Изобретение относится к аналитической химии и может быть использовано для автоматического или экспресс-анализа технологических растворов, оборотных и сточных вод цветной металлургии и других отраслей промышленности.
Известен способ вольтамперометрического определения перманганат-ионов на пиролитическом графитовом электроде (см. статью: Miller F.J., Zittel H.E. Voltammetry of Ce(IV), Mn(VII), Cr(VI), and V(V) with the pyrolytic graphite electrode // J. Electroanal. Chem. 1964. 7. P. 116-122).
Сущность известного способа состоит в вольтамперометрическом определении ионов МnО- 4 на пиролитическом графитовом электроде в деарированной
1М H2SО4 по высоте катодной волны при Е1/2=-0,95 В (отн. н.к.э.).
Способ осуществляют следующим образом.
Приготовленный на фоне 1 М Н2SО4 анализируемый раствор помещают в электрохимическую ячейку и деарируют потоком аргона. Регистрируют вольтамперную кривую на пиролитическом графитовом электроде с площадью рабочей поверхности 0,28 см2 в интервале напряжений от 1,1 до 0,8 В со скоростью катодной развертки потенциала 0,1 В/мин. Измерения производят относительно насыщенного каломельного электрода, связанного с ячейкой через электролитический мостик, заполненный насыщенным раствором КNО3. Концентрацию перманганат-ионов в растворе определяют по высоте катодной волны (величине диффузионного тока), характеризующейся величиной E1/2=-0,95 В. Диапазон измеряемой концентрации ионов Mn(VII) от 1 до 10-4М/л. Каждое измерение производят на новом или тщательно очищенном пиролизитовом графитовом электроде.
Недостатком известного способа является очень плохая воспроизводимость результатов анализа, связанная с образованием при восстановлении перманганат-ионов двуокиси марганца, адсорбирующейся на поверхности рабочего электрода. Колебания величины диффузионного тока объясняются авторами известного способа изменениями толщины слоя МnО2 на поверхности пиролизитового графитового электрода. Для достижения хотя бы минимальной воспроизводимости необходимо тщательно чистить рабочий электрод после каждого измерения (восстановления перманганат-ионов) путем промывки хромовой кислотой с последующей обработкой аммиачной водой.
Известен также способ вольтамперометрического определения перманганат-ионов на золотом электроде (см. статью: Huber С.О. Voltammetric determination of permanganate at the gold electrode // Analytical Chemistry. 1964. V. 36. 9. Р. 1873-1874).
Способ осуществляется следующим образом.
Анализируемую пробу обрабатывают хлорной или азотной кислотой таким образом, чтобы при проведении измерений концентрация кислоты поддерживалась в пределах от 1 до 3 N. При анализе органических проб производят мокрое озоление смесью хлорной и азотной кислот. Раствор, содержащий ионы марганца (II), нагревают с добавлением периодата калия и кипятят в течение 5-10 мин до полного окисления Мn2+ до МnO- 4. Для предотвращения восстановления перманганата водой, характерного в некоторой степени для разбавленных проб, в раствор вводят избыток периодата калия. После разбавления раствора до требуемого объема производят вольтамперометрическое измерение концентрации. Анализ осуществляют в ячейке, представляющей собой 150 мл лабораторный стакан. В качестве рабочего используют конвекционный золотой электрод, вращающийся со скоростью 600 об. /мин с помощью синхронного электродвигателя. Конструкция электрода исключает утечку электролита на контактные проводники.
Для предотвращения восстановления перманганата хлорид-ионами в качестве электрода сравнения используют ртутно-сульфатный электрод, связанный с ячейкой с помощью солевого мостика, заполненного 1N раствором хлорной кислоты. Измерения производят после добавления в вольтамперометрическую ячейку 20,0 мл 1 или 3N хлорной или азотной кислоты в качестве фонового электролита (фон предварительно кипятят с периодатом калия для удаления способных к восстановлению примесей сопутствующих веществ). Вращающийся золотой электрод предварительно поляризуют при потенциале +2,5 В (отн. н.к.э.) в течение 15 с, а затем при потенциале 0,0 В в течение 5 с. После чего выдерживают рабочий микроэлектрод при потенциале +0,80 В примерно 1 минуту до снижения остаточного тока до 0,1 мкА, вводят в ячейку аликвотную часть анализируемого раствора (1-10 мл) и измеряют величину тока, пропорционального концентрации перманганат-ионов. Затем вводят в электрохимическую ячейку добавку стандартного раствора МnO- 4 и по приросту тока рассчитывают концентрацию марганца в анализируемом растворе. Раствор, используемый для стандартных добавок, готовят путем восстановления основного стандартного раствора перманганата сульфитом натрия, с последующим кипячением для удаления остатков Nа2SO3 и окислением до перманганата по методике, используемой при подготовке проб к анализу.
Недостатком известного способа является нестабильность поверхности вращающегося золотого электрода вследствие ее окисления перманганатом. Состав (природа) поверхности рабочего электрода, а следовательно, измеряемый ток, изменяются во времени. Это вызывает необходимость применения метода стандартных добавок при проведении аналитических измерений. Достаточно сложная методика пробоподготовки и самой процедуры вольтамперометрического измерения затрудняют автоматизацию анализа и снижают его оперативность.
Наиболее близким по технической сущности к заявляемому является способ полярографического определения перманганат-ионов на ртутном капельном электроде (см. статью: Сонгина О.А., Рождественская З.Б. Полярографическое восстановление иона перманганата на платиновом и ртутном электродах // Журнал аналитической химии. 1956. Т.П. Вып.6. С. 717-722).
Способ осуществляют следующим образом.
Сернокислый раствор (0.1-12 н. H2SО4), содержащий 10-3-10-4 N ионов МnО- 4, обескислороживают в течение 20-60 мин путем продувки водорода, а затем полярографируют на ртутно-капельном электроде, регистрируя катодную волну, площадка предельного диффузионного тока которой лежит в области потенциалов от +0,1 до -1,0 В (отн. н.к.э.). Концентрацию перманганат-ионов в анализируемом растворе определяют по высоте полярографической волны.
Известный способ имеет ряд недостатков, наиболее существенным из которых является неселективность анализа. Авторами экспериментально доказано, что регистрируемая волна обусловлена восстановлением ионов ртути, образующихся при окислении металлической ртути перманганатом. В этой связи аналогичная волна регистрируется на ртутно-капельном электроде в присутствии любого достаточно сильного окислителя, например бихромата калия. Таким образом, при наличии в анализируемом растворе нескольких окислителей полярографом будет сниматься суммарная волна, обусловленная восстановлением катионов ртути, образовавшихся под воздействием всех присутствующих в растворе окислителей. Кроме того, растянутая по шкале потенциалов площадка предельного тока (от +0,1 до -1,0 В) регистрируемой катодной волны неизбежно приводит к снижению селективности полярографического анализа из-за возможности восстановления на ртутном капельном электроде целого ряда ионов сопутствующих веществ. Например, по данным работы: Пац Р.Г., Васильева Л.Н. Методы анализа с использованием полярографии переменного тока. М.: Металлургия, 1967. С. 100-101, потенциалы восстановления ионов Cu2+, Sn2+, Pb2+, Tl2+, Cd2+ на ртутном электроде в фоновом электролите 1М H2SO4 соответственно равны: -0,2; -0,4; -0,51; -0,61; -0,73 В (концентрация элементов 10-4 М).
Высота волны восстановления ионов ртути, образующихся при взаимодействии металлической ртути с ионами перманганата, существенно зависит от концентрации серной кислоты. Это приводит к необходимости стабилизации концентрации фонового электролита строго на заданном уровне во избежание снижения точности полярографического определения перманганат-ионов.
К числу обязательных условий полярографического анализа ионов МnО- 4 на ртутном электроде согласно известному способу относится обескислороживание контролируемого раствора перед регистрацией полярографической волны. Это не только усложняет процедуру анализа, но и значительно снижает его оперативность.
Указанные недостатки известного способа практически исключают возможность его применения для оперативного избирательного контроля перманганат-ионов в сложных по составу технологических растворах, оборотных и сточных водах предприятий цветной металлургии.
Целью предлагаемого изобретения является повышение избирательности, точности и оперативности анализа.
Поставленная цель достигается тем, что согласно способу вольтамперометрического определения перманганат-ионов в растворах сульфата цинка на ртутном индикаторном электроде, включающем регистрацию катодной вольтамперной кривой, полярографирование ведут на фоне 2М NH4Cl+2М NH4OH+2•10-3 М диметилглиоксима +2•10-3 М ЭДТА (этилендиаминтетрауксусная кислота) в диапазоне напряжений от -0,70 до -1,10 В, а концентрацию перманганат-ионов измеряют по высоте пика при потенциале -0,98±0,01 В.
В результате проведения патентных исследований не было выявлено каких-либо известных способов вольтамперометрического определения перманганат-ионов, предусматривающих полярографирование на фоне 2М NH4Cl+2М N4UOH+2•10-3 М диметилглиоксима + 2•10-3 М ЭДТА в диапазоне напряжений от -0,70 до -1,10 В, с измерением концентрации перманганат-ионов по высоте пика при потенциале -0,98±0,01 В, т.е. имеющих признаки, сходные с признаками, отличающими заявляемый способ от прототипа.
