В изобретении предлагаются новые нейтральные ингибиторы эндопептидазы и способы их использования. Соединения согласно изобретению полезны, например, в качестве сердечно-сосудистых агентов общей формулы I
R3-S-CH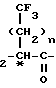 NH-X где Х представляет собой
NH-X где Х представляет собой
-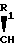 -COOH, -(CH2)2-COOH, -(CH2)
-COOH, -(CH2)2-COOH, -(CH2)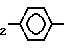 OH,
OH,
-(CH2) , -
, - ,
,
-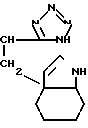 или
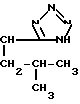
или 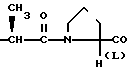 OH- , R1 представляет собой прямой или разветвленный низший алкил, содеpжащий 1-4 атома углерода
OH- , R1 представляет собой прямой или разветвленный низший алкил, содеpжащий 1-4 атома углерода
-CH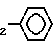 , -CH
, -CH OH,
OH,
-CH OH , -CH
OH , -CH , -CH
, -CH ,
,
-CH2-COOH или -CH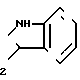 ; R3 представляет собой водород; n = 0.
; R3 представляет собой водород; n = 0.
Это изобретение в самом широком аспекте касается нейтральных ингибиторов эндопептидазы формулы I и способов лечения застойной сердечной недостаточности, пониженного кровяного давления и формирования диуреза и мочеиспускания путем введения фармацевтической композиции, содержащей эти ингибиторы.
Неожиданно было обнаружено, что трифторметилзамещенные соединения формулы I значительно более активны в качестве нейтральных ингибиторов эндопептидазы, чем меркаптан и меркаптоалканоильные соединения, не включающие трифторметила.
Термин низший алкил, использованный для определения различных символов, относится к радикалам с прямолинейными или разветвленными цепями, имеющими до 7 атомов углерода, кроме специально заявленных. Предпочтительными низшими алкильными группами являются радикалы с прямолинейными или разветвленными цепями, содержащие до 4 атомов углерода.
Соединения формулы I могут быть получены путем взаимодействия карбоновой кислоты формулы II:
R3-S-CH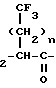 OH с аминопромежуточным соединением формулы III:
OH с аминопромежуточным соединением формулы III:
H2N - X
Промежуточное соединение формулы III может быть использовано в качестве свободного основания или соли гидрохлорида. Карбоновая кислота формулы II является предпочтительной, легко превращаемой в активированную форму, например, в хлорангидрид кислоты, смешанный ангидрид и т.д.
Предпочтительно, чтобы аминопромежуточное соединение формулы III вначале обрабатывалось бис(триметилсилил) трифторацетамидом или бис(триметилсилил)ацетамидом в апротоновом растворителе, например, тетрагидрофуране или ацетонитриле с последующим добавлением соединения формулы II, например, хлорангидрида кислоты.
Альтернативно реакцию предпочтительно проводить в присутствии органического основания, например, триэтиламина. В качестве и реагента, и основания могут быть использованы два эквивалента аминопромежуточного соединения формулы III.
В тех случаях, когда аминопромежуточное соединение III не содержит свободной карбоновой кислоты, реакция взаимодействия кислоты II и амина III может быть проведена с растворимым в воде карбодиимидом, например, [1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид].
Полученные продукты, где Х содержит -COR2, который является сложным эфиром, могут быть гидролизованы путем обработки основанием, например, гидроокисью натрия для получения соединения, где R2 является гидроксигруппой и R3 - водород, поскольку имеет место сопутствующий гидролиз существующей ацетилтиогруппы. Производные S-ацила формулы I могут быть обработаны основанием, например, гидрохлоридом аммония, гидроксиамином, гидроксилинатрием и т.п. для получения производных меркаптана (т.е. соединения формулы I, где R3 = H) известными способами.
Предпочтительными соединениями формулы I в настоящем изобретении являются соединения, в которых Х представляет
- H
H  -
-
-(CH2)2-COOH, - -
- H, -(CH2)
H, -(CH2)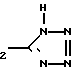
-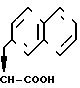 или -
или - ; R3 = H и n = 0
; R3 = H и n = 0
Как показано выше, соединения формулы I содержат асимметричные центры, как указано знаком в формуле I. Дополнительный асимметричный центр присутствует в эфирных продуктах.
Таким образом, соединения формулы I могут существовать в зеркальной изомерной или диастереомерной форме или в виде их смеси. В указанных процессах в качестве исходных материалов могут использоваться рацематы, изомеры или диастереомеры. При получении диастереомерных продуктов они могут выделяться либо традиционными способами, либо хроматографией, либо фракционной кристаллизацией.
Соединения формулы I ингибируют активность нейтральной эндопептидазы (ЕС 3.4.24.11) и мембраносвязанной цинковой металлопептидазы, обнаруженной во многих тканях, включая мозг и почки. Нейтральная эндопептидаза гидролизует пептидные связи, которые находятся на аминном конце гидрофобных аминокислотных остатков.
Не ограничивая объема изобретения до специальной теории или механизма взаимодействия, заявители указывают, что ингибирование нейтральной эндопептидазы, по их мнению, снижает инактивацию экзогенно-введенных или эндогенных предсердных натрийуретных пептидов. Таким образом, соединения формулы I полезны при лечении гипертензии (артериальной гипертонии), сердечной недостаточности, почечной недостаточности или цирроза печени.
Диурез, натрийурез и снижение кровяного давления происходят в организме млекопитающего, например, человека, путем введения от около 1 до около 100 мг на кг массы тела в день, предпочтительно от около 1 до около 50 на кг массы тела в день одного или более ингибиторов нейтральной эндопептидазы формулы I или фармацевтически приемлемых солей. Ингибиторы нейтральной эндопептидазы формулы I предпочтительно вводятся перрорально, но могут вводиться парентерально, например, подкожно, внутримышечно и внутривенно. Ежедневная доза может вводиться единовременно или 2 или 4 раза в день.
Ингибиторы нейтральной эндопептидазы формулы I могут вводиться в комбинации с другими агентами, снижающими кровяное давление. Например, ингибиторы нейтральной эндопептидазы формулы I могут быть объединены для двойного введения с ингибитором ангиотензивного трансформируемого энзима АТЭ, таким как каптоприл, эофеноприл, фозиноприл, аналаприл, лизинопропил и т.д. Такие комбинации используют при массовом отношении пептидазы к АТЭ от около 1:10 до около 10:1.
Нейтральные ингибиторы эндопептидазы формулы I могут вводиться в комбинации с человеческим ANF (антинуклеарный фактор) 99-126. Такая комбинация содержала бы ингибитор формулы I в количестве 1-100 мг/кг массы тела и человеческий ANF 99-126 в количестве 0,001-0,1 мг на кг массы.
Нейтральные ингибиторы эндопептидазы формулы I или их формацевтически приемлемые соли могут вводиться также млекопитающим, например, человеку для ингибирования разложения эндогенной опиодид пентапептидазы [Met5-] энкефалин [Tyr-Gly-Gly-Phe-Met] и [Leu5] энкефалин [Tyr-Gly-Gly-Phe-Leu] в головном мозге или в периферических тканях.
По этой причине соединения, согласно изобретению, полезны в терапии, например, астмы, воспалений, боли, эпилепсии, эффективных расстройств, слабоумия и старческой спутанности сознания, ожирения и желудочно-кишечных расстройств (в частности, диареи и синдрома легко возбудимого кишечника), модуляции секреции желудочной кислоты и лечения гиперренинаемии и лейкемии. Соединения согласно изобретению и их фармацевтически приемлемые соли могут вводиться пациентам перрорально или парентерально в эффективном количестве при ежедневной дозе в количестве от 0,1 до 25 мг соединения на кг массы пациента. Введение может быть одноразовым или в 2-4 приема в день.
Кроме того, соединения формулы I, где Х представляет -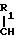 -COOH , также проявляют свойство ингибирования активности ангиотензивного измененного энзима. Поэтому эти соединения формулы I являются двойными ингибиторами и могут быть использованы в терапии, когда известна полезность ингибиторов ангиотензивного измененного энзима, например каптоприла [1]. Сюда может относиться сердечно-сосудистая терапия, например лечение гипертензии, сердечной недостаточности, снижение пред- и постишемической аритмии миокарда и фибрилляции и т.д., а также улучшение познавательной функции, лечение депрессии и тревоги и т.д.
-COOH , также проявляют свойство ингибирования активности ангиотензивного измененного энзима. Поэтому эти соединения формулы I являются двойными ингибиторами и могут быть использованы в терапии, когда известна полезность ингибиторов ангиотензивного измененного энзима, например каптоприла [1]. Сюда может относиться сердечно-сосудистая терапия, например лечение гипертензии, сердечной недостаточности, снижение пред- и постишемической аритмии миокарда и фибрилляции и т.д., а также улучшение познавательной функции, лечение депрессии и тревоги и т.д.
Ингибиторы формулы I и другие фармацевтически приемлемые ингредиенты могут быть объединены для описанного фармацевтического использования. Приемлемые композиции для перрорального введения могут быть в виде таблеток, капсул, эликсиров, а для парентерального введения - в виде стерильных растворов и суспензий. Около 10-50 мг активного ингредиента соединяется с физиологически приемлемым носителем, наполнителем, связующим, консервантом, стабилизатором, корригентом и т.д. в единичной дозе, причем приемлемым в фармацевтической практике способом.
Ниже приведены примеры, иллюстрирующие изобретение, температура указана по стоградусной шкале.
П р и м е р 1. 1-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-аланин.
А. N-[2-[ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-аланин.
Аланин в количестве 445 мг, 5 ммоля суспендировался в 5 мл метиленхлорида, добавлялось 4 мл бис(триметилсилил)трифторацетамида и полученный раствор перемешивался в течение 3 ч при комнатной температуре. Добавлялось 4 мл диметилформамида и смесь перемешивалась всю ночь. Весь материал переходил в раствор. Затем добавлялся 1 г (4,26 ммоля) 2-трифторметил-3-ацетилтиопропионилхлорида и 2 мл тетрагидрофурана и смесь перемешивалась всю ночь при комнатной температуре. Затем реакционная смесь концентрировалась на вращающемся испарителе. Остаток распределялся между этилацетатом и 5%-ным тисульфатом калия. Этилацетатный раствор промывался 5%-ным бисульфатом калия и рассолом и концентрировался на 400 мл колонне Мерке с силикагелем с использованием смеси 40:1:1 метиленхлорида:метанола:уксусной кислоты в качестве элюента, в результате получали соединение А, указанное в названии, в виде желтого масла в количестве 960 мг. При тонкослойной хроматографии Rf = 0,45 (20:1:1 метиленхлорид:метанол:уксусная кислота).
В. 1-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-аланин.
Соединение А, указанное в названии, в количестве 960 мг (3,3 ммоля) перемешивалось при 0оС в атмосфере аргона с 2,2 мл концентрированного гидрохлорида аммония и 4,7 мл воды в течение 10 мин, а затем добавлялся 5%-ный бисульфат калия (100 мл).
Полученный раствор экстрагировался этилацетатом. Общий этилацетатный экстракт промывался 5% -ным тисульфатом калия и рассолом, высушивался над сульфатом магния и концентрировался до получения масла, которое затем хроматографировалось на 200 мл колонне Мерка с силикагелем с использованием смеси 40: 1: 1 метиленхлорид:метанол:уксусная кислота в качестве элюента с получением смеси диастереомеров соединения А, указанного в названии, в виде белого твердого продукта в количестве 720 мг с точкой плавления 106-107оС. При тонкослойной хроматографии Rf - 0,40 (20:1:1 метиленхлорид:метанол:уксусная кислота), [α]D = -27,1o (C = 0,73, метанол).
Вычислено, %: C 34,28; H 4,11; N 5,71; F 23,24; S 13,07; SH 13,49.
C7H10.
Найдено, %: C 34,52; H 4,25; N 5,76; F 23,20; S 12,76; SH 13,74.
П р и м е р 2. N-[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-фенилала-нин, изомер А.
Перемешанная суспензия L-фенилаланина в количестве 7,60 г (46 ммоля) в 45 мл сухого ацетонитрила охлаждалась в атмосфере аргона до 0оС и к ней добавлялось 24,7 мл, 82 ммолей бис(триметилоилил)трифторацетамида. Реакционная смесь перемешивалась и медленно нагревалась до комнатной температуры.
После 4 ч практически вся аминокислота переходила в раствор, который был светло-желтого цвета. По каплям в течение 45 мин при 5оС к раствору добавлялся растворенный в 10 мл ацетонитрила 2-трифторметил-3-ацетилтиопропионилхлорид в количестве 4,69 г (20 ммоля), после чего реакционная смесь нагревалась до комнатной температуры и перемешивалась всю ночь. Затем смесь выпаривалась при разряжении с получением светло-желтого масла. Масло распределялось между водой (75 мл) и этилацетатом (100 мл). Вся смесь отфильтровывалась для удаления белого осадка (фенилаланина) и органический слой отделялся. Водная фаза еще раз экстрагировалась 50 мл этилацетата. Объединенный этилацетатный экстракт промывался рассолом, высушивался над безводным сульфатом магния и выпаривался с получением неочищенной смеси диастереомеров в виде светло-желтого порошка. 2 г продукта подвергалось тонкослойной хроматографии на силикагеле (125:1 силикагель:соединение) с использованием в качестве элюента смеси этилацетат:гексаны:уксусная кислота при соотношении 100:100:1, при этом выход составил: 0,370 г быстроэлюирующего диастереомера (изомер В).
Оставшиеся 18,1 г неочищенного материала подвергались тонкой хроматографии аналогичным образом, при этом выход составил: 2,67 г изомера А и 2,92 г изомера В. Общий выход изомера А составил 3,04 г в виде белого кристаллического порошка с точкой плавления 183-185оС, при тонкослойной хроматографии Rf = 0,30 (200:100:1 этилацетат:гексаны:уксусная кислота). [α]D = -98,8o (C = 1,00, метанол).
Вычислено, %: С 49,58; H 4,44; N 3,86; F 15,69; S 8,82.
C15H16F3NO4S.
Найдено, %: C 49,63; H 4,35; N 3,70; F 15,35; S 8,81.
П р и м е р 3. N-[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-фенилала- нин, изомер В.
Неочищенная смесь диастереомеров, полученных в примере 3, в количестве 20,1 г повергалась тонкослойной хроматографии на силикагеле (125:1 силикагель:соединение) с использованием в качестве элюента смеси:этилацетат:гексаны: уксусная кислота при соотношении 100:100:1 с получением медленно элюирующего изомера (изомер В) в количестве 0,38 г и 2,92 г.
Указанные 2,92 г измельчались в порошок, посредством гексанов, содержащих 10% этилацетата, затем белый кристаллический порошок отфильтровывался с получением 0,72 г материала. Фильтрат концентрировался с получением 2,1 г материала, который подвергался тонкослойной хроматографии, как указано выше, с получением приблизительно 1,3 г материала, который соединялся с 0,38 г и 0,72 г и выпаривался из метанола с получением 2,20 г изомера В в виде белого кристаллического порошка с точкой плавления 134-136оС, при тонкослойной хроматографии Rf = 0,14 (200:100:1 этилацетат:гексаны:уксусная кислота), [α]D = +118o (c = 1,00, метанол).
Вычислено, %: C 49,58; H 4,44; N 3,86; F 15,69; S 8,82.
Найдено, %: C 49,58; H 4,33; N 3,78; F 15,44; S 8,54.
В примерах 5 и 6 описаны способы получения единичных диастереомеров, изомеров А и В, относящихся к смеси диастереомеров, указанных в примере 1.
П р и м е р 4. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-фенилаланин, изомер А.
Суспензия соединения изомера А, указанного в названии, например из примера 3, в количестве 500 мг, 1,37 ммоля в 0,9 мл концентрированного гидрохлорида аммония и 2 мл воды перемешивалась при комнатной температуре в атмосфере аргона в течение 5-6 мин, в течение этого времени получали прозрачный раствор. Затем раствор обрабатывался 50 мл 5%-ного раствора гидросульфата калия и полученный водный раствор экстрагировался четырьмя 25 мл порциями этилацетата. Весь этилацетатный экстракт высушивался над безводным сульфатом магния и выпаривался с получением 0,43 г неочищенного продукта.
Тонкослойной хроматографией на силикагеле (125:1 силикагель:соединение) с CH2Cl2:CH3OH:HOA с при соотношении 40:1:1 получалось 370 мг продукта.
Аналогично другие 2,3 г (6,3 ммоля) изомера А соединения из примера 3 диацетилировались и очищались тонкослойной хроматографией с выходом 2,00 г продукта. Эти два продукта объединялись и полученный твердый продукт измельчался в порошок при помощи гексанов и нескольких капель эфира. Растворитель сливался, а твердый продукт высушивался в вакууме с получением 1,79 г изомера в виде белого кристаллического порошка с т.пл. 133-134,5оС, при тонкослойной хроматографии Rf = 0,30 (40:1:1 метиленхлорид:метанол:уксусная кислота): [α]D = -8,1o (C = 1,00, метанол).
Вычислено, %: C 48,59; H 4,39; N 4,36; F 17,74; S 9,98; SH 10,29.
C13H14F3NO3S.
Найдено, %: C 48,47; H 4,26; N 4,30; F 17,49; S 10,34; SH 10,57.
П р и м е р 5. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-фенилаланин, изомер В.
Суспензия указанного в названии изомера В соединения из примера 4 в количестве 2,05 г, 5,64 ммоля в 3,6 мл концентрированного гидрохлорида аммония и 8,0 мл воды перемешивалась в атмосфере аргона в течение 7-10 мин. Прозрачный светло-желтый раствор подкислялся водным 5% гидросульфатом калия до рН 2 и экстрагировался четырьмя порциями по 50 мл этилацетата. Весь органический экстракт промывался 50 мл рассола, высушивался над безводным сульфатом магния и растворитель удалялся при пониженном давлении с получением 1,87 г желтого твердого продукта на силикагеле (130:1 силикагель:соединение) с использованием в качестве элюента CH2Cl2:CH3OH:HOA при соотношении 50: 1:1 получалось 1,7 г изомера В в виде белого твердого с т.пл. 148-150оС при тонкослойной хроматографии Rf = 0,29 (40:1:1 метиленхлорида:метанол:уксусная кислота), [α]D = +49,2o (C = 1,00, метанол).
Вычислено, %: C 48,59; H 4,39; N 4,36; F 17,74; S 9,98; SH 10,29.
C13H14F3NO3S.
Найдено, %: C 48,43; H 4,10; N 4,21; F 17,42; S 10,27; SH10,09.
П р и м е р 6. N-[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-триптофан, изомер А.
Перемешанная суспензия L-триптофана (4,8 г, 20 ммоля) в 50 мл сухого ацетонитрила в атмосфере аргона охлаждалась до 0-5оС и затем к ней добавлялся бис(триметилсилил)трифторацетамид (5,3 мл, 20 ммол). Реакционная смесь перемешивалась и нагревалась до комнатной температуры. Через 3 ч раствор охлаждался до 5оС и через 45 мин постепенно добавлялся раствор 2-трифторметил-3-ацетилтиопропионилхло-рида в количестве 4,69 г, 20 ммоля в 7 мл ацетонитрила. Реакционная смесь перемешивалась всю ночь, постепенно нагреваясь до комнатной температуры. Растворитель выпаривался и желтый маслянистый осадок распределялся между этилацетатом (50 мл) и водой (35 мл). Затем водная фаза экстрагировалась этилацетатом (3х50 мл) и объединенный этилацетатный экстракт промывался рассолом, высушивался MgSO4 и выпаривался с получением желтого маслянистого остатка в количестве 14,15 г. Часть продукта в количестве 12,25 г подвергалась тонкослойной хроматографии на силикагеле (120:1 силикагель:соединение) с использованием смеси этилацетат:гексаны:уксусная кислота при соотношении 150:100:1 с получением быстроэлюирующего диастереомера (изомер А) в количестве 1,9 г 4,2 г обоих диастереомеров и 1,5 г медленноэлюирующего диастереомера (изомер В). При дальнейшем элюировании смесью этилацетат:гексаны:уксусная кислота при соотношении 200:100:1 дополнительно получалось 170 мг изомера В. Смешанные фракции еще раз подвергались хроматографии при тех же условиях с получением 1,65 г изомера А и 1,76 г изомера В.
1,9 г и 1,65 г изомера А объединялись и перекристаллизовывались из толуола с получением 2,8 г не совсем белого кристаллического порошка. Дополнительные 360 мг изомера А) из тонкослойной хроматографии 1,5 г неочищенного продукта), перекристаллизовывались из толуола с получением 260 г белого кристаллического порошка; общий выход изомера А составил 3,06 г, точка плавления 140-143оС при тонкослойной хроматографии Rf = 0,46 (300:100:1 этилацетат:гексаны:уксусная кислота) [α]D = -96,0o (C = 1,00, метанол).
Вычислено, %: C 50,74; H 4,26; N 6,96; F 14,16; S 7,97.
C17H17F3N2O4S.
Найдено, %: C 50,47; H 4,04; N 6,86; F 13,82; S 8,18.
П р и м е р 7. N-[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-триптофан, изомер В.
3,62 г объединенного изомера В из примера 7 перекристаллизовывалось из толуола с получением 2,56 г изомера В в виде порошка белого цвета с точкой плавления 148-151оС, при тонкослойной хроматографии Rf = 0,35 (300:100:1 этилацетат:гексаны:уксусная кислота), [α]D = +110,6o (C = 1,00, метанол).
Вычислено, %: С 50,51; H 4,29; N 6,93; F 14,10; S 7,93.
C17H17F3N2O4S˙ 0,1H2O.
Найдено, %: C 50,67; H 4,08; N 6,62; F 13,72; S 7,82.
П р и м е р 8. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-триптофан, изомер А.
Суспензия продукта из примера 7 в количестве 2,6 г, 6,5 ммоля в 4,25 мл концентрированной гидроокиси аммония и 9,3 мл воды перемешивались при комнатной температуре в атмосфере аргона в течение 12-15 мин. Полученный раствор подкислялся 200 мл 5% бисульфата калия и экстрагировался этилацетатом (5х50 мл). Весь этилацетатный экстракт промывался рассолом, высушивался (MgSO4) и выпаривался с получением 2,4 г светло-желтого порошка. Этот материал объединялся с дополнительным количество 80 мг соединения и подвергался тонкослойной хроматографии на 300 г силикагеля с использованием смеси метиленхлорид: метанол:уксусная кислота при соотношении 40:1:1 с получением 2,3 г белого твердого продукта. Этот твердый продукт превращался в порошок при помощи толуола, содержащего приблизительно 5% этилацетата, при этом при фильтрации собиралось 1,25 г белого кристаллического порошка, фильтрат выпаривался до полного высушивания и остаток измельчался аналогичным образом с получением дополнительного количества 560 г материала, который объединялся с первоначальным количеством 1,25 г с получением 1,81 г изомера А в виде белого кристаллического порошка с температурой плавления 148,5-150,5оС, при тонкослойной хроматографии Rf = 0,48 (300:100:1 этилацетат:гексаны:уксусная кислота), [α]D = -10,3o (C = 1,00, метанол).
Вычислено, %: C 49,99; H 4,20; N 7,78; F 15,82; S 8,90; SН 9,18.
C15H15F3N2O3S.
Найдено, %: С 49,98; H 3,97; N 7,54; F 15,42; S 9,12; SH 9,38.
П р и м е р 9. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-триптофан, изомер В.
Суспензия продукта из примера 8 в количестве 2,41 г 5,99 ммоля и 3,75 мл концентрированного гидрохлорида аммония и 8,25 мл воды перемешивались при комнатной температуре и атмосфере аргона в течение 12-15 мин. Полученный раствор подкислялся 200 мл 5%-ного бисульфата калия и экстрагировался этилацетатом (5х50 мл). Объединенный этилацетатный экстракт промывался рассолом, высушивался (MgSO4) и выпаривался с получением 2,3 г желтого смолистого твердого вещества. Проводилась тонкослойная хроматография на 315 г силикагеля с использованием смеси метиленхлорид:метанол:уксусная кислота при соотношении 40:1:1 с получением 2,1 г светло-желтого стекловидного остатка, который измельчался в порошок посредством толуола, содержащего приблизительно 5% этилацетата с получением 1,55 г изомера В, указанного в названии в виде белого кристаллического порошка с т.пл. 144-147оС, при тонкослойной хроматографии Rf = 0,44 (300:100:1 этилацетат:гексаны:уксусная кислота [α]D = +35,8o (C = 1,00, метанол).
Вычислено, %: C 49,74; H 4,23; N 7,74; F 15,74; S 8,85; SH 9,13.
C15H15F3N2O3S˙0,1H2O.
Найдено, %: C 50,05; H 3,99; N 7,45; F 15,36; S 9,07; SH 9,34.
П р и м е р 10. 3-[[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил] амино]-пропио- новая кислота.
Перемешанная суспензия β -аланина (1,34 г, 15 ммоля) в 30 мл сухого ацетонитрила в атмосфере аргона охлаждалась до 0-5оС и к ней добавлялось 8,0 мл, 30 ммоля бис(триметилсилил)трифторацетамида. Реакционная смесь постепенно нагревалась до 6-7оС в течение часа, за это время вся аминокислота переходила в раствор. Затем 3,52 г, 15 ммоль 2-трифторметил-3-ацетилтиопропионилхлорида растворялось в 6 мл ацетонитрила и по каплям добавлялось к смеси при 5оС в течение 45 мин, после чего реакционная смесь перемешивалась и нагревалась до комнатной температуры. Через 1,5 ч растворитель выпаривался, остаток в виде желтой жидкости распределялся между водой (50 мл) и этилацетатом (50 мл) и органический слой отделялся. Водная фаза затем экстрагировалась этилацетатом (3х40 мл) и объединенный этилацетатный экстракт промывался рассолом, высушивался над безводным сульфатом магния и выпаривался с получением 6,7 г желтого твердого вещества. Тонкослойной хроматографией на силикагеле (100:1 силикагель:соединение) с использованием смеси метиленхлорид:метанол:уксусная кислота при соотношении 40:1 в качестве элюента получали 1,62 г материала, который гомогенизировался тонкослойной хроматографией, также как 0,65 г материала, который был почти гомогенный, и 1,16 г материала, который не очищался, не содержит в большей степени требуемый продукт. 0,65 г и 1,62 г продукта измельчались в порошок посредством смеси гексаны:этилацетат (4:1) 2,21 г рацемического продукта в виде белого кристаллического порошка с температурой плавления 114-116оС, при тонкослойной хроматографии Rf = 8,44 (20:1:1 метиленхлорид:метанол:уксусная кислота).
C9H13F3NO4S.
Вычислено, %: C 37,63; H 4,21; N 4,88; F 19,84; S 11,66.
Найдено, %: C 37,62; H 4,12; N 4,83; F 19,79; S 11,38.
П р и м е р 11. [3-[[2-(Меркаптометил)-3,3,3-трифтор-I-оксопропил]амино]-пропионовая кислота.
Суспензия соединения, указанного в названии примера 11, в количестве 2,08 г 7,24 ммоль в 4,2 мл концентрированной гидроокиси аммония и 9,3 мл воды перемешивалась при комнатной температуре в атмосфере аргона в течение 7-10 мин, за это время образовывался прозрачный раствор. Затем раствор подкислялся до рН 2 приблизительно 200 мл 5% гидросульфата калия и экстрагировался этилацетатом (5х50 мл). Объединенный этилацетатный экстракт промывался 50 мл рассола, высушивался над безводным сульфатом магния и выпаривался с получением 1,74 г не совсем белого твердого вещества.
Тонкослойной хроматографией на силикагеле (120:1 силикагель:соединение) с использованием смеси метиленхлорид:метанол:уксусная кислота при соотношении 40: 1: 1 в качестве элюента получалась 1,5 г соединения. Аналогично из 1,2 неочищенной фракции того же исходного материала (номинально 4,2 ммоля) деацилированием и тонкослойной хроматографией получалось еще 0,45 г требуемого продукта. Две фракции продукта объединялись с получением 1,95 г соединения, указанного в названии, рацемического соединения в виде белого кристаллического порошка с т.пл. 108-110оС, при тонкослойной хроматографии Rf = 0,40 (20:1:1 метиленхлорид:метанол:уксусная кислота).
Вычислено, %: C 34,28; H 4,11; N 5,71; F 23,24; S 13,07; SH 13,49.
C7H10F3NO3S.
Найдено, %: C 34,36; H 4,04; N 5,58; F 22,94; S 13,07; SH 13,64.
П р и м е р 12. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-аспарагино-вая кислота.
а) N-[2-[(ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-аспарагиновая кислота.
Перемешанная суспензия L-аспарагиновой кислоты в количестве 2,66 г, 20 ммоля в 35 мл сухого ацетонитрила в атмосфере аргона охлаждалась до 0оС и затем добавлялся бис(триметилсилил)трифторацетамид в количестве 10,62 мл, 40 ммоля. Реакционная смесь перемешивалась и постепенно нагревалась до комнатной температуры всю ночь, затем 5 мл диметилформамида добавлялось к смеси и она перемешивалась в течение 2 ч. Полученный прозрачный светло-желтый раствор охлаждался до 50oС и к нему по каплям добавлялось 4,69 г, 20 ммоль 2-трифторметил-3-ацетилтиопропионилхлорида, растворенного в 6 мл ацетонитрила. Реакционная смесь перемешивалась и нагревалась до комнатной температуры всю ночь, а затем растворитель выпаривался. Желтый, имеющий консистенцию сиропа, остаток распределялся между водой (50 мл) и этилацетатом (50 мл) и органический слой отделялся. Затем водная фаза экстрагировалась этилацетатом (3х50 мл) и объединенный этилацетатный экстракт промывался рассолом, высушивался и выпаривался с выходом 12,2 г светло-желтого остатка. Тонкослойной хроматографией на силикагеле (125:1 силикагель:соединение) с использованием в качестве элюента смеси этилацетат:уксусная кислота при соотношении 15: 1 получалось 5,25 г основной фракции, которая была в виде светло-желтого липкого твердого продукта, и 0,63 г фракции, которая практически гомогенизировалась при тонкослойной хроматографии, общего количества 5,88 г соединения, указанного в названии, в виде смеси диастереомеров.
в) N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил]-L-аспарагиновая кислота.
Суспензия продукта, полученного в разделе а/ в количестве 4,68 г, 14,1 ммоль в 8,25 мл концентрированной гидроокиси аммония и 20,6 мл воды перемешивалось при комнатной температуре в атмосфере аргона приблизительно в течение 10 мин. Полученный раствор подкислялся 400 мл 5%-ного гидросульфата калия и экстрагировался этилацетатом (4х75 мл). Объединенный этилацетатный экстракт промывался рассолом, высушивался (MgSO4) и выпаривался с получением 3,41 г светло-желтого твердого продукта. Тонкослойной хроматографией на 350 г силикагеля элюированием смесью метиленхлорид:метанол:уксусная кислота при соотношении 15:1:1 получали 2,4 г светло-желтого твердого продукта, который измельчался в порошок метиленхлоридом, содержащим несколько капель этилацетата и метанола, и получалось 2,1 г соединения, указанного в названии (приблизительно 1:1 смесь диастереомеров) в виде белого кристаллического порошка с т.пл. 180-183оС, при тонкослойной хроматографии Rf = 0,31 (10:1:1 метиленхлорид:метанол:уксусная кислота), [α]D = -1,9o (C = 1,00, метанол).
Вычислено, %: C 32,70; H 3,60; N 4,77; F 19,40; S 10,92; SH 11,26.
С8Н10F3NO5S˙0,25H2O.
Найдено, %: C 32,64; H 3,20; N 4,70; F 19,23; S 10,85; SH 11,15.
П р и м е р 13. N-[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-( β )-нафтиилаланин, изомер А.
Перемешанная суспензия L-нафтилаланина в количестве 1,92 г, 8,92 мл в 50 мл ацетонитрила охлаждалась в атмосфере аргона до 0-5оС и к ней добавлялся бис(триметилсилил)трифторацетамид (4,75 мл, 17,84 ммол). Реакционная смесь перемешивалась и медленно нагревалась до комнатной температуры. Через 1,5 ч к ней добавлялось дополнительно 2,3 мл бис(триметилсилил)трифторацетамида (8,7 ммоля), в результате получался прозрачный раствор. Реакционная смесь охлаждалась до 5оС, и по каплям добавлялся 2-трифторметил-3-ацетилтиопропионилхлорид (2,09 г, 8,92 ммоль), растворенный в 6 мл ацетонитрила, затем смесь медленно нагревалась до комнатной температуры. Через час реакционная смесь выпаривалась с образованием желтого твердого остатка, распределенного между водой (50 мл) и этилацетатом (75 мл), и органический слой сепарировался.
Водный слой экстрагировался дополнительным количеством этилацетата (3х45 мл), после чего общий этилацетатный экстракт промывался рассолом, высушивался (MgSO4) и этилацетат удалялся под вакуумом, с получением светло-желтого твердого остатка. Этот неочищенный материал подвергался тонкослойной хроматографии на 700 г силикагеля и элюированием смесью этилацетат: гексаны: уксусная кислота при соотношении 100:100:1, получалось 1,58 г быстроизолирующего изомера А и 1,56 г медленноэлюирующего изомера В. 300 мг изомера А кристаллизовалось из смеси гексаны:этилацетат (2:1) с выходом 160 мг указанного в названии изомера в виде белых длинных игольчатых кристаллов, температура плавления 162-164оС, при тонкослойной хроматографии Rf = 0,29 (100: 50: 1 этилацетат: гексан:уксусная кислота), [α]D = -79,2o (C = 1,00, метанол).
Вычислено, %: C 55,20; H 4,39; N 3,39; F 13,79; S 7,76.
C19H18F3NO4S.
Найдено, %: C 55,02; H 4,15; N 3,29; F 13,60; S 7,54.
П р и м е р 14. N-[2-[(Ацетилтио)метил)-3,3,3-трифтор-1-оксопропил]-L-( β )-нафтилаланин, изомер В.
Изомер В продукта из примера 14 вновь подвергался тонкослойной хроматографии с использованием смеси этилацетат:уксусная кислота (200:1,5) и этилацетат: гексаны:уксусная кислота (100:50:1), в качестве элюента для удаления изомера А, являющегося примесью, и получения 1,4 г указанного в названии изомера В в виде белого твердого продукта с т.пл. 166-169оС, при тонкослойной хроматографии Rf = 0,15 (100:50:1 этилацетат:гексан:уксусная кислота), [α]D = +116,3o (C = 1,00, метанол).
Вычислено, %: С 55,20; H 4,39; N 3,39; F 13,76; S 7,76.
C19H18F3NO4S.
Найдено, %: C 54,96; H 4,39; N 3,35; F 13,75; S 7,83.
П р и м е р 15. (S)-α-[[3,3,3-Трифтор-2-(меркаптометил)-]-оксопропил] амино]-2- нафталенпропионовая кислота, изомер А.
Суспензия продукта из примера 14 (2,01 г, 4,86 ммоль) в 3,2 мл концентрированного гидроксида аммония и 8,0 мл воды перемешивалась при комнатной температуре в атмосфере аргона в течение 8 мин. Прозрачный раствор подкислялся 5%-ным бисульфатом калия до рН около 2 и экстрагировался этилацетатом (4х50 мл). Общий этилацетатный экстракт промывался рассолом, высушивался (MgSO4) и выпаривался с получением 1,72 г светло-желтого воскообразного твердого продукта. Очистку продукта производили тонкослойной хроматографией на силикагеле (150:1 силикагель:вещество) с элюированием смесью этилацетат: гексаны: уксусная кислота (100:50:1) с получением 1,42 г белого кристаллического продукта, после обработки которого толуолом и высушивании в вакууме получилось 1,24 г указанного в названии изомера в виде белого твердого продукта с температурой плавления 165-167оС, при тонкослойной хроматографии Rf = 0,26 (100:50:1 этилацетат:гексан:уксусная кислота), [α]D = +31,9o (C = 1,00, метанол).
Вычислено, %: C 54,98; H 4,34; N 3,77; F 15,35; S 8,63; SH 8,90.
C17H16F3NO3S.
Найдено, %: C 55,17; H 4,29; N 3,63; F 15,42; S 8,68; SH 9,18.
П р и м е р 16. (S)-α-[[3,3,3-трифтор-2-(меркаптометил)-метоксопропил] амино]-2-нафталенпропи оновкислота, изомер В.
Раствор продукта из примера 15 в 10 мл метанола обрабатывался раствором гидроксиламингидрохлорида (0,552 г, 7,94 ммоль) в 7,9 мл 1н. гидроокиси натрия и перемешивался при комнатной температуре. Реакция завершалась через 35 мин, судя по тонкослойной хроматографии. Реакционная смесь выпаривалась, полученное белое твердое вещество помещалось в 25 мл воды и экстрагировалось этилацетатом (4х35 мл). Общий органический экстракт промывался 50 мл рассола, высушивался (MgSO4) и выпаривался с получением 1,4 г белого твердого продукта. Тонкослойной хроматографией проводили очистку продукта на силикагеле (180: 1 силикагель:соединение), элюируя смесью этилацетат:гексаны: уксусная кислота (100:50:1) с получением 1,24 г указанного в названии изомера В в виде белого кристаллического порошка с т.пл. 172-179оС при тонкослойной хроматографии Rf = 0,19 (100:50:1 этилацетат:гексан:уксусная кислота), [α]D = +71,3o (C = 1,00, метанол).
Вычислено, %: C 54,98; H 4,34; N 3,77; F 15,35; S 8,63; SH 8,90.
C17H16F3NO3S.
Найдено, %: C 55,02; H 4,30; N 3,55; F 15,18; S 8,64; SH 9,09.
П р и м е р 17. N-[2-[Ацетилтио метил]-3,3,3-трифтор-1-оксопропил]-L-лейцин, изомер А.
Перемешанная суспензия L-лейцина (2,62 г, 20 ммоль) в 35 мл сухого ацетонитрила в атмосфере аргона охлаждалась до 0-5оС и к ней добавлялся бис(триметилсилил)-трифторацетамид (10,62 мл, 40 ммоль). Реакционная смесь перемешивания медленно нагревалась до комнатной температуры. Через 2 ч добавлялось еще 3,5 мл (13 ммоль) бис(триметилсилил)трифторацетамида, в результате получался прозрачный раствор лимонного цвета после дополнительного перемешивания в течение 1 ч при комнатной температуре. Затем реакционная смесь охлаждалась до 0-5оС и к ней через 45 мин по каплям добавлялся 2-трифторметил-3-ацетилтиопропионилхлорид (4,69 г, 20 ммоль), растворенный в 8 мл ацетонитрила. После перемешивания в течение 1,5 ч при 0-5оС до комнатной температуры тонкослойная хроматография показала завершение реакции. Реакционная смесь выпаривалась при пониженном давлении с получением желтого маслянистого остатка, который распределялся между этилацетатом (75 мл) и водой (50 мл). Затем водная фаза экстрагировалась этилацетатом (4х50 мл), общий экстракт промывался 75 мл рассола, высушивался (MgSO4) и выпаривался с получением 10,3 г желтого полутвердого остатка. 0,68 г этого неочищенного материала подвергалось тонкослойной хроматографии на 75 г силикагеле при элюировании смесью этилацетат:гексаны:уксусная кислота при соотношении 100: 100:1 с выходом 0,15 г быстроэлюирующего диастереомера (изомер А) и 0,12 г медленноэлюирующего диастереомера (изомер В). Оставшиеся 9,6 г неочищенного материала подвергались тонкослойной хроматографии аналогичным образом (силикагель, 1300 г), было получено 2,5 г изомера А, 2,7 г смеси обоих диастереомеров и 1,5 г изомера В.
Смешанные фракции еще раз хроматографировались при тех же условиях с получением 0,5 г изомера А и 1,25 г изомера В. Обе порции 2,5 г и 0,5 г изомера А объединялись, измельчались в порошок посредством гексанов, содержащих несколько капель этилацетата, фильтровались и высушивались и получалось 2,57 г указанного в названии изомера А в виде белого кристаллического порошка с температурой плавления 150-152oС, при тонкослойной хроматографии Rf = 0,40 (100:50:1 этилацетат:гексан:уксусная кислота), [α]D = -146,9o (C = 1,00, метанол).
Вычислено, %: C 43,76; H 5,51; N 4,25; F 17,31; S 9,73;
C12H18F3NO4S.
Найдено, %: C 43,79; H 5,50; N 3,96; F 17,10; S 9,81.
П р и м е р 18. N-[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-лейцин, изомер В.
Часть изомера В из примера 18 (2,61 г) дважды превращалась в порошок посредством гексанов, содержащих несколько капель этилацетата, и один раз смесью гексаны:эфир (3:1), а затем фильтровалась и высушивалась с получением 1,6 г указанного в названии изомера В в виде белого кристаллического порошка с т.пл. 100-113оС, при тонкослойной хроматографии Rf = 0,27 (100:50:1 этилацетат:гексаны:уксусная кислота), [α]D = +109,6o (C = 1,00, метанол).
Bычислено, %: C 43,78; H 5,51; N 4,25; F 17,31; S 9,78.
C12H18F3NO4S.
Найдено, %: C 43,80; H 5,56; N 3,92; F 17,03; S 9,42.
П р и м е р 19. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-лейцин, изомер А.
Суспензия продукта из примера 18 (2,3 г, 6,98 ммоль) в 4,45 мл концентрированной гидроокиси аммония и 10 мл воды перемешивались при комнатной температуре в атмосфере аргона в течение 8 мин. Полученный прозрачный раствор подкислялся 5%-ным бисульфатом калия до рН 2 и экстрагировался 100 мл этилацетата и затем дополнительно 3х75 мл. Общий этилацетатный экстракт промывался 100 мл рассола, высушивался (MgSO4) и выпаривался с получением 1,94 г не совсем белого твердого продукта. Тонкослойной хроматографией на 200 г силикагеля элюированием смесью этилацетат:гексаны:уксусная кислота при соотношении 100:100:1 получалось 1,72 г указанного в названии изомера А в виде белого кристаллического порошка с т.пл. 143-145оС, при тонкослойной хроматографии Rf = 0,38 (100:50:1 этилацетат:гексан:уксусная кислота) [α]D = -62,2o (C = 1,00, метанол).
Вычислено, %: C 41,80; H 5,61; N 4,88; F 19,84; S 11,16; SH 11,51.
С10Н16F3NO3S.
Найдено, %: C 41,89; H 5,62; N 5,12; F 19,51; S 11,05; SH 11,78.
П р и м е р 20. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-лейцин, изомер В.
Суспензия продукта из примера 19 (1,3 г, 3,95 ммоля) в 2,52 мл концентрированной гидроокиси аммония и 5,6 мл воды перемешивались при комнатной температуре в атмосфере аргона в течение 8 мин. Прозрачный раствор подкислялся 5%-ным раствором бисульфата калия (120 мл) до рН 2 и экстрагировался 75 мл этилацетата, а затем 3х40 мл этилацетата. Общий этилацетатный экстракт промывался 50 мл рассола, высушивался (MgSO4) и выпаривался с получением 1,04 г светло-желтого твердого продукта. Этот материал соединялся с 35 мл продукта, полученного аналогичным образом. Объединенный неочищенный материал очищался тонкослойной хроматографией на 200 г силикагеля с элюированием смесью этилацетат:гексаны:уксусная кислота при соотношении 100:100:1 с выходом 0,93 г, указанного в названии изомера В в виде белого кристаллического порошка с температурой плавления 132-134оС при тонкослойной хроматографии Rf = 0,33 (100:50:1 этилацетат:гексан:уксусная кислота) [α]D = +4,8o (C = 1,00, метанол).
Вычислено, %: С 41,80; H 5,61; N 4,88; F 19,84; S 11,61; SH 11.51.
C10H16F3NO3S.
Найдено, %: C 41,83; H 5,66; N 5,04; F 19,67; S 11,13; SH 11,79.
П р и м е р 21. 2-(Ацетилтиометил)-3,3,3-трифтор-N-[2-(4-гидроксифенил)-этил]-про-панамид.
В перемешанный раствор 4-(2-аминоэтил)-фенола (4,60 г, 3,6,2 ммоль) в 250 мл сухого метиленхлорида, находящегося при -10оС добавлялся раствор 2-трифторметил-3-ацетилтиопропионилхлорида (4,24 г, 18,1 ммоль) в 50 мл сухого метиленхлорида и затем реакционная смесь перемешивалась 3 раза 1н. хлористоводородной кислотой и органический слой высушивался над безводным сульфатом магния, отфильтровывался и концентрировался в вакууме с выходом 3,5 г желтого масла. Неочищенный материал подвергался тонкослойной хроматографии на 400 мл колонне силикагеля ЛПС-1 с использованием смеси 2:1 гексан/этилацетат в качестве элюента, при выделении получали 1,6 г указанного в названии соединения в виде прозрачного масла. При тонкослойной хроматографии Rf = 0,12 (4:1 EtOAc:CH2Cl2).
Вычислено, %: C 49,61; H 4,88; N 4,13; F16,82; S 9,46.
C14H16F2NO3S˙1,1H2O
Найдено, %: C 49,38; H 4,77; N 4,34; F 16,58; S 9,51.
П р и м е р 22. 2-(Меркаптометил)-3,3,3-трифтор-N-[2-(4-гидроксифенил)-этил]про-панамид.
Раствор указанного в названии примера 22 соединения в 20 мл метанола обрабатывался раствором 1н. гидроокиси аммония (9 мл, 9,0 ммолей) и затем перемешивался при комнатной температуре в течение 3 ч. Реакционная смесь разделялась между эфиром и 1н. хлористоводородной кислотой и слои разделялись. Органический слой промывался 1н. хлористоводородной кислотой и рассолом, высушивался над безводным сульфатом магния, отфильтровывался и концентрировался в вакууме с выходом 1,4 г стеклообразного желтого твердого продукта. Неочищенный материал подвергался с использованием в качестве элюента смеси метиленхлорид:метанол при соотношении 97:3, и получалось 0,74 г указанного в названии соединения в виде белого твердого продукта с т.пл. 98-100оС.
Вычислено, %: C 48,84; H 4,85; N 4,75; F 19,32; S 10,87; SH 11,23.
Найдено, %: C 48,97; H 4,63; N 4,62; F 18,96; S 10,76; SH 10,93.
П р и м е р 23. 2-[(Ацетилтио)метил]-3,3,3-трифтор-N-[2-(1Н-тетразол-5-илэтил]пропанамид.
А. (N)-t-бутоксикарбонил-3-аминопропионитрил.
3-Аминопропионитрил формат (1,28 г, 10 ммоль) растворялся в 40 мл абсолютного этанола путем добавления диизопропилэтиламина (3,48 мл, 20 ммолей) при 0оС. После перемешивания в течение 10 мин добавлялся ди-трет-бутилдикарбонат (2,18 г, 10 ммолей) и реакционная смесь перемешивалась при комнатной температуре в течение 48 ч, концентрировалась под вакуумом и остаток растворялся в этилацетате (200 мл). Этилацетатный раствор промывался водой, насыщался бикарбонатом натрия, высушивался над сульфатом натрия и концентрировался в вакууме, в результате получалось указанное в названии вещество в виде твердого продукта (1,68 г) с т.пл. 44-45оС.
В. 5-((N)-t-Бутоксикарбонил-2-аминоэтил)-тетразол.
Три-n-бутилоловоанид и указанное в названии соединение А нагревались до 115оС в 100 мл ксилола в течение 48 ч.
Реакционная смесь концентрировалась под вакуумом остаток растворялся в 2: 8 (20: 6:11 пиридин:уксусная кислота:вода):этилацетат. Добавлялся силикагель (25 г) и смесь перемешивалась при комнатной температуре всю ночь.
Реакционная смесь отфильтровывалась и подушка из двуокиси кремния тщательно промывалась указанным растворителем 2:8, (20:6:11) пиридин:уксусная кислота: вода). Фильтрат концентрировался в вакууме и остаток концентрировался из толуола (2х).
Продукт затвердевал при добавлении изопропилового эфира. Продукт фильтровался с выходом 1,88 г указанного в названии соединения В, т.пл. 121-126оС.
С. 5-(2-аминоэтил)тетразолгидрохлорид.
Указанное в названии соединение В (1,88 г, 8,8 ммоля) растворялось в 20 мл этилацетата, насыщенного HCl. Реакционная смесь перемешивалась в течение 3 ч и полученный осадок отфильтровывался с выходом соединения С, указанного в названии, (1,04) в виде твердого продукта с т.пл. 120-129оС (разложение).
Д. 2-[(Ацетилтио)-метил] -3,3,3-трифтор-N-[2-(1H-тетразол-5-ил)этил] пропанамид.
Указанное в названии соединение С (0,92 г, 6,15 ммоля) суспендировалось в 15 мл сухого ацетонитрила в атмосфере аргона. К нему добавлялся бис(триметилсилил)трифторацетамид (5,2 мл, 19,6 ммоля) и смесь перемешивалась до получения прозрачного раствора (2 ч).
Реакционная смесь охлаждалась до 5оС, к ней по каплям добавлялся 2-трифторметил-3-ацетилтиопропионилхлорид (1,44 г, 6,14 ммоля), смесь перемешивалась в течение 2 ч при 5-10оС. Затем реакционная смесь концентрировалась под вакуумом, растворялась в этилацетате (200 мл) и этилацетатный раствор промывался водой (3 х), буферным раствором с рН 4,02, рассолом (2 х), высушивался над сульфатом натрия и концентрировался в вакууме. Добавление изопропилового эфира вызывало затвердение продукта. Твердый продукт отфильтровывался и высушивался (1,38 г), т.пл. 157-160оС.
Вычислено, %: C 34,72; H 3,89; N 22,50; F 18,31; S 10,30.
C9H12F3N5O2S.
Найдено, %: C 34,41; H 3,80; N 21,39; H 18,64; S 10,34.
П р и м е р 24. 3,3,3-Трифтор-2-(меркаптометил)-N-[2-(1H-тетразол-5-ил)этил]пропанамид.
Указанное в названии примера 26 соединение (1,0 г, 3,2 ммоль) растворялось при 0оС в атмосфере аргона в 2,5 мл 1:1 концентрированного раствора NH4OH:H2O.
Реакционная смесь перемешивалась при 0оС в течение 5 мин и затем подкислялась 5н. HCl до рН 6 при 0оС. Продукт экстрагировался этилацетатом, содержащим 5% метанола (3 х) порции по 200 мл. Органический слой промывался рассолом, высушивался над сульфатом натрия и концентрировался в вакууме. Остаток подвергался хроматографии через 50 г колонну Мерка с силикагелем с использованием (3:3:94) смеси метанол:уксусная кислота:метиленхлорид. Соответствующие фракции объединялись и концентрировались, выход составил 0,56 г указанного в названии продукта, т.пл. 182-184оС.
Вычислено, %: C 30,95; H 3,81; N 25,78; F 20,98; S 11,80; SH 12,17.
С7H10N5OSF3˙0,13H2O.
Найдено, %: C 31,15; H 3,66; N 25,53; F 20,58; S 11,83; SH 12,21.
П р и м е р 25. N-[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-норлейцин, изомер А.
Бис(триметилсилил)трифторацетамид (16 мл, 60 ммоля) добавлялся к суспензии L-норлейцина (2,62 г, 20 ммоля) в 40 мл ацетонитрила при 0оС. Суспензия нагревалась до комнатной температуры и перемешивалась в течение 2,5 ч, к этому времени практически весь твердый продукт растворялся. Реакционная смесь вновь охлаждалась до 0оС и по каплям через 20 мин добавлялся раствор 2-трифторметил-3-ацетилтиопропионилхлорида (4,7 г, 20 ммоль) в 10 мл ацетонитрила. После перемешивания при комнатной температуре в течение 18 ч реакционная смесь выливалась в 200 мл воды и полученная смесь экстрагировалась этилацетатом (500 мл). Затем добавлялся рассол, чтобы разбавить эмульсию, которая образовывалась при экстракции. Водный слой экстрагировался еще раз этилацетатом (100 мл) и соединенные органические слои промывались рассолом, высушивались (MgSO4) и концентрировались с получением маслянистого остатка, который предварительно абсорбировался на целите. Неочищенный продукт и целитная смесь погружались в колонну на 8х30 см силикагеля и элюировались следующим образом: 5 л гексан:этилацетат:уксусная кислота (750: 250: 25), 2 л гексан:этилацетат:уксусная кислота (700:300:25) 2 л гексан:этилацетат, уксусная кислота (500:500:25). Чистые наименее полярные реакции концентрировались и совместно выпаривались из гептана с получением белого твердого продукта, который дважды перекристаллизовывался из этилацетата/гексана. При сушке в глубоком вакууме при комнатной температуре в течение 18 ч получали 1,95 г указанного в названии изомера А в виде белого порошка с температурой плавления 176-178оС, при тонкослойной хроматографии Rf = 0,30 (этилацетат:гексан:уксусная кислота 30:70:1).
[α]D = -133,3oC (С = 0,6, метанол).
Вычислено, %: C 43,76; H 5,51; N 4,25; F 17,31; S 9,73.
C12H18F3NO4S.
Найдено, %: C 43,76; H 5,45; N 4,35; F 17,57; S 9,67.
П р и м е р 26. N-[2[-(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил-L-норлейцин, изомер В.
В способе, раскрытом в примере 29, чистые более полярные фракции концентрировались с получением белого твердого продукта, который перекристаллизовывался дважды из этилацетата/гексана. Сушкой в глубоком вакууме получали 1,953 г указанного в названии изомера В с т.пл. 123-125оС, при тонкослойной хроматографии Rf = 0,19 (этилацетат:гексан:уксусная кислота, 30:70:1), [α]D = +123o (c = 0,63, метанол).
Вычислено, %: C 43,76; H 5,51; N 4,25; F 17,31; S 9,73.
C12H18F3NO4S.
Найдено, %: C 43,97; H 5,45; N 4,29; F 17,13; S 9,89.
П р и м е р 27. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-норлейцин, изомер А.
Концентрированный гидроксид аммония (3 мл) добавлялся к суспензии продукта из примера 29 в 12 мл обедненной кислородом воды. Реакционная смесь, которая стала гомогенной менее, чем за 1 мин, перемешивалась в течение 15 мин. За это время рН достигало 1,5 при добавлении бисульфата калия и полученная водная смесь экстрагировалась этилацетатом (2 х 150 мл). Все органические слои промывались рассолом, высушивались (MgSO4) и концентрировались с получением маслянистого осадка. Этот осадок подвергался тонкослойной хроматографии на 5 х 12 см колонне силикагеля с использованием смеси гексан:этилацетат:уксусная кислота, 65:35:1.
Чистые фракции концентрировались и остаток выпаривался из гептана несколько раз с получением белого твердого продукта. После измельчения в порошок гексаном и сушки в течение 18 ч под глубоким вакуумом получали 1,175 г указанного в названии изомера А в виде белого твердого продукта с т.пл. 96-98оС, при тонкослойной хроматографии Rf = 0,38 (этилацетат:гексан: уксусная кислота 40:60:2), [α]D = -43,2о (С = 0,60, метанол).
Вычислено, %: C 41,37; H 5,67; N 4,83; F 19,84; S 11,16; SH 11,51.
C10H16F3NO3S.
Найдено, %: C 41,68; H 5,62; N 4,52; F 19,53; S 10,89; SH 11,78.
П р и м е р 28. N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-норлейцин, изомер В.
Концентрированный гидроксид аммония (3 мл) добавлялся к суспензии продукта из примера 30 в 12 мл обедненной кислородом воды при комнатной температуре. Реакционная смесь становилась гомогенной в течение 1 мин. После перемешивания в течение 15 мин реакционная смесь подкислялась до рН 1,5 концентрированным бисульфатом калия. Эта подкисленная смесь экстрагировалась этилацетатом (2 х 150 мл). Объединенные органические слои промывались рассолом (100 мл), высушивались (MgSO4) и концентрировались с получением светло-желтого остатка. Этот остаток подвергался тонкослойной хроматографии на 5 х 12 см колонне силикагеля с использованием этилацетат: гексан: уксусной кислоты 40:60:1 для элюирования.
Чистые фракции концентрировались и выпаривались совместно из гептана с получением бесцветного твердого продукта, который измельчался в порошок посредством гексана. Фильтрацией и сушкой получалось 1,16 г белого порошка. Этот материал перекристаллизовывался дважды из этилацетата/гексана с получением 632 мг указанного в названии изомера В в виде бесцветных иголок с т. пл. 131-133оС, при тонкослойной хроматографии Rf = 0,26 (этилацетат: гексан: уксусная кислота 40:60:2), [α]D = +21,3о (С = 0,71, метанол).
Вычислено, %: C 41,53; H 5,65; N 4,84; F 19,71; S 11,09; SH 11,44.
C10H16F3NO3S˙0,1H2O
Найдено, %: C 41,55; H 5,61; N 4,85; F 19,14; S 10,99; SH 13,66.
П р и м е р 29. (S)-N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]-L-изолей-цин.
а) N-[2-[2(ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-L-изолейцин.
Бис(триметилсилил)трифторацетамид (16 мл, 60 ммолей) добавлялся к суспензии L-изолейцина (2,62 г, 20 ммолей) в 40 мл ацетонитрила при 0оС. Суспензия нагревалась до комнатной температуры и перемешивалась в течение 2 ч. За это время приблизительно одна треть твердого продукта растворялась. Затем добавлялось дополнительное количество бис(триметилсилил)трифторацетамида (4 мл, 15 ммоль) и смесь перемешивалась в течение 45 мин. Смесь охлаждалась до 0оС и через 15 мин по каплям добавлялся 2-трифторметил-3-ацетилтиопропионил (4,7 г, 20 ммоль) в виде раствора в 10 мл ацетонитрила. После перемешивания в течение 18 ч при комнатной температуре получалась реакционная смесь в виде прозрачного светло-желтого раствора. Добавлялось приблизительно 150 мл воды и полученная смесь экстрагировалась этилацетатом (2 х 150 мл). Все органические слои объединялись и промывались рассолом (100 мл), высушивались (MgSO4) и концентрировались с получением маслянистого остатка, который предварительно адсорбировался на целите для хроматографии. После нескольких хроматографий, измельчения и перекристаллизаций получалось 1,41 г (R)-N-[2-[(ацетилтио-метил]-3,3,3-трифтор-1-оксопропил]-L-изолейцина с т. пл. 81-85оС, [α] D = -105,5о (С = 0,38, метанол) и 1,56 г (S)-N-[2-[(ацетилтио)метил] -3,3,3-трифтор-1-оксопропил]-α-изолейцина с температурой плавления 111-112оС, [α]D = +122,6о (С = 0,38, метанол).
b) (S)-N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил]-L-изолейцин.
Концентрированный гидроксид аммония (3 мл) добавлялся к суспензии (S) диастереомере продукта из части а) (1,5 г, 4,55 ммоля) в 12 мл обедненной кислородом воды. Растворение проходило через несколько секунд и реакционная смесь перемещалась в атмосфере аргона в течение 5 мин. Затем смесь подкислялась до рН 1,5 бисульфатом калия и экстрагировалась этилацетатом (2 х 150 мл). Объединенные органические слои промывались рассолом, высушивались (MgSO4) и концентрировались. Остаток хроматографировался на 5 х x15 колонне силикагеля и элюированием смесью гексан: этилацетат: уксусная кислота 75: 25: 1. Чистые фракции концентрировались и выпаривались из гептана с получением 1,285 г белого твердого продукта (тот твердый продукт перекристаллизовывался три раза с получением 0,834 г указанного в названии (R) диастереомера с т. пл. 130-131оС, при тонкослойной хроматографии Rf = 0,43 (этилацетат: гексан: уксусная кислота 40:60:1), [α]D = +35,8о (С = 0,45, метанол).
Вычислено, %: C 41,55; H 5,65; N 4,85; F 19,72; S 11,09; SH 11,44.
C10H16F3NO3S˙0,10H2O
Найдено, %: C 41,74; H 5,62; N 4,76; F 19,33; S 10,95; SH 13,47.
П р и м е р 30. (R)-N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил]-L-изолейцин.
Концентрированный гидроксид аммония (2,6 мл) добавлялся к суспензии (R) диастереомера продукта из примера 33а (1,3 г, 3,95 ммоля) в 10 мл воды. Растворение происходило в течение 1 мин и реакционная смесь перемешивалась в атмосфере аргона в течение 5 мин при комнатной температуре.
Затем смесь подкислялась до рН 1,5 бисульфатом калия и экстрагировалась этилацетатом (2 х 100 мл). Объединенные органические слои промывались рассолом, высушивались (MgSO4) и концентрировались. Остаток хроматографировался на 5 х 25 см колонне силикагеля при элюировании смесью гексан: этилацетат: уксусная кислота 75:25:1. Чистые фракции концентрировались и выпаривались из гептана, выход составил 1,01 г белого твердого продукта. Этот продукт требовал очень широкого и тщательного измельчения в порошок гептаном, гексаном, пентаном для удаления следов этилацетата. После окончательной фильтрации этот материал высушивался при глубоком вакууме при 100оС с выходом 0,79 г указанного в названии (R) диастереомере в виде белого твердого продукта с температурой плавления 140-142оС при тонкослойной хроматографии Rf = 0,39 (этилацетат: гексаны: уксусная кислота 40:60:2), [α]D = -37,3о (С = 0,55, метанол).
Вычислено, %: C 41,80; H 5,61; N 4,88; F 19,84; S 11,16; SH 11,51.
C10H16F3NO3S.
Найдено, %: C 42,18; H 5,67; N 5,02; F 19,36; S 11,48; SH 12,29.
П р и м е р 31. (S, R)-3[[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]амино]-5-метилгексанов ая кислота.
Раствор 3-[[(2,2-диметилэтил)окси] /-карбонил]-амино-5-метилгексановая кислота, метиловый эфир (полученный по схеме Аридт/Айстерт, Ондетти и др. -Журнал медицинской Химии, том 18, стр.761, 1975) в количестве 6,26 г, 20,3 ммоль в 100 мл метанола обрабатывался 1н. раствором гидроокиси натрия (40 мл, 40 ммоль) и затем перемешивался при комнатной температуре в течение 3 ч. Затем реакционная смесь концентрировалась под вакуумом и остаток помещался в воду. Водный раствор промывался один раз этилацетатом, затем подкислялся 6н. HCl, а затем экстрагировался этилацетатом (4х). Органические экстракты объединялись, высушивались (MgSO4), фильтровались и концентрировались в вакууме с получением 5,68 г 3-[[(2,2-диметилэтил)-окси]карбонил] амино-5-метилгексановой кислоты в виде прозрачного масла.
Раствор этой кислоты (0,63 г, 2,57 ммоль) в этилацетате (5 мл) обрабатывался HCl (3,2 М раствора в этилацетате, 25 мл, 80 ммолей) и перемешивался при комнатной температуре в течение 3 ч. Реакционная смесь барботировалась N2 и затем концентрировалась в вакууме с получением белого твердого продукта, который измельчался этиловым эфиром, выход составил 0,37 г 3-амино-5-метилгексановой кислоты, гидрохлорид, температура плавления 125-126оС.
Раствор этой кислоты, гидрохлорид (2,7 г, 14,9 ммоля) в сухом ацетонитриле, находящийся при температуре около 0оС, обрабатывался диизопропиламином (2,6 мл, 14,9 ммоля) и бис(триметилсилил)трифторацетамидом (8 мл, 30,1 ммоля) и перемешивался при 0оС в течение 30 мин. Затем в течение 15 мин по каплям прибавлялся раствор 2 трифторметил-3-ацетилтиопропионилхло-рида (3,46 г, 14,7 ммоль) в 16 мл сухого ацетонитрила и раствор нагревался до комнатной температуры всю ночь. Реакционная смесь концентрировалась в вакууме и остаток помещался в этилацетат и промывался 1н. HCl (3 х), а затем рассолом. Органический слой высушивался (MgSO4), отфильтровывался и подвергался тонкослойной хроматографии на 1800 мл силикагеля с элюированием смесью гексан: этилацетат:уксусная кислота при соотношении 2:1:0,01 с выделением 2,33 г изомера S, R как наиболее перемещающегося изомера. Затем неочищенный материал измельчался несколько раз в порошок смесью гексан: этилацетат и окончательное обрабатывался обеспечивающим углем с выходом 1,7 г указанного в названии (S, R) изомера с температурой плавления 102-103оС, при тонкослойной хроматографии Rf = 0,34 (гексан, этилацетат, уксусная кислота 100:100:1 [α]D = (c = 0,78, метанол).
Вычислено, %: C 44,78; H 5,95; N 4,02; F 16,35; S 9,20.
C13H20F3NO4˙0,3H2O
Найдено, %: С 45,02; H 5,82; N 3,78; F 16,62; S 9,51.
П р и м е р 32. (S,R)-3-[[2-(ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]амино]-3- метилгексановая кислота.
Фракции, содержащие медленно перемещающийся изомер, после тонкослойной хроматографии примера 35, объединялись и концентрировались в вакууме, в результате получалось желтое масло, которое разбивалось несколько раз гексаном:этилацетатом и окончательно обрабатывалось обесцвечивающим углем с выходом 1,2 г указанного в названии (S,S) изомера в виде белого твердого продукта с т. пл. 124-125оС, при тонкослойной хроматографии Rf = 0,78 (гексан: этилацетат: уксусная кислота 100:100:1), [α]D = +98,4o (C = 0,89, метанол).
Вычислено, %: C 45,28; H 5,89; N 4,06; F 16,53; S 9,30.
C13H20F3NO4S˙0,08H2O.
Найдено, %: C 45,35; H 5,98; N 3,99; F 16,35; S 9,18.
П р и м е р 33. (S,R)-3-[[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил] амино]-5- метилгексановая кислота.
Суспензия изомера (S,R) продукта из примера 35 (1,0 г, 2,9 ммоля) в воде, находящаяся при температуре около 0оС в атмосфере аргона, обрабатывалась гидроксидом аммония (5 М раствор, 10 мл, 50 ммолей) и перемешивалась в течение 5 мин. Реакция гасилась при 0оС 140 мл 10%-ного бисульфата калия и затем экстрагировалась этилацетатом (4% ). Органический экстракт объединялся, высушивался (MgSO4) отфильтровывался и концентрировался в вакууме с выходом 1,0 г желтого твердого продукта. Неочищенный продукт реакции подвергался тонкослойной хроматографии на 200 мл силикагеле с использованием элюирующего раствора гексан:этилацетат (2:1), выделялось 0,61 г указанного в названии (S,R) изомера в виде белого твердого продукта с т.пл. 155-156оС, при тонкослойной хроматографии Rf = 0,19 (гексан:этилацетат:уксусная кислота 100:100:1), [α]D = -55,5o (C = 0,89, метанол).
Вычислено, %: C 43,84; H 6,02; N 4,65; F 18,92; S 10,64; SH 10,97.
C11H18F3NO3S.
Найдено, %: C 43,81; H 6,08; N 4,79; F 18,52; S 10,62; SH 11,37.
П р и м е р 34. (S,S)-3-[(3,3,3-Трифтор-2-меркаптометил-1-оксопропил] амино]-5-ме- тилгексановая кислота.
Суспензия изомера (S,S) из примера 36 (0,91 г, 2,7 ммоля) в воде, находящаяся при 0оС в атмосфере аргона обрабатывалась гидроксидом аммония (5М раствор, 10 мл, 50 ммолей) и перемешивалась в течение 5 мин. Реакция гасилась при 0oC 140 мл 10%-ного бисульфата калия и экстрагировалась этилацетатом (4 х). Органические экстракты объединялись, высушивались, фильтровались и концентрировались в вакууме с выходом 1,0 г в виде желтого твердого продукта. Неочищенный продукт измельчался в порошок гексаном:этилацетатом с выходом 0,78 г указанного в названии изомера (S,S) в виде не совсем белого твердого продукта с температурой плавления 155-156оС, при тонкослойной хроматографии Rf = 0,18 (этилацетат:гексан:уксусная кислота 100:100:1), [α] D = +26,1o (C = 0,8, 1, метанол).
Вычислено, %: C 43,84; H 6,02; N 4,65; F 18,92; S 10,64; SH 10,97.
C11H18F3NO3S.
Найдено, %: C 44,26; H 6,26; N 4,62; F 18,34; S 10,22; SH 10,71.
П р и м е р 35. (S,R)-3,3,3-трифтор-2-(меркаптометил)-N-[3-метил-1-(1Н-тетразол- 5-ил) бутил]пропанамид.
а) N-[(Фенилметокси)карбонил]-L-лейцинамид.
К раствору -N-[(фенилметокси)карбонил]-L-лейцина (3 г, 11,3 моля) в 60 мл метиленхлорида при -20оС в атмосфере аргона добавлялся N-метил-морфолин (1,24 мл, 11,3 ммоль), затем изобутилхлорформат (1,47 мл, 11,3 ммоля). Полученная суспензия перемешивалась при -20оС в течение 15 мин, в течение которого добавлялся раствор аммония (метанола) (около 5,6 М, 20 мл, 113 ммоль). Через 30 мин при -20оС реакция гасилась водой и смесь нагревалась до комнатной температуры. Слои сепарировались и водный слой экстрагировался метиленхлоридом (2 х). Соединенные органические экстракты высушивались (MgSO4) и выпаривались с получением 2,8 г указанного в названии соединения А.
в) N-[(фенилметокси)карбонил]-L-лейциннитрил.
К раствору указанного в названии соединения А (9,25 г, 219,86 ммоль) в 100 мл безводного тетрагидрофурана при комнатной температуре в атмосфере аргона добавлялся метоксикарбонилсульфамоилтриэтиламмоний гидроксид внутренняя соль (11,83 г, 49,65 ммоля) в виде пяти равных порций в течение 2 ч. Реакционная смесь подавалась на тонкослойную хроматографическую колонну, элюирование осуществлялось смесью гексаны:этилацетат (50:40), получалось 4,6 г указанного в названии соединения в виде масла. При тонкослойной хроматографии Rf = 0,57 (этилацетат:гексан 1:1), [α]D = -49,7o (C = 0,92, метанол).
с) (S)-[3-Метил-1-(1Н-тетразол-5-ил)бутил] карбамановая кислота, фенилметильный эфир.
Раствор указанного в названии соединения В (2,8 г, 12,10 ммоля) и трибутилоловоазид (6,1 г, 18,14 ммоля) в 100 мл ксилола перемешивались при 110оС в течение 2 ч. Смесь охлаждалась до комнатной температуры и подавалась непосредственно на тонкослойную хроматографическую колонну с элюированием 1 л смеси гексаны:этилацетат (60:40), затем 2 л (60:40) гексаны: этилацетат плюс 2% уксусная кислота, получалось 2,23 г указанного в названии соединения в виде густого масла.
d) (S)-L-(2-метилпропил)-1Н-тетразол-5-метанамин, гидрохлорид.
Суспензия указанного в названии соединения С (2,84 г, 9,81 ммоля) и 100 мл метанола, содержащего ацетилхлорид (1,14 мл, 19,62 ммоля) и 20% палладий на угольном катализаторе 0,28 г, перемешивалась в атмосфере водорода (баллон) при комнатной температуре. Через 4 ч добавлялось дополнительное количество 20% палладия на угольном катализаторе (0,28 г) и ацетилхлорид (1,14 мл). Через 22 ч реакционная смесь отфильтровывалась и выпаривалась. После измельчения гексанами получалось 1,88 г указанного в названии соединения D в виде белой пены.
е) (S,R)-2-[(ацетилтио)метил]-3,3,3-трифтор-N-[метил-1-(Н-тетразол-5-ил) бутил]-пропанамид.
К раствору указанного в названии соединения D (1,73 г, 9,9 ммоля) в 22 мл ацетонитрила при 0оС в атмосфере аргона добавлялся бис(триметилсилил)трифторацетамид (7,67 мл, 31,36 ммоля) и полученный мутный раствор перемешивался при комнатной температуре в течение 2 ч. Реакционная смесь охлаждалась вновь до 0оС, за это время добавлялся 2-трифторметил-3-ацетилтиопропионилхлорид (2,1 г, 9,8 ммоля) в ацетонитриле (6 мл).
После перемешивания светло-желтого раствора в течение 2 ч при 0оС реакция гасилась путем переливания реакционной смеси в воду и затем экстрагировалась этилацетатом (2 х 250 мл). Объединенные органические экстракты подвергались тонкослойной хроматографии (900 мм х 10 дюйм - 60:40 гексаны: этилацетат + 2% уксусной кислоты, 90 мм х 7 дюйм - 1:1 гексаны:этилацетат + 0,5% уксусной кислоты, 90 мм х 12 дюйм - 1:1 гексаны:этилацетат + 0,5% уксусной кислоты, 50 мм х 10 дюйм - 1:1 гексаны:этилацетат + 0,5% уксусной кислоты) с получением после перекристаллизации из этилацетата/гексанов 1,10 г указанного в названии изомера (S,R)E с т.пл. 188-191оС, при тонкослойной хроматографии Rf = 0,22 (гексаны/этилацетат + 0,5% уксусная кислота), [α]D = 163,8o (C = 0,53, метанол) и 1,15 г (S,S)-2-[(ацетилтио)метил]-3,3,3-трифтор-N-[3-метил-1Н-тетразол-5-ил) бутил] пропанамида] с температурой плавления 180-184оС, при тонкослойной хроматографии Rf = 0,16 (гексаны) этилацетат + 0,5% уксусной кислоты).
[α]D = +60,6o (C = 0,51, метанол).
f) (S,R)-3,3,3-трифтор-2-(меркаптометил)-N-[3-метил-1-(1Н-тетразол-5-ил) бутил]пропанамид.
К суспензии изомера (S,R) указанного в названии соединения Е (0,98 г, 2,8 ммоль), в 5,65 мл дегазированной воды при комнатной температуре в атмосфере аргона добавлялась смесь концентрированной NH4OH (1,85 мл) и вода (1,85 мл).
Через две минуты реакция гасилась насыщенным бисульфатом калия (рН 1,5) и реакционная смесь экстрагировалась этилацетатом. Органические экстракты высушивались и очищались тонкослойной хроматографией (5 мм х 6 дюйм, 1:1 гексаны: этилацетат + 0,5% уксусной кислоты с получением 0,72 г белого твердого продукта. Этот материал измельчался гептаном и высушивался при глубоком вакууме при 100оС с получением 0,7 г указанного в названии продукта (S,R) с т.пл. 189-195оС (разложение), при тонкослойной хроматографии Rf = 0,18 (1:1 этилацетат:гексан + 0,5% уксусная кислота), [α]D = -97,7o (C = 0,57, метанол).
Вычислено, %: C 39,02; H 5,29; N 21,88; F 17,80; S 10,01; SH 10,33.
C10H16F3N5OS˙0,1CH3COOC2H5.
Найдено, %: C 39,25; H 5,35; N 21,54; F 17,60; S 10,17; SH 10,60.
П р и м е р 36. (S,S)-3,3,3-Трифтор-2-(меркаптометил)-N-[3-метил-1-(1Н-тетразол-5-ил) бутил]пропанамид.
К суспензии (S,S)-2-[(ацетилтио)-метил]-3,3,3-трифтор-N-[3-метил-1-(1Н-тетразол-5- ил) бутил]пропанамида (полученного в примере 39 с, 1,034 г, 2,95 ммоля) в 6 мл дегазированной воды при комнатной температуре в атмосфере аргона добавлялась смесь концентрированной NH4OH (1,95 мл) и вода (1,95 мл).
Через 2 мин реакция гасилась насыщенным бисульфатом калия (рН 1,5) и экстрагировалась этилацетатом. Органические экстракты высушивались и очищались тонкослойной хроматографией (50 мм х 6 дюйм, 1:1 гексаны:этилацетат + 0,5% уксусная кислота) с получением 0,76 г белого твердого продукта.
Этот материал объединялся с 25 мг продукта из предыдущего опыта и перекристаллизовывался из этилацетата (гексанов с получением 0,69 г указанного в названии (S,S) изомера в виде белого твердого продукта с т.пл. 183-184оС, при тонкослойной хроматографии Rf = 0,14 (1:1 гексаны:этилацетат + 0,5% уксусной кислоты), [α]D = -39,8o (C = 0,57, метанол).
Вычислено, %: C 38,58; H 5,18; N 22,50; F 18,31; S 10,30; SH 10,62.
Найдено, %: C 38,70; H 5,27; N 22,43; F 17,67; S 10,28; SH 11,52.
П р и м е р 37. (S)-3-гидрокси-N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил] -L-тирозин.
а) (S)-3-гидрокси-N-[2-(ацетилтио)метил] -3,3,3-трифтор-1-оксопропил] -L-тирозин.
Бис(триметилсилил)трифторацетамид (16 мл, 60 ммоля) добавлялся к суспензии 3 гидрокси-L-тирозина (2,37 г, 12 ммоля) в 25 мл ацетонитрила при 0оС. Суспензия перемешивалась в течение 6 ч при комнатной температуре, а затем охлаждалась до 0оС. Добавлялся раствор 2-трифторметил-3-ацетилтио-пропионилхлорида (2,81 г, 12 ммоль) в 5 мл ацетонитрила и реакционная смесь перемешивалась при комнатной температуре в течение 20 ч. Затем реакционная смесь выливалась в приблизительно 130 мл 1н. HCl, двухфазная смесь перемешивалась при комнатной температуре в течение 30 мин. Затем смесь экстрагировалась этилацетатом (2 х 200 мл) и объединенный органический слой промывался водой (200 мл), затем рассолом (100 мл), высушивался (MgSO4) и концентрировался с получением маслянистого остатка. Проводилось несколько хроматографий и получалось 1,71 г (R)-3-гидрокси-N-[2-[(ацетил-тио)метил]-3,3,3-трифтор-1-оксопропил] -L-тирозин, = -6,5о (С = 0,15, метанол), который выделялся в виде белой пены, и 1,63 г (S)-3-гидрокси-N-[2-[(ацетилтио)метил] -3,3,3-трифтор-1-оксо- пропил] -L-тирозин с т.пл. 175-184оС, [α]D= = +106,3 (C = 0,49, метанол), в виде не совсем белого аморфного твердого продукта.
b) (S)-3-гидрокси-N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил] -L-тирозин.
Концентрированный гидроксид аммония (2,5 мл) добавлялся к суспензии (S) изомера продукта указанного в названии А (1,4 г, 3,54 ммоля) в обедненной кислородом воде при комнатной температуре. После перемешивания мутной реакционной смеси в течение 1 мин к ней добавлялся насыщенный раствор бисульфата калия до достижения рН около 1,5. Подкисленная смесь экстрагировалась этилацетатом (2 х 125 мл) и объединенные органические слои промывались рассолом, высушивались (MgSO4) и концентрировались. Осадок хроматографировался на 5 х 12 см колонне силикагеля с использованием в качестве движущейся фазы смеси этилацетат:гексан:уксусная кислота 55:45:2.
Самые чистые фракции концентрировались с получением 997 мг белой пены, которая повторно хроматографировалась на 5 х x12 см колонне силикагеля с использованием в качестве движущейся фазы этилацетат:гексан:уксусная кислота. Самые чистые фракции концентрировались с получением пенообразного остатка, который окончательно выпаривался из толуола, гептана, метиленхлорида и гексана. Материал тщательно измельчают в порошок гептаном и пентаном и всю ночь высушивают под глубоким вакуумом при комнатной температуре с получением 780 мг указанного в названии (S) изомера в виде аморфного твердого продукта с температурой плавления 103-107оС (разложение). [α]D = -52,5 (C = 0,55, метанол).
Вычислено, %: C 43,28; H 4,14; N 3,88; F 15,80; S 8,89; SH 9,17.
C3H14F3NO2S˙0,42H2O.
Найдено, %: C 43,51; H 4,40; N 3.65; F 15,63; S 8,90; SH 9,67.
П р и м е р 38. (R)-3-Гидрокси-N-[3,3,3-трифтор-(2-меркаптометил)-1-оксопропил] -L-тирозин.
Концентрированный гидроксид аммония (2,6 мл) добавлялся к суспензии (R)-3-гидрокси-N-[2-[(ацетилтио)метил]3,3,3-три-фтор-1-оксопропиля] -L-тирозина (1,5 г, 3,8 ммоль) из примера 45а в обедненной кислородом воде при комнатной температуре. После перемешивания в течение 1 мин к смеси добавлялся насыщенный раствор бисульфата калия до достижения рН около 1,5. Подкисленная смесь экстрагировалась этилацетатом (2 х 125 мл) и объединенные органические слои промывались рассолом, высушивались (MgSO4) и концентрировались. Остаток хроматографировался на 5 х 15 см колонне силикагеля с использованием продвигающей фазы этилацетат:гексан:уксусная кислота 55:45: 2. Чистые фракции концентрировались и выпаривались совместно из гексана с получением твердого продукта, который измельчался в порошок гексаном и перекристаллизовывался из изопропанол/гексана. Кристаллический порошок измельчался в тонкую пудру и перемешивался в гептане в течение 3 ч для удаления удержанного изопропанола. Затем материал отфильтровывался, промывался гексаном и высушивался при 95оС при глубоком вакууме в течение 24 ч с получением 586 мг указанного в названии изомера (R) в виде белого порошка с т.пл. 176-177оС, [α]D = +2,5o (C = 0,32, метанол).
Вычислено, %: C 43,85; H 4,05; N 3,93; F 16,00; S 9,00; SH 9,20.
C13H14F3NO5S˙0,16H2O.
Найдено, %: C 43,99; H 4,22; N 3,79; F 15,60; S 8,87; SH 10,5.
П р и м е р 39. (S,S)-L-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил] -амино] циклогексанированная кислота.
а) N-[2-[Ацетилтио/метил]-3,3,3-трифтор-1-оксопропил]-L-циклогексилаланин.
Бис(триметилсилил)трифторпентамид (12,2 мл, 45,6 ммоля) добавлялся к суспензии L-циклогексиламина (2,6 г, 15,2 ммоля) в 35 мл ацетонитрила при 0оС. Суспензия перемешивалась в течение 6 ч при комнатной температуре. Значительное количество твердого продукта оставалось. После охлаждения реакционной смеси до 0оС по каплям добавлялся -2-трифторметил-3-ацетилтиопропионилхлорид в виде раствора в 10 мл ацетонитрила.
После перемешивания в течение 24 ч при комнатной температуре реакционная смесь была в виде прозрачного желтого раствора. Реакционная смесь распределялась между этилацетатом (150 мл) и 1н. HCl (150 мл). Водный слой экстрагировался дополнительным количеством этилацетата. Объединенные органические слои промывались водой (2 х 400 мл) и рассолом (200 мл), высушивались (MgSO4) и концентрировались до маслянистого остатка.
После ряда хроматографий и нескольких перекристаллизаций получалось 1,7 г (R)-N-[2(ацетилтио)метил]-3,3,3-трифтор-1-оксопропил]-N-циклогексилаланин с температурой плавления 142-145оС, [α]D = -135,1o (C = 0,37, метанол) и 2,19 г (S)-N-[2-(ацетилтио/-метил]-3,3,3-трифтор-1-оксопропил]-L-циклогексиланин с т.пл. 106-109оС.
[α]D = +97,2o (C = 0,36, метанол).
b) (S,S)-α-[[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил]амино]циклогексан-про панокислота.
Концентрированная гидроокись аммония (3,1 мл) добавлялась к суспензии указанного в названии изомера (S), продукта из части (е), (1,734 г, 4,69 ммоля) в воде, обедненной кислородом, при комнатной температуре. Растворение происходило почти мгновенно и реакционная смесь перемешивалась в течение 10 мин при комнатной температуре. Величина рН приблизилась к 1,5 посредством насыщенного раствора бисульфата калия и смесь экстрагировалась этилацетатом (200 мл). Органический слой промывался рассолом, высушивался (MgSO4) и концентрировался, остаток хроматографировался на колонне 5 х 15 см силикагеля.
Элюирование осуществлялось смесью гексан:этилацетат:уксусная кислота 80:20:2, концентрированием чистых фракций и выпариванием из гептана получали белый твердый продукт, который измельчался в порошок гексаном.
Этот твердый продукт высушивался под глубоким вакуумом в течение 18 ч с получением 1,28 г указанного в названии изомера с т.пл. 141-143оС, [α]D = +13,6o (C = 0,85, метанол).
Вычислено, %: C 47,70; H 6,16; N 4,28; F 17,41; S 9,79; SH 10,10.
C13H20F3NO3S.
Найдено, %: C 47,61; H 6,15; N 4,12; F 17,24; S 9,49; SH 11,77.
П р и м е р 40. (S,R)-α-[[3,3,3-трифтор-2-(меркаптометил-1-оксопропил] амино]-циклогексанпроп ановкислота.
Концентрированная гидроокись аммония (2,6 мл) добавлялась к суспензии (R)-N-[2[(ацетилтио)метил]-3,3,3-трифтор-1-оксо-пропил]-L-циклогексилаланина , полученного так, как указано в примере 47а, в 10,5 мл обедненной кислородом воды при комнатной температуре. Растворение осуществлялось практически мгновенно и полученный раствор перемешивался в течение 5 мин.
За этом время реакционная смесь подкислялась до рН 1,5 насыщенным раствором бисульфата калия. Полученная суспензия экстрагировалась этилацетатом (около 250 мл). Органический слой промывался рассолом, высушивался (MgSO4) и концентрировался с получением не совсем белого твердого продукта. Материал хроматографировался на 2,5 х 20 см колонне силикагеля с использованием в качестве мобильной фазы этилацетата:гексана, уксусной кислоты 80:20:2. После концентрации чистых фракций, выпаривания из гептана и измельчения в порошок гексаном получалось 858 мг указанного в названии изомера в виде белого твердого продукта с т.пл. 116-118 [α]D = -47,2o (C = 0,65, метанол).
Вычислено, %: C 47,69; H 6,16; N 4,28; F 17,41; S 9,79; SH 10,10.
C13H20F3NO3S.
Найдено, %: C 47,89; H 6,29; N 4,25; F 16,76; S 9,66; SH 10,30.
П р и м е р 41. (R)-N-[2-(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил] -L-тирозин.
Cуспензия L-тирозина (2,72 г, 15 ммолей) в 50 мл сухого ацетонитрила в атмосфере аргона охлаждалась до 0-5оС и к ней добавлялся бис(триметилсилил)трифторрацетамид (12 мл, 45 ммолей).
Реакционная смесь перемешивалась и постепенно нагревалась до комнатной температуры. Через 2 ч добавлялось 3 мл диметилформамида и смесь перемешивалась всю ночь при комнатной температуре. Поскольку реакционная смесь представляла прозрачную суспензию, к ней добавлялось еще 2 мл диметилформамида и после перемешивания в течение 2 ч при комнатной температуре добавлялось еще 3 мл бис(триметилсилил)трифторацетамида, получался через 2,5 ч прозрачный раствор.
Реакционная смесь охлаждалась до 0-5оС и по каплям добавлялся 2 трифтор-3-ацетилтиопропионилхлорид (3,52 г, 15 ммолей), растворенный в 7 мл ацетонитрила, и смесь постепенно нагревалась до комнатной температуры. Реакционная смесь выпаривалась до получения желтого сиропообразного остатка, распределялась между этилацетатом (100 мл) и водой (100 мл).
Водный слой затем экстрагировался дополнительным количеством этилацетата (4 х 75 мл) и объединенный этилацетатный экстракт промывался рассолом (75 мл), высушивался (MgSO4) и выпаривался с получением 10,5 г желтого твердого остатка. Остаток соединялся с 2,5 г дополнительного неочищенного материала, полученного в другом опыте. Тонкослойной хроматографией этих 13 г неочищенного материала на силикагеле (120:1 силикагель:соединение) c использованием в качестве элюента этилацетат:гексаны:уксусная кислота 100:100:1 получали 2,29 г быстроэлюирующего диастереомера (изомер А), 2,31 г смеси обоих стереомеров и 1,88 г медленно элюирующего диастереомера (изомер В).
Часть 2,29 г измельчалась в порошок этилацетатом (гексанами (1:1) с получением 1,25 г чистого изомера А и 0,83 г маточного раствора, содержащего оба изомера. Аналогичной обработкой 1,88 г материала этилацетатa:гексанами (1: 2) получалось 1,2 г чистого изомера В и 0,38 г маточного раствора, который содержал преимущественно изомер В. Кристаллизацией фракции 2,31 г из этилацетат:гексанов (1:1) получалось 0,8 г чистого изомера А и 1,5 г смешанных изомеров А и В. Объединенные смешанные фракции 2,71 г вновь хроматографировались дважды при тех же условиях и получалось 0,41 г чистого изомера А и 1,14 г чистого изомера В. Порции 0,8 г, 1,25 г, 0,41 г чистого изомера А объединялись и вначале измельчались гексанами, содержащими несколько капель этилацетата, и затем перекристаллизовывались из этилацетат:гексаны (1:1) с получением 1,6 г указанного в названии изомера (R) в виде белого кристаллического твердого продукта с температурой плавления 202-204оС, [α]D = -85,7 (C = 1,0, метанол).
Вычислено, %: C 47,49; H 4,25; N 3,69; F 15,02; S 8,45.
C15H16F3NO5S.
Найдено, %: C 47,55; H 4,16; N 3,62; F 14,70; S 8,25.
П р и м е р 42. (S)-N-[2-[(Ацетилтио)метил]-3,3,3-трифтор-1-оксопропил] -L-тирозин.
Порции 1,2 г и 1,14 г очищенного изомера В из примера 49 объединялись и измельчались в порошок гексаном, содержащим несколько капель этилацетата, с получением 2,2 г указанного в названии соединения (S) изомера в виде белого твердого продукта с т.пл. 170-173оС, [α]D = +11,8o (C = 1,00, метанол).
Вычислено, %: C 47,49; H 4,25; N 3,69; F 15,02; S 8,45;
C15H16F3NO5S.
Найдено, %: C 47,49; H 4,2; N 3,67; F 14,67; S 8,27.
П р и м е р 43. (R)-N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил]тирозин.
Суспензия изомера (R) из примера 49 (1,66 г, 4,4 ммоля) и 2,8 мл концентрированной гидроокиси аммония и 6,2 мл воды перемешивалась при комнатной температуре в атмосфере аргона в течение 7 мин. Полученный прозрачный раствор подкислялся 5% -ным раствором бисульфата калия до рН 1,5 и экстрагировался 75 мл этилацетата, а затем дополнительно 3 х 50 мл этилацетата.
Объединенный этилацетатный экстракт промывался рассолом (75 мл), высушивался (MgSO4) и выпаривался с выходом 1,48 г твердого кристаллического продукта. Очистка тонкослойной хроматографией неочищенногго продукта на силикагеле (240:1 силикагель:гексаны:уксусная кислота 100:100:1 позволяла получить 1,32 г указанного в названии изомера (R) в виде белого кристаллического продукта с т.пл. 204-206оС, [α]D = +5,2o (C = 1,0, метанол).
Вычислено, %: C 46,29; H 4,18; N 4,15; F 16,90; S 9,50; SH 9,80.
Найдено, %: C 46,32;H 4,02; N 4,04; F 16,60; S 9,52; SH 9.86.
П р и м е р 44. (S)-N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил]-L-тирозин.
Cуспензия изомера (S) из примера 50 (2,05 г, 5,4 ммоля) в 3,5 мл концентрированной гидроокиси аммония и 7,6 мл воды перемешивались при комнатной температуре в атмосфере аргона в течение 7 мин. Прозрачный раствор подкислялся 150 мл 5%-ного раствора бисульфата калия и экстрагировался 75 мл этилацетата, а затем дополнительно 4 х 50 мл этилацетата. Объединенный этилацетатный экстракт промывался рассолом (100 мл), высушивался (MgSO4) и выпаривался при пониженном давлении, получилось 1,95 г не совсем белого твердого продукта. Тонкослойной хроматографией на 400 г силикагеля с использованием в качестве элюента смеси этилацетат:гексаны:уксусная кислота 100:100:1 получалось 1,56 г указанного в названии изомера (S) в виде белого кристаллического продукта с температурой плавления 184-187оС [α]D = + 59,0о (С = 1,0, метанол).
Bычислено, %: C 46,29; H 4,18; N 4,15; F 16,90; S 9,50; SH 9,80.
C13H14F3NO4S.
Найдено, %: C 46,38; H 4,15; N 4,12; F 16,73; S 9,37; SH 9,44.
П р и м е р 45. (S,S)-3,3,3-трифтор-2-(меркаптометил)-N-[2-(1Н-индол-3-ил)-1 (1Н-тетразол-5-ил)этил]-пропанамид.
a) (S)-L-(1H-тетразол-3-ил/)-1H-индол-3-этанамин, гидрохлорид.
К раствору N-[(1,1-диметилэтокси)-карбонил]-L-триптофана (7,74 г, 25,42 ммоля) в метиленхлориде (150 мл) при -20оС в атмосфере аргона добавлялся N-метилморфолин (2,79 мл, 25,42 ммоля), затем изобутилхлорформат (3,31 мл, 25,42 ммоля).
Полученная суспензия перемешивалась при -20оС в течение 15 мин, за это время добавлялось 5,6М NH3/метанол (45 мл, 252 ммолей). Через 30 мин при -20оС реакция гасилась водой и нагревалась до комнатной температуры. Слои разделялись и водный слой экстрагировался дважды метиленхлоридом.
Объединенные органические экстракты высушивались и выпаривались с получением 8,0 г белого твердого продукта. Перекристаллизацией из этилацетата/гексанов получалось 6,6 г N-[1,1-диметилэтокси)карбонил]-L-триптофанамида.
К раствору триэтиламина (10,5 мл, 75,16 ммоля) в бензоле (40 мл) при 10оС в атмосфере аргона добавлялся хлорсульфонилметилкарбамат (6,5 г, 27,58 ммол), растворенный в бензоле (70 мл) в течение 30 мин. Реакционная смесь перемешивалась дополнительно в течение 30 мин при комнатной температуре, после чего добавлялся N-[(1,1-диметилэтокси)-карбонил]-L-трипто- фенамид (5,7 г, 18,79 ммоля), растворенный в безводном тетрагидрофуране (100 мл), через 10 мин. Полученная смесь перемешивалась в течение часа и затем поступала на тонкослойную хроматографию в колонну (50 мм х 6 дюйм, элюирование раствором 60: 40 гексаны:этилацетат) с получением 4,22 г N-[(1,1-диметилэтокси)карбонил]-L-триптофе- нилнитрила в виде белого твердого продукта, при тонкослойной хроматографии Rf = 0,47 (1:1 этилацетат:гексаны), [α]D = -85,8o (C = 0,57, метанол).
Раствор этого нитрильного соединения (2,42 г, 8,48 ммоля) и трибутилоловоазид (4,25 г, 12,72 ммоля) в ксилолах (20 мл) перемешивался при 90оС в течение 1 ч. Затем смесь охлаждалась до комнатной температуры и подавалась непосредственно на тонкослойную хроматографическую колонну (50 мм х 8 дюйм, элюирование 2 л 60:40 гексаны:этилацетат + 2%-ная уксусная кислота и 1 л 3: 1 этилацетат:гексаны + 2%-ная уксусная кислота), и получалось 1,91 г (S-[2-(1H-индол-3-ил)-1-(1Н-тетразол-5-ил)этил]-карбаминовая кислота, 1,1-диметилэтил эфира в виде белого твердого продукта. При тонкослойной хроматографии Rf = 0,09 (60:40 гексаны:этилацетат + 2% уксусная кислота), [α]D = +2,23o (C = 0,71, метанол).
Этилацетат (30 мл) насыщался HCl в газообразном состоянии при 0оС, при этом добавлялся анизол (7,4 мл) затем (S)-[2-(H-индол-3-ил)-1-(1Н-тетразол-5-ил)этил] кар- баминовая кислота, 1,1-диметилэтил эфир (1,24 г, 3,77 ммол), растворенный в этилацетате (7 мл). Полученный раствор перемешивался при 0оС в течение 1 ч, тонкослойная хроматография показала, что исходного материала не осталось.
Реакционная смесь перемешивалась и выдавливалась метиленхлоридом и эфиром, и окончательно выкачивалась в количестве 0,95 г в виде (S)-L-(1H-тетразол-5-ил)-1Н-индол-3-этанамин, гидрохлорида в виде светлокоричневого твердого продукта.
b) (2S)-трифторметил-3-ацетилтиопропионовая кислота, эфедриновая соль.
2-трифторметил-3-ацетилтиопропио- новая кислота (7,18 г, 33,2 ммоля) в эфире (100 мл) обрабатывалась (1S, 2R)-(+)-эфедрином (2,73 г, 16,6 ммоля) в эфире (100 мл) и потом она отстаивалась в течение 18 ч. Затем реакционная смесь фильтровалась с получением 5,37 г твердого продукта, который перекристаллизовывался из этилацетата (4 х 40 мл) с получением 3,47 г (2S)-трифторметил-3-ацетилтиопропионовая кислота, эфедриновая соль, т.пл. 139-140оС, [α]D = +100,0o (C = 1,00, метанол).
с) (S, S)-2[(ацетилтио)метил] -3,3,3-трифтор-N-[2-(1H-индол-3-ил)-1- (1Н-тетразол-5-ил)этил]пропанамид.
Эфедриновая соль продукта из раздела b) (2,2 г, 5,74 ммоля) встряхивалась в делительной воронке с этиловым эфиром (50 мл) и водой (50 мл), содержащей 1н. HCl (5,74 мл). Органический слой отделялся, высушивался (MgSO4), выпаривался и смешивался в азеотропную смесь с метиленхлоридом.
Полученная свободная карбоновая кислота растворялась в метиленхлориде (14 мл), к которой был добавлен оксалилхлорид (0,5 мл, 5,74 ммол), а затем осторожно по каплям диметилформамид (0,05 мл). Полученная смесь перемешивалась при комнатной температуре в течение 2 ч. В это время к суспензии указанного в названии продукта А (1,73 г, 5,74 ммоля) в ацетонитриле (30 мл) при 0оС в атмосфере аргона добавлялся бис(триметилоилил)трифторацетамид (6,3 мл, 24,12 ммоля) и полученный мутный раствор перемешивался при комнатной температуре в течение 2 ч. Затем этот раствор охлаждался до -25оС и за это время к нему по каплям в течение 15 мин добавлялся (2S)-трифторметил-3-ацетил-тиопропионовая кислота хлорид. После перемешивания светло-желтого раствора в течение 2 ч при -25оС до -15оС реакция гасилась путем слива в воду (75 мл) и смесь экстрагировалась этилацетатом (2 х 75 мл).
Объединенные органические экстракты промывались рассолом (75 мл), высушивались (Na2SO4) и выпаривались с получением твердого продукта. Тонкослойной хроматографией (50 мМ х 8 дюйм в колонне при элюировании 2 л 1:1 гексан: этилацетат + 1% уксусная кислота, 1 л 3;1 этилацетат:гексаны + 2% уксусная кислота, 1 л 4:1 этилацетат:гексаны +2%-ная уксусная кислота) получалось после перекристаллизации из этилацетат/гексанов 1,52 г указанного в названии изомера (S, S) в виде белого твердого продукта с т.пл. 208-213оС (разложение), при тонкослойной хроматографии Rf - 0,13 (60:40 гексаны:этилацетат +2% уксусная кислота), [α]D = -163,8o (C = 0,53, метанол).
d) (S,S)-3,3,3-трифтор-2-(меркаптометил)-N-[2-(1H-индол-3-ил) -1-(1Н-тетразол-5-ил)этил)]-пропанамид.
К суспензии указанного в названии продукта С (1,14 г, 2,67 ммол) в дегазированной воде (5,3 мл) при комнатной температуре добавлялась смесь концентрированной NH4OH (1,76 мл) и вода (1,76 мл). Через 2 мин реакция гасилась насыщенным бисульфатом калия (рН 1,5) и полученный твердый продукт отфильтровывался и очищался измельчением смесью этилацетата (этиловый эфир) гексаны с получением 0,73 г (S,S) изомера в виде белого твердого продукта с т.пл. 218-223оС (разложение), при тонкослойной хроматографии Rf = 0,13 (60: 40 гексаны) этилацетат + 2%-ная уксусная кислота), [α]D = +54,8o (C = 0,54, метанол).
Вычислено, %: C 46,87; H 3,93; N 21,86; F 14,83; S 8,34; SH 8,60.
C15H15F3N6O5S.
Найдено, %: C 47,22; H 3,80; N 21,71; F 14,99; S 8,05; SH 12,35.
П р и м е р 46. (S,R)-3,3,3-Трифтор-2-(меркаптометил)-N-[2-1Н-индол-3-ил-1-(1Н-тетразол-5-ил ) этил]пропанамид.
а)(2R)-трифторметил-3-ацетилтиопро- пионовая кислота, эфедриновая соль.
Фильтрат, полученный в примере 53b, выпаривался, обрабатывался водным HCl и затем экстрагировался в этилацетате, высушивался и выпаривался с получением 3,8 г масла. Это масло растворялось в эфире (40 мл) и объединялось с (1R, 2S)-(-)-эфедрином (2,9 г, 17,6 ммоля), растворенным в эфире (40 мл) и выстаивалось в течение 16 ч. Твердый продукт отфильтровывался и перекристаллизовывался из этилацетата (2 х 40 мл) с получением 4,0 г (2R)-трифторметил-3-ацетилтиопропионовая кислота, эфедриновой соли с т.пл. 138-139оС, [α]D = -100,3o (С = 1,0, метанол).
b) (S,R)-2-[(ацетилтио)метил]-3,3,3-трифтор-N-[2-(1Н-индол-3-ил)-1-(1Н- тетразол-5-ил)этил]пропанамид.
Эфедриновая соль указанного в названии продукта А (1,53 г, 4,0 ммол) встряхивалась в разделительной воронке с этиловым эфиром (50 мл) и водой (50 мл), содержащей 1н. HCl (4,0 мл). Органический слой отделялся, высушивался (MgSO4), выпаривался и переводился в азеотропное состояние метиленхлоридом. Полученная свободная карбоновая кислота растворялась в метиленхлориде (10 мл), к которому был добавлен оксалилхлорид (0,35 мл, 4,0 ммол), а затем осторожно по каплям добавлялся диметилформамид (0,035 мл). Полученная смесь перемешивалась при комнатной температуре в течение двух часов. В это же время к суспензии (S)-α-(1H-тетразол-5-ил)-1Н-индол-3-этанамина гидрохлорида (1,73 г, 5,74 ммол) в ацетонитриле (21 мл) при 0оС в атмосфере аргона добавлялся бис(триметилсилил)трифторацетамид (4,4 мл, 16,8 ммол) и полученный мутный раствор перемешивался при комнатной температуре в течение 2 ч. Затем этот раствор охлаждался до -25оС и по каплям к нему добавлялся раствор хлорангидрида в течение 15 мин. После перемешивания светло-желтого раствора в течение 2 ч при -25оС до -15оС реакция гасилась путем сливания в воду (50 мл) и реакционная смесь экстрагировалась этилацетатом (2 х 50 мл). Объединенные органические экстракты промывались рассолом 50 мл) высушивались (Mg2SO4) и выпаривались с получением твердого продукта. Тонкослойной хроматографией (50 мл х 8 дюйм элюированием смесью 2 л 1:1 гексаны:этилацетат + 1%-ная уксусная кислота, 1 л 3:1 этилацетат:гексаны: + 1% -ная уксусная кислота) получалось после перекристаллизации из этилацетатгексанов 0,68 г указанного в названии белого твердого продукта В. маточные растворы выпаривались и вновь хроматографировались (25 мм х 6 дюйм, элюирование 2 л 1:1 гексаны:этилацетат + 1%-ная уксусная кислота) с получением после перекристаллизации из этилацетат/гексанов дополнительного количества 0,26 г белого твердого продукта с общим выходом 0,96 г указанного в названии продукта В с т.пл. 206-213оС (разложение), при тонкослойной хроматографии Rf = 0,23:60:40 гексаны:этил-ацетат: + 2%-ная уксусная кислота), [α]D = -93,9o (C = 0,52, метанол).
с) (S, R)-3,3,3-трифтор-2-(меркаптометил)-N-[2-(1Н-индол-3-ил)-1-(1Н- тетразол-3-ил)этил]пропанамид.
К суспензии указанного в названии продукта В (S,R) (0,9 г, 2,1 ммоля) в дегазированной воде (4,2 мл) при комнатной температуре в атмосфере добавлялась смесь концентрированного NH4OH (1,39 мл) и воды (1,39 мл). Через 2 мин реакция гасилась насыщенным бисульфатом калия (рН 1,5) и полученная смесь экстрагировалась в этилацетате. Органический слой промывался водой и рассолом, высушивался и выпаривался с получением твердого продукта, который очищался тонкослойной хроматографией (50 мм х 7 дюйм с элюированием смесью 1: 1 этилацетат:гексаны +1%-ная уксусная кислота). Полученный материал перекристаллизовывался из этилацетат/гексана с получением 0,59 г указанного в названии (S, R) продукта в виде белого твердого продукта с т.пл. 206-213оС (разложение), при тонкослойной хроматографии Rf = 0,11 (60:40 гексаны: этилацетат + 2%-ная уксусная кислота), [α]D = -38,6o (С = 0,42, метанол).
Вычислено, %: C 46,66; H 3,97; N 21,76; F 14,76; S 8,30; SH 8,56.
C15H15F3N6OS˙0,1H2O.
Найдено, %: C 46,93; H 3,80; N 21,38; F 15,06; S 8,38; SH 15,40.
П р и м е р 47. (S)-1-[N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил] -L-аланил]-L- пролин.
а) (S)-1-[N-[3,3,3-трифтор-2-[(ацетилтио)-метил]-1-оксопропил]-L-алинил -L-пролин.
Эфедриновая соль продукта из примера 53 (b) 1,64 г, 4,3 ммоля) распределялась между этилацетатом и водным HCl (6 ммолей). Органический слой промывался водой и рассолом, , высушивался (MgSO4) и концентрировался в вакууме с получением 0,94 г свободной кислоты. Материал растворялся в метиленхлориде (10 мл) при комнатной температуре и обрабатывался оксихлоридом (400 л, 573 мг, 4,51 ммол) и одной каплей диметилформамида. Через 1,5 ч этот раствор добавлялся к раствору L-аланил-L-пролин/бис(триметилсилил)трифторацетамида в ацетонитриле (полученному из L-аланил-L-пролина, тозилата (1,54 г, 4,3 ммол), диизопропилэтиламина (0,75 мл 554 мг, 4,3 ммол) и бис(триметилсилил)трифторацетамида (2,3 мл, 2,21 г, 8,6 ммол) в 15 мл ацетонитрила, перемешивался в течение 2 ч при 0оС до комнатной температуры с получением гомогенного раствора и перемешивался всю ночь. Летучие компоненты удалялись вакуумом и остаток, растворенный в этилацетате, промывался 10%-ным бисульфатом калия, водой и рассолом. Органическая фракция высушивалась (MgSO4) и концентрировалась в вакууме с получением масла. Тонкослойной хроматографией на 250 мл силикагеля элюированием этилацетат: гексан: метанол: уксусная кислота (10:6:2:0,2) получалось 1,2 г указанного в названии продукта А в виде пены, при тонкослойной хроматографии Rf = 0,29 (этилацетат:гексан:метанол:уксусная кислота 10:4:2:0,2).
b) (S)-1-[N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил] -L-аланил] -пролин.
Указанный в названии продукт А (1,2 г, 3,12 ммоль) в атмосфере аргона при 0оС обрабатывался гидроокисью аммония (7 мл 5 М раствора, полученного разбавлением концентрированной гидроокиси аммония дегазированной водой). Через 10 мин реакция гасилась 10%-ным бисульфатом калия (50 мл). Смесь экстрагировалась этилацетатом и органическая фракция промывалась водой и рассолом, высушивалась (MgSO4) и концентрировалась в вакууме с получением 1,0 г твердого продукта. Тонкослойной хроматографией на силикагеле (200 мл) и элюированием смесью этилацетат:гексан:метанол:уксусная кислота (10:6:2: 0,2) получался пенообразный продукт. Измельчением гексаном (содержащим несколько процентов этилацетата) получалось 750 мг указанного в названии (S) изомера, продукта с т.пл. 100-110оС, при тонкослойной хроматографии Rf = 0,37 (этилацетат:гексан:метанол:уксусная кислота:10:6:2:0,2), [α]D = -65,8o (C = 0,8, метанол).
Вычислено, %: C 41,53; H 5,09; N 8,07; F 16,42; S 9,24; SH 9,53.
C12H17F3N2O4S˙0,25H2O.
Найдено, %: C 41,75; H 4,87; N 7,85; F 16,41; S 9,24; SH 11,24.
П р и м е р 48. (R)-1-[N-[3,3,3-трифтор-2-(меркаптометил)-1-оксопропил] -L-аланил] -L-пролин.
(а) (R)-1-[N-[3,3,3-трифтор-2-[(ацетилтио)метил]-1-оксопропил]-L-аланил] -L-пролин.
Эфедриновая соль продукта из примера 54 (а) (1,66 г, 4,35 ммоля) превращалась в свободную кислоту и затем в хлорангидрид и взаимодействовала с раствором L-аланил-L-пролин/бис(триметилсилил)-трифтора- цетамида в ацетонитриле, как указано в примере 59(а), с получением 1,2 г указанного в названии продукта А в виде пены, при тонкослойной хроматографии Rf = 0,37 (этилацетат:гексан:метанол:уксусная кислота 12:6:2:0,2).
b) (R)-1-[N-[3,3,3-Трифтор-2-(меркаптометил)-1-оксопропил] -L-аланил] -L-пролин.
Указанный в названии продукт А (1,25 г, 3,25 ммол) обрабатывался гидроокисью аммония в соответствии с примером 59(b) выход после хроматографической очистки и измельчения гексаном (содержащим несколько процентов этилацетата) составил 740 мл изомера (R), указанного в названии продукта с т. пл. 100-105оС, при тонкослойной хроматографии Rf = 0,37 (этилацетат:гексан: метанол:уксусная кислота 12:6:2:0,2), [α]D = -124,2o (C = 1,1, метанол).
Bычислено, %: C 41,53; H 5,09; N 8,07; F 16,42; S 9,25; SH 9,53.
Найдено, %: C 41,75; H 4,95; N 8,01; F 16,59; S 9,36; SH 11,01.
П р и м е р 49. 1000 таблеток, каждая из которых содержит следующие ингредиенты, мг:
N-[[3,3,3-Трифтор-2-
(меркаптометил)-1-
оксопропил]-L-трип- тофан, изомер А 100 Кукурузный крахмал 50 Желатин 7,5
Авицел/микрокристал- лическая целлюлоза/ 25 Стеарат магния 2,5 готовились из достаточного количества компонентов путем смешивания продуктов из примера 9, кукурузного крахмала с водным раствором желатина. Затем смесь высушивалась и измельчалась в порошок. Затем примешивались с дроблением авицел и стеарата магния. Смесь прессовалась на прессе для изготовления таблеток и формировались 1000 таблеток, каждая из которых содержит 100 мг активного ингредиента. Аналогичным образом могут быть получены таблетки, содержащие 50 мг активного ингредиента. Аналогично могут быть приготовлены таблетки, содержащие 50 мг или 100 мг продукта из любого примера 1-8 или 10-83.
П р и м е р 50. Две желатиновые капсулы номер 1 заполнялись смесью, солстоящей из следующих ингредиентов, мг:
N-[3,3,3-Трифтор-2-
(меркаптометил)-1-
оксопропил]-L-лейцин, изомер В 100 Стеарат магния 7 Лактоза 193
Аналогичным образом могут быть приготовлены капсулы содержащие 100 мг продукта из примеров 1-20 и 22-83.
Биологические испытания.
Мочегонные свойства соединений по иону Na+ определяли в соответствии со следующей методикой. Эксперименты были поставлены на женских взрослых особях обезьян. После анестезии животным вводили катетеры Фоли непосредственно в мочевой канал и затем помещали животных в специальные стулья, оснащенные приспособлениями для ограничения движений. В течение опыта мочу собирали в коллектор, а мочевой пузырь промывали 5 мл дистиллированной воды с интервалами в 30 мин.
Испытуемые соединения (100 мкмоль/кг) вводили внутривенно и взятие проб продолжали в течение 3 ч с 30-минутными интервалами. Содержание Na+измеряли ион-селективным методом, а скорость выделения рассчитывали по стандартной формуле. Данные приведены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ АКТИВНОСТЬ АНГИОТЕНЗИНА II, И СПОСОБ ЛЕЧЕНИЯ ГИПЕРТОНИИ У МЛЕКОПИТАЮЩИХ | 1991 |
|
RU2111208C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2026296C1 |
| СЕРУЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МЕВИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2041205C1 |
| ПУРИНИЛ- ИЛИ ПИРИМИДИНИЛПРОИЗВОДНЫЕ МЕТИЛЕНЦИКЛОПЕНТИЛА | 1991 |
|
RU2037496C1 |
| Способ получения производных пролина | 1979 |
|
SU1115668A3 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА ИЛИ БЕНЗОТИАЗЕПИНА | 1989 |
|
RU2090562C1 |
| 7-ОКСАБИЦИКЛОГЕПТИЛЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ АМИДЫ ИЛИ ИХ СТЕРЕОИЗОМЕРЫ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ ТРОМБОКСАНА | 1991 |
|
RU2015980C1 |
| БИФЕНИЛЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ХИНОЛИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ | 1993 |
|
RU2122540C1 |
| Способ получения производных пролина или их основных солей | 1979 |
|
SU1066460A3 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАСТЕРЕОИЗОМЕРНОЙ ПАРЫ ФОСФИНИЛУКСУСНОЙ КИСЛОТЫ | 1991 |
|
RU2044740C1 |

Использование: в химии сероорганических веществ, в частности в синтезе нейтрального ингибитора эндопептидазы. Сущность изобретения: продукт - производные трифторметилмеркаптана ф-лы 1:  , где X - низший алкил, возможно замещенный гидроксифенилом или тетразолом, карбокси-низший алкил, возможно замещенный нафталеном, карбокси-низший-циклоалкил, 1-тетразолил-2-индолилэтил или остаток аминокислоты. Реагент 1: хлорангидрид кислоты ф-лы 2:
, где X - низший алкил, возможно замещенный гидроксифенилом или тетразолом, карбокси-низший алкил, возможно замещенный нафталеном, карбокси-низший-циклоалкил, 1-тетразолил-2-индолилэтил или остаток аминокислоты. Реагент 1: хлорангидрид кислоты ф-лы 2:  . Реагент 2: амин ф-лы 3: H2N-X . Условия реакции: в присутствии амина или в растворимом в воде карбодиимиде. Промежуточный продукт-соединение ф-лы 4:
. Реагент 2: амин ф-лы 3: H2N-X . Условия реакции: в присутствии амина или в растворимом в воде карбодиимиде. Промежуточный продукт-соединение ф-лы 4: 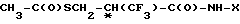 . Реагент 4: основание. 1 табл.
. Реагент 4: основание. 1 табл.
СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТРИФТОРМЕТИЛМЕРКАПТАНА формулы
HS-CH2-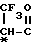 -NH-X
-NH-X
где X - низший алкил, возможно замещенный гидроксифенилом или тетразолом; карбоксинизший алкил, возможно замещенный нафталеном; карбоксинизший циклоалкил, 1-тетразолил-2-индолилэтал или остаток аминокислоты, выбранный из группы: l-аланин, l-фенилаланин, l-аспарагин, l-лейцин, l-норлейцин, l-изолейцин, l-аланил - l-пролин или l-тирозин,
отличающийся тем, что хлорангидрид формулы
CH3--- -S-CH2-
-S-CH2- -Cl
-Cl
подвергают взаимодействию с амином формулы
H 2 N - X,
где X имеет указанные значения,
или его хлористоводородной солью и полученное при этом промежуточное соединение формулы
CH3--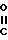 -S-CH2-
-S-CH2-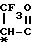 -NH-X
-NH-X
где X имеет указанные значения,
обрабатывают основанием.
| Мошковский М.Д | |||
| Лекарственные средства, М.:Медицина, 1986, ч.1, стр.469. |
