1
(21)4202958/23-04
(22)22.07.87
(31)8617986; 8701832
(32)23.07.86; 28.01,87
(33)GB
(46) 07.03.90. Бюл. f, 9
(71)Империал Кемикал Индастриз ПЛС (GB)
(72)Бриан Рой Холлоувей, Ральф Хауе, Балбир Синг Рао и Дональд Стриблинг (GB)
(53)547.298.1 .07(088.8)
(56)ЕР (Г 171760, кл. С 07 С 103/34, опублик. 19.02.86.
(54)СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ АМИДА ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ КИСЛОТНО-АДДИТИВНЫХ СОЛЕЙ
(57)Изобретение относится к получению производных амида ф-лы I
O -OCH2CH(OH)CH2NHCH7CH70- @OCH7COM
где М - R -- Н или F; R2 - циклоалкильная группа , С. -алкенил или -алкил, который
может быть замещен одной гидроксиль- ной, карбамоильной, С -С -алкоксилъ- ной, фенильной или хлорфенильной группой; , метил или этил или R и R5 вместе с N образуют группу пипери- дино, пирролидино, морсЬолино или 1,2,3,4-тетрагидроизохинол 2-ил, или их фармацевтически приемлемых кислотно-аддитивных солей, которые обладают свойством стимулировать тер- могенез теплокровных животных и могут быть использованы в медицине или ветеринарии. Цель - разработка способа попучечия новых более активных соединений. Получение ведут реакцией эфира ф-лы I, где R,, - указано выше; М - С -С4-алкоксигруппа, или его оптически активной Лормы с амином ф-лы HNR2R3, где R2 и R, указаны, с последующим ваделением продукта, полученного в форме энантиомера или оптически активной формы в виде свободного основания, или с переводом его в фармацевтически приемлемую кислотно-аддитивную соль реакцией с соответствующей кислотой. 3 табл.
с SS
(Л
ел
Јь С Ј j
05








| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения производных феноксиуксусной кислоты или их фармакологически приемлемых солей в форме рацемата или оптически-активного изомера | 1986 |
|
SU1632370A3 |
| АРОМАТИЧЕСКИЕ СОЕДИНЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1996 |
|
RU2182574C2 |
| 4-АМИНОПИРИДИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1994 |
|
RU2126388C1 |
| Способ получения производных аминопропанола или их солей | 1980 |
|
SU1243622A3 |
| Способ получения спирозамещенных производных глутарамида или их фармацевтически допустимых солей | 1987 |
|
SU1612996A3 |
| Способ получения 3-(4-аминоэтоксибензоил) бензо( @ )тиофенов или их солей | 1982 |
|
SU1155157A3 |
| Способ получения производных бензамида | 1988 |
|
SU1627082A3 |
| ПРОИЗВОДНЫЕ АРОИЛПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1998 |
|
RU2258702C2 |
| ЗАМЕЩЕННОЕ КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ, СПОСОБ ФАРМАКОЛОГИЧЕСКОГО ВОЗДЕЙСТВИЯ, СПОСОБ ИНГИБИРОВАНИЯ 5-ЛИПОКСИГЕНАЗЫ, ИНГИБИРОВАНИЯ ПРОДУКЦИИ ЛИПИДНЫХ ПЕРОКСИДОВ ИЛИ СНИЖЕНИЯ УРОВНЯ САХАРА В КРОВИ | 1998 |
|
RU2196141C2 |
| ПРОИЗВОДНЫЕ ПИПЕРАЗИНА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ИНГИБИРОВАНИЯ РВОТЫ | 1999 |
|
RU2258068C2 |
Изобретение относится к получению производных амида ф-лы I @ -OCH2CH(OH)CH2NHCH2CH2O- @ -OCH2COM, где M-NR2R3
R1-H или F
R2 - циклоалкильная группа C3-C5, C3-C4-алкенил или C1-C4-алкил, который может быть замещен одной гидроксильной, карбамоильной, C1-C4-алкоксильной, фенильной или хлорфенильной группой
R3-H, метил или этил или R2 и R3 вместе с N образуют группу пиперидино, пирролидино, морфолино или 1,2,3,4-тетрагидроизохинол-2-ил, или их фармацевтически приемлемых кислотно-аддитивных солей, которые обладают свойством стимулировать термогенез теплокровных животных и могут быть использованы в медицине или ветеринарии. Цель - разработка способа получения новых более активных соединений. Получение ведут реакцией эфира ф-лы I, где R1 - указано выше
M-C1-C4-алкоксигруппа, или его оптически активной формы с амином ф-лы HNR2R3, где R2 и R3 - указаны, с последующим выделением продукта, полученного в форме энантиомера или оптически активной формы в виде свободного основания, или с переводом его в фармацевтически приемлемую кислотно-аддитивную соль реакцией с соответствующей кислотой. 1 табл.
Изобретение относится к органической химии, а именно к способу получеRi ОН @-OCH2CHCH2NHCH2CH20- grOCH2CONR2R:
где R, - водород или фтор;
R- - циклоалкильная группа C-j-Cg-, С5-С(.-алкенил или С,-С4-алния новых производных амида общей формупы
(I)
LM
кил, который молет быть замещен одной гидроксильной, карбамоильной, С(-С -алкок
сильной, фенильной или хлор- фенильной группой; R - водород, метил или этил или R и R вместе образуют группу пиперидине, пирроли- дино, морфолино или 1,2,3,4- -тетрагидроизохинол 2-ил, или их фармацевтически приемлемых кислотно-аддитивных солей, которые обладают свойством стимулировать термо- генез теплокровных животных и могут быть использованы в медицине или ветеринарии.
