Предлагается новый способ получения полиоксистероидов прегнанового ряда общей формулы 1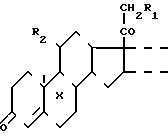
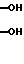 где Х - водород или фтор;
где Х - водород или фтор;
R1 и R2 - гидроксильная группа или один из R1 и R2 - гидроксильная группа, а другой - водород;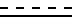 двойная или насыщенная связь между С1 и С2,
двойная или насыщенная связь между С1 и С2,
обладающих ценными фармакологическими свойствами.
Обычно полиоксистероиды прегнанового ряда формулы 1 получают гидролизом их сложных эфиров, имеющих 16 α, 17 α - защищенную в виде кеталя диольную группу, щелочными агентами, например гидроокисями и алкоголятами щелочных металлов [1], [3].
Для получения целевых продуктов 16α, 17 α - защитную кетальную группу далее удаляют кислотным гидролизом в муравьиной кислоте при нагревании [2]. Выход целевых продуктов составляет 60-65%.
Недостатком известного способа является низкий выход целевых продуктов вследствие образования побочных продуктов, что требует дополнительной очистки полученных соединений.
Целью изобретения является увеличение выхода целевых полиоксистероидов прегнанового ряда.
Цель достигается новым способом, заключающимся в том, что 16 α, 17 α - циклоборатные производные сложных эфиров общей формулы 1, где Х имеет указанные значения, а R1 и R2 - ацетилоксигруппа или один из R1 и R2 - ацетилоксигруппа, а другой водород или гидроксил, подвергают щелочному гидролизу с помощью гидроокисей щелочных металлов при 0-25оС в токе азота в среде низшего спирта и воды с последующим кислотным гидролизом полученного продукта обработкой водными растворами минеральных кислот при рН - 3-4. После чего целевой продукт выделяют. Для ускорения процесса щелочного гидролиза циклоборатные производные сложных эфиров формулы 1 желательно обрабатывать избыточным количеством щелочи для ускорения процесса гидролиза.
Преимущество описываемого способа заключается в том, что способ позволяет получить целевые полиоксистероиды с высоким выходом 76-94,6%.
П р и м е р 1. 9 α-фтор-11 β , 16 α ,17α,21-тетраокси-1,4-прегнадиендион-3,20(триамиционолон).
Суспензию 2,04 г (4,27 ммоль) 11,21-диацетата 9 α-фтор-11 β,16 α,17 α , 21-тетраокси-1,4-прегнадиендиона-3,20 в смеси 6,5 мл метанола и раствора 2,02 г (0,53 ммоль) 10-водного тетрабората натрия в 3 мл воды и 0,043 г (1,07 ммоль) гидроокиси натрия в 2 мл воды, свободных от кислорода, нагревают в токе азота до 40-45оС и перемешивают 10 мин. Образовавшийся раствор охлаждают до 0оС, после чего приливают раствор 0,78 г (0,19 ммоль) гидроокиси натрия в 15 мл метанола, свободного от кислорода. Реакционную массу перемешивают при 0-2оС в токе азота в течение 24 ч, подкисляют 10%-ной серной кислотой до рН 3-4, после чего медленно приливают 100 мл охлажденной дистиллированной воды. Выпавший осадок фильтруют, промывают водой до нейтральной реакции и сушат в вакууме при 40оС над пятиокисью фосфора.
Получают 1,31 г (76,5%) 9 α-фтор-11β,16α,17α ,21-тетраокси-1,4-прегнадиендиона-3,20 с т. пл. 250-251оС после очистки из смеси ацетонпетролейный эфир т. пл. 256-257оС. [α]D20 = +60o(C = 1, ДМФА), + 73,6о (С = 0,174, ацетон).
ИК-спектр, см-1:3383 (оксигруппы), 1700 (20-кето), 1665 (3-кето), 1615 (Δ 4 -3-кето), 1605 (Δ1 -3-кето).
УФ-спектр: λмаксСНзОН = 238 нм, ε238 = 14800.
Найдено, %: С 64,06; Н 6,65.
