1
Изобретение относится к способу получения новых производных аспарги- новой кислоты общей формулы
R
м
H N-CH-CO --KH-C-NH-CO-R
СИ, R
где Я - низший ялкил или низший оксиалкил,
R - водород или низший апкил и, R - разветвленный алкил С., циклоалкил С -т низший апкилциклоалкил, дицикло- (низший)алкил, 2,2,4,4-тет- раметилтиетан или фенил. Указанные производные формулы I могут быть использованы в качестве подслащивателей в пищевой промьшшен- ности.
Целью изобретения является способ получения производных М-(Ь-аспаргкп)
О4
-N-(1-ациламино)-алкиламино - новых подслащивателей с улучшенными свойствами.
Изобретение иллюстрируется следующими примерами.
Пример 1. М-(1.-аспаргил)-Ы- -циклопентакарбонил-R-l, t-лиаминоэта ( I, R-CHi,R -H, К -циклопен- тил) .
А. Растворяют 20 г (0,225 моль) П-аланина в 400 мл диметилформамида, обрабатывают 26,8 г (0,250 моль) хлортриметилсилана и перемешивают смесь при комнатной температуре до получения гомогенного раствора (приблизительно 45 мин). Тем временем растворяют 72 г (0,200 моль) Ы -бен- зилоксикарбонил-ft-бензил-Ь-аспарги- новой кислоты в смеси (1:1) диметилформамида и тетрагидрофурана (880мл) охлажденной до -15 С, и обрабатывают 22,4 г (0,200 моль) N-метилморфолина и 26,2 мл (0, 200 моль) изобутилхлор- формата. После 8-минутной активации при -15°С приливают к полученному раствору предварительно охлажденный раствор силилового эфира D-аланина,
В. Растворяют 2,2 г (5,1 ммоль) продукта раздела Б в 50 мл ацетонит- рила. Раствор разбавляют равным объемом воды. Затем добавляют 2,4 г (5,6 ммоль) иодбензол-бис-(трифтор- ацетата). Реакционную смесь перемешивают при комнатной температуре в
35
затем по каплям - 22,4 мл (0,200 моль) N-метилморфолина, обеспечивая поддер- ЗО течение 4 ч (прозрачный раствор обра- жание температуры реакции -15 С.Раст- зуется примерно через 2 ч). Раствор вору дают нагреваться до комнатной температуры (медленно и при перемешивании) в течение нескольких часов перед подкислением до рН 1-2 (при охлаждении) водным раствором соляной кислоты. Прибавляют хлороформ,разде- ля4от фазы, водньй слой повторно экстрагируют хлороформом. Объединенные органические экстракты промывают 1 н. Q соляной кислотой (три раза), насьпцен- ным водным раствором хлористого натрия и сушат над сульфатом магния. После выпаривания растворителя при
зуется
выпаривают, остаток вновь растворяют в водной соляной кислоте и лиофилизи- руют. Получают с количественным лы- ходом Ы-(Н -бензил-оксикарбонил-/ - -6п11зил-Ь-аспаргшО-К-1. 1-диамино- зтан в виде солянокислой соли,которую используют без дальнейшей очистки.
Г, Растворяют 2,95 г (5,1 ммоль) продукта раздела В в 50 мл тетрагидрофурана, добавляют 1,5 г (10,6 ммоль) циклопентанкар онилхлорида, затем 2,5 г (25 ммоль; биклрбоната калия и 50 мл воды. Смесь перемешивают при
пониженном давлении маслянистый оста- , комнатной температуре, Через 2,5 ч поток тщательно растирают с эфиром.Отфильтровывают полученный твердый продукт и сушат в вакууме. Получают 67 г Ы бензш1оксикарбонил /3-бензил- -Ь-аспаргил-В-аланина с т.пл. 158- , который является гомогенным по данным тех.
Б. Растворяют продукт раздела А (64 г, 0,150 моль) в 660 мл диметилформамида, охлаждают до -15°С и обрабатывают 16,5 мл (0,150 моль) N-ме- тилморфолина и 19,5 мл (0,150 моль) иэобутилхлорформата. После активации в течение 5 мин при -15 С добавляют
лучают прозр.1чный расгпор,. Но данные тех показыв.гют, чго реакция еще не завершена, следовательно, добавляют вторую порцию 1,5 г циклопентанкар- бони,ггхлорида (10,6 ммоль), 2 г (20 ммоль) бикарбоната калия. Процесс повторяют через 15 мин. Через 20 мин добавляют этилацетат и воду, Фазы разделяют, водную фазу повторно экстрагируют этилацетатом. Объединенные органические фазы промывают два раза 1 М бикарбонатом натрия,три раза - 2 н. соляной кислотой, зате снова два раза 1 М бикарбонатом нат1Д94862
34 г (0,225 моль) аммонийной соли 1-оксибеняотриазола в виде твердого продукта, смесь перемегт{вают 15 мин при -15 с. После медленного нагрева в течение 4 ч до комнатной температуры добавляют хлороформ и поду, разделяют фазы, водную фазу повторно экстрагируют хлороформом. Объединенные органические экстракты три раза промывают 1 н. соляной кислотой, три раза - насыщенным водным раствором бикарбоната натрия, насыщенным раствором хлористого натрия и сушат над сульфатом магния. Растворитель выпаривают при пониженном давлении, твердый остаток перекристаллизовывают из этилацетата (гексана). Получают 50 г Ы -бенэилоксикарбонил-р-бен- зил-Ь-аспаргил-Г)-алпниламида с т.пл. 170-171 С, .которьй является гомогенным по данным тех.
В. Растворяют 2,2 г (5,1 ммоль) продукта раздела Б в 50 мл ацетонит- рила. Раствор разбавляют равным объемом воды. Затем добавляют 2,4 г (5,6 ммоль) иодбензол-бис-(трифтор- ацетата). Реакционную смесь перемешивают при комнатной температуре в
течение 4 ч (прозрачный раствор обра- зуется примерно через 2 ч). Раствор
течение 4 ч (прозрачный раствор обра- зуется примерно через 2 ч). Раствор
зуется
выпаривают, остаток вновь растворяют в водной соляной кислоте и лиофилизи- руют. Получают с количественным лы- ходом Ы-(Н -бензил-оксикарбонил-/ - -6п11зил-Ь-аспаргшО-К-1. 1-диамино- зтан в виде солянокислой соли,которую используют без дальнейшей очистки.
Г, Растворяют 2,95 г (5,1 ммоль) продукта раздела В в 50 мл тетрагидрофурана, добавляют 1,5 г (10,6 ммоль) циклопентанкар онилхлорида, затем 2,5 г (25 ммоль; биклрбоната калия и 50 мл воды. Смесь перемешивают при
лучают прозр.1чный расгпор,. Но данные тех показыв.гют, чго реакция еще не завершена, следовательно, добавляют вторую порцию 1,5 г циклопентанкар- бони,ггхлорида (10,6 ммоль), 2 г (20 ммоль) бикарбоната калия. Процесс повторяют через 15 мин. Через 20 мин добавляют этилацетат и воду, Фазы разделяют, водную фазу повторно экстрагируют этилацетатом. Объединенные органические фазы промывают два раза 1 М бикарбонатом натрия,три раза - 2 н. соляной кислотой, зате снова два раза 1 М бикарбонатом нать1
рия и, наконец, иасьпцеиным раствором хлористого натрия. Сушат над сульфатом магния. Раствор фильтруют, выпаривают при пониженном давлении,остаток тщательно растирают с эфиром. Получают 1,5 г N-(N -бензилоксикар- боншт-Л-бензил-Ь-аспаргил)-N -цикло- пентакарбонил К-1,1-диаминометана в виде твердого кристаллического вещества, которое является гомогенным по данным тех. ЯМР-спектр продукта соответствует означенной структуре.
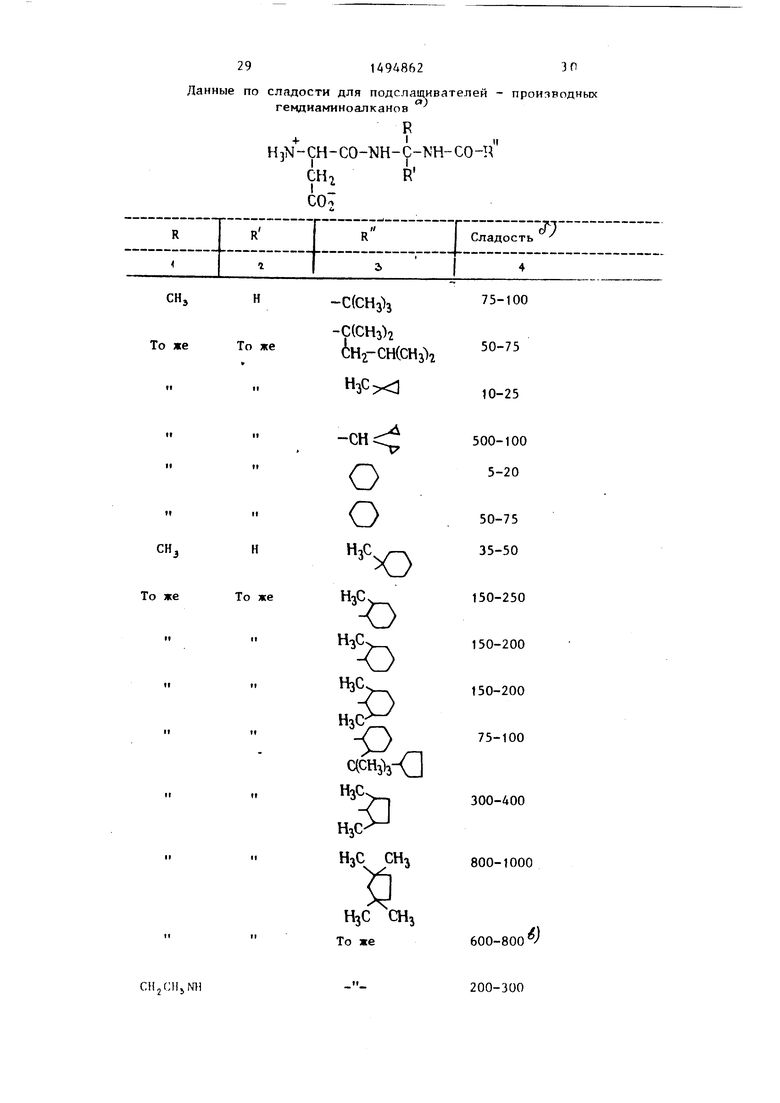
Д. Гидрируют 1,5 г продукта раздела Г (0,03 ммоль) в 50 мл ледяной уксусной кислоты на 10%-ном палладии на угле (приблизительно 0,2 г) при давлении 40 фунт/дюйм (2,812 кг/см в течение ночи. Отфильтровывают катализатор, промывают ледяной уксусной кислотой и лиофилизируют фильтрат.Полученный в результате порошок снова растворяют в воде и повторно дважды лиофилизируют. Получают с количественным выходом Ы-(Ь-аспаргил-)-К - -циклопентакарбонил-t,1-диаминоэтан, с т.пл. 220 С (раэлож). Сладость 75-100 X сахароза.
