Предлагаемое изобретение относится к области аналитической химии и может быть использовано для экспресс-анализа технологических растворов, оборотных и сточных вод цветной металлургии
Известен способ фотоколориметрического определения никеля в цинковом купоросе с помощью офурилдиоксима, в
, котором после перевода пробы в раствор и связывания мешающих элементов в нимм- лексы с лимонной кислотой проводят определение никеля в слабощелочной среде при рН9 путем измерения интенсивности окраски растворов эго комплекса с фурилдиоксимом в хлороформе в области длин волн 413-435 НМ.
Недостатком известного способа является чрезвычайно сложная пробоподготов- ка, включающая в частности операции подкисления пробы соляной кислотой, обработки перекисью водорода, выпаривания с последующим растворением солей при нагревании. При этом выпавшие осадки гидрата окиси цинка подвергают растворению с помощью азотной или соляной кислоты. К полученному раствору добавляют растворы лимонокислого натрия, сернистого аммония и er-фуриодиоксима и нейтрализуют аммиа 4 VJ
4 О О СЛ
ком до рН9 по универсальной индикаторной Бумаге. После 15-минутной выдержки раствор переводят в делительную воронку, добавляют воду и хлороформ и экстрагируют в течение 2 мин. После расслаивания хлороформный раствор переводят в следующую воронку, промывают встряхивая с аммиаком. Причем эту операцию проводят 2-3 раза с новыми порциями аммиака до полного обесцвечивания аммиачного раствора. Отмытый хлороформный раствор измеряют оптическую плотность раствора на фотоколориметре, а концентрацию никеля определяют по калибровочному графику,
Столь длительная и трудоемкая пробо- подготовка, предусматривающая использование многих, в том числе токсичных, реактивных, резко снижает оперативность анализа и приводит к его значительному удорожанию.
Известен также способ фотометрического анализа, основанный на экстракции никеля с тиазолилазакрезолом (ТАК) в аммиачной среде. При концентрации 0.01-2 ММН/ЮН образуется комплекс, экстрагирующийся хлороформом, максимум светопог- лощения которого наблюдается при 610 НМ с Молярным коэффициентом погашения 31200. Закон Вера соблюдается при концентрации никеля 0.1-2 мкг/нл.
Способ осуществляется следующим образом. Аликвотную часть анализируемого раствора в 5 нл переносят в делительную воронку, прибавляют 0,4 нл 40%-ного тарт- рата натрия - калия и доводят 2NH40H объем водной фазы до 10 мл. Затем добавляют 1мл 1 10 М хлороформного раствора ТАК и 9 мл, содержимое воронки встряхивают четыре раза в течение минуты с интервалом в 5 мин. Оптическую плотность измеряют на фотоколориметре ФЭК-56 со светофильтром N 8 в кюветах с I 1 см. ,В качестве раствора сравнения используют экстракт реагента. Содержимое никеля находят по калибровочному графику.
Недостатком данного способа является низкая селективность контроля никеля в присутствии мешающих примесей. Так предельно допустимые соотношения концентраций ионов в анализируемом рас-творе
составляют: Ј- 500, - 500, §
CNICNICNI
380. Это практически исключает возможность применения известного способа фотометрического анализа для определения никеля в растворах сульфата цинка, загрязненных медью и кадмием (например, в верхнем сливе нейтрального сгустителя
гидрометаллургического производства цинкового купороса).
Известен способ полярографического определения никеля и меди в продуктах автоклавного производства на фоне 0,1 MNH/jCI + 0,1 MNHMOH после отделения железа фторидом натрия (см. статью: Л.Н.Васильева, Н.Б.Коган, З.Л.Юстус. Исследование условий полярографического оп0 ределения никеля и меди в продуктах автоклавного производства. Заводская лаборатория. Том XXXIX, № 8, 1973 г., с. 924- 926). Использование слабоаммиачного раствора обеспечивает возможность конт5 роля никеля после отделения железа фторидом натрия. Преимуществом электролита 0,1 MNH/iCI + O. является возможность создать в растворе концентрацию ионов NH4 , достаточную для образования
0 аммиаката никеля. Одновременно создается малая концентрация ОН. - при которой не происходит разрушения комплекса FeFee . После связывания железа фторидом возможно определение меди и никеля.
