Исследования в области аллергических реакций легкого показали, что производные арахидоновой кислоты, образованные действием липоксигеназ, соотносятся с различными болезненными состояниями. Некоторые из этих метаболитов арахидоновой кислоты классифицированы как члены семейства эйкозантетраеновых кислот, названных лейкотриенами. Полагают, что три из этих соединений являются основными компонентами вещества, которое ранее называли медленно реагирующим веществом анафилаксии (SRS-A) и обозначены как лейкотриены C4, D4 и E4 (LDTC4, LTD4 и LTE4, соответственно).
Другой метаболит арахидоновой кислоты лейкотриен B4 (LTB4) является предвоспалительным липидом, вовлечены в патогенез псориаза, артрита, хронических заболеваний легких, острых респираторных расстройств, шока, астмы, воспалительных заболеваний пузыря и других воспалительных процессов характеризующихся инфильтрацией и активированием нейтрофильных гранулоцитов и других предшествующих воспалению клеток. Активированные таким образом нейтрофильные гранулоциты освобождают разрушающие ткани ферменты и реакционноспособные химические соединения, вызывающие воспаление. Антагонизм LTB4 поэтому должен обеспечить новый терапевтический подход к лечению этих состояний.
Целью изобретения является разработка новых химических средств, которые являются избирательными антагонистами лейкотриена B4 и которые могут быть использованы терапевтически при лечении воспалений и аллергических расстройств, таких, как астма, при которых лейкотриены, по-видимому, являются причинными медиаторами.
Это изобретение предусматривает соединения формулы I:
или их фармацевтически приемлемые основно-аддитивные соли,
где
R1 представляет собой алкил с 1-5 углеродными атомами, алкенил с 2-5 углеродными атомами, алкинил с 2-5 углеродными атомами, алкокси с 1-4 углеродными атомами, (C1-C4-алкил)-тио, галоид, фенил, замещенный радикалом R2;
каждый из R2 и R3 независимо друг от друга является водородом, галоид, гидрокси, алкил с 1-4 атомами углерода, алкокси с 1-4 углеродными атомами, (C1-C4-алкил)-S(O)q-, трифторметилом или ди_(C1-C4-алкил)-амино;
X представляет собой -O-, -S-, -C(=O)- или CH2-;
Y является -O- или -CH2- или взятые вместе -X-Y- означает -CH=CH- или -C=C-;
Z является прямой или разветвленной цепью алкилиденила с 1-10 углеродными атомами;
A представляет собой связь, -O-, -S-, -CH=CH- или CRaRb, где Ra и Rb каждый независимо друг от друга является водородом, алкилом с 1-5 углеродными атомами или R7-замещенным фенилом, или взятые вместе с атомом углерода, к которому они присоединены, образуют циклоалкильное кольцо с 4-8 углеродными атомами; R4 является R6 группой формулы: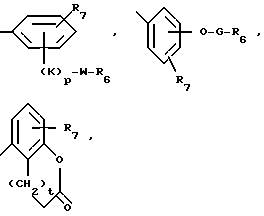

каждый R6 независимо является COOH, 5-тетразолилом, -CON(Rg)2 или -CONHSO2R10;
каждый R7 является водородом, C1-C4-алкилом, C2-C5-алкенилом, C2-C5-алкинилом, бензилом, метокси, -W-R6, -T-G-R6, (C1-C4-алкил)-T-(C1-C4-алкилиденил)-O- или гидрокси;
R8 представляет собой водород или галоид,
каждый R9 независимо представляет собой водород, фенил или алкил с 1-4 углеродными атомами, или взятые вместе с атомом азота образуют группу морфолино, пиперидино, пиперазино или пирролидино;
R10 представляет собой алкил с 1-4 углеродными атомами или фенил;
R11 является R2, -W-R6 или -T-G-R6;
Каждый W является связью или двухвалентным углеводородным остатком с прямой или разветвленной цепью с 1-8 углеродными атомами;
каждый G является двухвалентным углеводородным остатком с прямой или разветвленной цепью с 1-8 углеродными атомами;
каждый T является связью, -CH2-, -O-, -NH-, -NHCO-, -C(=O)- или -S(O)q-;
K представляет собой -C(=O)- или -CH(OH)-;
каждое q независимо является 0, 1 или 2;
p равно нулю или единице и t равно нулю или единице;
при условии, что, если X является -O- или -S-, то Y не может -O-;
при условии, что, если A представляет собой -O- или -S-6 то R4 не является R6;
при условии, что, Z и A оба не могут быть связью, если Y является -O-;
при условии, что, если A представляет собой -O- или -S- и Z является связью, то Y не может быть -O-;
при условии, что Z не может быть связью, если Y является -O-,
и при условии, что W не может быть связью, если p равно нулю.
Далее изобретение предусматривает способ лечения острых аллергических состояний, таких как воспаление или астма, включающий введение эффективного количества соединения формулы I.
Изобретение предусматривает также фармацевтическую композицию, которая содержит в качестве активного ингредиента соединение согласно изобретение, как определено выше, в сочетании с фармацевтически приемлемым носителем активного ингредиента.
Предусмотрены также промежуточные соединения для получения соединений формулы I. Такие соединения определяются формулой II:
где
R1 представляет собой алкил с 1-5 углеродными атомами, алкенил с 2-5 углеродными атомами, C2-C2-алкинил, алкокси с 1-4 углеродными атомами (C1-C4-алкил)-тио, гало или R2замещенный фенил;
каждый R2 и R3 независимо друг от друга представляют собой водород, гало, окси, алкил с 1-4 углеродными атомами, алкокси с 1-4 углеродными атомами, (C1-C4-алкил)-S(O)q-, трифторметил или ди-(C1-C3-алкил)-амино;
X является -O-, -S-, -C(=O)- или -CH2-;
Y представляет собой -O- или -CH2-;
или взятые вместе -X-Y-являются -CH=CH- или -C=C-;
Z представляет собой прямую или разветвленную цепь алкилиденила с 1-10 углеродными атомами;
A является связью, -O-, -S-, -CH=CH- или CRaRb-,
где Ra и Rb независимо друг от друга являются водородом, C1-C5-алкилом или R7-замещенным фенилом, или взятые вместе с атомом углерода, к которому они присоединены, образуют циклоалкильное кольцо с 4-8 углеродными атомами; является
является  , группой формул:
, группой формул: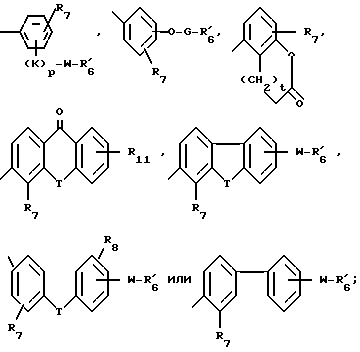
каждый  независимо является -COOR или -CN;
независимо является -COOR или -CN;
каждый R7 представляет собой водород, алкил с 1-4 углеродными атомами, алкенил с 2-5 углеродными атомами, алкинил с 2-5 углеродными атомами, бензил, метокси, -W-  , -T-G-
, -T-G-  , (C1-C4-алкил-T-(C1-C4-алкилиденил)-O- или гидроксигруппу:
, (C1-C4-алкил-T-(C1-C4-алкилиденил)-O- или гидроксигруппу:
R8 является водородом или атомом галогена;
каждый R9 независимо представляет собой водород, фенил, алкил с 1-4 углеродными атомами или взятые вместе с атомом азота, образуют группу морфолино, пиперидино, пиперазино или пирролидино;
R10 представляет собой алкил с 1-4 углеродными атомами или фенил;
R11 является R2, -W-  , или -T-G-R6 :
, или -T-G-R6 :
R представляет собой алкил с 1-6 углеродными атомами:
каждый W является связью или двухвалентным гидрокарбильным остатком с прямой или разветвленной цепью с 1-8 углеродными атомами;
каждый G представляет собой двухвалентный гидрокарбильный остаток с прямой или разветвленной цепью с 1-8 углеродными атомами;
каждый T представляет собой связь, -CH2-, -O-, -NH-, -NHCO-, -C(=O)- или -S(O)q-;
K является -C(=O)- или -CH(OH)-;
каждое q независимо равно нулю, единице или двум;
p равно нулю или единице и t равно нулю или единице,
при условии, что, если X является -O- или -S-6 то Y не может быть -O-;
при условии, что Z и A оба не могут быть связью, если Y является -O-;
при условии, что, если A является -O- или -S-, то  не может быть
не может быть  ;
;
при условии, что если A является -O- или -S- и Z является связью, то Y не может быть -O-;
при условии, что W не может быть связью, если p равно нулю.
Изобретение касается новых органических соединений, полезных при лечении состояний и заболеваний, связанных с избыточным выделением лейкотриена B4. Предпочтительной группой соединений являются соединения формулы Ia:
и их фармацевтически приемлемые основно-аддитивные соли.
Особенно предпочтительны соединения, в которых R2 является гало, в частности атом фтора. Предпочтительными R1-заместителями являются пропил и особенно этил.
Предпочтительные заместители Z включают алкилидены с 1-4 углеродными атомами, в частности -CH2CH2 и -CH2CH2CH2CH2-. Предпочтительные группы A включают -O-, -CH2-, CH-(R7-замещенный фенил)- и -C-(CH3)2-. Предпочтительные группы R4 включают -COOH, 5-тетразолил, или моно-, ди- или трициклическую группу, как показано выше, в которой имеется по меньшей мере одна кислотная группа, присоединенная к кольцу, такая как -W-COOH, -T-G-COOH или соответствующие производные тетразола. Предпочтительной частью молекулы W является связь или алкилиден с прямой цепью с 1-4 углеродными атомами; предпочтительной частью молекулы G является алкилиден с прямой цепью с 1-4 углеродными атомами. Предпочтительно, что R5 или R7 был алкил с 1-4 углеродными атомами, особенно н-пропил.
Особенно предпочтительными группами являются те группы, в которых A является -CH(R7 - замещенный фенилом)- и R4 является -COOH или 5-тетразолилом. Также предпочтительны те соединения, в которых A является -O- и R4 - группа формулы: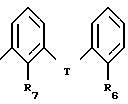
Предпочтительными аспектами этой субструктуры являются те соединения, в которых R7 представляет собой алкил с 1-4 углеродными атомами, особенно н-пропил, и R6 является группой -W-COOH. Особенно предпочтительны те соединения, в которых T означает -O- или -S- и W является связью.
Следующие определения касаются различных терминов, использованных в описании. Термин "алкил с 1-6 углеродными атомами" относится к алифатическим радикалам с прямой или разветвленной цепью с 1-6 углеродными атомами, таким как метил, этил, пропил, изопропил, н-бутил, втор-бутил, трет-бутил, н-пентил, 2,2-диметилпропил, гексил и т.п. Под это определение подпадает также термин "алкил с 1-3 углеродными атомами", "алкил с 1-4 углеродными атомами" и "алкил с 1-5 углеродными атомами". Термин "алкенил с 2-5 углеродными атомами" относится к прямым и разветвленным алифатическим радикалам с 2-5 углеродными атомами, содержащим одну двойную связь, таким как -CH=CH2, -CH2CH= CH2, -CH2CH2CH= CH2, -CH2C(CH3)= CH2, -CH2CH=C(CH3)2 и т.п. Термин "алкинил с 2-5 углеродными атомами касается прямых и разветвленных алифатических остатков с 2-5 углеродными атомами, содержащих одну тройную связь, таких как -C= C, -CH2-C=C, -CH2CH2C=CH, -CH2C=CCH3и т.п. Термин "алкокси с 1-4 углеродными атомами" относится к метокси, этокси, пропокси, изопропокси, бутокси, вторбутокси и трет-бутокси. Термин "гало" касается атома фтора, хлора, брома и иода.
Термин "алкилиден с 1-10 углеродными атомами" касается двухвалентного радикала, производного от алкана с 1-10 углеродными атомами, такого как -CH2-, -CH(CH3)-, -CH(C2H5)-, -CH2CH2-, -CH2CH(CH3)-, -CH(CH3)CH2-, -CH(CH3)CH(CH3)-, -CH2C(CH3)2-, -CH2CH(C2H5)-, -CH2CH2CH2-, -CH(CH3)CH2CH2-,
-CH2CH(CH3)CH2-, -CH2CH(C2H5)CH2-, -C(CH3)2CH2CH2-, -CH(CH3)CH2CH(CH3)-,
-CH2CH2CH2CH2-, -CH2C(CH3)2CH2CH2-, -CH2C(CH3)2CH2-, -CH2CH2CH(C2H5)CH2-,
-CH2CH2CH2CH2CH2-, //-CH2C(CH3)2CH2CH2-, -CH2(CH3)2CH2-,
-CH2CH2CH(C2H5)CH2-, / -CH(CH3)CH2CH2CH2CH2-, -CH2CH2CH2CH2CH2CH2-,
-(CH2)10- и т. п. Под это определение подпадают термины "алкилиден с 1-4 углеродными атомами" и "алкилиден с 2-4 углеродными атомами.
Термин "циклоалкил с 4-8 углеродными атомами" касается циклоалкильного кольца с 4-8 углеродными атомами, такого как циклобутил, циклопентил, 4,4-диметилциклогексил, циклогептил, циклооктил и т.п.
Термин "гидрокарбильный двухвалентный остаток с прямой или разветвленной цепью с 1-8 углеродными атомами" касается двухвалентных радикалов, производных от прямого или разветвленного алкана, алкена или алкина с 1-8 углеродными атомами. В зависимости от разветвления и количества углеродных атомов, как могут оценить химики-органики6 такая часть молекулы может содержать одну, две или три двойных или тройных связи или сочетание обеих. Как таковой этот термин может расцениваться как алкилиденовая группа в приведенном выше определении, содержащая от 1 до 8 углеродных атомов и от одной до трех двойных или тройных связей, или сочетание обеих, ограниченное как указано в предыдущем предложении.
Изобретение включает фармацевтически приемлемые основно-аддитивные соли соединений формулы I. Такие соли включают таковые, производные от неорганических оснований, таких как гидроокиси аммония и щелочных и щелочноземельных металлов, карбонаты, бикарбонаты и т.п., а также соли, производные от основных органических аминов, таких как алифатические и ароматические амины, алифатические диамины, оксиалкиламины и т.п. Такие основания, полезные при получении солей согласно изобретению, включают, таким образом, гидроокись аммония, карбонат калия, бикарбонат натрия, гидроокись кальция, метиламин, диэтиламин, этилендиамин, циклогексиламин, этаноламин и т.п. Особенно предпочтительны формы солей калия и натрия. Это изобретение включает как формы моносолей, т.е. когда соединение формулы I находится в отношении 1:1 с основанием, как описано ранее, так и форму дисолей в тех случаях, когда соединение формулы I имеет две кислотные группы. В дополнение это изобретение включает любые сольватные формы соединений формулы I или их солей, таких как сольваты этанола, гидраты и т.п.
Следует иметь в виду, что в соединениях, имеющих разветвленные алкильные, алкилиденовые или гидрокарбильные функциональные группы, и в соединениях, несущих двойные или тройные связи, могут существовать различные стереоизометрические формы. Это изобретение не ограничивается каким-либо конкретным стереоизомером, но включает все возможные отдельные изомеры и их смеси. Термин "5-тетразолил" относится к обоим таутомерам, т.п. (1H)-5-тетразолилу и (2H)-5-тетразолилу.
Соединения согласно изобретению могут быть получены стандартными, известными в данной области методами. Например, тетразольные соединения формулы I (в которых по меньшей мере один R6 является 5-тетразолилом) могут быть получены из соответствующих промежуточных соединений формулы II, в которых соответствующая группа R6 является нитрилом, любым из многообразных стандартных методов. В общем нитрил взаимодействует с азидным реагентом в инертном растворителе. Предпочтительные условия предполагают использование азида лития или аммония в диметилформамиде, азида натрия в диглиме и солянокислом N, N-диметилэтаноламине, или азида три-н-бутилолова в нереакционноспособном растворителе, таком как диметоксиэтан или тетрагидрофуран. При последних условиях реакционную смесь обычно нагревают до температуры образования флегмы реакционной смеси или близкой к ней. Превращение обычно завершается при этих условиях за два-три дня. Другие рабочие условия реакции включают взаимодействие нитрила формулы II с задом щелочного металла, таким как азид натрия, хлористым аммонием и (необязательно) хлористым литием в нереакционноспособном растворителе с высокой температурой кипения, таком как N,N-диметилформамид (ДМФ), предпочтительно при температуре примерно от 60 до 125oC. Альтернативно вместо азида щелочного металла, хлористого аммония, хлористого лития и диметилформамида могут быть использованы азид три-н-бутилолова, или азид тетраметилгуанидиния в растворителе, таком как тетрагидрофуран, диметоксиэтан, диэтоксиэтан или тому подобном.
Аналогично кислоты согласно изобретению (формула I, в которой по меньшей мере один R6 является -COOH) получают из промежуточных соединений формулы II, в которых соответствующая группа  является -COOR или -CN. Гидролиз таких сложных эфиров или нитрилов может быть осуществлен в любых многообразных кислотных или основных условиях, предпочтительно в водных условиях. Предпочтительные методы включают использование гидроокиси лития в смеси растворителя ацетон-вода, гидроокиси натрия в диоксане или гидроокиси калия или карбоната калия в смеси растворителя метанол-вода. В предшествующих условиях гидролиз обычно заканчивается в течение 12-18 ч в интервале температур 20-30oC, тогда как последняя реакция обычно завершается в течение 1 ч при 20-30oC.
является -COOR или -CN. Гидролиз таких сложных эфиров или нитрилов может быть осуществлен в любых многообразных кислотных или основных условиях, предпочтительно в водных условиях. Предпочтительные методы включают использование гидроокиси лития в смеси растворителя ацетон-вода, гидроокиси натрия в диоксане или гидроокиси калия или карбоната калия в смеси растворителя метанол-вода. В предшествующих условиях гидролиз обычно заканчивается в течение 12-18 ч в интервале температур 20-30oC, тогда как последняя реакция обычно завершается в течение 1 ч при 20-30oC.
Обычно предпочитают в соединениях, содержащих как нитрильную, так и эфирную функциональные группы, преобразовать нитрильную группу в тетразолил до гидролиза сложного эфира.
Промежуточные соединения формулы II могут быть получены целым рядом методов синтеза, как известно специалисту в данной области, в зависимости от конкретного желаемого соединения. Для соединений, в которых один из X или Y является -O-, вообще применима схема I.

где
один из -X-E и -Z-B является -OH и другой является -CH2-L, где L является легко удаляемой группой, такой как гало, особенно атом хлора, брома или иода, и R'' является окси или предпочтительно защищенной оксигруппой, такой как бензилокси.
Реакцию по схеме I обычно проводят с использованием эквимолярных количеств двух реагентов, хотя и другие соотношения, отличные от эквимолярных количеств, полностью работоспособны. Реакцию лучше всего проводить в нереакционноспособном растворителе, таком как кетоны, особенно ацетон или метилэтилкетон, или диметилформамид и в присутствии основания, предпочтительно гидрида щелочного металла или карбоната щелочного металла, предпочтительно карбоната калия. Особенно, если L является атомом хлора, то для повышения скорости реакции может быть использован катализатор, такой как йодид калия или натрия. Реакция может быть проведена в интервале температур от комнатной температуры до точки кипения реакционной смеси, при этом первая температура предпочтительна.
В предпочтительном случае, когда оксигруппа защищена, защитную группу удаляют с последующим процессом присоединения, как описано выше. Как известно специалистам в данной области, средства для снятия защиты оксигруппы будут зависеть от выбора применяемой защитной группы. В предпочтительной ситуации, когда используют бензильную группу, бензильную группу удаляют каталитической гидрогенизацией, например, в присутствии 10% палладия на угле в этилацетате, чтобы получить желаемый фенол. Обычно эту операцию проводят перед обращением  в R4, однако, как известно специалистам, эту последовательность можно перевернуть в зависимости от вовлеченных в реакцию функциональных групп. Таким образом, присоединение, как замечено выше, может при определенных обстоятельствах, известных в данной области, сначала включать превращение группы
в R4, однако, как известно специалистам, эту последовательность можно перевернуть в зависимости от вовлеченных в реакцию функциональных групп. Таким образом, присоединение, как замечено выше, может при определенных обстоятельствах, известных в данной области, сначала включать превращение группы  (например, нитрила) в группу R4 (например, 5-тетразолил) с последующим снятием защиты фенола.
(например, нитрила) в группу R4 (например, 5-тетразолил) с последующим снятием защиты фенола.
Аналогичные серии реакций можно найти в схеме II.

где
Q является атомом брома, хлора, иода, метилсульфонилом, толилсульфонилом или аналогичной удаляемой группой, и A' представляет собой -O- или -S. Аспекты этой схемы реакции и все вариации ее в общем те же, что и рассмотренные выше в связи со схемой I.
Другой путь получения промежуточных соединений формулы II можно найти в схеме реакции III.

где
D представляет собой B(OH)2, атом брома или хлора и Bn является бензилом или родственной защитной группой.
В приведенной выше схеме промежуточный бромистый фенил (3A, D является бромом) может быть обращен в соответствующую борную кислоту (3A, D = B(OH)2) несколькими методами. В одном методе бромистый фенил сначала обрабатывают алкиллитиевым реактивом, таким как трет-н-бутиллитий в нереакционноспособном растворителе, с последующим взаимодействием с триалкилборатом, таким как триизопропилборат, и гидролизом водной кислотой, такой как разбавленная хлористоводородная кислота. Альтернативно производное лития (3A, D = литий) сначала может быть подвергнуто взаимодействию с силилирующим реагентом, таким как хлористый триметилсилил, чтобы получить промежуточное соединение, в котором D является триметилсилилом; взаимодействие этого промежуточного соединения с трехбромистым бором с последующей обработкой метанолом и водной кислотой аналогично приводит к получению желаемой фенилборной кислоты /(3A, D является B(OH)2/.
Реакцию диарильного соединения, описанную в связи с приведенной выше схемой, можно затем осуществить взаимодействием по существу эквимолярных количеств бората фенила (3A, D является B(OH)2) с бромистым фенилом 3B в присутствии тетракис-(трифенилфосфин)-палладия (O) и водного карбоната натрия в смеси этанола и бензола. Если взаимодействие проводить при повышенной температуре, такой как температура обратного холодильника для реакционной смеси, то реакция обычно заканчивается в течение 2-18 ч.
Другой метод диарильного присоединения может быть осуществлен взаимодействием одного из двух промежуточных соединений бромистого фенила 3A или 3B с трет-бутиллитием в нереакционноспособном растворителе, таком как тетрагидрофуран, с последующей обработкой хлористым цинком для получения соответствующего промежуточного соединения, в котором функциональная группа брома обращена в группу - ZnCl. Этот реактив затем подвергают взаимодействию с другим промежуточным соединением брома (или хлора) в присутствии тетракис-(трифенилфосфин)-палладия (O) для получения желаемого соединения формулы II.
Другие варианты и сочетания химических реакций могут быть также применены для получения соединений согласно изобретению. Например, одна серия реакций показана на схеме IV, приведенной в конце описания; эта последовательность приведена для тех соединений, в которых X является -O-, но как известно специалистам в области органической химии, аналогичные превращения могут быть применимы и к другим вариантам значений X,где  представляет собой алкил с 1-4 углеродными атомами и Bn означает бензил или аналогичную защитную группу фенола.
представляет собой алкил с 1-4 углеродными атомами и Bn означает бензил или аналогичную защитную группу фенола.
На схеме IV 2,5-диметоксибензойную кислоту (4A) сначала обращают в соответствующий галоидангидрид, например, хлорангидрид обработкой хлористым тионилом в хлористом метилене, который затем подвергают взаимодействию с 2-амино-2-метил-1-пропанолом. Последующая обработка, например, хлористым тионилом обеспечивает защиту карбоновой кислоты как 5,5-диметил-2-оксазолина 4B. Обработкой этого промежуточного соединения соответственно замещенных фениловым реактивом Гриньяра в растворителе, таком как тетрагидрофуран, получают промежуточное дифенильное соединение 4C. Оксазолин превращают в соответствующий альдегид 4D последовательной обработкой иодистым метилом, борогидридом натрия в этаноле и хлористоводородной кислотой в растворителе, таком как тетрагидрофуран. Обработка альдегида 4D окислителем, таким как мета-хлорнадбензойная кислота в хлористом метилене дает фенол 4E. Фенол защищают бензилом или аналогичной защитной группой при обработке бромистым бензилом (или подобным реактивом) в растворителе, таком как диметилформамид и в присутствии кислотного акцептора, такого как карбонат калия. Полученное промежуточное соединение 4F затем ацилируют реагентом  -COCI или аналогичным реактивом в присутствии кислоты Льюиса, такой как хлористое олово (4) в растворителе, таком как хлористый метилен, при этом предпочтительны охлаждающие температуры в интервале от -20oC до 0oC. Полученное ацилированное промежуточное соединение 4G затем дважды деблокируют до дифенила 4H обработкой реактивом, таким как треххлористый бор в растворителе, таком как хлористый метилен. Бензил (или аналогичную защитную группу) замещают таким же образом, как описано выше, чтобы получить промежуточное соединение 41, которое затем алкилируют, как описано ранее реагентом
-COCI или аналогичным реактивом в присутствии кислоты Льюиса, такой как хлористое олово (4) в растворителе, таком как хлористый метилен, при этом предпочтительны охлаждающие температуры в интервале от -20oC до 0oC. Полученное ацилированное промежуточное соединение 4G затем дважды деблокируют до дифенила 4H обработкой реактивом, таким как треххлористый бор в растворителе, таком как хлористый метилен. Бензил (или аналогичную защитную группу) замещают таким же образом, как описано выше, чтобы получить промежуточное соединение 41, которое затем алкилируют, как описано ранее реагентом  -A-Z-CH2-L (см. схему I), особенно если L является атомом хлора или иода, для получения промежуточного соединения 4J. Бензильную группу удаляют каталитической гидрогенизацией, например, в присутствии 10% палладия на угле в этилацетате для получения фенола 4K. Восстановление ацильной части молекулы соединения 4K, например, обработкой триэтилсиланом в трифторуксусной кислоте в растворителе, таком как четыреххлористый углерод приводит к получению соответствующего промежуточного соединения формулы II, которое может быть дальше преобразовано, как описано ранее.
-A-Z-CH2-L (см. схему I), особенно если L является атомом хлора или иода, для получения промежуточного соединения 4J. Бензильную группу удаляют каталитической гидрогенизацией, например, в присутствии 10% палладия на угле в этилацетате для получения фенола 4K. Восстановление ацильной части молекулы соединения 4K, например, обработкой триэтилсиланом в трифторуксусной кислоте в растворителе, таком как четыреххлористый углерод приводит к получению соответствующего промежуточного соединения формулы II, которое может быть дальше преобразовано, как описано ранее.
Родственная последовательность представлена на схеме V, приведенной в конце описания: как и прежде, эта последовательность приведена для тех соединений, в которых X является -O-, но, как известно специалистам в области органического синтеза, подобные превращения применимы и к другим вариантам значений для X.
Бромистый диметоксифенил формулы 5A обращают в соответствующий дифенил формулы 5B обработкой подходящей борной кислотой в стандартных условиях. В дополнение к стандартным условиям использование катализатора, такого как хлористый бис-(трифенилфосфин)-никель с соответствующим арильным реактивом Гриньяра в тетрагидрофуране при температуре обратного холодильника или в диэтиловом эфире представляет собой альтернативный метод проведения этой конденсации. Затем дифенил 5B может быть ацилирован как описано выше, для получения соединения 5C, которое затем деблокируют треххлористым бором, как показано выше, чтобы получить фенол 5D. Таким же способом, как описано ранее, фенол может быть алкилирован реактивом  -A-Z -CH2-L (см. схему I), особенно если L является атомом хлора или иода, чтобы получить соединение 5E, которое, в свою очередь, восстанавливают до соединения 5F. Деметилирование соединения 5F для получения соответствующего фенола формулы II осуществляют обработкой триэтилатом натрия в диметилформамиде при повышенной температуре (например, 90-100oC). Альтернативно неметилирование может быть проведено обработкой трехбромистым бором в растворителе, таком как хлористый метилен.
-A-Z -CH2-L (см. схему I), особенно если L является атомом хлора или иода, чтобы получить соединение 5E, которое, в свою очередь, восстанавливают до соединения 5F. Деметилирование соединения 5F для получения соответствующего фенола формулы II осуществляют обработкой триэтилатом натрия в диметилформамиде при повышенной температуре (например, 90-100oC). Альтернативно неметилирование может быть проведено обработкой трехбромистым бором в растворителе, таком как хлористый метилен.
Вариация процесса по схеме V описана в схеме VI, приведенной в конце описания: как и прежде, эта последовательность приведена для тех соединений, в которых X является -O-, но, как известно специалистам в области органического синтеза, аналогичные преобразования могут быть применены к другим вариантам значений для X.
Промежуточное соединение формулы 4C (из схемы IV) деметилируют трехбромистым бором в хлористом метилене способом, аналогичным процессу превращения соединения 4C в соединение формулы 4H. Полученный фенол 6A имеет нетронутую защитную группу оксазолина и его алкилируют соответствующим реагентом  -A-Z-CH2-L (см. схему I), особенно, если L является атомом хлора или иода, в растворителе, таком как диметилформамид, необязательно в присутствии кислотного акцептора, такого как карбонат калия. Полученный продукт-соединение формулы 6B обращают затем в бензальдегид 6C, как описано выше при получении соединения 4D из 4C, окисляют до соответствующего фенола 6D, как описано выше при превращении соединения 4D в 4E, обращают в защищенный фенол 6E группой, такой как бензил, как описано при превращении соединения формулы 4E в 4F, и ацилируют для получения соединения 4I таким же путем, что и при обращении соединения 4F в 4G. С промежуточного соединения в 4I сначала может быть снята защита и затем оно может быть окислено, как предусмотрено в схеме IV; альтернативно операции могут быть реверсированы, т.е. соединение 4I может быть восстановлено до промежуточного соединения 6F обработкой триэтилсиланом и трифторуксусной кислотой в четыреххлористом углероде, и затем проводят снятие защиты для получения желаемого промежуточного соединения формулы II.
-A-Z-CH2-L (см. схему I), особенно, если L является атомом хлора или иода, в растворителе, таком как диметилформамид, необязательно в присутствии кислотного акцептора, такого как карбонат калия. Полученный продукт-соединение формулы 6B обращают затем в бензальдегид 6C, как описано выше при получении соединения 4D из 4C, окисляют до соответствующего фенола 6D, как описано выше при превращении соединения 4D в 4E, обращают в защищенный фенол 6E группой, такой как бензил, как описано при превращении соединения формулы 4E в 4F, и ацилируют для получения соединения 4I таким же путем, что и при обращении соединения 4F в 4G. С промежуточного соединения в 4I сначала может быть снята защита и затем оно может быть окислено, как предусмотрено в схеме IV; альтернативно операции могут быть реверсированы, т.е. соединение 4I может быть восстановлено до промежуточного соединения 6F обработкой триэтилсиланом и трифторуксусной кислотой в четыреххлористом углероде, и затем проводят снятие защиты для получения желаемого промежуточного соединения формулы II.
Иной вариант химических операций обобщен на схеме VII, приведенной в конце описания. Снова эту последовательность приводят для тех соединений, в которых X является -O-; в дополнение общая схема приведена для соединений, в которых Ra является R7-замещенным фенилом, Rb является водородом и  является -CN; как очевидно для специалистов в области органического синтеза, аналогичные преобразования будут применимы для других вариантов значений X, Ra, Rb и
является -CN; как очевидно для специалистов в области органического синтеза, аналогичные преобразования будут применимы для других вариантов значений X, Ra, Rb и  .
.
Галоидное промежуточное соединение формулы 7A подвергают взаимодействию с фенолом 7B таким же образом, как описано выше для схемы I. Полученное соединение 7C бромируют реагентом, таким как N-бромсукцинимид в растворителе, таком как хлористый метилен с получением бромистого промежуточного соединения 7D. Это промежуточное соединение затем подвергают взаимодействию с подходящим боратом фенила и тетракис-(трифенилфосфин)-палладием (O), как описано выше для схемы III, с получением продукта присоединения 7E. Соединение 7E затем может быть преобразовано в промежуточные соединения и конечные продукты согласно настоящему изобретению описанными ранее способами, то есть гидролизом, взаимодействием с азидом, дебензилированием и так далее. Вариации этой последовательности будут также очевидны, например, эта серия превращений может быть осуществлена с использованием реагента 7B, в котором группа R1 заменена группой 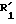 -CO, как показано на схемах IV, V и VI; полученное промежуточное соединение 7C или более поздние промежуточные соединения 7D или 7E могут быть затем восстановлены для получения соединений, в которых группа R1 является
-CO, как показано на схемах IV, V и VI; полученное промежуточное соединение 7C или более поздние промежуточные соединения 7D или 7E могут быть затем восстановлены для получения соединений, в которых группа R1 является  -CH2-.
-CH2-.
Другой вариант, применяющий другие предшественники некоторых более предпочтительных соединений, в общем виде представлен на схеме VIII, приведенной в конце описания, хотя два примера таких преобразований показаны с применением феноксисоединения, но понятно, что такой химизм реакций применим к другим арильным группам и боковым цепям,где Px является защитной группой.
На схеме VIII соединение 8A является конкретным осуществлением соединения 2A (схема II), в котором R'' является -OH. Фенол защищен с получением соединения 8B (аналог соединения 2A, в котором R'' является защищенной оксигруппой). Одной предпочтительной защитной группой для последующих превращений является триметилсилилэтоксиметильная группа (SEM), которая может быть введена при обработке соединения 8A триметилсилилэтоксиметилхлоридом в присутствии диизопропилэтиламина в растворителе, таком как хлористый метилен. Другой полезной группой является алканоильная группа, такая как ацетил, которая может быть просто введена при обработке соединения 8A алканоильным ангидридом (например, уксусным ангидридом) в растворителе, таком как хлористый метилен и предпочтительно в присутствии триалкиламина, такого как триэтиламин.
Защищенное промежуточное соединение 8B затем подвергают взаимодействию с подходящим предшественником-промежуточным соединением таким же образом, как описано схем I и II выше, с получением продукта присоединения 8C. В приведенном для иллюстрации примере на схеме VIII используют защищенный оксибензальдегид, альдегидная часть которого защищена как циклический ацеталь. В примере на схеме VIII, в котором присоединяют фенол, Q предпочтительно является группой хлора, которая обработана каталитическим количеством иодида щелочного металла для облегчения реакции.
Полученное промежуточное соединение 8C может быть затем превращено в функционализованное соединение согласно изобретению обработкой производным малоновой кислоты. Например, после обработки ацеталя 8C разбавленной хлористоводородной кислотой и тетрагидрофураном обработка полученного бензальдегида метилмалоновой кислотой в солянокислом пиридинии и толуоле дает в результате 2-метилпропеновую кислоту 8D. В случае триметилсилилэтоксиметильной защитной группы снятие защиты фенола фтористым тетрабутиламмонием в тетрагидрофуране дает конечный продукт этого изобретения. Аналогично применение малоновой кислоты с последующим гидролизом соединения 8C дает акриловую кислоту 8E; в случае, когда защитной группой является алканоильная часть молекулы, например, ацетил, обработка соединения 8E карбонатом калия в метаноле и воде дает соответствующий фенол согласно этому изобретению.
Многие промежуточные соединения для получения некоторых групп R4 (или  ) более предпочтительных соединений согласно изобретению известны в данной области. Многие группы R4 и
) более предпочтительных соединений согласно изобретению известны в данной области. Многие группы R4 и  , который являются простыми дифениловыми эфирами, дифенилтиоэфирами и дифениламинами, могут быть получены одним из ряда путей синтеза, однако имеется один общепринятый путь - это синтез Ульмана, при котором, например, фенол конденсируют с йод- или бромбензолом в присутствии пиридина, карбоната калия и медной бронзы с получением соответствующего простого дифенилового эфира. Йодистую медь (I) и трет-бутилат калия можно применять вместо медной бронзы и карбоната калия. В общем эти реакции дают низкий выход и трудны для разработки особенно в крупномасштабном производстве.
, который являются простыми дифениловыми эфирами, дифенилтиоэфирами и дифениламинами, могут быть получены одним из ряда путей синтеза, однако имеется один общепринятый путь - это синтез Ульмана, при котором, например, фенол конденсируют с йод- или бромбензолом в присутствии пиридина, карбоната калия и медной бронзы с получением соответствующего простого дифенилового эфира. Йодистую медь (I) и трет-бутилат калия можно применять вместо медной бронзы и карбоната калия. В общем эти реакции дают низкий выход и трудны для разработки особенно в крупномасштабном производстве.
Изобретение включает предпочтительный способ получения дифениловых соединений формулы XI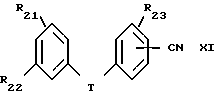
в которой
R21 представляет собой водород, алкокси с 1-4 углеродными атомами, алкил с 1-4 углеродными атомами, трифторметил, -COOR25 или гало;
R22 представляет собой водород, алкокси с 1-4 углеродными атомами, алкил с 1-4 углеродными атомами, трифторметил или гало;
R23 представляет собой водород, алкокси с 1-4 углеродными атомами, алкил с 1-4 углеродными атомами, нитро, трифторметил, необязательно замещенное фенокси, необязательно замещенное фенилтио или пирролидино, и
T представляет собой -O-, -S- или -NR20-, где R20 является водородом или C1-C3-алкилом и R25-алкил с 1-4 углеродными атомами, при условии, что цианогруппа находится во 2- или 4-положении фенильного кольца относительно точки присоединения к T,
включающий взаимодействие производного фенола, тиофенола или анилина формулы XII:
в которой
T, R21 и R22 имеют те же значения, которые определены для них выше, с производным фторбензонитрила формулы XIII:
где
R23 имеет значения, определенные для него ранее, и цианогруппа находится во 2- или 4-положении фенильного кольца относительно группы фтора, в апротонном растворителе в присутствии сильного основания.
В условиях, описанных в этом разделе, реакция сочетания происходит почти с количественным выходом и поэтому предполагает экономически выгодный процесс реакции с высоким выходом, в котором не используют медные реактивы. Установлено, что реакция ограничена фторбензонитрилами, как определено выше в формуле XIII, и не работает на сравнимых бромированных, хлорированных, иодированных или метоксианалогах таких фторпроизводных. Более того, только 2- и 4-фторбензонитрилы, по-видимому, работают в этой последовательности реакций; 3-фторбензонитрилы, по-видимому, не взаимодействуют, сложные эфиры карбоновых кислот не работают вместо функциональной группы нитрила.
Следующие определения касаются различных терминов, использованных в этом разделе. "C1-C4-алкил" относится к прямым или разветвленным алифатическим радикалам с 1-4 углеродными атомами, таким как метил, этил, пропил, изопропил, н-бутил, втор-бутил и трет-бутил. Термин "C1-C4-алкокси" касается метокси, этокси, пропокси, изопропокси, бутокси, втор-бутокси, и трет-бутокси. Термин "гало" касается атома фтора, хлора, брома и иода.
Термин "необязательно замещенный" фено лили тиофенол касается соответственно фенольной или тиофенольной группы, которая незамещена или замещена одной или двумя группами, выбранными из группы, включающей гало, алкил с 1-4 углеродными атомами и алкокси с 1-4 углеродными атомами.
Соединения формулы XI являются полезными промежуточными соединениями для получения лейкоториеновых антагонистов, охарактеризованных в описании, в которых R23 является метокси и могут быть деметилированы, чтобы получить соответствующий фенол; этот фенол сопрягают с замещенным алкилгалогенидом для получения нитрилового предшественника многих описанных здесь антагонистов лейкотриена. Нитриловая функциональная группа может быть затем гидролизована или обработана производным азида для получения соответствующей карбоновой кислоты или тетразольного производного этого ряда. Такие соединения, как сообщалось, являются антагонистами лейкотриена B4.
В реакции согласно изобретению два реагента формул XII и XIII могут взаимодействовать в присутствии сильного основания. Предпочтительно использовать приблизительно равные молярные количества обоих реактивов, хотя и другие отношения допустимы. Применяемым сильным основанием может быть гидрид натрия, трет-бутилат калия или тому подобные, но предпочтителен покрытый фтористым калием глинозем, иначе рассматриваемый как смесь фтористого калия и окиси алюминия. Преимущество этого последнего реактива заключается в том, что он может быть эффективно удален после реакции. Типично количество применяемого основания составляет один-два весовых эквивалента относительно реактива XII. Предпочтительным основанием является 37-40% фтористый калий на глиноземе. При приготовлении этого реагента могут быть использованы основные, нейтральные или кислые глиноземы, при этом первые два предпочтительны. Эти реактивы имеются также в продаже.
Реакцию лучше всего проводить в инертном апротонном растворителе. Хотя комнатные температуры являются рабочими, предпочтительно, чтобы последовательность реакций осуществлялась при повышенной температуре, типично примерно от 80oC до температуры образования флегмы реакционной смеси. Такие растворителя включают глимы, в том числе глим, диглим и триглим, нереакционноспособные карбитолы, такие как диэтилкарбитол, нереакционноспособные целлосольвы, такие как диметилцеллосольв, диэтилцеллосольв и тому подобные, и другие органические растворители с точной кипения по меньшей мере 80oC, такие как бензол, диоксан, пиридин, и толуол. По причинам не совсем очевидным диметилформамид, по-видимому, не является предметом выбора в качестве растворителя. Однако предпочтительным растворителем является ацетонитрил и реакцию лучше всего проводить при нагревании реакционной смеси до температуры обратного холодильника, которая обычно составляет 85-90oC. Вообще количество растворителя не является критическим и составляет обычно приблизительно 10-30 мл применяемого растворителя на каждый грамм реактива XII.
В дополнение к растворителю, основанию и реактивам XII и XIII предпочтительно применять катализатор для облегчения проведения взаимодействия. Такие катализаторы включают катализаторы переноса фаз, которые обычно признаны как способные переносить нуклеофил из водной фазы в органическую фазу. См., например, "Advanced Organic Chemistry", Jerry March, Ed. (3rd Edition, John Wiley and Sons, Inc., 1985) p. 320-322. Подходящие катализаторы переноса фаз включают соли четвертичного аммония или фосфония, простые эфиры крауны и криптанды. Предпочтительное место среди катализаторов переноса фаз занимают соли четвертичного аммония, в частности, галогениды тетраалкиламмония, такие как бромистый тетрабутиламмоний, и в особенности простые краунэфиры. Если используют предпочтительное основание, т.е. фтористый калий на глиноземе, то наиболее эффективными являются те простые краунэфиры, которые способны принимать ион калия; в этом отношении особенно полезен 18-краун-6.
Количество применяемого катализатора не является критическим, однако приблизительно 0,05-0,2 молярных эквивалента по отношению к реагенту XII, по-видимому, является оптимальным. Обычно мы находим, что 0,1 эквивалента достаточно без существенных экономических издержек.
Реакцию обычно проводят в течение длительного периода времени в зависимости от применяемой температуры. При комнатной температуре и в присутствии катализатора реакция может быть в значительной степени завершена через несколько дней. Если катализатор не применяют, то реакция может длиться по меньшей мере неделю до существенного завершения, особенно при комнатной температуре. Однако при предпочтительных условиях применения катализатора и повышенных температурах, таких как 90o, большинство реакций обычно заканчивается в течение 24 ч.
Реакцию "разрабатывают" в соответствии с применяемыми условиями. Если применяют нерастворимое основание, такое как смесь фтористого калия и окиси алюминия, то реакционную смесь фильтруют в горячем состоянии и растворитель удаляют в вакууме. Остаток затем может быть растворен в органическом растворителе, в котором растворим конечный продукт, обычно это этилацетат или хлористый метилен. Если используют катализатор, то обычно органический раствор промывают либо водой для удаления катализатора, или, если используют простой краун-эфир6 раствором хлористого натрия для лучшего удаления краун-эфира. Конечный продукт может быть очищен, если желают, любым из известных в данной области стандартных методов, таких как дистилляция, вакуумная дистилляция, хроматография, кристаллизация и т.п.
Промежуточные соединение XII и XIII и любые другие необходимые реагенты, потребные для завершения процесса согласно изобретению, являются либо имеющимися в продаже, известными из литературных источников, или могут быть получены известными в данной области методами.
Упомянутые выше промежуточные соединения и любые другие необходимые реагенты либо имеются в продаже, известны из литературных источников или могут быть получены методами, известными в данной области, как описано более детально ниже. Следует иметь в виду, что возможны различные взаимопревращения различных соединений и промежуточных соединений согласно изобретению. Например, карбоновая кислота может быть этерифицирована стандартными средствами или обращена в галоидангидрид, который затем взаимодействует с амином формулы (R9)2NH или H2NSO2R10 для получения соответствующего амида. Аналогично сложные эфиры, амиды и нитрилы могут быть гидролизованы до карбоновой кислоты средствами, описанными ранее. Нитрилы могут быть также гидролизованы до первичного амида обработкой водным основанием.
В дополнение могут быть использованы предшественники некоторых функциональных групп R1 в синтезе либо различных "половинок" молекулы, после чего "половинки" сочетают (например, схемы I и II). Например, в соединении-предшественнике, в котором R1 является алкенилом, двойная связь может быть окислена перкислотой до соответствующего эпоксидного промежуточного соединения, которое в условиях каталитической гидрогенизации может быть преобразовано в оксиалкильное производное. Восстановлением предшественника, в котором R1 является алканоильной группой, также получают аналог карбинола. Гидрогенизация алкенового производного или дальнейшее восстановление карбинола дает соединение согласно изобретению в котором R1 является алкилом.
Тиопроизводные и промежуточные соединения согласно изобретению (q равно нулю) могут быть преобразованы в соответствующие сульфоксидные (q равно 1) соединения обработкой мягким окислителем, таким как перекись водорода в метаноле, метахлорнадбензойная кислота в хлористом метилене при 0oC, или периодатом щелочного металла в водном спирте. Соответствующие сульфоны (q равно 2) получают из тио- или сульфоксидных соединений обработкой сильным окислителем, таким как перекись водорода в уксусной кислоте или тетра-хлорнадбензойная кислота в хлористом метилене при 20-30oC.
В дополнение различные соединения формулы I могут быть получены из других соединений, предшественников или промежуточных соединений формулы I стандартными методами, такими как гидролиз, этерификация, алкилирование, окисление, восстановление и т.п., как хорошо известно специалистам в данной области. Приведенные выше схемы иллюстрируют более традиционные методы получения соединений согласно этому изобретению. Разумеется, что могут быть эффективно применены различные сочетания этих химических операций и других, вообще известных в органической химии; конкретная последовательность любых таких превращений и взаимопревращений будет оценена специалистами в области органической химии ввиду многообразия функциональных групп, присутствующих в выбранном соединении. Например, тетразольная группа может быть защищена группой, такой как трифенилметил; другая реакция может быть проведена на оставшейся части молекул и трифенилметильную группу удаляют обработкой разбавленной кислотой, чтобы получить незащищенный тетразол. Другие вариации этого и родственных превращений будут очевидны специалистам в данной области.
Следующие получения и примеры иллюстрируют далее получение промежуточных соединений и соединений согласно изобретению. Примеры приведены только для иллюстрации и не ограничивают объема изобретения. Точки плавления определены на аппарате Thomas-Hoover и не корректировались. Спектры ядерного магнитного резонанса (ЯМР) определены на спектрометре GE QE-300. Все химические сдвиги даны в частях на миллион ( δ ) относительно тетраметилсилана. Химические сдвиги ароматических протонов в образцах хинолина в диметилсульфоксиде ДМСО-d6 зависят от концентрации. Следующие сокращения приняты для обозначения сигналов от образцов: с - синглет, д - дублет, т - триплет, кв - квартет, ш - широкий, м - мультиплет. Инфракрасные спектры определены на спектрометре DXIOFT-IR Nicolet. Масс-спектральные данные получены на спектрометре CEC-21-110, используя условия электронного соударения (EI), на спектрометре MAT-731, используя условия свободной десорбции (FD) или на спектрометре YGZAB-3F, используя условия бомбардировки быстрыми атомами. Хроматографию на силикагеле проводили с использованием градиентов этилацетат-гексан, если не указано особо. Обращенно-фазовую хроматографию проводили на геле MCI CHP20P, используя градиент смеси ацетонитрил-вода или метанол-вода6 если не указано особо. Тетрагидрофуран отгоняли из смеси натрий-бензофенон непосредственно перед использованием. Все реакции проводили в атмосфере аргона при перемешивании, если не указано иначе. Там, где структуры подтверждались спектром поглощения ИК, протонным ядерным магнитным резонансом или масс-спектральным анализом, то соединение обозначено как "ИК", "ЯМР" или "МС" соответственно.
Получение 1. Метиловый эфир 5-(3-оксифеноси)-пентановой кислоты.
Смесь 11 г резорцина, 8,8 г метилового эфира 5-бромпентановой кислоты и 13,8 г карбоната калия в 150 мл диметилформамида нагревали в масляной ванне при 90oC 24 ч. Реакционную смесь охлаждали, разбавляли водой и экстрагировали этилацетатом. Органическую фазу промывали водой, промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали указанное в названии примера промежуточное соединение с 34%-ным выходом, ЯМР.
Получения 2-3. Следующие соединения получали способом, согласно получению 1: 5-(3-оксифенокси)-пентаннитрил, 42 %-ный выход, ЯМР.
Этиловый эфир 5-(3-оксифенокси)-пентановой кислоты, 55% выход, ЯМР.
Получение 4. N,N-диметил-4-(3-оксифенил)-бутанамид.
Смесь 3,7 г метилового эфира 4-(3-оксифенил)-пентановой кислоты и 40 мл 40% диметиламина в воде перемешивали 25 ч. Смесь подкисляли 5 н. хлористоводородной кислотой и экстрагировали дихлорметаном. Органическую фазу промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью этиловый эфир-метанол, и получали 1,61 г целевого промежуточного соединения, ЯМР.
Получение 5. Этиловый эфир 3-[2-окси-6-(4-метоксикарбонилбутилокси)-фенил]-пропионовой кислоты.
Смесь 3,1 г метилового эфира 4-(3-оксифенокси)-пентановой кислоты, 0,7 г триметилуксусной кислоты и 2,4 г этилового эфира ортоакриловой кислоты в 50 мл толуола нагревали с обратным холодильником 2 ч. Смесь охлаждали и затем перемешивали с 25 мл 1 н. хлористоводородной кислоты 2 ч. Органическую фазу промывали насыщенным бикарбонатом натрия, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали 1,13 г (25% выход) желаемого целевого промежуточного соединения. ЯМР.
Получения 6-10. Следующие соединения получали способом, описанным в получении:
этиловый эфир 3-(2-окси-6-(4-этоксикарбонилбутилокси)-фенил)-пропионовой кислоты, ЯМР,
этиловый эфир 3-(2-оксифенил)-пропионовой кислоты, 84% выход, ЯМР,
этиловый эфир 3-/2-окси-6-(4-цианобутилокси)-фенил/-пропионовой кислоты, 17 %-ный выход, ЯМР,
этиловый эфир 3-/2-окси-6-(4-диметиламинокарбонилбутилокси)-фенил/-пропионовой кислоты, 14 %-ный выход, ЯМР,
этиловый эфир 3-/2-окси-6-метоксифенил/-пропионовой кислоты, 30%-ный выход, ЯМР.
Получение 11. 3-метокси-1,2-дигидронафталин
Смесь 1,4 г 2-тетрадона, 1,5 мл метилового эфира ортомуравьиной кислоты и несколько кристаллов моногидрата паратолуолсульфокислоты в 75 мл бензола перемешивали 22 ч и затем выпаривали с вакууме. Остаток хроматографировали на флорисиле (Florisil® ), элюируя смесью гексан-этиловый эфир, и получали 1,15 г (75%) желаемого целевого промежуточного соединения, ЯМР.
Получение 12. 3,8-диметокси-1,2-дигидронафталин.
Целевое соединение получали из 5-метокси-2-тетралона способом, описанным в получении 11, с 60 %-ным выходом, ЯМР.
Получение 13. Метиловый эфир 3-(2-формилфенил)-пропионовой кислоты.
Суспензию 4,5 г 3-метокси-1,2-дигидронафталина, 1,0 г бикарбоната натрия, 20 мл метанола и 80 мл дихлорметана перемешивали и охлаждали в ванне со смесью сухой лед-ацетон. Быструю струю озона барботировали в суспензию до появления устойчивого видимого синего окрашивания. Смесь продували азотом и добавляли 3,5 мл метилсульфида. Смесь перемешивали в ванне со смесью лед-ацетон 1 ч и затем 2 ч при комнатной температуре. Органический раствор промывали водой, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир и получали 5,0 г (90%) желаемого целевого промежуточного соединения, ЯМР.
Получение 14. Метиловый эфир 3-(2-формил-6-метоксифенил)-пропионовой кислоты.
Целевое соединение, указанное в названии получения 14, получали способом, описанным в получении 13, с 90%-ным выходом. ЯМР.
Получение 15. 2-метил-2-циано-(2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-гептан
Раствор 1,5-г 2-метил-2-циано-7-(2-этил-4-бром-5-бензилоксифенокси)-гептана и 0,5 г тетракис-(трифенилфосфин)-палладия (0) в 70 мл бензола перемешивали с 15 мл 2,0 м карбоната натрия. Добавляли раствор 1,1 г 4-фторфенилборной кислоты в 15 мл этанола. Смесь нагревали с обратным холодильником 16 ч. Смесь охлаждали и разбавляли этилацетатом. Органическую фазу промывали насыщенным раствором хлористого аммония, промывали насыщенным хлористым натрием, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали 1,44 г (93%) желаемого целевого промежуточного соединения, ЯМР.
Получение 16. 2-метил-2-циано-7-(2-этил-4-(3-фторфенил)-5-бензилокси-фенокси/-гептан
Целевое соединение, указанное в назначении примера, получали способом по получению 15, с 91%-ным выходом. ЯМР
Получение 17. 2-метил-2-(1Н-тетразол-5-ил)-7-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-гептан.
Смесь 1,44 г 2-метил-2-циано-7-(2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-гептана, 4,1 г гидрохлорида триэтиламина и 1,95 г азида натрия в 40 мл диметилформамида нагревали в масляной ванне при 125oC в течение 17 ч, добавляя дополнительно 4 г гидрохлорида триэтиламина и 2 г азида натрия по истечении 5 ч. Смесь охлаждали, разбавляли водой, подкисляли 1,0 н. хлористоводородной кислотой и экстрагировали этилацетатом. Органическую фазу промывали водой, промывали насыщенным хлористым натрием, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью дихлорметан-метанол, и получали 1,12 г (72%) желаемого целевого продукта, ЯМР.
Получение 18. 2-метил-2-(1Н-тетразол-5-ил)-7-/2-этил-4-(3-фторфенил) -5-бензилоксифенокси)-гептан.
Целевое соединение, указанное в названии примера, получали способом, описанным в получении 17, с 75% выходом, ЯМР.
Пример 1. 2-метил-2-(1Н-тетразол-5-ил)-7-/2-этил-4-(4-фторфенил) -5-гидроксифенокси/-гептан
Смесь 1,1 г 2-метил-2-(1Н-тетразол-5-ил)-7-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси)-гептана, 1 г 10 %-ного палладия на угле и 200 мл этанола гидрогенизировали на аппарате под давлением 2,5-3,0 кг/см2 в течение 2 ч. Смесь фильтровали и фильтрат выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью дихлорметанметанол, и получали 750 мг (84%) желаемого целевого соединения. МС, ЯМР.
Кристаллизацией из смеси диэтиловый эфир-гексан получали продукт с точкой плавления 135-137oC: при кристаллизации из толуола точка плавления была 142-143oC.
Элементный анализ для C23H29FN4O2:
Вычислено,%: C 66,97; H 7,09; N 13,58
Найдено,%: C 67,18; H 6,91; N 13,50.
Пример 2. 3-Метил-2-(1Н-тетразол-5-ил)-7-(2-этил-4-(3-фторфенил) -5-оксифенокси/-гептан.
Целевое соединение, указанное в названии примера, получали из соответствующего нитрилового предшественника с 73% выходом, повторяя процесс по примеру 1, ЯМР.
Получение 19. 3-/2-этил-4-(4-фторфенил)-6-бензилоксифенокси/-пропилхлорид
Целевое промежуточное соединение получали со 100 %-ным выходом из хлористого 3-(2-этил-4-бром-5-бензилоксифенокси)-пропила способом, описанным в получении 15, ЯМР.
Получения 20-24. Следующие ниже соединения получали способом, изложенным в получении 1, используя хлористый 3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропил, смешанный с иодистым калием в качестве алкилирующего реагента.
Этиловый эфир 3-{ 2-[3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/ -пропокси]-фенил}-пропионовой кислоты, 40 %-ный выход, ЯМР.
Этиловый эфир 3-{ 2-[3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/ -пропокси] -6-(4-этоксикарбонилбутилокси)-фенил} -пропионовой кислоты, выход 56%, ЯМР.
Этиловый эфир 3-{ 2-[3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/ -пропокси]-6-(4-цианобутилокси)-фенил}-пропионовой кислоты, выход 52%, ЯМР.
Этиловый эфир 3-{ 2-[3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/ -пропокси/-6-(4-диметиламинокарбонилбутилокси)-фенил} -пропионовой кислоты, 24%-ный выход, ЯМР.
Этиловый эфир 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси) -пропокси/-6-метоксифенил}-пропионовой кислоты, 68 %-ный выход, ЯМР.
Получение 25. Этиловый эфир 3-{2-/3-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-пропокси/-6-/4-(1Н-тетразол-5-ил)-бутилокси/-фенил} -пропионовой кислоты.
Целевое соединение получали с 45 %-ным выходом из этилового эфира 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-6-(4-цианобутилокси/-фенил}-пропионовой кислоты способом, описанным в получении 17, ЯМР.
Получение 26. 3-{ -/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/ -пропокси/-фенил}-пропионовая кислота.
Раствор этилового эфира 3-(2-/3-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-пропокси/-фенил/-пропионовой кислоты (375 мг) в 25 мл этанола смешивали с 5 мл 5,0 н. гидроокиси натрия и перемешивали 16 ч. Смесь разбавляли 1,0 н. хлористоводородной кислотой и экстрагировали смесью дихлорметан-изопропиловый спирт в отношении 3:1. Органическую фазу промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия, выпаривали в вакууме и получали желаемое целевое соединение с 93%-ным выходом, ЯМР.
Получения 27-30. Следующие соединения получали из соответствующих сложных эфиров способом по получению 26.
3-{2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-пропокси/ -6-(4-карбоксибутилокси)-фенил}-пропионовая кислота, выход 20%, ЯМР.
3-{2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-пропокси/ -6-метоксифенил}-пропионовая кислота, выход 80%, ЯМР.
3-{2-/3-/2-этил-4-(4-фторфенил)-5-бенизлоксифенокси/-пропокси/-6- (4-диаметиламинокарбонилбутилокси)фенил}-пропионовая кислота, выход 41%, ЯМР.
3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-бенизлоксифенокси/-пропокси/-6- /4-(1Н-тетразол-5-ил)-бутилокси/-фенил}-пропионовая кислота, выход 30%, ЯМР.
Примеры 3-7. Следующие соединения получали из соответствующих бензилокси-предшественником способом по примеру 1.
3. 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-6- (4-диметиламинокарбонилбутилокси)-фенил}-пропионовая кислота, выход 42%, ЯМР.