Целью изобретения является разработка доступного способа получения соединений формулы (I), которые обладают высокой термогенной активностью.
В приведенных примерах также, как и в других случаях, проводят все операции при комнатной температуре, т.е. 18-26°С, упаривание под уменьшенным давлением на роторном испарителе хроматографию на кизельгуре фирмы Мерк (марка 7734)полученном из Е-Мер ка, Дармштадт, ФРГ, спектры ядерного магнитного резонанса (ЯМР) снимают на приборе 200 МГц в dg-ДМСО в качестве растворителя с использованием тетраметилсилана (ТМС) в качестве внутреннего стандарта, для протонов приведены данные в шкале дельта (в. миллионных долях) относительно ТМС, все кристаллические конечные продукты имеют удовлетворительный микроанализ и ЯМР-спектры.
Пример 1. Смесь метил-2-И- (2-гидрокси-З-феноксипропил)- -амино этокси феноксиацетат (0,38 г) в метаноле (20 мл) и 33%-ного (мае./об.) раствора метиламина в этаноле (10 мл) выдерживают при комнат
ЯМР: 3,08 (д. д., 1Н, СНСН2Ш) 3,26 (д.д., 1Н, ); 3,36 ( 2HS NHCH2CHa); 3,7 (с., ЗН, СОгСН 4,0 (д., 2Н, ОСНаСН); 4,25 (м., ОСН2, СНОН-); 4,74 (с., 2Н, ОСНа 6,8-7,05 (м., 7 ароматических пр нов); 7,31 (м., 2 ароматических
ной температуре в течение 3 ч. Раствор испаряют, а осадок перекристалли- 45 тона)
зовывают из этилацетата, получая N- Гидрохлорид (1,9 г) переносят -метил 2-п-Г2- ((2-гидрокси-З-фенокси- пропил)амино этокси феноксиацетамид
(0,24 г), т.пл. 115°С.
Найдено, %: С 63,9; Н 70,0; N 7,3,
C,
Вычислено, %: С 64,2; Н 7,0; N 7,5.
ЯМР: 1,84 (уширенньй с., 1Н, NH); 2,68 (м., 5Н, CH(OH)CHZNH + NHCH3); 2,86 (т., 2Н, МНСНгСН20); 3,90 (м., 5Н, ОСНг-СН3ОН,+ ОСН2СН2); 4,38 (с., 2Н, ОСН2СО); 4,80 (уширенный с., 1Н, ОН); 6,80-7,00 (MO, 7 ароматичес50
55
смесь 5%-ного (мас./об„) раствор гидрокарбоната натрия (50 мл) и д хлорметана (50 мл). Органический сушат (MgSO) и растворитель упа
ют. Оставшийся твердый остаток п кристаллизовьшают из метанола, п чая 2-П-|2- (2-гидрокси-З-фенокс
Iпил)амино этокси1феноксиацетат (1,67 г), т.пл. 116-118°С.
Исходное производное пропилам получают следующим образом.
А. Перемешиваемую смесь 2-(проксифенокси)этиламина (4,0 г) и
0
5
ких Н); 7,25 (м., 2 ароматических Н); 7,80 (уширенный с., 1Н, CONH).
Исходные вещества получают следующим образом.
Смесь N-бензил-Н-(2-Г -гидроксифе- ноксиэтил)-2-гидрокси-З-феноксипро- пиламина (4,0 г), метилбромацетата (1,56 г), безводного карбоната калия (1,7 г) и иодида калия (0,05 г) перемешивают при кипячении в сухом ацетоне (50 мл) в течение 24 ч. Реакционную смесь охлаждают, осадок..отфильтровывают, растворитель упаривают. Остаток- метил-2-п -{2-(Н-бензил-2-гиД - рокси-3-феноксипропил)амино этоксиТ- -феноксиацетата растворяют в метаноле (90 мл) и уксусной кислоте (30 мл). Полученный раствор гидрогенизируют в присутствии 10% (мае./мае.) палладия на угле (0,4 г) при 20 бар и 60°С в течение 48 ч. Смесь охлаждают, твердый остаток удаляют фильтрованием, растворитель упаривают. Оставшееся масло растворяют в метаноле и обрабатывают раствором эфира, насыщенного хлороводородом. Выпавший твердый осадок дважды перекристаллизов ывают из метанола, получая гтздрохлорид метил- -2-П- 2 С(2-гидрокси-3-феноксипропил) амино этоксиЯ-феноксиацетата (0,22 г), т.пл. 170°С.
Найдено, %: С 58,2; Н 6,3; N 3,6; С1 8,8.
0
CWHWNC106
Вычислено, %: С 58,3; Н 6,3; N 3,4; С1 8,6.