П р и м е р 2. N-(L-acпapгил)- -Ы -триметилацетил-К-1,1-диаминоэтан (формула I, R-CH,,R -H, (СНз)з).
А. Получают 5,2 г (12 ммоль) N-(N -бензилоксикарбонил- Р -бензил- -Ь-аспаргил)-К-1,1-диаминоэтана солянокислой соли, как описано в примере 1 (раздел в), суспендируют его в 50 мл воды при комнатной температуре. Добавляют 6 г (60 ммоль) бикарбоната калия, затем 1,5 мл (12ммоль) пивалоилхлорида, растворенного в 50 мл ацетонитрила.Гомогенную реакционную смесь перемешивают в течение 3 ч при комнатной температуре. По данным тех реакция на этом не завершена, поэтому добавляют 0,6 мл соляной кислоты и 5 г бикарбоната калия. Реакционную смесь перемешивают еще час. Затем раствор разбавляют 500 мл этилацетата и три раза экстрагируют 1 н. соляной кислотой, три раза - насьш1енным водным раствором бикарбоната натрия и один раз - на- сьш5енным раствором хлористого натрия Органическую фазу сушат над сульфатом магния, фильтруют и выпаривают при пониженном давлении. Остаток кристаллизуют из этилацетата/гексана Получают 4,8 г N-(N -бензилоксикарбонил- | -бензил-Ь-аспаргил)-М три4862
мети;и1 етил- - I , 1-диаминоэтана,который является гомогенньтм по данным тех, т.пл. 66-69 с. ЯМР-спектр продукта соответствует означенной структуре.
Б. Растворяют 4 г продукта раздела Л R 150 мл ледяной уксусной кислоты и гидрируют в течение ночи при
0 давлении 40 фунт/дюйм (2,812 кг/см ) ,5 г 10%-ного палладия на угле. Отфильтровывают катализатор, промывают ледяной уксусной кислотой, фильтрат лиофилизируют. Полученный поро5 шок снова растворяют в воде и повторно дважды лиофилизируют. Получают с количественным выходом М-(Ь-аспаргил)- -N -триметилацетил-R-l , 1 -диаминоэтан , т.пл.150 С. Сладость 75-100 х саха0 роза.
1
П р и м е р 3. М-(Ь-аспаргил)-Ы - -(2-норборнанкарбонил)-К-1,1-диаминоэтан (формула I, R-CH-,R -H, -норборнил).
А. Суспендируют 10,7 г (25 ммоль) N -бензилоксикарбонил-/3-бензил-L- -аспа-ргил-В-аланиламида (пример 1, раздел в) в 200 мл. смеси (1:1) ацето0 нитрил:вода и добавляют 12 г(28ммоль) иодбензол-бис(трифторацетата).Реакционную смесь перемешивают при комнатной температуре в течение 4 ч (гомогенный раствор получается через 2 ч), а затем обрабатывают 10 г (63 ммоль) норборнан-2-карбоксилхло- рида и 12 г (120 ммоль) бикарбоната калия. После двухчасового перемешивания при Комнатной температуре
п тех показывает завершенность реакции, продукт экстрагируют и обрабатывают по обычной методике.После сушки над сульфатом магния органическую фазу выпаривают досуха при понис женном давлении, остаток кристаллизуют из этилацетата/гексана.Получают 10,3 г Ы-(Н -бензилоксикарбонил-/3- -бензил-Ь-аспаргил)-N - ( 2-норборнан- Kap6oHmi)-R-1,1-диаминоэтана; т.пл.
Q 127-130°С, который является гомогенным по данным тех. ЯМР-спектр подтверждает означенную структуру.
Б, Гидрируют 9 г продукта из раздела А обычным способом в 200 мл ледяной уксусной кислоты на 10%-ном палладии иа угле. После нескольких лиофилизаций из воды получают с количественным выходом N-(L-acnaprHJi)- -N -(2-нopбopнaнкapбoнил)-R-1,1-
5
5
-диаминоэтан, т.пл. 1 77-1 78 с.Сладость 75-100 X сахароза.
П р и м е р 4, Ы-(Ь-аспаргил)- -N -( 1-адамантанкарбонил)-К-1 , 1-диаминоэтан (формула I, R-CH.,, R -H, R -l-адамантил) .
А. Обрабатывают 8,54 г (20 ммоль) N -бензилоксикарбонилбензил- -ас- паргил-П-аланиламида (пример 1,раз- дел Б) иодбензол-бис(трифторацетатом) используя методику, описанную в примере 3 (раздел А), Полученный в результате раствор обрабатывают 6 г (30 ммоль) 1-адамантанкарбонилхлори- да и 15 г (150 ммоль) бикарбоната калия, перемешивают 4 ч при комнатной температуре. После обычной обработки получают сырой продукт в виде масла, которое очищают хроматографией на си- ликагеле, элюируя смесью (3:1, объем: : объем) хлороформа/гексана. Получают 2,5 г N-(N -бензилоксикарбонил-р-бен зил-Ь-аспаргиЛ)(1-адамантанкар- бонил)-R-1,1-диаминоэтана в виде мае- ла, который является гомогенным по данным тех. ЯМР-спектр продукта соответствует указанной структуре,
Б. Гидрируют 2,0 г продукта раздела А обычным способом в 50 мл ледяной уксусной кислоты на 10%-ном палладии на угле. После нескольких Лиофилиза- ций из воды получают с количественным выходом К-(Ь--аспаргил)-Ы -(1-ада- мантанкарбонил)-R-1,1-диаминоэтан; т.пл.174-175 с. Сладость 5-15 х X сахароза.
П р и м е р 5. Н-(Ь-аспаргил)-к - -(1-мeтилциклoпpoпaнкapбoнил)-R -1,1- диаминоэтан (формула I, R-CH,,R -Н, Я -1-метш1ЦИклопропш1).
А. Обрабатывают 10,7 г (25 ммоль) N -бензилоксикарбонил-/ -бензил-L- -аспаргил-р-аланиламида (пример 1, раздел Б) иодбензол-бис-(трифторацета том) по методике, описанной в примере 3 (раздел А). Полученный в результате раствор обрабатывают 20 г (200 ммоль) бикарбоната калия, затем 3,65 г (35 ммоль) 1-метилциклопропан- карбонилхлорида и еще двумя аликво- тами по 2 г каждая через 3 ч. После завершения реакции по данным ТСХ реакционную смесь обрабатывают обычным способом. Продукт кристаллизуют из этилацетата/гексана.Получают 8 г N- (М° -бензнлоксикарбоннп- /Ь -бензил- -Ь-аспаргип)-м -(1-метилциклопропан- , карбонил)-К-1,1-диаминозтана, кото
5 0 5
0 -
0
с
0
5
рый является гомогенным по данным тех; т.пл. 120-123 0, ЯМР-спектр продукта соответствует означенной структуре.
Б. Гидрируют 7 г продукта раздела А обычным способом в 200 мл ледяной уксусной кислоты на 10%-ном палладии на угле. После нескольких лио- филизаций из воды получают с количественным выходом Ы-(Ь-аспаргил)-м -(1- -мeтилциклoпpoпaнкapбoнил)-R-1,1- -диаминоэтан , т.пл. 134-135 С, Сладость 10-25 X сахароза.
П р и м е р 6, N-(L-acпapгил)-N - -(тpимeтилциклoгeкcaнкapбoнил)-R-1,1- -диаминоэтан (формула 1, R-CH,, R -H, R -триметилциклогексил).
A.Охлаждают раствор 35 г (0,277 моль) 2,6-диметилциклогексана в 200 мл эфира до -78 С и обрабатывают двукратным избытком раствора метил- магнийбромида в эфире (2,В М, 198 мл). ТГосле 3-часового перемешивания при -78 С реакционную смесь нагревают до О С и осторожно закаливают водой и рассолом. Отделяют органический слой, сушат над сульфатом магния и выпаривают эфир при пониженном давлении, получают 32,2 г 1.2,6-триметилгекса- нола.