4.3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил} -пропионовая кислота, выход 15%, ЯМР, МС.

5. 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил} -пропионовая кислота, выход 40%, ЯМР, МС.
Элементный анализ для C31H35FO8:
Вычислено,%: C 67,14; H 6,36,
Найдено,%: C 66,52; H 6,54.

6. 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/ -6-метоксифенил}-пропионовая кислота, выход 16%, ЯМР, МС.

7. 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-6-/- (1Н-тетразол-5-ил)бутилокси/-фенил}-пропионовая кислота, выход 34%, ЯМР, МС.

Получение 31. Метиловый эфир 3-{2-/4-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-1-(1-бутенил)/-6-метоксифенил}-пропионовой кислоты.
К раствору 900 мг иодистого 3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропилтрифенилфосфония в 10 мл метилсульфоксида и 60 мл тетрагидрофурана, охлажденного в ванне со смесью сухой лед-ацетон, добавляли 1,5 л 1,6 М раствор н-бутиллития в гексане. Раствор нагревали до -5oC в течение 30 мин и добавляли раствор 225 мг метилового эфира 3-/(2-формил-6-метокси)-фенил/-пропионовой кислоты в 3 мл тетрагидрофурана. Раствор перемешивали при -5oC в течение 45 мин и нагревали до комнатной температуры. Смесь разбавляли водой, подкисляли 1,0 н. хлористоводородной кислотой и экстрагировали этилацетатом. Органическую фазу промывали водой, промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали 130 мг (23%) желаемого промежуточного целевого соединения, указанного в названии примере, ЯМР.
Получение 32. Метиловый эфир 3-{2-/4-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-(1-бутенил)/-фенил} -пропионовой кислоты.
Целевое соединение получали из метилового эфира 3-(2-формилфенил)-пропионовой кислоты способом, описанным в получении 31, с 60%-ным выходом, ЯМР.
Пример 8. Метиловый эфир 3-{2-/4-/2-этил-4-(4-фторфенил)-5-оксифенокси/-(1-бутенил)/фенил}-пропионовой кислоты.
Целевое соединение, указанное в названии примера, получали способом по получению 31, используя соответствующую дебензилированную соль Виттига и дополнительный эквивалент н-бутиллития, выход 28%, ЯМР.
Получения 33-34. Следующие соединения получали из их соответствующих сложных эфиров способом по приготовлению 26:
3-{2-/4-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-(1-бутенил)/-фенил} -пропионовая кислота, выход 100%, ЯМР, МС.
3-{ 2-/4-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-(1-бутенил)/ -6-метоксифенил}-пропионовая кислота, выход 100%, ЯМР, МС.
Пример 9. 3-{ 2-/4-/2-этил-4-(4-фторфенил)-5-оксифенокси/-(1-бутенил)/ -фенил}-пропионовая кислота.
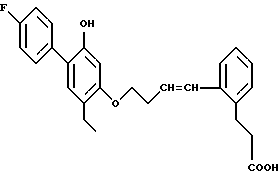
Целевое соединение получали из соответствующего сложного эфира со 100%-ным выходом способом, описанным в получении 26. ЯМР. Отдельные цис- и транс-изомеры были выделены и каждый показал по существу такую же активность, против лейкотриена B4, какая определена для смеси.
Примеры 10-11. Следующие соединения получали из соответствующих бензилокси-предшественников способом, описанным в примере 1.
10. 3-{2-/4-/2-этил-4-(4-фторфенил)-5-оксифенокси)-бутил/-фенил}-пропионовая кислота, выход 61%, ЯМР, МС.
Анализ для C27H29FO4;
Вычислено,%: C 74,29; H 6,70
Найдено,%: C 74,55; H 6,81.

11. 3-{ 2-/4-/2-этил-4-(4-фторфенил)-5-оксифенокси/-бутил/-6-метоксифенил} -пропионовая кислота, выход 75%, ЯМР МС.

Получение 35. 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/ -пропокси/-6-оксифенил}-пропионовая кислота.
Целевое соединение получали из 5-оксибензо-1-пиран-2-она способом, изложенным в получении 1, с 50%-ным выходом, ЯМР.
Получение 36. 5-{ 3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси}-бензо-1-пиран-2-он.
Раствор 1,2 г 5-оксибензо-1-фуран-2-она в 75 мл тетрагидрофурана и 25 мл метилсульфоксида обрабатывали 300 г гидрида натрия (60% в минеральном масле). После перемешивания в течение 10 мин добавляли раствор 1,1 эквивалента иодистого 3-(2-этил-4-(4-фторфенил)-5-бензилоксифенил)-пропила в 10 мл тетрагидрофурана. Раствор перемешивали 19 ч, разбавляли 0,1 н. хлористоводородной кислотой и экстрагировали этилацетатом. Органическую фазу промывали насыщенным хлористым натрием, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали 1,80 г (47%) желаемого целевого промежуточного соединения, ЯМР.
Получение 37. Метиловый эфир 3-{2-/3-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-пропокси/-6-оксифенил}-пропионовая кислота.
К раствору 1,8 г 5-{3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси} -бензо-1-пиран-2-она в 30 мл смеси тетрагидрофурана и метанола в отношении 1: 1 добавляли 40 мл 0,06 М раствора метилата натрия в метаноле. Смесь перемешивали 18 ч, разбавляли водой, подкисляли 1,0 н. хлористоводородной кислотой и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали 1,8 г (100%) желаемого целевого промежуточного соединения, ЯМР.
Примеры 12-13. Следующие соединения получали способом по примеру 1 из соответствующих бензилокси-предшественников.
12. Метиловый эфир 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-6-оксифенил} -пропионовой кислоты, выход 85%, ЯМР.
13. 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-6-оксифенил} -пропионовая кислота, выход 30%, ЯМР, МС.

Получение 38. Метиловый эфир 3-{2-/3-/2-этил-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-6-(4-бутилокси)-фенил}-пропионовой кислоты.
Целевое соединение получали из метилового эфира 3-{2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-6-оксифенил} -пропионовой кислоты, используя иодистый н-бутил и процесс получения 36 с 70%-ным выходом, ЯМР.
Пример 14. 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-6-(4-бутилокси)-фенил} -пропионовая кислота.

Целевое соединение, указанное в названии примера, получали из метилового эфира 3-{2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-6-(4-бутилокси)-фенил} -пропионовой кислоты, повторяя последовательно процесс получения 26 и способ по примеру 1, с 78%-ным выходом, ЯМР, МС.
Пример 15. 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-6- (4-метилтиобутилокси)-фенил}-пропионовая кислота.

A. Получение метилового эфира 3-{2-/3-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-пропокси/-6-(4-хлорбутилокси)-фенил}-пропионовой кислоты.
Целевое соединение, указанное в пункте A, получали с 90%-ным выходом из целевого соединения по получению 37, используя бромистый 4-хлорбутил и процесс по получению 36. ЯМР.
B. Получение метилового эфира 3-{2-/3-/2-этил-4-(4-фторфенил) -5-оксифенокси/-пропокси/-6-(4-хлорбутилокси)-фенил}-пропионовой кислоты.
Целевое соединение получали из метилового эфира 3-{2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-5- (4-хлорбутилокси)-фенил} -пропионовой кислоты с 60%-ным выходом, используя процесс по примеру 1, ЯМР.
C. Получение метилового эфира 3-{2-/3-/2-этил-4-(4-фторфенил) -5-оксифенокси/-пропокси/-6-(4-метилтиобутилокси/-фенил}-пропионовой кислоты.
Раствор 420 мг метилового эфира 3-{2-/3-/2-хтил-4-(4-фторфенил)-5-оксифенокси/-пропокси-6-(4-хлорбутилокси)-фенил} -пропионовой кислоты в 10 мл тетрагидрофурана добавляли к 70 мл 0,14 М раствора метан-меркаптила натрия в диметилформамиде. Смесь перемешивали 2 ч, разбавляли водой и экстрагировали этилацетатом. Органическую фазу промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия и упаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали желаемое целевое промежуточное соединение с 98%-ным выходом, ЯМР.
D. Получение 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/ -6-(4-метилтиобутилокси)-фенил}-пропионовой кислоты.
Целевое соединение получали с 94% выходом из метилового эфира 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-6- (4-метилтиобутилокси)-фенил}-пропионовой кислоты способом, изложенным в получении 26, ЯМР, МС.
Пример 16. 3-{ 2-(3-/2,4-ди-(4-фторфенил)-5-оксифенокси/пропокси/-6- (4-карбоксибутокси)-фенил}-пропионовая кислота.

A. Получение этилового эфира 3-{2-/3-/2,4-ди-(4-фторфенил) -5-бензилоксифенокси/-пропокси/-6-(4-этоксикарбонилбутилокси)-фенил} -пропионовая кислота.
Целевое промежуточное соединение получали из этилового эфира 3-/2-окси-6-(4-этоксикарбонилбутилокси)-фенил/-пропионовой кислоты и иодистого 3-/2,4-ди-(4-фторфенил)-5-бензилоксифенокси/-пропила, руководствуясь процедурой по получению 1, ЯМР.
B. Получение 3-{2-/3-/2,4-ди-(4-фторфенил)-5-бензилоксифенокси/ -пропокси/-6-(4-карбоксибутокси)-фенил}-пропионовой кислоты.
Целевое промежуточное соединение получали выделением со 100% выходом в виде масли из этилового эфира 3-{2-/3-/2,4-ди-(4-фторфенил) -5-бензилоксифенокси/-пропокси/-6-(4-этоксикарбонилбутокси)-фенил} -пропионовой кислоты, следуя процессу получения 26, ЯМР.
C. Получение 3-{2-/3-/2,4-ди-(4-фторфенил)-5-оксифенокси/-пропокси/-6-(4-карбоксибутокси)-фенил}-пропионовой кислоты.
Целевое соединение получали с 44%-ным выходом из 3-{2-/3-/2,5-ди-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-6-(4-карбоксибутокси)-фенил} -пропионовой кислоты, следуя процессу по примеру 1, ЯМР, МС.
Получение 39. 7-(2-ацетил-5-бензилоксифенокси)-2-метил-2-н-пентилгептаннитрил.
Целевое промежуточное соединение получали из 2-окси-5-бензилоксиацетофенона и 2-метил-2-н-пентил-7-иодогептаннитрила, следуя процедуре получения 36.
Получение 40. 2-метил-2-н-пентил-7-(2-этил-5-бензилоксифенокси)-гептаннитрил.
7-(2-ацетил-5-бензилоксифенокси)-2-метил-2-н-пентилгептаннитрил растворяли в четыреххлористом углероде, содержавшем 6 эквивалентов триэтилсилана. После добавления 60 эквивалентов трифторуксусной кислоты раствор перемешивали 24 ч и затем выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали целевое соединение в виде масла с 90%-ным выходом, ЯМР.
Получение 31. 2-Метил-2-н-пентил-7-(2-этил-4-бром-5-бензилоксифенокси)-гептан-нитрил.
2-Метил-2-н-пентил-7-(2-этил-5-бензилоксифенокси)-гептаннитрил и 1,1 эквивалента N-бромсукцинимида в четыреххлористом углероде перемешивали 1,5 ч, промывали водным тиосульфатом натрия, промывали насыщенным хлористым натрием, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали желаемое целевое промежуточное соединение с 60%-ным выходом в виде масла, ЯМР.
Получение 42. 2-Метил-2-н-пентил-7-(2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-гептаннитрил.
Целевое промежуточное соединение получали из 2-метил-2-н-пентил-7-(2-этил-4-бром-5-бензилоксифенокси)-гептаннитрила с 94 %-ным выходом в виде масла, следуя процедуре получения 15, ЯМР.
Получение 43. 6-Метил-6-(1Н-тетразол-5-ил)-11-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-ундекан.
Целевое соединение выделяли в виде масла с 42%-ным выходом взаимодействием 2-метил-2-н-пентил-7-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-гептаннитрила согласно процедуре получения 17, ЯМР.
Пример 17. 6-Метил-6-(1Н-тетразол-5-ил)-11-(2-этил-4-(4-фторфенил) -5-оксифенокси/-ундекан.

Целевое соединение получали из 6-метил-6-(1Н-тетразол-5-ил)-11-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-ундекана с 68% выходом, используя процесс по примеру 1, ЯМР, МС.
Элементный анализ для C27H37FN4O2:
Вычислено,%: C 69,20; H 7,96; N 11,96.
Найдено,%: C 69,50; H 8,22; N 12,00.
Пример 18. N, N-диметил-3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/ -пропокси/-фенил}-пропионамид.

Раствор 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил} -пропионовой кислоты и нескольких эквивалентов хлористого тионила в дихлорметане выдерживали при комнатной температуре 3 ч и затем выливали в перемешиваемый раствор 40% диметиламина в воде. Органический слой промывали водной хлористоводородной кислотой, промывали насыщенным хлористым натрием, сушили над сульфатом натрия и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя этилацетатом, и получали желаемое целевое соединение, ЯМР, МС.
Пример 19. N-Метансульфонил-3-{2-/3-/2-этил-4-(4-фторфенил) -5-оксифенокси/-пропокси/-фенил}-пропионамид.

Раствор хлорангидрида получали как и в примере 18 в тетрагидрофуране и добавляли к суспензии 10 эквивалентов -ли-тиометансульфонамида в тетрагидрофуране при -5oC. Смесь нагревали до комнатной температуры, разбавляли водной хлористоводородной кислотой и экстрагировали этилацетатом. Органический раствор сушили и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью дихлорметан-метанол, и получали желаемое целевое соединение с 37%-ным выходом, ЯМР.
Пример 20. N-фенилсульфонил-3-{2-/3-/2-этил-4-(4-фторфенил) -5-оксифенокси/-пропокси/-фенил}-пропионамид
Целевое соединение получали способом по примеру 19, используя N-литиобензолсульфонамид. Продукт выделяли препаративной на С18 обращенно-фазовой жидкостной хроматографией высокого давления, ЯМР.
Получение 44. 4-(2,4-диметоксифенил)-фторбензол.
Целевое промежуточное соединение представляло собой масло, полученное с 85%-ным выходом из 2,4-диметоксибромбензола способом по получению 15, ЯМР.
Получение 45. 2,4-диметокси-5-(4-фторфенил)-ацетофенон.
Раствор 4-(2,4-диметоксифенил)-фторбензола в дихлорметане при -5oC обрабатывали 2 эквивалентами хлористого олова и 1,5 эквивалентами хлористого ацетила. Раствор перемешивали 2 ч без охлаждения. Органический раствор сушили, выпаривали в вакууме и получали целевое промежуточное соединение в виде масла с 97%-ным выходом, ЯМР.
Получение 46. 2,4-Диметоксибутирофенон.
Целевое соединение выделяли в виде масла с 81%-ным выходом и- 4-(2,4-диметоксифенил)-фторбензола, используя процесс по получению 45, ЯМР.
Получение 47. 2-окси-4-метокси-5-(4-фторфенил)-ацетофенон.
Дихлорметановый раствор 2,4-диметокси-5-(4-фторфыенил)-ацетофенона обрабатывали 1,2 эквивалентами треххлористого бора при 0oC в течение 15 мин. Органический раствор промывали водой, сушили, выпаривали в вакууме и получали желаемое промежуточное соединение с 96%-ным выходом, ЯМР.
Получение 48. 2-окси-4-метокси-5-(4-фторфенил)-бутирофенон.
Целевое соединение, указанное в названии примера, получали с 97%-ным выходом из 2,4-диметоксибутирофенона способом по получению 47, ЯМР.
Получение 49. 2-(3-хлорпропокси)-4-метокси-5-(4-фторфенил)-ацетофенон.
Целевое соединение выделяли в виде масла с 53%-ным выходом из 2-окси-4-метокси-5-(4-фторфенил)-ацетофенона и бромистого 3-хлорпропила способом по получению 36, ЯМР.
Получение 50. 2-(3-хлорпропокси)-4-метокси-5-(4-фторфенил)-бутирофенон.
Целевое соединение выделяли в виде масла с 64%-ным выходом из 2-окси-4-метокси-5-(4-фторфенил)-бутирофенона и бромистого хлорпропила способом по получению 36, ЯМР.
Получение 51. 1-/2-(3-хлорпропокси)-4-метокси-5-(4-фторфенил)-фенил/-бутан.
Целевое соединение, указанное в названии примера, выделяли в виде масла 89% выходом из 2-(3-хлорпропокси)-4-метокси-5-(4-фторфенил)-бутирофенона, используя способ по получению 40, ЯМР.
Получение 52. Этиловый эфир 3-{2-/3-/2-бутил-4-(4-фторфенил) -5-метоксифенокси/-пропокси/-фенил}-пропионовой кислоты.
Целевое промежуточное соединение получали из 1-/2-(3-хлорпропокси)-4-метокси-5-4-фторфенил)-фенил/-бутана и этилового эфира 3-(2-окси)-фенилпропионовой кислоты способом по получению 36: соединение представляло собой масло, полученное с 80 %-ным выходом, ЯМР.
Пример 21. 3-{2-/3-/2-бутил-4-(4-фторфенил)-5-оксифенокси) -пропокси/-фенил}-пропионовая кислота.

Этиловый эфир 3-{ 2-/3-/2-бутил-4-(4-фторфенил)-5-метокси-фенокси/ -пропокси/фенил} -пропионовой кислоты в дихлорметане обрабатывали 2 эквивалентами трехбромистого бора при -75oC. Смесь перемешивали без охлаждения 18 ч, промывали водой, сушили и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью дихлорметанметанол, и получали желаемое целевое соединение с 56%-ным выходом, ЯМР.
Пример 21 - альтернативный синтез. 3-{2-/3-/2-бутил-4-(4-фторфенил) -5-оксифенокси/-пропокси/-фенил}-пропионовая кислота.
Раствор этилового эфира 3-{2-/3-/2-бутил-4-(4-фторфенил)-5-метоксифенокси/ -пропокси/-фенил} -пропионовой кислоты и 5 эквивалентов этантиолята натрия в диметилформамиде нагревали при 110oC 2 ч, охлаждали, разбавляли водной хлористоводородной кислотой и экстрагировали этилацетатом. Органический раствор промывали водой, сушили и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя этилацетатом, и получали желаемое целевое соединение, указанное в названии примера, с 66%-ным выходом, ЯМР.
Получение 53. Этиловый эфир 3-/2-(4-иодобутокси)-фенил/-пропионовой кислоты.
Целевое соединение получали с 85%-ным выходом из этилового эфира 3-(2-оксифенил)-пропионовой кислоты и бромистого 4-хлорбутила способом по получению 36, с последующей обработкой иодистым натрием: продукт представлял собой масло, ЯМР.
Получение 54. Этиловый эфир 3-/2-/4-(2-ацетил-5-бензилоксифенокси) -бутокси/-фенил-пропионовой кислоты.
Целевое промежуточное соединение выделяли в виде масли с 71 %-ным выходом из этилового эфира 3-/2-(4-иодобутокси)-фенил/-пропионовой кислоты способом по получению 36, ЯМР.
Получение 55. Этиловый эфир 3-/2-/4-(2-этил-5-бензилоксифенокси)-бутилокси)-фенил/-пропионовой кислоты.
Целевое промежуточное соединение выделяли в виде масла и 85%-ным выходом из этилового эфира 3-{ 2-/4-(2-ацетил-5-бензилоксифенокси)-бутилокси/-фенил-пропионовой кислоты способом по получению 40, ЯМР.
Получение 56. Этиловый эфир 3-/2-/4-(2-этил-4-бром-5-бензилоксифенокси) -бутилокси/-фенил/-пропионовой кислоты.
Целевое соединение выделяли в виде масла с 85%-ным выходом из этилового эфира 3-/2-/4-(2-этил-5-бензилоксифенокси)-бутилокси/-фенил/-пропионовой кислоты способом, описанным в получении 41, ЯМР.
Получение 57. Этиловый эфир 3-{2-/4-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-бутилокси/-фенил}-пропионовой кислоты.
Указанное в названии примера промежуточное соединение выделяли из этилового эфира 3-/2-/4-(2-этил-4-бром-5-бензилоксифенокси)-бутилокси/-фенил/-пропионовой кислоты способом по получению 15, ЯМР.
Пример 22. Этиловый эфир 3-{2-/4-/2-этил-4-(4-фторфенил)-5-оксифенокси/-бутилокси/-фенил}-пропионовой кислоты.
Указанное в названии примера промежуточное соединение выделяли из этилового эфира 3-{2-/4-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/ -пропокси/-фенил}-пропионовой кислоты способом по примеру 1, ЯМР.
Пример 23. 3-{2-/4-/2-этил-4-(4-фторфенил)-5-оксифенокси/-бутилокси/-фенил}-пропионовая кислота.

Целевое соединение получали с 72%-ным выходом способом по получению 26, используя в качестве реагента этиловый эфир 3-{2-/4-(2-этил-4-(4-фторфенил)-5-оксифенокси/-бутилокси/-фенил}-пропионовой кислоты, ЯМР.
Получение 58. Метиловый эфир 4-(3-аллилоксифенокси)-бензойной кислоты.
Соединение, указанное в названии примера, получали с 96%-ным выходом в виде масли из метилового эфира 4-(3-оксифенокси)-бензойной кислоты и бромистого аллила способом по получению 36, ЯМР.
Получение 59. Метиловый эфир 4-(2-аллила-3-оксифенокси)-бензойной кислоты и метиловый эфир 4-(4-аллил-3-оксифенокси)-бензойной кислоты.
Раствор метилового эфира 4-(3-аллилоксифенокси)-бензойной кислоты в N, N-диметиланилине нагревали при 190oC в течение 19 ч, охлаждали, разбавляли этилацетатом, промывали водной хлористоводородной кислотой, сушили и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали смесь указанных в названии примера соединений в отношении 40:60 в виде масла с 92%-ным выходом, ЯМР.
Получение 60. Метиловый эфир 4-/2-(3-оксипропил)-3-оксифенокси/бензойной кислоты и метиловый эфир 4-/3-окси-4-(3-оксиропил)-фенокси/-бензойной кислоты.
Тетрагидрофурановый раствор целевых соединений по получению 59 обрабатывали 3,4 эквивалента 9-борабициклононана в течение 16 ч. Смесь охлаждали до -5oC, обрабатывали 50 эквивалентами водного ацетата натрия и затем 5,0 эквивалентами перекиси водорода. Смесь перемешивали без охлаждения 6 ч, разбавляли водным тиосульфатом натрия, подкисляли хлористоводородной кислотой и экстрагировали этилацетатом. Органический раствор сушили и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью дихлорметан-метанол, и получали смесь целевых соединений с количественным выходом, ЯМР.
Получение 61. Метиловый эфир 3-(2-окси-6-/4-(метоксикарбонил)-фенокси/-фенил/-пропионовой кислоты и метиловый эфир 3-/2-окси-4-/4-(метоксикарбонил)-фенокси/-фенил/-пропионовой кислоты.
Раствор целевых соединений по получению 60 в ацетоне при -5oC обрабатывали большим избытком реактивы Джонеса и затем перемешивали без охлаждения 1,5 ч. Избыток окислителя разлагали изопропанолом и смесь экстрагировали этилацетатом. Органический раствор сушили и выпаривали в вакууме. Остаток растворяли в метаноле, содержащем несколько капель серной кислоты, нагревали с обратным холодильником 5 ч, охлаждали, концентрировали в вакууме, разбавляли этилацетатом, промывали водным бикарбонатом натрия, сушили и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали смесь целевых соединений в виде масла с 70%-ным выходом, ЯМР.
Получение 62. Метиловый эфир 3-{2-/3-/2-этил-4-(4-фторфенил) -5-бензил-оксифенокси)-пропокси/-6-/4-(метоксикарбонил)-фенокси/-фенил} -пропионовой кислоты и метиловый эфир 3-{2-/3-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-пропокси/-4-(4-метоксикарбонил)-фенокси/-фенил} -пропионовой кислоты.
Взаимодействие соединений по получению 61 и йодистого 3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропила способом, изложенным в получении 36, давало смесь целевых соединений, указанных в названии настоящего примера, в отношении 40:60 в виде масла с 30%-ным выходом, ЯМР.
Пример 24. Метиловый эфир 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/ -пропокси/-6-/4-метоксикарбонил)-фенокси/-фенил} -пропионовой кислоты и метиловый эфир 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/ -пропокси/-4-(метоксикарбонил)-фенокси/-фенил}-пропионовой кислоты.
Указанные в названии примера сложные эфиры получали из соединений по получению 62, способом по примеру 1, в форме смеси целевых соединений, в виде масла, со 100%-ным выходом, ЯМР.
Примеры 25 и 26. 3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/ -6-(4-карбоксифенокси)-фенил}-пропионовая кислота.

и 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-4- (4-карбоксифенокси)-фенил}-пропионовая кислота
Гидролиз целевых соединений по примеру 24 способом по получению 26 давал смесь целевых соединений, которую разделяли препаративной обращенно-фазовой жидкостной хроматографией высокого давления на колонке С18.
25. 3{ -2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси)-пропокси/-6- (4-карбкосифенокси)-фенил}-пропионовая кислота, выход 49%, ЯМР.
26. 3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-4- (4-карбоксифенокси)-фенил}-пропионовая кислота, 34% выход, ЯМР.
Получение 63. 3,3-диметил-3-{2-/3-/2-этил-4-(4-фторфенил )-5-бензилоксифенокси/-пропокси/-фенил}-пропионовая кислота.
Раствор 4,4-диметилбензофуран-2-она в диметилсульфоксиде обрабатывали 2,0 эквивалентами гидроокиси калия в течение 16 ч. Добавляли раствор двух эквивалентов иодистого 3-/2-этил-4-(4-фторфенил)-5-бензлиоксифенокси/-пропила в смеси диметилсульфоксида и тетрагидрофурана и перемешивали 1 ч, разбавляли водной хлористоводородной кислотой и экстрагировали этилацетатом. Органический раствор промывали водой, сушили и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью гексан-этиловый эфир, и получали желаемое целевое промежуточное соединение, указанное в названии примера, в виде масла с 29%-ным выходом, ЯМР.
Также выделено было целевое соединение, этерифицированное иодистым алкилом, масло, 50%-ный выход, ЯМР.
Пример 27. 3,3-диметил-3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/фенил}-пропионовая кислота.
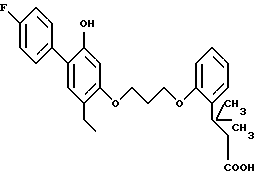
Целевое соединение получали с 75%-ным выходом из 3,3-диметил-3- {2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-пропокси/-фенил} -пропионовой кислоты способом по примеру 1, ЯМР.
Получение 64. 2-циано-2метил-3-{2-/3-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси/-пропокси/фенил}-пропан.
Целевое промежуточное соединение выделяли в виде масла с 77%-ным выходом из 2-циано-2-метил-3-(2-окси)-фенилпропана и иодистого 3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропила способом по получению 36, ЯМР.
Получение 65. 2-Метил-2-(1Н-тетразол-5-ил)-3-{2-/3-/2-этил-4-(4-фторфенил) -5-бензилоксифенокси)-пропокси/-фенил}-пропан.
Целевое промежуточное соединение выделяли в виде масла и 77%-ным выходом из 2-циано-2-метил-3-{ 2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/ -пропокси/-фенил}-пропана способом по получению 17, ЯМР.
Пример 28. 2-Метил-2-(1Н-тетразол-5-ил)-3-{2-/3-/2-этил-4-(4-фторфенил) -5-оксифенокси/-пропокси/фенил}-пропан.
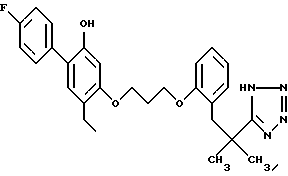
Дебензилирование 2-метил-2-(1Н-тетразол-5-ил)-3-{ 2-/3-/2-этил-4- (4-фторфенил)-5-бензлиоксифенокси/-пропокси, -фенил} -пропана, проведенное способом по примеру 1, давало целевое соединение с 76%-ным выходом, ЯМР.
Получение 66. 2-/3-/2-этил-4-(4-фторфенил)-5-бензилокси/-пропокси/-бензальдегид
Целевое промежуточное соединение получали из салицилового альдегида с 71%-ным выходом способом по получению 36, ЯМР.
Получение 67. (±)-2,2-диметил-3-окси-3-{2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-фенил}-пропаннитрил.
Раствор 1,3 мл изобутиронитрила в 100 мл толуола при -5oC обрабатывали одним эквивалентом диизопропиламида лития и затем нагревали до комнатной температуры. Добавляли раствор 690 мг целевого соединения по получению 66 в 10 мл толуола и суспензию перемешивали 1 ч, выливали в разбавленную водную хлористоводородную кислоту и экстрагировали этилацетатом. Органическую фазу промывали, сушили и выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя дихлорметаном, и получали целевое промежуточное соединение с 91%-ным выходом, ЯМР.
Получение 68. 2-Метил-2-(1Н-тетразол-5-ил)-3-окси-3-{2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси/фенил}-пропан.
Раствор 715 мг целевого соединения по получению 67 и 8 мл азида три-н-бутилолова нагревали при 95oC в течение 92ч, охлаждали, разбавляли 5 мл тетрагидрофурана, 25 мл ацетонитрила и 10 мл уксусной кислоты и затем перемешивали 3,5 ч. Раствор хорошо промывали гексаном и затем выпаривали в вакууме. Остаток хроматографировали на силикагеле, элюируя смесью дихлорметан-метанол, и получали целевое соединение с 87%-ным выходом, ЯМР.
Пример 29. 3-Метил-2-(1Н-тетразол-5-ил)-3-окси-3-{2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил}-пропан.

Указанное в названии примера целевое соединение с 67%-ным выходом получали из целевого соединения по получению 68, способом по примеру 1, ЯМР, МС.
Элементный анализ для C28H31FN4O4:
Вычислено,%: C 66,34; H 6,17; N 11,06.
Найдено,%: C 66,41; H 6,34 N 11,07.
Получение 69. 4-(2,4-Диметокси-5-бромфенил)-фторбензол.
Целевое промежуточное соединение получали из 4-(2,4-диметоксифенил)-фторбензола и 97%-ным выходом способом по получению 41, ЯМР.
Получение 70. 4-(2-Метил-4-окси-5-бромфенил)-фторбензол и 4-(2-окси-4-метокси-5-бромфенил)-фторбензол.
Каждое из целевых соединений выделяли с 35 %-ным выходом из преобразованного целевого соединения по получению 69 способом по примеру 21, ЯМР.
Получение 71. Хлористый 3-/4-(4-фторфенил)-2-бром-5-метоксифенокси)-пропил.
Целевое промежуточное соединение получали из 4-(2-метокси-4-окси-5-бромфенил)-фторбензола и хлористого 3-бромпропила с 84 %-ным выходом способом по получению 36, ЯМР.
Получение 72. Этиловый эфир 3-{2-/3-/2-бром-4-(4-фторфенил) -5-метоксифенокси/-пропокси/фенил}-пропионовой кислоты.
Целевое промежуточное соединение получали из хлористого 3-/4-(4-фторфенил)-2-бром-5-метоксифенокси/-пропила и этилового эфира 3-(2-оксифенил)-пропионовой кислоты с 76%-ным выходом способом по получению 36, ЯМР.
Примеры 30-31. 3-{2-/3-/2-бром-4-(4-фторфенил)-5-оксифенокси/-пропокси/ -фенил}-пропионовая кислота.

3-{ 2-/3-/2-этилтио-4-94-фторфенил)-5-оксифенокси/-пропокси/-фенил} -пропионовая кислота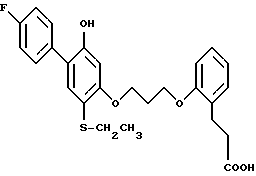
Целевые соединения, указанные в названии примера, получали из целевых соединений по получению 72 способом по примеру 21 (альтернативный синтез).
30. 3-{ 2-/3-/2-бром-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил}-пропионовая кислота, выход 18%, ЯМР.
Элементный анализ для C24H22BrFO5:
Вычислено,%: C 58,91; H 4,53; Br 16,33.
Найдено,%: C 59,17; H 4,72; Br 16,48.
31. 3-{ 2-/3-/2-этилтио-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил} -пропионовая кислота, выход 10%, ЯМР.
Пример 32. Метиловый эфир 3-{2-окси-3-(4-метоксикарбонилбутил)-6-/3- /2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил}-пропионовой кислоты.
Смесь 250 мг этилового эфира 3-2-окси-3-(4-метоксикарбонилбутил) -6-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил-пропионовой кислоты, 0,1 г 10% палладия на угле и несколько капель концентрированной серной кислоты в 125 мл метанола гидрогенизировали в аппарате Парра под давлением 3,1 кг/см2 в течение 18 ч. Смесь фильтровали и фильтрат выпаривали в вакууме, получая целевое промежуточное соединение с 80%-ным выходом, ЯМР.
Пример 33. 5-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-8-(4-карбоксибутил)-дигидрокумарин.

