Изобретение относится к высокоэффективной жидкостной хроматографии (ВЭЖХ), в частности к получению химически модифицированных сорбентов для ВЭЖХ, и может быть использовано для определения низкомолекулярных гидрофобных примесей (например, лекарств) в биологических жидкостях.
Известен способ получения сорбента, заключающийся в последовательной сорбции на поверхности упакованного в хрома- тографическую колонку силикагеля с привитыми октадецилдиметилхлорсилиль- ными группами, бычьего сывороточного альбумина (БСА) и белков плазмы крови из фосфатного буферного раствора (рН 7,4).
Недостатком указанного способа является невысокая стабильность и однородность слоя БСА, что приводит к изменению хроматографических свойств сорбента в процессе эксплуатации, особенно при использовании градиентного элюирования. Кроме того, в указанном способе затруднено получение сорбента с однородной по длине колонки поверхностью, особенно при использовании частиц силикагеля малого размера.
Перечисленных недостатков лишен способ получения сорбента, заключающийся в последовательной обработке кремнезема с диаметром пор 6-10 нм. у-глицидоксип- ропилтриэтоксисиланом в водном буферном растворе, активирующим веществом (п-толуолсульфохлоридом), пептидом (используют глицил-1-лейцин) и ферментом (па- паином), причем после обработки
XI
оо оо
N О СО
кремнезема у-глицидоксипропилтриэток- сисиланом, производится раскрытие эпок- сидных групп в кислой среде (рН 1,5) при кипячении в течение 1 ч. Однако указанный сорбент весьма трудно получить с воспроизводимыми хроматографическими характеристиками (например, количество сорбируемого гемоглобина) из-за многостадийное™ модифицирования его поверхности. Кроме того, в слое модифицирующих внутреннюю поверхность привитых групп, имеются вторичные аминогруппы, что обусловливает отличие такого модифицирующего слоя от модифицирующего слоя ODS-сорбентов, обычно применяемых для анализа лекарственных препаратов и их метаболитов в очищенных от белков пробах биологических жидкостей методом обра- щенно-фазной ВЭЖХ. Это приводит к изме- нению порядка выхода некоторых лекарственных препаратов по сравнению с порядком выхода при использовании традиционных методик с ODS-сорбентами.
Целью изобретения является упрощение получения сорбента и улучшение воспроизводимости его хроматографических свойств.
Поставленная цель достигается предлагаемым способом получения сорбента, заключающемся в последовательной обработке кремнезема с диаметром пор 6- 10 нм алкилсиланом, раствором белка с молекулярной массой 40-90 тыс. дальтон и глутаровым альдегидом.
В качестве белков с молекулярной массой 40-90 тыс. дальтон используют, например овальбумин, бычий или человеческий (ЧСА) сывороточный альбумин, что соответствует размеру белковых глобул 9-15 нм в изоэлектрической точке данного белка.
Использование белков с меньшими размерами и молекулярной массой, например, цитбхрома С, резко снижает коэффициент емкости, а использование белков с большей молекулярной массой, например, фибриногена (330 тыс. дальтон), не позволяет проводить анализ лекарственных препаратов в плазме крови, поскольку некоторые из содержащихся в плазме белков имеют размеры меньшие, чем образующиеся в результате обработки сорбента окна во внешнем защитном гидрофильном слое из белковых глобул.
Отличие предлагаемого способа состоит в том, что в качестве модификатора используется алкандиметилхлорсилан, в качестве биологических молекул овальбумин, БСА или ЧСА, с последующим добавле- нием глутарового альдегида, причем обработка сорбента белком и глутаровым
альдегидом проводится в буферном растворе вблизи изоэлектрической точки используемого белка.
Пример 1. 2,5 г силикагеля Si-300 (Лахема, ЧСФР) со средним диаметром пор 10 нм и частицами диаметром 15 мкм с гид- роксилированной поверхностью помещают в абсолютный толуол, добавляют 0,4 мл мор- фолина и нагревают на песчаной бане до
0 140°С при медленном перемешивании механической мешалкой. В суспензию заливают 2 мл свежеперегнанного октадецилдиметилхлорсилана и перемешивают смесь при той же температуре в тече5 нии 2 ч. Затем продукт последовательно отмывают на стеклянном фильтре абс. толуолом, продажным толуолом, ацетоном смесью ацетона и воды (60 : 40), ацетоном и эфиром, а затем высушивают.
