Изобретение относится к новому соединению 11,21-диацетату (22RS)-16 α , 17α -бутилидендиокси-11 β , 21-дигидрокси- прегна-1,4-диен-3,20-диону (диацетату будесонида) формулы I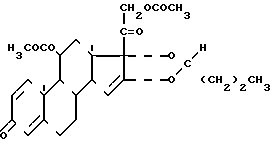 в качестве промежуточного продукта для синтеза эффективного гормонального бронхолитика будесонида (22R,S-16 α , 17α -бутилидендиокси-11 β , 21-дигидрокси-1,4, -прегнадиен-3,20-диона).
в качестве промежуточного продукта для синтеза эффективного гормонального бронхолитика будесонида (22R,S-16 α , 17α -бутилидендиокси-11 β , 21-дигидрокси-1,4, -прегнадиен-3,20-диона).
Цель изобретения - новый доступный промежуточный продукт для синтеза будесонида, который позволит упростить способ получения эффективного бронхолитика будесонида, исходя из преднизолона, с высоким выходом будесонида.
Диацетат будесонида синтезирован ацетализацией 11,21-диацетата 16 α -оксипреднизолона н-масляным альдегидом в растворе хлористого метилена с HClO4 в качестве катализатора.
Выход диацетата будесонида составил 96,5%, считая на диацетат 16 α -оксипреднизолона. При этом диацетат 16 α -оксипреднизолона получен по модифицированному методу Паничи, предусматривающему не избирательное ацилирование 17-оксигруппы, а исчерпывающее ацилирование преднизолона.
По литературным данным 11,21-диацетат 16 α -оксипреднизолона может быть получен из преднизолона с общим выходом 55,8%.
П р и м е р 1. Получение триацетата преднизолона.
К охлажденной до 5-7оС смеси 125 мл уксусной кислоты и 56 мл (60,6 г, 0,59 моль) уксусного ангидрида прикапывают 8,1 мл (12,1 г, 0,069 моль) 57% хлорной кислоты. Температура поднимается до +10оС. Охлаждают смесь до 5о и порционно загружают 25 г (0,069 моль) преднизолона. Поднимают температуру реакционной массы до 18-20оС, перемешивают ≈ 40 мин, отбирают пробу на определение конца реакции ацетилирования методом ТСХ на пластинах силуфол в системе хлороформ - ацетон 10:1 (по объему). Реакцию проводят до отсутствия пятна преднизолона (Rf 0,02) и образующегося в ходе реакции промежуточного диацетата преднизолона (Rf0,32); Rf триацетата преднизолона 0,59. По окончании реакции выливают реакционную смесь в охлажденный до ≈ 5оС раствор 200 мл 25%-ного водного аммиака в 2,5 л воды, перемешивают 1 ч и выдерживают 1 ч без перемешивания. Осадок фильтруют, промывают водой, высушивают до постоянной массы.
Получают 33,44 г ( ≈ 99%) 11,17, 21-триацетата преднизолона; т.пл. 158-170о; [ α ]D20+70,8o (Cl, хлороформ).
П р и м е р 2. Получение 11,21-диацетат 11 β ,21-дигидроксипрегна-1,4,16-триен-3,20-диона.
Смесь 25 г (0,051 моль) триацетата преднизолона, 12,5 г (0,127 моль) свежеплавленного ацетата калия в 250 мл диметилсульфоксида перемешивают 15 мин в токе азота при комнатной температуре. Нагревают до 82-85оС, перемешивают при этой температуре 45 мин в атмосфере азота и отбирают пробу на определение конца реакции методом ТСХ на пластинах силуфол в системе эфир-изопропиловый спирт 10:1 (по объему). Реакцию проводят до отсутствия пятна триацетата преднизолона (Rf 0,45). Пятно с Rf 0,38 соответствует Δ 16-соединению. По окончании реакции реакционную массу охлаждают до 30оС и выливают в 2,5 л охлажденной до 3-5оС воды. Перемешивают 2.ч, осадок фильтруют, промывают водой, сушат до постоянной массы.
Получают 21,03 г (96%) технического 11,21-диацетат 11 β,21- дигидроксипрегна-1,4,16-триен-3,20-диона, т.пл. 195-220оС.
Очистка технического 11,21-диацетата 11 β ,21-дигидроксипрегна -1,4, 16-триен-3,20-диона. 20 г технического продукта растворяют при 40-45оС в 900 мл ацетона, прибавляют 5 г активированного угля и кипятят 30 мин. Суспензию фильтруют горячей через угольную подушку, уголь промывают горячим ацетоном (180 мл в 3 приема). Фильтрат упаривают в вакууме до объема ≈ 60 мл и оставляют при 3-5оС на 16-20 ч. Осадок фильтруют, промывают охлажденным ацетоном, сушат до постоянной массы. Получают 14,8 г (74%) продукта; т. пл. 235-240оС. Литературные данные: т.пл. 225-230оС.
П р и м е р 3. Получение 11,21-диацетата 16 α -оксипреднизолона.
15 г (0,0352 моль) очищенного целевого соединения примера 2 растворяют при 35-40оС в 840 мл ацетона. Охлаждают до температуры -3 до -5оС и быстро приливают при интенсивном перемешивании сначала 3,1 мл (3,73 г, 0,081 моль) муравьиной кислоты, затем свежеприготовленный, охлажденный до 5оС раствор 6,67 г (0,042 моль) перманганата калия в 150 мл воды и 30 мл ацетона. Реакционную смесь интенсивно перемешивают 5-6 мин, прибавляют раствор 1,05 г тиосульфата натрия в 3 мл воды. Охлаждение прекращают и после 10-минутного перемешивания отстаивают реакционную массу ≈ 30 мин. Отфильтровывают осадок двуокиси марганца, промывают ацетоном, присоединяя промывку к основному фильтрату. Фильтрат упаривают в вакууме до полного прекращения отгона ацетона и выдерживают остаток 2 часа при 3-5оС. Осадок фильтруют, промывают водой, сушат до постоянной массы.
