(54) СПОСОБ ПОЛУЧЕНИЯ ПЛЕВРОМУТИЛИНОВЫХ ГЛИКОЗВДНЫХ ПРОИЗВОДНЬК
I
Изобретение относится к способу получения новых производных плевромутилина, обладающих ценными фармакологическими свойствами.
Цель изобретения - получение новых соединений, расширяющих арсенал средств воздействия на живой организм.
Поставленная цель достигается путем синтеза, основанного на извест-. ной. реакции 10 , гликозидных производных общей формулы
O-C-CH -R
.2.
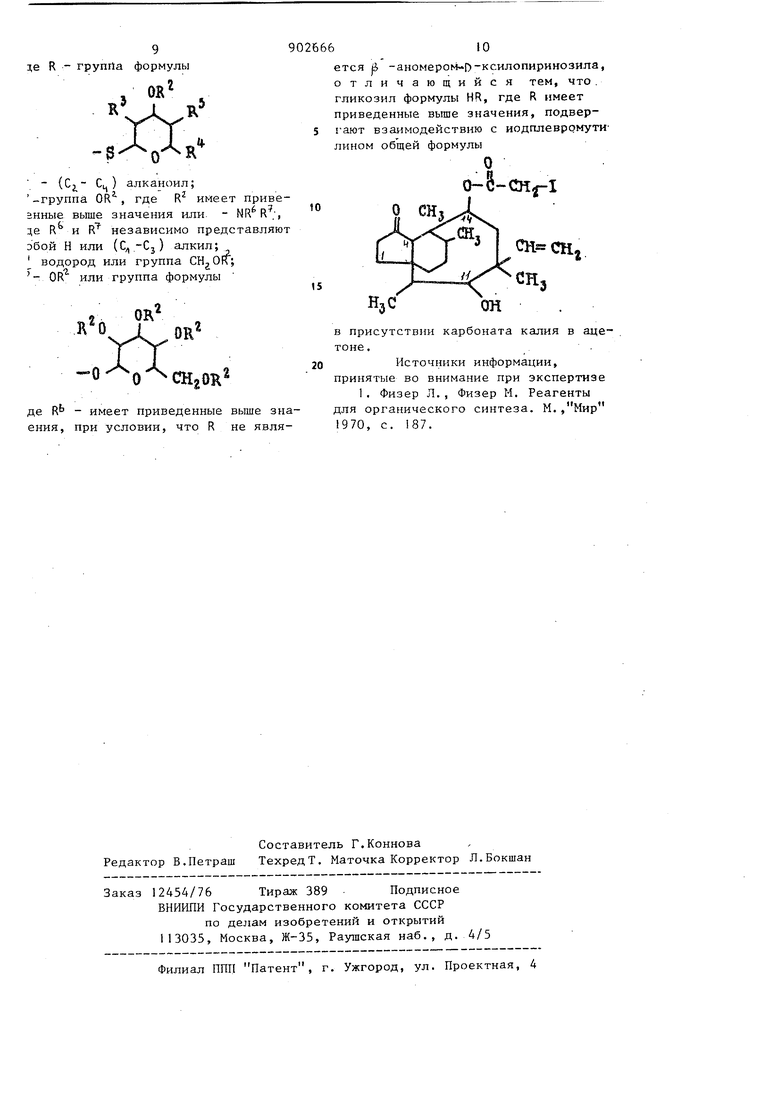
где R - группа формулы 5 5
rV
6
где R - (Cj- Сц) алканоил; R - OR, где R имеет приведенные выше значения или MR R, где R и R независимо представляют собой Н или (Сд-Сз) алкил; ,, Н или группа CHj OR; R - OR или группа формулы
СНгОК
где имеет приведенные вьше значения, при условии, что R не является |5 ганомером D - ксилопирано3V, который заключается во взаимд действии гликозила формулы HR, где R имеет приведенные выше значения, с илевронутилином формулы О СН Cli СН, в присутствии карбоната калия в аце топе. Целевой продукт вьщеляют известными приемами. Предпочтительным вариантом осущестлления способа является добавление раствора иодплевромутилина в ацетоне к раствору производного пер -0-ацилмеркаптосахара. Карбонат каЛИЯ добавляют к смеси при перемешиван ии и перемешивание продолжают. Реакционную смесь экстрагируют мети ендихлоридом и экстрагирующий растворитель удаляют путем выпаривания. Пример i. Получение 4-деокси 14- ( 2,3,4,6- тетра-0-ацетил- Р- D - глюкопиранозил) тиоацетокси мутилина. Раствор иодплевромутилина в коли честве 577,3 кг в ацетоне (2 мл) до бавляют к раствору, состоящему из 2,3,4,6-тетра-О-ацетил- ji- D - глюк пиранозил меркаптана (428,8 мг) в2 мл ацетона. Раствор KjCOj в количестве 168,4 мг в 1 мл воды добавляют к перемешиваемой реакционной смеси и полученный в результате рас вор подвергают перемешиванию при комнатной температуре в течение 30 мин. Полученную реакционную смесь далее выливают в деионизированную воду (25 мл) и водньй раствор экстрагируют хлористым метиленом. Получен ный экстракт сушат безводным сульфа том натрия,, фильтруют и выпаривают под- вакуумом до получения сухого пр дукта. Остаток подвергают сушке в глубоком акууме в течение 1,5ч с, получегшем 989,7 мг 14 - деокси-1 ,4,6 - тетра-0-ацетил-р-D глюкопиразонил) тиоацетокси мутили на, М 724. ЯМР: 4 CHg при 0,7(д) 088 (д), 1,17 (с1, 1,94 LC). Пример 2. А. Получение гидробромида 2,3,4-три-О-ацетил-1-тиоуроний- |3 - 0-ксилопиранозы. 