1
Изобретение относится к технике лабораторного контроля и, в частности, к кулонометрическому Определению плутония в двуокиси; во фторидах, карбидах, нитридах, азотнокислых и солянокислых растворах.
Известны способы кулонометрического определения плутония, в частности способ кулонометрического титрования плутония (VI) до плутония (IV) при постоянном токе электрогенерируемыми ионами железа (II) с использованием потеициометрической системы индикации с двумя платино-родиевыми электродами, поляризуемыми током 0,1 ма.
По этому способу взятую на анализ пробу азотнокислого раствора плутония упаривают с серной кислотой, окисляют плутоний до шестивалентного состояния нагреванием с перманганатом калия, избыток которого потом восстана;вливают при нагревании формальдегидом или азидом натрия. Перед внесением подготовленной пробы раствора плутония (VI) в кулонометрическую ячейку к находящемуся в ней фоновому электролиту (0,6 М по f 62(504)3, -- 2,5 м по и 1 М по HzPOit) приливают сначала небольшое количество раствора бихромата калия и оттитровывают его электрогенерируемыми ионами железа (II) до устойчивого значения потенциала ниже 0,52 в.
Затем в ячейку вносят подготовленную пробу и плутоний (VI) оттитрОВывают генерируемыми ионами железа (II) до установления той же величины потенциала, что и при титровании бихромата калия. Так как в фоновом электролите указанного выше состава скорость реакции восстановления плутония (VI) до плутония (IV) электрогенерируемыми ионами железа (И) недостаточно велика и
вследствие этого потенциал индикаторного электрода устанавливается медленно (особенно вблизи эквивалентной точки), то для установления потенциала ток генерации в процессе титрования отключают на 3 мин через каждые 0,1 мин, а вблизи конечной точки титрования - через каждые 0,03 мин. Зная ток генерации и время, затраченное на титрование плутония (VI) до плутония (IV) в пробе, рассчитывают содержание его в анализируемом продукте. При титровании 2 мг плутония относительная погрешность составляет ±1 %.
Однако известный способ кулонометрического титрования плутония (VI) до плутония
(IV) характеризуется тем, что из-за медленного установления потенциала индикаторного электрода ток генерации в процессе титрования приходится периодически отключать. Полезное время, затрачиваемое на генерацию
ионов железа (И), составляет всего 3%
от общего времени, затрачиваемого на весь процесс титрования. Это приводит к увеличению времени на выполнение анализа, к увеличению погрешности в определении времени, затрачиваемого на генерацию ионов железа (И), а следовательно, и к снижению точности и чувствительности определения плутония.
Целью изобретения является повышение точности и эксцресности анализа.
Для этого точки эквивалентности онределяют путем регистрации двух интервалов времени, каждый от заданного значения диффузионного тока до внесения пробы до такого же значения диффузионного тока после ее титрования. При титровании электрорегеиируемыми ионами для установления эквивалентных точек титрования используют амперметрическую систему индикации с одним поляризуемым электродом; ток генерации и самопишущий потенциометр, на ленте которого записывается изменение диффузионного тока / во время t, в процессе титрования не выключаются, а отсчет времени ti и fe производится от двух точек /i и /2, расположенных за эквивалентной точкой на кривой диффузионного тока до внесения пробы, до двух точек с теми же значениями-/i и /2 на-кри-вой диффузионного тока после титрования пробы и по данным этих замеров алгебраическим путем рассчитывают время t, затраченное на титрование плутония (VI), а затем и количество плутония в пробе.
Для проведения титрования по предложенному способу подобран такой состав фонового электролита по серной кислоте (4- 5 моль/л) и по фосфорной кислоте (2- 4 моль/л), в котором плутоний (VI) при комнатной температуре с большой скоростью (даже вблизи эквивалентной точки) количественно восстанавливается электрогенерируемыми ионами железа (II) до четырехвалентного состояния, а для удаления из приготовленного для анализа хлорно-кислого раствора плутония (VI) продуктов разрушения хлорной кислотой (хлора и его окислов), мешающих определению плутония, подобран селективный восстановитель - сульфосалициловая кислота.
