Область техники, к которой относится изобретение
Изобретение относится к области фармакологической химии, и в частности, относится к способу химического синтеза филлирина.
Преимущества и практическая значимость способа химического синтеза филлирина заключаются в следующем: легко получить сырьевой материал, катализаторы для гликозилирования являются дешевыми и легкодоступными, затраты на производство существенно снижаются, и его можно применять для промышленного производства.
Предшествующий уровень техники
Плоды форзиции являются сухими плодами Форзиции пониклой (сем. Маслиновые) (Forsythia suspensa (Thunb.) Vahl (Oleaceae)), которая растет главным образом в провинциях Хенань, Шаньси, Шаньдун и других районах Китая, а также провинциях Хубэй, Хэбей, Сычуань и Ганьсу. Форзицию обычно применяют для лечения простуды, фурункулов и язв, туберкулезного лимфаденита, инфекции мочевых путей, и т.д. Главным ингредиентом плодов форзиции является филлирин с антивирусным, антибактериальным, антиоксидантным эффектом, способностью к удалению свободных радикалов, противоопухолевым эффектом и другими фармакологическими эффектами. В настоящее время имеются отчеты о многочисленных исследованиях по экстракции филлирина из натуральной форзиции; источники медицинских растений все больше истощаются, и содержание эффективного ингредиента становится относительно низким; таким образом, химический синтез филлирина может значительно снизить затраты, повысить выход, и играет роль в защите растительных ресурсов.
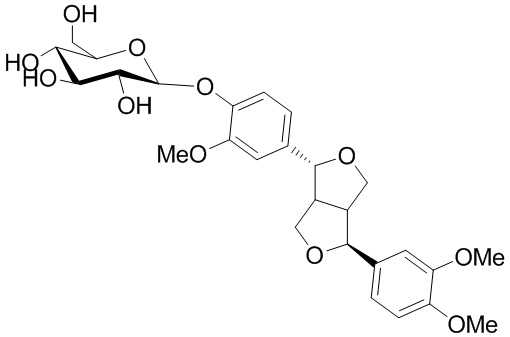
Филлирин
Химический синтез филлирина разработан в 2014, Fan Hongyu et al., с применением 1-бромо-тетра-О-ацетил-альфа-D-глюкозы и филлигенина для проведения гликозилирования, катализируемого катализатором фазового переноса, и основания, и с применением натрия метоксида для снятия защиты для генерации филлирина [Fan Hongyu, Fu Li, «Synthesis and Structure Characterization of Phillyrin», Liaoning Chemical Industry, 2014, 43, 241-243 («Синтез и структурная характеристика филлирина»)]; однако, выход синтеза в этом способе является относительно низким, необходимы пента-ацетил-бета-D-глюкоза и 33% бромоводородная кислота для бромирования с целью получения 1-бромо-тетра-О-ацетил-альфа-D-глюкозы, и бромоводородная кислота не облегчает работы из-за коррозии.
Сущность изобретения
Изобретение обеспечивает способ синтеза филлирина, нацеленный на решение технических проблем в существующем способе химического синтеза филлирина. Способ по настоящему изобретению позволяет преодолеть недостатки предшествующего уровня техники с высоким выходом синтезированного продукта филлирина. Способ по настоящему изобретению характеризуется простой операцией и технологическим процессом, коротким периодом получения, высоким содержанием филлирина в синтезированном продукте, высоким выходом и явно сниженными затратами на производство филлирина; и применим для периодического процесса и промышленного производства.
Для целей настоящего изобретения в одном аспекте изобретение обеспечивает способ химического синтеза филлирина, включающий следующие этапы:
1) растворение акцептора гликозила филлигенина и донора гликозила в первом органическом растворителе, и проведение гликозилирования для получения тетраацил-филлирина;
2) растворение тетраацил-филлирина во втором органическом растворителе, затем добавление натрия метоксида для деацилирования, добавление кислотного регулятора рН для доведения значения рН реакционной смеси до нейтрального, и проведение очистки для получения филлирина;
где температура гликозилирования на этапе (1) составляет 0-20°С, предпочтительно 0-10°С, и более предпочтительно 0°С; а время реакции гликозилирования на этапе (1) составляет 4-15 часов, предпочтительно 8-10 часов, и более предпочтительно 10 часов.
В частности, 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидат применяют в качестве донора гликозила; дихлорметан, трихлорметан, 1,2-дихлорэтан или толуол, предпочтительно дихлорметан, применяют в качестве первого органического растворителя.
В частности, 2,3,4,6-тетра-О-ацетил-D-глюкопиранозил трихлорацетимидат или 2,3,4,6-тетра-О-бензоил-D-глюкопиранозил трихлорацетимидат применяют в качестве 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидата.
При этом молярное отношение 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидата к филлигенину составляет 1,0-5,0:1.
В частности, во время гликозилирования из настоящего изобретения, применяемое количество донора гликозила 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидата является малым, выход продукта гликозилирования является низким, побочные продукты возрастают при повышении используемого количества, а молярное отношение донора гликозила 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидата к филлигенину предпочтительно составляет 1,0-5,0:1.
В частности, после этого акцептор гликозила филлигенин и донор гликозила растворяют в органическом растворителе для гликозилирования в присутствии катализатора.
При этом в качестве катализатора применяют кислоту Льюиса.
