f
Изобретение относится к аналитической химии, а именно к способам экстракционно-фотометрического определения ксантогенатов в водных растворах.
Известен способ определения ксантогенатов в водных растворах путем обработки анализируемой пробы раствором сульфата никеля при рН 4,5-5,0, экстракции толуолом с последующим фотометрированием экстракта С11.
Недостатками этого способа являются малая чувствительность (нижняя граница определяемых содержаний 0,025 мг/л) и невозможность определения ксантогенатов в присутствии меди.
Наиболее близким к изобретению по технической г-ущности и достигаемым результатам является способ определения ксантогенатов в водных растворах путем последовательного добавления к анализируемой пробе раствора цианида калия, хлороформа, сульфата никеля, цианидов меди, хлорида натрия, отделения хлороформеннго слоя с последующим спектрофото.метрированием в ультрафиолетовой области спектра L2J .
Недостатком такого способа также является малая чувствительность (нижняя граница определяемых содержаний 0,5 мг/л). I
Цель изобретения - повышение чуствительности способа.
Поставленная цель достигается тем, что согласно способу определения ксантогенатов в водных растворах путем обработки анализируемой пробы раствором соли двухвалентной меди и органическим растворителем, экстрат обрабатывают 9-12 М раствором соляной кислоты, воднтй слой отделяют, устанавливают в нем концентрацию кислоты 10 -6 М,добавля 0,5-7,0%-ный раствор диэтилдитиокарбамината свинца в хлороформе.
Пример Id. Определяемый компонент - этиловый ксантогенат.
В делительную воронку вместимостью 1 л помещают 500 мл водного модельного раствора 1 химический состав которого приведен в таблице (рН 4,5). Добавляют 10 мл раствора двуххлористой-меди, содержащего 5 мг/мл меди (II), 10 мл четыреххпористого углерода и энергично
37212
встряхивают в течение 1 мин. Органи- ческий слой отфильтровывают через фильтр синяя лента, в который предварительно помещают порцию без5 водного сульфата натрия (массой 1 г) в чистую сухую делительную воронку вместимостью 50 мл. К органическому слою добавляют 5 мл 9 М соляной кислоты и энергично встряхивают в
О течение 30 Cj а органическую фазу (т.е. нижний слой) отбрасывают. К водному раствору добавляют 5 мл воды, т.е. в отделенном водном слое устанавливают концентрацию кислоты
15 ,4,5 М. В делительную воронку вводят 3 мл 7%-ного раствора диэтилдитиокарбамината свинца в хлороформе и энергично встряхивают в течение 30 с. После расслаивания органичес20 кую фазу сливают в кювету с толщиной слоя 10 мм и измеряют оптическую плотность на фотоэлектроколориметре при 436 нм против раствора контрольного опыта, проведенного че5 рез все стадии анализа.
Содержание этилового ксантогена- та находят по градуировочному графику, для построения которого в ряд делительных воронок вместимостью 200 мл помещают по 100 мл воды и затем вводят О; 0,5; 1,0 и 2,0 свежеприготовленного водного раствора этилового ксантогената, содержащего 10 мкг/мл. Далее добавляют 10 мл
растврра двуххлористой меди, содержащей 5 мг/мл меди (II), 10 мп четыреххлористого углерода и энергично встряхивают в течение 1 мин. Органический слой отфильтровывают через фильтр синяя лента, в который предварительно помещают порцию безводного сульфата натрия (массой 1 г), в чистую сухую делительную воронку вместимостью 50 мп. К органическому
слою добавляют 5 мп 9 М соляной кислоты и энергично встряхивают в течение 30 с. К водному раствору добавляют 5 МП воды (органический слой отбрасывают), вводят 3 мл 7%-ного
раствора диэтилдитиокарбамината свинца в хлороформе и энергично встряхивают в течение 30 с. После расслаивания органическую фазу сливают в кювету с толщиной слоя 10 мм и измеряют оптическую плотность на фотоэлектроколориметре при 436 нм против нулевого члена шкалы. По найденным значениям оптических плотностей и
соответствующим им количествам этилового ксантогената строят градуировечный график в координатах: мкг этилового ксантогената - оптическая плотность раствора.