Б. Добавляют по каплям раствор 32,2 г (0,226 моль) 1,2,6-триметил- пиклогексанола, 46 г (1 моль) 98%-ной муравьиной кислоты и охлажденной на льду смеси 3 мл 98%-ной муравьиной кислоты и 270 мл (4,86 моль) 90-ной серной кис;1ты (раствор при этом интенсивно вспенивается). После перемешивания еще в течение чася реакционную смесь выливают на 2 кг измельченного льда и обрабатывают, как описано в примере 10 (разде.ч А).Выход триметилциклогекслнкарбоновой кислоты составляет 29,9 г.
B,Осторожно прибавляют 29,9 (0,176 ммоль) продукта раздела Б
к избытку (65 мл) тионилхлорида.смесь перемешивают всю ночь при комнатной температуре. Отгоняют тионилхлорид при пониженном давлении. Получают 25,5 г триметилциклогексанкарбонил- хлорида, который используют без дальнейшей очистки.
Г, Обрабатывают 10,7 г (25 ммоль) N -бензилоксикарбонил- /i -бензшт-L- -аспаргил-D-аланиламида (пример 1, раздел Б, иодбензол-бис-(трифтораце- татом) ао методике, описанной в примере 3 (раздел А) . Получеиньп в результате раствор обрабатывают 20 г (220 ммоль) бикарбоната калия н 6,15 г (30 ммоль) триметилциклогек- санкарбонилхлорнда, а через 30 мин - второй порцией (3 г). Через 3 ч (по данным тех реакция завершена) реакционную смесь обрабатывают обычным способом. Продукт кристаллизуют из этилацетата/гексана. Получают 8,6 г N-(N -бензилоксикарбонил-/5-бензил- -Ь-аспаргил)-М -(триметилциклогек- санкарбонил)-К-1,1-диаминоэтана,который является гомогенным по AaHHbr t тех. ЯТ-ГР-спектр продукта соответствует означенной структуре.
Д. Гидрируют 8 г продукта раздела Г (обычным способом) в 200 мл ледяной уксусной кислоты на 10%-ном палладии на угле. После нескольких лиофилизаций из воды получают с количественным выходом Ы-(Ь-аспаргил)- -N -триметилциклогексанкарбонил-R- -1,1-диаминоэтан, Сладость 25-50 х X сахароза.
Пример. N-(L-acпapгил)-N - -(1,1-дициклопропилацетил)-К-1,1- -диаминоэтан (формула 1, R-CH,R -Н R -дициклопропилметил) .
А. Суспендируют 116 г (0,325 моль) метилтрифенилфосфонийбромида в 600 мл сухого эфира, охлажденного до -10°С, и обрабатывают раствором н-бутилли- тия в гексане (2,2 М, 175 мл).Смесь перемешивают 5 мин перед добавлением раствора 35,6 г (0,325 моль) ди- циклопропилкетона в 100 мл эфира, предварительно охлажденного до О С. Суспензии дают нагреться до комнатно температуры и перемешивают затем еще 2 ч. Добавляют 1000 мл воды (сначла маленькими порциями) и перемешивают смесь до растворения осадка. Отделяют органический слой, промывают водой, сушат над сульфатом магния выпаривают растворитель при пониженном давлении. Остаток, содержащий твердый трифенилфосфиноксид, который отд еляют от масла промычают малым количеством эфира. Объединенные эфирные (органические остатки) фракционируют. Получают 6,5 г дициклопропил- этилена; т.кип. 130°С/760 мм, который является чистым по данным газово хроматографии (ГХ).
Б. Растворяют 19 г (0,176 моль) дициклопропилэтилена в 100 мл сухого тетрагилрофурана в трехгорлой колбе
0
5
0
5
0
5
0
5
0
5
в атмпс(Ьере азота и обрабатывают бо- ран-тетрагидрофураном в тетрагидро- фуране (1М, 210 мл). Смесь перемешивают А ч при комнатной температуре перед добавлением (с осторожностью) 60 мл 3 н. гидроокиси натрия (возможно вспенивание).Когда добавление окончено, по каплям добавляют 60 мл водной 30%-ной перекиси водорода со скоростью, достаточной для поддержания кипения. После завершения добавления смесь кипятят с обратным холодильником еще 30 мин, охлаждают, водный слой насыщают хлористым натрием. Разделяют слои. Органический слой сушат над сульфатом магния, выпаривают при пониженном давлении. Получают с количественным выходом 2,2-ди- циклопропилэтанол, который является чистым по данным ГХ (продукт также может быть перегнан)i т.кип. 99°С/ /25 мм.
В. Растворяют 16 г (0,127 моль) продукта раздела Б в 300 мл эфира, добавляют раствор к смеси 60 г бихромата калия, растворенных в 120 мл концентрированной серной кислоты, и 600 мл ледяной воды. Реакционную смесь, которая немедленно становится темной, перемешивают в течение часа при комнатной температуре. Затем отделяют органический слой, три раза промывают водой, сушат над сульфатом магния и отгоняют эфир при пониженном давлении. Остаток перегоняют. Получают 10,3 г 1,1-ди- циклопропилуксусной кислоты, т.кип. 130-141°С/25 мм, которая является чистой по данным ГХ.,
Г. Растворяют 10 г .(0,071 моль) продукта раздела В в 25 мл сухого тетрагидрофурана и обрабатывают избытком (25 мл) тионилхлорида. После перемешивания смеси при кoмнatнoй температуре в течение часа, превращение в хлорангидрид по данным ГХ, завершено. Отгоняют растворитель и избыток тионилхлорида при пониженном давлении. Получают с количественным выходом 1,1-дициклопропилацетилхло- рид, который используют без дополнительной очистки.
Д. Обрабатывают 8,54 г (20 ммоль) N -бензилоксикарбонил-| -бензил- -Ь-аспаргил-В-аланиламида (пример 1, раздел Б) иодбензол-бис-(трифтораце- татом) по методике, описанной в примере 3 (раздел А). Полученный в ре
111Д9
зультате раствор обрабатывают 16 г (160 ммоль) бикарбоната калия, затем добавляют по каплям 4,7 г (30 ммоль) 1,1-дициклопропилацетилхлорида.Почти сразу же образуется осадок. Реакционную смесь перемешивают при комнатной температуре в течение часа,затем добавляют воду и хлороформ. Разделяют фазы и промьгаают органический слой (три раза) насьпценным водным раствором бикарбоната натрия. После сушки над сульфатом магния отгоняют растворитель при пониженном давлении. Твердый остаток перекристаллизовывают из этилацетата. Получают 6,5 г l4-(N - -бензилоксикарбонил -бензил-L-ac- паргил)-N -(1,1-дициклопропилацетил)- -R-1,1-диаминоэтана, который является гомогенным по данным ТСХ, т.пл. ZOO-ZOI C, ЯМР-спектр продукта соответствует означенной структуре.
Е. Гидрируют 4 г продукта раздела Д обычным способом в 200 мл ледяной уксусной кислоты на палладии на угле.После нескольких лиофилиза- ций из воды кристаллизуют остаток из этанола/воды.Получают 1,0 г N-(L-acnap гил-)-Ы -(1,1-дициклопропипацетил)- -R-1,1-диаминоэтана ; т.пп. 209-210 С Сладость 500-700 х сахароза.
Примере. N-(L-аспаргил;- -N -(2,5-диметилциклопентакарбонил)- -К-1,1-диамяноэтан (формула I, R-CHj, R -H, R -димeтилциклoпeнтшl) .
,А, Растворяют 16 г (0,7 моль) металлического натрия в 500 мл абсолют кого этанола в атмосфере аргона при охлалщении, необходимом дпя поддержания температуры ниже 70 С. Раствор охлаждают и добавляют по каплям; 54,3. г (0,362 моль) перегнанного ди- этилмалоната (при охлаждении, если необходимо),затем одну порцию 85 г (0,348 моль) 2,5-дибромгексана,Реакционную смесь перемешивают всю ночь при комнатной температуре, затем кипятят с обратным холодильником в течение 2 ч. Смесь концентрируют примерно до половины объема при пониженном давлении, добавляют 500 мл воды и экстрагируют три раза по 200 мл эфира. Объединенные экстракты сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Остаток фракционируют в вакууме.Получают 35 г диэтилового эфира 2,5-ди- метилциклопентан-1,1-дшсарбоновой
12
0
5
0
5
0
5
0
кислоты, который является гомогенным по ланным ТСХ.
Б. 35 г (0,145 моль) продукта раздела А добавляют к раствору 55 г гидроокиси калия в 300 мл абсолютного этанола. Смесь кипятят с обратным холодильником всю ночь. Ракционную смесь выпаривают при пониженном давлении, остаток растворяют в 500 мл воды. Водный раствор экстрагируют 200 мл зтилацетата, подкисляют до рН1 (конц.НС) и три раза экстрагируют эфиром порциями по 200 мл. Объединенные экстракты промьгеают 1 н. соляной кислотой и сушат над сульфатом натрия. Раствор выпаривают при пониженном давлении, оставшееся место тщательно растирают с пентаном, чтобы вызвать кристаллизацию. Отфильтровывают продукт и сушат в вакууме. Получают 10,5 г 2,5-диметилциклопен- тан-1,1-дикарбоновой кислоты, которая является гомогенной по данным ГХ.
В. Нагревают 10,5 г (56 ммоль) продукта раздела Б до 230 С в токе аргона в течение 1,5 ч. Остаток растворяют в тетрагидрофуране, обесцвечивают (Норит А) и отгоняют растворитель при пониженном давлении. Оставшееся масло кристаллизуется при стоянии. Получают 6,3 г 2,5-диметил- циклопентакарбоновой кислоты (т.пл. 45°С), которая является чистой по данным ГХ.