В частности, один или несколько из С3-С9 галоацетамидов, С2-С8 силил-фторгидрокарбонил-сульфоната, С1-С6 серебра фторгидрокарбонил-сульфоната и бора трифторид-этерата, предпочтительно N-йодосукцинимид, серебра трифторметансульфонат, триметилсилил-трифлат или бора трифторид-этерат, и более предпочтительно серебра трифторметансульфонат, триметилсилил-трифлат или бора трифторид-этерат применяют в качестве катализатора на основе кислоты Льюиса.
При этом молярное отношение катализатора на основе кислоты Льюиса к донору гликозила 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидату составляет 1:1,0-10,0.
Низкое применяемое количество катализатора на основе кислоты Льюиса приводит к разрушению донора гликозила 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидата, и к снижению выхода; высокое применяемое количество катализатора на основе кислоты Льюиса приводит к разрушению донора гликозила тетраацил филлирина и к снижению выхода.
В частности, молярное отношение катализатора на основе кислоты Льюиса к донору гликозила 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидату предпочтительно составляет 1:5,0-10,0, более предпочтительно 1:5-6, и еще более предпочтительно 1:5.
В частности, затем акцептор гликозила филлигенин и донор гликозила растворяют в органическом растворителе для гликозилирования под защитой инертного газа и в присутствии катализатора.
При этом инертным защитным газом является азот, аргон или гелий, предпочтительно азот.
В частности, затем акцептор гликозила филлигенин и донор гликозила растворяют в органическом растворителе для гликозилирования под защитой инертного газа и в присутствии катализатора и молекулярного сита.
При этом в качестве молекулярного сита применяют алюмосиликатное молекулярное сито или алюмосиликатный порошок.
В частности, в качестве алюмосиликатного молекулярного сита применяют алюмосиликатное молекулярное сито 3Å-5Å типа, предпочтительно алюмосиликатное молекулярное сито 4Å типа.
В частности, применяемое количество молекулярного сита удовлетворяет требованию, чтобы массовое отношение молекулярного сита к филлигенину было 1-10:1, предпочтительно 2:1.
В частности, способ также включает этап (1А) гашения гликозилирования посредством гасящего агента перед растворением тетраацил-филлирина во втором органическом растворителе.
При этом триметиламин, триэтиламин или натрия тиосульфат применяют в качестве гасящего агента.
В частности, применяемое количество гасящего агента удовлетворяет требованию, чтобы молярное отношение гасящего агента к кислоте Льюиса составляло 1:1-3, предпочтительно 1:1-1,5, и более предпочтительно 1:1.
При этом смешанный раствор дихлорметана и метанола применяют в качестве второго органического растворителя на этапе (2).
В частности, объемное отношение дихлорметана к метанолу в смешанном растворе дихлорметана и метанола составляет 1:1-10, предпочтительно 1:2.
При этом молярное отношение натрия метоксида к 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидату составляет 1:300-500, предпочтительно 1:375-500.
В частности, время деацилирования составляет 4-12 часов, предпочтительно 4 часа.
В частности, уксусную кислоту, пропионовую кислоту или соляную кислоту, предпочтительно уксусную кислоту, применяют в качестве кислого регулятора рН.
В частности, значение рН реакционной смеси доводят до 6-7.
Схема химической реакции химического синтеза филлирина в настоящем изобретении является следующей.
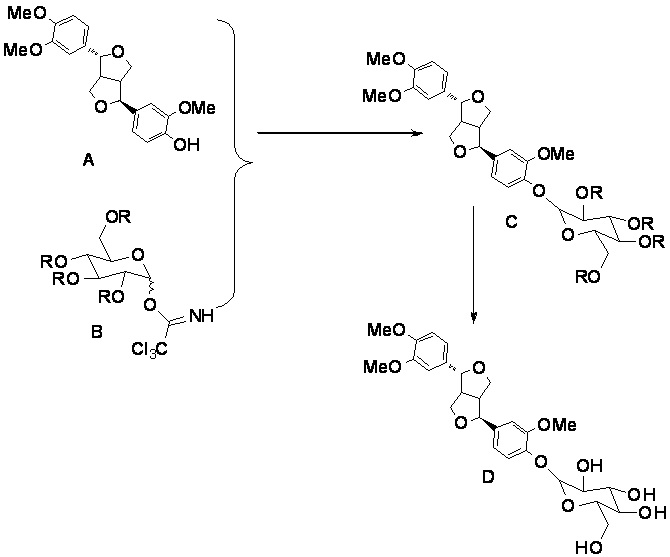 ,
,
где структурная формула А представляет собой филлигенин; структурная формула В представляет собой 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидат; структурная формула С представляет собой тетра-ацилфиллирин; структурная формула D представляет собой филлирин.
Преимущества и практическая значимость способа химического синтеза филлирина заключаются в следующем: легко получить сырьевой материал, катализаторы для гликозилирования являются дешевыми и легкодоступными, затраты на производство существенно снижаются, и его можно применять для промышленного производства.
Подробное описание изобретения
Изобретение далее описано посредством следующих примеров, однако эти примеры являются просто иллюстрациями настоящего изобретения и не должны рассматриваться как ограничивающие объем настоящего изобретения. Далее, реагенты и сырьевые материалы в примерах могут быть получены коммерческим путем, если они отсутствуют, можно использовать указания по органическому синтезу, указания по применению лекарств и инструкции от производителей соответствующей аппаратуры и реагентов.
