ел
со
00 ОС
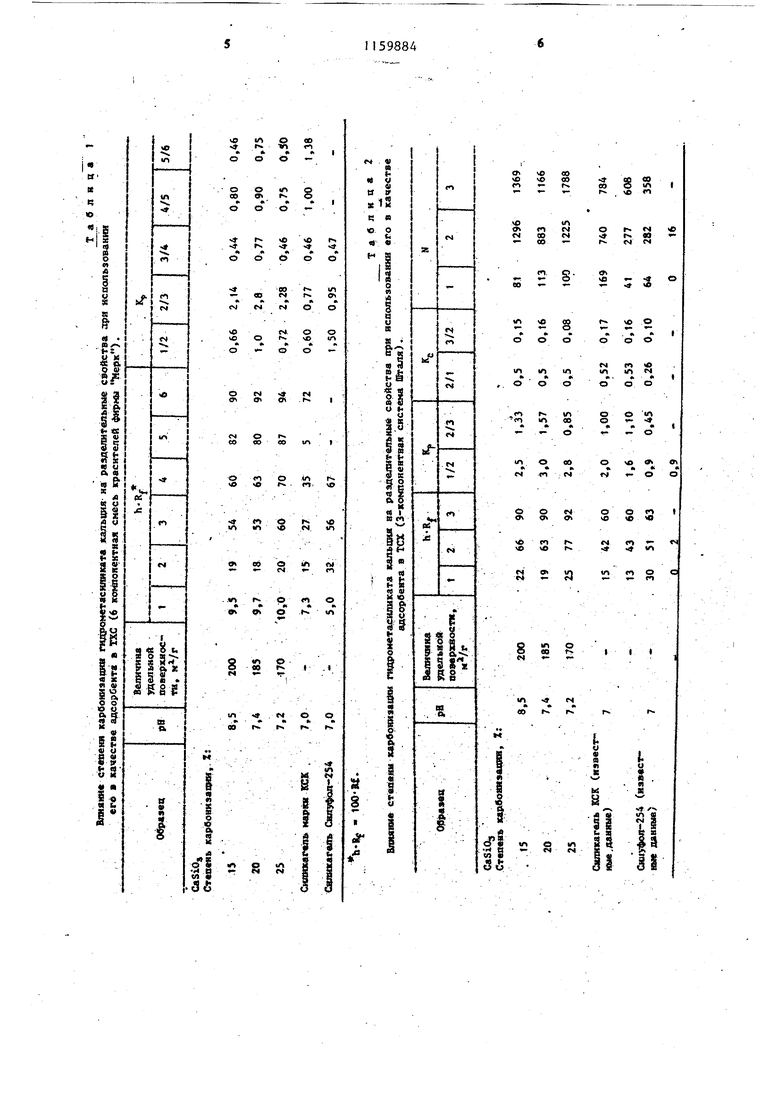
1 Изобретение относится к произво ству силикатных материалов, в част ности неорганическим адсорбентам для тонкослойной хроматографии (тех). Цель изобретения - повьшение ра делительной способности при исполь зовании гидросиликата кальция в ка честве сорбента для тонкослойной хроматографии. Пример 1, Берут 300 мл щелочносиликатного раствора (SiOj 126,9 г/л,- Naj.0(jeu,, 137,8 г/л) подо ревают до 85°С и при непрерывном перемешивании приливают 300 мл 2М раствора . перемешивание продолжают 60 мин, затем продукт реа1гдии, представляющий собой оса- док белого цвета, выдерживают в ма точном растворе при в течение 24 ч, после чего осадок отделяют от маточного раствора, отмьшают от солей натрия и фильтруют. Отмытый осадок, представляющий собой гидро метасиликат кальция (SiOi:CaO 1,0т в количестве 130 г с влажностью 60% переводят в состояние пульпы, отношение Ж (вода):Т 3:1, через которую при 50 С и непрерывном перемешивании пропускают ток газа, COi со скоростью 3 л/ч в течение 10 мин. Полученный карбонизированный до 15% степени карбонизации гидрометасиликат кальция (эквивале но 34% содержание CaGO,) фильтруют сушат при 25 С до 20% содержания влаги, а затем при до 5% содержания остаточной влаги. 0,12 г гидрометасиликата кальци карбонизированного до 15% степени карбонизации (S 200 MVr, VCP 1,0 CMVr, - 200A, но Р водной вытяжки 0,2) суспендируют в 7 мп дистиллированной воды в течение 3 мин, сус пензию ровным слоем накатывают н поверхность стеклянной пластинки, высушивают препарат при 25°С в течение 1 ч, а затем активируют при 110 С в течение 30 мин. Аналогичен процесс получения и последующего использования в ТСХ силикатного материала, полученного при применении в качестве кальдийсодержащего реагента CaCOj (63,45 CaCOj в 300 мл воды) или СаО (35,5 г СаО в 300 мл воды). Степен карбонизации 15%. 842 Пример 2. Берут 300 мл щелочносиликатного раствора (SiOj. 126,9 г/л, Na2.0o6m, 137,8 г/л) подогревают до 85 С и при непрерывном перемешивании приливают 300. мл 2М раствора СаСЙ. перемешивание продолжают 60 мин, затем продукт реакции, представляющий собой осадок белого цвета, выдерживают в маточном растворе при 38°С в течение 22 ч, после чего осадок отделяют от маточного раствора, отмывают от солей натрия и фильтруют. Отмытый осадок, представляюищй собой гидрометасиликат кальция ( 1:10) в количестве 135 г с влажностью 63% переводят в состояние пульпы, отношение Ж (вода), через которую при и непрерывном перемешивании пропускают ток газа COj со скоростью 7 л/ч в течение 30 мин. Полученный карбонизированный до 25% степени карбонизации гидрометасиликат кальция (экБивалентно 57% содержания CaCOj) фильтруют, сушат при 20°С до 30% содержания влаги, а затем при 115°С до 6% содержания остаточной влаги. 