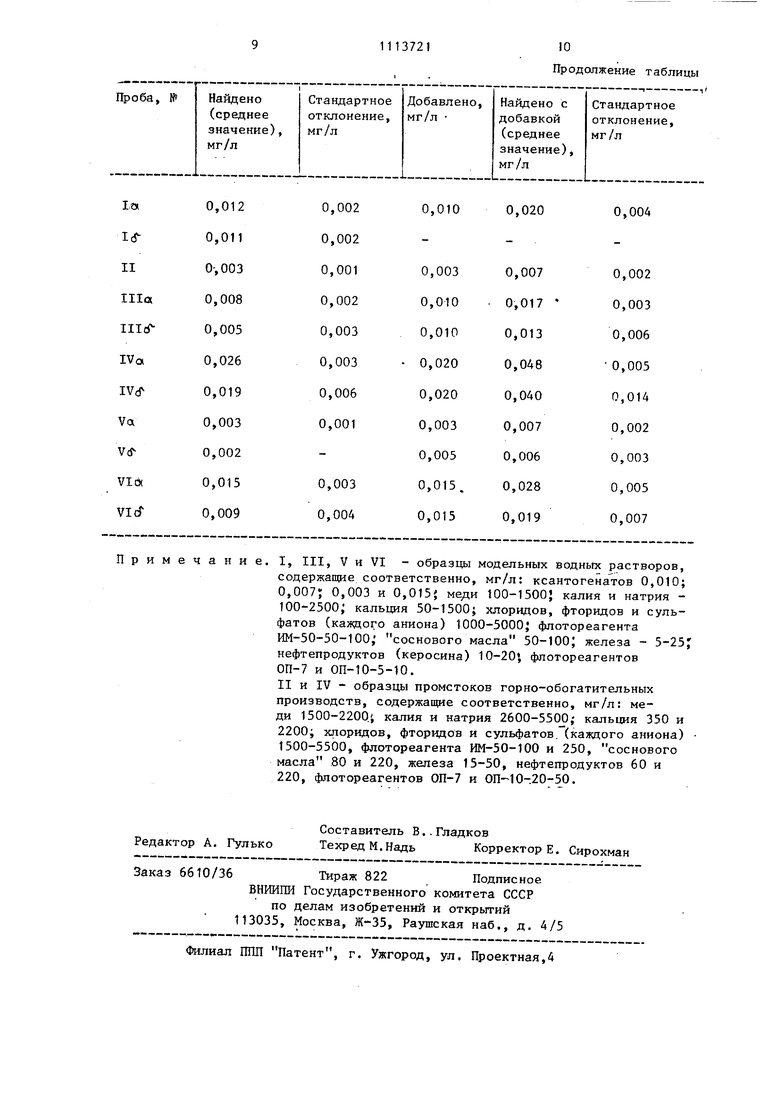
Для оценки воспроизводимости результата анализа раствора I процедуру анализа повторяют не менее 8 раз. Среднее значение результата 0,012 кг/л. Стандартное отклонение, т.е. среднеквадратичная погрешность 0,002 мг/л. Для проверки правильности результата анализа вводят добавку этилового ксантогената,равную 0,010 мл/л. Раствор с добавкой вновь анализируют (как описано). Результат а.напиза пробы с добавкой равен 0,025 мг/л, а его стандартное отклонение - 0,ОП4 мг/л.
Пример 1 d Определяемый компонент - этиловый ксантогенат.
В делительную воронку вместимостью 1,5л помещают 1000 мл водного модельного раствора I, химический состав которого приведен в таблице. Добавляют 10 мл раствора двуххлористой меди, содержащего 5 мг/л меди (II), 10 мл четыреххлористого углерода и энергично встряхивают в течение 1 мин. Органический слой отфильтровывают через фильтр синяя лента, в который предварительн помещают порцию безводного сульфата натрия (массой 1 г), в чистую сухую воронку вместимостью 50 мл. К органическому слою добавляют 5 мл 10 М соляной кислоты и энергично встряхивают в течение 30 с, а органическую фазу отбрасывают. К водному слою прибавляют по каплям 10%-ньй раство
едкого натрия (при интенсивном перемешивании палочкой)до установления рН 1-1,5, а затем прибавлением (по каплям) 1%-ного раствора щелочи устанавливают рН Д,5, т.е. в отдельном водном слое устанавливают концентрацию кислоты 1П м. В делительную воронку вводят 3 мл 1%-ного раствора диэтилдитиокарбамината свинца в хлороформе и энергично встряхивают в течение 30 с. После расслаивания органическую фазу сливают в кювету с толщиной слоя 10 мм и измеряют оптическую плотность на фотоэлектроколориметре при 436 нм против раствора контрольного опыта, проведенного через все стади анализа.
Содержание этилового ксантогената находят по градуировочному графику, построе1тие которого описано в примере 1Q Среднее значение концентрации ксантогената 0,011 мг/л (из
10 параллельных определений), а стандартное отклонение - 0,002 мг/л.
Пример 2. Определяемый компонент - технический бутиловый
ксантогенат.