ЯМР: 3,08 (д. д., 1Н, СНСН2Ш); 3,26 (д.д., 1Н, ); 3,36 (т., 2HS NHCH2CHa); 3,7 (с., ЗН, СОгСН3); 4,0 (д., 2Н, ОСНаСН); 4,25 (м., ЗН, ОСН2, СНОН-); 4,74 (с., 2Н, ОСНаСО); 6,8-7,05 (м., 7 ароматических протонов); 7,31 (м., 2 ароматических протона)
Гидрохлорид (1,9 г) переносят
0
5
смесь 5%-ного (мас./об„) раствора гидрокарбоната натрия (50 мл) и ди- хлорметана (50 мл). Органический слой сушат (MgSO) и растворитель упарива ют. Оставшийся твердый остаток пере- кристаллизовьшают из метанола, получая 2-П-|2- (2-гидрокси-З-феноксипроIпил)амино этокси1феноксиацетат (1,67 г), т.пл. 116-118°С.
Исходное производное пропиламина получают следующим образом.
А. Перемешиваемую смесь 2-(пгид роксифенокси)этиламина (4,0 г) и бензальдегида (5,0 г) в метаноле (50 мл) охлаждают льдом и прибавляют натрий- боргидрид (2,0 г) порциями в течение 1 ч. После перемешивания в течение 18 ч растворитель упаривают. Остаток распределяют между 2М соляной кислотой (200 мл) и этилацетатом (100 мл). Кислотный слой отделяют, доводят до щелочной реакции действием карбоната калия и затем экстрагируют этилацетатом. Экстракты сушат (MgS04) и упаривают. Оставшееся масло растворяют в этилацетате и пропускают через сухой
10
и растворитель vnapuBaiOT. Оставшееся масло растворяют в этилацетате. Затем через раствор пропускают сухой хлоро- водород до прекращения выделения t осадка. Твердый осадок отделяют фильтрованием и перекристаллизовывают из смеси метанола и этилацетата, получая гидрохлорид N-бензил-2(п-гидроксифе- нокси)этиламина (0S9 г), т.пл. 182 - 184°С.
П р и м е р 2. Процедуру, описанную в примере 1, повторяют с использованием (-)-энантиомерной формы
хлороводород до тех пор, пока не вы- 15 метил-2-Ц- 2-(2-гидрокси-3-фенокси
пропил)амино этоксич Леноксиацетата (Z) в количестве 0,66 г с получением соответствующей оптически активной формы Ы-метил-2-И-|2- (2-гидрокси-З- 20 -феноксипропил)-амино этоксиjфенокси- ацетамида (.0,50 г), т.пл. 114-1 О -8,1° (С 0,97, этанол).
Исходный материал (Z) получают следующим образом.
25 Смесь метил-2-п-{2-(2-гидрокси- -3-феноксипропил)амино этоксиТфенок- сиацетата (0,92 г), моногидрата (-)- -ди-п-толилвинной кислоты (0,99 г) в метаноле (15 мл) упаривают при ки- Зо пячении, получая конечный объем 5мл. Прибавляют метилацетат (0 мл) и смесь вновь упаривают до объема 5 мл. Эту обработку повторяют еще раз. Смесь оставляют при комнатной температуре в течение 18 ч. Образовавшееся твердое вешество собирают и пе- рекристаллизовывают из метанола и этилацетата с образованием (-)-ди-ft- -толилт ар трата ()-метил-2-м-4(2-гид- рокси-3-феноксипропил)амино этокси феноксиаисетата (0,337 г), т.пл. 46- 148°С; М -80,3° (С 0,97S метанол) .
(-)-Ди-п-толилтартрат (-)-метил-2- 45 -{2- (2-гидрокси-З-Феноксипропил) амино этоксиуфеноксиацетата (0S33 г) распределяют между 5%-ным (мае./об.) раствором гидрокарбоната натрия (10 мл) и дихлорметаном (10 мл). Ор- 5Q ганический слой упаривают, сушат
(MgSO), растворитель упаривают. Оставшееся твердое вещество (0.148 г), т.пл. 114-1 16°С, -7,8й (С 0,97, дихлорметан). растворяют в ,-г метилацетате. Газообразный сухой хлористый водород пропускают через раствор, пока не прекратится выпадение осадка. Осадок отделяют и перекристаллизовывают из метанола и метилацепадет весь осадок. Выпавший осадок собирают и перекристаллизовывают из метанола и этилацетата, получая гидрохлорид М-бензил-2-(П-гидроксифенок- си)этиламина (2,3 г), т.пл. 182 - 184°С.
Б. Гидрохлорид N-бензил-2-(И-гид- .роксифенокси)этиламина (3,5 г) встряхивают с 1М раствором гидроксида натрия (20 мл) и дихлорметаном {20 мл). Органический слой отделяют и промывают водой (10 мл), сушат (MgS04) и растворитель упаривают, получая в виде масла К-бензил-2-(п-гидроксифенок- си)-этиламин.
В. Смесь М-бензил-2-(ц-гидроксифе- нокси)-этиламина (2,5 г) и 1,2-эпок- си-3-феноксипропана (1,54 г) в про- пак-2-оле (50 мл )нагревают при кипении в течение 72 ч. Растворитель удаляют упариванием, получая М-бензил-К -(2-л-гидроксифеноксиэтил)-2-гидрок- си-3-феноксипропиламин в виде масла, достаточно чистого, что показано методом тонкослойной хроматограЛии(ТСХ) (используют гранулы силикагеля и 5%- ный метанол в дихлорметане как элю- ент), и которое используют далее без очисткио
Исходный гидрохлорид №-бензил 2- -(М-гидроксифенил)-этиламина может быть также получен по следующему методу.