5 К недостаткам известного способа полярографического анализа относится сложная и длительная пробоподготовка и низкая селективность. Так способ обеспечивает проведение анализа никеля при соотноше0 нии Zn:NI 100:1 и Cu:Ni 100:1. что совершенно недостаточно для контроля никельсодержащих растворов гидрометаллургического производства цинкового купороса и электролизного цинка.
5 Наиболее близким по технической сущности и достигаемому результату к заявляемому является способ полярографического определения никеля в стоках гальванического производства на фоне аммиачного
0 буферного раствора, содержащего сульфо- салициловую кислоту.
Определение производят по полярографической волне восстановления ионов никеля до металла на ртутном капельном
5 электроде в растворе
0.6MNH4 ОН + 0.3MNH4CI + + 0,02МНОз5(НО)СбН3СООН 2Н20, Использование аммиачного буферного электрслита, содержащего сульфосалици0 ловую кислоту обеспечивает возможность количественного определения ионов никеля в присутствии ионов меди, кадмия и цинка. Способ осуществляется следующим образом.
5 Пробу сточных вод помещают в делительную воронку на объемом 500 или 1000 мл, добавляют 3 капли раствора метилового рранжевого и подкисляют раствором 0.5 MHCI до появления красного цвета. Эту операцию не производят, если раствор был
предварительно подкислен. Затем добавляют твердый ацетат аммония до приобретения раствором желтой окраски и добавляют 5 мм 1 %-ного раствора ДДТС-Na (диэтилди- тиокарбоната натрия). После чего дважды экстрагируют с четыреххлористым углеродом (с порциями ССМпо 10мл). Выпаривают связанный с органическими соединениями ССЦ и добавляют по 5 мл концентрированной азотной и хлорной кислоты. Кипятят пробу в стаканчике под часовым стеклом в течение 10 мин. Удаляют избыток кислоты путем выпаривания в меньшем объеме, разбавляют водой и снова выпаривают. Доводят объем до 5 мл водой, добавляют 2 мл 96%-ного этилового спирта и нагревают до кипения. Переносят пробу в мерную колбу на 25 мл и доводят объем раствора до метки водой (раствор 1).
В мерную колбу на 10 мл помещают примерно 40 мл сульфосалициловой кислоты, вводят с помощью пипетки 5 мл раствора 1, добавляют 4 мл аммиачного буферного электролита (0,6 MNH/jOH + 0,3 MNH4CI) и 0,1 мл 0.5% раствора крахмала. Затем до- бавляют 40 нг сульфита натрия и доводят объем до метки водой. Если после тщательного перемешивания появится осадок, пробу необходимо подвергнуть центрифугированию со скоростью 12000 об/мин в течение 3-х минут, а очищенный раствор использовать для полярографического анализа. При этом помещают часть раствора в полярографическую ячейку и регистрируют волны (классические, полярог- ранные) меди, кадмия, никеля и цинка в диапазоне потенциалов от - 0,15 до - 1.60В относительно насыщенного каломельного электрода.
Концентрацию никеля в растворе опре- деляют по калибровочному графику.
Недостатком известного способа является низкая избирательность определения никеля в присутствии ионов цинка и кадмия, связанная с близостью потенциалов восста- новления указанных катионов на фоне
0,6 ММНлОН + 0,3 MNH4CI +
+ 0,02 МНОз5(НО)СеНзСООН 2Н20 Потенциалы полуволн восстановления ионов Cd2+; Ni2+; Zn2+ на данном фоне соот- ветственно равны -0,76В; - 1,05В и- 1,32В (отм. Н.К.Э).
Причем, возможности повышения селективности анализа в существенной мере ограничены из-за невозможности использо- вания ваналитических целях высокоизбирательных методов переменнотоковой полярографии. Это связано с необратимостью восстановления ионов никеля на ртутном электроде при игпользовании аммиачного буферного электролита с добавлением сульфосалициловой кислоты.
Кроме того, существенным недостатком известного способа является сложная и длительная пробоподготовка, включающая, в частности, операции кипячения, выпаривания и экстрагирования при проведении которых возможны количественные потери контролируемого вещества. Применение при подготовке проб к анализу концентрированных минеральных кислот и высокотоксичного четыреххлористого углерода вызывает необходимость тщательного соблюдения правил техники безопасности и создания специальных условий при проведении аналитических процедур. Все это в существенной мере осложняет использование известного способа для оперативного контроля содержания никеля в растворах сульфата цинка непосредственно в промышленных условиях предприятий цветной металлургии.
