Область техники, к которой относится изобретение
Настоящее изобретение обеспечивает гидроксиалкилтиадиазольное соединение или его фармацевтически приемлемую соль, обладающее превосходной антибактериальной активностью, и также превосходное с точки зрения безопасности. Кроме того, настоящее изобретение обеспечивает фармацевтическую композицию, содержащую гидроксиалкилтиадиазольное соединение, его полиморф или фармацевтически приемлемую соль в качестве фармацевтически активного ингредиента. В частности, настоящее изобретение обеспечивает гидроксиалкилтиадиазольное соединение, его полиморфную форму или фармацевтически приемлемую соль, подходящее для лечения и/или профилактики инфекционных заболеваний.
Уровень техники
Существует несколько грамположительных видов организмов, которые вызывают заболевания у человека. Наиболее распространенные организмы включают виды Staphylococcus, Streptococcus, Enterococcus, Clostridium, Bacillus, Corynebacterium и Listeria. Инфекции с распространенными грамположительными организмами стали более проблематичными для лечения из-за растущей тенденции к резистентности к антибиотикам.
Примеры таких трудноизлечимых резистентных бактерий включают метициллин-резистентный Staphylococcus aureus (MRSA), пенициллин-резистентный Streptococcus pneumoniae (PRSP) и ванкомицин-резистентный Enterococcus (VRE).
Staphylococcus aureus может вызывать ряд заболеваний, таких как акне, импетиго, чирей, флегмона фолликулит, фурункулы, карбункулы, синдром ошпаренной кожи, пневмония, менингит, остеомиелит, эндокардит, синдром токсического шока и сепсис. S. aureus является одной из наиболее распространенных причин внутрибольничных инфекций. Streptococcus pneumoniae может вызывать многие виды инфекций, такие как внебольничная пневмония, бронхит, ринит, острый синусит, воспаление среднего уха, конъюнктивит, менингит, бактериемия, сепсис, остеомиелит, септический артрит, эндокардит, перитонит, перикардит, флегмона и абсцесс головного мозга. Enterococcus может вызывать инфекции мочевыводящих путей, бактериемию, эндокардит, дивертикулит и менингит.
Инфекция Clostridium difficile (CDI) является еще одной проблемной грамположительной бактериальной инфекцией. Смертность от CDI увеличилась из-за распространения гипервирулентного штамма NAP1/027. Современные методы лечения приводят к более чем 23% рецидива и имеют ограничения в отношении этого вирулентного штамма.
Haemophilus influenzae, грамотрицательная бактерия, может вызывать многие виды инфекций, включая, но не ограничиваясь ими, ушные инфекции, бактериемию, внебольничные респираторные инфекции, пневмонию и острый бактериальный менингит.
Лечение бактериальных инфекционных заболеваний становится все более сложным и дорогостоящим из-за развития устойчивости к существующим антибиотикам, распространения гипервирулентных штаммов и отсутствия более эффективных новых антибактериальных средств.
На основании вышеизложенных фактов, авторы настоящего изобретения поняли, что должен существовать новый класс антибактериального средства, имеющий новый механизм действия. После исчерпывающего исследования изобретатели настоящего изобретения обнаружили новые соединения, нацеленные на субъединицу GyrB ДНК-гиразы и/или субъединицу ParE топоизомеразы IV, и, следовательно, готовы удовлетворить потребности миллионов пациентов во всем мире.
В разработке антибиотиков, имеющих новый механизм действия, известны в данной области синтетические ингибиторы, нацеленные на субъединицу GyrB ДНК-гиразы. Например, в WO 2005/026149, WO 2006/087543, WO 2006/087544, WO 2006/087548, WO 2006/092599, WO 2006/092608, WO 2008/152418, WO 2008/020222, WO 2008/020227, WO 2008/020229, WO 2010/013222, WO 2010/067123 и WO 2010/067125 описаны производные пиррола, обладающие антибактериальной активностью. В 2007/071965 описаны бициклические гетероароматические соединения. В WO 2014/57415 описаны соединения на основе хинолина. Эти соединения имели проблемы с недостаточной активностью, низкой растворимостью в воде и токсичностью. Кроме того, ни в одной из цитируемых ссылок не раскрываются производные имидазола.
В WO 2009/084614, включенной в настоящее описание в качестве ссылки в полном объеме, описаны производные имидазола. Соединения, раскрытые в WO 2009/084614, обладают хорошими свойствами, например, достаточной антибактериальной активностью in vitro и отсутствием цитотоксичности. Однако соединение No. 150, имеющее тиадиазольный заместитель, имело проблему неэффективности на животных моделях инфекции, поэтому оно не пригодно для использования у человека.
Соединение No. 150 (WO 2009/084614)
Кроме того, растворимость является низкой по сравнению с соединениями, раскрытыми ниже в этой заявке на патент для пероральной абсорбции.
Неожиданно гидроксиалкилтиадиазольные соединения по настоящему изобретению показали не только достаточную антибактериальную активность in vitro, отсутствие цитотоксичности, хорошую растворимость в воде для пероральной абсорбции, но также превосходную эффективность и безопасность и, следовательно, пригодны для использования у человека.
Таким образом, настоящее изобретение дает большую надежду на то, что новый антибиотик справится с серьезной глобальной проблемой здравоохранения из-за проблемных бактерий, вызывающих бактериальные инфекции, например, но не ограничиваясь ими, внебольничные респираторные инфекции, внутрибольничные инфекции, инфекции мочевыводящих путей и инфекции Clostridium difficile.
Сущность изобретения
Задачи, решаемые изобретением
Как понятно авторам настоящего изобретения, существует потребность в новом антибиотике, обладающем новым механизмом действия, который проявляет сильную антибактериальную активность не только против чувствительных бактерий, но также и против резистентных бактерий и в то же время обладает превосходной растворимостью для пероральной абсорбции и профилем безопасности, пригодными для использования человеком.
Средства для решения задач
В результате интенсивных исследований авторы настоящего изобретения обнаружили, что соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль, ингибирует субъединицу GyrB ДНК-гиразы и/или субъединицу ParE топоизомеразы IV. В частности, соединения по настоящему изобретению проявляют сильную антибактериальную активность и обладают превосходной растворимостью и профилем безопасности, пригодными для использования человеком.
Таким образом, в одном аспекте настоящее изобретение обеспечивает:
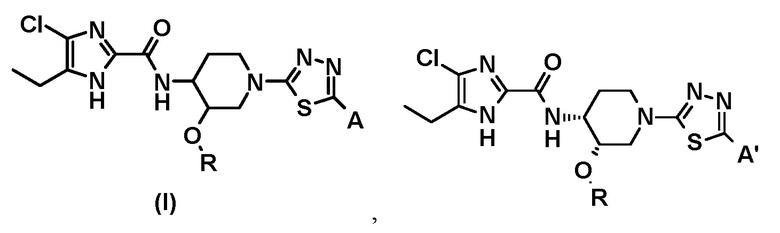
[1] Соединение, представленное общей формулой (I), или его фармацевтически приемлемую соль;
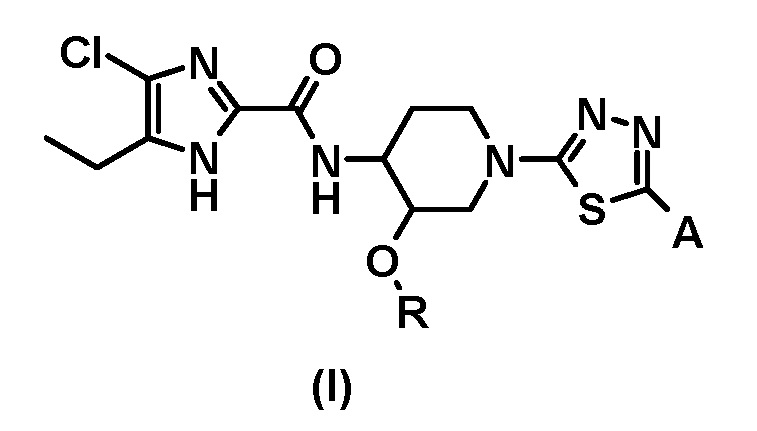
где R представляет собой (C1-C3) алкил, и
A представляет собой следующие формулы:
 или
или 
при условии, что в общей формуле (I), например, включены таутомеры с водородом в различных положениях имидазольного кольца.
[2] Соединение или его фармацевтически приемлемую соль по пункту [1], где соединение общей формулы (I) имеет следующие структуры:
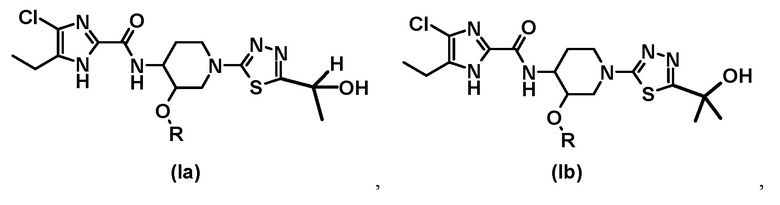
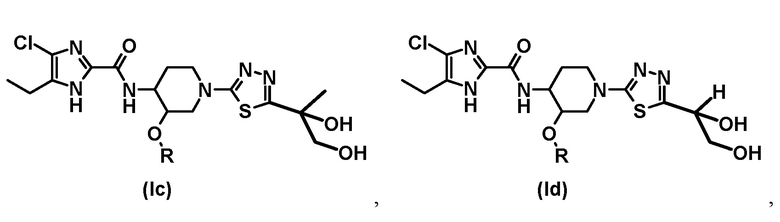
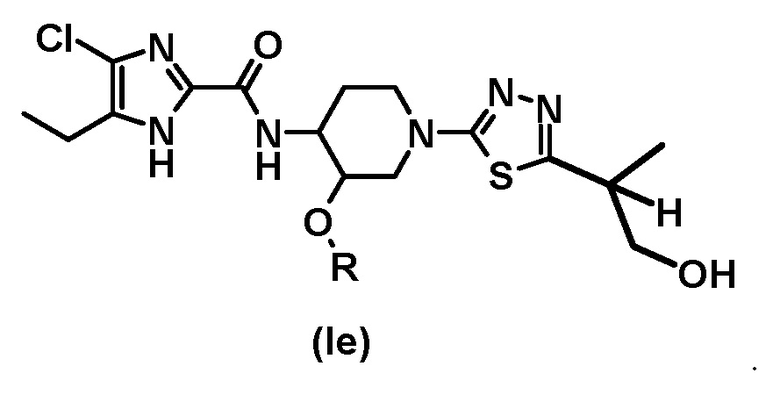
[3] Соединение или его фармацевтически приемлемую соль по пункту [1], где соединение общей формулы (I) имеет следующую структуру:
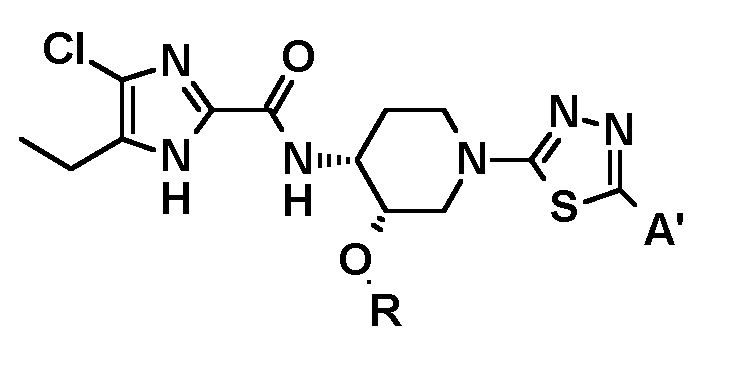
где R представляет собой (C1-C3) алкил, и
A' представляет собой следующие формулы:
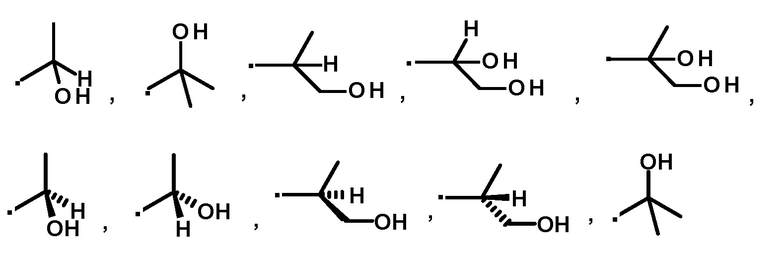
 или
или  .
.
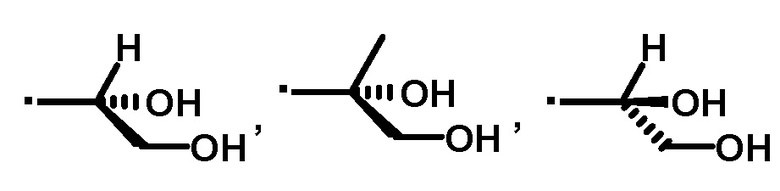
[4] Соединение или его фармацевтически приемлемую соль по пункту [3], где A' представляет собой следующие формулы:
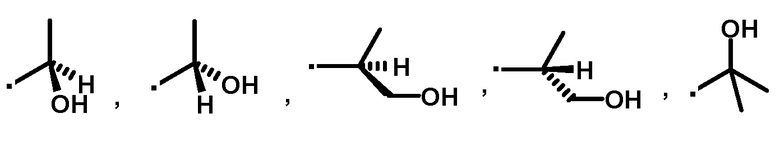
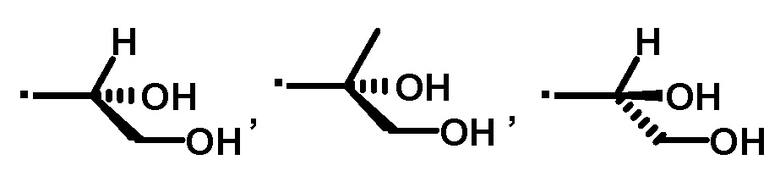 или
или 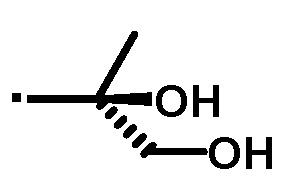
[5] Соединение или его фармацевтически приемлемую соль по пункту [1] или [3], где соединение общей формулы (I) имеет следующую структуру:
[6] Соединение или его фармацевтически приемлемую соль по пункту [1] или [4], где соединение общей формулы (I) имеет следующие структуры:
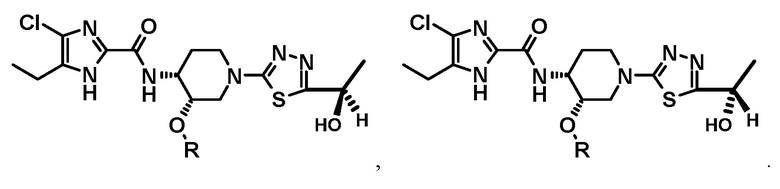
[7] Соединение или его фармацевтически приемлемую соль по пункту [1] или [4], где соединение общей формулы (I) имеет следующие структуры:
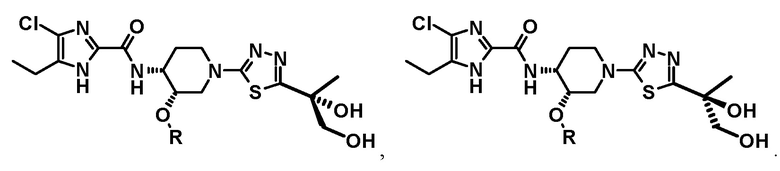
[8] Соединение или его фармацевтически приемлемую соль по пункту [1] или [4], где соединение общей формулы (I) имеет следующие структуры:
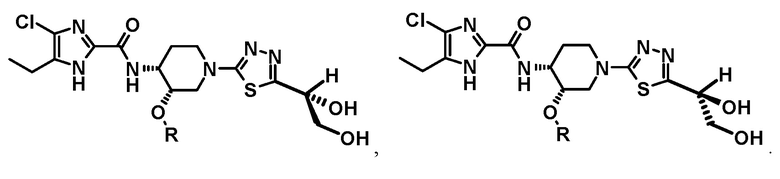
[9] Соединение или его фармацевтически приемлемую соль по любому из пунктов [1]-[8], где R представляет собой метил.
[10] Соединение или его фармацевтически приемлемую соль по любому из пунктов [1]-[8], где R представляет собой этил.
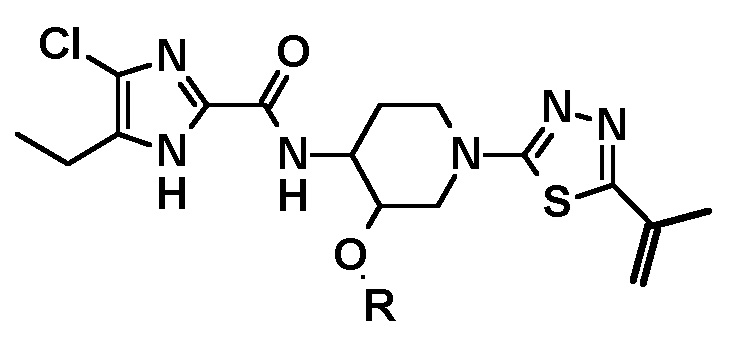
[11] Соединение или его фармацевтически приемлемую соль по любому из пунктов [1]-[10], где соединение выбрано из:
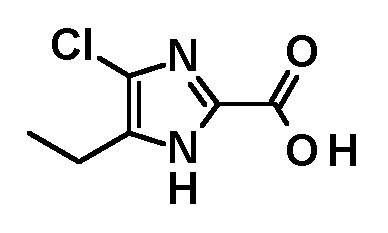
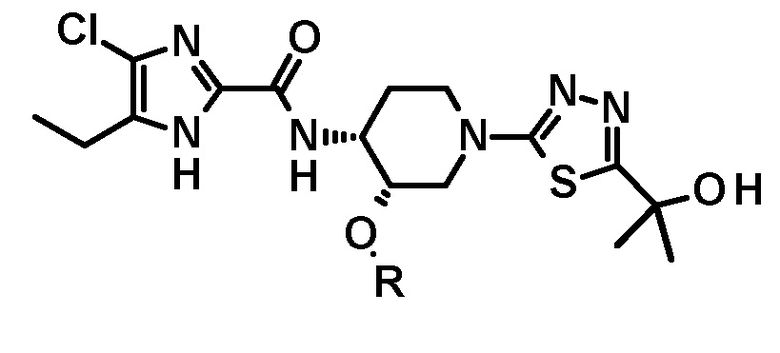
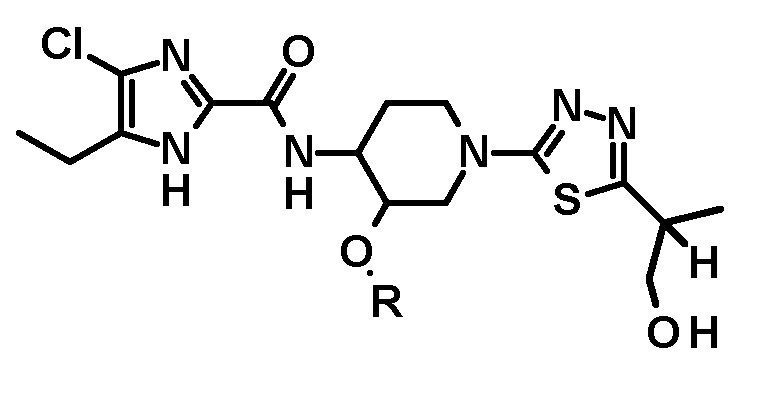
4-хлор-N-{(3S,4R)-3-этокси-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида (Соединение No. 1),
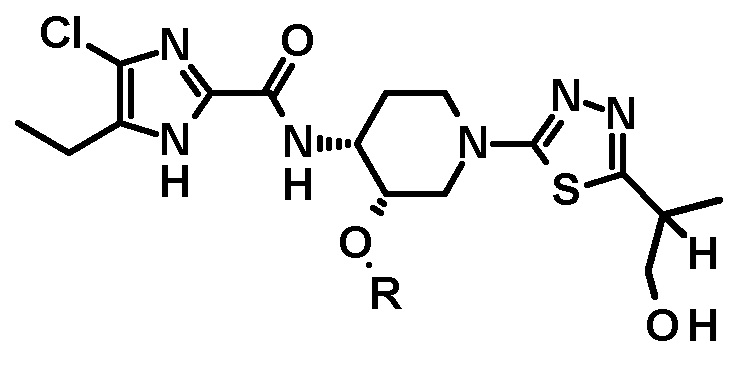
4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида (Соединение No. 2),
4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида (Соединение No. 3),
4-хлор-5-этил-N-[(3S,4R)-1-{5-[(1R)-1-гидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида (Соединение No. 4),
4-хлор-5-этил-N-[(3S,4R)-1-{5-[(1S)-1-гидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида (Соединение No. 5),
4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида (Соединение No. 6),
4-хлор-N-{(3S,4R)-3-этокси-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида (Соединение No. 7),
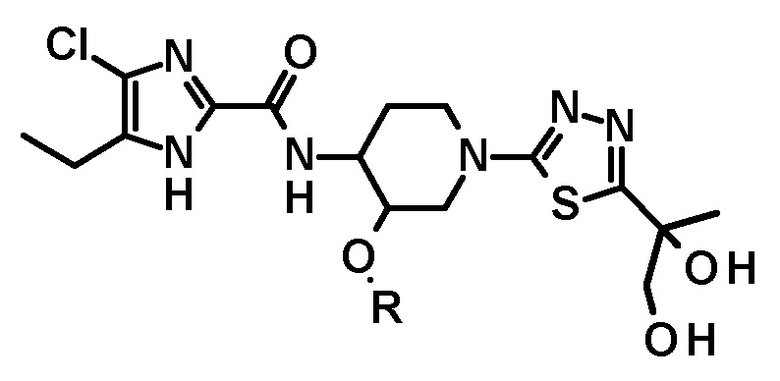
4-хлор-N-[(3S,4R)-1-{5-[(1S)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (Соединение No. 8),
4-хлор-N-[(3S,4R)-1-{5-[(1R)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (Соединение No. 9),
4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (Соединение No. 10),
4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (Соединение No. 11).
[12] Кристаллическую 2/3 гидратную форму 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида, характеризующуюся спектром порошковой рентгеновской дифракции (XRD), по существу, в соответствии с картиной, показанной на фиг. 2.
[13] Безводную кристаллическую форму 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида, характеризующуюся спектром порошковой рентгеновской дифракции (XRD), по существу, в соответствии с картиной, показанной на фиг.5.
[14] Кристаллическую форму 4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида, характеризующуюся спектром порошковой рентгеновской дифракции (XRD), по существу, в соответствии с картиной, показанной на фиг. 8.
[15] Кристаллическую форму 4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида, характеризующуюся спектром порошковой рентгеновской дифракции (XRD), по существу, в соответствии с картиной, показанной на фиг. 9.
[16] Безводную кристаллическую форму 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида.
[17] Фармацевтическую композицию, содержащую терапевтически эффективное количество соединения или его фармацевтически приемлемой соли по любому из пунктов [1]-[16] в качестве его активного ингредиента.
[18] Фармацевтическую композицию по пункту [17], где указанную фармацевтическую композицию вводят для лечения или профилактики бактериальных инфекционных заболеваний.
[19] Фармацевтическую композицию по пункту [18], где указанные бактериальные инфекционные заболевания вызваны грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus, Clostridium, Bacillus, Corynebacterium и Listeria.
[20] Фармацевтическую композицию по пункту [18], где указанные бактериальные инфекционные заболевания вызваны резистентными бактериями, выбранными из метициллин-резистентного Staphylococcus aureus (MRSA), пенициллин-резистентного Streptococcus pneumoniae (PRSP) и ванкомицин-резистентного Enterococcus (VRE).
[21] Фармацевтическую композицию по пункту [18], где указанные бактериальные инфекционные заболевания выбраны из акне, импетиго, чириев, флегмона фолликулита, фурункулов, карбункулов, синдрома ошпаренной кожи, пневмонии, менингита, остеомиелита, эндокардита, синдрома токсического шока, бронхита, ринита, острого синусита, воспаления среднего уха, конъюнктивита, бактериемии, сепсиса, остеомиелита, септического артрита, перитонита, перикардита, флегмона, абсцесса головного мозга, инфекций мочевыводящих путей, инфекций Clostridium difficile, обыкновенных угрей, гонореи, газовой гангрены, пищевого отравления, тетануса, ботулизма, диареи, псевдомембранозного колита, токсического мегаколона и перфорации толстой кишки.
[22] Фармацевтическую композицию по пункту [18], где указанные бактериальные инфекционные заболевания выбраны из внебольничных респираторных инфекций, внутрибольничных инфекций или инфекций Clostridium difficile.
[23] Способ лечения бактериального инфекционного заболевания у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения или его фармацевтической соли по любому из пунктов [1]-[16].
[24] Способ по пункту [23], где указанные бактериальные инфекционные заболевания вызваны грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus, Clostridium, Bacillus, Corynebacterium и Listeria.
[25] Способ по пункту [23], где указанные бактериальные инфекционные заболевания вызваны резистентными бактериями, выбранными из метициллин-резистентного Staphylococcus aureus (MRSA), пенициллин-резистентного Streptococcus pneumoniae (PRSP) и ванкомицин-резистентного Enterococcus (VRE).
[26] Способ по пункту [23], где указанные бактериальные инфекционные заболевания выбраны из акне, импетиго, чириев, флегмона фолликулита, фурункулов, карбункулов, синдрома ошпаренной кожи, пневмонии, менингита, остеомиелита, эндокардита, синдрома токсического шока, бронхита, ринита, острого синусита, воспаления среднего уха, конъюнктивита, бактериемии, сепсиса, остеомиелита, септического артрита, перитонита, перикардита, флегмоны, абсцесса головного мозга, инфекций мочевыводящих путей, инфекций Clostridium difficile, обыкновенных угрей, гонореи, газовой гангрены, пищевого отравления, тетануса, ботулизма, диареи, псевдомембранозного колита, токсического мегаколона и перфорации толстой кишки.
[27] Способ по пункту [23], где указанные бактериальные инфекционные заболевания выбраны из внебольничных респираторных инфекций, внутрибольничных инфекций или инфекций Clostridium difficile.
[28] Соединение или его фармацевтически приемлемую соль по любому из пунктов [1]-[16] для использования в качестве фармацевтического средства для лечения бактериальных инфекционных заболеваний.
[29] Применение по пункту [28], где указанные бактериальные инфекционные заболевания вызваны грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus, Clostridium, Bacillus, Corynebacterium и Listeria.
[30] Применение по пункту [28], где указанные бактериальные инфекционные заболевания вызваны резистентными бактериями, выбранными из метициллин-резистентного Staphylococcus aureus (MRSA), пенициллин-резистентного Streptococcus pneumoniae (PRSP) и ванкомицин-резистентного Enterococcus (VRE).
[31] Применение по пункту [28], где указанные бактериальные инфекционные заболевания выбраны из акне, импетиго, чириев, флегмона фолликулита, фурункулов, карбункулов, синдрома ошпаренной кожи, пневмонии, менингита, остеомиелита, эндокардита, синдрома токсического шока, бронхита, ринита, острого синусита, воспаления среднего уха, конъюнктивита, бактериемии, сепсиса, остеомиелита, септического артрита, перитонита, перикардита, флегмоны, абсцесса головного мозга, инфекций мочевыводящих путей, инфекций Clostridium difficile, обыкновенных угрей, гонореи, газовой гангрены, пищевого отравления, тетануса, ботулизма, диареи, псевдомембранозного колита, токсического мегаколона и перфорации толстой кишки.
[32] Применение по пункту [28], где указанные бактериальные инфекционные заболевания выбраны из внебольничных респираторных инфекций, внутрибольничных инфекций или инфекций Clostridium difficile.
[33] Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV для использования при лечении бактериальных инфекционных заболеваний, имеющий структуру общей формулы (I), его полиморфная форма, гидрат или фармацевтически приемлемая соль:
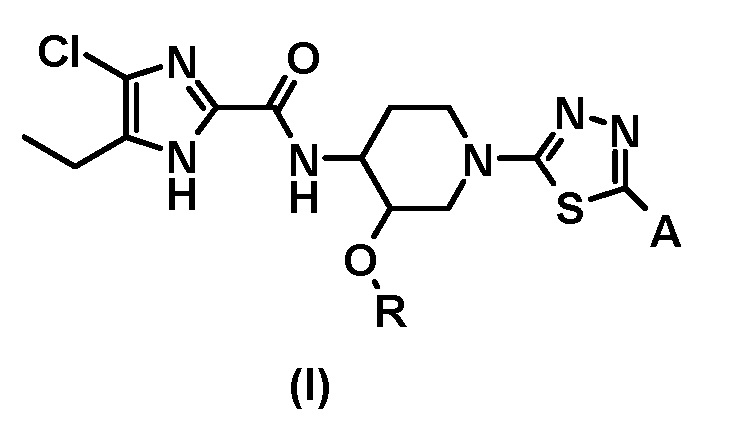
где R представляет собой (C1-C3) алкил, и
A представляет собой следующие формулы:
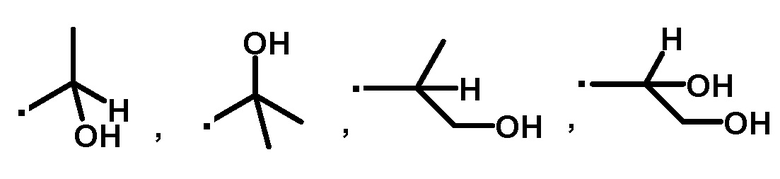 или
или 
при условии, что в общей формуле (I), например, включены таутомеры с водородом в различных положениях имидазольного кольца.
[34] Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV по пункту [33], где соединение выбрано из:
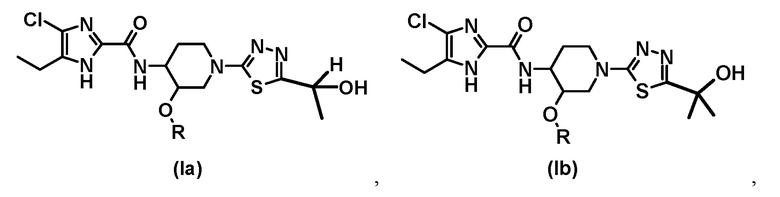
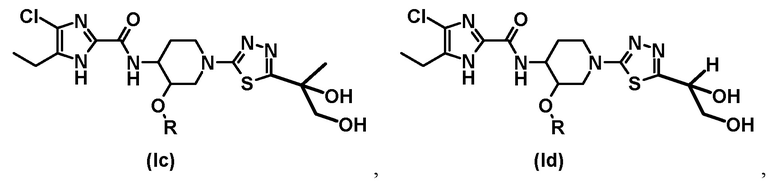
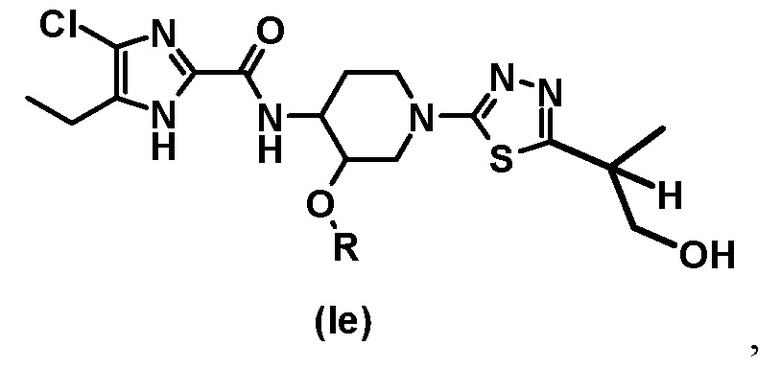
где R представляет собой (C1-C3) алкил.
[35] Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV по пункту [33], где соединение представлено:
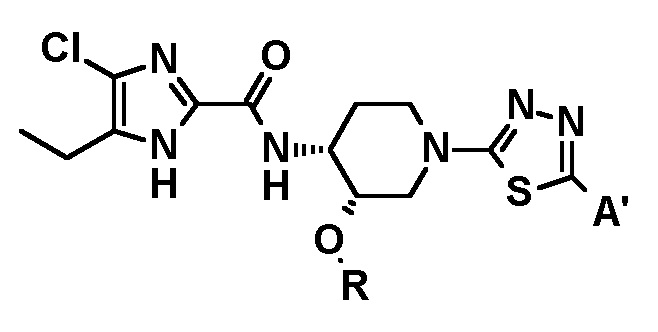
где R представляет собой (C1-C3) алкил, и
A' представляет собой следующие формулы:
 или
или 
[36] Соединение или его фармацевтически приемлемая соль по пункту [35], где A' представляет собой следующие формулы:
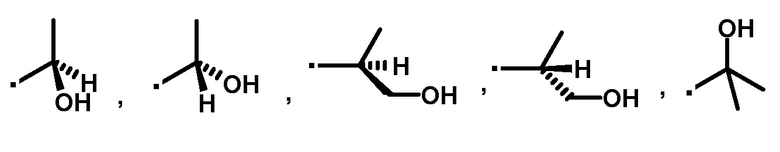
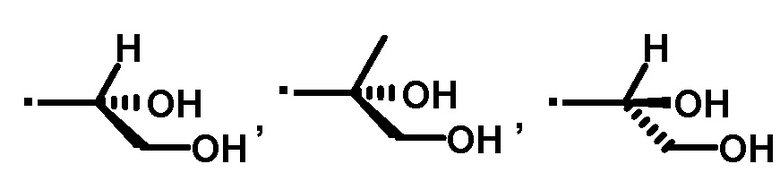 или
или 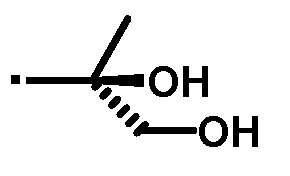
[37] Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV по любому из пунктов [33]-[36], где указанные бактериальные инфекционные заболевания вызваны грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus, Clostridium, Bacillus, Corynebacterium и Listeria.