Г. Растворяют 6,3 г (48 ммоль).. продукта.раздела В в 100 мл смеси (1:1, объем/объем) тетрагидрофурана и тионилхлорида. Смесь перемешивают час при комнатной температуре. Раствор упаривают при пониженном давле- НИИ. Получают с количественным выходом 2,5 -диметилциклопентанкарбонил- хлорид, которьп используют без дополнительной очистки.
Д. Обрабатывают 8,6 г (20 ммоль) N -бензилоксикарбонил-/5 -бензил-L- -аспаргш1-В-аланиламида (пример 1, раздел Б) иодбензол-бис-(трифтораце- татом), используя методику, описанную в примере 3 (раздел А). Полуденный в результате раствор обрабатьша- ют 20 г (200 ммоль) бикарбоната калия, затем 4,8 г (30 ммоль) 2,5-ди- метилциклопентанкарбоксилхлорида,добавляемого по каплям в течение 5 мин. Продукт почти сразу же осаждается. Перемешивание продолжают еще 2 ч при комнатной температуре. Реакцион
3
иую смесь обрабатывают обычиьси способом. Продукт кристаллизуется во время сушки конечных экстрактов над сульфатом натрия, no3Tot-fy раствор нагревают до кипения, фильтруют горячим и промывают сульфатом натрия в этилацетате. Фильтрат выпаривают при пониженном давлении и кристаллизуют остаток из этилацетата/гексана Получают 6,1 г N-(N -бензилоксикарб нил- fi -бензил-Ь-аспаргил)-Н -2,5-(д метилциклопентанкарбонил)-1,1-ди- аминоэтана, который является гомогенным по данным тех, т.пл. 193-195 ЯМР-спектр соответствует означенной структуре продукта.
Е. Гидрируют 5,5 г продукта раздла Д обычным способом в 200 мл ледяной уксусной кислоты на 10%-ном палладии на угле. После нескольких лио филизаций из воды твердый остаток п рекристаллизовыв-ают из этанола/воды Получают 2,6 г N-(L-acпapгил)-N- -(2,5-диметилциклопентанкарбонил)- -R-1,1-диаминоэтана; т.пл.208-209 С Сладость 300-400 х сахароза.
П р и м е р 9. N-(L-acnapr -ui)- -N -(2,2,5,6-тетраметилциклопентан- карбонил)-R-1,1-диаминоэтан (формула I, R-CH , Я -Н, ,2,5,5-тетраJч
метилциклопентил).
А. Добавляют 144 г (3,0 моль) 50%-ной дисперсии в масле гидрида натрия в трехгорлую колбу емкостью 3 л, снабженную обратным холодильником, механической мешалкой.и вводом азота. Через колбу пропускают умереный ток азота. Загружают 1,5 л сухо тетрагидрофурана. Одновременно малыми порциями (20-40 мл) к перемешиваемой суспензии добавляют растворы 53,6 г (0,64 моль) циклопентанона в 350 мл сухого тетрагидрофурана и 285 МП (3,0 моль) диметилсульфата в 120 мл того же растворителя так, чтбы поддерживалось легкое выделение водорода. Реакционную смесь охлаждают, поддерживая температуру ниже 40 С. По окончании добавления (несколько часов) реакционную смесь кипятят с обратным холодильником в течение 2 ч. После охлаждения медленн добавляют 100 мл третичного бутано для разрушения избытка гидрида, зат 1 л воды (вначале осторожно). Затем реакционную смесь 2 ч кипятят с обрным холодильником для разргушения избытка диметилсульфата. Охлаждают,
0
5
4862
0
5
0
14
разделяют слои, промывают органичес- кий слой насьпценным раствором хлористого натрия, сушат над сульфатом натрия. Выпаривают растворитель при пониженном давлении, остаток фракционируют в вакууме. Получают 59 г 2,2,5,5- -тетраметилциклопентанона, т.кип. 55°С/20 мм.
Б. Обрабатывают раствор 30 г (0,215 моль) 2,2,5,5-тетраметилцикло- пентанона в 50 мл эфира в атмосфере азота 100 мл 3 М раствора метилмагний- бромида. Реакционную смесь перемеши-- вают Ьсю ночь при комнатной температуре, затем добавляют по каплям 65 мл насыщенного раствора хлористого аммония в воде. Смесь перемешивают 10 мин, декантируют эфирный раствор, твердый остаток тщательно растирают с эфиром. Эфирные экстракты сушат над сульфатом натрия и выпаривают при пониженном давлении. Получают 30 г сырого 1,2,2,5,5-пентаметил- циклопентанола, который используют без дальнейшей очистки.
В. Растворяют 30 г сырого продукта раздела Б в 150 мл пиридина.Раствор охлаждают до 0°С и обрабатывают, добавляя по каплям 20 мл тионилхло- рида (0,28 моль). Температуру поддерживают ниже 5 С. Реакционную смесь перемешивают всю ночь, фильтруют, добавляют эфир и воду. Разделяют фазы. Органическую фазу промывают два раза (по 200 мл) водой и сушат над сульфатом натрия. Выпаривают растворитель при пониженном давлении.Получают 10,8 г 1-метилен-2,2,5,5-тетра- метилциклопентана, который является чистым по данным ГХ.
Г. Растворяют 10,8 г (78 ммоль) продукта раздела В в 100 мл сухого тетрагидрофурана и обрабатывают в атмосфере азота 1 М боран-тетрагидро- фурана в 100 мл тетрагидрофурана. Реакционную смесь перемешивают всю ночь при комнатной температуре и обрабатывают 40 мл 3 н. водного раствора гидроокиси натрия, затем добавляют по каплям 40 мл 30%-ной перекиси водорода со скоростью, достаточной для поддержания слабого кипения. Смесь кипятят с обратным холодильником еще в течение часа, добавляют хлористый натрий до насыщения.Смесь охлаждают до комнатной температуры при перемешивании. Разделяют фазы, органическую фазу сушат над суль0
0
5
151
фатом натрия и выпаривают при пониженном давлении. Получают с количественным выходом сырой 2,2,5,5-тетра- метилциклопентилметанол, который используют без дополнительной очистки,
Д. Растворяют продукт раздела Г в 300 мл эфира и добавляют к раствору 45 г (0,15 ммоль) бихромата калия в 90 мл (1,7 моль) концентрированной серной кислоты и 450 мл воды.Смесь перемешивают при комнатной температуре в течение 3 ч. Затем разделяют фазы, органический слой промывают насыщенным раствором хлористого натрия и сушат над сульфатом натрия. Растворитель упаривают при пониженном давлении, остаток разгоняют в вакууме. Получают 6,6 г 2,2,5,5-тет- раметилциклопентанкарбоновой кисло
ты, которая является гомогенной по данным ГХ.
Е, Растворяют 6,5 г (38 ммоль) продукта раздела Д в 100 мл тетрагид- рофурана и обрабатывают, добавляя по каплям 21) мл тионилхлорида (270 ммоль) . Раствор кипятят 2 ч с обратньм холодильником, выпаривают при пониженном давлении, остаток разгоняют в вакууме. Получают 4,8 г 2,2,5,5- -тетраметилциклопентанкарбонштхлори- да, т.кип. 65-75 С/4 мм.
Ж. Обрабатывают 10,7 г (25 ммоль) Ы -бензилоксикарбонил-р)-бензил-1-ас- паргил-В-аланиламида (пример 1,раздел Б) иодбензол-бис-(трифторацетатом) по методике примера 3 (раздел А), Полученный в результате раствор выпаривают почти досуха при пониженном давле
нии, добавляют воду и большой избы
ток концентрированной соляной кислоты. Смесь снова упаривают досуха. Растворяют твердый остаток в 20 мл 4,4 М НС1/(Иоксан. Раствор упаривают досуха, остаток снова растворяют в диоксане (100 мл)и лиофилизируют.Получают 10,2 г N-N -бензилоксикарбо- нил- -бензил-(Ь-аспаргил)-Е-1,1- , -диаминоэтана в виде солянокислой со- Ли, которая является гомогенной по данным тех.
3. Растворяют 6,6 г (15 ммоль) продукта раздела Ж в 150 мл-сухого тет- рагидрофурана и обрабатывают 3,1 г (15 ммоль) 2,2,5,5-тетраметилцикло- пентанкарбонилхлорида (раздела Е), затем 4,2 мл (30 ммоль) триэтиламина. Реакционную смесь перемешивают в течение 1 часа при комнатной темпера
16
0
5
5
туре, добавляют этилацетат. Продукт обрабатывают обычным способом. При кристаллизации из этилацетата/гексана получают 6,0 г N-N -бензилоксикарбо- нил- |3-бензил-(Ь-аспаргнп)-Ы -(2, 2- 5,5-тетраметилциклопентанкарбонил)- -R-1,1-диаминоэтана, которьй является гомогенным по данным ТСХ; т.пл.122- 125°С, ЯМР-спектр соответствует означенной структуре.
И. Гидрируют 5,5 г продукта раздела 3 обычным способом в 200 мл ледяной уксусной кислоты на 10%-ном палладии на угле. После лиофилизации (несколько раз) из воды твердый остаток кристаллизуют из этанол/гексана. Получают 2,0 г Ы-(Ь-аспаргил)-М - (2,2,5,5-тетраметилциклопентанкарбо0 нил)-К-1,1-диаминоэтана, т.ш1.171- 172 с. Соединение является гомогенным по данным высокоэффективной жидкостной хроматографии под давлением (HPLc). Условия: Лихросорб RP-18,
5 линейный градиент 24-33% ацетонитри- ла в 0,01 М триэтиламмонийфосфата, рН 4,5, скорость потока 1 мл/мин, время удерживания 12,31 мин. Сладость 800-1000 х сахароза.