Целью изобретения является повышение избирательности анализа.
Поставленная цель достигается тем, что согласно способу вольтамперометрическо- го определения концентрации никеля в растворах сульфата цинка, включающему наложение линейно-изменяющегося потенциала на ртутный индикаторный электрод и регистрацию вольтамперной кривой, в контролируемый раствор вводят 100-200 г/л металлической цинковой пыли, перемешил вают полученную пульпу в течение 30-60 с, фильтруют, добавляют к фильтрату раствор роданида калия и проводят регистрацию на фоне (1,5-2) MKCNS в интервале потенциалов от - 0,4 до 0,8В, а концентрацию никеля определяют по высоте пика при потенциале (-0.64±0.05)В.
В современной гидрометаллургии для очистки растворов сульфата цинка от примесей тяжелых цветных металлов широко используется метод цементации металлическим цинком в основе которого лежит электрохимическая реакция
Ме2+ + Zn - Me + Zn2+
При этом металл - примесь переходит из ионного состояния в металлическое, а цинк из металлического в ионное.
Причем, если медь и кадмий выделяются из раствора легко, то никель осаждается значительно труднее. Такое различие электрохимического поведения ионов тяжелых металлов в процессе цементации может быть использовано для освобождения контролируемых растворов сульфата цинка от примесей меди и кадмия, мешающих воль- тамперометрическому определению ионов никеля.
После серии специально проведенных экспериментов были выбраны условия процесса цементации при которых достигается высокая эффективность осаждения меди и одновременно полностью сохраняется исходная концентрация ионов никеля.
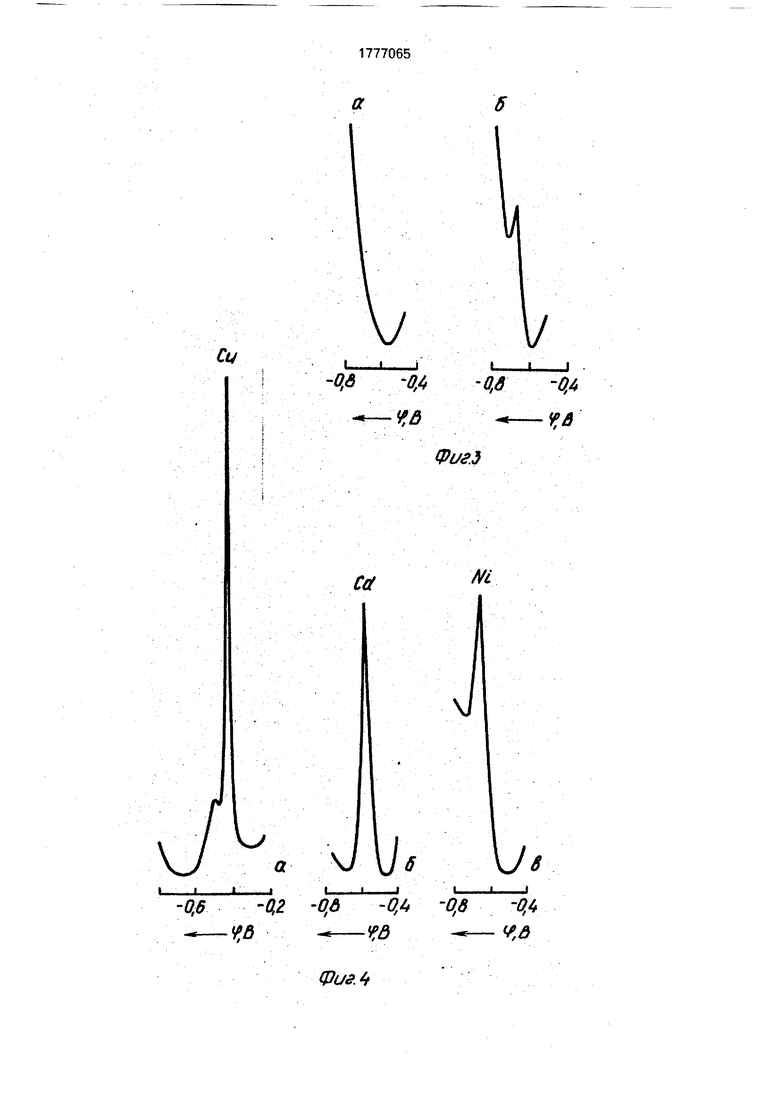
На фиг.1 представлены дифференциальные импульсные кривые, иллюстрирующие влияние процесса цементации на определение ионов никеля в растворах сульфата цинка:
а - концентрация ионов Zn ; Ni Си ; соответственно равны 100000; 10; 0 и О мг/л. Раствор сульфата цинка не подвергался цементации,
б - концентрация ионов Zn24; Ni 4; Си 4; соответственно равны 100000; 10; 0 и О мг/л. Раствор сульфата цинка подвергался цементации,
в - концентрации ионов Zn24: Ni24 соответственно равны 100000; 10; 4000 и 2000 мг/л. Раствор подвергался цементации,
г - концентрации ионов Zn24; Ni24 соответственно равны 100000; 0; 4000 и 2000 мг/л. Раствор сульфата цинка подвергался цементации.
