Изобретение относится к приготов лению сорбентов и может быть исполь зовано для разделения и анализа веществ методом хроматографии. Известно использование в качеств сорбентов для хроматографии диатоми тов с физически сорбированным слоем неподвижной жидкой , например гексадекана или сквалана 11. Однако эти сорбентьГ имеют недостаточную термическую стабильность, растворимость неподвижной фазы в элюенте. Наиболее близким к предлагаемому по технической сущности и достигаемому результату является способ получения сорбента для хроматографии, содержащего химически привитые алкильные группы, который состоит в обработка кремнезема алкилхлорсиланами(|2}. Сорбенты, представлякяцие собой кремнезем с химически привитыми алкильными группами, обладают более высокой термической стабильностью, привитый органический слой не смыва |СЯ органическими элементами и устой чив в широком интервале рН. Недостатками известного способа являются необходимость использовани труднодоступных реактивов, сложность и дороговизна техниче ского решения. Цель изобретения - удешевление способа синтеза сорбента и оптимизация свойств химически связанной неподвижной фазы. Поставленная цель достигается тем, что согласно способу получения сорбента на первой стадии кремнезем обрабатывают гексадецилтрихлорсиланом в среде абсолютного органического растворителя при нагревании до температуры кипения, затем экстрагируют последовательно абсолютным неполярньЕМ, полярным растворителями и смесью вода-зталон и дополнительно силилируют поверхность триметилхлорсиланом, гексаметилдисилазаном или их смесью. Первую стадию можно вести в присутствии пиридина или третичного амина. Пример. Юг с илохрома с удельной поверхностью 115 предварительно высушенного под вакуумом в течение 5 ч при 180°С, помеща эт в колбу объемом 250 мл и приливают 100 мл абсолютного ксилола. Затем добавляют 3 мл гексадецилтрихлорсилана и 5 МП сухого пиридина. Реакционную смесь перемешивают в течение
8 ч при 1400С. Продукт реакции промывают сухим бензолом, ацетоном (порциями по 300 ма), затем 500 мл смеси подкисленной воды с эталоном и 500 мл воды. Сорбент сушат в течение 3 ч под вакуумом при TCNffiepaтурё кипения смеси. Полученный сорбент последовательно экстрагируют сухими бензоло и эфиром и сушат под вакуумом при 180°С втечение 3
Пример 2. В условиях примера 1 первую стадию проводят без пиридина в течение 18 ч.
П римерЗ. В условиях приме ра 1 вместо силохрома применяют силикагель марки КСК-2 с удельной поверхностью 250-300 и 10 мл гексадецилтрихлорсилана.
Пример4, В условиях примера 3J вместо силикагеля марки КСК-2 используют силикагель марки С-3.
п р и м е р 5. Исследования на дериватографе показали, что сорбен устойчив вплоть до температуры на воздухе и. до температуры, в атмосфере аргона.
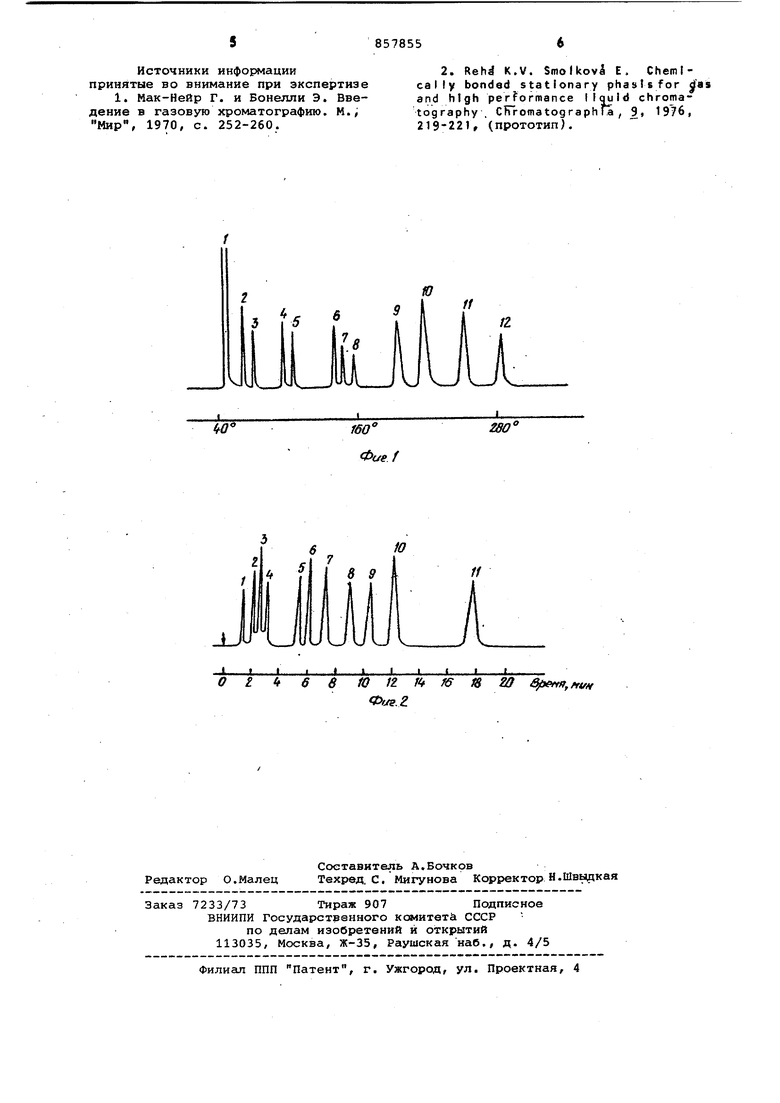
Пример. На фиг. 1 представлена хроматограмма, полученная на газовом хроматографе Цвет 4. Разделение проводят на колонке из стекла длиной 50 см и внутренним диаметром 4 мм. Сорбент-силохрсм С-80 с привитой Qj -фазой, размер чтиц 0,16-0,20 мм, газ-носитель азот, расход 45 мл/мин, программирование температуры осуществляют от 40 до со скоростью . На хроматограмме изображены следующие пики: 1 - растворитель (ацетонэтанол) , 2 - бензол, 3 - октен-1,