Перманганат-ион в хлоридно-аммиачной среде не дает самостоятельной полярографической волны, однако, как нами установлено, при дополнительном введении в состав фонового электролита диметилглиоксима при проведении анализа методом дифференциальной импульсной полярографии (ДИП) на стационарном ртутно-капельном электроде (СРКЭ) регистрируется четко выраженный пик, пропорциональный концентрации Mn(VII) в анализируемом растворе.
В щелочной среде перманганат-ион восстанавливается до двуокиси марганца по реакции:
MnO4 -+4OH--->MnO2+2H2O+O2. (1)
Есть основания полагать, что МnO2 при взаимодействии с диметилглиоксимом образует комплекс типа:
Полученные экспериментальные данные свидетельствуют о том, что комплекс двуокиси марганца с диметилглиоксимом обратимо восстанавливается на СРКЭ в хлоридно-аммиачной среде, в то время как Mn(VII) в этих условиях не дает аналитического сигнала.
На фиг. 1 представлены катодная (1) и анодная (2) ДИП кривые Mn(VII) 10 мг/л на фоне 0,8М NH4Сl+0,8М NH4ОH+2•10-3 М ДМГ.
На фиг.2 представлены ДИП кривые Mn(VII) на фоне 2М NН4ОН+2М NH4Cl+2•10-3 М ДМГ, снятые до и после введения в анализируемый раствор Mn(ll)
1 - фон
2 - СMn(VII) =10 мг/л; СMn(II) =0;
3 - СMn(VII) =10 мг/л; СMn(II) =40 мг/л.
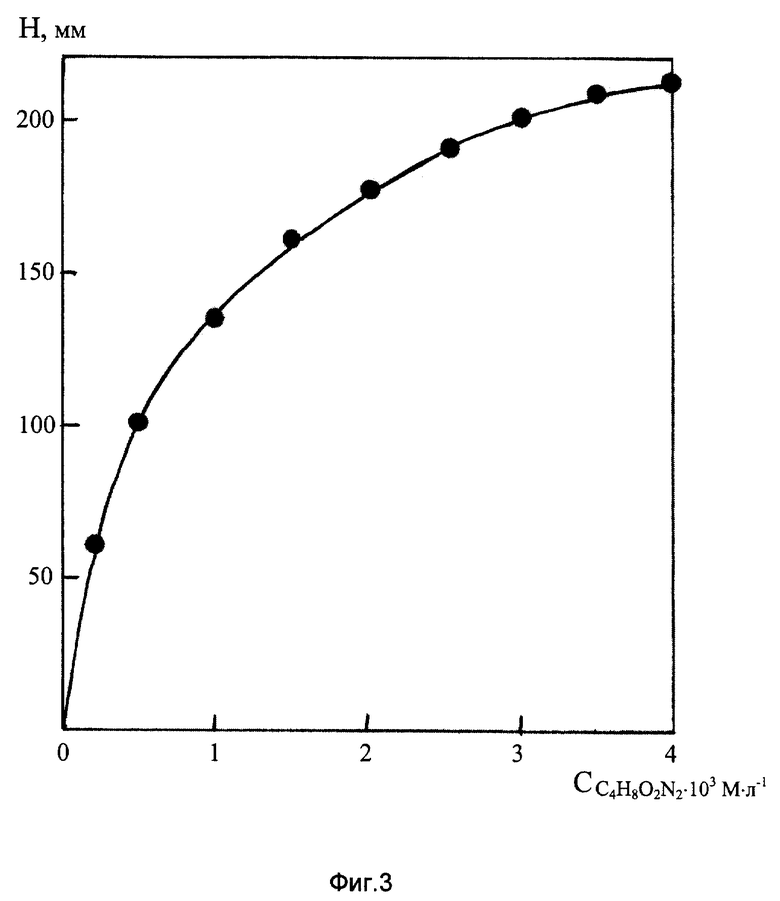
На фиг. 3 представлена зависимость высоты ДИП пика Mn(VII) 10 мг/л на фоне 2М NH4Cl + 2М NH4OH от концентрации диметилглиоксима.
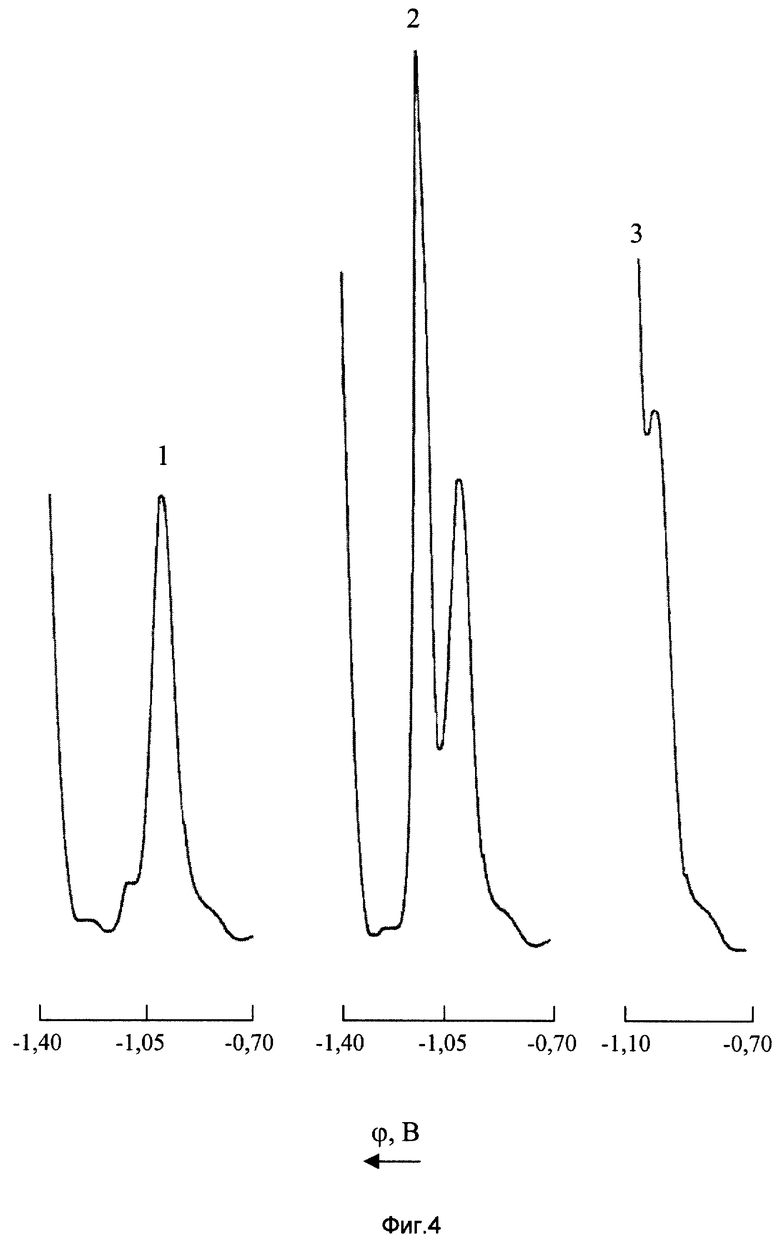
На фиг. 4 показано влияние концентрации цинка на полярографическое определение марганца (VII) на фоне 0,1М NH4Cl+0,1М NH4OH + 2•10-3 М ДМГ
1 - CMn(VII)=10 мг/л; CZn(II)=0;
2 - CMn(VII)=10 мг/л; CZn(II)=10 мг/л
3 - СMn(VII)=10 мг/л; CZn(II)=100 мг/л.