Полярограммы зарегистрированы на разработанном СКФ ОНТК СоюзЦМА вольтамперометрическом анализаторе АЖЭ-11 с рабочим электродом типа висящая капля.
Как видно из фиг.1а, б., на которой изображены дифференциальные импульсные полярограммы никеля (10 мг/л) в растворе сульфата цинка (100 г/л Zn24), снятые соответственно до и после агитации с цинковой пылью, ионы NI металлическим цинком не осаждаются.
Было установлено (см. фиг.1 в), что высота пика никеля (10 мг/л) остается неизменной при введении в подвергаемый обработке цинковый раствор сульфата цинка ионов меди (4000 мг/л) и кадмия (2000 мг/л). При этом на вольтамперной кривой подвергшегося цементации раствора сульфата цинка с аналогичным содержанием меди и кадмия (см. фиг.1г) не было зафиксировано каких-либо следов указанных элементов, что свидетельствует о высокой эффективности процесса очистки;
В результате проведенных экспериментов было показано, что предельно допустимые соотношения ионов тяжелых цветных металлов в растворах сульфата цинка, подвергаемых цементации в целях последующего волыамперометрического анализа на содержание никеля, составляют, соответственно
Сси
200 и
Ccd
1000. Этообеспе0
CNICNI
чивает возможность контроля наиболее сложных по составу технологических растворов, в том числе верхнего слива нейтрального сгустителя (ВСНС) цинковых заводов, содержащего до 1000 мг/л ионов Cd24 и до 2000 мг/л ионов Си24,
При этом для проведения процесса це0 ментации в оптимальном режиме необходимо поддерживать удельный расход цинковой пыли в пределах 100 - 200 г/л, а перемешивание пульпы производить в течение 30-60 с, При расходе цинковой пыли
5 ниже 100 г/л и времени агитации менее 30 с в растворах ВСНС, содержащих до 2 г/л ионов Си24 и 1 г/л ионов Cd24, после цементации обнаруживались следовые количества этих металлов. Увеличение расхода цинковой пыли свыше 200 г/л, а длительность агитации сверх 60 с не приводит к сколь-нибудь заметному улучшению степени очистки растворов сульфата цинка от примесей меди и кадмия. В связи с чем
5 выход за верхний предел удельного расхода цинковой пыли приводит к неоправданному перерасходу данного реагента. Кроме того, с увеличением расхода цинковой пыли несколько ухудшаются условия последующей
0 фильтрации проагитированной пульпы. Нецелесообразно также увеличение длительности перемешивания раствора с цинковой
пылью свыше 60с ввиду связанных с этим потерь времени на предварительную пробо- подготовку.
Данные граничные значения удельного расхода цинковой пыли и длительности процесса цементации были установлены при скорости вращения мешалки равной 200 об/мин. Однако, как показали исследования, данный метод очистки растворов сульфата цинка от примесей тяжелых металлов в незначительной степени зависит от интенсивности перемешивания пульпы. Таким об5 разом, использование метода цементации для пробоподготовки контролируемых растворов обеспечивает практически полное их освобождение от .примесей меди и кадмия и тем самым позволяет свести аналитиче0 скую задачу к обеспечению необходимой селективности вольтамперометрического определения никеля в присутствии избыточных количеств цинка.
Проведение исследования показали,
5 что наибольшая избирательность анализа может быть достигнута при использовании в качестве фонового электролита роданида калия. Установлено, что на фоне (1,5-2) MKCNS возможно прямое вольтамперомет5
0
рическое определение никеля при соотношении концентрации ионов в растворе Czn
См
1000.