Использованпе амперометрической системы индикации (более чувствительной но сравнению с потенциометрической) в сочетании со способом непрерывного титрования от двух точек в фоновом электролите предложенного состава обеспечивает достижение целей изобретения. Так например, увеличение скорости реакции восстановления плутония (VI) до плутония (IV) электрогенерируемыми ионами железа (II) в фоновом электролите указанного состава обеспечивает получение кривых / (t) с четко выраженным перегибом в точках эквивалентности, что позволяет с большой точностью определять значения величины t по результатам замера времени (i и 4) в двух точках. На точность определения величины tg таким способом не влияет различие в наклоне кривых, обусловленное разбавлением фонового электролита в ячейке за счет внесения пробы раствора, изменением скорости перемешивания раствора в ячейке, изменением активности индикаторного электрода в процессе титрования и другими факторами. Использование селективного восстановителя для удаления продуктов разрушения хлорной кислоты существенно упрощает
технику проведения анализа и приводит к сокращению времени на его выполнение.
Следующие примеры иллюстрируют предложенный способ.
Титрование проводится на кулонометрической установке с системами генерации и индикации. Индикаторный ток записывают при помощи а1втоматичес1кого потенциометра. Объем кулонометрической ячейки мл. Пример 1. Онределение плутония в азотнокислых растворах.
а) Окисление плутония до шестивалентного состояния.
Навеску азотнокислого раствора плутония (1-25 мл PU) переносят во взвещенную мерную колбочку емкостью 25-50 мл, приливают-5-10 мл - 60%-ной хлорной кислоты, нагревают раствор до выделения пустых паров HClOi и продолжают дымление 15-20 мин. Затем раствор охлаждают, приливают 15-
30 мл М. НС1О4 и 0,5-1 мл 1%-ного раствора сульфосалициловой кислоты, разбавляют - 1 М HClOi до метки, перемешивают, взвешивают и часть его переносят в сухую весовую пипетку.
б) Подготовка установки для титрования. Включают потенциостат, питание индикаторной системы и устанавливают потенциал индикатор юго электрода (1,05-1,15) в. Кулонометрическую ячейку промывают
,5 М H2SO4 и начинают пропускать через нее аргон со скоростью 10-15 л/час, затем в ячейку вносят 0,2-0,4 мл 0,01 Н водного раствора -бихромата калия, 3-4 мл 0,3 м раствора сульфата железа (III) в 4 М H2SO4 и
10 М H2S04 и 10 М НзРО4 в таких количествах, чтобы концентрация каждой из них в конечном объеме раствора ( мл после внесения пробы раствора плутония (VI) составляла соответственно 4-5 моль/л и 2-
4 моль/л.
в) Титрование и расчет результатов анализа.
Продолжая продувку аргона, включают самопишущий потенциометр и ток генерации
0,2-2 ма. Когда бихромат будет оттитрован к /днф, за счет появления в растворе избытка ионов Fe (II) возрастет по сравнению с фоновым током /ф на 10 делений по шкале самописца /ь включают первый секундомер на
20 делений /г -второй, затем сразу же через воронку вносят в ячейку из весовой пипетки 1-2 г раствора пробы плутония (VI) и смывают воронку 1 мл 4-5 М НС1О4. При внесении раствора плутония (VI) в ячейку
/диф. резко упадет почти до /ф и останется
постоянным, пока весь плутоний (VI) не восстановится до четырехвалентного состояния, а когда /дцф. за счет ноявления в растворе избытка ионов железа (II) снова возрастет до /1, останавливают .первый секундомер (отмечают время ti, сек) до /2 - второй (отмечают время 2 сек. Время (tg, сек), затраченное на титрование плутония, рассчитывают по формуле
.1 J А - // f
сек
fe - ti/.,/, (
Зная значение величины , ток генерации (/2 на, вес раствора, взятого на титрование (т, 2), рассчитывают концентрацию плутония по формуле
Ср„, мг1г .
96497.2m
Для титрования следующей навески раствора плутония в ячейку, не меняя в ней фонового электролита, вносят ,2 мл 0,01 Н раствора бихромата калия и все остальные операции проводят точно так же, как это описано выше.
Коэффициент вариации, полученный при статистической обработке 20 параллельных определений плутония в одном и том же азотнокислом растворе по предложенному способу составляет ±0,2% относительных, если на титрование берут 1-2 мг плутония, и ±2% относительных, если на титрование берут 20-40 мкг плутония.
Пример 2. Определение плутония по предложенному способу в двуокиси и фторидах этого элемента.
При определении плутония в двуокиси п фторидах операцию растворения этих продуктов совмещают с операцией окисления плутония до шестивалентного состояния. Для этого 20-100 мг продукта помещают в коническую кварцевую колбочку емкостью 50 мл, приливают 5-6 мл концентрированной HNOs 6- 10 мл 50-70%-НОЙ ПС1О4, закрывают колбу стеклянной воронкой и кипятят раствор на электрической плитке до выделения густых наров НС1О4. Дымление с HCU нродолжают до полного растворения продукта. Далее раствор охлаждают, количественно перепосят во взвешенную мерную колбу емкостью 50 мл, разбавляют - 1 М ПС1О4 до 15-30 мл, приливают 0,5-1 мл 1%-.ного раствора сульфосалициловой кислоты, разбавляют до метки - 1 М ПС1О4, перемешивают и взвешивают. Все последующие операции по определению плутония в приготовленном растворе проводят точно так же, как это указано в примере 1.
Коэффициент вариации при определении плутония в двуокиси и фторидах составляет ±0,2% относительных.
Онределению плутония по предложенному способу не мешают большие количества урана, железа, алюминия и целый ряд других элементов, кроме хрома и нептуния, которые
так же, как п плутоний, окисляют кипящей до высших валентных состояний, а затем восстанавливают генерируемыми ионами железа (II).
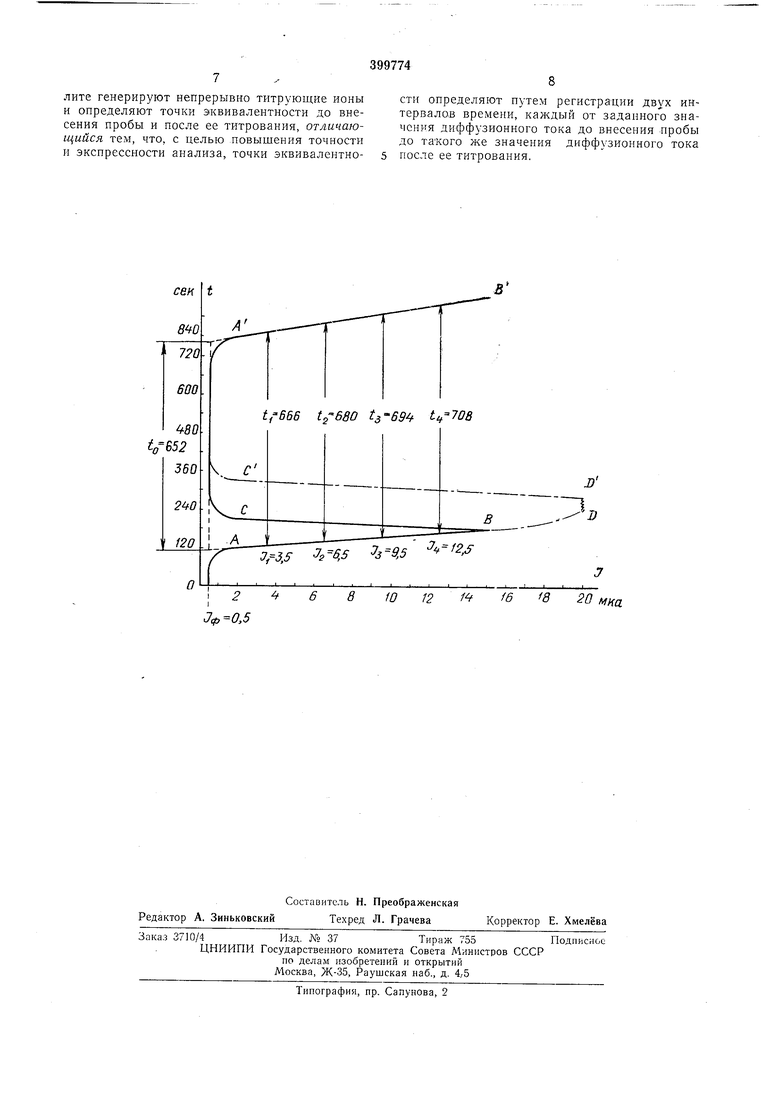
Способ пояснен чертежом.