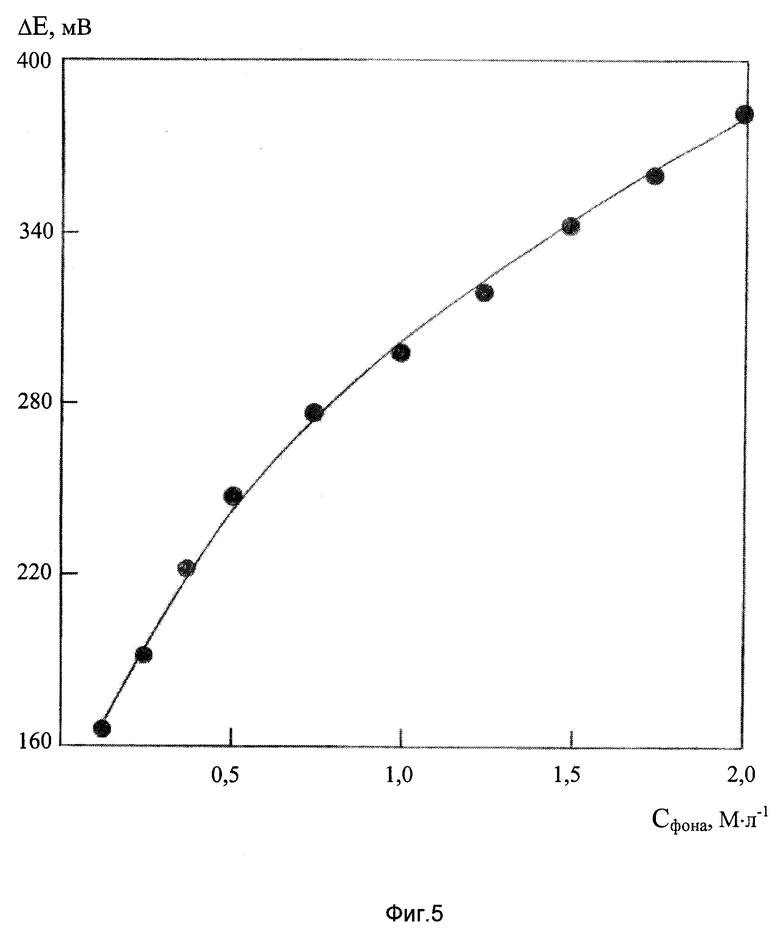
На фиг.5 представлена зависимость разности потенциалов ΔЕ, мВ ДИП пиков Mn(VII) 10 мг/л и Zn((II)) 10 мг/л от концентрации хлоридно-аммиачного фонового электролита. 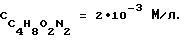
На фиг.6 представлена зависимость высоты катодных пиков Mn(VII) 10 мг/л (кривая 1) и Zn((II)) 10 мг/л от концентрации хлоридно-аммиачного фонового электролита. 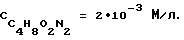
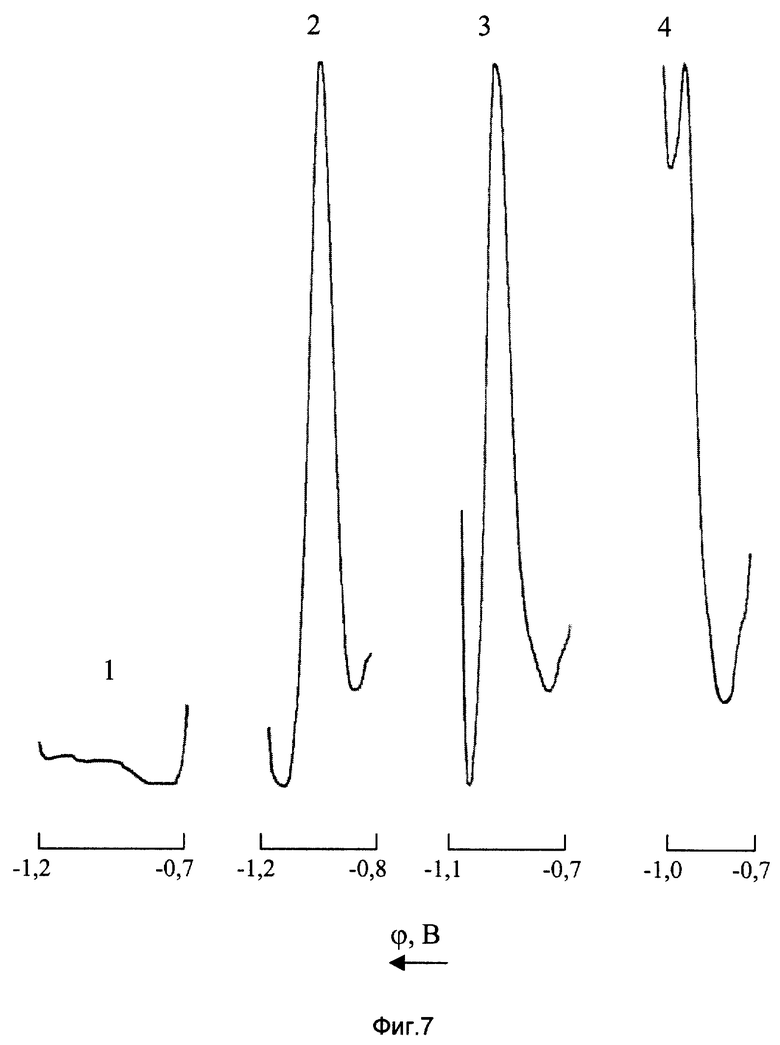
На фиг. 7 показано влияние концентрации цинка на полярографическое определение марганца (VII) на фоне 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ
1 - фон
2 - СMn(VII)=2 мг/л; CZn(II)=0;
3 - СMn(VII)=2 мг/л; CZn(II)=12000 мг/л
4 - CMn(VII)=2 мг/л; CZn(II)=16000 мг/л.
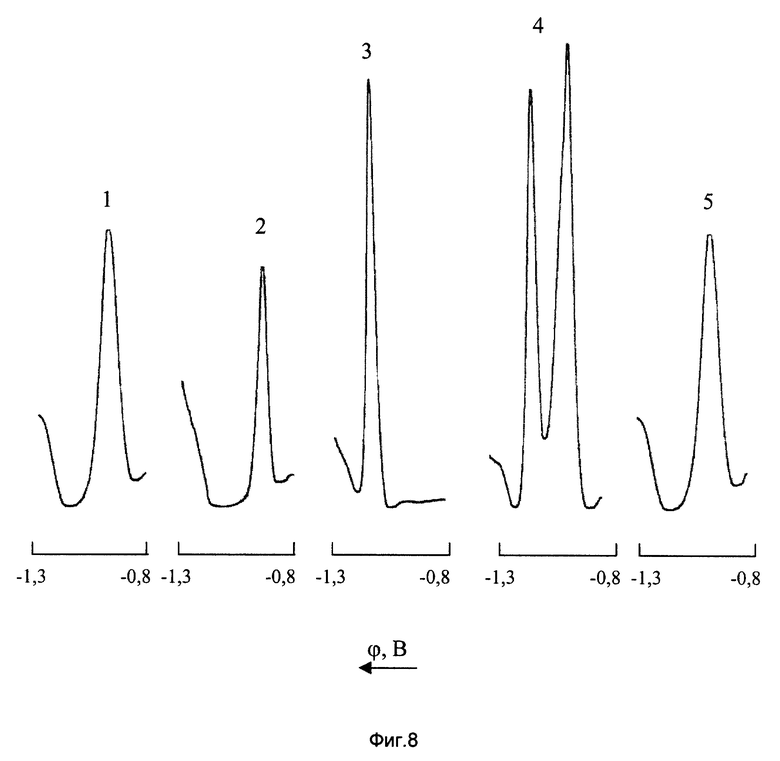
На фиг. 8 показаны ДИП кривые марганца, никеля и кобальта на фоне 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ до и после введения в анализируемый раствор ЭДТА
1 - CMn(VII)=10 мг/л; СЭДТА=0;
2 - СNi(II)=2 мг/л; СЭДТА=0;
3 - CCo(II)=0,4 мг/л; СЭДТА=0;
4 - CMn(VII)=10 мг/л; СNi(II)=2 мг/л; CCo(II)=0,4 мг/л; СЭДТА=0;
5 - СMn(VII)=10 мг/л; СNi(II)=2 мг/л; CCo(II)=0,4 мг/л; СЭДТА=2•10-3 М/л
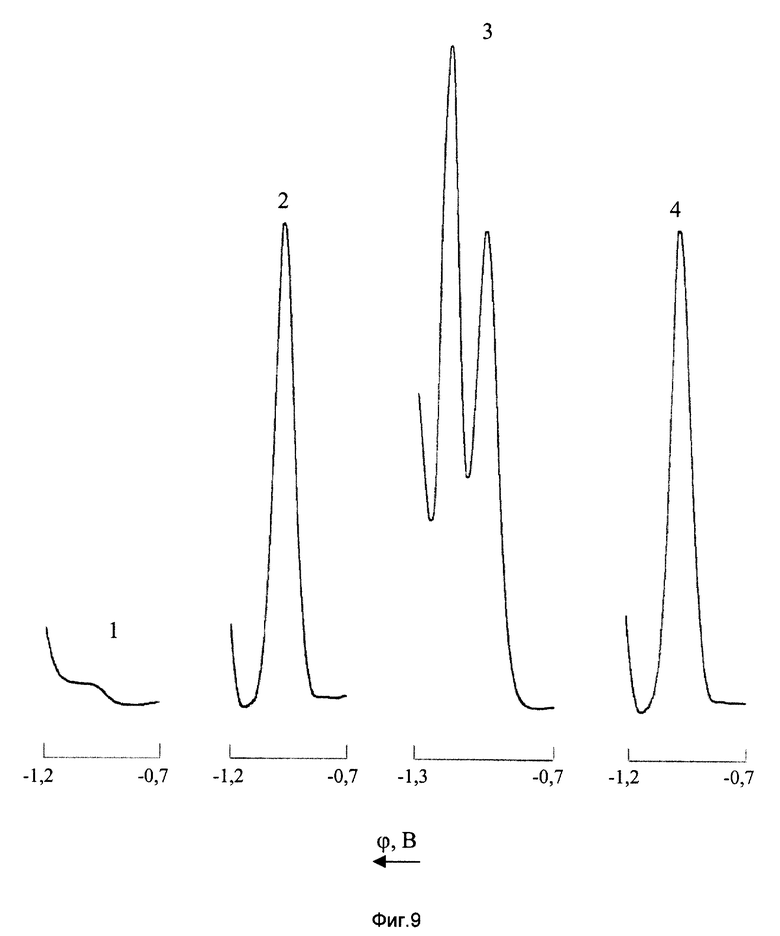
На фиг. 9 показаны ДИП кривые марганца в присутствии железа на фоне 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ до и после введения в анализируемый раствор ЭДТА
1 - фон
2 - CMn(VII)=10 мг/л; СFe(II)=0; СЭДТА=0;
3 - CMn(VII)=10 мг/л; СFe(II)=20 мг/л; СЭДТА=0;
4 - СMn(VII)=10 мг/л; СFe(II)=20 мг/л; СЭДТА=2•10-3 М/л.
