Настоящее изобретение относится к гликозидам, их солям и фармацевтическим композициям, содержащим такие гликозиды в качестве активных ингредиентов. Более того, изобретение обеспечивает способ профилактики, лечения или облегчения симптомов острых или хронических воспалительных расстройств дыхательных путей у млекопитающих, включая астму и патологии, связанные с астмой.
УРОВЕНЬ ТЕХНИКИ
Воспаление представляет собой многоступенчатый каскадный процесс, который может быть предметом потенциального терапевтического вмешательства. Вкратце, воспаление вызывает инфильтрацию иммунологически компетентных клеток (например, эозинофилов, тучных клеток, активированных T-лимфоцитов) в месте повреждения, где они, вместе с резидентными клетками, высвобождают биологически активные медиаторные вещества (например, гистамин, протеазы, множество цитокинов и хемокинов), которые увеличивают проницаемость близких кровеносных сосудов, привлекают и стимулируют соседние клетки. Измененная проницаемость сосудов приводит к образованию жидкого экссудата в месте повреждения с последующим притоком реактивных лейкоцитов и их потенциальным оттоком в поврежденную область. (Для описания смотрите, Trowbridge and Emling, Inflammation: A Review of the Process Quintessence Pub. Co., 1997). Секреция коллагена и слизи и пролиферация резидентных клеток (гладкомышечные и эпителиальные клетки или фибробласты, стимулированные высвобожденными медиаторами) устанавливают степень выраженности патологических изменений (например, обструкции дыхательных путей) и участвуют в их развитии.
Воспаление связано со множеством легочных состояний, включая, например, эндогенную или экзогенную бронхиальную астму, любое воспалительное заболевание легких, острый или хронический бронхит, легочные воспалительные реакции, вторичные по отношению к хроническому бронхиту, хронические обструктивные заболевания легких, пневмосклероз, а также любые заболевания легких, при которых могут играть роль лейкоциты, включая, но не ограничиваясь, идиопатический пневмосклероз и любые другие аутоиммунные заболевания легких. Астма является одной из наиболее частых форм легочного воспаления, поражающей крупные и мелкие дыхательные пути легких. Ей страдает от 5% до 10% населения, что приводит к предполагаемым 27 миллионам визитов пациентов, 6 миллионам потерянных рабочих дней, и 90,5 миллиона дней ограниченной активности в год. Степень заболеваемости и смертности от астмы увеличивается во всем мире (Plaut and Zimmerman, "Allergy and Mechanisms of Hypersensitivity" in Fundamental Immunology. 3rd Ed, Paul (ed.), Raven Press, New York, NY, at 1399 (1993)).
Обычное лечение от астмы основано на четком избегании всех инициирующих аллергенов, чего по существу трудно достичь, и на терапевтических схемах, основанных на фармакологических агентах, имеющих неблагоприятные побочные эффекты и субоптимальные фармакокинетические свойства. β2-адренергические агонисты, используемые для лечения бронхоспазма, не оказывают эффекта на воспаление дыхательных путей или гиперреактивность бронхов (Palmer et al, New Engl. J. Med. 331:1314 (1994)). Также систематическое или длительное применение β2-адренергических агонистов связано с недостаточным контролем астмы, увеличением гиперреактивности дыхательных путей в ответ на аллерген и сниженной защитой от бронхоконстрикции (Bhagat et al., Chest 108:1235 (1995)). Более того, считается, что хроническое использование β2-адренергических агентов отдельно посредством подавления β2-адренергических рецепторов, будет ухудшать гиперреактивность бронхов. Теофиллин (противоастматический метилксантин) характеризуется существенной вариабельностью всасывания и выведения. Кортикостероиды, хотя являются относительно безопасными у взрослых пациентов, являются токсичными для детей, приводя к подавлению надпочечников и снижению костной плотности и роста (Woolock et al, am. Respir. Crit. Care Med. 153:1481 (1996)). Кромолин, используемый для предотвращения астматических эпизодов, эффективен для предотвращения астматической реакции только при введении перед воздействием (Volcheck et al, Postgrad Med. 104(3): 127 (1998)). Антигистаминные средства периодически предотвращают или останавливают аллергические астматические эпизоды, особенно у детей, но часто являются только частично эффективными, так как гистамин является только одним из множества медиаторов, связанных с воспалением (Cuss, "The Pharmacology of Antiasthma Medications", in Asthma as an Inflammatory Disease. O'Byrne, Ed., Dekker, Inc., New York, at 199 (1990)) and O'Byrne, "Airway Inflammation and Asthma", in Asthma as an Inflammatory Disease. O'Byrne, Ed., Dekker, Inc., New York, NY, 143 (1990)).
Следовательно, существующие возможности лекарственных средств имеют ряд недостатков. В общем, обычные агенты имеют относительно небольшую продолжительность действия и могут быть частично или полностью неэффективными при введении после возникновения антигенной стимуляции. Более того, из-за серьезных побочных эффектов, связанных с применением агентов, таких как β2-адренергические агонисты и кортикостероиды, терапевтические границы диапазона безопасности таких агентов являются относительно узкими, и за пациентами, использующими такие агенты, необходимо тщательно наблюдать (смотрите, например, WO 94/06783, WO 99/06025, патенты США №№ 5690910 и 5980865). В последнем клиническом исследовании ингаляционных кортикостероидов у детей 5-11 лет возникало только временное улучшение функции дыхательных путей с астмой после первого года лечения, с уменьшением до наблюдаемого при использовании плацебо в течение последующих 3 лет (The Childliood Asthma Management Program Research Group, N. Engl. J. Med., 343:1054 (2000)). Такое снижение наилучшим образом может быть объяснено перестроечными изменениями (характерным свойством астмы), возникающими в дыхательных путях, которые являются устойчивыми к кортикостероидам (Davies, Curr. Opin. Allergy Clin. Immunol, 1:67 (2001)).
Из соответствующей литературы известно, что определенные смеси полисульфатированных дисахаридов, имеющих структуру, тесно связанную с таковой согласно настоящему изобретению, и которые были синтезированы обработкой азотистой кислотой таких естественных продуктов, как, например, гепарин или сульфат гепарина, с последующим восстановлением боргидридом и последующим сульфатированием частично очищенных образцов (US 5690910; US 5980865 и WO 02/083700) - проявляли противовоспалительный эффект на различных моделях астмы.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым гликозидам и способам получения таких соединений и фармацевтических композиций, содержащих такие соединения, с четко определенной химической структурой, которые имеют более благоприятные фармакологические свойства и меньше нежелательных побочных эффектов, чем известные противоастматические средства. Изобретение, кроме того, относится к способам лечения пациентов, нуждающихся в лечении, включающим введение новых гликозидов и композиций указанных гликозидов указанным пациентам.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с упомянутыми выше фактами изобретение относится к новым полисульфатированным гликозидам формулы (I),
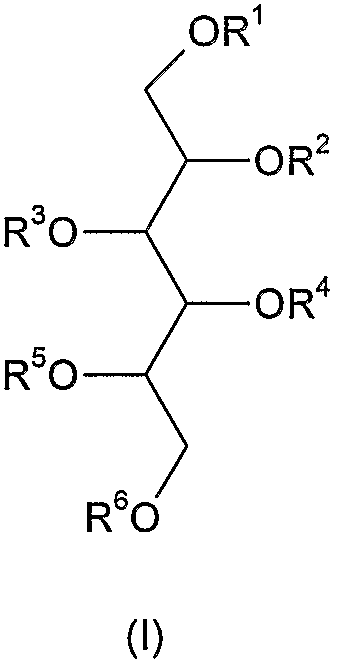
где R1, R2, R3, R4, R5 и R6 независимо друг от друга означают -H, C1-4 алкил, -SO3H, сульфатированную или несульфатированную гликозильную или сульфатированную или несульфатированную дигликозильную группу, при условии, что, по меньшей мере, один из R1-R6 представляет собой сульфатированную или несульфатированную гликозильную или сульфатированную или несульфатированную дигликозильную группу - а также их возможные изомеры и фармацевтически приемлемые соли. Термин "фармацевтически приемлемые соли" включает, например, соли щелочных металлов и соли щелочноземельных металлов и любых других фармацевтически приемлемых противоиона или противоионов, связанных с одной или более сульфатными группами в молекуле.
Так как все четыре вторичных атома углерода сахарного спирта представляют собой хиральные центры, очевидно, все возможные стереоизомеры (аллит, галактит, идит, маннит, глюцит и талит), а также их D- и L-энантиомеры охватываются формулой (I). Термин "изомер" в настоящем описании включает все такие соединения и их варианты в соединении формулы (I).
Значением сульфатированной гликозильной группы может быть молекула пентопиранозы или гексопиранозы с конфигурацией по выбору, одна или более гидроксильных групп которой присутствуют в виде O-сульфатного сложного эфира, и компонент сахара прикреплен к агликону его аномерным атомом углерода посредством α- или β-связи. Несульфатированная гликозильная группа содержит все гидроксильные группы или их защищенные варианты. Несульфатированные соединения являются применимыми в качестве промежуточных соединений для получения сульфатированных соединений, указанных в настоящем описании.
Значением полисульфатированной дигликозильной группы может быть молекула пентопиранозы или гексопиранозы с конфигурацией по выбору, одна из гидроксильных групп которой является гликозилированной дополнительной молекулой пентопиранозы или гексопиранозы с конфигурацией по выбору, и все гидроксильные группы образованного таким образом дигликозильного компонента присутствуют в виде O-сульфатных сложных эфиров, а сахарный компонент прикреплен к агликону своим аномерным атомом углерода посредством α- или β-связи.
Все возможные стереоизомеры (арабино-, ликсо-, рибо- и ксило-) включены в структуру пентоз, а также их D- и L-энантиомеров. Подобным образом все возможные стереоизомеры (алло-, альтро-, галакто-, глюко-, гуло-, идо-, манно- и талло-) включены в структуру гексоз, а также их D- и L-энантиомеров. Термин "изомер" включает все такие соединения и их варианты в соединение формулы (I).
Значением C1-4 алкильной группы являются группы метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил предпочтительно метильная группа.
Соли щелочных металлов и соединений по изобретению означают соли Na, К или Li, тогда как соли щелочноземельных металлов предпочтительно представляют собой соли Мg и Са.
Такие соединения формулы (I), а также их соли щелочных металлов и щелочноземельных металлов, где значением R1 является полисульфатированная гликозильная или дигликозильная группа, а значением R2-R6 является -SO3Н, представляют собой предпочтительную группу соединений по изобретению.
Такие соединения формулы (I), а также их соли щелочных металлов и щелочноземельных металлов, где значением R1, R2, R4, R5 и R6 является -SO3H, а значением R3 является полисульфатированная гликозильная или дигликозильная группа, представляют собой дополнительную предпочтительную группу соединений по изобретению.
Такие соединения формулы (I), а также их соли щелочных металлов и щелочноземельных металлов, где значением R1, R2, R3, R5 и R6 является -SO3H, а значением R4 является полисульфатированная гликозильная или дигликозильная группа, представляют собой дополнительную предпочтительную группу соединений по изобретению.
Такие соединения формулы (I), а также их соли щелочных металлов и щелочноземельных металлов, где значением R1 и R3 является полисульфатированная гликозильная группа, а значением R2, R4, R5 и R6 является -SO3H, представляют собой другую предпочтительную группу соединений по изобретению.
Такие соединения формулы (I), а также их соли щелочных металлов и щелочноземельных металлов, где значением R1 и R6 является полисульфатированная гликозильная или дигликозильная группа, а значением R2, R3, R4 и R5 является -SO3H, представляют собой другую предпочтительную группу соединений по изобретению.
Такие соединения формулы (I), а также их соли щелочных металлов и щелочноземельных металлов, где значением R1 является полисульфатированная гликозильная или дигликозильная группа, значением R3 и R4 является C1-4 алкильная группа, тогда как значением R2, R5 и R6 является -SO3H, представляют собой другую предпочтительную группу соединений по изобретению.
Такие соединения формулы (I), а также их соли щелочных металлов и щелочноземельных металлов, где значением R1 и R6 является полисульфатированная гликозильная или дигликозильная группа, значением R3 и R4 является C1-4 алкильная группа, тогда как значением R2 и R5 является -SO3H, представляют собой другую предпочтительную группу соединений по изобретению.
Особенно предпочтительными представителями соединений формулы (I) по настоящему изобретению являются - без ограничения - следующие:
нона-калиевая соль 2,3,4,5,6-пента-О-сульфато-1-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-маннита,
нона-калиевая соль 1,2,3,4,5-пента-O-сульфато-6-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола,
нона-калиевая соль 2,3,4,5,6-пента-О-сульфато-1-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-глюцитола,
нона-калиевая соль 1,2,4,5,6-пента-O-сульфато-3-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола,
нона-калиевая соль 1,2,3,5,6-пента-О-сульфато-4-О-(2,3,4,6-тетра-О-сульфато-α-D-глюкопиранозил)-D-глюцитола,
нона-калиевая соль 1,2,3,5,6-пента-О-сульфато-4-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-глюцитола,
нона-калиевая соль 1,2,3,5,6-пента-О-сульфато-4-О-(2,3,4,6-тетра-О-сульфато-β-D-галактопиранозил)-D-глюцитола,
додека-калиевая соль 2,4,5,6-тетра-О-сульфато-1,3-бис-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-глюцитола,
додека-калиевая соль 2,4,5,6-тетра-О-сульфато-1,6-бис-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-маннита,
октадека-калиевая соль 2,4,5,6-тетра-O-сульфато-1,6-бис-O-(2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозил)-D-маннита,
додека-калиевая соль 2,3,4,5,6-пента-О-сульфато-1-О-(2,3,4,2',3',4',6'-гепта-О-сульфато-β-гентиобиопиранозил)-D-маннита,
гепта-калиевая соль 3,4-ди-O-метил-2,5,6-три-O-сульфато-1-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-маннита,
дека-калиевая соль 3,4-ди-О-метил-2,5-ди-О-сульфато-1,6-бис-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-маннита,
дека-калиевая соль 3,4-ди-O-метил-2,5,6-три-О-сульфато-1-O-(2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозил)-D-маннита,
гексадека-калиевая соль 3,4-ди-О-метил-2,5-ди-О-сульфато-1,6-бис-О-(2,3,6,2',3',4',6'-гепта-О-сульфато-β-лактозил)-D-маннита,
окта-калиевая соль 2,3,4,5,6-пента-О-сульфато-1-О-(2,3,4-три-О-сульфато-α-D-арабинопиранозил)-D-маннита,
окта-калиевая соль 2,3,4,5,6-пента-О-сульфато-1-О-(2,3,4-три-О-сульфато-β-D-ксилопиранозил)-D-маннита,
додека-калиевая соль 2,4,5,6-тетра-O-сульфато-1,6-бис-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)галактитола,
нона-натриевая соль 1,2,4,5,6-пента-О-сульфато-3-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-глюцитола.
Соединения формулы (I) по настоящему изобретению могут быть синтезированы из соединений формулы (II)
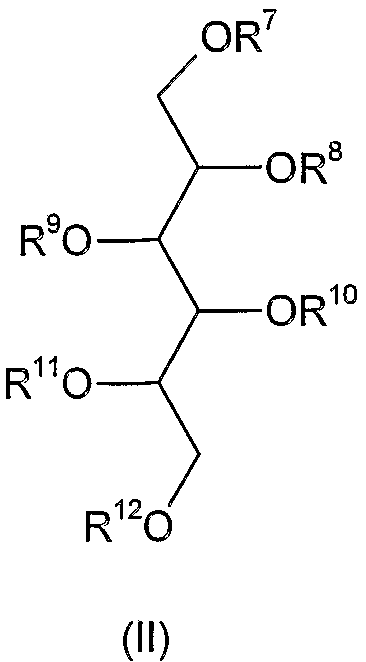
где R7, R8, R9, R10, R11 и R12 независимо друг от друга означают атом водорода, С1-4 алкильную, гликозильную или дигликозильную группу и, по меньшей мере, один из R7-R12 представляет собой гликозильную или дигликозильную группу;
преобразованием его свободных гидроксильных групп в сульфатные сложные эфиры с использованием известных способов.
Триоксид серы или его аддукт, образованный с органическим основанием (например, триэтиламином или пиридином) или с диметилформамидом, может быть использован для получения O-сульфатных сложных эфиров.
Необязательно монофункциональные кислые сложные эфиры, полученные вышеуказанными способами, могут быть преобразованы в соли, например, с ацетатами щелочных металлов или щелочноземельных металлов. После очистки соли могут быть получены лиофилизацией, осаждением или кристаллизацией.
Некоторые из соединений формулы (II), используемые в виде исходных веществ в вышеуказанных процессах для синтеза соединений формулы (I) по настоящему изобретению, могут быть синтезированы, например, следующими, известными способами.
Такие соединения формулы (II), где один из R7 и R8 представляет гликозильную группу и другой представляет собой атом водорода, а также значениями R9-R12 является атом водорода, могут быть синтезированы, например, с использованием соединения формулы (III) или (IV)
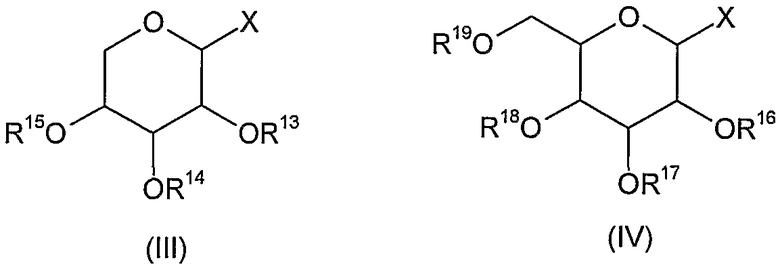
где X означает атом галогена, трихлорацетимидат или фенилтиогруппу и R13-R19 представляют собой алифатический или ароматический сложный эфир или группу простого эфира,
в качестве молекулы-донора, и соединения формулы (V)
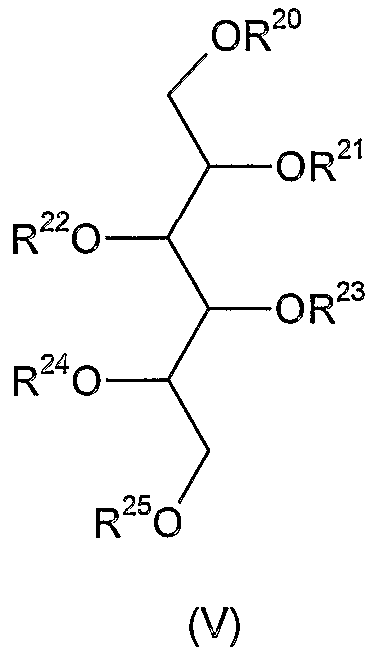
где R20 и R21 означают атомы водорода, R22-R25 представляют собой защитные группы типа простого эфира, в качестве акцептора, и гликозилирование проводят в присутствии соответствующих активаторов. Затем защитные группы отщепляют от полученного таким образом соединения формулы (V), где R22-R25 означают защитную группу типа простого эфира, тогда как один из R20 и R21 представляет собой защищенную гликозильную группу и другая является атомом водорода.
В соответствии с другим способом соединение формулы (V) используют в вышеуказанной реакции, в которой R20 и R22 означают атом водорода, тогда как R21, R23, R24 и R25 представляют собой защитные группы типа простого эфира, затем защитные группы отщепляют от полученного таким образом соединения формулы (V), где R21, R23, R24 и R25 являются защитными группами типа простого эфира, R20 представляет собой защищенную гликозильную группу и R22 является атомом водорода.
Такие соединения формулы (II), где R7 и R9 означают гликозильную группу, тогда как R8, R10, R11 и R12 представляют собой атом водорода, могут быть синтезированы, например, проведением гликозилирования в соответствии со способом b), но с использованием молекулы-донора в избытке, и защитные группы отщепляют от образованного таким образом соединения общей формулы (V), где R20 и R22 представляют собой защищенные гликозильные группы и R21, R23, R24 и R25 являются защитными группами типа простого эфира.
Такие соединения формулы (II), где один из R7 и R8 представляет собой дигликозильную группу, другой является атомом водорода, а R9-R12 означают атомы водорода, могут быть синтезированы, например, с использованием соединения формулы (VI) или (VII)

где X означает атом галогена, трихлорацетимидат или фенилтиогруппу и R26-R32 представляют собой алифатический или ароматический сложный эфир или группу простого эфира,
в качестве молекулы-донора, и соединения формулы (V), где R20 и R21 означают атом водорода, R22-R25 представляют собой защитные группы типа простого эфира, в качестве акцептора, и гликозилирование проводят в присутствии соответствующих активаторов. Затем защитные группы отщепляют от полученного таким образом соединения общей формулы (V) - где R22-R25 являются защитными группами типа простого эфира, тогда как один из R20 и R21 представляет собой защищенную гликозильную группу и другой является атомом водорода.
Такие соединения формулы (II), где R7 означает дигликозильную группу, могут быть синтезированы, например, с использованием соединения формулы (V), где R20 и R25 являются атомами водорода, R21 и R24 представляют собой защитные группы типа сложного эфира, тогда как R22 и R23 являются защитными группами типа простого эфира, в качестве акцептора в вышеуказанной реакции, затем защитные группы отщепляют от полученного таким образом соединения формулы (V), где R21 и R24 представляют собой защитные группы типа сложного эфира, тогда как R22 и R23 представляют собой защитные группы типа простого эфира, R20 представляет собой защищенную дигликозильную группу и значением R25 является атом водорода.
Такие соединения формулы (II), где R7 и R12 означают дигликозильную группу, и R8-R11 представляют собой атом водорода, могут быть синтезированы, например, с использованием соединения формулы (VI) или (VII), где X означает атом галогена, трихлорацетимидат или фенилтиогруппу и R26-R32 представляют собой алифатический или ароматический сложный эфир или группу простого эфира; в качестве молекулы-донора в избытке, и соединения формулы (V), где R20 и R25 означают атом водорода, R21 и R24 представляют собой защитные группы типа сложного эфира, тогда как R22 и R23 представляют собой защитные группы типа простого эфира, в качестве акцептора, и гликозилирование проводят в присутствии соответствующих активаторов. Затем защитные группы отщепляют от полученного таким образом соединения формулы (V) - где R21 и R24 представляют собой защитные группы типа сложного эфира, R22 и R23 представляют собой защитные группы типа простого эфира, тогда как R20 и R25 являются защищенными дигликозильными группами.
В вышеуказанных реакциях гликозилирования, соли ртути или серебра, диэтиловый этерат трифтористого бора, N-йодсукцинимид и трифторметансульфоновая кислота или смесь двух последних также могут использоваться в качестве активаторов.
Отщепление защитных групп может проводиться кислым гидролизом или восстановлением в присутствии катализатора в случае простых эфиров и ацеталей, тогда как в случае сложных эфиров может использоваться метод Цемплена (транс-эстерификация, катализируемая основанием) или гидролиз в присутствии основания.
Сокращения и выражения, используемые в описании:
Ac = ацетил
Bz = бензоил
Me = метил
Ph = фенил
NIS = N-йодсукцинимид
ТfОН = трифторметансульфоновая кислота
Как используется в настоящем описании, формы единственного числа также охватывают формы множественного числа терминов, к которым они относятся, если только из содержания явно не следует другое. Например, ссылка на "модулятор" включает смесь модуляторов.
Как используется в настоящем описании, или в промежуточной фразе, или в тексте формулы изобретения, термины "включает" и "включающий" должны интерпретироваться как имеющие открытое значение. А именно термины должны интерпретироваться однозначно с фразами "имеющий, по меньшей мере" или "включающий, по меньшей мере". При использовании в контексте способа термин "включающий" означает, что способ включает, по меньшей мере, указанные стадии, но может включать дополнительные стадии. При использовании в контексте соединения или композиции термин "включающий" означает, что соединение или композиция включает, по меньшей мере, указанные свойства или компоненты, но также может включать дополнительные свойства или компоненты.
Термин "около" используется в настоящем описании для обозначения приблизительно, в области, ориентировочно или примерно. Когда термин "около" используют в связи с областью числовых значений, он определяет, что указывается диапазон с расширенными границами выше и ниже указанной области числовых значений. В общем, термин "около" используется в настоящем описании для модификации области числовых значений выше и ниже указанного значения с отклонением 20%.
Как используется в настоящем описании, если особенно не указано иначе, слово "или" используется с "включительным" смыслом "и/или" и не "исключающим" смыслом "или/или."
Как используется в настоящем описании, термины "лечение" или "излечение" используют для указания снижения, облегчения, профилактики, ингибирования развития и/или обращения симптомов состояния. Состояния для лечения способами и композициями по изобретению включают любое состояние, характеризующееся или включающее острые и хронические воспалительные расстройства дыхательных путей. Следовательно, термины "воспалительное расстройство" или "воспалительные расстройства дыхательных путей" охватывают любые воспалительные заболевания легких, включая астму, эндогенную или экзогенную бронхиальную астму, обострение хронического бронхита, аллергический ринит, легочные воспалительные и структурные реакции, вторичные по отношению к хроническому бронхиту, хронические обструктивные заболевания легких, пневмосклероз. Изобретение также применимо для состояний легких, при которых могут играть роль лейкоциты и перестройка дыхательных путей, включая, без ограничения перечисленными, идиопатический пневмосклероз и любое другое аутоиммунное заболевание легких.
Под "астмой" подразумевают состояние аллергического происхождения, симптомы которого включают непрерывную или пароксизмальную одышку, сопровождаемую хрипами, ощущением сдавления в грудной клетке, и частыми приступами кашля или затрудненного дыхания. Под "патологией, связанной с астмой" подразумевают состояние, чьи симптомы являются преимущественно воспалительными по природе со связанным бронхоспазмом. Следовательно, и астма, и патологии, связанные с астмой, характеризуются симптомами, которые включают сужение дыхательных путей, обусловленное различными степенями сокращения (спазма) гладких мышц, отеком слизистой, включая таковую верхних дыхательных путей и слизистой оболочки в просвете бронхов и бронхиол. Неограниченные характерные примеры "патологий, связанных с астмой" включают неастматические состояния, характеризующиеся гиперреактивностью дыхательных путей (например, хронический бронхит, эмфизема, муковисцидоз и респираторный дистресс-синдром).
Композиции и способы, указанные в настоящем описании, являются примерными для астмы. Однако изобретение не должно рассматриваться как ограниченное таким определенным заболеванием легких. Астма дает преимущество тщательного исследования и обеспечивает несколько приемлемых моделей для оценки изобретения. Известно, что сенсибилизация и введение аллергена ведет к гиперреактивности дыхательных путей на различные агонисты. Следовательно, ацетилхолин, известный как спазмогенный агент, способен вызывать более сильные сокращения мышечных клеток в тканях, полученных из трахеи умерщвленных животных (которые были сенсибилизированы для провокации гиперреактивности дыхательных путей), чем от контрольных животных после введения аллергена (смотрите, например, Tokuoka et at, Br. J. Pharmacol. 134:1580 (2001); Nakata et al, Int. Immunol. 13:329 (2001); Emala и Hirshman, Monogr. Allergy 33:35 (1996)).
Наиболее явной характеристикой астмы является бронхоспазм, или сужение дыхательных путей. У пациентов с астмой развивается выраженное сокращение гладких мышц крупных и мелких дыхательных путей, увеличенная продукция слизи, и увеличенное воспаление (Plaut и Zimmerman, выше). Воспалительный ответ при астме является типичным для тканей, покрытых слизистой оболочкой и характеризуется расширением кровеносных сосудов, выпотом плазмы, привлечением воспалительных клеток, таких как нейтрофилы, моноциты, макрофаги, лимфоциты и эозинофилы, к месту воспаления, и высвобождением воспалительных медиаторов местными тканевыми клетками (например, тучными клетками или эпителиальными клетками дыхательных путей) или миграцией воспалительных клеток (Hogg, "Pathology of Asthma", in Asthma as an Inflammatory Disease. O'Byrne (ed.), Marcel Dekker, Inc., New York, NY, at 1 (1990)). Астму могут запускать ряд причин, таких как аллергические реакции, вторичный ответ на инфекции, производственные или профессиональные воздействия, потребление определенных химических веществ или лекарственных веществ, физические нагрузки (Hargreave et al, J. Allergy Clin. Immunol. 83:1013 (1986)).
Соединения формулы (I) в соответствии с изобретением также были обнаружены как эффективные в снижении продукции слизи эпителиальными клетками бронхов и ингибировании пролиферации гладкомышечных клеток, опосредованной факторами роста.
Увеличение гиперреактивности бронхов (AHR), признак более тяжелой формы астмы, может быть индуцировано и антигенными и неантигенными раздражителями дыхательных путей. Ответ поздней фазы и постоянная гиперреактивность при астме, индуцированной аллергеном, ассоциированы с привлечением лейкоцитов и особенно эозинофилов в воспаленную ткань легких (Abraham et al, Am. Rev. Respir. Dis. 138:1565 (1988)). Эозинофилы высвобождают несколько медиаторов воспаления, включая 15-HETE, лейкотриен C4, PAF, катионные белки, эозинофильную пероксидазу.
Термины "антиген" и "аллерген" используют взаимозаменяемо для описания таких молекул, как пыль или пыльца, которые могут вызывать аллергические и/или индуцировать астматические симптомы у пациента, страдающего от астмы. Таким образом, пациент с астмой "при воздействии" аллергена или антигена подвергается воздействию достаточного количества аллергена или антигена, чтобы вызвать астматический ответ. Было обнаружено, что соединения формулы (I) по изобретению эффективны для лечения AHR после сенсибилизации яичным белком и воздействия антигена.
Биологическая активность соединений формулы (I) по настоящему изобретению на различных животных моделях продемонстрирована ниже:
Модель 1
Изучение эффекта вводимых местно полисульфатированных гликозидов на гиперреактивность дыхательных путей ex vivo
Воспаление дыхательных путей может вести к гиперреактивности бронхов, что является характерным признаком астмы.
Коричневых Норвежских (BN) крыс активно сенсибилизировали яичным белком (OA) посредством подкожной инъекции 0,5 мл гелевой смеси OA/Al(OH)3 (2 мг OA + 10 г Al(OH)3/100 мл физиологического раствора) в день 1 с последующими подкожными инъекциями (10 мг OA + 10 г Al(OH)3/100 мл физиологического раствора), вводимыми в дни 14 и 21. В день 28 животные получали соединение, описанное в примере 4, интратрахеально (дозу 0,01, 0,1 или 1,0 мг/кг) за 2 часа до воздействия антигена. Раздражение антигеном проводили ингаляцией распыленного яичного белка (1% раствор антигена, вводимый в системе для ингаляций TSE в течение 1 часа). Животных умерщвляли через 48 часов после воздействия антигена, затем удаляли трахеи в кювету для органов. Иссеченным трахеям позволяли уравновеситься в течение 30 минут до измерения кривых спазмогенного ответа трахеи на ацетилхолин (Ach).
Как показано в таблице 1 раздражение яичным белком сенсибилизированных животных в этой модели вызывало существенную гиперреактивность трахеи на ацетилхолин, когда ответ на спазмогенный агент определяли через 48 часов после воздействия антигена. Соединение, описанное в примере 4, в дозе 0,01 мг/кг, возвращало это повышение обратно почти до контрольного уровня.
Эффект воздействия антигена
и интратрахеального предварительного лечения
соединением примера 4
на сокращение трахеи в ответ на ацетилхолин у BN крыс
* -log M ацетилхолина (Ach), вызывающего 50% сокращение по отношению к контролю (среднее ± SEM)
** Сокращение при максимальной концентрации Ach по отношению к контролю (среднее ± SEM)
Модель 2
Исследование эффекта полисульфатированных гликозидов на продукцию слизи эпителиальными клетками дыхательных путей, стимулированную аллергеном
У сенсибилизированных животных антигенная стимуляция приводила к продукции слизи эпителиальными клетками дыхательных путей, что является характерным свойством аллергической астмы.
Сенсибилизированным BN крысам вводили интратрахеально различные (0,01-1,0 мг/кг) дозы соединения, описанного в примере 4, за два часа перед антигенной стимуляцией, с использованием подобного протокола, описанного в модели 1. Легкие получали через 48 часов после стимуляции и фиксировали в 8% фосфатном буферном формальдегиде. Затем образцы обрабатывали для гистохимии обычным образом. Сечения толщиной 5 мкм окрашивали реагентами периодической кислоты Шиффа (PAS) и докрашивали гематоксилином-эозином. На сечениях все эпителиальные клетки дыхательных путей подсчитывали в цельном препарате с увеличением 400×. Количество PAS(+) [продуцирующих слизь] эпителиальных клеток выражали по отношению к общему количеству эпителиальных клеток.
Как показано в таблице 2, воздействие аллергена стимулировало продукцию слизи эпителиальными клетками дыхательных путей (контроль vs. воздействие). Соединение существенно снижало количество PAS(+), продуцирующих слизь клеток в применяемой более высокой дозе.
Эффект антигенного воздействия и интратрахеального лечения соединением примера 4 на продукцию слизи
эпителиальными клетками дыхательных путей,
индуцированную аллергеном у BN крыс
* - количество PAS(+) клеток, как процент от общего количества клеток (среднее ± SEM)
Модель 3
Изучение эффекта полисульфатированных гликозидов на степень периваскулярного отека, развивающегося в астматической легочной ткани
У сенсибилизированного животного воздействие антигена, в результате развития воспалительного процесса, увеличивает проницаемость кровеносных сосудов, приводя к экссудации плазмы вокруг периферической части сосудистого русла.
Сенсибилизированные BN крысы получали лечение интратрахеально различными дозами (0,01-1,0 мг/кг) соединения, описанного в примере 4, за два часа до воздействия антигена, с использованием подобного протокола, описанного в модели 1. Легкие получали через 48 часов после воздействия и фиксировали в 8% фосфатном буферном формальдегиде. Затем образцы обрабатывали для гистохимии обычным образом. Сечения толщиной 5 мкм окрашивали реагентами периодической кислоты Шиффа (PAS) и докрашивали гематоксилином-эозином. На сечениях 5 мкм определяли площадь соединительной ткани вокруг сосудистого русла и выражали как соотношение к площади соответствующего кровеносного сосуда как такового.
Как показано в таблице 3, воздействие аллергена вызывает отек вокруг сосудистой сети, степень которого была существенно снижена даже при наименьшей дозе исследуемого соединения.
Эффект воздействия антигена и интратрахеального лечения соединением по примеру 4 на степень развития астмы у BN крыс
* - площадь отека относительно площади сосудистого русла (среднее ± SEM)
Модель 4
Антагонистический эффект полисульфатированых гликозидов к рецептору IP-3
Гликозиды по настоящему изобретению, в зависимости от их химической структуры, ингибируют связывание инозитол-1,4,5-трифосфата (IР3) с его рецептором в препаратах микросомальных мембран. Так как IP3 является молекулой-мессенджером, играющей известную роль в активации различных клеток, вмешательство в эту функцию может объяснить антиастматический эффект таких полисульфатированных гликозидов.
Антагонистический к IP3 эффект полисульфатированных гликозидов определяли с использованием препаратов мембран мозжечка крыс в соответствии с Worley et al. (JBC 262, 12132, 1987). Как показано в таблице 4, все соединения, описанные в примерах 1-16, обладают различной антагонистической к IP3 активностью.
Антагонистический к рецептору IP-3 эффект
полисульфатированных гликозидов
Фармацевтические композиции по настоящему изобретению предназначены для применения у любого млекопитающего, которое может испытывать пользу от способов по изобретению. Основным таким млекопитающим является человек, хотя изобретение не является настолько ограниченным и применимо для ветеринарного использования. Следовательно, в соответствии с изобретением "млекопитающее" или "нуждающееся млекопитающее" включает человека, а также млекопитающих, отличных от человека, особенно домашних животных, включая, без ограничения, кошек, собак и лошадей.
Термин "терапевтически эффективное количество" используют для обозначения лечения в дозировках, эффективных для достижения терапевтического результата. Более того, специалист должен принимать во внимание, что терапевтически эффективное количество соединения по изобретению может быть снижено или увеличено точным подбором и/или введением более одного соединения по изобретению или введением соединения по изобретению с другим противоастматическим соединением (например, кортикостероидом). Следовательно, изобретение обеспечивает способ адаптации введения/лечения с определенными нуждами, специфичными для данного млекопитающего. Как показано в следующих примерах, терапевтически эффективное количество может быть легко определено, например, эмпирически, начиная с относительно низких количеств и пошаговым увеличением с одновременной оценкой благоприятного эффекта. Клинические изменения, значимые для оценки терапевтического эффекта лечения, в соответствии с изобретением включают уменьшение характерных симптомов и признаков астмы и связанной патологии (например, одышки, хрипов, кашля, гиперчувствительности бронхов, перестройки дыхательных путей) и улучшение исследований функции легких. Это основывается на симптомах пациента и наблюдениях врача.
Как используется в настоящем описании, перечисление числовых значений для переменных предназначено для обозначения, что изобретение может быть осуществлено с переменными, равными любому значению в рамках. Следовательно, для переменной, которая является по существу дискретной, переменная может быть равной любому целому значению числовой области, включая конечные точки диапазона. Подобным образом для переменной, которая является по существу непрерывной, переменная может быть равной любому реальному значению числового диапазона, включая конечные точки диапазона. В качестве примера переменная, которая описана как имеющая значения между 0 и 2, может быть 0, 1 или 2 для переменных, которые являются по существу дискретными, и может быть 0,0, 0,1, 0,01, 0,001 или любым другим реальным значением для переменных, которые являются по существу непрерывными.
Для местного введения ингаляцией, например, предусматриваемые терапевтически эффективные количества составляют от около 0,1 мкг/кг/день до около 1000 мкг/кг/день при введении системно (например, пероральном введении). В варианте осуществления изобретения, при системном введении терапевтически эффективные количества составляют от около 0,5 мкг/кг/день до около 200 мкг/кг/день.
Дозированные формы и частота их введения будет зависеть от обычных факторов, обычно рассматриваемых специалистом в области техники для получения терапевтически эффективного количества, как обсуждается выше, у данного животного. Следовательно, специалист должен рассматривать состояние, подвергаемое лечению, определенное вводимое соединение по изобретению, пути введения и другие клинические факторы, такие как возраст, масса тела и состояние млекопитающего, а также комфорт и приверженность пациента к лечению (комплаентность).
Специалист в области техники понимает, что количество введений соединений по изобретению будет варьироваться от пациента к пациенту на основании определенного медицинского состояния в любое заданное время.
Когда возможно (например, для лечения астмы), соединение по этому аспекту изобретения может вводиться перед, во время или после воздействия на млекопитающее антигена. Кроме того, время введения соединения по изобретению в отношении воздействия антигена будет варьироваться от млекопитающего к млекопитающему в зависимости от определенной ситуации. Специалист в области техники может оптимизировать введение тщательным мониторингом пациента при изменении времени и/или порядка введения соединения по изобретению. Следовательно, необходимо понимать, что млекопитающее не должно страдать от воспаления легких для получения пользы от изобретения. Соединение по изобретению может вводиться профилактически пациентам, предрасположенным к развитию астмы и/или патологии, связанной с астмой. Например, пациенту с аллергией на пыльцу можно вводить соединение по изобретению (например, пероральным введением) на ежедневной основе и/или перед входом в область, богатую пыльцой (например, сад). Также пациенту только с семейным анамнезом астматических приступов можно вводить соединения по изобретению профилактически - для предотвращения или ингибирования начала такого астматического приступа.
На основании вышеуказанных фактов настоящее изобретение также обеспечивает способ лечения острых и хронических воспалительных расстройств дыхательных путей млекопитающих, включая астму и патологии, связанные с астмой. Такой способ включает введение млекопитающему, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I).
Соединения по изобретению оптимально рецептируют в фармацевтически приемлемом носителе с любым из хорошо известных фармацевтически приемлемых носителей, включая разбавители и вспомогательные вещества (смотрите Remington's Pharmaceutical Sciences. 18th Ed., Gennaro, Mack Publishing Co., Easton, PA 1990 и Remington: The Science and Practice of Pharmacy. Lippincott, Williams & Wilkins, 1995). Тогда как тип фармацевтически приемлемого носителя/растворителя, используемого в создании композиций по изобретению, будет варьироваться в зависимости от типа введения композиции млекопитающему; обычно фармацевтически приемлемые носители являются физиологически инертными и нетоксичными. Рецептуры композиций по изобретению могут содержать более одного типа соединений по изобретению, а также любые другие фармакологически активные ингредиенты, применимые для лечения определенного подвергаемого лечению воспаления легких. Такие соединения могут включать, без ограничения, антагонисты β-адренорецепторов: албутерол, метапротеренол, левалбутерол, пирбутерол, сальметерол, битолтерол; глюкокортикоиды: беклометазон, триамцинолон, флунизолид, будесонид, флутиказон; антагонисты рецепторов лейкотриенов и ингибиторы синтеза лейкотриенов: зафирлукаст, монтелукаст, зилеутин; другие противоастматические средства: кромолин, недокромил, теофиллин; антихолинергические агенты: ипратропиум, окситропиум, тиотропиум; антигистаминные антагонисты рецепторов H1: дифенгидрамин, пириламин, прометазин, лоратадин, хлорциклизин, хлорфенирамин, фексофенадин и адренокортикостероиды.
Композиции по изобретению могут вводиться стандартными путями (например, пероральным, ингаляционным, ректальным, назальным, местным, включая буккальный и сублингвальный, или парентеральным, включая подкожный, внутримышечный, внутривенный, внутрикожный, трансдермальный, и интратрахеальный). Кроме того, могут быть добавлены полимеры в соответствии со стандартными методиками в области техники для замедленного высвобождения заданного соединения.
Композиции, подходящие для ингаляционного введения, включают композиции, которые могут подаваться ингаляционными устройствами, известными в области техники. Такие композиции могут включать носители, такие как порошок и аэрозоли. Настоящее изобретение охватывает жидкие и порошкообразные композиции, подходящие для распыления и интрабронхиального применения, или аэрозольные композиции, вводимые посредством аэрозольного дозирующего устройства ("MDI"). Особенно предпочтительные предусматриваемые устройства описаны в патенте США № 5447150.
Активный ингредиент может быть рецептирован в водном фармацевтически приемлемом ингаляционном носителе, таком как, например, изотонический солевой раствор или бактериостатическая вода и другие типы носителей, которые хорошо известны в области техники. Растворы вводят посредством насоса или запускаемого сдавлением распыляющего устройства, или любыми другими обычными средствами, вызывающими или позволяющими необходимое дозирование количества жидкой композиции для ингаляций в легкие пациента.
Порошкообразные композиции, содержащие противовоспалительные соединения по настоящему изобретению включают, в качестве иллюстрации, фармацевтически приемлемые порошкообразные препараты активного ингредиента, тщательно перемешанные с лактозой или другими инертными порошками, приемлемыми для интрабронхиального введения. Порошкообразные композиции могут вводиться посредством устройства, включая, без ограничения, аэрозольное устройство, или заключенными в ломкую капсулу, которая может быть вставлена пациентом в устройство, которое прокалывает капсулу и выдувает порошок стабильным потоком.
Аэрозольные композиции для применения в способе по изобретению обычно включают пропелленты, поверхностно-активные вещества и сорастворители и могут быть заполнены в аэрозольные контейнеры, которые закрыты подходящим дозирующим клапаном.
Для перорального введения противовоспалительные композиции по изобретению могут быть представлены в виде отдельных единиц, таких как капсулы, каплеты, гелевые капсулы, облатки, пилюли или таблетки, каждая содержащая заранее определенное количество активного ингредиента в виде порошка или гранул; в виде раствора или суспензии в водной жидкости или неводной жидкости; или в виде жидкой эмульсии типа масло-в-воде или эмульсии типа вода-в-масле и в виде болюса и др. Альтернативно, введение композиции по всем аспектам настоящего изобретения может осуществляться посредством жидких растворов, суспензий или эликсиров, порошков, пастилок, микронизированных частиц и осмотических систем доставки.
Рецептура композиций по настоящему изобретению, подходящих для назального введения, где носитель является твердым веществом, включает крупный порошок, имеющий размер частиц, например, в диапазоне от 20 до 500 микрон, который вводят посредством вдыхания через нос, т.е. быстрой ингаляцией через носовые ходы из контейнера с порошком, подносимого близко к носу. Подходящие композиции, где носителем является жидкость, для введения, например, посредством назального спрея, аэрозоля или назальных капель, включают водные или масляные растворы соединений по изобретению. Полужидкие композиции, такие как назальный гель, также являются подходящими.
Рецептуры композиций, подходящих для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, стабилизаторы, буферы, бактериостатики и растворы, которые делают композицию изотоничной с кровью предназначенного реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители.
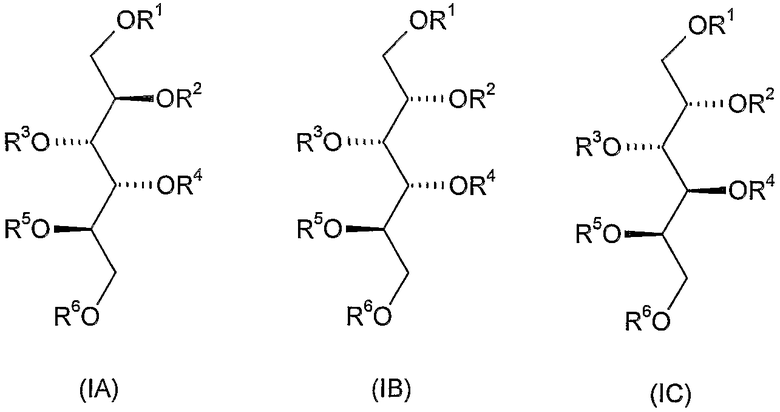
Способ для синтеза соединений общей формулы (IA), (IB) и (IС), которые являются стереохимически определенными представителями соединений общей формулы (I) по настоящему изобретению, проиллюстрированы следующими примерами.
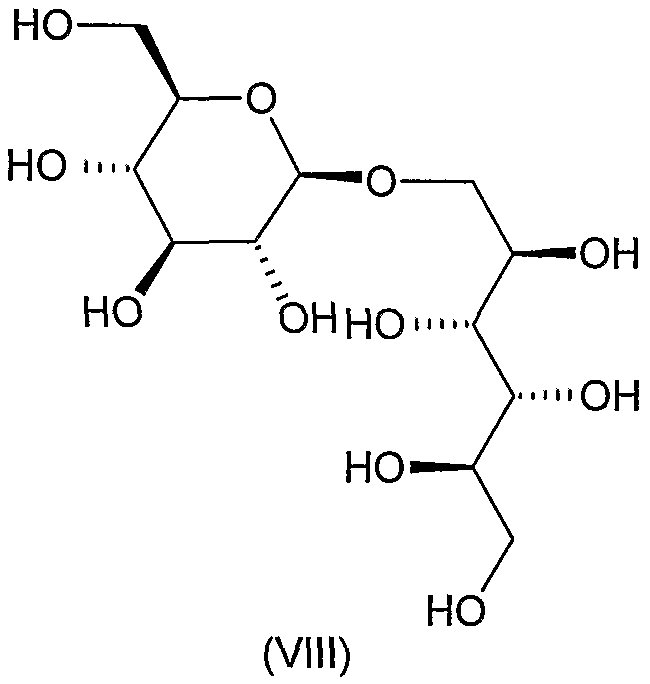
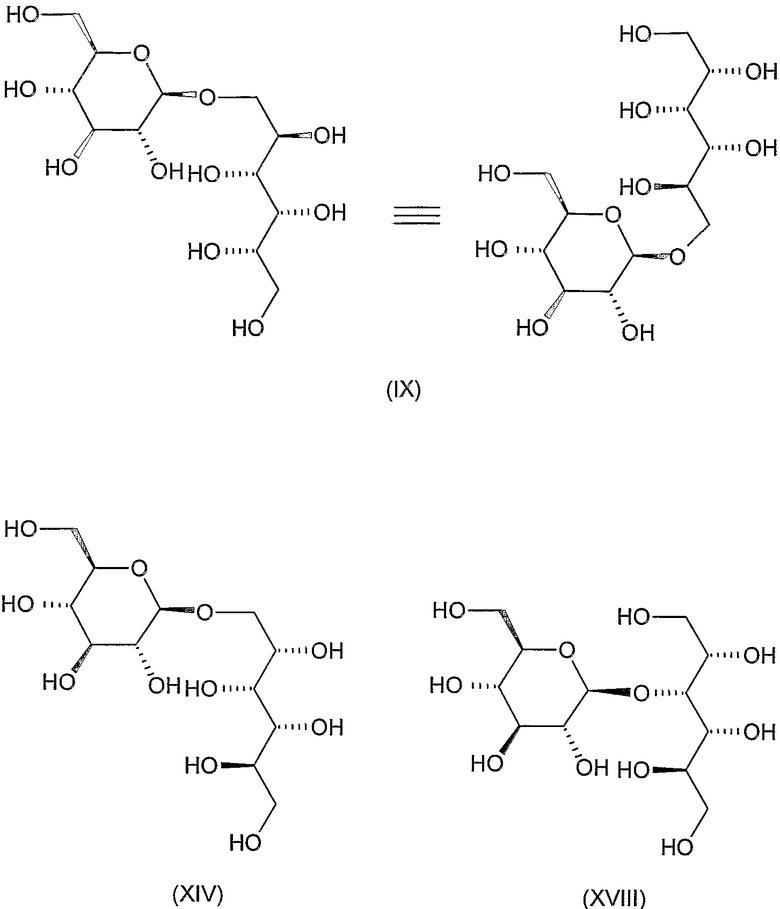
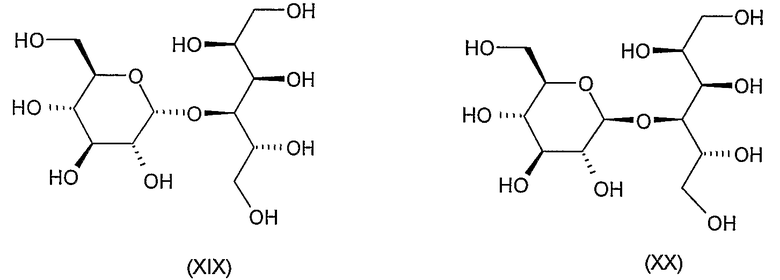
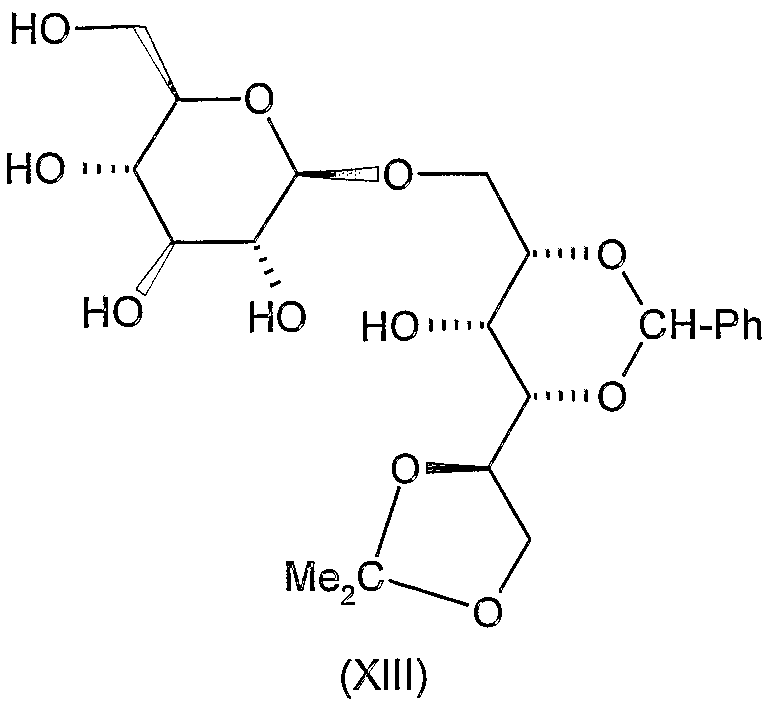
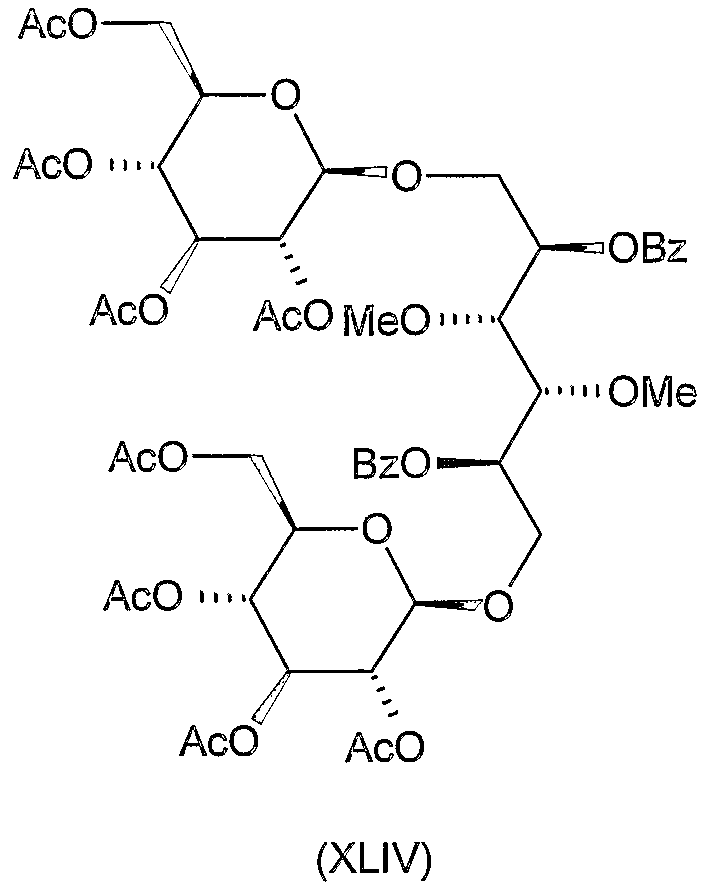
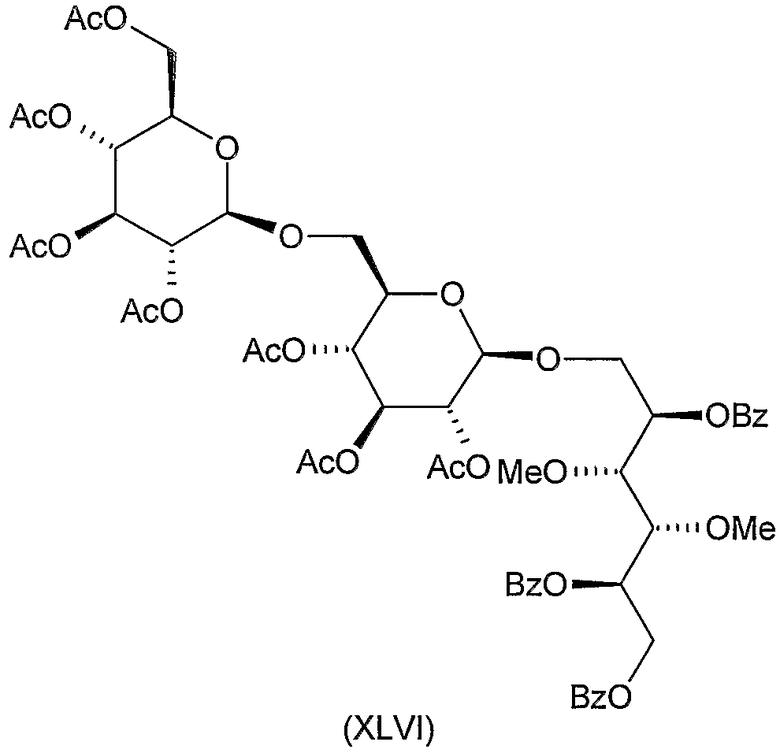
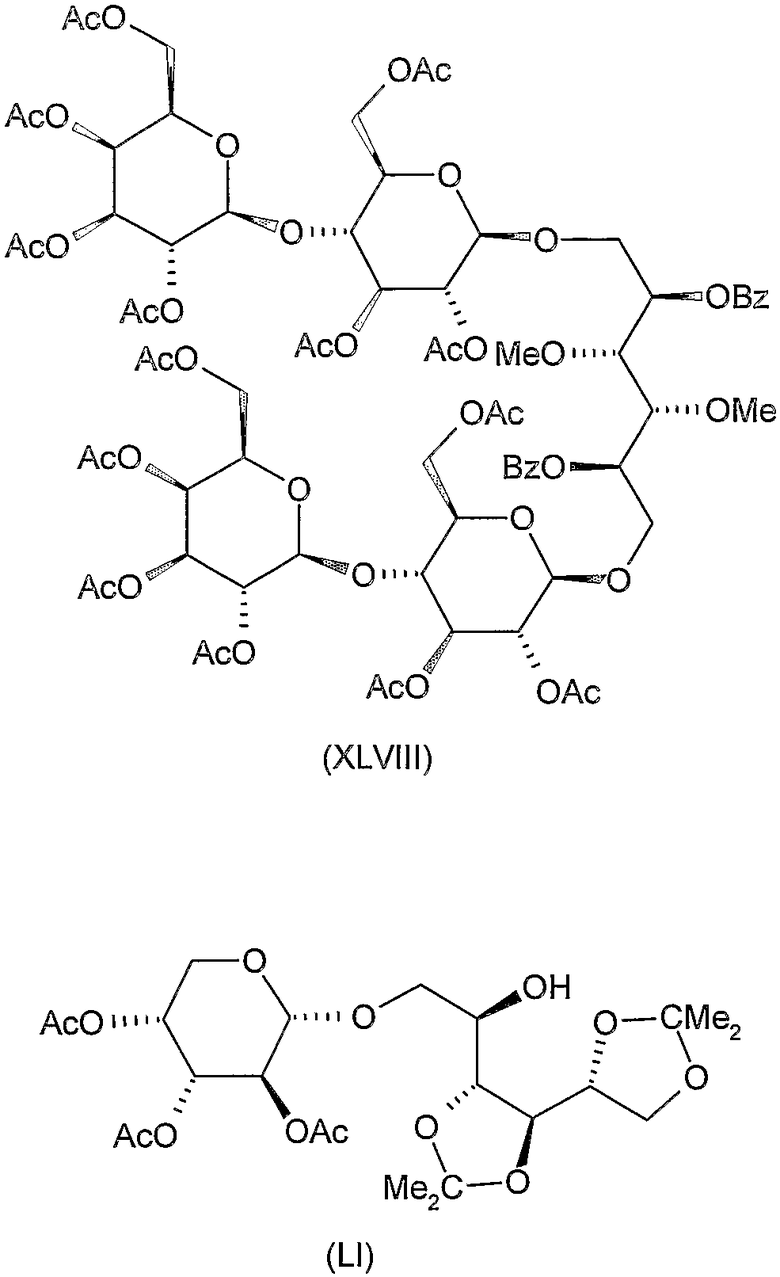
Соединения формул VIII, IX, XIV, XVIII, XIX, XX, XXI, XXIV, XXIX, XXXIII, XXXVII, XLIII, XLV, XLVII, IL, LIII, LVII и LXI, используемые в качестве исходных соединений в примерах, являются конкретными, стереохимически определенными, изомерно чистыми представителями формулы (II). Их химические структуры продемонстрированы ниже:
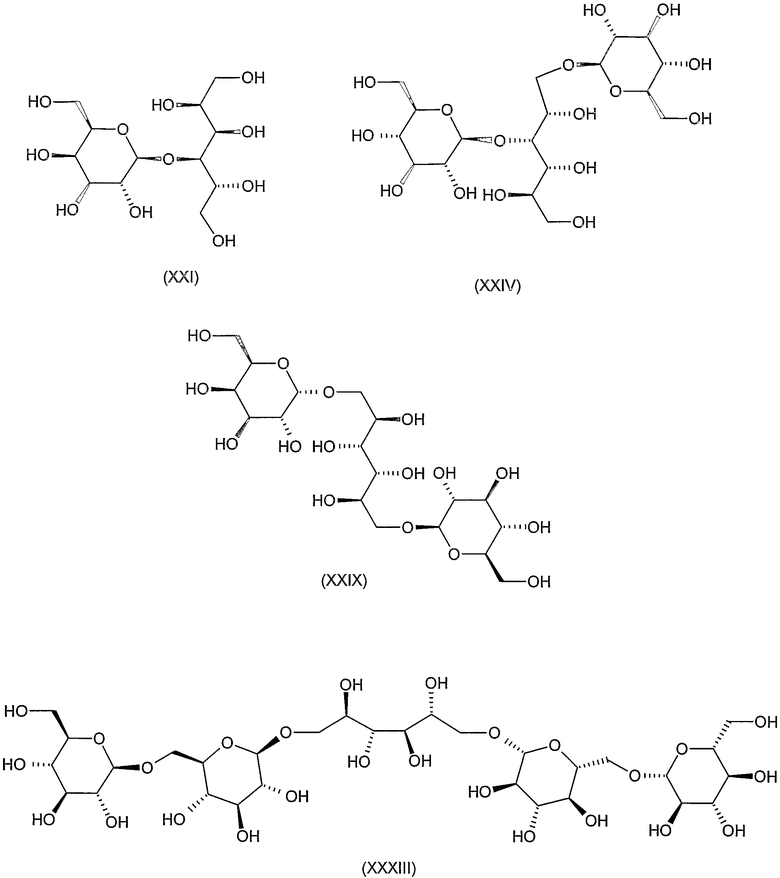
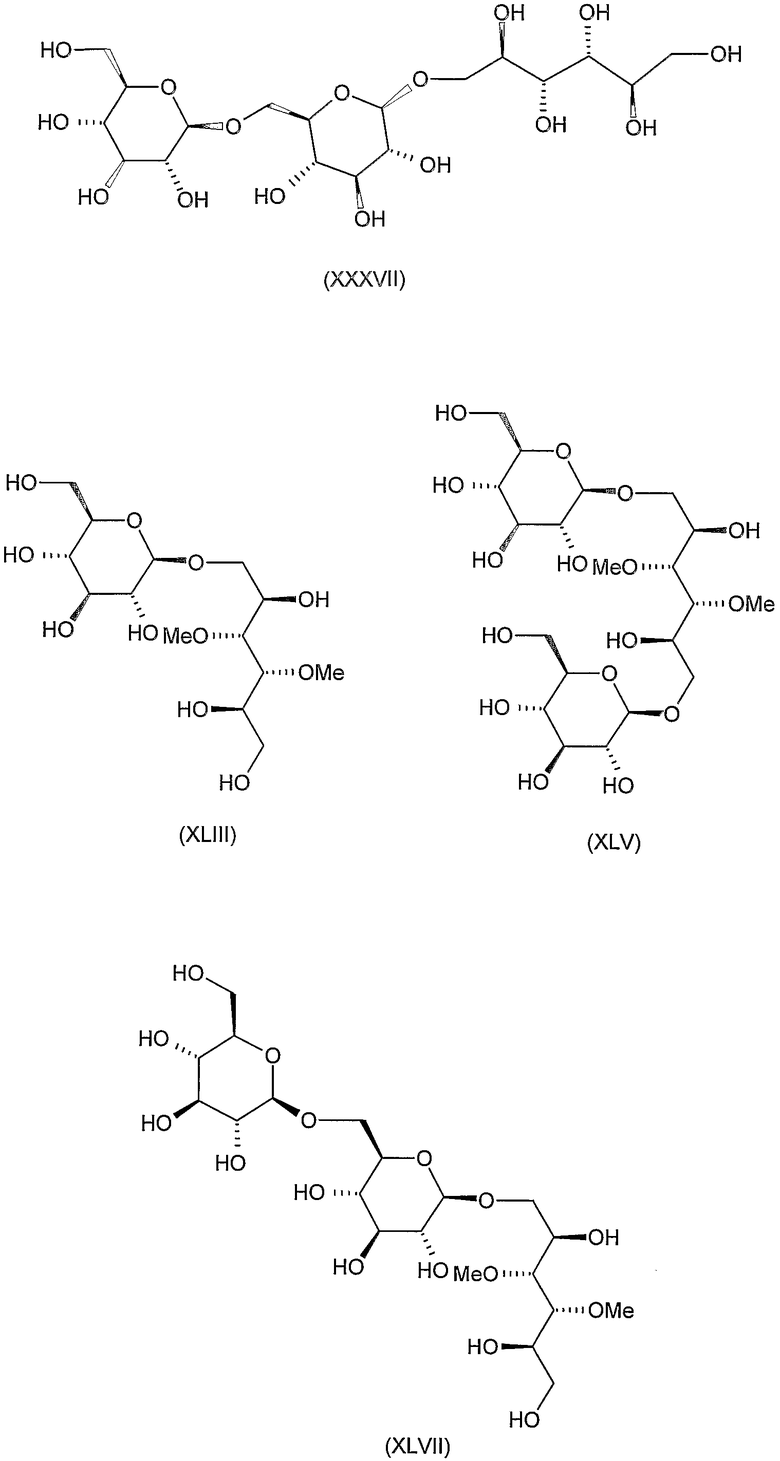

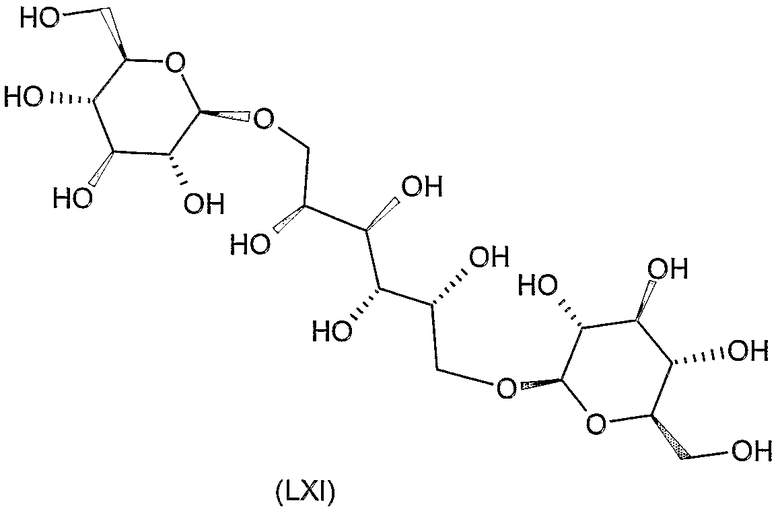
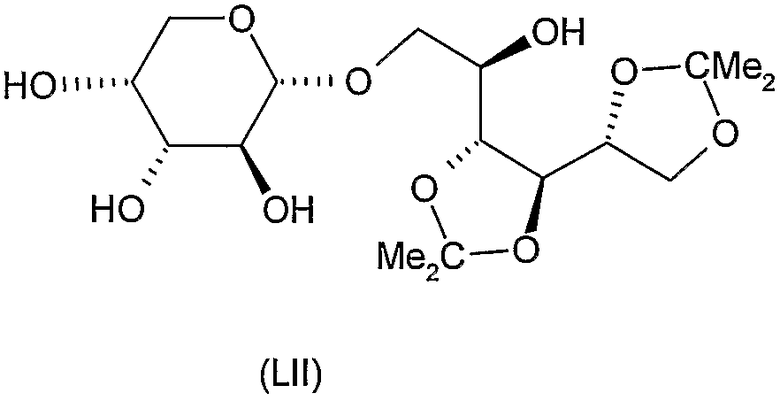
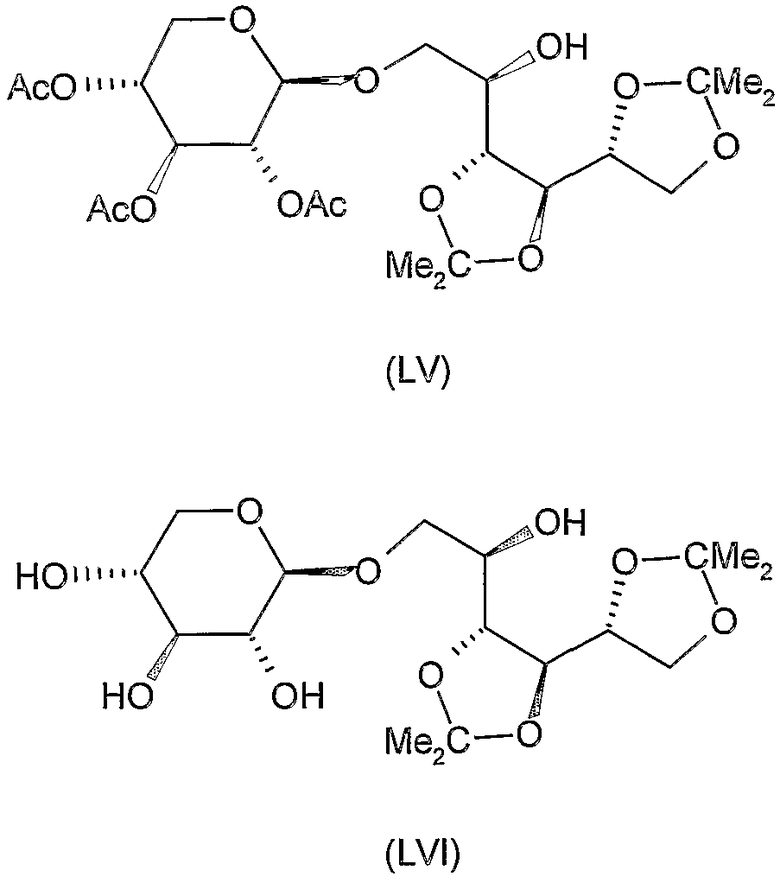
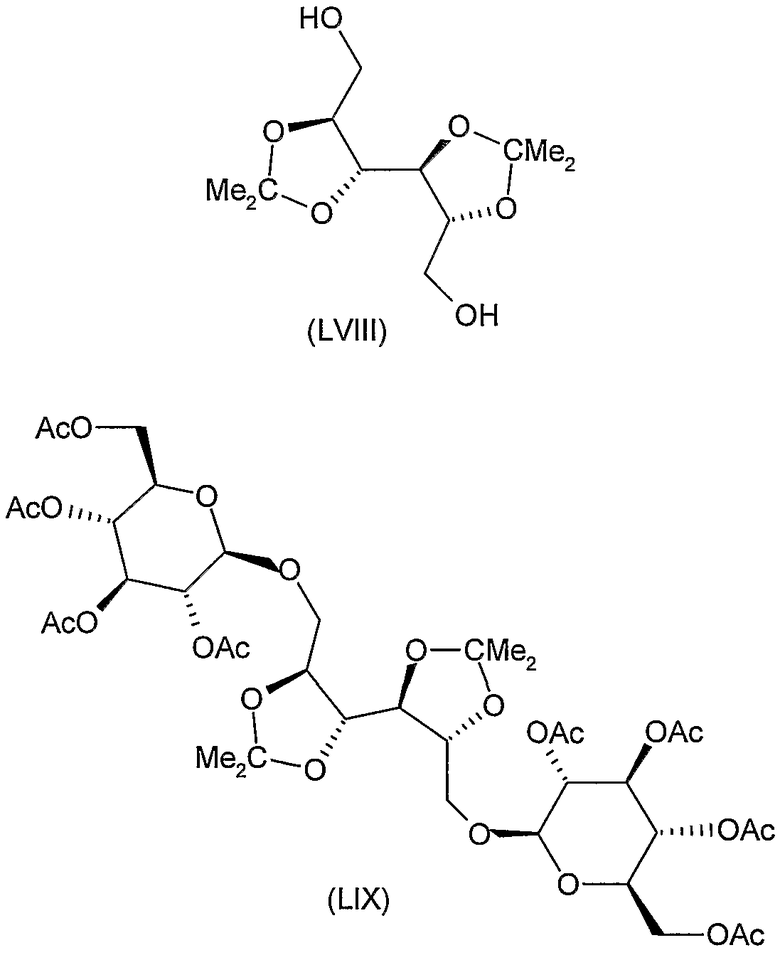
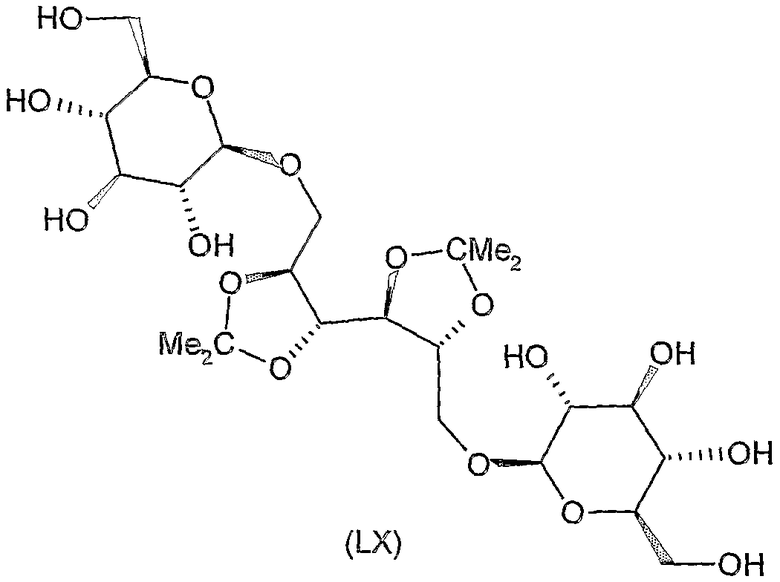
Соединения формул XI, XII, XIII, XV, XVI, XVII, ХХII, XXIII, XXV, XXVI, XXVII, XXVIII, XXXI, ХХХII, XXXIV, XXXV, XXXVI, XXXVIII, XLI, XLII, XLIV, XLVI, XLVIII, LI, LII, LV, LVI, LVIII, LIX и LX, которые используют в синтезе новых исходных веществ формулы (II), являются конкретными, стереохимически определенными, изомерно чистыми представителями формулы (V). Их химические структуры продемонстрированы ниже:




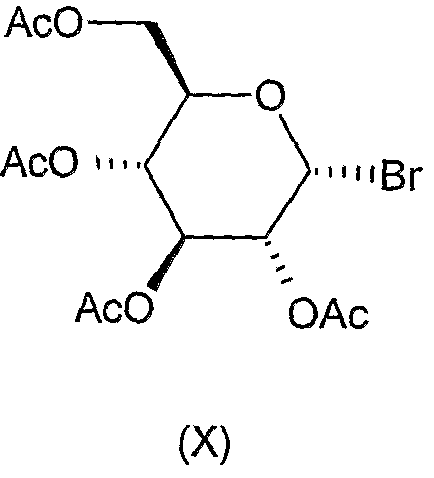
Молекулы-доноры, используемые в реакциях гликозилирования, являются или коммерчески доступными, например соединение формулы (X)
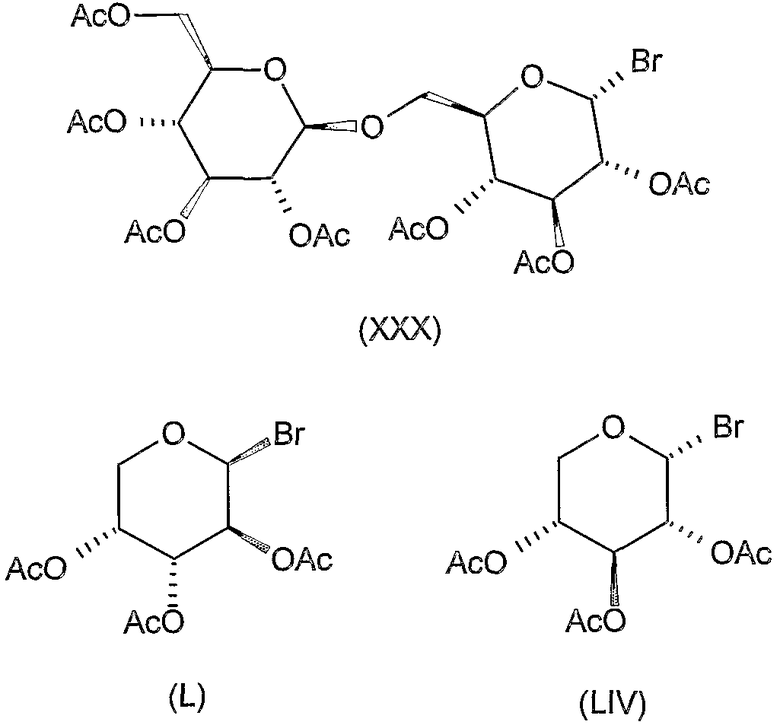
или могут быть синтезированы известными способами (смотрите экспериментальную часть), например соединения формул (XXX), (L) и (LIV).
Значения Rf, данные в примерах, определяли тонкослойной хроматографией с использованием силикагеля (DC-Alufolien Kieselgel 60 F254, Merck, Darmstadt) и следующих смесей растворителей:
(A) Этилацетат - гексан 1:1
(B) Этилацетат - гексан 1:2
(C) Этилацетат - гексан 2:1
(D) Этилацетат - гексан 3:1
(E) Этилацетат - метанол 1:1
(F) Этилацетат - метанол 3:1
(G) Этилацетат - метанол 5:1
Области определяли или в УФ-свете, или орошением планшетов 1:1 смесью 0,1 M KMnO4 - 1 M H2SO4 с последующим нагреванием до 200°C. Колоночную хроматографию проводили на Kieselgel 60. Оптическое вращение измеряли при 20°C. Спектры ЯМР записывали спектрометром Bruker Avance 500 МГц с использованием Me4Si в качестве внутреннего стандарта. Распределение протонов было основано на экспериментах COSY, 2D и селективных ID TOCSY, а также селективных ID NOESY. Многообразие спектров 13C получали из экспериментов DEPT. Связь между идентифицированными протонами и протонированными углеродами наблюдали посредством экспериментов HMQC и HMBC.
В случае реакций ацилирования, проводимых в присутствии пиридина, "обычная обработка" означает, что если продукт не становится кристаллическим после вливания реакционной смеси в ледяную воду, его экстрагируют органическим растворителем, органический слой промывают водой, 1 M ледяным водным раствором серной кислоты до постоянной кислотности, водой, 5% водным раствором бикарбоната натрия и водой, сушат, фильтруют и растворитель выпаривают в вакууме.
Исходное вещество для соединения формулы (XIV) синтезируют, например, следующим способом:
Стадия a)
2,4-O-бензилиден-5,6-O-изопропилиден-D-глюцитол (XI)
К перемешиваемой суспензии 27 г (0,1 моль) 2,4-O-бензилиден-D-глюцитола (L. Vargha, Ber. 68 (1935) 18-24) в 150 мл диметилформамида добавляли при комнатной температуре 20 мл (0,26 моль) 2,2-диметоксипропана и 100 мг п-толуолсульфоновой кислоты. После перемешивания в течение 10 минут получали чистый раствор и добавляли 1 мл триэтиламина через 1 час. Реакционную смесь концентрировали, остаток растворяли в хлороформе, нерастворимое исходное вещество отфильтровывали, фильтрат концентрировали и остаток перекристаллизовывали из 200 мл бензола для получения 14 г (45%) указанного в заголовке соединения. Т.пл. 178°C, Rf 0,4 (растворитель С).
Стадия b)
1-O-(2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозил)-2,4-O-бензилиден-5,6-O-изопропилиден-D-глюцитол (XII)
К раствору 3,1 г (10 ммоль) продукта формулы (XI), полученного на предыдущей стадии, в 50 мл ацетонитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 4,5 г (11 ммоль) ацетобром-D-глюкозы (X) и 3 г (12 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 100 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель C) для получения 2,7 г (42%) указанного в заголовке соединения, Rf 0,7, [α]D +5° (c 1, хлороформ).
Стадия c)
1-O-(β-D-глюкопиранозил)-2,4-O-бензилиден-5,6-O-изопропилиден-D-глюцитол (ХIII)
К раствору 4,1 г продукта формулы (XII), полученного на предыдущей стадии, в 40 мл метанола добавляли 0,5 мл 2 M раствора метоксида натрия в метаноле при комнатной температуре. Через 2 часа ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (растворитель G) для получения 2,2 г (73%) указанного в заголовке соединения, Rf 0,6, [α]D -8° (c 1, хлороформ).
Стадия d)
1-O-(β-D-глюкопиранозил)-D-глюцитол (XIV)
К перемешиваемому раствору 3 г продукта формулы (ХIII), полученного на предыдущей стадии, в 80 мл метанола добавляли 3 мл воды, 1 мл уксусной кислоты и 2 г 10% катализатора Pd/C и смесь гидрогенизировали при атмосферном давлении. Когда в соответствии с ТЖХ реакция была завершенной (~4 часа), катализатор отфильтровывали, фильтрат концентрировали, остаток растворяли в 20 мл 0,05 M серной кислоты и перемешивали при 60°C в течение 90 минут. Охлажденный раствор нейтрализовали добавлением ионообменной смолы, отфильтровывали, концентрировали до объема 15 мл и лиофилизировали для получения 1,9 г (86%) указанного в заголовке соединения, Rf 0,1 (растворитель E), [α]D -105° (c 1, вода).
Исходное вещество формулы (XVIII) может быть синтезировано, например, следующим способом:
Стадия a)
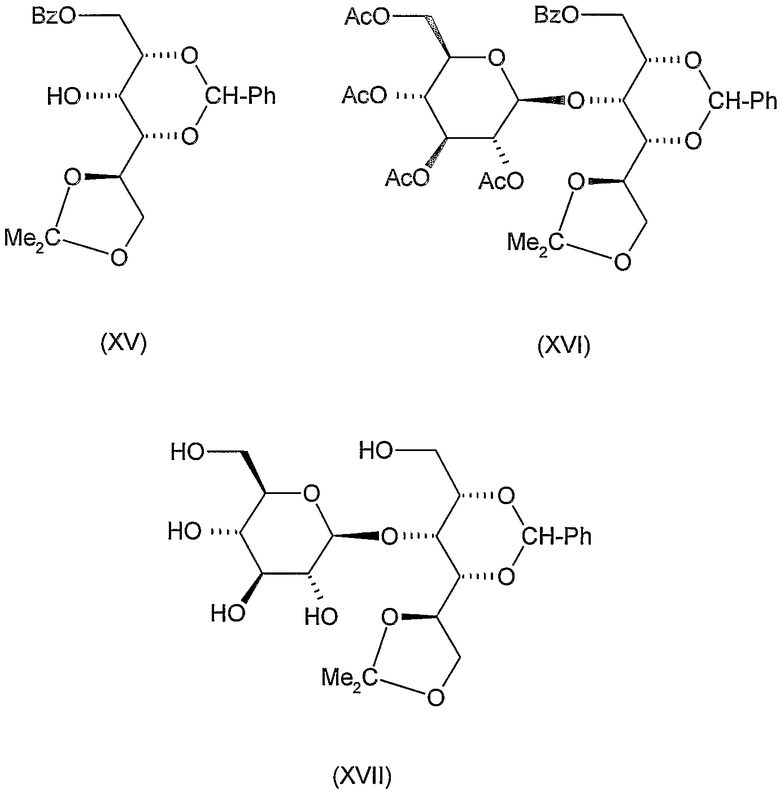
2,4-O-бензилиден-1-O-бензоил-5,6-О-изопропилиден-D-глюцитол (XV)
К перемешиваемому раствору 3,1 г (10 ммоль) 2,4-О-бензилиден-5,6-О-изопропилиден-D-глюцитола (L. Vargha, Ber, 68 (1935) 18-24 и 1377-1384) в 10 мл пиридина добавляли 1,3 мл (11 ммоль) бензоилхлорида по каплям при -20°C. Реакционную смесь перемешивали при той же температуре в течение 15 минут и при комнатной температуре в течение 30 минут, затем обрабатывали обычным образом. Остаток, полученный после концентрирования органической фазы, очищали колоночной хроматографией (растворитель B) для получения 2,5 г (60%) указанного в заголовке соединения в виде бесцветного сиропа, Rf 0,6, [α]D -24° (c 1, хлороформ).
Стадия b)
3-О-(2,3,4,6-тетра-О-ацетил-β-D-глюкопиранозил)-2,4-О-бензилиден-1-О-бензоил-5,6-О-изопропилиден-D-глюцитол (XVI)
К перемешиваемому раствору 2,5 г (6 ммоль) продукта формулы (XV), полученного на предыдущей стадии, в 20 мл ацетонитрила добавляли 5 г молекулярного сита (4 Е), смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 2,5 г (6 ммоль) ацетoбром-D-глюкозы (X) и 1,6 г (6,5 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 40 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель A) для получения 2,45 г (55%) указанного в заголовке соединения, Rf 0,5,
[α]D -4° (c 1, хлороформ).
Стадия c)
3-О-(β-D-глюкопиранозил)-D-глюцитол (XVIII)
К раствору 4,46 г (6 ммоль) продукта формулы (XVI), полученного на предыдущей стадии, в 20 мл метанола добавляли 0,2 мл 2 M раствора метоксида натрия в метаноле при комнатной температуре. Через 1 час, когда в соответствии с ТЖХ деацилирование завершалось, ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток, который содержал деацилированный продукт (XVII) и метилбензоат, растворяли в 50 мл метанола, добавляли 3 мл воды, 1 мл уксусной кислоты и 2 г 10% катализатора Pd/C и смесь гидрогенизировали при атмосферном давлении. Когда в соответствии с ТЖХ реакция завершалась (~15 часов), катализатор отфильтровывали, фильтрат концентрировали, остаток растворяли в смеси хлороформа и воды и растворяли. 1,5 мл серной кислоты добавляли к водной фазе и перемешивали при 60°C в течение 1 часа. Охлажденный раствор нейтрализовали добавлением ионобменной смолы, отфильтровывали, концентрировали до объема 15 мл и лиофилизировали для получения 2 г (97%) указанного в заголовке соединения в виде гигроскопичного порошка, которое использовали на следующей стадии без последующей очистки. [α]D -14° (c 1, вода).
Исходное вещество формулы (XXIV) может быть синтезировано, например, следующим способом:
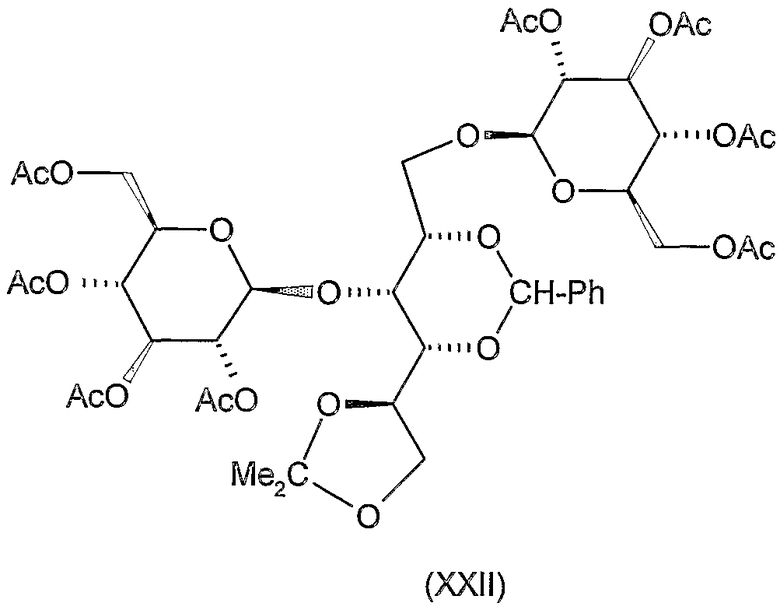
Стадия a)
1,3-бис-О-(2,3,4,6-тетра-О-ацетил-β-D-глюкопиранозил)-2,4-О-бензилиден-5,6-О-изопропилиден-D-глюцитол (XXII)
К раствору 6,2 г (20 ммоль) 2,4-O-бензилиден-5,6-O-изопропилиден-D-глюцитола (XI) в 200 мл ацетoнитрила добавляли 24 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 18 г (44 ммоль) ацетoбром-D-глюкозы (X) и 12 г (48 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 400 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток перекристаллизовывали из 150 мл метанола для получения 1,8 г (9,3%) указанного в заголовке соединения. Т.пл. 168-170°C, Rf 0,7 (растворитель С), [α]D -5° (c 1, хлороформ).
Стадия b)
1,3-бис-O-(β-D-глюкопиранозил)-2,4-O-бензилиден-5,6-О-изопропилиден-D-глюцитол (ХХIII)
К раствору 2,65 г продукта формулы (ХХII), полученного на предыдущей стадии, в 30 мл метанола добавляли 0,5 мл 2 M раствора метоксида натрия в метаноле при комнатной температуре. Через 2 часа ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали для получения 1,7 г (97%) указанного в заголовке соединения, Rf 0,3 (растворитель F), [α]D -8° (c 1, хлороформ).
Стадия c)
1,3-бис-О-(β-D-глюкопиранозил)-D-глюцитол (XXIV)
К перемешиваемому раствору 1,7 г продукта формулы (ХХIII), полученного на предыдущей стадии, в 50 мл метанола добавляли 3 мл воды, 1 мл уксусной кислоты и 1 г 10% катализатора Pd/C и смесь гидрогенизировали при атмосферном давлении. Когда в соответствии с ТЖХ реакция завершалась (~4 часа), катализатор отфильтровывали, фильтрат концентрировали, остаток растворяли в 20 мл 0,05 M серной кислоты и перемешивали при 60°C в течение 90 минут. Охлажденный раствор нейтрализовали добавлением ионобменной смолы, отфильтровывали, концентрировали до объема 15 мл и лиофилизировали для получения 1,25 г (92%) указанного в заголовке соединения. Rf 0,1 (растворитель E),
[α]D -20° (c 1, вода).
Исходное вещество формулы (ХХIХ) может быть синтезировано, например, следующим способом:
Стадия a)
2,5-ди-О-бензоил-3,4-О-изопропилиден-1,6-ди-О-тритил-D-маннит (XXV)
К перемешиваемому раствору 11,1 г (50 ммоль) 3,4-O-изопропилиден-D-маннита (T. Horvath и L. Vargha, Carbohydr. Res., 16 (1971) 253-259) в 50 мл пиридина добавляли 33,4 г (120 ммоль) тритилхлорида при комнатной температуре. Через 2 дня 14 мл бензоилхлорида добавляли по каплям к реакционной смеси ниже 20°C. Через 2 часа реакционную смесь вливали в ледяную воду, воду сливали с осажденного липкого вещества и остаток кристаллизовали с 400 мл этанола. Кристаллическое вещество отфильтровывали, промывали этанолом и сушили. Полученный таким образом продукт перекристаллизовывали из 2,5-кратного этилацетата для получения 36,1 г (79%) указанного в заголовке соединения. Т.пл. 165-167°C, [α]D +7° (c 1, хлороформ).
Стадия b)
2,5-ди-O-бензоил-3,4-O-изопропилиден-D-маннит (XXVI)
К раствору 30 г (33 ммоль) продукта формулы (XXV), полученного на предыдущей стадии, в 300 мл диоксана добавляли 50 мл 0,1 M серной кислоты и раствор перемешивали при 90°C в течение 6 часов. Охлажденный раствор нейтрализовали добавлением ионобменной смолы, отфильтровывали и концентрировали. Остаток очищали колоночной хроматографией (растворитель A) и полученный сырой продукт перекристаллизовывали из эфира-гексана для получения 3,3 г (23,6%) указанного в заголовке соединения. Т.пл. 97-99°C, Rf 0,45, [α]D -20° (c 1, хлороформ).
Стадия c)
1,6-бис-О-(2,3,4,6-тетра-О-ацетил-β-D-глюкопиранозил)-2,5-ди-О-бензоил-3,4-О-изопропилиден-D-маннит (XXVII)
К раствору 3 г (7 ммоль) продукта формулы (XXVI), полученного на предыдущей стадии, в 65 мл ацетонитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 6,5 г (16 ммоль) ацетобром-D-глюкозы (X) и 4,4 г (18 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 130 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель C) и полученный сырой продукт перекристаллизовывали из этанола для получения 2,75 г (36%) указанного в заголовке соединения. Т.пл. 193-195°C, Rf 0,4,
[α]D -28° (c 1, хлороформ).
Стадия d)
1,6-бис-O-(β-D-глюкопиранозил)-D-маннит (XXIX)
К перемешиваемому раствору 2,6 г (2,4 ммоль) продукта формулы (XXVII), полученного на предыдущей стадии, в 40 мл метанола добавляли 0,5 мл 2 M раствора метоксида натрия в метаноле при комнатной температуре. Через 1 час ионы натрия удаляли добавлением катионно-обменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток растворяли в воде и экстрагировали хлороформом с целью удаления метилбензоата. Водный раствор, содержащий соединение формулы (XXVIII) концентрировали до объема 15 мл и добавляли 1,5 мл 1 M серной кислоты. Раствор перемешивали при 60°C в течение 90 минут, затем охлаждали и нейтрализовали добавлением ионобменной смолы. Отфильтрованный раствор лиофилизировали для получения 1,15 г (95%) указанного в заголовке соединения в виде аморфного порошка. [α]D -23,4° (c 1, вода).
Исходное вещество формулы (XXXIII) может быть синтезировано, например, следующим способом:
Стадия a)
1,6-бис-O-(2,3,4,2',3',4',6'-гепта-O-ацетил-β-гентиобиопиранозил)-2,5-ди-O-бензоил-4,5-O-изопропилиден-D-маннит (XXXI)
К перемешиваемому раствору 1,72 г (4 ммоль) 2,5-дибензоил-3,4-O-изопропилиден-D-маннита (XXVI, описанного выше) в 60 мл ацетонитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 6 г (8,6 ммоль) ацетобромгентиобиозы (XXX) (K. Takiura, S. Honda, T. Endo, K. Kakehi. Chem. Pharm. Bull. 20 (1972) 438-442) и 4,4 г (18 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 120 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель D) и полученный сырой продукт перекристаллизовывали из этанола для получения 1,3 г (22%) указанного в заголовке соединения. Т.пл. >220°C, Rf 0,5, [α]D -16° (c 1, хлороформ).
Стадия b)
1,6-бис-O-(β-D-гентиобиопиранозил)-D-маннит (ХХХIII)
К раствору 1,3 г (0,78 ммоль) продукта формулы (XXXI), полученного на предыдущей стадии, в 25 мл метанола добавляли 0,25 мл 2 M раствора метоксида натрия в метаноле и реакционную смесь перемешивали при 45°C в течение 2 часов. После охлаждения ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток растворяли в воде и экстрагировали хлороформом с целью удаления метилбензоата. Водный раствор, содержащий соединение формулы (ХХХII), концентрировали до объема 15 мл и добавляли 1,5 мл M серной кислоты. Раствор перемешивали при 60°C в течение 90 минут, затем охлаждали и нейтрализовали добавлением ионобменной смолы. Отфильтрованный раствор лиофилизировали для получения 0,65 г (~100%) указанного в заголовке соединения в виде аморфного порошка. [α]D -3,5° (c 1, вода).
Исходное вещество формулы (XXXVII) может быть синтезировано, например, следующим способом:
Стадия a)
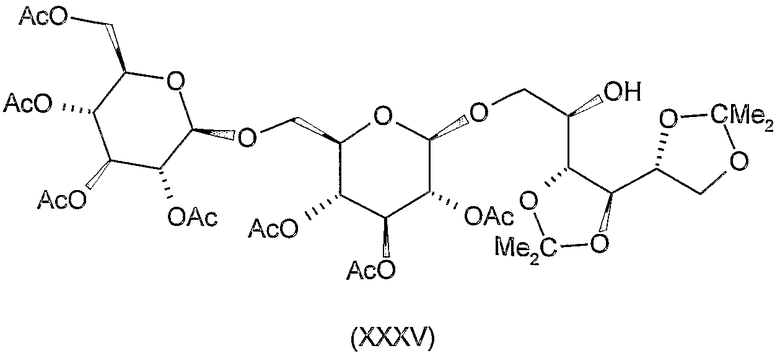
1-O-(2,3,4,2',3',4',6'-гепта-О-ацетил-β-гентиобиопиранозил)-3,4:5,6-ди-O-изопропилиден-D-маннит (XXXV)
К раствору 2,4 г (9,2 ммоль) 1,2:3,4-ди-O-изопропилиден-D-маннита (XXXIV) (L.F. Wiggins, J. Chem. Soc (1946) 13-14) в 60 мл ацетонитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 6,3 г (9 ммоль) ацетобромгентиобиозы (XXX) (K. Takiura, S. Honda, T. Endo, K. Kakehi. Chem. Pharm. Bull. 20 (1972) 438-442) и 2,5 г (10 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 120 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель C) для получения 4,1 г (52%) указанного в заголовке соединения. Rf 0,4, [α]D +2° (c 1, хлороформ).
Стадия b)
1-O-β-гентиобиопиранозил-3,4:5,6-ди-O-изопропилиден-D-маннит (XXXVI)
К перемешиваемому раствору 3,9 г (4,4 ммоль) продукта формулы (XXXV), полученного на предыдущей стадии, в 50 мл метанола добавляли 0,5 мл 2 M раствора метоксида натрия в метаноле при комнатной температуре. Через 2 часа ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (растворитель F) для получения 1,6 г (62%) указанного в заголовке соединения. Rf 0,4, [α]D -5,5° (c 1, вода).
Стадия c)
1-O-β-гентиобиопиранозил-D-маннит (XXXVII)
Раствор 1,4 г (2,4 ммоль) продукта формулы (XXXVI), полученного на предыдущей стадии, в 20 мл 0,01 M серной кислоты перемешивали при 60°C в течение 1,5 часа. Охлажденный раствор нейтрализовали добавлением ионобменной смолы, отфильтровывали и лиофилизировали для получения 1,15 г (95%) указанного в заголовке соединения. [α]D -17° (c 1, вода).
Исходное вещество формулы (XLIII) может быть синтезировано, например, следующим способом:
Стадия a)
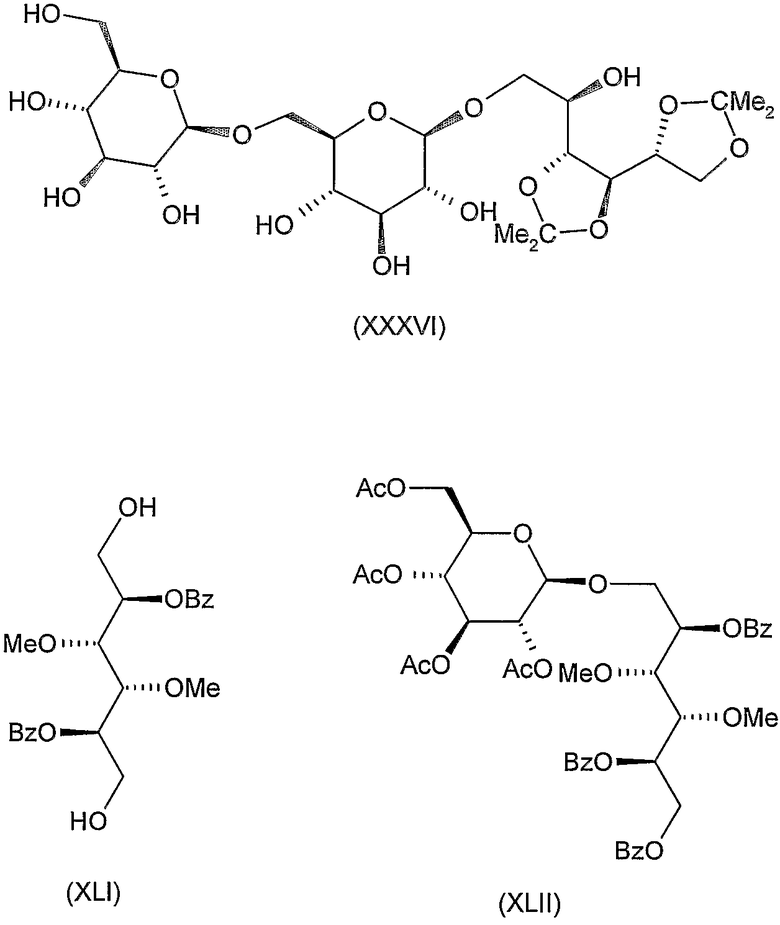
2,5-ди-O-бензоил-3,4-ди-O-метил-1,6-ди-O-тритил-D-маннит (XL)
К перемешиваемому раствору 6,3 г (20 ммоль) 3,4-ди-O-метил-D-маннита (XLI) (J. Kuszmann, Carbohydr. Res., 71 (1979) 123-134) в 60 мл пиридина добавляли 20,1 г (72 ммоль) тритилхлорида. Реакционную смесь выдерживали при комнатной температуре в течение 2 дней, затем добавляли 8,4 мл бензоилхлорида по каплям к перемешиваемому и охлаждаемому раствору. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем вливали в ледяную воду, экстрагировали дихлорметаном и органический слой обрабатывали обычным образом. Остаток, полученный после концентрирования, растворяли в 150 мл горячего этанола, охлаждали, осажденный продукт отфильтровывали и промывали этанолом. Полученный таким образом продукт растворяли в 40 мл этилацетата и добавляли 120 мл этанола. Осажденный продукт отфильтровывали и промывали этанолом для получения 20,25 г (75%) указанного в заголовке соединения. Т.пл. 128-130°C, [α]D +45° (c 1, хлороформ).
Стадия b)
2,5-ди-O-бензоил-3,4-ди-O-метил-D-маннит (XLI)
К перемешиваемому раствору 20 г продукта формулы (XL), полученного на предыдущей стадии, в 300 мл горячей уксусной кислоты добавляли 100 мл воды небольшими порциями и смесь перемешивали при 80-90°C в течение 30 минут. После охлаждения осажденный тритиловый спирт отфильтровывали и фильтрат экстрагировали хлороформом. Органический слой промывали водой, 5% водным раствором бикарбоната натрия, водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель E) для получения 7,0 г (75,5%) указанного в заголовке соединения в виде сиропа. Rf 0,2, [α]D +33° (c 1, хлороформ).
Стадия c)
1-O-(2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозил)-2,5,6-три-O-бензоил-3,4-ди-O-метил-D-маннит (XLII)
К перемешиваемому раствору 6,3 г (15 ммоль) 2,5-ди-O-бензоил-3,4-ди-O-метил-D-маннита (XLI), полученного на предыдущей стадии, в 65 мл ацетoнитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 6,2 г (15 ммоль) ацетoбром-D-глюкозы (X) и 4,2 г (16 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 130 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток растворяли в 50 мл пиридина и 4 мл бензоилхлорида добавляли по каплям к перемешиваемому раствору при комнатной температуре. Через 2 часа реакционную смесь вливали в ледяную воду, экстрагировали дихлорметаном и обрабатывали обычным образом. Осадок, полученный при концентрировании, очищали колоночной хроматографией (растворитель А) для получения 3,5 г (27%) указанного в заголовке соединения в виде сиропа. Rf 0,6, [α]D 0° (c 1, хлороформ).
Стадия d)
1-O-(β-D-глюкопиранозил)-3,4-ди-O-метил-D-маннит (XLIII)
К перемешиваемому раствору 3,3 г (2,87 ммоль) продукта формулы (XLII), полученного на предыдущей стадии, в 40 мл метанола добавляли 0,5 мл 2 M раствора метоксида натрия и реакционную смесь нагревали в колбе с обратным холодильником в течение 2 часов. После охлаждения ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток растворяли в воде и экстрагировали хлороформом с целью удаления метилбензоата. Водный раствор концентрировали до объема 20 мл и лиофилизировали для получения 1,4 г (97%) указанного в заголовке соединения в виде аморфного порошка. [α]D +10° (c 1, вода).
Исходное вещество формулы (XLV) может быть синтезировано, например, следующим способом:
Стадия a)
1,6-бис-O-(2,3,4,6-тетра-O-ацетил-β-D-глюкопиранозил)-2,5-ди-O-бензоил-3,4-ди-O-метил-D-маннит (XLIV)
К перемешиваемому раствору 3,34 г (8 ммоль) 2,5-ди-O-бензоил-3,4-ди-O-метил-D-маннита (XLI, описанного выше) в 65 мл ацетонитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 8,2 г (20 ммоль) ацетoбром-D-глюкозы (X) и 5,5 г (22 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 130 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель C) и полученный сырой продукт перекристаллизовывали из этанола для получения 3,4 г (39%) указанного в заголовке соединения. Т.пл. 138-140°C, Rf 0,35, [α]D +38° (c 1, хлороформ).
Стадия b)
1,6-бис-O-β-D-глюкопиранозил-3,4-ди-O-метил-D-маннит (XLV)
К перемешиваемому раствору 3,1 г (2,87 ммоль) продукта формулы (XLIV), полученного на предыдущей стадии, в 40 мл метанола добавляли 0,5 мл 2 M раствора метоксида натрия в метаноле при комнатной температуре. Через 2 часа ионы натрия удаляли добавлением катионообменой смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток растворяли в воде и экстрагировали хлороформом с целью удаления метилбензоата. Водный раствор концентрировали до объема 15 мл и лиофилизировали для получения 1,56 г (~100%) указанного в заголовке соединения в виде аморфного порошка. [α]D -4° (c 1, вода).
Исходное вещество формулы (XLVII) можеут быть синтезировано, например, следующим способом:
Стадия a)
1-O-(2,3,4,2',3',4',6'-гепта-O-ацетил-β-гентиобиопиранозил)-2,5,6-три-O-бензоил-3,4-ди-O-метил-D-маннит (XLVI)
К перемешиваемому раствору 5,0 г (12 ммоль) 2,5-ди-O-бензоил-3,4-ди-O-метил-D-маннита (XLI, описанного выше) в 70 мл ацетонитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 8,4 г (12 ммоль) ацетoбромгентиобиозы (XXX) (K. Takiura, S. Honda, T. Endo, K. Kakehi. Chem. Pharm. Bull. 20 (1972) 438-442) и 3,3 г (12 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 140 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток растворяли в 50 мл пиридина и добавляли 4 мл бензоилхлорида по каплям к перемешиваемому раствору при комнатной температуре. Через 2 часа реакционную смесь вливали в ледяную воду, экстрагировали дихлорметаном и обрабатывали обычным образом. Остаток, полученный при концентрировании, очищали колоночной хроматографией (растворитель A) для получения 5,2 г (38%) сырого продукта, который перекристаллизовывали из 10-кратного метанола для получения указанного в заголовке соединения. Т.пл. 166-168°C, Rf 0,6, [α]D +8° (c 1, хлороформ).
Стадия b)
1-O-β-гентиобиопиранозил-3,4-ди-O-метил-D-маннит (XLVII)
К перемешиваемому раствору 2,3 г (2,03 ммоль) продукта формулы (XLVI), полученного на предыдущей стадии, в 40 мл метанола добавляли 0,5 мл 2 M раствора метоксида натрия и реакционную смесь нагревали в колбе с обратным холодильником в течение 2 часов. После охлаждения ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток растворяли в воде и экстрагировали хлороформом с целью удаления метилбензоата. Водный раствор концентрировали до объема 20 мл и лиофилизировали для получения 1,05 г (97%) указанного в заголовке соединения в виде аморфного порошка. [α]D -11° (c 1, вода).
Исходное вещество формулы (IL) может быть синтезировано, например, следующим способом:
Стадия a)
1,6-бис-O-(2,3,6,2',3',4',6'-гепта-O-ацетил-β-лактозил)-2,5-ди-O-бензоил-3,4-ди-O-метил-D-маннит (XLVIII)
К перемешиваемому раствору 3,15 г (7,5 ммоль) 2,5-ди-O-бензоил-3,4-ди-O-метил-D-маннита (XLI, описанного выше) в 100 мл ацетoнитрила добавляли 14 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 12 г (17,25 ммоль) ацетoбромлактозы (C.S. Hudson, J.M. Johnson, J. Am. Chem. Soc. 37 (1915) 1270-1275) и 5 г (20 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 200 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель C) для получения 4,2 г (34%) указанного в заголовке соединения. Rf 0,2, [α]D -1,5° (c 1, хлороформ).
Стадия b)
1,6-бис-O-β-лактозил-3,4-ди-O-метил-D-маннит (IL)
К перемешиваемому раствору 4,2 г (2,4 ммоль) продукта формулы (XLVIII), полученного на предыдущей стадии, в 50 мл метанола добавляли 1,0 мл 2 M раствора метоксида натрия в метаноле и реакционную смесь нагревали в колбе с обратным холодильником в течение 2 часов. После охлаждения ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток растворяли в воде и экстрагировали хлороформом с целью удаления метилбензоата. Водный раствор концентрировали до объема 15 мл и лиофилизировали для получения 1,9 г (92%) указанного в заголовке соединения в виде аморфного порошка, [α]D +28° (c 1, вода).
Исходное вещество формулы (LIII) может быть синтезировано, например, следующим способом:
Стадия a)
1-O-(2,3,4-три-O-ацетил-α-D-арабинопиранозил)-3,4:5,6-ди-O-изопропилиден-D-маннит (LI)
К перемешиваемому раствору 3,9 г (15 ммоль) 1,2:3,4-ди-O-изопропилиден-D-маннита (XXXIV) (L.F. Wiggins, J. Chem. Soc (1946) 13-14) в 60 мл ацетонитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 5 г ацетобром-D-арабинозы (L) (M. Barczai-Martos и F. Korosy, Nature. 165 (1950) 369) и 4 г (16 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 120 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель C) для получения 3,1 г (40%) указанного в заголовке соединения. Rf 0,5, [α]D -2° (c 1, хлороформ).
Стадия b)
1-O-α-D-арабинопиранозил-3,4:5,6-ди-O-изопропилиден-D-маннит (LII)
К перемешиваемому раствору 2,9 г (5,6 ммоль) продукта формулы (LI), полученного на предыдущей стадии, в 30 мл метанола добавляли 0,3 мл 2 M раствора метоксида натрия в метаноле при комнатной температуре. Через 2 часа ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (растворитель G) для получения 1,3 г (59%) указанного в заголовке соединения. Rf 0,5, [α]D +8° (c 1, вода).
Стадия c)
1-O-α-D-арабинопиранозил-D-маннит (LIII)
Раствор 1,15 г (2,92 ммоль) продукта формулы (LII), полученного на предыдущей стадии, в 20 мл 0,05 M серной кислоты перемешивали при 60°C в течение 1,5 часа. Охлажденный раствор нейтрализовали добавлением ионообменной смолы, отфильтровывали и лиофилизировали для получения 0,9 г (98%) указанного в заголовке соединения. [α]D -8°(c 1, вода).
Исходное вещество формулы (LVII) может быть синтезировано, например, следующим способом:
Стадия a)
1-О-(2,3,4-три-О-ацетил-β-D-ксилопиранозил)-3,4:5,6-ди-О-изопропилиден-D-маннит (LV)
К перемешиваемому раствору 4,2 г (16 ммоль) 1,2:3,4-ди-O-изопропилиден-D-маннита (XXXIV) (L.F. Wiggins, J. Chem. Soc (1946) 13-14) в 65 мл ацетoнитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли 6,5 г (19 ммоль) ацетoбром-D-ксилозы (LIV) (M. Barczai-Martos и F. Korosy, Nature. 165 (1950) 369) и 5,5 г (22 ммоль) Hg(CN)2 и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 130 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток очищали колоночной хроматографией (растворитель C) для получения 4,8 г (56%) указанного в заголовке соединения. Rf 0,6, [α]D -22° (c 1, хлороформ).
Стадия b)
1-O-β-D-ксилопиранозил-3,4:5,6-ди-O-изопропилиден-D-маннит (LVI)
К перемешиваемому раствору 4,6 г (8,84 ммоль) продукта формулы (LV), полученного на предыдущей стадии, в 50 мл метанола добавляли 0,3 мл 2 M раствора метоксида натрия при комнатной температуре. Через 2 часа ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток очищали колоночной хроматографией (растворитель F) для получения 2,2 г (63%) указанного в заголовке соединения. Rf 0,6, [α]D +1,5° (c 1, вода).
Стадия c)
1-O-β-D-ксилопиранозил-D-маннит (LVII)
Раствор 2,0 г (5,08 ммоль) продукта формулы (LVI), полученного на предыдущей стадии, в 20 мл 0,05 M серной кислоты перемешивали при 60°C в течение 1,5 часа. Охлажденный раствор нейтрализовали добавлением ионообменной смолы, отфильтровывали и лиофилизировали для получения 1,35 г (85%) указанного в заголовке соединения. [α]D -21,5° (c 1, вода).
Исходное вещество формулы (LXI) может быть синтезировано, например, следующим способом:
Стадия a)
1,6-бис-О-(2,3,4,6-тетра-О-ацетил-β-D-глюкопиранозил)-2,3:4,5-ди-О-изопропилиденгалактитол (LIX)
К перемешиваемому раствору 2,62 г (10 ммоль) 2,3:4,5-ди-О-изопропилиденгалактитола (LVIII) (R.M. Horm, W.D. Maclay, C.S. Hudson J. Am. Chem. Soc 61 (1939) 2438) в 60 мл ацетонитрила добавляли 7 г молекулярного сита (4 Е) и смесь перемешивали при комнатной температуре в течение 30 минут. Затем 8,5 г (21 ммоль) ацетобром-D-глюкозы (X) и 5,0 г (20 ммоль) Hg(CN)2 добавляли и смесь перемешивали при комнатной температуре в течение 20 часов. Затем реакционную смесь отфильтровывали и фильтрат разводили 120 мл хлороформа, промывали 5% водным раствором бикарбоната натрия, 10% водным раствором бромида калия и водой, сушили и концентрировали. Остаток перекристаллизовывали из 5-кратного этанола для получения 4,9 г (53%) указанного в заголовке соединения. Т.пл. 164-166°C, [α]D -25° (c 1, хлороформ).
Стадия b)
1,6-бис-О-(β-D-глюкопиранозил)галактитол (LXI)
К перемешиваемому раствору 4,65 г (5,04 ммоль) продукта формулы (LIX), полученного на предыдущей стадии, в 50 мл метанола добавляли 0,5 мл 2 M раствора метоксида натрия в метаноле при комнатной температуре. Через 2 часа ионы натрия удаляли добавлением катионообменной смолы, смесь отфильтровывали и фильтрат концентрировали. Остаток (LX) растворяли в 40 мл 0,05 M серной кислоты и раствор перемешивали при 60°C в течение 1,5 часа. Охлажденный раствор нейтрализовали добавлением ионообменной смолы, отфильтровывали и лиофилизировали для получения 2,5 г (98%) указанного в заголовке соединения. [α]D -29° (c 1, вода).
ПРИМЕРЫ
Пример 1
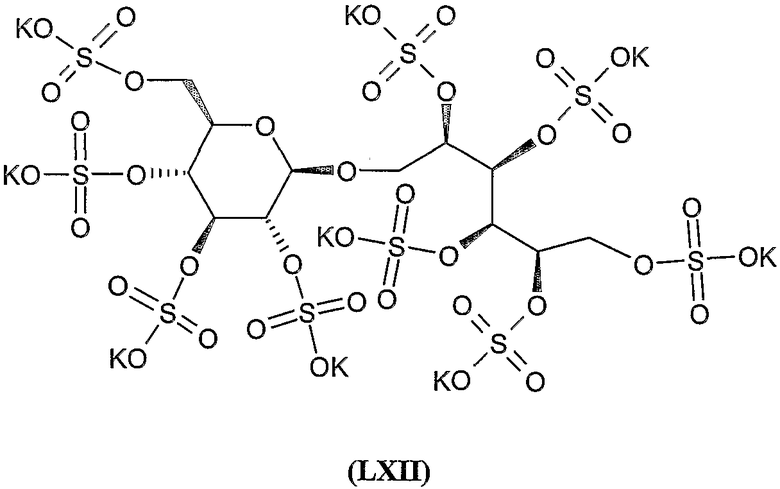
Нона-калиевая соль 2,3,4,5,6-пента-О-сульфато-1-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-маннита (LXII) (IA, R 1 - тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил, R 2 = R 3 = R 4 = R 5 = R 6 = SO 3 K)
5,1 г (48%, 30 ммоль) комплекса триоксида серы - диметилформамида суспендировали в 5 мл сухого диметилформамида при перемешивании, смесь охлаждали до -20°C и 0,69 г (2 ммоль) 1-O-β-D-глюкопиранозил-D-маннита (VIII, Lindberg, Acta Chim, Scand. 7 (1953) 1218) в 5 мл диметилформамида постепенно добавляли с такой скоростью, чтобы поддерживать температуру ниже
-15°C. Через 15 минут температуру смеси повышали до -5°C и поддерживали такой в течение 45 минут. Впоследствии реакционную смесь снова охлаждали до -20°C и 1 мл этанола постепенно добавляли с такой скоростью, чтобы поддерживать температуру ниже -15°C. Затем реакционную смесь вливали в перемешиваемый и охлажденный (-5°C) раствор 5 г ацетата калия и 40 мл метанола. Осадок отфильтровывали и промывали 3×40 мл метанола. Твердый остаток растворяли в 40 мл воды и pH раствора доводили до 8 с помощью M раствора гидроксида калия, затем концентрировали до объема 20 мл и охлаждали до +4°C. Кристаллы отфильтровывали и промывали холодной водой для получения 2,3 г (78%) указанного в заголовке соединения. Т.пл. >220°C; [α]D +10° (c 1, вода). C12H15O38S9K9 Рассчитано: С 10,22; H 1,06; S 20,45; К 24,30. Обнаружено: С 9,95; H 1,27; S 20,27; К 23,92.
Пример 2
Нона-калиевая соль 1,2,3,4,5-пента-О-сульфато-6-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-глюцитола (LXIII) (IB, R 6 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 1 = R 2 = R 3 = R 4 = R 5 = SO 3 K)

Указанное в заголовке соединение (LXIII) получали в соответствии со способом, описанным в примере 1, с использованием 6-O-β-D-глюкопиранозил-D-глюцитола (IX, M.L. Wolfrom and T.G. Gardner, J. Am Chem. Soc 65 (1943) 750-752) в качестве исходного вещества. Т.пл. >220°C, выход 76%, [α]D -6° (c 1, вода). C12H15O38S9K9 Рассчитано: С 10,22; H 1,06; S 20,45; К 24,30. Обнаружено: С 10,17; H 1,27; S 20,37; К 24,70.
Пример 3
Нона-калиевая соль 2,3,4,5,6-пента-O-сульфато-1-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола (LXIV) (IB, R 1 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 2 = R 3 = R 4 = R 5 = R 6 = SO 3 K)
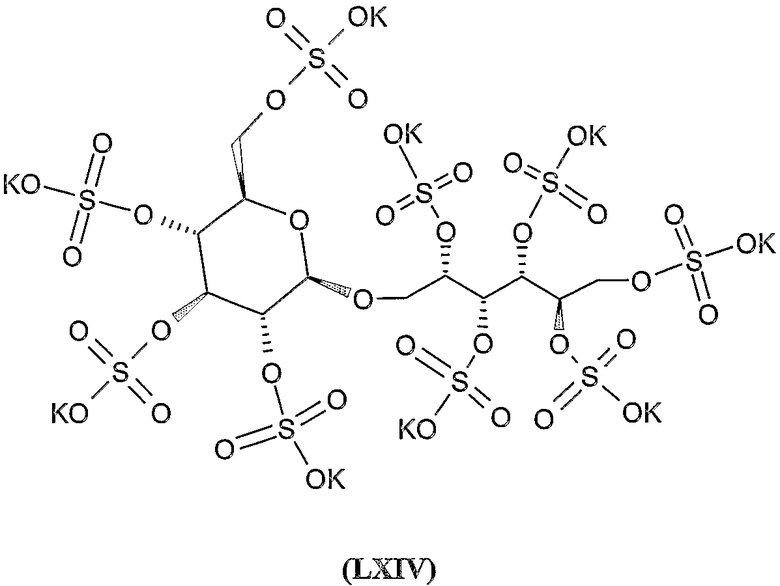
Указанное в заголовке соединение (LXIV) получали в соответствии со способом, описанным в примере 1, с использованием 1-O-β-D-глюкопиранозил-D-глюцитола (XIV) в качестве исходного вещества. Т.пл. >220°C, выход 90%, [α]D -3,5° (c 1, вода). C12H15O38S9K9 Рассчитано: С 10,22; H 1,06; S 20,45; К 24,30. Обнаружено: С 10,09; H 1,35; S 20,20; К 29,70.
Пример 4
Нона-калиевая соль 1,2,4,5,6-пента-O-сульфато-3-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола (LXV) (IB, R 3 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 1 = R 2 = R 4 = R 5 = R 6 = SO 3 K)
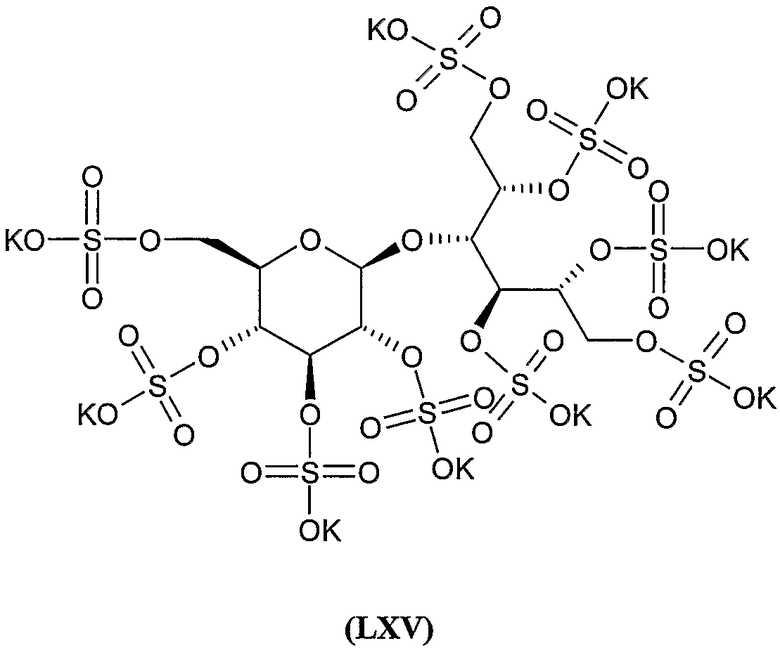
Указанное в заголовке соединение (LXV) получали в соответствии со способом, описанным в примере 1, с использованием 3-O-β-D-глюкопиранозил-D-глюцитола (XVIII) в качестве исходного вещества. Т.пл. >220°C, выход 87%, [α]D +5° (c 1, вода). C12H15О39S9K9 Рассчитано: С 10,22; H 1,06; S 20,45; К 24,30. Обнаружено: С 10,10; H 1,45; S 20,31; К 24,67.
Пример 5
Нона-калиевая соль 1,2,3,5,6-пента-O-сульфато-4-O-(2,3,4,6-тетра-O-сульфато-α-D-глюкопиранозил)-D-глюцитола (LXVI) (IB, R 4 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-α-D-глюкопиранозила, R 1 = R 2 = R 3 = R 5 = R 6 = SO 3 K)
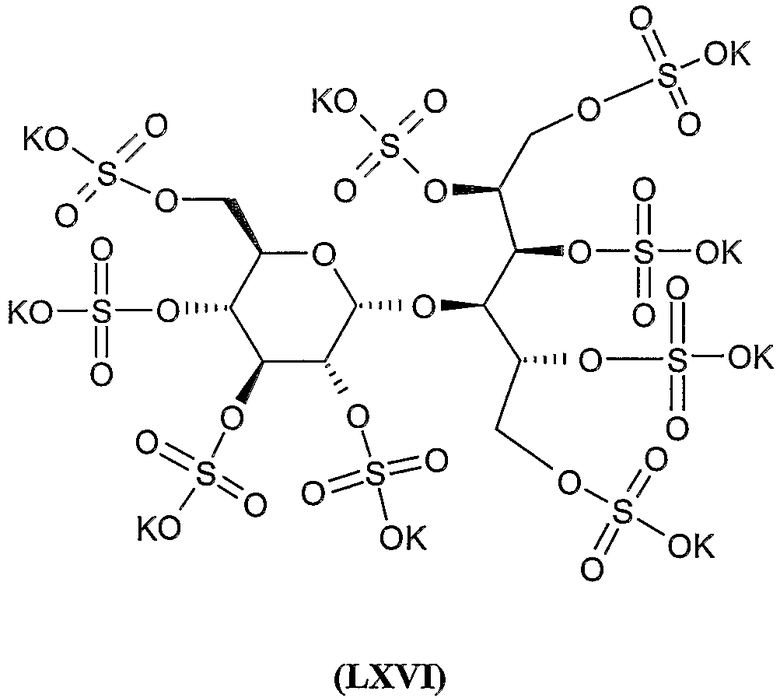
Указанное в заголовке соединение (LXVI) получали в соответствии со способом, описанным в примере 1, с использованием 4-O-α-D-глюкопиранозил-D-глюцитола (ХIХ, M.L. Wolfrom et al., J. Am. Chem. Soc. 62 (1940) 2553; E. Dryselius et al. Acta Chem. Scand. 11 (1957) 663-667) в качестве исходного вещества. Т.пл. >220°C, выход 73%,
[α]D +40° (c 1, вода). С12H15O39S9K9 Рассчитано: С 10,22; H 1,06; S 20,45; К 24,30. Обнаружено: С 9,84; H 1,40; S 19,98; К 23,99.
Пример 6
Нона-калиевая соль 1,2,3,5,6-пента-О-сульфато-4-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-глюцитола (LXVII) (IB, R 4 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-α-D-глюкопиранозила, R 1 = R 2 = R 3 = R 5 = R 6 = SO 3 K)
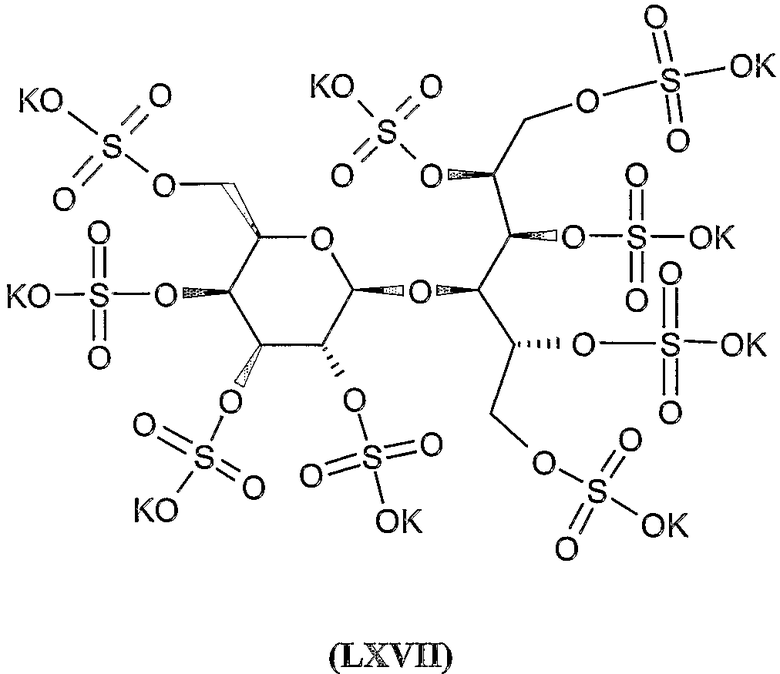
Указанное в заголовке соединение (LXVII) получали в соответствии со способом, описанным в примере 1, с использованием 4-O-β-D-глюкопиранозил-D-глюцитола (XX, M.L. Wolfram et al., J. Am. Chem. Soc. 74 (1952) 1105) в качестве исходного вещества. Т.пл. >220°C, выход 41%, [α]D +5° (c 1, вода). C12H15О39S9K9 Рассчитано: С 10,22; H 1,06; S 20,45; К 24,30. Обнаружено: С 9,89; H 1,42; S 19,99; К 23,87.
Пример 7
Нона-калиевая соль 1,2,3,5,6-пента-О-сульфато-4-О-(2,3,4,6-тетра-O-сульфато-α-D-галактопиранозил)-D-глюцитола (LXVIII) (IB, R 4 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-α-D-галактопиранозила, R 1 = R 2 = R 3 = R 5 = R 6 = SO 3 K)
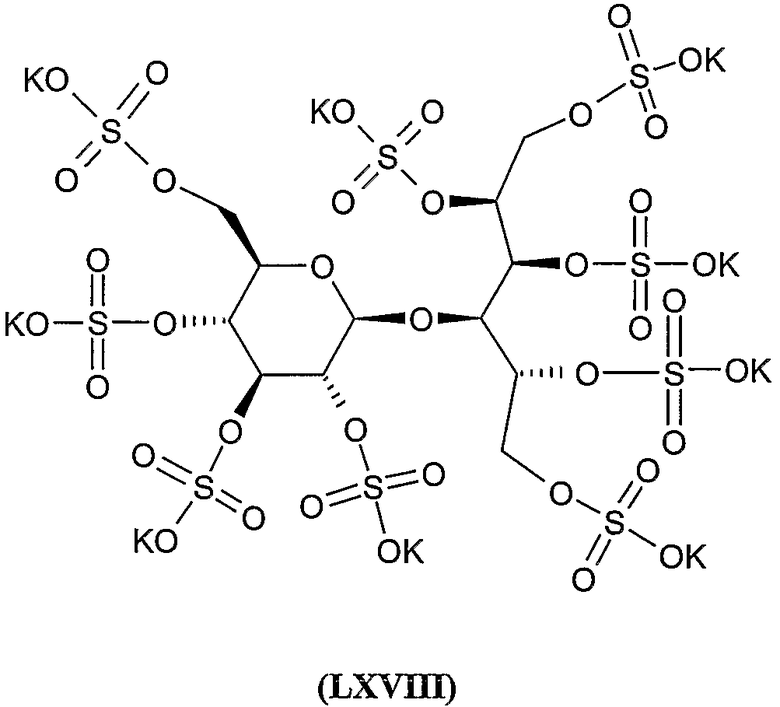
Указанное в заголовке соединение (LXVIII) получали в соответствии со способом, описанным в примере 1, с использованием 4-O-β-D-галактопиранозил-D-глюцитола (XXI, W.J. Whelan и K. Morgan, Chem. и Ind. (1955) 1449-1450) в качестве исходного вещества. Т.пл. >220°C, выход 40%, [α]D 0° (c 1, вода). C12H15O39S9K9 Рассчитано: С 10,22; H 1,06; S 20,45; К 24,30. Обнаружено: С 9,99; H 1,29; S 19,88; K 23,87.
Пример 8
Додека-калиевая соль 2,4,5,6-тетра-O-сульфато-1,3-бис-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола (LXIX) (IB, R 1 = R 3 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 2 = R 4 = R 5 = R 6 = SO 3 K)

Указанное в заголовке соединение (LXIX) получали в соответствии со способом, описанным в примере 1, с использованием 1,3-бис-O-β-D-глюкопиранозил-D-глюцитола (XXIV) в качестве исходного вещества. Т.пл. >220°C, выход 82%, [α]D -6,5° (c 1, вода). C18H22O52S12K12 Рассчитано: С 11,24; H 1,15; S 19,99; К 24,38. Обнаружено: С 11,01; H 1,32; S 19,07; К 23,99.
Пример 9
Додека-калиевая соль 2,3,4,5-тетра-O-сульфато-1,6-бис-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-маннита (LXX) (IA, R 1 = R 6 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 2 = R 3 = R 4 = R 5 = SO 3 K)
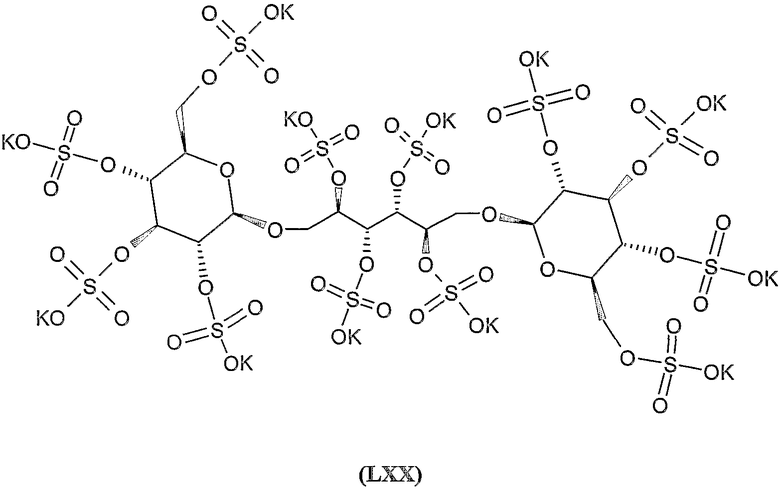
Указанное в заголовке соединение (LXX) получали в соответствии со способом, описанным в примере 1, с использованием 1,6-бис-O-β-D-глюкопиранозил-D-маннита (XXIX) в качестве исходного вещества. Т.пл. >220°C, выход 83%, [α]D +1,5° (c 1, вода). C18H22O52S12K12 Рассчитано: С 11,24; H 1,14; S 19,98; К 24,35. Обнаружено: С 10,93; H 1,55; S 19,26; К 23,99.
Пример 10
Октадека-калиевая соль 2,3,4,5-тетра-O-сульфато-1,6-бис-O-(2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозил)-D-маннита (LXXI) (IA, R 1 = R 6 = гептакалиевая соль 2,3,4,2',3',4',6'-гепта-O-β-гентиобиопиранозила, R 2 = R 3 = R 4 = R 5 = SO 3 K)
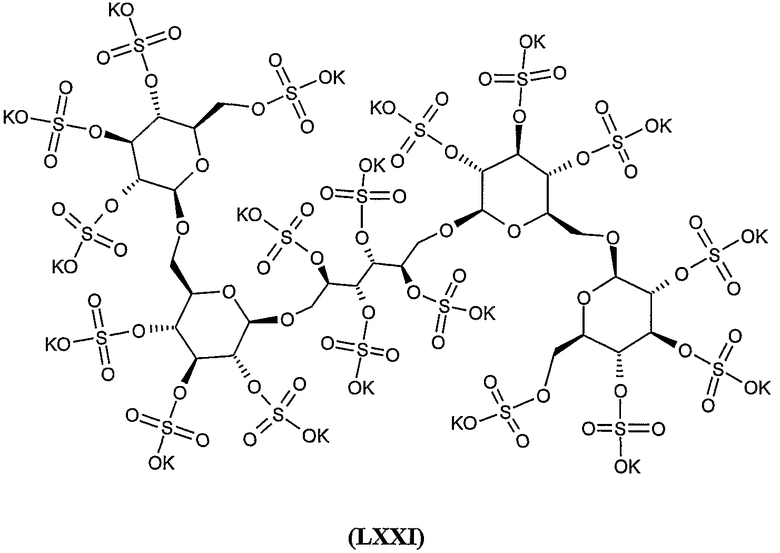
Указанное в заголовке соединение (LXXI) получали в соответствии со способом, описанным в примере 1, с использованием 1,6-бис-O-β-гентиобиопиранозил-D-маннита (ХХХIII) в качестве исходного вещества, с отличием, что добавляли 20 мл этанола к очень тиксотропному водному раствору, концентрированному до 20 мл, затем смесь охлаждали до +4°C. Осажденный продукт отфильтровывали и промывали этанолом. Т.пл. >220°C, выход 99%, [α]D +0° (c 1, вода). C30Н36O80S18K18 Рассчитано: С 12,18; H 1,33; S 19,51; К 23,80. Обнаружено: С 11,88; H 1,65; S 19,92; К 24,16.
Пример 11
Додека-калиевая соль 2,3,4,5,6-пента-O-сульфато-1-O-(2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозил)-D-маннита (LXXII) (IA, R 1 = гептакалиевая соль 2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозила, R 2 = R 3 = R 4 = R 5 = R 6 = SO 3 K)
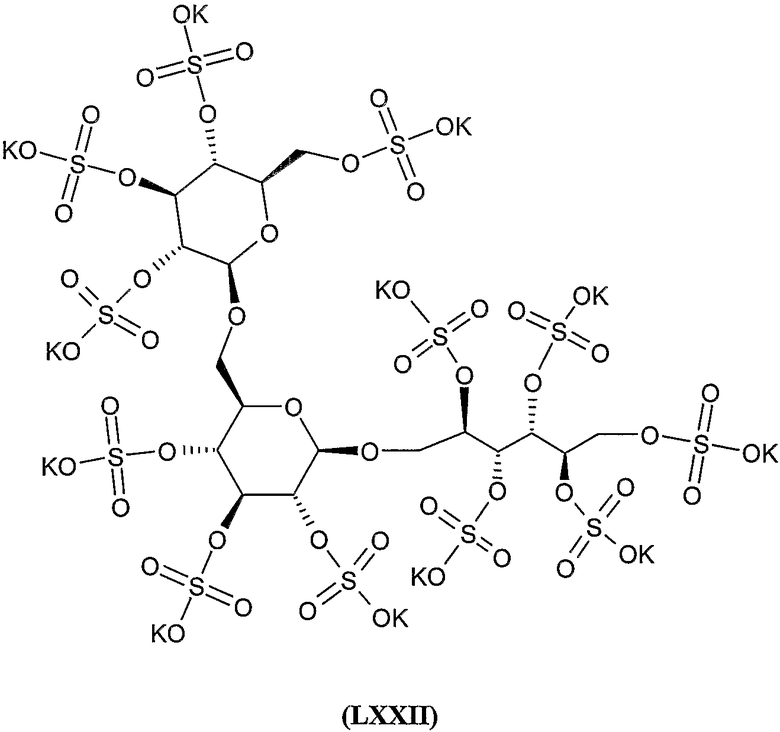
Указанное в заголовке соединение (LXXII) получали в соответствии со способом, описанным в примере 1, с использованием 1-O-β-гентиобиопиранозил-D-маннита (XXXVII) в качестве исходного вещества. Т.пл. >220°C, выход 79%, [α]D +6° (c 1, вода). C18Н22O52S12K12 Рассчитано: С 11,24; H 1,14; S 19,98; К 24,38. Обнаружено: С 10,98; H 1,51; S 19,37; К 24,02.
Пример 12
Гептакалиевая соль 3,4-ди-O-метил-2,5,6-три-O-сульфато-1-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-маннита (LXXIII) (IA, R 1 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 3 = R 4 = Me, R 2 = R 5 = R 6 = SO 3 K)
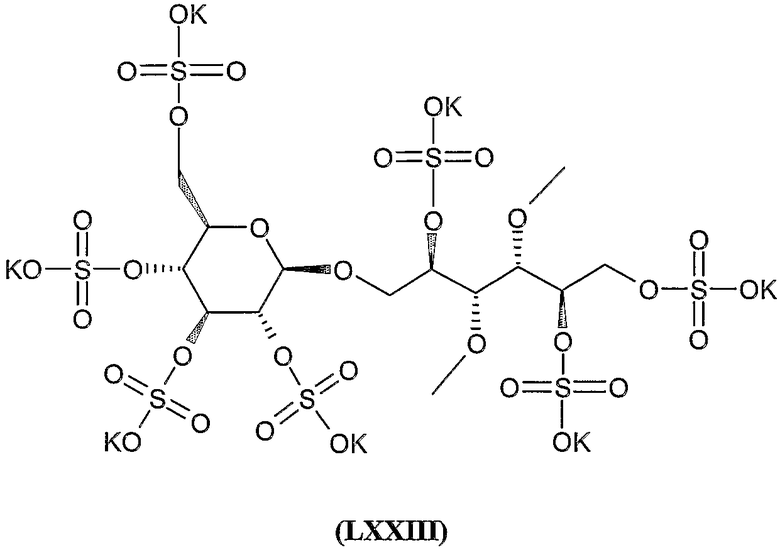
Указанное в заголовке соединение (LXXIII) получали в соответствии со способом, описанным в примере 1, с использованием 1-O-β-D-глюкопиранозил-D-маннита (XLVI) в качестве сходного вещества, с отличием, что сырой продукт, который осаждали метанолом и отфильтровывали, растворяли в воде и pH полученного таким образом раствора доводили до 8 с помощью 1 н. раствора гидроксида калия. После этого 3 мл 1 н. водного раствора ацетата стронция добавляли к раствору, до окончания образования осадка (SrSO4). Осадок отфильтровывали и фильтрат вносили на колонку, нагруженную смолой CHELX 100 (калиевая форма) (10 мл), с целью удаления ионов стронция. Колонку элюировали дистиллированной водой и элюат концентрировали. Остаток обрабатывали этанолом, отфильтровывали и промывали этанолом. Т.пл. >220°C, выход 94%, [α]D 0° (c 1, вода). C14H21O32S7K7 Рассчитано: С 14,02; H 1,76; S 18,71; К 22,82. Обнаружено: С 14,06; H 2,02; S 18,31; К 22,67.
Пример 13
Дека-калиевая соль 3,4-ди-О-метил-2,5-ди-О-сульфато-1,6-бис-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)-D-маннита (LXXIV) (IA, R 1 = R 6 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 3 = R 4 = Me, R 2 = R 5 = SO 3 K)
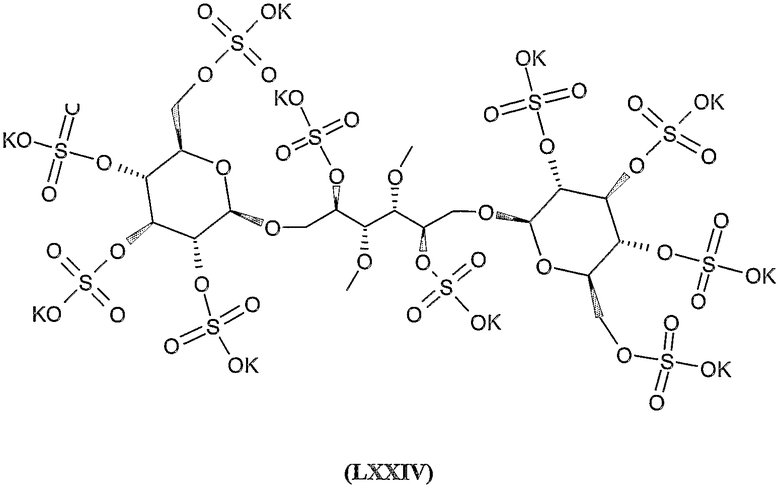
Указанное в заголовке соединение (LXXIV) полученное в соответствии со способом, описанным в примере 1, с использованием 1,6-бис-О-β-D-глюкопиранозил-3,4-ди-О-метил-D-маннита (XLVIII) в качестве исходного вещества. Т.пл. >220°C, выход 85%,
[α]D -6,5° (c 1, вода). C20H28O46S10K10 Рассчитано: С 14,00; H 1,63; S 18,68; К 22,78. Обнаружено: С 13,85; H 1,99; S 18,06; К 21,97.
Пример 14
Дека-калиевая соль 3,4-ди-О-метил-2,5,6-три-О-сульфато-1-О-(2,3,4,2',3',4',6'-гепта-О-сульфато-β-гентиобиопиранозил)-D-маннита (LXXV) (IA, R 1 = гептакалиевая соль 2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозила, R 3 = R 4 = Me, R 2 = R 5 = R 6 = SO 3 K)

Указанное в заголовке соединение (LXXV) получали в соответствии со способом, описанным в примере 1, с использованием 1-O-β-гентиобиопиранозил-3,4-ди-O-метил-D-маннита (XLVII) в качестве исходного вещества. Т.пл. >220°C, выход 99%,
[α]D -5° (c 1, вода). C20H28O46S10K10 Рассчитано: С 14,00; H 1,64; S 18,68; К 22,78. Обнаружено: С 13,87; H 1,99; S 19,24; К 22,43.
Пример 15
Гексадека-калиевая соль 3,4-ди-O-метил-2,5-ди-O-сульфато-1,6-бис-O-(2,3,6,2',3',4',6'-гепта-O-сульфато-β-лактозил-D-маннита (LXXVI) (IA, R 1 = R 6 = гептакалиевая соль 2,3,6,2',3',4',6'-гепта-O-сульфато-β-лактозила, R 3 = R 4 = Me, R 2 = R 5 = SO 3 K)
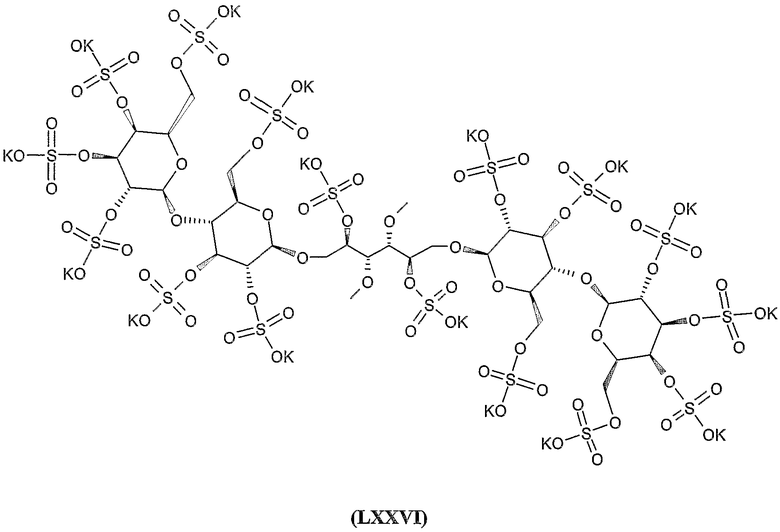
Указанное в заголовке соединение (LXXVI) получали в соответствии со способом, описанным в примере 1 с использованием 1,6-бис-лактопиранозил-3,4-диметил-D-маннита (LII) в качестве исходного вещества. Т.пл. >220°C, выход 44%, [α]D +2° (c 1, вода). С32Н42О74S16K16 Рассчитано: С 13,98; H 1,53; S 18,65; К 22,75. Обнаружено: С 13,51; H 1,73; S 17,98; К 21,15.
Пример 16
Октакалиевая соль 2,3,4,5,6-пента-О-сульфато-1-О-(2,3,4-три-О-сульфато-α-D-арабинопиранозил)-D-маннита (LXXVII) (IA, R1 = трикалиевая соль 2, 3, 4-три-O-сульфато-α-D-арабинопиранозила, R2=R3=R4=R5=R6=SO3K)
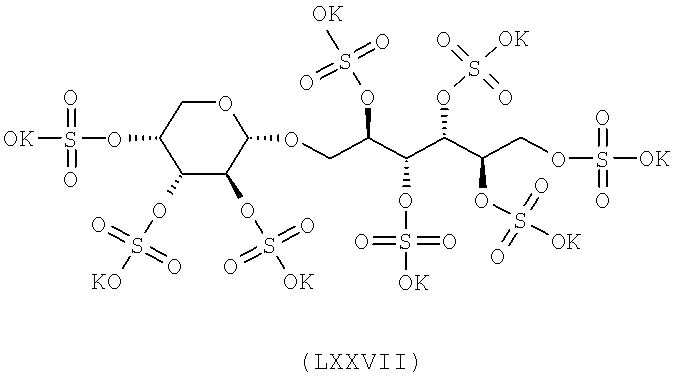
Указанное в заголовке соединение (LXXVII) получали в соответствии со способом, описанным в примере 1 с использованием 1-O-α-D-арабинопиранозил-D-маннита (LV) в качестве исходного вещества. Т.пл. >220°С, выход 99%, [α]D +19° (с 1, вода). С11Н14O34S8К8 Рассчитано: С 10,49; Н 1,12; S 20,36; К 24,83. Обнаружено: С 10,06; Н 1,40; S 19,56; К 24,04.
Пример 17
Октакалиевая соль 2,3,4,5,6-пента-О-сульфато-1-О-(2,3,4-три-О-сульфато-β-D-ксилопиранозил)-D-маннита (LXXVIII) (IA, R 1 = трикалиевая соль 2,3,4-три-O-сульфато-β-D-ксилопиранозила, R 2 = R 3 = R 4 = R 5 = R 6 = SO 3 K)
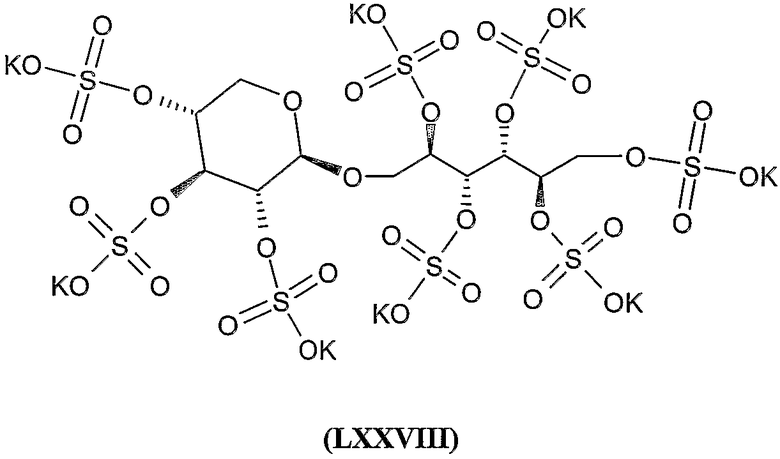
Указанное в заголовке соединение (LXXVIII) получали в соответствии со способом, описанным в примере 1, с использованием 1-O-β-D-ксилопиранозил-D-маннита (LVII) в качестве исходного вещества. Т.пл. >220°C, выход 85%, [α]D -6° (c 1, вода). C11H14O32S8K8 Рассчитано: С 10,49; H 1,12; S 20,36; К 24,83. Обнаружено: С 10,05; H 1,29; S 19,98; К 14,52.
Пример 18
Додека-калиевая соль 2,3,4,5-тетра-О-сульфато-1,6-бис-О-(2,3,4,6-тетра-О-сульфато-β-D-глюкопиранозил)галактитола (LXXIX) (IС, R 1 = R 6 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 2 = R 3 = R 4 = R 5 = SO 3 K)
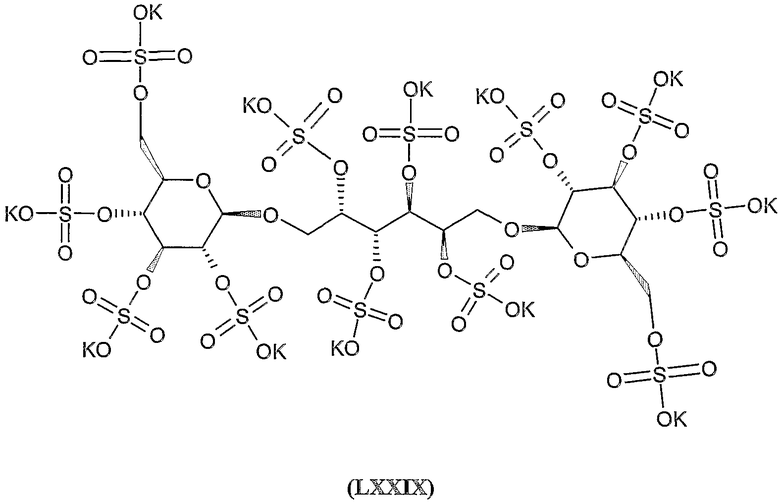
Указанное в заголовке соединение (LXXIX) получали в соответствии со способом, описанным в примере 1, с использованием 1,6-бис-глюкопиранозилгалактитола (LXII) в качестве исходного вещества. Т.пл. >220°C, выход 83%, [α]D -10° (c 1, вода). C18Н22О52S12K12 Рассчитано: С 11,24; H 1,15; S 19,99; К 24,38. Обнаружено: С 10,98; H 1,35; S 19,28; К 24,07.
Пример 19
Нона-натриевая соль 1,2,4,5,6-пента-O-сульфато-3-О-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола (LXXX) (IB, R 3 = тетракалиевая соль 2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозила, R 1 = R 2 = R 4 = R 5 = R 6 = SO 3 Na)
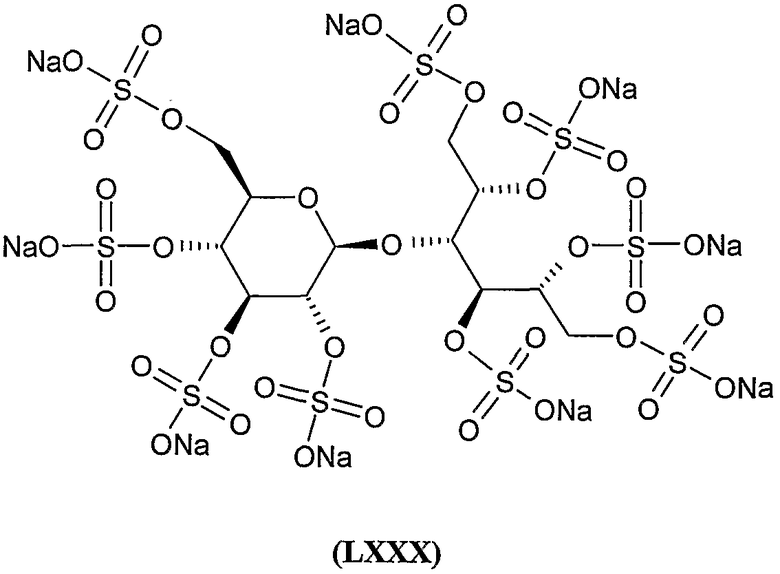
5,1 г (48%, 30 ммоль) комплекса триоксида серы - диметилформамида суспендировали в 5 мл сухого диметилформамида при перемешивании, смесь охлаждали до -20°C и постепенно добавляли 0,52 г (1,5 ммоль) 3-О-β-D-глюкопиранозил-D-глюцитола (XVII) в 5 мл диметилформамида с такой скоростью, чтобы поддерживать температуру ниже -15°C. Смесь перемешивали при 5°C в течение 1 часа. После этого реакционную смесь снова охлаждали до -15°C и постепенно добавляли 1,5 мл этанола с такой скоростью, чтобы поддерживать температуру ниже -10°C. Затем реакционную смесь вливали в перемешиваемый и охлаждаемый (0°C) раствор 5 г ацетата натрия и 40 мл метанола. Осадок отфильтровывали и промывали 3×40 мл метанола. Твердый остаток растворяли в 30 мл воды и pH раствора доводили сначала до 8 с помощью 1 M раствора гидроксида натрия, затем 3 мл 1 M водного раствора ацетата стронция добавляли к раствору. Через 30 минут осадок отфильтровывали и промывали холодной водой. Фильтрат вносили на колонку, нагруженную смолой CHELX 100 (натриевая форма) (15 мл) с целью удаления ионов стронция. Колонку элюировали дистиллированной водой и элюат концентрировали. Остаток обрабатывали этанолом, отфильтровывали и промывали этанолом для получения 1,9 г (99%) указанного в заголовке соединения. Т.пл. >220°C; [α]D +1,5° (c 1, вода). C12H15О38S9Na9 Рассчитано: С 11,41; H 1,20; S 22,85; Na 24,39. Обнаружено: С 11,74; H 1,57; S 22,25; Na 16,09.
Эквиваленты
Тогда как заявленное изобретение описано подробно и со ссылками на его специфические варианты осуществления, понятно, что обычный специалист в области техники может осуществить различные уточнения и модификации не выходя за пределы сущности и объема настоящего изобретения. Следовательно, например, специалист в области техники понимает или может установить, с использованием не более чем обычных экспериментов, множество эквивалентов специфических веществ и методик, описанных в настоящем описании. Такие эквиваленты рассматриваются в объеме настоящего изобретения и охватываются следующей формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ СПИРОКЕТАЛЕЙ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННОГО СРЕДСТВА ПРОТИВ ДИАБЕТА | 2006 |
|
RU2416617C2 |
| ПРОИЗВОДНЫЕ 1-ТИО-D-ГЛЮЦИТОЛА | 2006 |
|
RU2387649C2 |
| С-ФЕНИЛ-1-ТИОГЛЮЦИТОЛЫ | 2007 |
|
RU2434862C2 |
| СИНТЕТИЧЕСКИЕ ПОЛИСАХАРИДЫ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩАЯ ИХ ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1997 |
|
RU2167163C2 |
| НОВОЕ ЦИКЛОГЕКСАНОВОЕ ПРОИЗВОДНОЕ, ЕГО ПРОЛЕКАРСТВО И ЕГО СОЛЬ И СОДЕРЖАЩЕЕ ИХ ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО ОТ ДИАБЕТА | 2005 |
|
RU2394015C2 |
| ПРОИЗВОДНЫЕ АРИЛ 5-ТИО-β-D-ГЛЮКОПИРАНОЗИДА И ТЕРАПЕВТИЧЕСКИЕ СРЕДСТВА ПРИ ДИАБЕТЕ, СОДЕРЖАЩИЕ ИХ | 2003 |
|
RU2322449C2 |
| ЧУВСТВИТЕЛЬНЫЕ К ГЛЮКОЗЕ КОНЪЮГАТЫ ИНСУЛИНА | 2014 |
|
RU2676307C2 |
| ПРОИЗВОДНЫЕ ГЕКСОЗЫ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ | 2015 |
|
RU2731563C2 |
| ПРОИЗВОДНОЕ ОЛИГОСАХАРИДОВ | 2004 |
|
RU2290408C2 |
| СПОСОБ ПОЛУЧЕНИЯ 6-O-[β-D-(2,3,4,6-ТЕТРА-O-АЦЕТИЛ)ГЛИКОПИРАНОЗИЛ]-d,l-α-ТОКОФЕРОЛОВ | 2007 |
|
RU2350620C1 |
Изобретение относится к группе соединений общей формулы (I), где R1, R2, R3, R4, R5 и R6 независимо друг от друга означают C1-4 алкил, -SO3H, полисульфатированную β-гликозильную или полисульфатированную дигликозильную группу, при условии, что, по меньшей мере, один из R1-R6 представляет собой полисульфатированную β-гликозильную или полисульфатированную дигликозильную группу, или их фармацевтически приемлемым солям, где гликозильная группа содержит молекулу пентопиранозы или гексопиранозы с конфигурацией по выбору, а дигликозильная группа содержит молекулу пентопиранозы или гексопиранозы с конфигурацией по выбору, одна гидроксильная группа которой гликозилирована другой молекулой пентопиранозы или гексопиранозы с конфигурацией по выбору. Изобретение относится также к фармацевтической композиции для использования в лечении острых или хронических воспалительных заболеваний дыхательных путей у млекопитающих на основе указанных соединений или их фармацевтически приемлемых солей. Кроме того, изобретение относится к применению указанных соединений или их фармацевтически приемлемых солей в получении лекарственного средства для лечения острых или хронических воспалительных заболеваний дыхательных путей у млекопитающих. 7 н. и 30 з.п. ф-лы, 4 табл.
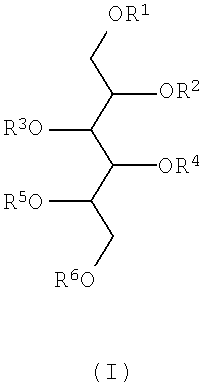
1. Соединение формулы (I)

где R1, R2, R3, R4, R5 и R6 независимо друг от друга означают C1-4 алкил, -SO3H, полисульфатированную β-гликозильную или полисульфатированную дигликозильную группу, при условии, что, по меньшей мере, один из R1-R6 представляет собой полисульфатированную β-гликозильную или полисульфатированную дигликозильную группу, или его фармацевтически приемлемая соль, где гликозильная группа содержит молекулу пентопиранозы или гексопиранозы с конфигурацией по выбору, а дигликозильная группа содержит молекулу пентопиранозы или гексопиранозы с конфигурацией по выбору, одна гидроксильная группа которой гликозилирована другой молекулой пентопиранозы или гексопиранозы с конфигурацией по выбору.
2. Соединение или соль по п.1, где R1 означает полисульфатированную β-гликозильную или полисульфатированную дигликозильную группу, a R2-R6 представляют собой -SO3H группы.
3. Соединение или соль по п.1, где R1, R2, R4, R5 и R6 означают -SO3H группы, а R3 представляет собой полисульфатированную β-гликозильную или полисульфатированную дигликозильную группу.
4. Соединение или соль по п.1, где R1 и R2 означают полисульфатированные β-гликозильные или полисульфатированные дигликозильные группы, a R2, R4, R5 и R6 представляют собой -SO3H группы.
5. Соединение или соль по п.1, где R1 и R6 означают полисульфатированные β-гликозильные или полисульфатированные дигликозильные группы, а R2, R3, R4 и R5 представляют собой -SO3Н группы.
6. Соединение или соль по п.1, где R1 означает полисульфатированную β-гликозильную или полисульфатированную дигликозильную группу, R3 и R4 представляют собой C1-4 алкильные группы, а R2, R5 и R6 являются -SO3H группами.
7. Соединение или соль по п.1, где R1 и R6 означают полисульфатированные β-гликозильные или полисульфатированные дигликозильные группы, R3 и R4 представляют собой C1-4 алкильные группы, а R2 и R5 являются -SO3H группами.
8. Соединение по п.1, выбранное из группы, состоящей из:
2,3,4,5,6-пента-O-сульфато-1-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-маннита,
1,2,3,4,5-пента-O-сульфато-6-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола,
2,3,4,5,6-пента-O-сульфато-1-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола,
1,2,4,5,6-пента-O-сульфато-3-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола,
1,2,3,5,6-пента-O-сульфато-4-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола,
1,2,3,5,6-пента-O-сульфато-4-O-(2,3,4,6-тетра-O-сульфато-β-D-галактопиранозил)-D-глюцитола,
2,4,5,6-тетра-O-сульфато-1,3-бис-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола,
2,3,4,5-тетра-O-сульфато-1,6-бис-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-маннита,
2,3,4,5-тетра-O-сульфато-1,6-бис-O-(2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозил)-D-маннита,
2,3,4,5,6-пента-O-сульфато-1-O-(2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозил)-D-маннита,
3,4-ди-O-метил-2,5,6-три-О-сульфато-1 -O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-маннита,
3,4-ди-O-метил-2,5-ди-O-сульфато-1,6-бис-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-маннита,
3,4-ди-O-метил-2,5,6-три-O-сульфато-1-O-(2,3,4,2',3',4',6'-гепта-O-сульфато-β-гентиобиопиранозил)-D-маннита,
3,4-ди-O-метил-2,5-ди-O-сульфато-1,6-бис-O-(2,3,6,2',3',4',6'-гепта-O-сульфато-β-лактозил)-D-маннита,
2,3,4,5,6-пента-O-сульфато-1-O-(2,3,4-три-O-сульфато-β-D-ксилопиранозил)-D-маннита,
2,3,4,5-тетра-O-сульфато-1,6-бис-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)галактитола,
или фармацевтически приемлемой соли любого из этих соединений.
9. Соединение или соль по п.1, в котором полисульфатированная β-гликозильная или полисульфатированная дигликозильная группа представляют собой полисульфатированную β-D-гликозильную или полисульфатированную β-D-дигликозильную группу.
10. Соединение или соль по п.9, где полисульфатированная β-D-гликозильная или полисульфатированная β-D-дигликозильная группы представляют собой полисульфатированную β-В-гликопентапиранозильную группу, полисульфатированную β-D-гликогексапиранозильную группу, полисульфатированную β-D-дигликопентапиранозильную группу или полисульфатированную β-D-дигликогексапиранозильную группу.
11. Соединение или соль по п.3, где R3 означает полисульфатированную β-гликозильную группу, а R1, R2, R4, R5 и R6 являются -SO3H группами.
12. Соединение или соль по п.11, где полисульфатированная β-гликозильная группа представляет собой полисульфатированную β-D-гликозильную группу.
13. Соединение или соль по п.12, где полисульфатированная β-D-гликозильная группа представляет собой полисульфатированную β-D-гликопентапиранозильную группу или полисульфатированную β-D-гликогексапиранозильную группу.
14. Соединение по п.1 в форме калиевой соли.
15. Соединение по п.8 в форме калиевой соли.
16. Соединение по п.10 в форме калиевой соли.
17. Соединение по п.16, где полисульфатированная β-гликозильная группа представляет собой полисульфатированную β-D-гликозильную группу.
18. Соединение по п.17, где полисульфатированная β-D-гликозильная группа представляет собой полисульфатированную β-D-гликопентапиранозильную группу или полисульфатированную β-D-гликогексапиранозильную группу.
19. Соединение по п.18, представляющее собой нонакалиевую соль 1,2,3,5,6-пента-O-сульфато-4-O-(2,3,4,6-тетра-O-сульфато-β-D-глюкопиранозил)-D-глюцитола.
20. Соединение по п.19 в кристаллической форме.
21. Соединение по п.18, представляющее собой нонакалиевую соль 1,2,3,5,6-пента-O-сульфато-4-O-(2,3,4,6-тетра-O-сульфато-β-D-галактопиранозил)-D-глюцитола.
22. Соединение по п.21 в кристаллической форме.
23. Фармацевтическая композиция для использования в лечении острых или хронических воспалительных заболеваний дыхательных путей у млекопитающих, содержащая терапевтически эффективное количество соединения или соли по п.1 и фармацевтически приемлемый носитель.
24. Применение соединения по п.1 или его фармацевтически приемлемой соли в получении лекарственного средства для лечения острых или хронических воспалительных заболеваний дыхательных путей у млекопитающих.
25. Применение по п.24, где фармацевтически приемлемая соль представляет собой калиевую соль.
26. Применение по п.24, где воспалительное заболевание дыхательных путей выбирают из группы, состоящей из астмы, аллергического ринита, эндогенной или экзогенной бронхиальной астмы, острого или хронического бронхита, хронических обструктивных заболеваний легких и пневмосклероза.
27. Применение по п.24, где воспалительное заболевание дыхательных путей представляет собой аллергическое воспалительное заболевание дыхательных путей.
28. Применение по п.27, где аллергическое воспалительное заболевание дыхательных путей выбрано из группы, состоящей из идиопатического пневмосклероза и аутоиммунного заболевания легких.
29. Применение по п.24, где указанное лекарственное средство вводится в виде одной или нескольких доз.
30. Фармацевтическая композиция для использования в лечении острых или хронических воспалительных заболеваний дыхательных путей у млекопитающих, содержащая терапевтически эффективное количество соединения по п.8 и фармацевтически приемлемый носитель.
31. Фармацевтическая композиция для использования в лечении острых или хронических воспалительных заболеваний дыхательных путей у млекопитающих, содержащая терапевтически эффективное количество соединения по п.20 и фармацевтически приемлемый носитель.
32. Фармацевтическая композиция для использования в лечении острых или хронических воспалительных заболеваний дыхательных путей у млекопитающих, содержащая терапевтически эффективное количество соединения по п.22 и фармацевтически приемлемый носитель.
33. Применение соединения по любому из пп.19-22 в получении лекарственного средства для лечения острых или хронических воспалительных заболеваний дыхательных путей у млекопитающих.
34. Применение по п.33, где воспалительное заболевание дыхательных путей представляет собой аллергическое воспалительное заболевание дыхательных путей.
35. Применение по п.33, где воспалительное заболевание дыхательных путей выбирают из группы, состоящей из астмы, аллергического ринита, эндогенной или экзогенной бронхиальной астмы, острого или хронического бронхита, хронических обструктивных заболеваний легких и пневмосклероза.
36. Применение по п.35, где воспалительное заболевание дыхательных путей представляет собой астму.
37. Применение по п.33, где воспалительное заболевание дыхательных путей выбрано из группы, состоящей из идиопатического пневмосклероза и аутоиммунных заболеваний легких.
| Прибор для очистки паром от сажи дымогарных трубок в паровозных котлах | 1913 |
|
SU95A1 |
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Способ получения глюкопиранозидо -1,6-маннита | 1976 |
|
SU665806A3 |