Вариант осуществления 1
1) Гликозилирование
Филлигенин (372 мг, 0,001 моль) и 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидат (738 мг, 0,0015 моль) вносили в 100 мл трехгорлую колбу, где молярное отношение филлигенина к 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидату составляло 1:1,5; в колбу добавляли 20 мл безводного дихлорметана и алюмосиликатного молекулярного сита 4Å типа (744 мг); затем инертный газ азот вводили в колбу для защиты инертным газом, затем перемешивали в течение 0,5 часа; после однородного перемешивания по каплям добавляли триметилсилил-трифлат в качестве катализатора на основе кислоты Льюиса (ТМСОТф, 0,06 мл, 0,312 ммоль), где молярное отношение катализатора на основе кислоты Льюиса к 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидату составляло 1:5, массовое отношение молекулярного сита к филлигенину составляло 2:1; полученную смесь подвергали гликозилированию в течение 10 часов при 0°С при перемешивании.
Реактивное промежуточное соединение, полученное при дегидрогенировании гидроксигрупп субстрата реакции филлигенина с кислотой Льюиса, может быть окислено при воздействии кислорода; возможность воздействия кислорода на промежуточное соединение устраняли путем защиты инертным газом для обеспечения нормальной реакции; поскольку при гликозилировании может образовываться вода, добавляли молекулярное сито с целью удаления полученной воды из реакции для обеспечения нормальной реакции; в то время как ТМСОТф может разрушаться водой, добавляли молекулярное сито для дополнительного обеспечения нормальной реакции.
2) Обработка гасящим агентом
Триэтиламин в качестве гасящего агента (0,312 ммоль) добавляли к реакционной смеси для гашения гликозилирования, где массовое отношение триэтиламина в качестве гасящего агента к триметилсилил-трифторметансульфонату (т.е. триметилсилил-трифлату) в качестве катализатора на основе кислоты Льюиса составляло 1:1; затем погашенную смесь для гликозилирования фильтровали с применением воронки Бюхнера, фильтрат концентрировали и очищали посредством колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3:2 (о/о)), для получения тетраацетил-филлирина.
3) Деацилирующая обработка
3.1) Тетраацетил-филлирин растворяли в 30 мл смеси дихлорметана и метанола, где объемное отношение дихлорметана к метанолу составляло 1:2, затем добавляли натрия метоксид (0,22 мг, 0,004 ммоль), где молярное отношение натрия метоксида к 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидату составляло 1:375, затем проводили реакцию деацилирования в течение 4 часов при перемешивании, затем добавляли уксусную кислоту в качестве регулятора рН для доведения значения рН полученной смеси из реакции деацилирования до 6.
Во время реакции деацилирования из настоящего изобретения добавляемый натрия метоксид не вызывает индуцируемого щелочными условиями разрушения гликозидных связей, и также служит в качестве основания для реакции деацилирования для удаления ацил-защитных групп, и таким образом, для стимуляции реакции гликозилирования. Время реакции деацилирования составляет по меньшей мере 4 часа, предпочтительно 4-12 часов.
В настоящем изобретении уксусную кислоту добавляют к смеси для деацилирования для доведения рН смеси и нейтрализации избытка метоксида натрия, терминации реакции; далее, благодаря умеренной активности, уксусная кислота не разрушает генерируемые гликозидные связи и может увеличить выход продукта.
3.2) Полученную смесь концентрировали под вакуумом с ротационным испарителем для удаления растворителей путем выпаривания, с последующей очисткой посредством колоночной хроматографии на силикагеле (элюент: хлороформ/метанол = 8:2 (о/о)); получали 400,5 мг белого твердого вещества филлирина с общим выходом 79,8%.
Белое твердое вещество имело точку плавления 181-183°С и является растворимым в хлороформе и метаноле. Оно имело те же самые физические свойства, как стандартная субстанция филлирина (полученная из Национального института контроля продуктов питания и лекарственных средств), сохраняло неизменную точку плавления после смешивания со стандартной субстанцией филлирина, и имело спектральные и масс-спектрометрические данные, согласующиеся с данными, отмеченными для филлирина в научной литературе; таким образом, это соединение идентифицируется как филлирин.
В соответствии с данными ВЭЖХ, указанными в Приложении VID к Первому тому Китайской Фармакопеи (КФ, 2000), чистота приготовленного филлирина составляет 99,5%; ЭРИ-МС, m/z [M-H] составляет 533, молекулярная масса составляет 534.
1H-ЯМР (600МГц, d6-ДМСО) δ: 7,66(br,1H,OH), 7,49(d,1H,J=8,43Гц), 7,21(br,2H), 7,14(s,1H), 7,13(s,1H), 7,01(d,1H.J=7,89Гц), 6,92(d,1H,J=8,12 Гц), 6,88(d,1H,J=8,34Гц), 6,54(br,1H), 5,60(d,1H,J=7,03Гц), 4,82(d,1H,J=5,92Гц), 4,54(d,1H,J=6,78Гц), 4,42(d,1H,J=11,43Гц), 4,25(m,4H), 4,13(d,1H,J=9,18Гц), 4,01(br,1H), 3,90(t,1H,J=8,72Гц), 3,75(dd,1H,J=8,99Гц,6,43Гц), 3,68(s,3H), 3,65(s,3H), 3,64(s,3H), 3,44(t,1H,J=8,72Гц), 3,27(m,1H), 2,82(q,1H,J=6,78Гц).