С21Н27FО6
Вычислено, %: C 63,94; Н 6,87,
П р и м е р 2. 16 α,17α ,21-триокси-4-прегнендион-3,20.
Суспензию 0,97 г (2,4 ммоль) 21-ацетата 16 α,17 α,21-триокси-4-прегнендиона-3,20 в смеси 3 мл этанола и раствора 0,114 г (0,3 ммоль) 10-водного тетрабората натрия в 1,9 мл воды и 0,024 г (0,06 ммоль) гидроокиси натрия в 1,3 мл воды, свободных от кислорода, нагревают в токе азота до 40-45оС и перемешивают 10 мин. Образовавшийся раствор охлаждают до 25оС, после чего приливают раствор 0,125 г (3,12 ммоль) гидроокиси натрия в 2 мл воды, свободной от кислорода. Реакционную массу перемешивают при 25оС в токе азота 10 мин и обрабатывают, как указано в примере 1.
Получают 0,82 г (94,6%) 16 α, 17 α -21-триокси-4-прегнендиона-3,20 с т. пл. 202-203оС после перекристаллизации из метанола т. пл. 225,5-226оС. [α] D20 = + 112о (С = 0,1, метанол).
ИК-спектр, см-1: 3400 (оксигруппы) 1720 (20-кето), 1675 (3-кето), 1616 (Δ4 -3-кето).
УФ-спектр, λмаксСНзОН = 241 нм, ε 241 = 16400.
Найдено, %: С 69,30; Н 8,29.
С21Н30О5
Вычислено, %: С 69,58; Н 8,33.
П р и м е р 3. Триамцинолон.
Суспензию 1 г (2,08 ммоль) 11,21-диацетата триамцинолона в смеси 4,4 мл изопропилового спирта и раствора 0,1 г (0,26 ммоль) тетрабората натрия 10-водного и 0,021 г (0,52 ммоль) гидроокиси натрия в 7 мл воды, свободных от кислорода, нагревают в токе азота до 40-45оС и перемешивают 10 мин. Образовавшийся раствор охлаждают до 0оС, после чего к нему приливают раствор 0,3 г (7,5 ммоль) гидроокиси натрия в 2,5 мл воды.
Реакционную массу нагревают до 30-35оС, перемешивают при 30-35оС в токе азота в течение 1,5 ч, охлаждают до 0оС и обрабатывают, как указано в примере 1.
Получают 0,63 г (76,5%) триамцинолона с т. пл. 253-254оС, после очистки ацетоном т. пл. 258-259оС; [α]D20 = + 67 (С = 1, ДМФА); ε1cм1% 381 (λмакс 238, метанол).
П р и м е р 4. Триамцинолон.
А. 16,17-Циклоборатный эфир 11,21-диацетата триамцинолона.
Раствор 1,216 г (2,54 ммоль) 11,21-диацетата триамцинолона и 1,9 г (27,3 ммоль) борного ангидрида в 15 мл метанола кипятят в течение 1 ч, после чего приливают к нему 15 мл воды. Выпавший осадок фильтруют, промывают 2 мл воды и сушат при 50оС.
Получают 1,215 г (95,2% ) 16,17-циклоборатного эфира 11,21-диацетата триамцинолона.
Б. Триамцинолон.
Суспензию 1,215 г (2,41 ммоль) 16,17-циклоборатного эфира 11,21-диацетата триамцинолона, полученного в п. А, в 14 мл метанола охлаждают в токе азота по температуры -10оС, после чего к ней приливают раствор 0,7 г (13 ммоль) метилата натрия в 6 мл метанола, свободного от кислорода. Реакционную массу перемешивают при температуре от -10 до -5оС в токе азота в течение 30 ч, подкисляют 10% -ной серной кислотой до рН 1-2 и обрабатывают, как указано в примере 1.
Получают 0,76 г (80%) триамцинолона с т. пл. 248-250оС, после очистки ацетоном т. пл. 257-258оС; [α]D20 = + 65 (С = 1, ДМФА); ε1см1% 382 (λмакс 238, метанол).
П р и м е р 5. Триамцинолон.