Целевое соединение получали из метилового эфира 3-{2-окси-3-(4-метоксикарбонилбутил)-6-/2-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил}-пропионовой кислоты способом по получению 26. Целевое соединение очищали препаративной обращенно-фазовой жидкостной хроматографией высокого давления и выделяли с 15%-ным выходом, ЯМР.
Получение 73. 1-Бензилокси-2-фенил-4-этил-5-(6-метил-6-цианогептилокси)-бензол
A. Получение 4-бензилокси-2-оксиацетофенона.
В сухой колбе с круглым дном в атмосфере азота растворяли 2,4-диоксиацетофенон (15,2 г, 100 ммоль) в метилэтилкетоне (400 мл) и диметилсульфоксиде (100 мл). К этому раствору добавляли бромистый бензил (17,0 г, 100 ммоль) и карбонат калия (27,6 г, 200 ммоль). Реакционную смесь нагревали с обратным холодильником и перемешивали в течение 15 ч. Метилэтилкетон удаляли в вакууме, раствор диметилсульфоксида разбавляли этилацетатом и несколько раз промывали рассолом. Органическую фазу собирали, сушили (сульфат магния), фильтровали, концентрировали и получали твердое вещество темного цвета. Твердое вещество перекристаллизовывали из смеси гексан-толуол и получали целевой бензиловый эфир в виде твердого вещества рыжевато-коричневого цвета (12,8 г, 55,7%), точка плавления 143-144,5oC, ЯМР.
(CDCl3) δ 12,77 (с, 1Н), 7,70 (д, 1Н, J=7 Гц), 7,3-7,5 (м, 5Н), 6,54 (д, 1Н, J=7 Гц), 6,53 (с, 1Н), 5,11 (с, 2Н), 2,58 (с, 3Н).
Элементный анализ для C15H12O3:
Вычислено,%: C 74,36; H 5,82
Найдено,%: C 74,52; H 5,97.
B. Получение 2-(6-метил-6-цианогептилокси)-4-бензил-оксиацетофенона.
К раствору 4-бензилокси-2-оксиацетофенона (9,65 г, 42 ммоль) в диметилформамиде (150 мл) добавляли подходящий хлористый алкил (6,86 г, 40 ммоль), карбонат калия (10,6 г, 77 ммоль) и иодистый калия (1,6 г, 9,6 ммоль). Перемешиваемую реакционную смесь нагревали до 90oC за 24 ч. Твердые вещества удаляли фильтрацией и диметилформамид удаляли в вакууме. Остаток очищали препаративной жидкостной хроматографией высокого давления, используя градиент от 5 до 20% этилацетата в гексане в течение 30 мин в качестве подвижной фазы и получали целевой простой эфир в виде прозрачного масла (12,1 г, 79,8%).
ЯМР (CDCl3), δ 7,85 (д, 1Н, J=7,4 Гц), 7,3-7,5 (м, 5Н), 6,60 (дд, 1Н, J= 7,4 и 1,8 Гц), 6,53 (д, 1Н, J=1,8 Гц), 5,12 (с, 2Н), 4,04 (т, 2Н, J=5,3 Гц), 2,61 (с, 3Н), 1,85-1,95 (м, 2Н), 1,5-1,6 (м, 6Н), 1,37 (с, 6Н): ИК-спектр (CHCl3): 2943, 2238, 1601 см-1: масс-спектр: (m/e) 379.
C. Получение 4-бензилокси-2-(6-метил-6-цианогептилокси)-этилбензола.
К раствору 2-(6-метил-6-цианогептилокси)-4-бензилоксиацетофенона (12,1 г, 31,6 ммоль) в четыреххлористом углероде (30 мл) добавляли трифторуксусную кислоту (44,4 г, 390 ммолей) и триэтиламин (21,8 г, 188 ммоль). Реакционную смесь перемешивали при комнатной температуре 1,5 ч, затем обрабатывали разбавлением этилацетатом и промывкой водным карбонатом натрия. Органическую фазу собирали, сушили (сульфат магния), фильтровали и концентрировали в вакууме. Остаток очищали препаративной жидкостной хроматографией высокого давления, используя градиент от 3 до 5% этилацетата в гексане в течение 15 мин, затем обтирая при 5 %-ной концентрации подходящие фракции, получали желаемой целевое соединение (10,6 г, 91,5%) в виде прозрачной жидкости.
Спектр ЯМР (CDCl3) δ : 7,35-7,5 (м, 5Н), 7,06 (д, 1Н, J=6,5 Гц), 6,53 (с, 1Н), 6,52 (дд, 1Н, J=6,5 и 2,0 Гц), 5,06 (с, 2Н), 3,96 (т, 2Н, J=5,3 Гц), 2,60 (кв, 2Н, J=6,3 Гц), 1,8-1,85 (м, 2Н), 1,5-1,6 (м, 6Н), 1,37 (с, 6Н), 1,20 (т, 3Н, J=6,3 Гц).
D. Получение 1-бром-2-бензилокси-4-(6-метил-6-цианогептилокси)-5-этилбензола
К перемешиваемому раствору 4-бензилокси-2-(6-метил-5-цианогептилокси)-этилбензола (10,6 г, 28,9 ммоль) в четыреххлористом углерода (125 мл) добавляли N-бромсукцинимид (6,0 г, 33,3 ммоля). Перемешивание продолжали 6 ч при комнатной температуре. Затем смесь разбавляли хлористым метиленом и промывали водой. Органическую фазу собирали, сушили (сульфат магния), фильтровали и концентрировали в вакууме. Остаток перекристаллизовывали из смеси гептан-этилацетат и получали целевое соединение бромистого арила (12,6 г, 97,8%) в виде бледножелтого твердого вещества.
ЯМР (CDCl3) δ : 7,35-7,5 (м, 5Н), 7,22 (м, 1Н), 6,50 (с, 1Н), 5,17 (с, 2Н), 3,90 (т, 2Н, J=5,3 Гц), 2,58 (кв, 2Н, J=6,3 Гц), 1,75-1,85 (м, 2Н), 1,50-1,65 (м, 6Н), 1,37 (с, 6Н), 1,18 (т, J=6,3 Гц, 3Н). ИК-спектр (CHCl3): 3020, 2981, 2946, 2238, 1662, 1600 см-1: Масс-спектр (m/e) 444, 445, 446.
E. Пример осуществления реакции диарильного сопряжения.
Метода A. В колбе с круглым дном растворяли соответствующий бромистый арил (один эквивалент) в бензоле. К этому раствору добавляли тетракис-(трефенилфосфин)палладия (Pd(PPh3)4 / (10 мол.%) и 2,0 М водного раствора карбоната натрия (10 эквивалентов). В отдельной колбе растворяли арилборную кислоту (два эквивалента) в этаноле. Раствор арилборной кислоты добавляли к раствору бромистого арила и смесь нагревали с обратным холодильником и перемешиванием 16 ч. Смесь разбавляли этилацетатом и промывали насыщенным водным раствором хлористого аммония. Органическую фазу собирали, сушили (сульфат магния), фильтровали и концентрировали. Остаток очищали тонкослойной хроматографией (6%-ный этилацетат в гексане) и получали желаемый диарил.
Метод B. Раствор соответствующего бромистого арила в тетрагидрофуране охлаждали до -78oC. К этому раствору добавляли третбутиллитий (два эквивалента). Реакционную смесь перемешивали при -78oC 30 мин, затем добавляли тетрагидрофурановый раствор хлористого цинка (один эквивалент). Смесь нагревали до комнатной температуры и перемешивали 15 мин. В отдельной колбе готовили раствор, содержащий соответствующий галоидный арил (один эквивалент) и Pd(PPh3)4 (10 мол.%) в тетрагидрофуране. Этот раствор добавляли к раствору арилцинка и смесь перемешивали при комнатной температуре 2-18 ч. Реакционную смесь разбавляли этилацетатом и промывали водным раствором хлористого аммония. Органическую фазу сушили (сульфат магния), фильтровали и концентрировали. Остаток очищали тонкослойной хроматографией (6%-ный этилацетат в гексане) и получали желаемый диарил.
F. Получение 1-бензилокси-2-фенил-4-этил-5-(6-метил-6-цианогептилокси) -бензола.
Это соединение получали с 75%-ным выходом по методу A. ЯМР-спектр (CDCl3), δ : 7,60 (д, 2Н, J=6,5 Гц), 7,3-7,5 (м, 8Н), 7,18 (с, 1Н), 6,59 (с, 1Н), 5,04 (с, 2Н), 3,95 (т, 2Н, J=5,3 Гц), 2,63 (5с. 2Н, J=6,3 Гц), 1,8-1,9 (м, 2Н), 1,5-1,65 (м, 6Н), 1,38 (с, 6Н), 1,25 (т, 3Н, J=6,3 Гц). ИК-спектр (CHCl3): 3013, 2977, 2943, 2238, 1611, 1488 см-1: масс-спектр (m/e), 439.
Элементный анализ для C30H35NO2:
Вычислено,%: C 81,59; H 7,99; N 3,17
Найдено,%: C 81,34; H 8,18; N 3,05.
Получения 74-80.
Следующие промежуточные соединения получали, следуя процессам, описанным в получении 73, применяя соответствующую арилборную кислоту или арилгалогенид.
1-Бензилокси-2-(4-метилфенил)-4-этил-5-(6-метил-6-цианогептилокси-бензол, выход 58% по методу A.
Спектр ЯМР (CDCl3) δ : 7,49 (д, 2Н, J=7,0 Гц), 7,3-7,4 (м, 5Н), 7,22 (д, 2Н, J=7,0 Гц), 7,15 (с, 1Н), 6,56 (с, 1Н), 5,05 (с, 2Н), 3,96 (т, 2Н, J=5,3 Гц), 2,64 (кв. 2Н, J=6,3 Гц), 2,41 (с, 3Н), 1,8-1,9 (м, 2Н), 1,5-1,65 (м, 6Н), 1,38 (с, 6Н), 1,22 (т, 3Н, J=6,3 Гц):
Спектр ИК (CHCl3): 3018, 2977, 2934, 2878, 1611, 1419, 1219 см-1: масс-спектр (m/e) 456.
Элементный анализ для C31H37NO2:
Вычислено,%: C 81,72; H 8,18; N 3,07
Найдено,%: C 81,92; H 8,41; N 3,13.
1-Бензилокси-2-(3-метилфенил)-4-этил-5-(6-метил-6-цианогептилокси)-бензол с 75%-ным выходом методом A.
ЯМР-спектр (CDCl3) δ : 7,29-7,44 (м, 8Н), 7,18 (с, 1Н), 7,16 (д, 1Н, J= 6,0 Гц), 6,60 (с, 1Н), 5,06 (с, 2Н), 4,00 (т, 2Н, J=5,3 Гц), 2,66 (кв, 2Н, J= 6,3 Гц), 2,42 (с, 3Н), 1,8-1,9 (м, 2Н), 1,55-1,65 (м, 6Н), 1,39 (с, 6Н), 1,22 (т, 3Н, J=6,3 Гц): ИК-спектр (хлороформ): 2944, 2238, 1612, 1504, 1471 см-1: масс-спектр (m/e) 455.
Элементный анализ для C31H37NO2:
Вычислено,%: C 81,72; H 8,18; N 3,07;
Найдено,%: C 81,48; H 8,22; N 3,17.
1-Бензилокси-2-(2-метилфенил)-4-этил-5-(6-метил-6-цианогептилокси)-бензол, выход 40%, по методу A.
Спектр ЯМР (CDCl3) δ : 7,2-7,5 (м, 9Н), 6,99 (с, 1Н), 6,57 (с, 1Н), 4,98 (с, 2Н), 3,99 (т, 2Н, J=5,3 Гц), 2,63 (кв, 2Н, J=6,3 Гц), 2,23 (с, 3Н), 1,8-1,9 (м, 2Н), 1,55-1,65 (м, 6Н), 1,37 (с, 6Н), 1,22 (т, 3Н, J=6,3 Гц).
ИК-спектр (хлороформ): 3026, 2943, 2238, 1613, 1455 см-1. Масс-спектр (m/e) 455.
Элементный анализ для C31H37NO2:
Вычислено,%: C 81,72; H 8,18; N 3,07
Найдено,%: C 81,49; H 7,95; N 3,00.
1-Бензилокси-2-(4-метоксифенил)-4-этил-5-(6-метил-6-цианогептилокси) -бензол, выход 82% по методу A.
ЯМР-спектр (CDCl3) δ : 7,53 (д, 2Н, J=7,0 Гц), 7,3-7,4 (м, 5Н), 7,14 (с, 1Н), 6,96 (д, 2Н, J=7,0 Гц), 6,57 (с, 1Н), 5,04 (с, 3Н), 3,97 (т, 2Н, J=5,3 Гц), 3,87 (с, 3Н), 2,64 (кв., 2Н, J=6,3 Гц), 1,8-1,9 (м, 2Н), 1,5-1,7 (м, 6Н), 1,38 (с, 6Н), 1,22 (т, 3Н, J=6,3 Гц):
ИК-спектр (хлороформ): 2971, 2942, 2238, 1610, 1496, 1245 см-1: масс-спектр (m/e) 472.
Элементный анализ для C31H37NO3:
Вычислено,%: C 78,95; H 7,91; N 2,97;
Найдено,%: C 78,67, H 7,99, N 2,81
1-Бензилокси-2-(3-метоксифенил)-4-этил-5-(6-метил-6-цианогептилокси)-бензол, 53% выход по методу A.
Спектр ЯМР (CDCl3) δ : 7,0-7,45 (м, 6Н), 7,15-7,20 (м, 3Н), 6,87 (дд, 1Н, J= 6 и 2 Гц), 6,58 (с, 1Н), 5,04 (с, 2Н), 3,99 (т, 2Н, J=5,3 Гц), 3,79 (с, 3Н), 2,64 (кв, 2Н, J=6,3 Гц), 1,8-1,9 (м, 2Н), 1,5-1,65 (м, 6Н), 1,38 (с, 6Н), 1,24 (т, 3Н, J=6,3 Гц), ИК-спектр (хлороформ): 2943, 2238, 1610, 1467, 1251 см-1: масс-спектр (m/e) 471.
Элементный анализ для C31H37NO3:
Вычислено,%: C 78,95; H 7,91; N 2,97
Найдено,%: C 77,12; H 7,83; N 3,32.
1-Бензилокси-2-(3-трифторметилфенил)-4-этил-5-(6-метил-6-цианогептилокси)-бензол, выход 55% по методу B.
ЯМР (CDCl3) δ : 7,88 (с, 1Н), 7,71 (д, 1Н, J=5 Гц), 7,3-7,5 (м, 7Н), 7,14 (с, 1Н), 6,60 (с, 1Н), 6,06 (с, 2Н), 4,01 (т, 2Н J=5,3 Гц), 2,64 (кв, 2Н, J=6,3 Гц), 1,8-1,9 (м, 3Н), 1,5-1,7 (м, 6Н), 1,38 (с, 6Н), 1,22 (т, 3Н, J= 6,3 Гц); ИК-спектр (хлороформ): 2944, 2872, 2238, 1612, 1507, 1333 см-1; масс-спектр (m/e) 509.
1-Бензилокси-2-(3-диметиламинофенил)-4-этил-5-(6-метил-6-цианогептилокси) -бензол, 94 %-ный выход по методу A.
Спектр ЯМР (CDCl3) δ : 7,54 (д, 2Н, J=7,0 Гц), 7,3-7,5 (м, 5Н), 7,16 (с, 1Н), 6,82 (с, 2Н, J=7,0 Гц), 6,55 (с, 1Н), 5,03 (с, 2Н), 3,95 (т, 2Н, J=5,3 Гц), 3,00 (с, 6Н), 2,62 (кв, 2Н, J=6,3 Гц), 1,8-1,9 (м, 2Н),1,5-1,7 (м, 6Н), 1,38 (с, 6Н), 1,22 (т, 3Н, J=6,3 Гц): ИК-спектр (хлороформ): 3009, 2944, 2868, 2238, 1612, 1498 см-1: масс-спектр (m/e) 484.
Элементный анализ для C32H40N2O2:
Вычислено,%: C 79,30; H 8,32; N 5,78
Найдено,%: C 77,04; H 8,32; N 5,67.
Примеры 34-41. Примерные способы дебензилирования (см. в конце описания).
К раствору простого арилбензилового эфира в этилацетате добавляли 10%-ный палладий на угле. Атмосферу реакции изменяли на газообразный водород (1 атм) и реакционную смесь перемешивали при комнатной температуре 2-48 ч. Дисперсию фильтровали через целит (Celite® ) и несколько раз промывали этилацетатом. Полученный раствор концентрировали в вакууме и очищали тонкослойной хроматографией (15%-ный этилацетат в гексане) и получали желаемый фенол.
34. 2-фенил-4-этил-5-(6-метил-6-цианогептилокси)-фенол, 79,4% выход. Спектр ЯМР (CDCl3) δ : 7,4-7,5 (м, 5Н), 7,03 (с, 1Н), 6,53 (с, 1Н), 5,22 (с, 1Н), 4,00 (т, 3Н, J= 5,3 Гц), 2,63 (кв, 2Н, J=6,3 Гц), 1,8-1,9 (м, 2Н), 1,55-1,65 (м, 6Н), 1,38 (с, 6Н), 1,21 (т, 3Н, J=6,3 Гц);
ИК-спектр (хлороформ): 3558, 3019, 2843, 2237, 1624, 1408, 1219 см-1: масс-спектр (m/e/ 351.
Элементный анализ для C23H29NO2:
Вычислено,%: C 78,63; H 8,26; N 3,99
Найдено,%: C 79,04; H 8,41; N 4,24
35. 2-(4-метилфенил)-4-этил-5-(6-метил-6-цианогептилокси)-фенол, 44,5% выход. ЯМР (CDCl3) δ/ : 7,35 (д. 2Н, J=7,0 Гц), 7,29 (д, 2Н, J=7,0 Гц), 7,00 (с, 1Н), 6,52 (с, 1Н), 5,21 (с, 1Н), 4,00 (т, 2Н, J=5,3 Гц), 2,62 (кв, 2Н, J=6,3 Гц), 2,42 (с, 3Н), 1,8-1,9 (м, 2Н), 1,5-1,7 (м, 6Н), 1,38 (с, 6Н), 1,21 (т, 3Н, J=6,3 Гц): ИК-спектр (хлороформ): 3554, 3020, 2843, 2237, 1624, 1587, 1497; масс-спектр (m/e) 365.
Элементный анализ для C24H31NO2:
Вычислено,%: C 78,86; H 8,55; N 3,83
Найдено,%: C 76,84; H 8,44; N 3,84.
36.2-(3-метилфенил)-4-этил-5-(6-метил-6-цианогептилокси)-фенол, 80,1%-ный выход. Спектр ЯМР (CDCl3) δ : 7,2 (м, 4Н), 7,01 (с, 1Н), 6,52 (с, 1Н), 5,30 (с, 1Н), 4,00 (т, 2Н, J=5,3 Гц), 2,61 (кв. 2Н, J=6,3 Гц), 2,43 (с, 3Н), 1,8-1,9 (м, 2Н), 1,6-1,5 (м, 6Н), 1,37 (с, 6Н), 1,20 (т, 3Н, J=6,3 Гц): ИК-спектр (хлороформ): 3553, 3023, 2977, 2943, 2872, 2237, 1625 см-1: масс-спектр (m/e) 365.
Элементный анализ для C24H30NO2:
Вычислено,%: C 79,08; H 8,30; N 3,84.
Найдено,%: C 78,89; H 8,84; N 3,92.
37. 2-(2-метилфенил)-4-этил-5-(6-метил-6-цианогептилокси)-фенол, выход 66,9%. Спектр ЯМР (CDCl3) δ : 7,2-7,4 (м, 4Н), 6,87 (с, 1Н), 6,51 (с, 1Н), 4,69 (с, 1Н), 4,00 (т, 2Н, J=5,3 Гц), 2,61 (кв, 2Н, J=6,3 Гц), 2,20 (с, 3Н), 1,8-1,9 (м, 2Н), 1,55-1,65 (м, 6Н), 1,38 (с, 1Н), 1,20 (т, 3Н, J=6,3 Гц); ИК-спектр (хлороформ): 3551, 3019, 2944, 2871, 2238, 1625, 1586, 1502 см-1: масс-спектр (m/e) 365.
Элементный анализ для C24H31NO2:
Вычислено,%: C 78,86; H 8,55; N 3,83.
Найдено,%: C 78,11; H 8,52; N 3,78.
38. 2-(4-метоксифенил)-4-этил-5-(6-метил-6-цианогептилокси)-фенол, количественный выход.
Спектр ЯМР (CDCl3) δ : 7,38 (д, 2Н, J=8,6 Гц), 7,15 (д, 2Н, J=8,6 Гц), 6,99 (с, 1Н), 6,52 (с, 1Н), 5,27 (с, 1Н), 3,99 (т, 2Н, J=6,2 Гц), 3,87 (с, 3Н), 2,62 (кв, 2Н, J=7,5 Гц), 1,8-1,9 (м, 2Н), 1,55-1,65 (м, 6Н), 1,37 (с, 6Н), 1,21 (т, 2Н, J=7,5 Гц): ИК-спектр (хлороформ): 3600, 3019, 2977, 2943, 2238, 1609, 1496, 1241 см-1: масс-спектр (m/e) 381.
Элементный анализ для C24H31NO3:
Вычислено,%: C 75,56; H 8,19; N 3,67
Найдено,%: C 75,38; H 8,32; N 3,50.
39. 2-(3-метоксифенил)-4-этил-5-(6-метил-6-цианогептилокси)-фенол, 72,3 %-ный выход.
Спектр ЯМР (CDCl3) δ : 7,40 (т, 1Н, J=6,8 Гц), 6,90-7,05 (м, 4Н), 6,52 (с, 1Н), 5,33 (с, 1Н), 4,00 (т, 2Н, J=5,3 Гц), 3,86 (с, 1Н), 2,62 (кв. 2Н, J=6,3 Гц), 1,8-1,9 (м, 2Н), 1,55-1,65 (м, 6Н), 1,38 (с, 6Н), 1,21 (т, 3Н, J= 6,3 Гц); ИК-спектр (хлороформ): 35550, 3023, 2943, 2238, 1597, 1485, 1287 см-1: масс-спектр (m/e) 382.
Элементный анализ для C24H31NO3:
Вычислено,%: C 75,56; H 8,17; N 3,67.
Найдено,%: C 73,95; H 8,08; N 2,59.
40. 2-(3-Трифторметилфенил)-4-этил-5-(6-метил-6-цианогептилокси)-фенол, выход 56,3%. Спектр ЯМР (CDCl3) δ : 7,75 (с, 1Н), 7,5-7,7 (м, 3Н), 7,03 (с, 1Н), 6,50 (с, 1Н), 5,09 (с. 1Н), 4,01 (т, 2Н, J=5,3 Гц), 2,62 (кв, 2Н, J=6,3 Гц), 1,8-1,9 (м, 2Н), 1,5-1,65 (м, 6Н), 1,38 (с, 6Н), 1,21 (т, 3Н, J=6,3 Гц), ИК-спектр (хлороформ): 2943, 2238, 1378, 1239 см-1; масс-спектр (m/e) 419, 420.
Элементный анализ для C24H28F3NO2:
Вычислено,%: C 68,72; H 6,73; N 3,34.
Найдено,%: C 68,72; H 7,02; N 3,38.
41. 2-(4-диметиламинофенил)-4-этил-5-(6-метил-6-цианогептилокси)-фенол, выход 38,5%.
Спектр ЯМР (CDCl3) δ : 7,32 (д, 2Н, J=7,3 Гц), 6,99 (с, 1Н), 6,85 (д, 2Н, J= 7,3 Гц), 6,52 (с, 1Н), 3,99 (т, 2Н, J=5,3 Гц), 3,01 (с, 6Н), 1,8-1,9 (м, 2Н), 1,5-1,6 (м, 6Н), 1,37 (с, 6Н), 1,20 (т, 3Н, J=6,3 Гц): ИК-спектр (хлороформ): 3600 3020, 2900, 2238, 1612, 1498 см-1: масс-спектр (m/e) 394, 394.
Элементный анализ для C25H34N2O2:
Вычислено,%: C 76,10; H 8,69; N 7,10.
Найдено,%: C 74,10; H 8,57; N 7,91.
Примеры 42-49. Примерные процессы тетразольного синтеза (см. в конце описания).
К раствору нитрила (один эквивалент) в диглиме добавляли гидрохлорид N, N-диметилэтаноламина (два эквивалента) и азид натрия (4 эквивалента). Суспензию нагревали до 140oC и перемешивали 8-72 ч. Смесь разбавляли хлористым метиленом и подкисляли хлористоводородной кислотой. Органическую фазу собирали, сушили (сульфат магния), фильтровали и концентрировали в вакууме. Полученный материал растворяли в этаноле и к этому раствору добавляли водную гидроокись натрия (4 эквивалента). Эту реакционную смесь перемешивали при комнатной температуре 30 мин, затем растворитель удаляли в вакууме. Для очистки остатка использовали систему обращенно-фазовой жидкостной хроматографии высокого давления, применяя в качестве подвижной фазы сначала воду, затем 40%-ную воду в метаноле. Желаемые фракции смешивали и концентрировали в вакууме. Затем остаток лиофилизировали и получали тетразол в форме его соли натрия.
92. 2-Фенил-4-этил-5-6-(2Н-тетразол-5-ил)-6-метилгептилокси)-фенол, натриевая соль, выход 34,3%.
Спектр ЯМР (ДМСО-d6) δ : 7,55 (д, 3Н, J=6,5 Гц), 7,35 (т, 2Н, J=6,5 Гц), 7,20 (т, 1Н, J=6,5 Гц), 6,98 (с, 1Н), 6,60 (с, 1Н), 3,82 (т, 2Н, J=5,3 Гц), 2,65 (кв, 2Н, J=6,3 Гц), 1,55-1,70 (м, 6Н), 1,25-1,35 (м, 8Н), 1,10 (т, 3Н, J= 6,3 Гц): ИК-спектр (бромистый калий): 3192, 2970, 2937, 1617, 1488, 1353, 1214 см-1: масс-спектр (m/e) 439.
Элементный анализ для C23H29N4NaO2:
Вычислено,%: C 59,87; H 7,16; N 12,25.
Найдено,%: C 60,28; H 7,45; N 12,07.
43. Динатриевая соль 2-(4-метилфенил)-4-этил-5-/6-метил-6 (2Н-тетразол-5-ил)-гептилокси/-фенола, выход 29,0%.
Спектр ЯМР (ДМСО-d6) δ : 7,40 (д, 2Н, J=6,0 Гц), 7,15 (д, 2Н, J=6,0 Гц), 6,95 (с, 1Н), 3,82 (т, 2Н, J=5,3 Гц), 2,45 (кв. 2Н, J=6,3 Гц), 2,32 (с, 3Н), 1,5-1,7 (м, 6Н), 1,2-1,4 (м, 8Н), 1,07 (т, 3Н, J=6,3 Гц), ИК-спектр (KBr): 2935, 1616, 1502, 1443 см-1: масс-спектр (m/e) 409.
Элементный анализ для C24H30N4Na2O2•1,5 H2O:
Вычислено,%: C 60,13; H 6,89; N 11,69.
Найдено,%: C 59,99; H 6,71; N 11,98.
44. 2-(3-Метилфенил)-4-этил-5-(6-метил-6-(2Н-тетразол-5-ил)-гептилокси) -фенола натриевая соль, выход 26,81.
ЯМР-спектр (ДМСО-d6) δ : 7,32 (с, 1Н), 7,30 (д, 1Н, J=6,3 Гц) 7,20 (т, 1Н, J=6,3 Гц), 7,00 (д, 1Н, J=6,3 Гц), 6,94 (с, 1Н), 6,61 (с, 1Н), 3,82 (т, 2Н, J= 5,3 Гц), 2,46 (кв, 2Н, J=6,3 Гц), 2,30 (с, 3Н), 1,5-1,7 (м, 6Н), 1,2-1,4 (м, 8Н) 1,10 (т, 3Н, J=6,3 Гц); ИК-спектр (KBr): 2935, 1616, 1486, 1140 см-1: масс-спектр (m/e) 453.
Элементный анализ для C24H30N4Na2O2•0,5H2O:
Вычислено,%: C 62,34; N 6,71; H 12,12.
Найдено,%: C 61,91; N 8,02; H 11,64.
45. 2-(2-метилфенил)-4-этил-5-(6-метил-6-(2Н-тетразол-5-ил)-гептилокси/ -фено, динатриевая соль, выход 34,9%.
Спектр ЯМР (ДМСО-d6) δ : 7,04-7,12 (м, 4Н), 6,71 (с, 1Н), 6,59 (с, 1Н), 3,83 (т, 2Н), J=5,3 Гц), 2,43 (кв, 2Н, J=6,3 Гц), 2,08 (с, 3Н), 1,55-1,7 (м, 4Н), 1,1-1,25 (м, 6Н), 1,08 (т, 3Н, J=6,3 Гц). ИК-спектр (KBr): 2935, 1617, 1486, 1326, 1243 см-1; масс-спектр (m/e) 431.
Элементный анализ для C24H30N4Na2O2• 2 H2O:
Вычислено,%: C 59,02; H 6,97; N 11,48.
Найдено,%: C 59,61; H 7,41; N 11,96.
46. 2-(4-Метоксифенил)-4-этил-5-/6-метил-6-(2Н-тетразол-5-ил)-гептилокси/ -фенола натриевая соль, выход 29,0%.
Спектр ЯМР (ДМСО-d6) δ : 7,43 (Д, 2Н, J=7,3 Гц), 6,91 (с, 1Н), 6,89 (д, 2Н, J=7,3 Гц), 6,57 (с, 1Н), 3,81 (т, 2Н, J=5,3 Гц), 3,74 (с, 3Н), 2,43 (кв, 2Н, J= 6,3 Гц), 1,7-1,9 (м, 6Н), 1,2-1,4 (м, 8Н), 1,06 (т, 3Н, J=6,3 Гц): ИК-спектр (KBr): 3009, 2969, 2937, 1609, 1497, 1242 см-1: масс-спектр (m/e) 425.
Элементный анализ для C24H30N4Na2O3•2 H2O:
Вычислено,%: C 57,15; H 6,75; N 11,11,
Найдено,%: C 57,63; H 7,57; N 10,20.
47. Натриевая соль 2-(3-Метоксифенил)-4-этил-5-/6-метил-6- (2Н-тетразол-5-ил)-гептилокси/-фенола, выход 15,9%.
Спектр ЯМР (ДМСО-d6) δ : 7,26 (т, 1Н, J=6, Гц), 7,05-7,10 (м, 2Н), 6,98 (с, 1Н), 6,80 (дд, 1Н, J=2,6 Гц), 6,60 (с, 1Н), 3,84 (т, 2Н, J=5,3 Гц), 3,76 (с, 3Н), 2,46 (кв, 2Н, J=6,3 Гц), 1,5-1,7 (м, 6Н), 1,2-1,4 (м, 8Н), 1,8 (т, 3Н, J= 6,3 Гц); ИК-спектр (KBr): 3416, 2961, 2936, 2869, 1608, 1487, 1140 см-1: масс-спектр (m/e) 469.
Элементный анализ для C24H30N4Na2O3•1,5 H2O:
Вычислено,%: C 58,18; H 6,67; N 11,31.
Найдено,%: C 58,40; H 7,73; N 10,69.
48. Двунатриевая соль 2-(4-трифторметилфенил)-4-этил-5-/6-метил-6-(2Н-тетразол-5-ил)-гептилокси/-фенила, выход 29%.
Спектр ЯМР (ДМСО-d6) δ : 7,8-7,9 (м, 2Н), 7,55-7,6 (м, 1Н), 7,55 (с, 1Н), 7,04 (с, 1Н), 6,65 (с, 1Н), 3,84 (т, 2Н, J=5,3 Гц), 2,48 (кв, 2Н, J=6,3 Гц), 1,7-1,9 (м, 6Н), 1,2-1,4 (м, 8Н), 1,06 (т, 3Н, J=6,3 Гц): ИК-спектр (KBr): 3412, 2965, 2937, 2870, 1617, 1336 см-1: масс-спектр (m/e) 507.
Элементный анализ для C24H27F3N4Na2O2•H2O:
Вычислено,%: C 54,96; H 5,53; N 10,69.
Найдено,%: C 55,26; H 6,23; N 10,10.
49. Двунатриевая соль 2-(3-диметиламинофенил)-4-этил-5-/6-метил-6- (2Н-тетразол-5-ил)-гептилокси/-фенола, 28,8% выход.
Спектр ЯМР (ДМСО-d6) δ : 7,36 (д, 2Н, J=7,3 Гц), 6,89 (с, 1Н), 6,71 (д, 2Н, J=7,3 Гц), 6,89 (с, 1Н), 6,71 (д, 2Н, J=7,3 Гц), 6,53 (с, 1Н), 3,81 (т, 2Н, J= 5,3 Гц), 2,45 (кв, 2Н, J=6,3 Гц), 1,5-1,7 (м, 6Н), 1,2-1,4 (м, 8Н), 1,0 (т, 3Н, J=6,3 Гц); ИК-спектр (бромистый калий): 3412, 2968, 2935, 2867, 1613, 1505 см-1: масс-спектр (m/e) 437, 438.
Элементный анализ для C25H33N5Na2O2•2H2O:
Вычислено,%: C 58,48; H 7,21; N 13,65.
Найдено,%: C 58,63; H 6,66; N 12,53.
Пример 50. 3-{5-/6(4-Фенил-5-окси-2-этилфенокси)-пропокси/-2-карбоксиметил -1,2,3,4-тетрагидронафталин-1(2Н)-он}-карбоновая кислота
A. Получение этилового эфира 3-(2-карбометоксиметил-6-окси-1,2,3,4- тетрагидронафталин-1(2Н)-он-5-ил)-пропионовой кислоты.
Смесь метилового эфира (6,5 г, 0,027 моля) 6-окси-1,2,3,4-тетрагидронафталин-1(2Н)-он-2-уксусной кислоты, 9,5 г (0,055 моля) триэтилового эфира ортоакриловой кислоты и 1,4 г (0,00137 моля) пивалиновой кислоты в 50 мл толуола нагревали для поддержания температуры обратного холодильника в течение 20 ч. После охлаждения реакционную смесь промывали водой, сушили (сульфат магния) и хроматографировали на силикагеле, элюируя градиентом (толуол в смеси толуол-этилацетат в отношении 9:1), и получали 6,5 г желтого масла. Это масло кристаллизовали из метанола и получали 3,9 г (40%) белого кристаллического твердого вещества. Часть этого твердого вещества 92,1 г) перемешивали в 20 мл этилацетата, содержащего 1 мл 1 н. хлористоводородной кислоты в течение 15 мин. Смесь промывали водой, сушили (сульфат натрия), концентрировали при пониженном давлении и получали желаемый целевой продукт, указанный в названии пункта A, с 93%-ным выходом. Спектр ЯМР (CDCl3) δ : 1,28 (т, 3), 1,93 (м, 1), 2,23-2,50 (м, 2), 2,72 (т, 2), 2,95-3,10 (м, 6), 3,75 (с, 3), 4,20 (кв, 2), 6,9 (д, 1), 7,95 (д, 1), 8,30 (с, 1).
B. Получение этилового эфира 3-{5-/6-(4-фенил-5-бензилокси-2-этилфенокси) -пропокси/-2-карбометоксиметил-1,2,3,4-тетрагидронафталин-1(2Н)-он} -пропионовой кислоты.
Смесь этилового эфира 3-(2-карбометоксиметил-6-окси-1,2,3,4- тетрагидронафталин-1(2Н)-он-5-ил)-пропионовой кислоты (0,67 г, 1,9 ммоля), 0,76 г (2,0 ммоль) 1-фенил-2-бензилокси-4-(3-хлорпропокси)-6-этилбензола, 0,2 г йодистого калия в 25 мл метилэтилкетона нагревали с обратным холодильником 20 ч. Реакционную смесь охлаждали, разбавляли этилацетатом, промывали водой и сушили (сульфат натрия). Целевое соединение выделяли хроматографией на силикагеле, элюируя толуолом, и получали 0,73 г (54%) указанного в пункте B промежуточного соединения. Спектр ЯМР (CDCl3) δ : 1,14 (т, 3), 1,23 (т, 3), 1,90 (м, 1), 2,23-2,60 (м, 8), 2,85-3,20 (м, 6), 3,75 (с, 3), 4,12 (м, 4), 4,25 (т, 2), 5,15 (с, 2), 6,55 (с, 1), 6,90 (д, 1), 7,15-7,55 (м, 11), 8,02 (д, 1).
C. Получение 3-{5-/6-(4-фенил-5-окси-2-этилфенокси)-пропокси/ -2-карбоксиметил-1,2,3,4-тетрагидронафтилан-1-(2Н)-он}-пропановая кислота.
Смесь 50 мл 90%-ного водного метанола, 0,36 г (6,45 ммоль) гидроокиси натрия и 0,73 г (1608 ммоль) этилового эфира 3-{5-/6-(4-фенил-5-бензилокси-2- этилфенокси)-пропокси/-2-карнбометоксиметил-1,2,3,4-тетрагидронафталин-1(2Н)он} -пропановой кислоты перемешивали при 25oC в течение 24 ч. Реакционную смесь разбавляли 100 мл воды, подкисляли 5 н. хлористоводородной кислотой и экстрагировали этилацетатом. Этот раствор сушили (сульфат натрия), концентрировали при пониженном давлении и получали 0,48 г (70 %-ный выход) масла. Это масло растворяли в 50 мл этилацетата и гидрогенизировали при давлении водорода 2 кг/см2 с 0,48 г 5%-ного палладия на угле в качестве катализатора. После отфильтровывания катализатора и концентрирования при пониженном давлении получали желаемый целевой продукт (0,31 г, выход 75%) хроматографией на смоле Р-18, элюируя смесью метанол-вода, точка плавления 72-76oC. Спектр ЯМР (CDCl3) δ : 1,10 (т, 3), 1,91 (м, 1), 2,25-2,65 (м, 6), 2,86-3,20 (м, 8), 4,21 (т, 2), 4,28 (т, 2), 6,58 (с, 1), 6,90 (д, 1), 7,02 (с, 1), 7,32-7,52 (м, 5), 8,03 (д,1).
Элементный анализ для C32H34O8:
Вычислено,%: C 70,32; H 6,27.
Найдено,%: C 70,13; H 6,31.
Пример 51. 3-{5-/6-(4-(4-фторфенил)-5-окси-2-этилфенокси/-пропокси/ -2-карбоксиметил-1,2,3,4-тетрагидронафталин-1(2Н)-он}пропановая кислота.
A. Этиловый эфир 3-{5-/6-/4-(4-фторфенил)-5-бензил-окси-2-этилфенокси/ -пропокси/-2-карбометоксиметил-1,2,3,4-тетрагидронафталин-1(2Н)-он} -пропановой кислоты.
Используя процесс, описанный для синтеза по примеру 50(B), подвергали взаимодействию 0,91 (2,72 ммоль) этилового эфира 3-(2-карбометоксиметил-6-окси -1,2,3,4-тетрагидронафталин-1(2Н)-он-5-ил)пропионовой кислоты и 1,09 г (2,73 ммоль) 1-(4-фторфенил)-2-бензилокси-4-(3-хлорпропокси)-6-этилбензола и получали 1,08 г целевого соединения с 57%-ным выходом. Спектр ЯМР (CDCl3) δ : 1,20 (м, 6), 1,90 (м, 1), 2,22-2,68 (м, 8), 2,90-3,20 (м, 6), 3,75 (с, 3), 4,12 (кв, 23), 4,22 (т, 2), 4,30 (т, с), 5,03 (с, 2), 6,63 (с, 1), 6,90 (д, 1), 7,03-7,55 (м, 10), 8,02 (д, 1).
B. Получение 3-{ 5-/6-/4-(4-фторфенил)-5-окси-2-этилфенокси-1(2Н)-он} пропионовая кислота.
Этиловый эфир 3-{ 5-/6-/4-(4-фторфенил)-5-бензилокси-2-этилфенокси/)-пропокси/-2-карбометоксиметил-1,2,3,4-тетрагидронафталин-1(2Н)-он} -пропановой кислоты подвергали взаимодействию по методу, использованному в примере 50(C), и получали 0,47 г (77%-ный выход) желаемого целевого продукта, точка плавления 68-72oC.
Спектр ЯМР (CDCl3) δ : 1,15 (т, 3), 1,91 (м, 1), 2,23-2,62 (м, 8), 2,85-3,18 (м, 6), 4,21 (т, 2), 4,28 (т, 2), 6,55 (с, 1), 6,88 (д, 1), 7,10 (т, 2), 7,41 (м, 2), 8,02 (д, 1).
Элементный анализ для C32H33FO8:
Вычислено,%: C 68,07; H 5,89.
Найдено,%: C 57,84; H 6,14.
Пример 52. 3-{4-/4-/4-(4-фторфенил)-5-окси-2-этилфенокси/-пропокси/-2-карбоксиметил-2,3-дигидроинден-1-(2Н)-он}-пропановая кислота.
A. Получение этилового эфира 3-/4-(2-карбометоксиметил)-5-окси-2,3-дигидроинден-1(2Н)-он/-пропановой кислоты.
Используя метод, описанный в примере 50(A), метиловый эфир 5-окси-2,3-дигидроинден-1(2Н)-он-2-уксусной кислоты (4,8 г, 21,8 ммоль) обращали в 1,6 г целевого промежуточного соединения с 76%-ным выходом. Спектр ЯМР (CDCl3) δ : 1,25 (т, 3), 2,50-2,80 (м, 4), 2,80-3,10 (м, 4), 3,36 (м, 1), 3,73 (с, 3), 4,15 (м, 2), 6,94 (д, 1), 7,58 (д, 1), 8,75 (с, 1).
B. Получение 3-{4-/5-(4-фторфенил)-5-бензилокси-2-этилфенокси/-пропокси/ -2-карбометоксиметил-2,3-дигидроинден-1(2Н)-он}-пропановой кислоты.
Используя метод, описанный в примере 50(B), 0,93 г (2,91 ммоль) этилового эфира 3-/4-(2-карбометоксиметил)-5-окси-2,3-дигидроинден-1 (2Н)-он/-пропановой кислоты и 1,16 г (2,9 ммоль) 1-(4-фторфенил)-2-бензилокси-4-(3-хлорпропокси)-6-этилбензола обращали в 0,58 г (30%-ный выход) целевого промежуточного соединения.
Спектр ЯМР (CDCl3) δ : 1,20 (м, 6), 2,15-3,10 (м, 12), 3,47 (м, 1); 3,73 (с, 3), 4,10 (кв, 2), 4,22 (т, 2), 4,32 (т, 2), 5,03 (с, 2), 6,62 (с, 1), 6,94-7,56 (м, 11), 7,70 (д, 1).
C. Получение 3-{ 4-/5-/4-(4-фторфенил)-5-окси-2-этилфенокси/-пропокси/ -2-карбоксиметил-2,3-дигидроинден-1-(2Н)-он}пропановой кислоты.
3-{4-/5-(4-фторфенил)-5-бензилокси-2-этилфеноски/-пропокси/ -2-карбометоксиметил-1,2-дигидроинден-1(2Н)-он} -пропановую кислоту (0,55 г, 0,8 ммоль) подвергали взаимодействию с использованием метода, описанного в примере 50(C), и получали 0,16 г (30%-ный выход) целевого соединения, точка плавления 78-81oC.
Спектр ЯМР (CDCl3) δ : 1,20 (т, 3), 2,30-3,05 (м, 12), 3,45 (м, 1), 4,22 (т, 2), 4,32 (т, 2), 6,55 (с, 1), 6,88-7,45 (м, 6), 7,70 (д, 1).
Элементный анализ для C31H31FO8:
Вычислено,%: C 67,63; H 5,67.
Найдено,%: C 67,44; H 5,92.
Пример 53. 3,3-Диметил-5-{3-(2-карбоксиэтил)-4-(4-фторфенил) -5-окси-2-этилфенокси/-пропокси/-фенил}-5-оксипентановая кислота.

A. Получение 5-(3-(2-карбоэтоксиэтил)-4-метоксифенил/-3,3-диметил -5-оксопентановой кислоты.
Хлористый алюминий (41,4 г, 0,3 моля) добавляли по частям в смеси 20,8 г (0,1 моль) этилового эфира 1-(2-метоксифенил)-пропановой кислоты и 14,2 г (0,1 моль) b, b-диметилглутаровому ангидриду в 250 мл хлористого метилена, охлажденного в ванне со смесью воды и льда. Перемешивание продолжали 4 ч, медленно нагревая за это время до 25oC. Смесь выливали в 500 г льда и 50 мл концентрированной хлористоводородной кислоты. Органический слой отделяли, промывали водой и сушили над сульфатом магния. После удаления растворителя при пониженном давлении получали 28 г неочищенной 5-/3-(2-карбэтоксиэтил)-4-метоксифенил/-3,3-диметил-2-оксопентановой кислоты.
B. Получение этилового эфира 5-/3-(2-карбэтоксиэтил)-4-оксифенил)-3,3-диметил-2-оксопентановой кислоты.
Неочищенную 5-/3-(2-карбоэтоксиэтил)-4-метоксифенил/-3,3-диметил-5-оксопентановую кислоту (0,08 моль) помещали в 100 г гидрохлорида пиридина и смесь нагревали при 180oC в масляной ванне 20 ч. После охлаждения смесь растворяли в воде, сильно подкисляли 2 н. хлористоводородной кислотой и три раза экстрагировали этилацетатом. Объединенный этилацетатный экстракт промывали водой и сушили (сульфат натрия). Растворитель удаляли при пониженном давлении и остаток растворяли в 200 мл этанола. Добавляли метансульфокислоту (0,5 мл) и смесь нагревали при температуре обратного холодильника 20 ч. После концентрирования в вакууме остаток растворяли в этилацетате, промывали водой, и сушили (сульфат натрия). Растворитель удаляли при пониженном давлении и целевой сложный эфир (19,1 г, 66%-ный выход) получали хроматографией на силикагеле, элюируя смесью толуол-этилацетат в отношении 13:3.
Спектр ЯМР (ДМСО-d6) δ : 0,94 (с, 6), 0,97 (т, 3), 0,98 (т, 3), 2,44 (с, 2), 2,56 (т, 2), 2,82 (т, 2), 2,97 (с, 2), 4,02 (м, 4), 6,85 (д, 1), 7,70 (м, 2), 10,36 (с, 1).
C. Получение этилового эфира 3,3-диметил-5-{3-(2-карбэтоксиэтил) -4-/4-(4-фторфенил)-5-бензилокси-2-этилфенокси/-пропокси} -оксопентановой кислоты.
Методом, описанным в примере 50(B), 1,6 г (4,4 ммоль) этилового эфира 5-/3-(2-карбэтоксиэтил)-4-оксифенил/-3,3-диметил-5-оксопентановой кислоты и 1,75 г (4,4 ммоль 1-а(4-фторфенил)-2-бензилокси-4-(3-хлорпропокси)-6-этилбензола обращали в 1,1 г (45%-ный выход) желаемого целевого промежуточного соединения.
Спектр ЯМР (CDCl3) δ/ : 1,20 (м, 15), 2,34 (т, 2), 2,61 (м, 4), 3,00 (т, 2), 3,07 (с, 2), 4,10 (м, 4), 4,20 (м, 2), 4,28 (т, 2), 5,02 (с, 2), 6,63 (с, 1), 6,91 (д, 1), 7,07 (м, 3), 7,30 (м, 5), 7,51 (м, 3), 7,81 (с, 1), 7,86 (д, 1).
D. Получение 3,3-диметил-5-/3-(2-карбоксиэтил)-4-/3-(4-фторфенил) -5-окси-2-этилфенокси/-пропокси/-фенил/-оксопентановой кислоты.
Используя метод, описанный в примере 50(C), 3,3-диметил-2-3- (2-карбэтоксиэтил)-4-/4-(4-фторфенил)-5-бензилокси-2-этилфенокси/ -пропокси-2-оксопентановой кислоты этиловый эфир (0,6 г 0,9 ммоль) обращали в 0,41 г (51%-ный выход) целевого соединения, точка плавления 52-55oC.
Спектр ЯМР (CDCl3) δ : 1,6 (м, 9), 2,45 (т, 2), 2,50-2,72 (м, 6), 3,02 (м, 4), 4,19 (т, 2), 4,28 (т, с), 6,55 (с, 1), 6,93 (д, 1), 7,00 (с, 1), 7,12 (м, 2), 7,42 (м, 2), 7,85 (с, 1), 7,90 (д, 1).
Элементный анализ для C33H37FO8:
Вычислено,%: C 68,26; H 6,42.
Найдено,%: C 67,60; H 6,63.
Пример 54. 7-/2-/(5-этил-2-окси-/1,1-дифенил/-4-ил)-пропокси/ -3,4-дигидро-9-пропил-2Н-1-бензопиран-2-карбоновая кислота (см. в конце описания).
A. Получение этилового эфира 7-окси-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты.
К раствору 225 мл абсолютного этанола в атмосфере аргона и при комнатной температуре добавляли 16,56 г металлического натрия в течение 1 ч. После того, как весь натрий был добавлен, реакционную смесь нагревали с обратным холодильником 1 ч, затем охлаждали до комнатной температуры. К раствору этилата натрия в течение 25 мин добавляли смесь 2,-4-диокси-3-проплацетофенона (34,82 г, 0,180 моль), диэтилоксилата (54,57 мл, 0,41 моль), абсолютного спирта (45 мл) и диэтилового эфира (45 мл). Полученную темнокаштановую смесь нагревали с обратным холодильником 2,5 ч и затем охлаждали до комнатной температуры. Реакционную смесь выливали приблизительно в 600 мл 1 н. хлористоводородной кислоты и затем несколько раз экстрагировали диэтиловым эфиром. Эфир удаляли в вакууме и полученную смолу растворяли в 135 мл этанола. К этому раствору добавляли затем 2,25 мл концентрированной серной кислоты. Эту смесь последовательно нагревали с обратным холодильником в течение 45 мин. Реакционную смесь охлаждали до комнатной температуры и этанол удаляли при пониженном давлении, получая коричневое твердое вещество. Это твердое вещество растворяли в этилацетате и промывали водой 1 раз, 2 раза насыщенным раствором бикарбоната натрия и 1 раз водой, затем сушили над сульфатом магния. После фильтрации и удаления растворителя получали 87 г коричневого твердого вещества, которое перекристаллизовывали из смеси этилацетат-петролейный эфир, Перекристаллизацией получали 24,07 г (48%-ный выход) целевого промежуточного соединения в виде светлокоричневого твердого вещества.
Тонкослойная хроматография: Rf=0,27 (40%-ная смесь этилацетата в гексане).
Спектр ЯМР (CDCl3) δ : 8,80 (шир. синглет, 1), 7,98 (д, 1, J=8,78 Гц), 7,13 (д, 1, J=8,78 Гц), 7,13 (с, 1), 4,47 (кв, 2, J=7,11 Гц), 2,96 (т, 2, J= 7,25 Гц), 1,73 (м, 2), 1,46 (т, 3, J=7,16 Гц), 1,02 (т, 3, J=7,11 Гц).
B. Получение этилового эфира 3,4-дигидро-7-окси-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты.
В сосуде Парра растворяли этиловый эфир 7-окси-8-пропил-2Н-1-бензопиран-2-карбогидрата (12,07 г, 0,44 моля) в 210 мл уксусной кислоты. К этому раствору добавляли 10%-ный палладий на угле (7,2 г катализатора) и в реактор подавали газообразный водород под давлением 3,7 кг/см2. Реакционную смесь перемешивали 23 ч. Катализатор удаляли фильтрацией через слой целита в воронке со спекшимся стеклом. Катализатор отмывали этилацетатом. Растворитель удаляли из фильтрата и полученное масло обращали в азеотропную смесь с толуолом и получали 12 г коричневого масла. Продукт очищали жидкостной хроматографией высокого давления на аппарате Waters Prep 500, снабженном ампулами с силикагелем, элюируя градиентом смеси этилацетат-гексан от 5 до 40% в течение 50 мин при скорости тока 250 мл/мин и отбирая 500 мл фракций. Очищенное целевое промежуточное соединение получали в виде розового масла (10 г, выход 86%).
Тонкослойная хроматография: Rf=0,50 (40% смесь этилацетат-гексан. Спектр ЯМР (CDCl3) δ : 6,73 (д, 1, J=8,20 Гц), 6,37 (д,1, J=8,30 Гц), 4,78 (шир. с. , 1), 4,75 (м, 1), 4,72 (м, 2), 2,68 (м, 4), 2,16 (м, 2), 1,60 (м, 2), 1,29 (т, 3, J=7,07 Гц), 0,99 (т, 3, J=7,34 Гц).
C. Получение 4-бензилокси-2-(3-хлор-1-пропилокси)-ацетофенона.
Суспензию гидрида натрия (80 мг 60%-ной масляной дисперсии, 2,0 ммоля) в 5 мл сухого диметилформамида перемешивали в атмосфере азота. К этой суспензии при комнатной температуре добавляли 2 мл раствора 4-бензилокси-2-оксиацетофенона (242 мг, 1,0 ммоль). Реакционную смесь перемешивали 25 мин при комнатной температуре. К образовавшемуся алкоголяту добавляли раствор 1-бром-3-хлорпропана (625 мг, 4 ммоль) в 3 мл диметилформамида с последующим добавлением 18-краун-6 (26 мг, 0,1 ммоль). Реакционную смесь перемешивали 19 ч при комнатной температуре, после чего ее разбавляли этилацетатом и 2 раза промывали насыщенным водным раствором хлористого натрия. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученный продукт хроматографировали на силикагеле, элюируя смесью 20 %-ного этилацетата в гексане, и получали целевой простой хлорпропиловый эфир (170 мг, 53%-ный выход).
Спектр ЯМР (CDCl3) δ : 7,84 (д, 1, J=9,0 Гц), 7,40 (м, 5), 6,51 (дд, 1, J= 3,0 и 9,0 Гц), 6,56 (д, 1, J=3,0 Гц), 5,12 (с, 2), 4,20 (т, 2, J=6,0 Гц), 3,78 (т, 2, J=6,0 Гц), 2,58 (с, 3), 2,30 (м, 2);
ИК-спектр (хлороформ): 3019, 1930, 1663, 1600, 1574, 1499, 1435 см-1: масс-спектр (свободная десорбция) (m/z) 318 (M+).
Элементный анализ для C18H19O3Cl:
Вычислено,%: C 76,82; H 6,01.
Найдено,%: C 76,56; H 5,99.
D. Получение 1-бензилокси-4-этил-3-(3-хлор-1-пропилокси)-бензола.
4-Бензилокси-2-(3-хлор-1-пропилокси-ацетофенон (1,0 г) растворяли в четыреххлористом углероде (3 мл). При комнатной температуре в атмосфере азота добавляли трифторуксусную кислоту (3 мл) и затем триэтилсилан (3 мл). После перемешивания в течение 1,5 ч, реакционную смесь разбавляли хлористым метиленом и органический слой несколько раз промывали насыщенным водным раствором бикарбоната натрия. Органический материал сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученный остаток очищали хроматографией на силикагеле, элюируя 1%-ной смесью этилацетата в гексане, и получали 0,620 г желаемого целевого соединения.
Спектр ЯМР (CDCl3) δ : 7,38 (м, 5), 7,04 (д,1, J=8,0 Гц), 6,72 (с, 1), 6,70 (дд, 1, J=8,0 Гц), 5,02 (с, 2), 4,07 (т, 2, J=7,0 Гц), 3,74 (т, 2, J= 7,0 Гц), 2,56 (кв, 2, J=8,0 Гц), 2,25 (м, 2), 1,16 (т, 3, J=8,0 Гц); ИК-спектр (хлороформ): 3011, 2958, 2912, 2876, 1612, 1505, 1455 см-1.
E. Получение 2-бензилокси-1-бром-5-этил-4-(3-хлор-1-пропилокси)-бензола.
К раствору 1-бензилокси-4-этил-3-(3-хлор-1-пропилокси)-бензола (39,5 г, 0,13 моль) в четыреххлористом углероде (500 мл) при комнатной температуре добавляли твердый N-бромсукцинимид (23,1 г, 0,13 моль). Раствор перемешивали при комнатной температуре 15 ч. Реакционную смесь разбавляли хлористым метиленом и промывали водой. Органический экстракт сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Неочищенный продукт хроматографировали на аппарате для жидкостной хроматографии высокого давления Waters Prep 500.
Желаемый целевой бромистый арил (27,1 г) получали с 55%-ным выходом.
Спектр ЯМР (CDCl3) δ : 7,50-7,22 (м, 7), 6,50 (с, 1), 5,13 (с, 2), 4,04 (т, 2, J= 8,0 Гц), 3,74 (т, 2, J=8,0 Гц), 2,53 (кв, 2, J=8,0 Гц), 2,22 (м, 2), 1,14 (т, 3, J=8,0 Гц), ИК-спектр (хлороформ): 3012, 2971, 1602, 1500, 1454 см-1: масс-спектр (свободная десорбция) (m/z) 384 (M+), 304.
Элементный анализ для C18H20O2BrCl:
Вычислено,%: C 56,45; H 5,25;
Найдено,%: C 56,55; H 5,36.
F. Получение этилового эфира 7-{3-/(2-бензилокси-1-бром-5-этил-4-ил) -окси/-пропокси}-3,4-дигидро-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты.
2-Бензилокси-1-бром-5-этил-4-(3-хлор-1-пропилокси)-бензол (5,09 г, 13,3 ммоль) растворяли в метилэтилкетоне (60 мл) и добавляли твердый йодистый натрий (20 г, 183 ммоль). Реакционную смесь нагревали с обратным холодильником в атмосфере аргона 18 ч. Реакционную смесь охлаждали до комнатной температуры, реакцию прекращали водой, затем 3 раза экстрагировали диэтиловым эфиром. Органические экстракты объединяли, сушили над сульфатом магния, фильтровали и получали 6,72 г желтого масла.
Раствор этилового эфира 3,4-дигидро-7-окси-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты (2,1 г, 8,1 ммоль) в диметилформамиде (5 мл) добавляли к суспензии гидрида натрия (324 мг, 8,1 ммоль, 60%-ная масляная дисперсия) в 10 мл сухого диметилформамида в атмосфере азота. После перемешивания реакционной смеси в течение 30 мин добавляли смесь иодистого алкила (3,8 г, 8,1 ммоль), приготовленного выше, и 18-краун-6 (110 мг, 0,4 ммоль). Реакционную смесь перемешивали 1,5 ч при комнатной температуре. Реакцию прекращали водой и затем несколько раз экстрагировали этилацетатом. Органический материал сушили над сульфатом магния, фильтровали и концентрировали в вакууме. Полученное вещество очищали хроматографией на силикагеле, элюируя 6%-ным этилацетатом в гексане, и получали 2,5 г (86%) желаемого целевого соединения.
Тонкослойная хроматография: Rf=0,61 (20%-ный этилацетат в гексане). Спектр ЯМР (CDCl3) δ : 7,60-7,30 (м, 6), 6,85 (д, 1, J=8,50 Гц), 6,55 (с, 1), 6,46 (д, 1, J=8,50 Гц), 5,13 (с, 2), 4,75 (м, 1), 4,40-4,10 (м, 6), 2,90-2,50 (м, 6), 2,40-2,10 (м, 4), 1,60 (м, 2), 1,30 (т, 3, J=7,40 Гц), 1,15 (т, 3, J= 7,50 Гц), 0,95 (т, 3, J=7,30 Гц): ИК-спектр(хлороформ): 3019, 2969, 1730, 1590, 1492 см-1: Масс-спектр (бомбардировка быстрыми атомами) (m/z) 611 (M+).
Элементный анализ для C33H39O6Br:
Вычислено,%: C 64,81; H 6,42; Br 13,07.
Найдено,%: C 65,10; H 6,52; Br 12,89.
G. Получение этилового эфира 7-{3-/(2-бензилокси-5-этил-/1,1'-дифенил/ -4-ил)окси/-пропокси} -3,4-дигидро-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты.
Этиловый эфир 7-{3-/(2-бензилокси-1-бром-5-этил-4-ил)-окси/-пропокси}-3,4-дигидро-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты (1,3 г, 2,24 ммолm) перемешивали в 4,0 мл бензола в атмосфере аргона. К этому раствору добавляли тетракис-(трифенилфосфин)-палладий (0) (0,40 г, 0,35 ммоля) и бикарбонат натрия (10 мл) 2 М водного раствора). К реакционной смеси добавляли этаноловый раствор (10 мл) фенилборной кислоты (1,3 г, 10,7 ммолm) и затем реакционную смесь нагревали с обратным холодильником 21 ч. Реакционную смесь охлаждали до комнатной температуры, реакцию прекращали насыщенным водным раствором хлористого аммония, разбавляли водой и затем экстрагировали этилацетатом. Органическую фазу сушили над сульфатом магния и фильтровали. Фильтрат концентрировали в вакууме и получали 1,3 г коричневого твердого вещества. Твердое вещество растворяли в смеси 20%-ного этилацетата в гексане и фильтровали через силикагель Merck 60, элюируя 500 мл 20% смеси этилацетата в гексане. Полученное желтое масло (1,0 г) очищали хроматографией на силикагеле, элюируя смесью 18%-ного этилацетата в гексане. Желаемый целевой сложный эфир (0,875 г, 64%-ный выход) получали в виде желтого масла.
Тонкослойная хроматография: Rf=0,65 (30% этилацетат-гексан)
Спектр ЯМР (CDCl3) δ : 7,72 (д, 2, J=7,13 Гц), 7,46 (м, 8), 7,29 (с, 1), 6,93 (д, 1, J=8,40 Гц), 6,75 (с, 1), 6,60 (д, 1, J=8,40), 5,13 (с, 2), 4,85 (м, 1), 4,31 (м, 6), 2,80 (м, 6), 2,35 (м, 4), 1,75 (м, 2), 1,39 (т, 3, J= 7,10 Гц), 1,35 (т, 1, J= 7,50 Гц), 1,11 (т, 3, J=7,33 Гц): ИК-спектр (хлороформ): 3010, 2965, 1769, 1612, 1400 см-1. Масс-спектр (бомбардировка быстрыми атомами) (m/z) 608 (M+), 519.
Элементный анализ для C39H44O6:
Вычислено,%: C 76,94; H 7,29.
Найдено,%: C 75,70; H 7,39.
H. Получение этилового эфира 7-{3-/(5-этил-2-окси-/1,1-дифенил/-4-ил) -окси/-пропокси}-3,4-дигидро-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты.
Газообразный водород барботировали через 10 мл раствора этилового эфира 7-{ 3-/(2-берзилокси-5-этил-/1,1-дифенил/-4-ил)-окси/-пропокси} -3,4-дигидро-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты (0,850 г, 1,4 ммоль) в этилацетате, содержащем 0,14 г 10%-ного палладия на угле в качестве катализатора. Над реакционной смесью поддерживали атмосферу водорода и смесь перемешивали 4 дня. Реакционную смесь фильтровали через целитный слой в воронке из спеченного стекла и катализатор отмывали этилацетатом. Растворитель удаляли из фильтрата и получали прозрачное масло. Масло очищали хроматографией на силикагеле, элюируя смесью 20%-ного этилацетата в гексане. Желаемый целевой фенол (0,354 г, 49%) получали в виде прозрачного масла.
Тонкослойная хроматография: Rf=0,32 (30% смесь этилацетата в гексане). Спектр ЯМР (CDCl3) δ : 7,51 (д, 4, J=4,43 Гц), 7,40 (м, 1), 7,08 (с, 1), 6,86 (д, 1, J=8,26 Гц), 6,60 (с, 1), 6,54 (Д, 1, J=8,29 Гц), 5,41 (с, 1), 4,80 (м, 1), 4,26 (м, 6), 2,73 (м, 6), 2,30 (м, 4), 1,65 (м, 2), 1,33 (т, 3, J=6,94 Гц), 1,26 (т, 3, J=7,39 Гц), 1,02 (т, 3, J=7,33 Гц).
Спектр ИК (хлороформ): 3026, 2967, 1749, 1612, 1488 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 519 (M++1), 518 (M+), 305.
Анализ для C32H38O6:
Вычислено,%: C 74,11; H 7,39.
Найдено,%: C 72,40; H 7,14.
I. Получение 7-{3-/(5-этил-2-окси-/1,1'-дифенил/-4-ил)-окси/-пропокси} -3,4-дигидро-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты.
Раствор этилового эфира 7-{ 3-/(5-этил-2-окси-/1,1-дифенил/-4-ил)-окси/-пропокси} -3,4-дигидро-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты (0,367 г, 0,71 ммоль) в 4 мл диоксана обрабатывали 1,10 мл 2 н. раствора гидроокиси натрия и перемешивали при комнатной температуре. Через 1,25 ч диоксан удаляли в вакууме при комнатной температуре и оставшийся водный раствор разбавляли водой, подкисленной до pH 1,0 5 н. хлористоводородной кислотой. Полученную суспензию экстрагировали этилацетатом. Органический экстракт сушили над сульфатом магния и фильтровали. Оставшееся белое твердое вещество перекристаллизовывали из смеси толуол-гексан. Целевую кислоту получали в виде белого кристаллического твердого вещества (0,245 г, 71%). Тонкослойная хроматография: Rf= 0,25 (10%-ная смесь метанола в хлористом метилене, полоса).
Спектр ЯМР (CDCl3) δ : 7,45 (м, 6), 7,02 (с, 1), 6,86 (д, 1, J=8,57 Гц), 6,56 (с, 1), 6,53 (д, 1, J=8,28 Гц), 5,30 (шир. с., 1), 4,78 (дд, 1, J=3,70 и 7,50 Гц), 4,20 (т, 2, J=6,02 Гц), 4,18 (т, 2, J=6,04 Гц), 2,69 (м, 8), 2,26 (м, 6), 1,55 (м, 2), 1,19 (т, 3, J=7,38 Гц), 0,96 (т, 3, J=7,31 Гц): ИК-спектр (KBr): 3426, 2959, 2870, 1718, 1615 см-1: масс-спектр (бомбардировка быстрыми атомами): (m/z) 491 (M++1), 490 (M+), 277.
Элементный анализ для C30H34O6:
Вычислено,%: C 73,45; H 6,99.
Найдено,%: C 73,53; H 6,82.
Пример 55. 8-Пропил-7-{3-/4-(4-фторфенил)-2-этил-5-5-оксифенокси/ -пропокси}-3,4-дигидро-2Н-1-бензопиран-2-карбоновая кислота.