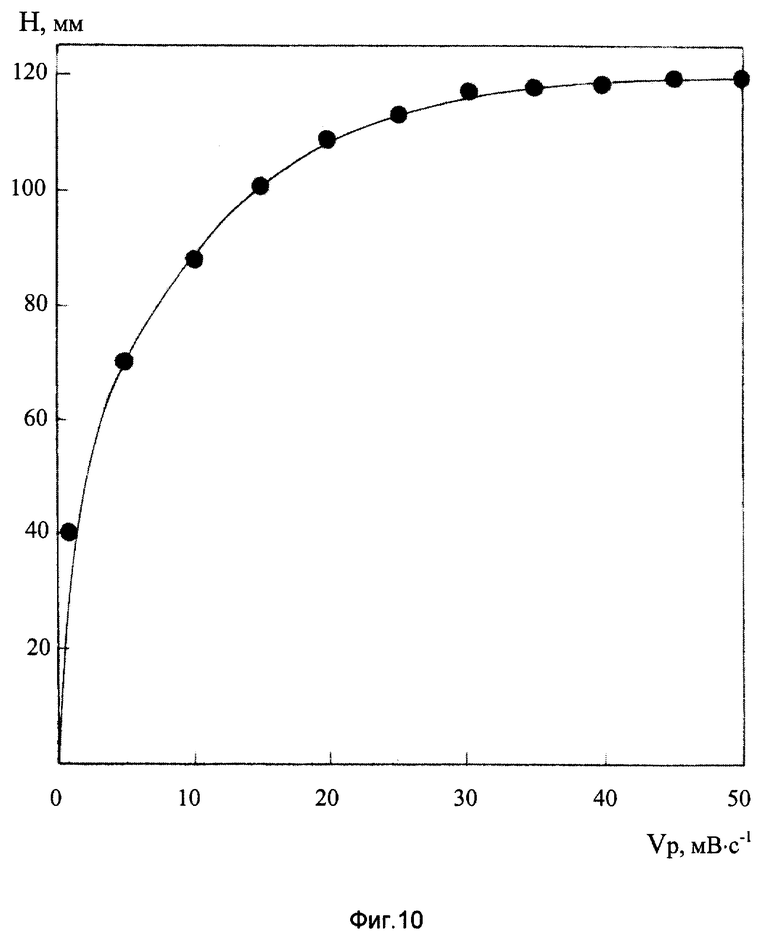
На фиг. 10 представлена зависимость высоты ДИП пика Mn(VII) 10 мг/л на фоне 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ+2•10-3 М ЭДТА от скорости развертки потенциала.
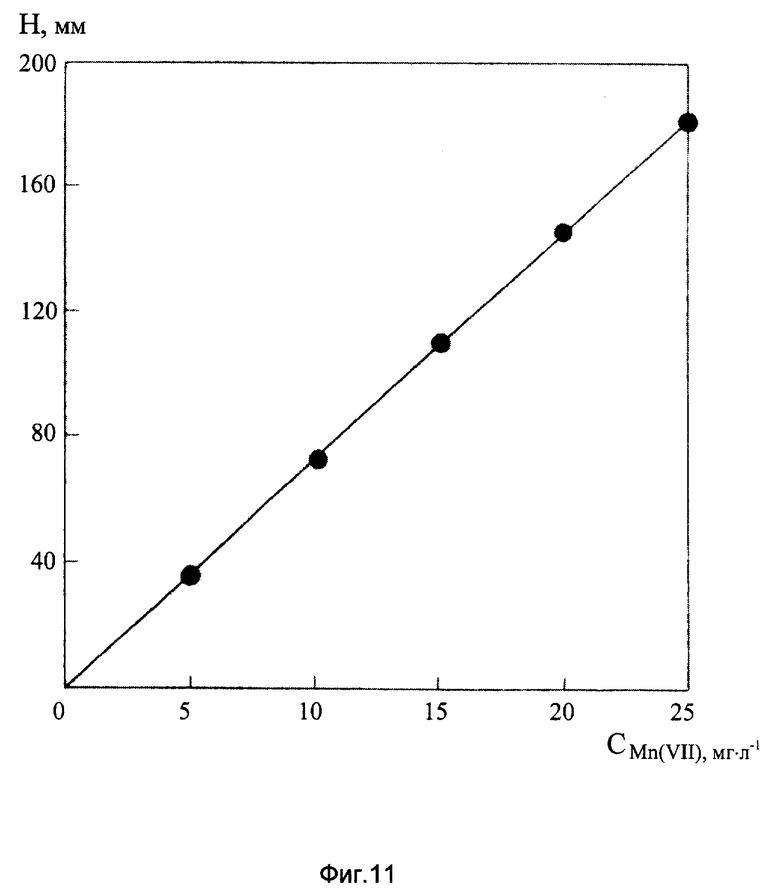
На фиг. 11 представлен градуировочный график для определения Mn(VII) на фоне 2М NH4Cl+2М NH4ОH+2•10-3 М ДМГ+2•10-3 М ЭДТА.
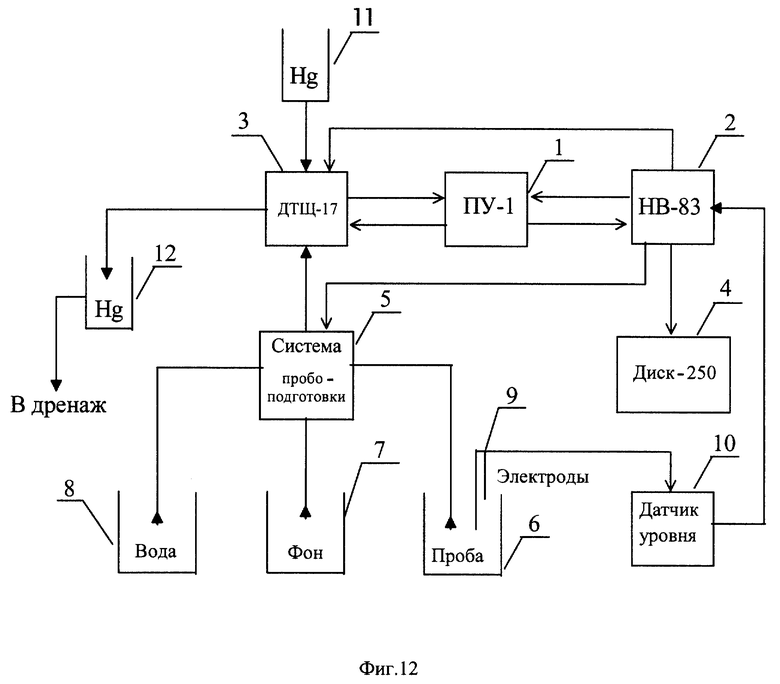
На фиг. 12 представлена структурная схема автоматического вольтамперометрического анализатора ионов Mn(VII) в нейтральных цинковых растворах.
Как видно из фиг.1, потенциалы катодного (ϕ
Измерения проводились на вольтамперометрическом анализаторе АЖЭ-11, изготовленном НПК "Югцветметавтоматика" и предназначенном для работы в режимах прямой и инверсионной дифференциальной импульсной полярографии на датчике со стационарным ртутным электродом (см. статью Боровков Г.А., Зарецкий Л. С. , Бабицкий Л.Б. Внедрение анализатора АЖЭ-11 на предприятиях свинцово-цинковой подотрасли // Цветная металлургия. 1987. 1, с. 49-51). Схема электрохимической ячейки 3-электродная (рабочий электрод типа "висящая" ртутная капля, электрод сравнения хлорсеребряный, вспомогательный электрод стеклоуглеродный). Длительность наложения прямоугольных импульсов 40 мс с периодом 80 мс. Остальные режимные параметры (амплитуда импульсного поляризующего напряжения, скорость развертки потенциала, начальное напряжение, амплитуда развертки) выбирались в ходе экспериментальных исследований. Анализатор АЖЭ-11 обеспечивает полную автоматизацию всех стадий измерительного процесса, включая пробоподготовку и подачу анализируемого раствора в электрохимическую ячейку; формирование "висящей" ртутной капли; регистрацию вольтамперной кривой и обсчет ее показателей с выдачей выходного сигнала в единицах концентрации контролируемого вещества; сброс и складирование отработанных ртутных капель и удаление проанализированного раствора в дренаж.
