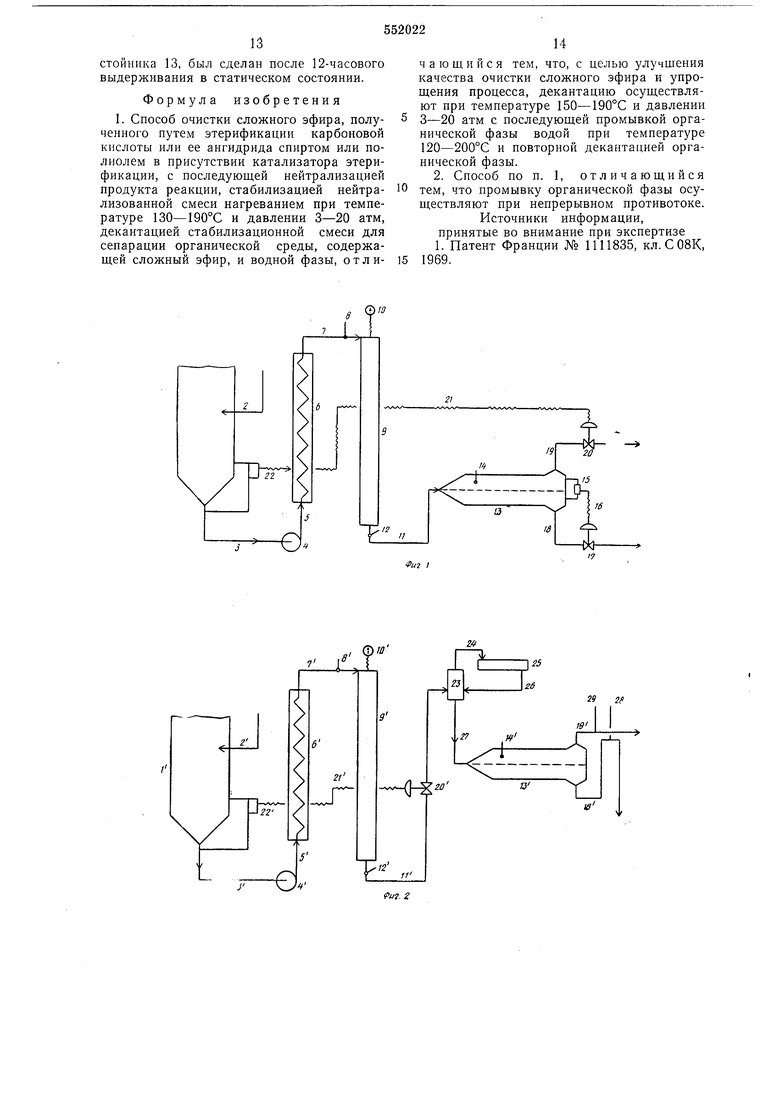
подается щелочной раствор для нейтрализации кислотности неочищенной смеси, полученной в результате этерификации.
Из колонны 1 смесь по линии 3, перемешиваясь насосом 4, по трубопроводу 5 поцается в обогреватель 6, который отрегулирован так, что поддерживает нужные температуры в различных элементах схемы (отсчет ведется по термометрам).
Из обогревателя 6 эмульгированная смесь по линии 7, имеющей термометр 8, подается в реактор 9, на котором смонтирован манометр 10. Величина давления, создаваемого насосом 4, на 2-5 бар иревышает давление пара смеси при температуре, показываемой термометром 8.
Смесь, которая подверглась стабилизации термообработкой, после реактора 9 по линии И с термометром 12 подается в горизонтальный цилнндрическо-копический отстойник 13 с термометром 14. Уровень межфазной иоверхности контролируется и регулируется регулятором 15 уровня, который имеет электрическую связь 16 с вентилем 17, находящимся на линии 18. Через линию 18 водная фаза выпускается так, что межфазовая поверхность удерживается на определенном уровне (см. фиг. 1, пунктирная линия).
По линии 18 водная фаза подается в холодильник (на схеме не показан), где она расширяется и при этом происходит конденсация образованных паров, в результате чего полезные продукты (например, остаточный спирт) восстанавливаются, либо подается в небольшую дистилляционную колонку (на фиг. 1 не показана), где водная фаза расширяется и выделяемые при переиаде давления калории используются для восстановления органических продук- тов.
Органическая фаза (верхний слой декантации) выпускается по трубопроводу 19. Вентиль 20, который имеет электрическую связь 21 с регулятором 22 уровпя жидкости, регулирует выпуск органической фазы в зависимости от уровня л идкости в основании колонны 1.
Органическая смесь по линии 19 подается к промывочному устройству для ее дополнительной промывки.
Таким устройством может служить противоточная колонна, содержащая соответствующую насадку. Процесс декантации непродолжителен и общий объем отстойника составляет 1/6 часового расхода смеси.
Опыт 1. В установке (см. фиг. 1) фталевый ангидрид подвергают периодической обработке этилом-2 гексановой кислоты в присутствии в качестве катализатора серной кислоты и циклогексана. Для этого продукт этерификации загружают в резервуар вместе со щелочным раствором, состоящим из 0,76 кг карбоната натрия и 27л чистой воды. Продукт этерификации имеет следующий состав, кг:
Диэтил-2 фталата капроновой кислоты30
Циклогексан12,4
2-Этил гексанол6,4
2-Этил кислой фталевой соли
капроновой кислоты0,15
2-Этил кислого сульфата
натрия капроновой кислоты 1,05
Смесь, перемешивая, нагревают до температуры кипения, при этом циклогексан,
образующий с водой азеотропную смесь, перегоняется, полученный дистиллат подвергается декантации, а водная фаза - продукт декантации подается врезеруар. Температура жидкости в резервуаре повыщается с 30 до 10ГС (приблизительно). После удаления циклогексана (процесс длится около 1 ч) перекрывают выход паров из резервуара и, чтобы перейти к процессу стабилизации, повышают температуру жидкости, находящейся в резервуаре до 180°С, затем, не прекращая механического перемещивания, в течение 1 ч выдерживают жидкость при указанной температуре и давлении в пределах 9,6-10 бар.
Затем прекращают нагрев и перемешивание и, дав жидкости отстояться в течение 5 мин, сливают водный слой (нижний), сразу же охлаждая его. И так за 5 мин сливают 26 л водной фазы, прозрачной с
желтоватым оттенком.
После охлаждения органической фазы в резервуаре до температуры 95°С ее подвергают при перемешивании двум последовательным промывкам водой при температуре 80-90°С (объем воды каждой промывки равен 90 л). Процесс декантации очень непродолжительный, как это можно судить по скорости слива: после 5-минутного отстаивания сливают 90 л водной фазы, фаза пмеет белый цвет.
Для получения конечного диэтил-2 фталата капроновой кислоты из промытой органической фазы из нее удаляют с парами воды избыточный 2-ЭТИЛ гексапол, затем производят сушку сложного эфира воздухом. В результате воздействия соляной кислоты на оси происходит гидролиз, позволяющий определить щелочность различных фаз. Необходимо отметить, что этот способ
не дает возможности определить дозировку сульфата натрия, а позволяет лишь сравнить различные пробы.
Также определяют электрическое удельное сопротивление конечного диэтил-2 фталата капроновой кислоты до и после 6-часового нагрева его до температуры 180°С.
Результаты этих различных определений приведены в табл. 1.
Таблица I


| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОЧИСТКИ сложных ЭФИРОВ | 1971 |
|
SU294320A1 |
| Способ получения фталатов | 1978 |
|
SU739063A1 |
| Способ получения сложных эфиров | 1982 |
|
SU1068418A1 |
| МАКРОМОНОМЕРЫ, СОДЕРЖАЩИЕ ПОЛИИЗОБУТЕНОВЫЕ ГРУППЫ, И ИХ ГОМО- И СОПОЛИМЕРЫ | 2017 |
|
RU2745788C2 |
| СПОСОБ ПОЛУЧЕНИЯ АЦЕТОНЦИАНГИДРИНА | 2007 |
|
RU2497805C2 |
| Способ получения алкил-или аллилпроизводных флуорена или фенантрена | 1980 |
|
SU925924A1 |
| СПОСОБ РЕАКЦИОННОЙ ЭКСТРАКЦИИ ЛЕВУЛИНОВОЙ КИСЛОТЫ | 2005 |
|
RU2391333C2 |
| СПОСОБ ПОЛУЧЕНИЯ МЕТАКРИЛОВОЙ КИСЛОТЫ | 2012 |
|
RU2602080C2 |
| СПОСОБ И УСТРОЙСТВО ДЛЯ ПОЛУЧЕНИЯ СЛОЖНЫХ АЛКИЛОВЫХ ЭФИРОВ МЕТАКРИЛОВОЙ КИСЛОТЫ | 2007 |
|
RU2486173C2 |
| СПОСОБ ПОЛУЧЕНИЯ СЛОЖНЫХ ДИЭФИРОВ ТЕРЕФТАЛЕВОЙ КИСЛОТЫ С ОБОГАЩЕНИЕМ ВОЗВРАТНЫМ СПИРТОМ | 2015 |
|
RU2665579C1 |
Начальная кислотность постоянно (всегда) выше 0,1 ммоль/л.
Водная фаза после 1-й декантации и процесса стабилизации обрабатывается диэтиловым эфиром для удаления из нее органических продуктов. Для определения приблизительных потерь органических веществ в водной фазе делают хроматографический анализ паров этилового растворителя. Результаты этого анализа приведены в табл. 2.
Таблица 2
Опыт 2. В процессе опыта смесь, подобную смеси, взятой в опыте 1, подвергают такой же обработке, как и в опыте 1 до процесса термической стабилизации включительно (процесс стабилизации осуществляется в резервуаре при температуре 180°С и давлении 9,6-10 бар в течение 1 ч). Затем содерл имое резервуара при непрерывном перемещивании быстро охлаждают до температуры 95°С. Прекратив охлаждение и перемещивание, смеси дают возможность отстояться в течение 10 мии и затем сливают водную фазу. Вначале сливают 15 л, декантация продолжается, и только спустя 15-20 мин получают 25 л фазы солевого раствора.
Проводят две промывки органической фазы чистой водой при тех же условиях, как и при опыте 1. Декантация, которая следует за 1-й промывкой, осуществляется очень медленно; после 5-минутной выдержки начинают слив, за 12 мин сливают 81 л и 90,8 л по истечении 20 мин. Декантация после 2-й промывки осуществляется так же быстро, как и при опыте 1.
Затем приступают к определению тех же параметров, что и в опыте 1. Полученные результаты приведены в табл. 3.
Опыт 3. В устройство (см. фиг. 1) загружают исходный продукт для этерификации, подобный тому, что использовался в опыте 1. В основании колонны 1 поступает в результате ретроградации около 1800 л смеси (при 20°С) следующего состава, кг/ч:
Диэтил-2 фталата капроно1600вой кислоты 2-Этил кислотного фталата
3
капроновой кислоты 2-Этил гексанол 128 2-Этил гексила кислого
сульфата
21
В основание колонны 1 в это же время вводится через патрубок 2 раствор из 29,4 кг/ч карбоната натрия, и 540 кг/ч воды.
Хорощо эмульгированная насосом 4 смесь подается под давлением 15 бар (отсчет по манометру 10) в обогреватель 6, где происходит ее нагрев до 183°С (отсчет по термометру 8). После реактора 9 смесь, имеющая на выходе температуру 179- 180°С (отсчет по термометру 12), подается в отстойник 13, полезный объем которого составляет 250 л, где выдерживается в течение 6 мин. После остывания до темпе Коэффициент разделения есть отношение щелочности водной фазы к щелочности органической фазы после каждого процесса декантации.
ратуры 100°С солевой раствор сливают по линии 18, пропускная способность которой 250 л/ч. Водная фаза светлая и после охлаждения до температуры окружающей среды принимает белый оттенок.
Органическая фаза сливается через линию 19 с пропускной способностью 1820 л/ч. После расширения и охлаждения до 98°С органическую фазу промывают в противотоке 900 л/ч горячей воды, промывку осуществляют в колонне с насадкой, имеющей две ступени, затем ее наТак же, как и в опыте 1, определяют содержание органических продуктов в водОпыт 4. В процессе опыта смесь, подобную смеси, взятой в опыте 3, подвергается обработке в установке (см. фиг. 2).
Таблица 3
правляют к другим установкам по очистке фталата.
Аналогично опыту 1 определяют щелочность водных фаз, сливаемых из отстойника 13 и из промывочной колонны. Практически остаточная щелочность органической фазы равна 0.
Результаты этих определений и измерений удельного электрического сопротивления конечного фталата и полихлорвиниловой пластины, пластифицированной этим фталатом, приведены в табл. 4.
Таблица 4
ной фазе, сливаемой по линии 18. Результаты анализа приведены в табл. 5.
Таблица 5
15 Условия обработки смеси аналогичны условиям опыта 3 до процесса стабилизации в реакторе включительно. На выходе
из реактора 9 слив смеси по линии И регулируется вентилем 20, управляемым регулятором 22. Затем смесь подается в расширительную камеру 23. Пары, которые там образуются, по линии 24 подаются в конденсатор 25, откуда конденсат по трубопроводу 26 вновь сливается в расширительную камеру.
Весь объем жидкости камеры 23 по трубопроводу 27 подается в отстойник 13, полезный объем которого составляет 500 л, где выдерживается в течение 13 мин. Трубопроводы 18 и 19, по которым сливаются водная и органическая фазы, имеют воздушники 28 и 29 соответственно, сообщаюш;иеся с атмосферой.
Декантация в отстойнике 13 осушествляется при 100-95°С. Производительность отстойника составляет 518 л/ч солевого раствора, сливаемого по трубопроводу 18. Органическая фаза по трубопроводу 19, пропускная способность которого 1822 л/ч, переливается в промывочную колонну, как и в опыте 3. Промывка ведется чистой водой, расход воды, подаваемой в противотоке, составляет 1650 л/ч, последуюшая обработка органической фазы происходит так же, как и в опыте 3.
Затем проводят анализы как и в опыте 3. Результаты измерений приведены в табл. 4 и 5.
Опыт 5. В установке (см. фиг. 1) подвергают этерификации фталевый ангидрид и тридеканол.
Смесь, подаваемая к насосу 4, имеет следующий состав, кг/ч:
Дитридециловый фталат6,95
Циклогексан1,03
Тридеканол0,53
Кислый тридециловый фталат 0,004 Тридецил кислого сульфата 0,11 Вода2,8
Карбонат натрия (сухой вес) 0,13 Эта смесь хорошо эмульгируется насосом 4, который нагнетает ее под давлением, равным 15 бар (отсчет по манометру 10), затем нагревается до 885°С (отсчет по термометру 8). После прохождения через реактор 9 смесь, нагретая до 181°С (отсчет по термометру 12), подается в отстойник 13, где она выдерживается в течение 10 мин (объем и расходы выражены при20°С).
Расход соляного раствора, сливаемого по трубопроводу 18 после процесса охлаждения, составляет 2,75 л/ч. При этом соляной раствор мутно-белый и не отстаивается.
Расход органической фазы, сливаемой по трубопроводу 19 после процесса охлаждения, составляет 9,25 л/ч. Затем органическая фаза подвергается двухразовой периодической промывке в резервуаре, оснащенном турбиной-смесителем, промывку производят дистиллированной водой при
80°С и при перемешивании в течение 10 мин. Процесс декантации после каждой промывки эффективен и непродолжителен (максимум 15 мин).
Результаты измерений, как и в предыдущих случаях, даны в табл. 6 и 7.
Опыт 6. Для проведения опыта используется установка (см. фиг. 2) небольшого размера с отстойником 13 емкостью 2 л. Вся установка имеет теплоизоляцию, как и при опыте 5. В такой установке подвергают обработке исходное сырье этерификации, как и при опыте 5, но при меньшем расходе последнего. Смесь, подаваемая к насосу 4, имеет следуюший состав, кг/ч: Дитридециловый фталат5,82
Циклогексан0,863
Тридеканол0,445
Кислый тридециловый
фталат0,0033
Тридецил кислого сульфата 0,092 Вода2,3
Карбонат натрия (сухой вес) 0,108 Условия обработки смеси аналогичны условиям опыта 5 до процесса термической стабилизации в реакторе включительно. После реактора 9 смесь, пройдя расширительный бачок и будучи охлажденной в элементах 20, 23 и 25 до 100+ГС, подается в отстойник 13, где выдерживается в течение 12 мин. Соляной раствор, сливаемый через трубопровод 18 при расходе 2 л/час, имеет белый цвет и очень медленно отстаивается (10-12 часов 64 мл органической фазы. Эта органическая фаза при 20°С имеет плотность 0,936, а анализ (хроматография) показывает, что она имеет следующий состав, вес. %:
Тридециловый фталат91
Тридексанол5
Циклогексан3
Вода1
Органическая фаза, сливаемая по линии
19, также имеет белый цвет, ее расход 8 л/час. Для промывки фазы невозможно использовать резервуар с турбиной-смесителем, как это было в опыте 5, так как смесь при равных объемах воды и органической фазы при 80-90°С образует стойкую эмульсию с большим слоем пены. Равномерно перемешивая две фазы смеси путем нагрева с обратным холодильником до температуры кипения, добиваются проведения первых двух промывок и соответствующих им процессов декантации. Можно добиться получения органической фазы с той же остаточной щелочностью, как и в опыте 5, но только путем 5-кратной промывки. Последние 3 промывки как в резервуаре с турбиной-смесителем, так и в резервуаре, где перемешивают путем нагрева с обратным холодильником. Анализ на содержание органических продуктов в водной фазе, сливаемой из ота
Я S
ч ю га
а S ч VO 14 Fстойника 13, бЫоП сделан после 12-часового выдерживания в статическом состоянии. Формула изобретения 1. Снособ очистки сложного эфира, полученного путем этерификации карбоновой кислоты или ее ангидрида спиртом или полиолем в присутствии катализатора этерификации, с последующей нейтрализацией продукта реакции, стабилизацией нейтрализованной смеси нагреванием при температуре 130-190°С и давлении 3-20 атм, декантацией стабилизационной смеси для сепарации органической среды, содержащей сложный эфир, и водной фазы, отли-г-€) чающийся тем, что, с целью улучщения качества очистки сложного эфира и упрощения процесса, декантацию осуществляют при температуре 150-190°С и давлении 3-20 атм с последующей промывкой органической фазы водой при температуре 120-200°С и повторной декантацией органической фазы. 2. Способ по п. 1, отличающийся тем, что промывку органической фазы осуществляют при непрерывном противотоке. Источники информации, принятые во внимание при экспертизе 1. Патент Франции № 1111835, кл. С08К, 1969.