0,12 г гидрометасиликата кальция, карбонизированного до 25% степени карбонизации (S 170 MVr, V 0,7 см5/г, d,p 150 1, SH,JO /S, - 1,0, pH водной вытяжки 7,.) суспендируют в 7 мл дистиллированной воды в течение 3 мин, суспензию ровным слоем накатывают на поверхность стеклянной пластинки , высуигивают препарат при 25°С в течение 1 ч, а затем активируют при 110 С в течение 30 мин. Аналогичен процесс получения и последующего использования в ТСХ силикатного материала, полученного при применении в качестве кальцийсодержащего реагента СаСО (63,45 г CaCOj в 300 мл воды) или СаО (35,5 г СаО в 300 мл воды). Степень карбонизации 25%. П р и м е р 3. Берут 300 мл щелочносиликатного раствора (SiO. .126,9 г/л, NaiOog 137,8 г/л)-, подогревают в реакторе до 85с и при непрерывном перемешивании приливают 300 МП 2М раствора СаС, перемешивание продолжают 60 мин. Затем продукт реакции, представляющий собой осадок белого цвета, вьщерживают в маточном растворе при 39С 3 в течение 23 ч, после чего осадок отделяют от маточного раствора, от мывают от солей натрия и фильтруют Отмытый осадок, представляющий собой гидрометасиликат кальция (Si07.:CaO 1:0,5) в количестве 140 г с влажностью 65% переводят в состояние пульпы, отношение Ж (вода),5:1, через которую при 55°С и непрерь1вном перемешивании пропускают ток газа COj. со скоростью 5 л/ч в течение 20 мин. Полученный карбонизированный до 20% степени карбониаации гидрометасили кат кальция (эквивалентно 50% содержания СаСО) фильтруют, сушат при 25 С до 25% содержания влаги, а затем при 120°С до5% содержания остаточной влаги, 0,12 .г гидрометасиликата кальция, карбонизированного.до 20% сте пени карбонизации (5ц 185 0,84 CMVr, d,,,, 180А, Т7 h ОЛ л.5 / А- 1ЙПЛ HjO Р водной В55ГГЯЖКИ 7,4) суспендируют в 7 мл дистиллир ванной воды в течение 3 мин, суспензию ровным слоем накатывают н поверхность стеклянной пластинки, высушивают препарат при 25°С в течение 1 ч, а затем активируют при 120°С в течение 30 мин. В д-абл. 1 и 2 приведены характ ристики разрешающей способности сорбента для ТСХ, полученного пред лагаемым Способом По сравнению с применяемыми в настоящее время сил гелем КСК-2 и пластинами Силуфол-2 Применение в качестве сорбента гид росиликата кальция, полученного по известному способу не позволяет ра делить компоненты системы Шталя и фирмы Мерк. 4 , 4 Из табл. 1 и 2 видно, что разделительная способность сорбента превосходит аналогичные характеристики для Силуфола-254 и не уступает силикагелю КСК-2. Пример 4. В условиях примера 1 синтезируют сорбент, проводя сйнёрезис при 60 С в течение 24 ч. Получают агрегированный порошок 5-200 мкм не пригодный для ТСХ. Пример 5. В условиях примера 1 сйнёрезис проводят при 25°С в течение 2 сут. Получают порошок, содержащий кристаллическую фазу. уменьшается .