Смесь ц - (2-бромэтокси)фенола (2,2 г), бензиламина (1,07 г) и три- этиламина (1,01 г) в этаноле (30 мл) нагревают при кипении в течение 18 ч. Растворитель упаривают, а остаток переносят в смесь 2М раствора хлористоводородной кислоты (100 мл) и этил- ацетата (50 мл). Кислотный слой отделяют, доводят до щелочной реакции карбонатом калия и затем экстрагируют этилацетатом„ Экстракты сушат (MgSO)
35
40
0
и растворитель vnapuBaiOT. Оставшееся масло растворяют в этилацетате. Затем через раствор пропускают сухой хлоро- водород до прекращения выделения t осадка. Твердый осадок отделяют фильтрованием и перекристаллизовывают из смеси метанола и этилацетата, получая гидрохлорид N-бензил-2(п-гидроксифе- нокси)этиламина (0S9 г), т.пл. 182 - 184°С.
П р и м е р 2. Процедуру, описанную в примере 1, повторяют с использованием (-)-энантиомерной формы
метил-2-Ц- 2-(2-гидрокси-3-фенокси
тата, получая гидрохлорид (-)-метил- -2-п-|(2-гидпокси-3-феноксипропил) амино этокси -феноксиацетат(0,092 г), т.пл. 156-15 7°С, -12,1° (С 1, О, метанол),
Примеры 3-16, Используют процедуру, которая описана в примере 1, но применяя подходящий амин формулы НМИгДз и проводя реакцию до ее завершения по данным ТСХ на силикаге- ле, получают следующие соединения фор мулы (I) с выходом 60-90%, выделенные либо в виде свободного основания, либо в виде солей - гидрохлорида или оксалата реакцией свободного основания с эфирным раствором хлороводородной или щавелевой кислоты и перекристаллизацией из указанных растворителей (см, табл. 1)„
Исходные сложные эфиры для примера 4 получены следующим образом.
Смесь резорцина (88 г)s 1,2-дибром этана (180 г) и гидроксида калия (44,8 г) перемешивают при кипячении в метаноле (600 мл) в течение 24 ч. Реакционную смесь охлаждают.
Выпавшее твердое вещество удаляют фильтрованием и фильтрат упаривают, получая 3 f2-бромэтокси)фенол в виде масла, которое оказывается достаточно чистым по данным ТСХ (используют гранулированньй еиликагель и 10%-ный (об0/об„) метанол в дихлорметане в качестве элюента) и используется далее без очистки,
Смесь 3-(2-бромэтокси)фенола 40 г и бензиламина (39,2 г) перемешивают при кипении в этаноле (800 мл) в течение 18 ч о Реакционную смесь охлаждают и растворитель упаривают. Оставшееся масло растворяют в этилацетате (200 мл). Раствор промывают 2М хлороводородной кислотой (100 мл) „ Водный слой подщелачивают твердым карбонатом калия и экстрагируют эфиром 2х100мл Экстракты эффективно промывают водой (50 мл) и насыщенным раствором хлорида натрия (50 мл), а затем сушат (MgSO). Сухой эфирный раствор обрабатывают раствором эфира, насыщенного хлороводородомо Выпавшее твердое вещество перекристаллизовывают дважды из смеси метанол/этилацетат, получая гидрохлорид Н бензил-2-(м-гидрокси- Фенокси)этиламина (19,2 г), т.пл. 148-149°С.
ЯМР; 3,2 (т,, 2Н, ); 4,2 (с. + т., 4Н, СНгО, NCH2P); 6,4 (м.,
0
5
5
0
5
50
55
3 ароматических Н); 7,1 (т., 1 ароматический Н); 7,3-7,8 (м., 5 ароматических Н).
Смесь гидрохлорида Ы-бензил-2-(м- -гидроксифенокси)этиламина (2,79 г), 1,2-эпокси-З-феноксипропана (1,5 г), безводного карбоната калия (2,0 г) нагревают при кипении в пропан-2-оле в течение 18 ч. Реакционную смесь охлаждают и растворитель упаривают, получая И-бензил-И-(2-М-гидроксифе- ноксиэтил)-2-гидрокси-3-феноксипро- пиламин в виде мясла, которое оказывается достаточно чистым по данным ТСХ (гранулированный еиликагель и 5%-ный метанол в дихлорметане в качестве элюента) и используется без очистки.
N-Бензил-N-(2-М-гидроксифенокси- этил)-2 -гидрокси-3-феноксипропиламин (1,6 г) вводят в реакцию с метилбром- ацетатом (0,5 г), безводным карбонатом калия (0,6 г) и иодидом калия (0,05 г) в ацетоне (80 мл), используя аналогичную процедуру, которая описана для исходного сложного эфира в примере 1 и с промежуточным выделением метил-2-М- 2-Јы-бензил-(2-гидрок си-3-феноксипропил) амино этокси 1 -фе- ноксиацетата (1,1 г). Получают гидрохлорид метил-2-М- 2- Ј(2-гидрокси-3- -феноксипропил)амино этокси фенокси- ацетата (0,35 г), т.пл. 164-167°С.
Найдено, %: С 58,0; Н 6,5; N 3,3; С1 8,7.