[38] Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV по любому из пунктов [33]-[36], где указанные бактериальные инфекционные заболевания вызваны резистентными бактериями, выбранными из метициллин-резистентного Staphylococcus aureus (MRSA), пенициллин-резистентного Streptococcus pneumoniae (PRSP) и ванкомицин-резистентного Enterococcus (VRE).
[39] Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV по любому из пунктов [33]-[36], где указанные бактериальные инфекционные заболевания выбраны из акне, импетиго, чириев, флегмона фолликулита, фурункулов, карбункулов, синдрома ошпаренной кожи, пневмонии, менингита, остеомиелита, эндокардита, синдрома токсического шока, бронхита, ринита, острого синусита, воспаления среднего уха, конъюнктивита, бактериемии, сепсиса, остеомиелита, септического артрита, перитонита, перикардита, флегмона, абсцесса головного мозга, инфекций мочевыводящих путей, инфекций Clostridium difficile, обыкновенных угрей, гонореи, газовой гангрены, пищевого отравления, тетануса, ботулизма, диареи, псевдомембранозного колита, токсического мегаколона и перфорации толстой кишки.
[40] Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV по любому из пунктов [33]-[36], где указанные бактериальные инфекционные заболевания выбраны из внебольничных респираторных инфекций, внутрибольничных инфекций или инфекций Clostridium difficile.
[41] Применение соединения, его полиморфной формы или фармацевтически приемлемой соли по любому из пунктов [1]-[16] для получения терапевтического средства для бактериальных инфекций.
[42] Применение по пункту [41], где указанные бактериальные инфекции вызваны грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus, Clostridium, Bacillus, Corynebacterium и Listeria.
[43] Применение по пункту [42], где резистентные бактерии выбраны из метициллин-резистентного Staphylococcus aureus (MRSA), пенициллин-резистентного Streptococcus pneumoniae (PRSP) и ванкомицин-резистентного Enterococcus (VRE).
[44] Применение по любому из пунктов [41]-[42], где указанные бактериальные инфекционные заболевания выбраны из акне, импетиго, чириев, флегмона фолликулита, фурункулов, карбункулов, синдрома ошпаренной кожи, пневмонии, менингита, остеомиелита, эндокардита, синдрома токсического шока, бронхита, ринита, острого синусита, воспаления среднего уха, конъюнктивита, бактериемии, сепсиса, остеомиелита, септического артрита, перитонита, перикардита, флегмоны, абсцесса головного мозга, инфекций мочевыводящих путей, инфекций Clostridium difficile, обыкновенных угрей, гонореи, газовой гангрены, пищевого отравления, тетануса, ботулизма, диареи, псевдомембранозного колита, токсического мегаколона и перфорации толстой кишки.
[45] Применение по пункту [44], где указанные бактериальные инфекционные заболевания выбраны из внебольничных респираторных инфекций, внутрибольничных инфекций или инфекций Clostridium difficile.
[46] Способ получения соединения следующей формулы, его стереоизомера или фармацевтически приемлемой соли:
в котором диметилирование проводят на карбонильном атоме углерода -C(=O)-O-R1 фрагмента соединения, имеющего следующую формулу, или его стереоизомера:
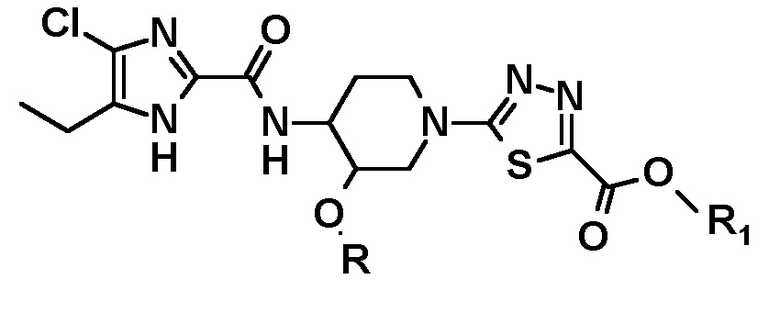
где R представляет собой (C1-C3) алкильную группу и R1 представляет собой алкильную группу.
[47] Способ по пункту [46], где диметилирование проводят с помощью реакции Гриньяра.
[48] Способ получения соединения следующей формулы, его стереоизомера или фармацевтически приемлемой соли:
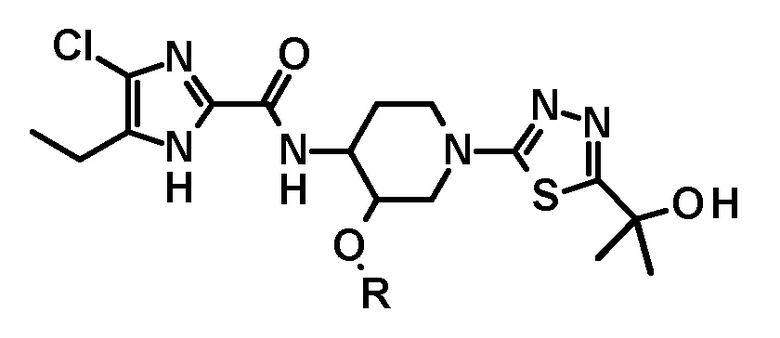
в котором соединение следующей формулы или его стереоизомер:
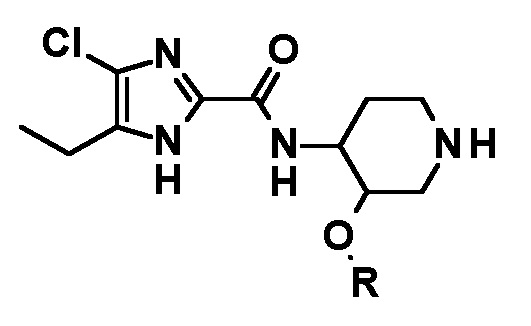
подвергают взаимодействию с соединением следующей формулы:
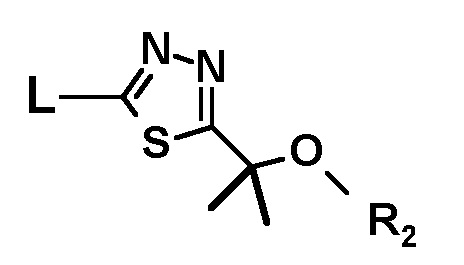
где L представляет собой уходящую группу, R2 представляет собой защитную группу для гидроксигруппы, и R имеет значения, определенные в пункте [46], с получением соединения следующей формулы или его стереоизомера:
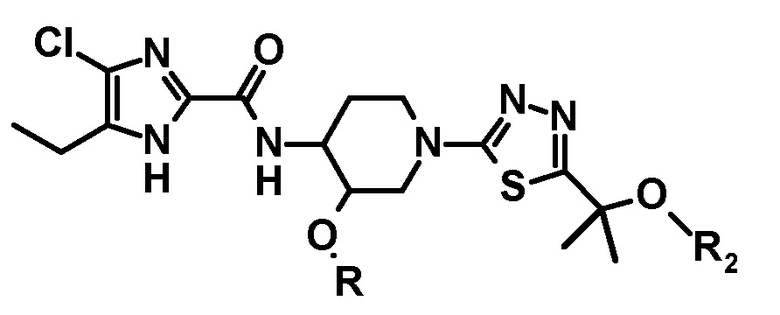
и затем защитную группу удаляют.
[49] Способ по пункту [48], где L представляет собой атом галогена.
[50] Способ по пункту [49], где L представляет собой атом брома.
[51] Способ получения соединения, имеющего следующую формулу, его стереоизомера или фармацевтически приемлемой соли:
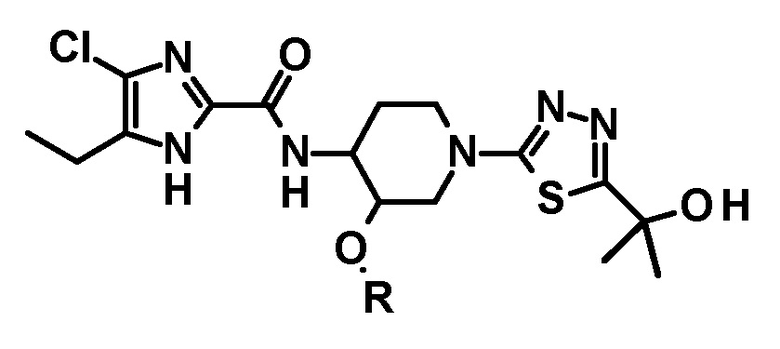
в котором соединение следующей формулы или его стереоизомер:
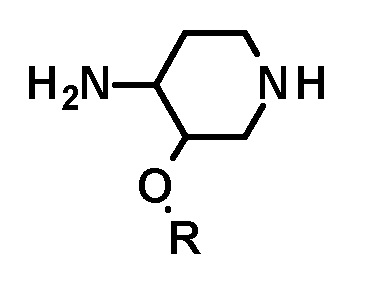
подвергают взаимодействию с соединением следующей формулы:
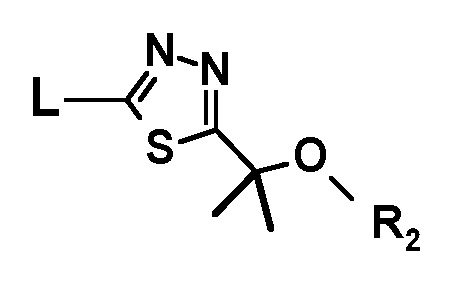
с получением соединения, имеющего следующую формулу, или его стереоизомера:
и указанное соединение подвергают взаимодействию с соединением, имеющим следующую формулу:
с получением соединения, имеющего следующую формулу, или его стереоизомера:
и затем защитную группу удаляют, где L, R и R2 имеют значения, определенные в пункте [48].
[52] Способ по любому из пунктов [48] 51], где R2 представляет собой алкильную группу, аралкильную группу или ацильную группу.
[53] Способ по пункту [52], где R2 представляет собой ацильную группу.
[54] Способ по любому из пунктов [48]-[51], где R2 выбран из группы метильной группы, трет-бутильной группы, бензильной группы, п-метоксибензильной группы, метоксиметильной группы, этоксиметильной группы, 2-тетрагидропиранильной группы, ацетильной группы, пивалоильной группы и бензоильной группы.
[55] Способ по пункту [54], где R2 выбран из группы ацетильной группы, пивалоильной группы и бензоильной группы.
[56] Способ по пункту [54], где R2 представляет собой бензоильную группу.
[57] Способ по любому из пунктов [46]-[56], где полученное таким образом соединение имеет следующую структуру:
 .
.
[58] Способ по любому из пунктов [46]-[57], где R представляет собой метильную группу или этильную группу.
[59] Способ по любому из пунктов [46]-[57], где R представляет собой метильную группу.
[60] Способ получения соединения следующей формулы, его стереоизомера или фармацевтически приемлемой соли:
в котором соединение следующей формулы или его стереоизомер:
восстанавливают с получением соединения следующей формулы или его стереоизомера:
и затем окисление проводят с указанным соединением с получением соединения следующей формулы или его стереоизомера:
и метилирование проводят на атоме углерода формильной группы указанного соединения.
[61] Способ по пункту [60], где восстановление проводят боргидридом.
[62] Способ по пункту [61], где восстановление проводят боргидридом натрия.
[63] Способ по любому из пунктов [60]-[62], где метилирование проводят с помощью реакции Гриньяра.
[64] Способ по любому из пунктов [60]-[63], где полученное таким образом соединение имеет структуру
 или
или 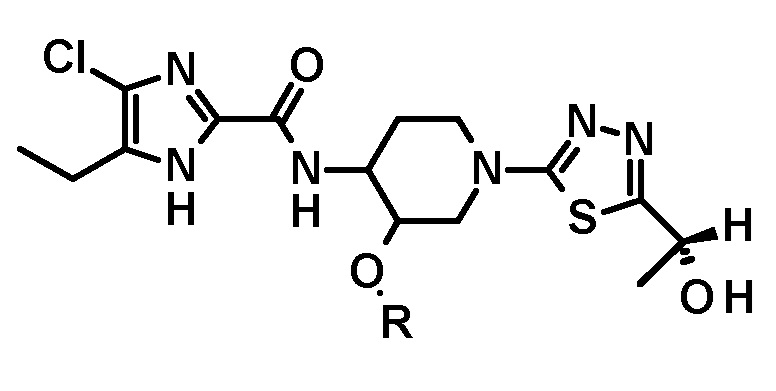 .
.
[65] Способ по любому из пунктов [60]-[64], где полученное таким образом соединение имеет структуру
 или
или 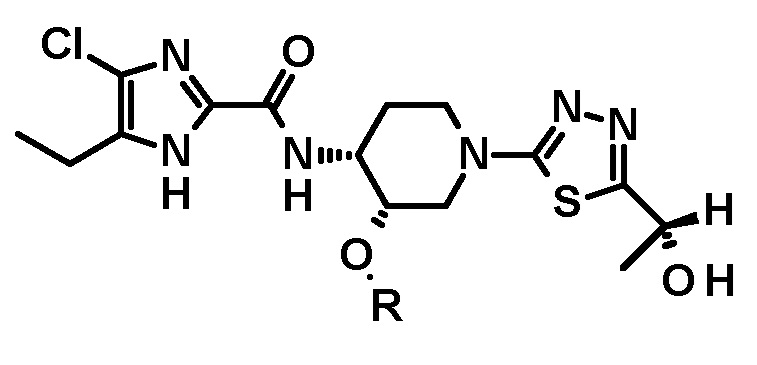 .
.
[66] Способ получения соединения следующей формулы, его стереоизомера или фармацевтически приемлемой соли:
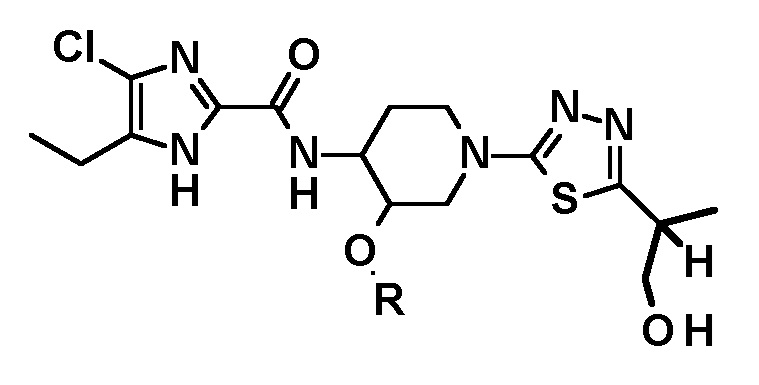
в котором соединение следующей формулы или его стереоизомер:
подвергают взаимодействию с бораном и затем обрабатывают пероксидом водорода.
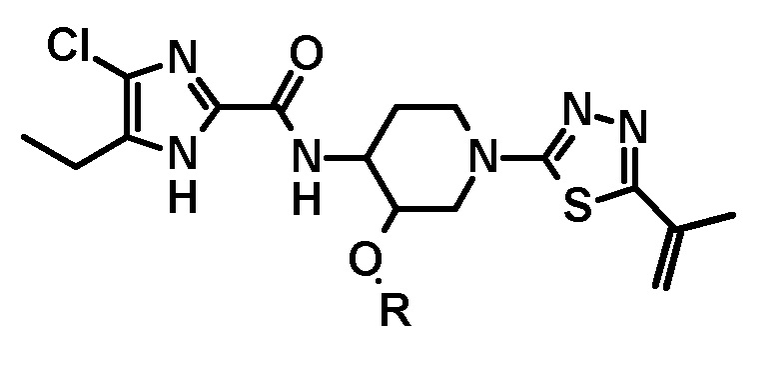
[67] Способ по пункту [66], где полученное таким образом соединение имеет структуру
 .
.
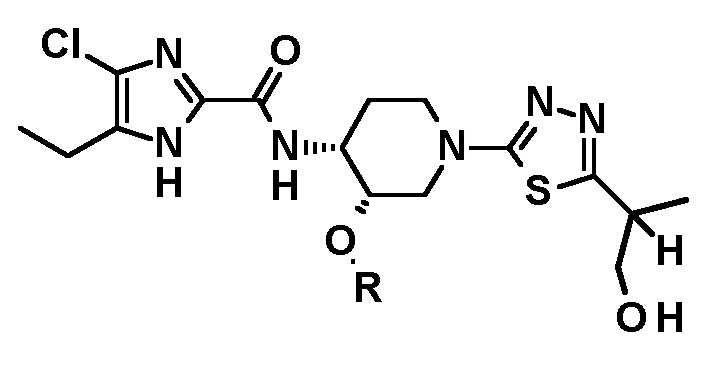
[68] Способ получения соединения следующей формулы, его стереоизомера или фармацевтически приемлемой соли:
в котором дигидроксилирование проводят на 5-этенильной группе 1,3,4-тиадиазольного фрагмента соединения следующей формулы или его стереоизомера:
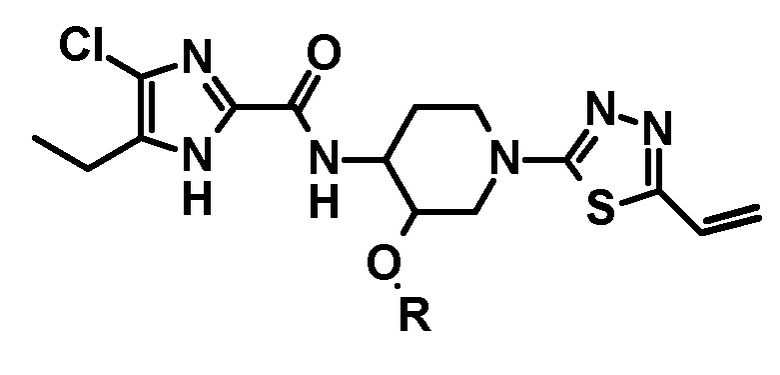 .
.
[69] Способ по пункту [68], где дигидроксилирование представляет собой асимметрическое дигидроксилирование.
[70] Способ по пункту [69], где асимметрическое дигидроксилирование представляет собой асимметрическое дигидроксилирование по Шарплессу.
[71] Способ по любому из пунктов [68] или [70], где полученное таким образом соединение имеет структуру
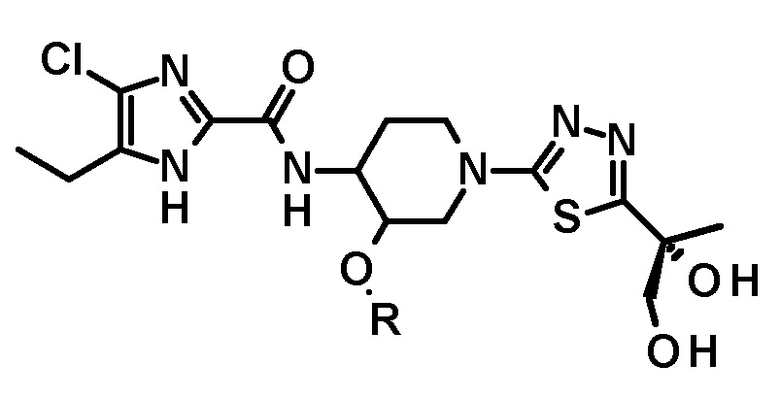 или
или 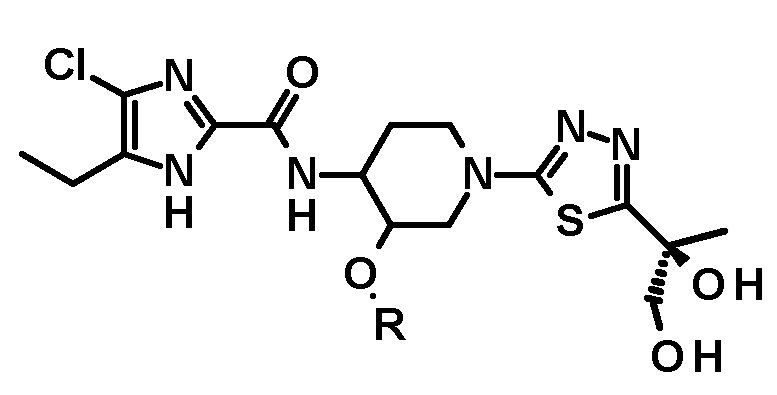 .
.
[72] Способ по любому из пунктов [68] или [70], где полученное таким образом соединение имеет структуру
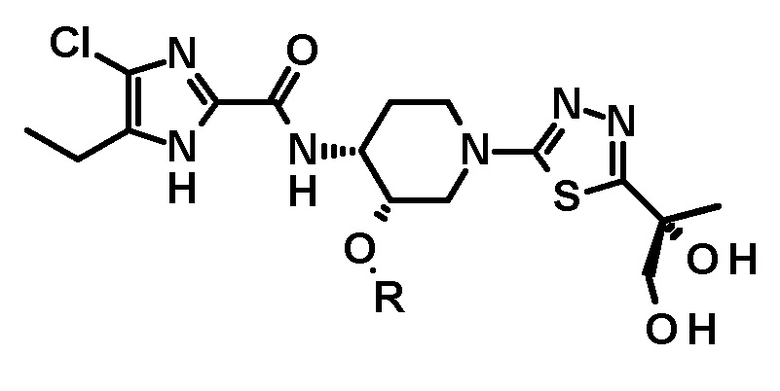 или
или 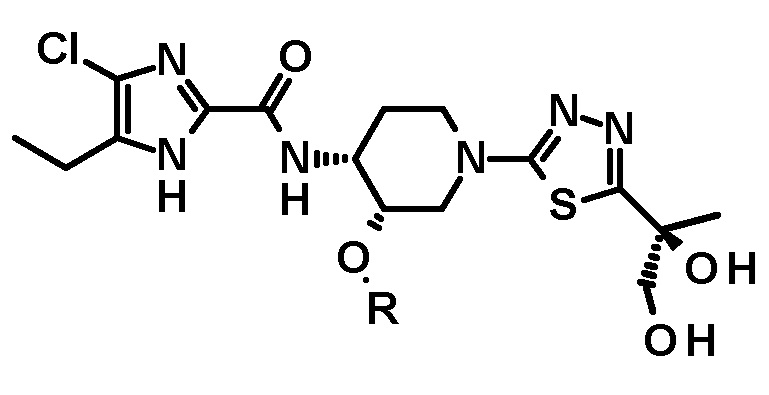 .
.
Краткое описание фигур
Различные аспекты настоящего изобретения проиллюстрированы со ссылкой на следующие чертежи.
Фигура 1: Эффективность в мышиной модели инфекции легких пенициллин-резистентного Streptococcus pneumoniae (PRSP).
Фигура 2: Картина порошковой рентгеновской дифракции (XRD) для кристаллического 2/3 гидрата 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида (Соединение No. 2).
Фигура 3: График термогравиметрического анализа (график TG/DTA) для кристаллического 2/3 гидрата 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида.
Фигура 4: Картина изменения массы для кристаллического 2/3 гидрата 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида.
Фигура 5: Картина порошковой рентгеновской дифракции (XRD) для безводной кристаллической формы 4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидрокси-1-метилэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида.
Фигура 6: График термогравиметрического анализа (график TG/DTA) для безводной кристаллической формы 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида.
Фигура 7: Картина изменения массы для безводной кристаллической формы 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида.
Фигура 8: Картина порошковой рентгеновской дифракции (XRD) для 4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида.
Фигура 9: Картина порошковой рентгеновской дифракции (XRD) для 4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида.
Вышеупомянутые аспекты и варианты осуществления и другие аспекты, объекты, признаки и преимущества настоящего изобретения будут очевидны из последующего подробного описания и прилагаемой формулы изобретения.
Подробное описание изобретения
Как используется в настоящем описании, используются следующие определения, если явно не указано иное.
Следует понимать, что, если прямо не указано иное, ʺсоединение общей формулы (I)ʺ относится к и включает любое и все соединения, описываемые формулой (I), их варианты, а также подроды, включая вс соли, их стереоизомеры. Следует также отметить, что формы единственного числа включают множественное число, если контекст явно не диктует иное.
В одном аспекте настоящего изобретения обеспечивается соединение общей формулы (I), его стереоизомер или фармацевтически приемлемая соль:
где R, A и A' имеют значения, определенные выше.
Соединение общей формулы (I) может иметь таутомеры с водородом в различных положениях имидазольного кольца. Все такие таутомеры входят в объем настоящего изобретения. Соединение общей формулы (I) включает следующие структуры:
 .
.
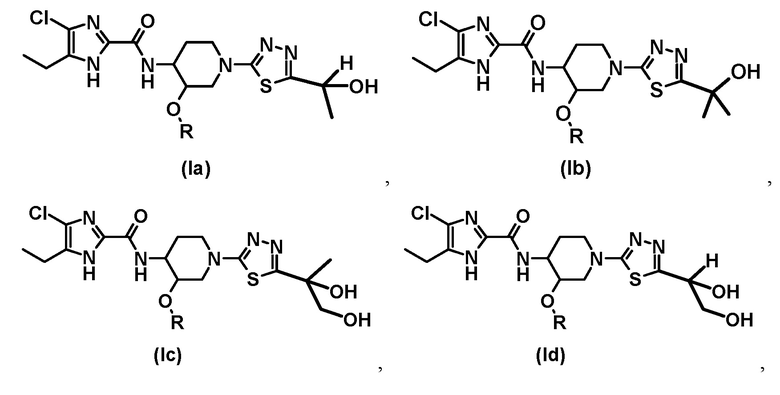
В предпочтительном варианте осуществления обеспечивается соединение формулы (Ia), его стереоизомер или фармацевтически приемлемая соль;
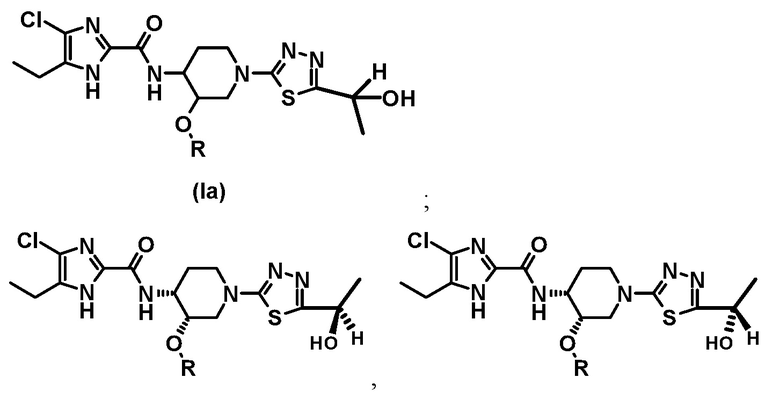
где R представляет собой (C1-C3) алкил, и включены таутомеры с водородом в различных положениях имидазольного кольца.
В другом предпочтительном варианте осуществления обеспечивается соединение формулы (Ib), его стереоизомер или фармацевтически приемлемая соль;
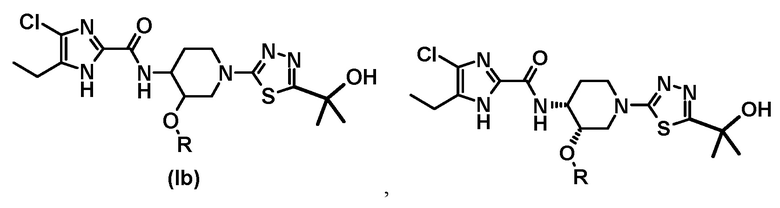
где R представляет собой (C1-C3) алкил, и включены таутомеры с водородом в различных положениях имидазольного кольца.
В другом предпочтительном варианте осуществления обеспечивается соединение формулы (Iс), его стереоизомер или фармацевтически приемлемая соль;
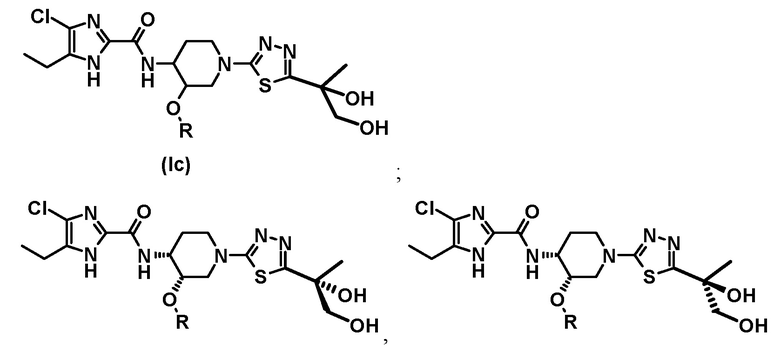
где R представляет собой (C1-C3) алкил; и включены таутомеры с водородом в различных положениях имидазольного кольца.
В другом предпочтительном варианте осуществления обеспечивается соединение формулы (Id), его стереоизомер или фармацевтически приемлемая соль;
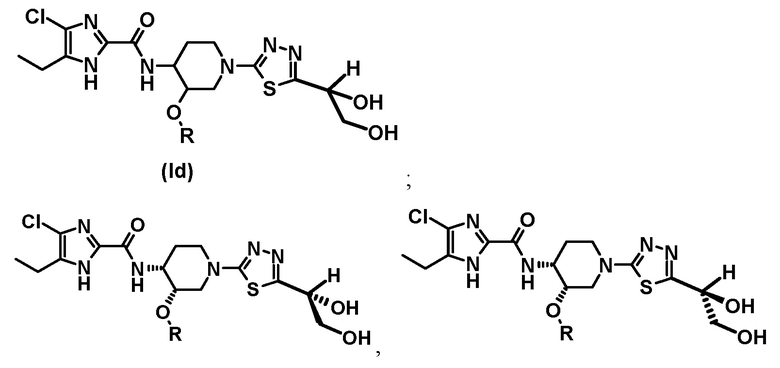
где R представляет собой (C1-C3) алкил; и включены таутомеры с водородом в различных положениях имидазольного кольца.
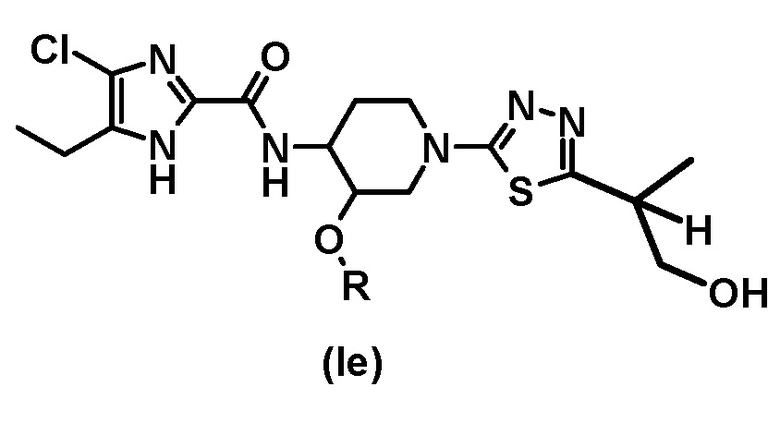
В другом предпочтительном варианте осуществления обеспечивается соединение формулы (Iе), его стереоизомер или фармацевтически приемлемая соль;
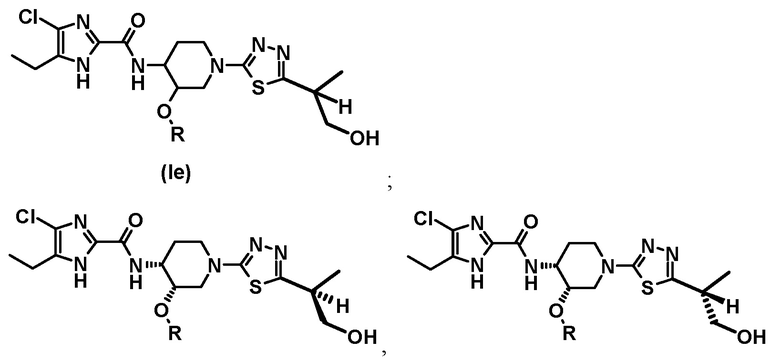
где R представляет собой (C1-C3) алкил; и включены таутомеры с водородом в различных положениях имидазольного кольца.
Настоящее изобретение предполагает включение в объем первого аспекта различные предпочтительные варианты осуществления для улучшения изобретения, как указано в разделе ʺуровень техникиʺ.
Например, в одном варианте осуществления обеспечивается соединение формулы (Iа), его стереоизомер, фармацевтически приемлемая соль, где R представляет собой метил или этил.
В другом варианте осуществления обеспечивается соединение формулы (Ib), его стереоизомер, фармацевтически приемлемая соль, где R представляет собой метил или этил.
В другом варианте осуществления обеспечивается соединение формулы (Ic), его стереоизомер, фармацевтически приемлемая соль, где R представляет собой метил или этил.