A. Получение этилового эфира 8-пропил-7-{3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси}-3,4-дигидро-2Н-1-бензопиран-2-карбоновой кислоты.
Тетракис-(трифенилфосфин)-палладий (0) (0,659 г, 0,6 ммоль) и водный раствор карбоната натрия (20 мл, 2 М раствор) добавляли к раствору этилового эфира 7-{ 3-/(2-бензилокси-1-бром-6-этил-4-ил)-окси/-пропокси} -3,4-дигидро-8-пропил-2Н-1-бензопиран-2-карбоновой кислоты (2,163 г, 3,5 ммоля) в 30 мл бензола в атмосфере аргона. Реакционную смесь нагревали с обратным холодильником 17 ч, затем охлаждали до комнатной температуры и экстрагировали этилацетатом. Органический экстракт сушили над сульфатом магния, фильтровали и растворитель удаляли в вакууме. Неочищенный продукт очищали хроматографией на силикагеле Waters Prep 500, элюируя градиентом от 5 до 20% этилацетата в смеси с гексаном в течение 50 мин. Желаемый целевой дифенил получали в виде прозрачного масла (1,722 г, 78%).
Спектр ЯМР (CDCl3) δ : 7,51 (м, 2), 7,32 (м, 5), 7,09 (м, 3), 6,83 (д, 1, J= 8,32 Гц), 6,62 (с, 1), 6,49 (д, 1, J=8,50 Гц), 5,02 (с, 2), 4,75 (дд, 1, J= 4,10 и 6,50 Гц), 4,22 (м, 6), 2,69 (м, 6), 2,25 (м, 4), 1,59 (м, 2), 1,30 (т, 3, J=7,10 Гц), 1,121 (т, 3, J=7,42 Гц), 0,96 (т, 3, J=7,33 гц).
ИК-спектр (хлороформ): 3019, 2968, 1745, 1611, 1495 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 627 (M++1), 626 (M+), 536.
Элементный анализ для C39H43O5F:
Вычислено,%: C 74,74; H 6,91; F 3,03.
Найдено,%: C 74,98; H 7,05; N 3,39.
B. Получение этилового эфира 7-{3-/4-(4-фторфенил)-2-этил-5-оксифеноски/-пропокси}-3,4-дигидро-2Н-1-бензопиран-2-карбоновой кислоты.
Газообразный водород барботировали в течение 10 мин через раствор этилового эфира 8-пропил-7-{3-(2-этил-4-(4-фторфенил)-5-бензилоксифенокси) -пропокси} -3,4-дигидро-2Н-1-бензопиран-2-карбоновой кислоты (1,610 г, 2,57 ммоль) в 30 мл этилацетата, содержащего 1,0 г 10%-ного палладия на угле в качестве катализатора. Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода 2 ч. Реакционную смесь фильтровали через слой целита в воронке со спеченным стеклом и катализатор отмывали этилацетатом. Растворитель удаляли из фильтрата и получали 1,242 г прозрачного масла. Масло очищали хроматографией на силикагеле Merck, элюируя смесью 20%-ного этилацетата в гексане. Желаемый целевой фенол получали с 74%-ным выходом (1,020 г) в виде белого твердого вещества.
Тонкослойная хроматография: Rf=0,35 (30% этилацетат-гексан) Спектр ЯМР (CDCl3) δ : 7,43 (м, 2), 7,16 (дд, 2, J=5,97 и 5,97 Гц), 6,908 (с, 1), 6,82 (д, 1 J= 8,44 Гц), 6,53 (с, 1). 6,46 (д, 1, J=9,43), 5,07 (с, 1), 4,76 (м, 1), 4,21 (м, 6), 2,67 (м, 6), 2,26 (м, 4), 1,58 (м, 2), 1,29 (т, 3, J=6,96 Гц), 1,91 (т, 3, J=7,35 Гц), 0,96 (т, 3, J=7,27 Гц)
ИК-спектр (бромистый калий): 3434, 2962, 2869, 1738, 1624, 1588, 1502 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 537 (M++1), 536 (M+).
Элементный анализ для C32H37O6:
Вычислено,%: C 71,62; H 6,95.
Найдено,%: C 71,63; H 7,06.
C. Получение 8-пропил-7-{3-/4-(4-фторфенил)-2-этил-5-оксифенокси/-пропокси}-3,4-дигидро-2Н-1-бензопиран-2-карбоновой кислоты.
Раствор этилового эфира 8-пропил-7-{3-/4-(4-фторфенил)-2-этил-5- оксифенокси/-пропокси} -3,4-дигидро-2Н-1-бензопиран-2-карбоновой кислоты (0,968 г, 1,8 ммоль) в 12 мл диран-2-карбоновой кислоты (0,968 г, 1,8 ммоль) в 12 мл диоксана обрабатывали гидридом натрия (2,71 мл, 2 н. раствор) и перемешивали при комнатной температуре. Через 2,5 ч перемешивания при комнатной температуре диоксан удаляли из реакционной смеси и оставшийся продукт разбавляли водой и подкисляли до pH 1,5 с помощью 5 н. хлористоводородной кислоты. Полученную молочно-белую суспензию смешивали, затем с этилацетатом и экстрагировали этилацетатом. Органический экстракт сушили над сульфатом магния, фильтровали, растворитель удаляли и получали белое твердое вещество (1,098 г). Твердое вещество перекристаллизовывали из смеси этилацетатгексан и получали указанную в названии примера кислоту в виде белых игловидных кристаллов (0,568 г, выход 62%).
Тонкослойная хроматография: Rf= 0,31 (10%-ный метанол в хлористом метилене). Спектр ЯМР (CDCl3) δ : 7,42 (м, 2Н), 7,15 (дд, J=8,68 Гц), 6,98 (с, 1), 6,85 (д,1, J=8,0 Гц), 6,53 (с, 1), 6,52 (д, 1, J=6,93 Гц), 4,77 (дд, 1, J= 3,63 и 7,43 Гц), 4,18 (м, 4), 2,70 (м, 6), 2,27 (м, 4), 1,56 (м, 2), 1,19 (т, 3, J= 7,42 Гц), 0,95 (т, 3, J=7,30 Гц); ИК-спектр (бромистый калий): 3421, 2959, 2871, 1706, 1615, 1500 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 509 (M++1), 508 (M+).
Элементный анализ для C30H35O6:
Вычислено,%: C 70,73; H 6,54.
Найдено,%: C 70,05; H 6,82.
Пример 56. 2-{3-/3-/(5-этил-2-окси-1/1,1-дифенил/-4-ил)-окси/-пропокси/-2-пропилфенокси}-пропановая кислота (см. в конце описания).
A. Получение 2-пропил-1,3-диметоксибензола.
1,3-Диметоксибензол (20 г, 145 ммоль) в 200 мл сухого тетрагидрофурана охлаждали до -10oC. К этому раствору при -10oC в течение 20 мин добавляли 7-бутиллитий (100 мл, 1,6 М раствор в гексане, 160 ммоль). Затем реакционную смесь перемешивали при 0oC в течение 2,5 ч. При 0oC в течение 15 мин медленно добавляли йодистый пропил (24,65 г, 145 ммоль). По завершении добавки реакционная смесь нагревалась до комнатной температуры и ее оставляли на ночь при перемешивании. После этого реакционную смесь нагревали с обратным холодильником 1,5 ч, затем охлаждали до комнатной температуры и реакцию прекращали льдом. Тетрагидрофуран удаляли в вакууме и полученный водный слой несколько раз экстрагировали диэтиловым эфиром. Органический экстракт сушили над сульфатом магния, фильтровали и получали после удаления растворителя прозрачное масло (26,11 г). Масло очищали вакуумной дистилляцией и получали целевое промежуточное соединение (24,0 г, выход 92%). Точка кипения 80-82oC при 10 мм рт.ст.
Спектр ЯМР (CDCl3) δ : 7,16 (т, 1, J=8,30 Гц), 6,58 (д, 2, J=8,30 Гц), 3,85 (с, 6), 2,67 (т,2, J=7,57 Гц), 1,56 (м, 2), 0,99 (т, 3, J=7,35 Гц).
B. Получение 2-пропил-1,3-диоксибензола.
Смесь твердого 1,3-диметокси-2-пропилбензола (33,70 г, 190 ммолей) и твердого гидрохлорида пиридина (150 г, 1,30 моля) нагревали до 180oC. Через 7,5 ч реакцию охлаждали до 110oC и медленно добавляли 50 мл воды. После охлаждения реакционной смеси до комнатной температуры ее разбавляли 100 мл воды и несколько раз экстрагировали этилацетатом. Этилацетатный экстракт один раз промывали 2 н. хлористоводородной кислотой и затем сушили над сульфатом магния. Фильтрация и удаление растворителя давали 38,5 г оранжевого твердого вещества. Целевое соединение перекристаллизовывали из дихлорметана и получали 11,86 г (41%) желтых кристаллов.
Спектр ЯМР (CDCl3) δ : 6,94 (т, 1, J=8,10 Гц), 6,40 (д, 2, J=8,10 Гц), 4,84 (с,2), 2,63 (т, 2, J=7,57 Гц), 1,62 (м, 2), 1,01 (т, 3, J=7,33 Гц).
C. Получение этилового эфира 2-(2-пропил-2-оксифенокси)-пропановой кислоты.
Гидрид натрия (1,08 г, 60%-ная масляная дисперсия, 27 ммолей) в атмосфере аргона промывали 15 мл сухого гексана. Надосадочный слой гексана удаляли посредством шприца. К гидриду натрия добавляли сухой тетрагидрофуран (60 мл) и при перемешивании при комнатной температура добавляли 2-пропил-1,3-диоксибензол (4,00 г, 27 ммоль) в форме 40 мл тетрагидрофуранового раствора. После перемешивания при комнатной температуре в течение 25 мин быстро добавляли этиловый эфир бромпропионовой кислоты (4,64 г, 26 ммолей). После перемешивания при комнатной температуре в течение 17 ч реакцию гасили насыщенным водным раствором хлористого аммония и тетрагидрофуран удаляли в вакууме. Полученную водную смесь несколько раз экстрагировали этилацетатом. Органический экстракт сушили над сульфатом магния. Фильтрация и удаление растворителя давали оранжевое масло. Масло очищали хроматографией на силикагеле Merck, элюируя 20%-ной смесью этилацетата в гексане. Желаемый целевой сложный эфир получали в виде белого твердого вещества (2,43 г, выход 36%).
Тонкослойная хроматография: Rf=0,47 (30% этилацетат-гексан) Спектр ЯМР (CDCl3) δ : 6,93 (дд, 1, J=8,00 Гц), 6,45 (д, 1, J=8,00 Гц), 6,30 (д, 1, J= 8,00 Гц), 5,77 (с, 1), 4,76 (кв, 1, J=6,786 Гц), 4,23 (кв, 2, J=7,02 Гц), 2,69 (м, 2), 1,63 (д, 3, J=6,70 Гц), 1,60 (м, 2), 1,28 (т, 3, J=7,50 Гц), 0,99 (т, 3, J=7,50 Гц); ИК-спектр (бромистый калий): 3435, 2955, 2872, 1733, 1600, 1500, 1465 см-1: масс-спектр (свободная десорбция) (m/z) 253 (M++1).
Элементный анализ для C14H20O4:
Вычислено,%: C 66,65; H 7,99.
Найдено,%: C 66,41; H 8,04.
D. Получение этилового эфира 2-{3-/3-/(2-бензилокси-1-бром-5-этил-4-ил- окси/-пропокси/-2-пропилфеноски}-пропановой кислоты.
Целевое соединение получали, используя этиловый эфир 2-(2-пропил-3-оксифенокси)-пропановой кислоты, как описано в примере 54(F). Целевое промежуточное соединение получали с 68% выходом (2,90 г) в виде прозрачного масла.
Тонкослойная хроматография: Rf = 0,47 (30%-ный этилацетат-гексан). Спектр ЯМР (CDCl3) δ : 7,56-7,37 (м, 6), 7,12 (т, 1, J=8,20 Гц), 6,62 (д, 1, J= 8,38 Гц), 6,50 (с, 1), 6,45 (д, 1, J=8,31 Гц), 5,16 (с, 2), 4,80 (кв, 1, J= 6,90 Гц), 4,26 (кв, 2, J=7,20 Гц), 4,18 (дд, 4, J=5,91 и 12,02 Гц), 2,80 (м, 2), 2,62 (кв, 2, J=7,47 Гц), 2,31 (м, 2), 1,69 (д, 3, J=6,70 Гц), 1,65 (м, 2), 1,30 (т, 3, J=7,20 Гц), 1,22 (т, 3, J=7,54 Гц), 1,03 (т, 3, J=7,35 Гц). ИК-спектр (хлороформ): 3015, 2967, 2930, 2780, 1752, 1595, 1500, 1464 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 599 (M+).
Элементный анализ для C32H39O6Br:
Вычислено,%: C 64,11; H 6,56; Br 13,33.
Найдено,%: C 64,01; H 6,56; Br 13,06.
E. Получение этилового эфира 2-{3-/3-/(2-бензилокси-5-)тил-/1,1'-дифенил/-4-ил)-окси/-пропокси/-2-пропилфенокси}-пропановой кислоты.
Сложный эфир получен из этилового эфира 2-{3-/3-/(2-бензилокси-1-бром-5- этил-4-ил)-окси/-пропил-2-пропилфенокси} -пропановой кислоты как описано в примере 54(G). Целевое промежуточное соединение получали с 47%-ным выходом в виде прозрачного масла.
Тонкослойная хроматография: Rf= 0,48 (30%-ная смесь этилацетата в гексане): Спектр ЯМР (CDCl3) δ : 7,10 (д, 2, J=8,06 Гц), 7,44 (м, 8), 7,27 (с, 1), 7,15 (т, 1, J=8,14 Гц), 6,782 (с, 1), 6,66 (д, 1, J=8,27 Гц), 6,48 (д, 1, J=8,27 Гц), 5,11 (с, 2), 4,83 (кв, 1, J=6,71), 4,28 (м, 6), 2,78 (м, 4), 2,38 (м, 2), 1,72 (д, 3, J=6,96 Гц), 1,69 (м, 2), 1,32 (м, 3, J=7,28 Гц), 1,31 (т, 3, J=7,30 Гц), 1,08 (т, 3, J=7,36 Гц): ИК-спектр (хлороформ): 3015, 2966, 2930, 2880, 1750, 1594, 1488, 1464 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 597 (M++1), 596 (M+).
Элементный анализ для C38H44O6:
Вычислено,%: C 76,48; H 7,43.
Найдено,%: C 76,42; H 7,52.
F. Получение этилового эфира 2-{3-/3-/(5-этил-2-окси-/1,1'-дифенил/-4-ил)-окси/-пропокси/-2-пропилфенокси}-пропановой кислоты.
Получен из этилового эфира 2-{3-/3-/(2-бензилокси-5-этил-/1,1'-дифенил/ил) -окси-/-пропокси/-2-пропилфенокси/-пропановой кислоты как описано в примере 54(H). Целевое промежуточное соединение получали с 53%-ным выходом как прозрачное масло.
Тонкослойная хроматография: Rf= 0,36 (30%-ный этилацетат в гексане): спектр ЯМР (CDCl3) δ : 7,43 (м, 5), 7,06 (д, 1, J=8,84 Гц), 6,56 (с, 1), 6,37 (д, 1, J=8,28 Гц), 5,20 (с, 1), 4,74 (кв, 1, J=6,73), 4,20 (м, 6), 2,71 (м, 2), 2,61 (кв, 2, J=7,58 Гц), 2,33 (т, 2, J=6,05 Гц), 1,61 (д, 3, J=6694 Гц), 1,58 (М, 2), 1,25 (т, 3, J=7,30 Гц), 1,19 (т, 3, J=7,30 Гц), 0,96 (т, 3, J= 7,35 Гц). ИК-спектр (хлороформ): 3558, 3029, 3011, 2964, 2935, 2873, 1745, 1625, 1593, 1488, 1464 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 507 (M++1), 506 (M+).
Элементный анализ для C31H38O6:
Вычислено,%: C 73,49; H 7,56.
Найдено,%: C 73,70; H 7,67.
G. Получение 2-{ 3-/3-/(5-этил-2-окси-/1,1'-дифенил)-4-ил)-окси/-пропокси/ -пропилфенокси}-пропановой кислоты.
Получали из этилового эфира 2-{3-/3-/(5-этил-2-окси-/1,1'-дифенил/-4-ил) -окси/-пропокси/-2-пропилфенокси} -пропановой кислоты, как описано в примере 54(I). Целевое соединение кристаллизовали из смеси толуол-гексан и получали белого туфа (0,582 г, 80%-ный выход).
Тонкослойная хроматография: Rf= 0,21 (10% метанол-хлористый метилен). ЯМР-спектр (CDCl3) δ : 7,45 (м, 5), 7,09 (т, 1, J=8,16 Гц), 7,03 (с, 1), 6,60 (д, 1, J=8,28 Гц), 6,56 (с, 1), 6,42 (д, 1, J=8,29 Гц), 4,79 (кв, 1, J= 7,00 Гц), 4,20 (м, 4), 2,70 (м, 2), 2,62 (кв, 2, J=7,49 Гц), 2,33 (т, 2, J= 6,00 Гц), 1,67 (д, 3, J=6,93 Гц), 1,56 (м, 2), 1,20 (т, 3, J=7,39 Гц), 0,96 (т, 3, J=7,30 Гц); ИК-спектр (KBr): 3381, 2964, 2871, 1707, 1515, 1594, 1461 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 479 (M++1), 478 (M+).
Элементный анализ для C29H34O6:
Вычислено,%: C 72,78; H 7,16.
Найдено,%: C 73,39; H 7,29.
Пример 57. Мононатриевая соль 2-(4-хлорфенил)-4-этил-5-/6-метил-6-(2Н-тетразол-5-ил)-гептилокси/-фенола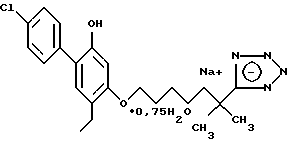
A. Получение 1-бензилокси-2-(4-хлорфенил)-4-этил-5-/6-метил-6- цианогептилокси/-бензола
Целевое промежуточное соединение получали из 1-бензил-окси-2-бром-4-этил-5-/6-метил-6-цианогептилокси/-бензола и 4-хлорфенилборной кислоты путем биарильного сопряжения по методу A (получение 73 (E)). Целевое промежуточное соединение получено в виде белого твердого вещества с 67%-ным выходом. Тонкослойная хроматография: Rf=0,51 (30%-ный этилацетат в гексане). Спектр ЯМР (CDCl3) δ : 7,52 (д, 2, J=8,95 Гц), 7,32 (м, 7), 7,12 (с, 1), 6,75 (с, 1), 5,05 (с, 2), 3,98 (т, 2, J=6,15 Гц), 2,63 (кв, 2, J=7,54 Гц), 1,85 (М, 2), 1,58 (шир. с, 6), 1,38 (с, 6), 1,22 (т, 3, J=7,49 Гц), ИК-спектр (бромистый калий): 2973, 2937, 2858, 1609, 1580, 1561, 1518, 1498 см-1: масс-спектр (свободная десорбция): (m/z) 476 (M++1).
Элементный анализ для C30H34NO2Cl:
Вычислено,%: C 75,69; H 7,20; N 2,94; Cl 7,45.
Найдено,%: C 75,95; H 7,29; N 2,78; Cl 7,68.
B. Получение 2-(4-хлорфенил)-4-этил-5-/6-метил-6-цианогептилокси/-фенола.
Получен из 1-бензилокси-2-(4-хлорфенил)-4-этил-5-/6-метил-6-цианогептил/-оксибензола каталитической гидрогенизацией, как описано в примере 54 (H), с 97%-ным выходом. Белое твердое вещество.
Тонкослойная хроматография: Rf=0,33 (30%-ный этилацетат в гексане). Спектр ЯМР (CDCl3) δ : 7,43 (м, 4), 7,00 (с,1), 5,11 (с, 1), 3,99 (т, 2, J=6,22 Гц), 2,62 (кв, 2, J=7,45 Гц), 1,87 (т, 2, J=6,64 Гц), 1,63 (шир. с, 6), 1,38 (с, 6), 1,20 (т, 3, J=7,63 Гц); ИК-спектр (бромистый калий): 3400, 2940, 2860, 2237, 1618, 1514, 1489, 1468 см-1: масс-спектр (свободная десорбция) (m/z) 385 (M+), 350.
Элементный анализ для C23H28NO2Cl:
Вычислено,%: C 71,58; H 7,31; N 3,63; Cl 9,19.
Найдено,%: C 71,73; H 7,55; N 3,87; Cl 9,11.
C. Получение 2-(хлорфенил)-4-этил-5-(6-метил-6-(2Н-тетразол-5-ил)-гептилокси/-фенола мононатриевой соли
Целевое соединение получали с 38%-ным выходом и 2-(4-хлорфенил)-4-этил-5-(6-метил-6-цианогептилокси/-фенола способом по примерам 43-49 в виде белого лиофилата.
Тонкослойная хроматография: Rf=0,26 (10%-ная смесь метанола в хлористом метилене). Спектр (ДМСО-d6) δ/ : 7,57 (д, 2, J=8,53 Гц), 7,37 (д, 2, J=8,38 Гц), 6,98 (с, 1), 6,67 (с, 1), 3,82 (т, 2, J=6,5 Гц), 1,20 (м, 5), 1,24 (с, 6), 1,07 (т, 3, J=7,42 Гц): ИК-спектр (бромистый калий): 2936, 1616, 3300 (шир. ), 1489 см-1: масс-спектр (бомбардировка быстрыми атомами) (m/z) 473 (M++1+2a).
Элементный анализ для C23H28N4O2ClNa•0,75 H2O:
Вычислено,%: C 59,42; H 6,40; N 12,06; Cl 7,53.
Найдено,%: C 59,57; H 6,43; N 11,89; Cl 7,08.
Пример 58. Мононатриевая соль 2-(3,5-дихлорфенил)-4-этил-5-/6-метил-6-(2Н-тетразол-5-ил)-гептилокси/-фенола.

A. Получение 1-бензилокси-2-(3,5-дихлорфенил)-4-этил-5-/6-метил-цианогептилокси/-бензола.
Целевой дихлордифенил получали из 1-бензилокси-2-бром-5-/6-метил-6-цианогептилокси/-бензола и 3,5-дихлорфенилборной кислоты биарильным сопряжением по методу A. Целевое соединение получали в виде твердого вещества с 54%-ным выходом.
Тонкослойная хроматография: Rf=0,47 (30% этилацетат-гексан). Спектр ЯМР (CDCl3) δ : 7,55 (д, 2, J=2,14 Гц), 7,37 (м, 6), 7,17 (с, 1), 6,63 (с, 1), 5,13 (с, 2), 4,04 (т, 2, J=6,22 Гц), 2,70 (кв, 2, J=7,64 Гц), 1,90 (м, 2), 1,63 (м, 4), 1,42 (с, 6), 1,29 (т, 3, J=7,44 Гц);
ИК-спектр (бромистый калий): 2972, 2941, 2859, 1612, 1583, 1484, 1387 см-1: масс-спектр (свободная десорбция): (m/z) 510 (M++1).
Элементный анализ для C30H33NO2Cl2:
Вычислено,%: C 70,58; H 6,52; N 2,74; Cl 13,89.
Найдено,%: C 70,58; H 6,60; N 2,58; Cl 13,54.
B. Получение 2-(3,5-дихлорфенил)-4-этил-5-(6-метил-6-цианогептилокси/-фенола.
Целевое соединение получали с 72%-ным выходом из 2-(3,5-дихлорфенил)-4-этил-5-/6-метил-6-цианогептилокси/-бензола каталитической гидрогенизацией, как описано в примере 54(H). Белое твердое вещество.
Тонкослойная хроматография: Rf= 0,37 (30%-ный этилацетат в гексане). Спектр ЯМР (CDCl3) δ : 7,36 (д, 2, J=1,3 Гц), 7,31 (с, 1), 6,97 (с, 1), 6,45 (с, 1), 3,97 (т, 2, J=6,26 Гц), 2,58 (кв, 2, J=7,49 Гц), 1,85 (м, 2), 1,56 (м, 6), 1,35 (с, 6), 1,17 (т, 3, J=7,49 Гц): ИК-спектр (бромистый калий): 3360, 2977, 2940, 2856, 2247, 1617, 1582, 1554, 1516, 1465 см-1; масс-спектр (свободная десорбция) (m/z) 419 (M+).
Элементный анализ для C23H27NO2Cl2:
Вычислено,%: C 65,71; H 6,47; N 3,33; Cl 16,88.
Найдено,%: C 65,58; H 6,44; N 3,15; Cl 17,05.
C. Получение мононатриевой соли 2-(3,5-дихлорфенил)-4-этил-5-/6-метил-6-(2Н-тетразол-5-ил)-гептилокси/-фенола.
Целевое соединение получали с 13%-ным выходом и 2-(3,5-дихлорфенил)-4-этил-5-/6-цианогетил/-оксифенола способом по примерам 42-49 и получали белый лиофилат.
Тонкослойная хроматография: Rf=0,26 (101 метанол-хлорметилен). ЯМР-спектр (ДМСО-d6) δ : 7,61 (д, 2, J=1,96 Гц), 7,39 (м, 1), 7,06 (с, 1), 6,66 (с, 1), 3,82 (м, 2), 3,38 (шир. с, 1), 2,44 (кв, 2, J=7,38 Гц), 1,59 (м, 3), 1,26 (м, 6), 1,24 (с, 6), 1,06 (т, 3, J=7,46 Гц); ИК-спектр (KBr): 3408, 2936, 1617, 1585, 1557, 1512, 1376 см-1: масс-спектр (бомбард. быстрыми частицами) (m/z) 485 (M+ +1 +Na), 463 (M+).
Элементный анализ для C23H27N4O2Cl2Na:
Вычислено,%: C 56,91; H 5,61; N 11,54; Cl 14,61.
Найдено,%: C 56,80; H 5,73; N 11,39; Cl 14,37.
Пример 59. Динатриевая соль 3-{2-/3-/(5-этил-2-окси/-1,1-дифенил/-4-ил)-окси/-пропокси/-1-дибензовуран/-пропановой кислоты (см. в конце описания).
A. Получение 3,3-диэтокси-2,3-дигидро-1Н-бензофуро-[3,2-f] [1]-бензопирана.
Раствор 2-оксидибензофурана (5,00 г, 27,2 ммоль), триэтилового эфира орто-акриловой кислоты (10,1 г, 54,3 ммоль) и триметилуксусной кислоты (1,39 г, 13,6 ммоль) в толуоле (100 мл) нагревали с обратным холодильником 18 ч. Смесь охлаждали до комнатной температуры и промывали 1 раз водой и один раз насыщенным раствором бикарбоната натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая оранжевое масло. Это продукт разбавляли гексаном и выдерживали при -20oC в течение 18 ч. Полученные кристаллы собирали вакуумной фильтрацией и получали 5,67 г (67%) желаемого целевого промежуточного соединения, точка плавления 64oC.
Спектр ЯМР (CDCl3) δ : 7,96 (д, 1Н, J=7,8 Гц), 7,57 (д, 1Н, J=8,0 Гц), 7,46 (т, 1Н, J=8 Гц), 7,35 (м, 2Н), 7,06 (д, 1Н, J=8,8 Гц), 3,82 (кв, 2Н, J= 7,2 Гц), 3,73 (кв, 2Н, J=6,8 Гц), 3,35 (т, 2Н, J=6,9 Гц), 2,29 (т, 2Н, J=7,0 Гц), 1,23 (т, 6Н, J= 7,1 Гц); масс-спектр (своб. десорбция) m/z 312 (р); ИК-спектр (хлороформ): 2982, 1494, 1476, 1451, 1434, 1251, 1090, 1054, 975 см-1.
Элементный анализ для C19H20O4:
Вычислено,%: C 73,06; H 6,45.
Найдено,%: C 72,81; H 6,72.
B. Получение этилового эфира 3-/1-(2-оксидибензофуран)/пропановой кислоты.
Смесь 3,3-диэтокси-2,3-дигидро-1Н-бензофуро-[3,2-f] [1] -бензопирана (3,50 г, 11,2 ммоль) и 10%-ной хлористоводородной кислоты (5 мл) в этилацетате (30 мл) перемешивали при комнатной температуре 1 ч. Полученную смесь промывали один раз водой, сушили над сульфатом натрия, концентрировали в вакууме и получали твердое вещество светло-коричневого цвета. Перекристаллизацией из смеси гексан-этилацетат получали 3,11 г (98%) желаемого целевого промежуточного соединения в виде не совсем белого кристаллического вещества, точка плавления 129-131oC.
Спектр ЯМР (CDCl3) δ : 7,88 (д, 1Н, J=7,7 Гц), 7,59 (д, 1Н, J=8,4 Гц), 7,47 (т, 1Н, J=7,2 Гц), 7,37 (д, 1Н, J=8,9 Гц), 7,36 (т, 1Н, J=6,6 Гц), 7,13 (д, 1Н, J=8,8 Гц), 7,13 (кв, 2Н, J=8,8 Гц), 3,43 (т, 2Н, J=5,8 Гц), 3,01 (т, 2Н, J=7,7 Гц), 1,23 (т, 3Н, J=7,2 Гц). Масс-спектр (свободная десорбция) m/e 284 (100, р), 256 (65), 238 (17): ИК-спектр (бромистый калия, см-1): 2985 (шир.), 1701, 1430, 1226, 1183, 1080.
Элементный анализ для C17H16O4:
Вычислено,%: C 71,82; H 5,67.
Найдено,%: C 71,90; H 5,43.
C. Получение 3-{ 2-/3-//5-этил-2-(фенилметокси)-/1,1'-дифенил/-4-ил/-окси/-пропокси/-1-дибензофуран}-пропановой кислоты этиловый эфир.
Этиловый эфир 3-/1-(2-оксидибензовурфн)/-пропановой кислоты (625 мг, 2,20 ммоль) растворяли в диметилформамиде (10 мл) и осторожно обрабатывали при комнатной температуре 95%-ным гидридом натрия (58 мг, 2,4 ммоль). После прекращения выделения газа добавляли 2-бензилокси-1-фенил-5-этил-4-(3-хлор-1-пропилокси)-бензол (836 мг, 2,20 ммоль) и полученную смесь перемешивали 18 ч. Смесь разбавляли эфиром и 1 раз промывали водой. Органический слой сушили над сульфатом натрия, концентрировали в вакууме и получали темное масло. Хроматографией на силикагеле (этилацетат-гексан) получали 200 мг (14%) желаемого целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 8,11 (д, 1Н, J=7,7 Гц), 7,57 (м, 3Н), 7,48 (т, 1Н, J= 7,3 Гц), 7,20-7,44 (м, 10Н), 7,17 (с, 1Н), 7,08 (д, 1Н, J=8,9 Гц), 6,67 (с, 1Н), 5,05 (с, 2Н), 4,29 (т, 2Н, J=7,2 Гц), 3,54 (т, 2Н, J=8,5 Гц), 2,67 (м, 4Н), 2,37 (т, 27, J=6,0 Гц), 1,21 (м, 6Н).
D. Получение двунатриевой соли 3-{2-/3-/(5-этил-2-окси-/1,1'-дифенил/-4-ил)-окси/-пропокси/-1-дибензофуран}-пропановой кислоты.
К продуваемому азотом раствору этилового эфира 3-{2-/3-//5-этил-2-(фенилметокси)-/1,1'-дифенил/-4-ил/-пропокси/-1-дибензофуран} -пропановой кислоты (200 мг, 0,418 ммоль) в смеси метанол-тетрагидрофуран в отношении 1: 1 (40 мл) добавляли 10%-ный палладий на угле (25 мг). Полученную суспензию гидрогенизировали под давлением 1 атм в течение 24 ч при комнатной температуре. Смесь фильтровали через тонкий слой флоризила ( Florisil® ) и фильтрат концентрировали в вакууме. Остаток растворяли в смеси метанол-тетрагидрофуран в отношении 1:1 (20 мл) и обрабатывали 5 н. раствором гидроокиси натрия (2 мл) при комнатной температуре в течение 24 ч. Полученную смесь экстрагировали 1 раз диэтиловым эфиром. Водный слой подкисляли 5 н. раствором хлористоводородной кислоты и 2 раза экстрагировали хлористым метиленом. Объединенные фракции хлористого метилена концентрировали в вакууме. Остаток растворяли в минимальном количестве 1 н. раствора гидроокиси натрия и очищали на смоле НР-20, получая 53 мг (30%) желаемого целевого соединения в виде рыхлого белого твердого вещества.
Спектр ЯМР (ДМСО-d6) δ/ : 8,12 (д, 1Н, J=6,9 Гц), 7,64 (д, 1Н, J=8,2 Гц), 7,37-7,57 (м, 5Н), 7,30 (м, 2Н), 7,14 (м, 2Н), 6,96 (с, 1Н), 6,93 (с, 1Н), 4,30 (т, 2Н, J=7,3 Гц), 4,14 (т, 2Н, J=5,4 Гц), 2,48 (м, 4Н), 2,23 (м, 4Н), 1,10 (т, 3Н, J=7,6 Гц); масс-спектр (бомбардировка быстрыми атомами) m/e 555, (88 Р +1), 533 (62): ИК-спектр (хлороформ): 3384, (шир.), 2969, 1566, 1428, 1257, 1181 см-1.
Элементный анализ для C32H28O6Na2:
Вычислено,%: C 69,31; H 5,09.
Найдено,%: C 69,51, H 5,39.
Пример 60. Моногидрат динатриевой соли 7-карбокси-9-оксо-3-/3- (2-этил-5-окси-4-фенилфенокси)-пропокси/-9Н-ксантен-4-пропановой кислоты.

Смесь 2-бензилокси-1-фенил-5-этил-4-(3-хлор-1-пропилокси)-бензола (749 мг, 1,97 ммоль), этилового эфира 7-карбоэтокси-3-окси-9-оксо-9Н-ксантен-4-пропановой кислоты (729 г, 1,97 ммоль), карбоната калия 91,36 г, 9,85 ммоль) и йодистого калия (33 мг, 0,20 ммоль) нагревали с обратным холодильником 24 ч. Добавляли диметилсульфоксид (2 мл) и нагревание продолжали 24 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и 1 раз промывали водой. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения твердого светло-коричневого вещества. Этот продукт растворяли в этилацетате (30 мл) и полученный раствор продували азотом. К этому раствору добавляли 10%-ный палладий на угле (120 мг) и полученную суспензию гидрогенизировали при давлении 1 атм. Раствор фильтровали, концентрировали в вакууме и получали бесцветное масло. Этот продукт растворяли в смеси метанол-тетрагидрофуран в соотношении 1:1 (30 мл) и обрабатывали 5 н. раствором гидроокиси натрия (2 мл) при комнатной температуре в течение 18 ч. Полученный раствор один раз экстрагировали диэтиловым эфиром и водный слой подкисляли 5 н. раствором хлористоводородной кислоты. Выпавший осадок собирали фильтрацией с всасыванием. Этот продукт обращали в динатриевую соль и очищали как описано выше в примере 59 (Д) для получения 390 мг (56%) желаемого целевого соединения в виде рыхлого белого твердого вещества.
Спектр ЯМР (ДМСО-d6) δ : 12,65 (с, 1Н, -OH), 8,65 (с, 1Н), 8,28 (дд, 1Н, J= 2,0 Гц), 8,01 (д, 1Н, J=8,9 Гц), 7,50 (м, 3Н), 7,29 (т, 2Н, J=7,8 Гц), 7,17 (м, 2Н), 6,93 (с, 1Н), 6,89 (с, 1Н), 4,26 (м, 4Н), 3,12 (м, 2Н), 2,47 (м, 2Н), 2,23 (м, 2Н), 1,10 (т, 3Н, J=7,4 Гц); масс-спектр (бомбардировка быстрыми атомами) m/e 627 (24, р), 605 (40), 583 (24), 331 (24), 309 (100): ИК-спектр (бромистый калий): 3419 (шир. ), 2962, 1612, 1558, 1443, 1390, 1277, 1084 см-1.
Элементный анализ для C34H28O9Na2•H2O:
Вычислено,%: C 63,34; H 4,69.
Найдено,%: C 63,36; H 4,50.
Пример 61. Полугидрат натриевой соли 2-{2-пропил-3-/3-(2-этил-5-окси-4-фенилфенокси)-пропокси/-фенокси}-бензойной кислоты (см. в конце описания).
A. Получение метилового эфира 2-(3-окси-2-пропилфенокси)-бензойной кислоты.
Смесь 1,3-диокси-2-пропибензлоа (75,0 г, 0,490 моль) метилового эфира 2-иодобензойной кислоты (129 г, 0,490 моль) медной бронзы (47,0 г, 0,740 моль) и карбоната калия (81,7 г, 0,592 моль) в сухом пиридине (1 л) тщательно дегазировали азотом, затем нагревали с обратным холодильником 6 ч. Смесь охлаждали до комнатной температуры, фильтровали, концентрировали в вакууме до получения темного остатка. Этот материал растворяли в этилацетате и пропускали через короткую (примерно 500 см3) колонку с флоризилом. Полученный раствор 2 раза промывали насыщенным раствором сульфата меди и концентрировали в вакууме. Остаток растворяли в хлористом метилене, промывали 0,5 н. раствором гидроокиси натрия и 1 раз промывали разбавленным раствором гидроокиси натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения прозрачного коричневого масла. Хроматография на силикагеле (смесь этилацетат-гексан) давала 45,4 г (32%) желаемого целевого промежуточного соединения, в виде белого твердого вещества, т.пл. 80oC.
Спектр ЯМР (CDCl3) δ : 7,92 (дд, 1Н, J=7,8 и 1,6 Гц), 7,42 (т, 1Н, J=8,4 Гц), 7,13 (т, 1Н, J=7,2 Гц), 6,97 (т, 1Н, J=8,1 Гц), 6,86 (д, 1Н, J=8,1 Гц) 6,62 (д, 1Н, J=8,0 Гц), 6,51 (д, 1Н, J=8,0 Гц), 5,65 (шир.с, 1Н, -OH), 3,88 (с, 3Н), 2,66 (т, 2Н, J=7,6 Гц), 1,62 (гекстет, 2Н, J=7,6 Гц), 0,96 (т, 3Н, J= 7,4 Гц); масс-спектр (свободная десорбция) m/e 286 (р): ИК-спектр (хлороформ): 3350 (шир), 2950, 1718, 1602, 1480, 1306, 1225, 1086, 981 см-1.
Анализ для C17H18O4:
Вычислено,%: C 71,31; H 6,34.
Найдено,%: C 71,53; H 6,37.
B. Получение полугидрата натриевой соли 2-{2-пропил-3-/3-(2-этил-5-окси-4-фенилфенокси)-пропокси/-фенокси}-бензойной кислоты.
Метиловый эфир-2-(3-окси-2-пропилфенокси)-бензойной кислоты (450 мг, 1,57 ммоль) алкилировали 2-бензилокси-1-фенил-5-этил-4-(3-хлор-1-пропилокси)-бензолом, дебензилировали и гидролизовали, как описано выше в примере 60, за исключением того, что не использовали диметилсульфоксид. Солеобразование и очистка как описано выше в примере 59 (Д) давали 200 мг (21%) желаемого целевого соединения в виде рыхлого белого твердого вещества.
Спектр ЯМР (ДМСО-d6) δ : 7,48 (д, 2Н, J=7,5 Гц), 7,42 (д, 1Н, J=7,2 Гц), 7,31 (т, 2Н, J=7,3 Гц), 7,18 (т, 1Н, J=7,5 Гц), 7,16 (т, 1Н, J=7,1 Гц), 6,98 (м, 3Н), 6,64 (Т, 2Н, J=7,2 Гц), 6,60 (1Н, с), 6,24 (д, 1Н, J=7,9 Гц), 4,15 (м, 2Н), 4,02 (м, 2Н), 2,61 (м, 2Н), 2,49 (м, 2Н), 2,16 (т, 2Н, J=5,5 Гц), 1,46 (текстет, 2Н, J= 6,6 Гц), 1,07 (т, 3Н, J=7,4 Гц), 0,82 (т, 3Н, J=7,4 Гц): масс-спектр (бомбардировка быстрыми атомами) m/e 549 (100, р+1), 526 (32), 295 (28) 252 (34), 227 (20), 213 (21): ИК-спектр (хлороформ): 3450 (шир.), 2974, 1602, 1586, 1461, 1393, 1240, 1113, 1048 см-1.
Элементный анализ для C33H32O6Na•0,5 H2O:
Вычислено,%: C 71,22; H 5,94.
Найдено,%: C 71,42; H 6,16.
Пример 62. Моногидрат двунатриевой соли 3-/3-(2-этил-5-окси-4-фенил-фенокси)-пропокси/-/1,1'-дифенил/-4-пропановой кислоты (см. в конце описания).
A. Получение этилового эфира 3-/(2-окси-4-фенил)фенил/-пропановой кислоты.
Смесь 3-фенилфенола (5,00 г, 29,4 ммоль), триэтилортоакрилата (10,9 г, 58,8 ммоль) и триметилуксусной кислоты (1,50 г, 14,7 ммоля) в толуоле (100 мл) нагревали с обратным холодильником 24 ч. Полученный раствор охлаждали до комнатной температуры и 1 раз промывали водой и 1 раз разбавленным раствором гидроокиси натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до образования бесцветного масла. Полученное твердое вещество растворяли в тетрагидрофуране (25 мл) и обрабатывали при комнатной температуре 1 н. раствором хлористоводородной кислоты (0,5 мл) в течение 5 мин. Смесь разбавляли эфиром, 1 раз промывали водой, концентрировали в вакууме и получали воскоподобное твердое вещество.
Спектр ЯМР (CDCl3) δ : 7,57 (м, 3Н), 7,44 (т, 2Н, J=7,1 Гц), 7,35 (д, 1Н, J= 7,3 Гц), 7,17 (м, 2Н), 4,19 (кв, 2Н, J=7,2 Гц), 2,97 (т, 2Н, J=6,8 Гц), 2,77 (т, 2Н, J=6,8 Гц), 1,27 (т, 3Н, J=5,4 Гц); масс-спектр (свободная десорбция) m/e 270 (р); ИК-спектр (хлороформ): 3328 (шир.), 3013, 1708, 1564, 1485, 1411, 1379, 1237, 1166 см-1.
Элементный анализ для C17H18O3:
Вычислено,%: C 75,53; H 6,71.
Найдено,%: C 75,80; H 6,60.
B. Получение моногидратной двунатриевой соли 3-/3-(2-этил-5-окси-4-фенилфенокси)-пропокси/-/1,1'-дифенил/4-пропановой кислоты.
Этиловый эфир /(2-окси-4-фенил)-фенил/-пропановой кислоты (354 мг, 1,31 ммоля) алкилировали 2-бензилокси-1-фенил-5-этил-4-(3-хлор-1-пропилокси)-бензолом, дебензилировали и гидролизовывали как описано выше в примере 60, за исключением того, что не применяли диметилсульфоксил. Солеобразование и очистка, как описано выше в примере 59(Д) давали 32 мг (5%) желаемого целевого соединения. Спектр ЯМР (ДМСО-d6) δ : 7,62 (д, 2Н, J=7,9 Гц), 7,53 (д, 2Н, J=8,0 Гц), 7,42 (т, 2Н, J=7,9 Гц), 7,25 (м, 3Н), 7,12 (м, 4Н), 6,93 (с, 1Н), 6,90 (с, 1Н), 4,26 (м, 2Н), 4,19 (м, 2Н), 2,79 (м, 2Н), 2,48 (м, 2Н), 2,18 (м, 4Н), 1,09 (т, 3Н, J=7,7 Гц); масс-спектр (бомбардировка быстрыми атомами) m/e 540 (51, р), 518 (76); ИК-спектр (хлороформ): 3480 (шир.), 2975, 1602, 1408, 1049 см-1.
Элементный анализ для C32H30O5Na2•H2O:
Вычислено,%: C 66,62; H 5,95.
Найдено,%: C 66,25; H 5,67.
Пример 63. Полуторагидрат двунатриевой соли 5-этил-4-{3-/2-пропил-2-/2- (2Н-тетразол-5-ил)-фенокси/-фенокси/-пропокси} -/1,1-дифенил/-2-ола (см. в конце описания).
A. Получение 3-(2-цианофенокси)-2-пропилфенола
Смесь 3-окси-2-пропилфенола (7,50 г, 49,3 ммоль), 2-бромбензонитрила (8,97 г, 49,3 (ммоль), медной бронзы (3,76 г, 59,2 ммоль) и карбоната калия (6,80 г, 49,3 ммоль) в пиридине (250 мл) нагревали с обратным холодильником 72 ч. Смесь охлаждали до комнатной температуры, фильтровали и концентрировали в вакууме. Остаток растворяли в этилацетате и промывали 1 раз водой и 3 раза насыщенным раствором сульфата меди. Органический слой сушили над сульфатом натрия и концентрировали в вакууме до получения темного масла. Хроматография на силикагеле давала белое твердое вещество. Сублимацией этого материала (дистиллятор колба к колбе, 200oC) удаляли избыток 3-окси-2-пропилфенола и получали 1,79 г (14%) желаемого целевого промежуточного соединения в виде не совсем белого кристаллического вещества, точка плавления 103-107oC.
Спектр ЯМР (CDCl3) δ : 7,68 (д, 1Н, J=8 Гц), 7,47 (т, 1Н, J=7 Гц), 7,12 (т, 1Н, 8 Гц), 7,10 (т, 1Н, J=8 Гц), 6,80 (д, 1Н, J=9 Гц), 6,71 (д, 1Н, J=9 Гц), 6,58 (д, 1Н, J=9 Гц), 4,95 (с, 1Н, -OH), 2,62 (т, 2Н, J=7 Гц), 1,60 (гекстет, 2Н, J= 6 Гц), 0,98 (т, 3Н, J=7 Гц); масс-спектр (свободная десорбция) m/e 253 (р): ИК-спектр (хлороформ): 3300 (шир), 2967, 2234, 1600, 1485, 1483, 1450, 1247, 1097, 980 см-1.
Элементный анализ для C16H15NO2:
Вычислено,%: C 75,87; H 5,97; N 5,53.
Найдено,%: C 75,09; H 5,88; N 5,58.
B. Получение полутора гидрата двунатриевой соли 5-этил-4-{3-/2-пропил-3-/2-(2Н-тетразол-5-ил)фенокси/-фенокси/-пропокси}-/1,1'-дифенил/-2-ола.
3-(2-Цианофенокси)-2-пропилфенол (1,66 г, 6,56 ммоля) алкилировали 2-бензилокси-1-фенил-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано выше в примере 60. Неочищенный продукт растворяли в смеси гексан-этилацетат и пропускали через короткую колонку силикагеля. Раствор концентрировали в вакууме и остаток растворяли в 2-метоксиэтаноле (50 мл). К этому раствору добавляли азид лития (1,38 г, 24,2 ммоль) и бромистый триэтиламмоний (1,30 г, 7,14 ммоль). Полученную смесь нагревали с обратным холодильником 48 ч, охлаждали до комнатной температуры и пропускали через короткую колонку силикагеля. Колонку промывали избытком этилацетата и объединенные промывочные жидкости концентрировали в вакууме. Полученный продукт дебензилировали как описано выше в примере 60. Неочищенный тетразол обращали в натриевую соль и очищали как описано выше в примере 59(Д) и получали 320 мг (8%) желаемого целевого соединения в виде рыхлого белого твердого вещества.
ИК-спектр (ДМСО-d6) δ : 7,81 (д, 1Н, J=7,7 и 1,5 Гц), 7,21 (м, 2Н), 7,11 (т, 1Н, J=7,3 Гц), 6,99 (м, 2Н), 6,76 (д, 1Н, J=8,1 Гц), 6,68 (д, 1Н, J=8,2 Гц), 6,56 (с, 1Н), 6,22 (д, 1Н, J=8,2 Гц), 4,16 (т, 2Н, J=5,8 Гц), 4,10 (т, 2Н, J= 5,9 Гц), 2,61 (т, 2Н, J=6,5 Гц), 2,22 (м, 2Н), 1,45 (гекстет, 2Н, J= 7,4 Гц), 1,08 (т, 3Н, J= 7,4 Гц), 0,79 (т, 3Н, J=7,3 Гц),: Масс-спектр (бомбардировка быстрыми атомами) m/e 595 (35, р+1), 574 (39), 573 (100), 551 (99): ИК-спектр (бромистый калий): 3418 (шир), 2962, 1577, 1458, 1243, 1229, 1147, 1117 см-1.
Элементный анализ для C33H32N4O4Na•1,5 H2O:
Вычислено,%: C 64,76; H 5,68; N 9,01.
Найдено,%: C 63,63; H 5,59; N 8,80.
Пример 64. 3-{9-/3-/3-(2-этил-5-окси-4-фенилфенокси)-пропокси/-9-оксо-9Н-ксантен/} -пропановой кислоты полугидрат натриевой соли (см. в конце описания).
A. Получение 3,3-диэтокси-2,3-дигидро-1Н,7Н-пирано-[2,3-с]-ксантен-7-она.
Смесь 3-окси-9-оксо-9Н-ксантена (3,00 г, 12,2 ммоль), триэтилового эфира орто-акриловой кислоты (5,26 г, 28,4 ммоль) и триметилуксусной кислоты (0,720 г, 7,06 ммоль) в толуоле (75 мл) нагревали с обратным холодильником 16 ч. Смесь охлаждали до комнатной температуры и разбавляли эфиром. Полученную смесь промывали 1 раз водой и 1 раз разбавленным раствором гидроокиси натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Перекристаллизацией остатка из смеси гексан-этилацетат получали 4,31 г (90%) желаемого целевого промежуточного соединения в виде белого кристаллического материала, точка плавления 156oC.
Спектр ЯМР (CDCl3) δ : 8,33 (дд, 1Н, J=8,0 Гц), 8,15 (д, 1Н, J=8,8 Гц), 7,69 (т, 1Н, J=6,9 Гц), 7,48 (д, 1Н, J=8,6 Гц), 7,37 (т, 1Н, J=7,7 Гц), 6,93 (д, 1Н, J=8,8 гц), 3,76 (м, 4Н), 3,11 (т, 2Н, J=6,9 Гц), 2,22 (т, 2Н, J=6,9 Гц), 1,23 (т, 6Н, J=7,1 Гц); масс-спектр (свободная десорбция) m/e 340 (р): ИК-спектр (хлороформ): 2980, 1650, 1662, 1606, 1466, 1437, 1230, 1089, 1045 см-1.
Элементный анализ для C20H20O5:
Вычислено,%: C 70,58; H 5,92.
Найдено,%: C 70,83; H 5,84.
B. Получение этилового эфира 3-/4-(3-окси-9-оксо-9Н-ксантен)/-пропановой кислоты.
3,3-диэтокси-2,3-дигидро-1Н, 7Н-пирано-[2,3-с] -7-он (3,40 г, 10,0 ммолей) растворяли в тетрагидрофуране (30 мл) и обрабатывали при комнатной температуре 1 н. раствором хлористоводородной кислоты (0,20 мл) в течение 1 ч. Реакционную смесь разбавляли этилацетатом и промывали 1 раз водой. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Остаток перекристаллизовывали из смеси гексан-этилацетат и получали 3,09 г (99%) желаемого целевого промежуточного соединения в виде белого микрокристаллического твердого вещества, точка плавления 181oC.
Спектр ЯМР (CDCl3) δ : 9,10 (с, 1Н, -OH), 8,34 (дд, 1Н, J=7,9 и 2,0 Гц), 8,17 (д, 1Н, J=8,8 Гц), 7,71 (т, 1Н, J=8 Гц), 7,50 (д, 1Н, J=8,0 Гц), 7,34 (т, 1Н, J=7,8 Гц), 7,03 (д, 1Н, J=8,1 Гц), 4,19 (кв, 2Н, 7,2 Гц), 3,22 (т, 2Н, J= 5,7 Гц), 2,90 (т, 2Н, J=6,6 Гц),1,25 (т, 3Н, J=7,3 Гц): масс-спектр (свободная десорбция) m/e 312 (р): ИК-спектр (хлороформ): 3260 (шир.), 3025, 1648, 1620, 1607, 1467, 1328, 1242 см-1.
Элементный анализ для C18H16O5:
Вычислено,%: C 69,22; H 5,16.
Найдено,%: C 69,13; H 5,22.
C. Получение этилового эфира 3-{4-/3-/3-/2-этил-4-фенил-5-(фенилметокси)-фенокси/-пропокси/-9-оксо-9Н-ксантен/}-пропановой кислоты.
Этиловый эфир 3-/4-(3-окси-9-оксо-9Н-ксантен)/-пропановой кислоты алкилировали 2-бензилокси-1-фенил-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано выше в примере 60, и получали неочищенный продукт в виде оранжевого масла. Хроматография на силикагеле давала 1,48 г (86%) желаемого целевого промежуточного соединения в виде белого твердого вещества, точка плавления 99-102oC. Спектр ЯМР (CDCl3) δ : 8,35 (д, 1Н, J=7,7 Гц), 8,28 (д, 1Н, J=8,9 Гц), 7,73 (т, 1Н, J=7 Гц), 7,54 (м, 3Н), 7,25-7,50 (м, 9Н), 7,18 (с, 1Н), 7,04 (д, 1Н, J=9,0 Гц), 6,66 (с, 1Н), 5,05 (с, 2Н), 4,39 (т, 2Н, J=6 Гц), 4,25 (т, 2Н, J= 5,6 Гц), 4,12 (кв, 2Н, J=7,1 Гц), 3,34 (т, 2Н, J=7,5 Гц), 2,64 (м, 4Н), 2,39 (т, 2Н, J=5,9 Гц), 1,20 (м, 6Н): масс-спектр (свободная десорбция) m/e 656 (100, р), 362 (9): ИК-спектр (хлороформ): 3000, 1727, 1652, 1618, 1604, 1466, 1434, 1276, 1149, 1087 см-1.
Элементный анализ для C42H40O7:
Вычислено,%: C 76,81; H 6,14.
Найдено,%: C 77,05; H 6,24.
D. Получение полугидрата натриевой соли 3-{4-/3-/3- (2-этил-5-окси-4-фенилфенокси)-пропокси/-9-оксо-9Н-ксантен/}-пропановой кислоты.
Дебензилирование, гидролиз, получение соли и очистку 3-{4-/3-/3- /2-этил-4-фенил-5-(фенилметокси)-фенокси/-пропокси/-9-оксо-9Н-ксантен/} -пропановой кислоты этилового эфира (1,24 г, 1,89 ммоля) проводили, как описано выше в примере 59, и получали 817 мг (73%) желаемого целевого соединения в виде белого рыхлого твердого вещества.
Спектр ЯМР (ДМСО-d6) δ : 8,48 (д, 1Н, J=8,0 Гц), 8,00 (д, 1Н, J=8,9 Гц), 7,80 (т, 1Н, J=7,2 Гц), 7,62 (д, 1Н, 8,3 Гц), 7,52 (д, 2Н, J=7,8 Гц), 7,16 (т, 2Н, J=8,7 Гц), 6,94 (с, 1Н), 6,86 (с, 1Н), 4,26 (м, 4Н), 3,10 (м, 2Н), 2,48 (кв, 2Н, J=7,3 Гц), 2,23 (м, 4Н), 1,09 (т, 3Н, J=7,6 Гц): масс-спектр (бомбардировка быстрыми атомами) m/e 583 (7, р), 561 (54), 539 (100): ИК-спектр (бромистый калий): 3410 (шир), 2961, 1605, 1433, 1278, 1147, 1087, 766, 699 см-1.
Элементный анализ для C33H28O7Na•0,5H2O:
Вычислено,%: C 67,00; H 4,94.
Найдено,%: C 67,26; H 5,12.
Пример 65. Двунатриевая соль 2-фтор-6-{2-пропил-3-/3-этил-5-окси-4-фенилфенокси/-пропокси/-фенокси}-бензойной кислоты (cм. в конце описания).
A. Получение метилового эфира 2-фтор-6-(3-окси-2-пропилфенокси)-бензойной кислоты.
Метиловый эфир 2-фтор-6-иодобензойной кислоты (13,1 г, 46,8 ммоля) подвергали реакции Ульмана, описанной выше в примере 61 (А). Этим процессом получали 3,10 г (22%) желаемого целевого промежуточного соединения в виде масла.
Спектр ЯМР (CDCl3) δ : 7,26 (м, 1Н), 7,03 (т, 1Н, J=8,1 Гц), 6,83 (т, 1Н, J=8,6 Гц), 6,65 (д, 1Н, J=8,0 Гц), 6,56 (д, 1Н, J=8,8 Гц), 6,53 (д, 1Н, J=7,6 Гц), 5,30 (шир.с, 1Н, -OH), 3,93 (с, 3Н), 2,59 (т, 2Н, J=7,3 Гц), 1,56 (гекстет, 2Н, J=7,6 Гц), 0,94 (т, 3Н, J=7,3 Гц).
B. Получение двунатриевой соли 2-фтор-6-{2-пропил-3-/3-(2-этил-5-окси-4-фенилфенокси)-пропокси/-фенокси}-бензойной кислоты.
Метиловый эфир 2-фтор-6-(3-окси-2-проилфенокси)-бензойной кислоты )0,660 г, 2,17 ммоля) алкилировали 2-бензилокси-1-фенил-5-этил-4-(3-хлор-1-пропилокси)-бензолом как описано выше в примере 60 и получали продукт в виде масла. Дебензилирование и гидролиз проведены как описано выше в примере 60. Образование соли и очистка, проведенные как описано выше в примере 59(Д) давали 468 мг (37%) желаемого целевого соединения в виде белого рыхлого твердого вещества.
Спектр ЯМР (ДМСО-d6) δ : 7,49 (д, 2Н, J=8,8 Гц), 7,32 (т, J=7,5 Гц, 2Н), 7,18 (т, J=7,4 Гц, 2Н), 7,18 (т, J=7,4 Гц, 1Н), 6,85-7,10 (м, 3Н), 6,74 (т, J=8,1 Гц, 2Н), 6,62 (с, 1Н), 6,42 (д, J=8,1 Гц, 1Н), 6,33 (д, J=8,2 Гц, 1Н), 4,13 (т, J=6,0 Гц, 2Н), 4,04 (т, J=5,8 Гц, 2Н), 2,40-2,63 (м, 4Н), 2,15 (м, 2Н), 1,41 (гекстет, J=7,3 Гц, 2Н), 1,07 (т, J=7,4 Гц, 3Н), 0,79 (т, J=7,2 Гц, 3Н): масс-спектр (бомбардировка быстрыми атомами) m/e 589 (16, р), 567 (100), 546 (30), 527 (15): ИК-спектр (хлороформ): 2975, 1601, 1456, 1395, 1115, 1047 см-1.
Элементный анализ для C33H31O6FNa2:
Вычислено,%: C 67,34; H 5,31; F 3,23.
Найдено,%: C 67,43; H 5,59; F 2,99.
Пример 66. Натриевая соль 2-{2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенокси}-бензойной кислоты (см. в конце описания).
A. Получение метилового эфира 2-{ 2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-фенокси}-бензойной кислоты.
Смесь 2-бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензола (20,0 г, 50,2 ммоль) и йодистого натрия (75,3 г, 502 ммоль) и 2-бутаноле (200 мл) нагревали с обратным холодильником 6 ч. Смесь разбавляли эфиром и 1 раз промывали водой. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения бесцветного масла. Этот продукт растворяли в диметилформамиде (100 мл) и обрабатывали метиловым эфиром 2-(3-окси-2-пропилфенокси)-бензойной кислоты (14,4 г, 50,2 ммоль) и карбонатом натрия (20,8 г, 151 ммоль) при комнатной температуре в течение 24 ч. Эту смесь разбавляли водой и 2 раза экстрагировали эфиром. Водный слой отделяли и снова экстрагировали 1 раз этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения желтого масла. Хроматографией на силикагеле получали 25,4 г (78%) желаемого целевого промежуточного соединения в виде бледно-желтого масла.
Спектр ЯМР (CDCl3) δ : 7,91 (д, J=7,8 Гц, 1Н), 7,54 (д, J=8,6 Гц, 1Н), 7,52 (д, J=8,5 Гц, 1Н), 7,25-7,43 (м, 6Н), 7,03-7,38 (м, 5Н), 6,84 (д, J=8,3 Гц, 1Н), 6,71 (д, J=8,1 Гц, 1Н), 6,63 (с, 1Н), 6,47 (д, J=8,1 Гц, 1Н), 5,03 (с, 2Н), 4,42 (т, J=5,7 Гц, 2Н), 4,21 (т, J=5,8 Гц, 2Н), 3,86 (с, 3Н), 2,69 (т, J= 7,8 Гц, 2Н), 2,64 (т, J=7,7 Гц, 2Н), 2,34 (квинтет, J=6,0 Гц, 2Н), 1,60 (гекстет, J= 5,0 Гц, 2Н), 1,22 (т, J=7,5 Гц, 3Н), 0,94 (т, J=7,5 Гц, 3Н): масс-спектр (свободная десорбция) m/e 648 (р): ИК-спектр (хлороформ): 2960, 1740, 1497, 1461, 1112 см-1.
Элементный анализ для C41H41O6F:
Вычислено,%: C 75,91; H 6,37.
Найдено,%: C 76,15; H 6,45.
B. Получение метилового эфира 3-{3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенокси}-бензойной кислоты.
Метиловый эфир 2-{2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-фенокси} -бензойной кислоты (33,0 г, 50,9 ммоль) дебензилировали, как описано выше в примере 60, и получали 27,3 г (96%) целевого промежуточного соединения в виде янтарного масла.
Спеитр ЯМР (CDCl3) δ : 7,90 (дд, J=7,8 и 1,7 Гц, 1Н), 7,42 (м, 3Н), 7,05-7,23 (м, 4Н), 6,99 (с, 1Н), 6,84 (д, J=8,1 Гц, 1Н), 6,70 (д, J=8,1 Гц, 1Н), 6,55 (с, 1Н), 6,46 (д, J=8,1 Гц, 1Н), 5,05 (с, 1Н, -OH), 4,23 (м, 4Н), 3,86 (с, 3Н), 2,68 (т, J= 7,4 Гц, 2Н), 2,62 (кв, J=7,5 Гц, 2Н), 2,36 (квинтет, J= 6,0 Гц, 2Н), 1,60 (гекстет, J=7,7 Гц, 2Н), 1,20 (т, J=7,6 Гц, 3Н), 0,94 (т, J=7,3 Гц, 3Н): масс-спектр (свободная десорбция) m/e 558 (р): ИК-спектр (хлороформ): 2965, 1727, 1603, 1496, 1458, 1306, 1112 см-1.
Элементный анализ для C34H35O6F:
Вычислено,%: C 73,10; H 6,31.
Найдено,%: C 73,17; H 6,42.
C. Получение натриевой соли 2-{2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/фенокси}-бензойной кислоты.
Метиловый эфир 2-{2-пропил-/3-/2-ээтил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенокси} -бензойной кислоты (21,5 г, 38,5 ммоля) гидролизовали как описано выше в примере 60. Кислоту обращали в натриевую соль и очищали как описано выше в примере 59(Д) и получали 16,7 г (77%) желаемого целевого продукта в виде белого аморфного твердого вещества.
Спектр ЯМР (ДМСО-d6) δ : 10,50 (шир.с, 1Н, -OH), 7,51 (м, 3Н), 7,20 (т, J= 7,4 Гц, 1Н) 7,13 (м, 2Н), 7,00 (м, 2Н), 6,95 (с, 1Н), 6,67 (дд, J=8,2 и 3,3 Гц, 2Н), 6,62 (с, 1Н), 6,26 (д, J=8,2 Гц, 1Н), 4,14 (т, J=5,8 Гц, 2Н), 4,02 (т, J= 5,7 Гц, 2Н), 2,л60 (т, J=6,8 Гц, 2Н), 2,47 (кв, J=7,3 Гц, 2Н), 2,16 (т, J= 5,9 Гц, 2Н), 1,45 (гекстет, J=7,5 Гц, 2Н), 1,07 (т, J=7,5 Гц, 3Н), 0,81 (т, J=7,4 Гц, 3Н): масс-спектр (бомбардировка быстрыми атомами) m/e 568 (38, р+1), 567 (100, р), 544 (86), 527 (77), 295 (65), 253 (45): ИК-спектр (KBr): 3407 (шир.), 2962, 1603, 1502, 1446, 1395, 1239, 1112 см-1.
Элементный анализ для C33H32O6FNa:
Вычислено,%: C 69,95; H 5,69; F 3,35.
Найдено,%: C 69,97; H 5,99; F 3,52.
Пример 67. Тригидрат двунатриевой соли 3-{4-/7-карбокси-9-оксо-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-9Н-ксантен/} -пропановой кислоты.
Этиловый эфир 3-{4-/7-карбометокси-9-оксо-3/-3/-2-этил-4(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-9Н-ксантен/}-пропановой кислоты (550 мг, 751 ммоль) дебензилировали и гидролизовали как описано выше в примере 60, за исключением того, что в аппарате Парра поддерживали давление водорода 2 атм. Кислоту обращали в натриевую соль и очищали как описано в примере 58(Д), и получали 242 мг (46%) желаемого целевого соединения в виде белого рыхлого твердого вещества.
Спектр ЯМР (ДМСО-d6) дельта: 8,65 (д, J=1,8 Гц, 1Н), 8,29 (дд, J=8,6 и 1,8 Гц, 1Н), 8,00 (д, J=8,9 Гц, 1Н), 7,52 (м, 3Н), 7,11 (м, 3Н), 6,92 (с, 1Н), 6,89 (с, 1Н), 4,26 (м, 4Н), 3,10 (м, 2Н), 2,48 (кв, J=7,2 Гц, 2Н), 2,21 (м, 4Н), 1,09 (т, J=7,5 Гц, 3Н); масс-спектр (ББА) m/e 645 (18, р), 624 (30, 623 (61), 601 (74), 309 (100), 307 (54): ИК-спектр (KBr): 3414 (шир.), 2926, 1609, 1391, 1276, 1101, 785 см-1.
Элементный анализ для C34H27O9FNa2•3 H2O:
Вычислено,%: C 58,61; H 4,74.
Найдено,%: C 58,34; H 4,34.
Пример 68. 3-{ 4-/9-оксо-3-/3-/2-этил-4-(4-фторфенил)-5-оксофенокси/ пропокси/-9Н-ксантен/}-пропановая кислота.