При этом нижний предел обнаружения никеля в растворах сульфата цинка, содержащего 100 г/л Zn2 , составляет 1 мг/л. Столь высокая разрешающая способность вольтамперометрического измерения была обеспечена за счет значительной разности потенциалов восстановления ионов никеля и цинка, достигающей 400 мВ.
На фиг.2 представлена дифференциальная импульсная полярограмма никеля (100 мг/л) и цинка (100 мг/л) на фоне 2 MKCNS, показывающая положение пиков восстановления ионов Nf2+ и Nn2+ на шкале потенциалов. На фиг.З соответственно приведена дифференциальная импульсная полярограмма никеля (1 мг/л) в растворе сульфата цинка на фоне 2MKCNS, иллюстрирующая возможности вольтамперометрического измерения ионов Ni + и Zn + на уровне нижнего предела обнаружения при соотношении
Czn CNJ
100000
а - концентрации ионов Zn + и со
ответственно равны 100000 и 0 мг/л
б - концентрации ионов Zn2+ и Nl2+ соответственно равны 100000 и 1 мг/л,
Оптимальная концентрация фонового электролита составляет 1,5-2 MKCNS.
Выход за указанные пределы не целесообразен, т.к. при концентрации фона менее 1,5 м снижается избирательность анализа никеля в присутствии избыточных содержаний цинка и, например, при проведении измерения в растворе 1 MKCNS предельно
л
допустимое соотношение -- 40000,
v Ni
концентрации фона более 2 MKCNS ведет к неоправданному перерасходу реактива без сколько-нибудь заметного повышения селективности контроля. Кроме того, было замечено, что при увеличении концентрации фонового электролита свыше 2 MKCNS происходит выпадение кристаллических осадков, загрязняющих электрохимическую ячейку и электродную систему датчика.
Таким образом, использование в заяв- ляемом способе вольтамперометрического измерения никеля в среде роданида калия в сочетании с предварительной обработкой анализируемого раствора металлической цинковой пылью создает все необходимые условия для проведения избирательного анализа в высококонцентрированных растворах сульфата цинка загрязненных примесями меди и кадмия.
5 10
15 0
5
Необходимость использования метода цементации для удаления из раствора мешающих примесей иллюстрируется с помощью фиг.4, на которой соответственно представлены дифференциальные импульсные полярограммы а - меди (20 мг/л). б - кадмия (40 мг/л) и в - никеля (100 мг/л) в растворе сульфата цинка (100 г/л Zn) на фо- не 2 MKCNS. Как видно из фиг.4, потенциалы пиков восстановления ионов NI2+ и Cd2+ соответственно равны (- 0,64) и (-0,61)8, т.е. практически совпадают. Двухступенчатое восстановление меди на этом фоне сопровождается появлением двух пиков при потенциалах (- 0,37) и (-0,50)8. Причем, из-за большой ширины второго пика определение низких концентраций никеля (5-10 мг/л) в присутствии значительных избытков меди (например, в растворах ВСНС содержащего до 2000 мг/л ионов Си24) становится невозможным.
Предлагаемый способ обеспечивает прямое вольтамперометрическое определение никеля в растворах сульфата цинка с нижним пределом обнаружения 1 мг/л при следующих соотношениях концентраций ионных компонентов:
Czn
CNI
100000;
Ccu
2000;
Ccd
1000.
См CNI
Способ может быть реализован на различных отечественных и зарубежных полярографических анализаторах.
Измерение производится в трехэлект- родной электрохимической ячейке. Электрод индикаторный - висящая ртутная капля, электрод сравнения хлор-серебряный, например, типа ЭВЛ-1МЗ, вспомогательный электрод - стеклоуглеродный. Скорость развертки потенциала -(5 - 10) мВ/с. Амплитуда импульсного напряжения 20-40 мВ.