Из представленных на фиг.2 вольтамперных кривых видно, что в присутствии избыточной концентрации Мn(II) в растворе потенциал, высота и форма ДИП пика Mn(VII) не изменяются. Таким образом, диметилглиоксим избирательно взаимодействует с перманганат-ионами в хлоридно-аммиачной среде с образованием электрохимически активного комплекса, восстанавливающегося на ртутном электроде при потенциале -0,98 В. Мn(II) в этих условиях не дает аналитического сигнала и не мешает определению Mn(VII), что создает возможность избирательного вольтамперометрического контроля перманганат-ионов в растворах, содержащих обе валентные формы марганца. Это, в частности, необходимо при проведении анализа технологических растворов цинкового производства.
Водорастворимые соединения марганца играют существенную роль в процессе гидрометаллургического производства цинка. Концентрация марганца в цинковом электролите изменяется в широких пределах: от 2 до 8 г/л. В количестве 3-5 г/л марганец необходим для образования на свинцовом аноде защитной пленки, препятствующей переходу свинца в раствор. Снижение содержания марганца в растворе (менее 3 г/л) вызывает разряд ионов Сl- на аноде и выделение хлора в атмосферу. При значительных концентрациях марганца в электролите выход цинка по току снижается, т.к. марганец усиливает вредное действие на электролиз таких примесей, как сурьма и мышьяк. Кроме того, при больших концентрациях марганца в цинковом растворе ускоряется накопление шлама в ваннах и на анодах (см. книгу: Лакерник М.М., Пахомова Г.Н. Металлургия цинка и кадмия. М.: Металлургия, 1969, 488 с.).
В технологических растворах цинкового производства марганец одновременно существует в нескольких валентных формах, между которыми происходят окислительно-восстановительные реакции, существенно влияющие на ход электролиза. Визуальным признаком протекания таких реакций является изменение оттенков цвета электролита от зеленого до красного (см. книгу: Снурников А.П. Гидрометаллургия цинка. М.: Металлургия, 1981. 384 с.). Контроль и поддержание на заданном экспериментально определенном уровне концентрации ионов Mn(II) и Mn(VII) в поступающем на электролиз растворе создает возможность оптимизации режимных параметров цинкового производства.
Не менее важной является информация об ионном составе технологических растворов при проведении процесса перманганатной очистки от больших концентраций кобальта (II), входящего в число примесей, оказывающих наиболее вредное воздействие на электролиз цинка (см. книгу: Лакерник М.М., Пахомова Г. Н. Металлургия цинка и кадмия. М.: Металлургия, 1969, 488 с.). Сущность метода очистки состоит в окислении ионами МnО- 4 кобальта (II) до кобальта (III) и осаждении последнего в виде гидроксида, легко удаляемого из технологического раствора в процессе сгущения и фильтрации.
Одним из необходимых условий оптимизации технологического процесса перманганатной очистки растворов сульфата цинка от кобальта является оперативный контроль остаточной концентрации ионов МnО- 4. Поддержание концентрации перманганат-ионов на заданном уровне обеспечивает требуемую степень извлечения Co(II) из цинкового электролита и, кроме того, этот показатель может быть использован для регулирования расхода КМnO4 в процесс очистки. Например, по данным опытно-промышленного цеха завода "Электроцинк", при переработке богатых по кобальту концентратов для снижения содержания Со(II) в очищенном цинковом растворе до 0,5 мг/л необходимо поддерживать остаточную концентрацию Mn(VII) на уровне 500-700 мг/л.
Основным методом оперативного контроля ионного состава гидрометаллургических растворов является вольтамперометрия (см. например, статьи: Bond A. M., Knight R.W. Flow-through cell for continual on-line monitoring of cadmium, copper, antimony and lead by anodic stripping voltammetry in highly dense zinc plant electrolyte // Analytical Chemistry. 1988. V.60. 21. P. 2445-2448; Geibler M., Maia R.D. Determination of cobalt in the presense of hight concentrations of zinc by differential pulse polarography // Fresenius Z. Anal. Chem. 1988. 330. 7. Р. 624-626; Боровков Г.А., Зарецкий Л.С. Автоматическое определение индия в технологических растворах завода "Электроцинк" // Заводская лаборатория. 1986. Т.52. 11. С. 7-10; Боровков Г.А., Джиоева Е. А. Автоматизированное вольтамперометрическое определение ионов никеля в технологических растворах // Цветные металлы. 1991. 7. С. 67-70; Боровков Г.А. , Бровкин С. А. Автоматический контроль содержания ионов меди и хлора в растворах сульфата цинка // Цветная металлургия. 1993. 10. С. 22-23). Различные варианты полярографии успешно применяются для автоматизированного контроля цинковых электролитов на содержание ионов Сu2+, Cd2+, Pb2+, Co2+, Mi2+, In3+, Sb3+, Сl-. Однако для определения перманганат-ионов в растворах сульфата цинка вольтамперометрические методы анализа до настоящего времени не использовались.
В аналитической практике цинкового производства наибольшее распространение получил пламенно-фотометрический метод определения марганца, основанный на измерении интенсивности излучения атомов Мn в пламени пропан-воздух. Содержание марганца в пробе вычисляют по градуировочному графику, построенному в координатах С-Н, где С-концентрация марганца, %, Н - высота "пика" линии марганца, мм. Линия марганца 403,3 нм (см. книгу: Руководство. Методы аналитического контроля в цветной металлургии. Том II. Производство свинца и цинка. Часть 2. Методы аналитического контроля в производстве цинка. M.: Цветметинформация, 1977. С. 127-128). Пламенно-фотометрический метод обеспечивает определение суммарного содержания марганца в пробах независимо от его валентных форм. Анализ осуществляется на дорогостоящем оборудовании, требующем высококвалифицированного обслуживания, что существенно увеличивает капитальные и эксплуатационные затраты.
Заявляемый способ позволяет преодолеть указанные трудности и создает необходимые условия для избирательного и надежного контроля перманганат-ионов в сложных по составу технологических растворах цинкового производства.
Как видно из фиг.3, чувствительность полярографического определения ионов МnО- 4 согласно заявляемому способу в значительной степени зависит от концентрации диметилглиоксима в анализируемом растворе, существенно возрастая при увеличении содержания C4H8O2N2 от 0,2•10- до 2•103 М/л. Дальнейшее повышение концентрации ДМГ не дает ощутимого прироста высоты ДИП пика марганца, поэтому в целях сокращения расхода реактива его содержание в контролируемом растворе поддерживалось на уровне 2•10-3 М/л.
Возможности полярографического определения Mn(VII) в хлоридно-аммиачном фоновом электролите, содержащем диметилглиоксим, существенно ограничиваются мешающим влиянием различного рода сопутствующих веществ и, в первую очередь, ионов Zn2+, Co2+, Ni2+, Fe2+. В таблице приведены характерные для большинства цинковых заводов данные об ионном составе цинковых растворов, поступающих на электролиз.
При проведении вольтамперометрического контроля Mn(VII) в технологических растворах цинкового производства наиболее актуальной является задача обеспечения необходимой селективности анализа в присутствии избыточных концентраций Zn2+.
В растворах NH4OH-NH4Cl, содержащих ПАВ, Zn2+ дает хорошо выраженную волну, использующуюся в аналитических целях (см. книгу: Крюкова Т.А., Синякова С.И., Арефьева Т.В. Полярографический анализ. М.: Госхимиздат, 1959. 772 с. ). Несмотря на то, что в хлоридно-аммиачной среде Zn(ll) электрохимически восстанавливается при более отрицательном потенциале, чем комплекс Mn(VII) с ДМГ, уже сравнительно незначительный избыток цинка приводит к резкому ухудшению селективности полярографического определения перманганат-ионов. Например, на фоне 0,1М NH4Cl+0,1М NH4OH+2•10-3 М ДМГ потенциалы ДИП пиков Mn(VII) и Zn(ll) соответственно равны -1,014 и -1,142 В. Из фиг.4 видно, что даже при равных концентрациях этих ионов в анализируемом растворе наблюдается заметное искажение ДИП пика марганца (кривая 2), а при соотношении концентраций (мг/л) [Zn]:[Mn]=10:1 полярографическое определение перманганат-ионов становится практически невозможным (кривая 3).
Известно (см., например, книгу Крюкова ТА., Синякова С.И., Арефьева Т.В. Полярографический анализ. М.: Госхимиздат, 1959. 772 с.), что потенциал полуволны цинка сдвигается к более отрицательным значениям с увеличением концентрации аммиака, что создает возможность повышения селективности определения ионов Mn(VII) в присутствии Zn(ll) за счет увеличения концентрации фонового электролита. Исследования показали, что увеличение концентрации фона от 0,25М NH4Cl+0.25М NH4OH до 2М NH4Cl+2М NH4OH потенциал ДИП пика Mn(VII) уменьшается от -1,01 до -0,98 В, а потенциал ДИП пика Zn(II) увеличивается от -1,20 до -1,36 В (концентрация ДМГ в растворе поддерживалась при проведении опытов на уровне 2•10-3 М/л). Как видно из фиг.5, с повышением концентрации фона разность потенциалов восстановления указанных ионов (ΔЕ, мВ) увеличивается, т. е. селективность определения марганца в присутствии цинка улучшается.