Пример 10. М-(ь-аспаргил)- -N -(2,6-димeтиJ:циклoгeкcaнкapбoнил)- -R-1,1-диаминоэтан, (формула I, R-СНз, R -H, ,6-диметилциклогек- сил).
А. Суспендируют 286 г (0,80 моль) метйлтрифенилфосфонийбромида в 1500 мл эфира и обрабатьшают 500 мл (0,80 моль) 1,6 М н-бутиллития в эфире, затем 50,4 г (0,40 моль) 2,6-диметилциклагексанона по методике,описанной в при- мере 7 (раздел Л). Разгоняют сырой продукт, Получают 24 г 1-метнпсн- -2,6-диметилциклогексана, т.кип.146- 154 0/760 мм.
Б. Растворяют 24 г (0,10 моль) продукта раздела А в 50 мл сухого тстра- гидрофурана и обрабатывают в атмосфере азота 250 м.п 1 М боран-тетрагидро- фурана в тетрагидрофуране. Реакционную смесь перемешивают всю ночь при комнатной температуре, обрабатывают 20 МП 3 н,водного раствора гидроокиси натрия, добавляя ее по каплям (возможно вспенивание), затем добавляют по каплям 20 мл 30«-ной водной перекиси водорода. Смесь кипятят с обратным холодильником в течение 30 мин, добавляют хлористый натрий до насыщения и охлаждают до комнатной тёмпе0
0
171
ратуры при перемешивании. Разделяют фазы, сушат органический слой сульфатом натрия и выпаривают при пониженном давлении. Получают с количествен- ным выходом 2,6-диметилциклогексилме- танол. Продукт очищают фракционированием в вакууме, т.кип. 187-210 С/ /760 мм.
-R-1,1-диаминоэтяна, которьп является гомогенным, по данным ТСХ,т.пл.1А6- 50°С. ЯМР-спектр продукта соответствует означенной структуре,
Е. Гидрируют 4 г продукта раздела Л по обычной методике в 150 мл ледяной уксусной кислоты на 10%-ном палладии на угле. После нескольких лио














| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения продукта присоединения дипептидного производного и аминокислоты | 1978 |
|
SU910117A3 |
| СПОСОБ ПОЛУЧЕНИЯ N-КАРБОКСИАНГИДРИДОВ ИЛИ N-ТИОКАРБОКСИАНГИДРИДОВ УРЕТАНЗАЩИЩЕННЫХ АМИНОКИСЛОТ | 1989 |
|
RU2007396C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2026296C1 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА ИЛИ БЕНЗОТИАЗЕПИНА | 1989 |
|
RU2090562C1 |
| Способ получения N-ацетилмурамилпептидов | 1979 |
|
SU1326197A3 |
| Способ получения производных 6-( -2ациламидо-2-фенил-ацетамидо) пеницилановой кислоты или их солей | 1976 |
|
SU622407A3 |
| Способ получения полипептидов или их солей | 1977 |
|
SU910116A3 |
| СЕЛЕКТИВНЫЕ ДИПЕПТИДНЫЕ ИНГИБИТОРЫ КАЛЛИКРЕИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ И СПОСОБ ЛЕЧЕНИЯ | 2003 |
|
RU2301225C2 |
| Способ получения производных аминокислот, их солей, рацематов или оптически-активных антиподов | 1976 |
|
SU648082A3 |
| Способ получения карбацефалоспориновых соединений | 1986 |
|
SU1575940A3 |
ИЗОБРЕТЕНИЕ КАСАЕТСЯ ЗАМЕЩЕННЫХ АМИНОВ ,В ЧАСТНОСТИ, ПОЛУЧЕНИЯ N-(L - АСПАРГИЛ) - N - (1-АЦИЛАМИНО)-АЛКИКАМИНОВ ОБЩЕЙ Ф-ЛЫ 1: H3N+ - CHX-C(O)-NH-CR1R2 - NH-C(O)R3 ГДЕ X-CH2C-O2
R1 - НИЗШИЕ АЛКИЛ ИЛИ ОКСИАЛКИЛ
R2 - H ИЛИ НИЗШИЙ АЛКИЛ
R3 - ИЗО-C3-10-АЛКИЛ, C5-7-ЦИКЛОАЛКИЛ, НИЗШИЙ АЛКИЛЦИКЛОАЛКИЛ, ДИЦИКЛО (НИЗШИЙ) АЛКИЛ, 2,2,4,4-ТЕТРАМЕТИЛТИЕТАН ИЛИ ФЕНИЛ, КОТОРЫЕ ИМЕЮТ СЛАДКИЙ ВКУС И МОГУТ БЫТЬ ИСПОЛЬЗОВАНЫ В ПИЩЕВОЙ ПРОМЫШЛЕННОСТИ. ЦЕЛЬ - СОЗДАНИЕ НОВЫХ ВЕЩЕСТВ УКАЗАННОГО КЛАССА С УЛУЧШЕННЫМИ СВОЙСТВАМИ. ИХ СИНТЕЗ ВЕДУТ ИЗ СООТВЕТСТВУЮЩИХ АМИНОКИСЛОТ Ф-Л YO-C(O)-CH2-CH(NHX)-C(O)OH И ZO-C(O)-CR1R2-N H2, ГДЕ X - ЗАЩИТНАЯ ГРУППА - БЕНЗИЛОКСИКАРБОНИЛ
Y - ЗАЩИТНАЯ ГРУППА - БЕНЗИЛ
Z - H ИЛИ ЗАЩИТНАЯ ГРУППА, КОТОРУЮ ЗАТЕМ УДАЛЯЮТ. ПОЛУЧЕННОЕ СОЕДИНЕНИЕ ОБРАБАТЫВАЮТ АММИАКОМ С ПОСЛЕДУЮЩЕЙ ПЕРЕГРУППИРОВКОЙ ГОФМАНА АМИДА В СООТВЕТСТВУЮЩУЮ МОНОАЦИЛИРОВАННУЮ ГЕМДИАМИНОАЛКАНОВУЮ СОЛЬ, КОТОРУЮ ОБРАБАТЫВАЮТ СООТВЕТСТВУЮЩИМ ХЛОРАНГИДРИДОМ КИСЛОТЫ, И УДАЛЕНИЕМ ЗАЩИТНЫХ ГРУПП X И Y. У НОВЫХ ВЕЩЕСТВ СТЕПЕНЬ СЛАДОСТИ ЗАВИСИТ В ОСНОВНОМ ОТ ПРИРОДЫ АЦИЛИРУЮЩЕЙ ГРУППЫ R3, ПРЕДПОЧТИТЕЛЬНЫ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ РАЗВЕТВЛЕННЫЕ МАССИВНЫЕ ГИДРОФОБНЫЕ ГРУППЫ, НАПРИМЕР ЦИКЛОАЛКИЛЬНЫЕ ИЛИ ГЕТЕРОЦИКЛОАЛКИЛЬНЫЕ. УРОВЕНЬ СЛАДОСТИ У ЛУЧШИХ ПРЕДСТАВИТЕЛЕЙ 600-800. 1 ТАБЛ.
В. Растворяют 20 г (0,14 моль) npo-4Q филизаций из воды кристаллизуют твер-, дукта раздела Б в 300 мл эфира, дый остаток из этанола/воды и получают 0,8 г N-(L-acпapгил)-N -(2,6- -диметилциклогексанкарбонил)-К-1,1-диамяноэтана. Сладость 150 - 200 х
добавляют к раствору 30 г (0,30 моль) бихромата калия в 175 мл концентрированной серной кислоты и 900 мл воды на ледяной бане. Смесь нагрева- 5 сахароза.
ют до комнатной температуры и пере- Пример 11. Ы-(Ь-аспаргил)- мешивают два дня. Разделяют фазы,ор- -N -(2,2,5,5-тетраметилциклопентанкар- ганическуто фазу промывают водой,сушат бонил)-К-1,1-диамино-2-оксиэтан (формула I, , R -H, R 2,2,5,5- 20 -тетраметилциклопентил).
А. Растворяют 5,0 г (25,6 ммоль) О-бензил-В-серина в 50 мл диметилнад сульфатом магния и упаривают при пониженном давлении. Остаток фракционируют. Получают 16,7 г 2,6- -диметшщиклогексанкарбоновой кислоты, которая является чистой по данным ГХ, т.кип. 145-148°С.
Г. Растворяют 16,7 г пррдукта раз- 25 перемешивают при комнатной темперадела В в 100 мл тетрагидрофурана и обрабатывают избытком (30 мл) тио- нилхлорида при комнатной температуре, После перемешивания при комнатной
туре до тех пор, пока не получат гомогенный раствор (приблизительно 1 ч). Одновременно растворяют 9,14 г (25,6 ммоль) N -бензилоксикарбонилтемпературе в течение часа отгоняют зо / -бензил-Ь-аспаргиновой кислоты
избыток тионилхлорида и растворитель при пониженном давлении. Получают с количественным выходом 2,6-диметил- циклогексанкарбонилхлорид, который используют без дальнейшей очистки.
в смеси (1:1) диметилформамида и тетрагидроф урана, охлажденной до ,обрабатывают 2,81 мл (25,6 ммоль) N-метилморфолина, затем 3,32 мл (25,6 ммоль) изобутилхлорформата.