13C-ЯМР(150MГц,d6-DMSO) δ: 50,65(C-9), 55,33(C-31), 56,04(C-32), 56,09(C-8), 56,12(C-11), 62,50 C-29), 70,20(C-12), 71,38(C-34), 71,43(C-13), 75,03(C-33), 78,69(C-10), 79,04(C-30), 82,43(C-2), 88,07(C-21), 102,53(C-25), 110,52(C-6), 111,20(C-3), 112,47(C-5), 116,37(C-27), 118,58(C-4), 119,22(C-23), 132,29(C-17), 136,40(C-20), 147,60(C-36), 149,09(C-38), 150,35(C-28), 150,38(C-24).
Вариант осуществления 2
1) Гликозилирование
Филлигенин (372 мг, 0,001 моль) и 2,3,4,6-тетра-О-бензоил-D-глюкопиранозил трихлорацетимидат (1,11г, 0,0015 моль) вносили в 100 мл трехгорлую колбу, где молярное отношение филлигенина к 2,3,4,6-тетра-О-бензоил-D-глюкопиранозил трихлорацетимидату составляло 1:1,5; в колбу добавляли 20 мл безводного дихлорметана и алюмосиликатного молекулярного сита 3Å типа (744 мг); затем инертный газ аргон вводили в колбу для защиты инертным газом, затем перемешивали в течение 0,5 часа; по каплям добавляли 80 мг (0,312 ммоль) серебра трифтормтансульфоната в качестве катализатора на основе кислоты Льюиса; где молярное отношение катализатора на основе кислоты Льюиса к 2,3,4,6-тетра-О-бензоил-D-глюкопиранозил трихлорацетимидату составляло 1:5, массовое отношение молекулярного сита к филлигенину составляло 2:1; полученную смесь подвергали гликозилированию в течение 8 часов при 10°С при перемешивании.
Молекулярное сито добавляли с целью удаления образующейся воды из реакции для обеспечения позитивного результата реакции.
2) Обработка гасящим агентом
Натрия тиосульфат в качестве гасящего агента (0,312 ммоль) добавляли к реакционной смеси для гашения гликозилирования, где массовое отношение натрия тиосульфата в качестве гасящего агента к триметилсилил-трифторметансульфонату в качестве катализатора на основе кислоты Льюиса составляло 1:1; затем погашенную смесь для гликозилирования фильтровали с применением воронки Бюхнера, фильтрат концентрировали и очищали посредством колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 2:1 (о/о)), для получения тетра-бензоил-филлирина.
3) Деацилирующая обработка
3.1) Тетра-бензоил-филлирин растворяли в 30 мл смеси дихлорметана и метанола, в которых объемное отношение дихлорметана к метанолу составляло 1:2, затем добавляли натрия метоксид (0,22 мг, 0,004 ммоль), где молярное отношение натрия метоксида к 2,3,4,6-тетра-О-бензоил-D-глюкопиранозил трихлорацетимидату составляло 1:375; затем проводили реакцию деацилирования в течение 4 часов при перемешивании, затем добавляли уксусную кислоту в качестве регулятора рН для доведения значения рН полученной смеси из реакции деацилирования до 7.
3.2) Полученную смесь концентрировали под вакуумом с ротационным испарителем для удаления растворителей путем выпаривания, с последующей очисткой посредством колоночной хроматографии на силикагеле (элюент: хлороформ/метанол = 8:2 (о/о)); получали 373,8 мг белого твердого вещества филлирина с общим выходом 70%.
Физико-химические характеристики, спектральные данные и масс-спектрометрические данные очищенного белого твердого продукта согласуются с данными, отмеченными для филлирина в научной литературе; таким образом, это соединение идентифицируется как филлирин.
Вариант осуществления 3
1) Гликозилирование
Филлигенин (372 мг, 0,001 моль) и 2,3,4,6-тетра-О-ацетил-D-глюкопиранозил трихлорацетимидат (1,23 г, 0,0025 моль) вносили в 100 мл трехгорлую колбу, где молярное отношение филлигенина к 2,3,4,6-тетра-О-ацетил-D-глюкопиранозил трихлорацетимидату составляло 1:2,5; в колбу добавляли 20 мл безводного дихлорметана и алюмосиликатного молекулярного сита 5Å типа (744 мг); затем инертный газ азот вводили в колбу для защиты инертным газом, затем перемешивали в течение 0,5 часа; после однородного перемешивания по каплям добавляли триметилсилил-трифлат в качестве катализатора на основе кислоты Льюиса (0,08 мл, 0,416 ммоль), где молярное отношение катализатора на основе кислоты Льюиса к 2,3,4,6-тетра-О-ацетил-D-глюкопиранозил трихлорацетимидату составляло 1:6, массовое отношение молекулярного сита к филлигенину составляло 2:1; полученную смесь подвергали гликозилированию в течение 10 часов при 0°С при перемешивании.
2) Обработка гасящим агентом
Триэтиламин в качестве гасящего агента (0,416 ммоль) добавляли к реакционной смеси для гашения гликозилирования, где массовое отношение триэтиламина в качестве гасящего агента к триметилсилил-трифторметансульфонату в качестве катализатора на основе кислоты Льюиса составляло 1:1; затем погашенную смесь для гликозилирования фильтровали с применением воронки Бюхнера, фильтрат концентрировали и очищали посредством колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3:2 (о/о)), для получения тетраацетил-филлирина.