Экспериментально установлено, что изменение концентрации фонового электролита практически не влияет на чувствительность определения Mn(VII) в присутствии Zn(II). Как следует из фиг.6, при повышении концентрации фона от 0.25М NH4Cl+0.25М NH4OH+2•10-3 М ДМГ до 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ высота ДИП пика марганца остается неизменной, а высота ДИП пика цинка возрастает не более чем в 1,7 раза.
Таким образом, высокая разрешающая способность полярографического определения перманганат-ионов в цинксодержащих растворах может быть достигнута только при использовании достаточно концентрированных фоновых электролитов. Оптимальным является фон 2М NH4Cl+2М NH4OH + 2•10-3 М ДМГ, в среде которого обеспечивается возможность прямого полярографического определения Mn(VII) с нижним пределом обнаружения 0,5 мг/л при соотношении [Zn]:[Mn]=7500:1. На фиг. 7 представлены ДИП пики марганца (VII) 2 мг/л на фоне 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ, до (кривая 2) и после введения в анализируемый раствор 12000 мг/л (кривая 3) и 16000 мг/л (кривая 4) цинка (II).
К числу сопутствующих веществ, оказывающих существенное влияние на полярографическое определение перманганат-ионов в хлоридно-аммиачной среде, содержащей диметилглиоксим, относятся Co(II) и Ni(II). Известно (см. книги: Бургер К. Органические реагенты в неорганическом анализе. М.: Мир, 1975. 272 с; Инцеди Я. Применение комплексов в аналитической химии. М.: Мир, 1979. 376 с.), что кобальт (II) и никель (II) образуют с диметилглиоксимом прочные комплексные соединения, каталитически действующие на выделение водорода на ртутном электроде. Наибольший каталитический эффект наблюдается при полярографировании диметилглиоксиматов никеля и кобальта на фоне хлоридно-аммиачных буферных растворов, где достигается чувствительность определения Co(II) и Ni(II) на уровне 1•10-8 М. Это позволяет использовать данный метод для контроля микроконцентраций никеля и кобальта в различных природных объектах, например в атмосферном воздухе (см. книгу: Манита М.Д., Салихджанова Р. М. -Ф., Яворская С.Ф. Современные методы определения атмосферных загрязнений населенных мест. М.: Медицина, 1980. 255 с.).
В фоновом электролите состава 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ потенциалы ДИП пиков Ni(II), Mn(VII) и Co(II) соответственно равны -0,95; -0,98 и -1,14 В. Из приведенных на фиг.8 вольтамперных кривых видно, что ДИП пики Ni(II) и Mn(VII) суммируются, а присутствие в анализируемом растворе кобальта (II) приводит к искажению левой нисходящей ветви суммарного пика марганца и никеля (кривая 4). Такая картина наблюдается при соотношении концентраций (мг/л) [Mn] : [Ni] :[Co]=25:5:1. Естественно, что при дальнейшем повышении концентрации ионов Ni2+ и Со2+ их мешающее влияние на определение МnO- 4 усиливается.
Маскирующее действие никеля и кобальта при полярографировании перманганат-ионов может быть устранено путем введения в контролируемый раствор ЭДТА, образующего с ионами Ni2+ и Со2+ устойчивые, электрохимически неактивные комплексы (см. книги: Бургер К. Органические реагенты в неорганическом анализе М.: Мир, 1975. 272 с.; Выдра Ф., Штулик К., Юлакова Э. Инверсионная вольтамперометрия. М.: Мир, 1980. 280 с.). Из представленной на фиг.8 кривой 5 видно, что введение в анализируемый раствор ЭДТА полностью нивелирует мешающее влияние никеля и кобальта при полярографическом определении МnО- 4. Экспериментально установлено, что в фоновом электролите состава 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ + 2•10-3 М ЭДТА вольтамперометрическое измерение Mn(VII) возможно в присутствии 5-кратных избытков Co(II) и 50-кратных избытков Ni(II).
Введение в состав фонового электролита ЭДТА позволило также устранить мешающее влияние ионов Fe(II). Известно, что в хлоридно-аммиачной среде ионы Fe2+ электрохимически восстанавливаются на ртутном электроде в электроотрицательной области потенциалов. Так, в фоновом электролите состава 1М NH4Cl + 1М NH4OH потенциал полуволны Fe(II) равен -1,52 В (см. книгу: Виноградова Е. Н. , Галлай З.А., Финогенова З.М. Методы полярографического и амперометрического анализа М.: Изд-во МГУ, 1960. 280 с.). Однако при введении диметилглиоксима в хлоридно-аммиачный фон железо (II) образует яркоокрашенный пурпурного цвета комплекс, полярографически восстанавливающийся при более положительном потенциале. Например, в среде фона 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ +2•10-3 М ЭДТА на СРКЭ регистрируется ДИП пик Fe(II) при потенциале -1,15 В.
Из фиг. 9 (кривая 3) видно, что в присутствии в анализируемом растворе железа (II) ДИП пик Mn(VII) искажается, однако введение в раствор ЭДТА полностью устраняет мешающее влияние Fe2+ (кривая 4). Исследования показали, что на фоне 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ + 2•10-3 М ЭДТА определению перманганат-ионов не мешает 10-кратный избыток железа (II), а при увеличении концентрации ЭДТА до 4-10-3 М измерение Mn(VII) возможно уже при 20-кратном избытке ионов Fe2+
К числу наиболее характерных компонентов ионного состава растворов гидрометаллургического производства цинка относятся также Cu(II), Cd(II), Sb(III), As(III). В хлоридно-аммиачном фоновом электролите, содержащем ДМГ, медь (II) восстанавливается при потенциале -0,35 В и не мешает полярографическому определению марганца (см. статью: Астафьева В.В., Прохорова Г.В., Салихджанова Р.М.-Ф. Изучение восстановления диметилглиоксиматов меди, никеля и кобальта с помощью вектор-полярографии //ЖАХ. 1976. Т. 31. Вып.2. С. 260-264). Присутствующие в цинковом электролите ионы Sb3+ в аммиачной среде гидролизуются и выпадают в осадок (см. книгу: Пац Р.Г., Васильева Л.Н. Методы анализа с использованием полярографии переменного тока. М.: Металлургия, 1967. 116 с.). Мышьяк (III) в хлоридно-аммиачных фоновых электролитах восстанавливается на ртутном электроде при потенциале -1,7 В (см. книгу: Немодрук А.А. Аналитическая химия мышьяка. М.: Наука, 1976. 244 с.) и полярографическому анализу марганца (VII) не мешает. Экспериментально установлено, что в фоновом электролите 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ+2•10-3 М ЭДТА не удается зарегистрировать ДИП пик кадмия, при концентрации последнего 250 мг/л. В очищенном цинковом растворе содержание Cd2+ обычно не превышает 1-2 мг/л (см., например, книгу: Снурников А.П. Гидрометаллургия цинка. М.: Металлургия, 1981. 384 с.).
Изучение влияния скорости катодной развертки потенциала (Vp) на чувствительность полярографического определения Mn(VII) на фоне 2М NH4Cl+2М NH4OH+2•10-3 М ДМГ+2•10-3 М ЭДТА (см. фиг.10) показало, что при изменении Vp от 0,5 до 10 мВ/с высота ДИП пика заметно возрастает; дальнейшее увеличение скорости развертки сказывается на чувствительности полярографического анализа марганца (VII) в значительно меньшей степени. Регистрация вольтамперных кривых со скоростью катодной развертки 10 мВ/с позволяет достичь высокой оперативности измерения при хорошей воспроизводимости и точности результатов анализа.
Градуировочная кривая Mn(VII) на фоне 2,0 М NH4Cl+2,0 М NH4OH+2•10-3 М ДМГ+2•10-3 М ЭДТА линейна в диапазоне концентраций 0-25 мг/л (фиг.11). Растворы, содержащие свыше 25 мг/л марганца (VII) перед анализом необходимо разбавлять.
Способ может быть реализован на различных отечественных и зарубежных полярографических анализаторах, обеспечивающих проведение измерений в режиме дифференциальной импульсной полярографии на стационарных ртутных электродах, в частности на приборах ПУ-1, ПЛС, PAR-174, PAR-384, PA-4, АЖЭ-11, АЖЭ-12, и других.
Способ осуществляется следующим образом.
В мерную колбу на 50 мл вводят пипеткой 0,5 мл анализируемого раствора, например нейтрального цинкового раствора, добавляют 25 мл фонового электролита 4 М NH4Cl+4М NH4OH+4•10-3 М ДМГ+4•10-3 М ЭДТА и доводят объем раствора в колбе до метки дистиллированной водой. Полученную смесь заливают в электрохимическую ячейку и снимают катодную кривую при линейно-изменяющемся во времени потенциале в интервале напряжений (-0,8) - (-1,2) В. Развертку потенциала производят со скоростью 10 мВ/с. Регистрируют серию из 3-5 кривых и, измеряя высоту катодных пиков при потенциале -0,98 В, определяют содержание Mn(VII) в растворе по градуировочному графику, построенному с учетом разбавления пробы в 100 раз. Общая длительность контроля одной пробы на содержание перманганат-ионов при ручной пробоподготовке не превышает 3-5 мин.