Д. Обрабатьгаают 10,7 г (25 ммоль) После 10-минутной активации прибавN -бензилоксикарбонил- -Ь-бензил-L- -аспаргил-В-аланиламида (пример 1, раздел Б) иодбензол-бис-(трифтораце- татом) по методике, описанной в примере 3 (раздел А). Полученный в результате раствор обрабатьшают 20 г (200 ммоль) бикарбоната калия, зате:м добавляют по каплям 6,1 г (35 ммоль) 2,6-диметш1циклогексанкарбонилхлори40
ляют предварительно рхлажденный раствор силилового эфира О-бензил-В-сери- на, затем (по каплям) - 2,81 мл (25,6 ммоль) N-метилморфолина, обеспечивая поддержание температуры реакции -15 С. Раствору дают возможность нагреться до кoм aтнoй температуры (медленно и при перемешивании)
,- „,. в течение 4 ч перед подкислением до
да в течение 2 мин. Реакционную смесь рН 1-2 (с охлаждением), используя перемешивают 3 ч при комнатной темпе- водную соляную кислоту. Добавляют ратуре, обрабатывают обычным спосо- хлороформ, разделяют фазы и повторно
бом. Отличие состоит в том, что продукт кристаллизуется из конечных экстрактов над сульфатом натрия, поэтому раствор нагревают до кипения, фильтруют горячим и промывают сульфатом натрия в зтилацетате. Фильтрат выпаривают при пониженном давлении и кристаллизуют остаток из этилацета- та. Получают 4,5 г N-(N -6eH3HnoKCH- карбонил-/1-бензш1-Ь-аспаргил)-Ы - -(2,6-диметилциклогексанкарбонил)-диамяноэтана. Сладость 150 - 200 х
сахароза.
формамида, обрабатывают 3,053 г
(28,1 ммоль) хлортриметилсилана.Смесь
туре до тех пор, пока не получат гомогенный раствор (приблизительно 1 ч). Одновременно растворяют 9,14 г (25,6 ммоль) N -бензилоксикарбонил / -бензил-Ь-аспаргиновой кислоты
в смеси (1:1) диметилформамида и тетрагидроф урана, охлажденной до ,обрабатывают 2,81 мл (25,6 ммоль) N-метилморфолина, затем 3,32 мл (25,6 ммоль) изобутилхлорформата.
ляют предварительно рхлажденный раствор силилового эфира О-бензил-В-сери- на, затем (по каплям) - 2,81 мл (25,6 ммоль) N-метилморфолина, обеспечивая поддержание температуры реакции -15 С. Раствору дают возмож
экстрагируют водный слой хлороформом. Объединенные Iорганические экстра кты
три раза промьгоают 1 н. соляной кислотой и насыщенным раствором хлористого натрия, сушат над сульфатом магния . Упаривают растворитель при пониженном давлении и кристаллизуют твердый остаток из этилацетата/гек- сана. Получают 11,0 г N -бензилоксикарбонил- А-бензил-Ь-аспаргнл-О-бен- зил-В-серина, т.пл.107-108 С,который.
1 9
является гомогенным по данным ТСХ, ЯМР-спектр продукта соответствует оз наченной структуре.
Б, Растворяют 10,0 г (18,72 ммоль) продукта раздела А в 100 мл дргметил- формамида, охлажденного до -15 С, и обрабатывают 2,05 мл (18,72 ммоль) N-метилморфолина, а затем 2,43 мл (18,72 ммоль) изобутилх1;орформата.Пос- Q ле 4-минутной активации при добавляют 3,13 г (20,5 ммоль) аммонийной соли 1-оксибенэотриазола в ви14948
де твердого продукта и перемешивают смесь 30 мин при -15 с, После нагрева до комнатной температуры при перемешивании в течение 4 ч добавляют хлороформ и воду, разделяют фазы,Водную фазу повторно экстрагируют хлоро30
формом. Объединенные органические фа- 20 обычным способом в 150 мл ледя- зы промывают три раза 1 н. соляной кислотой, насыщенным водным раствором бикарбоната натрия (три раза), насыщенным водным раствором хлористого натрия и сушат над сульфатом магния. Отгоняют растворитель при пониженном давлении, твердый остаток кристаллизуют из этилацетата/гексана.Получают 7,4 г N -бензилоксикарбонил- - |Ь -бензил-Ь-аспаргил-О-бензил-П-се- рнламида (т.пл. 150 С), который является гомогенным по данным ТСХ. ЯМР-спектр продукта соответствует указанной структуре,
В. Растворяют 5,33 г (10 ммоль) продукта раздела Б в 10 мл ацетонит- рила и разбавляют раствор равным объемом воды.Затем добавляют 4,8 г (11,2 ммоль) иодбензол-бис-(трифтор ацетата) и перемешивают реакционную смесь при комнатной температуре в течение 5 ч. Раствор выпаривают при пониженном давлении, снова растворя ют остаток, в 4 н. безводном НС1/ди- океан и лиофилизируют раствор Полу35 „oi
ной уксусной кислоты на 10%-ном палладии на угле. После лиофилизации и релиофилизации из воды несколько раз получают с количественным выхо- 25 дом К-(Ь-аспаргил)-Ы -(2,2,5,5-тет- раметилциклопентанкарбонил)-К-1,1- -диамино-2-оксиэтан (т.пл. 174-176 С, с разл.). Сладость 500-400 х сахароза.
П р и М е р 12. Н-(Ь-аспаргил)- -N -(2,2, 5,5--тетраметилциклопентан- карбонил)-S-1,1-диаминоэтан (формула I, R-H, R -СНз,к -2,2,5,5-тетра метилциклопентил),
А. Растворяют 1,70 г (5 ммоль) Н бензилоксикарбонил- /З-бензил-Ь- -аспаргиновой кислоты в 100 мл тетрагидрофурана, раствор охлаждают до -15°С и обрабатьшают 0,5 мл (5 ммоль) N-метилморфолина. После 10-минутной активации при -15 С добавляют предварительно охлажденный раствор 0,75 г (6 ммоль) солянокислого L-аланинами- да и 50 мл диметилформамида, затем 0,66 мл (6 ммоль) N-метилморфолина. Раствору дают нагреться до комнатной температуры и перемешивают всю ночь., Затем добавляют воду и хлороформ,разделяют фазы, водную фазу повторно экстрагируют хлороформом. Объединенные органические фазЫ промывают три раза 1 н, соляной кислотой, насыщенным раствором хлористого натрия и сушат над сульфатом магния. Раствор филь руют, фильтрат выпаривают при пониженном давлении, остаток кристаллизуют из этилацетата/гексана.Получают 2,0 г N --бeнзшloкcикapбoнил-/ - -бензил-Ь-аспаргил-П-аланиламида
40
45
чают с количественным выходом солянокислый N-(N -бензилоксикарбонил- - Р)- бензил-Ъ-аспаргил)-К-1,1-диами- но-2-оксиэтан, который используют без дополнительной очистки.
Г; Растворяют продукт раздела В в 50 мл тетрагидрофурана, добавляют 3,30 мл (30 ммоль) N-метилморфолина, затем 3,1 г (16 ммоль) 2,2,5,5-тет- раметилциклопентанкарбонштхлорида. Смесь перемешивают 4 ч при комнатной температуре. Прибавляют этилацетат и воду, разделяют фазы. Водную фазу повторно экстрагируют этилацетатом.
Q 4862 20
Объединенные органические фазы два раза промывают 1 М раствором бикарбоната натрия, три раза - 2 н. соляной кислотой, два раза - снова 1 М бикарбонатом натрия, затем насыщенным раствором хлористого натрия и сушат над сульфатом магния. Раствор фильтруют, фильтрат выпаривают при пониженном давлении, остаток кристаллизуют из этилацетата/гексана.Получают 4,0 г Ы-(К -бензилоксикарбонил- - Ь-бензил-(Ь-аспаргил)-Н -2,2,5,5-тетраметилциклопентанкарбонил-1.1- Диамино-2-оксиэтана (т.пл.90-93 С), который является гомогенным по данным ТСХ. ЯМР-спектр продукта соответствует приведенной структуре.
Д.Гидрир гТот 3,8 г продукта разде0
0 обычным способом в 150 мл ледя-
5 „oi
ной уксусной кислоты на 10%-ном палладии на угле. После лиофилизации и релиофилизации из воды несколько раз получают с количественным выхо- 5 дом К-(Ь-аспаргил)-Ы -(2,2,5,5-тет- раметилциклопентанкарбонил)-К-1,1- -диамино-2-оксиэтан (т.пл. 174-176 С, с разл.). Сладость 500-400 х сахароза.
П р и М е р 12. Н-(Ь-аспаргил)- -N -(2,2, 5,5--тетраметилциклопентан- карбонил)-S-1,1-диаминоэтан (формула I, R-H, R -СНз,к -2,2,5,5-тетра метилциклопентил),
А. Растворяют 1,70 г (5 ммоль) Н бензилоксикарбонил- /З-бензил-Ь- -аспаргиновой кислоты в 100 мл тетрагидрофурана, раствор охлаждают до -15°С и обрабатьшают 0,5 мл (5 ммоль) N-метилморфолина. После 10-минутной активации при -15 С добавляют предварительно охлажденный раствор 0,75 г (6 ммоль) солянокислого L-аланинами- да и 50 мл диметилформамида, затем 0,66 мл (6 ммоль) N-метилморфолина. Раствору дают нагреться до комнатной температуры и перемешивают всю ночь., Затем добавляют воду и хлороформ,разделяют фазы, водную фазу повторно экстрагируют хлороформом. Объединенные органические фазЫ промывают три раза 1 н, соляной кислотой, насыщенным раствором хлористого натрия и сушат над сульфатом магния. Раствор филь руют, фильтрат выпаривают при пониженном давлении, остаток кристаллизуют из этилацетата/гексана.Получают 2,0 г N --бeнзшloкcикapбoнил-/ - -бензил-Ь-аспаргил-П-аланиламида
0
5
0
5
10
15
211494862
(т.пл.180-180,), который является- гомогенным по данным ТСХ. ЯМР-спектр продукта соответствует указанной структуре,
Б. Растворяют 1,0 г (4,4 моль) продукта раздела А в 25 мл ацетонит- рила, разбавляют равным объемом воды и обрабатывают 2,13 г (5 ммоль) иодбензол-бис-(трифторацетата).После перемешивания раствора при комнатной температуре в течение 5 ч продукт обрабатывают по методике примера 17 (раздел В), получают с количественным выходом солянокислый N-(N -бeнзил- оксикарбонил- | -бензил-Ь-аспаргил)- -S-1,1-диаминоэтан, который используют без дальнейшей очистки.