3) Деацилирующая обработка
3.1) Тетраацетил-филлирин растворяли в 30 мл смеси дихлорметана и метанола, в которых объемное отношение дихлорметана к метанолу составляло 1:2, затем добавляли натрия метоксид (0,337 мг, 0,00625 ммоль), где молярное отношение натрия метоксида к 2,3,4,6-тетра-О-ацетил-D-глюкопиранозил трихлорацетимидату составляло 1:400, затем проводили реакцию деацилирования в течение 4 часов при перемешивании, затем добавляли уксусную кислоту в качестве регулятора рН для доведения значения рН полученной смеси из реакции деацилирования до 6.
3.2) Полученную смесь концентрировали под вакуумом с ротационным испарителем для удаления растворителей путем выпаривания, с последующей очисткой посредством колоночной хроматографии на силикагеле (элюент: хлороформ/метанол = 8:2 (о/о)); получали 384,4 мг белого твердого вещества филлирина с общим выходом 72%.
Физико-химические характеристики, спектральные данные и масс-спектрометрические данные очищенного белого твердого продукта согласуются с данными, отмеченными для филлирина в научной литературе; таким образом, это соединение идентифицируется как филлирин.
Вариант осуществления 4
1) Гликозилирование
Филлигенин (372 мг, 0,001 моль) и 2,3,4,6-тетра-О-ацетил-глюкопиранозил трихлорацетимидат (492,6 мг, 0,0025 моль) вносили в 100 мл трехгорлую колбу, где молярное отношение филлигенина к 2,3,4,6-тетра-О-ацетил-глюкопиранозил трихлорацетимидату составляло 1:1; в колбу добавляли 20 мл безводного дихлорметана и алюмосиликатного молекулярного сита 4Å типа (744 мг); затем инертный газ азот вводили в колбу для защиты инертным газом, затем перемешивали в течение 0,5 часа; по каплям добавляли комплекс бора и трифторид-этилового эфира в качестве катализатора на основе кислоты Льюиса (0,025 мл, 0,2 ммоль), где молярное отношение катализатора на основе кислоты Льюиса к 2,3,4,6-тетра-О-ацетил-глюкопиранозил трихлорацетимидату составляло 1:5, массовое отношение молекулярного сита к филлигенину составляло 2:1; полученную смесь подвергали гликозилированию в течение 10 часов при 0°С при перемешивании.
2) Обработка гасящим агентом
Триэтиламин в качестве гасящего агента (0,2 ммоль) добавляли к реакционной смеси для гашения гликозилирования, где массовое отношение триэтиламина в качестве гасящего агента к бора трифторид-этерату в качестве катализатора на основе кислоты Льюиса составляло 1:1; затем погашенную смесь для гликозилирования фильтровали с применением воронки Бюхнера, фильтрат концентрировали и очищали посредством колоночной хроматографии на силикагеле (элюент: петролейный эфир/этилацетат = 3:2 (о/о)), для получения тетраацетил-филлирина.
3) Деацилирующая обработка
3.1) Тетраацетил-филлирин растворяли в 30 мл смеси дихлорметана и метанола, где объемное отношение дихлорметана к метанолу составляло 1:2, затем добавляли натрия метоксид (0,11 мг, 0,002 ммоль), где молярное отношение натрия метоксида к 2,3,4,6-тетра-О-ацетил-D-глюкопиранозил трихлорацетимидату составляло 1:500, затем проводили реакцию деацилирования в течение 12 часов при перемешивании, затем добавляли уксусную кислоту в качестве регулятора рН для доведения значения рН полученной смеси из реакции деацилирования до 7.
3.2) Полученную смесь концентрировали под вакуумом с ротационным испарителем для удаления растворителей путем выпаривания, с последующей очисткой посредством колоночной хроматографии на силикагеле (элюент: хлороформ/метанол = 8:2 (о/о)); получали 400,5 мг белого твердого вещества филлирина с общим выходом 75%.
Физико-химические характеристики, спектральные данные и масс-спектрометрические данные очищенного белого твердого продукта согласуются с данными, отмеченными для филлирина в научной литературе; таким образом, это соединение идентифицируется как филлирин.
Опытный пример: анализ противовирусной активности филлирина
1. Анализ противовирусной активности in vitro
1.1. Материалы для анализа
(1) Лекарственные средства: следующие лекарственные средства растворяли в очищенной воде, фильтровали, стерилизовали, разделяли и хранили при 4°С до применения.
1) Филлирин: белое твердое вещество, поставляемое Dalian Fusheng Natural Drug Development Co., Ltd. Чистота: 99,1%, определенная посредством ВЭЖХ системы, оснащенной УФ детектором и испарительным детектором светорассеяния (ELSD) с применением способа нормирования площадей пиков;
2) Рибавирин для инъекций: бесцветная прозрачная жидкость, произведенная Henan Runhong Pharmaceutical Co., Ltd., серия №1206261; регистрационное удостоверение №H19993553; использован в качестве лекарственного средства для положительного контроля в дозе 100 мг/мл в этом анализе;
3) Осельтамивир фосфат, поставляемый Национальным институтом контроля фармацевтических и биологических продуктов; серия №101096-200901; использован в качестве лекарственного средства для положительного контроля в дозе 100 мг на инъекцию в этом анализе.
(2) Клеточный штамм.
Штамм клеток Vero (клетки почки африканской зеленой мартышки) предоставлен Колледжем основных медицинских наук Цзилиньского университета.