В другом варианте осуществления обеспечивается соединение формулы (Id), его стереоизомер, фармацевтически приемлемая соль, где R представляет собой метил или этил.
В другом варианте осуществления обеспечивается соединение формулы (Ie), его стереоизомер, фармацевтически приемлемая соль, где R представляет собой метил или этил.
В соответствии с конкретным вариантом осуществления настоящего изобретения обеспечивается конкретное соединение формулы (I), которое выбрано из:
4-хлор-N-{(3S,4R)-3-этокси-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-[(3S,4R)-1-{5-[(1R)-1-гидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-[(3S,4R)-1-{5-[(1S)-1-гидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида,
4-хлор-N-{(3S,4R)-3-этокси-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-N-[(3S,4R)-1-{5-[(1S)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-N-[(3S,4R)-1-{5-[(1R)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида,
его фармацевтически приемлемая соль.
В предпочтительном варианте осуществления обеспечивается кристаллическая 2/3 гидратная форма 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида, обозначенная как форма I, характеризующаяся спектром порошковой рентгеновской дифракции (XRD), по существу, в соответствии с картиной, показанной на фиг. 2.
Форма I дополнительно характеризуется i) графиком термогравиметрического анализа (график TG/DTA), по существу, в соответствии с графиком, показанным на фиг.3, и ii) изменением массы по существу в соответствии с картиной, показанной на фиг.4.
В другом варианте осуществления обеспечивается безводная кристаллическая форма 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида, обозначенная как форма II, характеризующуюся спектром порошковой рентгеновской дифракции (XRD), по существу, в соответствии с картиной, показанной на фиг. 5.
Форма II дополнительно характеризуется i) графиком термогравиметрического анализа (график TG/DTA), по существу, в соответствии с графиком, показанным на фиг.6, и ii) изменением массы по существу в соответствии с картиной, показанной на фиг.7.
В другом варианте осуществления обеспечивается кристаллическая форма 4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида, обозначенная как форма A, характеризующаяся спектром порошковой рентгеновской дифракции (XRD), по существу, в соответствии с картиной, показанной на фиг. 8.
В другом варианте осуществления обеспечивается кристаллическая форма 4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида, обозначенная как форма B, характеризующаяся спектром порошковой рентгеновской дифракции (XRD), по существу, в соответствии с картиной, показанной на фиг. 9.
В другом варианте осуществления обеспечивается безводная кристаллическая форма 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида.
В другом предпочтительном варианте осуществления форма I, форма II, форма A и форма B дополнительно характеризуются выраженными пиками XRD, как представлено в таблице 1.
XRD (CuK, λ=1,54Å, скорость сканирования=20°/минута*)
В предпочтительном варианте осуществления Форма I характеризуется основными пиками XRD при 8,38, 10,90, 14,50, 14,94, 19,82 и 24,68 (2θ).
В другом предпочтительном варианте осуществления Форма II характеризуется основными пиками XRD при 13,20, 14,94, 16,08, 17,76, 19,46 и 24,82 (2θ).
В другом предпочтительном варианте осуществления Форма A характеризуется основными пиками XRD при 15,70, 17,06, 19,10 и 21,92 (2θ).
В еще одном предпочтительном варианте осуществления Форма B характеризуется основными пиками XRD при 13,13, 14,27 и 19,10 (2θ).
В настоящем изобретении должно быть понятно, что соединение общей формулы (I) или его соль может иногда проявлять явление таутомерии, и формулы и фигуры в настоящем описании могут представлять только одну из возможных таутомерных форм. Следует понимать, что настоящее изобретение охватывает любую таутомерную форму, которая ингибирует субъединицу GyrB ДНК-гиразы и/или субъединицу ParE топоизомеразы IV и не ограничивается только одной из таутомерных форм, используемых в формулах или фигурах. Следует понимать, что формулы и фигуры в настоящем описании могут представлять только одну из возможных таутомерных форм, и настоящее описание охватывает не только формы, которые могут быть показаны в формулах, но также и все возможные таутомерные формы соединений, показанных в формулах. То же самое относится и к названиям соединения.
Соединение по настоящему изобретению, представленное общей формулой (I), или его фармацевтически приемлемая соль, если его оставить на воздухе или перекристаллизовывать, может поглощать воду для связывания с адсорбированной водой или с образованием гидрата. Такие водосодержащие соединения и соли также охватываются настоящим изобретением.
Соединение по настоящему изобретению, представленное общей формулой (I), имеет основную группу, поэтому его «фармацевтически приемлемая соль» может быть образована путем взаимодействия соединения с кислотой.
Термин «фармацевтически приемлемый», как используется в настоящем описании, относится к соединению формулы (I) или его фармацевтической композиции, подходящему для введения животным, предпочтительно человеку, одобренному органом государственного регулирования и контроля, таким как Европейское агентство по лекарственным средствам (EMEA), Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) или любым другим Национальным Органом Государственного регулирования и контроля.
Предпочтительные примеры соли по настоящему изобретению включают, но не ограничиваясь ими, гидрохлорид, гидробромид, гидроиодид, нитрат, перхлорат, сульфат, фосфат, метансульфонат, трифторметансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат, ацетат, малат, фумарат, сукцинат, цитрат, аскорбат, тартрат, оксалат, малеат и соли аминокислоты, такие как соль глицина, соль лизина, соль аргинина, соль орнитина, глутамат и аспартат.
Соединение, представленное общей формулой (I), или его фармацевтически приемлемая соль имеет асимметрический атом углерода в молекуле, поэтому включены стереоизомеры с конфигурацией R или S. Каждый из этих стереоизомеров и все смеси стереоизомеров произвольных соотношениях также охватываются настоящим изобретением. Такие стереоизомеры могут быть получены, например, путем синтеза соединения (I) с использованием соответствующих разрешающих агентов или путем оптического расщепления синтезированного соединения (I) с помощью обычного оптического расщепления или способа разделения или диастереоселективного синтеза по желанию.
Соединение по настоящему изобретению, представленное общей формулой (I), или его фармацевтически приемлемая соль, включает оптические изомеры. Каждый из этих оптических изомеров и все смеси этих оптических изомеров также охватываются настоящим изобретением.
Соединение по настоящему изобретению, представленное общей формулой (I), или его фармацевтически приемлемая соль, включает стереоизомеры, основанные на типе замещения в положении 3 или 4 пиперидинового кольца. Например, в общей формуле (I) цис-изомер является предпочтительным, как показано ниже:
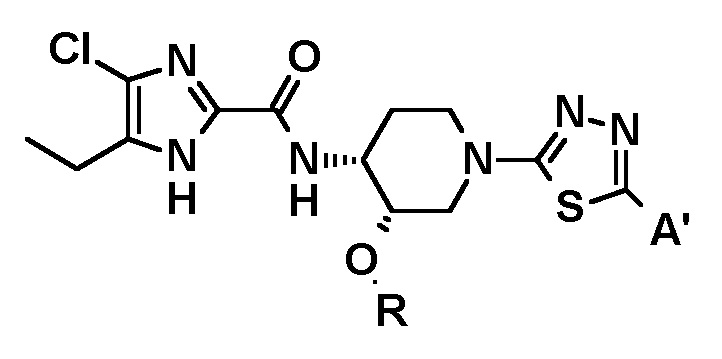
Настоящее изобретение включает предпочтительные изомеры, но не ограничивается ими,
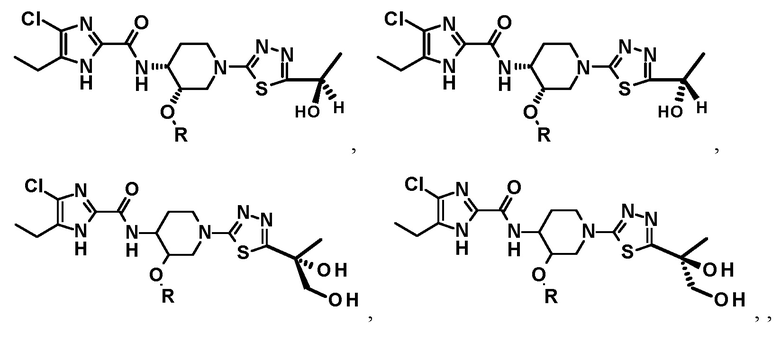
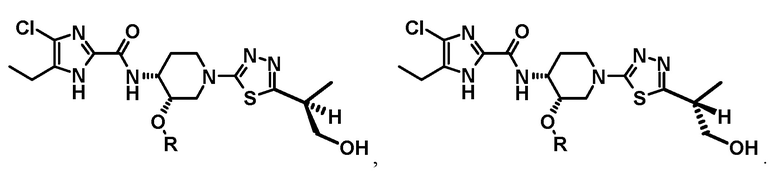
Некоторые соединения, как показано выше, показали полиморфные свойства, поэтому следует понимать, что настоящее изобретение охватывает полиморфную форму в дополнение к каждой рацемической, оптически активной, стереоизомерной форме или их смесям, которая ингибирует субъединицу GyrB ДНК-гиразы и/или субъединицу ParE топоизомеразы IV.
В другом варианте осуществления настоящее изобретение относится к аморфному твердому веществу, как показано в примерах, изложенных ниже, например, примеры 1, 2, 3 и 4. Другие полиморфные формы, например, кристаллические формы примеров 1, 2, 3 и 4, также включены в объем настоящего изобретения.
Оптически активную форму можно получить способами, известными в данной области, например, i) расщеплением рацемической формы методами перекристаллизации, ii) синтезом из оптически активных исходных материалов, iii) хиральным синтезом, iv) ферментативными разделениями, v) биоконверсией или vi) хроматографическим разделением с использованием хиральной неподвижной фазы. Аналогичным образом может быть использован любой способ, известный в данной области, для измерения ингибирующего действия для субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV, включая способ, описанный ниже.
Далее предоставляется фармацевтическая композиция, содержащая соединение общей формулы (I), его стереоизомер, полиморфную форму или фармацевтически приемлемую соль.
Соединение по настоящему изобретению отдельно или в форме фармацевтической композиции обычно можно использовать для профилактики или лечения бактериальных инфекций у животных, включая людей. Таким образом, для лечения и профилактики может потребоваться подходящая лекарственная форма. Подходящие лекарственные формы будут зависеть от применения или способа введения. Методики и лекарственные формы обычно можно найти в The Science and Practice of Pharmacy, 21st edition, Lippincott, Williams and Wilkins, Philadelphia, Pa., 2005 (включены в настоящее описание в качестве ссылки).
Таким образом, в другом аспекте настоящее изобретение относится к фармацевтической композиции для использования при лечении бактериальных инфекций у теплокровного животного, такого как человек, где композиция содержит соединение формулы (I), его стереоизомер, полиморфную форму или фармацевтически приемлемую соль вместе с фармацевтически приемлемым эксципиентом или носителем.
Фармацевтическая композиция по настоящему изобретению может быть в форме, пригодной для перорального применения (например, таблетки, пастилки, твердые или мягкие капсулы, водные или масляные суспензии, эмульсии, диспергируемые порошки или гранулы, сиропы и эликсиры), местного применения (например, кремы, мази, гели и водные или масляные растворы или суспензии), введения ингаляционным способом (например, мелкозернистые порошки и жидкие аэрозоли), введения методом аэрации (например, порошки, полученные распылением) или парентерального введения (например, стерильные водные или масляные растворы для внутривенного, подкожного или внутримышечного введения и суппозитории для ректального введения).
Фармацевтическую композицию по настоящему изобретению можно получить обычными методиками с использованием обычных фармацевтических эксципиентов, хорошо известных в данной области. Таким образом, композиции, предназначенные для перорального применения, могут содержать, например, один или несколько краситель(ей), подсластитель(ей), корригент(ов) и/или консервант(ов).
Примеры фармацевтически приемлемых эксципиентов, подходящих для приготовления таблеток, включают, но не ограничиваются ими, инертные разбавители (например, лактозу, карбонат натрия, фосфат кальция и карбонат кальция); гранулирующие средства и разрыхлители (например, кукурузный крахмал и альгиновая кислота); связующие вещества (например, крахмал); смазывающие вещества (например, стеарат магния, стеариновая кислота и тальк); консерванты (например, этил-п-гидроксибензоат и пропил-п-гидроксибензоат); и антиоксиданты (например, аскорбиновая кислота).
Таблетки, полученные таким образом, могут быть без покрытия или покрыты для изменения их распадаемости и последующей кишечной абсорбции активного ингредиента, или для улучшения их стабильности и/или внешнего вида. В обоих случаях могут быть использованы обычные покрывающие средства и методики, хорошо известные в данной области техники.
Фармацевтические композиции, предназначенные для перорального применения, могут быть в форме твердой желатиновой капсулы. В этом случае активный ингредиент смешивают с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином. Альтернативно, для использования в качестве мягкой желатиновой капсулы, активный ингредиент смешивают с водой или маслом, например, арахисовым маслом, жидким парафином или оливковым маслом.
Водные растворы обычно содержат активный ингредиент в тонкоизмельченной порошкообразной форме вместе с одним или несколькими суспендирующими агентами (например, натрий карбоксиметилцеллюлоза, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагантовая камедь и гуммиарабик); и диспергирующим агентом(ами) или смачивающим агентом(ами) (например, лецитин, продукты конденсации алкиленоксидов и жирных кислот, такие как полиоксиэтиленстеарат), продукты конденсации этиленоксида и длинноцепочечных алифатических спиртов (например, гептадекаэтиленоксицетанол), продукты конденсации этиленоксида и неполных сложных эфиров, полученных из жирных кислот и гекситов (например, полиоксиэтиленсорбитмоноолеата), продукты конденсации этиленоксида и длинноцепочечных алифатических спиртов (например, гептадекаэтиленоксицетанол), продукты конденсации этиленоксида и неполных сложных эфиров, полученных из жирных кислот и гекситов (например, полиоксиэтиленсорбитмоноолеат) и продукты конденсации этиленоксида и неполных сложных эфиров, полученных из жирных кислот и ангидридов гексита (например, полиэтиленсорбитанмоноолеат).
Водные растворы могут также содержать один или несколько консервант(ов) (например, этил п-гидроксибензоат и пропил п-гидроксибензоат), антиоксидант(ов) (например, аскорбиновую кислоту), краситель(ей), корригент(ов) и/или подсластитель(ей) (например, сахароза, сахарин и аспартам).
Масляные суспензии могут быть получены суспендированием активного ингредиента в растительном масле (например, арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле) или минеральном масле (например, жидком парафине). Масляные суспензии могут также содержать загуститель, такой как пчелиный воск, твердый парафин или цетиловый спирт. Для обеспечения приемлемых пероральных препаратов к ним могут быть добавлены такой подсластитель(и) и корригент(ы), как описано выше. Эти композиции могут храниться с помощью добавления к ним антиоксиданта, такого как аскорбиновая кислота.
Диспергируемые порошки и гранулы, подходящие для получения водных суспензий путем добавления воды, обычно содержат активный ингредиент вместе с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или несколькими консервантом(ами). Подходящие диспергирующие или смачивающие агенты и суспендирующие агенты являются такими, как описано выше. Кроме того, в них могут содержаться дополнительные эксципиенты, такие как подсластители, корригенты и красители.
Кроме того, фармацевтические композиции по настоящему изобретению могут быть в форме эмульсии вода-в-масле. Масляная фаза может быть растительным маслом (например, оливковым маслом или арахисовым маслом) или минеральным маслом (например, жидким парафином) или любой их смесью. Подходящими эмульгирующими агентами могут быть, например, встречающиеся в природе смолы (например, гуммиарабик и трагакантовая камедь), встречающиеся в природе фосфатиды (например, соя и лецитин), сложные эфиры или неполные сложные эфиры, полученные из жирных кислот и ангидридов гексита (например, сорбитан моноолеат), и продукты конденсации неполных сложных эфиров и этиленоксида (например, полиоксиэтиленсорбитанмонолаурат). Эмульсии могут также содержать подсластитель, корригент и консервант.
Сиропы и эликсиры могут быть приготовлены вместе с подсластителем, таким как глицерин, пропиленгликоль, сорбит, аспартам или сахароза, и могут содержать средство, уменьшающее раздражение, консервант, корригирующее вещество и/или краситель.
Фармацевтическая композиция может быть в виде стерильного состава для инъекций. Составы для инъекций могут быть получены в соответствии с известными методиками с использованием одного или нескольких подходящих диспергаторов или смачивающих агентов и суспендирующих агентов, описанных выше.
Кроме того, стерильные составы для инъекций могут представлять собой стерильные растворы или суспензии для инъекций в нетоксичном, парентерально приемлемом разбавителе или растворителе, например, растворы 1,3-бутандиола.
Фармацевтические композиции для применения при введении ингаляционным способом могут быть в виде обычного находящегося под давлением аэрозоля, который приспособлен для подачи активного ингредиента или в виде аэрозоля, содержащего мелко измельченное твердое вещество, или в виде аэрозоля, содержащего капельки жидкого вещества. Могут использоваться обычные аэрозольные пропелленты, такие как летучие фторированные углеводороды или углеводороды. Аэрозольное устройство соответствующим образом регулируют для распределения постоянного количества активного ингредиента.
Дополнительную информацию о готовых формах композиций читатель может получить в главе 25.2 тома 5, Comprehensive Medicinal Chemistry (Corwin Hansch; editor in chief; Pergamon Press, 1990).
Количество активного ингредиента, содержащегося вместе с одним или несколькими эксципиентом(ами) для получения одной дозированной лекарственной формы, необходимо варьировать в зависимости от подвергаемого лечению субъекта и от конкретного пути введения. Например, обычно предполагается, что препарат, предназначенный для перорального введения человеку, содержит, например, от 0,5 мг до 2 г активного ингредиента, составленного вместе с подходящим и удобным количеством наполнителя(ей). В этом контексте количество эксципиентов может варьироваться в пределах, но не ограничиваясь ими, от 5 до 98 мас.% от общей массы композиции. Обычно стандартная лекарственная форма составляет приблизительно от 1 мг до приблизительно 500 мг активного ингредиента. За дополнительной информацией о способах введения и схемах дозировки читатель может обратиться к главе 25.3 тома 5, Comprehensive Medicinal Chemistry (Corwin Hansch; editor in chief; Pergamon Press, 1990).
Фармацевтические композиции по настоящему изобретению могут также содержать в дополнение к описанному в настоящем документе соединению одно или несколько известных средства (средств), выбранных из клинически полезных антибактериальных средств, например, но не ограничиваясь ими, макролид (например, эритромицин, телитромицин, диритромицин, рокситромицин, кларитромицин, азитромицин или фидаксомицин), хинолон (например, ципрофлоксацин, норфлоксацин, левофлоксацин, моксифлоксацин или ситафлоксацин), β-лактам (например, амоксициллин, цефалексин, цефаклор, цефуроксим, цефдалоксим, цефепим, цефтобипрол или цефетризол), аминогликозиды (например, гентамицин, неомицин или стрептомицин) и карбапенемы (например, меропенем или имипенем) и/или другие противоинфекционные средства (например, противогрибковые триазолы и амфотерицин). Другое активное фармацевтическое средство, которое может быть использовано в комбинации с соединениями по настоящему изобретению, включает метронидазол и/или ванкомицин. Другое активное средство можно вводить совместно с соединением по настоящему изобретению одновременно, непрерывно или отдельно. Использование таких активных средств может повысить терапевтическую эффективность фармацевтической композиции по настоящему изобретению.
Как описано выше, величину дозы, необходимой для терапевтического или профилактического лечения конкретного состояния, необходимо варьировать в зависимости от подвергаемого лечению субъекта, пути введения и тяжести заболеваний, подлежащих лечению. Предпочтительно, суточная доза используется в диапазоне от 1 до 50 мг/кг. Однако суточную дозу обязательно определяют для изменения в зависимости от описанного выше. Таким образом, оптимальная доза может быть определена любым врачом общей практики, который проводит лечение пациента.
В конкретном варианте осуществления обеспечивается фармацевтическая композиция для лечения или профилактики бактериальных инфекционных заболеваний.
В другом конкретном варианте осуществления обеспечивается фармацевтическая композиция для лечения или профилактики бактериальных инфекционных заболеваний, вызванных грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus, Clostridium, Bacillus, Corynebacterium и Listeria.
В другом конкретном варианте осуществления обеспечивается фармацевтическая композиция для лечения или профилактики бактериальных инфекционных заболеваний, вызванных резистентными бактериями, выбранными из метициллин-резистентного Staphylococcus aureus (MRSA), пенициллин-резистентного Streptococcus pneumoniae (PRSP) и ванкомицин-резистентного Enterococcus (VRE).
В еще одном конкретном варианте осуществления обеспечивается фармацевтическая композиция для лечения или профилактики бактериальных инфекционных заболеваний, выбранных из акне, импетиго, чириев, флегмона фолликулита, фурункулов, карбункулов, синдрома ошпаренной кожи, пневмонии, менингита, остеомиелита, эндокардита, синдрома токсического шока, бронхита, ринита, острого синусита, воспаления среднего уха, конъюнктивита, бактериемии, сепсиса, остеомиелита, септического артрита, перитонита, перикардита, флегмоны, абсцесса головного мозга, инфекций мочевыводящих путей, инфекций Clostridium difficile, газовой гангрены, пищевого отравления, тетануса, ботулизма, диареи, псевдомембранозного колита, токсического мегаколона и перфорации толстой кишки.
Как описано, соединение общей формулы (I) имеет терапевтическое применение и может быть использовано для лечения или профилактики бактериальных инфекций.
Таким образом, настоящее изобретение в своем другом аспекте обеспечивает способ лечения или профилактики бактериальной инфекции у пациента, включающий стадии введения указанному пациенту терапевтически эффективного количества соединения общей формулы (I), его стереоизомера, полиморфной формы, гидрата или фармацевтически приемлемой соли, или фармацевтической композиции, содержащей их.
В соответствии с другим аспектом настоящее изобретение обеспечивает соединение, представленное общей формулой (I), или его стереоизомер, полиморфную форму или фармацевтически приемлемую соль, которое предназначено для использования при лечении бактериальных инфекций у пациента.
В соответствии с еще одним аспектом настоящее изобретение обеспечивает способ ингибирования субъединицы GyrB бактериальной ДНК и/или субъединицы ParE топоизомеразы IV у пациента, нуждающегося в антибактериальном лечении. Этот способ включает введение эффективного количества соединения, представленного общей формулой (I), или его стереоизомера, полиморфной формы или фармацевтически приемлемой соли пациенту.
Еще один аспект настоящего изобретения обеспечивает соединение, представленное общей формулой (I) или его стереоизомер, полиморфную форму или фармацевтически приемлемую соль, которое предназначено для использования в качестве фармацевтического средства для получения антибактериального эффекта у пациента.
Еще один аспект настоящего изобретения обеспечивает соединение, представленное формулой (I), или стереоизомер, полиморфную форму или фармацевтически приемлемую соль, которое предназначено для использования в качестве фармацевтического средства для ингибирования субъединицы GyrB бактериальной ДНК-гидразы и/или субъединицы ParE топоизомеразы IV у пациента.
В одном конкретном варианте осуществления обеспечивается соединение формулы (I) или стереоизомер, полиморфная форма или фармацевтически приемлемая соль, которое предназначено для использования в качестве фармацевтического средства для лечения бактериальных инфекций у пациента.
В соответствии с еще одним аспектом настоящее изобретение обеспечивает использование соединения, представленного общей формулой (I), или стереоизомера, полиморфной формы или фармацевтически приемлемой соли при получении фармацевтического средства, используемого для ингибирования субъединицы GyrB бактериальной ДНК-гидразы и/или субъединицы ParE топоизомеразы IV у пациента.
В конкретном варианте осуществления настоящее изобретение обеспечивает использование соединения, представленного общей формулой (I), или стереоизомера, полиморфной формы или фармацевтически приемлемой соли при получении фармацевтического средства, используемого для лечения бактериальных инфекций у пациента.
В соответствии с другим аспектом настоящее изобретение обеспечивает соединение, представленное общей формулой (I), или стереоизомер, полиморфную форму или фармацевтически приемлемую соль, которое предназначено для использования для получения антибактериального эффекта у пациента.
В соответствии с еще одним аспектом настоящее изобретение обеспечивает соединение, представленное общей формулой (I), или стереоизомер, полиморфную форму или фармацевтически приемлемую соль, которое предназначено для использования для ингибирования субъединицы GyrB бактериальной ДНК-гидразы и/или субъединицы ParE топоизомеразы IV у пациента.
В соответствии с конкретным вариантом осуществления настоящее изобретение обеспечивает соединение, представленное общей формулой (I), или стереоизомер, полиморфную форму или фармацевтически приемлемую соль, которое предназначено для использования для лечения бактериальных инфекций у пациента.
Как используется в настоящем описании термин «терапевтически эффективное количество» относится к количеству соединения по настоящему изобретению, когда его вводят пациенту для лечения или профилактики бактериальных инфекций, которое является достаточным для осуществления такого лечения или профилактики.
Как используется в настоящем описании термин «пациент» относится к субъекту, такому как человек, страдающему от бактериальных инфекций, как определено ниже, и который нуждается в терапевтическом вмешательстве для лечения и/или профилактики таких бактериальных инфекций.
Как используется в настоящем описании термин «бактериальные инфекции» относится к инфекциям, вызванным грамположительными и грамотрицательными бактериями, включая устойчивые бактерии. Наиболее распространенные организмы включают виды Staphylococcus, Streptococcus, Enterococcus, Clostridium, Bacillus, Corynebacterium, Haemophilus и Listeria. Заболевания, вызванные указанными бактериями, включают, но не ограничиваются ими, акне, импетиго, чиреи, флегмона фолликулит, фурункулы, карбункулы, синдром ошпаренной кожи, пневмонию, менингит, остеомиелит, эндокардит, синдром токсического шока, бронхит, ринит, острый синусит, воспаление среднего уха, конъюнктивит, бактериемия, сепсис, остеомиелит, септический артрит, перитонит, перикардит, флегмону, абсцесс головного мозга, инфекции мочевыводящих путей, инфекции Clostridium difficile, обыкновенные угри, гонорею, газовую гангрену, пищевое отравление, тетанус, ботулизм, диарею, псевдомембранозный колит, токсический мегаколон и перфорацию толстой кишки.
В другом варианте осуществления настоящего изобретения обеспечивается способ лечения инфекционных заболеваний, особенно вызванных патогеном, выбранным из метициллин-резистентного Staphylococcus aureus (MRSA), пенициллин-резистентного Streptococcus pneumoniae (PRSP) и ванкомицин-резистентного Enterococcus (VRE) и Clostridium difficile.
MRSA представляет собой бактерию, которая устойчива к обычным антибиотикам, подобным пенициллину. Это может вызвать инфекции кожи, кровотока и операционной раны и пневмонию. Соединения, описанные в настоящем документе, превосходят линезолид в отношении антибактериальной активности in vitro, эффективности и частоты резистентности.
Инфекция Clostridium difficile (CDI) представляет собой кишечное заболевание, вызванное анаэробной бактерией C. difficile, которая колонизирует в толстой кишке. C. difficile продуцирует спору и токсины, которые ответственны за его патогенез. Клинические симптомы, вызванные CDI, представляют собой диарею и боль в животе и в тяжелых случаях псевдомембранозный колит, токсический мегаколон и смерть. Частые рецидивы также очень распространены даже после успешного лечения из-за образования спор. Случаи CDI растут во всем мире. Смерть, связанная с CDI, увеличилась из-за распространения гипервирулентного штамма NAP1/027 в США и Европе.
Соединения по настоящему изобретению активны против гипервирулентных штаммов NAP1/027, следовательно, обеспечивают возможность лечения бактериальных инфекций, таких как MRSA и CDI.
Соответственно, настоящее изобретение обеспечивает соединения для использования при лечении инфекций MRSA, внебольничных респираторных инфекций, инфекций Clostridium difficile и их клинических симптомов, таких как диарея, псевдомембранозный колит, токсический мегаколон, перфорация толстой кишки и сепсис.
В другом варианте осуществления настоящего изобретения обеспечивается способ лечения инфекционных заболеваний, вызванных MRSA.
В другом варианте осуществления настоящего изобретения обеспечивается способ лечения CDI.
В другом варианте осуществления настоящего изобретения обеспечивается способ лечения инфекционных заболеваний, вызванных PRSP и VRE.
Haemophilus influenzae, грамотрицательные бактерии, могут вызывать многие виды инфекций, включая, но не ограничиваясь ими, ушные инфекции, бактериемию, внебольничные респираторные инфекции, пневмонию и острый бактериальный менингит. Неожиданно было обнаружено, что соединения по настоящему изобретению очень активны против этого патогена и, следовательно, могут быть использованы для лечения указанных инфекций, вызванных Haemophilus influenzae.
Соответственно, настоящее изобретение относится к соединениям для использования при лечении таких заболеваний, как внебольничные респираторные инфекции, пневмония, бактериемия и острый бактериальный менингит, вызванных Haemophilus influenzae.
Propionibacterium acnes, грамположительный условно-патогенный микроорганизм кожи человека, который предпочитает анаэробные условия роста и участвует в патогенезе акне, может вызвать кожное заболевание, такое как обыкновенные угри, которое наиболее часто ассоциируется с инфекцией P. acnes. Кроме того, P. acnes были связаны с эндокардитом искусственных и нативных аортальных клапанов, инфекциями роговицы и послеоперационным эндофтальмитом. Она также была признана источником инфекции при фокальных внутричерепных инфекциях и различных инфекциях при шунтировании для обеспечения оттока спинномозговой жидкости. Неожиданно было обнаружено, что соединения по настоящему изобретению очень активны против этого патогена и, следовательно, могут быть использованы для лечения указанных инфекций, предпочтительно, обыкновенных угрей.
Neisseria gonorrhoeae, грамотрицательная бактерия, может вызвать гонорею, которая является наиболее распространенным заболеванием, связанным с N. gonorrhoeae. Кроме того, N. gonorrhoeae также может вызывать конъюнктивит, фарингит, проктит, уретрит, простатит или орхит. Неожиданно было обнаружено, что соединения по настоящему изобретению очень активны против этого патогена и, следовательно, могут быть использованы для лечения указанных инфекций, предпочтительно, гонореи.
В одном конкретном варианте осуществления обеспечиваются соединения для использования при лечении заболеваний, но не ограничиваясь ими, внебольничные респираторные инфекции, внутрибольничные инфекции, инфекции мочевыводящих путей, обыкновенные угри, гонорея и инфекции Clostridium difficile.
Далее будут представлены общие способы получения соединения по настоящему изобретению.
В общем случае, соединение по настоящему изобретению можно получить, с помощью следующей общей схемы и экспериментальных методик, описанных ниже, и/или с помощью дополнительных или альтернативных известных способов и методик в сочетании со знаниями среднего специалиста в данной области. Следует понимать, что способы, изложенные в следующей общей схеме, предназначены для иллюстративных целей и не должны истолковываться как ограничивающие объем раскрытия.
Таким образом, в другом аспекте настоящее изобретение обеспечивает способы синтеза для получения соединения, представленного общей формулой (1), или его фармацевтически приемлемой соли.
На схеме 1 представлен общий способ получения соединений по настоящему изобретению.
Схема 1
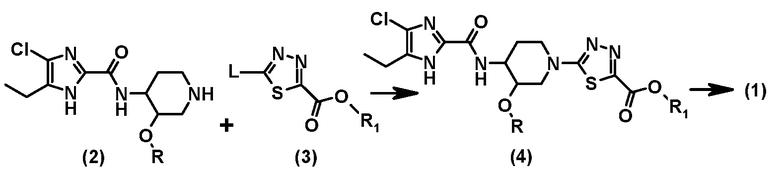
Соединение общей формулы (1) можно получить, следуя стадиям схемы 1. На первой стадии реакцию нуклеофильного замещения соединения формулы (2) с (3) (где L представляет собой подходящую уходящую группу, такую как галоген, выбранный из фтора, хлора, брома или йода; R1 представляет собой алкильную группу) осуществляют при нагревании в подходящем растворителе, таком как диметилформамид, в присутствии основания, такого как диизопропилэтиламин, с получением соединения формулы (4). На второй стадии сложноэфирная группа промежуточного соединения формулы (4) превращается в соединение общей формулы (I):
(a) путем алкилирования (реакция Гриньяра), когда А в общей формуле (I) представляет собой
в подходящем растворителе, таком как тетрагидрофуран, в присутствии метил металлического соединения, такого как метилмагний бромид (в тетрагидрофуране), при или ниже 20°С, более предпочтительно при или ниже 0°C.
(b) путем восстановления, окисления с последующим алкилированием, когда А в общей формуле (I) представляет собой
где восстановление соединения формулы (4) проводят при комнатной температуре в подходящем растворителе, таком как метанол, в присутствии восстанавливающего средства, такого как борогидрид натрия, с получением спиртового промежуточного соединения, которое при окислении при комнатной температуре в подходящий растворителе, таком как метиленхлорид, в присутствии окислителя, такого как диоксид марганца, дает альдегидное промежуточное соединение, которое, в заключение, подвергают алкилированию (реакция Гриньяра), как описано выше.
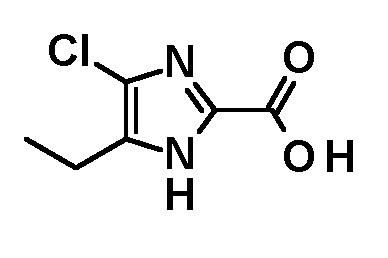
Соединение, представленное формулой (3), является коммерчески доступным, уже известным в литературе или синтезированным стандартными способами синтеза, хорошо известными в данной области.