Получают 14,57 г (90% от теоретического) диацетата 16 α - оксипреднизолона; т. пл. 188-203оС. После перекристаллизации из ацетона, т.пл. 192-202оС.
Найдено, %: С 64,89; Н 7,02.
С25Н32О8.
Вычислено, %: С 65,2; Н 6,96.
П р и м е р 4. Получение диацетата будесонида.
К раствору 20 г (0,0435 моль) диацетата 16-оксипреднизолона в 200 мл хлористого метилена прибавляют 5,9 мл (4,7 г, 0,0652 моль) перегнанного н-масляного альдегида и 3 мл (4,5 г, 0,026 моль) 57% хлорной кислоты. Перемешивают 1,5 ч при комнатной температуре и отбирают пробу на конец реакции методом ТСХ на пластинах силуфол в системе бензол-ацетон 3:2 (по объему). Реакцию проводят до отсутствия пятна диацетата 16 α -оксипреднизолона (Rf 0,59); Rf диацетата будесонида 0,85. По окончании реакции прибавляют 100 мл воды и 10%-ный раствор углекислого калия до нейтральной реакции водного слоя. Отделяют органический слой, содержащий целевой продукт. Водную фазу экстрагируют хлористым метиленом, который присоединяют к органическому слою. Объединенный органический слой промывают водой, пропускают через угольную подушку ( ≈ 5 г угля) и полностью упаривают. Остаток обрабатывают гексаном.
Получают 21,6 г (96,5%) 11,21-диацетата будесонида; т.пл. 118-128оС. После перекристаллизации из смеси гексан-ИПС; т.пл. 150-160оС; [ α ]D20 +108о (СО,5 хлороформ).
Найдено, %: С 67,3; Н 7,57.
С29Н38О8.
Вычислено, %: С 67,7; Н 7,39.
ИК-спектр, см-1: 1720, 1750, 1230, 1710, 1670, 1620, 1600.
П р и м е р 5. Получение будесонида (технического продукта).
Подготовку культуры Coryn mediolanum проводят в известных условиях.
В лабораторный ферментер с 3,4 л среды ВК-4К, содержащей культуру Coryn mediolanum, вносят 24 г 11,21-диацетата будесонида в 50 мл воды (нагрузка стероида 7 г/л). Ферментацию проводят при 33оС с перемешиванием (700 об/мин) и аэрацией (0,2 л/л/мин) в течение 6-7 ч до отсутствия на хроматограмме (пластина силуфол) пятна 11-ацетата будесонида (Rf 0,68) в системе бензол - ацетон 3:2.
Культуральную жидкость экстрагируют этилацетатом (3х3 л), экстракт упаривают в вакууме.
Получают 20,1 г ( ≈ 100%) технического будесонида, т.пл. 180-200оС.
П р и м е р 6. Очистка технического будесонида.
5 г технического будесонида растворяют при кипении в 300 мл этилацетата, охлаждают до 20-25оС, прибавляют 1 г активированного угля и перемешивают 1 ч. Суспензию фильтруют через угольную подушку (1,5 г), уголь промывают 25 мл этилацетата. Фильтрат упаривают до остаточного объема ≈ 5 мл, охлаждают до 3-5оС и выдерживают при этой температуре 2 ч. Осадок фильтруют, промывают охлажденным этилацетатом, высушивают до постоянной массы.
Получают 3,5 г (70%) будесонида в виде белого кристаллического порошка, т. пл. 224-236оС; [ α ]D20+101,8o (C 0,28, CH2Cl2). По литературным данным т.пл. 221-232оС. [ α ]D25 +98,9о (С 0,28; CH2Cl2).
Общий выход будесонида составляет ≈43%, считая на преднизолон.
Таким образом, описываемый в качестве промежуточного продукта диацетат будесонида формулы I позволяет получать будесонид из преднизолона с более высоким выходом ( ≈ на 20%) и более коротким путем (5 стадий вместо 7) по сравнению с применением известного промежуточного продукта.
Изобретение касается стероидных веществ, в частности 11,21-диацетата (22RS)- 16α , 17α - бутилидендиокси- 11β,21-дигидроксипрегна-1,4-диен-3,20-диона в качестве промежуточного продукта в синтезе будесонида. Цель - создание нового полупродукта для синтеза эффективного бронхолитика с высоким выходом. Синтез полупродукта ведут ацетализацией 11,21-диацетата 16α - оксипреднизолона масляным альдегидом в растворе CH2Cl2 в присутствии HClO4 (катализатор). Выход 96,5%; т.пл. 118 - 128°С, после перекристаллизации 150 - 160°С; [α]
11,21-Диацетат (22 RS) - 16α , 17 α -бутилидендиокси - 11 β , 21 - - дигидроксипрегна-1,4-диен-3,20-дион формулы 1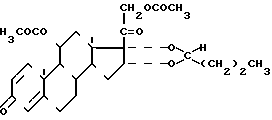
в качестве промежуточного продукта в синтезе будесонида.
| Демченко Б.И | |||
| и др | |||
| Хим | |||
| фарм | |||
| ж., 1979, N 3, p.76-82. |