A. Получение этилового эфира 3-{ 4-/9-оксо-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-9Н-ксантен/}-пропановой кислоты.
2-Бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензол (0,593 г, 1,49 ммоль) обращали в соответствующий иодид и подвергали взаимодействию с этиловым эфиром 3-4-(3-окси-9-оксо-9Н-ксантен)-пропановой кислоты как описано выше в примере 66(А). Неочищенное соединение перекристаллизовывали из смеси гексан-этилацетат и получали 610 мг желаемого целевого промежуточного соединения в виде не совсем белого твердого кристаллического вещества, точка плавления 115oC.
Спектр ЯМР (CDCl3) δ : 8,34 (дд, J=7,9 и 1,6 Гц, 1Н), 8,27 (д, J=8,9 Гц, 1Н), 7,73 (т, J=7,0 Гц, 1Н), 7,52 (м, 1Н), 7,39 (т, J=7,9 Гц, 1Н), 7,31 (м, 5Н), 7,01-7,13 (м, 4Н), 6,64 (с, 1Н), 5,04 (с, 2Н), 4,39 (т, J=6,0 Гц, 2Н), 4,24 (т, J= 5,8 Гц, 2Н), 4,10 (кв, J=7,1 Гц, 2Н), 3,32 (т, J=7,6 Гц, 2Н), 2,64 (м, 4Н), 2,39 (квинтет, J=5,9 Гц, 2Н), 1,19 (м, 6Г); масс-спектр (СД) m/e 674 (р): ИК-спектр (хлороформ): 2973, 1727, 1653, 1618, 1604, 1497, 1466, 1275, 1146, 1087 см-1.
Элементный анализ для C42H39O7F:
Вычислено,%: C 74,76; H 5,83; F 2,82.
Найдено,%: C 74,49; H 5,72; F 2,65.
B. Получение 3-{ 4-/9-оксо-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-9Н-ксантен/}-пропановой кислоты.
Этиловый эфир 3-{4-/9-гоксо-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-9Н-ксантен/} -пропановой кислоты (500 мг, 0,742 ммоль) дебензилировали и гидролизовали как описано выше в примере 60. Перекристаллизация (толуол-этилацетат) давал 278 мг (67%) целевого соединения в форме белого кристаллического вещества, точка плавления 205oC.
Спектр ЯМР (ДМСО-d6) δ : 12,38 (шир.с, 1Н, -COOH), 9,36 (с, 1Н, -OH), 8,13 (дд, J=7,9 и 1,6 Гц), 1Н), 8,07 (д, J=8,9 Гц, 1Н), 7,82 (т, J=8,7 Гц, 1Н), 7,63 (д, J=8,4 Гц, 1Н), 7,38-7,52 (м, 2Н), 7,08-7,30 (м, 4Н), 6,97 (с, 1Н), 6,55 (с, 1Н), 4,37 (т, J=6,1 Гц, 2Н), 4,13 (т, J=6,0 Гц), 2Н), 3,15 (т, J= 8,2 Гц, 2Н), 2,48 (м, 4Н), 2,28 (квинтет, J=3,7 Гц, 2Н), 1,06 (т, J=7,5 Гц, 3Н): масс-спектр (СД) м/е 556 (р): ИК-спектр (хлороформ): 2974, 1711, 1652, 1618, 1604, 1498, 1466, 1434, 1277, 1146, 1088 см-1.
Анализ для C33H29O7F:
Вычислено,%: C 71,21; H 5,25.
Найдено,%: C 71,14; H 5,23.
Пример 69. 3-{2-/1-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-4-(5-оксо-5-морфолинопентанамидо)-фенил} -пропановая кислота (см. в конце описания).
A. Получение 5-оксо-5-морфиолнопентановой кислоты.
Смесь глутарового ангидрида (28,8 г, 253 ммоль) и морфолина (20,0 г, 230 ммоль) в ксилоле (500 мл) нагревали с обратным холодильником 45 мин. Смесь концентрировали в вакууме и получали целевое промежуточное соединение с количественным выходом в виде светло-оранжевого масла, которое кристаллизовалось при выстаивании, точка плавления 81-83oC.
Спектр ЯМР (CDCl3) δ : 9,80 (шир.с., 1Н, -OH), 3,67 (м, 4Н), 3,61 (м, 2Н), 2,44 (м, 4Н), 1,98 (квинтет, J=6 Гц, 2Н), масс-спектр (с.д.) м.е. 202 (р): ИК-спектр (хлороформ): 3100 (шир.), 3020, 1711, 1635, 1439, 1272, 1237, 1116, 1033 см-1.
B. Получение 4-(5-оксо-5-морфолинопентанамидо)-фенола.
5-Оксо-5-морфолинопентановую кислоту (20,0 г, 99,8 ммоль) растворяли в хлористом метилене (250 мл) и тщательно обрабатывали хлористым оксалилом (15,2 г, 200 ммоль) при комнатной температуре. После того как выделение газа убывало (примерно через 1 ч), смесь концентрировали в вакууме. Остаток растворяли в свежей порции хлористого метилена (50 мл) и в течение 2 ч по каплям добавляли суспензию 4-аминофенола (9,88 г, 99,8 ммоль) и триэтиламина (11,1 г, 110 ммоль), охлажденную до 0oC. После перемешивания в течение 2 ч смесь 2 раза промывали водой. Водный слой снова экстрагировали тремя свежими порциями этилацетата. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения темного масла. Хроматография на силикагеле (этилацетат-гексан) давала 9,70 г (34%) целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 8,58 (шир.с, 1Н), 7,24 (д, J=8,0 Гц, 2Н), 6,70 (д, J=8 Гц, 2Н), 3,55 (м, 6Н), 3,38 (м, 2Н), 2,32 (кв, J=6 Гц, 4Н), 1,0 (м, 2Н).
C. Получение 2,2-диэтокси-3,4-дигидро-6-(5-оксо-5-морфолинопентанамидо)-2Н-1-бензопирана.
4-(5-Оксо-5-морфолинопентанамидо)-фенол (3,00 г, 10,3 ммоль) обращали в желаемое целевое промежуточное соединение, как описано выше в примере 59 (A). Перекристаллизацией (этилацетат-гексан) получали 3,51 г (81%) желаемого целевого промежуточного соединения в виде белого кристаллического твердого вещества, точка плавления 141-143oC.
Спектр ЯМР (CDCl3) δ : 7,88 (шир.с, 1Н, -NH), 7,40 (д, J=2,4 Гц, 1Н), 7,15 (дд, J= 8,7 и 2,6 Гц, 1Н), 6,83 (д, J=8,7 Гц, 1Н), 3,70 (м, 3Н), 3,51 (м, 2Н), 2,86 (т, J=6,8 Гц, 2Н), 2,48 (м, 4Н), 2,07 (4Н, м), 1,20 (т, J=7,1 Гц, 6Н); масс-спектр (СД) м/е 421 (р+1, 14), 420 (р, 100): ИК-спектр (хлороформ): 3010, 1629, 1116, 1086, 1048 см-1.
Элементный анализ для C22H32N2O6:
Вычислено,%: C 62,84; H 7,67; N 6,66.
Найдено,%: C 62,64; H 7,38; N 6,47.
D. Получение этилового эфира 3-{2-/2-окси-4-(5-оксо-5-морфолинопентанамидо/}-пропановой кислоты.
К раствору 2,2-диэтокси-3,4-дигидро-6-(5-оксо-5-морфолинопентанамидо)-2Н-1-бензопирана (1,33 г) в тетрагидрофуране (25 мл) добавляли 1 н. водную хлористоводородную кислоту (0,15 мл). Смесь перемешивали 1 ч при комнатной температуре, затем разбавляли водой и 3 раза экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и получали не совсем белое твердое вещество. Перекристаллизация из смеси гексан-этилацетат давала 1,03 г (83%) желаемого целевого сложного эфира, точка плавления 77-79oC.
Спектр ЯМР (CDCl3) δ : 7,98 (шир.с, 1Н, -NH), 7,35 (д, J=2,5 Гц, 1Н), 7,14 (дд, J=8,5 и 2,5 Гц, 1Н), 6,82 (д, J=8,5 Гц, 1Н), 4,14 (кв, J=7,1 Гц, 2Н), 3,64 (м, 6Н), 3,50 (м, 2Н), 2,87 (т, J=6,6 Гц, 2Н), 2,69 (т, J=6,6 Гц, 2Н), 2,4Q (т, J=6,9 Гц, 2Н), 2,43 (т, J=7,7 Гц, 2Н), 2,02 (м, 2Н), 1,24 (т, J= 7,2 Гц, 3Н); масс-спектр (СД) м/е 393 (р); ИК-спектр (хлороформ): 3350 (шир.), 3020, 1629, 1503, 1234, 116 см-1.
Элементный анализ для C20H28N2O5:
Вычислено,%: C 61,21; H 7,19; N 7,14.
Найдено,%: C 61,10; H 7,18; N 7,14.
E. Получение этилового эфира 3-{ 2-/1-/2-этил-4-(4-фторфенил)-5- (фенилметокси)-фенокси/-пропокси/-4-(5-оксо-5-морфолинопентанамидо)-фенил} -пропановой кислоты.
2-Бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензол (1,00 г, 2,51 ммоль) обращали в соответствующий иодид и подвергали взаимодействию с этиловым эфиром 3-{2-/1-окси-4-(5-оксо-5-морфолинопентанамидо)-фенил/}-пропановой кислоты (980 мг, 2,51 ммоль), как описано выше в примере 66(A). Неочищенный продукт очищали хроматографией на силикагеле и получали 800 мг (32%) целевого промежуточного сложного эфира в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 9,66 (шир.с, 1Н, -NH), 7,50 (м, 2Н), 7,25-7,42 (м, 7Н), 7,16(т, J=8,9 Гц, 2Н), 7,04 (с, 1Н), 6,89 (д, J=8,8 Гц, 1Н), 6,81 (с, 1Н), 5,10 (с, 2Н), 4,20 (т, J=6,1 Гц, 2Н), 4,12 (т, J=5,8 Гц, 2Н), 3,96 (кв, J=7,2 Гц, 2Н), 3,52 (м, 4Н), 3,40 (м, 4Н), 2,76 (т, J=7,3 Гц, 2Н), 2,48 (м, 6Н), 2,28 (м, 4Н), 2,18 (квинтет, J=7 Гц, 3Н), 1,07 (м, 6Н): Масс-спектр (СД) м/е 755 (р); ИК-спектр (хлороформ): 3420 (шир.), 2975, 1604, 1500, 1234, 1145, 1117, 1042 см-1.
Анализ для C44H51N2O8F:
Вычислено,%: C 70,01; H 6,81; N 3,71.
Найдено,%: C 69,77; H 6,89; N 3,77.
F. Получение 3-{2-/1-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-4-(5-оксо-5-морфолинопентанамидо)-фенил}-пропановой кислоты.
Этиловый эфир 3-{ 2-/1-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-4-(5-оксо-5-морфолинопентанамидо)-фенил} -пропановой кислоты (800 мг, 1,06 ммоля) подвергали дебензилированию и гидролизу, как описано выше в примере 60. Эта процедура давала 450 мг (46%) целевого продукта в виде не совсем белого кристаллического вещества, точка плавления 78-80oC.
Спектр ЯМР (CDCl3)дельта:8,48 (шир.с, 1Н, -NH), 7,45 (м, 3Н), 7,20 (с, 1Н), 7,06 (м, 2Н), 6,99 (с, 1Н), 6,75 (д, J=8,8 Гц, 1Н), 6,56 (с, 1Н), 4,10 (м, 4Н), 3,58 (м, 6Н), 3,35 (м, 2Н), 2,88 (т, J=7,5 Гц, 2Н), 2,55 (м, 4Н), 2,37 (м, 4Н), 2,24 (т, J=5,5 Гц, 2Н), 1,92 (м, 2Н), 1,16 (т, J=7,5 Гц, 3Н); масс-спектр (СД) м/е 638 (р+1, 63), 637 (р, 100): ИН-спектр (хлороформ): 2973, 1618, 1502, 1235, 1146, 1117 см-1.
Элементный анализ для C35H41N2O8F:
Вычислено,%: C 66,02; H 6,49; N 4,40.
Найдено,%: C 66,29; H 6,72; N 4,26.
Пример 70. Гидрат двунатриевой соли 2-фтор-6-{2-пропил-3-/3-/2-этил-5- окси-4-(4-фторфенил)-фенокси/-пропокси/-фенокси}-бензойной кислоты.

A. Получение метилового эфира 2-фтор-6-{2-пропил-3-/3-4-бром-2-этил-5-(фенилметокси)-фенокси/-пропокси/-фенокси}-бензойной кислоты.
Метиловый эфир 2-фтор-6-(3-окси-2-пропилфенокси)-бензойной кислоты (1,84 г, 4,80 ммоля) алкилировали 2-бензилокси-1-бром-5-этил-4-(3-хлор-1-пропилокси)-бензолом как описано выше в примере 60 и получали неочищенный продукт в виде масла. Хроматография на силикагеле давала 2,05 г (66%) очищенного целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ/ : 7,49 (д, J=7,1 Гц, 2Н), 7,20-7,35 (м, 5Н), 7,14 (т, J=8,2 Гц, 1Н), 6,82 (т, J=8,5 Гц, 1Н), 6,73 (д, J=8,3 Гц, 1Н), 6,60 (д, J= 8,3 Гц, 1Н), 6,53 (с, 1Н), 6,52 (д, J=8,5 Гц, 1Н), 5,13 (с, 2Н), 4,20 (т, J= 6,0 Гц, 2Н), 4,13 (т, J=6,0 Гц, 2Н), 3,92 (с, 3Н), 2,58 (м, 4Н), 2,3 (квинтет, J= 6,0 Гц, 2Н), 1,51 (гекстет, J=7,6 Гц, 2Н), 1,16 (т, J=7,9 Гц, 3Н), 0,90 (т, J=7,3 Гц, 3Н).
B. Получение гидрата двунатриевой соли 2-фтор-6-{2-пропил-3-/3-/2-этил-5-окси-4-4-фторфенил)-фенокси/-пропокси/-фенокси}-бензойной кислоты.
К раствору метилового эфира 2-фтор-6-{2-пропил-3-/3-/4-бром-2-этил-5-(фенилметокси)-фенокси/-пропокси/-фенокси} -бензойной кислоты (1,77 г, 2,72 ммоль) в бензоле (12 мл) добавляли тетракис-(трифенилфосфин)-палладий (0) (0,33 г, 0,30 ммоль) и 2,0 М водный карбонат натрия 94 мл). К этой смеси добавляли раствор 4-фторфенилборной кислоты (4,10 г, 8,16 ммоля) в этаноле (5 мл). Полученную смесь нагревали с обратным холодильником 4 ч, затем охлаждали до комнатной температуры. Смесь разбавляли этилацетатом и встряхивали. Органический слой один раз промывали водой и один раз 1 н. водной гидроокисью натрия, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и получали масло. Дебензилирование и гидролиз проводили как описано выше в примере 60. Солеобразование и очистка, проведенные как описано выше в примере 59 (Д), давали 403 мг (25%) желаемого целевого соединения в виде рыхлого белого твердого вещества.
Спектр ЯМР (ДМСО-d6) δ : 9,83 (шир.с, 1Н), 7,50 (м, 2Н), 6,96-7,16 (м, 4Н), 6,96 (с, 1Н), 6,74 (т, J=8,4 Гц, 2Н), 6,57 (с, 1Н), 6,40 (д, J=8,3 Гц, 1Н), 6,35 (д, J=8,3 Гц, 1Н), 4,16 (т, J=5,7 Гц, 2Н), 4,05 (т, J=5,5 Гц, 2Н), 2,40-2,58 (м, 4Н), 2,18 (квинтет, J=4,1 Гц, 2Н), 1,41 (гекстет, J=7,4 Гц, 2Н), 1,07 (т, J=7,5 Гц, 3Н), 0,80 (т, J=7,3 Гц, 3Н): масс-спектр (ББА) м/е 586 (р+1, 35), 585 (р, 100), 562 (33), 313 (30): ИК-спектр (хлороформ): 3300 (шир.), 2967, 1616, 1455, 1398, 115 см-1.
Элементный анализ для C33H31O6F2Na•H2O:
Вычислено,%: C 65,77; H 5,52.
Найдено,%: C 65,81; H 5,41.
Пример 71. 4-Фтор-2-{2-пропил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси)-пропокси/-фенокси}-бензойная кислота.

A. Получение метилового эфира 4-фтор-2-(3-окси-2-пропилфенокси)-бензойной кислоты.
К раствору 2-пропилрезорцинола (10,0 г, 65,7 ммоль) в пиридине (120 мл) добавляли трет-бутилат калия (7,0 г, 62,5 ммоль) при комнатной температуре и с перемешиванием. К этой смеси добавляли смесь метилового эфира 2-бром-4-фторбензойной кислоты (7,60 г, 32,6 ммоль) и йодистую медь (1) (12,5 г, 65,7 ммоль) в пиридине (120 мл). Полученную смесь мягко нагревали с обратным холодильником в течение 4 ч. Реакцию охлаждали до комнатной температуры и перемешивали 18 ч. Смесь концентрировали в вакууме и полученный продукт растворяли в этиловом эфире. Раствор промывали 1 раз 5 н. хлористоводородной кислотой. Водный слой 1 раз экстрагировали свежим этиловым эфиром и объединенные органические слои 2 раза промывали 5 н. водной гидроокисью аммония. Органический слой промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Хроматография на силикагеле полученного остатка давала 4,45 г (15%) желаемого промежуточного соединения в виде светло-коричневого твердого вещества, точка плавления 92-94oC.
Спектр ЯМР (CDCl3) δ : 7,95 (м, 1Н), 7,04 (т, J=9,5 Гц, 1Н), 6,79 (т, J= 9,0 Гц, 1Н), 6,65 (Д, J=9,5 Гц, 1Н), 6,50 (м, 2Н), 5,25 (шир.с, 1Н, -OH), 3,88 (с, 3Н), 2,60 (т, J=8,7 Гц, 2Н), 1,55 (гекстет, J=7,8 Гц, 2Н), 0,92 (т, J=7,8 Гц, 3Н); масс-спектр (СД) м/е 305 (р+1, 40), 304 (р, 100): ИК-спектр.
Элементный анализ для C17H17O4F:
Вычислено,%: C 67,10; H 5,63.
Найдено,%: C 67,32; H 5,78.
B. Получение метилового эфира 4-фтор-2-[2-пропил-3-/3-/4-(4-фторфенил)-2-этил-5-(фенилметокси)-фенокси/-пропокси/-фенокси]-бензойной кислоты.
Метиловый эфир 4-фтор-6-(3-окси-2-пропилфенокси)-бензойной кислоты (0,534 г, 1,75 ммоль) алкилировали 2-бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано е в примере 66(A), и получали неочищенный продукт в виде масла. Очистка хроматографией на силикагеле давала 640 мг (55%) желаемого целевого промежуточного соединения в виде белого твердого кристаллического вещества, точка плавления 77-70oC.
Спектр ЯМР (CDCl3) δ : 7,95 (т, J=7,8 Гц, 1Н), 7,53 (м, 2Н), 7,32 (м, 4Н), 7,03-7,20 (м, 3Н), 6,77 (м, 2Н), 6,62 (с, 1Н), 6,55 (д, J=8 Гц, 1Н), 6,50 (д, J= 9 Гц, 1Н), 5,05 (с, 2Н), 4,25 (м, 4Н), 3,89 (с, 3Н), 2,65 (м, 4Н), 2,34 (квинтет, J=6 Гц, 4Н), 1,55 (гекстет, J=6 Гц, 2Н), 1,22 (т, J=7 Гц, 3Н), 0,92 (т, J= 7 Гц, 3Н); масс-спектр (СД) м/е 666 (р): ИК-спектр (хлороформ): 2960, 1730, 1600, 1499, 1461, 1268, 1110 см-1.
Элементный анализ для C44H40O6F2:
Вычислено,%: C 73,86; H 6,05.
Найдено,%: C 73,17; H 6,44.
C. Получение метилового эфира 4-фтор-2-[2-пропил-3-/3-/4-(4-фторфенил)-2-этил-5-оксифенокси/-пропокси/фенокси]-бензойной кислоты.
Метиловый эфир 4-фтор-2-[2-пропил-3-(3-/4-(4-фторфенил)-2-этил-5-(фенилметокси)-фенокси/-пропокси/-фенокси] -бензойной кислоты (590 мг) растворяли в этилацетате (25 мл), содержащем 10%-ный палладий на угле (118 мг), и гидрогенизировали при двух атмосферах 18 ч. Смесь фильтровали через целит и концентрировали в вакууме до получения масла. Очистка сырого продукта хроматографией на силикагеле давала 400 мг (79%) целевого промежуточного соединения в виде стекла.
ЯМР-спектр (CDCl3) δ : 7,97 (т, J=7,8 Гц, 1Н), 7,44 (м, 2Н), 7,17 (м, 3Н), 7,03 (с, 1Н), 6,79 (м, 2Н), 6,45-6,63 (м, 3Н), 5,38 (шир.синглет, 1Н, -OH), 4,22 (м, 4Н), 3,92 (с, 3Н), 2,65 (м, 4Н), 2,35 (квинтет, J=5 Гц, 2Н), 1,57 (гекстет, J=7 Гц, 2Н), 1,24 (т, J=7,8 Гц, 3Н), 0,95 (т, J=7,8 Гц, 3Н). Масс-спектр (СД) м/е 578 (р+2, 50), 577 (р+1, 900), 576 (р, 100): ИК-спектр (хлороформ): 3563 (шир. ), 2965, 1722, 1604, 1585, 1461, 1267, 1251, 1152, 1110 см-1.
Элементный анализ для C34H34O6F2:
Вычислено,%: C 70,82; H 5,94.
Найдено,%: C 71,12; H 5,96.
D. Получение 4-фтор-2-[2-пропил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-фенокси]-бензойной кислоты.
Метиловый эфир 4-фтор-2-[2-пропил-3-/3-/4-(4-фторфенил) -2-этил-5-оксифенокси/-пропокси/-фенокси] -бензойной кислоты (350 мг) гидролизовали как описано выше в примере 60 и получали 310 мг (91%) желаемого целевого продукта в виде белого твердого вещества, точка плавления 62-64oC.
Спектр ЯМР (CDCl3) δ : 8,21 (т, J=7,8 Гц, 1Н), 7,35 (м, 2Н), 7,10-7,30 (м, 3Н), 7,97 (с, 1Н), 6,84 (м, 2Н), 6,63 (д, J=6,8 Гц, 1Н), 6,52 (с, 1Н), 6,41 (д, J= 9 Гц, 1Н), 5,10 (шир.с, 1Н, -OH), 4,23 (м, 4Н), 2,57 (м, 4Н), 2,34 (квинтет, J=5 Гц, 2Н), 1,50 (гекстет, J=6 Гц, 2Н), 1,17 (т, J=7,8 Гц, 3Н), 0,88 (т, J= 7,8 Гц, 3Н): масс-спектр (СД) м/е 564 (р+2, 30), 562 (р, 100): ИК-спектр (хлороформ): 3379, 2963, 1699, 1607, 1500, 1268, 1247, 1146, 1110, 839 см-1.
Элементный анализ для C33H32O6F2:
Вычислено,%: C 70,45; H 5,73.
Найдено,%: C 70,15; H 5,81.
Пример 72. 2-[2-пропил-3-/5-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пентокси/-фенокси]-бензойная кислота ( см. в конце описания).
A. Получение 2-(5-хлорпентокси)-4-(фенилметокси)-ацетофенона
Смесь 2-окси-4-(фенилметокси)-ацетофенона (15,5 г, 64 ммоль), карбоната калия (8,83 г, 64,0 ммоль) и диметилсульфоксида (15 мл) в 2-бутаноле (145 мл) перемешивали при комнатной температуре 30 мин. Добавляли 1-бром 5-хлорпентан (11,9 г, 64,0 ммоль) и полученную смесь нагревали с обратным холодильником 18 ч. Реакционную смесь охлаждали, разбавляли водой и экстрагировали этилацетатом. Органический слой один раз промывали насыщенным раствором хлористого натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения восковидного твердого вещества. Очистка хроматографией на силикагеле (смесь этилацетат-гексан) давала 16,1 г (73%) целевого промежуточного соединения в виде белого твердого вещества, точка плавления 76-77oC.
Спектр ЯМР (CDCl3) δ : 7,85 (д, J=8,7 Гц, 1Н), 7,43 (м, 5Н), 6,59 (д, J= 8,5 Гц, 1Н), 6,53 (с, 1Н), 5,11 (с, 2Н), 4,05 (т, J=6 Гц, 2Н), 3,61 (т, J= 6,0 Гц, 2Н), 2,60 (с, 3Н), 1,90 (м, 4Н), 1,69 (м, 2Н); масс-спектр (сд) м/е 348 (р+2, 65), 346 (р, 100): ИК-спектр (хлороформ): 3025, 1662, 1598, 1268, 1184, 1139, 1027 см-1.
B. Получение 2-(5-хлорпентокси)-4-(фенилметокси)-этилбензола
К раствору 2-(5-хлорпентокси)-4-(фенилметокси)-ацетофенона (15,0 г, 43,2 ммоль) в трифторуксусной кислоте (33,3 мл), при 0oC добавляли по каплям триэтилсилан (11,0 г, 95,1 ммоль). Полученную смесь перемешивали при 0oC 2,5 ч, затем обрабатывали избытком насыщенного раствора бикарбоната натрия. Смесь экстрагировали эфиром. Органический слой промывали один раз насыщенным раствором хлористого натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения желтого масла. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 10,45 г (73%) целевого промежуточного соединения в виде бледножелтого масла.
Спектр ЯМР (CDCl3) δ : 7,20-7,55 (м, 5Н), 7,08 (д, J=9,7 Гц, 1Н), 6,53 (С, 1Н), 6,51 (д, J=8,7 Гц, 1Н), 5,05 (с, 2Н), 3,95 (т, J=6,8 Гц, 2Н), 3,60 (т, J= 6,8 Гц, 2Н), 2,59 (кв, J= 7,8 Гц, 2Н), 1,75-1,95 (м, 4Н), 1,69 (квинтет, J= 6 Гц), 1,18 (т, J=7,8 Гц, 3Н); масс-спектр (сд) м/е: ИК-спектр (хлороформ): 2937, 1613, 1587, 1505, 1289, 1258, 1172, 1132, 1028 см-1.
Элементный анализ для C20H25O2Cl:
Вычислено,%: C 72,12; H 7,57.
Найдено,%: C 71,24; H 7,64.
C. Получение 3-бром-6-(5-хлорпентокси)-4-(фенилметокси)-этилбензола.
Смесь 2-5-хлорпентокси)-4-(фенилметокси)-этилбензола (10,0 г, 31,0 ммоль) и N-бромсукцинимида (5,35 г, 30,1 ммоля) в четыреххлористом углероде (100 мл) слегка нагревали 2 ч, затем перемешивали при комнатной температуре 18 ч. Смесь последовательно промывали водой, 1 н. водным раствором тиосульфата натрия и насыщенным раствором хлористого натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения белого твердого вещества. Перекристаллизацией из гексана получали 10,0 г (81%) желаемого промежуточного соединения, указанного в пункте C, в виде белого кристаллического твердого вещества, точка плавления 54-55oC.
Спектр ЯМР (CDCl3) δ : 7,50 (м, 2Н), 7,25-7,46 (м, 4Н), 6,48 (с, 1Н), 5,15 (с, 2Н), 3,91 (т, J=6,0 Гц, 2Н), 3,58 (т, J=6,0 Гц, 2Н), 2,55 (кв, J=7 Гц, 2Н), 1,85 (м, 4Н), 1,65 (м, 2Н), 1,16 (т, J=7,8 Гц, 3Н); масс-спектр (сд) м/е 414 (р+2, 25), 412 (р, 100), 410 (р - 2, 85): ИК-спектр (хлороформ): 2950, 1602, 1501, 1450, 1370, 1300, 1163 см-1.
Элементный анализ для C20H24O2BrCl:
Вычислено,%: C 58,34; H 5,87.
Найдено,%: C 58,31; H 6,04.
D. Получение 6-(5-хлорпентокси)-2-(4-фторфенил)-4-(пентилметокси)-этилбензола.
3-Бром-6-(5-хлорпентокси)-4-(фенилметокси)-этилбензол (8,80 г, 26,4 ммоля) подвергали взаимодействию с 4-фторфенилборной кислотой как описано выше в примере 70(B). Очистка хроматографией на силикагеле (этилацетат-гексан) с последующей перекристаллизацией из гексана давали 7,04 г (77%) целевого промежуточного соединения в виде белого твердого вещества, точка плавления 55-56oC.
Спектр ЯМР (CDCl3) δ : 7,54 (м, 2Н), 7,33 (м, 5Н), 7,11 (м, 3Н), 6,59 (с, 1Н), 5,07 (с, 2Н), 3,99 (т, J=6 Гц, 3Н), 3,62 (т, J=6 Гц, 2Н), 2,65 (кв, J= 8 Гц, 2Н), 1,90 (м, 4Н), 1,70 (м, 2Н), 1,14 (т, J=8 Гц, 3Н); ИК-спектр (хлороформ): 2938, 1613, 1497, 1143, 1027 см-1.
Элементный анализ для C26H28O2ClF:
Вычислено,%: C 73,14; H 6,61.
Найдено,%: C 72,91; H 6,69.
E. Получение метилового эфира 2-[2-пропил-2-/5-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пентокси/-фенокси]-бензойной кислоты.
Метиловый эфир 2-(3-окси-2-пропилфенокси)-бензойной кислоты (2,0 г, 6,99 ммоль) алкилировали 6-(5-хлорпентокси)-2-(4-фторфенил)-4-(фенилметокси)-этилбензолом, как описано выше в примере 66(A) и получали неочищенный продукт в виде масла. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 3,90 г (83%) целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 7,94 (д, J=8 Гц, 1Н), 7,5 (м, 2Н), 7,35 (м, 6Н), 7,11 (м, 5Н), 6,85 (д, J=9 Гц, 1Н), 6,70 (д, J=9 Гц, 1Н), 6,60 (с, 1Н), 6,48 (д, J= 9 Гц, 1Н), 5,07 (с, 2Н), 4,08 (т, J=5 Гц, 2Н), 4,03 (т, J=5 Гц, 2Н), 3,89 (с, 3Н), 2,70 (м, 4Н), 1,95 (м, 4Н), 1,76 (м, 2Н), 1,62 (м, 2Н), 1,24 (м, J= 7 Гц, 3Н), 0,95 (т, J=7 Гц, 3Н); масс-спектр (СД) м/е (р+1, 65), 676 (р, 100): ИК-спектр (хлороформ): 2965, 1740, 1604, 1497, 1461, 1306, 1111.
Элементный анализ для C42H45O6F:
Вычислено,%: C 76,31; H 6,70.
Найдено,%: C 76,24; H 6,83.
F. Получение 2-[2-пропил-3-/5-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пентокси/-фенокси]-бензойной кислоты.
Метиловый эфир 2-[2-пропил-3-/5-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пентокси/-фенокси] -бензойной кислоты (3,60 г, 5,32 ммоль) подвергали дебензилированию и гидролизу как описано выше в примере 60. Вещество выделяли вакуумной фильтрацией в виде белого кристаллического вещества, точка плавления 65oC (разлагается).
Спектр ЯМР (CDCl3)дельта : 8,25 (дд, J=7,9 и 1,7 Гц, 1Н), 7,44 (м, 3Н), 7,18 (м, 4Н), 6,97 (с, 1Н), 6,80 (д, J=8,2 Гц, 1Н), 6,75 (д, J=8,5 Гц, 1Н), 6,65 (д, J=8,1 Гц, 1Н), 6,54 (с, 1Н), 5,15 (шир.с, 1Н, -OH), 4,10 (т, J=6,1 Гц, 2Н), 4,05 (т, J=5,6 Гц, 2Н), 2,61 (м, 4Н), 1,93 (м, 4Н), 1,75 (м, 2Н), 1,54 (гекстет, J= 7,4 Гц, 2Н), 1,18 (т, J=7,3 Гц, 3Н), 0,89 (т, J=7,3 Гц, 3Н); масс-спектр (СД) м/е 572 (р): ИК-спектр (хлороформ): 3350 (шир), 2965, 1739, 1605, 1496, 1455, 1238, 1108 см-1.
Элементный анализ для C35H37O6F:
Вычислено,%: C 73,41; H 6,51.
Найдено,%: C 73,13; H 6,59.
Пример 73. Полутора гидрат 2-[2-пропил-3-/4-/2-этил-5-окси-4- (4-трифторфенил)-фенокси/-бутокси/-фенокси]-бензойной кислоты.

A. Получение 2-(4-хлорбутокси)-4-(фенилметокси)-ацетофенона.
2-Окси-4-(фенилметокси)-ацетофенон (9,20 г, 37,9 ммоля) алкилировали 1-бром-4-хлорбутаном, как описано выше в примере 72 (A). Неочищенный продукт очищали хроматографией на силикагеле (этилацетат-гексан) и получали 7,70 г (61%) желаемого целевого соединения в виде белого твердого вещества, точка плавления 58-60oC.
Спектр ЯМР (CDCl3) δ : 7,83 (д, J=9 Гц, 1Н), 7,33-7,47 (м, 5Н), 6,59 (дд, J= 9,2 Гц, 1Н), 6,53 (д, J=2 Гц, 1Н), 5,10 (с, 2Н), 4,05 (т, J=5 Гц, 2Н), 3,62 (т, J=5 Гц, 2Н), 2,57 (с, 3Н), 2,02 (м, 4Н); масс-спектр (СД) м/е 339 (р+1, 50), 333 (р, 28), 332 (р-1, 100); ИК-спектр (хлороформ): 3013, 1663, 1599, 1267, 1184, 1027 см-1.
Элементный анализ для C19H21O3Cl:
Вычислено,%: C 68,57; H 6,36.
Найдено,%: C 68,77; H 6,60.
B. Получение 2-(4-хлорбутокси)-4-(фенилметокси)-этилбензола.
2-(4-хлорбутокси)-4-(фенилметокси)-ацетофенон (3,50 г, 10,5 ммоля) восстанавливали, как описано выше в примере 72(B). Очистка хроматографией на силикагеле (этилацетатгексан) давала 2,60 г (79%) желаемого целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 7,13-7,55 (м, 5Н), 7,08 (д, J=8,9 Гц, 1Н), 6,54 (м, 2Н), 5,07 (с, 2Н), 3,99 (д, J=5,7 Гц, 2Н), 3,65 (т, J=6,0 Гц, 2Н), 2,65 (кв, J= 7,5 Гц, 2Н), 2,00 (м, 4Н), 1,22 (т, J=7,5 Гц, 3Н): масс-спектр (СД) м/6: ИК-спектр (хлороформ): 2966, 1613, 1506, 1289, 1171, 1132, 1028 см-1.
Элементный анализ для C19H23O2Cl:
Вычислено,%: C 71,57; H 7,27.
Найдено,%: C 71,78; H 7,40.
C. Получение 3-бролм-6-(4-хлорбутокси)-4-(фенилметокси)-этилбензола.
2-(4-хлорбутокси)-4-(фенилметокси)-этилбензол (2,50 г, 7,84 ммоля) бромировали, как описано выше в примере 72(C). Перекристаллизация неочищенного продукта из гексана давала 2,52 г (81%) желаемого целевого соединения, точка плавления 65-66oC. Спектр ЯМР (CDCl3) δ : 7,50 (д, J=8 Гц, 2Н), 7,34-7,48 (м, 3Н), 7,32 (с, 1Н), 6,49 (с, 1Н), 5,15 (с, 2Н), 3,92 (т, J=5,6 Гц, 2Н), 3,64 (т, J=569 Гц, 2Н), 2,55 (кв, J=7,5 Гц, 2Н), 1,97 (м, 4Н), 1,15 (т, J= 7,5 Гц, 3Н): масс-спектр (СД) м/е 398 (р, 100), 396 (р-2, 70): ИК-спектр (хлороформ): 2967, 1602, 1501, 1455, 1389, 1285, 1163, 1060 см-1.
Элементный анализ для C19H22O2BrCl:
Вычислено,%: C 57,38; H 5,57.
Найдено,%: C 57,27; H 5,62.
D. Получение 6-(4-хлорбутокси)-2-(4-фторофенил)-4-(фенилметокси)-этилбензола.
3-бром-6-(4-хлорбутокси)-4-(фенилметокси)-этилбензол (2,30 г, 26,4 ммоля) подвергали взаимодействию с 4-фтор-фенилборной кислотой как описано выше в примере 70(B). Очистка хроматографией на силикагеле (этилацетат-гексан) с последующим растиранием с метанолом давала целевой промежуточный продукт в виде белого твердого вещества, точка плавления 48-49oC.
Спектр ЯМР (CDCl3) δ : 7,55 (м, 2Н), 4,03 (т, J=5,3 Гц, 2Н), 3,68 (т, J= 5,3 Гц, 2Н), 2,67 (кв, J=7,5 Гц, 2Н), 2,02 (м, 4Н) 1,24 (т, J=7,5 Гц, 3Н): масс-спектр (СД) м/е 412 (р): ИК-спектр.
Элементный анализ для C25H26O2ClF:
Вычислено,%: C 72,72; H 6,35.
Найдено,%: C 72,59; H 6,46.
E. Получение 2-[2-пропил-3-/4-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-бутокси/-фенокси] -бензойной кислоты метилового эфира.
Метиловый эфир 2-(3-окси-2-пропилфенокси)-бензойной кислоты (1,40 г, 4,84 ммоль) алкилировали 6-(4-хлорбутокси)-2-(4-фторфенил)-4-(фенилметокси)-этилбензолом как описано выше в примере 66(A), и получали неочищенный продукт в виде масла. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 2,40 г (75%) целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 7,93 (дд, J=6,2 и 1,7 Гц, 1Н), 7,54 (м, 2Н), 7,25-7,45 (м, 6Н), 7,13 (м, 5Н), 6,88 (д, J=8,8 Гц, 1Н), 6,70 (д, J=8,8 Гц, 1Н), 6,63 (с, 1Н), 6,50 (д, J=8,3 Гц, 1Н), 5,07 (с, 2Н), 4,12 (м, 4Н), 3,89 (с, 3Н), 2,68 (м, 4Н), 2,09 (м, 4Н), 1,63 (гекстет, J=7,4 Гц, 2Н), 1,15 (т, J= 7,4 Гц, 3Н), 0,97 (т, J=7,3 Гц, 3Н): масс-спектр (СД) м/е 663 (р+1, 35), 662, (р, 100): ИК-спектр (хлороформ): 3470, 2950, 1760, 1740, 1461, 1305, 1135, 1071 см-1.
Элементный анализ для C42H43O6F:
Вычислено,%: C 76,11; H 6,54.
Найдено,%: C 76,36; H 6,65.
F. Получение полтора гидрата метилового эфира 2-[2-пропил-3-/4-/2-(4-фтофренил)-5-(фенилметокси)-фенокси/-бутокси/фенокси]-бензойной кислоты.
Метиловый эфир 2-[2-пропил-3-/4-/2-этил-4-(4-фторфенил)-5- (фенилметокси)-фенокси/-бутокси/-фенокси] -бензойной кислоты (2,20 г, 3,32 ммоль) подвергали дебензилированию и гидролизу как описано выше в примере 60. Этот процесс давал 1,00 г (85%) целевого соединения в виде белого твердого вещества, точка плавления 65-68oC.
Спектр ЯМР (CDCl3)дельта : 8,26 (дд, J=6,0 и 1,8 Гц, 1Н), 7,43 (м, 3Н), 7,12-7,29 (м, 4Н), 6,99 (с, 1Н), 6,81 (д, J=8 Гц, 1Н), 6,75 (д, J=8,2 Гц, 1Н), 6,65 [(д, J=8 Гц, 1Н), 6,53 (с, 1Н), 5,08 (шир. синглет, 1Н, -OH),4,12 (м, 4Н), 2,63 (м, 4Н), 2,08 (м, 4Н), 1,55 (гекстет, J=7,4 Гц, 2Н), 1,20 (т, J= 7,3 Гц, 3Н), 0,90 (т, J=7,4 Гц, 3Н), масс-спектр (ДС) м/е 559 (р+1, 57), 558 (р, 100): ИК-спектр (хлороформ): 3350 (шир.), 2950, 1739, 1625, 1496, 1455, 1237, 1108 см-1.
Элементный анализ для C34H35O6F•1,5H2O:
Вычислено,%: C 69,73; H 6,43.
Найдено,%: C 69,74; H 6,54.
Пример 74. 2-[2-(2-метилпропил)-3-/3-/2-этил-5-окси-4-(4-фторфенил) -фенокси/-пропокси/-фенокси]-бензойная кислота.