(3) Вирусные штаммы
1) Вирус гриппа, вирус парагриппа и респираторно-синцитиальный вирус (РСВ) являются коммерческими штаммами, поставляемыми Институтом вирусологии Китайской академии профилактической медицины;
2) Штамм вируса Коксаки В3 (CVB3) получен из США и хранился в нашем научно-исследовательском отделе;
3) Штамм вируса Коксаки А16 (СохА16) и штамм энтеровируса ЕV71 были предоставлены Сендайским национальным госпиталем из Японии, и хранились в нашем научно-исследовательском отделе;
4) Штамм аденовируса (AdV) был предоставлен Педиатрическим исследовательским отделом Первого госпиталя Медицинского университета Нормана Бетьюна.
5) Вирус простого герпеса I типа (HSV-1) был получен из Института контроля фармацевтических и биологических продуктов Министерства здравоохранения.
(4) Основные приборы и реагенты
Кабинет биологической безопасности BHC-1300 II A/B3, AIRTECH
CO2 инкубатор MCO-18AIC, SANYO
Инвертированный микроскоп CKX41, OLYMPUS
Электронные аналитические весы AR1140/C, DHAUS
Среда для культивирования DMEM, HyClone
Эмбриональная телячья сыворотка HyClone
Трипсин Gibco
МТТ Sigma
ДМСО был получен от Tianjin Beilian Fine Chemicals Development Co., Ltd.
1.2. Методы анализа
(1) Приготовление клеток
Клетки Vero культивировали в субкультуре в течение 1-2 суток до формирования слоя. Затем культуры подвергали обработке трипсином после проявления выраженных границ и явной трехмерной ориентации и диоптрии. Расщепление прекращали после появления игольчатых отверстий на клеточной поверхности, затем клетки диспергировали в нескольких миллилитрах среды, подсчитывали, затем разбавляли примерно до 5×107 клеток/литр с DMEM, содержащей 10% эмбриональной телячьей сыворотки, и переносили на 96-луночный планшет для культивирования до образования монослоя.
(2) Анализ токсичности лекарства
Анализ цитотоксичности: лекарства разбавляли в соответствии концентрациями, указанными в Таблице 1-1 для анализа цитотоксичности.
Таблица 1-1. Справочная таблица по разведениям лекарств (единица: г/л)
Различные концентрации лекарств, разбавленных вышеуказанной средой для культивирования (DMEM, содержащей 2% эмбриональной телячьей сыворотки), добавляли по каплям к монослою клеток Vero с 0,2 мл на лунку, и для каждой концентрации лекарства добавляли в шести повторностях в 6 лунок, соответственно. Кроме того, 6 лунок использовали для нормального контроля (без лекарств), а другие 6 лунок использовали в качестве пустого контроля (только среда). Клетки выращивали при 37°С в инкубаторе при 5% CO2. ЦПЭ визуализировали под инвертированным микроскопом и регистрировали ежедневно. Спустя 72 часа добавляли 20 мкл МТТ раствора (5 мг/мл) в каждую лунку, и инкубировали в течение 4 часов. Культуральную среду в каждой лунке удаляли аспирацией, добавляли 100 мкл ДМСО в каждую лунку. Затем культуру встряхивали в течения 5 минут, измеряли значение ОП при 492 нм для подсчета доли выживших клеток. Долю выживших клеток анализировали с применением пробит-регрессионной модели с применением статистического программного обеспечения SPSS 18.0, и определяли максимальную нетоксичную концентрацию (TC0) и медианную токсичную концентрацию (TC50) лекарств против клеток Vero.
(3) Определение TCID50 для каждого вируса
Проводили 10-кратные серийные разведения для каждого вируса для получения разведений 10-1, 10-2, 10-3, 10-4, 10-5 и 10-6. Каждую из лунок с 6 повторностями на 96-луночном планшете для культивирования, содержащую монослой клеток Vero, инокулировали 100 мкл разбавителя для каждого разведения в последовательности при установке нормального контроля клеток. Планшеты инкубировали в течение 2 часов при 37°C в 5% CO2, затем удаляли раствор вируса, и добавляли 100 мкл среды для культивирования клеток в каждую лунку для последующей инкубации при 37°C в 5% CO2. Цитопатический эффект оценивали под микроскопом от 3 суток, и результаты определяли и регистрировали на 7-8-е сутки. Титр вирусов подсчитывали методом Карбера с максимальным титром разбавления, обеспечивающим положительный цитопатический эффект в 50% лунок с клетками в качестве конечной точки.
Уравнение: LogTCID50=XM+

TCID50: доза 50% инфицирования гистиоцитов
ХМ: логарифм наибольшей концентрации разбавления вируса
d: логарифм коэффициента разведения (кратное число)
Σpi: сумма процента цитотоксического эффекта для каждого разведения
(4) Влияние лекарств на вирус-индуцированную цитотоксичность
Культуральную среду в планшетах, покрывающую монослой клеток, удаляли аспирацией, и клетки инокулировали агрессивными вирусами в дозе 100TCID50 с последующей инкубацией при 37°C в инкубаторе при 5% CO2 в течение 2 часов, а затем добавляли определенную концентрацию (максимальную нетоксичную концентрацию или около того) каждого лекарственного средства. Каждую концентрацию использовали в 6 повторностях в 6 лунках с 200 мкл на лунку. Рибавирин для инъекций и осельтамивир фосфат служили в качестве групп положительного контроля, в то время как нормальную контрольную группу (без вируса и без лекарства) и группу контроля вирусов (с добавлением вируса, но без лекарства) использовали для оценки эффекта лекарств в отношении вирус-индуцированного цитопатического эффекта (ЦПЭ). Спустя 72 часа определяли значение ОП при длине волны 492 нм с применением МТТ-колориметрического метода, и рассчитывали уровень антивирусного эффекта (ER%) лекарства. Использовали дисперсионный анализ (ANOVA) со статистическим программным обеспечением SPSS 18.0 для определения достоверной разницы антивирусной эффективности лекарств из различных групп.
