1
Изобретение относится к области определения объема и внутренней поверхности пор, в частности с применением адсорбции. Оно может быть использовано при отработке оптимальных технологий получения пористых адсорбентов и катализаторов с нужной пористой структурой, при изучении пористой структуры адсорбентов и катализаторов, а также при отборе эффективных адсорбентов и катализаторов для определенных областей их применения.
Известен способ определения пористости адсорбентов путем последовательного их насыщения газами с различными критическими размерами молекул 1.
Недостатком способа, основанном на ситовых свойствах пористых тел и на использовании молекул рабочих газов в качестве молекулярных щупов, является его трудоемкость и недостаточная информативность. Для определения пористости нужна целая серия адсорбционных экспериментов с различными рабочими газами, отличающимися размерами молекул. Кроме того, эти способы являются косвенными - mm не дают непрерывного распределения пор по размерам, а в связи с дискретностью критических размеров молекул рабочих газов получается лишь информация об объемах пор с размерами большими некоторого данного.
Никаких сведений о форме пор этот способ не дает.
Известен способ определения пористой структуры катализаторов электронно-микроскопичеоким методом, заключаюшнйся в исследовании нескольких реплик, снимаемых с поверхности произвольного сечения катализатора, при этом для увеличения контрастности, реплики оттеняются
золотом 2.
Однако получение контрастности в этом способе требует дорогостоящего материала, затрудняет регенерацию образца для последующих анализов, и, следовательно,
возможно загрязнение пор.
Целью изобретения является повышение точности и экспрессности способа.
Поставленная цель достигается тем, что в качестве контрастирующего вещества
берут аргон, насыщение им адсорбирующего материала ведут при температуре 40- 150°К и давлении не выше 10 мм рт. ст., затем понижают давление аргона до 10 мм рт. ст., после чего производят
съем1ку исследуемой поверхности в проходящем пучке электронов. Предпочтительным является проведение насыщения адсорбента при температуре 78°К.
В качестве рабочего газа берется аргон.
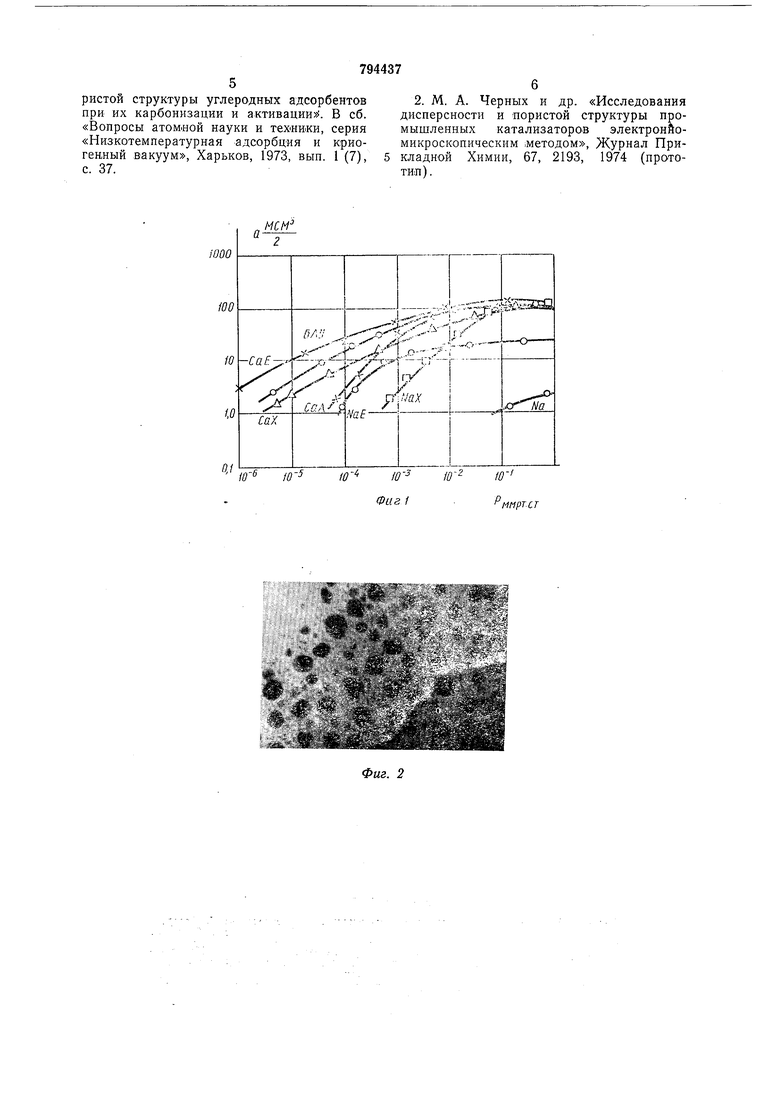
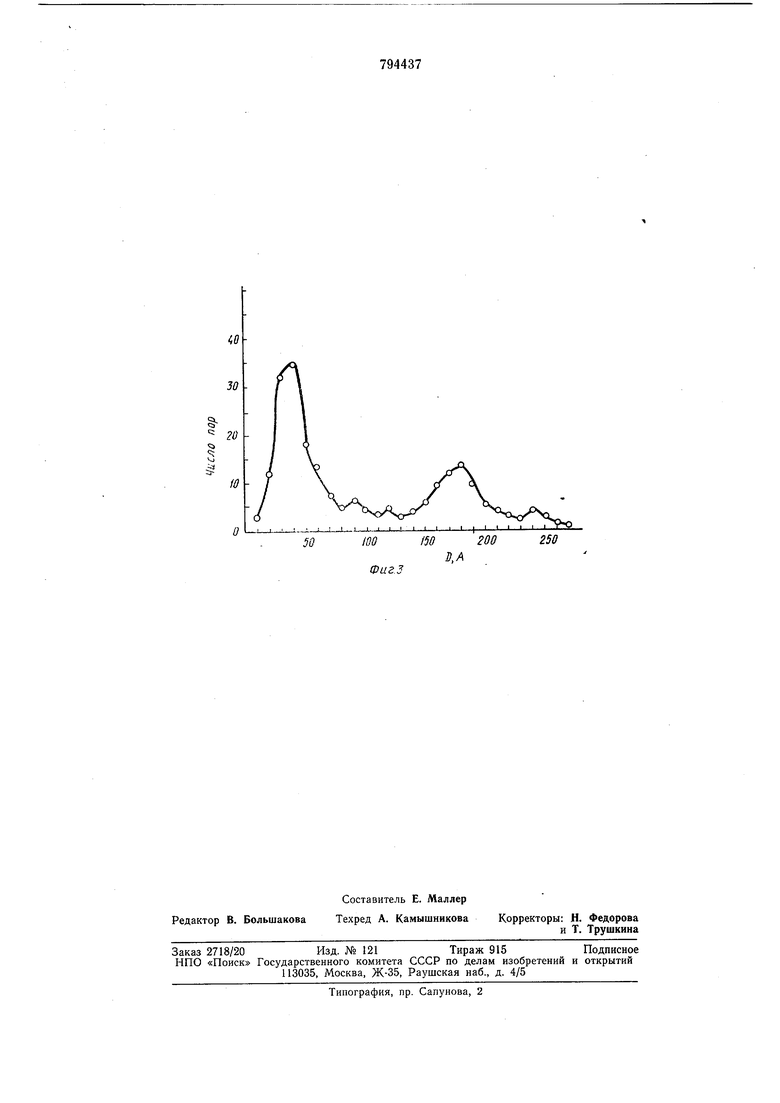
Основания для выбора аргона следующие: 1.Его молекулярный вес 40, т. е. втрое больше, чем у атомов матрицы углеродных адсорбентов, поэтому, поры, заполненные аргоном существенно сильнее ноглощают электроны, чем углеродная матрица. Этим обеспечивается хороший контрастируюш,ий эффект. 2.Критический диаметр атомов аргона , лч сравнительно невелик (3,67 А). Он меньще, чем критические диаметры частиц других рабочих газов и пикнометрических жидкостей, использующихся в способах порометрии, основанных на применении молекулярных щупов. Используя аргон в качестве рабочего газа можно с его помощью контрастировать все поры с диаметром больше 3,67 А, т. е. подавляющую часть пористой системы обычных микропористых адсорбентов. 3. Обладая температурой кипения 87,3°К, аргон в сравнительно легко достигаемом интервале температур 100°К обеспечивает высокие заполнения большинства известных адсорбентов (см. фиг. 1, на которой изображены изотермы адсорбции аргона на различных адсорбентах). Высокие заполнения пористой системы адсорбента необходимы для ее хорошего контрастирования при электронной микроскопии на просвет. Насыщение аргоном исследуемого адсорбента введут при давлении 10 мм рт. ст. Как |Видно из изотерм фиг. 1, при меньщих давлениях заполнение адсорбента рабочим газом существенно ниже, чем при 10 мм рт. ст., а дальнейшее повышение давления нецелесообразно, т. к. изотермы имеют форму кривых с насыщением и при давлениях выше 10-2 у,м рт ст. увеличение заполнения происходит крайне медленно. Температура адсорбента в процессе насыщения его рабочим газом должна находиться в пределах 40-150°К. При температурах «иже 40°К аргон, находящийся под давлением 10- мм рт. ст. конденсируется в твердую фазу. Создавая твердую корку на поверхности адсорбента, охлажденного до температур ниже 40°К, он не будет проникать внутрь пористой системы. При температурах выше 150°К (150°К - критичеекая точка) степень заполнения адсорбента аргоном невелика из-за высокой интенсивности десорбционных процессов. Наиболее просто обеспечиваемая температура из указанного интервала - это 78°К - температура кипения жидкого азота. После насыщения адсорбента аргоном давление в колонне электронного микроскопа снижается до ЫО мм рт. ст. (верхний предел рабочего давления электронного микроскопа по паспорту). На фиг. 1 приведены изотермы адсорбции аргона на различных адсорбентах при 78° К, где а -удельная эффективная поверхность адсорбента при нормальных условиях; на фиг. 2 приведен электронном-икроскопический снимок от среза углеволокнистого микроаористо го адсорбента при температуре 78°К. Отчетливо виден контрастирующий эффект от адсорбированного газа. Поры, заполненные гадом, видны на позитиве в виде отчетливых темных пятен на фоне более светлой углеродной матрицы. Обработка снимка позволяет получить данные о форме пор и их распределении по размерам. На фиг. 3 приведена кривая распределения диаметра пор по размерам, полученная в результате обмера и обсчета электронномикроскопического снимка, приведенного на фиг. 2. Отчетливо видна бидисперсная структура пористой системы с двумя максимума распределения - при диаметрах 40 А н 180 А. Способ осуществлялся следующим образом. В качестве -адсорбента брали углеродные нити 5 мг. Регенерацию производили «агревом непосредственно в объектодержателе, в камере объектива прибора, в вакууме при давлении тор проходящим электронным пучком повыщенной плотности тока 300 МКА в течение 5 минут. Затем производили .насыщение адсорбента рабочим газом (аргоном) при температуре 78°К. При этом давление только в колоине прибора повышали не выше 2-10-2 тор. Далее производили откачку колонны прибора до давления тор и производили съемку микроснимков. Полученные микроснимки (см. фиг. 2) обрабатывали на полуавтоматическом координатном устройстве. Формула изобретения Способ определения пористости адсорбирующих материалов, заключающийся в нанесении койтрастирующего вещества на поверхность Материала с последующим анализом материала с помощью электронного микроскопа, отличающийся тем, что, с целью повыщения точности « экспрессивности способа, в качестве контрастирующего .вещества берут аргон, насыщение им адсорбирующего материала ведут при- температуре 40-150°К и давлении не выше мм рт. ст., затем -понижают давление аргова до 10 мм рт. ст., после чего производят съемку исследуемой поверхности в проходящем пучке электронов. Источники информации, принятые во внимание при экспертизе 1. К. Г. Бреславец, В. С. Коган. «Криовакуумная адсорбция как метод исследования процессов изменения ультрамикропо56
ристой структуры углеродных адсорбентов2. М. А. Черных и др. «Исследования
при их карбонизации и активации ;:. В сб.дисперсности и пористой структуры нро«Вопросы атом.ной науки и , сериямышленных катализаторов электронйо«Низкотемпературная адсорбция и крио-микроскопическим методом, Журнал Пригеиныи вакуум, Харьков, 1973, вып. 1 (7),5 кладной Химии, 67, 2193, 1974 (прото - -тип).
794437

| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ОПРЕДЕЛЕНИЯ РЕАКЦИОННОЙ ПОВЕРХНОСТИ УГЛЕРОДНЫХ МАТЕРИАЛОВ | 2010 |
|
RU2447423C1 |
| СПОСОБ ПОЛУЧЕНИЯ АДСОРБЕНТА - МОЛЕКУЛЯРНОГО СИТА ДЛЯ СЕЛЕКТИВНОЙ АДСОРБЦИИ АЗОТА И АРГОНА | 2003 |
|
RU2297276C2 |
| СПОСОБ ПОЛУЧЕНИЯ НАНОСТРУКТУРИРОВАННОГО УГЛЕРОДНОГО МАТЕРИАЛА | 2002 |
|
RU2206394C1 |
| Способ селективной адсорбции кислорода | 1980 |
|
SU1028349A1 |
| РАЗДЕЛЕНИЕ И ХРАНЕНИЕ ТЕКУЧИХ СРЕД С ИСПОЛЬЗОВАНИЕМ ITQ-55 | 2015 |
|
RU2672424C2 |
| РАЗДЕЛЕНИЕ И ХРАНЕНИЕ ТЕКУЧИХ СРЕД С ИСПОЛЬЗОВАНИЕМ ITQ-55 | 2015 |
|
RU2675874C2 |
| РАЗДЕЛЕНИЕ И ХРАНЕНИЕ ТЕКУЧИХ СРЕД С ИСПОЛЬЗОВАНИЕМ ITQ-55 | 2015 |
|
RU2674121C2 |
| Способ получения упорядоченного массива углеродных нанотрубок при использовании молекул-координаторов, развития в полученных супрамолекулярных структурах вторичной пористости и материал, полученный этим способом | 2017 |
|
RU2714350C2 |
| Блочный нанопористый углеродный материал для аккумулирования природного газа, метана и способ его получения | 2016 |
|
RU2625671C1 |
| Селективация адсорбентов для разделения газов | 2013 |
|
RU2648074C2 |
§S
50
т200
100 ФигЗ
S,A