C2oH26NC10e
Вычислено, %: С 58,3; Н 6,4; N 3,4; С1 8,6.
ЯМР: 3,1 (д.д., 1Н, СНСНгЖ); 3,25 (д.д., 1Н, ); 3,4 (т., 2HS NHCH4CH4)} 3,7 (с., ЗН, С02СН3); 3,9-4,1 (м., 2Н, ОСНгСН); 4,2-4,4 (м., ЗН, ОСН2СНОН-); 4,78 (с., 2Н, ); 5,98 (д., 1Н, СНОН); 6,5- 6,7 (м., 3 ароматических Н); 6,9-7,0 (с„9 3 ароматических Н); 7,1-7,4 (м., 3 ароматических Н); 9,1 (с., 2Н, №ф.
Примеры 17, и 18. Используя процедуру, которая описана для примеров 3-16, но исходя из метил-2- (3-о-фторфенокси-2-гидрокси- пропил)амино этоксиЯфеноксиацетата, получают соединения с выходом приблизительно 80-90%:
К-метил 2-И- 2-Ј(3-о-фторФенокси- -2-гидроксипропил)амино этокси фенок- сиацетамид в виде гидрохлорида, т.пл.
168-169CC (перекристаллизация из смеси метанола и этилацетата ) - пример 1
И-пиперидино-2-ц- 2- (3-0 -фторфе- нокси-2-гидроксипропил) амино этокси феноксиацетамид в виде гидрохлорида, т.пл. 144-146°С (перекристаллизация из смеси эфира и метанола) - пример 18.
Необходимые исходные метиловые
эфиры получают следующим образом.
Смесь N-бензил- 2- ( гидроксисЪенок- си)этиламингидрохлорид (см. пример 1) (5,6 г), 1,2-эпокси-3-9-фторфенокси- пропан (3,6 г) и безводный карбонат калия (2,7 г) нагревают при кипении в пропан-2-оле (100 мл) в течение 24 ч. Реакционную смесь охлаждают, твердый осадок удаляют фильтрованием, из фильтрата упаривают раствори- тель„ Оставшееся масло очищают хроматографией на силикагеле, элюируя 1%- ным (мае./мае.) метанолом в дихлорэтане, получая N-6eH3Kn-N-(2-n-raflpOK сифеноксиэтил)-3-0-фторфенокси-2-гид- роксипропиламин в виде бесцветного масла.
ЯМР: 2,27-3,15 (м., 4Н, CHINCHг); 3,8 (д.д., 2Н, NCH4P)j 3,9-4,2 (м., 5Н, .o -F-PhOCHz); 6,7 (с,, 4 ароматических протона); 6,8-7s (м., 4 ароматических протона Н)j 7S3 (м., 5Н, ).
Смесь 1 -бензил-М-(2-П-гидроксиФе- ноксиэтил)-З-О-фторфенокси-2-гидрок- сипропиламина (5,4 г), метилбромаце- тата (2,0 г), безводного карбоната калия (1,79 г) ииодида калия (0,05 г) перемешивают при кипении в сухом ацетоне (80 мл) в течение 24 ч. Реакци- онную смесь охлаждают, твердый осадок удаляют фильтрованием и растворитель упаривают. Остаток растворяют в ди- хлорметане (40 мл) и промывают эффективно 10%-ным (мае./об.) раствором бикарбоната натрия (20 мл) и водой (20 мл), затем сушат (MgSO) и растворитель удаляют упариванием. Масло (6,18 г) очищают хроматографировани- ем на силикагеле, элюируя 1%-ным
(об./об.) метанолом в дихлорметане, получая метил-2-П-{2-(К-бензил-3-0- -фторфенокси-2-гидроксипропил)амино этокстфеноксиацетата в виде бесцветного масла. Это масло растворяют в метаноле (100 мл) и перемешивают с обесцвечивающим активированным углем (1 г) в течение 1 ч. Уголь удаляют фильтрованием и фильтрат подвергают
гидрогенизации в присутствии бензил- хлорида (0,71 г) и 10% (мае./мае.) палладия на угле в течение 2 ч при атмосферном давлении. Катализатор удаляют Фильтрованием и растворитель из фильтрата упаривают. Оставшееся твердое вещество перекристаллизовыва ют дважды из смеси метанола и безводного эфира, получают гидрохлорид ме- тил-2-П-{2- Ц З-о -фторйенокси-2-гидро сипропил)амино этокси Феноксиацетата (0,55 г), т.пл. 120-122°С.
Найдено, %: С 55,7; Н 5,9; N 3,2; С1 8,3.
CMH25NC1F06
Вычисл ено, %: С 55,9; Н 5,9; N 3,3; С1 18,2.
ЯМР: 3,1 (д.д., 1Н, CHCH2NH); 3,27 (м., под сигналом от НО, 1Н, СНСН NH) 3,41 (т., 2Н, NHCH2CHa); 4,71 (с., ч 2Н, ОСН200); 5,93 (д., Ш, СНОН) ; 6,8-7,0 (м., 5 ароматических Н); 7,1- 7,3 (м., 3 ароматических Н); 9,12 (уширенный с., 2Н, NHp .
