклероза, ревматоидного артрита, остеоартрита, нейродегенеративных заболеваний, включая болезни Альцгеймера и Паркинсона, катаракты, заболеваний, связанных со старением. 1 з.п. ф-лы, 4 ил., 4 табл., 18 пр.
1. Азопроизводные 5-аминосалициловой кислоты общей формулы (I)
 ,
,
где R1 = CH3, H и другие фармацевтически приемлемые катионы, такие как Na, K, а R2 = R3 = H, или R2 и R3 вместе образуют заместитель вида СR5R6, где R5 = H, CH3 и R6 = H, CH3 и другие линейные алкильные заместители, такие как C2H5, н-C3H7, н-C7H15, н-C8H17;
R4 = Et3NH, H и другие фармацевтически приемлемые катионы органической и неорганической природы, такие как K, NH4.
2. Азопроизводные 5-аминосалициловой кислоты по п. 1, обладающие антигликирующей активностью – ингибируют образование конечных продуктов гликирования.
| Применение азопроизводных фенилсульфокислот в качестве ингибиторов образования конечных продуктов гликирования | 2016 |
|
RU2628605C1 |
| Ингибиторы образования конечных продуктов гликирования на основе азопроизводных фенилсульфокислот | 2016 |
|
RU2634594C1 |
| A | |||
| D | |||
| Strelnik et al, Inhibitors of the Formation of Advanced Glycation End Products Based on Pyridoxine Azo Derivatives, Russian Journal of General Chemistry, Vol | |||
| Домовый номерной фонарь, служащий одновременно для указания названия улицы и номера дома и для освещения прилежащего участка улицы | 1917 |
|
SU93A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Передвижной дровокольный станок | 1913 |
|
SU522A1 |