Фоновый электролит состава 4М NH4Cl+4М NH4OH+4•10-3 М ДМГ+4•10-3 М ЭДТА готовят из исходных растворов: 5М NH4Cl+5М NH4OH; 0,1 М ДМГ (спиртовой раствор); 0,1М ЭДТА. Как показали специально проведенные экспериментальные исследования, фоновый электролит сохраняет свои свойства в течение 1-2 месяцев.
При проведении анализа в автоматическом режиме смешение пробы с фоновым электролитом и подачу раствора в ячейку вольтамперометрического датчика производят с помощью трехкамерных дозирующих устройств мембранного типа ДЗЖ-4, выполненных с использованием коррозионностойких материалов (см. статью: Боровков Г.А., Зарецкий Л.С. Автоматическое вольтамперометрическое определение индия в технологических растворах завода "Электроцинк" // Заводская лаборатория. 1986. Т. 52. 11. С. 7-10).
Пример. Количественное определение перманганат-ионов в очищенном растворе сульфата цинка выщелачивательного цеха завода "Электроцинк", содержащем 0,5÷2,5 мг/л кобальта; 0,2÷1,0 мг/л никеля; 0,1÷0,2 мг/л меди; 0,2÷0,3 мг/л сурьмы; 0,01÷0,05 мг/л мышьяка; 0,5÷1,0 мг/л кадмия; 30÷50 мг/л железа; 500÷1500 мг/л марганца (VII); 2000÷8000 мг/л марганца (II); 130000÷140000 мг/л цинка.
Отбирают пипеткой 0,5 мл анализируемого цинкового раствора в мерную колбу на 50 мл, добавляют 25 мл фонового электролита 4М NH4Cl+4М NH4OH+4•10-3 М ДМГ+4•10-3 М ЭДТА и доливают колбу до метки дистиллированной водой. Полученную смесь переносят в электрохимическую ячейку датчика со стационарным ртутным электродом и регистрируют ДИП кривую марганца (VII) в интервале напряжений (-0,8) - (-1,2) В со скоростью развертки 10 мВ/с. Измеряют высоту пика при потенциале -0,98 В и определяют содержание Mn(VII) в анализируемом растворе по градуировочному графику. Всего снимают не менее трех ДИП пиков, при этом концентрацию марганца (VII) в пробе цинкового раствора рассчитывают по среднему арифметическому всех результатов анализа.
Заявляемый способ вольтамперометрического определения кобальта может быть реализован с помощью имеющихся технических средств, например анализаторов АЖЭ-11 и АЖЭ-12 (см. статьи: Г.А. Боровков, О.В. Щербич. Автоматизация контроля ионного состава сточных вод завода "Рязцветмет" // Заводская лаборатория. 1991. Т. 57. С. 9-12 и Г.А. Боровков, О.Г. Виноградов, В.В. Зеленский и др. Опыт разработки и внедрения автоматических анализаторов ионного состава флотационных пульп.// Цветные металлы. 1990. 9. С. 108-112).
Структурная схема автоматического вольтамперометрического анализатора АЖЭ-11, предназначенного для контроля ионов марганца (VII) в очищенных цинковых растворах, показана на фиг.12.
Анализатор состоит из полярографа ПУ-1 (1), блока управления НВ-83 (2), электрохимического преобразователя (вольтамперометрического датчика) ДТЩ-17 (3) со стационарным ртутно-капельным электродом, вторичного регистрирующего прибора "Диск-250" (4), системы пробоподготовки (5), выполненной на базе мембранных дозаторов жидкости и гидравлически связанной с приемной емкостью для пробы цинкового раствора (6), емкостью с фоновым электролитом (7), емкостью с водой (8) и проточной ячейкой электрохимического преобразователя (3). В приемной емкости (6) установлена электродная система (9) контактного датчика уровня (10), подключенного к блоку управления (2). Электрохимический преобразователь (3) гидравлически связан с ртутным резервуаром (11) и ловушкой для отработанной ртути (12).
Отбор и доставка контролируемых растворов к месту установки анализатора осуществляется с помощью центробежного насоса, работающего в сочетании с рукавным динамическим фильтром (не показаны). Отфильтрованная проба поступает самотеком в приемную емкость (6). Включение центробежного насоса производится автоматически по команде с блока управления (2) либо вручную технологическим персоналом выщелачивательного цеха.
Важнейшим узлом анализатора является электрохимический преобразователь с рабочим стационарным ртутно-капельным электродом, представляющим собой капиллярный дозатор ртути с клапаном игольчатого типа. Размеры формируемой ртутной капли определяются геометрическими параметрами капилляра и длительностью включения электромагнитного привода клапана, в момент открытия которого обеспечивается свободный исток ртути из связанного с атмосферой резервуара. Сброс капли осуществляется принудительно с помощью механической лопатки с электромагнитным приводом. Наличие в электрохимическом преобразователе электрического двигателя обеспечивает также использование лопатки в качестве мешалки анализируемого раствора.
С целью предотвращения капиллярных шумов, связанных с проникновением анализируемого раствора внутрь рабочего электрода, а также во избежание отрыва ртутной капли при механических воздействиях, например при сильных вибрациях, торец (исток) стеклянного капилляра защищен вваренной в него платиновой втулкой. Необходимые свойства капилляру такой конструкции придают предварительной электрохимической амальгамацией платиновой втулки. При этом капля удерживается на торце капилляра не только за счет сил поверхностного натяжения, но и вследствие химического взаимодействия ртути с покрывающим платину слоем амальгамы.
Схема электрохимической ячейки трехэлектродная (электрод сравнения хлорсеребряный, электрод вспомогательный - стеклоуглеродный). При работе анализатора в режиме дифференциальной импульсной полярографии весь измерительный процесс производится на одной ртутной капле.
Основу автоматической системы пробоподготовки составляют дозирующие устройства ДЗЖ-4, обеспечивающие по заданной программе отбор пробы из приемной емкости, разбавление ее водой, смешение с фоновым электролитом, подачу подготовленного к анализу раствора в проточную ячейку вольтамперометрического датчика с последующим сбросом в дренаж через ловушку отработанной ртути. Дозаторы выполнены из химически стойких материалов и представляют собой мембранные насосы с регулируемой производительностью, снабженные электрическими приводами.
Блок управления программирует работу дозаторов системы пробоподготовки, электромагнитных приводов роста и сброса ртутной капли, а также мешалки электрохимического преобразователя, включение развертки потенциала полярографа и привода диаграммной ленты регистрирующего прибора. Входящее в состав блока управления вычислительное устройство обсчитывает показатели вольтамперной кривой (измеряет высоту ДИП пика марганца) и выдает электрический сигнал на вторичный прибор, отградуированный в единицах концентрации контролируемого вещества.
Анализатор может работать в непрерывном или дискретном режимах измерения. Дискретность анализа определяется режимом работы системы пробоотбора и прободоставки.
Контроль проб очищенного цинкового раствора на содержание марганца заявляемым способом осуществляется следующим образом.
После заполнения фильтратом цинкового раствора приемной емкости (6) до уровня, установленного электродной системой (9), с выхода контактного датчика (10) поступает сигнал на включение блока управления (2). Программа блока управления начинается с команды на включение дозаторов жидкости системы пробоподготовки (5), осуществляющих отбор пробы из приемной емкости (6), разбавление ее в 50 раз водой, дозируемой из емкости (8), и смешение разбавленной пробы в соотношении 1:1 с фоновым электролитом состава 4М NH4Cl+4M NH4OH+4•10-ЗM ДМГ+4•10-3M ЭДТА, хранящимся в емкости (7). Подготовленная к анализу проба закачивается в проточную ячейку электрохимического преобразователя (3). При этом анализируемый раствор обновляется за счет вытеснения предыдущей пробы в дренаж через пороговое отверстие в боковой стенке ячейки. Отработанная ртуть удаляется в ловушку (12) через встроенный в днище ячейки затвор, препятствующий свободному истечению контролируемого раствора в дренажную линию.
Таким образом, в результате всех стадий пробоподготовки на анализ поступает разбавленная водой и обработанная фоновым электролитом проба цинкового раствора. После обновления раствора в ячейке электрохимического преобразователя (3) дозаторы жидкости системы пробоподготовки (5) выключаются и производится полярографическое определение марганца (VII). При этом в соответствии с программой блока управления (2) осуществляется сброс отработанной и формирование свежей ртутной капли, включение развертки потенциала на поля-рографе (1) и регистрация катодного ДИП пика. Полярографический анализ проводится в среде 2М NH4Cl+2M NH4ОH+2•10-3М ДМГ + 2•10-3 ЭДТА, выходные сигналы с полярографа поступают на вычислительное устройство блока управления, обрабатывающее (обсчитывающее) вольтамперную кривую и выдающее выходной сигнал на вторичный регистрирующий прибор (4), отградуированный в единицах концентрации ионов Mn(VII).