Пример 19. Используя процедуру, которая описана в примере 2, (-)-энантиомерный феноксиацетат (Z) (0,6 г) превращают в Ы-(2 гидрокси- этил)-2-п-|2- (2 гидрокси 3-Фенокси- пропил)амино этокси феноксиацетамид (0,32 г), т.пл. 111-113°С,25 7,1 (С 0,99, этанол), используя этанол- амин вместо метиламина,
Примеры 20-27, Используя процедуру, аналогичную описанной в примере 1, но используя подходящий амин Формулы и проводя реакцию до полного завершения по данным ТСХ на силикагеле, получают следующие соединения Формулы (I) (заместитель . находится в положении 4 кольца) с выходом 55 - 85%, выделенные в виде свободных оснований и после перекристаллизации из указанных растворителей (см. табл„ 2).
Пример 28. Гидрид натрия (0,132 г, 60%-ная (мае,/мае.) суспензия в минеральном масле) прибавляют в п -Ј2- С(2тидрокси-3-фенокси- пропил) амино этоксиТ-фенолу (1,0 г) s растворенному в сухом диметилформами- де (ДМФА) (50 мл). Полученную суспензию перемешивают приблизительно в течение 30 мин до получения прозрачного раствора. Прибавляют раствор N- -фенил-2-хлорацетамида (0,559 г) в сухом ДМФА (20 мл) и смесь перемешивают в течение 18 ч. Смесь затем выливают в воду (150 мл). Полученную смесь экстрагируют дихлорметаном (2x100 мл). Экстракты промывают водой (6x100 мл), затем сушат (MgS04J и растворитель упаривают. Остаток перекристаллизовывают из этилацетата, получая №-фенил-2-1 -|2- (2-гидрокси- -3-фейоксипропил)амино этокси фенок- сиацетамид (0,365 г), т.пл. 119-121°С. Найдено, %: С 68,7; Н 6,5; N 6,3.
CWH4BN205
Вычислено, %: С 68,8; N 6,4; Н 6,4.
Последний имеет удовлетворитель- ный ЯМР-спектр.
Исходные фенолы получают следующим образом.
Смесь гидрохлорида П-(2-аминоэток- си)фенола (1,89 г), триэтиламина (1,01 г) и 1,2-эпокси-З-феноксипропа- на (1,5 г) нагревают при кипении в течение 24 ч. Реакционную смесь охлаждают и упаривают растворитель. Остаток переносят в смесь дихлорме- тана (100 мл) и 10%-ного (мае./об.) раствора карбоната калия. Органический слой отделяют,.сугаат (MgSO), растворитель упаривают. Оставшееся масло растворяют в этилацетате и про- пускают через сухой хлороводород до полного осаждения осадка. Осадок отделяют и перекристаллизовывают из метанола и этилацетата, получая гидрохлорид п-Ј2-(2-гидрокси-3-фенокси- пропил)амино этокси фенола (0,53 г), т.пл. 171-172°С.
Найдено, %: С 60,3; Н 6,7; N 4,0; С1 10,6.
С„ H NClO-v
Вычислено, %: С 60,1; Н 6,5; N 4,1; С1 10,5.
Соответствующий гидрохлорид (1,5 г полученный согласно описанной выше процедуре, переносят в смесь 5%-ного (мае./об.) водного раствора гидрокарбоната натрия (15 мл) и дихлорметана (15 мл). Органический слой отделяют сушат (MgSO(j), растворитель удаляют упариванием, получая h (2-гидрок- си-3-феноксипропиламино)этокси фенол в виде густого сиропа (1,1 г), ко.то- рый используют без дальнейшей очист- ки.
№-Фенил-2-хлорацетамид получают следующим образом.
Смесь анилина (9,3 г) и триэтиламина (10,1 г) в дихлорметане (40 мл прибавляют по каплям за 1 ч в охлаждаемый льдом раствор хлорацетилхлори- да (11,2 г) в дихлорметане (40 мл). Смесь далее перемешивают в течение 18 ч. Органическую фазу отделяют фильтрованием, промывают водой (3x50 мл), ее высушивают (MgSO) растворитель удаляют упариванием, получая N-фенил- -2-хлорацетамид (6,1 г) в виде белого твердого вещества, т.пл. 128-129°С, который далее используют без дальнейшей очистки.
Пример 29. Используя проце- ДУРУ которая описана в примере 28, но используя И-пропил-2-хлорацетамид (0,447 г) вместо Ы-фенил-2-хлорацет- амида, получают К-пропил-2 п-{2- 2- -гидрокси-3-феноксипропил)амино эток- си}феноксиацетамид (0,47 г), т.пл. 105-107 С (перекристаллизация из этил ацетата) , фактически идентичный во всех отношениях веществу, полученному в примере 16.
Исходный 1 1-пропил-2-хлорацетамид получают как К-фенил-2-хлорацетамид, т.е. реакцией пропиламина с хлораце- тилхлоридом, в виде масла, которое используют без специальной очистки.
Соединения формулы (I) обладают термогенными свойствами и могут использоваться в лечении ожирения и/или родственных болезней метаболической дисфункции, такой как диабет mellitus особенно зрелого возраста. В некоторых случаях соединения формулы (I) могут иметь значение в модификации телесных композиций путем увеличения катаболизма жира животных, производителей мяса, таких как крупный рогатый скот, свиньи, овцы, козы и/или кролики о
Термогенный эффект соединений Формулы (I) может быть продемонстрирован с использованием одного или более следующих стандартных тестов.
