Изобретение относится к способам получения новых трициклических соединений, которые являются избирательными антагонистами к лейкотриенам и могут быть использованы в качестве терапевтического средства при лечении воспалительных и аллергических заболеваний, таких, как астма, где указанные лейкотриены, по всей вероятности, играют роль каузальных медиаторов.
Исследования в области аллергических реакций на легких показали, что производные арахидоновой кислоты, образующиеся при воздействии липоксигеназы, являются ответственными за различные болезненные состояния. Некоторые метаболиты указанной арахидоновой кислоты классифицируются как члены семейства эйкозатетраеновых кислот, называемых лейкотриенами. В настоящее время три из указанных веществ считают главными компонентами так называемой медленно реагирующей субстанции анафилаксии (SPS - А).
Лейкотриен В4 (LТВ4) является липидом, который способствует развитию воспалительных заболеваний и участвует в патогенезе псориаза, артритов, хронических легочных заболеваний, воспалительных заболеваний кишечного тракта и других воспалительных заболеваний, характеризующихся инфильтрацией и агрегацией нейтрофильных лейкоцитов. Агрегированные таким образом нейтрофильные лейкоциты способствуют высвобождению тканеразрушающих ферментов и реакционноспособных химических соединений, вызывая тем самым воспалительные состояния. Поэтому, получение антагониста к L ТВ4 позволяет создать новое терапевтическое средство для лечения указанных заболеваний.
Изобретение относится к способам получения соединений формулы I HOOC где Y является -СО-, или -СН2-;
где Y является -СО-, или -СН2-;
Y является связью или -О-;
р = 1-16; и
Z является -Н или -G-Q, где G - простая связь, или -СН= СН-;
а Q-фенил, замещенный С1-С3-алкокси группой.
Предпочтительными соединениями являются соединения формулы Iа HOOC и их фармацевтически приемлемые основные аддитивные соли, где: р' является числом от 4 до 12.
и их фармацевтически приемлемые основные аддитивные соли, где: р' является числом от 4 до 12.
Следует отметить, что если G представляет собой -СН = СН-, то соединения изобретения могут существовать в виде различных стереоизомеров. Изобретение не ограничено конкретными стереоизомерами и включает в себя все возможные отдельные изомеры и их смеси.
Соединения изобретения могут быть получены гидролизом соединения формулы II:  где Y, A, р и Z имеют указанные значения, а R - C1 - C4-алкил.
где Y, A, р и Z имеют указанные значения, а R - C1 - C4-алкил.
Гидролиз сложных эфиров формулы II может быть осуществлен в кислотных или основных условиях, а предпочтительно в водных условиях. Предпочтительный способ включает в себя использование гидроокиси калия в смеси воды с метанолом или этанолом. При указанных предпочтительных условиях гидролиз, в основном, завершается приблизительно за 1 ч при 20-30оС. Полученный в результате продукт является производным ксантона формулы 1.
Промежуточные соединения II являются либо коммерчески доступными продуктами, хорошо известными специалистам, либо они могут быть получены стандартными способами.
Приведенные примеры иллюстрируют получение промежуточных соединений и соединений изобретения, однако, при этом они не ограничивают объема изобретения. Соединения, структуры которых были подтверждены ИК-анализом, протонным ядерным магнитным резонансом или масс-спектральным анализом, помечены соответствующими аббревиатурами: "ИК", "ЯМР" или "МС".
П р и м е р 1. 5-Карбокси-3-(децилокси)-9-оксо-9Н-ксантен-2-пропановая кислота.
А. Получение 2-(3-метоксифенокси)бензонитрила
Смесь 100 г 2-бромобензонитрила, 68,1 г 3-метоксифенола, 35 г порошкообразной меди, и 75,8 г карбоната калия нагревали в сосуде с обратным холодильником в 3 л пиридина в течение 6 дней. Затем горячую реакционную смесь фильтровали, охлаждали до комнатной температуры и концентрировали в вакууме. К полученному остатку медленно добавляли 1,5 л концентрированной соляной кислоты. Затем добавляли этилацетат и воду и слои разделяли. Органический слой промывали несколько раз водой, осушали сульфатом натрия и концентрировали в вакууме. Остаток несколькими порциями очищали с помощью жидкостной хроматографии высокого давления на силикагеле, элюируя в градиенте: гексан - 10% этилацетата в гексане, и в результате получали 53,4 г целевого промежуточного соединения в виде маслянистого продукта, который после некоторого периода выдерживания кристаллизуется.
Вычислено, % : C 74,65; H 4,92; N 6,22.
C14H11NO2
Найдено, % : C 74,95; H 5,17; Н 6,24.
В. Получение 2-(3-метоксифенокси)бензойной кислоты.
Смесь 48,2 г промежуточного соединения примера 1А и 20 г гидроокиси калия в смеси этанола и воды нагревали в сосуде с обратным холодильником в течение ночи. После охлаждения до комнатной температуры раствор концентрировали в вакууме. После чего добавляли в этилацетат и воду, слои разделяли, и водный слой подкисляли. Подкисленный водный слой экстрагировали этилацетатом, а органический слой осушали и концентрировали в вакууме. Остаток кристаллизовали из смеси этилацетата и гексана и получали 18,81 г целевого промежуточного соединения, т. пл. 129-131оС.
Вычислено, % : C 68,85; H 4,95
C14H12O4
Найдено, % : С 68,57; H 4,02.
С. Получение 2-(2-гидроксифенокси)бензойной кислоты
15 г 2-(2-метоксифенокси)бензойной кислоты нагревали при 180-185оС вместе с 150 г гидрохлорида пиридина в течение 3 ч. После охлаждения до комнатной температуры добавляли воду и полученную смесь размешивали в течение ночи при комнатной температуре. Затем реакционую смесь фильтровали, а водный слой несколько раз экстрагировали этилацетатом. Объединенные слои этилацетата осушали и концентрировали в вакууме. После кристаллизации остатка из смеси этилацетата и гексана получали 6,88 г целевого промежуточного соединения, т. пл. 147-149оС.
Вычислено, % : С 67,82; Н 4,38.
C13H10O4
Найдено, % : С 67,55; Н 4,59.
1. Получение 2-(3-гидроксифенокси)бензойной кислоты сложного эфира.
Раствор 17,8 г 2-(3-гидроксифенокси)бензойной кислоты в 250 мл этанола и 1 мл серной кислоты нагревали в сосуде с обратным холодильником в течение двух дней. После охлаждения смесь концентрировали в вакууме. Остаток распределяли между этилацетатом и водой. Органический слой осушали и концентрировали в вакууме. Полученный остаток очищали с помощью жидкостной хроматографии высокого давления, элюируя 0-30% этилацетата в гексане (в градиенте). Подводящие фракции объединяли и концентрировали в вакууме, в результате чего получали 16,78 г целевого промежуточного соединения в виде маслянистого продукта.
Вычислено, % : С 69,76; Н 5,46.
С15Н14О4
Найдено, % : С 69,77, Н 5,68.
Е. Получение 2-[3-(2-пропенилокси)фенокси] бензойной кислоты сложного эфира.
Смесь 16,27 г промежуточного соединения примера 11, 7,56 г аллилбромида, 8,7 г карбоната калия, 500 мг иодида калия и 500 мл метилэтилкетона нагревали в сосуде с обратным холодильником в течение 4 дней. После охлаждения до комнатной температуры и фильтрации, фильтрат промывали водой, осушали и концентрировали в вакууме, в результате чего получали 17,6 г целевого промежуточного соединения в виде маслянистого продукта, который использовали в следующей стадии без очистки.
Вычислено, % : С 72,47; Н 6,08
С18Н18О4
Найдено, % : С 72,61; Н 6,10.
F. Получение 2-[3-гидрокси-2-(пропенил)-фенокси] -бензойной кислоты этилового сложного эфира и 2-[3-гидрокси-4-(2-проценил)фенокси] бензойной кислоты этилового сложного эфира.
17,6 г промежуточного соединения примера 1Е нагревали в течение 3 ч при 200оС. После охлаждения до комнатной температуры, остаток очищали с помощью жидкостной хроматографии высокого давления на силикагеле, элюируя 0-10% этилацетатом в гексане (в градиенте). Затем подходящие фракции объединяли и концентрировали в вакууме, в результате чего получали 12,5 г смеси изомеров целевых промежуточных соединений, которые использовали в следующей стадии без разделения.
Вычислено, % : С 72,47; Н 6,08.
С18Н18О4
Найдено, % : С 72,23. Н 6,11.
G. Получение сложного этилового эфира 2-[3-гидрокси-4-(3-гидроксипропил)фенокси] бензойной кислоты и сложного этилового эфира 2-[3-гидрокси-2-(3-гидроксипропил)фенокси] бензойной кислоты
Смесь примера 1F (6,86 г) растворяли в 200 мл безводного тетрагидрофурана. К полученной смеси добавляли 70 мл 0,5 М раствора 9-ВВN в тетрагидрофуране. Смесь размешивали в течение ночи при комнатной температуре, и в течение этого периода времени добавляли 10 мл 0,5 М 9-ВВN. Затем смесь размешивали еще 1 ч, после чего добавляли ацетат натрия и перекись водорода. Затем полученную смесь размешивали еще 1 ч, после чего слои разделяли и органический слой высушивали и концентрировали в вакууме. Остаток очищали с помощью жидкостной хроматографии высокого давления на силикагеле, элюируя 0-50% этилацетата в гексане с использованием техники градиентного элюирования. Подходящие фракции объединяли и концентрировали в вакууме, в результате чего получали 1,93 г 2-(3-гидроксипропил)-изомера и 2,86 г 4-(3-гидроксипропил)изомера целевых соединений. Оба продукта являются маслянистыми веществами. 2-[3-гидрокси-2-(3-гидроксипропил)фенокси] безойной кислоты сложный этиловый эфир.
Вычислено, % : С 68,35; Н 6,37.
С18Н20О5
Найдено, % : С 68,13; Н 6,62.
2-[3-Гидрокси-4-(3-гидроксипропил)фенокси] бензойной кислоты сложный этиловый эфир.
Вычислено, % : С 68,34; H 6,37.
С18Н20О5
Найдено, % : С 68,35. Н 6,39.
Н. Получение сложного этилового эфира 2-[3-(децилокси)-4-(3-гидроксипропил)фенокси] бензойной кислоты
Смесь 600 мг 2-[3-гидрокси-4-(3-гидроксипропил)фенокси] бензойной кислоты сложного этилового эфира, 0,4 мл децилиодида, и 0,26 г карбоната калия в 50 мл метилэтилкетона перемешивали в течение ночи при температуре перегонки. После охлаждения до комнатной температуры, смесь фильтровали и фильтрат концентрировали в вакууме. После очистки остатка с помощью колоночной хроматографии на силикагеле (элюент: 20% этилацетата в гексане) получали 510 мг целевого промежуточного соединения в виде маслянистого продукта, МС, ИК, ЯМР.
1. Получение сложного этилового эфира 2-(децилокси)-4-[2-(этоксикарбонил)бенокси] бензолпропионовой кислоты
К раствору 490 мг сложного этилового эфира 2-[3-децилокси)-4-(3-гидроксипропил)фенокси] бензойной кислоты в простом эфире добавляли 1 мл реагента Джонса (раствор хромовой кислоты). После размешивания полученной смеси в течение 1 ч добавляли дополнительное количество простого эфира и полученный раствор промывали раствором бисульфита натрия. Органический слой осушали и концентрировали в вакууме. К остатку добавляли этанол и несколько капель серной кислоты. Смесь нагревали с обратным холодильником в течение ночи, охлаждали до комнатной температуры и концентрировали в вакууме, а полученный остаток очищали с помощью колочной хроматографии, элюируя 5% этилацетата в гексане. После объединения подходящих фракций получали 110 кг целевого промежуточного соединения в виде маслянистого продукта МС, ИК, ЯМР.
J. Получение 5-(этоксикарбонил)-3-гидрокси-9-оксо-9Н-ксантен-2-пропионовой кислоты
К раствору 0,7 г 2-(децилокси)-4-[2-(этоксикарбонил)фенокси] бензолпропионовой кислоты этилового сложного эфира в метиленхориде при комнатной температуре добавляли 0,187 г хлорида алюминия, а затем 0,122 мл оксалилхлорида. После чего смесь перемешивали приблизительно в течение 1 ч и выливали в соляную кислоту со льдом. Затем смесь размешивали еще около 1 ч, после чего слои разделяли, органический слой высушивали и концентрировали в вакууме. Полученный продукт использовали в следующей стадии без очистки.
К. Получение сложного этилового эфира 5-этоксикарбонил-3-(децилокси)-9-оксо-9Н-ксантен-2-пропионовой кислоты
Фенол примера 1J обрабатывали 0,23 мл децилиодида и 0,147 г карбоната калия в 50 мл метилэтилкетона в течение ночи при температуре перегонки. После охлаждения до комнатной температуры, продукт концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле, элюируя 10% этилацетата в гексане. Целевые фракции объединяли и концентрировали, в результате чего получали 141 мг целевого промежуточного соединения, т. пл. 61-63оС.
Вычислено, % : С 70,97; Н 7,69;
С31Н40О7
Найдено, % : С 71,28; Н 7,81.
L. Получение 5-карбокси-3-(децилокси)-9-оксо-9Н-ксантан-2-пропионовой кислоты
Промежуточное соединение сложного диэфира примера 1К (130 г) размешивали со смесью этанола/воды и гидроокиси калия в течение 2 ч. Смесь концентрировали в вакууме и добавляли этилацетат и воду. Слои разделяли и водный слой подкисляли. Целевой продукт осаждали из раствора кислоты и выделяли путем фильтрации. После кристаллизации из смеси этилацетата и гексана получали 60 мг целевого продукта, т. пл. 180-182оС.
Вычислено, % : С 69,21; Н 6,88
С27Н32О7
Найдено, % : С 69,43; Н 6,92.
П р и м е р 2. 5-Карбокси-3-(децилокси)-9-оксо-9Н-ксантен-4-пропионовая кислота
А. Получение 2-[3-децилокси-2-(3-гидроксипропил)фенокси] бензойной кислоты сложного этилового эфира
Способом, аналогичным описанному в примере 1Н, получали соответствующее промежуточное соединение (с выходом 84,1% ) из сложного этилового эфира 2-[3-гидрокси-2-(3-гидроксипропил)-фенокси] бензойной кислоты, ИК, МС, ЯМР.
В. Получение сложного этилового эфира 2-(децилокси)-6-[2-(этоксикарбонил)фенокси] бензолпропионовой кислоты
В соответствии с процедурой примера 1J из соответствующего гидроксипропилового промежуточного соединения получали целевой продукт с выходом 23,6% , в виде маслянистого вещества.
Вычислено, % : С 72,27; Н 8,49.
С30Н42О6
Найдено, % : С 72,50; Н 8,38.
С. Получение сложного этилового эфира 5-(этоксикарбонил)-3-гидрокси-9-оксо-9Н-ксантен-4-пропионовой кислоты
В соответствии с процедурой примера 1J из промежуточного соединения примера 2В получали 1,12 г целевого продукта. Полученный продукт использовали в следующей стадии без дополнительной очистки.
D. Получение сложного этилового эфира 5-(этоксикарбонил)-3-децилокси-9-оксо-9Н-ксантен-4-пропионовой кислоты
Используя процедуру примера 1К из фенольного промежуточного соединения примера 2С получали соответствующее промежуточное соединение с выходом 26,1% и т. пл. 69-70оС.
Вычислено, % : С 70,97; Н 7,69
С31Н40О7
Найдено, % : С 70,93; Р 7,64
Е. Получение 5-карбокси-3-(децилокси)-9-оксо-9Н-ксантен-4-пропионовой кислоты
С помощью описанной выше процедуры примера 1L, из промежуточного соединения сложного диэфира примера 2D получали целевой продукт с выходом 43,3% и т. пл. > 210оС.
Вычислено, % : С 69,21; Н 6.88
С27Н32О7
Найдено, % : С 69,02; Н 6,89.
П р и м е р 3. 7-Карбокси-3-децилокси-9-оксо-9Н-ксантен-2-пропионовая кислота
А. Получение сложного этилового эфира 4-[3-(2-пропенилокси)-фенилокси] бензойной кислоты.
С помощью процедуры, описанной в примере 1Е, из сложного этилового эфира 4-(3-гидроксифенокси)бензойной кислоты получали целевое промежуточное соединение в виде маслянистого продукта (выход 83,4% ).
Вычислено, % : С 72,47; Н 6,08.
С18Н18О4
Найдено, % : С 72,43; Н 6,27.
В. Получение сложного этилового эфира 4-[3-гидрокси-4-(2-пропенил)фенокси] бензойной кислоты и сложного этилового эфира 4-[3-гидрокси-2-(2-пропенил)-фенокси] бензойной кислоты
Используя основную процедуру, описанную в примере 1F, 21,2 г пропенилокси - соединение примера 3А нагревали при 180оС приблизительно в течение 4 ч, и получали 8,01 г 4-пропенилового изомера и 4,87 г 2-пропенилового изомера.
4-[3-Гидрокси-4-(2-пропенил)фенокси] бензойной кислоты сложный этиловый эфир, маслянистый продукт
Вычислено, % : С 72,47; Н 6,08
С18Н18О4
Найдено, % : С 72,54; Н 6,26.
4-[3-Гидрокси-2-(2-пропенил)фенокси] бензойной кислоты сложный этиловый эфир, т. пл. 85-88оС
Вычислено, % : С 72,47; Н 6,08
С18Н18О4
Найдено, % : С 72,69; Н 6,22.
С. Получение сложного этилового эфира 4-[3-(децилокси)-4-(2-пропенил)фенокси] бензойной кислоты
В соответствии с основной процедурой, описанной в примере 1Н, 7,11 г сложного этилового эфира 4-[3-гидрокси-4-(2-пропенил)фенокси] бензойной кислоты подвергали реакции с 6,38 г децилиодида в присутствии 3,3 кг карбоната калия и получали в результате 5,02 г целевого промежуточного соединения в виде маслянистого продукта.
Вычислено, % : С 76,68; Н 8,73.
С28Н38О4
Найдено, % : С 76,80; Н 8,77.
D. Получение сложного этилового эфира 4-[3-(децилокси)-4-(3-гидроксипропил)фенокси] бензойной кислоты
В соответствии с процедурой примера 1G, 5,74 г 4-[3-(децилокси)-4-(2-пропенил)фенокси] бензойной кислоты подвергали реакции окисления, и получали в результате целевое гидроксипропиловое промежуточное соединение в виде маслянистого продукта, МС, ИК, ЯМР.
Вычислено, % : С 73,65; Н 8,83;
С28Н40Р5
Найдено, % : С 70,96; Н 9,51.
Е. Получение сложного этилового эфира 2-(децилокси)-4-[4-(этоксикарбонил)фенокси] бензолпропионовой кислоты
В соответствии с процедурой примера 1J, 3,01 г промежуточного соединения примера 3J окисляли и превращали в соответствующий сложный этиловый эфир, в результате чего получали 750 мг целевого промежуточного соединения в виде маслянистого продукта. МС, ИК, ЯМР.
Вычислено, % : С 72,26; Н 8,49
С30Н42О6
Найдено, % : С 71,66; Н 8,20.
F. Получение сложного этилового эфира 7-(этоксикарбонил)-3-гидрокси-9-оксо-9Н-ксантен-2-пропионовой кислоты
В соответствии с процедурой примера 1J, 390 мг сложного диэфира примера 3Е превращали в 200 мг соответствующего ксантона. Продукт идентифицировали с помощью МС, ИК, ЯМР.
G. Получение сложного этилового эфира 7-(этоксикарбонил)-3-(децилокси-9-оксо-9Н-ксантен-2-пропионовой кислоты
Из фенола примера 3G получали целевое промежуточное соединение с выходом 50,4% , МС, ИК, ЯМР.
Н. Получение 7-карбокси-3-(децидокси)-9-оксо-9Н-ксантен-2-пропионовой кислоты
В соответствии с процедурой примера 1L, из 130 мг соответствующего сложного диэфира получали 94,6 мг (т. пл. > 210оС) целевого соединения.
Вычислено, % : С 69,21; Н 6,88
С27Н32О7
Найдено, % : С 69,05; Н 6,97
П р и м е р 4. 7-Карбокси-3-(децилокси)-9-оксо-9Н-ксантен-4-пропионовая кислота
А. Получение сложного этилового эфира 4-[3-(децилокси)-2-(3-гидроксипропил)фенокси] бензойной кислоты
В соответствии с процедурой примера 1G, из сложного этилового эфира 4-[3-гидрокси-2-(3-гидроксипропил)фенокси] бензойной кислоты получали целевой продукт с выходом 67,1% .
Вычислено, % : С 73,65; Н 8,83
С28Н40О5
Найдено, % : С 73,56; Н 8,61.
В. Получение сложного этилового эфира 2-(децилокси)-6-[4-(этоксикарбонил)фенокси] бензолпропионовой кислоты
В соответствии с основной процедурой примера 1L из соответствующего гидроксипропилового промежуточного соединения получали целевое промежуточное соединение с выходом 61,9% , МС, ИК, ЯМР.
С. Получение сложного этилового эфира 7-(этоксикарбонил)-3-гидрокси-9-оксо-9Н-ксантен-4-пропионовой кислоты
В соответствии с процедурой примера 1J, из 540 мг соответствующего промежуточного соединения бисарилового простого эфира получали целевое промежуточное соединение, МС, ИК, ЯМР.
D. Получение сложного этилового эфира 7-(этоксикарбонил)-3-децилокси-9-оксо-9Н-ксантен-4-пропионовой кислоты
В соответствии с процедурой примера 1К, из фенолового предшественника получали целевое промежуточное соединение, с выходом 64,7% в виде маслянистого продукта, ИК, МС, ЯМР.
Е. Получение 7-карбокси-3-децилокси-9-оксо-9Н-ксантен-4-пропионовой кислоты
В соответствии с процедурой примера 1L, из 160 мг соответствующего сложного диэфира получали 119 мг целевого продукта, т. пл. > 210оС.
Вычислено, % : С 69,21; Н 6,88
С27Н32О7
Найдено, % : С 69,41; Н 6,71
П р и м е р 5. 5-Аллил-6-гидрокси-9-оксо-9Н-ксантен-2-карбоновой кислоты сложный этиловый эфир
А. Получение сложного диэтилового эфира 4-(3-метоксифенокси)-1,3-бензолдикарбоновой кислоты
Следуя основной процедуре примера 1А, 3,49 г сложного диэтилового эфира 4-бромо-1,3-бензолдикарбоновой кислоты и 1,43 г 3-метоксифенола подвергали взаимодействию в присутствии измельченной в порошок металлической меди и получали в результате 1,66 г целевого промежуточного соединения в виде маслянистого продукта.
Вычислено, % : С 66,27; Н 5,85
С18Н20О6
Найдено, % : С 66,50; Н 6,03
В. Получение 4-(3-метоксифенокси)-1,3-бензолкарбоновой кислоты
Следуя основной процедуре примера 18, из соответствующего сложного диэфира получали целевой продукт с выходом 79,6% , т. пл. 232-234оС.
Вычислено, % : С 62,50; Н 4,20
С15Н12О6
Найдено, % : С 62,71; Н 4,32.
С. Получение 6-метокси-9-оксо-9Н-ксантен-2-карбоновой кислоты
К раствору 17,25 г пентоксида фосфора в 17 мл метансульфоновой кислоты в атмосфере азота добавляли 15,4 г простого бисарилового эфира, полученного в соответствии с процедурой примера 5 B. Смесь размешивали в течение ночи при комнатной температуре, а затем выливали в лед. После размешивания в течение 1 ч, водный слой экстрагировали этилацетатом. Органический слой осушали и концентрировали в вакууме. После кристаллизации из метанола получали 12,7 г целевого промежуточного соединения, т. пл. > 250оС.
Вычислено, % : С 66,67; Н 3,93
С15Н40О5
Найдено, % : С 66,47; Н 3,93
D. Получение 6-гидрокси-9-оксо-9Н-ксантен-2-карбоновой кислоты
Следуя основной процедуре примера 1С, 12,7 г метоксисоединения примера 5С нагревали при 190оС в присутствии гидрохлорида пиридина и получали в результате 19,6 г целевого фенола. ЯМР.
Е. Получение сложного этилового эфира 6-гидрокси-9-оксо-9Н-ксантен-2-карбоновой кислоты
В соответствии с процедурой примера 1D, 10,6 г фенолового промежуточного соединения примера 5D нагревали в сосуде с обратным холодильником в присутствии смеси этанола с водой и серной кислоты в течение 13 дней. После обработки реакционной смеси аналогично описанному в указанном примере способу получали 8,16 г целевого сложного эфира, т. пл. > 250оС.
Вычислено, % : С 67,60; Н 4,26
С16Н12О5
Найдено, % : С 67,51; Н 3,32.
F. Получение сложного этилового эфира 9-оксо-6-2-пропенилокси 9Н-ксантен-2-карбоновой кислоты
К раствору 7,84 г промежуточного соединения примера 5Е в 200 мл диметилформамида добавляли 1,1 г 60% дисперсии в минеральном масле гидрида натрия. После размешивания в течение 1 ч добавляли 2,38 мл аллилбромида и полученную реакционную смесь размешивали в течение ночи при 65оС. После охлаждения до комнатной температуры, добавляли этилацетат и раствор промывали насыщенным раствором хлорида натрия. Органический слой осушали и концентрировали в вакууме. Остаток очищали с помощью хроматографии высокого давления на силикагеле, элюируя градиентом 5-30% этилацетата в гексане, в результате чего получали 5,4 г целевого промежуточного соединения, т. пл. 133-135оС.
Вычислено, % : С 70,36; Н 4,97
С19Н16О5
Найдено, % : С 70,62; Н 5,02.
G. Получение сложного этилового эфира 5-аллил-6-гидрокси-9-оксо-9Н-ксантен-2-карбоновой кислоты
В соответствии с процедурой примера 1F, 5,4 г промежуточного соединения примера 5F нагревали при 190оС. Часть продукта очищали путем растирания с горячим этилацетатом, получая целевое соединение, т. пл. 126-129оС.
Вычислено, % : С 70,36; Н 4,97
С19Н16О5
Найдено, % : С 70,31. Н 5,02.
Полученное промежуточное соединение затем может быть подвергнуто алкилированию, окислению и гидролизу в целях получения предпочтительных соединений формулы Iа.
П р и м е р 6. 7-Карбокси-3-{ [6-(4-метоксифенил)-5-гексенил] окси} -9-оксо-9Н-ксентен-4-пропионовая кислота
А. Получение 3,3-диэтокси-2,3-дигидро-7-оксо-1Н, 7Н-пирано[2,3-c] ксантен-9-карбоновой кислоты
Раствор 1,0 г сложного метилового эфира 6-гидрокси-9-оксо-9Н-ксантен-2-карбоновой кислоты и 1,37 г триэтилортакрилата (полученного в соответствии с способом Steller, Syurthesis 207 (1973) в 25 мл толуола нагревали в сосуде с обратным холодильником в течение ночи. Продукт охлаждали до комнатной температуры, кристаллизовали из раствора и выделяли путем фильтрации. После перекристаллизации из смеси этилацетата и гексана получали 1,1 г целевого промежуточного соединения, т. пл. 191-193оС.
Вычислено, % : С 66,32. Н 5,57,
С22Н22О7
Найдено, % : С 66,54; Н 5,72.
В. Получение сложного этилового эфира 7-метоксикарбонил-3-гидрокси-9-оксо-9Н-ксантен-4-пропионовой кислоты
"Ортолактон" примера 6А растворяли в 20 мл этилацетата. Затем добавляли 5 мл 10% -ного раствора соляной кислоты. Через 2 ч слои разделяли. Органический слой промывали насыщенным раствором хлорида натрия, высушивали сульфатом магния, и концентрировали в вакууме. После перекристаллизации из этилацетата получали 680 мл целевого промежуточного соединения. т. пл. > 215оС. ИК, МС, ЯМР.
Вычислено, % : С 64,86; Н 4,90
С20Н18О7
Найдено, % : С 66,13; Н 5,27.
С. Получение сложного этилового эфира 7-метоксикарбонил-6-{ [6- (4-метоксифенил)-5-гексенил] окси} -9-оксо-9Н-ксантен-4-пропионовой кислоты
Раствор 861 мг 6-(4-метоксифенил)-5-гексенилового спирта растворяли в 20 мл диэтилового эфира. После чего добавляли 0,42 мл триэтиламина, а затем 0,23 мл метансульфонилхлорида. Полученную смесь размешивали в течение 1 ч, а затем добавляли воду и слои разделяли, при этом органический слой осушали и концентрировали в вакууме. К остатку добавляли небольшое количество метилэтилкетона, и полученный раствор добавляли к суспензии 2 г карбоната калия и 680 мг фенолового промежуточного соединения примера 6В в метилэтилкетоне. Полученную смесь нагревали в сосуде с обратным холодильником в течение ночи, а затем охлаждали до комнатной температуры. После чего добавляли воду и слои разделяли, при этом органический слой осушали и концентрировали в вакууме. Полученный маслянистый продукт очищали с помощью жидкостной хроматографии высокого давления, используя технику градиентного элюирования (40-60% этилацетата в гексане). Соответствующие фракции объединяли и концентрировали в вакууме. Белый остаток кристаллизовали из смеси этилацетата и гексана и получали целевое промежуточное соединение, т. пл. 107-109оС.
Вычислено, % : С 70,83; Н 6,30
С33Н34О8
Найдено, % : С 71,04; H 6,13.
D. Получение 7-карбокси-3-{ [6-(4-метоксифенил)-5-гексенил] окси} -9-оксо-9Н-ксантен-4-пропионовой кислоты
В соответствии с процедурой примера 1L из 240 мг сложного диэфира примера 6С получали 160 мг целевого продукта, т. пл. > 215оС.
Вычислено, % : С 69,76; Н 5,46,
С30Н28О8
Найдено, % : С 69,99; Н 5,64.
П р и м е р 7. 7-Карбокси-3{ [6-(4-метоксифенил)гексил] окси} -9- оксо-9Н-ксантен-4-пропионовая кислота
А. Получение сложного этилового эфира 7-метоксикарбонил-3-{ [6-(4- метоксифенил)гексил] окси} -9-оксо-9Н-ксантен-4-пропионовой кислоты
Раствор 630 мг 7-метоксикарбонил-3-{ [6-(4-метоксифенил)-5- гексенил] окси} -9-оксо-9Н-ксантен-4-пропионовой кислоты сложного этилового эфира в этилацетате подвергали гидрованию в присутствии 10 мг 5% -ного палладированного угля. После завершения поглощения водорода, раствор фильтровали, концентрировали в вакууме и остаток кристаллизовали из смеси этилацетата и гексана, в результате чего получали 580 мг целевого промежуточного соединения т. пл. 108-109оС.
Вычислено, % : С 70,70; Н 6,47,
С33Н36О8
Найдено, % : С 70,46. Н 6,64.
В. Получение 7-карбокси-3-{ [6-(4-метоксифенил)гексил] окси} -9- оксо-9Н-ксантен-4-пропионовой кислоты
В соответствии с процедурой, описанной в примере 1L, из 500 мг соответствующего сложного диэфира получали 360 мг целевого продукта, т. пл. 210оС.
Вычислено, % : С 69,47; Н 5,84,
С30Н30О6
Найдено, % : C 69,29; Н 5,90.
П р и м е р 8. 7-Карбокси-3-децилокси-9-оксо-9Н-флоурен-2-пропионовая кислота.
А. Получение 2-(2,4-диметоксифенил)-4,4-диметилоксазолина
К раствору 26,2 г 2-амино-2-метил-1-пропанола в метиленхлориде при 0оС по капле добавляли раствор 27,4 г 2,4-диметоксибензоилхлорида в 30 мл метиленхлорида. Смесь доводили до комнатной температуры и размешивали в течение ночи. Затем смесь фильтровали и фильтрат дважды промывали 1 н. соляной кислотой и один раз водой, высушивали и концентрировали в вакууме. К полученному маслянистому остатку добавляли избыточное количество тионилхлорида. Смесь размешивали в течение 1 ч, а затем выливали в диэтиловый простой эфир. Твердое вещество выделяли путем фильтрации и промывали диэтиловым эфиром. Полученный твердый продукт добавляли к 1 н. гидроокиси натрия и смесь размешивали. Водную смесь экстрагировали этилацетатом, а органический слой отделяли, высушивали и концентрировали в вакууме. После дистилляции полученного желтого маслянистого продукта при 165-170оС и давлении 0,02 мм, получали 17,8 г целевого промежуточного продукта.
В. Получение 2-(4-бромфенил)-4,4-оксалолина
В соответствии с процедурой, описанной в примере 8А, 26,15 г 4-бромбензоилхлорида и 22,8 г 2-амино-2-метил-1-пропанола подвергали реакции взаимодействия и получали в результате 20,67 г целевого промежуточного соединения в виде бесцветного маслянистого продукта, который затем использовали без перегонки.
С. Получение 2-(4,4-диметилоксазолин-2-ил)-5-метокси-4'-(4,4- диметилоксазолин-2-ил)бифенила
Получали реагент Гриньяра, используя 5,4 г бромо-соединения примера 8В и 0,72 г металлического магния в тетрагидрофурана. Указанный реагент добавляли к раствору 2 г диметокси-соединения примера 8А в тетрагидрофуране. Реакционную смесь размешивали в течение ночи, а затем выливали в холодный водный раствор хлорида аммония. Полученную смесь три раза экстрагировали диэтиловым эфиром. Органические слои объединяли, осушали и концентрировали в вакууме. Остаток очищали с помощью жидкостной хроматографии высокого давления на силикагеле, используя технику градиентного элюирования (20-70% этилацетата в гексане). Соответствующие фракции объединяли и концентрировали в вакууме, в результате чего получали светло-оранжевый маслянистый продукт, который после выдерживания кристаллизовался. После перекристаллизации из гексана получали 2,6 г целевого промежуточного соединения в виде бесцветного твердого вещества. ЯМР.
D. Получение 2,4'-дикарбокси-5-метокси-бифенила.
2,5 г диоксазолина примера 8С нагревали в сосуде с обратным холодильником в течение ночи в присутствии 4,5 н. соляной кислоты. Раствор охлаждали, обрабатывали раствором гидроокиси натрия, и полученный маслянистый продукт экстрагировали этилацетатом. Органический слой промывали водой и концентрировали в вакууме, в результате чего получали 1,63 г целевого промежуточного соединения. ЯМР.
Е. Получение 2-карбокси-6-метокси-9-оксо-9Н-флуорена
Смесь 2,5 г пентоксида фосфора и 25 мл метансульфоновой кислоты размешивали в атмосфере азота в течение ночи. К этой смеси добавляли 2,3 г 2,4'-дикарбокси-5-метоксибифенила. Реакционную смесь размешивали в течение 6 ч при комнатной температуре, добавляли при этом метансульфоновую кислоту для достаточного размешивания смеси. После чего смесь нагревали до 40оС в течение 1 ч, охлаждали и выливали в ледяную воду. Полученный желто-зеленый осадок выделяли путем фильтрации, промывали водой, высушивали и получали в результате 1,6 г целевого промежуточного соединения.
F. Получение 2-этоксикарбонил-6-гидрокси-9-оксо-9Н-флуорена.
1,6 г метокси - соединения примера 8Е суспендировали в смеси 20 мл 48% бромистоводородной кислоты и 40 мл уксусной кислоты. Смесь нагревали в сосуде с обратным холодильником в течение 2 дней, затем охлаждали и выливали в лед. Полученное твердой вещество выделяли путем фильтрации, и смесь добавляли в этанол. После чего добавляли серную кислоту и смесь нагревали в сосуде с обратным холодильником. Через три дня смесь охлаждали и добавляли равное количество воды. Затем смесь экстрагировали этилацетатом. Объединенные органические экстракты промывали водой, высушивали и концентрировали в вакууме. Остаток эстрагировали этилацетатом, добавляли 5 г двуокиси кремния, и полученную смесь концентрировали в вакууме. Насыщенный диоксид кремния загружали в колонку с двуокисью кремния и элюировали 50 % этилацетата в гексан. Соответствующие фракции объединяли и концентрировали в вакууме, ЯРМ показал присутствие целевого продукта и около 20% сложного метокси-эфира. Выход составил 1,3 г.
G. Получение 2-этоксикарбонил-6-аллилокси-9-оксо-9Н-флуорена.
1,3 г фенола примера 8F растворяли в 25 мл диметилформамида. К полученному раствору добавляли 210 мг 60% гидрида натрия в масле. После размешивания смеси в течение 1 ч, добавляли 0,46 мл аллилбромида. Полученную смесь нагревали до 60оС. Через 1 ч смесь охлаждали и выливали в 1 н. соляную кислоту со льдом. Полученный ярко-желтый осадок выделяли путем фильтрации, промывали водой и высушивали, в результате чего получали 1,58 г целевого продукта, который содержал небольшое количество метоксипримеси из предыдущей реакции (как было установлено с помощью ЯМР-анализа). Полученный продукт непосредственно использовали в следующей стадии.
Н. Получение 2-аллил-3-гидрокси-7-этоксикарбонил-9-оксо-9Н-флуорена.
1,57 г продукта примера 8G растворяли в 25 мл диэтилаланилина и нагревали в течение 12 ч при 200-210оС. После охлаждения к смеси добавляли этилацетат. Органический слой промывали разбавленной соляной кислотой и концентрировали в вакууме. Остаток очищали с помощью жидкостной хроматографии высокого давления на силикагеле, используя технику градиентного элюирования (15-40% этилацетата в гексане). Соответствующие фракции объединяли и получали продукт, который был идентифицирован как целевое соединение.
1. Получение 2-аллил-3-децилокси-7-этоксикарбонил-9-оксо-9Н-флуорена
К 280 мг фенольного соединения примера 8Н в 25 мл диметилформамида добавляли 40 мг 60% гидрида натрия в масле. После размешивания в течение 30 минут при комнатной температуре добавляли децилиодид (0,21 мл). Затем реакционную смесь нагревали до 60оС и размешивали в течение ночи, после чего охлаждали и выливали в лед. Затем добавляли воду и этилацетат и слои разделяли. Органический слой дважды промывали водой, высушивали сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали желтый маслянистый продукт, который при выдерживании медленно отверждался. Выход составлял 348 мг. ЯМР.
I. Получение 2-(3-гидроксипропил)-3-децилокси-7-этокси-карбонил -9-оксо-9Н-флуорена
К 100 мг аллилового промежуточного соединения примера 1L в 15 мл тетрагидрофурана в атмосфере азота добавляли 0,93 мл 0,5 М раствора 9-борабицикло [3,3,1] нонана в тетрагидрофуране. После размешивания в течение ночи, добавляли 3,9 мл 3 М раствора ацетата натрия. Затем, после энергичного размешивания добавляли 0,72 мл 30% раствора перекиси водорода. После размешивания в течение 3 ч, слои разделяли. Органический слой промывали водой, высушивали сульфатом магния, фильтровали и концентрировали в вакууме. Полученный маслянистый остаток очищали с помощью тонкослойной хроматографии, элюируя 30% этилацетат в гексане. После выделения соответствующего слоя получали 62 мг целевого промежуточного соединения. ЯМР.
К. Получение 7-этоксикарбонил-3-децилокси-9-оксо-9Н- флуорен-2-пропионовой кислоты.
Раствор 170 мг 2-(3-гидроксипропил)-3-децилокси-7-этоксикарбонил -9-оксо-9Н-флуорена в диэтиловом эфире обрабатывали 0,75 мл реагента Джонса (раствор хромовой кислоты). Реакционную смесь перемешивали в течение ночи, затем выливали в воду и слои отделяли. Органический слой промывали водой, высушивали сульфатом магния, фильтровали и концентрировали в вакууме, в результате чего получали 90 мг продукта, который непосредственно использовали для гидролиза. ЯМР.
1. Получение 7-карбокси-3-децилокси-9-оксо-9Н-флуорен-2- пропионовой кислоты.
90 мл продукта, описанного выше примера 8К растворяли в 2 мл метаноле, содержащего две капли воды. К раствору добавляли 22,4 мг гидроокиси калия. Раствор размешивали в течение 4 ч, после чего растворитель удаляли выпариванием, а к твердому остатку добавляли воду. Водный слой промывали этилацетатом, а затем обрабатывали концентрированной соляной кислотой для доведения рН приблизительно до 1. Полученный желтый твердый остаток выделяли путем фильтрации. После перекристаллизации из этилацетата получали 51 мг целевого продукта, т. пл. 218-220оС.
Вычислено, % : С 71,66; Н 7,13.
С27Н32О6
Найдено, % : С 73,19; Н 6,99.
Соединения формулы 1 могут быть использованы для лечения заболеваний, связанных с избыточным высвобождением лейкотриена В4. Такими заболеваниями являются аллергические реакции немедленного типа. В соответствии с этим, следующим предметом настоящего изобретения является способ лечения аллергических состояний немедленного типа, таких как воспаления или астма, который заключается во введении эффективного количества соединения формулы 1.
Термин "избыточное высвобождение" лейкотриена В4 относится к количеству лейкотриена, достаточному для того, чтобы вызвать опрделенное состояние, ассоциируемое с указанным количеством. Количество лейкотриена, которое может рассматриваться как избыточное, зависит от ряда факторов, включая количество лейкотриена, требуемое для стимулирования определенного состояния, и тип млекопитающего. В связи с этим следует отметить, что успех в лечении млекопитающего, страдающего аллергическими состояниями, связанными с избыточным высвобождением лейкотриена, с помощью введения соединения формулы 1 может быть оценен посредством регресса или предупреждения симптомов указанного состояния.
Ингибирование связывания 3Н - L ТВ4 с периферическими нейтрофилами человека.
Эффективность соединений в ингибировании связывания лейкотриена В4 со специфическим рецептором на мембране человеческих нейтрофилов измерялась путем использования анализа, являющегося адаптацией метода связывания радиоактивного лиганда, разработанного Coldman и Goetz1, J. Immunol, 129, 1600 (1982). Аналогичные методы были разработаны и другими исследователями см. , например, Kneisle, и др. , J. Exp. Med, 157, 628, (1983) и Liu, и др. , Prostaglandis, 28, 937, (1984).
Клетки, использованные в анализе, выделяли с помощью стандартной техники центрифугирования на Fi coll-Hypaque, седиментации декстраном 70 и гипотонического лизиса. При этом использовали следующую методику. Из местного донорского пункта получали свежеприготовленные лейкоцитарные пленки от двух индивидуумов. Эти клетки смешивали и разбавляли до 484 мл фосфатно-буферным раствором, содержащим гепарин (10 ед/мл) и термо-инактивированную сыворотку теленка (5% ). Полученную смесь разделяли на 20 мл аликвоты, которые наносили слоями на верхнюю часть Fi. coll-Paque (12 мл). После чего полученный материал центрифугировали при 500 г в течение 40 мин при комнатной температуре. Полученный верхний слой тромбоцитов и мононуклеарных клеток отбрасывали, а нижний слой, содержащий эритроциты и нейтрофилы оставляли. После чего добавляли буфер (1 мл на 4 мл нижнего слоя) и суспензию смешивали. На каждый миллилитр этой смеси добавляли 0,33 мл 6% Макродекса. После размешивания клетки оставляли на 1 ч при 37оС для осаждения. Полученный осадок эритроцитов отбрасывали, а надосадочную жидкость, обогащенную нейтрофилами, центрифугировали при 500 г в течение 10 мин при 4оС. Эритроциты, еще присутствующие в полученном клеточном осадке, подвергали лизису путем инкубирования клеток с 5-8 мл охлажденной льдом дистиллированной водой в течениe 30-45 с. После чего объем разводили до 50 мл путем добавления охлажденного льдом буфера и клетки ресуспендировали. Затем суспензию центрифугировали при 300 г в течение 10 мин при 4оС. И наконец, клетки ресуспендировали в буфере для анализа при плотности клеток 2 х 107 кл/мл. Указанный буфер содержал сбалансированный солевой раствор Хэнкса и 0,1% oвальбумин (рН 7,3). В результате описанной процедуры выделения получали клеточные препараты, содержащие ≥ 90% нейтрофилов и обладающие жизнеспособностью ≥ 90% .
Анализ на связывание с радиоактивным лигандом проводили путем инкубирования нейтрофилов (1 х 107 клеток) с 0,1-0,2 нМ 3Н - L ТВ4(уд. акт. 150-220 Кюри/мМ) и испытуемого соединения (1 х 10-5 М и 1 х 10-6 М) в течение 10 мин при температуре 4оС. После чего определяли количество связанного 3Н - L ТВ4 и сравнивали со связанным количеством, полученным при отсутствии испытуемого соединения. Анализ проводили в пробирках для микроцентрифуги путем добавления сначала 10 мкл испытуемого соединения, растворенного в ДМСО, затем 20 мкл 3Н - L ТВ4, растворенных в буфере для анализа, и наконец, 500 мкл клеточной суспензии. После завершения 10-минутной инкубации, добавляли 300 мкл смеси дибутил- и динонилфталата (7: 2) и пробирки центрифугировали в микроцентрифуге в течение 2 мин. Радиоактивность связанного с клетками осадка измеряли с помощью сцинтилляционной спектроскопии. При этом были проведены коррекции на неспецифическое связывание 3H - L ТВ4. Резлуьтаты анализа представлены в таблице.
Изобретение также относится к фармацевтическим композициям, содержащим в качестве активного ингредиента соединение настоящего изобретения, определенное выше, в сочетании с фармацевтически приемлемым носителем.
Соединения или композиции изобретения могут быть введены перорально, ректально, посредством местного применения, парентерально, например, путем инъекции или путем непрерывного или перемежающегося внутриартериального вливания, в виде различных лекарственных форм, таких, как таблеток, драже, подъязычных таблеток, сашэ, крахмальных облаток, эликсиров, гелей, суспензий, аэрозолей, мазей, например, содержащих от 1 до 10 мас. % активного соединения, мягких и жестких желатиновых капсул, суппозиториев, суспензий и растворов для инъекций в физиологически приемлемой среде, и стерильных порошков, адсорбированных в материале носителя, предназначенных для приготовления инъецируемых растворов. Для указанных целей композиции настоящего изобретения могут быть изготовлены в виде одноразовых лекарственных форм, при этом каждая разовая стандартная доза предпочтительно содержит от около 5 до около 500 мг (в случае ингаляций или парентерального введения от 5 до 50 мг, и от 25 до 500 мг в случае перорального или ректального введения) соединения формулы I. Могут быть введены дозы приблизительно от 0,5 до 300 мг/кг в день, а предпочтительно от 0,5 до 20 мг/кг в день активного ингредиента, хотя совершенно очевидно, что количество соединения или соединений формулы 1, необходимое для введения пациенту, может быть определено лечащим врачом в зависимости от степени заболевания, вида соединения, и способа его введения, поэтому указанные выше предпочтительные дозы не ограничивают объем настоящего изобретения.
Композиции изобретения содержат, по меньшей мере, одно соединение формулы 1, смешанное с носителем, или разбавленное носителем, или инкапсулированное посредством приемлемого носителя в капсулу, сашэ, облатку, бумагу или другую форму, или в одноразовую форму, такую, как ампула. Носителем или разбавителем может быть твердый, полутвердый или жидкий материал, который служит связующим, наполнителем или средой для активного лекарственного компонента.
Примерами разбавителей или носителей, которые могут быть использованы в фармацевтических композициях настоящего изобретения, являются лактоза, декстроза, сахароза, сорбит, маннит, пропиленгликоль, вазелиновое масло, белый мягкий парафин, каолин, коллоидальная двуокись кремния, микрокристаллическая целлюлоза, силикат кальция, кремнозем, поливинилпирролидон, цетостеариловый спирт, крахмал, модифицированные крахмалы, аравийская камедь, фосфат кальция, какао-масло, этоксилированные сложные эфиры, арахисовое масло, масло-какао, альгинаты, трагакант, желатина, сироп, метилцеллюлоза, полиоксиэтиленсорбитанмонолаурат, этиллактат, метил- или пропилгидроксибензоат, сорбитантриолеат, сорбитансесквиолеат и олеиловый спирт, а примерами пропеллентов являются трихлоромонофторометан, дихлородифторометан и дихлоротетрафтороэтан. При изготовлении таблеток могут быть также введены замасливатели во избежание склеивания и связывания порошкообразных ингредиентов в пресс-формах и штампах таблетирующей машины. Для этих целей могут быть использованы, например, стеараты алюминия, магния или кальция, тальк или минеральное масло.
Предпочтительными фармацевтическими формами изобретения являются капсулы, таблетки, суппозитории, инъецируемые растворы, кремы и мази. Особенно предпочтительными являются препараты дли ингаляций, например, аэрозоли, и препараты для перорального введения.
Ниже приводятся примеры композиций, в которых в качестве активного ингредиента может быть использовано любое соединение настоящего изобретения. Указанные примеры лишь иллюстрируют осуществление настоящего изобретения, но не ограничивают его объема.
П р и м е р 9. Жесткие желатиновые капсулы изготавливали с использованием следующих ингредиентов.
Количество
(мг/капсула)
7-Карбокси-3-
{ [6-(4-метоксифенил)-
5-гексенил] окси} -
9-оксо-9Н- ксантен- 2-пропионовая кислота 250 Крахмал 200 Стеарат магния 10
Указанные ингредиенты смешивали и полученной смесью наполняли желатиновые капсулы в количестве 460 мг каждую.
П р и м е р 10. Таблетки изготавливали с использованием следующих ингредиентов:
Количество
(мг/таблетка)
7-Карбокси-3-
{ [6-(4-метоксифенил)-
5-гексенил] окси} -
9-оксо-9Н- флуорен- 2-пропионовая кислота 250 Микрокристаллическая целлюлоза 400 Коллоидальный диоксид кремния 10 Стеарат магния 5
Указанные компоненты смешивали и прессовали в таблетки, масса каждой из которых составляла 665 мг.
П р и м е р 11. Аэрозольный раствор изготавливали в использованием следующих компонентов:
Мас. %
7-Карбокси-3-
{ [6-(4-метилтиофенил)-
гексил] окси} -9-оксо-
9Н-флуорен - 2-пропионовая кислота 0,25 Этанол 30,00 Пропеллент 11 10,25 трихлорофторометан Пропеллент 12 29,75 дихлородифторометан Пропеллент 114 29,75
дихлоротетрафлуороктан
Активное соединение растворяли в этаноле и полученный раствор добавляли к пропелленту 11, охлаждали до -30оС, и переносили в заливочный клапан. После чего нужное количество подавали в баллон, а затем подавали смесь пропеллентов 12 и 114 путем холодного наполнения или наполнения под давлением. Затем к баллону подсоединяли клапанные устройства.
П р и м е р 12. Таблетки, каждая из которых содержала 60 мг активного ингредиента, изготавливали следующим образом:
7-Карбокси-3-
{ [6-(4-метилсульфонилфенил)-
гексил] окси} -9-оксо-
9Н-ксантен-2- пропионовая кислота 60 мг Крахмал 45 мг
Микрокристаллическая целлюлоза 35 мг Поливинилпирролидин 4 мг
10% раствор в воде
Натрийсодержащий
карбоксиметиловый крахмал 4,5 мг Стеарат магния 0,5 мг Тальк 1 мг Итого: 150 мг
Активный ингредиент, крахмал и целлюлозу пропускали через сито N 45 меш (США) и тщательно смешивали. Затем с полученными порошками смешивали раствор поливинилпирролидона и смесь пропускали через сито N 14 меш (США). Полученные таким образом гранулы высушивали при 50-60оС и пропускали через сито N 18 меш (США). После чего к полученным гранулам добавляли натрийсодержащий карбоксиметиловый крахмал, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш (США), и после смешивали, гранулы спрессовывали на таблетирующей машине, получая в результате таблетки, каждая из которых имела вес 150 мг.
П р и м е р 13. Капсулы, каждая из которых содержала 80 мг лекарственного компонента, изготавливали следующим образом: 7-Карбокси-3- (децилокси)-9-оксо- 9Н-ксантен-4- пропионовая кислота 80 мг Крахмал 59 мг Микрокристаллическая целлюлоза 59 мг Стеарат магния 2 мг Итого: 200 мг
Активный ингредиент, целлюлоза, крахмал и стеарат магния смешивали, пропускали через сито N 45 меш (США), и полученной смесью наполняли жесткие желатиновые капсулы, каждая из которых содержала 200 мг смеси.
П р и м е р 14. Суппозитории, каждый из которых содержал 225 мг активного ингредиента, изготавливали следующим образом: 7-Карбокси-3- { [6-(4-метоксифенил)- 5-гексенил] окси} - 9-оксо- 9Н-ксантен- 4-пропионовая кислота 225 мг Глицериды ненасыщенных или насыщенных жирных кислот До 2,000 мг
Активный игредиент пропускали через сито N 60 меш (США) и суспендировали в глицеридах жирных кислот, предварительно расплавленных при минимальной температуре. Затем смесь выливали в суппозиторные формы номинальной емкостью 2 г и оставляли для охлаждения.
П р и м е р 15. Суспензии, каждая из которых содержала 50 мг активного компонента на дозу 5 мл, приготавливали следующим образом: 7-Карбокси-3- { [6-(4-метоксифенил) гексил] окси} -9-оксо- 9Н- ксантен-4-пропионовая кислота 50 мг Натриевая карбоксиметилцеллюлоза 50 мг Сахар 1 мг Метилпарабен 0,05 мг Пропилпарабен 0,03 мг Ароматизатор по желанию Краситель по желанию Очищенная вода до 5 мл
Активный компонент пропускали через сито N 45 меш (США) и смешивали с натриевой карбосиметилцеллюлозой, сахаром и с частью воды для получения суспензии. Парабены, ароматизатор и краситель растворяли и разбавляли некоторым количеством воды, и, размешивая, добавляли к суспензии. Затем добавляли воду для получения нужного объема. (56) Патент США N 4945099, кл. C 07 D 457/04, 1988.

Использование: в медицине в качестве избирательных антагонистов к лейкотриену B4 и используемых при лечении воспалительных и аллергических заболеваний. Сущность изобретения: продукт соединения ф-лы 1 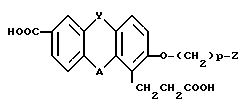 , где Y = -CO- или CH2; А - простая связь или -О-, p = 1 - 16, Z = Н или -GO, где G - простая связь или -CH = CH, а Q - фенил, замещенный C1-C3 - алкоксигруппой. Реагент 1: соед. ф-лы II
, где Y = -CO- или CH2; А - простая связь или -О-, p = 1 - 16, Z = Н или -GO, где G - простая связь или -CH = CH, а Q - фенил, замещенный C1-C3 - алкоксигруппой. Реагент 1: соед. ф-лы II  где Y, А, p и Z имеют указанные значения, R-C1-C4-алкил. Реагент 2: гидролизующий агент. Соединения формулы 1, ингибируют лейкотриен B4 в концентрации 10 -6M на 23 - 99% . 1 табл.
где Y, А, p и Z имеют указанные значения, R-C1-C4-алкил. Реагент 2: гидролизующий агент. Соединения формулы 1, ингибируют лейкотриен B4 в концентрации 10 -6M на 23 - 99% . 1 табл.
СПОСОБ ПОЛУЧЕНИЯ ТРИЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ общей формулы
где Y - CO-или - CH2-группа,
A - простая связь или O-группа,
p = 1 - 16, целое число;
Z - водород или G - Q, где G-простая связь или - CH = CH-;
Q - фенил, замещенный C1 - C3-алкоксигруппой,
отличающийся тем, что осуществляют гидролиз соединения общей формулы
где Y, A, p и Z имеют указанные значения;
R - C1 - C4-алкил.