ER% = (среднее значение ОП в группе с применением лекарств - среднее значение ОП в контрольной группе с вирусом)/(среднее значение ОП в группе контроля клеток - среднее значение ОП в контрольной группе с вирусом)×100%.
1.3. Результаты
(1) TCID50 для каждого вируса
Вирус парагриппа: LogTCID50=-2+0,5-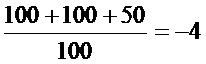
Вирус гриппа: LogTCID50=-2+0,5-
CVB3: LogTCID50=-2+0,5-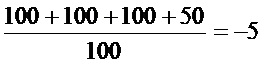
HSV-1: LogTCID50=-2+0,5-
AdV: LogTCID50=-2+0,5-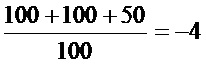
RSV: LogTCID50=-2+0,5-
CoxA16: LogTCID50=-2+0,5-
EV71: LogTCID50=-2+0,5-
(2) Определение токсичности лекарств
1) Определение цитотоксичности лекарств
Максимальная не-цитотоксичная концентрация (TC0), медианная токсичная концентрация (TC50) каждого лекарства против клеток Vero, и концентрации лекарств, используемых в антивирусном анализе, показаны в Таблице 1-2.
Таблица 1-2. Результаты анализа цитотоксичности лекарств (единица: г/л)
концентрация
2) Результаты защитного влияния лекарств на вирус-индуцированную цитотоксичность
Результаты анализа антивирусной эффективности и одностороннего дисперсионного анализа (ANOVA) показаны в Таблице 1-3.
Таблица 1-3. Статистические данные антивирусной эффективности лекарств (ЕR%).
Результаты в Таблице 1-3 показывают, что ингибирующие эффекты филлирина в отношении вируса гриппа, вируса парагриппа, респираторно-синцитиального вируса (RSV), вируса Коксаки (CVB3), вируса Коксаки A16 (CoxA16), энтеровируса EV71, аденовируса (AdV) и вируса простого герпеса I типа (HSV-1) были достоверными. При этом ингибирующие эффекты в отношении вируса гриппа, вируса парагриппа и вируса простого герпеса I типа (HSV-1) были сопоставимыми с противовирусными эффектами лекарств положительного контроля, таких как рибавирин и осельтамивир фосфат (Тамифлю); а ингибирующие эффекты в отношении вируса Коксаки В3 (CVB3), вируса Коксаки A16 (CoxA16), энтеровируса EV71 и аденовируса (AdV) были более выраженными, чем у лекарств положительного контроля, таких как рибавирин и осельтамивир фосфат (Тамифлю).
2. Анализ противовирусной активности in vivo
2.1. Материалы для анализа
(1) Экспериментальные животные
Куньминские мыши были получены из Центра лабораторных животных Исследовательского центра здравоохранения Нормана Бетьюна Цзилиньского университета. Лабораторные животные №10-5219.
(2) Экспериментальные приборы
Прибор для количественной ПЦР: 7300, ABI;
Прибор для ПЦР: ES-60J, Shenyang Longteng Electronic Weighing Instrument Co., Ltd.;
Электронные аналитические весы: FA1004, Shenyang Longteng Co., Ltd.
CO2 инкубатор: HG303-5, Нанкинский завод экспериментальных приборов;
Ламинарно-потоковый бокс: SW-CJ-IF, Suzhou Antai Air Tech Co., Ltd.;
Инвертированный микроскоп: CKX41, Olympus Instrument;
-80°C ультра-низкотемпературный морозильник: TECON-5082, Австралия;
Водяная баня с осциллятором: ГЦS-H, Harbin Donglian Electronic technology Development Co., Ltd.;
Ридер для микропланшетов: TECAN A-5082, Австралия;
Спектрофотометр: модель 7550; Япония.
2.2. Экспериментальные методы
(1) Изучение влияния филлирина на пневмонию, индуцированную вирусом гриппа и вирусом парагриппа
1) Разделение экспериментальных животных на группы
140 куньминских мышей в возрасте 4 недели было взято для выполнения двух анализов. Получали 140 мышей и произвольно распределяли на 14 групп (n=10 в каждой группе) для определения легочного индекса и уровня ингибирования легочного индекса после применения филлирина у мышей, инфицированных вирусом гриппа и вирусом парагриппа.
2) Способ инфицирования
В стакан (200-300 мл) помещали кусок адсорбирующей ваты, и наливали на него подходящее количество этилового эфира до увлажнения адсорбирующей ваты. Стакан с адсорбирующей ватой переворачивали вверх дном, и помещали в него мышь для анестезии. После проявления у мыши крайнего возбуждения и явной слабости, их помещали на спину и инфицировали назально с 15 LD50 вируса гриппа и вируса парагриппа по 0,03 мл на ноздрю. В группе нормального контроля суспензию вирусов заменяли физиологическим солевым раствором.