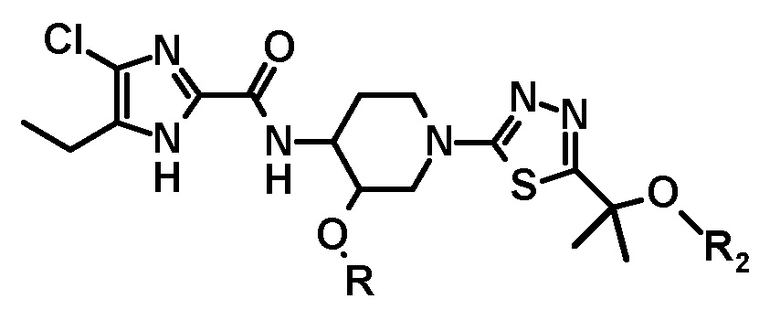
Соединение, представленное формулой (2), может быть получено следующей последовательностью реакций, как показано ниже:
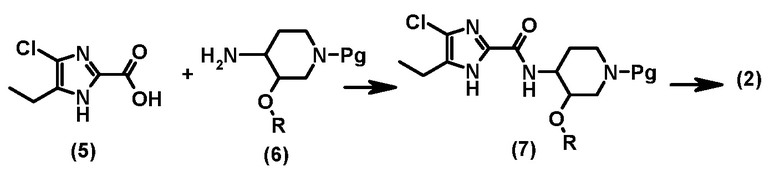
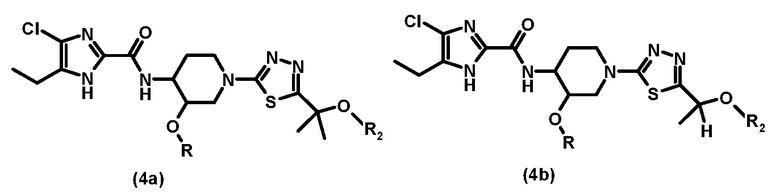
Промежуточное соединение формулы (2) может быть получено реакцией конденсации с последующим удалением защитной группы. Во-первых, соединение имидазола формулы (5) конденсировали с соединением формулы (6) (где R имеет значения, определенные выше, и Pg представляет собой защитную группу, такую как трет-бутилоксикарбонил, метоксикарбонил, этоксикарбонил или п-метоксибензил) в присутствие подходящего конденсирующего реагента для образования пептидной связи, известного в данной области, или связующего агента, такого как 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (EDCI). Такая реакция конденсации иногда проводится в присутствии катализатора, такого как 1-гидроксибензотриазол (HOBT) или диметиламинопиридин, а иногда в присутствии основания, такого как триэтиламин или диизопропилэтиламин, в подходящем растворителе, таком как дихлорметан, тетрагидрофуран, N,N-диметилацетамид и диметилформамид, и в диапазоне температуре от -40°C до 80°C.
На второй стадии соединение формулы (7) подвергают реакции удаления защитной группы в подходящем растворителе, таком как метанол, в присутствии кислоты, такой как хлористый водород в этилацетате.
Соединение имидазола формулы (5) известно в литературе (WO 2009/084614). Соединение формулы (6) является коммерчески доступным или известным в литературе. Оно также может быть синтезировано в соответствии со способом, описанным в данной области, например, WO 2006/087543.
В некоторых случаях оптически чистое соединение формулы (6) может быть получено с помощью следующих способов, описанных ниже.
Метилированное производное соединения, представленного формулой (3), такой как (3а), диметилированное соединение или (3b), монометилированное соединение, предпочтительно используют в качестве синтетических промежуточных соединений для соединения (I).
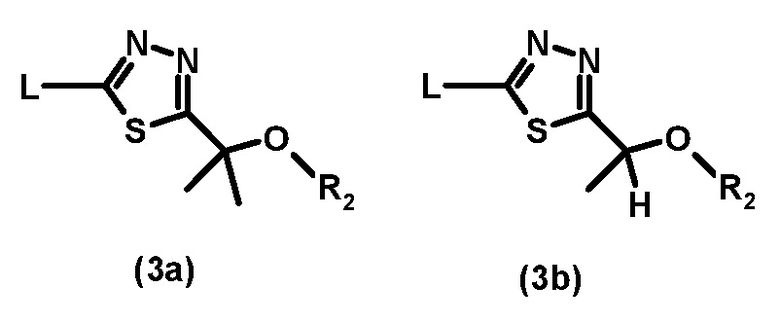
В этих условиях непосредственно получают метилированное соединение формулы (4), такое как (4а) или (4b), что позволяет избежать стадии алкилирования, как показано на схеме 1 выше.
Использование соединения (3а) или (3b) является предпочтительным, поскольку это позволяет избежать загрязнения карбонильной примеси на стадии алкилирования соединения формулы (4).
Более конкретно, бромпроизводное (3а) или (3b), где L в формуле (3а) или (3b) представляет собой бром, предпочтительно используют для реакции. Бромпроизводное (3а) или (3b) получают реакцией соответствующего аминопроизводного соединения (3а) или (3b), где L в формуле (3а) или (3b) представляет собой -NH2, бромированием соединения диазония, полученного взаимодействием нитрита натрия с аминосоединением (3а) и (3b) в соответствии с известным способом. Аминопроизводное соединения (3а) или (3b) получают взаимодействием гидразинкарботиоамида и 2-(R2-O-)-2-метилпропионовой кислоты или 2-(R2-O-)-пропионовой кислоты в присутствии фосфорного хлорирующего средства, такого как оксихлорид фосфора, пентахлорид фосфора, трихлорид фосфора и тому подобное. Любой растворитель, который не мешает реакции, может быть использован в этой реакции, и примерами являются эфир, такой как диоксан, 1,2-диметоксиэтан; углеводород, такой как бензол, толуол, ксилол; галогенированный углеводород, такой как хлороформ, 1,2-дихлорэтан; сложный эфир, такой как этилацетат, пропилацетат, бутилацетат. Что касается (R2-O-)- фрагмента сложного эфира 2-(R2-O-)-2-метилпропионовой кислоты или сложного эфира 2-(R2-O-)-пропионовой кислоты, то этот фрагмент предпочтительно представляет собой группу, полученную из защиты гидроксигруппы 2-гидроксипропионового эфира с помощью некоторой защитной группы для гидроксигруппы. Такая защитная группа для гидроксигруппы может быть выбрана из тех, которые известны в данной области; примерами являются алкильная группа, такая как метильная группа, трет-бутильная группа; аралкильная группа, такая как бензильная группа, п-метоксибензильная группа; ацильная группа, такая как ацетильная группа, пивалоильной группа, бензоильная группа. Что касается ((R2-O-)- фрагмента, предпочтительно использовать ацилоксигруппу и более предпочтительно использовать бензоилоксигруппу. Депротекцию защитной группы R2 можно осуществлять известным способом, соответствующим защитной группе, фактически выбранной для получения гидроксигруппы. Взаимодействие бромпроизводного (3а) или (3b) с соединением (2) осуществляют согласно описанному выше способу.
Предпочтительно соединение формулы (1) получают в соответствии со схемой 2 с использованием бромопроизводного соединения (3а) или (3b), где L означает бром.
Схема 2
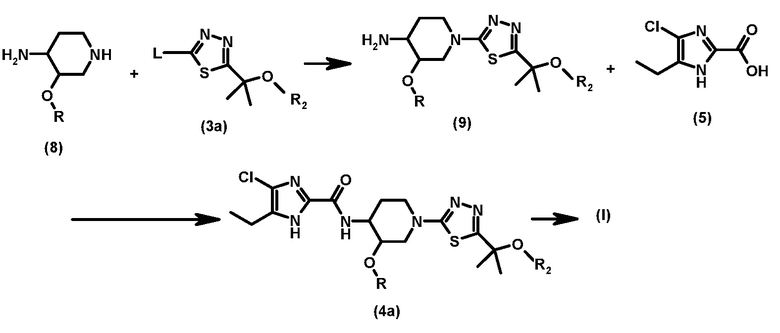
Согласно этому способу, тиадиазольный фрагмент вводят перед введением имидазольного фрагмента. Соединение формулы (9), в частности, диметилированное производное, получают взаимодействием соединения (3а) и соединения (8). Полученное соединение (9), особенно диметилированное производное, получали в виде твердой соли карбоновой кислоты и такую соль карбоновой кислоты очищали способом, хорошо известным в соответствующей области, таким как мокрый способ или перекристаллизация. Что касается соли соединения (9) с карбоновой кислотой, предпочтительным примером является соль пропионовой кислоты. Это является очень выгодным, так как соединение высокой чистоты (9) может быть получено для использования в качестве синтетического промежуточного продукта, что может обеспечить получение высокой чистоты соединения формулы (I). Свободную форму соединения (9) можно получить известным способом, таким как обработка соли соединения (9) основанием. Взаимодействие соединения (3а) и соединения (8) осуществляют с помощью условия, описанного выше.
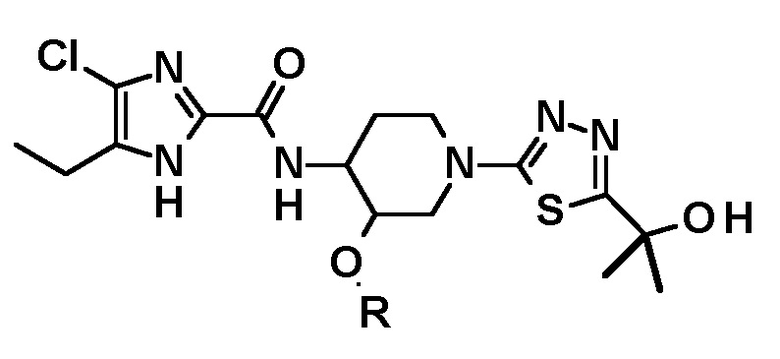
Соединение (9), диметилированное производное, может быть превращено в соединение (4а) взаимодействием соединения (9) и 4-хлор-5-этил-1H-имидазол-2-карбоновой кислоты (5). Это взаимодействие может быть достигнуто при условии, описанном ниже. Удаление R2 из соединения (4а) дает соединение (I). Это удаление может быть достигнуто известным способом в соответствии с R2, используемой защитной группой.
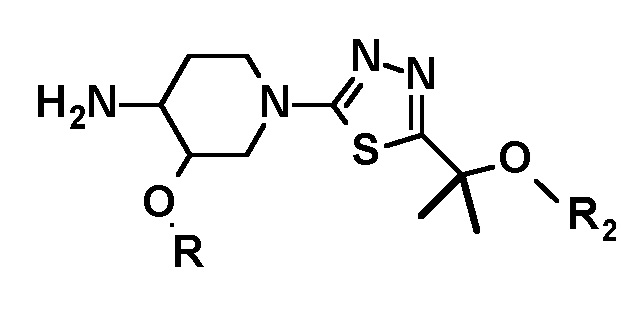
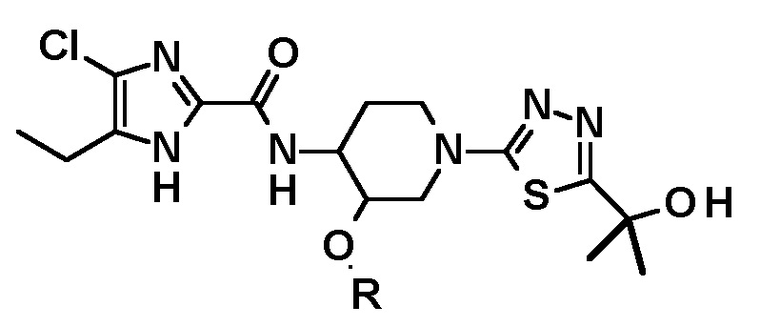
Способы, описанные в настоящем документе, предполагают предпочтительно включение следующих вариантов осуществления, например, в отношении получения производного 5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ила, имеющего следующую формулу, или его стереоизомера
обеспечивается способ, который включает диметилирование на карбонильном атоме углерода -C(=O)-O-R1 соединения следующей формулы, или его стереоизомера:
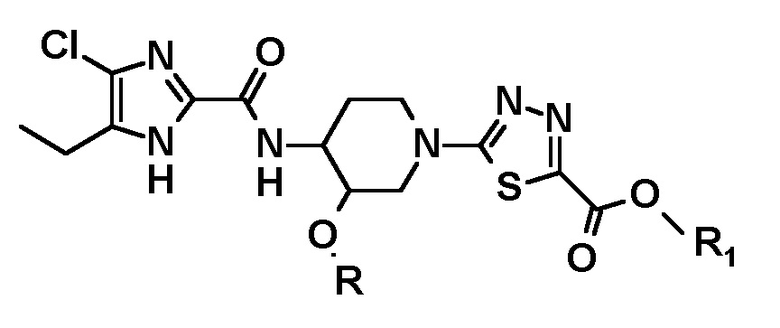
где, R представляет собой (C1-C3) алкильную группу и R1 представляет собой алкильную группу.
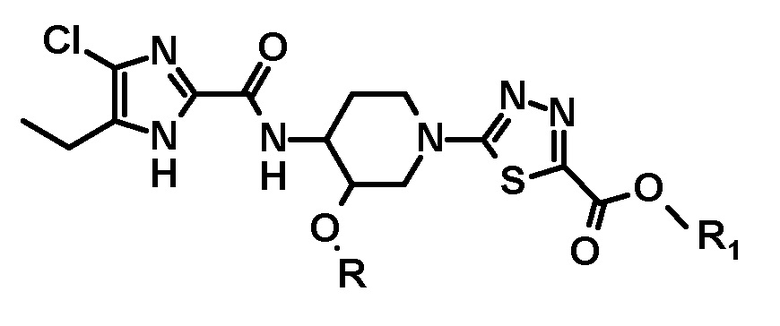
В другом варианте осуществления обеспечивается способ получения соединения следующей формулы или его стереоизомера
который включает стадии:
i) осуществления взаимодействия соединения следующей формулы или его стереоизомера:
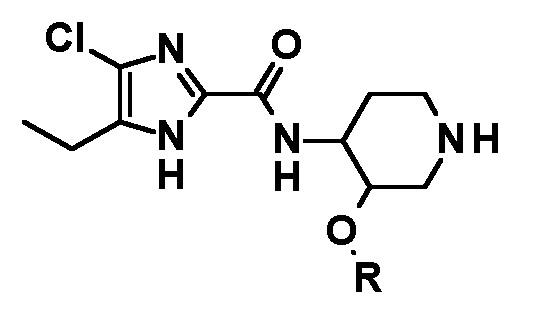
с соединением следующей формулы:
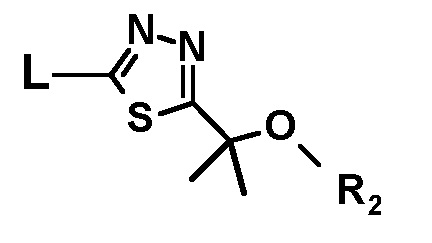
где L представляет собой уходящую группу, R2 представляет собой защитную группу для гидроксигруппы, и R представляет собой (C1-C3) алкильную группу, с получением соединения следующей формулы или его стереоизомера
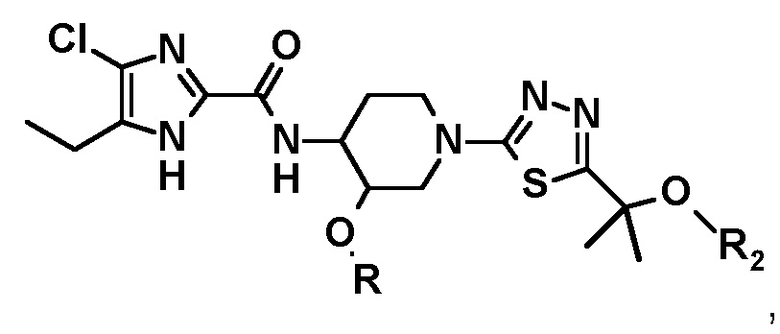 и
и
ii) удаление защитной группы у соединения, полученного на стадии i).
В другом варианте осуществления обеспечивается способ получения соединения следующей формулы или его стереоизомера:
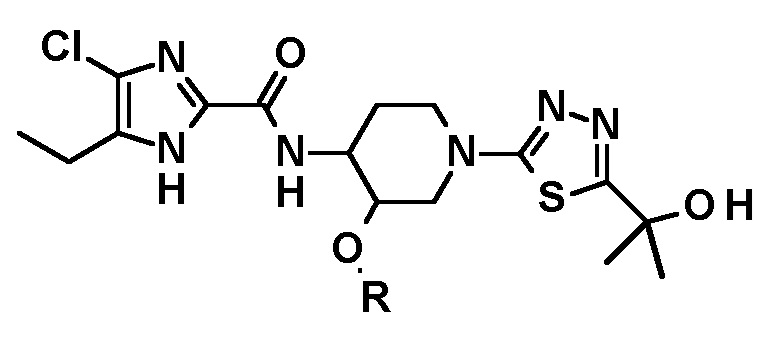
который включает стадии:
i) осуществления взаимодействия соединения следующей формулы или его стереоизомера:
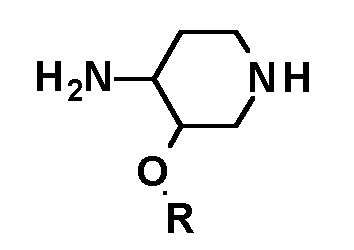
с соединением следующей формулы:
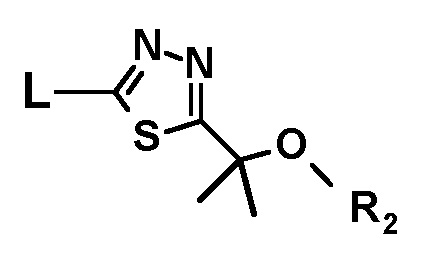
с получением соединения следующей формулы или его стереоизомера
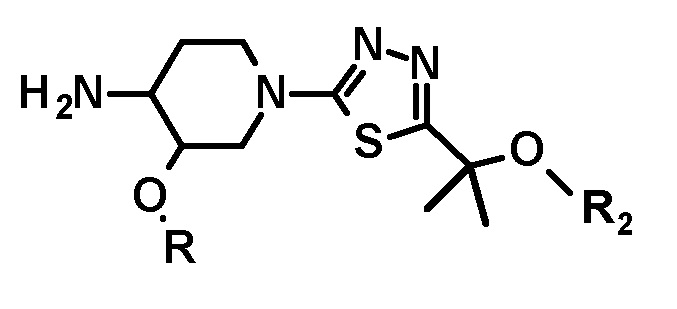
ii) соединение, полученное на стадии i) подвергают взаимодействию с соединением следующей формулы:
с получением соединения следующей формулы или его стереоизомера:
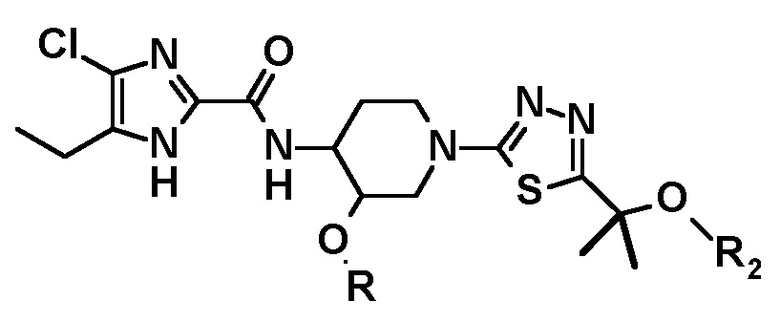
и затем
iii) удаления защитной группы у соединения, полученного на стадии ii), где L, R и R2 имеют значения, определенные выше.
В предпочтительном варианте осуществления соединение, полученное любым из способов, описанных выше, имеет следующую структурную формулу:
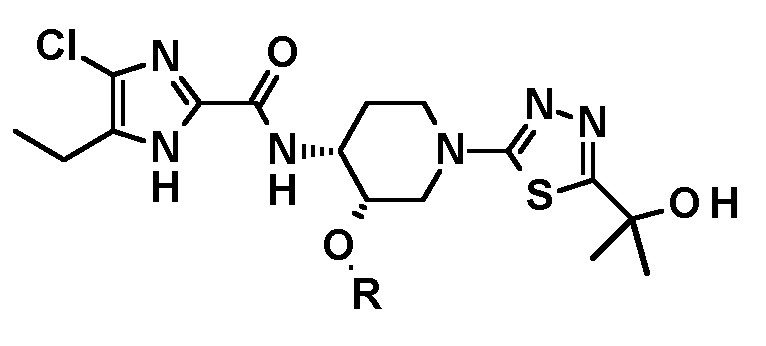
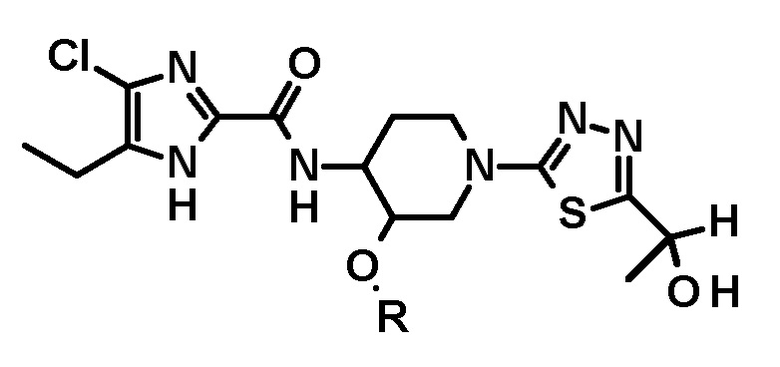
Относительно получения производного 5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ила, имеющего следующую формулу, или его стереоизомера
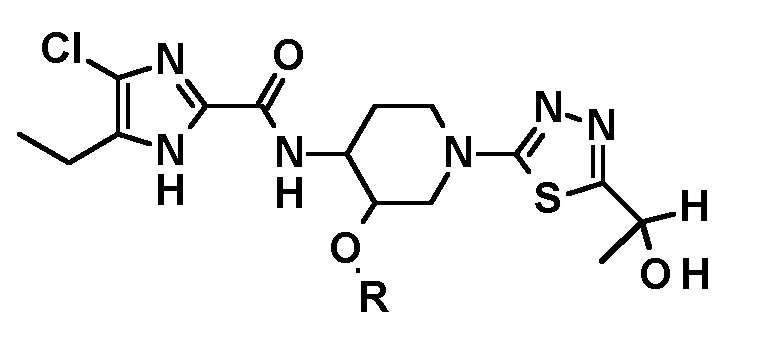
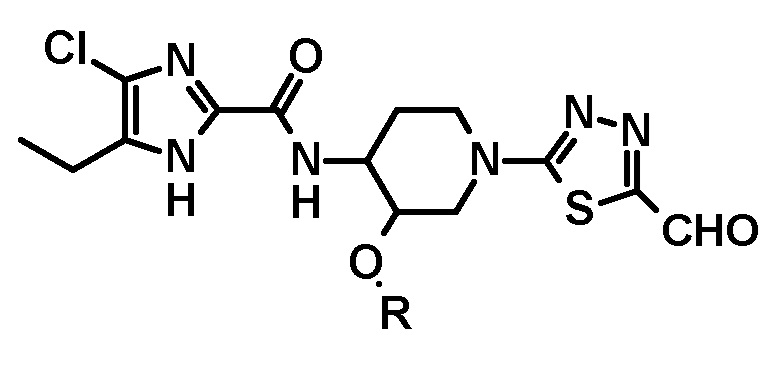
обеспечивается способ, который включает стадии:
i) восстановление соединения следующей формулы или его стереоизомера:
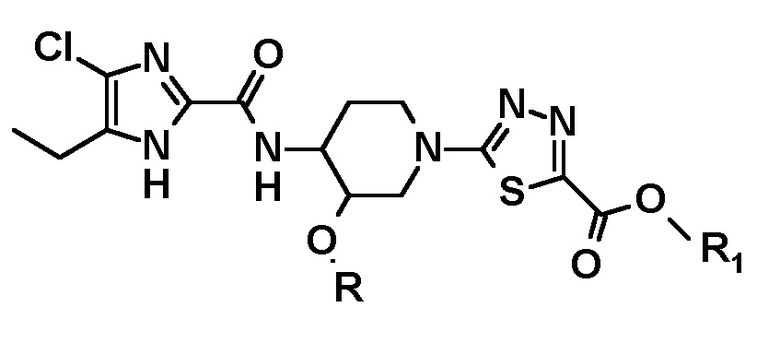
в присутствии подходящего восстанавливающего средства, такого как боргидрид натрия, с получением соединения следующей формулы или его стереоизомера:
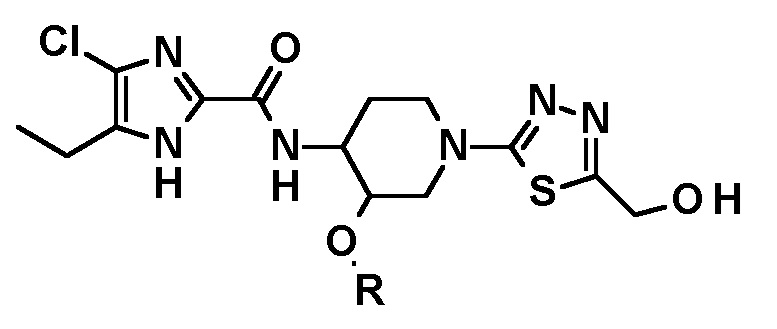
i) окисление соединения, полученного на стадии i), с использованием подходящего окислителя для получения соединения следующей формулы или его стереоизомера:
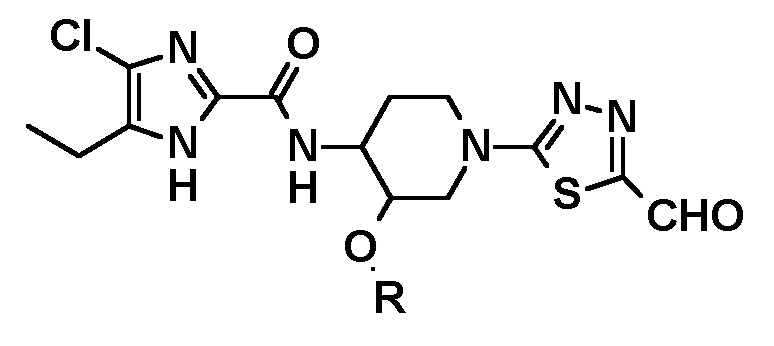
и
iii) метилирование на атоме углерода формильной группы реагентом Гриньяра.
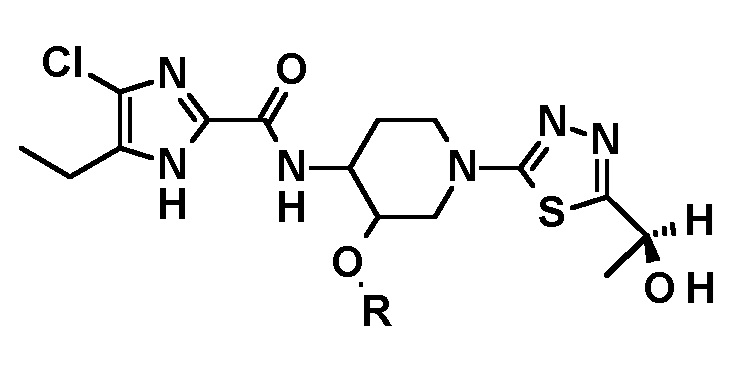
В предпочтительном варианте осуществления соединение, полученное следующими способами, описанными выше, имеет следующую структурную формулу:
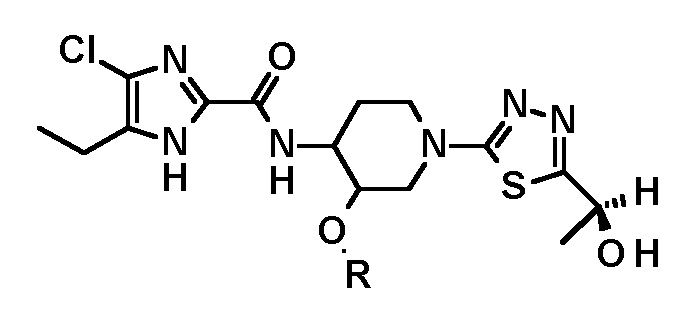 или
или 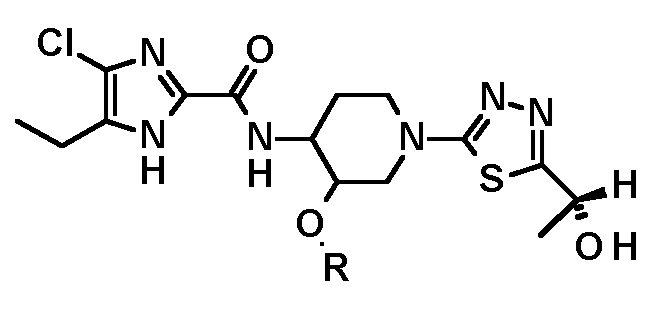 ;
;
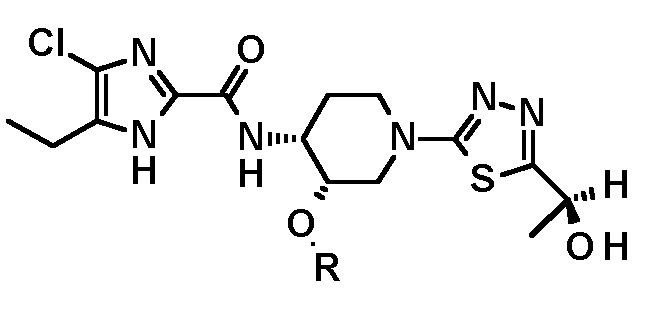 или
или 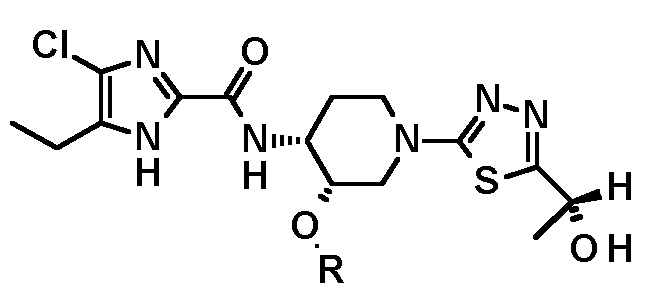 .
.
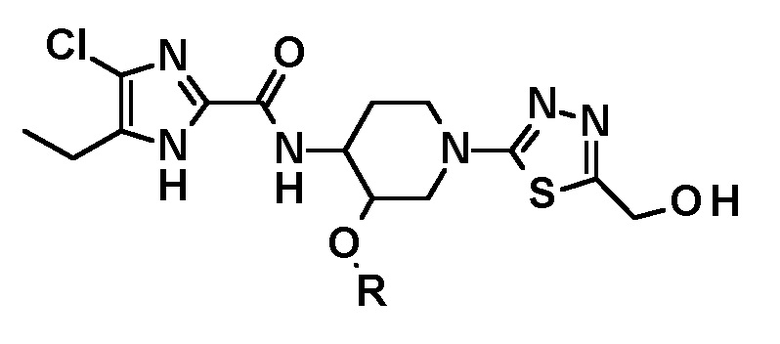
Относительно получения производного 5-(1-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ила, имеющего следующую формулу, или его стереоизомера
обеспечивается способ, который включает осуществление взаимодействия соединения следующей формулы или его стереоизомера
с бораном, с последующей обработкой перекисью водорода.
В предпочтительном варианте осуществления соединение, полученное по вышеуказанному способу, имеет структурную формулу:
 .
.
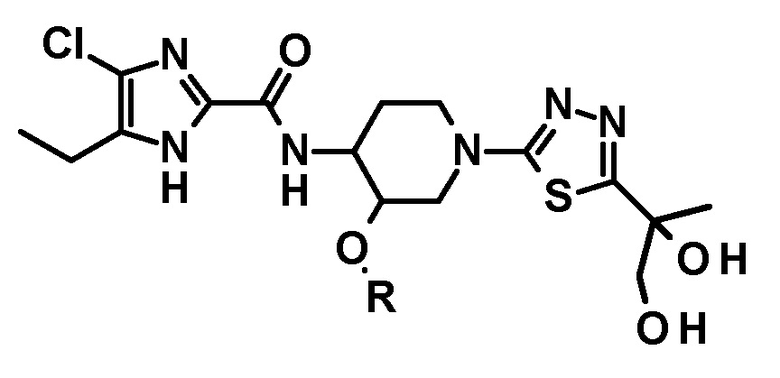
Относительно получения производного 5-[1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ила, имеющего следующую формулу, или его стереоизомера:
обеспечивается способ, который включает дигидроксилирование соединения (на 5-этенильной группе 1,3,4-тиадиазола) следующей формулы или его стереоизомера
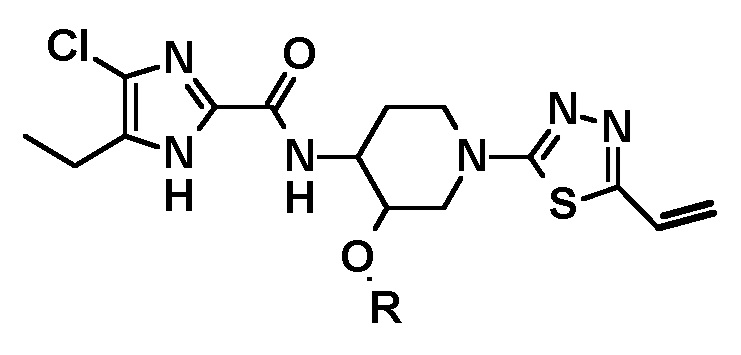
где дигидроксилирование представляет собой асимметрическое дигидроксилирование по Шарплессу.