В, Растворяют продукт раздела Б в 100 мл тетрагидрофурана, добавляют 1,1 мл (Ю ммоль) N-метилморфоли- на, затем 1,25 г (6,5 ммоль) 2,2,5,5- -тетраметилциклопентанкарбонилхлори- да и перемешивают смесь 5 ч при комнатной температуре. Затем добавляют этилацетат и воду, разделяю т фазы, водную фазу повторно экстрагируют этилацетатом. Объединенные органические фазы промывают два разя 1 М раствором бикарбоната натрия, затем на- сьпценным раствором хлористого натрия и сушат сульфатом магния. Раствор фильтруют, фильтрат выпаривают при пониженном давлении, кристаллизуют остаток из этилацетата/гексана. Полу20
25
30
22
A.Суспендируют 20 г (0,194 моль) й -аминоизомасляной кислоты в 400 NU тетрагидрофурана, обрабатывают раствор фосгеном в толуоле (3 М, 160 мл) Смесь всю ночь нагревают при 65 С. Полученный в результате прозрачный раствор выпаривают при пониженном да лении, повторно растворяют в тетра- гидрофуране и снова выпаривают. Полу чают N-карбоксиангидрил oi-аминоизома ляной кислоты в виде плотного масла которое используют без дальнейшей очистки.
Б. Растворяют продукт раздела А в 200 мл тетрагидрофурана, охлаждают до -20 С и обрабатывают избытком газ образного аммиака. Раствору дают мед ленно нагреться до комнатной темпера туры, а затем выпаривают досуха при пониженном давлении. Твердый остаток зкстрагируют этилацетатом в экстракторе Сокслета в течение 3 ч. Получен ный раствор фильтруют, продукт остав ляют кристаллизоваться. Получают 10 6 -аминоизоОутирамида в виде твердог кристаллического продукта (т.ш1.115- 118 С), который является гомогенным по данным ТСХ. ЯМРгспектр продукта соответствует указанной структуре.
B.Растворяют 24,2 г (67 ммоль) N -бензилоксикарбонил- -бензил-L- -аспаргиновог кислоты в 300 мл сухого диметилформамида, раствор охлаж40
дают до -20 С и обрабатьгеают 14,5 г
чают 1,3 г N-(№ -бeнзилoкcикapбoннп- (71 ммоль) дициклогексилкарбодиими- - |Ь-бензил-Ь-аспаргил)-N -(2,2,5,5- -тетраметилциклопентанкарбонил-5-1,1- -диаминоэтана (т.пл. 129-131 С), который является гомогенным по данным ТСХ. ЯМР-спектр продукта соответствует указанной структуре.
Г, Гидрируют продукт раздела В обычным способом в 50 мл ледяной уксусной кислоты на 10%-яом палладии на угле. После лиофилизации и нескольких повторных лиофилизации из воды получают с количественным выходом (L-acnapгил)(2,2,5,5-тетраме- тилциклопентанкарбонил)-5-1,1-диаМи- ноэтан . (т.пл. 174-176 С, с разложением) . Соединение является гомогенным по данным HPLc (условия - см, пример 14, время удержания 10,27 мин). Сладость 600 - 800 х сахароза.
П р и М е р 13. N-(L-acnaprHn)45
да. После 30-минутной активации при этой температуре добавляют заранее охлажденный раствор 6,9 г (67 ммоль) об-аминоизобутирамида в 125 мл диметилформамида. Смеси дают нагреться до комнатной температуры. После перемешивания в течение двух дней смесь выпаривают досуха при пониженном давлении. Остаток очищают флаш- хроматографией на силикагеле, элюи- руют с последовательным градиентом хлороформ/гексан 3:1 (объем/объем), хлороформом, а затем хлороформом/ме танолом (95:5, объем/объем). После 50 элюирования получают 10,0 г N -бен- зилоксикарбонил- -бензил-L-acnap- гил-об-аминоизобутирамида,который яв ляется гомогенным по данным ТСХ. Я ТР-спектр продукта соответствует/ук занной структуре.
55
-N -(циклопентанкарбонил)-2,2-диами- нопропан (формула I, , R гшклопентил).
0
5
4862
0
5
0
22
A.Суспендируют 20 г (0,194 моль) й -аминоизомасляной кислоты в 400 NUT тетрагидрофурана, обрабатывают раст| вор фосгеном в толуоле (3 М, 160 мл). Смесь всю ночь нагревают при 65 С. Полученный в результате прозрачный раствор выпаривают при пониженном давлении, повторно растворяют в тетра- гидрофуране и снова выпаривают. Получают N-карбоксиангидрил oi-аминоизомас- ляной кислоты в виде плотного масла, которое используют без дальнейшей очистки.
Б. Растворяют продукт раздела А в 200 мл тетрагидрофурана, охлаждают до -20 С и обрабатывают избытком газообразного аммиака. Раствору дают медленно нагреться до комнатной температуры, а затем выпаривают досуха при пониженном давлении. Твердый остаток зкстрагируют этилацетатом в экстракторе Сокслета в течение 3 ч. Полученный раствор фильтруют, продукт оставляют кристаллизоваться. Получают 10 г 6 -аминоизоОутирамида в виде твердого кристаллического продукта (т.ш1.115- 118 С), который является гомогенным по данным ТСХ. ЯМРгспектр продукта соответствует указанной структуре.
B.Растворяют 24,2 г (67 ммоль) N -бензилоксикарбонил- -бензил-L- -аспаргиновог кислоты в 300 мл сухого диметилформамида, раствор охлаж
(71 ммоль) дициклогексилкарбодиими-
да. После 30-минутной активации при этой температуре добавляют заранее охлажденный раствор 6,9 г (67 ммоль) об-аминоизобутирамида в 125 мл диметилформамида. Смеси дают нагреться до комнатной температуры. После перемешивания в течение двух дней смесь выпаривают досуха при пониженном давлении. Остаток очищают флаш- хроматографией на силикагеле, элюи- руют с последовательным градиентом хлороформ/гексан 3:1 (объем/объем), хлороформом, а затем хлороформом/метанолом (95:5, объем/объем). После элюирования получают 10,0 г N -бен- зилоксикарбонил- -бензил-L-acnap- гил-об-аминоизобутирамида,который является гомогенным по данным ТСХ. Я ТР-спектр продукта соответствует/указанной структуре.
Г. Растворяют 5,0 г (11 ммоль) npd- дукта раздела В в 30 мл ацетонитрнпа, раствор разбавляют рапным объемом
воды и обрабатывают 5,16 г (12 ммоль) иодбензол-бис-(трифторацетата).Реакционную смесь перемешивают 7 ч при комнатной температуре до завершения реакции (по данным ТСХ). Раствор выпаривают при пониженном давлении, остаток растворяют в 100 мл диоксана и 3 мл концентрированной соляной кис- ,лоты и лиофилизируют. Повторяют про- цесс. Получают с количественным выходом солянокислый N-(N -бeнзилoкcикap- бонил- р -бензил-Т -(аспаргил)-2,2-ди аминопропан, который используют без дополнительной очистки,
Д. Растворяют продукт раздела Г в 100 мл тетрагидрофурана и обрабатывают 2,5 г (24 ммоль) триэтиламина, затем 1,75 г (13,2 ммоль) циклопен- танкарбонилхлорида. Реакционную смесь перемешивают 5 ч при комнатной температуре, фильтруют. Фильтрат выпаривают при пониженном давлении. Остаток очищают хроматографией на силикаге- ле, Получают Н-(К -бензилоксикарбо- НИЛ- 1-бензил-Ь-аспаргил)-ыЦцикло- пентанкарбонил)-2,2-диаминопропан, который является гомогенным по данным ТСХ, ЯМР-спектр продукта соответствует указанной структуре,
Е, Гидрируют продукт раздела Л обычным способом в 100 мл ледяной уксусной кислоты на 10%-ном палладии на угле. После лиофилизации и нескольких повторных лиофилизации из воды получают с количественным выхо- дом Ы-(Ь-аспаргил)-N -(циклопентан- карбонил-2,2-диаминопропан).Сладость 50-100 X сахароза.
Пример 14. Ы-(1-аспаргил)- -N -(2,2,5,5-тетраметилциклопентан карбонил)-К-1,1-диаминопропан,(соединение I, ,CH j, ,,2,5,5- -тетраметилциклопентил).