Суспензию 5 г (10,4 ммоль) 11,21-диацетата триамцинолона в смеси 23,9 мл изопропилового спирта и раствора 0,5 г (1,3 ммоль) тетрабората натрия 10-водного и 0,105 г (2,6 ммоль) едкого натра в 25,15 мл воды, свободных от кислорода, нагревают в токе азота до 40-45оС и перемешивают 10-15 мин. Образовавшийся раствор охлаждают до 0оС, после чего к нему приливают раствор 2,02 г едкого натра (4,7 ммоль) в 12,65 мл воды. Реакционную массу перемешивают при 0-2оС в течение 5 ч в токе азота и обрабатывают, как указано в примере 1.
Получают 4,00 г (97,3%) триамцинолона с т, пл, 244-245оС, после очистки ацетоном т. пл. 251,5оС; [α]D20 + 66o (С = 1, ДМФА), ε1см1%382 (λмакс 238, метанол).
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ЭФИРОВ ПОЛИОКСИСТЕРОИДОВ ПРЕГНАНОВОГО РЯДА | 1976 |
|
SU668288A1 |
| Способ получения 16-ненасыщенныхСТЕРОидОВ пРЕгНАНОВОгО РядА | 1979 |
|
SU819119A1 |
| СПОСОБ ПОЛУЧЕНИЯ 6,16 а, 17 а- ТРИМЕТИЛ- А*^ -прЕГНАДИЕНДИОНА-3,20 | 1969 |
|
SU257500A1 |
| 11,21-ДИАЦЕТАТ (22RS)-16α,17α -БУТИЛИДЕНДИОКСИ- 11β,21 -ДИГИДРОКСИПРЕГНА-1,4-ДИЕН-3,20-ДИОН В КАЧЕСТВЕ ПРОМЕЖУТОЧНОГО ПРОДУКТА В СИНТЕЗЕ БУДЕСОНИДА | 1989 |
|
SU1660379A1 |
| СПОСОБ ПОЛУЧЕНИЯ 11 β -ГИДРОКСИСТЕРОИДОВ | 1989 |
|
SU1656870A1 |
| СПОСОБ ПОЛУЧЕНИЯ 6-ХЛОР-16а, 17а-ДИМЕТИЛ-Л-^''' ПРЕГНАДИЕНДИОНА-3,20 | 1969 |
|
SU233662A1 |
| Способ получения 11 @ -гидроксистероидов прегнанового ряда | 1983 |
|
SU1447826A1 |
| 11β -АЦИЛОКСИ- 17α -ГИДРОПЕРОКСИ- 16α -МЕТИЛПРЕГНАНЫ, ОБЛАДАЮЩИЕ МЕСТНОЙ ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 1991 |
|
RU2030421C1 |
| Способ получения 9 @ ,11 @ -оксидо-17 @ ,21-диокси-1,4-прегнадиендиона-3,20 | 1979 |
|
SU937460A1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 19-НОРПРОГЕСТЕРОНА | 1989 |
|
RU2009146C1 |
СПОСОБ ПОЛУЧЕНИЯ ПОЛИОКСИСТЕРОИДОВ ПРЕГНАНОВОГО РЯДА общей формулы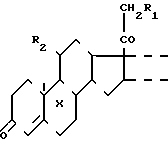
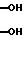
где X - водород или фтор;
R1 и R2 - гидроксильная группа или один из R1 и R2 гидроксильная группа, а другой - водород;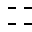

- двойная или насыщенная связь между C1 и C2,
отличающийся тем, что, с целью повышения выхода целевого продукта, 16α , 17α - циклоборатные производные соединения указанной общей формулы, где R1 и R2 - ацетилоксигруппа или один из R1 и R2 - ацетилоксигруппа, а другой водород или гидроксид, подвергают щелочному гидролизу с помощью оснований с последующим кислотным гидролизом полученного продукта обработкой водными растворами минеральных кислот и выделением целевого продукта.
| Allen W | |||
| S., Bernstein S.J | |||
| Am | |||
| Chem | |||
| Soc, 78, 1909, 1956. |