0 Полученный сорбент промывают 0,1 М натрийфосфатным буферным раствором с рН 5,15 и помещают в реакционный сосуд, снабженный мешалкой и нагревают на водяной бане до 40°С. 0,2 г человеческого сы5 вороточного альбумина (ЧСА) (Реанал, Венгрия) растворяют в 15 мл того же буферного раствора и заливают в реакционный сосуд с сорбентом. Полученную суспензию перемешивают при указанной температуре
0 Зч.
Полученный сорбент с адсорбированным ЧСА промывают на стеклянном фильтре 0,1 М натрийфосфатным буферным раствором (рН 5,15), суспендируют в 20 мл
5 того же буферного раствора и добавляют 60 мл 5% водного раствора глутарового альдегида. Суспензию перемешивают при 40°С 6 ч. Затем сорбент окончательно промывают на стеклянном фильтре сперва 0,1 М на0 трийфосфатным буферным раствором рН 5,15, затем 0,05 М натрийфосфатным буферным раствором с рН 7,0, и 40% раствором изопропилового спирта в 0,05 М натрийфос- фатном буферном растворе с рН. 7,0.
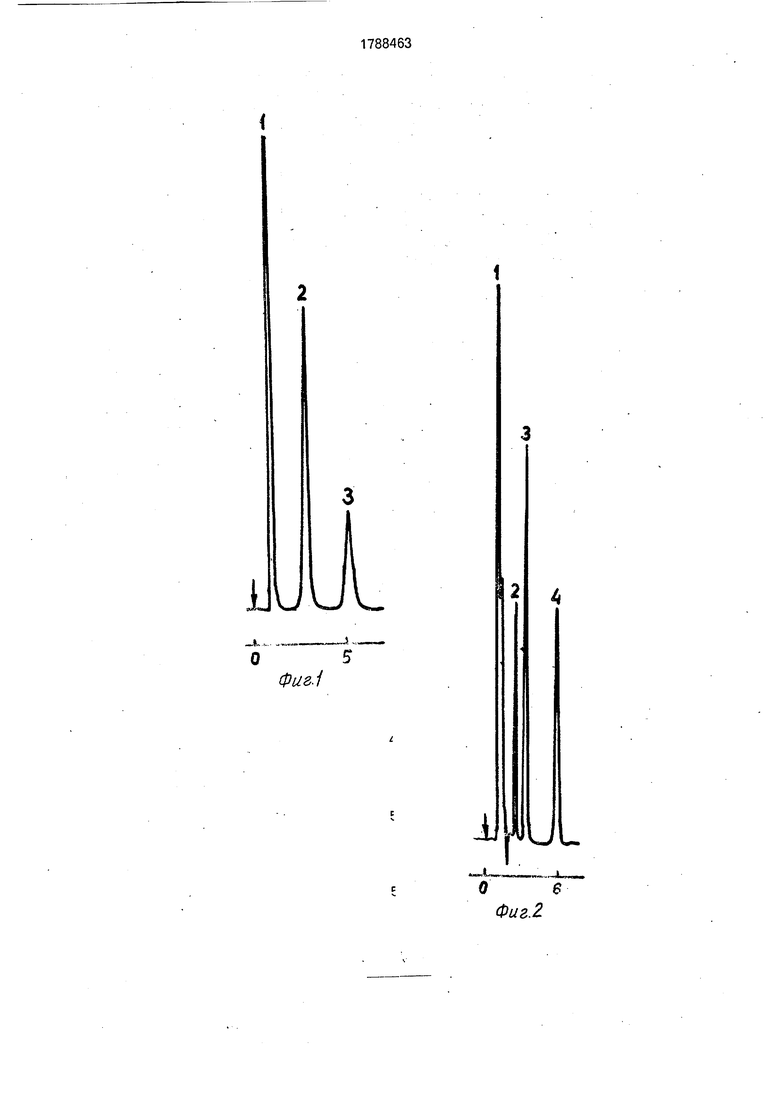
5 На фиг. 1 приведено хроматографиче- ское разделение лекарственных препаратов в человеческой плазме при прямом вводе на колонку с Силасорбом Si-ЗОО (размер частиц 15 мкм), обработанным по предлагаемому
0 способу, размер колонки 150 х 4 мм, На хро- матограмме: 1 - белки человеческой плазмы, 2 - теофилин, 3 - кофеин. Подвижная фаза 0,02 М натрийфосфатный буферный раствор рН 6,50 - ацетонитрил (90 : 10),
5 расход 1 мл/мин, детектор - УФ (254 нм), обьем образца 20 мкл.
Пример 2. Синтез сорбента проводят так же, как в примере 1. Отличие состоит в том, что в качестве исходного кремнезема используют силикагель КСК-Г с размером
частиц 10 мкм, а в качестве алкилсилана используют гексадецилдиметилхлорсйлан.