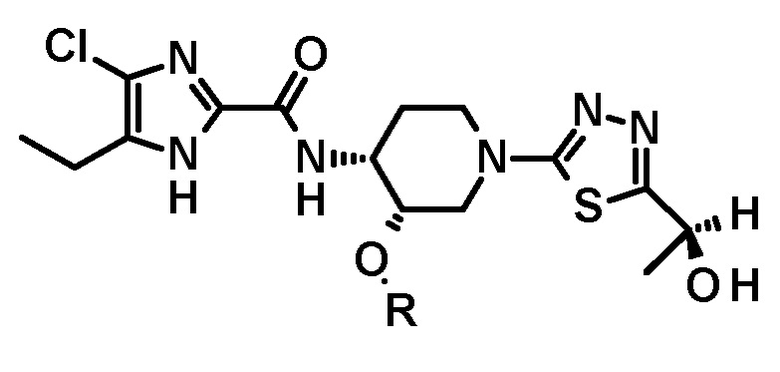
В предпочтительном варианте осуществления соединение, полученное в соответствии с вышеупомянутым способом, имеет структурную формулу:
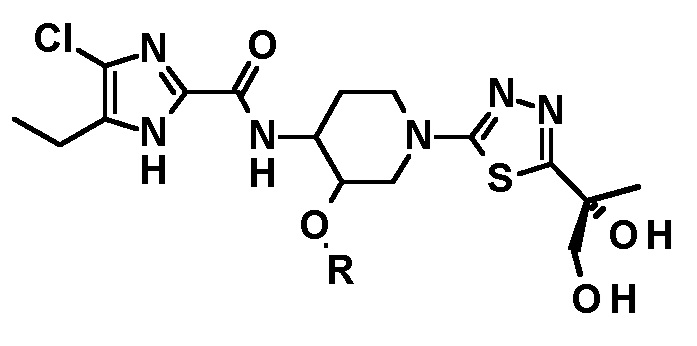 или
или 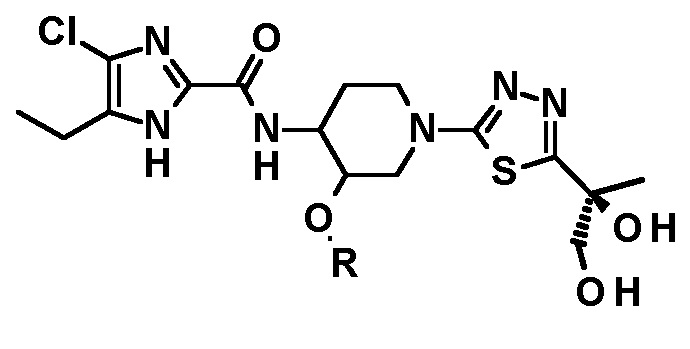 ;
;
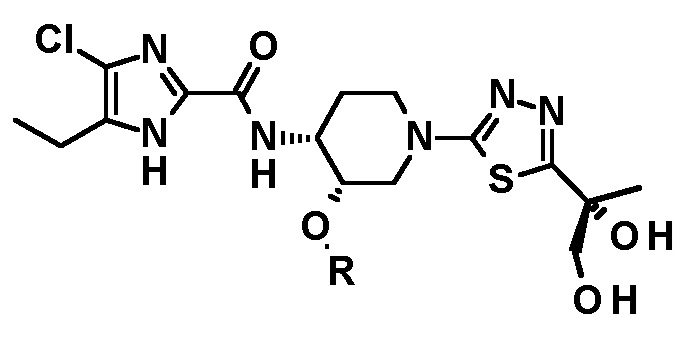 или
или 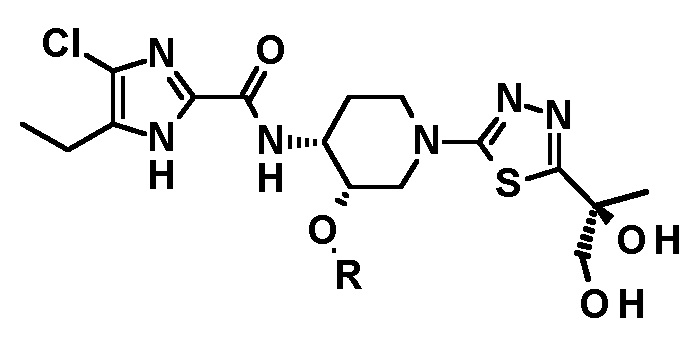 .
.
Методы синтеза, обычно используемые обычными специалистами в области органической химии для получения фармацевтически приемлемых солей, входят в объем настоящей патентной заявки.
Квалифицированные специалисты в области органической химии могут предположительно получить необходимые исходные материалы и продукты, используя справочные документы, описанные ниже, примеры, описанные в них, примеры, описанные ниже. Когда исходные материалы, необходимые для таких методик, как описано выше, не являются коммерчески доступными, они могут быть получены при помощи методик, выбранных из стандартных технологий органической химии, аналогичных синтезу структурно подобных соединений, и технологий, аналогичных методикам способов, описанных выше или в примерах.
Следует отметить, что многие исходные вещества для способов синтеза являются коммерчески доступными и/или широко представлены в научных документах или могут быть получены из коммерчески доступных продуктов при помощи надлежащего использования методов синтеза, описанных в научных документах. В качестве общего руководства по условиям реакции или реагентам, см. Advanced Organic Chemistry, Vol. 4 (Jerry March, ed., published by John Wiley and Sons, 1992).
В некоторых вариантах осуществления следует понимать, что вместо восстанавливающего средства, растворителя, защитных групп, литийорганических реагентов и основания, дополнительно указанных в одном или нескольких описанных в настоящем документе способах, любое другое восстанавливающее средство, растворитель, защитный агент, литийорганические реагенты и основание, как описано в настоящем документе, также может быть использовано.
Могут использоваться обычные защитные группы в соответствии со стандартными способами (в качестве иллюстрации, см. T.W. Greene, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991).
Примеры защитной группы, подходящей для аминогруппы, включают ацильные группы, такие как алканоильные группы (например, ацетил), алкоксикарбонильные группы (например, метоксикарбонил, этоксикарбонил и трет-бутоксикарбонил), арилметоксикарбонильные группы (например, бензилоксикарбонил) и ароильные группы (например, бензоил) или п-метоксибензил.
Условия удаления защиты для защитных групп будут неизбежно варьировать в зависимости от выбора защитных групп. Так, например, ацильные группы, такие как алканоильные или алкоксикарбонильные группы или ароильные группы, могут быть удалены, например, путем гидролиза с соответствующим основанием, таким как гидроксид щелочного металла (например, гидроксид лития или гидроксид натрия). Альтернативно, алкоксикарбонильные группы (например, трет-бутоксикарбонильная группа) могут быть удалены, например, путем обработки подходящей кислотой, такой как хлористоводородная кислота, серная кислота, фосфорная кислота или трифторуксусная кислота. Арилметоксикарбонильные группы (например, бензилоксикарбонильная группа) могут быть удалены, например, путем обработки водородом в присутствии катализатора на основе палладия (например, активированный уголь) или путем обработки кислотой Льюиса, например, бор трис(трифторацетат).
Примеры защитных групп, подходящих для карбоксильной группы, включают эстерифицированные заместители, например, метильную, этильную, трет-бутильную и бензильную группы.
Считается, что условия снятия защиты для защитных групп изменяются в зависимости от выбора защитных групп.
Так, например, метилэфирная или этилэфирная группа может быть удалена, например, гидролизом подходящим основанием, таким как гидроксид натрия. Например, трет-бутилэфирная группа может быть удалена, например, обработкой органической кислотой, такой как трифторуксусная кислота. Бензилэфирная группа может быть удалена, например, гидрогенолизом в присутствии катализатора на основе палладия (например, активированный уголь).
Эти защитные группы могут быть удалены на любой подходящей стадии синтеза с использованием общепринятых методов, хорошо известных в области химии. Альтернативно, защитные группы могут быть удалены на последующих стадиях реакции или во время обработки. Удаление любой защитной группы и образование фармацевтически приемлемой соли находится в пределах компетенции обычных специалистов в области органической химии с использованием стандартных методов.
Когда требуется оптически активная форма соединения по настоящему изобретению, эту форму можно получить, подвергая оптически активный исходный материал (например, образованный асимметрической дериватизацией на соответствующей стадии реакции) любому из описанных выше методов; или путем разделения рацемической формы настоящего соединения или промежуточного соединения с использованием стандартных способов; или путем разделения диастереоизомера, если образуется, путем хроматографии. Кроме того, ферментативные методы также могут быть полезны при получении оптически активного соединения и/или промежуточного соединения.
Аналогичным образом, когда требуется чистый диастереомер соединения по настоящему изобретению, этот изомер может быть получен путем подвергания очищенной диастереомерной смеси в качестве исходного материала любому из описанных выше методов или путем разделения смеси диастереомера или промежуточных продуктов с использованием стандартных способов.
Выход показан только в качестве примера и не всегда является достижимым максимальным значением.
Структура конечного продукта по настоящему изобретению обычно определяли с помощью ЯМР (относится к спектрe протонного магнитного резонанса) и/или масс-спектра (метод ESI, метод APCI или метод FAB). Химический сдвиг в спектре протонного магнитного резонанса (1H-ЯМР) выражается в миллионных долях в нижнем магнитном поле (δ шкала) относительно тетраметилсилана в качестве внутреннего стандарта, и константа взаимодействия (J) и кратность пиков обозначается следующим образом (с, синглет; д, дублет; дд, дублет дублетов; дт, триплет дублетов; т, триплет, кв, квартет; м, мультиплет; шир, широкий). Данные катионов и данные анионов включаются в масс-спектр по мере необходимости методом ESI, методом APCI или методом FAB. Измерение проводили при угле вращения 589 нм (25°C).
Каждое промежуточное соединение очищали и структурно определяли до уровня, необходимого на последующих стадиях (чистоту оценивали с помощью ТСХ или ЯМР, чтобы соответственно определить идентичность по масс-спектру или спектру ЯМР).
В обозначениях названий соединений следует понимать, что цис (±) или транс (±) при использовании означает рацемическую смесь цис- или транс-изомеров, и (-) или (+), когда она упоминается, означает один энантиомер, как в R, R или S, S.
В качестве восстанавливающего средства, если не указано иное, может быть использовано гидрогенизованное комплексное соединение, борсодержащее соединение, такое как борогидрид натрия, триацетоксиборгидрид натрия или цианоборгидрид натрия. Кроме того, предпочтительно использовать каталитическое восстановление с использованием металлического катализатора, такого как палладированный уголь, никель Ренея, оксид платины, гидроксид палладия или палладиевая чернь.
В соответствии с настоящим изобретением растворители, если не указано иное, включают полярные и неполярные растворители, хорошо известные в данной области, включая полярные апротонные и полярные протонные растворители. Примеры полярных растворителей включают, но не ограничиваются ими, метанол, этанол, изопропиловый спирт, трет-бутанол, н-бутанол, уксусную кислоту, муравьиную кислоту или воду, или апротонный растворитель, такой как тетрагидрофуран, ацетонитрил, диоксан, метиленхлорид, диметилсульфоксид, ацетон, N,N-диметилформамид, N,N-диметилацетамид, этилацетат, 1,2-диметоксиэтан, 1,2-дихлорэтан, хлороформ или пиридин. Полярный растворитель также включает смесь воды с любым из вышеуказанных или смесь любых двух или более из вышеуказанных растворителей. Примеры неполярных растворителей включают, но не ограничиваются ими, толуол, бензол, хлорбензол, ксилолы и гексаны.
Основание, если не указано иное, включает, но не ограничивается ими, гидроксид калия, гидроксид натрия, карбонат калия, карбонат натрия, карбонат цезия, карбонат магния, карбонат бария, метиламин, триэтиламин, диизопропилэтиламин или пиридин.
В некоторых вариантах осуществления настоящее изобретение охватывает изотопно меченые соединения общей формулы (I). Все изотопы любого конкретного атома или элемента, как указано, рассматриваются в рамках настоящего изобретения. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению, включают, но не ограничиваются ими, изотопы водорода (например, 2H или 3H), углерода (например, 13C или 14C), азота (например, 13N или 15N), кислорода (например, 15O, 17O или 18O), фосфора (например, 32P или 33P), серы (например, 35S), галогена (например, 18F, 36Cl, 123I или 125I). В предпочтительном варианте осуществления настоящее изобретение обеспечивает дейтериевые (D или 2H) соединения общей формулы (I). Изотопно меченые соединения формулы (I) могут быть получены по следующей общей схеме и методами с использованием изотопно меченых реагентов. Изотопно меченые по настоящему изобретению могут быть полезны в анализах распределения в тканях соединений и/или субстратов. Такие применения изотопно меченых соединений хорошо известны специалистам в данной области техники и поэтому находятся в пределах объема настоящего изобретения.
Иногда могут использоваться следующие сокращения.
ТСХ означает тонкослойную хроматографию; DMF означает N,N- диметилформамид; THF означает тетрагидрофуран; HOBT означает 1-гидроксибензотриазол; EDCI означает 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид; DMSO-D6 означает дейтерированный диметилсульфоксид; CDCl3 означает дейтерированный хлороформ; CD3OD означает дейтерированный метанол; FAB означает ионизацию при бомбардировке быстрыми атомами; ESI означает ионизацию в электроспрее; APCI означает химическую ионизацию при атмосферном давлении.
Экспериментальные процедуры
Далее в настоящем документе, настоящее изобретение подробно описано со ссылкой на примеры и примеры испытаний, но объем настоящего изобретения не ограничивается ими. Любая модификация в способах, описанных в настоящем документе, другие способы синтеза и модификация могут быть использованы или адаптированы. Все такие модификации и альтернативные способы находятся в пределах сущности и объема настоящего приложения.
(Пример 1) Синтез 4-хлор-N-{(3S,4R)-3-этокси-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида
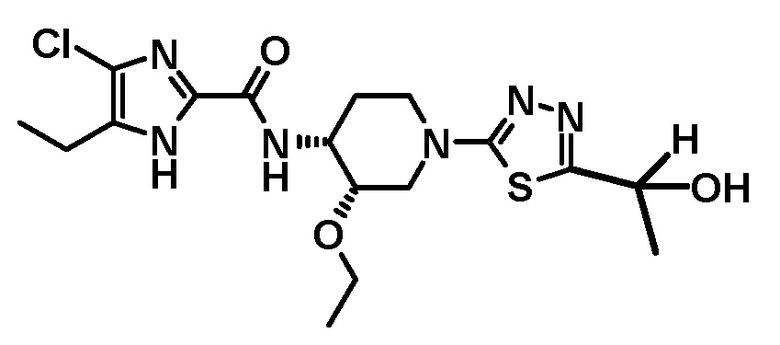
(Иллюстративное соединение No. 1)
Стадия 1: Синтез трет-бутил (3R,4S)-4-{[(бензилокси)карбонил]амино}-3-этоксипиперидин-1-карбоксилата
трет-Бутил цис(±)-4-{[(бензилокси)карбонил]амино}-3-этоксипиперидин-1-карбоксилат (200 г, 528 ммоль), синтезированный способом, описанным в литературе (WO 2009/084614), подвергали оптическому разделению с использованием колонки с оптически активной фазой (CHIRALPAK IC®, растворитель для элюирования: гексан/2-пропанол=80/20 (об./об.)).
Соединение с первым элюируемым пиком (96,7 г) и соединение со вторым элюируемым пиком (94,8 г) получали в виде бесцветных маслянистых веществ.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 7,33-7,38 (5H, м), 5,17-5,24 (1H, м), 5,10 (2H, с), 4,28-4,40 (1H, м), 4,00-4,18 (1H, м), 3,68-3,76 (2H, м), 3,39-3,42 (1H, м), 3,32 (1H, дкв, J=8,71, 7,34 Гц), 2,71-2,77 (2H, м), 1,62-1,78 (2H, м), 1,45 (9H, с), 1,14-1,18 (3H, м).
Стадия 2: Синтез трет-бутил (3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-этоксипиперидин-1-карбоксилата
10% Палладий на угле (1,3 г) добавляли к раствору соединения с первым элюируемым пиком, полученного на стадии 1, (3,58 г, 9,47 ммоль) в метаноле (50 мл), и смесь перемешивали в атмосфере водорода при комнатной температуре в течение 1 часа и 40 минут. Палладий на угле фильтровали через целит и фильтрат концентрировали при пониженном давлении с получением неочищенного трет-бутил (3S,4R)-4-амино-3-этоксипиперидин-1-карбоксилата в виде бесцветного маслянистого вещества.
4-Хлор-5-этил-1H-имидазол-2-карбоновую кислоту (1,82 г, 10,4 ммоль), синтезированную способом, описанным в литературе (WO 2009/084614), WSC (4,79 г, 25 ммоль) и HOBt (1,76 г, 13 ммоль) добавляли к раствору неочищенного трет-бутил (3S,4R)-4-амино-3-этоксипиперидин-1-карбоксилата, полученного выше, в диметилацетамиде (50 мл), и смесь перемешивали при 70°C в течение 1 часа. Реакционный раствор разбавляли этилацетатом, промывали водой 3 раза и насыщенным солевым раствором и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, и полученный остаток очищали при помощи колонки с силикагелем (этилацетат-гексан) с получением указанного в заголовке соединения (3,4 г, 90%) в виде бесцветного аморфного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 11,92 (1H, с), 7,55 (1H, с), 4,05-4,52 (3H, м), 3,60-3,70 (1H, м), 3,41-3,49 (1H, м), 3,27 (1H, дт, J=8,71, 6,42 Гц), 2,71-2,86 (2H, м), 2,69 (2H, кв, J=7,79 Гц), 1,85-1,94 (1H, м), 1,57-1,67 (1H, м), 1,47 (9H, с), 1,26 (3H, т, J=7,79 Гц), 0,92-0,99 (3H, м).
Стадия 3: Синтез этил 5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-этоксипиперидин-1-ил]-1,3,4-тиадиазол-2-карбоксилата
Раствор хлорида водорода/этилацетата (4 н., 30 мл) добавляли к раствору трет-бутил (3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-этоксипиперидин-1-карбоксилата (3,4 г, 8,5 ммоль), полученного на стадии 2, в дихлорметане (20 мл), и смесь оставляли стоять при комнатной температуре в течение 30 минут. Растворитель выпаривали при пониженном давлении с получением неочищенного 4-хлор-N-[(3S,4R)-3-этоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид гидрохлорида в виде бесцветного аморфного твердого вещества.
Раствор неочищенного 4-хлор-N-[(3S,4R)-3-этоксипиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамид гидрохлорида, полученного выше, этил 5-бром-1,3,4-тиадиазол-2-карбоксилата (2,01 г, 8,5 ммоль) и бикарбоната натрия (1,68 г, 20 ммоль) в диметилацетамиде (40 мл) перемешивали при 70°C в течение 1 часа. Реакционный раствор разбавляли этилацетатом, промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, и полученный остаток очищали при помощи колонки с силикагелем (этилацетат-гексан) с получением указанного в заголовке соединения (3,75 г, 97%) в виде бесцветного твердого вещества.
1H-ЯМР (400 МГц, DMSO-D6) δ ppm: 7,67 (1H, д, J=8,59 Гц), 4,35 (2H, кв, J=7,16 Гц), 4,26-4,17 (2H, м), 4,05-3,97 (1H, м), 3,70-3,40 (5H, м), 2,55 (2H, кв, J=7,45 Гц), 1,95-1,88 (1H, м), 1,72-1,68 (1H, м), 1,31 (3H, т, J= 7,16 Гц), 1,14 (3H, т, J=7,45 Гц), 1,03 (3H, т, J=7,16 Гц).
Стадия 4: Синтез 4-хлор-N-{(3S,4R)-3-этокси-1-[5-(гидроксиметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида
Боргидрид натрия (0,5 г, 13,3 ммоль) добавляли к раствору этил 5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-этоксипиперидин-1-ил]-1,3,4-тиадиазол-2- карбоксилата, полученного на стадии 3 (0,95 г, 2,08 ммоль), в метаноле (30 мл), и смесь перемешивали при комнатной температуре в течение 40 минут. Добавляли воду и экстрагировали этилацетатом. Затем концентрирование при пониженном давлении, и полученный остаток очищали при помощи колонки с силикагелем с получением указанного в заголовке соединения (0,75 г, 87%) в виде бесцветного аморфного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 11,12 (1H,с), 7,50 (1H, д, J=8,75 Гц), 4,88 (2H, д, J=5,73 Гц), 4,36-4,33 (1H, м), 4,26-4,21 (1H, м), 3,93-3,88 (1H, м), 3,73 (1H, дкв, J=8,88, 7,15 Гц), 3,63-3,61 (1H, м), 3,44 (1H, дкв, J=8,88, 7,15 Гц), 3,31-3,18 (2H, м), 2,74 (1H, т, J=5,73 Гц), 2,70 (2H, кв, J=7,45 Гц), 2,15-2,10 (1H, м), 1,80-1,76 (1H, м), 1,27 (3H, т, J=7,45 Гц), 1,16 (3H, т, J=7,16 Гц).
Стадия 5: Синтез 4-хлор-N-{(3S,4R)-3-этокси-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида
Диоксид марганца (3,5 г) добавляли к раствору 4-хлор-N-{(3S,4R)-3-этокси-1-[5-(гидроксиметил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида (0,7 г), полученному на стадии 4, в THF (20 мл), и смесь перемешивали при комнатной температуре в течение 1 часа. После фильтрации через целит, реакционный раствор концентрировали при пониженном давлении с получением неочищенного 4-хлор-N-[(3S,4R)-3-этокси-1-(5-формил-1,3,4-тиадиазол-2-ил)пиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (0,68 г, 97%) в виде бесцветного аморфного твердого вещества.
Метилмагния бромид (1,1 моль/л раствора THF, 17 мл, 19 ммоль) добавляли при охлаждении льдом к раствору неочищенного 4-хлор-N-[(3S,4R)-3-этокси-1-(5-формил-1,3,4-тиадиазол-2-ил)пиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (0,68 г), полученного выше, в THF (30 мл), и смесь перемешивали в течение 1 часа. К этому добавляли насыщенный водный раствор хлорида аммония с последующей экстракцией этилацетатом и органический слой промывали насыщенным солевым раствором. Остаток, полученный концентрацией при пониженном давлении, очищали при помощи колонки с силикагелем (этилацетат/метанол) с получением указанного в заголовке соединения (0,40 г, 75%) в виде бесцветного аморфного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 11,65 (1H, с), 7,55 (1H, д, J=9,16 Гц), 5,13 (1H, кв, J=6,11 Гц), 4,34-4,31 (1H, м), 4,25-4,22 (1H, м), 3,91-3,89 (1H, м), 3,72 (1H, дкв, J=8,88, 7,16 Гц), 3,62 (1H, с), 3,44 (1H, дкв, J=8,88, 7,16 Гц), 3,29-3,17 (3H, м), 2,71 (2H, кв, J=7,64 Гц), 2,16-2,11 (1H, м), 1,79-1,76 (1H, м), 1,62 (3H, д, J=6,11 Гц), 1,26 (3H, тд, J=7,64, 2,28 Гц), 1,16 (3H, тд, J=7,16, 1,72 Гц).
(Пример 2) Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
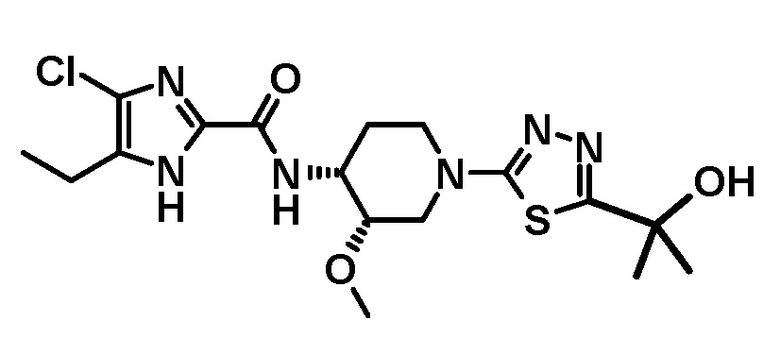
(Иллюстративное соединение No. 2)
Стадия 1: Синтез трет-бутил цис(±)-4-{[(бензилокси)карбонил]амино}-3-метоксипиперидин-1-карбоксилата
Диизопропилэтиламин (26,4 г, 204 ммоль) добавляли к раствору трет-бутил цис(±)-4-амино-3-метоксипиперидин-1-карбоксилата (39,2 г, 170 ммоль) в дихлорметане (400 мл), раствор бензилхлорформиата (43,5 г, 255 ммоль) в дихлорметане (80 мл) добавляли по каплям при охлаждении льдом, и смесь дополнительно перемешивали в течение 1 часа. Реакционный раствор промывали водой и 10% водным раствором хлорида натрия и концентрировали при пониженном давлении. Остаток очищали при помощи хроматографии на силикагеле (растворитель для элюирования: гексан/этилацетат= 100/0, 100/15, 100/30), и полученный продукт дополнительно отверждали, используя смешанный растворитель этилацетат/гексан с получением указанного в заголовке соединения (40,0 г, 65%) в виде белого твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 7,29-7,40 (5H, м), 5,10 (2H, с), 5,04-5,16 (1H, м), 4,26-4,50 (1H, м), 3,91-4,22 (1H, м), 3,37 (3H, с), 3,25-3,37 (1H, м), 2,60-2,90 (2H, м), 1,63-1,76 (2H, м), 1,46 (9H, с).
Масс-спектр (ESI): m/z 365 (M+H)+.
Стадия 2: трет-бутил (3S,4R)-4-{[(бензилокси)карбонил]амино}-3-метоксипиперидин-1-карбоксилат
трет-Бутил цис(±)-4-{[(бензилокси)карбонил]амино}-3-метоксипиперидин-1-карбоксилат (100 г, 274 ммоль), полученный на стадии 1, подвергали оптическому разделению с использованием колонки с оптически активной фазой (CHIRALCEL OJ-H®, растворитель для элюирования: гексан/2-пропанол=90/10 (об./об.)). Соединение с первым элюируемым пиком (49,5 г) и соединение со вторым элюируемым пиком (48,9 г) получали в виде бесцветных маслянистых веществ.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 7,29-7,40 (5H, м), 5,10 (2H, с) 5,04-5,16 (1H, м), 4,26-4,50 (1H, м), 3,91-4,22 (1H, м), 3,37 (3H, с), 3,25-3,37 (1H, м), 2,60-2,90 (2H, м), 1,63-1,76 (2H, м), 1,46 (9H, с).
Масса (ESI): m/z 365 (M+H)+.
Стадия 3: Синтез трет-бутил (3S,4R)-4-амино-3-метоксипиперидин-1-карбоксилата
10% Палладий на угле (108 г) добавляли к раствору соединения со вторым элюируемым пиком, полученного на стадии 2, (230 мг, 0,631 ммоль) в метаноле (7 мл), и смесь перемешивали в атмосфере газообразного водорода при комнатной температуре в течение 1 часа и 30 минут. Реакционный раствор, из которого катализатор удаляли фильтрованием, концентрировали при пониженном давлении с получением указанного в заголовке соединения (141 мг, 97%) в виде бесцветного маслянистого вещества.
Стадия 4: Синтез трет-бутил (3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-карбоксилата
Такую же операцию, что и в примере 1 (Стадия 2), проводили с использованием трет-бутил (3S,4R)-4-амино-3-метоксипиперидин-1-карбоксилата (224,4 мг, 0,96 ммоль), полученного на стадии 3 выше, 4-хлор-5-этил-1H-имидазол-2-карбоксилата (140 мг, 0,80 ммоль), синтезированного способом, описанным в литературе (WO 2009/084614), EDCI (440 мг, 2,29 ммоль) и HOBT (110 мг, 0,81 ммоль) с получением указанного в заголовке соединения (222,4 мг, 72%) в виде белого твердого вещества.
1H-ЯМР (400МГц, CDCl3) δ ppm: 10,90 (1H, шир.с), 7,45 (1H, шир.с), 4,19-4,35 (3H, м), 3,41 (3H, с), 3,32-3,39 (2H, м), 2,72-2,89 (1H, м), 2,68 (2H, кв, J=7,57 Гц), 1,79-1,91 (1H, м), 1,61-1,69 (1H, м), 1,47 (9H, с), 1,26 (3H, т, J=7,57 Гц).
Стадия 5: Синтез этил 5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-карбоксилата
Раствор хлорида водорода/этилацетата (4 н, 3 мл) добавляли к раствору трет-бутил (3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-карбоксилата (1,0 г), полученного на стадии 4, в этилацетате (3 мл), и смесь оставляли стоять при комнатной температуре в течение 30 минут. Растворитель выпаривали при пониженном давлении с получением неочищенного 4-хлор-N-[(3S,4R)-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид гидрохлорида в виде бесцветного аморфного твердого вещества.
Суспензию неочищенного 4-хлор-N-[(3S,4R)-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид гидрохлорида, полученного выше, этил 5-бром-1,3,4-тиадиазол-2-карбоксилата (0,66 г) и бикарбоната натрия (1,05 г) в DMF (40 мл) перемешивали при 70°C в течение 3 часов. Реакционный раствор разбавляли этилацетатом, промывали водой и насыщенным солевым раствором, и сушили над безводным сульфатом магния. Растворитель выпаривали при пониженном давлении, и полученный остаток очищали при помощи колонки с силикагелем (этилацетат-гексан) с получением указанного в заголовке соединения (1,4 г) в виде бесцветного твердого вещества.
Масса (ESI): m/z 443 (M+H)+.
Стадия 6: Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидрокси-1-метилэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
Метилмагний бромид (1,12 моль/л раствора THF, 10 мл, 11 ммоль) добавляли при охлаждении льдом к раствору этил 5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-карбоксилата, полученного на стадии 5, (0,35 г, 0,79 ммоль) в THF (15 мл), и смесь перемешивали в течение 40 минут. К этому добавляли насыщенный раствор хлорида аммония с последующей экстракцией этилацетатом, и органический слой промывали насыщенным солевым раствором. Остаток, полученный концентрацией при пониженном давлении, очищали при помощи колонки с силикагелем (этилацетат/метанол) с получением указанного в заголовке соединения (0,40 г, 88%) в виде бесцветного аморфного твердого вещества.
1H-ЯМР (400 МГц,CDCl3) δ: 11,29 (1H, с), 7,53 (1H, д, J=9,16 Гц), 4,48-4,45 (1H, м), 4,27-4,22 (1H, м), 3,85-3,82 (1H, м), 3,52 (1H, с), 3,42 (3H, с), 3,31-3,24 (1H, м), 3,15-3,12 (1H, м), 2,92 (1H, с), 2,70 (2H, кв, J=7,64 Гц), 2,12-2,07 (1H, м), 1,81-1,79 (1H, м), 1,68 (6H, д, J= 2,29 Гц), 1,26 (3H, т, J=7,64 Гц).
(Пример 3) Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
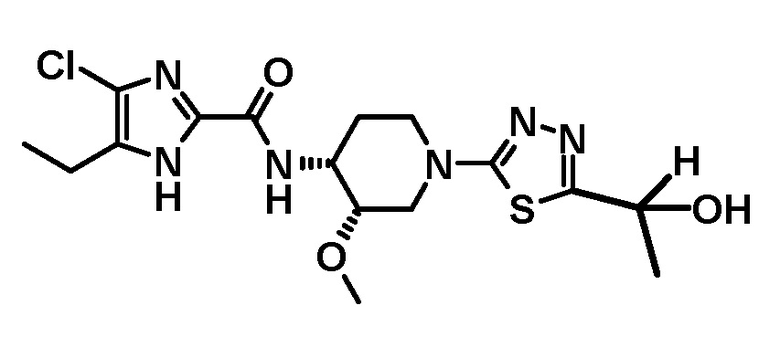
(Иллюстративное соединение No. 3)
Стадия 1: Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(гидроксиметил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
Боргидрид натрия (1,9 г) добавляли в пяти разделенных частях к раствору этил 5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-карбоксилата, полученного в примере 2, стадия 5 (1,4 г) в метаноле (50 мл) при охлаждении льдом, и смесь перемешивали. Реакционный раствор концентрировали при пониженном давлении и воду добавляли к остатку, экстрагировали этилацетатом и сушили над безводным сульфатом натрия. После концентрирования при пониженном давлении остаток очищали с помощью колонки с силикагелем с получением указанного в заголовке соединения (1,0 г) в виде аморфного твердого вещества.