В делительную воронку вместимостью 2 л помещают 1000 мл очищенного промстока обогатительной фабрики (рН 4, образец II). Добавляют
15 мл раствора двуххлористой меди, содержащее 5 мг/мл меди (II), 10 мл четыреххлористого углерода и энергично встряхивают в течение 1 мин. Органический слой отфильтровьгоают
через фильтр синяя лента, в который предварительно помещают порцию безводного сульфата натрия (массой 1 г), в чистую сухую делительную воронку вместимостью 50 мл. К органическому слою добавляют 3 кл
12,05 М соляной кислоты и энергично встряхивают в течение 30 с. После расслаивания фаз нижний слой отбрасывают, а к водному раствору добавляют 3 мл воды, т.е. устанавливают кислотность 6 М по НСР Далее в делительную воронку вводят 3 мл 0,5%-кого раствора диэтилдитиокарбам1 ната свинца в хлороформе
и энергично встряхивают в течение 30 с. После расслаивания органическую фазу количественно переносят в сухой цилиндр для колориметрирования (диаметр 6 мм, высота 150 мм).
Интенсивность окраски сравнивают на белом фоне с интенсивностью окраски шкалы сравнения, наблюдая окраску сверху вниз.
Содержание бутилового ксантогената находят по шкале сравнения, для построения которой в ряд делительных воронок вместимостью 200 мл помещают
по 100 мл воды и затем вводят
О, 0,J 0,2, 0,4 и 0,7 мл свежеприготовленного водного раствора бутилового ксантогената, содержащего 10 мкг/мл, и поступают далее, как описано в данном примере. Концентрация ксантогената в образце II равна 0,003 кг/л (среднее значение из 10 параллельных результатов), а стандартное отклонение - 0,001 мг/л. контроля правильности результата анализа вводят добавку бутилового ксантогената, равную 0,003 кг/л. Раствор с добавкой вновь анализируют (как описано), Результат анализа пробы с добавкой равен 0,007 мг/л, а его стандартное отклонение 0,002 мг/л. Пример 3. Определяемый компонент - бензиловый ксантогенат; В делительную воронку вместимостью 2 л помещают 1500 мл водного модельного раствора |11, химический состав которого подобен сбросному раствору флотационной установки обогатительной фабрики (рН 2,5). Далее добавляют 20 мп раствора двуххлористой ме,ци, содержащего 5 мг/мл меди (II), 10 МП четыреххлористого углерода и энергично встряхивают в течение 1 мин. Органический слой отфильт ровывают через фильтр синяя лента в который предварительно помещают порцию безводного сульфата натрия (массой 1 г), в чистую сухую воронку вместимостью 50 мл. К органическому слою добавляют 3 мл 10,5 М соля ной кислоты и энергично встряхивают в течение 30 с. После расслаивания фаз нижний слой отбрасывают, а к водному раствору добавляют 3 мл воды т.е. устанавливают кислотность 5,3 М по HCt. Далее в делительную воронку вводят 3 мл 3%-ного раствора диэтилдитиокарбамината свинца в хлороформе и энергично встряхивают в течение 30 с. После расслаивания органическую фазу сливают в кювету с толщиной слоя 10 мм и измеряют оптическую плотность на фотоэлектроколориметре при 436 нм против раствора контрольного опыта, проведенного через все стадии анализа. Содержание бензилового ксантоген та находят по градуировочному графику, для построения которого в ряд делительных воронок вместимостью 200 МП помещают по 100 мл воды и за тем вводят 0; 0,5, 1,0 и 2,0 мл свежеприготовленного водного раство бензилового ксантогената, содержаще 10 мкг/мл, и поступают далее, как описано в данном примере. Концентра ция ксантогената в о&разце III равна 0,008 мг/л (среднее значение из 8 параллельньгх результатов), а стан дартное отклонение - 0,002 мг/л. Контроль правильности результата проводят, как в примерах 1 и 2.Результаты этих опытов приведены в таблице (см. Ilia). Аналогичным образом анализируют растворы III -IV, где определяемыми компонентами являются бензиловый, изопропиловый и смеси ксантогенатов. Эти результаты сведены в таблицу, а химический состав растворов приведен в приложении к ней. Из таблицы видно, что предлагаемый способ позволяет с достаточно хорошей воспроизводимостью и правильностью определять микроколичества ксантогенатов в водных растворах, имеющих сложный химический состав. Эффективность способа существенно зависит от концентрации соляной кислоты при обработке экстракта.Переход микроколичеств меди (II), эквивалентных ксантогенатам, в солянокислый раствор, т.е. выход меди (II) в окрашенное фотометрируемое соединение, является количественным при кислотности 9 М и выше (насколько позволяет крепость продажной концентрированной кислоты), т.е. до 12 М. С Другой стороны, кислотность водного раствора, взаимодействующего с 0,5-7,0%-ным раствором диэтилдитиокарбамината свинца, не должна превышать 6 М НС2, так как в более кислой среде уменьшается выход окрашенного фотометрируемого соединения за счет ухудшения условий взаимодействия меди (II) с диэтилдитиокарбаминатом свинца. Уменьшение кислотности водного раствора, вплоть до слабокислой среды (рН 4-5), практически не уменьшает выхода окрашенного фотометрируемого соединения. Однако нейтрализация кислого реэкстракта путем введения щелочи или аммиака повышает трудоемкость анализа, поскольку добавлять растворы щелочи или аммиака следует малыми порциями (желательно по каплям) из-за возможности гидролиза микроколичеств меди. Поэтому следует устанавливать кислотность ниже 6 М НС1 соответствующим разбавлением реэкстракта водой. Если концентрация диэтилдитиокарбамината свинца в хлороформе берется менее 5 мг/мл, т.е. О,5%,то его, взаимодействие с микроколичествами меди при кислотности растворов близкой к 6 М НС4 ухудшается (см.таблицу
71
примеры Illot, IVrf, Vf и VI(J), т.е. ухудшаются метрологические параметры методики (результаты становятся заниженными на 30-60 отн.% и ухудшается воспроизводимость).Увеличение концентрации более 70 мг/мл т.е. 7%, ограничено растворимостью диэтилдитиокарбамината свинца в хлороформе.