На фиг. 2 приведено хроматографиче- ское разделение лекарственных препаратов в человеческой плазме при прямом вводе на колонку с силикагелем КСК-Г, обработанным по предлагаемому способу. Размер колонки и условия хроматографиче- ского разделения такие же, как в примере 1. На хроматограмме: 1 - белки плазмы человеческой крови, 2 - ацетилсалициловая кислота, 3 - теофиллин, 4 - кофеин. Объем образца 20 мкл.
Пример 3. Синтез сорбента проводят так же, как в примере 1. Отличие состоит в том, что в качестве алкилсилана используют октилдиметилхлорсилан.
На фиг. 3 представлено хроматографи- ческое разделение лекарственных препаратов в человеческой плазме при прямом вводе пробы на колонку с сорбентом, полученным по предлагаемому способу. Условия хроматографического разделения, размер колонки и обозначения как в примере 2.
Пример 4, Синтез сорбента проводят так же, как в примере1. Отличие состоит в том, что в качестве исходного кремнезема используют силикагель С-3 с размером частиц 7,5 мкм, а в качестве белка - овальбу- мин.
Хроматографические характеристики сорбента соответствуют хроматографиче- ским характеристикам сорбента по примеру 2.
Пример 5. Синтез сорбента проводят так же, как в примере 1, Отличие состоит в том, что в качестве исходного кремнезема используют силикагель Силасорб Si-600 с размером частиц 10 мкм, а в качестве белка бычий сывороточный альбумин.
Хроматографические характеристики сорбента соответствуют хроматографиче- ским характеристикам сорбента по примеру 2.
Пример 6. (Прототип). 10 гсиликагеля Силасорб Si-ЗОО (ЧССР) со средним диаметром пор 10 нм и частицами размером 15 мкм с гидроксилированной поверхностью помещают в 0,1 М натрий -ацетатный буферный раствор (рН 5,65) и нагревают на водяной бане до 95° С при медленном перемешивании механической мешалкой. Суспензию заливают 15 мл перегнанного у -глици- доксипропилтриэтоксисилана и перемешивают смесь при той же температуре 1 ч. Затем продукт отмывают на стеклянном фильтре водой и кипятят при перемешивании 1 ч в 0,01 М соляной кислоте для образования диольных групп вместо исходных
эпоксидных. Полученный диольный сорбент промывают водой до нейтрального рН, промывают ацетоном, эфиром и 200 мл абсолютного диоксана. Помещают сорбент в реакционный сосуд, снабженный мешал-, кой.
10 г предварительно очищенного и высушенного п-толуолсульфохлорида раство0 ряют в 10 мл абсолютного диоксана и, добавив 15 мл абсолютного миридина, заливают в реакционный сосуд с промытым в диоксане сорбентом. Выдерживают 1 ч при комнатной температуре, осторожно пере5 мешивая. Полученную активированную гли- кофазу промывают на стеклянном фильтре 350 мл диоксана для удаления непрореагировавших остатков реакционной смеси, сушат и промывают карбонатным буферным
0 раствором (0,15 М, рН 8,5).
0,6 г глицин-1-лейцина растворяют в 10 мл того же буферного раствора и добавляют туда же активированную гликофазу. Выдерживают при осторожном перемешивании и
5 при комнатной температуре 2 ч, после чего модифицированный кремнезем отмывают на стеклянном фильтре от непрореагировавшего дипептида и обрабатывают 1 М трис-HCI буферным раствором (рН 8,8) в те0 чении 1 ч.
0,5 гидрохлорида цистеина растворяют в 10 мл дистиллированной воды и доводят рН до 5,3 0,1 М КОН, Помещают в приготовленный раствор тщательно отмытый водой
5 модифицированный кремнезем и снова доводят рН до 5,3. Прибавляют 5 мл раствора, содержащего 0,5 г папаина, и выдерживают при перемешивании 1,5 ч при комнатной температуре. Полученный таким образом
0 сорбент промывают на стеклянном фильтре водой, 300 мл этанола и сушат.
На фиг. 4 приведена хроматограмма разделения лекарственных препаратов в человеческой плазме при прямом вводе пробы
5 на колонку с сорбентом, полученным г© предлагаемому способу, Условия хроматографического разделения, размеры колонки и обозначения как в примере 1.
В табл. 1 представлены сравнительные
0 данные по хроматографическим характеристикам колонок с сорбентами, полученными по заявляемому способу и способу-прототипу. Хроматографические испытания проводили в условиях, описанных в примере 1.