1H-ЯМР (400 МГц,CDCl3) δ ppm: 11,14 (1H, с), 7,53 (1H, д, J=9,03 Гц), 4,88 (2H, с), 4,44-4,48 (1H, м), 4,23-4,27 (1H, м), 3,85-3,87 (1H, м), 3,51 (1H, с), 3,41 (3H, с), 3,28-3,31 (1H, м), 3,14-3,18 (1H, м), 2,80 (1H, шир.с), 2,70 (2H, кв, J=7,57 Гц), 2,05-2,12 (1H, м), 1,78-1,81 (1H, м), 1,26 (3H, т, J=7,69 Гц).
Масса (ESI): m/z 401 (M+H)+.
Стадия 2: Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
Диоксид марганца (0,42 г) добавляли к раствору 4-хлор-5-этил-N-{(3S,4R)-1-[5-(гидроксиметил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида (0,1 г), полученного на стадии 1, в THF (5 мл), и смесь перемешивали при комнатной температуре в течение ночи. После фильтрации через целит, реакционный раствор концентрировали при пониженном давлении с получением неочищенного 4-хлор-5-этил-N-[(3S,4R)-1-(5-формил-1,3,4-тиадиазол-2-ил)-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида в виде аморфного твердого вещества.
Метилмагний бромид (1,1 моль/л раствора THF, 6 мл) добавляли при охлаждении льдом к раствору неочищенного 4-хлор-5-этил-N-[(3S,4R)-1-(5-формил-1,3,4-тиадиазол-2-ил)-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида в THF (5 мл), и смесь перемешивали. Добавляли воду и 1 моль/л хлористоводородной кислоты, с последующей экстракцией этилацетатом, и органический слой промывали насыщенным солевым раствором. Остаток, полученный концентрацией при пониженном давлении, очищали при помощи колонки с силикагелем (этилацетат/метанол) с получением указанного в заголовке соединения (0,057 г) в виде аморфного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 11,12 (1H, шир.с), 7,47-7,57 (1H, м), 5,07-5,18 (1H, м), 4,41-4,51 (1H, м), 4,19-4,31 (1H, м), 3,79-3,91 (1H, м), 3,51 (1H, шир.с), 3,41 (3H, с), 3,23-3,34 (1H, м), 3,15 (1H, шир. д, J=14,65 Гц), 2,91 (1H, шир.с), 2,70 (2H, кв, J=7,52 Гц), 2,02-2,15 (1H, м), 1,75-1,84 (1H, м), 1,60-1,64 (3H, м), 1,26 (3H, т, J=7,52 Гц).
Масса (ESI): m/z 415 (M+H)+.
(Пример 4) Синтез 4-хлор-5-этил-N-[(3S,4R)-1-{5-[(1R)-1-гидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида, и
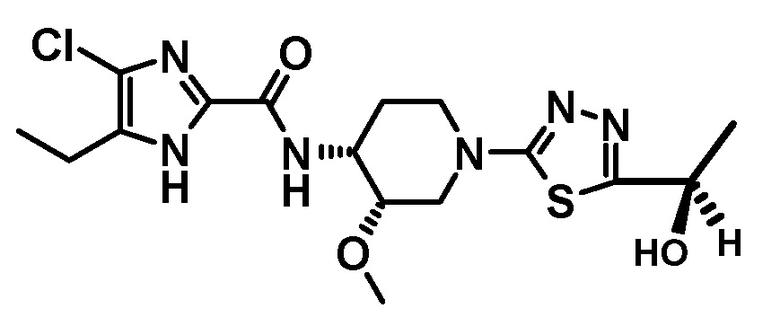
(Иллюстративное соединение No. 4)
4-хлор-5-этил-N-[(3S,4R)-1-{5-[(1S)-1-гидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида
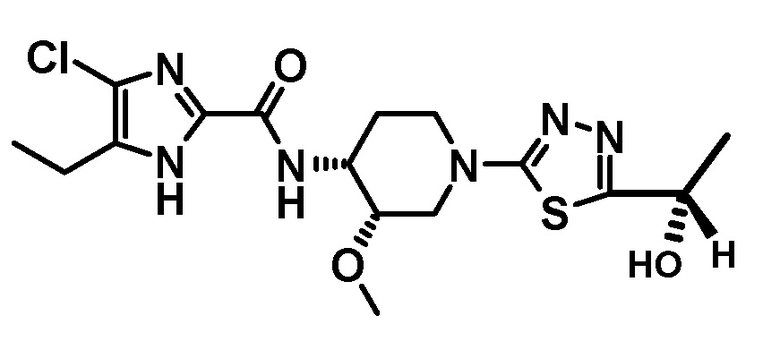
(Иллюстративное соединение No. 5)
4-Хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамид, полученный в примере 3 (стадия 2), (приблизительно 1:1 смесь диастереомеров, 53,2 г) подвергали диастереомерному разделению с использованием колонки с оптически активной фазой (CHIRALPAC IC®, растворитель для элюирования: гексан/2-пропанол=80/20 (об./об.)). Соединение с первым элюируемым пиком (23,1 г) и соединение со вторым элюируемым пиком (23,0 г) получали в виде светлых некристаллических твердых веществ, соответственно.
Соединение с первым элюируемым пиком представляет собой соединение No 4 и соединение со вторым элюируемым пиком представляет собой соединение No 5.
В соединении No 4 конфигурацию гидроксигруппы определяли как R-конфигурацию, поскольку данное соединение ацетилировали в условиях реакции стереоселективного ацетилирования с использованием липазы PS Amano SD/изопропенилацетата.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 11,21 (1H, шир.с), 7,53 (1H, д-подобный, J=9 Гц), 5,13 (1H, кв, J=6,5 Гц), 4,48 (1H, шир.д, J=13 Гц), 4,26 (1H, м), 3,88 (1H, шир.д, J=13 Гц), 3,52 (1H, шир.с), 3,42 (3H, с), 3,32 (1H, тд, J=13, 3 Гц), 3,19 (1H, шир. д, J=ca. 13 Гц), 2,70 (2H, кв, J=7,6 Гц), 2,09 (1H, квд, J=13, 4 Гц), 1,80 (1H, дкв-подобный, J=13, ca. 4 Гц), 1,62 (3H, д, J=6,59 Гц), 1,24 (3H, т, J=7,6 Гц).
IR (KBr)см-1: 2973, 1650, 1532, 1437, 1261, 1116, 1069.
Масса (ESI): m/z 415 (M+H)+.
Масс-спектр высокого разрешения (ESI): m/z C16H24ClN6O3S ((M+H)+), рассчитанный; 415,13191, наблюдаемый; 415,13209.
В соединении No 5 конфигурацию гидроксигруппы определяли как S-конфигурацию, так как настоящее соединение почти не ацетилируется в условиях стереоселективного ацетилирования с использованием липазы PS Amano SD/изопропенилацетата.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 11,25 (1H, шир.с), 7,55 (1H, д-подобный, J=9 Гц), 5,13 (1H, кв, J=6,5 Гц), 4,50 (1H, шир.д, J=13 Гц), 4,26 (1H, м), 3,86 (1H, шир.д, J=13 Гц), 3,52 (1H, шир.с), 3,42 (3H, с), 3,32 (1H, тд, J=13, 3 Гц), 3,19 (1H, шир. д, J=ca. 13 Гц), 2,70 (2H, кв, J=7,6 Гц), 2,09 (1H, квд, J=13, 4 Гц), 1,81 (1H, дкв-подобный, J=13, ca. 4 Гц), 1,62 (3H, д, J=6,59 Гц), 1,26 (3H, д, J=7,69 Гц).
IR (KBr)см-1: 2973, 1650, 1532, 1437, 1262, 1116, 1069.
Масса (ESI): m/z 415 (M+H)+.
Масс-спектр высокого разрешения (ESI): m/z C16H24ClN6O3S ((M+H)+), рассчитанный; 415,13191, наблюдаемый; 415,13129.
(Пример 5) Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
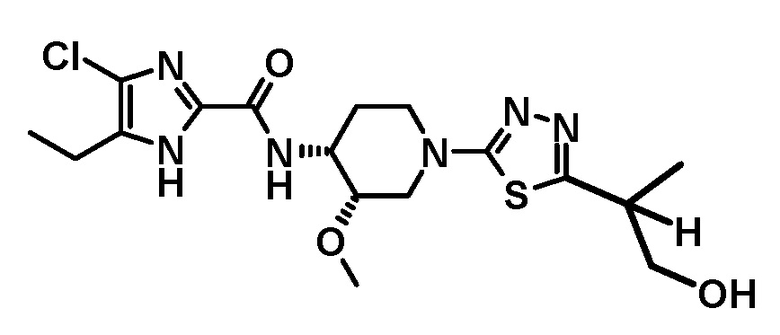
(Иллюстративное соединение No. 6)
Стадия 1: Синтез 4-хлор-5-этил-N-{(3S,4R)-3-метокси-1-[5-(проп-1-ен-2-ил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1H-имидазол-2-карбоксамида
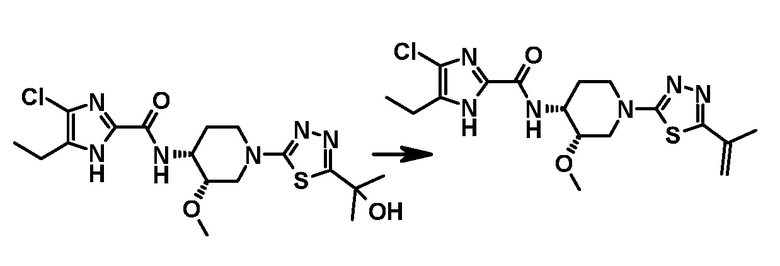
4-Хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамид (1,0 г, 2,3 ммоль) растворяли в сухом толуоле (50 мл) при комнатной температуре с последующим добавлением п-толуолсульфоновой кислоты (39 мг, 0,23 ммоль, 0,1 эквив.). Реакционную смесь перемешивали при 80°C в течение ночи. После завершения реакции, реакционную смесь разбавляли этилацетатом и промывали водой. Органический слой отделяли и концентрировали в вакууме. Неочищенную реакционную смесь подвергали колоночной хроматографии (метанол-дихлорметан, 5%) с получением указанного в заголовке соединения (575 мг) в виде бледно-коричневого твердого вещества.
Масса (ESI): m/z 411,11 (M+H)+.
Стадия 2: Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
4-Хлор-5-этил-N-{(3S,4R)-3-метокси-1-[5-(проп-1-ен-2-ил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1H-имидазол-2-карбоксамид, полученный на стадии 1 (200 мг, 0,486 ммоль), растворяли в сухом THF (10 мл) при комнатной температуре. Раствор охлаждали до 0°С с последующим добавлением раствора комплекса боран-тетрагидрофуран (1,944 ммоль, 2,0 мл, 1M). Полученную реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Затем к реакционной смеси добавляли раствор гидроксида натрия (0,5 н, 1,5 мл), воду (0,5 мл) и пероксид водорода (30% мас./об., 0,5 мл), перемешивали при комнатной температуре в течение 5 часов. После завершения реакции, реакционную смесь разбавляли этилацетатом (50 мл) и промывали водой (20 мл ×2). Органический слой отделяли и концентрировали в вакууме. Неочищенную реакционную смесь подвергали колоночной хроматографии (метанол-дихлорметан, 3%) с получением указанного в заголовке соединения (100 мг) в виде белого твердого вещества.
1H-ЯМР (400 МГц, MeOD) δ ppm: 4,28-4,20 (м, 2H), 3,91-3,88 (м, 1H), 3,70 (д, 2H, J=5,96 Гц), 3,58 (шир.с, 1H), 3,43 (с, 3H), 3,34-3,30 (м, 1H), 3,27-3,20 (м, 2H), 2,31 (кв, 2H, J=15,1 Гц, 7,6 Гц), 2,07-1,90 (м, 1H), 1,82-1,76 (м, 1H), 1,33 (дд, 3H, J=7,0 Гц, 1,4 Гц), 1,19 (т, 3H, J=15,1 Гц).
Масса (ESI): m/z 428,89 (M+H)+.
(Пример 6) Синтез 4-хлор-N-{(3S,4R)-3-этокси-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида
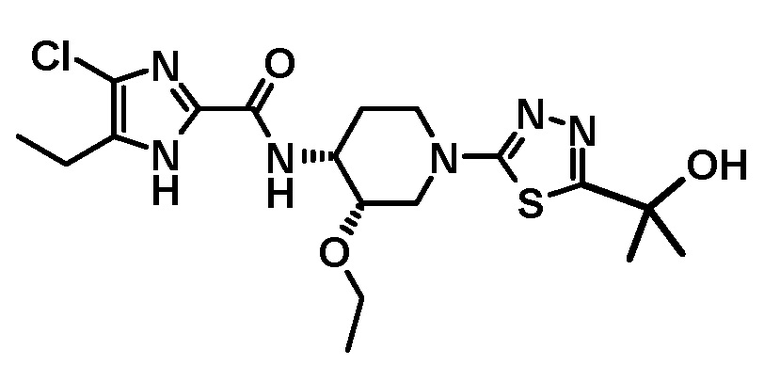
(Иллюстративное соединение No. 7)
Указанное в заголовке соединение получали аналогичным способом, описанным в примере 2, используя трет-бутил цис(±)-4-амино-3-этоксипиперидин-1-карбоксилат на стадии 1 вместо трет-бутил цис(±)-4-амино-3-метоксипиперидин-1-карбоксилата.
1H-ЯМР (400 МГц, MeOD) δ ppm: 4,24-4,17 (м, 2H), 3,96-3,93 (м, 1H), 3,77-3,63 (м, 3H), 3,53-3,47 (м, 1H), 3,36 (д, 1H, J=1,64 Гц), 2,62 (кв, 2H, J=15,1 Гц, 7,6 Гц), 2,12-2,02 (м, 1H), 1,85-1,75 (м, 1H), 1,59 (д, 6H, J=3,4 Гц), 1,20 (т, 3H, J= 7,6 Гц), 1,13 (т, 3H, J=6,9 Гц).
Масса (ESI): m/z 442,96 (M+H)+.
(Пример 7) Синтез 4-хлор-N-[(3S,4R)-1-{5-[(1S)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида и
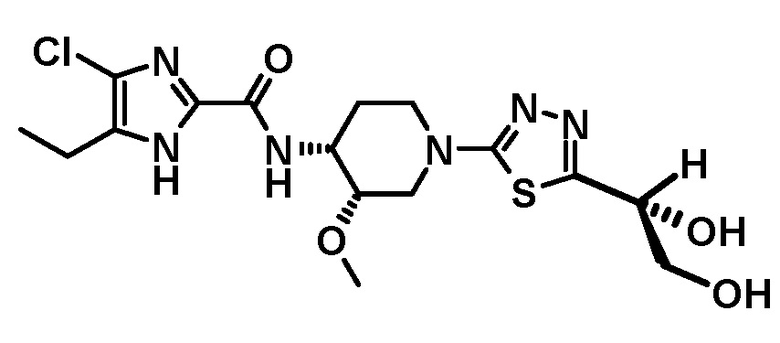
(Иллюстративное соединение No. 8)
4-хлор-N-[(3S,4R)-1-{5-[(1R)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида
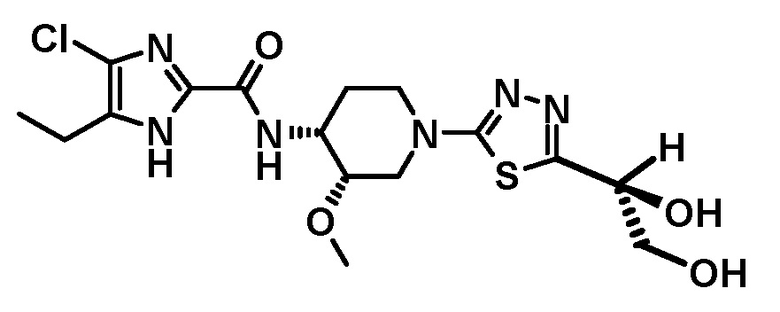
(Иллюстративное соединение No. 9)
Стадия 1: Синтез N-[(3S,4R)-1-(5-бром-1,3,4-тиадиазол-2-ил)-3-метоксипиперидин-4-ил]-4-хлор-5-этил-1H-имидазол-2-карбоксамида
Гидрохлоридную соль указанного в заголовке соединения (1,9 г) в виде белого твердого вещества получали аналогичным образом, как описано в примере 2, стадия 5, используя трет-бутил (3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-карбоксилат (2 н, 5,17 ммоль), этилацетат (20 мл), раствор хлористоводородной кислоты (20 мл, 4 н в диоксане).
К раствору этого соединения (0,1 г, 0,31 ммоль) в ацетонитриле (5 мл), добавляли диизопропилэтиламин (0,15 мл, 0,93 ммоль) и 2,5-дибром-1,3,4-тиадиазол (0,11 г, 0,46 ммоль). Реакционную смесь перемешивали при 80°C в течение 4 часов. Реакционную смесь разбавляли этилацетатом (50 мл) и водой (5 мл) и перемешивали в течение 10 минут. Органический слой отделяли, промывали насыщенным солевым раствором и концентрировали. Неочищенный продукт очищали при помощи колоночной хроматографии (этилацетат в гексане, 30%) с получением указанного в заголовке соединения (84 мг) в виде не совсем белой смолы.
Масса (ESI): m/z 451,07 (M+H)+.
Стадия 2: Синтез 4-хлор-N-[(3S,4R)-1-(5-этенил-1,3,4-тиадиазол-2-ил)-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида
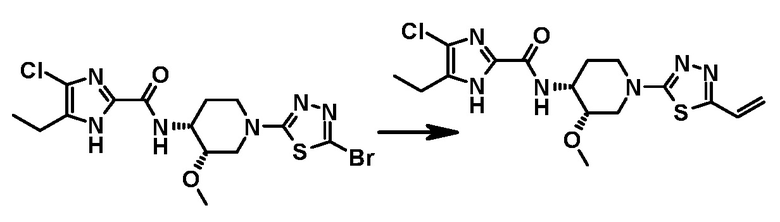
К раствору N-[(3S,4R)-1-(5-бром-1,3,4-тиадиазол-2-ил)-3-метоксипиперидин-4-ил]-4-хлор-5-этил-1H-имидазол-2-карбоксамида (0,08 г, 0,18 ммоль) в DMF (5 мл), добавляли винилтрибутилолова (0,17 мл, 0,53 ммоль) и бис-трифенилфосфинпалладий дихлорид (0,025 г, 0,03 ммоль). Реакционную смесь перемешивали при 90°C в течение 4 часов. Реакционную смесь разбавляли этилацетатом (50 мл) и водой (5 мл) и перемешивали в течение 10 минут. Органический слой отделяли, промывали насыщенным солевым раствором и концентрировали. Неочищенный продукт очищали при помощи колоночной хроматографии (этилацетат в гексане, 40%) с получением указанного в заголовке соединения (26 мг) в виде не совсем белой смолы.
Масса (ESI): m/z 397,14 (M+H)+.
Стадия 3: Синтез 4-хлор-N-[(3S,4R)-1-{5-[(1S)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида и 4-хлор-N-[(3S,4R)-1-{5-[(1R)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида
К раствору AD-mix-β® (741 мг, 1,4 г/ммоль) в трет-бутаноле и воде (3 мл каждый), добавляли метансульфонамид (0,05 г, 0,53 ммоль) и реакционную смесь перемешивали при 0°C в течение 20 минут. 4-Хлор-N-[(3S,4R)-1-(5-этенил-1,3,4-тиадиазол-2-ил)-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид (0,21 г, 0,53 ммоль; полученный на стадии 2) добавляли к реакционной смеси и смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь гасили раствором сульфита натрия и реакционную смесь разбавляли этилацетатом (100 мл) и водой (5 мл). Органический слой отделяли, промывали насыщенным солевым раствором и концентрировали. Неочищенный продукт очищали при помощи колоночной хроматографии (метанол в дихлорметане, 10%) с получением 4-хлор-N-[(3S,4R)-1-{5-[(1S)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (105 мг) в виде белого твердого вещества.
1H-ЯМР (400 МГц, MeOD) δ ppm: 4,30-4,20 (м, 2H), 3,93-3,81 (м, 2H), 3,75-3,71 (м, 1H), 3,60-3,55 (шир с, 1H), 3,44 (с, 3H), 3,40-3,30 (м, 3H), 2,64 (кв, 2H, J=14,8 и 7,6 Гц), 2,04-1,98 (м, 1H), 1,83-1,78 (м, 1H), 1,23 (т, 3H, J=7,6 Гц).
Масса: m/z 430,84 (M+H)+.
Аналогично, 4-хлор-N-[(3S,4R)-1-{5-[(1R)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид (32 мг) получали с использованием AD-mix-α® (247 мг, 1,4 г/ммоль), метансульфонамида (0,02 г, 0,17 ммоль) и 4-хлор-N-[(3S,4R)-1-(5-этенил-1,3,4-тиадиазол-2-ил)-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (0,07 г, 0,17 ммоль).
1H-ЯМР (400МГц, MeOD) δ ppm: 4,30-4,20 (м, 2H), 3,94-3,81 (м, 2H), 3,76-3,71 (м, 1H), 3,60-3,56 (шир с, 1H), 3,44 (с, 3H), 3,40-3,30 (м, 3H), 2,64 (кв, 2H, J=15,2 и 7,6 Гц), 2,04-1,98 (м, 1H), 1,81-1,79 (м, 1H), 1,21 (т, 3H, J=7,6 Гц).
Масса: m/z 430,88 (M+H)+.
(Пример 8) Синтез 4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида
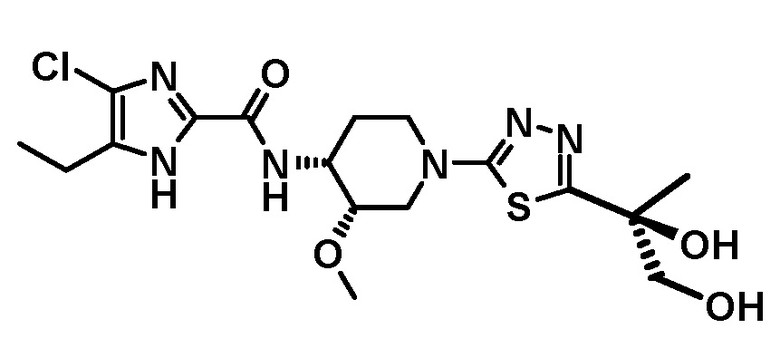
(Иллюстративное соединение No. 10)
К раствору AD-mix-α® (20,4 г, 1,4 г/ммоль) в трет-бутаноле и воде (300 мл каждый), добавляли метансульфонамид (1,38 г, 14,6 ммоль) и реакционную смесь перемешивали при 0°C в течение 20 минут. 4-Хлор-5-этил-N-{(3S,4R)-3-метокси-1-[5-(проп-1-ен-2-ил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1H-имидазол-2-карбоксамид, полученный в примере 5, стадия 1 (6,0 г, 14,6 ммоль) добавляли к реакционной смеси и смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь гасили раствором сульфита натрия и реакционную смесь разбавляли этилацетатом (500 мл) и водой (50 мл). Органический слой отделяли, промывали насыщенным солевым раствором и концентрировали. Неочищенный продукт очищали при помощи колоночной хроматографии (10% метанол в дихлорметане) с получением 4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (3,6 г) в виде не совсем белого аморфного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 11,89 (с, 1H), 7,64 (д, 1H, J=8,96 Гц), 4,37 (д, 1H, J=13,84 Гц), 4,30-4,20 (м, 1H), 4,13 (д, 1H, J=11,4 Гц), 3,80 (д, 1H, J=13,28 Гц), 3,69 (д, 1H, J=11,36 Гц), 3,50 (с, 1H), 3,37 (с, 3H), 3,22 (дт, 1H, J=13,2, 3,0 Гц), 3,11 (д, 1H, J=13,24 Гц), 2,66 (кв, 2H, J=15,16, 7,56 Гц), 2,15-2,05 (м, 1H), 1,80-1,70 (м, 2H), 1,54 (с, 3H), 1,23 (т, 3H, J=7,56 Гц).
Масса: m/z 445,10 (M+H)+.
(Пример 9) Синтез 4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид)
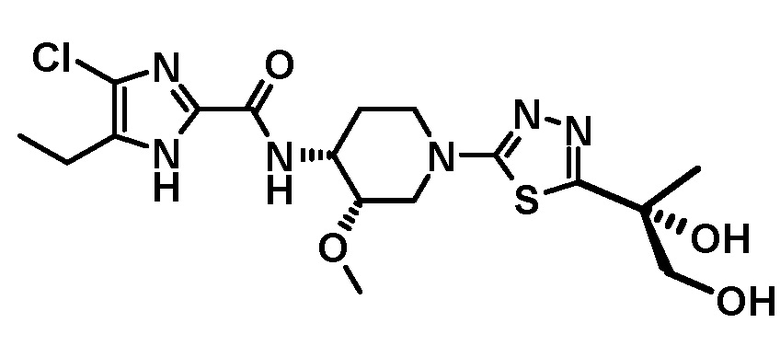
(Иллюстративное соединение No. 11)
К раствору AD-mix-β® (17,08 г, 1,4 г/ммоль) в трет-бутаноле и воде (75 мл каждый) добавляли метансульфонамид (1,16 г, 12,2 ммоль) и реакционную смесь перемешивали при 0°C в течение 20 минут. 4-Хлор-5-этил-N-{(3S,4R)-3-метокси-1-[5-(проп-1-ен-2-ил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-1H-имидазол-2-карбоксамид, полученный в примере 5, стадия 1 (5,0 г, 12,2 ммоль) добавляли к реакционной смеси и смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь гасили раствором сульфита натрия и реакционную смесь разбавляли этилацетатом (400 мл) и водой (30 мл). Органический слой отделяли, промывали насыщенным солевым раствором и концентрировали. Неочищенный продукт очищали при помощи колоночной хроматографии (10% метанол в дихлорметане) с получением указанного в заголовке соединения (3,1 г) в виде аморфного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 11,79 (шир с, 1H), 7,62 (д, 1H, J=8,96 Гц), 4,40-4,35 (м, 1H), 4,30-4,20 (м, 2H), 4,13 (с, 1H), 3,85-3,80 (м, 1H), 3,70-3,60 (м, 2H), 3,37 (с, 3H), 3,23 (дт, 1H, J=12,9, 2,9 Гц), 3,11 (д, 1H, J=12,92 Гц), 2,66 (кв, 2H, J=15,2, 7,56 Гц), 2,15-2,05 (м, 1H), 1,80-1,75 (м, 2H), 1,54 (с, 3H), 1,23 (т, 3H, J=7,56 Гц).
Масса: m/z 445,11 (M+H)+.
(Пример 10) Синтез кристаллической 2/3 гидратной формы 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида (форма I)
Добавляли воду (100 мкл) к 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамиду (примерно 20 мг), и смесь перемешивали при 40°C в течение примерно 3 недель. Твердое вещество собирали и сушили при комнатной температуре в течение ночи с получением кристаллического 2/3 гидрата указанного в заголовке соединения, обозначенного как форма I.
Форма I, полученная таким образом, характеризовалась i) дифракционной картиной, полученной из анализа с использованием порошковой рентгеновской дифракции (CuK, λ=1,54Å, скорость сканирования=20°/мин) (фиг.2) и ii) графиком термогравиметрического анализа (график TG/DTA), полученным при измерении образца (3 мг) при скорости нагрева приблизительно 10°C/минута (фиг. 3). Соединение показало, что потеря массы соответствовала 2/3 гидрату (теоретическое количество: 2,7%). Кроме того, была показана картина изменения массы образца (фиг.4), которая наблюдалась при постепенном изменении относительной влажности. Изменение массы этого соединения оставалось в пределах 0,3%, изменение массы было небольшим и не показывало изменения массы из-за абсорбции или десорбции влаги.
(Пример 11) Синтез безводной кристаллической формы 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида (Форма II)
4-Хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамид сушили при пониженном давлении при 40°C в течение ночи, затем к соединению (2 г) добавляли толуол (10 мл), смесь перемешивали путем магнитного воздействия в течение примерно 3 дней с периодическим перемешиванием с помощью шпателя. Полученное таким образом твердое вещество сушили под воздухом с получением безводной кристаллической формы указанного в заголовке соединения, обозначенного как форма II.
Дифракционная картина, полученная из анализа с использованием порошковой рентгеновской дифракции (CuK, λ=1,54Å, скорость сканирования=20°/мин) для этого ангидрата, показана на фиг.5.
График термогравиметрического анализа (график TG/DTA), полученный при измерении образца (3 мг) при скорости нагрева приблизительно 10°С/мин, изображен на фигуре 6. Потеря массы соответствует уровню снижения адсорбированной воды, и это сильно указывает на то, что соединение является ангидратом. Кроме того, график изменения массы образца показан на фигуре 7, которое наблюдалась при постепенном изменении относительной влажности. Это соединение по существу не показало изменения массы в результате абсорбции или десорбции влаги.
(Пример 12) Синтез безводной кристаллической формы 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида (форма III)
Толуол (100 мкл) добавляли к 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамиду (приблизительно 20 мг), и смесь перемешивали при 40°C в течение примерно 3 недель. Твердое вещество собирали и сушили при комнатной температуре в течение ночи с получением безводной кристаллической формы указанного в заголовке соединения, обозначенного как форма III.
(Пример 13) Синтез кристаллической формы 4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (форма A)
Загружали 4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид (225 г) с ацетонитрилом (4,5 л) и смесь нагревали до кипения с обратным холодильником в течение 1 часа. Смеси давали остыть до комнатной температуры и перемешивали при той же температуре в течение ночи. Нерастворимые вещества фильтровали и полученный прозрачный раствор оставляли перемешиваться при комнатной температуре в течение 2 дней. Получали твердое вещество, которое фильтровали, промывали ацетонитрилом (750 мл) и сушили в сушильном шкафу при 60°С в течение ночи с получением кристаллической формы указанного в заголовке соединения (125 г), обозначенного как форма А.
Картина порошковой рентгеновской дифракции кристаллической формы указанного в заголовке соединения показана на фиг.8.
(Пример 14) Синтез кристаллической формы 4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида (форма B)
4-Хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид (10 мг) помещали в небольшой флакон. Этот флакон помещали в больший флакон, содержащий этилацетат (4 мл), который закрывали и герметизировали с использованием тефлона. Эту систему выдерживали при 40°С в течение 4 дней с получением кристаллов указанного в заголовке соединения, обозначенная как форма B. (метод Растворитель-Пар).
Картина порошковой рентгеновской дифракции кристаллической формы указанного в заголовке соединения показана на фиг.9.
(Пример 15) Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
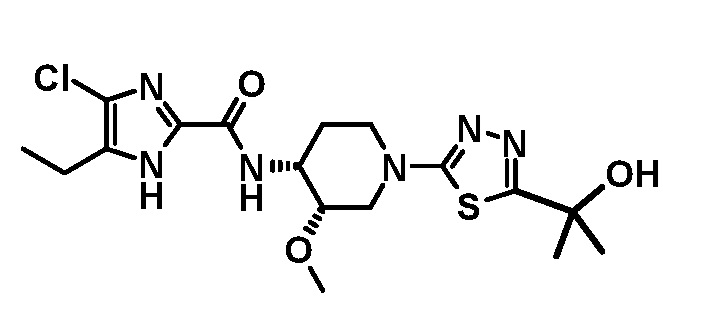
(Иллюстративное соединение No. 2)
Стадия 1: Синтез 2-(5-амино-1,3,4-тиадиазол-2-ил)пропан-2-ил бензоата
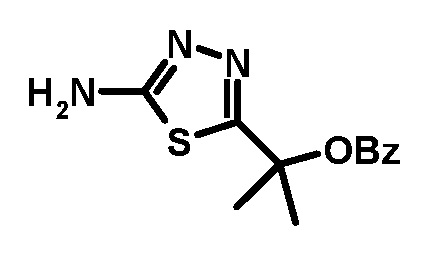
К 1,4-диоксановому раствору (100 мл) гидразинкарботиоамида (10 г, 109,72 ммоль), добавляли 2-(бензоилокси)-2-метилпропановую кислоту (22,85 г, 109,72 ммоль) и оксихлорид фосфора (25,24 г, 164,58 ммоль). Смесь нагревали до 50-55°C и перемешивали при этой температуре в течение 8 часов. Реакционную смесь охлаждали ниже 5°C, и затем к этому добавляли этилацетат (50 мл) и воду (70 мл), pH смеси доводили до 8 с помощью 8 М водного раствора гидроксида калия. К смеси, добавляли этилацетат (50 мл), тетрагидрофуран (20 мл) и воду (10 мл) и органический слой собирали. Затем органический слой промывали 10% водным раствором хлорида натрия (30 мл), органический раствор концентрировали при пониженном давлении до примерно 100 мл объема. К концентрату добавляли этилацетат (50 мл), затем смесь концентрировали при пониженном давлении до примерно 100 мл объема. К концентрату добавляли гептан (50 мл), затем смесь охлаждали ниже 5°C и перемешивали при этой температуре в течение 1 часа для получения суспензионной смеси. Твердое вещество в суспензии собирали фильтрованием и промывали холодной смесью этилацетат/гептан (2/1; 20 мл). Твердое вещество сушили при 50°С в течение более 10 часов с получением 19,98 г (69,2%) указанного в заголовке соединения в виде бесцветного твердого вещества.
1H-ЯМР (400 МГц, DMSO-d6) δ ppm: 7,26-7,22 (м, 2H), 7,01-6,95 (м, 1H), 6,88-6,81 (м, 2H), 6,57-6,48 (шир.с, 2H), 1,90 (с, 6H).