A. Получение 2-(2-метилпропил)-1,3-диметоксибензола.
К раствору 1,3-диметоксибензола (38,0 г, 272 ммоля) в тетрагидрофуране (380 мл) при 0oC добавляли 1,6 М раствор дибутиллития в гексане (188 мл, 299 ммоль). Полученную смесь перемешивали при 0oC 2 ч. Добавляли 1-йодо-2-метилпропан (50,0 г, 272 ммоля) и реакционную смесь нагревали до комнатной температуры, затем нагревали с обратным холодильником 36 ч. Смесь охлаждали до комнатной температуры, разбавляли насыщенным раствором хлористого аммония и 2 раза экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 13,8 г (26%) целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 7,22 (т, J=9 Гц, 1Н), 6,33 (д, J=10 Гц, 2Н), 3,89 (с, 6Н), 2,66 (д, J=9 Гц, 2Н), 2,0 (гептет, J=8 Гц, 1Н), 1,00 (д, J=8 Гц, 6Н): ИК-спектр (хлороформ): 2959, 1593, 1474, 1261, 1133, 1075 см-1.
B. Получение 2-(2-метилпропил)-1,3-диоксибензола.
2-(2-метилпропил)-1,3-диметоксибензол (18,0 г, 92,8 ммоля) расплавляли с гидрохлоридом пиридиния (90 г) и перемешивали при 180oC 8 ч. Смесь охлаждали до комнатной температуры, разбавляли водой и 2 раза экстрагировали этилацетатом. Органическую фазу промывали разбавленной водной хлористоводородной кислотой, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (эфир-гексан) давала 15,0 г (98%) целевого промежуточного соединения в виде светло-желтого масла.
Спектр ЯМР (CDCl3) δ : 6,97 (т, J=9 Гц, 1Н), 6,43 (д, J=10 Гц, 2Н), 5,68 (с, 2Н, -OH), 2,59 (д, J=9 Гц, 2Н), 2,03 (гептет, J=8 Гц, 1Н), 1,00 (д, J=8 Гц, 6Н); масс-спектр (СД) м/е 166 (р): спектр ИК (хлороформ): 3603, 3349 (шир), 2959, 1601, 1466, 1298, 1104, 987 см-1.
Элементный анализ для C10H14O2:
Вычислено,%: C 72,26; H 8,49.
Найдено,%: C 72,37; N 8,75.
C. Получение метилового эфира 2-/3-окси-2-(метилпропил)-фенокси/-бензойной кислоты.
2-(2-метилпропил)-1,3-диоксибензол (14,5 г, 87,3 ммоль) подвергали реакции Ульмана с метиловым эфиром 2-йодобензойной кислоты как описано выше в примере 61(A). Очистка хроматографией на силикагеле (эфир-гексан) давала 3,11 г (12%) желаемого целевого промежуточного соединения в виде светло-желтого масла.
Спектр ЯМР (CDCl3) δ : 7,91 (д, J=8 Гц, 1Н), 7,23 (т, J=8 Гц, 1Н), 7,16 (т, J= 8 Гц, 1Н), 6,99 (т, J=8 Гц, 1Н), 6,86 (д, J=9 Гц, 1Н), 6,63 (д, J=9 Гц, 1Н), 6,39 (д, J=9 Гц, 1Н), 5,42 (шир.с., 1Н, -OH), 3,84 (с, 3Н), 2,58 (д, J= 9 Гц, 2Н), 2,08 (гептет, J=8 Гц, 1Н), 0,99 (д, J=8 Гц, 6Н): масс-спектр (СД) м/е 300 (р). ИК-спектр (хлороформ): 3625, 3360 (шир.), 2950, 1718, 1602, 1453, 1306, 1235, 1107, 910 см-1.
Элементный анализ для C18H20O4:
Вычислено,%: C 71,98; H 6,71.
Найдено,%: C 72,19; H 6,86.
D. Получение метилового эфира 2-[2-(2-метилпропил)-3-/3-/2-этил-5- (фенилметокси)-4-(4-фторфенил)-фенокси/-пропокси/-фенокси] -бензойной кислоты.
Метиловый эфир 2-/3-окси-2-(2-метилпропил)-фенокси/-бензойной кислоты (750 мг, 2,51 ммоль) алкилировали 2-бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано выше в примере 66 (A), и получали неочищенный продукт в виде масла. Очистка хроматографией на силикагеле (эфир-гексан) давала 620 мг (35%) целевого промежуточного соединения в виде не совсем белого твердого вещества, точка плавления 82-84oC.
Спектр ЯМР (CDCl3) δ : 7,99 (д, J=8 Гц, 1Н), 7,62 (т, J=7 Гц, 2Н), 7,38 (м, 6Н), 7,18 (м, 5Н), 6,90 (д, J=9,0 Гц, 1Н), 6,78 (д, J=9 Гц, 1Н), 6,71 (с, 1Н), 6,53 (д, J=9 Гц, 1Н), 5,09 (с, 2Н), 4,27 (м, 4Н), 3,91 (с, 3Н), 2,70 (м, 4Н), 2,39 (квинтет, J=8 Гц, 2Н), 2,10 (гептет, J=8 Гц, 1Н), 1,30 (т, J= 9 Гц, 3Н), 1,00 (д, J=8 Гц, 6Н); масс-спектр (СД) м/е 663 (р+1, 42), 662 (р, 100): ИК-спектр (KBr): 3425, 2959, 2864, 1733, 1604, 1580, 1500, 1447, 1246, 1080, 837 см-1.
Элементный анализ для C42H43O6F:
Вычислено,%: C 76,11; H 6,54.
Найдено,%: C 76,20; H 6,83.
E. Получение 2-[2-(2-метилпропил)-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-фенокси]-бензойной кислоты.
Метиловый эфир 2-[2-(2-метилпропил)-3-/3-/2-этил-5-(фенилметокси)-4-(4-фторфенил)-фенокси/-пропокси/-фенокси] -бензойной кислоты (600 мг, 0,986 ммоль) подвергали дебензилированию в условиях, описанных выше в примере 71(C). Гидролиз полученного сложного эфира, проведенный как описано выше в примере 60, давал 250 мг (57%) целевого продукта в виде не совсем белого твердого вещества, точка плавления 48-49oC.
Спектр ЯМР (CDCl3) δ/ : 8,25 (д, J=9 Гц, 1Н), 7,44 (м, 3Н), 7,20 (м, 4Н), 7,05 (с, 1Н), 6,85 (д, J=9 Гц, 1Н), 6,76 (д, J=9 Гц, 1Н), 6,64 (д, J=9 Гц, 1Н), 6,59 (с, 1Н), 5,32 (шир. синглет, 1Н, -OH), 4,28 (м, 4Н), 2,63 (кв, J=8 Гц, 2Н), 2,52 (д, J=8 Гц, 2Н), 2,38 (квинтет, J=8 Гц, 2Н), 1,96 (гептет, J= 8 Гц, 1Н), 1,23 (т, J=9 Гц, 3Н), 0,98 (д, J=8 Гц, 6Н); масс-спектр (СД) м/е 559 (р+1, 39), 558 (р, 100): ИК-спектр (хлороформ): 3350 (шир.), 2958, 1699, 1604, 1457, 1222, 1112, 1062, 838, 756 см-1.
Элементный анализ для C34H35O6F:
Вычислено,%: C 73,10; H 6,31.
Найдено,%: C 73,32; H 6,50.
Пример 75. Гидрат 2-[2-бутил-3-/3-/2-этил-5-окси-4-(4-фторфенил) -фенокси/-пропокси/-фенокси]-бензойной кислоты.

A. Получение 2-бутил-1,3-диметоксибензола.
1,3-диметоксибензол (15,0 г, 109 ммоль) алкилировали 1-йодобутаном, как описано выше в примере 74(A), за исключением того, что конечную реакционную смесь не нагревали с обратным холодильником. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 15,0 г (71%) целевого промежуточного соединения в виде желтого масла.
ЯМР-спектр (CDCl3) δ : 7,18 (т, J=8,2 Гц, 1Н), 6,59 (д, J=9,7 Гц, 2Н), 3,84 (с, 6Н), 2,70 (т, J= 8,7 Гц, 2Н), 1,50 (гекстет, J=6 Гц, 2Н), 1,44 (квинтет, J=6 Гц, 2Н), 0,98 (т, J=8,2 Гц, 3Н); масс-спектр (СД) м/е 194 (р).
B. Получение метилового эфира 2-(3-окси-2-бутилфенокси)-бензойной кислоты.
2-Бутил-1,3-диметоксибензол (14,98 г, 77,6 ммоль) деметилировали как описано выше в примере 74 (B) и получали 19 г неочищенного продукта в виде коричневого масла. Раствор этого масла (15 г) и трет-бутилата калия (9,70 г, 86,5 ммоль) в пиридине (150 мл) добавляли ко второму раствору метилового эфира 2-иодобензойной кислоты (11,9 г,180 ммоль) и иодистой меди (1) (17,3 г, 91,0 ммоль) в пиридине (150 мл). Полученную смесь нагревали с обратным холодильником 36 ч. Смесь охлаждали до комнатной температуры, разбавляли водой и 3 раза экстрагировали диэтиловым эфиром. Объединенные эфирные фракции фильтровали через слой целита, 1 раз промывали 5 н. водной хлористоводородной кислотой, 1 раз 2 н. водной гидроокисью натрия и снова фильтровали через целит. Полученный раствор сушили над сульфатом магния, фильтровали и выпаривали в вакууме. Хроматография на силикагеле (этилацетат-гексан) давала 3,02 г (11%) целевого промежуточного соединения в виде желтого масла.
Спектр ЯМР (CDCl3) δ : 7,91 (д, J=8 Гц, 1Н), 7,31 (t, J=8 Гц, 1Н), 7,14 (т, J= 8 Гц, 1Н), 6,97 (т, J=9 Гц, 1Н), 6,83 (д, J=8 Гц, 1Н), 6,59 (д, J=8 Гц, 1Н), 6,39 (д, J=8 Гц, 1Н), 5,04 (широкий синглет, 1Н, -OH), 3,83 (с, 3Н), 2,66 (т, J=9 Гц, 2Н), 1,54 (квинтет, J=5 Гц, 2Н), 1,35 (гекстет, J=5 Гц, 2Н), 0,91 (т, J=8 Гц, 3Н); масс-спектр (электронное соударение) м/е 300 (р, 34), 225 (100), 213 (42), 197 (53), 107 (38): ИК-спектр (паста): 3410, 2926, 1709, 1600, 1463, 1234, 1107, 1090, 992 см-1.
Элементный анализ для C18H20O4:
Вычислено,%: C 71,98; H 6,71.
Найдено,%: C 70,82; H 6,67.
C. Получение метилового эфира 2-[2-бутил-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-фенокси]-бензойной кислоты.
Метиловый эфир 2-(3-окси-2-бутилфенокси)-бензойной кислоты (700 мг, 1,76 ммоль) алкилировали 2-бензилокси-1-(4-(фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано выше в примере получения 66(A), и получали неочищенный продукт в виде масла. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 700 мг (60%) целевого промежуточного соединения в виде желтого масла.
Спектр ЯМР (CDCl3) δ : 7,91 (д, J=9 Гц, 1Н), 7,58 (М, 2Н), 7,38 (м, 6Н), 7,18 (м, 5Н), 6,88 (д, J=10 Гц, 1Н), 6,76 (д, J=9 Гц, 1Н), 6,68 (с, 1Н), 6,47 (д, J= 9 Гц, 1Н), 5,09 (с, 2Н), 4,25 (м, 4Н), 3,91 (с, 3Н), 2,72 (м, 4Н), 2,40 (квинтет, J=5 Гц, 2Н), 1,60 (гекстет, J=5 Гц, 2Н), 1,38 (м, 2Н), 1,24 (т, J= 8 Гц, 3Н), 0,99 (т, J=8 Гц, 3Н): ИК-спектр (хлороформ): 3024, 1717, 1602, 1465, 1453, 1306, 1234, 1086, 1014 см-1.
Элементный анализ для C42H43O6F:
Вычислено,%: C 76,11 H 6,54.
Найдено,%: C 75,82; H 6,50.
D. Получение гидрата 2-[2-бутил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-фенокси]-бензойной кислоты.
Метиловый эфир 2-[2-бутил-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметилокси)-фенокси/-пропокси/-фенокси]-бензойной кислоты (690 мг, 1,04 ммоль) подвергали дебензилированию в условиях, описанных выше в примере 71 (C). Гидролиз полученного сложного эфира, как описано в примере 60, давал 114 мг (30%) целевого продукта в виде не совсем белого твердого вещества, точка плавления 62-64oC.
Спектр ЯМР (ДМСО-d6) δ : 12,75 (шир. синглет, 1Н, -COOH), 9,60 (шир. с., 1Н, -OH), 7,69 (д, J= 7,3 Гц, 1Н), 7,50 (м, 2Н), 7,35 (т, J=7,3 Гц, 1Н), 7,00-7,18 (м, 4Н), 6,96 (м, 2Н), 6,56 (с, 1Н), 6,31 (д, J=8,2 Гц, 1Н), 4,17 (т, J=5,1 Гц, 2Н), 4,09 (т, J=5,4 Гц, 2Н), 2,58 (т, J=7,3 Гц, 2Н), 2,48 (м, 2Н), 2,21 (квинтет, J=5,0 Гц, 2Н), 1,37 (гекстет, J=6,8 Гц, 2Н), 1,21 (м, 2Н), 1,06 (т, J=7,4 Гц, 3Н), 0,74 (т, J=7,1 Гц, 3Н): спектр ЯМР (СД) м/е 559 (р+1, 55), 558 (р, 100): ИК-спектр (бромистый калий): 3350 (шир.), 2963, 2933, 1738, 1605, 1497, 1461, 1455, 1236, 118 см-1.
Элементный анализ для C34H35O6F•H2O:
Вычислено,%: C 70,81; H 6,47.
Найдено,%: C 71,19; H 6,52.
Пример 76. 2-[2-(фенилметил)-3-(3-/2-этил-5-окси-4-(4-фенилфтор)-фенокси/-пропокси/-фенокси]-бензойная кислота.

A. Получение 2-(фенилметил)-1,3-диметоксибензола.
1,3-Диметоксибензол (75,0 г, 391 ммоль) алкилировали бромистым бензилом как описано выше в примере получения 74(A), за исключением того, что конечную реакционную смесь не нагревали с обратным холодильником. Очистка хроматографией на силикагеле (эфир-гексан) давала 18,8 г (8%) промежуточного соединения в виде белого твердого вещества, точка плавления 53-55oC.
Спектр ЯМР (CDCl3) δ : 7,15-7,37 (м, 6Н), 6,62 (д, J=10 Гц, 2Н), 4,12 (с, 2Н), 3,87 (с, 6Н); масс-спектр (СД) м/е 229 (р+1, 17), 228 (р, 100): ИК-спектр (KBr): 2925, 2839, 1594, 1476, 1435, 1259, 1197, 1106, 700 см-1.
Элементный анализ для C15H16O2:
Вычислено,%: C 78,92; H 7,06.
Найдено,%: C 79,21; H 7,33.
B. Получение 2-(фенилметил)-1,3-диоксибензола
2-(Фенилметил)-1,3-диметоксибензол (15,0 г, 65,8 ммоль) деметилировали как описано выше в примере 74(B). Очистка хроматографией на силикагеле (этилацетатгексан) давала 7,76 г (60%) целевого промежуточного соединения в виде не совсем белого твердого кристаллического вещества, точка плавления 81-83oC.
Спектр ЯМР (CDCl3) δ : 7,18-7,23 (м, 5Н), 7,01 (т, J=9 Гц, 1Н), 6,43 (д, J= 10 Гц, 2Н), 5,38 (шир.с, 2Н, -OH), 4,18 (с, 2Н): масс-спектр (СД) м/е 201 (р+1, 23), 200 (р, 100): спектр ИК (KBr): 3505 (шир), 1618, 1464, 1360, 1292, 1183, 1012, 739 см-1.
Элементный анализ для C13H12O2:
Вычислено,%: C 77,98; H 6,04.
Найдено,%: C 77,69; H 5,99.
C. Получение метилового эфира 2-/3-окси-2-(фенилметил)-фенокси/-бензойной кислоты.
2-(Фенилметил)-1,3-диоксибензол (14,5 г, 87,3 (ммоля) подвергали реакции сочетания по Ульману с метиловым эфиром 2-иодобензойной кислоты как описано выше в примере получения 61(A). Очистка хроматографией на силикагеле (этилацетат-гексан) давала 900 мг (7%) целевого промежуточного соединения в виде белого кристаллического вещества, точка плавления 79-81oC.
Спектр ЯМР (CDCl3) δ : 7,93 (д, J=9 Гц, 1Н), 7,35 (м, 3Н), 7,27 (м, 2Н), 7,13 (м, 2Н), 7,04 (д, J=9 Гц, 1Н), 6,83 (д, J=9 Гц, 1Н), 6,63 (д, J=9 Гц, 1Н), 6,41 (д, J=9 Гц, 1Н), 5,43 (шир.с., 1Н, -OH), 4,14 (с, 2Н), 3,79 (с, 3Н); масс-спектр (сд) м/е 335 (р+1, 23), 334 (р, 100): ИК-спектр (KBr): 3327 (шир.), 1687, 1598, 1453, 1315, 1233, 1008, 754 см-1.
Элементный анализ для C21H18O4:
Вычислено,%: C 75,43; H 5,43.
Найдено,%: C 75,21; H 5,57.
D. Получение метилового эфира 2-[2-(фенилметил)-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-фенокси]-бензойной кислоты.
Метиловый эфир 2-/3-окси-2-(Фенилметил)-фенокси/-бензойной кислоты (840 мг, 2,51 ммоля) алкилировали 2-бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом как описано выше в примере 66(A). Очисткой хроматографией на силикагеле (этилацетат-гексан) получали 680 мг (40%) желаемого целевого промежуточного соединения в виде стекла.
Спектр ЯМР (CDCl3) δ : 8,01 (д, J=8 Гц, 1Н), 7,65 (м, 3Н), 7,40 (м, 8Н), 7,15-7,30 (м, 8Н), 6,88 (д, J=10 Гц, 1Н), 6,80 (д, J=10 Гц, 1Н), 6,63 (с, 1Н), 6,48 (д, J=9 Гц, 1Н), 5,09 (с, 2Н), 4,34 (т, 7 Гц 2Н), 4,22 (с, 2Н), 4,20 (т, J=7 Гц, 2Н), 3,84 (с, 3Н), 2,77 (кв, J=8 Гц, 2Н), 2,40 (квинтет, J= 8 Гц, 2Н), 1,38 (т, J=9 Гц, 3Н): масс-спектр (СД) м/е 698 (р+1, 48), 697 (р, 100): ИК-спектр (хлороформ): 3015, 2975, 1717, 1604, 1496, 1453, 1306, 1081 см-1.
Элементный анализ для C45H41O6F:
Вычислено,%: C 77,57; H 5,93.
Найдено,%: C 77,80; H 6,08.
E. Получение 2-[2-(фенилметил)-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-фенокси]-бензойной кислоты.
Метиловый эфир 2-[2-(фенилметил)-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)фенокси/-пропокси/-фенокси] -бензойной кислоты (660 мг, 0,947 ммоль) подвергали дебензилированию и гидролизу в условиях, описанных в примере получения 60. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 450 мг (80%) желаемого промежуточного соединения в виде стекла.
Спектр ЯМР (CDCl3) δ : 8,16 (дд, J=7,8 и 1,8 Гц, 1Н), 7,43 (м, 2Н), 7,35 (м, 1Н), 7,05-7,32 (м, 9Н), 7,02 (с, 1Н), 6,86 (д, 8,4 Гц, 1Н), 6,66 (д, J= 8,4 Гц, 1Н), 6,61 (д, J=8,2 Гц, 1Н), 6,46 (с, 1Н), 4,28 (т, J=4,6 Гц, 2Н), 4,10 (т, J= 4,1 Гц, 2Н), 4,08 (с, 2Н), 2,64 (кв, J=7,5 Гц, 2Н), 2,33 (квинтет, J= 5,1 Гц, 2Н), 1,22 (т, J=7,5 Гц, 3Н); масс-спектр (СД) м/е 593 (р, 100), 592 (р - 1, 89); ИК-спектр (хлороформ): 3375 (шир.), 3020, 2970, 1738, 1605, 1496, 1068 см-1.
Элементный анализ для C37H33O6F:
Вычислено,%: C 74,98; H 5,61.
Найдено,%: C 75,21; H 5,72.
Пример 77. 2-[2-пропил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-фенокси]-фенилуксусной кислоты.
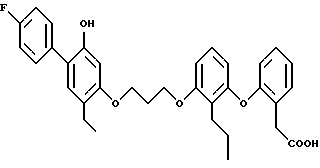
A. Получение метилового эфира 2-(3-окси-2-пропилфенокси)-фенилуксусной кислоты.
1,3-Диокси-2-пропилбензол (6,07 г, 39,9 ммоль) подвергали реакции Ульмана с метиловым эфиром 2-йодофенилацетатом, как описано выше в примере получения 61(A). Очистка неочищенного продукта хроматографией на силикагеле (этилацетат-гексан) давала 1,27 г (11%) целевого соединения в виде желтого масла.
Спектр ЯМР (CDCl3) δ : 7,34 (д, J=9 Гц, 1Н), 7,23 (т, J=8 Гц, 1Н), 7,08 (т, J= 8 Гц, 1Н), 6,97 (т, J=8 Гц, 1Н), 6,77 (д, J=8 Гц, 1Н), 6,58 (д, J=8 Гц, 1Н), 3,68 (д, J=9 Гц, 1Н), 3,75 (с, 2Н), 3,66 (с, 3Н), 2,63 (т, J=6 Гц, 2Н), 1,61 (гекстет, J=6 Гц, (2Н), 0,97 (т, J=7 Гц, 3Н): масс-спектр (СД) м/е 300 (р): ИК-спектр (хлороформ): 3350 (шир.), 3020, 2962, 1736, 1455, 1236, 1107, 982 см-1.
B. Получение метилового эфира 2-[2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-фенокси]-фенилуксусной кислоты.
Метиловый эфир 2-(3-окси-2-пропилфенокси)-фенилуксусной кислоты (750 мг, 2,5 ммоль) алкилировали 2-бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом как описано в примере 66(A). Очистка хроматографией на силикагеле (этилацетат-гексан) давала 750 мг (45%) целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 7,53 (м, 2Н), 7,25-7,40 (м, 6Н), 7,19 (т, J=8 Гц, 2Н), 7,04-7,17 (м, 4Н), 6,72 (д, J=8,7 Гц, 1Н), 6,69 (д, J=8,2 Гц, 1Н), 6,62 (с, 1Н), 6,45 (д, J=8,2 Гц, 1Н), 5,03 (с, 2Н), 4,22 (м, 4Н), 3,75 (с, 2Н), 3,66 (с, 3Н), 2,65 (м, 4Н), 2,34 (квинтет, J=6,0 Гц, 2Н), 1,54 (гекстет, J= 7,4 Гц, 2Н), 1,21 (т, J=7,5 Гц, 3Н), 0,93 (т, J=7,3 Гц, 3Н), масс-спектр (СД) м/е 663 (р+1, 57), 622 (р, 100): ИК-спектр (хлороформ): 2975, 1750, 1602, 1496, 1454, 1231, 1116 см-1.
Элементный анализ для C42H43O6F:
Вычислено,%: C 76,11; H 6,54.
Найдено,%: C 76,36; H 6,71.
C. Получение 2-[2-пропил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-фенокси]-фенилуксусной кислоты.
Метиловый эфир 2-[2-пропил-3-/3-/2-этил-4-(4-фтофренил)-5-(фенилметокси)-фенокси/-пропокси/-фенокси] -фенилуксусной кислоты (630 мг, 1,10 ммоль) подвергали дебензилированию и гидролизу в условиях, описанных в примере 60. Очистка хроматографией на силикагеле давала 320 мг (60%) целевого соединения в виде стекла.
Спектр ЯМР (CDCl3) δ : 7,48 (м, 2Н), 7,33 (д, J=7,4 Гц, 1Н), 7,00-7,30 (м, 6Н), 6,76 (д, J=7,8 Гц, 1Н), 6,73 (д, J=8,1 Гц, 1Н), 6,57 (с, 1Н), 6,52 (д, J=8,2 Гц, 1Н), 4,25 (м, 4Н), 3,82 (с, 2Н), 2,78 (м, 4Н), 2,38 (квинтет, J= 5,9 Гц, 2Н), 1,60 (гекстет, J=7,5 Гц, 2Н), 1,25 (т, J=7,5 Гц, 3Н), 0,95 (т, J=7,4 Гц, 3Н): масс-спектр (СД) м/е 559 (р+1, 65), 558 (р, 100): ИК-спектр (хлороформ): 3570, 2966, 2934, 2873, 1714, 1582, 1496, 1463, 1230, 1116 см-1.
Элементный анализ для C34H35O6F:
Вычислено,%: C 73,10; H 6,31.
Найдено,%: C 73,24; H 6,41.
Пример 78. 2-[2-пропил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-бензоил]-бензойная кислота ( см. в конце описания).
A. Получение 2-/3-(аллилокси)-бензоил/-бензойной кислоты
К раствору 3-(аллилокси)-бромбензола (15,0 г, 70,5 ммоль) в тетрагидрофуране (750 мл) при -70oC добавляли 1,6 М н-бутиллития 944,1 мл, 70,5 ммоль). После перемешивания в течение одного часа добавляли раствор фталевого ангидрида (11,4 г, 77,0 ммоль) в тетрагидрофуране (100 мл, предварительно охлажденном до -70oC) в течение 1 ч. Смесь разбавляли насыщенным раствором хлористого аммония и экстрагировали диэтиловым эфиром. Органический слой 3 раза промывали 1 н. раствором гидроокиси натрия и объединенные водные слои экстрагировали свежей порцией диэтилового эфира. Водный слой подкисляли до pH 3 водной хлористоводородной кислотой и 3 раза экстрагировали свежим диэтиловым эфиром. Объединенные органические слои промывали 1 раз водой, 1 раз насыщенным раствором хлористого натрия, фильтровали и концентрировали в вакууме до получения не совсем белого твердого вещества. Перекристаллизация из смеси эфир-гексан давала 10,3 г (52%) целевого промежуточного соединения в виде белого кристаллического вещества, точка плавления 109oC.
Спектр ЯМР (CDCl3) δ : 8,20 (д, J=8 Гц, 1Н), 7,65 (т, J=8 Гц, 1Н), 7,60 (т, J= 8 Гц, 1Н), 7,30-7,45 (м, 3Н), 7,28 (д, J=8 Гц, 1Н), 7,20 (д, J=8 Гц, 1Н), 6,02 (м, 1Н), 5,35 (д, J=16 Гц, 1Н), 5,30 (д, J=11 Гц, 1Н), 4,55 (д, J= 6 Гц, 2Н); масс-спектр (СД) м/е 283 (р+1, 27), 282 (р, 100).
Элементный анализ для C17H14O4:
Вычислено,%: C 72,33; H 5,00.
Найдено,%: C 72,07; H 5,22.
B. Получение метилового эфира 2-/3-(аллилокси)-бензоил/-бензойной кислоты.
Раствор 2-/3-(аллилокси)-бензоил/-бензойной кислоты (900 мг, 31,9 ммолm) в метаноле (100 мл) насыщали газообразным хлористым водородом. Полученный раствор перемешивали при комнатной температуре 18 ч. Реакционную смесь концентрировали в вакууме и разбавляли диэтиловым эфиром. Полученный раствор последовательно промывали насыщенным раствором бикарбоната натрия, водой и насыщенным раствором хлористого натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Полученное бледно-желтое масло отверждалось при выдерживании и получали 9,45 г (100%) желаемого целевого продукта в виде белого твердого вещества, точка плавления 50-52oC.
Спектр ЯМР (CDCl3) δ : 8,05 (д, J=7,8 Гц, 1Н), 7,65 (т, J=8 Гц, 1Н), 7,56 (т, J=8 Гц, 1Н), 7,40 (м, 2Н), 7,32 (т, J=8 Гц, 1Н), 7,22 (д, J=8 Гц, 1Н), 7,14 (д, J=8 Гц, 1Н), 6,08 (м, 1Н), 5,40 (д, J=16 Гц, 1Н), 5,30 (д, J= 11 Гц, 1Н), 4,78 (д, J=4 Гц, 2Н), 3,62 (с, 3Н); масс-спектр (СД) м/е 297 (р+1,40), 296 (р, 100(: ИК-спектр.
Элементный анализ для C18H16O4:
Вычислено,%: C 72,46; H 5,44.
Найдено,%: C 72,75; H 5,58.
C. Получение метилового эфира 2-[3-окси-2-/3-(1-пропенил)бензоил]-бензойной кислоты и метилового эфира 2-[3-окси-4-/3-(1-пропенил)/-бензоил]-бензойной кислоты.
Метиловый эфир 2-/3-аллилокси)-бензоил/-бензойной кислоты (6,70 г, 20,2 ммоля) нагревали около 175oC в течение 30 ч. Смесь продукта охлаждали до комнатной температуры и очищали хроматографией на силикагеле (смесь хлористый метилен-этилацетат в отношении 95: 5) и получали 3,62 г (54%) метилового эфира 2-[3-окси-2-/3-(1-пропенил)/-бензоил]-бензойной кислоты и 1,44 г (21%) метилового эфира 2-[3-окси-4-/3-(1-пропенил)/-бензоил]-бензойной кислоты в виде белых твердых веществ.
Метиловый эфир 2-[3-окси-2-/3-(1-пропенил)/-бензоил]-бензойной кислоты, точка плавления 107-109oC.
Спектр ЯМР (CDCl3) δ : 7,91 (дд, J=7,8 Гц, и 2,2 Гц, 1Н), 7,43-7,63 (м, 3Н), 7,08 (м, 1Н), 7,02 (д, J=8 Гц, 1Н), 6,80 (дд, J=8,0 и 2,0 Гц, 1Н), 6,15 (м, 1Н), 5,42 (широкий синглет, 1Н, -OH), 5,23 (д, J=16 Гц, 1Н), 5,16 (д, J= 11 Гц, 1Н), 3,81 (д, J=6 Гц, 2Н), 3,68 (с, 3Н; масс-спектр (СД) м/е 297 (р+1, 40), 296 (р, 100), 278 (45): ИК.
Элементный анализ для C18H16O4:
Вычислено,%: C 72,96; H 5,44.
Найдено,%: C 73,26; H 5,54.
Метиловый эфир 2-[3-окси-4-/3-(1-пропенил)/-бензоил]-бензойной кислоты, точка плавления 149-140oC. Спектр ЯМР (CDCl3) δ : 8,08 (дд, J=7,9 и 3,1 Гц, 1Н), 7,63 (т, J= 8 Гц, 1Н), 7,55 (т, J=8 Гц, 1Н), 7,40 (д, J=8,0 Гц, 1Н), 7,35 (с, 1Н), 7,16 (с, 2Н), 6,00 (м, 1Н), 5,62 (широкий синглет, 1Н, -OH), 5,15 (м, 2Н), 3,65 (с, 3Н), 3,47 (д, J=5 Гц, 2Н); масс-спектр (СД) м/е 297 (р + 1, 20), 296 (р, 100): ИК-спектр.
Элементный анализ для C18H16O4:
Dычислено,%: C 72,96; H 5,44.
Yайдено,%: C 73,11, H 5,50.
D. Получение метилового эфира 2-[2-/3-(1-пропенил)/-3-/3-/2-этил-5-(фенилметокси)-4-(4-фторфенил)-фенокси/-пропокси/-бензоил] -бензойной кислоты, точка плавления 149-140oC. Спектр ЯМР (CDCl3) δ : 8,08 (дд, J=7,9 и 3,1 Гц, 1Н), 7,63 (т, J=8 Гц, 1Н), 7,55 (т, J=8 Гц, 1Н), 7,40 (д, J=8 Гц, 1Н), 7,35 (с, 1Н), 7,16 (с, 2Н), 6,00 (м, 1Н), 5,62 (широкий синглет, 1Н, -OH), 5,15 (м, 2Н), 3,65 (с, 3Н), 3,47 (д, J=5 Гц, 2Н); масс-спектр (СД) м/е 297 (р+1, 20), 286 (р, 100): ИК-спектр.
Элементный анализ для C18H16O4:
Выделено,%: C 72,96; H 5,44.
Найдено,%: C 73,11; H 5,50.
D. Получение метилового эфира 2-[2-/3-(1-пропенил)/-3-/3-/2-этил-5-(фенилметокси)-4-(4-фторфенил)-фенокси/-пропокси/-бензоил] -бензойной кислоты.
Метиловый эфир 2-[3-окси-2-/3-(1-пропенил)/-бензоил]-бензойной кислоты (520 мг, 1,75 ммоль) алкилировали 2-бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано в примере получения 66(A). Перекристаллизация неочищенного продукта из смеси эфир-гексан давала 750 мг (65%) желаемого целевого промежуточного соединения в виде белого твердого вещества, точка плавления 90-91oC.
Спектр ЯМР (CDCl3) δ/ : 7,91 (м, 1Н), 7,53 (м, 4Н), 7,45 (м, 1Н), 7,32 (м, 5Н), 7,02-7,22 (м, 5Н), 6,85 (д, J=8,0 Гц, 1Н), 6,61 (с, 1Н), 6,10 (м, 1Н), 5,04 (д, J=16 Гц, 1Н), 5,03 (с, 2Н), 4,99 (д, J=11 Гц, 1Н), 4,23 (м, 4Н), 3,77 (д, J= 7 Гц, 2Н), 3,66 (с, 3Н), 1,64 (д, J=6 Гц, 2Н), 2,37 (квинтет, J= 6 Гц, 2Н), 1,19 (т, J=8 Гц, 3Н); масс-спектр (СД) м/е 659 (р + 1,44), 658 (р, 100).
Элементный анализ для C42H39O6F:
Вычислено,%: C 76,58; H 5,97.
Найдено,%: C 76,79; H 6,09.
E. Получение 2-[2-пропил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-бензоил]-бензойной кислоты.
Метиловый эфир 2-[2-/3-(1-пропенил)/-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-бензоил] -бензойной кислоты (318 мг, 0,483 ммоль) подвергали гидрогенизации в условиях, описанных в примере получения 71 (C). Гидролиз полученного сложного эфира, как описано в примере получения 60, и очистка хроматографией на силикагеле (этилацетат-гексан) давали 150 мг (56%) целевого соединения в виде стекла.
Спектр ЯМР (ДМСО-d6)дельта : 10,15 (широкий синглет, 1Н, -OH), 7,84 (м, 1Н), 7,49 (м, 2Н), 6,98-7,23 (м, 5Н), 6,96 (с, 1Н), 6,62 (д, J=7,2 Гц, 1Н), 6,59 (с, 1Н), 4,18 (т, J= 5,3 Гц, 2Н), 2,85 (м, 2Н), 2,49 (м, 2Н), 2,20 (квинтет, J=5,2 Гц, 2Н), 1,57 (гекстет, J=5 Гц, 2Н), 1,08 (т, J=7,3 Гц, 3Н), 0,90 (т, J=7,2 Гц, 3Н): ИК-спектр- масс-спектр.
Элементный анализ для C34H33O6F:
Вычислено,%: C 73,36; H 5,98.
Найдено,%: C 69,71; H 5,90.
Пример 79. 2-[/2-пропил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-фенил/-метил]-бензойная кислота.

A. Получение метилового эфира 2-/(3-окси-2-пропилфенил)-метил/-бензойной кислоты.
Смесь метилового эфира 2-/3-окси-2-/3-(1-пропенил)/бензоил/-бензойной кислоты (3,00 г, 10,1 ммоль), концентрированной серной кислоты (1 мл) и 5% палладия на угле (1,5 г) в метаноле (95 мл) гидрогенизировали при 4 атм в течение 18 ч. Смесь концентрировали в вакууме до объема приблизительно 30 мл, фильтровали и насыщали газообразным хлористым водородом. Полученную смесь перемешивали 18 часов, затем концентрировали в вакууме. Остаток растворяли в диэтиловом эфире и промывали насыщенным раствором бикарбоната натрия. Водный слой промывали обратной струей свежего диэтилового эфира. Объединенные органические слои промывали насыщенным раствором хлористого натрия, сушили, фильтровали и концентрировали в вакууме, получая 2,60 г (90%) целевого промежуточного соединения в виде оранжевого масла.
Спектр ЯМР (CDCl3) δ : 7,97 (д, J=7 Гц, 1Н), 7,38 (т, J=7 Гц, 1Н), 7,28 (т, J= 7 Гц, 1Н), 7,02 (м, 2Н), 6,70 (д, J=7 Гц, 1Н), 6,54 (д, J=7 Гц, 1Н), 5,20 (широкий синглет, 1Н, -OH), 4,45 (гекстет, J=7 Гц, 2Н), 0,92 (т, J=8 Гц, 3Н): масс-спектр (СД) м/е 285 (р + 1, 23), 284 (100): ИК-спектр.
B. Получение метилового эфира 2-[/2-пропил-3-/3-/2-этил-5-(фенилметокси) -4-(4-фторфенил)-фенокси/-пропокси/-фенил/-метил]-бензойной кислоты.
Метиловый эфир 2-/(3-окси-2-пропилфенил)-метил/-бензойной кислоты (2,00 г, 4,68 ммоль) алкилировали 2-бензилокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано выше для получения по примеру 66(A). Перекристаллизация неочищенного продукта из гексана давала 1,72 г (38%) целевого промежуточного соединения в виде твердого белого вещества, точка плавления 83-84oC.
Спектр ЯМР (CDCl3) δ : 7,94 (д, J=8 Гц, 1Н), 7,53 (м, 2Н), 7,25-7,40 (м, 7Н), 7,05-7,15 (м, 4Н), 7,00 (д, J=7 Гц, 1Н), 7,81 (д, J=7 Гц, 1Н), 6,62 (с, 1Н), 6,58 (д, J=7 Гц, 1Н), 5,02 (с, 2Н), 4,42 (с, 2Н), 4,21 (м, 4Н), 3,88 (с, 3Н), 2,54-2,68 (м, 4Н), 2,32 (квинтет, J=6 Гц, 2Н), 1,50 (гекстет, J=6 Гц, 2Н), 1,21 (т, J=8 Гц, 3Н), 0,96 (т, J=8 Гц, 3Н); масс-спектр (СД) м/е 648 (р+1, 40), 647 (р, 100): ИК-спектр (хлороформ): 2964, 1718, 1603, 1497, 1459, 1143.
Элементный анализ для C42H43O5F:
Вычислено,%: C 77,99; H 6,70.
Найдено,%: C 79,47; H 6,76.
C. Получение 2-[/-пропил-2-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-фенил/-метил]-бензойной кислоты.
Метиловый эфир 2-[/2-пропил-3-/3-/2-этил-5-(фенилметокси)-4- (4-фторфенил)-фенокси/-пропокси/-фенил/-метил] -бензойной кислоты (1,50 мг, 2,32 ммоля) подвергали дебензилированию в условиях, описанных для получения по примере 71(C). Гидролиз полученного сложного эфира, как описано в примере получения 60 с последующей перекристаллизацией неочищенного продукта из смеси эфир-гексан давали 860 мг (68%) желаемого целевого соединения в виде белого твердого вещества, точка плавления 150-151oC.
Спектр ЯМР (CDCl3) δ : 8,11 (дд, J=7,3 и 0,8 Гц, 1Н), 7,45 (м, 2Н), 7,30 (т, J= 7 Гц, 1Н), 6,95-7,25 (м, 5Н), 6,81 (д, J=8,0 Гц, 1Н), 6,58 (д, J=7,4 Гц, 1Н), 6,52 (с, 1Н), 4,50 (с, 2Н), 4,21 (м, 4Н), 2,62 (м, 4Н), 2,35 (квинтет, J= 6,0 Гц, 2Н), 1,46 (гекстет, J=7,6 Гц, 2Н), 1,18 (т, J=7,5 Гц, 3Н), 0,93 (т, J= 7,3 Гц, 3Н): масс-спектр (СД) м/е 543 (р+1, 40), 542 (р, 100): ИК-спектр (хлороформ): 3400 (шир. ), 2966, 1696, 1603, 1496, 1459, 1238, 1146, 1111 см-1.
Элементный анализ для C34H35O5F:
Вычислено,%: C 75,26; H 6,50.
Найдено,%: C 75,26; H 6,62.
Пример 80. 2-[2-пропил-2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-тиофенокси]-бензойная кислота (см. в конце описания).
A. Получение 2-бромфенилдисульфида.
К смеси 2-бромтиофенола (20,0 г, 106 ммоль) и 2 н. раствора гидроокиси натрия (100 мл) в диэтиловом эфире (400 мл) добавляли твердый йод (13,4 г, 53,0 ммоль) по частям. Смесь перемешивали при комнатной температуре 1 ч, после чего эфирный слой отделяли. Водный слой экстрагировали свежей порцией эфира и объединенные эфирные слои 1 раз промывали водой, 1 раз насыщенным раствором хлористого натрия, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и получали 17,2 г (43%) промежуточного соединения в виде белого твердого вещества, точка плавления 95-97oC.
Спектр ЯМР (CDCl3) δ : 7,52 (м, 4Н), 7,25 (т, J=9,7 Гц, 2Н), 7,06 (т, J= 9,7 Гц, 2Н), масс-спектр (СД) м/е 380 (р+4, 20), 379 (р+3, 30), 378 (р+2, 85), 376 (р, 100), 374 (р-2,75): ИК-спектр.
Элементный анализ для C12H8Br2S2:
Вычислено,%: C 38,32; H 2,14.
Найдено,%: C 38,61; H 2,13.
B. Получение 2-(3-(аллилокси)-тиофенокси/-бромбензола.
К раствору 3-(аллилокси)-бромбензола (8,20 г, 38,7 ммоль) в тетрагидрофуране (600 мл) при -74oC добавляли 1,6 М н-бутиллития (24,2 мл, 38,7 ммоль). После перемешивания 30 мин этот раствор канюлировали в раствор 2-бромфенилдисульфида (16,0 г, 42,5 ммоль) в тетрагидрофуране (160 мл) при -74oC. Полученную смесь нагревали до комнатной температуры, затем разбавляли насыщенным раствором хлористого аммония и фильтровали. Водный слой 3 раза экстрагировали диэтиловым эфиром и объединенные органические слой 2 раз промывали водой, 1 раз насыщенным раствором хлористого натрия, сушили над сульфатом натрия, фильтровали, концентрировали в вакууме и получали желтое масло. Очистка хроматографией на силикагеле давала 9,40 г (76%) целевого промежуточного соединения в виде светло-желтого масла.
Спектр ЯМР (CDCl3) δ : 7,58 (д, J=7 Гц, 1Н), 7,27 (т, J=7 Гц, 1Н), 7,17 (т, J= 7 Гц, 1Н), 6,85-7,15 (м, 5Н), 6,04 (м, 1Н), 5,41 (д, J=14 Гц, 1Н), 5,30 (д, J= 10 Гц, 1Н), 4,52 (д, J=4 Гц, 2Н): масс-спектр (СД) м/е 322 (р, 100), 320 (р, 75); ИК-спектр (бромистый калий): 3223 (шир), 1688, 1345, 1161, 1013, 678 см-1.
Элементный анализ для C15H13OBrS:
Вычислено,%: C 56,09; H 4,08.
Найдено,%: C 56,31; H 4,22.
C. Получение метилового эфира 2-/3-(аллилокси)-тиофенокси/-бензойной кислоты.
К раствору 2-/3-(аллилокси)-тиофенокси/-бромбензола (9,00 г, 28,0 ммоль) в тетрагидрофуране (175 мл) при -78oC по каплям добавляли 1,6 М 7-бутиллития (19,2 мл, 30,8 ммоль). После перемешивания в течение 15 мин раствор насыщали газообразной двуокисью углерода, что приводило к образованию плотного геля. Добавляли тетрагидрофуран (50 мл) и полученную смесь нагревали до комнатной температуры. Смесь разбавляли насыщенным раствором хлористого аммония. Водный слой экстрагировали 1 раз диэтиловым эфиром и объединенные органические слои концентрировали в вакууме. Остаток растворяли в порции свежего эфира и экстрагировали 1 н. водной гидроокисью натрия. Водный слой промывали свежей порцией эфира и подкисляли водной хлористоводородной кислотой. Полученный водный слой экстрагировали свежей порцией эфира. Органический слой промывали насыщенным раствором хлористого натрия, фильтровали и концентрировали в вакууме. Неочищенную кислоту растворяли в метаноле (125 мл) и полученный раствор насыщали газообразным хлористым водородом. После перемешивания в течение 18 ч реакционную смесь концентрировали в вакууме, остаток растворяли в эфире и полученный раствор промывали насыщенным раствором бикарбоната натрия. Водный слой экстрагировали обратной струей свежего эфира и объединенные органические слои промывали 1 раз водой, 1 раз насыщенным раствором хлористого натрия, сушили над сульфатом натрия, фильтровали и концентрировали в вакууме. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 4,80 г (68%) желаемого целевого промежуточного соединения в виде бледно-желтого масла.
Спектр ЯМР (CDCl3) δ : 7,99 (дд, J=7,8 Гц, 1Н), 7,33 (т, J=7 Гц, 1Н), 7,25 (т, J=7 Гц, 1Н), 7,15 (м, 3Н), 7,00 (ДД, J=8,7 и 2,8 Гц, 1Н), 6,88 (д, J= 8 Гц, 1Н), 6,04 (М, 1Н), 5,42 (д, J=14 Гц, 1Н), 5,30 (д, J=11 Гц, 1Н), 4,53 (д, J=3,9 Гц, 2Н), 3,97 (с, 3Н); масс-спектр (СД) м/е 301 (р + 1, 25), 300 (р, 100): ИК-спектр (хлороформ): 3025, 1712, 1590, 1463, 1437, 1254, 1060 см-1.
Элементный анализ для C17H16O3S:
Вычислено,%: C 67,98; H 5,37.
Найдено,%: C 67,86; H 5,29.
D. Получение метилового эфира 2-[3-окси-2-/3-(1-пропенил)/-тиофенокси] -бензойной кислоты и метилового эфира 2-[3-окси-4-/3-(1-пропенил)/-тиофенокси]-бензойной кислоты.
Метиловый эфир 2-/3-(аллилокси)-тиофенокси/-бензойной кислоты (5,40 г, 15,0 ммоль) нагревали около 175oC в течение 29 ч. Полученную смесь охлаждали до комнатной температуры и очищали хроматографией на силикагеле (хлористый метилен), получая 2,22 г (41%) метилового эфира 2-/3-окси-2-/3-(1-пропенил)/-тиофенокси/-бензойной кислоты и 1,46 г (27%) метилового эфира 2-[3-окси-4-/3-(1-пропенил)/тиофенокси] -бензойной кислоты в виде белых твердых веществ.
Метиловый эфир 2-[3-окси-2-/3-(1-пропенил)/-тиофенокси]-бензойной кислоты, точка плавления 72-74oC.
Спектр ЯМР (ДМСО-d6) δ : 9,79 (с, 1Н), -OH), 7,89 (д, J=8 Гц, 1Н), 7,33 (т, J=7 Гц, 1Н), 7,09-7,23 (м, 2Н), 6,94 (м, 2Н), 6,62 (дд, J=7 и 1 Гц, 1Н), 4,70-4,83 (м, 2Н), 3,86 (с, 3Н), 3,37 (д, J=5 Гц, 2Н); масс-спектр (СД): м/е 301 (р + 1, 21), 300 (р, 100): ИК-спектр (хлороформ): 3595, 3350 (шир.), 3029, 3010, 2954, 1711, 1420, 1436, 1273, 1146, 1060 см-1.
Элементный анализ для C17H16O3S:
Вычислено,%: C 67,98; H 5,37.
Найдено,%: C 68,28; H 5,41.
Метиловый эфир 2-[3-окси-4-/3-(1-пропенил)/-тиофенокси]-бензойной кислоты, точка плавления 96-97oC.
Спектр ЯМР (ДМСО-d6) δ : 9,78 (с, 1Н, -OH), 7,89 (д, J=8 Гц, 1Н), 7,40 (т, J=7 Гц, 1Н), 7,12-7,25 (м, 2Н), 6,93 (с, 1Н), 6,91 (д, J=8 Гц, 1Н), 6,81 (д, J=8 Гц, 1Н), 5,87 (м, 1Н), 5,00-5,12 (м, 2Н), 3,85 (с, 3Н), 3,30 (д, J=4 Гц, 2Н); масс-спектр (СД) м/е 301 (р + 1, 45), 300 (р, 100): ИК-спектр (хлороформ): 3595, 3300 (шир.), 3029, 3010, 2954, 1711, 1436, 1310, 1255, 942 см-1.
Элементный анализ для C17H16O3S:
Вычислено,%: C 67,98; H 5,37.
Найдено,%: C 68,04; H 5,47.
E. Получение метилового эфира 2-[2-/3-(1-пропенил)/-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-тиофенокси] -бензойной кислоты.
Метиловый эфир 2-[3-окси-2-/3-(1-пропенил)/-тиофенокси]-бензойной кислоты (2,00 г, 6,66 ммоль) алкилировали 2-бензилокси-1-(4-фтофренил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано выше в примере получения 66(A). Очисткой на силикагеле (гексан-диэтиловый эфир) получали 2,90 г (66%) желаемого промежуточного соединения в виде белого твердого вещества, точка плавления 76-77oC.
Спектр ЯМР (CDCl3) δ : 8,03 (дд, J=7,6 Гц, 1Н), 7,52 (м, 2Н), 7,17-7,40 (м, 8Н), 6,98-7,18 (м, 5Н), 6,71 (д, J=7,9 Гц, 1Н), 6,62 (с, 1Н), 5,87 (м, 1Н), 5,03 (с, 2Н), 4,83-4,95 (м, 2Н), 4,21 (т, J=7 Гц, 2Н), 3,98 (с, 3Н), 3,62 (д, J= 6,3 Гц, 2Н), 2,64 (кв, J=7,5 Гц, 2Н), 2,33 (квинтет, J=5,8 Гц, 2Н), 1,22 (т, J=7,5 Гц, 3Н); масс-спектр (СД) м/е 664 (р + 2, 40), 663 (р + 1, 70), 662 (р, 100): ИК-спектр (хлороформ): 3011, 2970, 2940, 2890, 1712, 1497, 1452, 1298, 1255, 1145, 1060 см-1.
Элементный анализ для C41H39O5FS:
Вычислено,%: C 74,30; H 5,93.
Найдено,%: C 74,46; H 6,13.
F. Получение метилового эфира 2-[2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/пропокси/-тиофенокси]-бензойной кислоты.
Метиловый эфир 2-[2-/3-(1-пропенил)/-3-/3-/2-этил-4-(4-фторфенил)-5-(фенилметокси)-фенокси/-пропокси/-тиофенокси] -бензойной кислоты (2,70 г, 4,07 ммоль) гидрогенизировали, как описано выше для получения по примеру 71 (C), и получали около 2,0 г масла. Раствор этого вещества (1,39 г) в хлористом метилене (25 мл) при -78oC обрабатывали 1 М трехбромистого бора (3,61 мл, 3,61 ммолm) и выдерживали при помешивании 1 ч. Реакционную смесь разбавляли водой и экстрагировали хлористым мтиленом. Органический слой промывали водой, сушили над сульфатом магния, фильтровали, концентрировали в вакууме и получали желтое масло. Очистка хроматографией на силикагеле давала 770 мг (47%) целевого промежуточного соединения в виде белого твердого вещества, точка плавления 105-106oC.
Спектр ЯМР (CDCl3) δ : 8,02 (дд, J=7,6 и 1,2 Гц, 1Н), 7,43 (м, 2Н), 7,07-7,30 (м, 8Н), 6,98 (м, 2Н), 6,71 (д, J=7,9 Гц, 1Н), 6,57 (с, 1Н), 5,10 (шир. с, 1Н, -OH), 4,24 (м, 2Н), 3,98 (с, 3Н), 2,83 (т, J=78 гц, 2Н), 2,65 (кв, J=7,5 Гц, 2Н), 2,36 (квинтет, J=5 Гц, 2Н), 1,52 (гекстет, J=6 Гц, 2Н), 1,21 (т, J=7,4 Гц, 3Н), 0,90 (т, J=7,5 Гц, 3Н); масс-спектр (СД) м/е 575 (р + 1, 20), 574 (р, 100): ИК-спектр.
G. Получение 2-[2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-тиофенокси]-бензойной кислоты.
Метиловый эфир 2-[2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-тиофенокси] -бензойной кислоты (700 мг, 1,22 ммоля) гидролизовали, как описано в примере получения 60, и получали 689 мг (100%) желаемого целевого промежуточного соединения в виде белого твердого вещества, точка плавления 153-155oC.
Cпектр ЯМР (CDCl3) δ : 8,13 (дд, J=8,2 и 0,9 Гц, 1Н), 7,32 (м, 2Н), 7,10-7,33 (6Н), 6,99 (м, 2Н), 6,72 (д, J=7,9 Гц, 1Н), 6,55 (с, 1Н), 4,90 (шир. синглет, 1Н, -OH), 4,24 (м, 4Н), 2,82 (т, J=6 Гц, 2Н), 2,63 (кв, J=7,5 Гц, 2Н), 2,34 (квинтет, J=6 Гц, 2Н), 1,51 (гекстет, J=7,5 Гц, 2Н), 1,18 (т, J=7,5 Гц, 3Н), 0,90 (т, J=7,5 Гц, 3Н); масс-спектр (СД) м/е 561 (р + 1, 20), 560 (р, 100): ИК-спектр (хлороформ): 2967, 1700, 1603, 1497, 1451, 1147, 1043 см-1.
Элементный анализ для C33H33O5FS:
Вычислено,%: C 70,69; H 5,93.
Найдено,%: C 70,43; H 5,97.
Пример 81. 2-[2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/фенилсульфинил]-бензойная кислота.
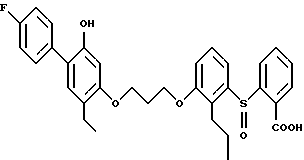
К раствору 2-[2-пропил-3-/3-/2-этил-4-(4-фтофренил)-5-оксифенокси/-пропокси/-тиофенокси] -бензойной кислоты (450 мг, 0,803 ммоль) в хлористом метилене (10 мл) при -78oC добавляли 85%-ный раствор мета-хлорнадоксибензойной кислоты (138 мг) в хлористом метилене (2 мл). Через 40 мин смесь концентрировали в вакууме. Очистка остатка хроматографией на силикагеле (95% хлороформа, 4,5% метанола и 0,5% уксусной кислоты) давала 380 мг (80%) целевого соединения в виде не совсем белого твердого вещества, точка плавления выше 100oC (разлагается).
Спектр ЯМР (CDCl3) δ : 8,53 (д, J=8 Гц, 1Н), 8,14 (д, J=8,0 Гц, 1Н), 7,93 (т, J= 8 Гц, 1Н), 7,63 (т, J=8 Гц, 1Н), 7,43 (м, 2Н), 7,13 (м, 2Н), 6,94-7,06 (м, 2Н), 6,88 (д, J=8 Гц, 1Н), 6,50 (Д, J=8 Гц, 1Н), 7,46 (с, 1Н), 6,38 (широкий синглет, 1Н, -OH), 4,15 (м, 4Н), 3,32 (м, 1Н), 3,08 (м, 1Н), 2,57 (кв, J=7,5 Гц, 2Н), 2,29 (квинтет, J=6 Гц, 2Н), 1,75 (м, 2Н), 1,17 (т, J= 7,5 Гц, 3Н), 1,05 (т, J=7,3 Гц, 3Н): масс-спектр (высокое разрешение): вычислено: 577.202642 (MH+), найдено: 577.203800: ИК-спектр (хлороформ): 2969, 1708, 1497, 1455, 1266, 1146, 1018 см-1.
Элементный анализ для C33H33O6FS:
Вычислено,%: C 68,73; H 5,77.
Найдено,%: C 67,54; H 5,69.
Пример 82. Гидрат 2-[2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/ -пропокси/-фенилсульфонил]-бензойной кислоты.