5 Наибольшие значения коэффициента емкости и селективности соответствуют сорбенту по примеру 1, наименьшие - прототипу (пример 6). Сорбенты по примерам 2-5 занимают промежуточное положение.
В табл. 2 приведены данные о количестве сорбируемого из пробы белка для раз
личных партий сорбентов, полученным по методикам, описанным в примере 1 и примере 6 (прототипе).
Из приведенных данных видно, что прототип позволяет получать работоспособный сорбент, но количества сорбируемого из пробы белка от партии к партии сильно меняется. Кроме того, как следует из результатов хроматографических исследований приведенных на фиг. 4 при анализе лекарственных препаратов в человеческой плазме при прямом вводе пробы на колонку с сорбентом, полученным по способу - прототипу (пример 6), белки человеческой крови и ацетилсалициловая кислота выходят из колонки общим пиком 1 не до конца разделенным с пиками теофиллина и кофеина (2 и 3 соответственно).
Этих недостатков лишены сорбенты, синтезированные по примерам 1-5. Как видно из представленных хроматограмм и табл. 1, во всех случаях наблюдается полное разделение компонентов пробы. Большее время удерживания ацетилсалициловой кислоты, теофиллина и кофеина позволяет при необходимости использовать более короткую хроматографическую колонку для выполнения анализа, чем при использовании сорбента, полученного по способу - прототипу. Данные о количестве сорбируемого из пробы белка для различных партий сорбентов, полученных по методике, описанной в примере 1, показывают хорошую воспроизводимость от партии к партии свойств доступной для белков пробы поверхности сорбента. Эти данные приведены в табл.2.
Как следует из результатов исследования, предложенный способ позволяет получать сорбенты, не уступающие по хроматографическим характеристикам прототипу и обладающие лучшей воспроизводимостью хроматографических свойств.
Еще одним достоинством предлагаемого способа является более простое получение сорбента по сравнению с прототипом. После обработки кремнезема кремнийорга- ническим модификатором, в способе прототипе продукт отмывают на стеклянном фильтре водой и кипятят при перемешивании 1 ч в 0,01 М соляной кислоте для образования диольных групп вместо исходных эпоксидных. Полученный диольный сорбент отмывают водой до нейтрального рН, промывают ацетоном, эфиром и 200 мл абсолютного диоксана. В нашем способе эти операции исключены и сразу после обработки кремнезема кремнийорганическим моди- фикатором, продукт отмывают на стеклянном фильтре.
Затем в способе-прототипе полученный сорбент помещают в реакционный сосуд, снабженный мешалкой, 10 г предварительно очищенного п-толуолсульфохлорида растворяют в 10 мл абсолютного диоксана и, добавив 15 мл абсолютного пиридина, заливают в реакционный сосуд с промытым в диоксане сорбентом. Выдерживают 1 ч при комнатной температуре, осторожно пере0 мешивая. Полученную активированную гли- кофазу промывают на стеклянном фильтре 350 мл диоксана для удаления непрореагировавших остатков реакционной смеси, сушат и промывают карбонатным буферным
5 раствором.
В предлагаемом нами способе, эти стадии исключены.
Далее в способе - прототипе и в предлагаемом способе сорбент обрабатывают
0 биологическими молекулами, отмывают на стеклянном фильтре и обрабатывают в способе - прототипе 1 М трис-HCI буферным раствором, а в предлагаемом спосрбе - водным раствором глутарового альдегида.
5- После этого, в предлагаемом нами способе, сорбент промывают на стеклянном фильтре и сушат.
В способе - прототипе 0,5 г гидрохлорида цистеина растворяют в 10 мл дистил0 лированной воды и доводят рН до 5,3. Прибавляют 5 мл раствора, содержащего 0,5 г папаина, и выдерживают при перемешивании 1,5 ч при комнатной температуре. Полученный таким образом сорбент промы5 вают на стеклянном фильтре водой, 300 мл этанола и сушат.
Как видно из приведенного сравнительного анализа, предлагаемый способ получения сорбента значительно проще способа,
0 описанного в прототипе. Сокращение таких операций, как обработка соляной кислотой для образования диольных групп вместо эпоксидных, отказ от активирования сорбента п-толуолсульфохлоридом е абсолют5 ном диоксане и обработка сорбента папаином в буферном растворе, не только упрощают способ, но и обеспечивают лучшую воспроизводимость хроматографических свойств сорбента от партии к партии.