Способ осуществляется следующим образом. К 50 мл анализируемого раствора, например ВСНС, содержащему до 1000 мг/л ионов кадмия и до 2000 мг/л ионов меди, добавляют 5-10 г/л металлической цинковой пыли. Полученную смесь перемешивают в течение 30-60 с с помощью механической мешалки и фильтруют на вакуум-воронке через бумажный фильтр. К 5 мл фильтрата добавляют 20 мл фонового электролита 2,5 MKCNS, а затем после перемешивания заливают в электрохимическую ячейку и полярографируют в интервале потенциалов (-0,4) - (-0,8)В, регистрируя при этом вольтамперную кривую (катодный пик) никеля. Содержание ионов никеля в анализируемом растворе определяют по высоте пика (величине предельного тока) при потенциале - 0,64В относительно хлорсереб- ряного электрода сравнения с использованием градуировочного графика или метода добавок стандартного раствора.
При этом общая длительность анализа, включая пробоподготовку, не превышает 3 мин. Использование разработанных СКФ Союзцветметавтоматика вольтамперо- метрических анализаторов типа АЖЭ-11, АЖЭ-12. АЖЭ-13, обеспечивает проведение всех стадий измерительного процесса в автоматическом режиме с выдачей выходного сигнала на вторичный прибор отградуированный в единицах концентрации контролирующего вещества (никеля).
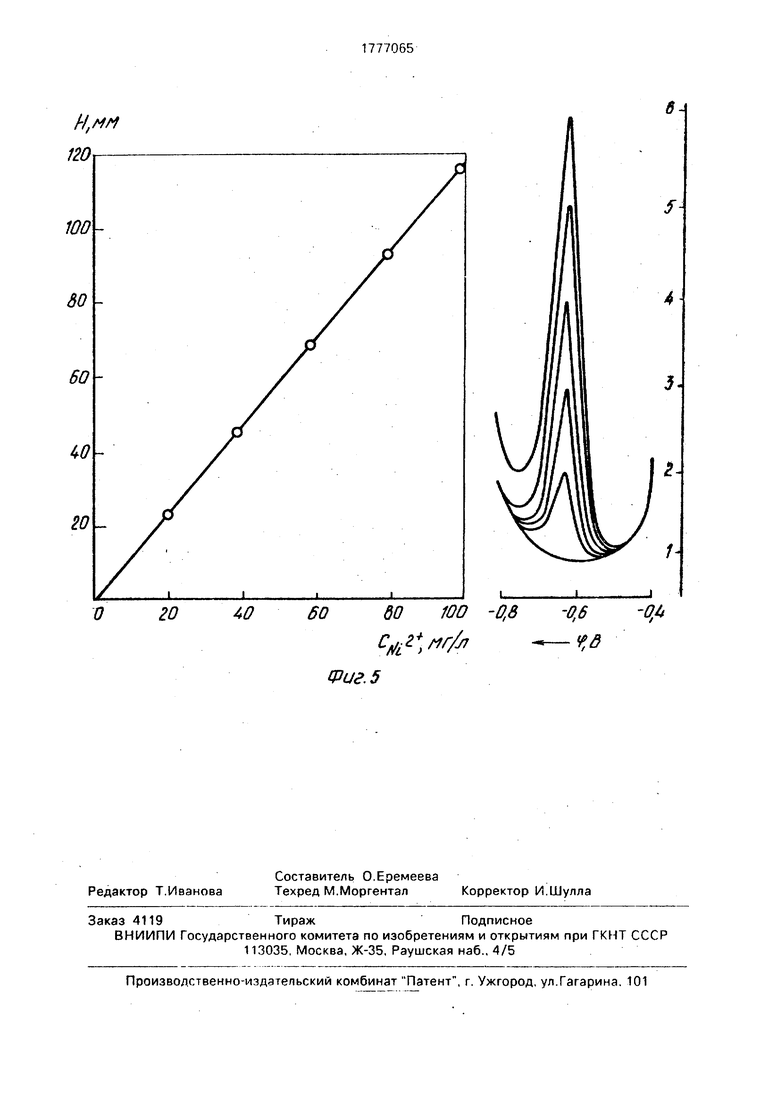
Количественная зависимость величины, тока восстановления (высоты пика) никеля от содержания его в анализируемом растворе сульфата цинка пропорциональна в диапазоне содержаний 0-100 мг/л.
На фиг.5 приведены градуировочный график для определения никеля в диапазоне концентрации 0-100 мг/л, и иллюстрирующие его дифференциальные импульсные полярограммы восстановления ионов NI2(1 - фон; 2 и 6 концентрации ионов никеля соответственно равны 20, 40, 60, 80 и 100 г/л). Таким образом заявляемый способ обеспечивает вольтамперометрический анализ растворов, содержащих 1-100 мг/л никеля. Растворы с концентрацией никеля превышающей 100 мг/л, перед анализом необходимо разбавлять. Нижний предел измерения может быть значительно расширен при использовании инверсионных методов вольтамперометрии.