2,3,4-Три-О-ацетил- оС- D-ксилопиранозил бромид в количестве 1,3 г ( ммоль), растворяют в 3 мл ацетона. К этому раствору добавляют тиомочевину в количестве 33Q мг (4,33 ммоль). После добавления дополнительного количества ацетона (около 3 мл) полученный раствор нагревают при дефлегмации (на масляной бане при 70С в течение примерно 20 мин) . Продукт кристаллизуют при охлаждении реакционной смеси на ледяной бане. Кристаллы отделяют фильтрацией, промывают минимальным количеством ацетона и сущат с получением 849 мг гидробромида, 2,3,4-три-О-ацетил-1-тиоуроний-Р- 0-ксилопиранозы; т.пл. 174175 С. В. Получение 2,3,4-три-О-ацетил-1-тио- - О-ксилопиранОзы. Воду (5 мл) и четыреххлористый углерод (5 мл) добавляют к гидробромиду 2,3,4-три-О-ацетил-I-тиоуроний-)3-0-ксилопиранозы в количестве 608,4 мг (1,466 ммоль), полученному на стадии А, и 218 мг (1,14 ммоль) NSjiSj O. Реакционную смесь нагревают при дефлегмации в течение 40 мин и затем охлаждают до комнатной температуры. Слой четыреххлористого углерода отделяют. Водный слой дважды промывают 10-милиметровыми порциями четыреххлористого углерода. Фракции объединяют, сушат над безводным сульфатом натрия, фильтруют и упаривают под вакуумом.-сПолучением 212,9 мг 2,3,4-три-О-ацетил-1-тио- -О-ксилопиранозы в виде желтого масла. Этот продукт подвергают дальнейшей очистке на колонке с силикагелем, получая 332,3 мг масла, которое кристаллизуют; т.пл. 117-122С . С. Получение .14-деокси-14-С С 2,3, 4 - три-О-ацетил- -О-ксилопиранозил) тиоацетоксиЗ 1 утилина. « 2,3,4-три-О-ацетил-1-тио- -Р-ксилопиранозу в 1,46 г (5 ммоль), полученную на стадии В, растворяют в 10 мл ацетона.Добавляют раствор 2,48 г (5,08 ммоль) иодплевромутилина в 10 мл ацетона. К перемешиваемой реакционной смеси добавляют растBOD 721 мг (5,19 ммоль) в 5мл воды. Полученный раствор пе ремешивают при комнатной темпера туре в течение 20 мин и далее выливают в деионизированную воду (100 мл). Этот раствор экстрагируют хлористым метиленом. Получен ный раствор сушат безводным сульфатом натрия, фильтруют упаривают досуха под вакуумом, а далее сушат пр глубоком вакууме в течение 8 ч с получением 3,8 г продукта в виде бело пены, которую кристаллизуют или из смеси диэтилового эфира и гексана или диэтилового эфира и этилацетата т.пл. 91-97°С. Масс-спектр; . ЯМР:4 CHj при 0,74 Сд) ,0,89(д) , 1,18(с) , 1,96 (.с) , Пример 3. Получение 14-деокси-14- (2, 3,4-три-0-ацетил-| - D-арабинопиранозил) тиоацетокси мутилина. 2,3,4-Три-О-ацетил-1-тио- -D-apaбинозу в количестве 3,177 г (0,0109 МОЛБ), полученную как описано в примере 2 (стадии А и в),растворенную в 20 мл ацетона,и 5,31г(0,0109 моль) иодплевромутилина, также растворенного в ацетоне (20 мл) , смешивают вместе. Добавляют 1,506 г , l/ff,0109 моль) KjCOj в 10 мл воды. Полученный раствор перемешивают при комнатной температуре в течение 30 мин и далее выпива1от в