0 Мы связываем это с возможностью точнее контролировать ход синтеза и с отказом от наиболее трудновоспроизводимой стадии обработки поверхности модифицированного дипептидом сорбента ферментом (папаи5 ном). В предлагаемом нами способе после обработки кремнезема кремнийорганическим модификатором и отмывки полученного модифицированного кремнезема, производится сорбция биологических молекул (белков с молекулярной массой 40-90
тыс. дальтон) и закрепление их на поверхности обработкой глутаровым альдегидом.
Таким образом, предлагаемый способ позволяет упростить получение сорбента, улучшить воспроизводимость его хроматог- рафических свойств.
Фор м у па изобретения Способ получения сорбента для разделения биологических жидкостей жидкостной хроматографией, включающий последовательную обработку кремнезема с диаметром пор 6-10 нм кремнийорганиче0
ским модификатором и биологическим веществом, отличающийся тем, что, с целью упрощения способа и повышения воспроизводительное™ хроматографиче- ских характеристик, в качестве кремнийор- ганического модификатора используют алкилсилан, в качестве биологического вещества белки с мол. м. 40-90 тыс. дальтон. при этом обработку ведут в буферном растворе с рН вблизи изоэлектрической точки соответствующего белка и сорбент дополнительно обрабатывают глутаровым альдегидом.




| название | год | авторы | номер документа |
|---|---|---|---|
| Способ получения сорбента для разделения биологических жидкостей жидкостной хроматографией | 1987 |
|
SU1478112A1 |
| Способ определения противосудорожных препаратов в биологических жидкостях | 1987 |
|
SU1476377A1 |
| Хроматографический сорбент для анализа низкомолекулярных соединений в биологических жидкостях | 2023 |
|
RU2818021C1 |
| СОРБЕНТ НА ОСНОВЕ КРЕМНЕЗЕМА | 1993 |
|
RU2040963C1 |
| СОРБЕНТ ДЛЯ УДАЛЕНИЯ ИММУНОГЛОБУЛИНОВ | 2008 |
|
RU2389022C2 |
| Способ приготовления сорбента" | 1977 |
|
SU671385A1 |
| СОРБЕНТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2001 |
|
RU2203730C1 |
| НАНОАЛМАЗНЫЙ СОРБЕНТ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2007 |
|
RU2352387C1 |
| СПОСОБ ПОЛУЧЕНИЯ, ВЫДЕЛЕНИЯ, ОЧИСТКИ И СТАБИЛИЗАЦИИ РЕКОМБИНАНТНОГО ГРАНУЛОЦИТАРНОГО КОЛОНИЕСТИМУЛИРУЮЩЕГО ФАКТОРА ЧЕЛОВЕКА, ПРИГОДНОГО ДЛЯ МЕДИЦИНСКОГО ПРИМЕНЕНИЯ, И ИММУНОБИОЛОГИЧЕСКОЕ СРЕДСТВО НА ЕГО ОСНОВЕ | 2004 |
|
RU2278870C2 |
| Анионообменный материал для разделения биологических макромолекул и способ его получения | 1976 |
|
SU867284A3 |

Использование: для определения низкомолекулярных гидрофобных примесей (например, лекарств) в биологических жидкостях. Способ получения сорбента для раз- деления биологических жидкостей жидкостной хроматографией включает последовательную обработку кремнезема с диаметром пор 6-10 нм кремнийорганиче- ским модификатором и биологическими молекулами, в качестве биологических молекул применяют молекулы белков с молекулярной массой 40-90 тыс. дальтон, обработку проводят в буферном растворе вблизи изоэлектрической точки используемых белков, образующиеся глобулы которых имеют диаметр 9-15 нм, а для закрепления биологических молекул на поверхности используют глутаровый альдегид. 2 табл. 4 ил.
Таблица
Таблица2
|„-.. -О5
Фие.1
1
««Ллют.
О6
Фиг.2.
О3
Фиг.З
О 2
Фиг.Ј
| Imai H | |||
| Yoshidu H., Masajima I | |||
| Anal, letters, 1983, v | |||
| Устройство для электрической сигнализации | 1918 |
|
SU16A1 |
| ЦЕНТРОБЕЖНАЯ ЗЕРНОСУШИЛКА | 1919 |
|
SU1106A1 |
| Yoshida H., Morita T.,Tomaib, v | |||
| Способ изготовления электрических сопротивлений посредством осаждения слоя проводника на поверхности изолятора | 1921 |
|
SU19A1 |
| Электромагнитное реле | 1922 |
|
SU466A1 |
| Способ получения сорбента для разделения биологических жидкостей жидкостной хроматографией | 1987 |
|
SU1478112A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