А, Растворяют в 50 мл диметилформ- амида 5,0 г (48,5 ммоль) D-(йб-амино- -н-масляной кислоты, обрабатывают 6,15мл (48,5 ммоль) хлортриметилсила- на. Смесь перемешивают 1 ч при комнатной температуре.Одновременно растворяют 15,73 г (45,1 ммоль) N -бен- зилоксикарбонил- |3-бензил-Ь-аспарги- новой кислоты в 50 мл диметилформами- да, охлаждают до и обрабатьша- ют 4,84 мл (44,1 ммоль) N-метилморфо- лина, затем 5,72 мл (44,1 ммоль) изобутилхлорформата.После 10-минутной активации добавляют предваритель- но охлажденный раствор силилового
Q
0 5
5
эфира D-o6-амино-н-масляной кислоты, затем второй эквивалент 4,84 мл (44,1 ммоль) Ы-метилморфолина,Раст- нору дают нагреться до комнатной температуры, перемешивают 4 ч, а затем подкисляют до рН 1-2 водной соляной кислотой. Добавляют хлороформ, разделяют фазы, водный слой повторно экстрагируют хлороформом. Объединенные органические фазы промывают три раза 1 н, соляной кислотой, насыщенным хлористым натрием и сушат сульфатом магния. Растворитель выпаривают при пониженном давлении и кристаллизуют остаток из этилацетата/гек- сана. Получают 13,3 г N -бензилокси- карбонил- -бензил-Ь-аспаргил-В-о - -амино-н-масляной кислоты (т,пл,150- 152 с), которая является гомогенной по данным ТСХ, ЯМР-спектр продукта соответствует указанной структуре,
Б, Растворяют 10,0 г (22,6 ммоль) продукта раздела А в 50 мл диметил- формамида, охлаждают до и обрабатывают 2,48 мл (22,6 ммоль) N-ме- тилморфолина, затем 2,93 мл (22,6 ммоль) изобутилхлорформата. После 4-минутной активации при -15 С добавляют 3,84 г (24,9 ммоль) аммонийной соли 1-оксибензтриазола в виде твердого продукта. Смесь перемешивают 45 мин при -15 С, затем дают нагреться до комнатной температуры медленно, при перемешивании в течение 4 ч, разбавляют водой .и хлороформом. Разделяют фазы, водную фазу повторно экстрагируют хлороформом. Объединенные органические экстракты промывают три раза 1н. соляной кислотой, три раза - насыщенным раствором бикарбоната натрия, насыщенным , раствором хлористого натрия и су-, шат над сульфатом магния. Растворитель выпаривают при пониженном давлении и перекристаллиэовывают твердый остаток из этилацетата/гексана. Получают 7,5 г N бeнзилoкcикapбoнил- - А - бензил-Ь-аспаргил-В-с -амино- -н-бутирамида (т,пл. 170-171°С), который является гомогенным по данным ТСХ. ЯМР-спектр продукта соответствует указанной структуре.
Б. Растворяют 5,0 г (11,3 ммоль) продукта раздела Б в водном ацетонцт- риле (1:1 по объему, 100 мл) и обрабатывают 5,85 г (13,6 ммоль) иодбензол-бис- (трифторацетата) , Реакционную смесь перемешивают 5 ч при комнатной температуре н выпаривают досуха при пониженном давлении. Остаток снова растворяют в 50 мл диоксана, добавляют избыток концентрированной водной соляной кислоты, раствор несколько раз повторно выпаривают и, наконец, лиофилизируют из диоксана,. Получают с количественным выходом солянокислый N- (М -бензилоксикарбо- нил- -бензил-Ь-аспаргил)-R-1,1-ди- аминопропан, который используют без дальнейшей очистки.
Г. Растворяют продукт раздела В в 25 мл тетрагидрофурана и обрабатывают 2,56 г (13,6 ммоль) 2,2,5,5- -тетраметилциклопентанкарбонилхлорида, затем 3,46 мл (24,9 ммоль) три- этиламина. Смесь перемешивают при комнатной температуре и контролируют реакцию с помощью ТСХ. По завершении реакции (примерно через 5 ч) добавляют этилацетат и воду. Разделяют фазы, водную фазу повторно экстрагируют этилацетатом. Объединенные органические фазы промывают два раза 1 М водным бикарбонатом натрия, три раза - 2 н. соляной кислотой, насыщенным раствором хлористого натрия и сушат над сульфатом магния. Растворитель выпаривают при пониженном давлении и перекристаллизовывают остаток из этилацетата/гексана. Получают 4,4 Ы-(Ы -бензилоксикарбонил- р-бензил- -L-аспаргил)-N -(2,2,5,5-те траметил- циклопентанкарбонил)-К-1,1-диамино- пропана, который является гомогенным по данным ТСХ. Я ГР-спектр продукта соответствует указанной структуре.
Д.Гидрируют 4,0 г продукта раздела Г обычным способом в 100 мл ледяной уксусной кислоты на 10%-ном палладии на угле. После лиофилизации и нескольких повторных лиофилизации из воды получают с количественным выходом N-(L-acпapгил)-N -(2,2,5,5-тет раметилциклопентанкарбонил)-К-1,1- диаминопропан (т.пл. 164 С, с разложением) . Сладость 200-300 х сахароза.
Как указывалось выше, синтезированные соединения могут быть использованы для получения подслащивателей в пищевой промьгашенности 1ЛЯ применения в пище, жевательной резинке, напитках и т.п.).
Кроме того, эти соединения, обес- печивах|щие высокую степень сладос0
5
0
5
0
5
0
5
0
5
ти, пьпынают особый интерес потому, что являются произиодньми аминокис;гот, а не пептидами, пслелствие чего степень безопасности, обеспечиваемая ими намного выше, чем у любого из известных синтетических подслащивателей .
Соединения,полученные предлагаемым способом являются высокостабильными. Они могут быть использованы в более широком интервале рН, чем такие соединения, как метиловый эфир Ь-аспаргил-Ь-фенилаланина, относя- пшйся к дипептидным подслащивателям. В условиях высокой температуры они также остаются стабильными.
Полученные соединения являются нек орийными в тех количествах,которые используются для подслащивания. Степень сладости их зависит от ряда факторов, наиболее важным из которых является природа ацилирую- щей группы R - предшественника кар-, боковой кислоты. Обычно предпочтение отдается разветвленным, массивным гидрофобным группам, но наиболее предпочтительньпчи являются цикло- алкильные и гетероциклоалкильные группы, содержащие алкильные замещающие группы рядом с карбонильной группой. Например, циклопентильная группа содержит геминальные диметильные заместители в 2-м и 5-м положениях кольца (см.таблицу). Замещение в 3-м и 4-м положениях кольца обычно не
дает высокого уровня .сладости.
t
Хотя, степень сладости соединений полученных по изобретению, в сравнении с сахарозой (считая вес на вес) в значительной мере зависит от заместителя R, все эти соединения обеспечивают преимущества как подслащивающие агенты благодаря тому, что все продукты их разложения являются совместимыми с человеческой физиологией, а именно уксусной кислотой и аминокислотами, а также благодаря их высокой стабильности как в твердой форме, так и в растворе.Кроме того, соединения по изобретению при использовании их с другими под- слащивателями, такими как сахарин, позволяют устранить нежелательное горькое послевкусие других подслащивателей.
Следовательно, соединения, полученные предлагаемым способом, могут быть
использованы для подслащивания съедобных материалов различных типов.
Формула изобретени
Способ получения N-(L-acпapгил)- -М-(1-ациламино)-алкиламинов общей формулы I
R
НЗМ -CH-CO-T H- -NH-CO-R
CH-i R
со
где R - низший алкил или низший оксиалкил,
R - водород или низший алкил и R - разветвленный С -алкил, Cg- Циклоалкил, низший ал- килциклоалкил, дицикло(низ- щий)алкил, 2,2,4,4-тетра- метилтиетан или фенил, отличающийся тем, что проводят взаимодействие защищенной аминокислоты формулы
X-NH-CH-COOH
сн сшо
где X является защищающей аминогруппу группой, такая, как бен- зилоксикарбонил, а У является защищающей карбоксил группой такая, как бензильная,
с производным аминокислоты формулы
H N-C-COOZ
R
где R и R имеют указанные выше знчения,
Z - водород или группа, защщающая карбоксил, для получения соединения формулы
R
X-NH-CH-CO-NH-C-COOZ СНг R GOOD
удаляют защищающую группу Z для получения соединения формулы
R X-MH-CH-CO-NH-(f-COOH
СН2
соо
R
которое обрабатывают ам1 иаком с получением соответствующего амида формулы
R
X-NH-CH-CO-NH-C-COMH, I I
CH
coo
R
где X,y,R и. R имеют указанные значения,
полученный амид перегруппировкой Гофмана превращают в соответствующую моноацилированнук) гемдиаминоалкановую соль формулы
R
Х- NH- CH-CO-NH- C-NnV II 3
СН2 R
соо-з
где R,R , X и Y имеют указанные значения, А - анион,
проводят взаимодействие этой соли с хлорангидридом кислоты формулы , где R имеет указанные значения, для получения соответствующего защищенного производного формулы
R
x-NH-cH- co-NH- C--NH-COR
CHi R
coo
50
в котором удаляют защищающие группы X и У.
по сладости для подслащивателей - производньсх гемдиаминоал каков
R
Н jN -CH- (p-NH- CO-I
R
СН, CO.
CHjClljN
R
200-300
31
а)Подслащиватели происходят из Ъ-аспаргил-К-гемдиаминоалканов,если нет других указаний
б)Относительно сахарозы
в)Происходят из 1,-аспаргш1-8-1,1-диаминоэтана
Составитель Л.Иоффе Редактор А.Маковская Техред М.Дидык Корректор Э.Лончакова
Заказ 4136/58
Тираж 352
ВНИИПИ Государственного комитета по изобретениям и открытиям при 1 КНТ СССР 113035, Москва, Ж-35, Раушская наб., д. 4/5
,,K..,.B...w.H.B . . .а..-.......,..... .
Производственно-издательский комбинат Патент, г.Ужгород, ул. Гяглрина,101
1494862
32 Продолжение таблицы
Подписное
| I.Cheni | |||
| ПРИБОР ДЛЯ ЗАПИСИ И ВОСПРОИЗВЕДЕНИЯ ЗВУКОВ | 1923 |
|
SU1974A1 |