воду (100 мл) Водный раствор экстрагируют три раза порциями по 50 мл хлористого метилена. Экстракты соединяют, сушат безводным сульфатом натрия и фильтруют; фильтрат упаривают под вакуумо получая неочищенный продукт в количестве 7,1 г. Этот продукт подвергают очистке методом жидкостной хроматографии с высокой разрешающей способностью, элюирование осуществляют с помощью смеси, состоящей из толуол а этилацетата в соотношении 1:1; ратворяющую систему подают со скоростью 250 мл/мин; собранные фракции имеют объем 250 мл. Содержание фракции определяют с помощью тонкослойной хроматографии на силикагеле,используя в качестве растворителя смес толуола и этилацетата (1:1) , в качестве детектора - иод.Фракции 17-22 содержат максимальное количество очищенного продукта. Эти фракции с образованием 4,989 г 14-дeoкcи-Ji4объединяют и упаривают под вакуумЪм - 2, 3,4-три-0-ацетш1-(- -арабинопиранозил)тиоацетокси мутилина. Г 653; ЯМР: 4 СН при 0,74 Са) , 0,89 eg) , 1,i6 Сс), 1,46 Сс). Выход 70%. Пример 4. Получение 14-де окси-14- (2,3,4-три-0-ацетил- -1арабинопира1фзил тиоацетокси мутилина. 4,636 г (0,0095 моль) иодплевро- мутилина подвергают взаимодействию с 2,3,4-три-0-ацетш1-1-тио-L-apaOH-нозой в количестве 2,76 г (0,0095 моль в соответствии с методикой, опи санной в примере 3, с получением 6,265 г неочищенного продукта, которьш очищают путем жидкостной хрома тографии с высокой разрешающей способностью, как описано в примере 3, при использовании градиентного растворителя от 4 л толуола до 4 л смеси, состоящей из этилацетата и толуола в соотношении 1:1. Получают . в результате 3,53 г 14-деокси-Ц2,3,4-три-0-ацетио-р-L-арабинопиранозил) тиоацетокси мутилина. М 652. ЯМР:4 СНз при 0,72(д) , 0,92 (о) , 1,17 (,с), 1,45 (с). Выход 54%. Пример 5. Получение 14-деокси- 14- (2,3,4,6-тетра-О-ацетил- -О-галактопиранозил) тиоацетоксиТ му- тилина. 9,27 г (о,019 моль) иодплевромутилина подвергают взаимодействию с 2,3,4, 6-тетра-О-ацетил- j5 - D гапактопиранозилмеркаптаном в количестве 6,96 г (05019 моль в соответствии с методикой, описанной в 1.римере 1, получая 14,14 г неочищенного пр одукта. Этот продукт очищают путем жидкостной хроматографии с высокой разрешающей способностью, как описано в примере 3, с использованием -градиентного растворителя от 4 л толуола до 4 л смеси толуоа и этилацетата в соотношении 1:1 8л), применяя тонкослойную хроматографию. Получают 3,99 г 14-деокси-14(2, 3,4,6-тетра-0-ацетил- -0-галактоиранозил)тиоацетокси мутилина. ЯМР: 0,74 (9) , 0,89(д) ., 1, 17 (с), 1,45 (с). Выход 29%. Пример 6. 