Стадия 2: Синтез 2-(5-бром-1,3,4-тиадиазол-2-ил)пропан-2-ил бензоата
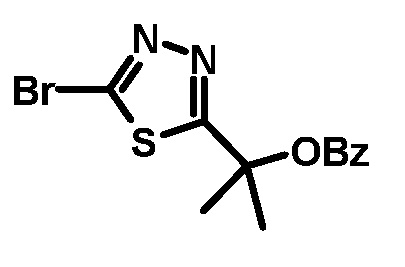
К раствору ацетонитрила (100 мл) 2-(5-амино-1,3,4-тиадиазол-2-ил)пропан-2-ил бензоата (10 г, 37,98 ммоль), добавляли воду (5 мл) и смесь охлаждали ниже 5°C, затем, к этому добавляли 48% бромистоводородную кислоту (25,62 г). К раствору добавляли водный раствор нитрита натрия (5,24 г/10 мл), поддерживая температуру ниже 10°С в течение более 2 часов, затем, смесь перемешивали при этой температуре в течение 30 минут. Потом смесь доводили до рН 3,4 с помощью 25% водного раствора гидроксида натрия, добавляли к этому этилацетат (50 мл) и 20% водный раствор сульфита натрия (50 мл), затем, смесь перемешивали при комнатной температуре в течение 30 минут. Органический слой собирали и промывали 20% водным раствором хлорида натрия (30 мл), органический слой концентрировали при пониженном давлении до примерно 20 мл объема. К концентрату добавляли 2-пропанол (60 мл) и активный уголь (1 г), и смесь перемешивали в течение 5 минут, затем уголь удаляли фильтрованием. Собранный уголь промывали 2-пропанолом (30 мл), затем объединенный фильтрат и промывные воды концентрировали при пониженном давлении до примерно 20 мл объема. К концентрату добавляли 2-пропанол (20 мл), затем, смесь охлаждали ниже 5°C. К этому добавляли аутентичный затравочный кристалл (0,1 г), полученный отдельно, и смесь перемешивали при этой температуре в течение 15 часов для получения суспензионной смеси. К суспензионной смеси добавляли воду (30 мл) и смесь перемешивали в течение 1 часа, затем, добавляли воду (30 мл) и смесь перемешивали в течение дополнительного 1 часа. Твердое вещество в суспензии собирали фильтрованием и промывали холодным водным 2-пропанолом (2-пропанол/вода=1/2, 30 мл). Твердое вещество сушили при 40°С в течение более 10 часов с получением 11,09 г (89,2%) указанного в заголовке соединения в виде желтоватого бесцветного твердого вещества.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 8,03-7,99 (м, 2H), 7,62-7,57 (м, 1H), 7,49-7,44 (м, 2H), 2,08 (с, 6H).
Стадия 3: Синтез 2-{5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-ил}пропан-2-ил бензоата
К диметилацетамидному раствору (90 мл) 4-хлор-5-этил-N-[(3S,4R)-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида (18 г, 62,77 ммоль) [полученного путем обработки его гидрохлорида, полученного на стадии 5, пример 2, с основанием], добавляли 2-(5-бром-1,3,4-тиадиазол-2-ил)пропан-2-ил бензоат (24,6 г, 75,32 ммоль) и трикалийфосфат (17,3 г), и смесь нагревали до 60°C, затем смесь перемешивали при этой температуре в течение 26 часов. Реакционную смесь охлаждали до комнатной температуры, и к смеси добавляли толуол (126 мл) и воду (90 мл), затем смесь перемешивали с получением трехслойной смеси. Нижний слой удаляли. Нижний слой остальной двухслойной смеси отделяли. К отделенному нижнему слою водного диметилацетамидного слоя добавляли толуол (126 мл), затем нижний слой удаляли. Органический слой объединяли с верхним слоем и это промывали 20% водным раствором хлорида натрия (54 мл) и органический слой концентрировали при пониженном давлении. Концентрированную смесь, содержащую указанное в заголовке соединение, использовали на следующей стадии без дополнительной очистки.
1H-ЯМР (400 МГц, CDCl3) δ ppm: 12,28 (с, 1H), 8,04-7,99 (м, 2H), 7,59-7,54 (м, 1H), 7,63-7,59 (м, 1H), 7,47-7,41 (м, 2H), 4,55-4,48 (м, 1H), 4,27-4,20 (м, 1H), 3,86-3,80 (м, 1H), 3,54-3,51 (м, 1H), 3,42 (с, 3H), 3,31-3,23 (м, 1H), 3,16-3,10 (м, 1H), 2,70 (кв, 2H, J=6,0 Гц), 2,17-2,06 (м, 1H), 2,03 (с, 3H), 2,03 (с, 3H), 1,83-1,77 (м, 1H), 1,26 (т, 3H, J=6,0 Гц).
Стадия 4: Синтез 4-хлор-5-этил-N-{(3S,4R)-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида
К толуольному раствору (72 мл) 2-{5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-ил}пропан-2-ил бензоата, полученного на предыдущей стадии, добавляли метанол (72 мл) и 28% метанольный раствор метоксида натрия (36 мл), и смесь перемешивали при комнатной температуре в течение 30 минут. К смеси добавляли толуол (90 мл), метанол (54 мл) и воду (90 мл), и смесь перемешивали, затем удаляли толуольный слой. К водному раствору метанола добавляли толуол (90 мл) и смесь доводили до рН 7,6 хлористоводородной кислотой и смесь перемешивали. Толуольный слой отделяли с получением толуольного слоя-1 и водный метанольный слой отделяли с получением водного метанольного слоя-1. К водному метанольному слою-1 добавляли толуол (90 мл), и смесь перемешивали. Толуольный слой отделяли с получением толуольного слоя-2 и водный метанольный слой отделяли с получением водного метанольного слоя-2. В объединенный толуольный слой -1 и -2 добавляли метанол (90 мл) и воду (54 мл), и смесь перемешивали. Водный метанольный слой отделяли и объединяли с водным метанольным слоем-2. Объединенный водный метанольный слой концентрировали при пониженном давлении до примерно 180 мл объема. К концентрату добавляли этилацетат (180 мл), затем добавляли хлористоводородную кислоту для доведения рН до 5,9. Смесь перемешивали и органический слой отделяли. Этот органический слой промывали 20% водным раствором хлорида натрия (54 мл), затем органический слой концентрировали при пониженном давлении до примерно 54 мл объема. К концентрату добавляли этилацетат (180 мл), затем смесь концентрировали при пониженном давлении до примерно 54 мл объема. К концентрату добавляли этилацетат (90 мл) и гептан (90 мл). Смесь нагревали до 45°C и перемешивали при этой температуре в течение 30 минут. Затем к этому добавляли гептан (54 мл), смесь перемешивали в течение 30 минут для получения суспензионной смеси. Суспензионную смесь охлаждали ниже 5°C и перемешивали в течение дополнительного 1 часа. Твердое вещество в суспензии собирали фильтрованием и собранное твердое вещество промывали холодной смесью этилацетат/гептан (1/1, 90 мл). Твердое вещество сушили при 50°C в течение более 10 часов, получая 21,42 г (79,6%) указанного в заголовке соединения в виде белого твердого вещества.
Альтернативно, 2-{5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-ил}пропан-2-ил бензоат
также получали следующим образом:
Стадия 1: Синтез трет-бутил (3S,4R)-3-метокси-4-{[(1R)-1-фенилэтил]амино}пиперидин-1-карбоксилат монобутандиоата
К раствору диметилацетамида (1000 мл) трет-бутил 3-метокси-4-оксопиперидин-1-карбоксилата (200 г, 872 ммоль), добавляли уксусную кислоту (100 мл), (1R)-1-фенилэтанамин (134 мл), диметилсульфоксид (5 мл) и 5% платина-оксид алюминия (10 г), и смесь нагревали до 60°C, затем смесь перемешивали при этой температуре в течение 8 часов в атмосфере водорода (0,3 MPa). Реакционную смесь охлаждали до комнатной температуры, затем платину-оксид алюминия удаляли фильтрованием. Собранную платину-оксид алюминия промывали толуолом (1000 мл), и к объединенному фильтрату добавляли толуол (1000 мл) и воду (1000 мл), затем смесь доводили до рН 6,9 с помощью 25% водного раствора гидроксида натрия. Смесь перемешивали и органический слой отделяли. К отделенному водному слою добавляли толуол (1000 мл), затем водный слой удаляли. Органический слой объединяли и промывали 2M водным раствором гидроксида натрия (1000 мл) и 20% водным раствором хлорида натрия (1000 мл), затем органический слой концентрировали при пониженном давлении до примерно 400 мл объема. К концентрату добавляли ацетонитрил (2000 мл), затем смесь концентрировали при пониженном давлении до примерно 400 мл объема. К концентрату добавляли ацетонитрил (1600 мл) и смесь нагревали до 50°C. Затем к смеси добавляли янтарную кислоту (113,3 г) и смесь перемешивали при этой температуре в течение 30 минут для получения суспензионной смеси. Суспензионную смесь охлаждали до комнатной температуры и перемешивали в течение дополнительного 1 часа и затем суспензионную смесь охлаждали ниже 5°C и перемешивали в течение дополнительного 1 часа. Твердое вещество в суспензии собирали фильтрованием и собранное твердое вещество промывали холодным ацетонитрилом (600 мл). Твердое вещество сушили при 50°С в течение более 10 часов с получением 329,3 г (83,4%) указанного в заголовке соединения в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ ppm: 7,42-7,37 (м, 2H), 7,35-7,30 (м, 2H), 7,26-7,21 (м, 1H), 4,16-4,06 (м, 1H), 4,03-3,95 (м, 1H), 3,90-3,64 (м, 1H), 3,46-3,30 (м, 1H), 3,38 (с, 3H), 2,80-2,40 (м, 3H), 2,38 (с, 4H), 1,45-1,35 (м, 2H), 1,38 (с, 9H), 1,26 (д, 3H, J=5,2 Гц)
Стадия 2: 2-{5-[(3S,4R)-4-амино-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-ил}пропан-2-ил бензоат монопропаноат
К суспензии трет-бутил (3S,4R)-3-метокси-4-{[(1R)-1-фенилэтил]амино}пиперидин-1-карбоксилат монобутандиоата (200 г, 441,9 ммоль) в этилацетате (1000 мл), добавляли 2M водный раствор гидроксида натрия (1000 мл). Смесь перемешивали в течение некоторого времени и водный слой удаляли, затем органический слой промывали 20% водным раствором хлорида натрия (1000 мл×2). Органический слой концентрировали при пониженном давлении до примерно 500 мл объема. К концентрату добавляли метанол (500 мл), затем смесь концентрировали при пониженном давлении до примерно 300 мл объема. К концентрату добавляли метанол (500 мл) и хлористоводородную кислоту (111,9 г), затем смесь нагревали до 55°C. Смесь перемешивали при этой температуре в течение 4 часов и охлаждали до комнатной температуры. К реакционной смеси добавляли воду (300 мл) и 25% водный раствор гидроксида натрия (177 г), затем смесь перемешивали и концентрировали при пониженном давлении до примерно 500 мл объема. К концентрату добавляли этилацетат (700 мл). Смесь перемешивали и органический слой отделяли. К отделенному водному слою добавляли этилацетат (700 мл), затем водный слой удаляли. Органический слой объединяли и промывали 20% водным раствором хлорида натрия (600 мл), затем органический слой концентрировали при пониженном давлении до примерно 500 мл объема. К концентрату добавляли метанол (500 мл), затем смесь концентрировали при пониженном давлении до примерно 300 мл объема с получением метанольного раствора (3S,4R)-3-метокси-N-[(1R)-1-фенилэтил]пиперидин-4-амина.
К метанольному раствору (3S,4R)-3-метокси-N-[(1R)-1-фенилэтил]пиперидин-4-амина. добавляли метанол (400 мл) и 5% палладий на угле (6,0 г), затем смесь нагревали до 55°C. Смесь перемешивали при этой температуре в течение 6 часов в атмосфере водорода (0,3 МПа). Реакционную смесь охлаждали до комнатной температуры, затем палладий на угле удаляли фильтрованием. Собранный палладий на угле промывали метанолом (200 мл) и объединенный фильтрат концентрировали при пониженном давлении до примерно 200 мл объема. К концентрату добавляли ацетонитрил (1000 мл), затем смесь концентрировали при пониженном давлении до примерно 200 мл объема с получением ацетонитрильного раствора (3S,4R)-3-метоксипиперидин-4-амина.
К ацетонитрильному раствору (3S,4R)-3-метоксипиперидин-4-амина, добавляли 2-(5-бром-1,3,4-тиадиазол-2-ил)пропан-2-ил бензоат (173,5 г), карбонат калия (73,3 г) и ацетонитрил (40 мл), затем смесь нагревали до 45°C и перемешивали при этой температуре в течение 22 часов. Реакционную смесь охлаждали до комнатной температуры, и к смеси добавляли этилацетат (1000 мл) и воду (300 мл). Смесь перемешивали и водный слой удаляли. Затем органический слой промывали 20% водным раствором хлорида натрия (300 мл), органический слой концентрировали при пониженном давлении до примерно 600 мл объема. К концентрату добавляли этилацетат (1000 мл) и смесь концентрировали при пониженном давлении до примерно 1000 мл объема. К концентрату добавляли этилацетат (400 мл) и пропионовую кислоту (32,7 г), затем смесь перемешивали при комнатной температуре в течение 1 часа, с получением суспензионную смесь. Затем к этому добавляли гептан (1000 мл), смесь перемешивали в течение 1 часа. Твердое вещество в суспензии собирали фильтрованием и промывали смесью этилацетат/гептан (1/1, 1000 мл). Собранное твердое вещество сушили при 50°С в течение более 10 часов с получением 144,0 г (72,3%) указанного в заголовке соединения в виде бесцветного твердого вещества.
Стадия 3: Синтез 2-{5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-ил}пропан-2-ил бензоата
К суспензии 2-{5-[(3S,4R)-4-амино-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-ил}пропан-2-ил бензоат монопропаноата (4,0 г, 8,88 ммоль) в смеси толуола (20 мл) и ацетонитрила (20 мл), добавляли 20% водный раствор хлорида натрия (12 мл), затем значение его рН доводили до 11 с помощью 25% водного раствора гидроксида натрия. Смесь перемешивали в течение некоторого времени и водный слой удаляли, затем органический слой промывали 20% водным раствором хлорида натрия (12 мл). К органическому слою добавляли 4-хлор-5-этил-1H-имидазол-2-карбоновую кислоту (1,70 г, 9,73 ммоль) и 1-гидроксибензотриазол моногидрат (1,36 г), и смесь охлаждали ниже 5°C. К смеси добавляли моногидрохлорид 1-этил-3-(3-диметиламинопропил)карбодиимида (2,55 г), и смесь перемешивали при этой температуре в течение 1,5 часов. К смеси добавляли толуол (20 мл) и воду (20 мл) и затем смесь перемешивали, водный слой удаляли. Органический слой промывали водой (20 мл), и органический слой концентрировали при пониженном давлении. Концентрат 2-{5-[(3S,4R)-4-{[(4-хлор-5-этил-1H-имидазол-2-ил)карбонил]амино}-3-метоксипиперидин-1-ил]-1,3,4-тиадиазол-2-ил}пропан-2-ил бензоата использовали для дальнейшего процесса модификации.
Биологический анализ
Можно использовать несколько различных анализов. В дополнение к анализу, упомянутому ниже, любой специалист в данной области будет знать другие анализы, которые могут быть использованы и может модифицировать анализ для конкретного применения. Такие анализы и их модификация находятся в пределах сущности и объема настоящего изобретения.
Пример испытания 1
Способ тестирования ингибирующей активности ферментов
(a) АТФ-гидролитическую активность GyrB и ParE измеряли путем корреляции расхода АТФ с относительной единицей света с использованием светоизлучающего реагента Kinase-Glo® в зависимости от количества АТФ.
В анализе гидролиза АТФ, в котором потребление АТФ коррелирует с относительной единицей света с использованием светоизлучающего реагента Kinase-Glo® в зависимости от количества АТФ, растворы испытуемого соединения серийно 4-кратно разведенные в диметилсульфоксиде:метаноле (9:1) добавляли при конечной концентрации 2% (об./об.). После смешивания с ферментом при конечной концентрации 40 ед/мл (в случае GyrB или ParE Streptococcus pneumoniae или ParE Haemophilus infuenzae) или ферментом при конечной концентрации 20 ед/мл (в случае GyrB Haemophilus infuenzae), 100 мM Tris-HCl (pH 7,5), 150 мM хлорид калия (KCl), 5 мM хлорид магния (MgCl2), 10 мM DTT, 2 мкM АТФ и 50 мкг/мл BSA добавляли к этому при конечной концентрации и смешивали. Реакционный раствор инкубировали при 30°С в течение 2 часов (2,5 часа в случае GyrB Haemophilus infuenzae). После завершения реакции реагент Kinase-Glo® добавляли в количестве, равном реакционному раствору, смешивали и давали взаимодействовать при комнатной температуре в течение 10 минут, а затем анализировали на относительную единицу света. В качестве контроля со скоростью ингибирования 0% реакцию проводили добавлением раствора диметилсульфоксид:метанол (9:1) и в качестве контроля со скоростью ингибирования 100% проводили реакцию с добавлением стерильной воды вместо фермента. Степень активности (%) рассчитывали путем деления значения, полученного путем вычитания относительной единицы света каждой реакции из относительной единицы света контроля со скоростью ингибирования 100% на величину, полученную путем вычитания относительной единицы света контроля со скоростью ингибирования 0% из относительной единицы света контроля со скоростью ингибирования 100%, и степень активности вычитали из 100 для определения скорости ингибирования гидролиза АТФ (%). Концентрацию ингибирования 50% (IC50) рассчитывали из реакции, проводимой в присутствии 8 различных концентраций тестируемого соединения.
Все соединения, раскрытые в настоящем описании, имели значение IC50 менее 0,05 мкг/мл для GyrB Streptococcus pneumoniae или Haemophilus influenzae.
(b) Потенциал соединений по настоящему изобретению против ДНК-гиразы измеряли ингибированием активности суперспирализации ДНК-гидразы Clostridium difficile. Анализы суперспирализации ДНК (30 мкл реакционной смеси) проводили в буфере, содержащем 15 мМ Tris-HCl, рН 7,5, 13% глицерина, 6 мМ MgCl2, 0,1 мг/мл BSA, 70 мМ KCl, 1 мМ дитиоэритрита, 400 нг ДНК-субстрата (релаксированной pBR322 ДНК) и различные концентрации тестируемых соединений и/или стандартов. Разведения тестируемых соединений и стандартов готовили в 20% диметилсульфоксиде (DMSO) и добавляли 3 мкл/реакцию. Реакцию начинали добавлением в реакционные смеси 2 мкл количества ДНК-гидразы Clostridium difficile, и реакциям давали возможность протекать в течение 60 минут при 37°C. Реакцию проводили добавлением 3 мкл 20% DMSO вместо тестируемого соединения в качестве полного контроля реакции, и другую реакцию проводили путем добавления стерильной воды вместо фермента как никакой контроль реакции.
Реакции останавливали добавлением 5 мкл 6×загрузочного красителя и анализировали электрофорезом через 1,0% агарозных гелей с 1×TAE подвижным буфером. Гели проводили при 60 В в течение 3 часов, окрашивали EtBr и визуализировали системой Gel Doc (BioRad). Интенсивность полос сверхспиральной ДНК определяли количественно, и IC50 ДНК-гидразы Clostridium difficile вычисляли с использованием Graph Pad Prism.
Все соединения, раскрытые в настоящем документе, имели значение IC50 менее 0,05 мкг/мл для ДНК-гидразы Clostridium difficile, например, соединения No. 1, 2, 4, 5, 10 и 11 имели значение IC50 0,0037, 0,0200, 0,0060, 0,0090, 0,0270 и 0,0120 мкг/мл, соответственно.
Пример испытания 2
(a) Метод тестирования чувствительности бактерий
Антибактериальную активность соединений по настоящему изобретению тестировали методом микродилюции бульона. Анализ проводили в соответствии с руководством CLSI по управлению процедурами тестирования на определение антимикробной чувствительности: "M7-A7 Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard - Seventh Edition (2006)".
Тестируемые штаммы выращивали на соответствующих агаровых пластинах в течение ночи. Бульон, содержащий тестируемое соединение, разведенное серийно 2-кратно, и разбавители, распределяли при 100 мкл в каждую лунку 96-луночного микропланшета. В качестве контроля роста также была приготовлена лунка с бульоном, не содержащим тестируемого соединения. Колонии, выращенные на пластинах, суспендировали в физиологическом растворе, доводили до 0,5 МакФарланда (OD625=0,08-0,10) с использованием колориметра и затем разбавляли 10-кратно физиологическим раствором. Бактериальную суспензию инокулировали при 4 мкл в каждую лунку микропланшета с использованием устройства для инокуляции бактериальной суспензии. Инокулированный микропланшет инкубировали при 35°C в течение ночи (около 20 часов). Состояние роста наблюдали невооруженным глазом, и минимальную концентрацию для ингибирования пролиферации бактерий определяли как минимальная ингибирующая концентрация (MIC).
MIC соединений, описанных в настоящем документе, для E. faecium, Streptococcus pneumoniae и Haemophilus influenzae, показаны в таблице 2, а для S. aureus в таблице 3.
ATCC 29212
ATCC 49619
ATCC 49247
(b) Метод тестирования чувствительности Clostridium difficile
Антибактериальную активность соединений по настоящему изобретению против клинических изолятов Clostridium difficile испытывали методом разведений в агаре. Анализ проводили в соответствии с руководством CLSI по управлению процедурами тестирования на определение антимикробной чувствительности ʺM11-A8, Methods for Antimicrobial Susceptibility Testing of Anaerobic Bacteria; Approved Standard - Eighth Edition. CLSI document (2012)ʺ.
Испытуемые штаммы распределяли на дополненных агаром для культивирования бруцелл (SBA) пластинах и выращивали при 36°C в анаэробных условиях в течение 42-48 часов. Разведения тестируемого соединения получали с бульоном Бруцелл и готовили SBA планшеты, содержащие разбавление тестируемого соединения. В качестве контроля анаэробного роста также готовили планшеты, не содержащие тестируемого соединения. Колонии, выращенные на пластинах, суспендировали с бульоном Бруцелла для корректировки мутности до 0,5 МакФарланда с использованием денситометра. Суспензию (1-2 мкл) тестируемых штаммов инокулировали на каждую агаровую поверхность с использованием устройств для инокуляции бактериальной суспензии. После анаэробной инкубации при 36°С в течение 42-48 часов, отсутствие и наличие роста каждого штамма исследовали невооруженным глазом. Определяли MIC, при котором наблюдается заметное снижение появления роста на тестовом планшете по сравнению с ростом на аэробных контрольных планшетах.
MIC соединений, описанных в настоящем документе, для Clostridium difficile показаны в таблице 3.
ATCC 43255
Пример испытания 3
Метод тестирования цитотоксичности
Линию клеток печени, HepG2, культивировали в каждую лунку 96-луночного микропланшета при 5000 клеток на лунку. Эти клетки обрабатывали тестируемыми соединениями или DMSO (1%) в течение 24 часов в условиях культивирования (37°C, 5% CO2). Амфотерицин B и аспирин были включены как положительный контроль и отрицательный контроль, соответственно. После обработки соединением, клетки промывали и инкубировали с раствором WST-8 в бессывороточной среде в течение 2 часов в условиях культивирования. Ячейки планшета считывали в Infinite M-1000 (Tecan), автоматическом планшет-ридере, для измерения абсорбции при 450 нм. Данные абсорбции обрабатывались вычитанием «холостого» и значение отрицательного контроля (аспирин в этом случае) также учитывалось для каждой концентрации. Рассчитывали снижение жизнеспособности клеток и вычисляли значение IC50 этих соединений для удобства сравнения.
Все соединения по настоящему изобретению имели IC50 более 100 мкг/мл.
Пример испытания 4
Метод испытания растворимости в воде
Раствор 10 ммоль/л тестируемого соединения готовили в DMSO и распределяли 100 мкл 10 ммоль/л исходных растворов DMSO в меченых стеклянных пробирках в двух экземплярах, один для первой жидкости японской фармакопеи (JP1) и второй для второй жидкости японской фармакопеи (JP2). После выпаривания DMSO из каждой пробирки в каждую пробирку добавляли 500 мкл жидкости JP1 и JP2, соответственно. Эти пробирки обрабатывали ультразвуком в течение 1 минуты и помещали на шейкер в течение 30 минут с интервалом 30 секунд каждые 5 минут. Пробирки помещали в темноту при комнатной температуре в течение 1 часа, и раствор фильтровали через мембранный фильтр. Фильтрат разбавляли 2-кратно и 10-кратно. Полученные тестовые растворы анализировали и количественно сравнивали со стандартами с использованием UPLC (стандартный препарат- 10 ммоль/л раствор в DMSO серийно разбавляли 50% водным раствором ацетонитрила для получения 2 растворов; 100 мкмоль/л стандартного раствора и 5 мкмоль/л стандартных растворов).
Пример испытания 5
Способ для тестирования оральной биодоступности (BA) у крыс и обезьян
Самцам крыс SD (7 недельного возраста), голодавшим в течение ночи, вводили в яремную вену раствор тестируемого соединения Каптизол® (3 животных) и раствор Лутрол® F68 вводили перорально (3 животных). Доза тестируемых соединений составляла 2 мг/кг массы тела крысы для обоих путей введения. Количество используемого Каптизола® составляло 0,36 мг/кг массы тела крысы. Количество используемого Лутрол® F68 составляло 0,02 мг/кг массы тела крысы. Значение ВА рассчитывали по следующей формуле с использованием значения интеграции (AUCiv(вв) (0-∞)) концентрации крови, экстраполированной от времени 0 до бесконечности после введения обоих тестируемых соединений.
Самцам Macaca fascicularis (3 или 4 летнего возраста), голодавшим в течение ночи, вводили в яремную вену раствор тестируемого соединения Каптизол® (3 животных) и раствор Лутрол® F68 вводили перорально (po) (3 животных). Доза обоих тестируемых соединений составляла 1 мг/кг массы тела обезьяны. Количество используемого Каптизола® составляло 0,18 мг/кг массы тела обезьяны. Количество используемого Лутрол® F68 составляло 0,02 мг/кг массы тела обезьяны. Значение ВА рассчитывали по следующей формуле с использованием значения интеграции (AUCiv (0-∞)) концентрации крови, экстраполированной от времени 0 до бесконечности после введения обоих тестируемых соединений.
BA(%)=[[(AUCpo(0-∞))/(доза po)]/[(AUCiv (0-∞))/(доза iv)]]×100
Чем выше значение BA для показанного соединения, тем выше оральная биодоступность.
Иллюстративные соединения имеют хорошую биодоступность (%) у крыс и обезьян, например, соединения No 2, 3 и 5 имеют 98, 51 и 60 у крыс, и соединения No 2, 3, 4 и 5 имеют 85, 100, 92 и 100 у обезьян, соответственно.
Пример испытания 6
(a) Метод для тестирования терапевтического эффекта с использованием мышиной модели инфекции легких Streptococcos pneumoniae
Штамм Streptococcos pneumoniae, культивированный с использованием бульона Тодда-Гевитта, собирали центрифугированием, суспендировали в физиологическом растворе и вводили назально в мышей CBA/JNCrlj (3-6-недельного возраста, Charles River Laboratories Japan Inc.: 4 мыши на группу) под анестезией смесью кетамин-ксилазин. Лекарственное средство вводили мышам дважды с интервалом от 6 до 10 часов. Количество бактерий в легком определяли в необработанной группе непосредственно перед первоначальным введением лекарственного средства (предварительный контроль) и в необработанную группу (пост-контроль) и в обработанной лекарственным средством группе на следующий день введение препарата и инфекции. В качестве показателя терапевтического эффекта использовали изменение количества бактерий в легких.
Все соединения примеров проявили терапевтический эффект в этом методе испытаний, например, на фиг. 1 показаны значения для соединений No. 1-5.
(b) Метод для тестирования терапевтического эффекта с использованием мышиной модели нейтропенической инфекции бедра или икры
После культивирования в бульонной среде (i) подходящим разбавителем бактериальной суспензии штамма Staphylococcus aureus или (ii) эквивалентной смесью подходящего разбавителя бактериальной суспензии штамма Enterococcus faecium и 10% муциновой суспензии использовали в качестве инокулята. Бактериальную суспензию инокулировали под анестезией смесью кетамин-ксилазин в бедро или икроножную мышцу швейцарских мышей-альбиносов или мышей ICR (4-6-недельного возраста), скомпрометированных путем введения циклофосфамида. Лекарственное средство вводили мышам 1-8 раз с интервалом от 3 до 24 часов. Количество бактерий в бедре или икре подсчитывали для необработанной группы непосредственно перед первоначальным введением лекарственного средства (предварительный контроль), для необработанной группы (пост-контроль) и для обработанной лекарственным средством группы на следующий день введения лекарственного средства и инфекции. В качестве показателя терапевтического эффекта использовали изменение количества бактерий в бедре или икре.
Иллюстративные соединения No 2, 4 и 5 продемонстрировали терапевтический эффект против Staphylococcus aureus, эквивалентный или превышающий чем линезолид этим методом испытаний.
(c) Метод тестирования терапевтического эффекта с использованием мышиной модели системной инфекции
После соответствующего разбавления бактериальной суспензии 1) штамма Staphylococcus aureus, 2) штамма Streptococcus pneumoniae и 3) штамма Enterococcus faecium, культивированного с использованием жидкой среды, добавляли 10% муциновую суспензию в эквивалентном количестве и внутрибрюшинно инокулировали в швейцарский мышей-альбиносов или мышей ddY (4-6-недельного возраста). Лекарственное средство вводили в эту модель инфекции дважды с интервалом от 4 до 6 часов. Подтверждены показатели выживаемости на 7-е сутки в необработанной группе и в группе с введенным лекарственным средством.
(d) Метод тестирования терапевтического эффекта в модели CDI хомячка
Эффективность тестируемых соединений in vivo оценивали в модели CDI хомячка, вызванной Clostridium difficile 2009155, штаммом NAP1/027. Сирийских хомячков (6-8-недельного возраста) примировали одной подкожной инъекцией клиндамицина (30 мг/кг) за 1-5 дней до заражения. Хомяков инфицировали через желудочный зонд 1 мл суспензии спор (2×104-3×106), приготовленной в PBS. Лекарственное средство вводили хомякам через 6 часов после инфицирования пероральным или подкожным путем с интервалом 24 часа в течение 5 дней. Смерть и выживание необработанной группы и обработанной лекарственным средством группы регистрировали один раз в день в течение 35 дней.
Все соединения, описанные в настоящем документе, показали терапевтический эффект в этом методе испытания, например, соединения No 1, 2, 5, 10 и 11 показали 100% выживаемость на 35 день при 0,3 мг/кг/день пероральным путем.
Пример испытания 7
Метод тестирования чувствительности P. acnes
Антибактериальную активность соединений по настоящему изобретению против клинических изолятов P. acnes тестировали методом разведений в агаре. Анализ проводили в соответствии с руководящим документом CLSI (Clinical and Laboratory Standards Institute, 2012 & 2016). Испытуемые штаммы распределяли на дополненных агаром для культивирования бруцелл (SBA) пластинах и выращивали при 36°C в анаэробных условиях в течение 42-48 часов. Разведения тестируемого соединения получали с бульоном Бруцелл и готовили SBA планшеты, содержащие разбавление тестируемого соединения. В качестве контроля анаэробного роста и для проверки аэробной контаминации также готовили планшеты, не содержащие тестируемого соединения. Колонии, выращенные на пластинах, суспендировали с бульоном Бруцелла для корректировки мутности до 0,5 МакФарланда с использованием денситометра. Суспензию (1-2 мкл) тестируемых штаммов инокулировали на каждую агаровую поверхность с использованием устройств для инокуляции бактериальной суспензии (многоточечный инокулятор). После анаэробной инкубации при 36°С в течение 42-48 часов, отсутствие и наличие роста каждого штамма на контрольных планшетах роста и планшетах с тестируемыми соединениями исследовали невооруженным глазом. Определяли MIC, при котором наблюдается заметное снижение появления роста на тестовом планшете по сравнению с ростом на аэробных контрольных планшетах.
ATCC 6921
Пример испытания 8
Метод тестирования чувствительности N. gonorrhoeae
Антибактериальную активность соединений по настоящему изобретению против клинических изолятов N. gonorrhoeae тестировали методом разведений в агаре [Clinical and Laboratory Standards Institute, 2016]. Испытуемые штаммы распределяли на чашки с шоколадным агаром и выращивали в течение 20-24 ч при 36°C в 5% диоксиде углерода. Разведения тестового соединения готовили с бульоном, и планшеты агара MIC готовили с использованием GC-агаровой основы, дополненной гемоглобином и 1% IsoVitaleX пластинами. Также были подготовлены контрольные планшеты роста, не содержащие тестируемого соединения. Колонии, выращенные на пластинах, суспендировали с бульоном Бруцелла для корректировки мутности до 0,5 МакФарланда с использованием денситометра. Суспензию (1-2 мкл) тестируемых штаммов инокулировали на каждую агаровую поверхность с использованием устройств для инокуляции бактериальной суспензии (многоточечный инокулятор). После анаэробной инкубации в течение 20-24 часов при 36°С в 5% диоксиде углерода, отсутствие и наличие роста каждого штамма на контрольных планшетах роста и планшетах с тестируемыми соединениями исследовали невооруженным глазом. Определяли MIC, при котором наблюдали заметное снижение появления роста на тестовом планшете по сравнению с ростом на аэробных контрольных планшетах.