Автоматическая регистрация вольтамперной кривой производится в интервале потенциалов от (-0,8) до (-1,2) В со скоростью катодной развертки потенциала 10 мВ/с. Измерение концентрации марганца (VII) в растворе осуществляется по амплитуде (высоте) ДИП пика при потенциале -0,98 В (относительно хлорсеребряного электрода сравнения).
По окончании измерения блок управления (2) выключает измерительные приборы анализатора. После поступления в приемную емкость (6) свежего фильтрата цинкового раствора по сигналу с контактного датчика уровня (10) цикл анализа полностью повторяется.
Таким образом, заявляемый способ позволяет максимально упростить пробоподготовку контролируемого раствора, исключив из нее все сложные для автоматизации операции, и может быть реализован на освоенных промышленностью анализаторах.
Испытания заявляемого способа анализа марганца (VII) проводились на промышленных пробах завода "Электроцинк", отобранных в процессе перманганатной очистки растворов сульфата цинка от ионов Со(11). В настоящее время контроль ионов МnО- 4 в выщелачивательном цехе не производится, что снижает эффективность очистки нейтрального раствора от кобальта и приводит к перерасходу дорогостоящего перманганата калия в процесс. В период проведения испытаний пробы отбирались на выходе фильтр-прессов процесса перманганатной очистки. По данным ОТК завода "Электроцинк" концентрация ионных компонентов в растворах, прошедших перманганатную очистку от ионов Со(II), в среднем изменяется в следующих пределах, мг/л: Zn(II) (130÷140)•103; Mn(VII) 500÷1200; Mn(II) 2000÷8000; Fe(II) 30÷50; Co(II) 0,5÷2,5; Cd(II) 0,5÷1,0; Ni(II) 0,2÷1,0; Sb(III) 0,2÷0,3; As(IIi) 0,01÷0,05.
Во время испытаний в качестве контрольного использовался метод стандартных добавок. Всего было проанализировано 32 пробы нейтрального цинкового раствора, концентрация ионов Mn(VII) в которых изменялась в пределах от 190 до 860 мг/л. При этом приведенная среднеквадратичная погрешность анализа марганца не превысила 1,92%.
Заявляемый способ может быть использован для автоматического или экспресс-анализа марганца (VII) в технологических растворах цинкового производства, а также в сточных и оборотных водах предприятий цветной металлургии.
Внедрение предлагаемого способа в аналитическую практику цинкового производства позволит значительно повысить оперативность контроля ионов Mn(VII), оптимизировать технологический процесс очистки растворов сульфата цинка от примесей кобальта и в конечном итоге улучшить технико-экономические показатели производства катодного цинка. Использование заявляемого способа для экомониторинга сложных по составу сточных вод обеспечит снижение выброса ионов Mn(VII) в открытые водоемы в зоне действия предприятий цветной металлургии.



Изобретение относится к аналитической химии и может быть использовано для автоматического или экспресс-анализа технологических растворов цинкового производства, а также сточных или оборотных вод предприятий цветной металлургии. Сущность: способ основан на вольтамперометрическом определении марганца (VII) в хлоридно-аммиачной среде в присутствии диметилглиоксима и ЭДТА. Диметилглиоксим переводит Мn(VII) в электрохимически активный комплекс, избирательно восстанавливающийся на ртутном электроде в присутствии избыточных концентраций Мn(VII) и Zn(II). Введение в анализируемый раствор ЭДТА позволяет устранить мешающее влияние ионов Ni(II), Co(II) и Fe(II) при полярографическом определении перманганат-ионов. Измерения производят на фоне 2М NH4Cl+2M NH4OH+2•10-3 M диметилглиоксима + 2•10-3 M ЭДТА методом дифференциальной импульсной полярографии. Вольтамперную кривую регистрируют в диапазоне напряжений от -0,8 до -1,2 В со скоростью развертки потенциала 10 мВ/с. Содержание Мn(VII) определяют по высоте катодного пика при потенциале -0,98В±0,01В (отн. х.с.э.). Технический результат изобретения заключается в повышении избирательности, точности и оперативности способа. 1 табл., 12 ил.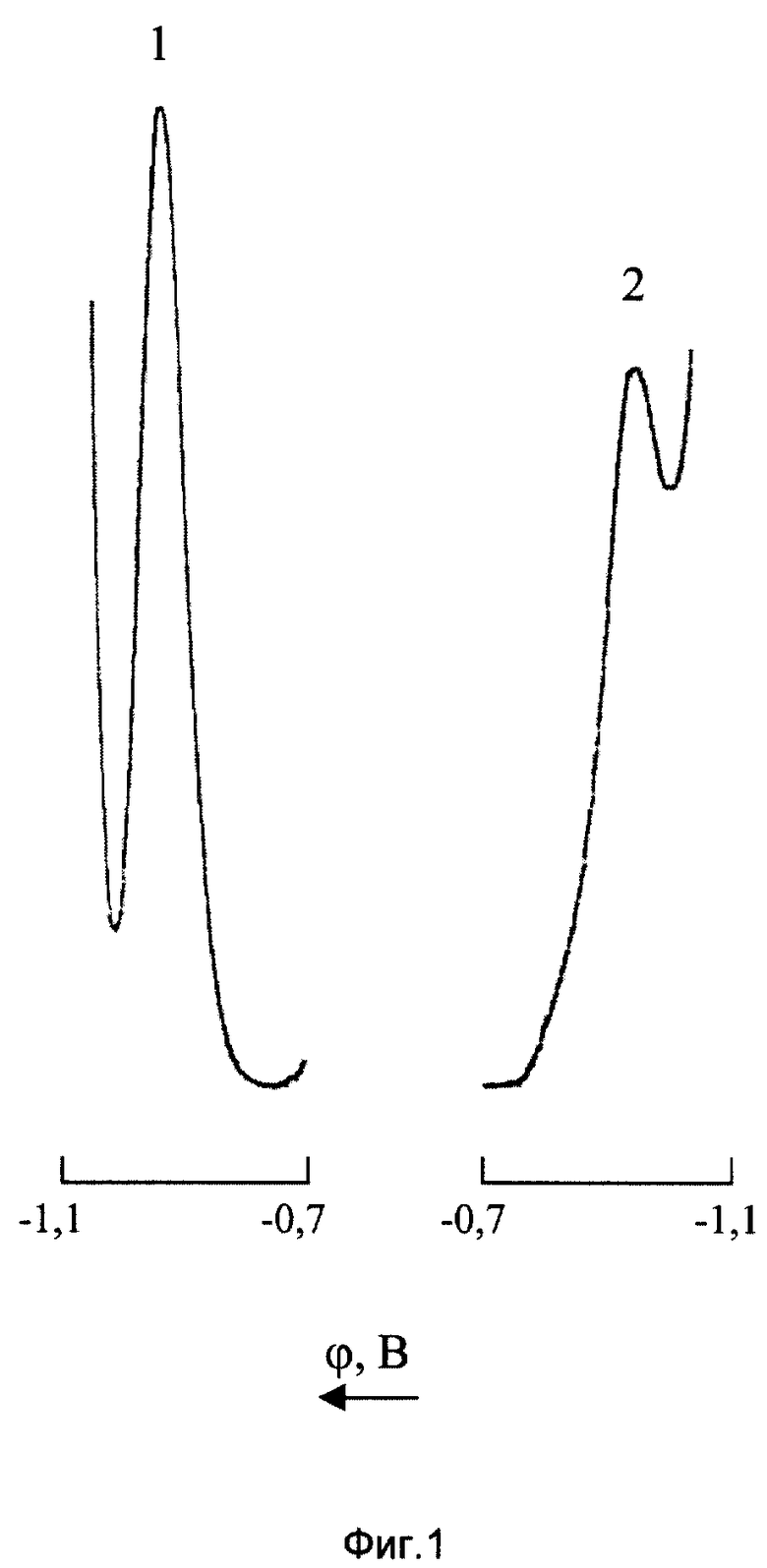
Способ вольтамперометрического определения концентрации перманганат-ионов в растворах сульфата цинка на ртутном индикаторном электроде, включающий регистрацию катодной вольтамперной кривой, отличающийся тем, что полярографирование ведут на фоне 2М NH4Cl+2M NH4OH+2•10-3 M диметилглиоксима +2•10-3 M ЭДТА в диапазоне напряжений от -0,7 до 1,10 В, а концентрацию перманганат-ионов измеряют по высоте пика при потенциале -0,98±0,01 В.
| Инверсионно-вольтамперометрический способ определения перманганат-ионов в водных растворах | 1987 |
|
SU1453303A1 |
| СОНГИНА О.А | |||
| и др | |||
| Полярографическое восстановление иона перманганата на платиновом и ртутном электродах //Журнал аналитической химии, 1956, т.11, вып.6, с | |||
| КОЛЬЦЕВОЙ ПОДПЯТНИК | 1923 |
|
SU717A1 |
| Huber C.O | |||
| Voltammetric determination of permanganate at the gold electrode // Anal | |||
| Chem, 1964, v | |||
| Коридорная многокамерная вагонеточная углевыжигательная печь | 1921 |
|
SU36A1 |
| Аппарат для подачи папирос в упаковочных машинах | 1924 |
|
SU1873A1 |