К раствору 2-[2-пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенилсульфинил] -бензойной кислоты (150 мг, 0,260 ммоль) в хлористом метилене (3,0 мл) при 0oC добавляли раствор 85%-ной мета-хлорнадоксибензойной кислоты (53 мг) в хлористом метилене (1 мл). Через 1 ч смесь нагревали до 4oC и перемешивали 18 ч. Смесь концентрировали в вакууме, очищали хроматографией на силикагеле (90% хлороформа, 9,5% метанола и 0,5% уксусной кислоты) и получали 90 мг (58%) целевого соединения в виде белого твердого вещества, точка плавления 80-90%
Спектр ЯМР (ДМСО-d6) δ : 7,88 (м, 2Н), 7,55-7,78 (м, 3Н), 7,50 (м, 2Н), 7,33 (м, 2Н), 7,04 (м, 2Н), 6,95 (с, 1Н), 6,51 (с, 1Н), 4,19 (т, J=4,8 Гц, 2Н), 4,05 (т, J=5,8 Гц, 2Н), 2,69 (м, 2Н), 2,44 (кв, J=5,8 Гц, 2Н), 2,19 (м, 2Н), 0,90-1,10 (м, 5Н), 0,71 (т, J=4,5 Гц, 3Н); масс-спектр (СД) м/е 595 (р + 2, 30), 594 (р + 1, 40), 593 (р, 100): ИК-спектр (хлороформ): 2966, 1730, 1603, 1497, 1299, 1146 см-1.
Элементный анализ для C33H33O7FS•Н2H:
Вычислено,%: O 64,90; C 5,78.
Найдено,%: H 64,89; C 5,67.
Пример 83. 0,4 гидрат двунатриевой соли 5-[3-/2-(1-карбокси)этил/-4-(4-фторфенил)-5-оксифенокси/-пропокси/фенил] -4-пентиновой кислоты (см. в конце описания).
A. Получение этилового эфира 3-(2-окси-5-йодофенил)-пропановой кислоты.
4-йодофенил (6,00 г, 27,3 ммоль) обрабатывали триэтиловым эфиром орто-акриловой кислоты как описано в примере получения 59(A). Неочищенный продукт растворяли в тетрагидрофуране (50 мл) и обрабатывали 1 н. водной хлористоводородной кислотой (0,3 мл) при комнатной температуре в течение 1 ч. Смесь разбавляли водой и экстрагировали этилацетатом. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения масла. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 1,64 г (19%) целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 7,60 (шир. синглет, 1Н, -OH), 7,40 (м, 2Н), 6,68 (д, J=9,0 Гц, 1Н), 4,17 (кв, J=7,1 Гц, 2Н), 2,85 (м, 2Н), 2,74 (м, 2Н) 1,26 (т, J=7,1 Гц, 3Н).
B. Получение этилового эфира 3-[2-/(1,1-диметилэтил)-диметилсилилокси/-5-йодофенил]-пропановой кислоты.
Смесь этилового эфира 3-(2-окси-5-иодофенил)-пропановой кислоты (1,64 г, 5,13 ммоль), хлористого трет-бутилдиметилсилила (772, г, 5,13 ммоль) и имидазола (700 мг, 10,3 ммоль) в тетрагидрофуране (30 мл) нагревали с обратным холодильником 18 ч. Смесь охлаждали до комнатной температуры, разбавляли эфиром и промывали насыщенным раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме до получения 2,02 (91%) целевого промежуточного соединения в виде масла.
Спектр ЯМР (CDCl3) δ : 7,46 (д, J=2,3 Гц, 1Н), 7,38 (дд, J=8,5 и 2,3 Гц, 1Н), 6,56 (д, J= 8,5 Гц, 1Н), 4,15 (кв, J=7,2 Гц, 2Н), 2,86 (т, J=7,5 Гц, 2Н), 2,56 (т, J=8,3 Гц, 2Н), 1,26 (т, J=7,1 Гц, 3Н), 1,02 (с, 9Н), 0,24 (с, 6Н): масс-спектр (СД) м/е 434 (р, 100): ИК-спектр (хлороформ): 2933, 1727, 1483, 1258, 1183, 1118, 1044, 918, 843 см-1.
C. Получение метилового эфира 5-[3-/2-(1-карбоэтокси)-этил/-4- (1,1-диметилэтил)-диметилсилилокси/-фенил]-4-пентиновой кислоты.
Смесь этилового эфира 3-[2-/(1,1-диметилэтил)-диметилсилилокси/-5-йодофенил] -пропановой кислоты (160 г, 3,68 ммоль), метилового эфира 4-пентиновой кислоты (412 мг, 3,68 ммоль), иодистой меди (I) (25 мг, 0,13 ммоль) и бис-(трифенилфосфин)-палладия (II) хлорида (20 мг, 0,028 ммоль) в диэтиламине (20 мл) перемешивали при комнатной температуре 18 ч. Реакционную смесь фильтровали и концентрировали в вакууме до получения темного масла. Очистка хроматографией на силикагеле давала 870 мг (58%) целевого промежуточного соединения в виде масла.
Спектр ЯМР (CDCl3) δ : 7,21 (д, J=2,1 Гц, 1Н), 7,13 (дд, J=8,3 и 2,1 Гц, 1Н0, 6,69 )д, J=8,3 Гц, 1Н), 4,14 (кв, J=7,1 Гц, 2Н), 3,73 (с, 3Н), 2,87 (т, J=7,4 Гц, 2Н), 2,72 (м, 2Н), 2,50-2,68 (м, 4Н), 1,25 (т, J=7,0 Гц, 3Н), 1,01 (с, 9Н), 0,24 (с, 0,6Н): масс-спектр (СД) м/е 419 (р + 1,26 ), 418 (р, 100): ИК-спектр (хлороформ): 3450 (широкая полоса), 3023, 1730, 1603, 1497, 1278, 1043, 842 см-1.
Элементный анализ для C23H34O5Si:
Вычислено,%: C 65,99; H 8,19.
Найдено,%: C 66,18; H 8,01.
D. Получение метилового эфира 5-[3-/2-(1-карбоэтокси)-этил/-4-оксифенил] -4-пентиновой кислоты.
Смесь метилового эфира 5-[3-/2-(1-карбоэтокси)-этил/-4/ (1,1-диметилэтил)-диметилсилилокси/-фенил] -4-пентиновой кислоты (3,70 г, 8,85 ммоль) и фтористого тетра-н-бутиламмония (2,50 г, 9,58 ммоль)-в тетрагидрофуране (20 мл) перемешивали при комнатной температуре 2 ч. Смесь разбавляли диэтиловым эфиром и два раза промывали водой. Органический слой сушили над сульфатом натрия, фильтровали, концентрировали в вакууме до получения коричневого масла. Очистка хроматографией на силикагеле (этилацетат-гексан) давала 1,10 г (41%) целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 7,62 (с, 1Н, -OH), 7,16 (м, 2Н), 6,80 (д, J=8 Гц, 1Н), 4,15 (кв, J=7,0 Гц, 2Н), 3,73 (с, 3Н), 2,85 (т, J=7 Гц, 2Н), 2,55-2,77 (м, 6Н), 1,24 (т, J=7,0 Гц, 3Н); масс-спектр (СД) м/е 305 (р + 1, 18), 304 (100): ИК-спектр (хлороформ): 3325 (шир.), 3028, 1733, 1500, 1379, 1233, 1167.
Элементный анализ для C17H20O5:
Вычислено,%: C 67,09; H 6,62.
Найдено,%: C 66,83; H 6,71.
E. Получение метилового эфира 5-[3-/2-(1-карбоэтокси)-этил/-4- /3-/2-этил-4-(4-фторффенил)-5-/2-(триметилсилил)-этоксиметокси/-фенокси/фенил] -4-пентиновой кислоты.
5-[3-/2-(1-карбоэтокси)-этил/-4-оксифенил]-4-пентиновой кислоты метиловый эфир (500 м, 0,942 ммоль) алкилировали 2-/(2-триметилсилил)-этокси/-метокси-1-(4-фторфенил)-5-этил-4-(3-хлор-1-пропилокси)-бензолом, как описано выше в примере получения 66(A). Очистка хроматографией на силикагеле (этилацетат-гексан) давала 320 мг (49%) целевого промежуточного соединения в виде бесцветного масла.
Спектр ЯМР (CDCl3) δ : 7,47 (м, 2Н), 7,27 (м, 2Н), 7,10 (м, 3Н), 6,83 (с, 1Н), 6,80 (д, J=8,3 Гц, 1Н), 5,15 (с, 2Н), 4,23 (м, 4Н), 4,13 (кв, J=7,2 Гц, 2Н), 3,74 (с, 3Н), 3,65 (т, J=8,3 Гц, 2Н), 2,93 (т, J=7,3 Гц, 2Н), 2,75 (м, 2Н), 2,58-2,68 (м, 6Н), 2,35 (квинтет, J=5,9 Гц, 2Н), 1,24 (т, J=7,1 Гц, 3Н), 1,20 (т, J= 7,3 Гц, 3Н), 0,00 (с, 9Н): масс-спектр (СД) м/е 637 (р, 100): ИК-спектр (хлороформ): 2972, 1731, 1605, 1498, 1234, 1058, 839 см-1.
Элементный анализ для C40H51O8Si:
Вычислено,%: C 67,96; H 7,27.
Найдено,%: C 68,19; H 7,28.
F. Получение 0,4 гидрата динатриевой соли 5-[3-/2-(1-карбокси)-этил/-4-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил] -пинтиновой кислоты.
Смесь метилового эфира 5-[3-/2-(1-карбоэтокси)-этил/-4-/3-/2-этил-4-(4-фторфенил)-5-/2-(триметилсилил)-этоксиметокси/-фенокси-пропокси/-фенил] -4-пентиновой кислоты (300 мг, 0,434 ммоль) и фтористого тетра-н-бутиламмония (465 мг, 1,78 ммоль( в тетрагидрофуране (20 мл) перемешивали при 40oC в течение 48 ч. Реакционную смесь охлаждали до комнатной температуры, разбавляли диэтиловым эфиром и промывали водой. Органический слой концентрировали в вакууме до получения масла. Гидролиз, солеобразование и очистка, как описано в получении по примеру 59 (D), давали 112 мг (45%) целевого соединения в виде не совсем белого твердого вещества, точка плавления 73-76oC.
Спектр ЯМР (ДМСО-d6) δ/ : 7,54 (м, 2Н), 7,04-7,21 (м, 4Н), 6,91 (с, 1Н), 6,84 (м, 2Н), 4,21 (т, J=7,2 Гц, 2Н), 4,05 (т, J=3,6 Гц, 2Н), 2,71 (т, J= 6,0 Гц, 2Н), 2,48 (м, 4Н), 2,13 (м, 6Н), 1,08 (т, J=7,4 Гц, 3Н); масс-спектр ББА) м/е 580 (р + 1, 6), 579 (р, 23): ИК-спектр (паста): 2925, 1565, 1502, 1464, 1377, 1241, 1148, 839 см-1.
Элементный анализ для C31H29O7FNa2•0,4 H2O:
Вычислено,%: C 63,56; H 5,13.
Найдено,%: C 63,68; H 4,96.
Пример 84. 1-фенил-1-(1Н-тетразол-5-ил)-6-(2-этил-4-(4-фторфенил)-5-оксифенокси)-гексан.

A. Получение 7-хлор-2-фенилгептаннитрила.
Диизопропиламин лития (0,1 моль) получали добавлением н-бутиллития в гексане (0,1 моль) к диизопропиламину (10,1 г, 0,1 моля) растворенному в толуоле, охлажденном до -78oC в атмосфере азота. К этому раствору добавляли цианистый бензил (11,7 г, 0,1 моля). Раствор перемешивали при -78oC в течение 1 ч, затем добавляли 5-хлор-1-бромпентан и раствор медленно нагревали в течение 2 ч до комнатной температуры. Мутный раствор перемешивали при комнатной температуре дополнительно 3 ч. Затем толуоловый раствор промывали водным раствором хлористого аммония (250 мл) и толуоловый слой отделяли и сушили над сульфатом магния. Раствор толуола выпаривали до масла, которое отгоняли колба в колбе при 10 мм рт.ст. и температура печи 120oC, чтобы удалить непрореагировавшие исходные материалы. Оставшееся масло было целевым соединением (16,8 г, 76%-ный выход), которое, как показал спектр ЯМР, содержало небольшой процент соответствующего бромида, но было чистым в других отношениях и использовалось как таковое, ЯМР.
B. Получение 2-фенил-7-(2-ацетил-5-бензилоксифенокси)-гептаннитрила.
2-Окси-2-бензилоксиацетофенон (2,42 г, 0,01 моль) растворяли в метилэтилкетоне (100 мл) и добавляли 7-хлор-2-фенил-гептаннитрил (2,21 г, 0,01 моль) с последующим добавлением тонкомолотого карбоната калия (5 г) и иодистого калия (1 г). Перемешиваемую суспензию нагревали с обратным холодильником в атмосфере азота 20 ч. Затем раствор фильтровали и выпаривали до масла, которое хроматографировали на колонке силикагеля, элюируя смесью эфир-гексан, и получали 2,8 8 (65,5%) целевого соединения в виде бесцветного масла, ЯМР.
C. Получение 2-фенил-7-(2-этил-5-бензилоксифенокси)-гептаннитрила
2-Ферил-7-(2-ацетил-5-бензилоксифенокси)-гептаннитрил, (1,4 г, 3,28 ммоль) растворяли в четыреххлористом углероде (100 мл) и добавляли трифторуксусную кислоту (10 мл) с последующим добавлением триэтилсилана (10 мл). Раствор выдерживали при комнатной температуре 6 ч. К этому времени спектр ЯМР реакционной смеси показал, что реакция не завершена, и дополнительно добавляли трифторуксусную кислоту (10 мл) и триэтилсилан (5 мл) и раствор оставляли на ночь. Затем раствор выпаривали досуха и остаток хроматографировали на колонке силикагеля, элюируя смесью эфир-гексан в отношении 1:1. Целевое соединение получали в виде масла с выходом 1,21 г (89%). Спектр ЯМР.
D. Получение 2-фенил-7-(2-этил-4-бром-5-бензилоксифенокси)-гептаннитрила.
2-фенил-7-(2-этил-5-бензилоксифенокси)-гептаннитрил (1,23 г, 3 ммоль) растворяли в четыреххлористом углероде (50 мл) и добавляли суспензию N-бромсукцинимида (543 мг, 3 ммоль). Затем раствор перемешивали при комнатной температуре. Через 40 мин из раствора выпадал осадок и через 1 ч реакция заканчивалась, как свидетельствовал анализ тонкослойной хроматографией. Суспензию фильтровали и выпаривали до масла, которое хроматографировали на колонке силикагеля, элюируя смесью эфир-гексан в отношении 1:1, и получали 1,21 г (82%-ный выход) целевого промежуточного соединения в виде бесцветного масла. Спектр ЯМР.
E. Получение 2-фенил-7-(2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-гептаннитрила.
2-Фенил-7-(2-этил-4-бром-5-бензилоксифенокси)-гептаннитрил (1,21 г, 2,46 ммоль) растворяли в бензоле (45 мл) и добавляли тетракис-(трифенилфосфин)-палладий (0) (284,3 г, 0,246 ммоля) с последующим добавлением раствора фторфенилборной кислоты (516 мг, 3,69 ммоль), в этаноле 915 мл). Добавляли 2 М водный раствор карбоната натрия 915 мл) и полученный оранжевый раствор нагревали с обратным холодильником 17 ч в атмосфере азота. Почти черную суспензию затем охлаждали и добавляли 10%-ный водный раствор аммиака (100 мл) и 3 раза экстрагировали дихлорметаном. Объединенные экстракты сушили сульфатом магния и выпаривало до получения масла, которое затем хроматографировали на колонке силикагеля, элюируя смесью гексан-эфир в отношении 1:1, для удаления трифенилфосфина. Целевое соединение получали в виде масла с 57,5% выходом (720 мг). Спектр ЯМР.
F. Получение 1-фенил-1-(1Н-тетразол-5-ил)-6-(2-хэтил-4-(4-фторфенил)-5-бензилоксифенокси)-гексана.
2-Фенил-7-(2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-гептаннитрил (700 мг, 1,38 ммоль) растворяли в диметилформамиде (20 мл), добавляли азид натрия (0,6 г) и гидрохлорид триэтиламина (1,2 г) и перемешиваемую суспензию нагревали при 110oC три дня. Затем суспензию добавляли к 1 М хлористоводородной кислоты (100 мл) и раствор 4 раза экстрагировали хлороформом. Объединенные хлороформные экстракты промывали водой и сушили сульфатом магния. После выпаривания раствора получали 680 мг (72%) целевого соединения в виде неочищенного масла, с которого затем непосредственно снимали защиту.
G. Получение 1-фенил-1-(1Н-тетразол-5-ил)-6-(2-этил-4-(4-фторфенил)-5-оксифенокси)-гептана
Неочищенный 1-фенил-1-(1Н-тетразол-5-ил)-6-(2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-гексан (150 мг, 0,27 ммоль) растворяли в этаноле (100 мл) и 5%-ный палладий на угле (0,5 г) добавляли к раствору под слоем углекислого газа. Затем суспензию гидрогенизировали при давлении 3,5 кг/см2 в течение 3 ч. Катализатор отфильтровывали, этанол выпаривали и получали масло, которое очищали обращенно-фазовой хроматографией на колонке с С18, элюируя смесью метанол-вода в отношении 9:1. Целевое соединение было вторым элюированным компонентом, полученным с выходом 88,5% (110 мг) в виде бесцветного масла после выпаривания растворителя, ЯМР, МС.
Элементный анализ для C27H29N4O2:
Вычислено,%: C 70,41; H 6,35; N 12,16.
Найдено,%: C 70,41; H 6,46; N 12,16.
Более полярный первым проявленный компонент был идентифицирован ЯМР-спектром как 1-фенил-1-(1Н-тетразол-5-ил)-6-(2-этил-5-оксифенокси)-гексан, выход 15 мг.
Пример 85. 1-(4-(карбоксиметокси)-фенил)-1-(1Н-тетразол-5-ил)-6- (2-этил-4-(4-фторфенил)-5-оксифенокси)-гексан.

A. Получение 7-хлор-2-(4-метоксифенил)-гептаннитрила.
7-хлор-2-(4-метоксифенил)-гептаннитрил получали с выходом 71% в виде бледно-желтого масла из 4-метоксибензилнитрила и 5-хлор-1-бромпентана, используя процедуру, описанную в примере 84(A), за исключением того, что вместо толуола использовали тетрагидрофуран по причине более высокой растворимости соли лития. Спектр ЯМР.
B. Получение 7-хлор-2-(4-оксифенил)-гептаннитрила.
7-Хлор-2-(4-метоксифенил)-гептаннитрил (4,0 г, 16,7 ммоль) растворяли в дихлорметане (100 мл) и перемешиваемый раствор охлаждали до 0oC. Избыток трехбромистого бора (5 мл) добавляли к раствору и реакционную смесь нагревали до комнатной температуры и оставляли на ночь при перемешивании. Затем раствор медленно добавляли к насыщенному водному раствору бикарбоната натрия (500 мл) и смесь 3 раза экстрагировали дихлорметаном. Объединенные дихлорметановые экстракты сушили и выпаривали до бледно-желтого масла (выход 3,15 г, 83,6%), которое использовали непосредственно в следующей реакции без очистки. Спектр ЯМР.
C. Получение 7-хлор-2-(4-этоксикарбонилметокси)-фенил)-гептаннитрила.
7-Хлор-2-(4-оксифенил)-гептаннитрил (1 г, 4,2 ммоль) растворяли в метилэтилкетоне (100 мл) и добавляли свежемолотый карбонат калия (5 г), чтобы получить взвесь. Добавляли избыток этилового эфира бромуксусной кислоты (1,4 г, 8,3 ммоль) и перемешиваемую суспензию нагревали с обратным холодильником 3 ч. Затем суспензию выливали в воду (200 мл) и экстрагировали три раза дихлорметаном. Объединенные дихлорметановые экстракты сушили над сульфатом магния и выпаривали до масла. Затем удаляли избыток сложного бромэфира разделением азеотропной смеси с толуолом и получали целевое промежуточное соединение с 98%-ным выходом (1,38 г) в виде бледно-желтого масла, которое было достаточно чистым по результатам спектрального анализа ЯМР, и использовали непосредственно в следующей реакции. Спектр ЯМР.
D. Получение 1-(4-этоксикарбонилметокси)-фенил)-1-циано-6- (2-ацетил-5-бензилоксифенокси)-гексана.
2-Окси-4-бензилоксиацетофенон (1,04 г, 4,3 ммоль) растворяли в диметилформамиде (50 мл) и добавляли 7-хлор-2-/4-(этоксикарбонилметокси)-фенил/-гептаннитрил (1,4 г, 4,3 ммоль). Затем добавляли йодистый калий (1,5 г) и суспензию перемешивали при комнатной температуре 2 ч. Добавляли карбонат калия (3 г) и перемешиваемую суспензию нагревали до 110oC за 16 ч в атмосфере азота. Затем суспензию добавляли к воде 9150 мл) и три раза экстрагировали хлороформом. Объединенные хлороформные экстракты сушили сульфатом магния и выпаривали до коричневого масла, которое хроматографировали на колонке силикагеля, элюируя смесью эфир-гексан в отношении 1,1. Основной компонент смеси выделяли в виде бледножелтых кристаллов (выход 270 мг, 14%) из смеси гексан-эфир, точка плавления 102-104oC. Спектр ЯМР. Другие 180 мг маслянистых кристаллов были выделены из маточных растворов.
E. Получение 7-йодо-2-/4-(этоксикарбонилметокси)-фенил/-гептаннитрила.
7-Хлор-2-/4-(этоксикарбонилметокси)-фенил/-гептаннитрил (3,34 г, 10 ммоль) растворяли в метилэтилкетоне (100 мл). Добавляли иодистый натрий (3 г) и перемешиваемую суспензию оставляли на ночь при нагревании с обратным холодильником. Раствор охлаждали, фильтровали и выпаривали до маслянистого твердого вещества. Масло растворяли в эфире и твердый иодид натрия отфильтровывали. Целевое соединение получали после выпаривания эфира, выход составил 4,25 г (100%). Неочищенный иодид использовали непосредственно в следующей реакции.
F. Получение 1-/4-(этоксикарбонилметокси)-фенил/-1-циано-6- (2-ацетил-5-бензилоксифенокси)-гексана.
2-Окси-4-бензилоксиацетофенон (2,42 г, 10 ммолей) растворяли в диметилформамиде (50 мл) и добавляли 7-йодо-2-/4-(этоксикарбонилметокси)-фенил/-гептаннитрил (4,3 г, 10 ммоль) с последующим добавлением карбоната калия (10 г) и перемешиваемую суспензию нагревали при 110oC в течение 24 ч. Затем реакционную смесь обрабатывали как описано в примере 85(Д) и получали 4,5 г (90%) целевого соединения, точка плавления 102-104oC. Спектр ЯМР, масс-спектр.
Элементный анализ для C32H35NO6:
Вычислено,%: C 72,57; H 6,66; N 2,65.
Найдено,%: C 72,84; H 6,65; N 2,48.
G. Получение 1-/4-(этоксикарбонилметокси)-фенил/-1-циано-6- (2-этил-5-бензилоксифенокси)-гексана.
1-/4-Этоксикарбонилметокси)-фенил/-1-циано-6-(2-ацетил-5-бензилокси)-феноксигексан обращали в целевое соединение, используя процесс, описанный в примере 84(C), выход 86%.
H. Получение 1-/4-(этоксикарбонилметокси)-фенил/-1-циано-6- (2-этил-4-бром-5-бензилоксифенокси)-гексана.
1-/4-Этоксикарбонилметокси)-фенил/-1-циано-6-(2-этил-5-бензилоксифенокси)-гексан (900 мг, 1,8 ммоль) бромировали с использованием процесса, описанного в примере 84 (D), за исключением того, что в качестве растворителя использовали дихлорметан и продукт хроматографировали, используя в качестве элюента смесь эфир-гексан в отношении 3:1. Выход после хроматографии составил 887 мг (83%). Спектр ЯМР.
I. Получение 1-/4-(Этоксикарбонилметокси)-фенил/-1-циано-6-(2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-гексана.
Целевое соединение получали из 1-/4-(этокси-карбонилметокси)-фенил/-1-циано-6-/2-этил-4-бром-5-бензилоксифенокси/-гексана (800 мг, 1,34 ммоль), используя процесс по примеру 84 (E), и получали 672 мг (82%) бесцветного масла, ЯМР.
J. Получение 1-/4-(этоксикарбонилметокси)-фенил/-1-(1Н-тетразол-5-ил)-6-/2-этил-4-/4-фторфенил)-5-бензилоксифенокси)-гексана.
1-/4-(Этоксикарбонилметокси)-фенил/-1-циано-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-гексан (670 мг, 1,09 ммоль) растворяли в диметилформамиде (20 мл). Добавляли соляно-кислый триэтиламин (1,3 г) и азид натрия (0,6 г) и перемешиваемую суспензию нагревали до 117oC за 24 ч. Добавляли дополнительно гидрохлорид триэтиламина (1,3 г) и азид натрия (0,6 г) и смесь нагревали при 117oC еще 16 ч. Затем реакционную смесь обрабатывали, используя процесс по примеру 84 (F), и получали 690 мг (97%) целевого промежуточного соединения в виде масла. Спектр ЯМР.
K. Получение 1-(4-этоксикарбонилметокси)-фенил/-1-(1Н-тетразол-5-ил)-6-/2-этил-4-(4-фторфенил)-5-оксифенокси/-гексана.
Целевое соединение получали из неочищенного 1-/4-(этоксикарбонилметокси)-фенил/-1-(1Н-тетразол-5-ил)-6-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-гексана (680 мг, 1,04 ммоль), используя процесс по примеру 84 (G), и получали 540 мг (92%) целевого промежуточного соединения в виде бесцветного масла, которое содержало некоторое количество этанола сольватации. Спектр ЯМР.
L. Получение 1-/4-(карбоксиметокси)-фенил/-1-(1Н-тетразол-5-ил)-6-/2-этил-4-(4-фтофренил)-5-оксифенокси/-гексана.
Неочищенный 1-/4-(этоксикарбонилметокси)-фенил/-1-(1Н-тетразол-5-ил)-6-/2-этил-4-(4-фторфенил)-5-оксифенокси/-гексан (50 мг) растворяли в этаноле (20 мл). Добавляли 1 М водного раствора карбоната натрия и полученный раствор перемешивали при комнатной температуре 3 ч. Устанавливали pH раствора, равным 2,0, используя 1М хлористоводородной кислоты и раствор 5 раз экстрагировали хлороформом. Объединенные хлороформные экстракты сушили сульфатом магния и выпаривали до масла, которое очищали обращенно-фазовой хроматографией на колонке С18, элюируя смесью метанол-вода в отношении 85:15:, и 0,1%-ной уксусной кислоты. По удалении растворителя получали 9,7 г целевого соединения в виде бесцветного масла. Масс-спектр, спектр ЯМР.
Пример 86. 1-/4-(диметиламинокарбонилметокси)-фенил/-1-(1Н-тетразол-5-ил)-6-(2-этил-4-94-фторфенил)-5-оксифенокси/-гексан.

1-/4-(этоксикарбонилметокси)-фенил/-1-(1Н-тетразол-5-ил) 7-(2-этил-4-(4-фторфенил)-5-оксифенокси)-гексан (100 мг, 0,18 ммоля) растворяли в этаноле (25 мл) и добавляли диметиламин, растворенный в 33%-ном этаноле (25 мл) и раствор выдерживали при комнатной температуре в герметично закрытой колбе 25 дней. Затем растворитель выпаривали и целевое соединение медленно кристаллизовалось из эфира, точка плавления 115-120oC, выход 36,6 г (36%). Спектры ЯМР, масс-спектр.
Элементный анализ для C31H36N5O4:
Вычислено,%: C 66,29; H 6,46; N 12,47.
Найдено,%: C 66,26; H 6,61; N 12,29.
Пример 87. 3-[2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил]-E-пропеновая кислота.

A. Получение 2-(2-оксифенил)-1,3-диоксолана.
2-Оксибензальдегид (12,2 г, 0,1 ммоль) растворяли в толуоле (125 мл). Добавляли этиленгликоль (12,4 г, 0,2 ммоль) с последующим добавлением приблизительно 30 мг пара-толуолсульфокислоты в качестве катализатора. Полученный раствор нагревали с обратным холодильником под ловушкой Dean-Stark. Через 2 ч добавляли дополнительно 10 мл этиленгликоля и смесь нагревали с обратным холодильником еще 2 ч. Затем толуол сливали с красной смолы и промывали водным бикарбонатом натрия. Затем толуоловый слой сушили сульфатом магния и выпаривали до бледно-желтого масла, которое кристаллизовали из смеси эфир-гексан, и получали целевое промежуточное соединение в виде белых кристаллов, точка плавления 68-69oC, выход 10,1 г (61%). Спектр ЯМР.
B. Получение хлористого 3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропила.
Хлористый 3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропил (0,5 г, 1,25 ммоль) растворяли в этилацетате (50 мл) и добавляли 10%-ный палладий на угле в качестве катализатора в инертной атмосфере двуокиси углерода. Суспензию гидрогенизировали при комнатной температуре и давлении 2,1 кг/см2 в течение 2 ч. Катализатор отфильтровывали и фильтрат выпаривали, получая масло, которое медленно кристаллизовалось как целевое промежуточное соединение, точка плавления 55-56oC, выход 380 мг (98%). Спектр ЯМР.
C. Получение хлористого 3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/-пропила.
Хлористый 3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропил (360 мг, 1,16 ммоль) растворяли в дихлорметане и к перемешиваемому раствору добавляли уксусный ангидрид (85 мкл, 1,16 ммоль) и триэтиламин (117 мг, 1,16 ммоль). Через 2 ч добавляли дополнительно 10% эквивалента уксусного ангидрида и триэтиламина и раствор перемешивали еще 2 ч при комнатной температуре. Дихлорметановый раствор промывали последовательно водным бикарбонатом натрия и 1 М хлористоводородной кислотой. Затем дихлорметановый раствор сушили сульфатом магния, выпаривали до масли и получали 400 мг (99%) целевого промежуточного соединения. Спектр ЯМР.
D. Получение иодистого 3-/2-этил-4-(4-фтофренил)-5-ацетоксифенокси/-пропила.
Хлористый 3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/пропил (400 мг, 1,14 ммоль) растворяли в метилэтилкетоне (50 мл) и добавляли иодистый натрий (2,5 г). Затем перемешиваемую суспензию нагревали с обратным холодильником 16 ч. Охлажденный раствор фильтровали, метилэтилкетон выпаривали и получали остаток, который снова растворяли в эфире. Эфирный раствор фильтровали и выпаривали до получения бледножелтого масла. Спектр ЯМР показал, что неочищенный материал представляет собой в основном требуемый продукт, плюс незначительное количество примесей: по причине нестабильности этого соединения его применяли непосредственно в следующей реакции.
E. Получение 2-[2-/3-/2-этил-4-4-фторфенил)-5-ацетоксифенокси/-пропокси/ -фенил]-1,3-диоксолана.
60%-ный гидрид натрия в масле промывали гексаном и суспендировали в сухом диметилсульфоксиде (50 мл) с перемешиванием в атмосфере азота. 2-(-оксифенил)-1,3-диоксолан (166 мг, 1,0 ммоль) растворяли в сухом тетрагидрофуране (10 мл) и добавляли к диметилсульфоксидному раствору, получая бледножелтый раствор. Через 20 мин при комнатной температуре добавляли иодистый 3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/-пропил (442 мг, 1 ммоль) в форме раствора в сухом тетрагидрофуране (10 мл). Еще через 2 ч при комнатной температуре реакционную смесь выливали в фосфатный буфер с pH 7,0 и смесь экстрагировали 5 раз эфиром. Объединенные эфирные экстракты промывали водой, сушили сульфатом магния и выпаривали до масла, которое хроматографировали на колонке силикагеля, элюируя смесью эфир-гексан в отношении 1:1. Целевое соединение выделяли в виде масла, которое на ЯМР спектре показало загрязнение исходным фенолом и побочным соединением, полученным потерей ацетильной группы и алкилированием, вызванным исходным йодидом. Эти примеси обычно не отделяют на этой стадии и частично очищенный материал передают на следующую операцию.
F. Получение 2-[]3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/-пропокси -бензальдегида.
2-[2-/3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/-пропокси/-фенил] -1,3-диоксолан (300 мг, 0,65 ммоль) растворяли в тетрагидрофуране (50 мл) и добавляли 1 моль хлористоводородной кислоты (10 мл). Полученный бесцветный раствор выдерживали при комнатной температуре 3 ч. Раствор выливали в водный раствор бикарбоната натрия и 3 раза экстрагировали эфиром. Объединенные эфирные экстракты сушили сульфатом магния и выпаривали до 270 мг масла. Материал содержал некоторое количество 2-оксибензальдегида, который удаляли пропусканием масла через короткую колонку с силикагелем, промываемую смесью эфир-гексан в отношении 1:1. Полученный материал все еще содержал альдегид упомянутого выше продукта алкилирования, образовавшегося в предыдущей реакции, но который не мог быть легко удален и очевиден на ЯМР-спектре. Неочищенный материал использовали затем на следующей реакции.
G. Получение 3-[2-/3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/-пропокси/-фенил]-E-пропеновой кислоты.
2-[3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/-пропокси] -бензальдегид (210 мг, 0,5 ммоль) растворяли в толуоле (25 мл) и добавляли пиридин (1 мл), пиперидин солянокислый (100 мг) и малоновую кислоту (1,0 г). Затем раствор нагревали с обратным холодильником 3 ч. В это время добавляли еще порцию малоновой кислоты (0,5 г) и раствор нагревали с обратным холодильником еще 1 ч. Охлажденный раствор три раза промывали эфиром и объединенные толуоловую и эфирные экстракты промывали 1 раз водой и сушили сульфатом магния. После выпаривания растворителя получали масло, которое являлось смесью двух соединений с одинаковыми значениями Rf. Соединения разделяли хроматографией на колонке силикагеля, элюированной смесью эфир-гексан в отношении 1:1 с содержанием 1,0% уксусной кислоты. Целевое соединение было более полярным соединением, полученным в виде стекла (выход 107 мг, 46%). Спектр ЯМР. Менее полярное соединение было идентифицировано как 3-[2-/3-/2-этил-4-(4-фторфенил)-5-/3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/-пропокси/-фенокси/ -пропокси-фенил] -E-пропеновая кислота по результатам масс- и ЯМР-спектров (выход 91 мг, в виде масла).
H. Получение 3-[2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси)-пропокси/-фенил]-E-пропеновой кислоты.
3-[2-/3-/2-этил-4-(4-фторфенил)-5-ацетоксифенокси/-пропокси/-фенил] -E-пропеновую кислоту (90 мг, 0,2 ммоль) растворяли в метаноле (10 мл). Добавляли 0,1 М водный раствор карбоната калия и раствор оставляли на ночь при перемешивании в атмосфере азота. Тонкослойная хроматография показала единственное пятно с тем же Rf, что и исходный материал, так что добавляли дополнительно 1,0 М раствор карбоната калия и раствор перемешивали еще 4 ч. Реакционную смесь выливали в 1 М хлористоводородную кислоту (50 мл) и смесь экстрагировали три раза хлороформом. Объединенные хлороформные экстракты сушили сульфатом магния и выпаривали, получая 63 мг целевого промежуточного соединения в виде масла, которое отверждалось до стекла. Масс- и ЯМР-спектры.
Пример 88. 3-[]2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил-2-метил-Е-пропеновая кислота.

A. Получение хлористого 3-/2-этил-4-(4-фторфенил)-5-/2-(триметилсилил)-этоксиметоксифенокси)-пропила.
Хлористый 3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропил (400 мг, 1,29 ммоль) растворяли в дихлорметане (25 мл) и раствор охлаждали до 0oC под азотом. Добавляли N,N-диизопропилэтиламин (832,0 мг, 6,45 ммоль) с последующим добавлением хлористого 2-(триметилсилил)-этоксиметила и полученную смесь нагревали до комнатной температуры в течение 1 ч. Затем реакционную смесь выливали в 1 М хлористоводородную кислоту и три раза экстрагировали дихлорметаном. Объединенные дихлорметановые экстракты сушили над сульфатом магния и дихлорметан выпаривали до получения масла. Это масло помещали затем в высокий вакуум на 48 ч при комнатной температуре для удаления примесей. Остаточно масло представляло собой целевое соединение, выход 490 мг (86%). Спектр ЯМР.
B. Получение йодистого 3-/2-этил-4-(4-фторфенил)-5-/2-(триметилсилил)-этоксиметоксифенокси/-пропила.
Целевое соединение получали из соответствующего хлорида используя процесс, описанный в примере 87 (D). Нестабильный иодид был идентифицирован спектром ЯМР и использован непосредственно в следующей реакции.
C. Получение 2-[2-/3-/2-этил-4-(4-фторфенил)-5-/2-(триметилсилил/-этоксиметоксифенокси/-пропокси/-фенил]-1,3-диоксолана.
Целевое соединение, указанное в названии части С, получали, используя общий процесс, описанный в примере 87(E), с выходом 86% в виде масла после хроматографии. Спектр ЯМР.
D. Получение 2-[3-/2-этил-4-(4-фторфенил)-5-/2-(триметилсилил) -этоксиметоксифенокси/-пропокси]-бензальдегида.
Целевое соединение получали из 2-[2-/3-/2-этил-4-(4-фторфенил)-5-/2- (триметилсилил)-этоксиметоксифенокси)-пропокси/-фенил] -1,3-диоксолана, используя общий процесс, описанный в примере 87(Г), выход 82% в виде масла. Спектр ЯМР.
E. Получение 3-[2-/3-/2-этил-4-(4-фторфенил)-5-/2-(триметилсилил)-этоксиметоксифенокси/-пропокси/-фенил]-2-метил-Е-пропеновой кислоты.
Целевое соединение получали, используя общий процесс, описанный в примере 87(C), за исключением того, что вместо малоновой кислоты использовали метилмалоновую кислоту. Было найдено, что неочищенный продукт являлся смесью целевого соединения плюс 5-оксианалог, образованный частичной потерей защитной группы триметилсилилэтоксиметила. Неочищенный продукт поэтому полностью лишали защиты в следующей реакции без дальнейшей очистки. Спектр ЯМР.
F. Получение 3-[2-/3-/2-этил-4-(4-фтофренил)-5-оксифенокси/-2-пропокси/ -фенил]-2-метил-Е-пропеновой кислоты.
Неочищенную 3-[2-/3-/2-этил-4-(4-фтофренил)-5-/2-(триметилсилил) -этоксиметоксифенокси/-пропокси/-фенил]-2-метил-Е-пропеновую кислоту (300 мг), содержащую некоторое количество целевого соединения, указанного в названии примера 88(F), растворяли в тетрагидрофуране (50 мл) и добавляли моногидрат фтористого тетрабутиламмония (2,0 г) в виде раствора также в тетрагидрофуране (20 мл). Полученный желтый раствор выдерживали при комнатной температуре 16 ч. Реакционную смесь выливали в 1М хлористоводородную кислоту и 3 раза экстрагировали эфиром. Объединенные эфирные экстракты сушили сульфатом магния и выпаривали досуха, получая масло. Это масло хроматографировали на колонке силикагеля, элюируя смесью эфир-гексан в отношении 1:1, содержащей 1,0% уксусной кислоты. Основным компонентом было целевое соединение с незначительными примесями. Масло очищали дальше на колонке с С18 обращенно-фазовой жидкостной хроматографией высокого давления, элюируя смесью метанол-вода в отношении 90: 30, содержащей 0,1% уксусной кислоты. Основной компонент был выделен и медленно кристаллизовался из смеси этиловый эфир-гексан, давая 110 мг желаемого целевого соединения, точка плавления 114oC. Спектры 2D-NOE подтвердили, что выделенный изомер был E-изомером. Спектр ЯМР, спектр-масс.
Элементный анализ для C27H27O5F:
Вычислено,%: C 71,98; H 6,04.
Найдено,%: C 72,06; H 6,21.
Пример 89. 5-[2-/2-/3-/2-этил-4-(4-фтофренил)-5-оксифенокси/-пропокси/-фенил/-этил]-1Н-тетразол.

A. Получение 3-[2-/3-/2-этил-4-(4-фторфенил)-5-бензил-оксифенокси/-пропокси/фенил]-пропилнитрила.
Хлористый 3-/2-этил-4-/4-фторфенил)-5-бензилоксифенокси/-пропил (199 мг, 0,5 ммоль) растворяли в метилэтилкетоне (50 мл), добавляли иодистый натрий (0,5 г) и полученную суспензию перемешивали при комнатной температуре 3 ч. Добавляли 3-(2-оксифенил)-пропилнитрил (73,5 мг, 0,5 ммоль) с последующим добавлением карбоната калия 91,0 г). Полученную суспензию нагревали с обратным холодильником в атмосфере азота 28 ч. Затем реакционную смесь выливали в воду (50 мл) и смесь экстрагировали 3 раза хлороформом. Объединенные экстракты сушили сульфатом магния и выпаривали до масла, которое хроматографировали на колонке силикагеля, элюируя смесью эфир-гексан в отношении 1: 1. Целевое соединение получено в виде бесцветного масла с выходом 131 мг (51%). Спектр ЯМР.
B. Получение 5-[2-/2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-фенил/-этил]-1Н-тетразола.
3-[2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси/-пропокси/-фенил] -пропилнитрил (120 мг, 0,24 ммоль) растворяли в диметилформамиде (20 мл) и добавляли азид натрия (0,6 г, 1,0 ммоль) и хлористый триэтиламмоний (1,37 г, 1,0 ммоль) и перемешиваемую смесь нагревали до 125oC за 24 ч под азотом. Дополнительно добавляли по 1 ммоль обоих реактивов - азида натрия и хлористого триэтиламмония. Еще после 24-ч нагревания добавляли другую аликвоту азида и гидрохлорида и смесь перемешивали конечные 6 ч. Затем реакционную смесь добавляли к 1 М хлористоводородной кислоте (100 мл) и смесь 3 раза экстрагировали хлороформом. Объединенные экстракты сушили сульфатом магния и выпаривали до получения масла, которое медленно становилось воскоподобным твердым веществом, не имеющим определенной точки плавления. Продукт идентифицировали как сольват диметилформамида целевого соединения. Спектр ЯМР.
C. Получение 5-[2-/2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенил/-этил]-1Н-тетразола.
5-[2-/2-/3-/2-этил-4-(4-фтофренил)-5-бенизлоксифенокси/-пропокси/-фенил/-этил] -1Н-тетразол (90 мг, 0,16 ммоль) растворяли в этаноле и в атмосфере углекислого газа добавляли катализатор - 10%-ный палладий на угле. Смесь гидрогенизировали при 2,1 кг/см2 и комнатной температуре 1 ч. Катализатор отфильтровывали и раствор выпаривали досуха, получая масло. Это масло очищали затем обращенно-фазовой жидкостной хроматографией высокого давления на колонке с С18, элюируя смесью метанол-вода в отношении 90:10, содержащей 0,01% уксусной кислоты. Целевое соединение выделяли в виде масла (выход 41 мг, 55%), содержащего 0,3 эквивалента уксусной кислоты. Спектр ЯМР, масс-спектр.
Пример 90. 3-[2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси)-пропокси/-4- (4-карбоксибутилокси)-фенил]-пропионовая кислота.

Таким же образом, как описано в примере 5, дебензилировали 3-[2-/3-/2-этил-4-(4-фторфенил)-5-бензилоксифенокси)-пропокси/-4- (4-карбоксибутилокси)-фенил] -пропионовую кислоту с получением целевого соединения с 20%-ным выходом. Спектр ЯМР.
Элементный анализ для C31H35FO8:
Вычислено,%: C 67,14; H 6,36.
Найдено,%: C 67,40; H 6,45
Пример 91. 5-[3-/4-(4-фторфенрил)-2-этил-5-оксифенокси/-пропокси]-3,4-дигидро-2Н-1-бензопиран-2-он.

Если метиловый эфир 3-[2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-6-оксифенил] -пропионовой кислоты (пример 12) гидролизовали в условиях получения 26, то в дополнение к желаемому продукту - 3-[2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-6-оксифенил] -пропионовой кислоте (пример 13), выделяли целевое соединение с 10%-ным выходом методом обращенно-фазовой жидкостной хроматографии высокого давления. Спектр ЯМР, масс-спектр.
Элементный анализ для C26H25FO5:
Вычислено,%: C 76,55; H 5,77.
Найдено,%: C 76,39; H 5,92.
Примеры 92-96. Следующие соединения получали из их соответствующих сложных эфиров способом по получению 26, используя вместо этанола метанол.
92. 3-[3-/3-/2-этил-4-(4-фторфенил)-5-оксифенилокси/пропокси/-фенил]-пропановая кислота, выход 10%, точка плавления 113-115oC.
Элементный анализ для C26H27FO5:
Вычислено,%: C 71,22; H 6,21.
Найдено,%: C 70,95; H 6,42.

93. Натриевая соль 3-[3-/3-/2-этил-4-(4-фтофренил)-5-оксифенилокси/-пропокси/-4-пропилфенил]-пропановой кислоты, выход 23%.
Элементный анализ для C29H32FNaO5:
Вычислено,%: C 69,31, H 6,42.
Найдено,%: C 69,35, H 6,83.