4- этаноламин, 5 - 2 - октанон,
б - 3 - хлорметилгентан, 7 - бензилхлорид, 8 - анилин, 9 - три-н-бутиламин, 10 - октановая кислота, 11 - . бромнафталин, 12 - н-октадекан.
Примерб. На фиг, 2 представлена хрсялатограмма , полученная на жидкостном хрсялатографе Цвет-304
Разделение сульфамидных препаратов проводят в следующих условиях: элюент 10 Т ZnSO,, 0,025% С -даена, 1% а,цетата аммония в смеси ацетонитрил, вода (35:65), колонка длиной 15 см и внутренним диаметром 4 мм, сорбент - силикагель КСК-2 с Ц -привитой фазой, диаметр частиц сорбента 1 О мкм. На хроматограмме (Фиг. 2 ) представлены следующиепики: 1 - .сульф ацетамид, 2 - сульфаниламид, 3 сульфабензамид, 4 - сульфизоксазол,
5- сульфахиноксалин, 6 - сульфапиридин, 7 - сульфадиазин, 8 - сульфамеразин, 9 - сульфамвтоксин, 10 - сульфатиазол и 11 - сульфадимегоксин.
П р и м е р 9. Анализ сорбентов на углерод показывает (таблица), что поверхностная плотность привитых алкильных радикалов не зависит от применяемого кремнезема и составляет 1,9-2,2 групп/нм.

| название | год | авторы | номер документа |
|---|---|---|---|
| Сорбент для хроматографии | 1980 |
|
SU928223A1 |
| Сорбент для хроматографии | 1979 |
|
SU928222A1 |
| Способ получения сорбента для хроматографии | 1980 |
|
SU936986A1 |
| Способ получения сорбента для хроматографии | 1979 |
|
SU857860A1 |
| Способ получения сорбента для жидкостной хроматографии | 1990 |
|
SU1775662A1 |
| Способ получения сорбента для разделения биологических жидкостей жидкостной хроматографией | 1987 |
|
SU1478112A1 |
| Сорбент для хроматографии оптических изомеров аминокислот | 1983 |
|
SU1132965A1 |
| Способ получения сорбента для жидкостной хроматографии | 1990 |
|
SU1721504A1 |
| Способ получения кремнезема с фосфонильными группами на поверхности | 1980 |
|
SU945156A1 |
| Способ получения сорбента для разделения биологических жидкостей | 1991 |
|
SU1788463A1 |
Пз приведенных примеров вчдно, чти предлагаемый способ синтеза дает возможность воспроизводимо получать ,углеводородное покрытие плотным монослоем (поверхностная плотность составляет 1,9-2,2 групп/нм). Дополнительные исследования показали также, что уменьшение длины цепи привитых углеводородных радикалов значительно снижает нагрузочную емкость сорбентов, а применение более длинных алкильных цепей приводит к усложнению синтеза, а следовательно, к его удорожанию.
Формула изобретения Способ получения сорбента для хроматографии обработкой кремнеземов
кремнийорганическими .соединениями в среде абсолютного органического растворителя при нагревании до температуры кипения последнего и дополнительного силилирования, о т л и ч аю щ и и с я тем, что, с целью удешевления способа синтеза и оптимизации свойств химически связанной неподвижной фазы, в качестве кремнийорганического соединения используют гексадецилтрихлорсилан.
Источники информации принятые во внимание при экспертизе 1. Мак-Нейр Г. и Бонелли Э. Введение в газовую хроматографию. М.; Мир, 1970, с. 252-260.
0
tIt
0 « S S Ю 1г Itt f6 j W eftef t,HUM
160 Фае f
11
III
Фиг,1