А. Крыс подвергают адаптации к холоду путем нахождения в холодной среде (4°С) в течение 10 дней, чтобы увеличить их способность к термогене- зу. Затем их переносят в термонейтральную среду (29°С). Спустя 3 ч измеряют внутреннюю температуру, чтобы установить основную точку отсчета показаний термометра, и тестовое вещество вводят подкожно или орально в виде раствора или суспензии в 0,45%- ном (масо/об.) водном хлориде натрия, 0,25%-ном (мае./об.) полисорбате 80. Через 1 ч вновь измеряют внутреннюю
температуру среды. Б ттом тесте соединение, которое вызывает статистически заметное повышение внутренней температуры на 0,4°С или более при подкожной дозе 15 мг/кг (или менее), рассматривается как обладающее значительной активностью. Тест действует как модель пониженного термогенеза, имеющего место при диете.
Б. Крыс подвергают адаптации к холоду в течение 4 дней, чтобы повысить их способность к термогенезу. Затем их переносят в теплую среду (23°С) и выдерживают в течение 2 дней На следующий день вводят тестовое вещество подкожно или орально как описано в пункте А.
Через 1 ч животных умерщвляют и удаляют внутрилопаточную бурую ади- позную ткань (ВАТ), ВАТ митохондрии приготовляют прсредством диФЛеренци- ального центрифугирования и определяют Р-связывание (Holloway et al. International Journal of obesity, 1984, 8, 295) как величину термогенной активации Каждый тест включает контроль, при котором вводят только разбавитель в виде раствора или суспензии, и положительный контроль, при котором вводят изопреналин (в виде сульфата) в количестве 1 мг/кг. Тестовое вещество дозируется по 091; 0,3; 1,0; 3,0 и 10 мг/кг и результаты выражаются в терминах влияния на связывание GDP, вызванное изопрена- лином. Из этих результатов доза () необходимая для получения 50%-ного эффекта изопреналина, рассчитывается методом линейного регрессионного ана- лиза. Соединения рассматриваются как активные в данном тестировании, если они вызывают значительное повышение связывания по сравнению с контрольны
ми опытами. Этот тест позволяет установить, что термогенные эЛФекты, наблюдаемые в тесте А проявляются в увеличении влияния ВАТ в большей степени, чем посредством неспецифического механизма или механизма токсичности.
В. Крыс адаптируют к термонейтральной среде (29°С) в течение 2 нед, чтобы уменьшить их способность ВАТ, вызывать non-shivering термогенез. В течение последних 3 дней животных подвергают тренировке с использовани ем аппарата для расчёта сердечной скорости с помощью электродов на подушечках стопы, соединенных с ECG-интегра
10
15
.
„ 25 ,Q 5. 35
45
50
55
тором, даюним постоянную заданную сердечную скорость. Тестовое соединение вводят подкожно при , указанной в тесте Б, и сердечную скорость определяют в течение 15-30 мин после дозирования. Процедуру затем повторяют в последующих тестах с использованием увеличивающихся многократных повторов ED50, указанных в тесте Б}до тех пор, пока сердечная скорость (HR) достигнет или превысит 500 ударов в минуту, что позволяет рассчитать дозу, необходимую для получение сердечной скорости 500 ударов в минуту ( -цоза) .
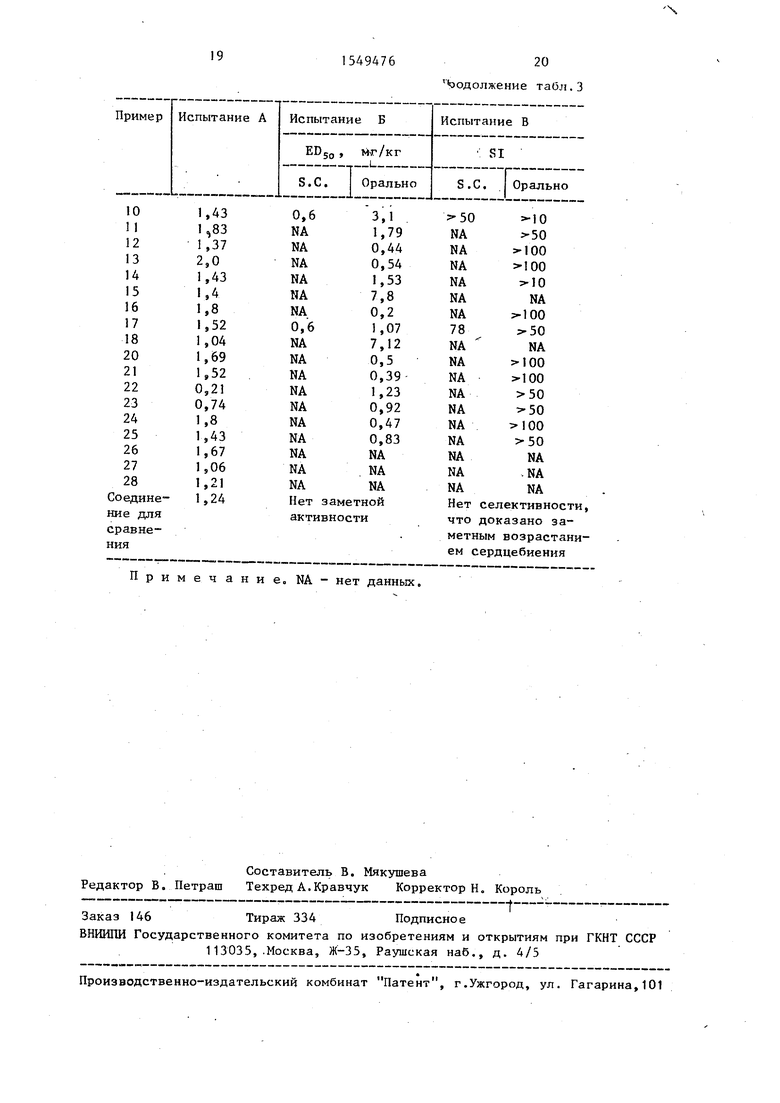
Отношение DSOO к EDbO в тесте Б может быть определено как индекс селективности (SI) и дает оценку селективности соедгиения для ВАТ как противопоказанного сердечно-сосудистой системе. Соединения, имеющие SI больше 1, рассматриваются как обладающие значительной селективностью. Неселективные соединения имеют SI меньше 1 (например, изопреналин 0,06).
Г. Крыс адаптируют к холоду при 4°С в течение 4 дней для увеличения их способности к термогенезу. Затем их выдерживают в теплой среде при 23 С в течение 2 дней. На следующий день определяют скорость основного метаболизма, используя аппарат с расходом циркулирующего в закрытой системе кислорода (описан Arundel и др Крыс затем подвергают обработке дозами (орально или подкожно) тестируемого соединения в количестве примерно 10 мг/кг в виде раствора или суспензии в 0,45%-ном (мае./об.) водном хлориде натрия, 0,25%-ном (.) полчсорбате 80. Затем определяют скорость метаболизма по крайней мере через 1 ч после введения дозы. Соединения считаются активными согласно этому тесту, если они вызывают заметное увеличение скорости метаболизма по сравнению с контрольными животными (Student s t-тест, р 0,5), которым вводят только раствор или суспензию связующего вещества.
В описанных тестах соединения формулы (I) в целом показывают эффекты следующего порядка без проявления явной токсичности:
тест А - увеличение внутренней температуры примерно на 0,5°С (или более) после подкожного введения 15 мг/кг;
15
тест Б - подкожное ЕП50 для GDP- связывания в ВАТ-митохондриях 0,01 - 10 мг/кг;
тест В - обнаруживает SI 50.
Для иллюстрации соединение, описанное в сопроводительном примере, обнаруживает следующий эффект в описанных тестах:
А - 2,25 С при подкожной дозе 10 мг/кг;
Б - подкожное , 0,133 мг/кг, оральное ED5o 1,18 мг/кг;
Я soo 3,3 мг/кг (подкожно), SI 10 (подкожно), SI 50 (орально) .
В противоположность этому известное структурно-родственное соединение №-метил-2-п- 2- (2-гидрокси-З-п- -гидроксифеноксипропил)амино этокси феноксиацетамид вызывает повышение температуры на 1,24°С в тесте А и не проявляет активности в тесте Б, но вызывает заметное повышение сердечной скорости (см. табл. 3).
154947616
В случае использования для получения термогенного эффекта у теплокровных животных, включая человека, сое-
, динение формулы (I) или подходящая фармацевтически приемлемая его соль должны быть введены так, чтобы ежедневно вводилась доза 0,002-20 мг/кг, в виде разовой дозы или отдельных
10 доз, как необходимо. Однако в данном процессе необходимо квалифицированно подбирать дозы, так как дозация должна варьироваться соответствующим образом, что зависит от жесткости усJ5 ловий в процессе лечения и от возраста и пола пациента и должно быть согласовано с известными принципами медицины.
Соединения формулы (I) могут ис2Q пользоваться для медицинских (или ветеринарных ) целей .
Формула изобретения
25 Способ получения производных амида общей формулы
R, он @-оснгснснгюнсн2снго-@-осн2сокргкз
де R R
водород или фтор; циклоалкильная группа С -С -алкенил или кил, который может быть замещен одной гидроксильной, карбамоильной, С -С4 алкок- сильной, фенильной или хлор- фенильной группой;
,1 ОН ;O -OCH2CHCH7NHCH1CH20где Rj имеет указанные значения
R,j - С -С -алкоксигруппа, или его оптически активную форму подвергают взаимодействию с амином формулы HNRj,R3, где R имеют указанные значения, с последующим выделением продукта, полученного в форме энантиомера или оптически активной формы, в виде свободного основания, или с переводом его в фармацевтичес-j ки приемлемую кислотно-аддитивную соль взаимодействием с соответствующей кислотой.
5
R-, - водород, метил или этил или R и R- вместе образуют . группу пиперидине, пирроли- дино, морфолино или 1,2,3,4- -тетрагидроизохинол-2-ил, или их фармацевтически приемлемых кислотно-аддитивных солей, отличающийся тем, что сложный эфир формулы
-OCH2COR
0
5
Приоритет по признакам
I
2,25 1,75 1,76 2,35 1,25 2,08 1,79 1,68
0,13 0,23 1,32 0,27 0,16 0,29 0,13 NA
Т а б л и ц а
Таблица
Таблица 3
100 100
NA 100 100 100 100
NA
50
10
74
100 100 100 10 100
19
154947620
оодолжение табл.3
П
римечание.КА- нет данных.