Использование вольтамперометриче- ского анализатора АЖЭ-11 обеспечивает проведение в автоматическом режиме всех стадий анализа, включая фильтрацию, смешение в фильтрате в заданном соотношении с фоновым электролитом, доставку полученной смеси в электрохимическую ячейку датчика, измерение содержания никеля с выдачей сигналов на вторичные приборы, отградуированные в единицах концентрации контролируемого вещества.
Предполагаемый способ легко реализуется с помощью легкодоступных, недорогостоящих реактивов с помощью стандартного оборудования. Раствор роданида калия неагрессивен, устойчив при длительном хранении, что в существенной мере снижает уровень требований к аппаратному оформлению и обеспечивает высокую эксплуатационную надежность дозирующих
устройств системы автоматической пробо- подготовки и вольтамперометрического датчика.
Способ может быть использован для автоматизированного вольтамперометрического определения никеля в гидрометаллургических растворах предприятий цветной металлургии. В особенности перспективного применения заявляемого способа для контроля непрерывных технологических процессов производства цинкового купороса и электролиза цинка.
Способ может быть также применен для определения никеля в сточных и оборотных водах предприятий цветной металлургии,
что позволит снизить выбросы токсичных веществ в окружающую среду за счет повышения оперативности и качества анализа на очистных сооружениях.
Формула изобретения
Способ вольтамперометрического определения концентрации никеля в растворах сульфата цинка, включающий наложение линейно изменяющегося потенциала на ртутный индикаторный электрод и регистрацию вольт-амперной кривой, отличающийся тем, что, с целью повышения избирательности анализа, в контролируемый раствор вводят 100-200 г/л металлургической цинковой пыли, перемешивают полученную пульпу в течение 30-60 с, фильтруют, добавляют к фильтру раствор роданида калия и проводят регистрацию на фоне 1,5 - 2,0 М KCNS в интервале потенциалов
-0,4 ... -0,8В, а концентрацию никеля определяют по высоте пика при потенциалах - 0,64 + 0,058.






Область применения, аналитическая химия, экспресс-анализ технологических растворов, оборотных и сточных вод цветной металлургии. Сущность изобретения: к анализируемому раствору, например 8СНС, добавляют металлическую цинковую пыль. Полученную смесь перемешивают 30-60 с с помощью механической мешалки и фильтруют на вакуум-воронке через бумажный фильтр, к 5 мл фонового электролита добавляют 2.5 М KCNS, а затем, после перемешивания, заливают в электролитическую ячейку и полярографируют в интервале потенциалов (-0,4 - (-0,8) В, регистрируя при этом вольт-амперную кривую (катодный пик) никеля. Содержание никеля в анализируемом растворе определяют по высоте пика (величине предельного тока) при потенциале -6,64 В относительно хлорсе- ребряного электрода сравнения с использованием градуировочного графика или метода добавок стандартного раствора. При этом общая длительность анализа, включая пробоподготовку, не превышает 3 мин. 5 ил. w Ё
IIiIiiI
0,6 -0,4 -0,4 -0,4 -0,8 -0,4 Q,& -0,4 - -#9--if Ј-
-1,0 0,8 -Ofb V,d
Фиг.г
jI
Фиг.1
Ni
i
ii
- -44 -0,6 -0,4
-У,
Фигз
-0,6 -0,2 -if Л
-ОА
фиг.Ь
ii
-о,в
-W 4,д
20 О
20406060 ЮО -0,8 -0,6
Фиг. 5
б
4
J.
/W
| Васильева Л.Н | |||
| и др Исследование условий полярографического определения никеля и меди в продуктах актоклавного производства | |||
| - Заводская лаборатория, т | |||
| XXXIX, № 8, 1973 г., с | |||
| ПНЕВМАТИЧЕСКИЙ НАСОС ДЛЯ ПОДЪЕМА ЖИДКОСТЕЙ ИЗ ГЛУБОКИХ КОЛОДЦЕВ | 1924 |
|
SU924A1 |
| I | |||
| Gollmowske и др | |||
| Chemlca analltyezna (PRL), 1974 г., t.19, s 1155-1164. | |||