Соединения, описанные в настоящем документе, показали очень хорошие значения MIC, например, соединения No 2 и 10 имели MIC 0,25 и 0,125, соответственно.
| название | год | авторы | номер документа |
|---|---|---|---|
| Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы ВИЧ, способы их получения и применения | 2019 |
|
RU2717101C1 |
| ПРОИЗВОДНЫЕ 1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-7-ОНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2013 |
|
RU2614418C2 |
| АЗОТСОДЕРЖАЩИЕ БИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2835672C1 |
| НОВОЕ ФОСФОРАМИДАТНОЕ ПРОИЗВОДНОЕ НУКЛЕОЗИДА И ЕГО ПРИМЕНЕНИЕ | 2014 |
|
RU2621709C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ ПИРИДИНЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756743C2 |
| АЗААДАМАНТАНОВЫЕ ПРОИЗВОДНЫЕ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2450002C2 |
| ПРОИЗВОДНЫЕ 1,6-ДИАЗАБИЦИКЛО[3.2.1]ОКТАН-7-ОНА И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ БАКТЕРИАЛЬНЫХ ИНФЕКЦИЙ | 2012 |
|
RU2570423C2 |
| АГЕНТЫ, ИНГИБИРУЮЩИЕ Р38 КИНАЗУ | 2010 |
|
RU2532376C2 |
| СОЕДИНЕНИЯ, КОМПОЗИЦИИ И СПОСОБЫ ДЛЯ ПОВЫШЕНИЯ АКТИВНОСТИ CFTR | 2015 |
|
RU2767460C2 |
| ЗАМЕЩЕННЫЕ ПИПЕРИДИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2021 |
|
RU2840772C1 |
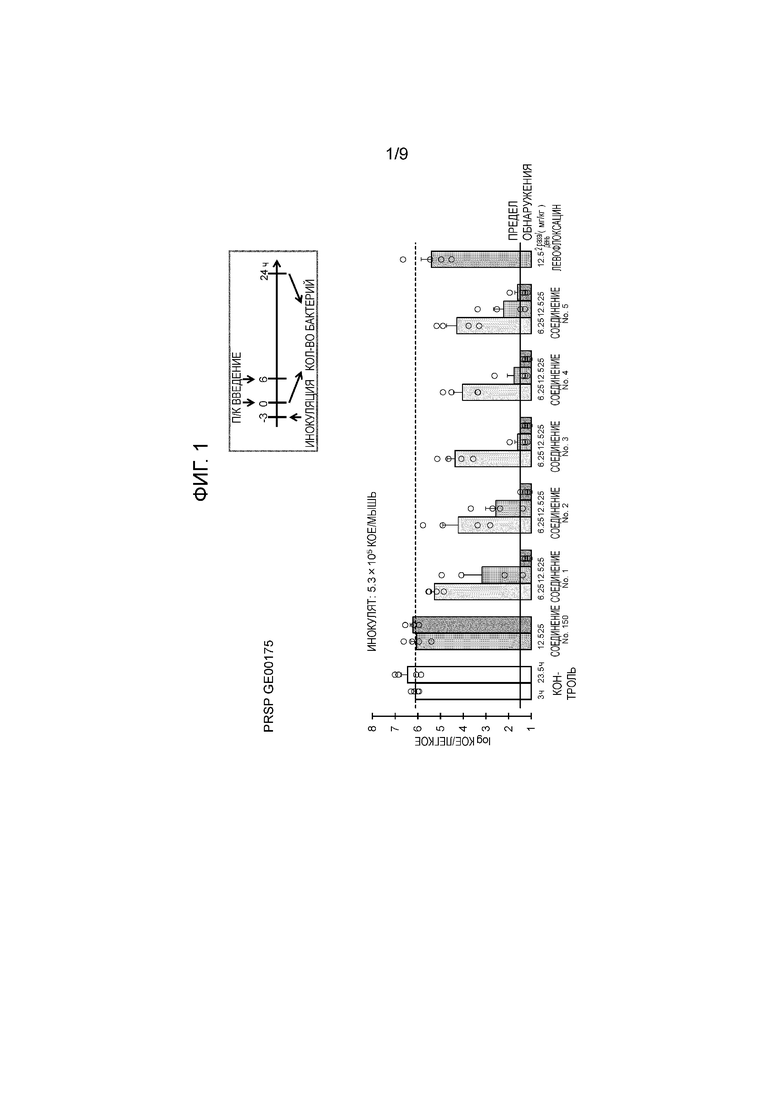
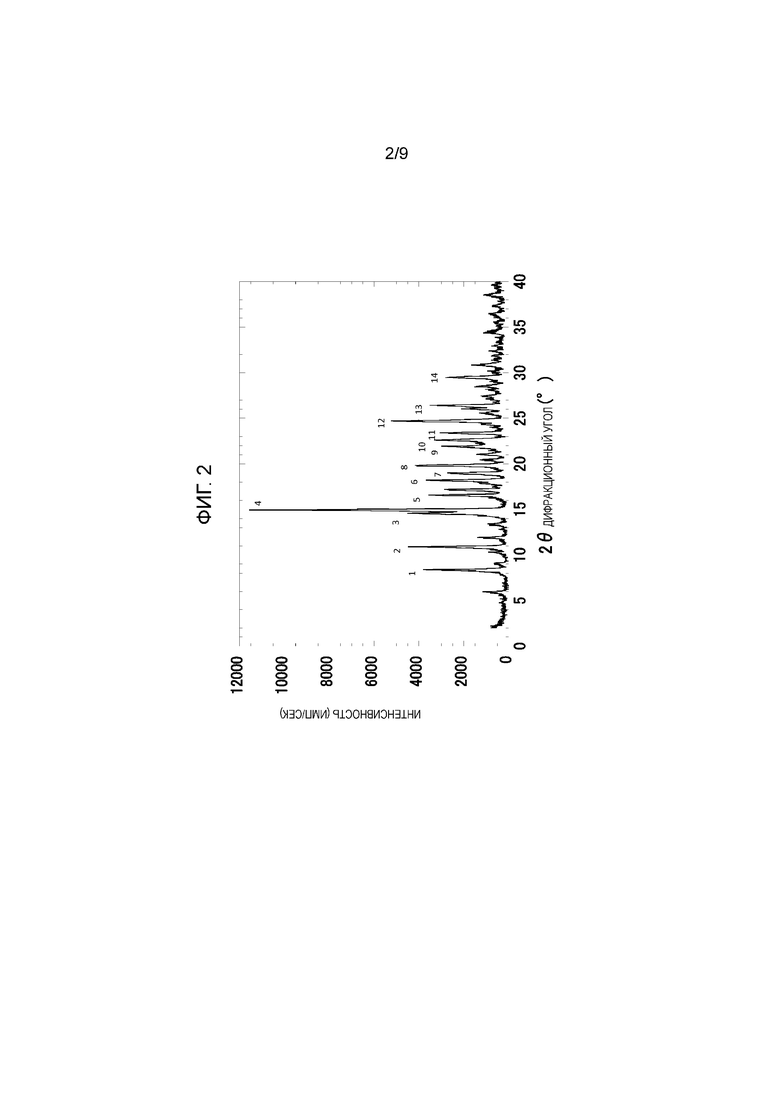
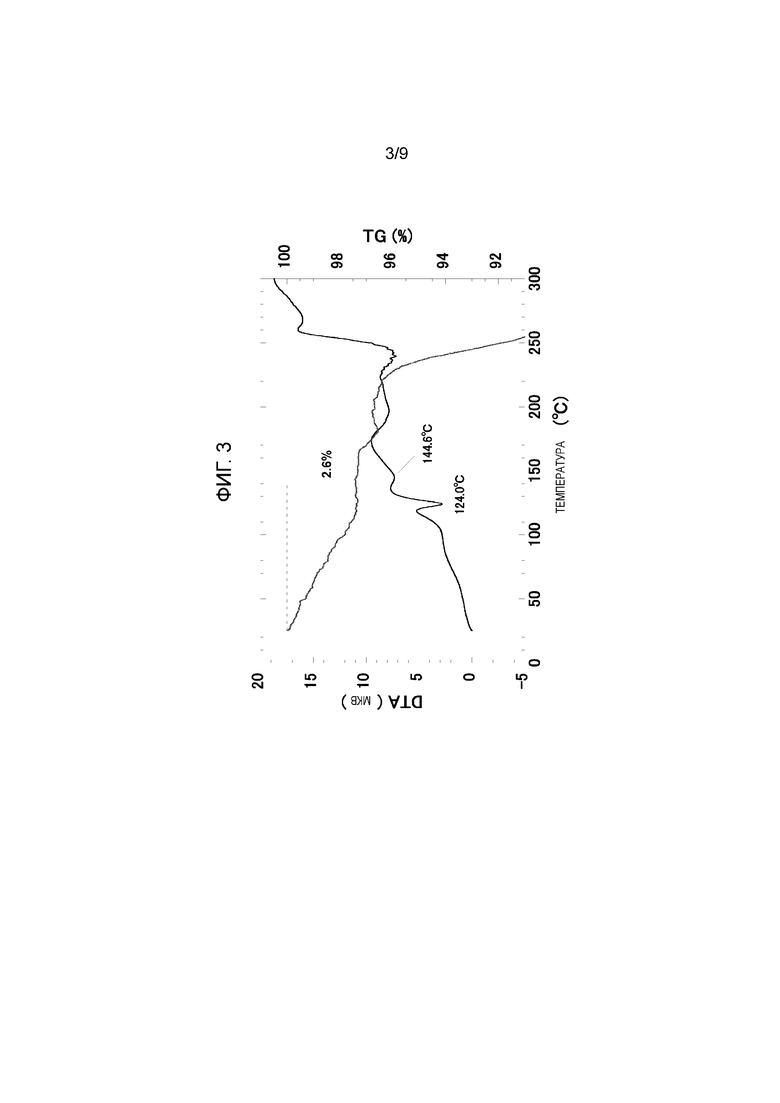

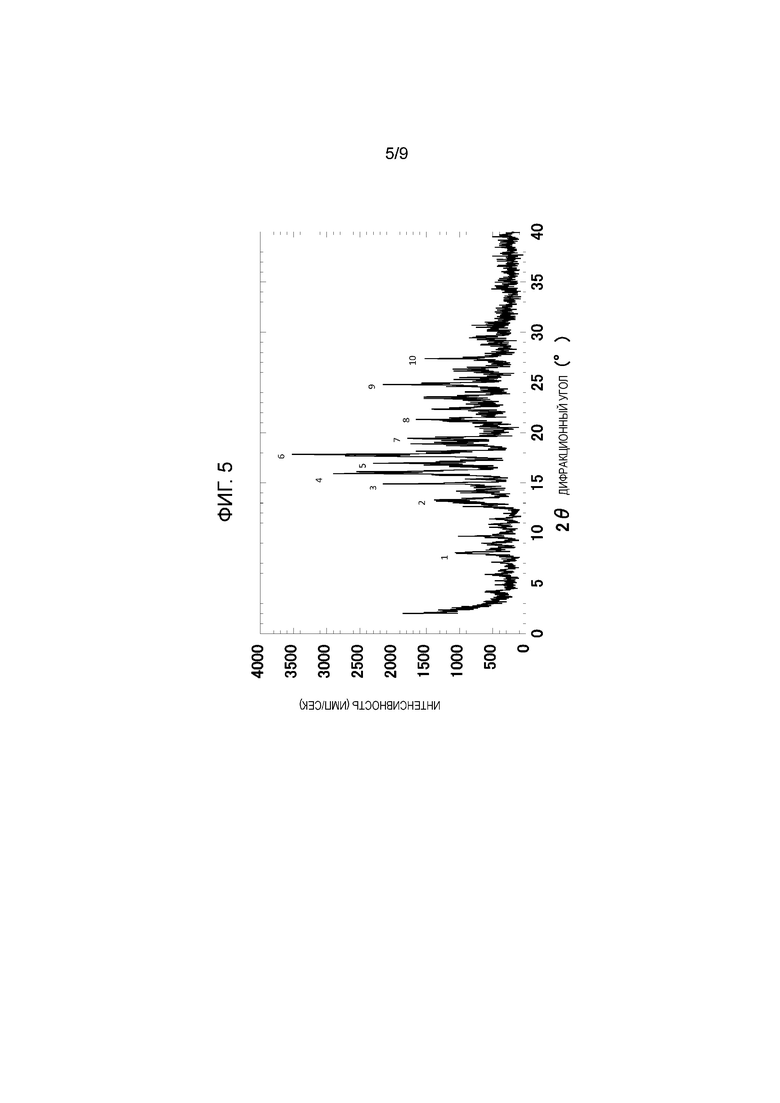
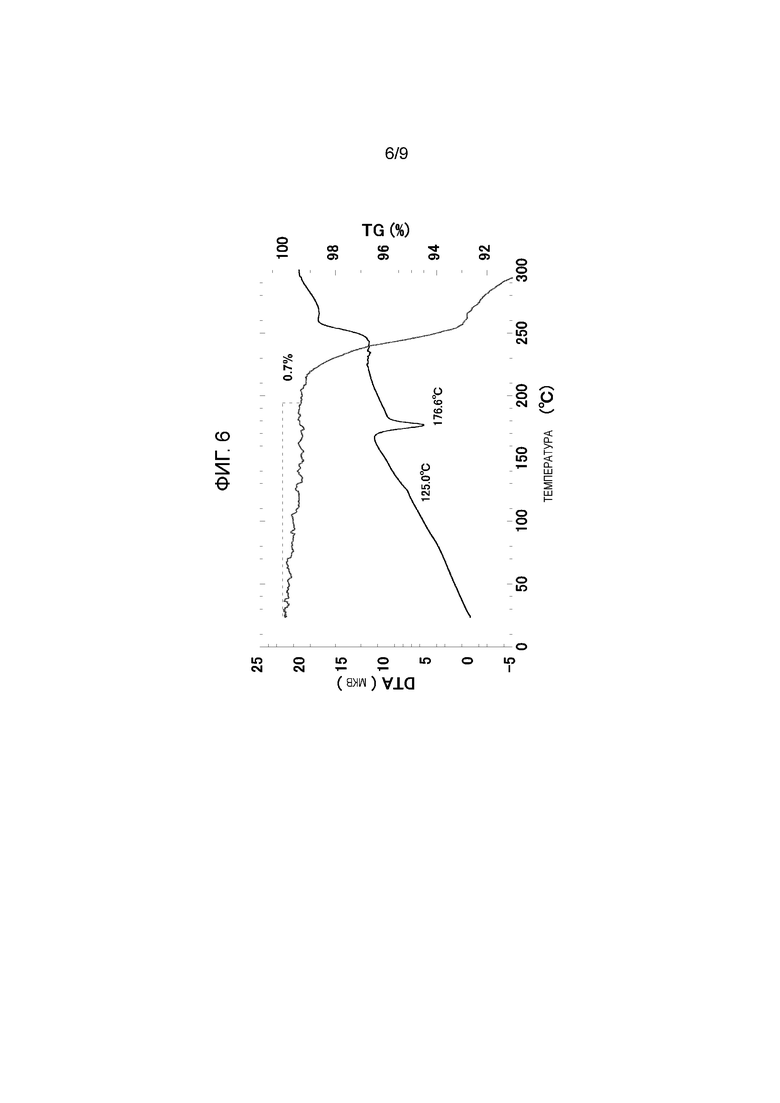
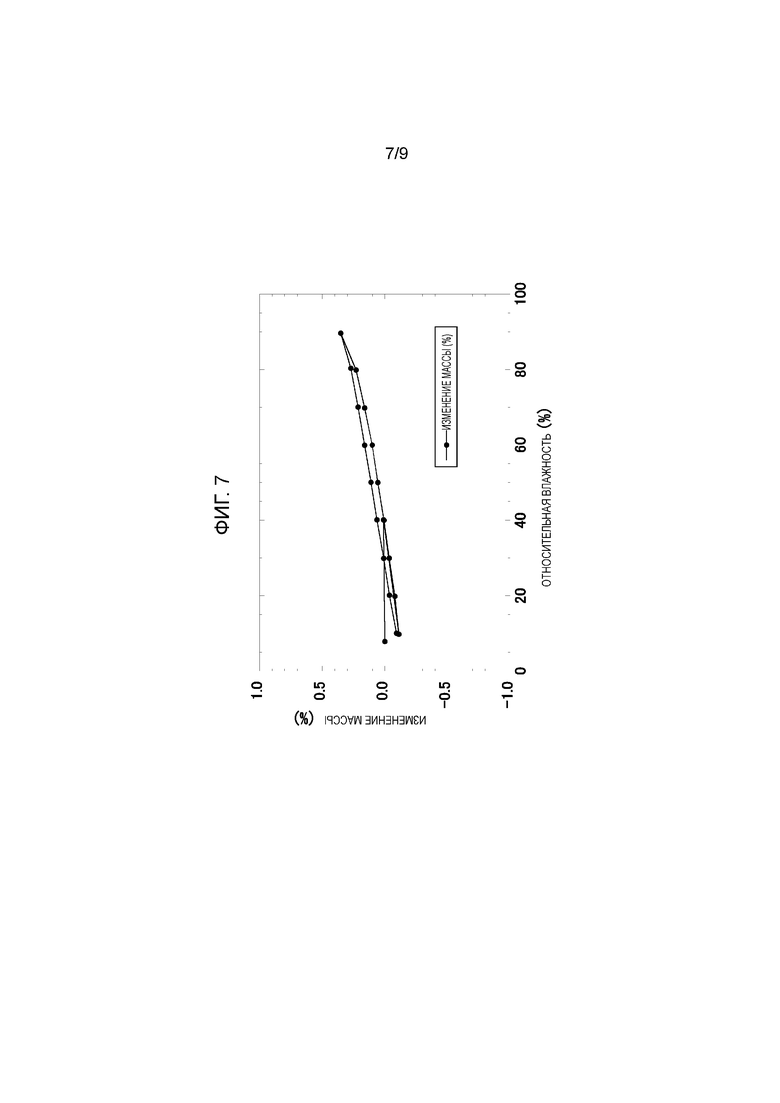
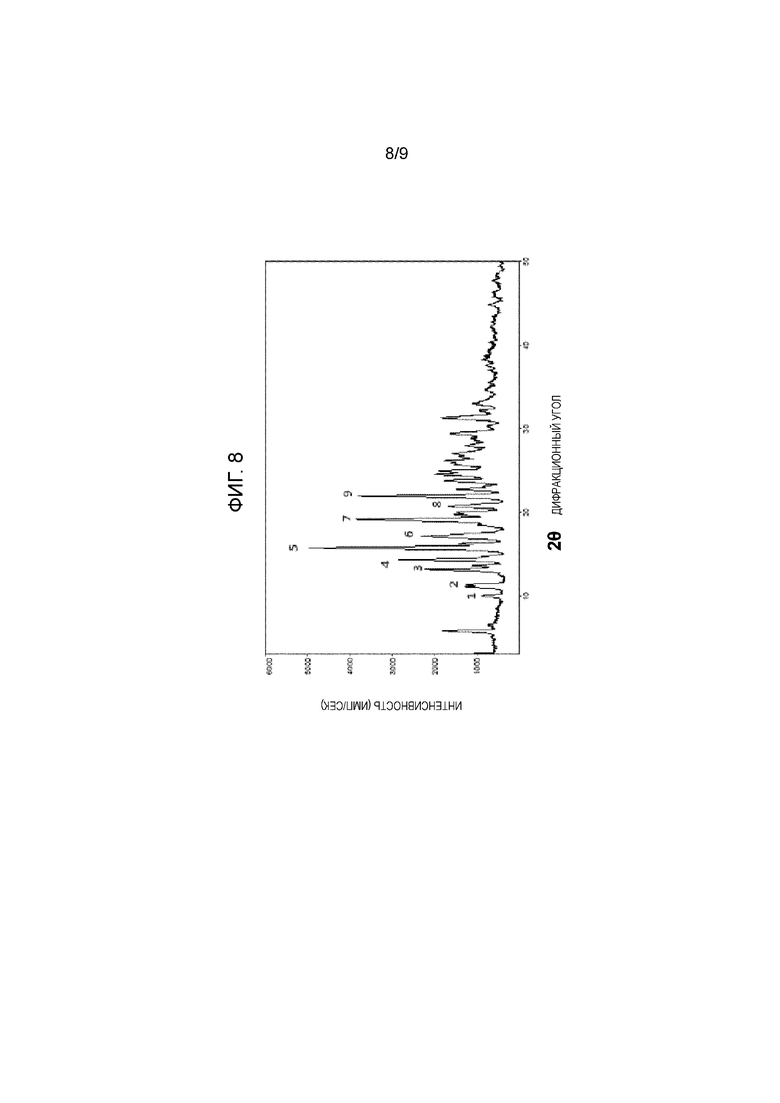
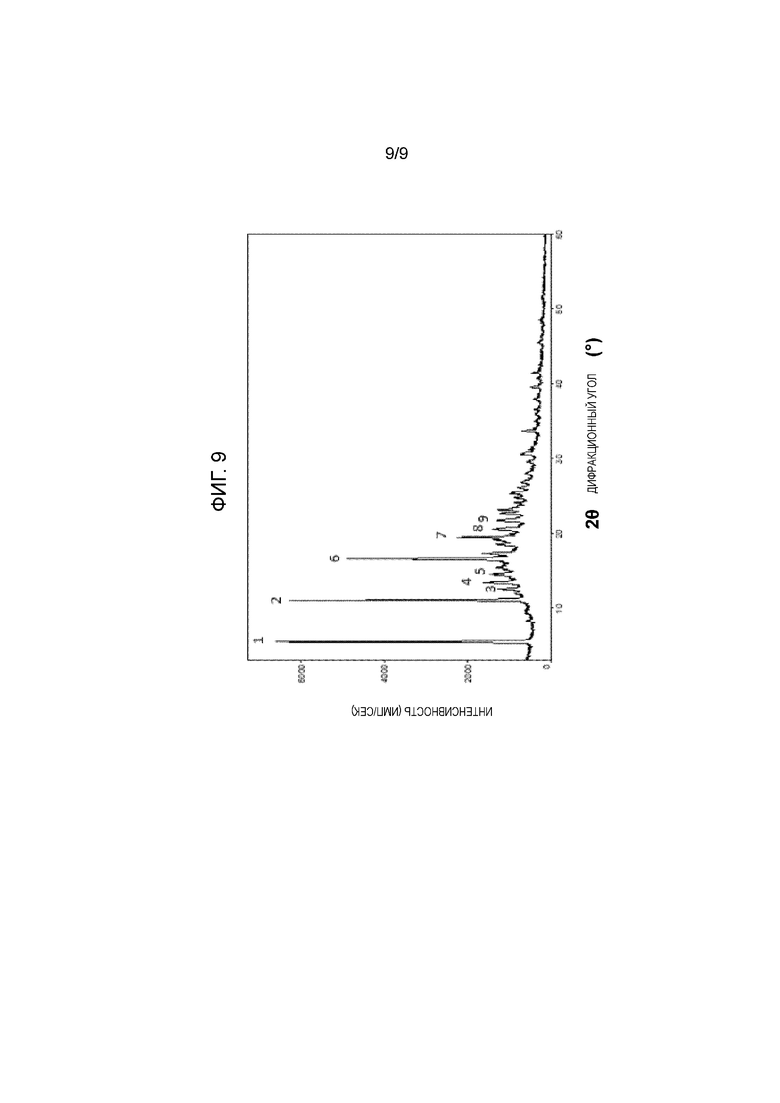
Изобретение относится к соединению общей формулы (I) или его гидрату, где R представляет собой (C1-C3) алкил, и A представляет собой следующие формулы:
 или
или 
при условии, что в общую формулу (I) включены таутомеры с водородом в различных положениях имидазольного кольца. Изобретение также относится к фармацевтической композиции для лечения бактериального инфекционного заболевания, где бактериальное инфекционное заболевание вызвано грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus и Clostridium, или грамотрицательными бактериями Neisseria gonorrhoeae, на основе указанного соединения. Технический результат – получено новое соединение, которое может найти применение в медицине для лечения бактериальных инфекционных заболеваний. 3 н. и 12 з.п. ф-лы, 9 ил., 5 табл., 15 пр.
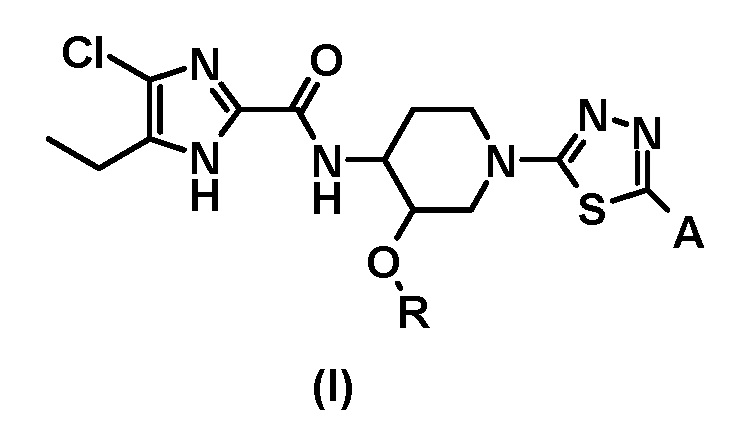
1. Соединение, представленное общей формулой (I), или его гидрат:
где R представляет собой (C1-C3) алкил, и
A представляет собой следующие формулы:
 или
или 
при условии, что в общую формулу (I) включены таутомеры с водородом в различных положениях имидазольного кольца.
2. Соединение по п.1, где соединение общей формулы (I) имеет следующую структуру:
где R представляет собой (C1-C3) алкил, и
A' представляет собой следующие формулы:
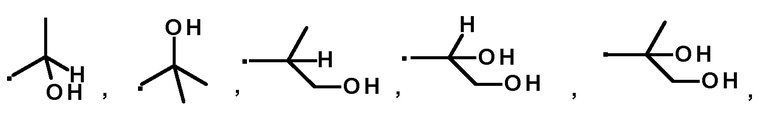
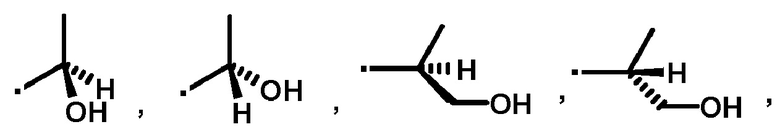
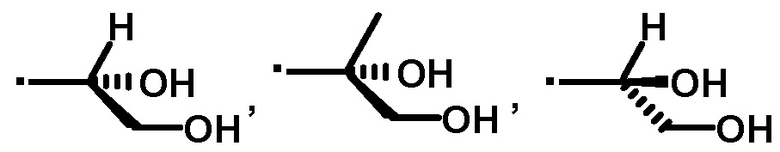 или
или 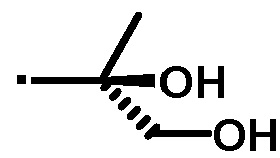 .
.
3. Соединение или его стереоизомер по п.1 или 2, где R представляет собой метил или этил.
4. Соединение по любому из пп. 1-3, где соединение выбрано из:
4-хлор-N-{(3S,4R)-3-этокси-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидрокси-1-метилэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксиэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-[(3S,4R)-1-{5-[(1R)-1-гидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-[(3S,4R)-1-{5-[(1S)-1-гидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-1H-имидазол-2-карбоксамида,
4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамида,
4-хлор-N-{(3S,4R)-3-этокси-1-[5-(2-гидроксипропан-2-ил)-1,3,4-тиадиазол-2-ил]пиперидин-4-ил}-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-N-[(3S,4R)-1-{5-[(1S)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-N-[(3S,4R)-1-{5-[(1R)-1,2-дигидроксиэтил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида,
4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамида.
5. Соединение по п.4, представляющее собой 4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидрокси-1-метилэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамид в виде кристаллической 2/3 гидратной формы.
6. Соединение по п.4, представляющее собой 4-хлор-5-этил-N-{(3S,4R)-1-[5-(1-гидрокси-1-метилэтил)-1,3,4-тиадиазол-2-ил]-3-метоксипиперидин-4-ил}-1H-имидазол-2-карбоксамид в виде кристаллического твердого вещества, в виде кристаллической 2/3 гидратной формы, обозначенной как форма I, характеризующаяся основными пиками порошковой рентгеновской дифракции при 8,38, 10,90, 14,50, 14,94, 19,82 и 24,68 (2θ), или в виде безводной формы, обозначенной как форма II, характеризующаяся основными пиками порошковой рентгеновской дифракции при 13,20, 14,94, 16,08, 17,76, 19,46 и 24,82 (2θ).
7. Соединение по п.4, представляющее собой 4-хлор-N-[(3S,4R)-1-{5-[(2R)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид в виде кристаллического твердого вещества, обозначенного как форма A, характеризующаяся основными пиками порошковой рентгеновской дифракции при 15,70, 17,06, 19,10 и 21,92 (2θ).
8. Соединение по п.4, представляющее собой 4-хлор-N-[(3S,4R)-1-{5-[(2S)-1,2-дигидроксипропан-2-ил]-1,3,4-тиадиазол-2-ил}-3-метоксипиперидин-4-ил]-5-этил-1H-имидазол-2-карбоксамид в виде кристаллического твердого вещества, обозначенного как форма B, характеризующаяся основными пиками порошковой рентгеновской дифракции при 13,13, 14,27 и 19,10 (2θ).
9. Фармацевтическая композиция для лечения бактериального инфекционного заболевания, где бактериальное инфекционное заболевание вызвано грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus и Clostridium, или грамотрицательными бактериями Neisseria gonorrhoeae, содержащая терапевтически эффективное количество соединения или его стереоизомера по любому из пп.1-8 в качестве активного ингредиента, вместе с фармацевтически приемлемым эксципиентом или носителем.
10. Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV, представленный общей формулой (I) как определено в п.1, для применения при лечении бактериального инфекционного заболевания.
11. Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV для применения по п.10, где соединение общей формулы (I) имеет следующую структуру:
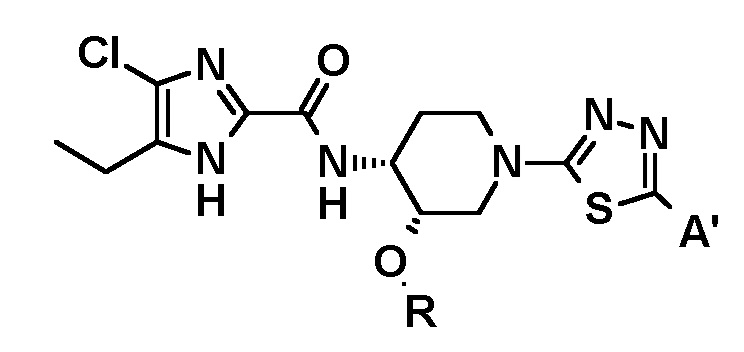
где R и A' определены в п.2.
12. Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV для применения по п.10 или 11, где указанные бактериальные инфекционные заболевания вызваны грамположительными бактериями, выбранными из видов рода Staphylococcus, Streptococcus, Enterococcus и Clostridium.
13. Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV для применения по любому из пп. 10-12, где указанное бактериальное инфекционное заболевание вызвано резистентными бактериями, выбранными из метициллин-резистентного Staphylococcus aureus (MRSA), пенициллин-резистентного Streptococcus pneumoniae (PRSP) и ванкомицин-резистентного Enterococcus (VRE).
14. Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV для применения по любому из пп.10-13, где указанное бактериальное инфекционное заболевание выбрано из акне, импетиго, чириев, флегмона фолликулита, фурункулов, карбункулов, синдрома ошпаренной кожи, пневмонии, менингита, остеомиелита, эндокардита, синдрома токсического шока, бронхита, ринита, острого синусита, воспаления среднего уха, конъюнктивита, бактериемии, сепсиса, остеомиелита, септического артрита, перитонита, перикардита, флегмона, абсцесса головного мозга, инфекций мочевыводящих путей, инфекций Clostridium difficile, обыкновенных угрей, гонореи, газовой гангрены, пищевого отравления, тетануса, ботулизма, диареи, псевдомембранозного колита и токсического мегаколона.
15. Ингибитор субъединицы GyrB ДНК-гиразы и/или субъединицы ParE топоизомеразы IV для применения по любому из пп.10-14, где указанное бактериальное инфекционное заболевание выбрано из внебольничных респираторных инфекций, внутрибольничных инфекций или инфекций Clostridium difficile.
| ОПОРНАЯ КОНСТРУКЦИЯ ДЛЯ АППАРАТА, В ЧАСТНОСТИ ДЛЯ ЭЛЕКТРИЧЕСКОГО АППАРАТА, И КОРПУС, СОДЕРЖАЩИЙ ТАКУЮ ОПОРНУЮ КОНСТРУКЦИЮ | 1999 |
|
RU2226322C2 |
| RU 2011127910 A, 20.01.2013. | |||