На чертеже приведены кривые / (t) для двух случаев титрования одного и того же количества плутония (VI). Для наглядности обе кривые помещены на одном графике. В
первом случае плутоний вносят в ячейку тогда, когда индикаторный ток при титровании бихромата калия достигнет точки В (кривая I (t) будет иметь вид АВСА В ). Во втором случае раствор нлутония вносят в точку Z) -
когда индикаторный ток становится настолько большим, что он не укладывается в шкалу п перо самопишущего потенциометра выписывает вертикальную прямую DD (кривая / (О будеть иметь вид ABDD С А В ).
Два отсчета времени во всех случаях рекомендуют производить от произвольно выбранных двух значений индикаторного тока (точки /ь /2, /3 и А), расположенных на прямолинейном участке АВ кривой / (/) титрования
бихромата калия перед внесением в ячейку раствора плутония, до таких же значений индикаторного тока (точки /i, Iz, 1я и fi), расположенных за эквивалентной точкой на прямолинейном участке А В кривой / (/) титрования плутония (VI).
В этом случае значенне величины 4 находится с высокой точностью по предложенной формуле, независимо от величины тока в ячейке перед внесением в нее плутония (VI).
Пиже приведены результаты расчета 4 из данных графика.
1. (, t,(f,-f,) 652
/2 /1
4()
2. 4 f.
652
3 - 1
Л3. 4 ,
(/, - () - 652 /,-/.
/2 /ф
4. 4 а (г., - /j) 1 652
5. f,f,-J(f,-i,) Q52
i 2
Из нрпведенных результатов следует, что расчетное значение величины U не изменяется, если в качестве начала отсчета и окончания отсчета двух значений времени t берутся два любых нроизвольно выбранных значения
/ соответственно на прямолинейных участках АВ и А В кривых / (t) титрования бихромата калия ( плутония (VI). Не меняется /э и от того, что внесение в ячейку плутония (VI) производится при различной величине /.
60
Предмет изобретения
Способ кулонометрического титрования с
амнерометрической системой индикации, за65 ключающийся в том, что в фоновом электролиге генерируют непрерывно титрующие ионы и определяют точки эквивалентности до внесения пробы и после ее титрования, отличающийся тем, что, с целью повышения точности и экспрессности анализа, точки эквивалентности определяют путем регистрации двух интервалов времени, каждый от заданного значения диффузионного тока до внесения пробы до такого же значения диффузионного тока после ее титрования.


| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ СОВМЕСТНОГО ОПРЕДЕЛЕНИЯ МАССОВОГО СОДЕРЖАНИЯ УРАНА И ПЛУТОНИЯ В РАСТВОРАХ КУЛОНОМЕТРИЧЕСКИМ МЕТОДОМ ПРИ ПОСТОЯННОЙ СИЛЕ ТОКА | 2017 |
|
RU2653090C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ТЕХНЕЦИЯ | 1997 |
|
RU2132552C1 |
| Устройство для кулонометрического титрования | 1972 |
|
SU443301A1 |
| СПОСОБ КУЛОНОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ АСКОРБИНОВОЙ КИСЛОТЫ В РАСТИТЕЛЬНОМ СЫРЬЕ И ПРЕПАРАТАХ ИЗ РАСТИТЕЛЬНОГО СЫРЬЯ | 2010 |
|
RU2464558C2 |
| СПОСОБ КУЛОНОМЕТРИЧЕСКОГО ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ОРГАНИЧЕСКИХ КИСЛОТ В РАСТИТЕЛЬНОМ СЫРЬЕ И ИХ ПРЕПАРАТАХ | 2010 |
|
RU2450265C2 |
| Способ кулонометрического титрования жидкостей электрогенерированным иодом и устройство для его осуществления | 1987 |
|
SU1578621A1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ИНТЕГРАЛЬНОЙ АНТИОКСИДАНТНОЙ ЕМКОСТИ БИОЛОГИЧЕСКИХ ЖИДКОСТЕЙ | 2002 |
|
RU2253114C2 |
| Способ кулонометрического определения галогенидов | 1982 |
|
SU1057837A1 |
| Способ оперативной оценки качества винодельческой продукции | 2016 |
|
RU2631489C1 |
| Способ определения активного кислорода в медьсодержащих высокотемпературных сверхпроводящих материалах | 1990 |
|
SU1730576A1 |
сек
600
tt-666 ty-BBo
80 tn-B52
6,5 3-9,5
Зф 0,5
j)
I)
8 fO J2 fS 8 20
мна