объем пор до 0,62 м/г, происходит агрегация частиц 5100 мкм. Сорбент не отвечает требованиям тех. . Пример 6. В условиях примера 1 синтезируют адсорбент и производят вьщерживание перед сушкой до различного содержания влаги. В табл. 3 приведены характеристики получающегося продукта. Из табл. 3 видно, что 30% содержание влаги в гидрометасиликате кальция перед сушкой при 120°С является предельно допустимой, так как при содержании влаги 30% имеет место термопаровое спекание скелета силикатного материала и комкование частиц порошкообразной системы. При содержании влаги 20% адсорбционно-структурные характеристики образцов гидрометасиликата кальция примерно одинаковые (ср. обр. 5, 7 и 8), поэтому более глубокое обезвоживание силикатного материала перед высокотемпературной сушкой нецелесообразно с точки зрения увеличения продолжительности сушки.
Таблица 3


| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения гидросиликатного продукта | 1980 |
|
SU903296A1 |
| Способ получения метасиликата кальция | 1980 |
|
SU929555A1 |
| Способ получения гидросиликатов | 1972 |
|
SU447362A1 |
| Способ получения одноступенчатых угольных реплик | 1986 |
|
SU1374087A1 |
| Способ получения метасиликата кальция | 1981 |
|
SU986852A1 |
| Способ получения силикатно-кальциевого продукта | 1980 |
|
SU981217A1 |
| Способ получения сорбента для газовой хроматографии | 1976 |
|
SU616589A1 |
| Способ получения окиси магния | 1978 |
|
SU781178A1 |
| Способ очистки себационовой кислоты | 1970 |
|
SU335933A1 |
| Способ очистки раствора силиката натрия от алюминия | 1983 |
|
SU1131829A1 |
1. СПОСОБ ПОЛУЧЕНИЯ гадРОСИЛИКАТА,КАЛЬЦИЯ, включающий смешивание щелочно-силикатного раствора с кальцийсодёржащим реагентом с образованием гидрогеля метасиликата кальция, фильтрование смеси., промывку и сушку продукта при 110120 С, отличающийся тем, что, с цепью повьппения разделительной способности при использовании гидросиликата кальция в качес-сзе сорбента для тонкослойной хроматографии, перед фильтрованием смесь вццерживают при 38-40с в течение 22-24 ч, а перед сушкой осуществляют карбониза1щю до 15-25% в пересчете на СО. и вьщерживают до остаточной влаги 20-30%. 2. Способ по п. 1, о т л и ч аю щи и с я тем, что карбонизацию осуществляют пропусканием углекис- . лого газа со скоростью 3-7 л/ч в те(Л чение tO-30 мин через пульпу при Ж:Т 
68 95 120 137 185
203 210 205
Сильное комкование
Комкование
Порошкообразная система
| Оглрблина И.П | |||
| Адсорбенты, их получение, свойства и применение | |||
| Л., Наука, 1978, с | |||
| Катодное реле | 1918 |
|
SU159A1 |
| Способ получения гидросиликатного продукта | 1980 |
|
SU903296A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
