В течение нескольких последних лет стало очевидным, что серотонин-(5-окситриптамин, сокращенно 5-НТ), высвобождаемый пресинаптическими клетками, прямо или косвенно связан с некоторым рядом физиологических явлений, включающих аппетит, память, депрессию, терморегуляцию, сон, сексуальное поведение, боязнь и галлюциногенное поведение (Glennon, R. A. J. Med. Chem. 30, 1 (1987).
Было установлено существование множественных типов 5-НТ-рецепторов. Эти рецепторы были классифицированы как 5-НТ1-, 5-НТ2 и 5-НТ3-рецепторы, при этом первый из названных рецепторов далее разделен на подклассы 5-НТ1A, 5-НТ1B, 5-НТ1C и 5-НТ1D.
Было показано, что некоторые 2-амино-1,2,3,4-тетра-гидронафталины (2- аминотетралины) и 3-аминохроманы обладают связывающей способностью 5-НТ1A-рецептора. Заявка на европейский патент N 385658, опубликованная 9 сентября 1990 г. описывает 2-аминотетралины, замещенные в 8-м положении, и 3-аминохроманы, замещенные в 5-м положении сульфидами, сульфоксидами и сульфонами. Эти соединения также описаны как обладающие способностью связывания 5-НТ1A-рецептора. Другой класс 2-аминотетралинов описан в заявке на европейский патент N 343830, опубликованной 29 ноября 1989 г. Эти соединения имеют пиперазиниловую или гомопиперазиниловую часть молекулы во втором положении и в отличие от предыдущих тетралинов проявляют ингибирование повторного усвоения серотонина в противовес к связывающей способности рецептора серотонина. В заявке на европейский патент N 399982, опубликованной 28 ноября 1990 г. описаны 2-аминотетралины, имеющие среди других заместителей в 8-м положении 5- или 6-членный арил, который может содержать один или два гетероатома, выбранных среди азота, кислорода или серы. В международной заявке по договору о патентной кооперации N 090/15047, опубликованной 13 декабря 1990 г. описаны 2-аминотетралины, замещенные в любом из положений 5, 6, 7 или 8 среди прочих и заместителем Het. Het описывается как пятиатомное гетероциклическое кольцо, содержащее азот, углерод и в некоторых случаях кислород.
В настоящее время открыт класс соединений, которые благодаря их исключительной 5-НТ1A-активности очень полезны при лечении, например, расстройств половой функции, страха, депрессии и нарушений питания, таких, как отсутствие аппетита.
Изобретение предусматривает новые промежуточные, используемые для получения замещенных в кольце 2-амино-1,2,3,4-тетрагидронафталинов и 3-аминохроманов, имеющих в 8-м положении тетрагидронафталина и в 5-м положении хромана определенный изоксазол-3-ильный или изоксазол-5-ильный заместитель.
Соединения проявляют частично сродство и антагонизм в отношении 5-НТ1A-рецептора.
Более конкретно, это изобретение касается соединения формулы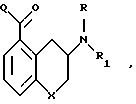
в которой R представляет C1-C4-алкил;
R1 C1-C4-алкил;
X является -CH2- или -O-;
Q означает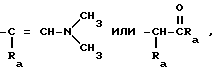
где каждый Ra означает независимо водород C1-C4 алкил или C1-4 алкокси.
В приведенных выше формулах термин "C1-C4-алкил" означает прямую или разветвленную алкильную цепь, имеющую от одного до четырех углеродных атомов. Такими C1-C4-алкильными группами являются метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил и трет-бутил.
Соединение изобретения используется для получения соединений, содержащих ароматические гетероциклические кольца, имеющие формулу
Предпочтительно ароматическое гетероциклическое кольцо является незамещенным. Однако кольцо может содержать водород, присутствующий на одном или обоих атомах углерода в кольцевой структуре замещенной любой из групп, выбранных из C1-C4-алкила, C1-C4-алкокси, C1-C4-тиоалкила, окси, амино, циано или фенила.
Множественные замещения включены в объем изобретения. Так, оба углеродных атома могут быть замещены, как описано ранее. Однако предпочтительно, если имеет место замещение в кольце, когда кольцо является однозамещенным.
Термин "C1-C4-алкил" имеет значения, определенные ранее.
Термин "C1-C4-алкокси" означает любую из группы, включающей заместители метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси. Термин "C1-C4-тиоалкил" означает любую из метилтио-, этилтио-, н-пропилтио-, изопропилтио-, н-бутилтио-, изобутилтио-, вторбутилтио и трет-бутилтиогруппу.
Хотя все соединения согласно изобретению полезны в качестве промежуточных для получения соединений, используемых для лечения различных расстройств, благодаря их способности модулировать функцию 5-НТ1A-рецептора у млекопитающих, однако некоторые соединения являются предпочтительными.
Так, R и R1 предпочтительно являются оба C1-C4-алкилом и более предпочтительно оба являются н-пропилом.
Следующие ниже соединения характеризуют далее соединения, входящие в объем изобретения:
2-этиламино-8-(1-оксо-3-(диметиламино)-проп-2-енил)-1,2,3,4- тетрагидронафталин,
2-(ди-н-пропиламино)-8-(1-оксо-2-метокси-3-(диметиламино)-проп- 2-енил)-1,2,3,4-тетрагидронафталин,
3-(дин-пропиламино)-5-(трет-бутоксикарбонилацетил)-хроман,
3-(ди-н-пропиламино)-5-(1-оксо-2-метил-3-(диметиламино)-проп-2- енил)-хроман и т.д.