В рекомендованных условиях определению не мешают более, чем 10 -кратные количества меди, щелочных и щелочноземельных элементов, хлоридов, фторидов и сульфатов, 10 -кратные количества флотореагента ИМ-50 (техническая смесь алкилгидроксамовых кислот) и соснового масла (техническое название флот ореагента, содержащего спирты и углероды терпеневого ряда), 10 -кратные
Результаты анализа образцов модельных водных растворов и промстоков горнообогатительного производства на содержание примеси ксантогенатов (п 8)
37218
количества железа, нефтепродуктов и поверхностно-активных реагентов типа ОП-7 и ОП-10.
Экспериментально найденная ниж5 няя граница определяемых содержаний, оцененная по 28-критерию (S стандартное отклонение результатов определения в наиболее чистых по ксантогенатам водных растворах),
равна 0,002 мг/л. Относительное стандартное отклонение единичного определения 0,2-0,3 (п 8) для концентраций, превьппающих в 2-5 раз минимально, определяемые.
Таким образом, предлагаемый способ позволяет в сравнении с известным существенно, т.е. не менее, чем в 250 раз, снизить нижние границы определяемых содержаний, а также упростить процесс анализа.



| название | год | авторы | номер документа |
|---|---|---|---|
| Способ определения флотореагента,содержащего алкилгидроксамовые кислоты,в водных растворах | 1981 |
|
SU983523A1 |
| Способ количественного определенияпРиМЕСи КСАНТОгЕНАТОВ B ВОдНыХРАСТВОРАХ | 1979 |
|
SU828034A1 |
| Способ определения высших изомерных карбоновых кислот в воде | 1984 |
|
SU1223099A1 |
| Способ определения трибутилфосфата в водном растворе | 1981 |
|
SU978026A1 |
| Способ получения трехфазных экстракционных систем | 1983 |
|
SU1158890A1 |
| Способ определения микропримесей в церии | 1979 |
|
SU787371A1 |
| СПОСОБ ИНВЕРСИОННОГО ВОЛЬТАМПЕРОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ МИКРОПРИМЕСЕЙ МЕДИ (II) И СУРЬМЫ (III) В ЦИНКОВОМ ЭЛЕКТРОЛИТЕ | 2004 |
|
RU2297626C2 |
| Способ количественного определения первичных алифатических аминов | 1979 |
|
SU883738A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ МЕДИ (I) | 2008 |
|
RU2374637C1 |
| Способ определения концентрации кадмия в пищевых продуктах | 1984 |
|
SU1278705A1 |
СПОСОБ ОПРЕДЕЛЕНИЯ КСАНТОГЕНАТОВ В ВОДНЫХ.РАСТВОРАХ путем обработки анализируемой пробы раствором соли двухвааентной меди и органическим растворителем с получением экстракта и последующего, его спектрофотометрирования, отличающийся тем, что, с целью повышения чувствительности способа, экстракт обрабатывают 9-12 М раствором соляной кислоты, водный слой отделяют, устанавливают в нем концентрацию кислоты 10 -6. М, добавляют 0,5-7,0%-ный раствор диэтилдитиокарбамината свинца в хлороформе. i (Л
| Лурье Ю.Ю., Рыбакова А.И | |||
| Химический анализ производственных сточных вод | |||
| М., Химия, 1974, с | |||
| Прибор для корчевания пней | 1921 |
|
SU237A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| С | |||
| Pohlandt, Е.В.Т | |||
| Cook, T.W | |||
| Steele, The determination of xanthogenates, Talanta, V | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| ПРИБОР ДЛЯ ЗАПИСИ ПРОЙДЕННОГО ПУТИ | 1923 |
|
SU1129A1 |