94. 3-[4-/3-/2-этил-4-(4-фторфенил)-6-оксифенилокси/-пропокси/-3-пропилфенил]-пропановая кислота, выход 69%, точка плавления 118-120oC.
Элементный анализ для C29H33FO5:
Вычислено,%: C 72,48; H 6,92.
Найдено,%: C 72,20; H 7,00.

95. 3-[3-/3-/2-этил-4-(4-фторфенил)-5-оксифенилокси/-пропокси/-2-пропилфенил]-пропановая кислота, выход 56%, точка плавления 125-127oC.
Элементный анализ для C29H33FO5:
Вычислено,%: C 72,48; H 6,92.
Найдено,%: C 72,67; H 7,05.

96. 3-[3-/3-(2-этил-5-оксифенилокси)-пропокси/-2-пропилфенил] -пропановой кислоты двунатриевая соль, выход 18%.
Элементный анализ для C29H32Na2O5:
Вычислено,%: C 68,76; H 6,37.
Найдено,%: C 68,00; H 6,46.

Пример 97. Полугидрат двунатриевой соли 2-[3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/-бензоил]-бензойной кислоты.

Целевое соединение, указанное в названии примера, гидролизовали из 400 мг соответствующего метилового эфира как описано выше в примере 60. Кислоту обращали в двунатриевую соль и очищали как описано выше для получения по примеру 59(D) и получали 170 мг (42%) целевого соединения в виде рыхлого белого твердого вещества.
Спектр ЯМР (ДМСО-d6) δ : 11,85 (С, 1Н, -OH), 7,82 (д, J=7,7 Гц, 1Н), 7,53 (м, 2Н), 7,28-7,42 (м, 4Н), 7,11 (м, 4Н), 6,99 (д, J=8,3 Гц, 1Н), 6,87 (м, 2Н), 3,99 (т, J=4,9 Гц, 2Н), 3,83 (т, J=3,9 Гц, 2Н), 2,42 (кв, J=7,3 Гц, 2Н), 1,82 (м, 2Н), 1,06 (т, J= 7,2 Гц, 3Н); масс-спектр (бомбардировка быстрыми атомами) м/е 559 (р + Na, 100), 537 (р): ИК-спектр (хлороформ): 3450 (шир.), 3021, 1601, 1370, 1226, 1048 см-1.
Элементный анализ для C31H26O6FNa2•0,5 H2O:
Вычислено,%: C 65,60; H 4,80.
Найдено,%: C 56,45; H 4,76.
Соединения формулы I должны быть полезны при лечении любого состояния, включая клинические случаи, которое характеризуется избыточным освобождением лейкотриена B4. Эти условия включают непосредственный тип аллергических реакций, такой как астма. Термин "избыточное освобождение" лейкотриена B4 касается количества лейкотриена, достаточного для того, чтобы вызывать состояние связанное с таким количеством. Количество лейкотриена, которое расценивается как избыточное, будет зависеть от многих факторов, включая количество лейкотриена, способное вызывать конкретное состояние, и от вида вовлеченного в процесс млекопитающего. Как может оценить специалист в данной области, успех лечения млекопитающих, страдающих от или подозрительных на состояния, характеризующиеся избыточным освобождением лейкотриена, соединениями формулы I будет определяться регрессией или предупреждением симптомов болезненного состояния. Эффективность соединений формулы I ингибировать связывание меченого тритием лейкотриена B4 легочных мембран морских свинок определяли следующим образом.
Тест на связывание радиоактивных лиганд меченого тритием лейкотриена B4 в легочных мембранах морских свинок.
Меченый тритием лейкотриен B4 (196-200 кюри/ммоль) приобретено т New England Nuclear (Boston, MA). Все остальные материалы получены от Sigma (St. Louis, MO). Инкубации проводили в полипропиленовых мини-пробирках в течение 45 мин при 30oC, содержащих 25 мг белка легочных мембран морских свинок (Saussy, et al. , Mol. Pharmacol, 39,72 (1991 г.) в буфере, содержащем 25 ммоль MOPS, 10 ммоль хлористого магния, 10 ммоль хлористого кальция, pH 6,5, приблизительно 140 pH меченого тритием лейкотриена B4 и замещенный лиганд или носитель (0,1% диметилсульфоксид в 1 ммоль карбоната натрия, конечная концентрация). Реакцию связывания заканчивали добавлением 1 мл охлажденного льдом промывочного буфера (25-ммольный трис-HCl, pH 7,5) с последующей немедленной вакуумной фильтрацией через стекловолоконные фильтры Whatman GF/C используя 48-местный инкубатор Brandel (Gaithersburg, MD). Фильтры три раза промывали по 1 мл промывочного буфера. Удержанную радиоактивность определяли жидким сцинтилляционным счетчиком при 50 %-ной эффективности счета, используя готовый белок плюс коктейль (Beckman, Fullerton, CA). Незамещенное связывание определяли в присутствии 1 ммоль лейкотриена B4, которое обычно составляло менее 10% от общего связывания. Данные анализировали, используя анализ линейной регрессии графиков в двойном логарифмическом масштабе значений между 10%- и 90%-ным контрольным связыванием, чтобы рассчитать ингибирующую концентрацию ИК50 и угловые коэффициенты (псевдо-барьерные коэффициенты). Полученные таким образом значения ИК50 корректировали с концентрацией радиоактивных лиганд (Cheng and Prusoff, Biochem, Pharmacol., 22, 3099, (1973), чтобы рассчитать значения показателя кислотности Кi. Представленные в таблице данные представляют среднее значение - Log Ki, иначе известного как pKi, для n экспериментов.
В дополнение некоторые соединения согласно изобретению, а именно соединения по примерам 42, 55 и 56, показали, что являются ингибиторами in vitro синовиальной и цитозольной фосфолипазы A2 человека (OLA2). Следовательно, соединения согласно изобретению, в частности, соединения, имеющие группы R4, как установлено в примерах 55 или 56, будут полезны для лечения состояний, таких как артрит, псориаз и астма, связанных с излишним образованием эйкозаноидов, которые образуются под действием фосфолипазы A2 на фосфолипиды мембран, такие как различные лейкотриены, простагландины, липоксины, оксиэйкозатетрановые кислоты и тромбоксаны.
Соединения или композиции согласно изобретению могут быть введены пероральным и ректальным путями, локально, парентерально, например, инъекцией и непрерывной или прерывистой внутриартериальной инфузией, в форме, например, таблеток, лепешек, подъязычных таблеток, облаток, эликсиров, гелей, суспензий, аэрозолей, мазей, например, содержащих от 1 до 10 мас.% активного соединения в подходящей основе, мягких и твердых желатиновых капсул, суппозиториев, растворов для инъекций и суспензий в физиологически приемлемой среде, и стерильно упакованных порошков, адсорбированных на материала подложки для приготовления растворов для инъекций. Для этих целей композиции согласно изобретению выгодно иметь в форме дозированной единицы, при этом предпочтительно, чтобы каждая дозированная единица содержала от 5 до 500 мг (от 5 до 50 мг в случае парентерального или ингаляционного введения и от 25 до 500 мг в случае перорального или гектального введения) соединения формулы I. Дозы примерно от 0,5 до 300 мг/кг в день, предпочтительно 0,5-20 мг/кг активного ингредиента, могут быть введены, хотя разумеется, вполне понятно, что количество соединения или соединений формулы I, которое действительно может быть введено, определяется врачом в свете всех относящихся к случаю обстоятельств, включая заболевание, которое подлежит лечению, выбор соединения, которое надлежит ввести, и выбор пути введения, и поэтому приведенные выше дозные интервалы не являются ограничивающими ни в коей мере объем изобретения.
Композиции согласно изобретению нормально содержат по меньшей мере одно соединение формулы I, смешанное с носителем или разбавленное носителем, или заключено или инкапсулировано в съедобный носитель в форме капсулы, облатки, саше, бумаги или другого контейнера или имеющегося в распоряжении контейнера, такого как ампула. Носитель или разбавитель может быть твердым, жидким или полутвердым материалом, который служит носителем или наполнителем или средой для активного лекарственного вещества. Некоторые примеры разбавителей или носителей, которые могут быть применены в фармацевтических композициях, согласно изобретению, включают лактозу, декстрозу, сахарозу, сорбит, маннит, пропиленгликоль, жидкий парафин, бесцветный мягкий парафин, каолин, коллоидная двуокись кремния, микрокристаллическую целлюлозу, силикат кальция, кремнезем, поливинилпирролидон, цетостеариловый спирт, крахмал, модифицированный крахмал, аравийскую камедь, фосфат кальция, масло какао, этоксилированные сложные эфиры, масло какао, арахисовое масло, альгинаты, трагакант, желатин, сироп, метилцеллюлозу, полиэтилен сорбитанмонолаурат, этиллактак, метиловый и пропиловый эфиры оксибензойной кислоты, сорбитантриолеат, сорбитансесквиолеат и олеиловый спирт и пропелленты, такие как трихлормонофторметан, дихлордифторметан и дихлортетрафторэтан. В случае таблеток может быть введена смазка, чтобы предотвратить слипание и связывание порошкообразных ингредиентов в красителях и на пуансонах машин для прессования таблеток. Для этих целей могут быть применены, например, стеараты алюминия, магния или кальция, тальк или минеральные масла.
Предпочтительными лекарственными формами изобретения являются капсулы, таблетки, суппозитории, растворы для инъекций, кремы и мази. Особенно предпочтительными являются композиции для ингаляции, такие как аэрозоль и лекарственные формы для перорального применения.
Поскольку все приведенные выше соединения показывают ингибирующую лейкотриен B4 активность in vitro, также установлено, что соединения, несущие одну кислотную группу (R6), являются гораздо более биологически активными при пероральном введении млекопитающим в сравнении с соединениями, несущими две такие кислотные группы. Таким образом, предпочтительным осуществлением изобретения при перорально введении соединений формулы I млекопитающим является введение соединений, несущих единственную кислотную функциональную группу R6.
В следующих ниже лекарственных формах могут быть применены в качестве активных ингредиентов соединения согласно изобретению. Примеры являются чисто иллюстративными и никоим образом не ограничивают объем изобретения.
Пример лекарственной формы 1.
Твердые желатиновые капсулы получали, используя следующие ингредиенты, количество (мг/капсула):
3-[2-/3-/2-этил-4-(4-фторфенил)-5-окси-фенокси/-пропокси/-6- (4-карбоксифенокси)-фенил]-пропановая кислота - 250
Крахмал - 200
Стеарат магния - 10
Перечисленные ингредиенты смешивали и заполняли твердые желатиновые капсулы по 460 мг в каждую.
Пример лекарственной формы 2.
Таблетки получали, используя следующие ингредиенты, количество (мг/таблетка):
1-/4-(карбоксиметокси)-фенил/-1-(1Н-тетразол-5-ил)-6-/2-этил-4- (4-фтофренил)-5-оксифенокси/-гексан - 250
Двуокись кремния, коллоидная - 10
Целлюлоза, микрокристаллическая - 400
Стеарат магния - 5
Компоненты смешивали и прессовали в форме таблеток весом 665 мг каждая.
Пример лекарственной формы 3.
Аэрозольный раствор готовили из следующих компонентов, вес.%:
3-[4-/7-карбокси-9-оксо-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-9Н-ксантен/]-пропановая кислота - 0,25
Этанол - 30,00
Пропеллент 11 (трихлорфторметан) - 10,25
Пропеллент 12 (дихлордифторметан) - 29,75
Пропеллент 114 (дихлортетрафторэтан) - 29,75
Активное соединение растворяли в этаноле и раствор добавляли к пропелленту 11, охлажденному до -30oC, и переносили в устройство для наполнения. Затем требуемое количество наливали в контейнер и далее заполняли предварительно смешанными пропеллентами 12 и 114 методом холодного заполнения или под давлением. Работу распределительных клапанов дозатора согласовывали с объемом контейнера.
Пример лекарственной формы 4.
Таблетки, содержащие 60 мг активного ингредиента каждая, готовили следующим образом, мг:
2-[2-пропил-3-/3-/2-этил-5-окси-4-(4-фторфенил)-фенокси/-пропокси/фенокси]-бензойной кислоты натриевая соль - 60
Крахмал - 45
Микрокристаллическая целлюлоза - 35
Поливинилпирролидон (10%-ный раствор в воде) - 4
Натриевая соль карбоксиметилкрахмала - 4,5
Стеарат магния - 0,5
Тальк - 1,0
Всего - 150,0
Активный ингредиент, крахмал и целлюлозу пропускали через сито N 45 меш США и тщательно перемешивали. Раствор поливинилпирролидона смешивали с полученными порошками, которые затем пропускали через сито N 14 меш США. Полученные таким образом гранулы сушили при 50-60oC и пропускали через сито N 18 меш США. Натрий-карбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенные через сито N 60 меш. США, добавляли затем к гранулам, которые после смешения прессовали на таблеточной машине, получая таблетки, весившие 150 кг каждая.
Пример лекарственной формы 5.
Капсулы, содержащие 80 мг медикамента, готовили следующим образом, мг:
5-[3-/2-(1-карбокси)-этил/-2-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/фенил]-4-пентиновая кислота - 80
Крахмал - 59
Микрокристаллическая целлюлоза - 59
Стеарат магния - 2
Всего - 200
Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивали, пропускали через сито N 45 меш США и смесью заполняли твердые желатиновые капсулы в количестве 200 мг в каждую.
Пример лекарственной формы 6.
Суппозитории, содержащие по 225 мг активного ингредиента каждый, получали следующим образом, мг:
3-[5-/6-/4-(4-фторфенил)-5-окси-2-этилфенокси/-пропокси/ -2-карбоксиметил-1,2,3,4-тетрагидронафталин-1(2Н)-он]-пропановая кислота - 225
Ненасыщенные или насыщенные глицериды жирных кислоты - До 2000
Активный ингредиент пропускали через сито N 60 меш США и суспендировали в глицеридах жирных кислот, предварительно расплавленных с использованием минимального нагрева, если необходимо. Затем смесь выливали в форму для литья суппозиториев с номинальной емкостью 2 г и охлаждали.
Пример лекарственной формы 7.
Суспензии, содержащие 50 мг медикамента в 5 мл дозе, получили следующим образом, мг:
2-[2-Пропил-3-/3-/2-этил-4-(4-фторфенил)-5-оксифенокси/-пропокси/-фенокси]-бензойная кислота - 50
Натриевая соль карбоксиметилцеллюлозы - 50
Сахар - 1 г
Метилпарабен - 0,05
Пропилпарабен - 0,03
Ароматическое вещество - По усмотрению
Краситель - По усмотрению
Очищенная вода - До 5 мл
Медикамент пропускали через сито N 45 меш США и смешивали с натриевой солью карбоксиметилцеллюлозы, сахаром и частью воды, чтобы получить суспензию. Парабены, ароматические и красящие вещества растворяли и разбавляли частью воды и добавляли при перемешивании. Затем добавляли воду до получения желаемого объема.
Следующие ниже примеры иллюстрируют способ получения промежуточных соединений, описанных выше.
Пример 98. Получение 2-(3-метоксифенокси)-бензонитрила.
Смесь 0,975 г 2-фторбензонитрила, 1,00 г 3-метоксифенола, 0,259 г бромистого тетрабутиламмония, 2,6 г смеси фтористого калия и окиси алюминия и 20 мл ацетонитрила нагревали при 90oC приблизительно 24 ч. Затем реакционную смесь фильтровали и промывали хлористым метиленом. Органический растворитель удаляли в вакууме и остаток растворяли в этилацетате. Органический раствор фильтровали через флорисил и подвергали экспресс-хроматографии, элюируя ступенчатым градиентом смеси 1% этилацетата в гексане и 2% этилацетата в гексане. Целевое соединение выделяли с 55%-ным выходом.
Пример 99. Получение 4-(3-метоксифенокси)-бензонитрила.
Следуя процессу по примеру 98, 5,00 г 3-метоксифенола6 5,12 г 4-фторбензонитрила 1,33 г бромистого тетрабутиламмония, 1360 г смеси фтористого калия и окиси алюминия и 50 мл ацетонитрила нагревали при 90-95oC, оставляя на ночь. Органическую фазу удаляли в вакууме выпариванием и остаток помещали в тетрагидрофуран. Добавляли 3 мл 5 н. гидроокиси натрия и смесь экстрагировали этилацетатом. Органический слой подвергали экспресс-хроматографии, используя ступенчатый градиент гексан, 3%-ный этилацетат в гексане и 4%-ный этилацетат в гексане. Целевой продукт выделяли с 53,8%-ным выходом.
Пример 100. Получение 2-3-метоксифенокси)-бензонитрила.
Повторяли реакцию по примеру 98, используя 5,12 г фенола, 5,0 г бензонитрила, 5,0 г смеси фтористый калий-окись алюминия, 1,33 г бромистого тетрабутиламмония и 25 мл ацетонитрила. Реакцию продолжали 2,5 дня. По истечении этого времени реакционную смесь фильтровали и фильтр тщательно промывали метанолом. Органическую фазу концентрировали в вакууме. Экспресс-хроматография 10 г полученного неочищенного соединения давала 8,82 г (94,9%) целевого продукта в виде желтого масла, которое кристаллизовалось при выдерживании, точка плавления 54-55oC.
Пример 101. Получение 2-(3-метоксифенокси)-бензонитрила.
Повторяли реакцию по примеру 100 без бромистого тетрабутиламмония. Оценка ЯМР-спектр неочищенной смеси показала, что имеет место отношение 2:1 желаемого продукта к исходному фенолу.
Пример 102. Получение 2-(3-метоксифенокси)-бензонитрила.
Повторяя процесс по примеру 98, 0,15 г бензонитрила, 0,16 г фенола, 0,15 смеси фтористого калия - окиси алюминия, 0,9 г бромистого тетрабутиламмония и 4 мл ацетонитрила оставляли на ночь при нагревании. Анализ реакционной смеси спектрометрией ЯМР к этому времени показал, что реакция закончилась.
Пример 103. Получение 2-феноксибензонитрила.
Смесь 3,00 г фенола, 3,86 г 2-фторбензонитрила, 3,86 г смеси фтористого калия и окиси алюминия, 1,0 г бромистого тетрабутиламмония и 25 мл ацетонитрила нагревали при 90oC 7 дней. Обработка реакционной смеси и хроматография с использованием 2%-ного этилацетата в гексане давали 5,87 г (94,3%) желаемого целевого соединения.
Пример 104. Получение 4-феноксибензонитрила.
Повторяли реакцию по примеру 103, используя 4-фторбензонитрил и нагревая 9 дней. Обработка смеси с использованием хроматографии, элюируя 1%-ным этилацетатом в гексане и 3%-ным этилацетатом в гексане, давала 5,6 г (90%) желаемого целевого соединения.
Пример 105. Получение 2-(4-бромфенокси)-бензонитрила.
Смесь 3,22 г 4-бромфенола, 2,25 г 2-фторбензонитрила, 2,25 г фтористого калия-окиси алюминия, 0,6 г бромистого тетрабутиламмония и 25 мл ацетонитрила нагревали 8 дней при 90oC. Обработкой обычным образом и хроматографией с 1 и 3%-ным этилацетатом в гексане получали 4,8 г (94%) желаемого целевого соединения в виде желтых кристаллов, точка пл. 66-68oC.
Пример 106. Получение 2-(3-метокси-2-пропилфенокси)-бензонитрила.
Смесь 0,9 г 3-метокси-2-пропилфенола, 0,266 г 2-фторбензонитрила, 0,9 г смеси фтористый калий-окись алюминия, 0,174 г бромистого тетрабутиламмония и 10 мл ацетонитрила нагревали при 90oC 2 дня. После обычной обработки и экспресс-хроматографии с 10-15%-ным хлористым метиленом в гексане получали желаемое целевое соединение с 67%-ным выходом.
Пример 107. Получение 2-(3-метоксифенокси)-бензонитрила.
Смесь 2,50 г 3)метоксифенола, 2,45 г 2-фторлбензонитрила, 0,5 г 18-краун-6, 2,5 г смеси фтористого калия и окиси алюминия и 35 мл ацетонитрила нагревали при 90oC 3 дня. Реакционную смесь фильтровали, промывали насыщенным раствором хлористого натрия и обрабатывали как прежде, получая 4,47 г (98,6%) желаемого целевого соединения.
Пример 108. Получение 4-(3-метоксифенокси)-бензонитрила.
Повторяли пример 107, используя 4-фторбенонитрил, и получали 4,9 г (99,1%) целевого продукта.
Пример 109. Получение 2-(3-метоксифенокси)-6-метоксибензонитрила.
Смесь 2,5 г 3-метоксифенола, 3,0 г 2-фтор-6- метоксибензонитрила, 2,5 г смеси фтористый калий - окись алюминия, 0,65 г бромистого тетрабутиламмония и 35 мл ацетонитрила нагревали при 90oC 3 дня. Смесь фильтровали, промывали водой, экстрагировали эфиром и получали 4,10 (99,2%) целевого соединения, указанного в названии примера. Кристаллизация из смеси гексан-этилацетат давала продукт с точкой плавления 94-95oC.
Пример 110. Получение 2-(3-метоксифенокси)-6-метоксибензонитрила
Повторяли пример 109, используя 2,0 г 3-метоксифенола, 2,43 г 2-фтор-6-метоксибензонитрила, 2,0 г смеси фтористого калия и окиси алюминия 0,4 г 18-краун-6 и 25 мл ацетонитрила. После нагревания в течение 1,5 дня при 90oC продукт фильтровали, промывали водой и насыщенным раствором хлористого калия и экстрагировали этилацетатом, получая 4,9 г (99,5%) целевого соединения, точка плавления 93-95oC.
Пример 111. Получение 2-(3-метоксифенокси)-4-нитробензонитрила.
Смесь 2,14 г 2-фтор-4-нитробензонитрила, 1,6 г 3-метоксифенола, 1,6 г смеси фтористого калия и окиси алюминия, 0,34 г 18-краун-6 и 35 мл ацетонитрила оставляли на ночь при нагревании при 90oC. Реакционную смесь фильтровали, промывали насыщенным раствором хлористого калия, экстрагировали этилацетатом и получали 3,36 г (96,5%) желаемого целевого соединения в виде желтого твердого вещества, точка плавления 90-92oC.
Пример 112. Получение 2-(3-метоксифенокси)-6-(4-метилфенилтио)-бензонитрила.
Смесь 1,20 г 3-метоксифенола, 2,35 г 2-фтор-6-(4-метилфенилтио)-бензонитрила, 1,20 г смеси фтористого калия и окиси алюминия, 0,255 г 18-краун-6- и 30 мл ацетонитрила оставляли на ночь при нагревании на 90oC. Обработка обычным порядком давала 3,32 г (98,9%) целевого продукта. Кристаллизацией из смеси гексан-этилацетат получали светло-коричневые и желтые кристаллы, точка плавления 107-109oC.
Пример 113. Получение N-метил-N-фенил-2-циано-5-нитроанилина.
Смесь 0,8 г N-метиланилина, 1,24 г 2-фтор-4-нитробензонитрила, 0,8 г смеси фтористого калия и окиси алюминия, 0,197 г 18-краун-6 и 15 мл ацетонитрила оставляли на ночь при нагревании при 90oC. Обработка обычным образом давала 1,85 г целевого соединения, которое перекристаллизовывали из смеси гексан-этилацетат, всего было получено 1,35 г светло-оранжевых кристаллов (71,4%), точка плавления 98-99oC.
Пример 114. Получение 2-(3-метоксифенокси)-3-хлорбенжонитрила.
Смесь 1,2 г 3-метоксифенола, 1,5 г 2-фтор-3-хлорбензонитрила, 0,255 г 18-краун-6, 1,2 г смеси фтористого калия и окиси алюминия и 20 мл ацетонитрила оставляли на ночь с нагреванием при 90oC. Обработка обычным путем давала 2,47 г (98,4%) целевого соединения в виде коричневого масла.
Пример 115. Получение N-фенил-2-цианоанилина.
Смесь 2,67 г 2-фторбензонитрила, 2,05 г анилина, 2,05 г смеси фтористый калий-окись алюминия, 0,581 г 18-краун-6 и 40 мл ацетонитрила нагревали при 90oC 4 дня. Хотя и было получено 4,03 г неочищенного продукта, но после перекристаллизации из смеси гексан-этилацетат выделено только 0,30 г желаемого целевого соединения, остальное представляло собой исходный материал.
Реакцию повторяли, используя 2,31 г анилина, 2,99 г фторбензонитрила, 2,31 г смеси фтористый калий - окись алюминия, 0,650 г 18-краун-6 и 30 мл ацетонитрила. После нагревания в течение 5 дней при 95oC никакого целевого продукта не было выделено.
Пример 116. Получение 2-(3-метоксифенилтио)-бензонитрила.
Смесь 2,31 г 3-метокситиофенола, 2,00 г 2-фторбензонитрила, 4,62 г смеси фтористый калий - окись алюминия, 0,434 г 18-краун-6 и 40 мл ацетонитрила оставляли на ночь с нагреванием при 90oC. Обработка реакционной смеси давала 3,96 г (99,7%) целевого продукта в виде желтого воскоподобного твердого вещества. Перекристаллизацией из смеси гексан-этилацетат получил светложелтые кристаллы, точка плавления 77-78oC.
Пример 117. Получение 2-(3-метокси-2-пропилфенокси)-бензонитрила.
Смесь 1,00 г 3-метокси-2-пропилфенола, 0,728 г 2-фторбензонитрила, 0,16 г 18-краун-6, 1, г смеси фтористый калий-окись алюминия и 25 мг ацетонитрила нагревали 3 дня при 90oC. Обработка обычным путем давала 1,58 г (98,7%) желаемого целевого продукта.
Пример 118. Получение 2-(3-метоксифенилотио)-6-(4-метилфенилтио)-бензонитрила.
Смесь 2,5 г 3-метоксибиофенола, 2,6 г 2-фтор-(4-метилфенилтио)-бензонитрила, 1,5 г смеси фтористый калий - окись алюминия, 0,283 г 18-краун-6 и 35 мл ацетонитрила нагревали при 90oC, оставляя на ночь. Обработка обычным путем давала 3,8 г (97,7%) целевого соединения в виде коричневого порошка, точка плавления 112-114oC.
Пример 119. Получение 2-(3-метоксифенокси)-6-(1-пирролидино)-бензонитрила.
Смесь 1,5 г 2-фтор-6-(1-пирролидино)-бензонитрила, 1,0 г 3-метоксифенола, 1,0 г смеси фтористый калий-окись алюминия, 0,129 г 18-краун-6 и 259 мл ацетонитрила нагревали при 90oC. После обычной обработки выделяли 2,29 г (98,2%) целевого соединения с точкой плавления 115-116oC.
Пример 120. Получение N-метил-N-фенил-2-цианоанилина.
Смесь 2,0 г N-метиланилина, 2,26 г 2-фторбензонитрила, 2,0 г фтористого калия и окиси алюминия, 0,491 г 18-краун-6 и 40 мл ацетонитрила нагревали два дня при 90oC. Не было выделено никакого продукта.
Пример 121. Получение 4-(4-метоксифенилтио)-бензонитрила.
Смесь 1,50 г 3-метокситиофенола, 1,295 г 4-фторбензонитрила, 1,5 г фтористого калия и окиси алюминия, 0,28 г 18-краун-6 и 35 мл ацетонитрила оставляли на ночь с нагреванием при 90oC. Реакционную смесь фильтровали, промывали хлористым калием и экстрагировали эфиром, получая 2,45 г (95%) целевого соединения в виде желтых кристаллов, точка плавления 86-87oC.
Пример 122. Получение 4-(3-метоксифенокси)-бензонитрила.
Реакционная смесь 1,5 г 3-меткосифенила, 1,46 г 4-фторбензонитрила, 0,319 г 18-краун-6, 1,5 г смеси фтористый калий-окись алюминия и 25 мл ацетонитрила после нагревания при 90oC в течение 4 дней давала 2,69 г (98,9%) желаемого целевого соединения.
Пример 123. Получение 2-(3-метокси-2-пропилфенокси)-бензонитрила.
Смесь 12,23 г 3-метокси-2-пропилфенола, 8,91 г 2-фторбензонитрила, 12,23 г смеси фтористый калий-окись алюминия, 1,94 г 18-крауна-6 и 150 мл ацетонитрила нагревали 2 дня при 90oC. Реакционную смесь промывали водой и насыщенным раствором хлористого калия, экстрагировали эфиром и получали неочищенный продукт, 20,0 г этого материала очищали хроматографией, элюируя 2% и 15% этилацетатом в гексане. После хроматографии получали 14 г (71,2%) желаемого целевого соединения.
Пример 124. Если в течение 5 дней нагревать при 90oC 3,0 г N-метиланилина, 3,4 г 2-фторбензонитрила, 3,0 г смеси фтористый калий-окись алюминия, 0,9 г бромистого тетрабутиламмония, то получали только исходные материалы.
Пример 125. Если повторять пример 98, заменяя ацетонитрил диметилформамидом, то выделяли только исходные материалы.
Пример 126. Получение метилового эфира 4-(2-цианофенокси)-бензойной кислоты.
Смесь 2,00 г метилового эфира 4-оксибензойной кислоты, 1,59 г 2-фторбензонитрила, 2,00 г фтористого калия и окиси алюминия, 0,347 г 18-крауна-6 и 40 мл ацетонитрила нагревали при 90oC 8 дней. Реакционную смесь фильтровали и фильтр тщательно промывали метанолом. Фильтрат промывали последовательно раствором хлористого калия, затем 1 н. гидроокисью натрия и экстрагировали диэтиловым эфиром. Эфир выпаривали в вакууме, 3,4 г остатка очищали экспресс-хроматографией, элюируя 2-3,5% этилацетатом в гексане, и получали 83%-ный выход целевого соединения.
Пример 127. Реакцию по примеру 126 проводили, используя 2,50 г фенола, 2,50 г смеси фтористого калия и окиси алюминия, 1,99 г нитрила, 0,515 г бромистого тетрабутиламмония и 40 мл ацетонитрила. После нагревания при 90oC в течение 7 дней были выделены только исходные материалы.
Экспрессия интегринов лейкоцитов и их миграция в воспаленную кожу. Действие нового антагониста рецептора LTB.
Интегрины играют важную роль в клеточно-клеточном и клеточно-матриксном взаимодействиях. CD11b является частью β2 - интегрина Mo-1 и играет важную роль в адгезии PMN к капиллярному эндотелию, и экстравазации в ранней стадии воспаления. Хемокин LTB является активным положительным регулятором экспрессии CD11b на PMN и усиливает их миграционные свойства. Также известно, что он положительно модулирует эпидермальную пролиферацию. Проведено двойное слепое контролируемое плацебо исследование LY293111 орального антагониста рецептора LTB4. Группа из 20 здоровых мужчин-добровольцев была произвольно разделена на 3 группы лечения, которые получали плацебо, 48 мг или 200 мг лекарства ежедневно в течение 7,5 дней. Оценка с помощью проточной цитометрии CD11b на PMN крови, in vitro стимулированных LTB4, была произведена до и после лечения. Биопсию кожи брали через 24 и 72 ч после накожного применения LTB4 до и после обработки лекарством. Эффекты 13/16 экспрессии PMN, круглых клеток (cycling cells), Т-лимфоцитов, клеток Лангерганса и кератина оценивались иммуногистохимически. Лекарство хорошо переносилось. Экспрессия CD11b на PMN значительно супрессировалась в обеих группах лечения по сравнению с группой плацебо. Супрессия была наиболее сильной в группе лечения высокими дозами. В группе плацебо существенной супрессии не было. В добавление к этому на коже имело место существенное подавление воспаления и гиперпролиферации. Миграция PMN в эпидермис выражено снижалась. В меньшей степени такой же эффект наблюдался в дерме. Количество круглых клеток (cycling cells), так же как и экспрессия кератина 13/10, снижалось в обеих группах лечения. Общий воспалительный инфильтрат уменьшался, так же как и количество периваскулярных Т-лимфоцитов и клеток Лангерганса. LY293111 оказался мощным ингибитором воспаления и пролиферации в коже, вызванных LTB4. Этот эффект, возможно, вызывается изменением экспрессии интегрина PMN.
CD11b является частью β2 -интегрина Mac-1 и играет важную роль в адгезии нейтрофилов. Лейкотриен B4 является активным положительным регулятором экспрессии CD11b на нейтрофилах и улучшает их миграционные свойства. Также известно, что он положительно модулирует эпидермальную пролиферацию. Мы провели контролируемое плацебо исследование LY293111, орального антагониста рецептора лейкотриена B4. Двадцать здоровых мужчин-добровольцев были произвольно разбиты на 3 группы лечения, получавших плацебо, 48 мг или 200 мг лекарства дважды в день в течение 10,5 дней. До и после лечения проводилась оценка с помощью проточной цитометрии CD11b на нейтрофилах периферической крови, стимулированных лейкотриеном B4 in vitro. В дополнение к этому, после накожного применения лейкотриена B4 проводилась биопсия кожи через 24 и 72 часа, до и после лечения. Действие на кожу оценивалось иммуногистохимически с использованием различных маркеров. Все наблюдавшиеся эффекты находились в зависимости от дозы. Положительная регуляция (up-regulation) CD11b на нейтрофилы периферической крови существенно подавлялась в обеих группах лечения по сравнению с группой плацебо, в коже отмечалась существенная супрессия воспаления и гиперпролиферации. Общий воспалительный инфильтрат уменьшался. Наблюдалось выраженное ингибирование миграции нейтрофилов в эпидермис. Сходный, но более слабый ответ наблюдался в дерме. Количество круглых клеток (cycling cells), также же как экспрессия супрабазального кератина 13/16, снижалось в обеих группах лечения. LY293111 оказался модным ингибитором кожного воспаления и гиперпролиферации, вызванных лейкотриеном B4. Мощный противовоспалительный эффект in vivo и тот факт, что это соединение не вызвало клинически значимых побочных эффектов, делает его интересным в будущем лекарством для лечения воспалительных состояний с преобладанием нейтрофилов.
Влияние антагониста рецептора лейкотриена B4 LY293111 на воспалительный ответ на провокацию аллергеном у больных атопической астмой.
Было показано, что лейкотриен B4 (LTB4) является хемоаттрактантом нейтрофилов и стимулом эозинофилов in vitro. В данном исследовании изучен эффект антагониста рецептора LTB4 LY293111 с использованием провокации аллергеном (allergen challenge, AC) в качестве модели астмы у 12 мужчин, страдающих атопической астмой, в двойной слепой контролируемой плацебо перекрестной пробе. В течение недели до этого испытуемым вводили дозу гистамина, подвергшегося PC20 часом раньше, через 3 ч после AC. ОФВ1 (FEV1) контролировался во время раннего астматического ответа (early asthmatic responce, EAR) и LAR (позднего астматического ответа). Каждому индивидуально проводилась бронхоскопия и BAl через 24 ч после АС. Действие плацебо и LY293111 на функцию легких, EAR или LAR, или на среднее геометрическое Log PC20 отсутствовало (плацебо, 0,68 [pre аллерген] 0.35 мг/мл [post]; LY293111, 0,71 мг/мл [pre] , 0.37 мг/мл [post]). В BAL были определенные изменения, согласующиеся с противовоспалительным эффектом. Последовавшее за BAL лечение LY293111 сократило уровень LTB4, C4, E4, эластазы, миелопероксидазы и цитокина IL8. Это достигало уровня значимости для LTB4 (средние концентрации BAL; плацебо 4.61±1.19 пг/мл, LY293111 2.222±0.222 пг/мл) и миелопероксидазы (p<0.05). Различия в уровнях цитокинов IL4,5 или GM-CSF отсутствовали. Анализ дифференциальных подсчетов клеток BAL выявил значительное снижение числа нейтрофилов (p<0.023). Изменение в числе эозинофилов или лимфоцитов отсутствовали. Данные результаты показывают влияние на миграцию нейтрофилов в воздухоносные пути вследствие АС. Несмотря на этот противовоспалительный эффект, измеримой физиологической пользы не было, и это ставит под вопрос роль нейтрофилов в патофизиологии экспериментального АС.

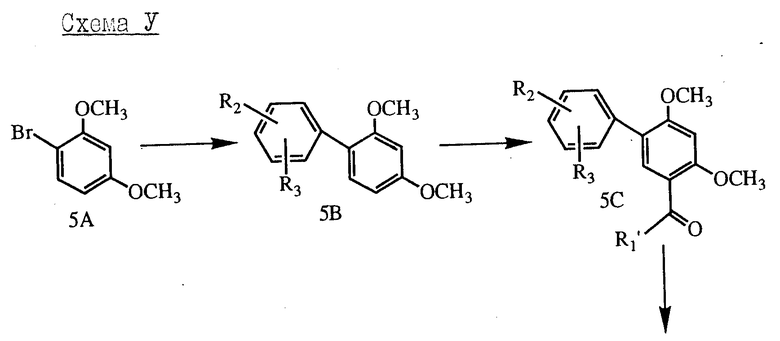

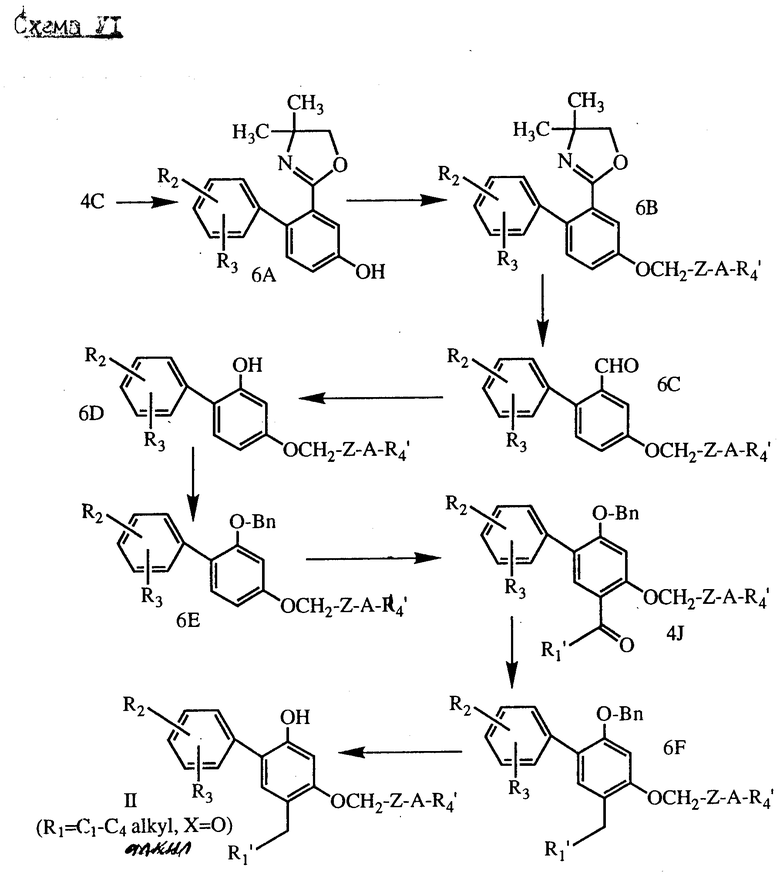





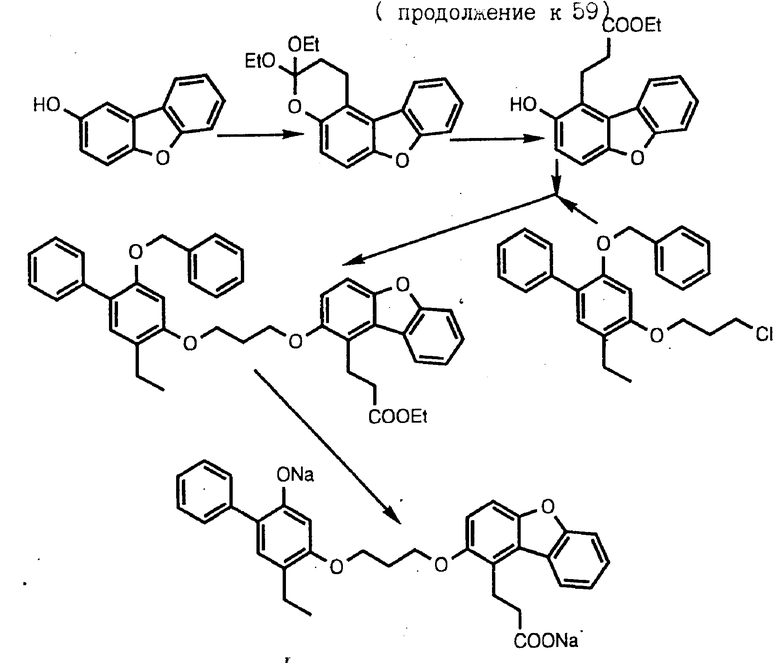
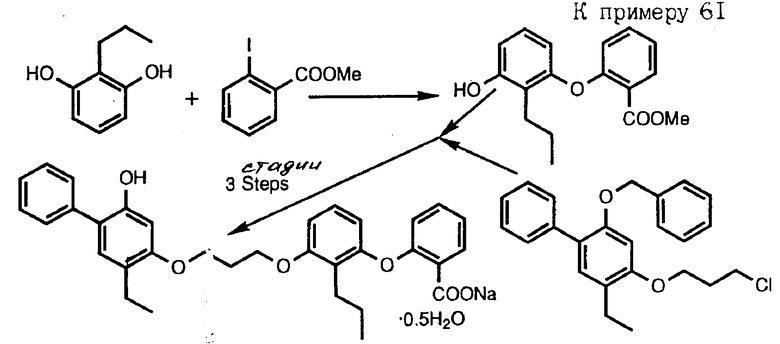


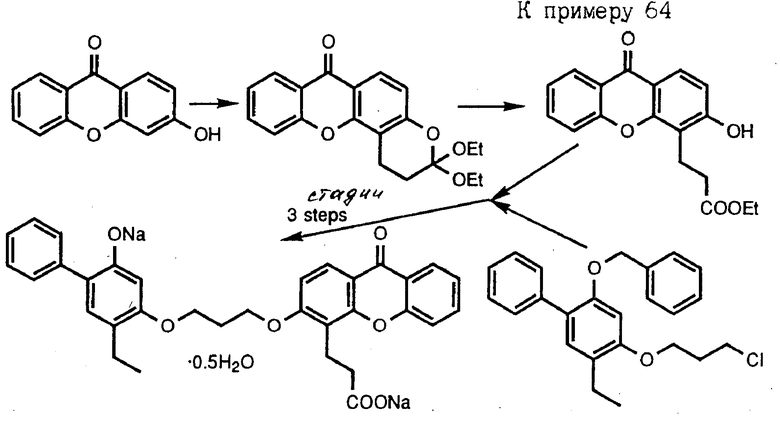


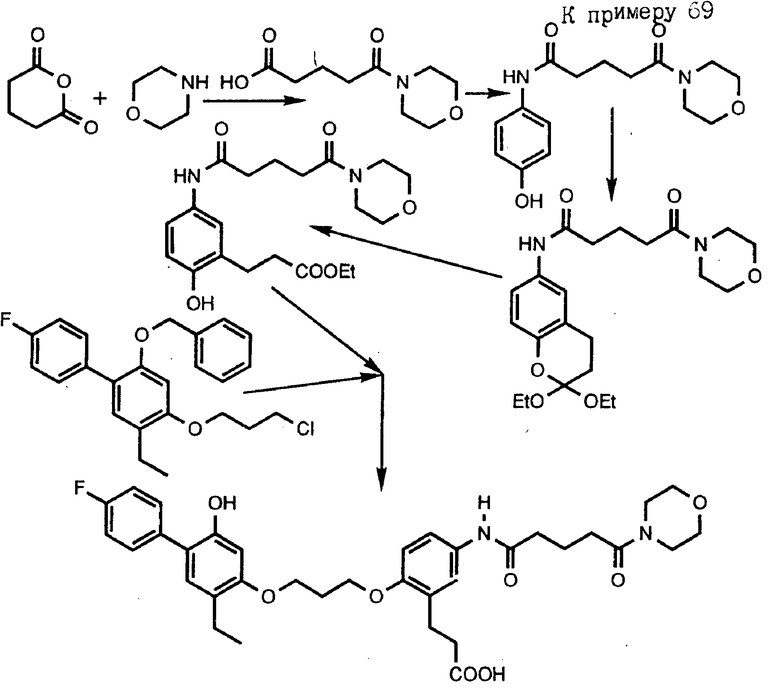



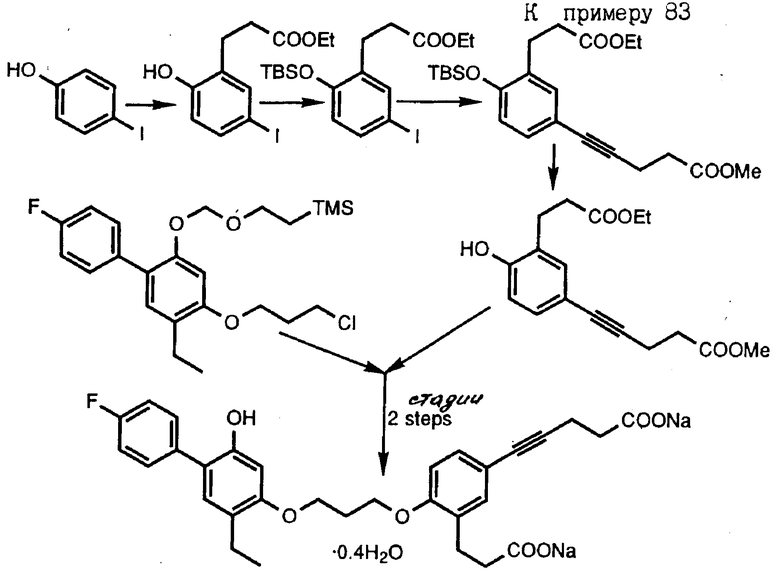


Антагонисты, имеющие замещенные фенилфенольные или замещенные фенольно-дифенильные структуры и их различные производные, являются специфическими антагонистами лейкотриенов. Описано их строение, использование и синтез. Раскрыты также фармацевтические композиции, используемые для лечения заболеваний и состояний, характеризующихся избыточным освобождением лейкотриена B4, одного из метаболитов арахидоновой кислоты. Первичные структуры антагонистов лейкотриена B4 представлены формулой
в которой R1, R2, R3, R4, X, Y, Z и A подробно охарактеризованы в описании к заявке на изобретение. Изобретение предусматривает также способ получения промежуточных соединений формулы XI
в которой R21, R22 и R23 и T имеют значения, указанные в заявке. 3 с. и 19 з.п. ф-лы, 1 табл.
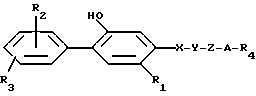
или их фармацевтически приемлемые аддитивные соли основания,
где R1 - C1 - C5-алкил, C2 - C5-алкенил, C2 - C5-алкинил, C1 - C4-алкокси, (C1 - C4-алкил)тио, галоид или R2 - замещенный фенил;
R2 и R3 каждый независимо - водород, галоид, гидрокси, C1 - C4-алкил, C1 - C4-алкокси, (C1 - C4-алкил)-S(O)q, трифторметил или ди-(C1 - C3-алкил)амино;
Х - -О-, -S-, -C(=О) или -CН2-;
Y - -О- или -CН2-, или, когда они взяты вместе, -Х - Y- представляет -CН=CН- или -C ≡ C-;
Z - C1 - C1 0-алкилиденил с прямой или разветвленной цепью;
A - связь, -О-, -S-, -CН=CН- или CRaRb, где Ra и Rb представляют каждый независимо водород, C1 - C5-алкил, или R7-замещенный фенил, или взятые вместе с атомом углерода, к которому они присоединены, образуют C4 - C8-циклоалкильное кольцо;
R4 представляет R6



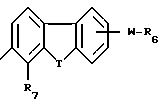
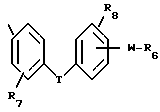

где R6 - -CООН, 5-тетразолил, -CON(R9)2 или -CONHSO2R1 0;
R7 - водород, C1 - C4-алкил, C2 - C5-алкенил, C2 - C5-алкинил, бензил, метокси, -W-R6, -T-G-R6, (C1 - C4-алкил)-Т- (C1 - C4-алкилиденил)-О- или гидрокси;
R8 - водород или галоид;
R9 независимо - водород, фенил, C1 - C4-алкил или взятые вместе с атомом азота образуют морфолино-, пиперидино,- пиперазино- или пирролидиногруппу;
R1 0 - C1 - C4-алкил или фенил;
R1 1 представляет R2, -W-R6 или -T-G-R6;
W - связь или двухвалентный углеводородный радикал с прямой или разветвленной цепью, содержащий 1 - 8 атомов углерода;
G - двухвалентный углеводородный радикал с прямой или разветвленной цепью, содержащий 1 - 8 атомов углерода;
Т - связь, -CН2-, -О-, -NH-, NHCO-, -C(=O)- или -S(O)
K - -C(=О)- или -CН(ОН)-;
q = 0, 1 или 2 независимо;
p = 0 или 1;
t = 0 или 1,
при условии, когда Х - -О- или -S-, Y не является -О-; Z и A оба не являются связью, когда Y - -О-; когда A - -О- или -S-, R4 не является R6; когда A - -О- или -S- и Z - связь, Y не является -О-; Z не является связью, когда Y - -О- и W не является связью, когда p = О.
и его фармацевтически приемлемые аддитивные соли с основанием.
Т - -О- или -S-.
где R6 - W-CООН.
или их фармацевтически приемлемые аддитивные соли основания, где
R1 - C1 - C5-алкил, C2 - C5-алкенил, C2 - C5-алкинил, C1 - C4-алкокси, (C1 - C4-алкил)тио, галоид или R2 - замещенный фенил;
R2 и R3 - каждый независимо водород, галоид, гидрокси, C1 - C4-алкил, C1 - C4-алкокси, (C1 - C4-алкил)-S(O)q, трифторметил, или ди-(C1 - C3-алкил)амино;
Х - -О-, -S-, -C(=О) или -CН2-;
Y - -О- или -CН2-, или когда они взяты вместе, -X-Y- представляет -CН= CН- или -C ≡ C- ;
Z - C1 - C1 0-алкилиденил с прямой или разветвленной цепью;
A - связь, -О-, S-, -CН=CН- или CRaRb-, где Ra и Rb представляют каждый независимо водород, C1 - C5-алкил или R7-замещенный фенил, или взятые вместе с атомом углерода, к которому они присоединены, образуют C4 - C8-циклоалкильное кольцо; R'4 представляет R'6


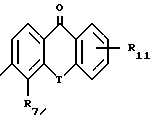


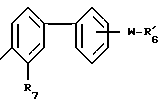
где
R'6 независимо -CООН, 5-тетразолил, CON(R9)2, -CONSO2R1 0, -COOR или CN;
R7 - водород, C1 - C4-алкил, C2 - C5-алкенил, C2 - C5-алкинил, бензил, метокси, -W-R'6, -T-G-R'6, (C1 - C4-алкил)-Т(C1 - C4-алкилиденил)-О- или гидрокси;
R8 - водород или галоид;
R9 независимо - водород, фенил, C1 - C4-алкил или взятые вместе с атомом азота образуют морфолино-, пиперидино-, пиперазино- или пирролидиногруппу;
R1 0 - C1 - C4-алкил или фенил;
R1 1 - R2, W-R'6, T-G-R'6
W - связь или двухвалентный углеводородный радикал с прямой или разветвленной цепью, содержащий 1 - 8 атомов углерода;
G - двухвалентный углеводородный радикал с прямой или разветвленной цепью, содержащий 1 - 8 атомов углерода;
Т - связь, -CH2-, -O-, -NH-, -NHCO-, -C(=O)- или -S(O)q;
K - -C(=О)- или -CН(ОН)-;
q = 0, 1 или 2 независимо;
p = О или 1;
t = О или 1,
при условии, когда Х - -О- или -S-, Y не является -О-; Z и A оба не являются связью, когда Y - -О-; когда A - -О или -S-, R'4 не является R'6, когда A - -О- или -S- и Z - связь, Y не является -О- и W не является связью, когда p = О, и при условии, что по крайней мере один R'6 - -COOR или CN.
16. Cоединение по п. 15, которое представляет собой C1 - C6-алкиловый эфир 2-[2-пропил-3-[3-[2- этил-4-(4-фторфенил)-5-гидроксифенокси]-пропокси] фенокси]-бензойной кислоты.
Т - -О- или -S-.
R'6 - -W-COOH.
| US, патент, 4816491, кл.C 07C 43/23, 1989. |