14-Деокси-14- (.2, 3,4-три-0-ацетил-6-1-ксилопирано зил тиоацетокси мутилин приготавлиают согласно методике, описанной примере 2, но в качестве исходно79I о материала используется L ксилоза. На заключительной стадии 31- г иодп.р-промутилина вводят в реакцию с 2,1, 4 три-0-ацетил-1-тио-р -Д. -ксило зой в количестве 9 г,получая39,6 г неочищенного продукта. Этот продукт подвергают очистке с помощью жидкост ной хроматографии с высокой разреша ющей способностью, как описано в при мере 3, но с использованием градиент но1о растворителя (8 л) от толуола до смеси толуола и этилацетата (7:3). Очищенные фракции кристаллизуют из смеси толуол-этилацетат, получая 21,05 г и-деокси-14-(,2,3,4-три-0-aцeтил-p-Lкcилoпиpaнoзилj тиоацеi; мутилена; т.пл. 210-213 С; + 653. ЯМР: 4 CHj при 0,73 (д) 0,88 (д), 1,77 (с), 1,46 LC). Выход, 49%. Пример 7. Получение 14-деокси-14- (2-деокси-2 (М,М-диметиламино -3,4, 6-три-0-ацетил-/3-р-глюкопиранозил) - тиоацетокси1мутилина. К раствору 10,8 г (0,05 моль) С-глюкозамингидрохлорида в 250 мл воды добавляют 250 мл 37%-ного водного раствора формальдегида и 5 г (10%)палладия на угле. Полученную смесь гидрируют до тех пор, нока не достигается теоретическое поглощение для превращения в 2-деокси-2(N,М-диметиламино)-0-глюкозамин. Катализатор удаляют методом фильтрации и фильтрат лиофилизуют. Полученный продукт ацетилируют уксусным ангидридом и пиридином, получая соответствующее тетра-0-ацетилпроизводное Это вещество превращают в 2деокси-2-(М,Ы-диметиламино)-3,4,6тpи-0-aцeтил-D-глюкoпиpaнoзилбpoмид, который далее переводят в соответствующее 1-меркаптопроизводное в соответствии с методикой, описанной в примере 2. Взаимодействие 2-деокси-2-(N,N-димeтилaминo)-3,4,6-три-Оацетил-1-тио-)-глюкопиранозы с иодплевромутилином. в соответствии с методикой, описанной в примере 2 (стадия С) дает целевой продукт; М 728. ЯМР:4 CHj при 0,70(.д)., 0,85 (д), 1,14 (с), 1,83 (с); CH(CH.j при0,91(д). Пример 8..Получение 14-деокси-14- (4-0-(2,3,4,6-тетра-О-ацетш1-1-D-глюкопиранозил)-2,3,6-три-Оацетил- Д-0-глюкопиранозип1 тиоацетоксиЗ мутилина. Указанное соединение получают в с методикой, описанной . соответствии в примере 2, за исключением того. что в качестве исходного материала используют окса-0-ацетат мальтозы. 8,6 г иодплевромутилина подвергают взаимодействию с 4-0-(2,3,4,6-тетра-0-ацетил-о(Е-глюкопиранозил) -2,3, 6-три-О-ацетил-1тиол- -0-глюкопиранозой в количестве 11,8 г, полученной в соответствии с методикой, описанной в примере 2, получая 17 г продукта в виде белой пены. Этот продукт подвергают дальнейшей очистке методом жидкостной хроматографии с высокой разрещающей способностью при использовании 8 л градиентного растворителя от этилацетата до смеси этилацетата и этанола (l : I) получая 1,12 г продукта. М 1012. ЯМР: 4 СН при 0,74 (g),0,90{g), 1,20 (с) , т 1,46 (с). Выход 6%. Новые плевромутилиновые гликоэидные производные являются ценными препаратами против грамположительных и грамотрицательных бактерий , анаэробных бактерий и микоплазмы. Предлагаемые плевромутилиновые гликозиды представляют собой относительно нетоксичные соединения; так, например, 50%-ная смертельная доза (LDso ) для 14-деокси-14-(р-Р-ксилопиранозил ) тиоацетокси} мутилина и 14-деокси-1.(. -Р-ксилопиранозил) тиоацетокси -19,20-дигидромутилина при вводе в брюшину мышей составляет более 1500 мг/кг живого веса, а 50%-ная смертельная доза (LDjo ) для 14-деокси-14-- { 3,4,6-три-0-ацетил-2-деокси-2- (оксиимино) - -Р-глюкопиранозил) оксиацетокси - 19,20 - дигидромутилина также при вводе в брюшину мышей составляет более 300 мг/кг живого веса. / Формула изобретения Способ получения плевромутилиновых гликозидных производных, общей формулы I 90266 9 т,е R - группа формулы rV - (Cj,- Ц) алканоил; -группа OR , где R имеет привеанные выше значения или. - NR R;, эе R и R независимо представляют эбой Н или (Сл-С, ) алкил; СН, 0#; водород или группа OR или группа формулы Л в прис тоне. принят для ор - имеет приведенные выше зна1970, при условии, что R не явля1. 6 ется jj -аномером-0 ксилопиринозила, отличающийся тем, что. гликозил формулы HR, где R имеет приведенные выше значения, подвергают взаимодействию с иодплевромутилином общей формулы , утствии карбоната калия в ацеИсточники информации, ые во внимание при экспертизе Физер Л., Физер М. Реагенты ганического синтеза. М.,Мир с. 187.



| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛИСУЛЬФАТИРОВАННЫЕ ГЛИКОЗИДЫ И ИХ СОЛИ | 2005 |
|
RU2413734C2 |
| ТЕТРАВАЛЕНТНЫЕ НЕОГЛИКОКОНЪЮГАТЫ С УГЛЕВОДНЫМ РАЗВЕТВЛЯЮЩИМ ЯДРОМ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2014 |
|
RU2575925C1 |
| Способ получения производных микофенольной кислоты | 1974 |
|
SU578006A3 |
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |
| ПРОИЗВОДНЫЕ СПИРОКЕТАЛЕЙ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ ДИАБЕТА | 2006 |
|
RU2416617C2 |
| ПРОИЗВОДНЫЕ АРИЛ 5-ТИО-β-D-ГЛЮКОПИРАНОЗИДА И ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА ПРИ ДИАБЕТЕ, СОДЕРЖАЩИЕ ИХ | 2003 |
|
RU2322449C2 |
| СПОСОБЫ ХИМИЧЕСКОГО СИНТЕЗА ФИЛЛИРИНА | 2014 |
|
RU2667917C2 |
| НОВЫЕ ПЕНТАСАХАРИДЫ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ | 1999 |
|
RU2193040C2 |
| СПОСОБ ПОЛУЧЕНИЯ N-ГЛИКОЗИДОВ ИНДОЛО[2,3-а]ПИРРОЛО[3,4-с]КАРБАЗОЛ-5,7-ДИОНОВ, ОБЛАДАЮЩИХ ЦИТОТОКСИЧЕСКОЙ И ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2009 |
|
RU2427585C9 |
| С-ФЕНИЛ-1-ТИОГЛЮЦИТОЛЫ | 2007 |
|
RU2434862C2 |