3) Способ применения и применяемые дозы
У каждой мыши применяли внутрижелудочно филлирин, рибавирин и осельтамивир фосфат за сутки до инфицирования. Филлирин применяли в наивысшей дозе 13 мг/кг, в средней дозе 6,5 мг/кг, или в низкой дозе 3,25 мг/кг, в то время как дозы лекарств положительного контроля рибавирина и осельтамивира фосфата составили 19,5 мг/кг и 58,5 мг/кг, соответственно. Применение осуществляли один раз в сутки в течение пяти последовательных дней. В группе вирусного контроля применяли физиологический солевой раствор того же самого объема.
4) Определение легочного индекса
На пятые сутки после применения лекарств у мышей предотвращали доступ к воде, и спустя 8 часов мышей взвешивали, а затем умерщвляли путем обескровливания при энуклеации глаз. Затем извлекали легкие после вскрытия грудины, дважды промывали физиологическим солевым раствором, затем удаляли влагу с поверхности фильтровальной бумагой и взвешивали. Легочный индекс и уровень ингибирования легочного индекса рассчитывали с помощью следующих уравнений:
Легочный индекс = Масса легкого мыши/Масса тела мыши × 100%
Уровень ингибирования легочного индекса = (Средний легочный индекс в группе модельной инфекции - Средний легочный индекс)/Средний легочный индекс в группе модельной инфекции × 100%.
2.3. Результаты экспериментов и анализ
После инфицирования мышей вирусом гриппа и вирусом парагриппа средние результаты легочного индекса показали, что филлирин в диапазоне от 3,25 до 13,0 мг/кг/сутки обеспечивает существенную защиту легочной ткани мыши при инфицировании вирусом гриппа и вирусом парагриппа с существенными снижениями легочных индексов. Результаты показаны в Таблицах 2-1 и 2-2.
Таблица 2-1. Легочные индексы и уровни ингибирования легочного индекса у мышей при применении филлирина после инфицирования вирусом гриппа (n=3).

По сравнению с группой вирусного контроля: *P<0,05,**P<0,01.
Таблица 2-2. Легочные индексы и уровни ингибирования легочного индекса у мышей при применении филлирина после инфицирования вирусом парагриппа (n=3).
По сравнению с группой вирусного контроля: *P<0,05.
2.4. Заключение
Результаты анализа противовирусной активности in vivo показали, что филлирин в диапазоне доз 3.25-13,0 мг/кг/сутки оказывает существенное ингибирующее влияние на вирусные эффекты у мышей, индуцированные вирусами гриппа и парагриппа, и может существенно снижать легочные индексы и титры гемагглютинации, и представляет достоверную разницу, по сравнению с группой вирусного контроля.
Изобретение относится к способу химического синтеза филлирина, который может быть применен в химической промышленности. Предложенный способ включает следующие этапы: (1) растворение акцептора гликозила - филлигенина и донора гликозила - 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидата в атмосфере инертного газа при добавлении катализатора и молекулярного сита в безводном дихлорметане или безводном трихлорметане для гликозилирования с получением тетраацил-филлирина, (2) растворение тетраацил-филлирина в органическом растворителе, добавление метоксида натрия для деацилирования, добавление кислотного регулятора рН для доведения значения рН реакционной смеси до нейтрального и проведение очистки для получения филлирина. Предложен новый эффективный способ химического синтеза филлирина. 5 з.п. ф-лы, 5 табл., 4 пр.
1. Способ химического синтеза филлирина, включающий следующие этапы:
(1) растворение акцептора гликозила - филлигенина и донора гликозила - 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидата в первом органическом растворителе под защитой инертного газа при добавлении катализатора и молекулярного сита для гликозилирования с целью получения тетраацил-филлирина, где первый органический растворитель представляет собой безводный дихлорметан или безводный трихлорметан;
(2) растворение тетраацил-филлирина во втором органическом растворителе, добавление метоксида натрия для деацилирования, добавление кислотного регулятора рН для доведения значения рН реакционной смеси до нейтрального и проведение очистки для получения филлирина.
2. Способ по п.1, характеризующийся тем, что кислоту Льюиса применяют в качестве катализатора.
3. Способ по п.1, характеризующийся тем, что молярное отношение катализатора к донору гликозила 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидату составляет 1:1-10.
4. Способ по п.1, характеризующийся тем, что в качестве молекулярного сита применяют алюмосиликатное молекулярное сито или алюмосиликатный порошок.
5. Способ по п.1 или 2, также включающий этап (1А) гашения гликозилирования гасящим агентом перед растворением тетраацил-филлирина во втором органическом растворителе.
6. Способ по п.1 или 2, характеризующийся тем, что на этапе (2) молярное отношение натрия метоксида к 2,3,4,6-тетра-О-ацил-D-глюкопиранозил трихлорацетимидату составляет 1:300-500.
| FAN Hongya et al | |||
| LIAONING CHEMICAL INDUSTRY, 2014, v | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| LI XIAODONG et al | |||
| JOURNAL OF SHENYANG PHARMACEUTICAL, 2011, v | |||
| Видоизменение прибора с двумя приемами для рассматривания проекционные увеличенных и удаленных от зрителя стереограмм | 1919 |
|
SU28A1 |
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |