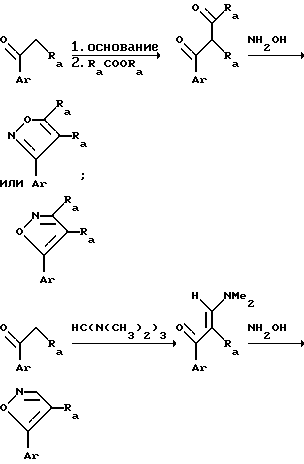
Соединения согласно изобретению могут быть получены способами, широко известными специалистам в данной области. Соединения, в которых X представляет собой -CH2-, могут быть синтезированы через 8-бром-2-тетралон. 8-Бром-тетралон затем подвергают восстановительному аминированию желаемым амином с получением желаемого промежуточного соединения 2-амино-8-бромтетралона. Промежуточное 8-бромсоединение затем используют в разнообразных реакциях для получения соединения согласно изобретению.
Соединения согласно изобретению, в которых X представляет собой кислород, получают как описано выше, но используя 5-замещенный 3-хроманон. Эта молекула может быть получена последовательностью реакций, начиная с мета-бромфенола. Коротко, мета-бромфенол обрабатывают бромистым аллилом в присутствии карбоната калия с получением простого аллил-3-бромфенилового эфира. Эфир обращают в 2-аллил-3-бромфенол нагреванием в присутствии N,N-диметиланилина. Фенол в реакции с этиловым эфиром хлоруксусной кислоты обращают в этиловый эфир 2-аллил-3-(карбоксиметокси)-бромбензола. При окислении с использованием озона и последующей обработкой в восстановительных условиях аллильную группу обращают в фомил-метильный заместитель, который затем окисляют далее с использованием реактива Джонеса в карбоксиметильный заместитель, при этом полученным продуктом является этиловый эфир (2-карбоксиметил-3-бром)-феноксиуксусной кислоты. Группу карбоновой кислоты этого соединения этерифицируют трет-бутилацетатом и концентрированной серной кислотой с получением этилового эфира 3-бром-2-(карбо-трет-бутоксиметил)-феноксиуксусной кислоты. В присутствии трет- бутилата калия сложный диэфир циклизуется с образованием 4-трет-бутоксикарбонил-5-бром-3-хроманона. При перемешивании при комнатной температуре в присутствии кислоты последний превращается в 5-бром-3-хроманон.
Соединения согласно изобретению могут быть получены рядом общих реакций. Общие схемы реакций представлены ниже. В них группы Ra и Rc имеют следующие значения:
Ra представляет собой водород, C1-C4-алкил; O(C1-C4-алкил);
Rc водород или C1-C4-алкил.
Соединения согласно изобретению имеют асимметрический углерод, представленный атомом углерода, помеченным звездочкой в следующей формуле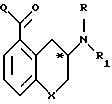
Таким образом каждое из соединений существует в форме отдельных d и l-стереоиэомеров, а также как рацемическая смесь таких изомеров.
Такие оптически активные изомеры могут быть получены из своих соответствующих оптически активных предшественников.
Один особенно полезный метод получения оптически активных изомеров осуществляют согласно изобретению через 8-замещенный-2-тетралон или 5-замещенный-З-хроманон. Любой из этих промежуточных продуктов может быть восстановительно алкилирован оптически активным α -фенетиламином, после чего полученную смесь диастереомеров разделяют известным методом, таким, как хроматография. Отщепление a -фенетильной части молекулы дает соответственно замещенный, оптически активный 2-амино-1,2,3,4-тетрагидронафталин или З-аминохроман. Условия, необходимые для удаления фенетильной части, относительно жесткие и могут привести к нарушению целостности ядра молекулы тетралина и хромана. Было установлено, что отщепление можно провести более легким и эффективным способом, требующим только мягких условий отщепления, если конкретным используемым a фенетиламином является пара-нитро- a -фенетиламин.
Отщепление пара-нитро- a -фенетильной части молекулы достигают восстановлением пара-нитрогруппы с последующим кислотно катализируемым сольволизом полученной пара-амино- a -фенетильной части молекулы. Восстановление нитрогруппы может быть осуществлено посредством многочисленных восстановителей, включая, например, треххлористый титан, алюмогидрид лития или смесь цинк-уксусная кислота, или каталитической гидрогенизацией. Сольволитическое отщепление имеет место, если хлористоводородная соль (или другая одноосновная соль) продукта восстановления обрабатываются водой или спиртом при комнатной температуре или в некоторых случаях при повышенных температурах. Особенно подходящим условием для удаления пара-нитро- a -фенетильной части является гидрогенизация монохлорида амина в метаноле над платиновым катализатором.
Как указывалось выше, соединениями, весьма полезными в качестве промежуточных соединений для получения соединений согласно изобретению, являются соответствующие 8-бромтетралины. Было установлено, что 8-бромсоединения в своей оптически активной форме невозможно получить, используя традиционную методологию, тогда как они могут быть получены, используя метод, описанный выше, с применением пара-нитро-фенетиламина.
Соединения, применяемые в качестве начальных исходных материалов в синтезе соединений согласно изобретению, широко известны и легко синтезируются стандартными методами, обычно применяемыми специалистами в данной области.
Эти примеры не расцениваются как ограничивающие объем изобретения ни в каком аспекте.
Пример 1. 2-ди-н-пропиламино-8-ацетил-1.2,3,4-тетрагидронафталин.
Раствор н-бутиллития (1,6 М в гексане, 15,1 мл 24,2 ммоля) добавляли к раствору 8-бром-2-ди-н-пропиламино-1,2,3,4-тетрагидронафталина (5,0 г, 16,1 ммоля) в тетрагидрофуране (50 мл) при -78oC, и реакционную, смесь перемешивали при -78oC 1 ч. Через реакционную смесь барботировали газообразную двуокись углерода при -78oC до исчезновения образовавшегося темно-фиолетового окрашивания. Добавляли метил-литий (1,4 М в эфире, 23 мл). Реакционную смесь перемешивали при -78oC -в течение 30 мин и нагревали до комнатной температуры. Реакцию перемешивали еще дополнительные 10 мин при комнатной температуре, и за это время утрачивалось розовое окрашивание. Добавляли дополнительные 10 мл раствора метиллития, и реакционная смесь снова приобретала розовую окраску. Через 15 мин розовая окраска исчезала и добавляли дополнительные 10 мл раствора метиллития. Реакционную смесь выливали на лед, подкисляли хлористоводородной кислотой и экстрагировали эфиром. Водный слой подщелачивали и экстрагировали хлористым метиленом. Основные экстракты сушили (сульфат натрия), концентрировали и получали 3,8 г продукта. Очисткой флэшхроматографией на силикагеле с использованием смеси гексан-эфир в отношении 2: 1, содержащей следы гидроокиси аммония, получали 2-ди-н-пропиламино-8-ацетил-1,2,3,4-тетрагидронафталин в виде желтого масла (2,7 г, 61%).
Пример 2. Получение 2-ди-н-пропиламино-8-(трет-бутоксикарбонил-ацетил)- 1,2,3,4-тетрагидронафталина.
К раствору 8-бром-2-ди-н-пропиламино-1,2,3,4-тетрагидро-нафталина (1,0 г, 3,22 ммоля) в тетрагидрофуране (50 мл) при -78oC добавляли раствор н-бутиллития в гексане (1,1 М, 4,4 мл, 1,5 эквивалента). Реакционную смесь перемешивали при -78oC 1 ч и через реакционную смесь барботировали газообразную двуокись углерода. Полученную смесь нагревали до комнатной температуры. После удаления летучих компонентов из реакционной смеси коричневое масло выливали в воду и промывали эфиром. Органическую фазу сливали, а водный слой концентрировали и растворяли в метаноле. Через раствор пропускали газообразный хлористый водород и реакционную смесь нагревали с обратным холодильником 3 ч. После охлаждения реакционную смесь выливали в воду (50 мл), подщелачивали с использованием кислого углекислого натрия (водный) и экстрагировали эфиром. Эфирный экстракт сушили над сульфатом магния, концентрировали и получали 1,0 г черного масла. Очисткой флэшхроматографией через колонку, элюируя смесью гексан-этилацетат в отношении 4:1, получали 440 мг 2-ди-н-пропиламино-8-(метоксикарбонил)-1,2,3,4-тетрагидронафталина.
Раствор LDA готовили из 17 ммолей 2,42 мл диизопропиламина и 17 ммолей (17 мл 1,0 М) н-бутиллития при -78oC. LDA-раствор нагревали до -20oC за 30 мин и снова охлаждали до -78oC перед добавлением 2,83 мл (20,96 ммолей) трет-бутилацетата. Через 10 минуют добавляли 440 мг (1,47 ммоля) 2-ди-н-пропиламино-8-метоксикарбонил- 1,2,3,4-тетрагидронафталина в 20 мл сухого тетрагидрофурана. Реакционную смесь нагревали до комнатной температуры и перемешивали три дня.
Полученную смесь выливали в воду (50 мл) и экстрагировали дихлорметаном (три раза по 50 мл). Экстракт сушили (сульфат магния), концентрировали и получали 1,8 г масла. Очистка тонкослойной колоночной хроматографией с элюированием 10%-ным метанолом в дихлорметане давала 160 мг 2-ди-пропиламино-8-(трет-бутоксикарбонилацетил)-1,2,3,4- тетрагидронафталин в виде желтого масла.
Пример 3. Получение соли малеиновой кислоты 2-ди-н-пропилами-но-8-(изоксазол-5-ил)-1,2,3,4-тетрагидронафталина.
Раствор 2-ди-н-пропиламино-8-ацетил-1,2,3,4-тетрагидронафталина (0,3 г, 1,1 ммоля), полученный как описано в примере 1, и трис-(диметиламино)-метана (0,32 г, 2,2 ммоля) в толуоле нагревали с обратным холодильником 5 ч и при температуре 60oC в течение 18 ч. Добавляли дополнительное аликвотное количество трис-(диметиламино)-метана (0,16 г, 1,1 ммоля) и реакционную смесь перемешивали при 60oC дополнительные 2 ч. Реакционную смесь концентрировали и получали 2-ди-н-пропиламино-8-[1-оксо-3- (диметиламино)-проп-2-енил] -1,2,3,4-тетрагидронафталин (0,39 г) в виде вязкого оранжевого масла.
Гидрохлорид гидроксиламина (0,32 г, 4,6 ммоля) добавляли к раствору 2-ди-н-пропиламино-8-[1-оксо-3-(диметиламино)-проп-2-енил]- 1,2,3,4-тетрагидронафталина (0,75 г, 2,29 ммоля) в уксусной кислоте (5 мл), и реакционную смесь перемешивали при комнатной температуре. Реакционную смесь концентрировали и остаток растворяли в воде. Раствор делали основным добавлением концентрированного раствора хлористого аммония и экстрагировали эфиром. Экстракт промывали рассолом, сушили сульфатом натрия, концентрировали и получали вязкое светло-оранжевое масло. Образовалась соль малеиновой кислоты. Кристаллизацией из смеси этанол-эфир получали целевое соединение, указанное в названии примера, в виде не совсем белых кристаллов (0,24 г), точка плавления 136-138oC. Перекристаллизацией этой соли из этанола получали бесцветные кристаллы (155 мг), точка плавления 139-141oC.
Элементный анализ:
Вычислено, C 66,65, H 7,29, N 6,76
Найдено, C 66,86; H 7,33; N 6,79.
Пример 4. 2-ди-н-пропиламино-8-(5-гидрооксиизоксазол-З-ил)-1,2,3,4- тетрагидронафталин.
2-ди-н-пропиламино-8-(трат-бутоксикарбонилацетил)-1,2,3,4- тетрагидронафталин (полученный как описано в примере 2), (1,0 г, 3,3 ммоля) растворяли в 25 мл метанола. Добавляли 10 эквивалентов гидрохлорида гидроксиламина (8,3 г, 33 ммоля) и реакционную смесь перемешивали при комнатной температуре 48 ч. Раствор фильтровали для удаления неиспользованного гидрохлорида гидроксиламина. Смесь концентрировали и три раза перекристаллизовывали из смеси метанол-этилацетат. Выделяли 30 мг целевого соединения.
Анализ масс-спектроскопии (РД) показал уточненную массу 314.
Пример 5. Получение соли малеиновой кислоты 2-ди-н-пропиламино-8- (4-метилизоксазол-5-ил)-1,2,3,4-тетрагидронафталина.
A) Получение 2-ди-н-пропиламино-8-пропионил-1,2,3,4-тетрагидронафталина.
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (8,5 г, 27,4 ммоля) растворяли в 80 мл тетрагидрофурана и охлаждали до -78oC, после чего добавляли 25,7 мл н-бутиллития (1,6 М в гексане). Смесь перемешивали при -78oC один час, после чего добавляли 2,4 мл (32,9 ммоля) пропиональде-гида. Смесь нагревали до комнатной температуры, затем выливали в воду и экстрагировали хлористым метиленом. Экстракт сушили над сульфатом натрия, выпаривали и получали 9,1 г желтого масла.
Масло вводили в колонку с силикагелем и элюировали смесью 3%-ного метанола в хлористом метилене, содержащем следы гидроокиси аммония. Нужные фракции объединяли и получали 6,5 г (82%) 2-ди-н-пропиламино-8-(1-оксипропил)-1,2,3,4-тетрагидронафталина в виде прозрачного масла.
Полученный выше продукт растворяли в 250 мл хлористого метилена и добавляли 17,0 г (78,7 ммоля) хлорхромата пиридиния с молекулярными ситами  Смесь перемешивали 3 ч при комнатной температуре, после чего добавляли 250 мл эфира и целит. Смесь вводили в короткую колонку силикагеля и элюировали эфиром. Добавляли метанол, чтобы растворить коричневый шлам, который выпал в осадок при добавлении эфира к реакционной смеси. Этот материал вводили в колонку и элюировали 10%-ным метанолом в хлористом метилене. Элюент концентрировали и получали коричневое масло, которое очищали далее колоночной хроматографией, используя в качестве растворителя смесь гексан-эфир в отношении 2:1, затем чистый эфир. Фракции, содержащие продукт, объединяли, концентрировали и получали 4,7 г 2-ди-н-пропиламино-8- пропионил-1,2,3,4-тетрагидронафталина.
Смесь перемешивали 3 ч при комнатной температуре, после чего добавляли 250 мл эфира и целит. Смесь вводили в короткую колонку силикагеля и элюировали эфиром. Добавляли метанол, чтобы растворить коричневый шлам, который выпал в осадок при добавлении эфира к реакционной смеси. Этот материал вводили в колонку и элюировали 10%-ным метанолом в хлористом метилене. Элюент концентрировали и получали коричневое масло, которое очищали далее колоночной хроматографией, используя в качестве растворителя смесь гексан-эфир в отношении 2:1, затем чистый эфир. Фракции, содержащие продукт, объединяли, концентрировали и получали 4,7 г 2-ди-н-пропиламино-8- пропионил-1,2,3,4-тетрагидронафталина.
B) Образование кольца 4-метил-изоксазол-5-ил.
2-Ди-Н-пропиламино-8-пропионил-1,2,3,4-тетрагидронафталин (1,5 г, 5,2 ммоля) растворяли в 50 мл толуола и добавляли 2,2 мл трис-(диметиламино)- метана. Смесь нагревали при 80oC, оставляя на ночь. Затем смесь выпаривали и остаток растворяли в уксусной кислоте. Добавляли гидрохлорид гидроксиламина (730 мг, 10,4 ммоля) и смесь оставляли на ночь при помешивании при комнатной температуре. Смесь выливали в воду, устанавливали pH 11 гидроокисью аммония и полученную смесь экстрагировали хлористым метиленом. Экстракт сушили над сульфатом натрия, выпаривали и получали 1,5 г оранжевого масла.
Масло помещали в колонку и элюировали смесью гексан-эфир в отношении 2: 1, при этом эфир содержал следы гидроокиси аммония. Подходящие фракции объединяли и получали 1,0 г (61,3%) свободного основания целевого соединения.
50 мг свободного основания превращали в соль малеиновой кислоты, перекристаллизовывали из смеси этанол-эфир и получали 55 мг белых кристаллов, точка плавления 118oC.
Элементный анализ для C24 H32N2O5.
Вычислено, C 67,27; H 7,53; N 6,54
Найдено, C 66,99; H 7,60; N 6,35.
Пример 6. Получение 2-ди-н-пропиламино-8-(4-этилизоксазол-5-ил)- 1,2,3,4-тетрагидронафталина.
A) Получение 2-ди-н-пропиламино-8-бутирил-1,2,3,4-тетрагидронафталина.
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (5,0 г, 16,1 ммоля) растворяли в 50 мл тетрагидрофурана и смесь охлаждали до -78oC, после чего добавляли 21,0 мл н-бутиллития (0,92 М в гексане). Смесь перемешивали 30 мин и добавляли 1,85 мл (21,0 ммоль) масляного альдегида. Смесь нагревали до комнатной температуры и оставляли на ночь при перемешивании, после чего ее выливали в воду и экстрагировали хлористым метиленом. Экстракт сушили над сульфатом натрия, выпаривали и получали 6,4 г остатка. Остаток вводили в колонну с силикагелем и элюировали смесью 2%-ного метанола в хлористом метилене, содержащем следы гидроокиси аммония. Подходящие фракции объединяли и получали 4,8 г 2-ди-н-пропил-амино-8-(1'-гидроксибутил) -1,2,3,4-тетрагидронафталина в виде вязкого масла.
Масло (4,0 г, 13,2 ммоля) растворяли в 200 мл хлористого метилена и добавляли молекулярные сита  (30 г). Смесь перемешивали и добавляли 10,0 г (46,2 ммоля) хлорхромата пиридиния; перемешивание продолжали 3 ч при комнатной температуре, после чего смесь выливали в слой силикагеля и последовательно элюировали эфиром и 3%-ным метанолом в хлористом метилене, содержащем следы гидроокиси аммония и выделяли продукт в виде коричневого масла.
(30 г). Смесь перемешивали и добавляли 10,0 г (46,2 ммоля) хлорхромата пиридиния; перемешивание продолжали 3 ч при комнатной температуре, после чего смесь выливали в слой силикагеля и последовательно элюировали эфиром и 3%-ным метанолом в хлористом метилене, содержащем следы гидроокиси аммония и выделяли продукт в виде коричневого масла.
Масло помещали в колонку силикагеля и элюировали смесью 3%-ного метанола в хлористом метилене, содержащем следы гидроокиси аммония. Подходящие фракции объединяли и получали масло, которое, будучи растворено в эфире, вызывало образование коричневого осадка. Осадок удаляли фильтрацией, фильтрат выпаривали и получали 3,0 г 2-ди-н-пропиламино-8-бути-рил-1,2,3,4-тетрагидронафталина в виде светло-коричневого масла.
B) Образование кольца 4-этил-изоксазол-5-ил.
Трет-бутилат натрия (0,82 г, 7,3 ммоля) суспендировали в 100 мл тетрагидрофурана. К смеси добавляли этиловый эфир муравьиной кислоты (1,0 г, 13,3 ммоля) и 2-ди-н-пропилами-но-8-бутирил-1,2,3,4-тетрагидронафталин (1,0 г, 3,3 ммоля). Полученную смесь оставляли на ночь при перемешивании при комнатной температуре. Добавляли гидроксиламин (1,2 г, 16,6 ммоля) с последующим добавлением достаточного количества воды для растворения твердого вещества. Полученную смесь, имеющую pH 6,0, перемешивали при комнатной температуре 20 ч, после чего ее выливали в воду и устанавливали pH 12 гидроокисью аммония. Затем смесь экстрагировали хлористым метиленом. Экстракт сушили над сульфатом натрия и выпаривали. Остаток растворяли в 100 мл толуола и добавляли 100 мг пара-толуолсульфокислоты. Затем смесь нагревали с обратным холодильником 1,5 ч после чего выливали в воду и экстрагировали хлористым метиленом. Экстракт хлористого метилена сушили над сульфатом натрия и выпаривали.
Остаток вводили в колонку с силикагелем и элюировали смесью гексан-эфир в отношении 2:1, при этом эфир содержал следы гидроокиси аммония. Подходящие фракции собирали и получали 0,9 г целевого соединения, указанного в названии примера. Масс-спектр (FD): 327 (100).
Пример 7. Получение соли малеиновой кислоты 2-дин-пропиламина-8- (3-метили зоксазол-5-ил)-1,2,3,4-тетрагидронафталина.
Трет-бутилат калия (450 мг, 4,0 ммоля) суспендировали в тетрагидрофуране и добавляли 0,7 мл (7,3 ммоля этилацетат и 0,5 г (1,8 ммоля) 2-ди-н-пропиламино-8-ацетил-1,2, 3,4-тетрагидронафталина (полученного как описано в примере 5) в тетрагидрофуране. Общее количество использованного тетрагидрофурана составляло 30 мл. Затем смесь оставляли при перемешивании при комнатной температуре на ночь, после чего добавляли 640 мг (9,2 ммоля) гидрохлорида гидроксиламина. Затем реакционную смесь перемешивали при комнатной температуре 64 ч. Смесь выливали в воду и устанавливали pH 12 гидроокисью аммония. Затем смесь экстрагировали смесью хлороформа и изопропилового спирта в отношении 3:1. Экстракт сушили над сульфатом натрия, выпаривали и получали 450 мг твердого вещества. Твердое вещество растворяли в толуоле, добавляли небольшое количество пара-толуолсульфокислоты и смесь нагревали с обратным холодильником 2 ч. Затем смесь выливали в воду, устанавливали pH 12 гидроокисью аммония и смесь экстрагировали хлористым метиленом. Экстракт хлористого метилена сушили над сульфатом натрия, выпаривали и получали 390 мг коричневого масла.
Масло вводили в колонку с кремнеземом и элюировали смесью 2%-ного метанола в хлористом метилене, содержащем следы гидроокиси аммония. Нужные фракции собирали и получали 210 мг (35% свободного основания целого соединения, указанного в названии примера}
Соединение обращали в соль малеиновой кислоты, которую перекристаллизовывали из смеси этанола и эфира и получали 200 мг целевого соединения, точка плавления 125,5-127,5oC. Масс-спектр (FD): 313 (100).
Вычислено, C 67,27; H 7,53; N 6,54
Элементный анализ для C24H31N2O5
Найдено, С 67,52; Н 7,29; Н 6,48.
Пример 8. Получение гидробромида 2-ди-н-пропиламино-8-(4-мето- ксиизоксазол-5-ил)-1,2,3,4-тетрагидронафталина.
2-Ди-н-пропиламино-8-бром-1,2,3,4-тетрагидронафталин (5,0 г, 16,1 ммоля) растворяли в 25 мл тетрагидрофурана и охлаждали до -78oC, после чего добавляли 3,22 мл н-бутиллития (1 М в гексане). Смесь выдерживали при -78oC полтора часа. Полученный раствор переносили посредством канюли в раствор метилового эфира метоксиуксусной кислоты (7,5 мл, 160 ммолей) в тетрагидрофуран при -78oC. Реакционную смесь оставляли при перемешивании при комнатной температуре на ночь, выливали в раствор кислого углекислого натрия и экстрагировали хлороформом. Экстракт сушили (сульфат натрия), концентрировали и получали 6,3 г неочищенного продукта.
Затем материал помещали в колонку для хроматографии и элюировали, используя 4%-ный метанол в хлористом метилене, содержащем следы гидроокиси аммония. Подходящие фракции объединяли и получали 1,4 г 2-ди-н-пропиламино-8-метоксиацетил-1,2,3,4-тетрагидронафталина.
Раствор 2-ди-н-пропиламино-8-метоксиацетил-1,2,3,4-тетрагидронафталина (1,0 г) и трис-(диметиламино)-метана (1,5 мл) в толуоле (25 мл) нагревали с обратным холодильником 1,5 ч. Реакционную смесь концентрировали и получали неочищенный 2-ди-н-пропиламино-8-(1-оксо-2-метокси-3- (диметиламино)-проп-2-енил)-1,2,3,4-тетрагидронафталин (1,2 г).
К раствору 2-ди-н-пропиламино-8-(1-оксо-2-метокси-3-(диметиламино)- проп-2-енил)-1,2,3,4-тетрагидронафталина (1,1 г) в метаноле добавляли гидрохлорид гидроксиламина (1,2 г) и реакционную смесь оставляли на ночь при перемешивании при комнатной температуре. Реакционную смесь концентрировали и остаток растворяли в толуоле. Добавляли пара-толуолсульфокислоту (660 мг) к полученному раствору и реакционную смесь нагревали с обратным холодильником 2 ч. Реакционную смесь концентрировали, и остаток растворяли в смеси воды и хлористого метилена. Эту смесь выливали в раствор бикарбоната натрия и полученную смесь экстрагировали хлористым метиленом. Экстракт сушили сульфатом магния, концентрировали и получали масло (600 мг). Очисткой тонкослойной хроматографией с использованием в качестве растворителя смеси эфир-гексан в отношении 1: 1 получали 160 мг свободного основания целевого соединения, указанного в названии примера. Соль хлористоводородной кислоты затем получали. Перекристаллизация из смеси метанол-эфир давала целевое соединение в виде белых кристаллов (86 мг). Т.пл. 178oC.
Вычислено, C 58,68; H 7,14 N 6,84
Найдено, C 58,88; H 7,23; N 6,60.
Пример 9. Получение З-ди-н-пропиламино-5-(изоксазол-5-ил)хромана.
A) Получение З-ди-н-пропиламино-5-бромхромана
Раствор З-кето-5-бромхромана (45 ммолей, 10,33 г), дипропиламина (90 ммолей, 9,0 г) и п-толуолсульфоновой кислоты (4,5 ммоля, 850 г) в 200 мл толуола нагревают до кипения с обратным холодильником, собирая воду в ловушку Дина Старка (Dean Stark trap). После нагревания в течение 5 ч при температуре дефлегмации удаляют ловушку Дина Старка и концентрируют реакционный раствор, получая коричневое масло.
Это масло растворяют в тетрагидрофуране, после чего добавляют 3,38 г (54 ммоля) цианборгидрида натрия. Затем реакционный раствор барботируют газообразным хлористым водородом с такой скоростью, чтобы температура раствора не превышала 45oC. Когда цвет реакционного раствора начинает светлеть, добавление хлористого водорода прекращают. После этого раствор перемешивают при комнатной температуре в течение 2 дней.
Через 2 дня реакционный раствор подщелачивают, используя раствор гидроксида натрия, и затем щелочной раствор экстрагируют хлористым метиленом. Экстракт сушат над сульфатом натрия и после этого концентрируют в вакууме, получая оранжевое масло. Это масло растворяют в растворе, состоящем из 200 мл тетрагидрофурана и 45 мл триэтиламина. Полученный раствор нагревают до кипения с обратным холодильником, поддерживая такую температуру в течение 30 мин, охлаждают до комнатной температуры и затем перемешивают при комнатной температуре в течение 3 дней. Через 3 дня реакционную смесь фильтруют через основный оксид алюминия и фильтрат концентрируют в вакууме, получая 15,3 г оранжевого масла. Это масло чистят способом хроматографии с мгновенным испарением, используя 8% -ный диэтиловый эфир в гексане (NH4OH) в качестве элюента и получая 10,77 г указанного выше в названии соединения.
B) Получение 3-ди-н-пропиламино-5-(1-оксиэтан)хромана.
К холодному (-78oC) раствору 4,95 г (15,9 ммоля) полученного выше соединения в 200 мл тетрагидрофурана добавляют 15,3 мл (20,6 ммоля) 1,35 М раствора н-бутиллития в гексане, полученный раствор перемешивают при -78oC в течение 20 мин, и затем добавляют 1,4 г (31,8 ммоля) ацетальдегида. Полученный раствор перемешивают при -78oC в течение 30 мин, нагревают до комнатной температуры и затем перемешивают при комнатной температуре еще 30 мин. По истечении этого времени реакционный раствор выливают в воду и полученный водный раствор экстрагируют раствором следующего состава: изопропанол/хлороформ 1:3. Экстракт сушат над сульфатом натрия и затем концентрируют в вакууме, получая желтое масло. Это масло чистят способом хроматографии с мгновенным испарением, используя в качестве элюента смесь 1:1 диэтиловый эфир/гексан (NH4OH) и получая 4,1 г указанного выше в названии соединения в виде светло-желтого масла.
C) Получение З-ди-н-пропиламино-5-ацетилхромана
К раствору 4,1 г (14,8 ммоля) полученного выше соединения в 120 мл ацетона добавляют 40 мл 2М раствора серной кислоты и 5 мл реагента Джонса (Jones Reagent), реагент Джонса и раствор серной кислоты добавляют с такой скоростью, чтобы температура реакции не превышала 30oC. Полученный раствор оставляют перемешивать при комнатной температуре в течение 1 ч. Через 1 ч реакцию гасят, добавляя изопропанол, и подщелачивают реакционный раствор, используя раствор гидроксида натрия. Этот щелочной раствор экстрагируют раствором следующего состава: изопропанол/хлороформ 1:3. Экстракт сушат над сульфатом натрия и концентрируют в вакууме, получая 7,32 г желтого масла. Это масло чистят способом хроматографии с мгновенным испарением, используя в качестве элюента смесь 1:3 диэтиловый эфир/гексан (NH4OH) и получая 3,48 г указанного выше в названии соединения в виде светло-желтого масла.
D) Получение 3-ди-н-пропиламино-5-(изоксазол-5-ил)хроман-гидробромида.
Раствор 500 мг (1,81 ммоля) полученного выше соединения и 540 мг (5,43 ммоля) трис-(диметиламино) метана в 20 мл толуола нагревают до кипения с обратным холодильником и перемешивают при этой температуре в течение 2 ч. Через 2 ч реакционный раствор разбавляют разбавленным раствором гидроксида натрия и затем экстрагируют раствором следующего состава: изопропанол/хлороформ 1: 3. Полученный экстракт сушат над сульфатом натрия и затем концентрируют в вакууме, получая темно-желтое масло.
Это масло растворяют в 10 мл уксусной кислоты и добавляют 480 мг твердого гидроксиламингидрохлорида. Полученный раствор перемешивают при комнатной температуре в течение 17 ч, по истечении этого времени раствор концентрируют в вакууме, получая 543 мг оранжевого масла. Это масло чистят способом хроматографии с мгновенным испарением, используя в качестве элюента смесь 1:1 диэтиловый эфир/гексан (NH4OH) и получая 482 мг бесцветного масла.
Это масло растворяют в диэтиловом эфире и полученный раствор обрабатывают бромистоводородной кислотой, получая белый клейкий осадок. Осадок извлекают и перекристаллизовывают из смеси этилацетат/гексан, получая 460 мг указанного в названии белого твердого вещества. Т.пл. 171-173oC. MS(FD): 300(100), 301(15)
Вычислено, C 56,70; H 6,61; N 7,35
Найдено, C 56,71; H 6,56; N 7,54.
Как отмечалось выше, соединения, полученные из промежуточных соединений согласно изобретению, обладают связывающей рецептор 5-НТ1A способностью.
Было показано, что различные физиологические функции являются объектом воздействия серотонергическими нервными системами мозга.
Как таковые соединения, полученные из промежуточных согласно изобретению, способны излечивать у млекопитающих различные состояния и расстройства, возникающие при посредничестве 5-НТ-рецепторов, такие, как половые расстройства, гипертония, расстройства аппетита, депрессия, алкоголизм, боль, старческое слабоумие, боязнь, желудочно-кишечные расcтройства и курение. Поэтому изобретение предусматривает также методы лечения упомянутых выше расстройств в приведенных выше дозах для воздействия на рецепторы 5-окситриптамина у млекопитающих.
Был проведен следующий эксперимент, показывающий способность соединений согласно изобретению связывать рецепторы 1A-серотонина. Участки, специфически маркированные меченым тритием 8-окси-2- дипропиламино-1,2,3,4-тетрагидронафталином (3H-8-OH-ДРАТ), были идентифицированы как рецепторы 5-НТ1A. Эта общая процедура представлена Wongetal J. Neural. Transm. 71:207-218 (1988).
Самцов крыс линии Sprague-Dawley (весом 110-150 г) из Harlan Industries (Cumberland, JN) кормили Purina Chow по потребности последние три дня перед использованием в исследованиях. Крыс умерщвляли обезглавливанием. Мозг быстро удаляли и рассекали кору головного мозга при 4oC.
Ткани мозга гомогенизировали в 0,32 М сахарозы. После центрифугирования при 1000g, в течение 10 мин и затем при 17000g в течение 20 мин осаждалась неочищенная синаптосомная фракция. Осадок суспендировали в 100 объемах 50 мМ трис-HCl, pH 7,4, инкубировали при 37oC в течение 10 мин и центрифугировали при 50000g в течение 10 мин. Процесс повторяли и конечный осадок суспендировали в охлажденном льдом 50-ммольном буфере трис-HCl с pH 7,4.
Связывание меченого тритием 8-окси-2-дипропиламино-1,2,3,4- тетрагидронафталина осуществляли описанным ранее методом (Wond et al. J. Neural. Transm. 64: 251-269 (1985). Коротко, синаптосомные мембраны, выделенные из коры головного мозга, инкубировали при 37oC в течение 10 мин в 2 мл 50 ммолей трис-HCl при pH 7,4, 10 ммолях паргилина, 0,6 ммоля аскорбиновой кислоты, 5 ммолях хлористого кальция, 2-на-номолях меченого тритием 8-окси-2-дипропиламино-1,2,3,4- тетрагидронафталина и 0,1-1000 нмолях испытываемого соединения. Связывание заканчивало фильтрованием образцов при пониженном давлении через фильтр из стекловолокна (CFB). Фильтры два раза промывали 5 мл охлажденного льдом буфера и помещали в сцинтилляционные сосуды с 10 мл сцинтилляционной жидкости PCS (Amesham/Searle). Радиоактивность измеряли жидким сцинтилляционным спектрометром. В отдельные пробы включали также немеченный тритием 8-окси-2-ди-пропил-амино-1,2,3,4-тетрагидронафталин в концентрации 10 микромолей для получения неспецифического связывания. Специфическое связывание меченого тритием 8-окси-2-дипропиламино-1,2, 3,4''-тетрагидронафталина определяли как разницу радиоактивности, связанной в отсутствии и в присутствии 10 микромолей немеченого 8-окси-2-дипропиламино-1,2,3,4-тетрагидронафталина.
Соединения согласно изобретению испытывали также на их влияние in vivo на уровни 5-оксииндолуксусной кислоты (5-HIAA) мозга и уровни кортикостерона сыворотки. Самцам крыс линии Sprague-Dawley, весившим 150-200 г, вводили подкожно или перорально водные растворы испытываемых соединений. Через 1 ч после обработки крыс обезглавливали и собирали исходящую из туловища кровь. Крови позволяли свернуться и затем центрифугировали для отделения сыворотки. Концентрацию кортикостерона в сыворотке определяли спектрофлюорометрическим методом Солема (Solem, J. H. Brinek-Johnsen, T. Scand. J. Clin. Inwesr. (Suppl. 80), 17,1, (]965).
Весь головной мозг обезглавленных крыс быстро удаляли, замораживали на сухом льду и хранили при -15oC. Концентрации 5-оксиндолуксусной кислоты (5-HIAA) измеряли жидкостной хроматографией с электрохимической регистрацией, как описано Fuller, R.W. Snoddy, H.D. Perry, K.W. Life Sci. 40, 1021 (1987).
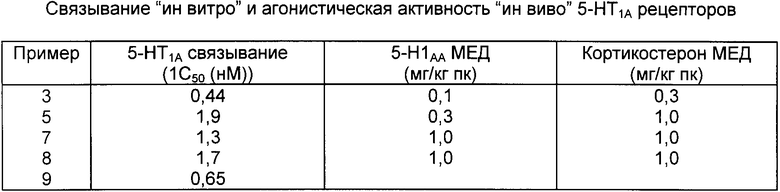
Результаты испытаний различных соединений согласно изобретению представлены в таблице. В таблице первая колонка представляет номера примеров испытываемых соединений, во второй колонке представлено количество испытываемых соединений, выраженное в наномолярной концентрации, потребной для ингибирования связывания меченого тритием 8-окси-2-дипропиламино-1,2,3,4-тетрагидронафталина на 50% выраженное как ингибирующая 50%-ная концентрация (ИН50), в третьей колонке представлена минимальная эффективная доза (МЭД) испытываемого соединения, введенного подкожно, для снижения уровней 5-HIAA мозга, в четвертой колонке представлена минимальная эффективная доза испытываемого соединения, введенного подкожно, потребная для повышения уровней кортико-стерона сыворотки. Результаты, показанные в колонке 3 указывают на 5-НТ1A агонистическую активность.
Изобретение предусматривает новые промежуточные соединения, используемые для получения замещенных в кольце 2-амино-1,2,3,4 тетрагидронафталинов и 3-аминохроманов, проявляющих связывающую активность в отношении рецептора 1А-серотонина. 2 з.п.ф-лы, 1 табл.

где R С1 - 4 алкил; R1 - C1 - 4 алкил;
Х СН2- или -О-;
Q представляет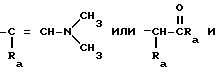
каждый Ra независимо водород, С1 - 4 алкил или С1 - 4 алкокси.
| WO, заявка, 90/15747, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 385658, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, заявка, 343830, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| EP, патент, 399982, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |

