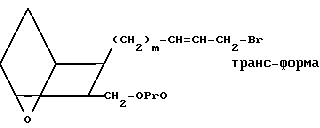
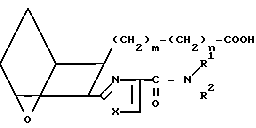
Изобретение относится к 7-оксабициклогептилзамещенным гетероциклическим амидам - аналогам простагландинов, которые являются антагонистами рецептора тромбоксана А2(ТХА2) или комбинированными антагонистами рецептора тромбоксана А2 - (ингибиторами тромбоксансинтазы, и используются, например, при лечении тромботической болезни и/или спазмов сосудов: обладают большой продолжительностью действия. Эти соединения имеют нижеследующую структурную формулу: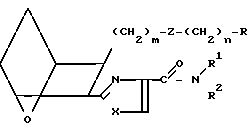
(I) включая все ее стереоизомеры, где m - 1, 2 или 3; n - 0, 1, 2, 3 или 4;
Z обозначает -(СН2)2- или -СН=СН-, при условии, что когда Z представляет собой -CH=CH-, n не равно 0;
R - карбоксил, его соль щелочного металла, карбокси (С1-С8)-алкил, CONHSO2(C1-C8)-алкил, CONHSO2-фенил; CONHSO2-бензил или СН2-5-тетразолил; Х - O, S или NH;
R1 - С1-С16-алкил, который может быть замещен С3-С8-циклоалкилом или фенилом, который может содержать атом галогена; С3-С8-циклоалкил; фенил, который может быть замещен галогеном;
R2 - водород или С1-С8-алкил, или R1 и R2, взятые вместе с соседним атомом азота, образуют 5-8-членное кольцо.
Таким образом, соединения формулы I включают соединения следующих типов: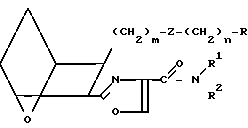 IA
IA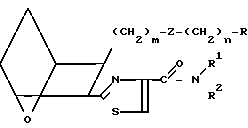 IB
IB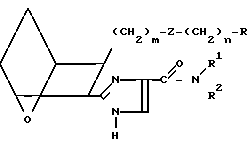 IC
IC
Предпочтительными являются те соединения формулы I, в которых Z представляет собой -СН=СН- в цис-конфигурации, m равно 1, n равно 2 или 3, R представляет собой CO2H, R1 представляет собой замещенный фенилалкил или циклогексилалкил, а R2 - водород или метил.
Предлагаемые соединения формулы I, в которых Z представляет собой -СН= СН- или -(СН2)2-, могут быть получены следующим образом.
Соединения, в которых Z представляет собой -СН=СН-, предпочтительно в цис-форме, а Х представляет собой O, получают, исходя из оксиметильного соединения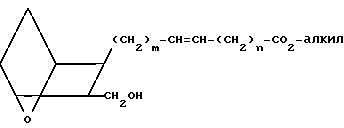
(II) (его получают, как описано в патенте США N 4143054), которое подвергают окислению по Джонсу, когда соединение II реагирует с реагентом Джонса (CrO3, растворенный или суспендированный в водном растворе серной кислоты, приготовляемый, как описано в книге: Физер и Физер, "Реагенты для органического синтеза", т. I, с. 142 (1967)), в присутствии ацетона, в инертной атмосфере, например в аргоне, при температуре от ≈- 10 до ≈20оС, с образованием соответствующей карбоновой кислоты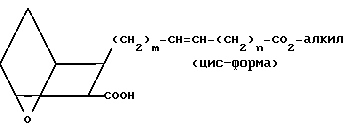
(III)
Затем в инертном органическом растворителе, например в тетрагидрофуране, проводят реакцию карбодиимидного связывания кислоты III с амидом A -
- -N
-N в присутствии дициклогексилкарбодиимида (DCC) или 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорида (WSC и 1-оксибензотриазола в инертной атмосфере, например в аргоне, используя мольное отношение A:III от ≈1,2:1 до ≈1:1, получая оксибисамид
в присутствии дициклогексилкарбодиимида (DCC) или 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорида (WSC и 1-оксибензотриазола в инертной атмосфере, например в аргоне, используя мольное отношение A:III от ≈1,2:1 до ≈1:1, получая оксибисамид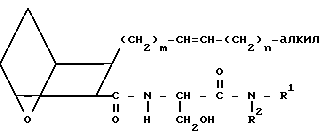
(IV)
Оксибисамид IV затем подвергают циклодегидратации, при которой раствор IV в инертном органическом растворителе, например в тетрагидрофуране, ацетонитриле или хлороформе, в инертной атмосфере, например в аргоне, обрабатывают трифенилфосфином и четыреххлористым углеродом в присутствии амина, например триметиламина, получая оксазолин.

(V)
В другом варианте оксибисамид IV обрабатывают сульфонилхлоридом, например метансульфонилхлоридом, и амином, например триэтиламином, с последующей обработкой карбонатом калия в ацетоне, получая оксазолин V.
Оксазолин V окисляют, обрабатывая диоксидом марганца или пероксидом никеля, предпочтительно последним, получая оксазол IA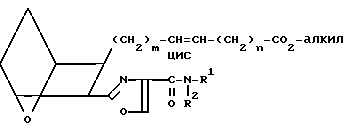
В другом варианте оксазол IA можно получить из кислоты III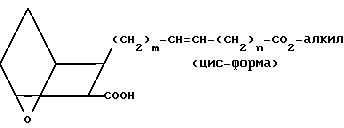 карбодиимидным связыванием, как это описано выше, с заменой A на A', получая VI,
карбодиимидным связыванием, как это описано выше, с заменой A на A', получая VI,
H2N -  A′
A′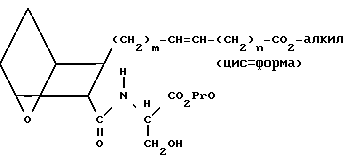
(VI) где Pro обозначает подходящую защищающую группу.
Оксиамид VI затем подвергают циклодегидратации и окислению, как это описано для IV и V, получая VII.
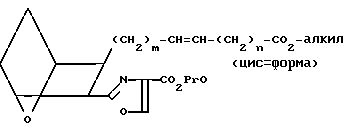
(VII)
Защищающая группа в соединении VII может быть удалена с образованием соответствующей кислоты VIII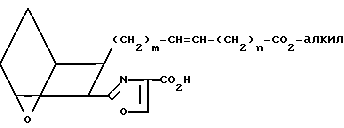
(VIII) которая при обработке избытком оксалилхлорида в присутствии инертного органического растворителя, например толуола, хлористого метилена или хлороформа, и, возможно, каталитического количества диметилформамида, при перемешивании в инертной атмосфере, например в аргоне, дает неочищенный хлорангидрид IX,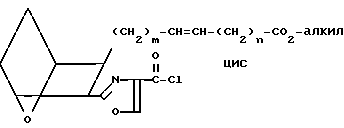
(IX) который обрабатывают солянокислой солью амина A"
HClH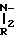 R1 A" в присутствии органического основания, например триэтиламина, в инертной атмосфере, например в аргоне, используя мольное отношение IX:A" от ≈0,5:1 до ≈1:1, предпочтительно от 0,8:1 до ≈1:1, получая IA'.
R1 A" в присутствии органического основания, например триэтиламина, в инертной атмосфере, например в аргоне, используя мольное отношение IX:A" от ≈0,5:1 до ≈1:1, предпочтительно от 0,8:1 до ≈1:1, получая IA'.

I A'
Соединения формулы I, в которых Z представляет собой трансизомер-СН= СН-, можно получить, исходя из оксиметильного соединения II, которое содержит двойную связь в цис-форме. Соединение II обрабатывают защищающим соединением, например трет-бутилдиметилсилилхлоридом или другой силильной защищающей группой, как описано выше, в присутствии имидазола или триэтиламина и инертного органического растворителя, например хлористого метилена или тетрагидрофурана, получая защищенное соединение X.
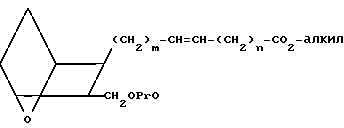
(X)
Раствор защищенного спирта Х в инертном органическом растворителе, например хлористом метилене или ацетоне, обрабатывают избытком озона при пониженной температуре от ≈- 78 до ≈- 60оС, с последующей обработкой избытком диметилсульфида (мольное отношение X:(CH3)2S - от ≈1:100 до ≈1:5), или трифенилфосфином, получая альдегид XI.
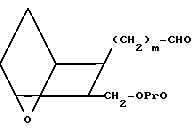
(XI)
Для заявляемых соединений, в которых Z представляет собой -СН=СН- в транс-форме, а n = 2, альдегид XI затем обрабатывают смесью бромида или хлорида лития и триметилфосфоноацетата и триэтиламина в инертном органическом растворителе, например, хлористом метилене или хлороформе, получая сложный эфир XII.
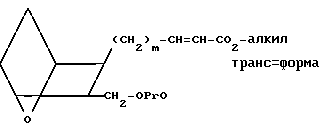
(XII)
Раствор сложного эфира XII в инертном органическом растворителе, например в тетрагидрофуране, простом эфире или диметоксиэтане, охлаждают до температуры от ≈- 78 до 0оС, и проводят его реакцию с диизобутилалюминийгидридом в ароматическом растворителе, например толуоле, в течение от ≈0,5 до ≈4 ч, получая спирт XIII.
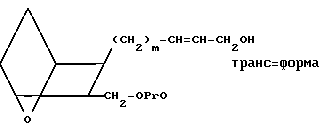
(XIII)
Спирт XIII обрабатывают бромтрифенилфосфонийбромидом (получаемым добавлением брома к трифенилфосфину в толуоле или в другом ароматическом растворителе в инертной атмосфере, например аргоне, при пониженной температуре от ≈ - 10 до ≈10оС) в присутствии пиридина и толуола, при пониженной температуре от ≈- 10 до ≈10оС, получая бромид XIV.
(XIV)
Алкиловый эфир уксусной кислоты, например трет-бутилацетат или этилацетат, обрабатывают раствором ДАЛ (диизопропиламид лития) в инертном органическом растворителе, например в тетрагидрофуране, при пониженной температуре от ≈- 78 до ≈- 60оС, в течение от ≈0,5 до ≈2 ч, с последующим добавлением раствора бромида XIV в инертном органическом растворителе, например тетрагидрофуране; в результате получают сложный эфир XV (в котором n = 2).
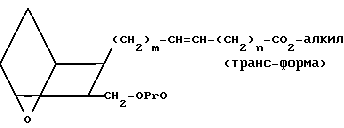
(XV)
В случае предлагаемых соединений, в которых Z представляет собой -СН= СН- в транс-форме, а n равно 1,3 или 4, проводят реакцию альдегида XI с фосфониевой солью формулы Р.
(C6H5)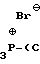 H2)n+1-CH2-OH (P)
H2)n+1-CH2-OH (P)
В присутствии сильного основания, например трет-амилата калия в толуоле или NaH/диметилсульфоксида, получая XIII',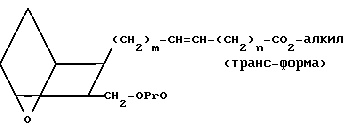
(XIII') который окисляют и этерифицируют, используя способы, известные специалистам в данной области техники, получая сложный эфир XV (где n = 1,3 или 4).
Затем со сложного эфира XV снимают защиту, обрабатывая XV в метаноле в инертной атмосфере, например аргоне, соляной кислотой в метаноле (получаемой добавлением ацетилхлорида к метанолу), получая спирт XVI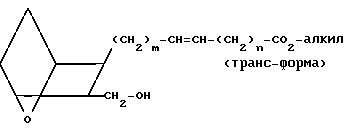
(XVI)
XVI можно затем использовать вместо II в качестве исходного вещества, следуя вышеописанному способу, получить кислоту IIIA,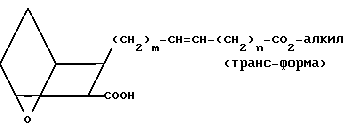
(III A) и затем получить заявляемое соединение IA" в транс-конфигурации.
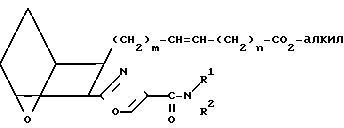
(IA'')
Заявляемые соединения IB, где Z представляет собой -СН= СН-, а Х представляет собой S, можно получить, исходя из кислоты III или IIIA, следующим образом.
Проводят реакцию кислоты III или IIIA с оксалилхлоридом, возможно, в присутствии каталитических количеств диметилформамида, в хлористом метилене, с получением соответствующего хлорангидрида кислоты, который амидируют, проводя реакцию с аммиаком, получая амид XVII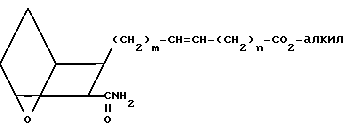
(XVII)
В другом варианте проводят реакцию кислоты III или IIIA с алкилхлорформиатом в присутствии амина, например триэтиламина, получая смешанный ангидрид, который амидируют, проводя реакцию с раствором аммиака в метаноле или с концентрированным водным раствором аммиака, получая амид XVII.
Затем амид XVII обрабатывают пентасульфидом фосфора или реагентом Лавессона [2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифос- фэтан-2,4-дисульфид], получая соответствующий тиоамид XVIII,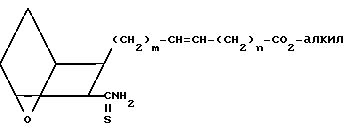
(XVIII) который обрабатывают бромпировиноградной кислотой
(Br-CH2-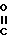 -CO2H) в полярном растворителе, например диметилформамиде, в присутствии слабого основания, например K2CO3, используя мольное отношение XVIII: бромпировиноградная кислота от ≈1:1 до ≈1:1,5, получая тиазолин XIX
-CO2H) в полярном растворителе, например диметилформамиде, в присутствии слабого основания, например K2CO3, используя мольное отношение XVIII: бромпировиноградная кислота от ≈1:1 до ≈1:1,5, получая тиазолин XIX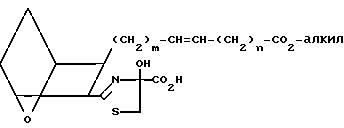
(XIX)
Затем тиазолин XIX дегидратируют, обрабатывая сульфонилхлоридом, например метансульфонилхлоридом, в присутствии основания, например триэтиламина, получая тиазоловую кислоту XX;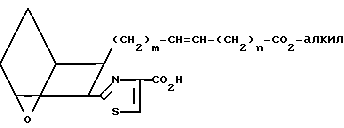
(XX)
Затем проводят реакцию карбодиимидного связывания этой кислоты с амином
H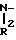 R1
R1
(A''') в присутствии DCC или WSC в инертной атмосфере, например аргоне, используя мольное отношение A''':XX от ≈1:1 до ≈2:1, получая амид IBa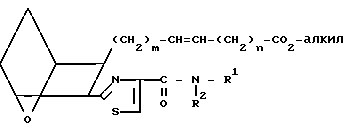
IBa
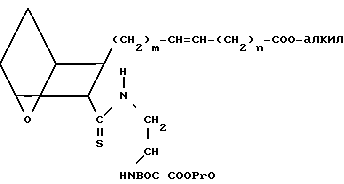
Предлагаемые соединения IC, в которых Х представляет собой Н, получают, исходя из кислоты III или IIIA, проводя реакцию связывания этой кислоты с амином B,
H2NCH2-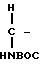 COOPrO
COOPrO
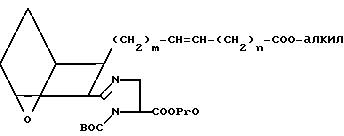
(B) где BOC представляет собой трет-бутилоксикарбонил, а PrO представляет собой защищающую группу, например, предпочтительно, -CH2CH2Si(CH3)3, в присутствии связывающего агента, например 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорида, 1-оксибензотриазола (ОВТ) и хлористого метилена, используя мольное отношение III или IIIA: B от ≈1,2:1 до ≈1:1, в течение от 12 до 90 ч. Полученный амид подвергают реакции тионирования, обрабатывая его реагентом Лавессона в присутствии бензола при температуре от ≈50 до около 75оС в течение от ≈ 1 до ≈ 4 ч, получая сложный эфир XXI.
(XXI)
Эфир XXI подвергают циклизации обработкой раствора XXI в инертном растворителе, например ацетонитриле, хлороформе или тетрагидрофуране, трифенилфосфином (используя мольное отношение XXI:трифенилфосфин от ≈0,8:1 до ≈1: 1) и четыреххлористым углеродом в присутствии амина, например триэтиламина или диизопропилэтиламина, получая имидазолин XXII.
(XXII)
Затем с имидазолина XXII снимают защиту, удаляя защищающую группу Pro, для чего используют соответствующие способы, например обработку трифторуксусной кислотой в присутствии хлористого метилена, и получают кислоту XXIII.
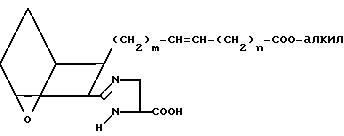
(XXIII)
Затем проводят реакцию связывания кислоты XXIII с амином A''' в присутствии амина, например пиридина или триметиламина, в инертной атмосфере, например аргоне, в присутствии связывающего агента, например WSC и ОБТ, и хлороформа, используя мольное отношение A''':XXIII от ≈0,8:1 до ≈1,2:1, получая амид XXIV.
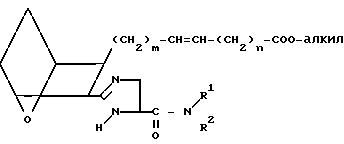
(XXIV)
Амид XXIV окисляют, обрабатывая окислителем, например диоксидом марганца, в присутствии инертного растворителя, например хлороформа, получая сложный эфир ICa.
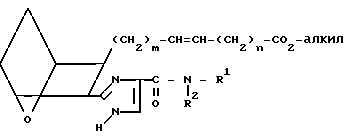
ICa
Сложные эфиры IA', IA", IBa и IСa можно превратить в соответствующие кислоты, а именно в I1,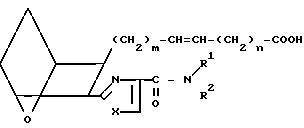
(I1) обрабатывая эти эфиры основанием, например гидроксидом лития, гидроксидом натрия или гидроксидом калия, получая соответствующие соли щелочных металлов, с последующей нейтрализацией кислотой, например разбавленной соляной или щавелевой кислотой, получая в результате предлагаемые кислотные соединения.
Соединения формулы I, в которых Z представляет собой -(СН2)2-, можно получить из кислоты I1, подвергнув ее гидрированию, с использованием для этой цели, например, катализатора гидрирования, такого как, например, палладий или древесный уголь, в присутствии инертного органического растворителя, например этилацетата (EtOAc) или уксусной кислоты (AcOH), получая в результате заявляемую кислоту I2.
I2
Заявляемые соединения, в которых R представляет собой CONHSO2R3, а именно I3,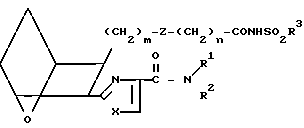
I3 получают, обрабатывая кислоту I1 или I2 сульфонамидом формулы С
H2N -R3
-R3
(C) в присутствии связывающего агента, например карбонилдиимидазола или WSC, в присутствии амина, например 4-диметиламинопиридина, в инертной атмосфере, например аргоне, используя мольное отношение C:I1 или I2 от ≈0,8:1 до ≈1,2:1, получая в результате сульфонамид I3.
Кислоты I1 или I2 можно превратить в соответствующие сложные эфиры, обрабатывая кислоты I1 и I2 соответствующими спиртами в присутствии кислотного катализатора с получением сложных эфиров.
Заявляемые соединения, в которых R представляет собой -СН2-5-тетразолил, а Z представляет собой -(СН2)2-, т.е. I4,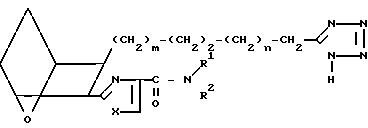
I4 получают, подвергая сложные эфиры IA',IA",IBa и ICa, или сложные эфиры кислоты I1, восстановлению гидридным реагентом, например боргидридом лития или боргидридом натрия, получая спирт XXV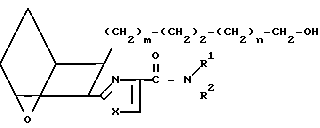
(XXV) который затем превращают в бромид обработкой трифенилфосфонийбромидом в инертном растворителе, например толуоле. Затем бромид превращают в нитрил XXVI обработкой цианидом щелочного металла в полярном растворителе, например смеси метанол/вода.
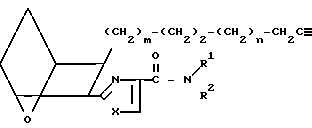

(XXVI)
Затем нитрил XXVI подвергают реакции циклоприсоединения, обрабатывая XXVIазидом натрия в присутствии хлорида аммония, диметилформамида и хлорида лития при температуре от ≈100 до ≈130оС, получая в результате I4.
Предлагаемые соединения, в которых R представляет собой -CH2-5-тетразолил, а Z - -CH=CH-, т.е. I5,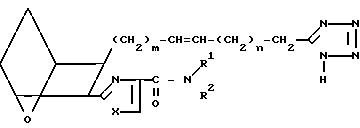
I5 получают, обрабатывая полуацеталь D (по-лученную, как это описано в патенте США N 4654356)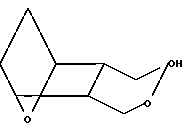
(D) реагентом Виттига формулы Е
(C6H5)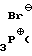 CH2)
CH2)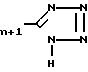
(E) в присутствии основания, например трет-бутоксида натрия или гидрида натрия - диметилсульфоксида, используя мольное отношение D:E от ≈1:1 до ≈0,2:1, получая оксиметильное соединение XXVII,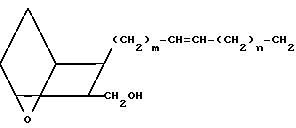
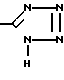
(XXVII) которое обрабатывают защищающим соединением F, где Pro-галоид, например бромметилметиловым эфиром, в присутствии основания, получая защищенный тетразол XXVIII,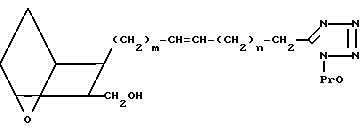
(XXVIII)
Защищенный тетразол XXVIII можно затем использовать вместо оксиметильного соединения II для получения различных соединений формулы XXIX, где Х представляет собой O,S или NH,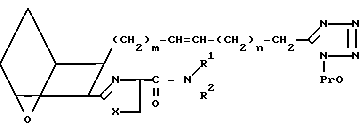
(XXIX) в котором защиту снимают обработкой водным раствором кислоты, например водным раствором соляной кислоты, получая предлагаемые соединения I5.
Соединения формулы I, в которых R представляет собой CONHR3a, где R3a не является атомом водорода, можно получить из соответствующей кислоты I6,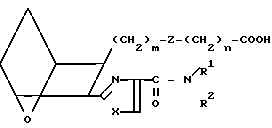
I6 обрабатывая кислоту I6(1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлори- дом в присутствии диметилформамида, 1-оксобензотриазола и органического основания, например триэтиламина, и амина G
HNHR3a (G) с образованием амида I7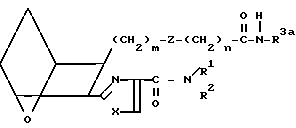
(I7) где R3a представляет собой низкий алкил, арил или аралкил.
Соединения формулы I, в которых R представляет собой CONH2 можно получить из соответствующей кислоты I6, используя вышеописанный способ для получения амида I7, за исключением того, что для получения амида I8вместо амина G используют хлорид аммония,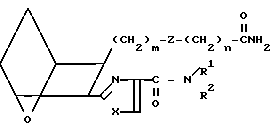
I8
Соединения формулы I, в которых R представляет собой CH2OH, можно получить из соответствующего сложного эфира I9,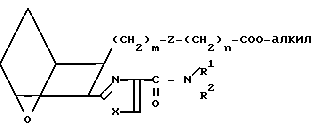
I9 который обрабатывают восстановителем, например боргидридом лития (LiBH4) в инертном растворителе, например диэтиловом эфире или тетрагидрофуране, получая спирт I10.
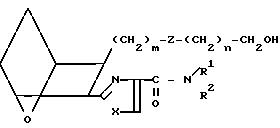
(I10)
Предлагаемые соединения имеют четыре асимметрических центра, которые в формуле I отмечены звездочками. Вместе с тем, должно быть очевидно, что каждая из вышеприведенных формул, не содержащая звездочек, также представляет все возможные стереоизомеры соответствующих соединений. Все различные стереоизомерные формы включены в объем изобретения.
Различные стереоизомерные формы заявляемых соединений, а именно цис-экзо, цис-эндо, все транс-формы и стереоизомерные пары, можно получить, используя исходные соединения и следуя способам, описанным в патенте США N 4143054. Примеры таких стереоизомеров приведены ниже.
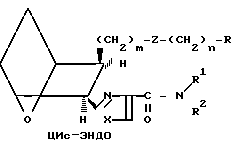
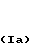
(Ia)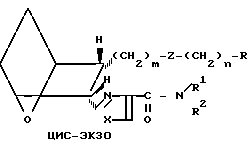

(Ib) 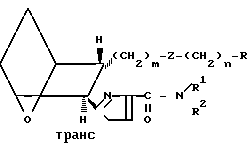

(Ic) 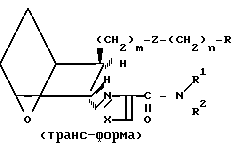
(Id)
В каждом из заявляемых соединений ядро из соображений удобства изображено следующим образом: Следует также учитывать, что ядро заявляемых соединений можно изобразить и следующим образом:
Следует также учитывать, что ядро заявляемых соединений можно изобразить и следующим образом:
Заявляемые соединения являются антагонистами рецептора тромбоксана, и поэтому могут использоваться в качестве ингибиторов процессов, опосредуемых рецептором тромбоксана. Термин "антагонист рецептора тромбоксана" включает в себя соединения, которые являются так называемыми антагонистами рецептора тромбоксана A2, антагонистами тромбоксана A2, антагонистами тромбоксана A2/эндопероксида простагландинов, антагонистами ТР-рецептора или антагонистами тромбоксана.
Заявляемые соединения являются также ингибиторами синтазы тромбоксана, и поэтому могут использоваться в качестве ингибиторов выработки тромбоксана.
Предлагаемые соединения могут использоваться в качестве ингибиторов функции тромбоцитов, т. е. для профилактики и лечения тромбоза сосудов, как полного, так и частичного, например тромбоза артерий, включая тромбоз коронарных артерий, артерий головного мозга, глазных артерий, артерий печени, брюшины, почек, периферических артерий или трансплантатов сосудов или органов, для профилактики и лечения острой стенокардии, приступов ишемической болезни, или перемежающейся хромоты. Они могут использоваться для профилактики тромбоза, вызванного повреждениями сосудов, нанесенными в ходе диагностических или терапевтических процедур, таких как, например, эндартеректомия или ангиография. Эти соединения могут использоваться для лечения или профилактики нарушений, связанных с расходом и/или активацией тромбоцитов, включая активацию тромбоцитов, их дисфункцию и/или потерю при экстракорпоральной циркуляции, использовании радиографических контрастных веществ, при тромботической тромбоцитопенической пурпуре, диссеминированной внутрисосудистой коагуляции, молниеносной пурпуре, при нарушениях, связанных с переливанием крови, или при гемолитическом уремическом синдроме, системной волчанке, вызванной циклоспорином почечной токсичности, легочной гипертензии, при побочных действиях диализа или при лечении аневризмы брюшной аорты. Эти соединения могут быть использованы при лечении тромбоза или закупорки вен, включая закупорку легочных вен, тромбоз глубоких вен, тромбоз вен печени и тромбоз вен почек.
Предлагаемые соединения можно использовать в качестве ингибиторов сужения артерий или вен. Соответственно, их можно использовать для профилактики сужения сосудов, связанного с острой стенокардией, хронической стенокардией и ее разновидностей, стенокардией Принцметаля, синдромом Рейнода, мигренью, спазмом коронарных артерий, артерий головного мозга, глаза, печени, брюшины, печени, периферических артерий или трансплантатов сосудов, а также с повреждениями сосудов в результате хирургического вмешательства или травмы. Гипертензия беременности, гепаторенальный синдром, а также легочная гипертензия являются дополнительными примерами связанных с сужением сосудов заболеваний, которые могут быть вылечены с применением заявляемых соединений.
Такие соединения могут использоваться в качестве ингибиторов бронхоспазма, т. е. повышенной реактивности дыхательных путей, аллергического бронхоспазма, астмы, а также бронхоспазматических реакций на воздействия окружающей среды, инфекций, ядов и на механические воздействия.
Предлагаемые соединения могут быть также использованы как ингибиторы ишемических и реперфузионных нарушений в различных тканях, включая сердечную мышцу, кожу, мозг, желчный пузырь, почку; при этом они могут быть использованы или в чистом виде, или в сочетании с другими веществами, способными восстанавливать кровоток. Например, эти соединения можно использовать для улучшения постишемической функции миокарда и для уменьшения размера инфаркта миокарда. Ишемия, вызванная пониженным кровотоком в процессе диагностических или терапевтических процедур, может быть устранена применением этих соединений; например, они ускоряют восстановление функций миокарда после шунтирования. Кроме того, они могут применяться при вызванных ушибами повреждениях тканей.
Заявляемые соединения могут использоваться для профилактики и лечения других состояний, включая ожоги, диабетическую ретиопатию, опухолевые метастазы и позднюю дискинезию. Эти соединения могут быть использованы для усиления действий диуретиков.
Кроме того, заявляемые антагонисты рецептора тромбоксана могут быть использованы с тромболитическими агентами, например t-PA, стрептокиназой, урокиназой, проурокиназой или анизоулированным активатором-комплексом плазминоген-стрептокиназа (APSAC) в течение первых 6 ч инфаркта миокарда. В таком случае тромболитический агент можно использовать в обычно применяемых количествах, например указанных в настольном врачебном справочнике для уменьшения постишемического повреждения миокарда.
Заявляемые соединения можно вводить оральным или парентеральным путем различным видам млекопитающих, подверженных указанным заболеваниям, например людям, кошкам, собакам и т.п., в эффективном количестве в пределах диапазона доз от ≈0,1 до ≈100 мг/кг, предпочтительно от ≈0,2 до ≈50 мг/кг, более предпочтительно от около 0,5 до ≈25 мг/кг (или от ≈ 1 до ≈2500 мг, предпочтительно от ≈5 до ≈2000 мг) один раз или 2-4 раза в день.
Заявляемые производные оксазола, а именно соединения формулы I, в которых Х представляет собой кислород, обладают особенно большой продолжительностью действия; эти соединения, если желательно, можно вводить в вышеуказанных дозах один раз в день, один раз в два дня или, если желательно, один раз в день дважды в неделю.
В табл. 1 представлены показатели активности в отношении ингибирования агрегации тромбоцитов при сравнении действия представителей известных соединений и заявленного, обозначенного как SQ 33 640. Результаты свидетельствуют об очевидном 10-кратном превышении активности последнего.
Кроме того, на чертеже показан график сравнения продолжительности действия для соединений SQ 33 640 и известного SQ 31 491, которое проявляет наиболее близкие свойства. Даже в том случае, когда доза SQ 33 640 составляла 1/50 дозы SQ 31 491 (0,2 мг/кг против 10 мг/кг). SQ 33 640 показало лучшую защиту от внезапного летального исхода (см.чертеж).
Описание методик испытания приведено ниже.
Агрегация человеческих тромбоцитов.
Венозную кровь отбирали из антикубитальной (локтевой) вены человека, не принимающего никаких лекарств в течение 2 недель. Кровь собирали во флаконы вместимостью 150 л, содержащие в качестве антикоагулянта цитрат натрия. PRP готовили центрифугированием крови, содержащей цитрат, в режиме 200g в течение 10 мин при 25оС. PRP определили центрифугированием PRP в течение 3 мин при 25оС.
Скопление тромбоцитов PRP изучали фотометрическим методом с использованием агрегометра Хронолог с линейным самописцем. 10 мм метанольного стандартного раствора пробы растворили в 0,5 мл PRP и проводили преинкубирование в течение 2,5 мин при 37оС до внесения агрегатора, а затем в течение 3 мин регистрировали величину оптического пропускания. Скорость увеличения последнего, которая в свою очередь является мерой скорости агрегации, определяли по наклону крутого участка кривой агрегации.
Летальный исход, вызываемый U - 46 619 у мышей.
Настоящий эффект определяли с помощью модифицированной методики Кохлера.
Самцам массой 20-25 г после предварительного суточного ограничения в пище вводили либо испытуемые соединения, либо растворитель (10%-ный этанол в солевом растворе) за 30 мин до обработки животных с применением U - 46 619 (2 мг/кг) внутривенно.
В указанных концентрациях летальный исход наблюдали у всех подопытных экземпляров. Продолжительность действия изучалась на животных, предварительно подвергнутых воздействию испытуемых соединений в концентрации 0,2 мг/кг, перорально перед противоопытом с применением U - 46619 (2 мг/кг) внутривенно в указанные промежутки времени.
Активное соединение можно применять в виде таблетки, капсулы, раствора или суспензии, содержащих от около 5 до около 500 мг на единицу дозировки соединения или смеси соединений формулы I, или применять наружным путем для заживления ран (0,01-5% по массе соединения формулы I, от 1 до 5 раз в день). Эти соединения можно смешивать обычными способами с физиологически пригодным наполнителем или носителем, связующим веществом, консервантом, стабилизатором, коpригентом и т.д., или с носителем для наружных препаратов, таким как, например, Plastibase (минеральное масло, загущенное полиэтиленом), как предусмотрено распространенной фармацевтической практикой. Кроме того, как следует из вышесказанного, определенные члены перечисленной группы вдобавок являются интермедиатами для других членов группы.
Заявляемые соединения можно также применять наружно для лечения заболеваний периферических сосудов; в этом случае они могут быть приготовлены в виде крема или мази.
Нижеследующие примеры иллюстрируют предпочтительные варианты осуществления настоящего изобретения. Если не оговорено особо, все значения температуры даны в градусах Цельсия.
П р и м е р 1. {1S-[1α ,2α (Z),3α ,4α ]}- 6-{3-[4-(4-циклогексилбутил)амино] карбо- нил-2-оксазолил}-7- оксабицикло[2.2.1-гептил-2]-4-гексеновая кислота
A.[1,1-диметилэтокси)карбонил]-N-(4- циклогексилбутил)-L-серинамид
К раствору 575 мг гидрохлорида 4-циклогексилбутиламина (3,0 ммоль), 615 мг трет-бутоксикарбонил-L-серина (3,0 ммоль, 1 экв.), 405 мг 1-оксибензотриазолгидрата (3,0 ммоль, 1,0 экв. ) и 387 мг диизопропилэтиламина (3,0 ммоль, 1 экв. ) в 10 мл сухого тетрагидрофурана (ТГФ) при перемешивании в атмосфере аргона при 0оС добавляют 618 мг 1,3-дициклогексилкарбодиимида (3,0 ммоль, 1,0 экв.) в виде одной порции. Медленно происходит образование осадка. По истечении одного часа смесь нагревают до комнатной температуры и перемешивают в течение 4 ч. После разбавления этилацетатом смесь фильтруют, и фильтрат промывают солевым раствором с рН = 1 (получен смешением воды, рассола и 1М водного раствора HCl). Затем дважды промывают 1М раствором NaHCO3, сушат над Na2SO4 и выпаривают, получая 1,1 г неочищенного целевого амида.
Тонкослойная хроматография (ТХ) (10%-ный водный NH3 в CH3OH, в CH2Cl2 - анизальдегид):
Солянокислый цикло- гексилбутиламин 0,27 Целевой амид 0,47
B. N-(4-циклогексилбутил)-L-серинамид
К раствору 1,1 г неочищенного амида (раздел A) в 4 мл CH2Cl2 при комнатной температуре добавляют 4 мл трифторуксусной кислоты. Смесь перемешивают в течение 4 ч. После выпаривания растворителя оставшуюся трифторуксусную кислоту удаляют в виде азеотропа с CHCl3 на роторном испарителе. Импульсная хроматография (150 г д иокси- да кремния, 10% [10%-ный водный NH3 в CH3OH] в CH2Cl2) дает, после азеотропной отгонки с толуолом и вакууми- рования, 495 мг чистого целевого амина в виде твердого белого вещества, Выход целевого амина составляет 68% в пересчете на гидрохлорид 4-циклогексилбутиламина.
ТХ (10% [10% -ный водный NH3 в CH3OH] в CH2Cl2 - анизальдегид): амид (раздел A) 0,47 целевой амид 0,17
13С ЯМР (67,8 МГц в CDCl3):
173,4, 64,6, 56,3, 39,1, 37,3, 36,9, 33,1, 29,6, 26,5, 26,2, 24,0.
С.[1S-[1α , 2α (Z), 3 α, 4α ]]-6-[3-(оксиметил)-7-оксабицикло[2.2.1]гептил-2]-4-гексе-новая кислота, метиловый эфир
К раствору 36,27 г (4аR-[4a α , 5β, 8β, 8 αβ)]-октагидро-5,8-эпокси-1Н-2-бензопиран-3-ола (полученного, как описано в патенте США N 4143054) (0,23 моль) и 3-карбоксипропилтрифенилфосфонийбро-мида (127,34 г, 0,37 моль) в 600 мл сухого ТГФ в атмосфере аргона при 3оС при механическом перемешивании по каплям добавляют в течение 1 ч раствор 370,6 мл трет-амилата калия (0,68 моль 1,8М толуольного раствора). Сначала температура реакции достигает максимального значения 8оС, затем ее снижают до 4оС для добавления остатка основания. После этого реакцию проводят при комнатной температуре в течение 90 мин. Реакционную емкость помещают в ледяную баню (0оС), и останавливают реакцию добавлением в течение 30 мин 152 мл ледяной уксусной кислоты. В вакууме удаляют растворители (в виде азеотропа с толуолом). Добавляют воду (640 мл) и 50 мл концентрированной HCl при рН 2,6. Разбавляют смесь 640 мл этилацетата, добавляют 149 г NaCl и несколько кристаллов 3-карбоксипропилтрифенилфосфонийбро-мида (затравка), затем интенсивно перемешивают в течение 15 мин. Осадок отделяют фильтрованием и промывают двумя порциями этилацетата (каждая порция - 320 мл). Отделяют этилацетатный слой, водный слой экстрагируют этилацетатом (2 раза по 200 мл); собранные этилацетатные слои сушат над MgSO4 и концентрируют. Добавляют 507 мл 5%-ного K2CO3, затем интенсивно перемешивают в течение 1 ч. Образования осадка не наблюдается. Реакционную смесь концентрируют до пастообразного состояния и суспендируют в 508 мл воды. При интенсивном перемешивании в течение нескольких часов образования осадка не происходит. Воду декантируют и остаток суспендируют в 200 мл 5% -ного водного раствора K2CO3. После интенсивного перемешивания взвесь твердого вещества отделя- ют фильтрованием и несколько раз промывают водой. Собранные водные слои 5 раз экстрагируют смесью толуол/эфир (1: 1, 230 мл на одну экстракцию). После охлаждения собранных водных слоев в ледяной бане до 0оС, добавляют концентрированную HCl до рН 2,5, затем экстрагируют один раз 460 мл и 2 раза 230 мл этилацетата. Собранные этилацетатные слои сушат над MgSO4 и выпаривают в вакууме, получая 49,74 г масла янтарного цвета. Растиранием с 330 мл эфира (комнатная температура, на протяжении ночи) удаляют из масла фосфорсодержащие побочные продукты. Эфирный раствор декантируют с темно-красного масла в делительную воронку, из которой сливают масло, захваченное при декантации (1,56 г). Упаривание эфирного раствора дает 43,08 г [1S-[1 α,2 α(Z),3 α ,4α ]]-6-(3-оксиметил)-7-оксабицикло[2.2.1]гептил-]-4-гексеновой кислоты в виде вязкого желтого масла.
ЯМР 1Н показывает, что мольное отношение продукт:трифенилфосфиноксид: эфир составляет 23:1:1,8 (в мас.%: 93:4,7-2,2). Отбрасывание эфира и трифенилфосфиноксида дает выход, равный 72,5% (40,06 г).
При комнатной температуре в атмосфере аргона к 80 мл метанола по каплям добавляют ацетилхлорид (5,20 мл, 0,073 ммоль). Раствор ацетилхлорида в метаноле добавляют (однократным добавлением) к раствору 42,98 г (0,18 моль) в 700 мл метанола. Перемешивают смесь в течение 3 ч. Добавляют триэтиламин (0,09 моль, 12,21 мл), в вакууме удаляют метанол и остаток разделяют между 300 мл этилацетата и 150 мл воды. После разделения слоев водный слой экстрагируют 150 мл этилацетата, собранные этилацетатные слои промывают рассолом, сушат над Na2SO4 и упаривают в вакууме, получая 43,06 г вязкого рыжевато-коричневого масла. Импульсная хроматография на 1350 г силикагеля (E. Merck Kieselgel 60) (меш 240-400, 75/25 эфир/гексан, затем эфир после того, как целевой продукт начинает вымываться из колонки) дает 35,74 г целевого сложного эфира в виде вязкого масла светло-желтого цвета, не содержащего (по данным ЯМР) трифенилфосфиноксида).
1Н ЯМР (CDCl3 стандарт тетраметилсилан - ТМС): δ 5,41-5,38, м (2Н); 5,49, д, I = =4,69 Гц (1Н); 4,22, д, I = 4,69 Гц (1Н); 3,73-3,69, м (1Н); 3,67, с. , (3Н); 3,60, м (1Н); 2,37, ш.с. (4Н); 2,12-1,99, м (3Н); 1,97-1,85, м (1Н); 1,72, м (2Н); 1,46, м (2Н).
13С ЯМР (CDCl3, стандарт 77,00):δ 173,50, 130,42, 128,63, 80,23, 79,22, 61,74, 51,49, 48,95, 46,45, 33,86, 29,69, 29,31, 25,94, 22,92
D. [1S-[1α , 2 α (Z),3 α ,4 α ]]-6-[3-(карбокси)-7-оксабицикло[2.2.1]-гептил-2-]-4-гексе- новая кислота, метиловый эфир
К раствору 2,43 г содержащего примеси спирта, полученного в разделе С (чистота 80% , 1,94 г, 7,6 ммоль спирта, примесь - трифенилфосфиноксид), в 40 мл ацетона, в атмосфере аргона при 0оС медленно добавляют 8 мл реагента Джонса (концентрация Cr" - 2,6 М). Красная окраска реагента сохраняется до окончания добавления. По- лученную смесь, содержащую осажденный компонент, перемешивают в течение 20 мин, после чего добавляют пропанол-2 для связывания избытка реагента. При 0оС при перемешивании добавляют 3М водный раствор NaHCO3 до растворения всех солей. Добавляют рассол, затем трижды экстрагируют смесь этилацетатом. После высушивания экстрактов над Na2SO4 и выпаривания растворителя проводят импульсную хроматографию (150 г диоксида кремния, 25-45% 5%-ной уксусной кислоты в этилацетате при градиенте гексана), которая, после азеотропной отгонки уксусной кислоты с толуолом, дает 1,91 г маслообразного продукта. Этот маслообразный продукт представляет собой содержащую примеси целевую кислоту (чистота 80%, 1,53 г целевого продукта, примесь - трифенилфосфиноксид), полученную с выходом 75%.
ТХ (50% [5% -ная уксусная кислота в этилацетате] в смеси гексананизальдегид): Спирт (раздел С) 0,33 Целевая кислота 0,35
13С ЯМР (67,8 МГц в CDCl3)
175,3, 173,1, 129,1, 128,8, 78,0, 78,0, 51,6, 51,1, 47,4, 33,5, 28,8, 28,5, 26,9, 22,5
E. [1S-[1α , 2 α, ( Z), 3α (R+),4α]]-6-[3-[[[2-[(4-циклогексилбутил)амино[1-(оксиметил)-2-оксоэтил] амино] карбонил]-7-оксабицикло[2.2.1] гептил-2]-4-гексеновая кислота, метиловый эфир
К раствору 733 мг содержащей примеси кислоты, полученной в разделе D (чистота 80%, 586 мг, 2,2 ммоль, 1,1 экв., примесь - трифенилфосфиноксид), в 4 мл сухого ТГФ в атмосфере аргона добавляют 356 мг 1,1-карбонилдиимидазола (2,2 ммоль, 1,1 экв.) и смесь выдерживают в течение 1 ч. Вследствие образования большого объема осадка добавляют 5 мл ТГФ и смесь осторожно нагревают, получая раствор (ТХ обнаруживает наличие стабильного ацилимидазола). После перемешивания в течение 30 мин добавляют раствор 495 мг амина, полученного в разделе B (2,0 ммоль), в 10 мл сухого ТГФ; при этом дополнительно используют еще 5 мл ТГФ для количественного переноса амина. ТХ гомогенной смеси, полученной после 1 ч перемешивания при комнатной температуре, показывает протекание очень медленной реакции. Поэтому ТГФ удаляют, пропуская через смесь в течение ночи аргон, до тех пор, пока ее объем не уменьшается до 2 мл и не выпадает осадок. При добавлении 5 мл ТГФ происходит растворение всего осадка. После 5 ч перемешивания смесь упаривают; импульсная хроматография (150 г диоксида кремния, от 50 до 100% этилацетата в градиенте гексана, затем от 0 до 10% CH3OH в градиенте этилацетата) дает 230 мг чистого целевого оксибисамида в виде масла. Выход целевого оксибисамида составляет 23%.
Также выделены изомерный аминоэфирамид (27%) и аддукт состава 2:1 (16% ). Эти побочные продукты можно с хорошим выходом превратить в целевой оксибисамид путем переэтерификации с использованием KCN в CH3OH при комнатной температуре, хотя аминоэфирамид может также претерпеть спонтанную изомеризацию.
ТХ (50% [5%-ная уксусная кислота в этилацетате] в смеси гексананизальдегид): Амин (раздел B) 0,00 Кислота (раздел D) 0,38 Ацилимидазол 0,18 Целевой оксибисамид 0,22 Аминоэфирамид 0,04 Аддукт 2:1 0,33
13С ЯМР (67,8 МГц в CDCl3):
173,3, 172,8, 170,4, 129,2, 129,0, 78,9, 78,8, 62,7, 54,0, 53,8, 51,3, 47,9, 34,9, 37,3, 33,6, 33,1, 29,5, 29,4, 28,6, 27,2, 26,4, 26,1, 24,0, 22,6
F. [1S-[1 α,2 α(Z),3α (R+),4 α]]-6-[3-[4-[[(4-циклогексилбутил)-амино] карбонил] -4,5- дигидро-2- оксазолил]-7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир
Методический подход описан M.J. Miller, P.G. Mattingly, M.A. Morrison, J.F. Kerwin, J.Am.Chem.Soc. 1980, 102, 7026.
К раствору 240 мг чистого оксибисамида, полученного в разделе Е (0,48 ммоль), в 3 мл сухого ТГФ, в атмосфере аргона при комнатной температуре добавляют 189 мг трифенилфосфина (0,72 ммоль, 1,5 экв.), 73 мг триэтиламина (0,72 ммоль, 1,5 экв.) и 89 мг CCl4 (0,58 ммоль, 1,2 экв.), и полученную смесь кипятят с обратным холодильником. Через 1 ч добавляют еще одну аликвоту CCl4 и триэтиламина, а через 2,5 ч - еще одну аликвоту этих реагентов. Спустя 2 ч добавляют еще по одной аликвоте CCl4 и триэтиламина и половину аликвоты (95 мг) трифенилфосфина. Спустя еще 2 ч ТХ свидетельствует о полном расходовании оксибисамида, полученного в разделе Е, и из первоначально бесцветной, однородной смеси выпадает белый осадок, который затем темнеет. После упаривания растворителя осуществляют импульсную хроматографию (диоксид кремния, 15% ацетона в толуоле), которая дает 190 мг чистого целевого оксазолина в виде масла. Выход оксазолина - 83%.
ТХ (20% ацетона в смеси гексан-анизальдегид): Оксибисамид (раздел Е) 0,07 Целевой оксазолин 0,29
13С ЯМР (67,8 МГц в CDCl3):
173,1, 171,2, 169,1, 129,3, 128,9, 79,0, 78,9, 69,6, 68,3, 51,3, 48,2, 46,3, 39,0, 37,4, 36,9, 33,7, 33,1, 29,6, 29,5, 28,7, 27,1, 26,5, 26,2, 24,0, 22,7
G.[1S-[1 α, 2 α(Z),3 α, 4α ]]-6-[3-[4-[[(4-циклогексилбутил)-амино]карбонил] -2-окса- золил]-7- оксабицикло[2.2.1]гептил-2-4-гексеновая кислота, метиловый эфир
Методический подход описан D.L. Evans, D.K. Minster, U. Jordis, S.M. Hecht, A.L. Mazzu. Jr., A.I.Meyers J.Org.Chem., 1979. 44, 497.
К раствору 190 мг чистого оксазолина, полученного в разделе F, (0,40 ммоль), в 10 мл CHCl3, добавляют 200 мг нетитрованного NiO2 и полученную неоднородную смесь перемешивают при комнатной температуре. Метод ТХ показывает, что в течение первого часа имеет место протекание реакции, однако затем реакция прекращается. Через 1 день добавляют пять дополнительных аликвот реагента до тех пор, пока реакция не завершится. Смесь разбавляют этилацетатом, после чего перемешивают с 3М водным раствором NaHSO3 до исчезновения черной окраски NiO2 и растворения большей части твердых компонентов. Смесь трижды экстрагируют этилацетатом, после чего экстракт сушат над Na2SO4 и упаривают. Импульсная хроматография (диоксид кремния, от 25 до 35% этилацетата в градиенте гексана) дает 90 мг чистого целевого оксазола в виде твердого вещества. Выход оксазола составляет 48%.
ТХ (100% -ный этилацетат - анизальдегид): Оксазолин (раздел F) 0,52 Целевой оксазол 0,81
13С ЯМР (67,8 МГц в CDCl3):
173,2, 163,8, 160,5, 140,4, 136,0, 129,4, 128,5, 79,5, 79,3, 51,4, 49,6, 46,6, 39,0, 37,4, 37,0, 33,7, 33,3, 29,8, 29,7, 28,9, 27,8, 26,6, 26,6, 26,3, 24,1, 22,7.
H. [1S-[1 α, 2 α, (Z),3 α ,4α ]]-6-[3-[4-[[(4-циклогексилбутил)-амино] карбонил] -2-окса-зо- лил] -7- оксабицикло[2.2.1] гептил-2]-4-гексеновая кислота.
К 90 мг чистого оксазола (0,19 ммоль), в 4 мл CH3OH, при комнатной температуре добавляют 2 мл 1М водного раствора NaOH. После перемешивания смеси в течение 1,3 ч добавляют 1М водный раствор HCl, снижая рН до 1. Смесь трижды экстрагируют этилацетатом. Экстракты сушат над Na2SO4 и выпаривают растворитель, получая неочищенную целевую кислоту. Импульсная хроматография (от 25 до 50%, 5%-ная уксусная кислота в этилацетате, в градиенте гексана) дает, после азеотропной отгонки уксусной кислоты с толуолом, 71 мг чистой целевой кислоты в виде твердого вещества. Выход целевой кислоты составляет 81%.
ТХ (50%, 5%-ная уксусная кислота в этилацетате, в смеси гексананизальдегид): Оксазол (раздел G) 0,43 Целевая кислота 0,25
13С ЯМР (67,8 МГц в CDCl3):
176,9, 163,9, 160,7, 140,9, 135,7, 129,5, 128,4, 79,5, 79,3, 49,6, 46,5, 39,1, 37,4, 36,9, 33,7, 33,2, 29,7, 29,7, 28,8, 27,8, 26,6, 26,2, 24,1, 22,5.
П р и м е р 2. [1S-[1 α, 2α (Z),3 α. ,4α ]]-6-[3-[4-[[(4-циклогексилбутил)метиламинокарбонил] -2-оксазолил] - 7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота.
A.N-[(1,1-диметилэтокси)карбонил]-O- (фенилметил(-L-серин, 2-(триметилсилил) этиловый эфир.
Методический подход описан P.Sieber, Helv.Chim.Acta (1977), 60, 2711
К раствору 20,7 г N-трет-бутоксикарбонил-O-бензил-(L)-серина (70 ммоль), 11,0 г пиридина (139 ммоль, 2,0 экв.) и 9,9 г 2-триметилсилилэтанола (84 ммоль, 1,2 экв.) в 50 мл сухого CH3CN, перемешиваемому при 0оС в атмосфере аргона, добавляют 15,8 г 1,3-дициклогексилкарбодиимида (76 ммоль, 1,1 экв.) в виде одной порции. Образуется осадок. Спустя 3 ч смесь нагревают до комнатной температуры и перемешивают в течение 12 ч. Добавляют раствор 1,4 г дигидрата щавелевой кислоты (11 ммоль, 0,15 экв.) в 3 мл диметилформамида (ДМФА) и перемешивают смесь в течение 1 ч перед фильтрованием. Осадок на фильтре промывают этилацетатом до полного отсутствия в фильтрате целевого эфира. Фильтрат дважды промывают 1М водным раствором HCl и рассолом, и дважды - водным раствором NaHCO3 (1М). Сушка над Na2SO4 и выпаривание дает 34,8 г неочищенного целевого сложного эфира (чистота 79%, содержание целевого продукта 27,6 г, примеси - растворитель и 2-триметилсилилэтанол) в виде масла; выход 100%.
ТХ (50%, 5%-ная уксусная кислота в этилацетате, в смеси гексананизальдегид):
N-трет-бутоксикар- бонил-O-бензил-(L)-cерин 0,40 Целевой сложный эфир 0,84
13С ЯМР (67,8 МГц в CDCl3): 170,5, 155,3, 137,5, 128,2, 127,6, 127,4, 79,5, 73,0, 69,9, 63,6, 54,0, 28,1, 17,2-1,7.
B. N-(1,1-диметилэтокси)карбонил] -L-се- рин, 2-(триметилсилил)этиловый эфир.
К раствору 34,8 г неочищенного эфира, полученного в разделе A (чистота 79% , 27,6 г 100%-ного эфира, примеси - растворитель и 2-триметилсилилэтанол, 70 ммоль), в 200 мл этилацетата и 300 мл уксусной кислоты, при комнатной температуре в атмосфере аргона добавляют 10 г катализатора Pd (10%) на угле. Затем полученную смесь перемешивают в атмосфере водорода в течение 4 дней. Метод ТХ свидетельствует о почти полном превращении. Смесь фильтруют через поликарбонатную мембрану, и после выпаривания растворителя оставшуюся уксусную кислоту удаляют азеотропной отгонкой с толуолом и CH2Cl2. Импульсная хроматография (750 г диоксида кремния, 10-30% этилацетата в градиенте гексана) дает 3,58 г чистого исходного вещества, выход 13%, и 16,49 г чистого целевого дебензилированного сложного эфира в виде масла. Выход целевого сложного эфира составляет 78%.
ТХ (25% этилацетата в смеси гексан-анизальдегид): Сложный эфир (раздел A) 0,61
Целевой сложный эфир (раздел B) 0,28
13С ЯМР (67,8 МГц в CDCl3): 170,9, 155,6, 79,9, 63,8, 63,0, 55,8, 28,1, 17,2-1,7.
С.Моногидрохлорид 2-(триметилсилил) этилового эфира L-серина.
Методический подход описан P. Sieber, R.H. Andreatta, K. Eliser, R. Kamber, B. Riniker, H. Rinsle. Пептиды: Труды Пятого американского симпозиума по пептидам, издатели: M.Goodman и I.Meienkofer, Halsted Press, Нью-Йорк (1977), с.543-545.
К раствору 10,4 г эфира, полученного в разделе B (34,1 ммоль), в 200 мл диэтилового эфира добавляют 40 мл раствора примерно 6,5М HCl (метанол/метилацетат, 260 ммоль). (Данный раствор приготовляют, добавляя 42 г ацетилхлорида (0,54 ммоль) по каплям к 35 г метанола (1,1 ммоль, перемешивая смесь при 0оС, а затем при комнатной температуре в течение 2 ч). Затем смесь перемешивают в течение 4 ч, при этом мед- ленно выделяется газ. При перемешивании при комнатной температуре осторожно малыми порциями добавляют 32,8 г NaHCO3 (390 ммоль), чтобы иметь возможность контролировать выделение газа при нейтрализации. Затем смесь фильтруют через стеклянный фильтр; осадок на фильтре промывают 50%-ным раствором метанола в диэтиловом эфире. После выпаривания растворители получают 8,06 г полужидкого неочищенного почти чистого целевого солянокислого амина. Это вещество может быть только частичной солью, но, принимая, что образуется чистая соль 1:1, получаем выход 98%.
ТХ (10%, 10%-ного концентрированного водного NH3H CH3OH, в смеси CH2Cl2 - анизальдегид): Сложный эфир (раздел B) 0,63 Амин (раздел С) 0,22
13С ЯМР (67,8 МГц в CDCl3): 171,3, 63,9, 61,9, 55,5, 17,1-1,8.
D. [1S-[1 α , 2α (Z),3 α(R*),4 α]]-6-[3-[[[1-(оксиметил)-2-оксо-2-[(триметилсилил)этокси] - этил] амино]карбонил]-7-оксабицикло-[2.2.1]гептил-2-]-4-гексеновая кислота, метиловый эфир.
К раствору 9,40 г кислоты, полученной в разделе D примера 1 (чистота 85% , 7,99 г, 29,8 ммоль), 8,04 г неочищенного, почти чистого солянокислого амина, полученного в разделе С (чистота 95%, 7,60 г, 31,6 ммоль, 1,06 экв. ), 4,43 г 1-оксибензотриазолгидрата (32,8 ммоль, 1,1 экв.), и 4,24 г диизопропилэтиламина (32,8 ммоль, 1,1 экв.) в 50 мл сухого ТГФ, перемешиваемому в атмосфере аргона при комнатной температуре, одной порцией добавляют примерно 6,8 г 1,3-дициклогексилкарбодиимида (33 ммоль, 1,1 экв.). Медленно образуется осадок. Спустя 16 ч растворитель испаряют, и импульсная хроматография (диоксид кремния, 30-100% этилацетата в градиенте гексана) дает масло, содержащее твердый компонент. Это вещество переносят в диэтиловый эфир, в котором твердый компонент не растворяется, и фильтруют смесь. Выпаривание дает 9,45 г почти чистого целевого амида (чистота 90%, 8,54 г 100% -ного целевого продукта) в виде светлого масла. Выход целевого амида составляет 63%.
ТХ (50%, 5%-ная уксусная кислота в этилацетате, в смеси гексананизальдегид): Кислота (раздел D примера 1) 0,42 Целевой амид 0,36
13С ЯМР (67,8 МГц в CDCl3): 173,5, 172,4, 170,3, 129,4, 129,1, 79,2, 79,1, 63,9, 63,2, 54,6, 54,5, 51,4, 47,8, 33,7, 29,7, 28,5, 27,3, 22,7, 17,2, - 1,7.
E. [1S-[1 α,2 α(Z),3 α (R*),4 α]]-6-[3-[4,5-дигидро-4-[[2-(триметилсилил)этокси] -кар-бонил] -2- оксазолил]-7-оксабицикло-[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору 8,3 г почти чистого амида, полученного в разделе )чистота 90%, 7,5 г, 16,6 ммоль), в 150 мл сухого CH3CN, в атмосфере аргона при комнатной температуре (баня с комнатной температурой) добавляют 13,1 г трифенилфосфина (50 ммоль, 3,0 экв.), 6,4 г диизопропилэтиламина (50 ммоль, 3,0 экв.), и 7,7 г CCl4 (50 ммоль, 3,0 экв.). После перемешивания в течение 2 ч к смеси добавляют 1М водный раствор NaHCO3, и трижды экстрагируют смесь хлористым метиленом. Экстракт сушат над Na2SO4 испаряют растворитель и проводят импульсную хроматографию (диоксид кремния, 20-50% этилацетата в градиенте гексана); получают 5,9 г почти чистого целевого оксазолина (чистота 90%, 5,3 г 100%-ного целевого продукта) в виде масла. Выход оксазолина составляет 73%.
ТХ (50% этилацетата в смеси гексан-анизальдегид): Амид (раздел D) 0,20 Целевой оксазолин 0,44
13С ЯМР (67,8 МГц в CDCl3): 172,7, 170,7, 169,0, 129,0, 128,8, 78,3, 78,3, 69,0, 67,5, 63,2, 50,9, 48,1, 46,0, 33,4, 29,3, 28,4, 26,7, 22,4, 16,8-2,0.
F. [1S-[1 α 2 α ( Z ),3α, 4 α ]]-6-[3-[4-[[2-(триметилсилил)этоксикарбонил]-2-оксазо-лил]-7- оксабицикло[2.2.1]гептил-2] -4-гексеновая кислота, метиловый эфир
Методический подход описан D.L. Evans, D.K. Minsten, U. Jordis, S.M. Hecht, A.L. Mazzu, Jr., и .A.I. Meyers, J. Org. Chem. (1979), 44, 497.
К раствору 5,1 г почти чистого оксазолина, полученного в разделе Е (чистота 90% , 4,6 г, 10,5 ммоль), в 50 мл CH2Cl2 добавляют 10,1 г нетитрованного NiO2 и полученную неоднородную смесь перемешивают при комнатной температуре. (Экзотермичность реакции приводит к некоторому саморазогреву смеси). Метод ТХ свидетельствует о том, что по истечении 1 ч реакция почти заканчивается. Затем добавляют дополнительную аликвоту NiO2 (2,0 г). По истечении 30 мин реакция завершается; добавляют 150 мл этилацетата. Для восстановления и растворения солей никеля добавляют 100 мл 3М водного раствора NaHSO3 и 200 мл 1М водного раствора тринатрийцитрата. При перемешивании все твердые компоненты растворяются, и смесь нагревается. Смесь дважды разделяют и экстрагируют этилаце- татом (ТХ показывает полную экстракцию целевого оксазола), затем экстракт сушат над Na2SO4 и выпаривают. Очистка 3,8 г неочищенного продукта методом импульсной хроматографии (150 г диоксида кремния, 20-75% этилацетата в градиенте гексана) дает 2,60 г чистого целевого оксазола в виде масла. Выход оксазола составляет 57%.
ТХ (50% этилацетата в смеси гексан-анизальдегид): Оксазолин (раздел Е) 0,34 Целевой оксазол 0,58
13С ЯМР (67,8 МГц в CDCl3): 172,9, 164,5, 161,0, 143,3, 133,0, 129,1, 128,4, 79,0, 78,9, 63,0, 51,1, 49,5, 46,8, 33,5, 29,5, 28,7, 27,6, 22,5, 17,2, 1,8.
G. [1S-[1 α , 2α (Z),3 α, 4 α ]-6-[3-(4-карбокси-2-оксазолил)-7-оксабицикло[2.2.1]геп- тил-2-4- гексеновая кислота, метиловый эфир.
Методический подход описан P. Sieber, R.H. Andreatta, K. Eliser, B. Kamber, B. Riniker and A. Rink
Пептиды: Труды Пятого американского симпозиума по пептидам, М., издатели: M. Goodman, I. Meienhofer, Нью-Йорк, (1977), с.543-545.
К раствору 3,1 г чистого оксазола, полученного в разделе (7,1 ммоль), в 20 мл сухого ДМФА в атмосфере аргона добавляют 12,0 г тетрабутиламмонийфторида на диоксиде кремния (Huka 13,9 ммоль, 1,96 экв.), и полученную неоднородную смесь перемешивают при комнатной температуре в течение 6 ч. Смесь разбавляют 20 мл 1%-ной трифторуксусной кислоты, 1%-ного метанола, 98%-ного этилацетата, и фильтруют, используя 40 мл этого же растворителя для промывания осадка на фильтре. Фильтрат выпаривают и трижды проводят азеотропную отгонку с толуолом для удаления ДМФА. Неочищенный продукт очищают на ионообменной смоле: после промывания колонки с 250 г AG 50 W-X8 (водородная форма) водой и затем 50%-ным CH3OH в воде до обесцвечивания элюента, сырой продукт растворяют в 40 мл 50%-ного метанола в воде, вводят в колонку и элюируют тем же растворителем. Получают 2,65 г почти чистой целевой оксазоловой кислоты (чистота 90%, 2,38 г 100%-ного целевого продукта) в виде масла. Выход оксазоловой кислоты составляет 100%.
ТХ (1% трифторуксусной кислоты, 1% метанола, 98% этилацетата - анизальдегид): Оксазол (раздел F) 0,46 Оксазоловая кислота 0,15
13С ЯМР (67,8 МГц в CDCl3): 175,0, 166,7, 163,8, 145,6, 134,2, 130,4, 129,7, 80,7, 52,0, 50,7, 47,7, 34,6, 30,5, 29,7, 28,9, 23,8.
Н. Моногидрохлорид N-метил-4-циклогексилбутиламина
К раствору 750 мг солянокислого 4-циклогексилбутиламина (3,9 ммоль) и 1,08 г триэтиламина (10,8 моль, 2,8 экв.) в 10 мл сухого ТГФ, перемешиваемому в атмосфере аргона при 0оС, добавляют 584 мг ClCO2C2H5(5,4 ммоль, 1,4 экв.). После нагревания до комнатной температуры неоднородную смесь перемешивают в течение 3 ч. После разбавления диэтиловым эфиром смесь промывают (дважды) 1М водным раствором HCl. Органический слой сушат над Na2SO4 и упаривают, получая 950 мг неочищенного промежуточного этилкарбамата в виде масла, содержащего примесь некоторого количества имида.
К раствору 950 мг неочищенного промежуточного этилкарбамата в 10 мл сухого ТГФ при перемешивании в атмосфере аргона при 0оС добавляют 950 мг литийалюминийгидрида (25 ммоль, 6,4 экв.). Происходит выделение газа. Затем смесь кипятят с обратным холодильником в течение 2 ч. После охлаждения до 0оС и введения 20 мл диэтилового эфира, осторожно добавляют 1,0 мл воды для связывания избытка гидрида. Смесь вновь нагревают до комнатной температуры и при интенсивном перемешивании добавляют сначала 1,0 мл 15%-ного водного раствора NaOH, а затем 3,0 мл воды. Смесь фильтруют, промывают осадок на фильтре (10% , 10%-ный концентрированный водный NH3 в CH3OH) в диэтиловом эфире, и фильтрат упаривают. Вещество подвергают упариванию в присутствии CH3OH несколько раз (для удаления NH3), и затем подкисляют концентрированным водным раствором HCl. Азеотропная отгонка воды с толуолом и CH3OH дает, после вакуумирования в высоком вакууме, 830 мг неочищенного солянокислого вторичного амина (80%-ная чистота, 660 мг, примесь - солянокислый третичный амин) в виде твердого вещества. Выход продукта составляет 83%; продукт используют без очистки.
ТХ (10% , 10%-ного концентрированного водного аммиака в CH3OH в смеси CH2Cl2 анизальдегид): 4-Циклогексилбутиламин 0,07
Промежуточный кар- бамат 0,89 Целевой амин 0,11
1. [1S-[1S, 2 α (Z), 3 α, 4 α]]-6-[3-[4-[[(4-циклогексилбутил)метиламино] карбонил] - 2-оксазолил]- 7-оксабицикло [2.2.1] гептил-2]-4-гексеновая кислота, метиловый эфир.
Образец содержащей примеси оксазоловой кислоты, полученной в разделе G (примесь - тетрабутиламмоний, образец находится в виде свободной кислоты, 0,20 ммоль), сушат азеотропной отгонкой с сухим ТГФ и толуолом (процедуру повторяют дважды, используя высокий вакуум). Полученное вещество переносят в 2 мл толуола, и при перемешивании неоднородной смеси при комнатной температуре в атмосфере аргона, добавляют 127 мг оксалилхлорида (1,0 ммоль, 5 экв. ). Происходит выделение газа. Спустя 1 ч добавляют еще 127 мг оксалилхлорида. Снова происходит выделение газа. Смесь перемешивают в течение ночи. Метод ТХ показывает полное превращение в хлорангидрид кислоты, носящий название: метиловый эфир [1S-[1α, 2 α(Z), 3 α, 4 α ]]-6-[3-[4-(хлоркарбонил)-2-оксазолил] -7-оксаби- цикло-[2.2.1] гептил-2]-4-гексеновой кислоты. Растворитель испаряют, затем добавляют и снова испаряют толуол для полного удаления оксалилхлорида.
К неочищенному хлорангидриду кислоты (раздел G) добавляют 4 мл CHCl3. Полного растворения вещества не происходит. При перемешивании в атмосфере аргона при комнатной температуре добавляют 100 мг содержащего примеси вторичного амина, полученного в разделе Н (чистота 80%, 80 мг, примесь - солянокислый третичный амин, 0,39 ммоль, 2 экв.), и 145 мг триэтиламина (1,4 ммоль, 7 экв.). После перемешивания при комнатной температуре в течение 1 ч смесь разбавляют этилацетатом, добавляют воду и дважды экстрагируют этилацетатом, после чего сушат экстракт над NaSO4и выпаривают. Получают 120 мг содержащего примеси целевого амида в виде смолы. Неочищенный продукт используют без дальнейшей очистки.
ТХ (1% трифторуксусной кислоты, 1% метанола, 98% этилацетата - анизальдегид):
Оксазоловая кислота (раздел G) 0,28
Промежуточный хлор- ангидрид кислоты 0,78 Амид (раздел I) 0,64
Y. [1S-[1 α , 2 α ( Z), 3 α, 4 α]-6-[3-[4-[[(4-циклогексилбутил)-метиламино] карбонил] - 2-оксазолил- 7-оксабицикло [2.2.1] гептил-2-]4-гексеновая кислота.
К 120 мг полученного в разделение I амида в 6 мл CH3OH при комнатной температуре добавляют 2 мл 1,0М водного раствора NaOH. После перемешивания смеси в течение 3 ч, добавляют 1М водный раствор HCl, снижая рН до 1. Затем проводят экстракцию этилацетатом (три раза). Экстракты сушат над Na2SO4 и после испарения растворителя получают неочищенную целевую кислоту. Импульсная хроматография (50-100%, 5% уксусной кислоты в этилацетате, в градиенте гексана) дает, после азеотропной отгонки уксусной кислоты с толуолом, 70 мг чистого целевого продукта в виде масла. Выход целевого продукта составляет 72% в расчете на оксазоловую кислоту, полученную в разделе G.
ТХ (50%, 5% уксусной кислоты в этилацетате, в смеси гексан-анизальдегид): Амид (раздел I) 0,36 Целевой продукт 0,20
13С ЯМР (67,8 МГц в CDCl3. Видны две конформации. Линии, относящиеся только к одной конформации, даны в скобках):
177,0, 163,1, (162,3), (162,0), 142,5, 136,4, 129,6, 128,5, 79,5, 79,4, (50,4), 49,7, (48,7), 46,7, 37,5, 37,0, (36,4), 34,1, 33,3, 29,7, 29,0, 27,9, (27,2), 26,6, 26,3, (24,2), (23,7), 22,9.
П р и м е р 3. [1S-[1 α, 2α (Z), 3α , 4 α]]-6-[3-[4-[(1-пирролидинил)карбонил] -2-оксазолил] -7-оксабицикло [2.2.1] гептил-2]-4-гексеновая кислота.
A. 1-[N-[(1,1-диметилэтокси)карбонил]-L-серил]пирролидин
К раствору пирролидина (1,11 г, 15,7 ммоль), трет-бутилоксикарбонил (БОК) - (L)-серина (3,22 г, 15,7 ммоль), 1-оксибензотриазолгидрата (2,12 г, 15,7 ммоль) и диизопропилэтиламина (2,73 г, 15,7 ммоль) в 30 мл ТГФ при перемешивании в атмосфере аргона добавляют 1,3-дициклогексилкарбодиимид (3,23 г, 15,7 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 17 ч и концентрируют в вакууме. Затем смесь разбавляют 200 мл этилацетата и отфильтровывают образовавшийся осадок. Осадок промывают этилацетатом (3 раза по 40 мл). Собранные фильтраты промывают 1 н. водным раствором НС (3 раза по 70 мл) и насыщенным раствором NaHCO3 (2 раза по 80 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют в вакууме. Продукт хроматографируют на 140 г силикагеля 60 (Merck), используя в качестве элюента 2%-ный CH3OH в CH2Cl2 и получают 1,64 г (41%) целевого амида.
ТХ: силикагель, 4% CH3OH в CH2Cl2, Rf = 0,24, Cl(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 169,4, 155,6, 79,7, 62,8, 53,5, 46,5, 45,9, 28,0, 28,0, 28,0, 25,7, 23,8.
B. [1S-[1 α , 2 α, (Z), 3 α (R*), 4 α] ]-6-[3-[[[(1-(оксиметил)-2-оксо-2-(1-пирролидинил)-этил-] амино] карбонил] -7-оксабицикло [2.2.1]-гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору амида, полученного в разделе A (0,96 г, 3,72 ммоль), в 20 мл сухого CH2Cl2 при перемешивании в атмосфере аргона при 0оС добавляют 5 мл трифторуксусной кислоты (ТФУК). Смесь перемешивают при 0оС в течение 2 ч и разбавляют 50 мл толуола. Полученную смесь концентрируют в вакууме. К раствору полученной соли амина с ТФУК, 1-оксибензотриазолгидрата (0,50 г, 3,73 ммоль) и 5 мл триэтиламина в 20 мл ДМФА при перемешивании добавляют раствор кислоты, полученной в разделе D примера 1 (1,00 г, 3,73 ммоль), в 10 мл ДМФА. Затем к этой смеси добавляют этил-3(3-диметиламино)-пропилкарбодиимид в виде его солянокислой соли. Реакционную смесь перемешивают при комнатной температуре в течение 19 ч и концентрируют в вакууме. Смесь разбавляют 400 мл этилацетата и промывают 1 н. раствором HCl (3 раза по 30 мл), 0,2 н. раствором NaOH (2 раза по 30 мл) и рассолом (1 раз, 60 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют в вакууме. Очистку проводят методом импульсной хроматографии на 50 г силикагеля 60 (Merck), используя в качестве элюента растворы 2%- и 4%-ного CH3OH в CH2Cl2 (по 0,4 л каждого раствора), и получают 320 мг (22%) целевого спирта.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2, Rf = 0,22, Cе(SO4)2
13С ЯМР (67,5 МГц, CDCl3): δ : 173,4, 172,2, 168,8, 129,3, 129,2, 78,9, 78,9, 63,5, 54,3, 52,2, 51,4, 48,0, 46,6, 45,9, 33,7, 29,5, 28,8, 27,2, 25,8, 14,0, 22,7.
C. [1S-[1 α, 2 α(Z), 3 α (R*), 4α ]]-6-[3-[4,5-дигидро-4-[(1-пирролидинил)карбонил]-2- оксазолил]-7- оксабицикло [2.2.1] гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору спирта, полученного в разделе B (305 мг, 0,75 ммоль), и диизопропилэтиламина (0,39 мл, 2,24 ммоль) в 10 мл CH2Cl2 при перемешивании в атмосфере аргона при 0оС добавляют метансульфонилхлорид (0,07 мл, 0,90 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 4 ч и концентрируют в вакууме. Неочищенный мезилат растворяют в 30 мл ацетона и соединяют с 0,60 г K2CO3. Полученную смесь кипятят с обратным холодильником в течение 4 ч, охлаждают до комнатной температуры, и разбавляют 100 мл ацетона. Осадок отфильтровывают и промывают ацетоном (3 раза по 40 мл). Фильтрат концентрируют в вакууме и хроматографируют на 50 г силикагеля 60 (Merck), используя в качестве элюента 2%-ный CH3OH в CH2Cl2 получают 210 мг (72%) целевого оксазолина.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2, Rf = 0,20, Ce(SO4)2.
13С ЯМР (67,5 МГц. CDCl3) δ: 173,4, 168,2, 167,7, 129,5, 129,1, 78,9, 78,9, 68,7, 67,5, 51,4, 48,4, 46,5, 46,4, 46,1, 33,9, 29,7, 28,8, 27,1, 25,9, 24,1, 22,8.
D. [1S-[1 α, 2 α (Z), 3 α, 4α ]]-6-[3-[4-[(1-пирролидинил)карбонил]-8-оксазолил] -7-окса-бицикло [2.2.1] гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе С оксазолина (200 мг, NiO2 0,51 ммоль) в 5 мл CH2Cl2 при перемешивании добавляют 200 мг. Смесь перемешивают при комнатной температуре в течение 3 ч, после чего добавляют еще 200 мг NiO2. Эту смесь перемешивают в течение еще 1 ч, и добавляют еще одну порцию в 200 мг NiO2. Полученную реакционную смесь перемешивают при комнатной температуре в течение 17 ч и разбавляют 80 мл этилацетата. К полученной в результате смеси добавляют 5 мл 3М раствора NaHSO3 и 40 мл 1М раствора лимоннокислого натрия. Отделяют органический слой, водный слой экстрагируют этилацетатом (3 раза по 70 мл). Собранные органические экстракты сушат над MgSO4, фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на 18 г силикагеля 60 (Merck), используя в качестве элюента 2%-ный CH3OH в CH2Cl2, получают 69,2 мг (35%) целевого сложного эфира.
ТХ: силикагель, 4% CH3OH в CH2Cl2, Rf = 0,24, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,2, 163,1, 160,3, 142,2, 137,1, 129,3, 128,7, 79,4, 79,1, 51,4, 49,6, 48,2, 46,7, 46,6, 33,7, 29,7, 28,9, 27,8, 26,4, 23,7, 22,7.
E. [1S- [1 α, 2 α (Z), 3 α, 4 α ]]-6-[3-[4-[(1-пирролидинил)карбонил] -2-оксазолил-7-ок- сабицикло [2.2.1] гептил-2]-4-гексеновая кислота.
К раствору полученного в разделе D сложного эфира (69,0 мг, 0,18 ммоль) и 2 мл воды в 12 мл ТГФ при перемешивании добавляют 2 мл 1 н. раствора LiOH. Полученную смесь в течение 10 мин продувают аргоном и перемешивают при комнатной температуре в течение 7 ч. Смесь подкисляют до рН 2, добавляя 1 н. HCl, и насыщают NaCl. Слой ТГФ отделяют, водный слой экстрагируют этилацетатом (4 раза по 60 мл). Собранные органические экстракты сушат над MgSO4, фильтруют и концентрируют в вакууме. Очистку проводят методом импульсной хроматографии на 10 г силикагеля 60 (Merck), используя в качестве элюента 10%-ный CH3OH в CH2Cl2, получают 26 мг (39%) целевой кислоты.
ТХ: силикагель, 6%-ный: CH3OH в CH2Cl2, Rf = 0,22, Ce(SO4)2.
13С ЯМР (67,5 МГц, ДМСО - d6) δ: 175,1, 163,2, 159,6, 142,2, 136,3, 130,5, 127,6, 78,8, 78,4, 48,8, 47,7, 46,3, 45,8, 29,3, 28,4, 27,5, 25,9, 23,3, 23,2, 22,8.
П р и м е р 4. [1S-[1 α, 2 α (Z), 3 , 4 α]]-6-[3-[4-[(циклогексиламино)карбонил] -2- оксазолил]- 7-оксабицикло [2.2.1] гепил-2] -4-гексеновая кислота.
A. [(1,1-Диметилэтокси)карбонил]-N-циклогексил-L-серинамид.
К раствору циклогексиламина (1,11 г, 15,7 ммоль), БОК-(L)-серина (3,22 г, 15,7 ммоль), 1-оксибензотриазолгидрата (2,12 г, 15,7 ммоль) и диизопропилэтиламина (2,73 мл, 15,7 ммоль) в 30 мл ТГФ при перемешивании в атмосфере аргона при 0оС добавляют 1,3-дициклогексилкарбодиимид (3,23 г, 15,7 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 18 ч. Смесь разбавляют 200 мл этилацетата и отфильтровывают осадок. Осадок промывают этилацетатом (3 раза по 40 мл). Собранные фильтраты промывают 1 н. водным раствором HCl (3 раза по 70 мл), и насыщенным раствором NaHCO3 (2 раза по 80 мл). Органический слой сушат над MgSO4, фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на 140 г силикагеля 60 (Merck), используя в качестве элюента 2%-ный CH3OH в CH2Cl2; получают 1,64 г (41%) целевого амида.
ТХ: силикагель: 4%-ный CH3OH в CH2Cl2; Rf = 0,24, Ce(SO4)2.
13C ЯМР (67,5 МГц, CDCl3) δ : 170,1, 156,1, 80,2, 62,7, 48,1, 32,6, 32,6, 28,1, 25,3, 24,5, 24,5, 24,5.
B. [1S-[1α , 2 α (Z), 3 α (R*), 4 α]]-6-[3-[[[(2-циклогексиламино)-1-(оксиметил)-2-оксоэ-тил] амино] карбонил]-7-оксабицикло [2.2.1]-гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе A амида (1,06 г, 3,73 ммоль) в 20 мл сухого хлористого метилена при перемешивании в атмосфере аргона при 0оС добавляют 5 мл ТФУК. Смесь перемешивают при 0оС в течение 2 ч и разбавляют 50 мл толуола. Полученную смесь концентрируют в вакууме. К раствору полученной соли амина с ТФУК, 1-оксибензотриазолгидрата (0,50 г, 3,73 ммоль) и 6 мл триэтиламина в 20 мл ДМФА при перемешивании добавляют раствор полученной в разделе D примера 1 кислоты (1,00 г, 3,73 ммоль) в 10 мл ДМФА. К полученной смеси затем добавляют солянокислую соль этил-3-(3-диметиламино)-пропилкарбодиимида (0,55 г, 3,73 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 19,5 ч и концентрируют в вакууме. Смесь разбавляют 400 мл этилацетата и промывают 1 н. раствором HCl (3 раза по 40 мл), 0,2 н. раствором NaOH (2 раза по 30 мл), насыщенным раствором NaHCO3 (1 раз, 30 мл) и рассолом (1 раз, 100 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистку осуществляют методом импульсной хроматографии на силикагеле 60 (60 г, Merck, используя в качестве элюента 2% -ный CH3OH в CH2Cl2 получают 690 мг (42%) целевого спирта.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,30, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,3, 173,0, 169,6, 129,4, 129,0, 79,1, 78,9, 62,7, 54,4, 53,7, 51,4, 48,2, 48,0, 33,7, 32,6, 32,6, 32,6, 29,5, 28,6, 27,5, 25,3, 24,6, 24,6, 22,7.
С. [1S-[1 α, 2 α (Z), 3 α, (R*), -6-[3-4-(циклогексиламино)-карбонил] -4,5"-дигидро-2-оксазолил-7- оксабицикло [2.2.1] гептил-2]-4-гексеновая кислота, метиловый эфир
К раствору полученного в разделе B спирта (680 мг, 1,56 ммоль) и диизопропилэтиламина (0,82 мл, 4,68 ммоль) в 20 мл хлористого метилена при перемешивании в атмосфере аргона при 0оС добавляют метансульфонилхлорид (0,13 мл, 1,63 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 1 ч и концентрируют в вакууме. Неочищенный мезилат растворяют в 20 мл ацетона и соединяют с 647 мг K2CO3. Полученную смесь нагревают при 54оС в течение 3,5 ч, охлаждают до комнатной температуры и разбавляют 100 мл ацетона. Осадок отфильтровывают и промывают ацетоном (3 раза по 50 мл). Фильтрат концентрируют в вакууме и хроматографируют на 50 г силикагеля 60 (Merck), используя в качестве элюента 2%-ный CH3OH в CH2Cl2 получают 540 мг (83%) целевого оксазолина.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2, Rf = 0,42, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,2, 170,4, 169,2, 129,5, 128,9, 79,1, 79,0, 69,7, 68,4, 51,4, 48,4, 47,8, 46,4, 33,8, 33,0, 32,8, 29,6, 28,9, 27,3, 25,4, 24,7, 24,7, 22,8.
D. [1S-[1 α, 2 α(Z), 3α , 4α ]]-6-[3-[4-[(циклогексиламино)карбонил]-2-оксазолил] -7- оксабицикло [2.2.1] гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе С оксазолина (530 мг, 1,26 ммоль) в 5 мл хлористого метилена при перемешивании добавляют 1,06 г NiO2. Реакционную смесь перемешивают при комнатной температуре в течение 3 ч, после чего добавляют еще 530 мг NiO2. Полученную смесь перемешивают при комнатной температуре в течение 1 ч и добавляют еще 530 мг NiO2. Эту смесь перемешивают в течение еще 80 мин и разбавляют 120 мин этилацетата. К полученной смеси добавляют 10 мл 3М раствора NaHSO3 и 70 мл раствора лимоннокислого натрия (1М). Отделяют органический слой, и экстрагируют водный слой этилацетатом (2 раза по 100 мл). Собранные органические экстракты промывают рассолом (1 раз, 100 мл) сушат (MgSO4), фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на 40 г силикагеля 60 (Merck), используя в качестве элюента 2% CH3OH в CH2Cl2; получают 370 мг (70%) целевого сложного эфира.
ТХ: силикагель, 4% CH3OH в CH2Cl2, Rf = 0,64, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ : 173,2, 163,7, 159,6, 140,6, 136,1, 129,4, 128,5, 79,5, 79,3, 51,4, 49,6, 47,8, 46,6, 33,7, 33,0, 33,0, 29,7, 28,8, 27,8, 25,4, 24,9, 24,9, 22,7.
E. [1S-[1 α, 2 α(Z), 3α , 4 α ]]-6-[3-[4-[(циклогексиламино)карбонил] -2-оксазолил]-7- оксабицикло [2.2.1] гептил-2]-4-гексеновая кислота.
К раствору полученного в разделе D сложного эфира (360 мг, 0,86 ммоль) и 10 мл воды в 60 мл ТГФ при перемешивании добавляют 10 мл 1 н. раствора LiOH. Полученную смесь в течение 10 мин продувают аргоном и перемешивают при комнатной температуре в течение 7 ч. Смесь подкисляют до рН 2, добавляя 1 н. раствор HCl и насыщают NaCl. Слой ТГФ отделяют; водный слой экстрагируют этилацетатом (4 раза по 80 мл). Собранные органические экстракты сушат (MgSO4) фильтруют и концентрируют в вакууме. Очистку проводят методом импульсной хроматографии на 40 г силикагеля 60 (Merck), используя в качестве элюента 4%-ный CH3OH в CH2Cl2 получают 300 мг (86%) целевой кислоты.
ТХ: силикагель, 6%-ный CH3OH в CH2Cl2; Rf = 0,32, Ce(SO4).
13С ЯМР (67,5 МГц, CDCl3) δ: 176,9, 163,9, 160,0, 141,0, 129,5, 128,5, 79,5, 79,5, 48,1, 46,6, 33,7, 32,9, 32,9, 29,7, 28,9, 27,9, 25,5, 24,9, 24,9.
П р и м е р 5. [1S-[1 α , 2α (Z), 3α , 4 α]] -6-[3-[[4-[[(2-циклогексилэтил)аминокарбо-нил] -2-оксазолил]-7- оксабицикло [2.2.1] гептил-2-]-4-гексеновая кислота.
A. [(1,1-диметилэтокси)карбонил-N-(2-циклогексилэтил)-L-серинамид.
К раствору 2-циклогексилэтиламина (2,00 г, 15,7 ммоль), БОК-(L)-серина (3,22 г, 15,7 ммоль), 1-оксибензотриазолгидрата (2,12 г, 15,7 ммоль), и диизопропилэтиламина (2,73 мл, 15,7 ммоль) в 40 мл ТГФ при перемешивании в атмосфере аргона при 0оС добавляют 1,3-дициклогексилкарбодиимид (3,23 г, 15,7 ммоль). Полученную смесь перемешивают при комнатной температуре и добавляют 10 мл ДМФА. Затем реакционную смесь перемешивают при комнатной температуре в течение 18 ч. Смесь разбавляют 300 мл этилацетата, и отфильтровывают осадок. Осадок промывают этилацетатом (3 раза по 40 мл). Собранный фильтрат промывают 1 н. раствором HCl (3 раза по 70 мл), и насыщенным раствором NaHCO3 (2 раза по 80 мл). Органический слой сушат (MgSO4) фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на силикагеле 60 (Merck) 160 г), используя в качестве элюента смесь гексан - диэтиловый эфир (1:4), получают 1,91 г (39%) целевого амида.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,34, Ce(SO4)2.
13С ЯМР (67,5, МГц, CDCl3) δ : 171,1, 80,3, 62,9, 37,3, 36,8, 35,2, 33,1, 33,1, 28,3, 28,3, 26,4, 26,4, 26,1.
B. [1S-[ 1 α, 2 α (Z), 3 α (R*), 4 α ]]-6-[3-[[[2-[2-(циклогексилэтил)амино] -1-(оксиметил)- 2-оксоэтил]- амино]карбонил]-7-оксабицикло [2.2.1] гептил-2-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе A амида (1,28 г, 3,73 ммоль) в 20 мл сухого хлористого метилена при перемешивании в атмосфере аргона при 0оС добавляют 5 мл ТФУК. Полученную смесь перемешивают при 0оС в течение 2 ч и разбавляют 50 мл толуола. Эту смесь концентрируют в вакууме. К раствору полученной соли амина с ТФУК, 1-оксибензотриазолгидрата (0,50 г, 3,73 ммоль) и 5 мл триэтиламина в 20 мл ДМФА при перемешивании добавляют раствор полученной в разделе D примера 1 кислоты (1,00 г, 3,73 ммоль) в 10 мл ДМФА. Затем к этой смеси добавляют солянокислую соль этил-3-(3-диметиламино)-пропилкарбодиимида (0,55 г, 3,73 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 17 ч и концентрируют в вакууме. Смесь разбавляют 400 мл этилацетата и промывают 1 н. раствором HCl (3 раза по 40 мл), 0,2 раство- ром NaOH (2 раза по 30 мл), насыщенным раствором NaHCO3 (1 раз, 30 мл) и рассолом (1 раз, 100 мл). Органический слой сушат (MgSO4), фильтруют и концентрируют в вакууме. Очистку осуществляют методом импульсной хроматографии на 60 г силикагеля 60 (Merck), используя в качестве элюента 2%-ной CH3OH в CH2Cl2; получают 1,00 г (54%) целевого спирта.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,30, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,4, 172,9, 170,4, 129,3, 129,0, 79,0, 78,9, 62,7, 54,2, 53,7, 51,4, 48,0, 37,3, 36,6, 35,3, 33,7, 32,9, 32,9, 29,5, 28,6, 27,3, 26,3, 26,0, 26,0, 22,7.
С. [1S-[1α ,2 α (Z),3α (R*),4 α ]]-6-[3-[4-[[(2-циклогексилэтил)амино] карбонил]-4,5-ди- гидро- 2-оксазолил]-7-оксабицикло[2.2.1]-гептил-2-4-гексеновая кислота, метиловый эфир. К раствору полученного в разделе B спирта (890 мг, 1,80 ммоль) и диизопропилэтиламина (0,94 мл, 5,40 ммоль) в 30 мл хлористого метилена при перемешивании в атмосфере аргона при 0оС добавляют метансульфонилхлорид (0,14 мл, 1,80 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 1 ч и концентрируют в вакууме. Неочищенный мезилат растворяют в 30 мл ацетона и соединяют с 0,77 г K2CO3. Эту смесь кипятят с обратным холодильником в течение 4 ч, охлаждают до комнатной температуры и разбавляют 100 мл ацетона. Осадок отфильтровывают и промывают ацетоном (3 раза по 50 мл). Фильтрат концентрируют в вакууме и хроматографируют на 60 г силикагеля 60 (Merck), используя в качестве элюента 25%-ный CH3OH в CH2Cl2 получают 480 мг (54%) целевого соединения.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,42, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,1, 171,2, 169,2, 129,4, 128,9, 79,1, 79,0, 69,6, 68,3, 51,4, 48,3, 46,3, 36,9, 36,8, 35,2, 33,7, 33,0, 32,9, 29,6, 28,8, 27,2, 26,3, 26,1, 26,1, 22,8.
D. [1S-[1α , 2α (Z),3 α, 4 α ]]-6-[3-[4-[[(2-циклогексилэтил)амино]карбонил] -2-окса-золил] -7- оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир. К раствору полученного в разделе С оксазолина (480 мг, 1,01 ммоль) в 20 мл хлористого метилена при перемешивании добавляют 480 мг NiO2. Реакционную смесь перемешивают при комнатной температуре в течение 1 ч, после чего добавляют еще 480 мг NiO2.
Смесь перемешивают при комнатной температуре еще 3 ч и добавляют еще 480 мг NiO2. Эту смесь перемешивают еще 15,5 часов, и добавляют еще одну порцию NiO2 (480 мг). Реакционную смесь перемешивают еще 1,5 ч и разбавляют 100 мл этилацетата. К полученной смеси добавляют 20 мл 3М раствора NaHSO3 и 30 мл 1М раствора лимоннокислого натрия. Отделяют органический слой и экстрагируют водный слой этилацетатом (2 раза по 100 мл). Собранные органические экстракты промывают рассолом (1 раз, 50 мл), сушат (MgSO4) фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на 45 г силикагеля 60 (Merck) используя в качестве элюента 2%-ный CH3OH в CH2Cl2 получают 200 мг (42%) целевого сложного эфира.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,60, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,1, 163,7, 160,4, 140,3, 136,0, 129,3, 128,4, 79,4, 79,3, 51,3, 49,6, 46,6, 36,9, 36,7, 35,2, 33,6, 33,0, 33,0, 29,7, 28,8 27,7, 26,4, 26,0, 26,0, 22,7.
Е. [1S-[1 α ,2 α(Z),3 α , 4 α]]-6-[3-[4-[[(2-циклогексилэтил)-амино]карбонил]-2-оксазолил]-7- оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота.
К раствору полученного в разделе D сложного эфира (190 мг, 0,40 ммоль) и 4 мл воды в 30 мл ТГФ при перемешивании добавляют 4 мл 1 н. раствора LiOH. Полученную смесь в течение 10 мин продувают аргоном и перемешивают при комнатной температуре в течение 5 ч. Смесь подкисляют до рН 2, добавляя 1 н. раствор HCl и насыщают NaCl. Слой ТГФ отделяют, водный слой экстрагируют этилацетатом (4 раза по 25 мл). Собранные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Очистку проводят методом импульсной хроматографии на 20 г силикагеля 60 (Merck), используя в качестве элюента 4%-ный CH3OH в CH2Cl2, получают 106,7 мг (59%) целевой кислоты.
ТХ: силикагель, 6%-ный CH3OH в CH2Cl2; Rf = 0,32, Ce(SO4)2.
13С ЯМР (67,5, МГц, CDCl3) δ : 176,9, 163,9, 160,7, 140,8, 135,7, 129,4, 128,4, 79,5, 79,4, 49,6, 46,5, 36,9, 36,8, 35,2, 33,7, 33,0, 33,0, 29,6, 28,8, 27,8, 26,4, 26,1, 26,1, 22,6.
П р и м е р 6. [1S-[1 α,2 α (Z),3α 4 α ]]-6-[3-[4-[[[2-(4-хлорфенил)-этил]амино]карбонил]-2-ок-сазолил]-7- оксабицикло[2.2.1]-гептил-4-гексеновая кислота.
A.[(1,1-диметилэтокси)карбонил]-N-[2- (4-хлорфенил)этил-L-серинамид.
К раствору 2-(4-хлорфенил)этиламина (2,44 г, 15,7 ммоль), БОК-(L)-серина (3,22 г, 15,7 ммоль), 1-оксибензотриазолгидрата (2,12 г, 15,7 ммоль) и диизопропилэтиламина (2,73 мл, 15,7 ммоль) в 30 мл ТГФ при перемешивании в атмосфере аргона при 0оС добавляют 1,3-дициклогексилкарбодиимид (3,23 г, 15,7 ммоль). Полученную смесь перемешивают при комнатной температуре и добавляют 10 мл ДМФА. Затем реакционную смесь перемешивают при комнатной температуре в течение 18 ч. Смесь разбавляют 200 мл этилацетата и отфильтровывают осадок. Осадок промывают этилацетатом (3 раза по 40 мл). Собранные фильтраты промывают 1 н. водным раствором HCl (3 раза по 70 мл) и насыщенным раствором NaHCO3 (2 раза по 80 мл). Органический слой сушат (MgSO4) фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на 160 г силикагеля 60 (Merck) используя в качестве элюента смесь гексан-диэтиловый эфир (1:4), получают 2,56 г (48%) целевого амида.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,34, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,1, 157,6, 139,2, 133,1, 131,4, 131,4, 129,4, 80,8, 63,3, 58,0, 41,7, 35,7, 34,7, 28,7, 28,7, 28,7.
B. [1S-[1 α ,2 α(Z),3 α (R*),4 α ]]-6-[3-[[[2-[[2-(4-хлорфенил)-этил] амино] -1-(оксиметил)-2- оксоэтил]амино]карбонил]-7-оксабицик-ло [ 2.2.1] гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе A амида (1,28 г, 3,73 ммоль) в 20 мл сухого хлористого метилена при перемешивании в атмосфере аргона при 0оС добавляют 5 мл ТФУК. Смесь перемешивают при 0оС в течение 2 ч и разбавляют 50 мл толуола. Полученную смесь концентрируют в вакууме. К раствору полученной соли амина и ТФУК, 1-оксибензотриазолгидрата (0,50 г, 3,73 ммоль) и 5 мл триэтиламина в 20 мл ДМФА при перемешивании добавляют раствор полученной в разделе D примера 1 кислоты (1,00 г, 3,73 ммоль) в 10 мл ДМФА. К полученной смеси затем добавляют солянокислый этил-3-(3-диметиламино)пропилкарбодии-мид (0,55 г, 3,73 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 17 ч и концентрируют в вакууме. Смесь разбавляют 400 мл этилацетата и промывают 1 н. раствором HCl (3 раза по 40 мл), 0,2 н. раствором NaOH (2 раза по 30 мл), насыщенным раствором NaHCO3 (1 раз, 30 мл) и рассолом (1 раз, 100 мл). Органический слой сушат (MgSO4), фильтруют и концентрируют в вакууме. Очистку проводят методом импульсной хроматографии на 60 г силикагеля 60 (Merck) используя в качестве элюента 2% -ный CH3OH в CH2Cl2получают 1,00 г (54%) целевого спирта.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,30, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ : 173,5, 172,9, 170,6, 137,2, 132,1, 130,0, 130,0, 129,3, 129,1, 128,5, 128,5, 79,0, 78,9, 62,7, 54,1, 53,9, 51,4, 48,0, 40,8, 34,8, 33,7, 29,5, 28,7, 27,3, 22,7.
C. [1S-[1α, 2 α (Z),3 α (R*),4 α]]-6-[3-[4-[[[2-(4-хлорфенил)-этил]-амино] карбонил]-4,5-ди- гидро- 2-оксазолил]-7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе B спирта (890 мг, 1,80 ммоль) и диизопропилэтиламина (0,94 мл, 5,40 ммоль) в 30 мл хлористого метилена при перемешивании в атмосфере аргона при 0оС добавляют метансульфонилхлорид (0,14 мл, 1,80 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 1 ч и концентрируют в вакууме. Неочищенный мезилат растворяют в 30 мл ацетона и совмещают с 0,77 г K2CO3. Полученную смесь нагревают с обратным холодильником в течение 4 ч, охлаждают до комнатной температуры и разбавляют 100 мл ацетона. Осадок отфильтровывают и промывают ацетоном (3 раза по 50 мл). Фильтрат концентрируют в вакууме и хроматографируют на 60 г силикагеля 60 (Merck) используя 2%-ный CH3OH в CH2Cl2 в качестве элюента; получают 480 мг (54%) целевого оксазолина.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,42, Ce(SO4)2.
13С ЯМР (67,5, МГц, CDCl3) δ : 171,6; 169,4; 169,3; 137,0; 132,3; 130,0; 130,0; 129,5; 129,0; 128,6; 128,6; 79,1; 79,0; 69,6; 68,3; 51,5; 48,2; 46,3; 40,0; 34,8; 33,8; 29,6; 28,9; 27,2; 22,8.
D.[1S-[1 α,2 α (Z),3α ,4 α]]-6-[3-[4-[[[2-(4-хлорфенил)этил]аминокарбонил] -2-оксазо- лил] -7- оксабицикло[2.2.1]гептил-2-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе С оксазолина (480 мг, 1,01 ммоль) в 20 мл хлористого метилена при перемешивании добавляют 480 мг NiO2. Реакционную смесь перемешивают при комнатной температуре в течение 1 ч, после чего снова добавляют 480 мг NiO2. Эту смесь перемешивают при комнатной температуре в течение 3 ч, и добавляют еще 480 мг NiO2. Полученную смесь перемешивают еще 15,5 ч и добавляют еще одну порцию NiO2 (480 мг). Реакционную смесь перемешивают в течение 1,5 ч и разбавляют 100 мл этилацетата. К полученной в результате смеси добавляют 20 мл 3М раствора NaHSO3 и 30 мл 1М раствора лимоннокислого натрия. Отделяют органический слой, и экстрагируют водный слой этилацетатом (2 раза по 100 мл). Собранные органические экстракты промывают рассолом (1 раз, 50 мл), сушат (MgSO4), фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на 45 г силикагеля 60 (Merck), используя в качестве элюента 2%-ный CH3OH в CH2Cl2; получают 200 мг (42%) целевого сложного эфира.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,60, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,2, 169,3, 160,6, 140,5, 137,2, 135,8, 132,2, 130,0, 130,0, 129,4,128,6, 128,6, 128,5, 79,5, 79,3, 51,4, 49,6, 46,6, 40,1, 35,2, 35,2, 33,7, 29,7, 28,9, 27,8, 22,7.
E.[1S-[1α,2 α(Z),3 α ,4 α]]-6-[3-[4-[[[2-(4-хлорфенил)этил]амино]карбонил]-2-оксазолил]-7- оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота.
К раствору полученного в разделе D сложного эфира (190 мг, 0,40 ммоль) и 4 мл воды в 30 мл ТГФ при перемешивании добавляют 4 мл 1 н. раствора LiOH. Полученную смесь продувают в течение 10 минут аргоном и перемешивают при комнатной температуре в течение 5 ч. Смесь подкисляют до рН 2, добавляя 1 н. HCl, и насыщают NaCl. Отделяют слой ТГФ, водный слой экстрагируют этилацетатом (4 раза по 25 мл). Собранные органические экстракты сушат (MgSO4) фильтруют и концентрируют в вакууме. Очистку проводят методом импульсной хроматографии на 20 г силикагеля 60 (Merck) используя в качестве элюента 4% -ный CH3OH в CH2Cl2 получают 106,7 мг (59%) целевой кислоты.
ТХ: силикагель, 6%-ный CH3OH в CH2Cl2; Rf = 0,32, Сe(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 176,9, 175,4, 164,0, 160,9, 140,9, 137,2, 130,1, 130,1, 129,4, 128,6, 128,6, 79,6, 79,5, 49,6, 46,6, 40,2, 35,1, 33,7, 29,7, 28,9, 27,9, 22,6.
П р и м е р 7. [1S-[1 α,2α (Z),3 α,4α ]]-6-[3-[4-[[[(4-хлорфенил)амино] карбонил]-2-окса-золил]-7-оксабицикло [2.2.1]-гептил-2]-4-гексеновая кислота.
A.[(1,1-диметилэтокси)карбонил]-N-(4-хлорфенил)-L-серинамид
К раствору 4-хлоранилина (2,00 г, 15,7 ммоль), ВОС-(L)-серина (3,22 г, 15,7 ммоль), 1-оксибензотриазолгидрата (2,12 г, 15,7 ммоль) и диизопропилэтиламина (5,40 мл, 31,2 ммоль) в 40 мл ДМФА при перемешивании в атмосфере аргона добавляют солянокислый этил-3(3-диметиламино)пропилкарбодиимид (2,31 г, 15,7 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 17 ч и концентрируют в вакууме. Смесь разбавляют 400 мл этилацетата и промывают 1 н. водным раствором HCl (3 раза по 70 мл), и насыщенным раствором NaHCO3 (2 раза по 70 мл). Органический слой сушат (MgSO4) фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на 80 г силикагеля 60 (Merck), используя в качестве элюентов смеси гексан-диэтиловый эфир состава 1:3 и 1:4 (по 1 л каждой смеси); получают 1,02 г (21%) целевого амида.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,34, Ce(SO4)2.
B. [1S-[1S, 2α (Z), 3α (R*), 4α ]]-6-[3-[[[2-[(4-хлорфенил)амино]-1-(оксиметил)-2-оксоэтил] амино] карбонил]-7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе A амида (1,02 г, 3,24 ммоль) в 12 мл сухого хлористого метилена при перемешивании в атмосфере аргона при 0оС добавляют 3 мл ТФУК. Смесь перемешивают при 0оС в течение 3 ч и разбавляют 50 мл толуола. Эту смесь концентрируют в вакууме. К раствору этой соли амина и ТФУК, 1-оксибензотриазолгидрата (0,50 г, 3,73 ммоль) и 5 мл триэтиламина в 20 мл ДМФА при перемешивании добавляют раствор полученной в разделе D примера 1 кислоты (1,00 г, 3,73 ммоль) в 10 мл ДМФА. Затем к полученной смеси добавляют солянокислый 1-(3-диметиламинопропил)-3-этилкарбодиимид. Реакционную смесь перемешивают при комнатной температуре в течение 17 ч и концентрируют в вакууме. Полученную смесь разбавляют 400 мл этилацетата и промывают 1 н. раствором HCl (3 раза по 30 мл), 0,2 н. раствором NaOH (2 раза по 30 мл) и рассолом (1 раз, 100 мл). Органический слой сушат (MgSO4) и концентрируют в вакууме. Очистку проводят методом импульсной хроматографии на 80 г силикагеля 60 (Merck), используя в качестве элюента 2%-ный CH3OH в CH2Cl2; получают 380 мг (22%) целевого спирта.
ТХ: силикагель; 4%-ный CH3OH в CH2Cl2; Rf = 0,31, Ce(SO4)2,
С. [1S-[1 α,2 α,(Z),3α (R*),4 α] ]-6-[3-[4-[[(4-хлорфенил)амино]карбонил]-4,5-дигидро-2-оксазолил]- 7-оксабицикло][2.2.1]-гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе B спирта (370 мг, 2,39 ммоль) и диизопропилэтиламина (0,42 мл, 2,39 ммоль) в 5 мл хлороформа при перемешивании в атмосфере аргона при 0оС добавляют метансульфонилхлорид (0,068 мл, 0,88 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 30 мин и концентрируют в вакууме. Неочищенный мезилат растворяют в 10 мл ацетона и совмещают с 0,70 г K2CO3. Эту смесь нагревают с обратным холодильником при температуре, близкой к температуре кипения, в течение 3 ч, охлаждают до комнатной температуры и разбавляют 100 мл ацетона. Осадок отфильтровывают и промывают ацетоном (3 раза по 30 мл). Фильтрат концентрируют в вакууме и хроматографируют на 35 г силикагеля 60 (Merck) используя в качестве элюента 2%-ный CH3OH в CH2Cl2, получая 210 мг (59%) целевого оксазолина.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,68, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) α: 173,2; 170,0; 169,6; 135,8; 129,5; 129,4; 128,9; 128,5; 120,9; 120,9; 79.2; 69,4; 68,7; 51,4; 48,3; 46,4; 33,7; 29,6; 28,8; 27,2; 22,8.
D. [1S-[1 α, 2 α (Z),3 α, 4α ]]-6[3-[4-[[(4-хлорфенил)амино]карбонил]-2-оксазолил] -7- оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе С оксазолина (200 мг, 0,45 ммоль) в 3 мл хлористого метилена при перемешивании добавляют 200 мг NiO2. Реакционную смесь перемешивают при комнатной температуре в течение 1,5 ч, после чего добавляют еще 200 мг NiO2. Эту смесь перемешивают при комнатной температуре в течение 1,5 ч, и добавляют еще 200 мг NiO2. Полученную смесь перемешивают еще 1,5 ч и вводят еще одну порцию (200 мг).
Реакционную смесь перемешивают в течение 2 ч и разбавляют 100 мл этилацетата. К полученной смеси добавляют 5 мл 3М NaHSO3 и 30 мл 1М лимоннокислого натрия. Органический слой отделяют; водный слой экстрагируют этилацетатом (2 раза по 200 мл). Собранные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Полученный продукт хроматографируют на 15 г силикагеля 60 (Merck), используя в качестве элюента 2%-ный CH3OH в CH2Cl2, получают 140 мг (70%) целевого сложного эфира.
ТХ: силикагель, 4%-ный CH3OH в CH2Cl2; Rf = 0,76, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 173,2, 164,2, 158,4, 141,4, 136,0, 135,9, 129,5, 129,3, 128,9, 128,9, 128,4, 121,1, 79,6, 79,4, 51,4, 49,6, 46,6, 33,7, 29,7, 28,9, 27,9, 22,8.
E. [1S-1 α, 2 α (Z),3 α , 4 α]]-6-[3-[4-[[(4-хлорфенил)амино]карбонил] -2-оксазолил]-7-оксабицикло [2.2.1]гептил-2]-4-гексеновая кислота.
К раствору полученного в разделе D сложного эфира (135 мг, 0,03 ммоль) и 3 мл воды в 15 мл ТГФ при перемешивании добавляют 3 мл 1 н. раствора LiOH. Полученную смесь в течение 10 минут продувают аргоном и перемешивают при комнатной температуре в течение 5 часов. Смесь подкисляют до рН = 2, добавляя раствор 1 н. HCl и насыщают NaCl. Отделяют слой ТГФ; водный слой экстрагируют этилацетатом (4 раза по 15 мл). Собранные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Очистку проводят методом импульсной хроматографии на 15 г силикагеля 60 (Merck) используя в качестве элюента 4% -ный СH3OH и CH2Cl2; получают 79,2 мг (61%) целевой кислоты.
ТХ: силикагель, 6%-ный CH3OH в CH2Cl2; Rf = 0,36, Ce(SO4)2.
13С ЯМР (67,5 МГц, CDCl3) δ: 179,0, 177,6, 164,2, 141,7, 136,0, 135,9, 129,4, 128,5, 121,2, 79,7, 79,5, 49,6, 46,6, 33,6, 32,3, 29,7, 28,9, 27,9, 22,6.
П р и м е р 8. [1S-[1 α,2 α (Z), 3 α ,4 α]]-6-[3-7-[4-[[[4-(4-хлорфенил)бутиламино] карбонил] -2-оксазолил] 7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота.
A. [1S-[1 α, 2 α(Z),3 α, 4 α]]-6-[3-[4-[[[4-(4-хлорфенил)бутил]амино] карбонил] -2-окса-золил] - 7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
0,21 г содержащего примеси соединения, полученного в разделе G примера 2 (чистота 50%, 0,11 г, 0,33 ммоль), перемешивают в 4 мл толуола и добавляют 0,1 мл (1,14 ммоль) оксазолилхлорида. Добавляют одну каплю ДМФА и перемешивают полученную смесь в течение 2 ч при комнатной температуре, после чего, по данным ТХ, реакция заканчивается. Реакционную смесь концентрируют в вакууме; остаток дважды повторно концентрируют из 2 мл толуола, получая оранжевое масло, а именно метиловый эфир [1S-[1 α , 2 α (Z),3 α ,4 α]]-6-(4-хлорфенил)-2-оксазолил)-7-оксабицикло[2.2.1] геп-тил-2] -4- гексеновой кислоты. К этому маслу добавляют около 3 мл хлороформа, а затем - 0,11 мл (0,08 г, 0,78 ммоль) триэтиламина и 0,13 г (0,71 ммоль) 4-(4-хлорфенил)бутиламина и полученную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь разбавляют этилацетатом и водой, отделяют органический слой, и водный слой дважды экстрагируют 20 мл этилацетата. Органические слои собирают вместе, промывают рассолом, сушат над MgSO4 и концентрируют в вакууме, получая 0,43 г, оранжевого масла. Это масло подвергают импульсной хроматографии (диоксид кремния, от 0 до 100% этилацетата в градиенте гексана), получая 0,26 г содержащего примеси целевого метилового эфира (чистота 50% , 0,13 г целевого продукта) в виде масла. Выход продукта составляет 78%.
13С ЯМР (67,8 МГц, CDCl3) δ: 172,8, 163,6, 160,3, 140,2, 135,7, 131,0, 129,4, 129,1, 128,2, 128,0, 79,2, 79,1, 51,1, 49,4, 46,3, 38,4, 34,4, 33,5, 29,4, 28,6, 28,1, 27,6, 22,5.
B. [1S-[1 α 2 α(Z),3 α,4α]]-6-[3-[4-[[[4-(4-хлорфенил)бутил]-амино] карбонил]-2-оксазолил]-7-оксабицикло[2.2.1]гептил-2]-4-гексе- новая кислота.
0,26 г содержащего примеси сложного эфира, полученного в разделе A. (чистота 50%, 0,13 г, 0,26 ммоль), перемешивают в примерно 30 мл 1 н. NaOH и 2 мл ТГФ в течение 8 ч при комнатной температуре. Реакционную смесь концентрируют для удаления ТГФ в течение 8 ч при комнатной температуре. Реакционную смесь концентрируют для удаления ТГФ и подкисляют до рН 2, добавляя концентрированную HCl. Добавляют этилацетат и отделяют органический слой. Водный слой дважды экстрагируют примерно 20 мл этилацетата. Органические слои собирают вместе, промывают насыщенным раствором NaCl сушат над MgSO4 и концентрируют в вакууме, получая 0,2 г светлого масла. Полученный продукт хроматографируют (диоксид кремния, 0,50% CH3OH, 0,25% CH3CO2H, 99,25% этилацетата), получая 0,10 г чистого продукта. Выход продукта составляет 79%. 13С ЯМР (67,8 МГц, CDCl3) δ : 178,4, 164,4, 161,4, 141,0, 140,4, 135,6, 131,3, 129,6, 129,3, 128,5, 128,3, 79,5, 79,4, 49,6, 46,5, 38,8, 34,6, 33,6, 29,6, 28,9, 28,4, 27,8, 22,5.
П р и м е р 9. [1S-[1 α,2 α (Z), 3 α ,4 α]]-6-[3-[4-[[(4-циклогексилбутил)амино] карбонил] -2- тиазолил]-7- оксабицикло[2.2.1]-гептил-2]-4-гексеновая кислота.
A. [1S-[1α, 2 α (Z),3 α,4 α]]-6-[3-(аминокарбонил)-7-оксабицикло[2.2.1] гептил-2]-4-гек- сеновая кислота, метиловый эфир.
К раствору полученной в разделе D примера 1 кислоты (2,45 г, 9,13 ммоль) в сухом бензоле (100 мл) по каплям в течение 10 мин добавляют оксалилхлорид (0,96 мл, 11 ммоль). После перемешивания в течение 5 ч реакционную смесь концентрируют и растворяют в сухом ТГФ (10 мл) и по каплям в течение 5 мин добавляют к раствору концентрированного гидроксида аммония (3 мл) в ТГФ (100 мл), имеющему температуру 0оС. Затем реакционную смесь концентрируют в вакууме. Оставшееся твердое вещество разделяют между этилацетатом (150 мл) и 0,25 М K2CO3 (25 мл). Водный слой экстрагируют этилацетатом (25 мл). Собранные органические слои сушат (Na2SO4) и концентрируют в вакууме. Остаток (2,2 г) суспендируют в кипящем простом эфире (100 мл). Добавляют этилацетат (около 10 мл) для получения раствора. Смесь концентрируют до около 50 мл на паровой бане, охлаждают до комнатной температуры, вносят затравку и вымораживают в течение ночи. Чистый целевой амид (1,19 г, 49% ) получают фильтрованием. Еще одну порцию целевого амида (145 мг) получают, концентрируя маточные растворы до 15 мл (после чего происходит кристаллизация продукта).
B. [1S-[1 α, 2 α(Z), 3 α , 4α]]-6-[3-(аминотиокарбонил)-7-оксабицикло[2.2.1]гептил-2]- 4-гексеновая кислота, метиловый эфир.
К раствору полученного в разделе A амида (267 мг, 0,999 ммоль) в сухом толуоле (10 мл) при температуре 60оС добавляют реагент Лавессона (222 мг, 0,55 ммоль). Реакционную смесь перемешивают при 60оС в течение 30 мин, разбавляют диэтиловым эфиром (50 мл) и промывают полунасыщенным раствором NaHCO3 (2 раза по 5 мл). Органический слой сушат (Na2SO4) и концентрируют в вакууме. Остаток пропускают через тонкий слой диоксида кремния, используя смесь 50% этилацетата-гексаны, и получают желтое твердое вещество (249 мг, 88%).
C. [1S-[1 α, 2 α(Z),3 α ,4 α]]-6-[3-(4-карбокси-2-тиазолил)-7-оксабицикло[2.2.1]гептил-2-] -4- гексеновая кислота, метиловый эфир.
К раствору полученного в разделе B тиоамида (400 мг, 1,41 ммоль) и порошкообразного безводного K2CO3 (390 мг, 2,82 ммоль) в сухом ДМФА (10 мл) несколькими порциями добавляют бромпировиноградную кислоту (содержание воды - 0,4 моль на 1 моль кислоты, 295 мг, 1,69 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 мин. По истечении этого времени добавляют еще 29,5 мг бромпировиноградной кислоты. По истечении еще одного часа растворитель удаляют в вакууме при температуре ниже 30оС. Остаток частично суспендируют, частично растворяют в хлористом метилене (10 мл). Добавляют триэтиламин (0,59 мл, 4,2 ммоль), затем по каплям добавляют метансульфонилхлорид (0,33 мл, 4,2 ммоль). После перемешивания в течение 5 мин реакционную смесь разбавляют диэтиловым эфиром (40 мл). Органический слой экстрагируют 0,5 М раствором K2CO3 (9 раз по 10 мл). Собранные органические слои подкисляют до рН 1,5, добавляя 6 н. раствор HCl и экстрагируют диэтиловым эфиром (6 раз по 25 мл). Затем органические слои сушат (Na2SO4) и концентрируют в вакууме. Выход кислоты составляет 212 мг (43%).
D. [1S-[1 α , 2 α (Z),3 α,4 α]]-6-[3-[4-[[(4-циклогексилбутил)-амино] карбонил] -2-тиазо- лил] -7- оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
К раствору полученной в разделе С кислоты (42,0 мг, 0,120 ммоль) в сухом ДМФА (1 мл) добавляют 1,1'-карбонилдиимидазол (20,3 мг, 0,125 ммоль). Реакционную смесь перемешивают в течение 1 ч. Затем добавляют раствор солянокислого циклогексилбутил)-амина (23,4 мг, 0,131 ммоль) и триэтиламина (0,020 мл, 0,14 ммоль) в сухом ДМФА (0,5 мл). Реакционную смесь перемешивают в течение 1 ч, и концентрируют, удаляя ДМФА. Остаток переносят в диэтиловый эфир (20 мл) и 0,5 н. раствор HCl (5 мл). Органический слой сушат (Na2SO4) и концентрируют в вакууме. Полученный амид (46,8 мг, 81%) хроматографируют (диоксид кремния: 50% этилацетат-гексаны), получая 37,1 мг (64% ) целевого сложного эфира в виде бесцветного масла.
E. [1S-[1 α ,2 α (Z),3 α ,4 α] ]-6-[3-[4-[[(4-(циклогексилбутил)амино] карбонил]-2-тиазолил]- 7- оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота.
К раствору полученного в разделе D амида в метаноле (1 мл) добавляют 2 н. KOH (0,3 мл). Реакционную смесь перемешивают в течение 2 ч. Вводят еще 0,3 мл KOH. Спустя еще 1 ч реакционную смесь концентрируют, удаляя метанол. Остаток растворяют в воде (1 мл) и доводят рН до 2, добавляя 1 н. HCl. Смесь экстрагируют метиленхлоридом (3 раза по 5 мл). Собранные органические слои сушат (Na2SO4) и концентрируют в вакууме, получая маслообразный продукт (34,7 мг, 96%).
П р и м е р 10. [1S-[1 α,2α (Z),3α ,4 α]]-6-[3-[5-[[(4-циклогексилбутил)амино] карбонил-1Н-имидазол-2-ил] - 7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
A. 2-{[(1,1-Диметилэтокси)карбонил]ами- но}-3-{[(фенилметокси)карбонил] амино}пропановая кислота.
N-α -БОК-аспарагин (18,0 г, 77,5 ммоль) добавляют к раствору бис (трифторацетокси) йодозилбензола (50,0 г, 116,3 ммоль) в смеси ДМФА - H2O (1: 1, 620 мл). Через 15 мин добавляют пиридин (12,5 мл, 155,1 ммоль). Темно-желтый раствор перемешивают в течение 16 ч. Образовавшийся бледно-желтый раствор концентрируют в вакууме при температуре ниже 40оС. Остаток разбавляют водой (600 мл), промывают диэтиловым эфиром (6 раз по 400 мл) и концентрируют в вакууме. Полученный неочищенный продукт растворяют в воде (100 мл). К раствору, добавляют 0,5М раствор Na2CO3, доводят рН до примерно 8. Затем добавляют ТГФ (100 мл). К этой смеси при интенсивном перемешивании по каплям добавляют бензилхлороформат (чистота 80%, согласно ЯМР-анализу; 19,8 г, 93,0 ммоль), растворенный в 50 мл ТГФ. Добавляют еще одну порцию раствора Na2CO3, чтобы поддержать значение рН в основной области (определяют по индикаторной бумаге на рН от 5 до 10). Спустя 1 ч добавление заканчивают и реакционную смесь концентрируют, удаляя ТГФ. Остаток экстрагируют диэтиловым эфиром (3 раза по 200 мл). рН водного слоя доводят до 2 добавлением концентрированной серной кислоты. Смесь экстрагируют диэтиловым эфиром (4 раза по 200 мл). Эти последние экстракты сушат (Na2SO4) и концентрируют в вакууме. Хроматографирование (80% этилацетата/гексаны, содержащие 1% уксусной кислоты), дает 4,23 г (16%) целевого продукта, содержащего небольшое количество примесей.
Rf (диоксид кремния, этилацетат + 0,5% уксусной кислоты) 0,27.
B. 2-(Триметилсилил)этил-2-{ [(1,1-диме-тилэтокси)карбонил]-амино}- 3-{ [(фенилметокси)карбонил]амино}пропанат.
К раствору полученной в разделе A кислоты (3,96 г, 11,7 ммоль) в сухом ТГФ (50 мл) при 0оС добавляют 1,1-карбонилдиимидазол (2,08 г, 12,9 ммоль). Через 1 ч добавляют 2-(триметилсилил)этанол (3,4 мл). Затем реакционную смесь выдерживают при 70оС в течение 1 ч. После охлаждения реакционную смесь концентрируют и хроматографируют (импульсный режим, диоксид кремния, диаметр 50 мм, 25% этилацетата - гексаны), получая 3,08 г (60%) целевого сложного эфира в виде прозрачного масла; Rf (диоксид кремния, 25% этилацетата - гексаны) 0,32.
С. 2-(Триметилсилил)этиловый эфир 3-амино-2-{ [(1,1-диметилэтокси)карбонил]ами- но}пропеновой кислоты.
К раствору полученного в разделе B Z-амина (2,89 г, 6,60 ммоль) в смеси пропанол-2/вода (65 мл/6 мл) добавляют формиат аммония (2,08 г, 33,0 ммоль) и затем суспензию Pd/C (10%, 0,5 г) в пропаноле-2 (4 мл). Смесь перемешивают при комнатной температуре в течение 1 ч. Реакционную смесь фильтруют через Celite и концентрируют в вакууме. Остаток переносят в 70 мл этилацетата и 30 мл насыщенного раствора NaCl содержащего 8 мл 5 н. NaOH. Далее водный слой экстрагируют этилацетатом (2 раза по 70 мл). Собранные органические слои сушат (Na2SO4) и концентрируют в вакууме, получая целевое соединение в виде масла (1,85 г, 92%).
и концентрируют в вакууме. Остаток переносят в 70 мл этилацетата и 30 мл насыщенного раствора NaCl содержащего 8 мл 5 н. NaOH. Далее водный слой экстрагируют этилацетатом (2 раза по 70 мл). Собранные органические слои сушат (Na2SO4) и концентрируют в вакууме, получая целевое соединение в виде масла (1,85 г, 92%).
Rf (диоксид кремния, 10% метанола/хлороформ) 0,75.
D. [1S-[1α,2 α(Z),3 α ,4 α ]]-6-[3-[[[2-[[(1,1-диметилэтокси)-карбонил] амино] -3-оксо-3-[2- триметилсилил)этокси]пропил]-амино]тиоксометил]-7-оксабицикло [2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
WSC (1,18 г, 6,15 ммоль) добавляют к раствору метилового эфира [1S-[1 α, 2 α (Z), 3α,4α ]]-6-[3-(карбокси)-7-оксабицикло[2.2.1]гептил-2]-4-гексеновой кислоты (1,65 г, 6,15 ммоль), соединения, полученного в разделе С (1,18 г, 6,08 ммоль) и ОБТ (831 мг, 6,15 ммоль) в хлористом метилене (60 мл). Реакционную смесь перемешивают в течение 16 ч, разбавляют хлористым метиленом (300 мл), экстрагируют (50 мл 1 н. HCl 1 раз, 50 мл насыщенного раствора NaHCO3 1 раз (50 мл воды, 1 раз), сушат (Na2SO4) и концентрируют в вакууме. Хроматографирование (импульсный режим, диоксид кремния, диаметр 50 мм, 40% этилацетата - гексаны, 2 л; 60% этилацетата, 1 л) дает маслообразный продукт в количестве 2,11 г (63% ); Rf (диоксид кремния, 10% метанола/хлороформ) 0,67.
Полученный таким образом амид (1,94 г, 3,50 ммоль) перемешивают при 65оС в бензоле (40 мл) с реагентом Лавессона (850 мг, 2,10 ммоль) в течение 1,5 ч. Реакционную смесь охлаждают и разбавляют диэтиловым эфиром (250 мл). Смесь промывают (2 раза по 50 мл 0,5 н. NaCO3), сушат (Na2SO4), концентрируют в вакууме и хроматографируют (импульсный режим, диоксид кремния, диаметр 50 мм, 25% этилацетата - гексаны), получая 1,61 г (80%) целевого соединения в виде масла; Rf (диоксид кремния, 25% этилацетата - гексаны) 0,14.
E. [1S-[1 α, 2α(Z), 3 α, 4 α]]-6-[3-[1-[(1,1-диметилэтокси)карбонил]-4,5-дигидро-5-[[2- (триметилсилил)этокси] карбонил-1Н-ими-дазол-2-ил]-7-оксабицикло[2.2.1] гептил-2]-4-гексеновая кислота, метиловый эфир.
Четыреххлористый углерод (3,00 мл, 31,2 ммоль) добавляют к раствору полученного в разделе D соединения (1,61 г, 2,82 ммоль), трифенилфосфина (2,22 г, 8,46 ммоль) и триэтиламина (1,18 мл, 8,46 ммоль) в сухом ацетонитриле (28 мл). Реакционную смесь перемешивают в течение 4 ч. Затем смесь разбавляют диэтиловым эфиром (125 мл), насыщенным раствором NaCl (125 мл) и водой (5 мл). Далее водную фазу экстрагируют простым эфиром (125 мл). Собранные органические слои сушат (Na2SO4) и концентрируют в вакууме. Остаток растирают в порошок последовательно с простым эфиром (20, 10 и 7 мл), каждый раз отбирая и концентрируя фильтрат для следующего цикла. Хроматографирование (импульсный режим, диоксид кремния, диаметр 50 мм, 50% этилацетата - гексаны) дает 1,09 г (72%) целевого соединения в виде масла; Rf (диоксид кремния, 50% этилацетата - гексаны) 0,29.
F. [1S-[1 α, 2 α (Z), 3 α, 4α ]]-6-[3-(5-карбокси-4,5-дигидро-1Н-имидазол-2-ил)-7-окса-бицикло [2.2.1]гептил-2]-4-гексеновая кислота, солянокислый метиловый эфир.
К раствору полученного в разделе Е соединения (1,09 г, 2,03 ммоль) в хлористом метилене (2 мл) добавляют ТФУК (4 мл). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч, разбавляют толуолом (40 мл) и концентрируют в вакууме. Остаток переносят в этилацетат (60 мл) и промывают насыщенным раствором NaHCO3 (2 раза по 5 мл). Органический слой сушат (Na2SO4) и концентрируют в вакууме, получая метиловый эфир [1S-[1α , 2 α(Z), 3 α, 4 α]]-6-[3-(5-карбокси-4,5-дигидро-1Н-имидазол-2-ил)-7-оксабицикло [2.2.1] гептил-2] -4-гексеновой кислоты (261 мг, 27%). рН водной фазы доводят до 2 и концентрируют ее в вакууме. Экстракция твердого продукта хлороформом (2 раза по 15 мл) с последующей концентрацией в вакууме дает целевое соединение (553 мг, 73%).
G. [1S-[1 α, 2 α (Z), 3 α, 4α ]]-6-[3-[5-[[(4-циклогексилбутил)амино] карбонил] -1Н-имида- зол-2-ил]- 7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
Смесь полученной в разделе F кислоты (516 мг, 1,38 ммоль), (4-циклогексилбутил)амина в виде солянокислой соли (345 мг, 1,80 ммоль), WSC (345 мл, 1,80 ммоль), ОБТ (243 мг, 1,80 ммоль) и триэтиламина (0,50 мл, 3,6 ммоль) в хлористом метилене (15 мл) перемешивают в течение 24 ч. Реакционную смесь разбавляют хлористым метиленом (50 мл) и промывают (2 раза по 10 мл насыщенного раствора NaHCO3), сушат (Na2SO4) и концентрируют в вакууме, получая желтое масло, которое представляет собой метиловый эфир [1S-[1 α, 2 α (Z), 3 α, 4α ]]-6-[3-[5-[[(4-циклогексилбутил)амино]карбонил]-4,5-дигид-ро-1Н- имидазол-2-ил]-7-оксабицикло[2.2.1]гептил-2]-4-гексеновой кислоты. Полученное масло растворяют в хлороформе (20 мл). Добавляют активный MnO2 (Aldrich, 1,0 г). Реакционную смесь перемешивают в течение 24 ч. Добавляют еще 0,5 г MnO2 и перемешивают смесь в течение 3 дней. Затем добавляют еще 0,5 г MnO2. Спустя 5 ч реакционную смесь фильтруют. Фильтр промывают хлороформом. Собранные фильтраты концентрируют в вакууме. Хроматографирование (импульсный режим, диоксид кремния, диаметр 25 мм, 2% метанола - хлороформ) дает 235 мг (36%) целевого сложного эфира в виде масла.
П р и м е р 11. [1S-[1 α, 2α (Z), 3α,4α ]]-6-[3-[4-[[(4-циклогексилбутил)амино] карбонил] -1Н- имидазол-2-ил]- 7-оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота.
К раствору полученного в примере 10 сложного эфира в метаноле (8 мл) добавляют 2 н. раствор KOH (4 мл). Реакционную смесь перемешивают при комнатной температуре в течение 4 ч. В вакууме удаляют метанол. Остаток переносят в хлористый метилен (25 мл) и рН доводят до 2 добавлением 1 н. раствора HCl. После встряхивания водный слой экстрагируют хлористым метиленом (25 мл). Собранные органические слои сушат (Na2SO4) и концентрируют в вакууме. Остаток переносят в хлористый метилен и интенсивно перемешивают с эфирным раствором HCl (4 мл) в течение 30 с. Смесь концентрируют в вакууме. Растирание с 10 мл этилацетата (от кипения до комнатной температуры) дает белое твердое вещество, которое собирают фильтрованием, промывают этилацетатом (5 мл) и сушат, получая целевую кислоту в виде твердого вещества белого цвета, 126,4 мг (52%); т.пл. 168-173оС.
1Н ЯМР (тетрадейтерометанол/дейтерохлороформ, 270 МГц) δ : 8,05 (с, 1Н), 5,23-5,36 (м, 2Н), 4,73 (с, 1Н), 4,41 (с, 1Н), 3,74 (д, I = 8 Гц, 1Н), 3,34-3,37 (м, 2Н), 0,84-2,44 (м, 28Н).
13С ЯМР (полная развязка 67,8 МГц, тетрадейтерометанол-дейтерохлороформ) δ : 175,4, 157,0, 147,8, 130,1, 127,3, 120,7, 80,0, 79,9, 44,9, 39,8, 37,4, 36,9, 33,5, 33,1, 29,1, 28,7, 28,1, 26,5, 26,2, 24,1, 22,6.
ИК (KBr): 3427 (м), 3232 (м), 3009 (м), 2923 (с), 2851 (м), 1730 (м), 1718 (м), 1651 (м), 1566 (м), 1448 (W), 1334 (W), 1189 (W) см-1.
LRMS (Cl, NH3, DEP 50-ионный спектр) m/z (отн. инт.) 458 (2), 184 (7), 173 (14), 172 (6), 169 (9), 168 (13), 167 (5), 157 (10), 156 (100), 155 (25), 154 (35), 162 (6).
Элементный состав
C26H40ClN3O4 ˙0,25 H2O
Вычислено, %: C 63,63; H 8,19; Cl 7,11; N 8,43.
Найдено, %: C 62,76; H 8,30; Cl 6,82; N 8,37.
П р и м е р 12. [1S-[1 α, 2α (Z, 3α , 4 α]]-N-(4-циклогексилбутил)-2-[2-[5-(1Н-тетразол-5- ил)-2-пентенил] -7- оксабицикло[2.2.1]-гептил-3]-4-оксазолкарбоксамид.
A. [1S-[1 α, 2 α (Z), 3 α, 4 α ]]-2-[5-(1Н-тетразол-5-ил)-2-пентенил]-7-оксабицикло[2.2.1] гептан-3-метанол.
К суспензии 1,0 ммоль [4aR (4a α, 5 β , 8 β, 8 a α)] октагидро-5,8-эпокси-1Н-2-бензопиран-3-ола, приготовленного в соответствии с патентом США N 4143054, и 1,4 ммоль 3-(тетразол-5-ил)пропилтрифенилфосфоний- бромида в тетрагидрофуране при 0оС по каплям добавляют 1,8М раствор KOt-амилата в толуоле (2,8 ммоль). Полученной смеси дают нагреться до комнатной температуры в течение ночи. Реакционную смесь закаливают добавлением уксусной кислоты. Реакционную смесь концентрируют в вакууме и очищают, хроматографируя на силикагеле с использованием в качестве элюентов смесей метанола с хлористым метиленом.
B. [1S-[1 α, 2 α (Z), 3 α, 4α ]]-2-[5-(1-метоксиметил-1Н-тетразол-5-ил)-2-пентенил]-7-оксабицикло [2.2.1]гептан-3-метанол.
Раствор 1,0 ммоль полученного выше тетразола в ТГФ охлаждают до 0оС и обрабатывают 1,0 ммоль триэтиламина и 1,0 ммоль бромметилметилового эфира. Полученную смесь перемешивают в течение 4 ч при комнатной температуре и затем разделяют между насыщенным водным раствором NaHCO3 и этилацетатом. Этилацетатный слой сушат, фильтруют и концентрируют в вакууме, получая целевой тетразол.
С. [1S-[1 α, 2α (Z), 3 α, 4 α]]-N-(4-циклогексилбутил)-2-[2-[5-(1-метоксиметил-1Н-тет- разол-5- ил)-2-пентенил]-7-оксабицикло[2.2.1]гептил-3] -4-оксазолкарбоксамид.
Синтез проводят по методике примера 1, за исключением того, что вместо сложного эфира раздела С примера 1 используют тетразол раздела B примера 12; при этом получают целевое соединение.
D. [1S-[1 α, 2α (Z), 3 α, 4 α ]]-N-(4-циклогексилбутил)-2-[2-[5-(1Н-тетразол-5-ил)-2- пентенил] - 7-оксабицикло[2.2.1]гептил-3]-4-оксазолкарбоксамид.
К раствору 1,0 ммоль полученного в разделе С тетразола в метаноле (25 мл) добавляют 1 каплю концентрированной HCl. Полученный раствор нагревают (кипятят) с обратным холодильником в течение 2 ч. После охлаждения реакционную смесь концентрируют в вакууме. Неочищенный продукт очищают жидкостной хроматографией высокого давления с обращенной фазой, используя смеси ацетонитрила с 0,02% -ным водным раствором H3PO4 в качестве подвижной фазы; получают целевой тетразол.
Примеры других соединений, соответствующих данному изобретению, которые можно получить, используя способы, приведенные в описании и примерах, включают в себя нижеследующие соединения, приведенные в табл.2.
П р и м е р 30. [1S-[1 α, 2α (Z),3α, 4 α]]- 6-[3-[4-[[(6-циклогексилгексил)-амино] карбо- нил]-2-оксазолил]-7- оксабицикло[2.2.1]-гептил-2-]-4-гексеновая кислота, метиловый эфир.
A. Сложный эфир бензолпентанола и метансульфоновой кислоты.
К раствору 5-фенилпентанола-1 (5,0 г, 30,4 ммоль) в 20 мл хлористого метилена при перемешивании в атмосфере аргона при -70оС добавляют сначала триэтиламин (4,0 г, 39,5 ммоль), а затем по каплям - метансульфонилхлорид (2,83 мл, 4,18 г, 36,5 ммоль).
Реакционную смесь медленно нагревают до комнатной температуры, добавляют воду, и экстрагируют реакционную смесь хлористым метиленом (50 мл). Водный слой еще два раза экстрагируют хлористым метиленом (25 мл). Органические слои собирают вместе и промывают рассолом, затем сушат над MgSO4 и концентрируют, получая 7,30 г (100%) целевого соединения в виде масла желтого цвета; Rf = 0,9 в смеси 50% гексана - этилацетат (УФ, Ce(SO4)2.
B. Бензолгексанонитрил.
К раствору полученного в разделе A соединения (7,30 г, 30,1 ммоль) в 70 мл этанола при перемешивании при комнатной температуре добавляют раствор KCN (9,81 г, 150,6 ммоль) в 29 мл воды. Полученную реакционную смесь перемешивают в течение 20 ч, затем трижды экстрагируют хлороформом (50 мл). Органические слои соединяют и промывают водой, затем рассолом, сушат над MgSO4 и концентрируют в вакууме, получая масло желтого цвета, содержащее целевое соединение и остатки соединения, полученного в разделе A. Это масло очищают методом импульсной хроматографии (смесь гексан-этилацетат состава 95:5); получают 2,16 г (41%) целевого продукта в виде масла.
13С ЯМР (CDCl3): δ 141,8; 128,0; 125,5; 119,5; 35,4; 30,5; 27,9; 24,9; 16,7.
С. Циклогексангексанамин.
К раствору полученного в разделе B соединения (2,16 г, 12,4 ммоль) в 100 мл уксусной кислоты добавляют PtO2 (0,60 г). Полученную смесь перемешивают при комнатной температуре в среде водорода под давлением 1 атмосфера. Через 20 ч реакционную смесь фильтруют, и фильтр дважды промывают этанолом. Фильтрат концентрируют до полужидкого состояния. Этот продукт перемешивают в гексане, и получают целевой продукт (2,00 г, 95%) после фильтрования в виде пухообразного твердого вещества белого цвета.
D. [1S-[1 α, 2 α (Z), 3 α, 4 α ]]-6-[3-[4-[[(6-циклогексилгексил)амино] карбонил] -2-ок-сазолил]-7- оксабицикло[2.2.1]гептил-2]-4-гексеновая кислота, метиловый эфир.
Образец оксазоловой кислоты, полученной в разделе G примера 2 (1,61 г, 4,8 ммоль), сушат азеотропной отгонкой в высоком вакууме с сухим ДМФА и толуолом. Полученное вещество (содержащее остаточный ДМФА) растворяют в 30 мл толуола, и при перемешивании при комнатной температуре в атмосфере аргона добавляют 2,9 г оксалилхлорида (23 ммоль). Происходит выделение газа. На дне емкости с реакционной смесью образуется темное масло. (Это масло представляет собой продукт реакции между ДМФА и оксалилхлоридом). Спустя 5 мин добавляют еще 2,9 г оксалилхлорида. Смесь перемешивают в течение ночи. Метод ТХ показывает, что произошла полная конверсия в хлорангидрид кислоты. Надосадочный слой отбирают пипеткой и переносят в сосуд, заполненный аргоном. Место дважды перемешивают с новыми порциями толуола, и отбирают надосадочный слой. Соединенные надосадочные слои выпаривают с хлороформом, получая 1,54 г (выход 91%) хлорангидрида кислоты, а именно, метилового эфира [1S-[1 α, 2 α (Z), 3α , 4α ]]-6-[3-[4-(хлоркарбонил)-2-оксазолил]-7-оксабицик-ло[2.2.1]гептил -2]-4-гексеновой кислоты.
К раствору полученного в разделе С амина (0,55 г, 3,0 ммоль) в 5 мл хлороформа при 0оС добавляют триэтиламин (0,3 г, 3,0 ммоль) и вышеуказанный хлорангидрид кислоты (0,53 г, 1,5 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 14 ч, затем разбавляют хлороформом и водой. Отделяют органический слой, водный слой дважды экстрагируют 20 мл хлороформа. Органические слои соединяют, промывают рассолом, сушат над MgSO4 и концентрируют. Импульсная хроматография (от 0 до 75% этилацетата в градиенте гексана) дает 0,49 г продукта в виде светлого масла (выход 65%). Rf = 0,5 в смеси гексан-этилацетат состава 1:1 (УФ, Ce(SO4)2.
13 С ЯМР (67,8 МГц, CDCl3) δ : 172,8, 163,5, 160,1, 140,1, 135,8, 129,1, 128,2, 79,2, 51,0, 49,4, 46,3, 38,7, 37,2, 37,0, 33,4, 33,0, 29,4, 29,2, 28,6, 27,5, 26,6, 26,3, 26,1, 22,5.
П р и м е р 31. [1S-[1 α, 2α (Z), 3α , 4 α]]-6-[3-[4-[[(6-циклогексилгексил)-аминокарбонил)-2-оксазолил)-7- оксабицикло[2.2.1]гепт-2-ил]-4-гексеновая кислота.
Раствор 0,49 г (0,97 ммоль) сложного эфира, полученного согласно примеру 30, в 8 мл 1 н. раствор гидроксида натрия и 8 мл тетрагидрофурана перемешивают в течение 18 ч, затем концентрируют в вакууме для удаления тетрагидрофурана и подкисляют до рН 1,5 посредством 1 н. соляной кислоты. К смеси добавляют этилацетат и отделяют органический слой. Водный слой дважды экстрагируют 20 мл этилацетата. Органические фракции объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме, получая в результате белое твердое вещество. Указанный продукт кристаллизуют из гексана и хлороформа, получая 0,40 г (84%) чистого продукта в виде белого твердого вещества.
Rf = 0,36 в 0,1% уксусной кислоты (4% метанола в этилацетате). Визуализация в УФ-излучении: сульфат церия Ce(SO4)2 [α ]Do = = + 30,2, с = 0,58 г/100 мл метанола, температура плавления 32-83оС.
П р и м е р 32. Метиловый эфир [1S-[1 α, 2α (Z), 3 α , 4α ]]-6-[3-(6-циклогексилгексил)амино] карбонил]-2-оксазолил-7- оксабицикло[2.2.1]-гепт-2-ил]-4-гексеновой кислоты.
К раствору 0,49 г (1,40 ммоль) хлорангидрида кислоты, полученного в соответствии с описанным в примере 2 способом (часть 1) и неизвестного количества соли Вильсмайера в 10 мл хлороформа, добавляют при перемешивании 0,28 г (2,80 ммоль) триэтиламина и 0,38 г (2,1 ммоль) 6-циклогексилгексиламина, полученного согласно примеру 30, часть С. Смесь перемешивают при комнатной температуре в течение 10 ч, после чего разбавляют этилацетатом и водой. Органический слой отделяют, а водный слой экстрагируют двумя поршнями по 20 мл этилацетата. Органические фракции объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют. Посредством хроматографии с испарением (градиент этилацетат-гексан, от 0 до 100%) получают 0,17 г указанного в заголовке продукта в виде прозрачного масла с выходом 25%. Rf = 0,46 в смеси этилацетат-гексан 1:1.
П р и м е р 33. [1S-[1 α, 2α (Z), 3α , 4 α]]-6-[3-[4-[[(6-циклогексил)-амино] карбонил] -2-ок-сазолоил] -7- оксабицикло[2.2.1]гепт-2-ил]-4-гексеновая кислота.
Раствор 0,17 г (0,33 ммоль) сложного эфира согласно примеру 32 в 5 мл 1 н. раствора гидроксида натрия и 2 мл тетрагидрофурана перемешивают при комнатной температуре в течение 10 ч, после чего концентрируют в вакууме для удаления тетрагид- рофурана и подкисляют концентрированной соляной кислотой до рН 1,5. К смеси добавляют этилацетат и отделяют органический слой. Водный слой дважды экстрагируют этилацетатом; объединяют органические фракции, промывают их рассолом, сушат над сульфатом магния и концентрируют в вакууме. В результате описанных операций получают прозрачное масло. Его хроматографируют с использованием смеси этилацетат-гексан (1:1) с 0,25% уксусной кислоты, получая в результате 0,13 г (78%) указанного в заголовке продукта в виде масла (содержит 2,5% цис-изомера соединения согласно примеру 31A). Rf = 0,36 в 4% метанола, 0,1% уксусной кислоты, 95,5% этилацетата (УФ-визуализация с использованием сульфата церия). [α]Do = + 55 в метаноле при концентрации 0,50 г/100 мл.
П р и м е р 34. Метиловый эфир (1 α, 2α (Z), 3 α, 4α (-5-3-[4-1-пирролидинилкарбонил)-2-оксазолил)-7-оксабицикло[2.2.1] гепт-2-ил]-4-гексеновой кислоты.
К раствору 0,18 г (0,50 ммоль) хлорангидрида кислоты, полученного согласно примеру 2 (часть 1), в 3 мл хлороформа добавляют при перемешивании в атмосфере аргона при 0оС 0,10 г (1,0 ммоль) триэтиламина и 0,07 г (1,0 ммоль) пирролидона. Смесь нагревают до комнатной температуры и перемешивают в течение 10 ч, после чего разбавляют этилацетатом и водой. Органический слой отделяют, а водный слой дважды экстрагируют 20 мл этилацетата. Органические слои объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют. Посредством хроматографии с испарением (градиент этилацетата в гексане, от 50 до 100%, затем 2% метанола в этилацетате) получают 0,13 г указанного в заголовке продукта в виде прозрачного масла, с выходом 68%; Rf = 0,6 в смеси этилацетат-метанол 9:1 (УФ-визуализация с сульфатом церия).
Данные ЯМР-С13 (67,8 МГц, дейтерохлороформ), δ : 172,9, 162,9, 160,0, 141,9, 136,9, 129,0, 128,5, 79,2, 78,9, 51,1, 49,4, 47,9, 46,5, 46,4, 33,5, 29,5, 28,7, 27,6, 26,1, 23,4, 22,5.
П р и м е р 35. (1 α, 2α (Z), 3α , 4α ]-6-[ 3-[4-(1-пирролидинилкарбонил)-2-оксазолил]- 7-оксабицикло [2.2.1]гепт-2-ил]-4-гексеновая кислота.
Раствор 0,13 г (0,33 ммоль) соединения согласно примеру 34 в 10 мл 1 н. раствора гидроксида натрия и 2 мл тетрагидрофурана перемешивают в течение 10 ч при комнатной температуре, после чего концентрируют в вакууме для удаления тетрагидрофурана и подкисляют до рН 1,5 посредством соляной кислоты. К смеси добавляют этилацетат и отделяют органический слой. Органические слои объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме. В результате получают прозрачное масло. Его хроматографируют с использованием этилацетата с 1% метанола, 0,25% уксусной кислоты (при 98,75% этилацетата), получая 0,12 г (92%) чистого продукта в виде масла; Rf = 0,2 в 1%-ном метаноле, 0,5% уксусной кислоты и 98,5% этилацетата, [α]Do = + 35,7 в метаноле при концентрации 2,40 г/100 мл.
П р и м е р 36. Метиловый эфир {1S-[1α , 2 α(Z), 3 α, 4 α] }-6-[3-[4-[(пропиламино)карбонил] -2-оксазолил] -7-оксабицикло [2.2.1]гепт-2-ил]-4-гексеновой кислоты.
К раствору 0,22 г (3,7 ммоль) пропиламина в 5 мл хлороформа добавляют при температуре 0оС в атмосфере аргона 0,162 г (0,46 ммоль) хлорангидрида кислоты, полученного согласно примеру 2, часть I. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 14 ч, после чего разбавляют хлороформом и водой. Органический слой отделяют и водный слой экстрагируют 2 порциями по 20 мл хлороформа. Органические слои объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют. После хроматографической обработки с испарениями (в градиенте этилацетата в гексане, от 0% до 75% ) получают 0,15 г указанного в заголовке продукта в виде прозрачного масла с выходом 85% . Rf = 0,29 в 4% метанола в этилацетате (УФ-визуализация, сульфат церия).
Спектр ЯМР-С13 (дейтерохлороформ), δ : 173,0, 163,7, 160,4, 140,2, 136,0, 129,2, 128,4, 79,3, 79,2, 51,2, 49,5, 46,5, 40,5, 33,6, 29,6, 28,7, 27,7, 22,7, 22,6, 11,2.
П р и м е р 37. [1S-[1 α, 2α (Z), 3α , 4 α]]-6-[3-[4-[(пропиламино)карбонил[-2-оксазолил] 7-оксабицикло [2.2.1]гепт-2-ил]-4-гексеновая кислота.
Раствор 0,15 г (0,39 ммоль) сложного эфира согласно примеру 36 в 8 мл 1 н. раствора гидроксида натрия и 8 мл тетрагидрофурана перемешивают в течение 18 ч, после чего концентрируют в вакууме для удаления тетрагидрофурана и подкисляют до рН 1,5 посредством 1 н. раствора соляной кислоты. К смеси добавляют этилацетат и отделяют органический слой. Водный слой экстрагируют 2 порциями этилацетата по 20 мл. Органические слои объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме. В результате получают прозрачное масло, которое кристаллизуют из смеси гексана и хлороформа, получая 0,14 г (выход 100%) чистого продукта в виде твердого вещества белого цвета; Rf = 0,41 в 0,1% уксусной кислоты, 4% метанола, 95,9% этилацетата (УФ-визуализация, сульфат церия). Температура плавления 117-118оС, [α]Do = + 42,12 при концентрации 4,7 г/100 мл метанола.
П р и м е р 38. Метиловый эфир [1S-[2 α, 2 α(Z), 3 α, 4 α]]-6-[3-[4-[[4-бутилфенил)амино] -карбонил]-2-оксазолил]-7- оксабицикло[2.2.1]гепт-2-ил]-4-гексеновой кислоты.
К раствору 0,189 г (1,27 ммоль) 4-бутиланилина в 5 мл хлороформа добавляют при 0оС в атмосфере аргона 0,17 г (0,48 ммоль) хлорангидрида кислоты, полученного согласно описанному в примере 2; часть 1 способу, и 0,145 г (1,44 ммоль) триэтиламина. Смесь нагревают до комнатной температуры и перемешивают в течение 14 ч, после чего разбавляют хлороформом и водой. Органический слой отделяют, а водный слой экстрагируют 2 порциями по 20 мл хлороформа. Органические слои объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют. После хроматографической обработки с испарением (в градиенте этилацетата в гексане, от 0 до 75% получают 0,16 г продукта, указанного в заголовке, в виде прозрачного масла (выход - 69% процента). Rf = 0,87 в 4% метанола в этилацетате (УФ-визулизация, сульфат церия).
Спектр ЯМР-С13 (дейтерохлороформ) δ: 173,1, 163,9, 158,2, 141,1, 139,0, 136,2, 129,4, 128,7, 128,4, 119,7, 79,4, 79,3, 51,3, 49,6, 46,6, 34,9, 33,6, 33,5, 29,7, 28,8, 27,8, 22,7, 22,1, 13,8.
П р и м е р 39. [1S-[1 α, 2α (Z), 3α , 4 α]]-5-[3-[4-[[(4-бутилфенил)амино] -карбонил] -2-ок- сазолил]-7- оксабицикло[2.2.1]гепт-2-ил]4-гексеновая кислота.
Раствор 0,16 г (0,34 ммоль) сложного эфира согласно примеру 38 в 1 н. растворе гидроксида натрия и 8 мл тетрагидрофурана перемешивают в течение 18 ч, после чего концентрируют в вакууме для удаления тетрагидрофурана и подкисляют до рН 1,5 посредством 1 н. раствора соляной кислоты. К смеси добавляют этилацетат и отделяют органический слой. Водный слой дважды экстрагируют порциями по 20 мл этилацетата. Органические экстракты объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме. Получают прозрачное масло, которое кристаллизуют из смеси гексана и хлороформа, получая 0,13 г (выход 87%) чистого продукта в виде твердого вещества желтого цвета. Rf = 0,47 в 0,1% уксусной кислоты, 4% метанола, 95,9% этилацетата (УФ-визуализация, сульфат церия). Температура плавления 113-114оС. [α ]Do = + 6,44, концентрация 0,59 г/100 мл метанола.
П р и м е р 40. Метиловый эфир (1S-[ 1α], 2 α(Z), 3 α, 4α [[-6-[3-[4-[(2,3-дигидро-1Н-индол-1-ил)карбонил] -2-оксазолил] -7- оксабицикло-[2.2.1] гепт-2-ил]-4-гексеновой кислоты.
Раствор 0,37 г (0,80 ммоль) сложного эфира согласно примеру 40 в 15 мл 1 н. раствора гидрата окиси натрия и 15 мл метанола перемешивают в течение 18 ч, после чего подкисляют до рН 2 посредством концентрированной соляной кислоты. К смеси добавляют этилацетат и отделяют органический слой. Водный слой дважды экстрагируют порциями по 20 мл этилацетата. Органические слои объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют в вакууме. Получают масло красного цвета, которое хроматографируют с использованием 0,1% раствора уксусной кислоты в градиенте этилацетата в гексане (от 0 до 100%), получая в результате 0,25 г (выход 75%) указанного в заголовке соединения в виде масла желтого цвета, Rf = 0,29 в 0,1% уксусной кислоты в 50% этилацетате в гексане (УФ-визуализация, сульфат церия). [α ]Do = + 31,2 в метаноле при концентрации 0,48 г/100 мл.
П р и м е р 41. [1S-[1 α, 2α (Z ) 3α , 4 α]]- 6-[3-[4-[[(4-циклогексилбутил)амино] карбо-нил]-2-оксазолил]-7 -оксабицикло[2.2.1]гепт-2-ил]-N-фенилсульфонил-4-ге- ксенамид.
К раствору хлоргидрата 1-(3-диметиламинопропил)-3-этилкарбодиимида (102 мг, 0,50 ммоль) и 66,7 г (0,50 ммоль) 4-диметиламинопиридина в 50 мл диметилформамида добавляют при перемешивании 80 мг (0,51 ммоль) бензолсульфонамида и затем 0,14 мл (1,00 ммоль) C2H5O3N и, наконец, 229 мг (0,30 ммоль) кислоты, полученной согласно описанному в примере 1. Реакционную смесь перемешивают при комнатной температуре в течение 18 ч, после чего концентрируют в вакууме. Остаток перераспределяют между 25 мл 1 н. раствора соляной кислоты и этилацетатом (4 порции по 40 мл). Объединенные экстракты в этилацетате промывают 30 мл рассола, сушат над сульфатом магния. Фильтруют и концентрируют в вакууме. Остаток хроматографируют на 30 г силикагеля-60 фирмы "Мерк", используя в качестве элюента 2% метанола в хлористом метилене и получая в результате 250 мг указанного в заголовке сульфонамида. Этот продукт перераспределяют между 60 мл этилацетата и водой (1 порция в 20 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая 210 мг (выход 70%) указанного в заголовке соединения, в виде твердого вещества. Температура плавления 154-156оС, данные тонкослойной хроматографии на силикагеле, 4% метанола - хлористый метилен; Rf = 0,45 (сульфат церия).
Спектр ЯМР-С13 указанного в заголовке соединения (дейтерохлороформ, 67,5 МГц) δ: 171,1, 170,5, 163,9, 161,1, 140,7, 139,0, 135,9, 133,5, 128,7, 128,2, 79,7, 79,7, 49,5, 46,4, 39,2, 37,4, 37,0, 35,9, 33,3, 33,3, 29,7, 28,8, 27,9, 26,6, 26,3, 26,3, 24,1, 22,3.
П р и м е р 42. [1S-[1 α, 2α (Z),3α , 4 α]]-6-[3-[4-[[(4-циклогексилбутил)-амино]карбонил]-2-оксазолил]-N- метилсульфонил-7-оксабицикло-[2.2.1] гепт-2-ил]-4-гексенамид
К раствору хлоргидрата 1-(3-диметиламинопропил)-3-этилкарбодиимида (102 мг, 0,50 ммоль) и 4-диметиламинопиридина (66,7 мг, 0,50 ммоль) в 50 мл диметилформамида добавляют 47,6 мг (0,50 ммоль) метансульфонамида и 0,14 мл (1,00 ммоль) (C2H5)3N, а затем - 229 мг (0,50 ммоль) кислоты согласно примеру 1. Реакционную смесь перемешивают в течение 18 ч при комнатной температуре и концентрируют в вакууме. Остаток перераспределяют между 25 1 н. раствора соляной кислоты и этилацетатом (4 порции по 40 мл). Объединенные экстракты в этилацетате промывают 30 мл рассола, сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток хроматографируют на 30 г силикагеля-60 фирмы "Мерк", используя в качестве элюента 2% метанола в хлористом метилене и получая в результате 200 мг указанного в заголовке сульфонамида. Полученный продукт перераспределяют между 60 мл этилацетата и 20 мл воды. Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая в результате 150 мг (выход 56%) указанного в заголовке соединения в виде твердого вещества. Температура плавления 140-142оС, результаты тонкослойной хроматографии (4% метанола - хлористый метилен); Rf = 0,40 (сульфат церия).
Спектр ЯМР -C13 указанного в заголовке соединения (дейтерохлороформ, 67,5 МГц) δ : 171,8, 163,9, 161,0, 140,8, 135,9, 128,8, 128,7, 79,7, 49,5, 46,4, 41,3, 39,2, 37,4, 37,0, 36,0, 33,3, 29,7, 28,8, 28,0, 26,6, 26,3, 24,1, 22,3.
П р и м е р 43. Метиловый эфир [1S-[1 α, 2 α (Z), 3 α, 4α ]]-7-[3-[4-[[(4-циклогексилбутил)амино] карбонил] -2-оксазолил] -7- оксабицикло[2.2.1] гепт-2-ил]-5-гептеновой кислоты.
A. [1S-[1 α, 2 α (Z), 3 α, 4 α]]-7-(3-карбокси)-7-оксабицикло-[2.2.1] -гепт-2-ил)-5-гепте- новая кислота.
К раствору метилового эфира [1S-[2 α, 2α (Z), 3 α, 4α ]]-7-(3-оксиметил)-7-оксабицикло[2.2.1] гепт-2-ил-5-гептеновой кислоты (1,60 г, 5,79 ммоль) в 100 мл ацетона добавляют при перемешивании при 0оС сульфат марганца MnSO4, обработанный реагентом Джонса (примерно 100 мг сульфата марганца, растворенного в 100 мл реагента Джонса) до появления красно-оранжевого цвета. Реакционную смесь перемешивают при 0оС в течение 30 мин и при комнатной температуре в течение 1,5 ч. Затем реакционную смесь гасят изопропиловым спиртом и концентрируют в вакууме. Остаток перераспределяют между 70 мл 3М раствора NaHSO3 и этилацетатом (4 порции по 100 мл). Объединенные экстракты в этилацетате промывают водой (1 порция 70 мл) и рассолом (1 порция 70 мл). Органический слой сушат над сульфатом магния и концентрируют в вакууме, получая 1,57 г (выход 93%) указанного в заголовке соединения, которое используют на следующей стадии без дополнительной очистки. Данные тонкослойной хроматографии (силикагель, 4% метанола/хлористый метилен):Rf = 0,24 (сульфат церия).
Спектр ЯМР-С13 указанного в заголовке соединения (дейтерохлороформ, 67,5 МГц) δ: 176,7, 174,0, 130,5, 128,3, 78,4, 78,2, 51,8, 51,3, 47,8, 33,3, 29,0, 28,7, 27,2, 26,6, 24,6.
B. [2-[(4-циклогексилбутил)амино)-1-оксиметил-2-оксоэтил]-карбаминовая кислотта, 1,1-диметиловый эфир.
К смеси хлоргидрата 4-циклогексилбутиламина (19,5 г, 102 ммоль), 1-оксибензотриазола, H2O (16,5 г, 122 моль) и ВОС-1-серина (25,0 г, 122 ммоль) в 400 мл диметилформамида добавляют при перемешивании в 0оС в атмосфере аргона последовательно 42,5 мл (305 ммоль) (C2H5)3N и 23,4 г (122 моль) хлоргидрата 1-(3-диметиламинопропил)-3-этилкарбодиимида. Реакционную смесь перемешивают при 0оС в течение 1 ч и при комнатной температуре в течение 18 ч. Затем смесь концентрируют в вакууме и разбавляют 800 мл этилацетата. Полученный раствор промывают 1 н. раствором соляной кислоты (3 порции по 120 мл), 0,2 н. раствором гидрата окиси натрия (2 порции по 100 мл), насыщенным раствором NaHCO3 (1 порция 100 мл) и рассолом (1 порция 150 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая 39,9 г указанного в заголовке амида с количественным выходом. Тонкослойная хроматография (силикагель, 4% метанола/хлористый метилен):Rf = 0,38 (сульфат церия).
Спектр ЯМР-С13 указанного в заголовке соединения (дейтерохлороформ, 67,5 МГц) δ: 171,2, 156,2, 90,3, 62,8, 39,6, 37,4, 37,0, 33,3, 29,6, 28,2, 26,6, 26,3, 24,0.
С. Метиловый эфир (1S-[1α,2 α(Z),3 α, 4α]]-7-[3-[[2-[(4-циклогексилбутил)амино] - 1-оксиметил-2-оксоэтил] амино]карбонил]-7-оксабицикло[2.2.1] гепт-2-ил]-5-гептеновой кислоты.
К раствору полученного в разделе B амида (1,90 г, 5,56 ммоль) в 20 мл хлористого метилена добавляют при перемешивании при 0оС 8 мл трифторуксусной кислоты. Полученную смесь перемешивают при 0оС в течение 3 ч. Затем смесь разбавляют 50 мл толуола и концентрируют в вакууме, получая соль амина и трифторуксусной кислоты. К раствору указанной соли амина и трифторуксусной кислоты, кислоты согласно разделу A (1,57 г, 5,57 ммоль) и 1-оксибензотриазола H2O (0,75 г, 5,57 ммоль) в 60 мл диметилформамида добавляют при перемешивании при 0оС в атмосфере аргона последовательно 3,88 мл (27,8 ммоль) (C2H5)3N и 1,07 г (5,57 ммоль) хлоргидрата 1-(3-диметиламинопропил)-3-этилкарбодиимида. Полученную смесь затем перемешивают при комнатной температуре в течение 16 ч и концентрируют в вакууме. Неочищенный продукт разбавляют 400 мл этилацетата и промывают 1 н. раствором соляной кислоты (3 порции по 70 мл), 0,2 н. раствором гидроксида натрия (2 порции по 50 мл) и насыщенным раствором NaHCO3 (1 порция 50 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток хроматографируют на 70 г силикагеля-60 фирмы "Мерк", используя 2% метанол в хлористом метилене в качестве элюента, получая в результате 1,74 г (выход 42%) указанного в заголовке спирта. Тонкослойная хроматография (силикагель, 2% метанола в хлористом метилене): Rf = 0,18 (сульфат церия).
Спектр ЯМР-С13 указанного в заголовке спирта дейтерохлороформ, 67,5 МГц) δ: 174,0, 173,2, 170,6, 130,6, 128,5, 79,2, 62,7, 54,4, 53,5, 51,4, 48,0, 39,5, 37,4, 37,0, 33,3, 33,3, 29,6, 28,6, 27,5, 26,6, 26,3, 26,3, 24,7, 24,1.
D. [1S-[1 α ,2 α (Z),3 α, 4 α]]-7-[3-[5-[[(4-циклогексилбутил)амино]карбонил] -4,5- дигидро-2- оксазол-2-ил]-7-оксабицикло-2.2.1]гепт-2-ил]-5-гептеновая кислота, метиловый эфир.
К раствору 1,10 г (2,17 ммоль) полученного согласно разделу С спирта и 0,61 мл (4,35 ммоль) (C2H5)N в 10 мл сухого хлористого метилена добавляют при перемешивании при 0оС 0,20 мл (2,61 ммоль) метансульфонилхлорида. Полученную смесь перемешивают при 0оС в атмосфере аргона в течение 1,5 ч и разбавляют 200 мл хлористого метилена. Реакционную смесь промывают насыщенным раствором NaHCO3 (1 порция 20 мл) и рассолом (1 порция 20 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Неочищенный продукт растворяют в 20 мл ацетона и добавляют 1,40 г карбоната калия K2CO3. Полученную смесь нагревают с обратным холодильником в течение 5 ч и охлаждают до комнатной температуры. Осадок отфильтровывают через слой Целита толщиной 51 мм и промывают ацетоном (4 порции по 50 мл). Фильтрат концентрируют в вакууме. Очистку производят методом хроматографии с испарением на 36 г силикагеля фирмы "Мерк" с использованием в качестве элюента 2% метанола в хлористом этилене, получая в результате 740 мг (выход 70% ) указанного в заголовке оксазолина. Тонкослойная хроматография (силикагель, 4% метанола в хлористом метилене) Rf = 0,48 (сульфат церия).
Спектр ЯМР-С13 указанного в заголовке соединения (дейтерохлороформ, 67,5 МГц) δ: 173,7; 171,2; 169,2; 130,5; 128,4; 79,0; 79,0; 69,6; 68,3; 51,3; 48,3; 46,3; 39,0; 37,4; 36,9; 33,3; 33,2; 29,7; 29,6; 28,8; 27,2; 26,6; 26,5; 26,2; 26,2; 24,6; 24,0.
E. Метиловый эфир [1S-[1 α,2α (Z).3 α,4 α]]-7-[3-4-[[(4-циклогексилбутил)амино] карбо- нил]-2-оксазолил]-7- оксабицикло-[2.2.1]гепт-2-ил]-5-гептеновой кислоты.
К раствору полученного согласно разделу D оксазолина (730 мг, 1,50 ммоль) в 15 мл сухого хлористого метилена добавляют при перемешивании 1,10 г оксида никеля NiO2. Полученную смесь перемешивают при комнатной температуре в течение 1 ч, после чего добавляют к ней 0,74 г NiO2. Смесь перемешивают при комнатной температуре в течение еще одного часа, и добавляют к ней вторую порцию в 0,74 г NiO2. Полученную смесь перемешивают при комнатной температуре в течение еще 1 ч, после чего разбавляют 80 мл этилацетата, 50 и 3М раствора NaHSO3 и 50 мл 1М раствора цитрата натрия. Водный слой отделяют и экстрагируют 3 порциями этилацетата по 120 мл. Объединенные органические экстракты сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток хроматографируют на 30 г силикагеля-60 фирмы "Мерк", используя 2% метанол в хлористом метилене в качестве элюента, получая в результате 410 мг (выход 56%) указанного в заголовке оксазола. Тонкослойная хроматография (силикагель, 2% метанола в хлористом метилене): Rf = 0,31 (сульфат церия).
Спектр ЯМР-С13 указанного в заголовке соединения (дейтерохлороформ, 67,5 МГц) δ : 173,7; 163,9; 160,5; 140,4; 136,1; 130,5; 128,0; 79,5; 79,3; 51,3; 49,7; 46,6; 39,0; 37,4; 37,0; 33,3; 33,3; 29,8; 28,9; 26,6; 26,3; 24,6; 24,1.
П р и м е р 44. [1S-[1 α,2α (Z),3α , 4α]]-7-[3-[4-[[(4-циклогексилбутил)-амино] карбо- нил] -2-оксазолил] -7- оксабицикло[2.2.1]гепт-2-ил]-5-гептеновая кислота.
К раствору 410 мг (0,84 ммоль) полученного в примере 44 оксазола в 20 мл метанола добавляют при перемешивании 5 мл 1 н. раствора гидроксида натрия. Полученную смесь перемешивают при комнатной температуре в течение 2,5 ч и концентрируют в вакууме. Указанную смесь перераспределяют между 10 мл 1 н. раствора соляной кислоты, насыщенного хлористым натрием, и этилацетатом (4 порции по 20 мл). Экстракты в этилацетате сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Очистку осуществляют посредством хроматографирования с испарением на 20 г силикагеля-60 фирмы "Мерк", используя 150 мл 2%-ного метанола в хлористом метилене с 1% уксусной кислоты для элюирования, и получая в результате 370 мг (выход 93%) чистой кислоты, указанной в заголовке. Температура плавления 123-125оС. Тонкослойная хроматография: силикагель, 6% метанола в хлористом метилене; Rf = 0,22 (сульфат церия).
Спектр ЯМР-С13 указанного в заголовке соединения (дейтерохлороформ, 67,5 МГц) δ : 177,8, 164,0, 160,9, 140,9, 135,8, 128,1, 79,4, 79,4, 49,8, 46,7, 39,2, 37,5, 37,0, 33,3, 33,3, 29,8, 27,8, 26,6, 26,5, 26,3, 26,3, 24,4, 24,2.
П р и м е р 45. Метиловый эфир [1S-[2 α, 2α(Z),3α,4α ]]-6-[3-[4-[[(7,7-ди-метилооктил)амино] карбонил]-2-оксазолил]-7- оксабицикло[2.2.1]-гепт-2-ил]-4-гексеновой кислоты.
A. 3,3-Диметилбутанол.
Раствор 9,4 мл (13,6 г, 107 ммоль) оксалилхлорида в 500 мл хлористого метилена получают в атмосфере аргона в колбе на 1000 мл и охлаждают его до -60оС К раствору добавляют по каплям в течение 10 мин раствор диметилсульфоксида (15,4 мл, 18,5 г, 235 ммоль) в 25 мл хлористого метилена. Реакционную смесь перемешивают в течение 10 мин и медленно добавляют к ней 10 г (98 ммоль) 3,3-диметил-1-бутанола. Перемешивание продолжают в течение дополнительных 20 мин, после чего добавляют к смеси 68,1 мл (49,5 г, 489 ммоль) (C2H5)3N и реакционной смеси дают нагреться до комнатной температуры. Затем к ней добавляют 50 мл воды, смесь разделяют и экстрагируют водный слой 1 порцией (30 мл) хлористого метилена. Органические слои объединяют, последовательно промывают 1%-ным водным раствором соляной кислоты, водой и насыщенным раствором NaHCO3, вновь водой и насыщенным раствором хлористого натрия и сушат над сульфатом магния. Отфильтрованный раствор концентрируют с использованием роторного испарителя, получая в результате 3,3 г (33%) указанного в заголовке соединения в виде желтого масла. Низкий выход возможно вызван высокой летучестью продукта. Спектр ЯМР-С13 (дейтерохлороформ, 67,8 МГц) δ: 203,3; 56,4; 31,0; 29,7.
B. (Z)-7,7-диметил-4-октеновая кислота.
К раствору 13,72 г (31,9 ммоль) 3-карбоксипропилтрифенилфосфонийбромида в 60 мл сухого тетрагидрофурана в атмосфере аргона добавляют при перемешивании при -15оС по каплям в течение 10 мин 1,72 н. К-трет-амилат в толуоле (32 мл, 57,9 ммоль). Окрашенную в оранжевый цвет смесь перемешивают в течение получаса. Затем к ней медленно добавляют 2,00 г (19,9 ммоль) альдегида согласно разделу A в виде раствора в 5 мл тетрагидрофурана. Реакционную смесь перемешивают в течение 1 ч при -15оС и при комнатной температуре - в течение 20 ч. Затем реакционную смесь гасят 12 мл уксусной кислоты, добавляемой по каплям, и концентрируют в вакууме. Остаток перераспределяют между 100 мл этилацетата и 100 мл воды. Водный слой дважды экстрагируют порциями по 100 мл этилацетата. Объединенные органические экстракты последовательно промывают 1%-ным водным раствором соляной кислоты, водой, насыщаемым раствором NaHCO3, водой и насыщенным раствором хлористого натрия, и сушат над сульфатом магния, после чего концентрируют в вакууме. Остаток хроматографируют, используя для элюирования 0,3% уксусной кислоты в градиенте этилацетата в гексане (от 1 до 100%), получая в результате 1,75 г (выход 51%) целевого продукта, указанного в заголовке.
Спектр ЯМР-С13 (дейтерохлороформ) δ : 179,8; 128,6; 128,4; 41,0; 34,1; 31,1; 29,2; 22,6.
C. 7,7-Диметилоктановая кислота.
К перемешиваемому раствору кислоты согласно разделу B (1,2 г, 7,04 ммоль) в 8 мл уксусной кислоты добавляют 0,2 г оксида платины. Реакционную смесь перемешивают в течение 14 ч при давлении водорода 1 атм (из баллона). Затем реакционную смесь фильтруют через слой Целита, а фильтрат концентрируют в вакууме. Остаток разбавляют 50 мл толуола и вновь концентрируют. Процесс повторяют еще раз, получая в результате 1,2 г (выход 100%) указанного в заголовке целевого продукта в виде масла.
D. 7,7-Диметилоктанамид.
К перемешиваемому раствору кислоты согласно разделу С (1,21 г, 7,02 ммоль) в 50 мл толуола добавляют 3 мл оксалилхлорида. Полученную реакционную смесь перемешивают в течение 1 ч при комнатной температуре в атмосфере аргона и концентрируют в вакууме. Остаток разбавляют 20 мл толуола и вновь концентрируют. Процесс повторяют для удаления следов оксалилхлорида. Остаток перемешивают при комнатной температуре под аргоном в 5 мл уксусной кислоты и добавляют к раствору 1,17 мл (8,43 ммоль) C2H5)3N и избыток 9М метанольного раствора аммиака (2 мл). После перемешивания в течение 16 ч реакционную смесь перераспределяют между 3 мл воды и 20 мл этилацетата. Водный слой экстрагируют еще 2 порциями по 20 мл этилацетата. Объединенные органические слои промывают раствором хлористого натрия, сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая полужидкий остаток. Указанный полужидкий продукт кристаллизуют посредством растирания с гексаном, получая в результате 0,5 г (выход 42%) указанного в заголовке целевого продукта в виде твердого вещества.
Спектр ЯМР-С13 (дейтерохлороформ) δ: 176,3; 43,9; 35,9; 30,2; 30,1; 29,3; 25,5; 24,2.
Е. 7,7-Диметилоктанамин.
К раствору полученного согласно разделу D амида (0,45 г, 2,62 ммоль) в 50 мл сухого эфира, перемешиваемому в атмосфере азота при 0оС, добавляют 0,11 г (2,88 ммоль) литийалюминийгидрида; при этом выделяется газ. Реакционную смесь перемешивают при комнатной температуре в течение 4 суток. При интенсивном перемешивании реакционную смесь гасят посредством добавления к ней 0,02 мл воды, 0,02 мл 15%-ного водного раствора гидрата окиси натрия, и 0,072 мл воды. При этом образуется белый осадок. После перемешивания в течение 0,5 ч, белый осадок отфильтровыва- ют, а фильтрат концентрируют в вакууме, получая 0,4 г (выход 89%) указанного в заголовке соединения в виде желтого масла. Это масло перекристаллизовывают посредством растирания с гексаном и хлороформом.
F. Метиловый эфир [1S-[1 α,2 α (Z),3 α, 4α] ]-6-[3-[4- ]](7,7-диметилоктил)амино] карбонил)- 2-оксазолил]-7-оксабицикло [2.2.1]гепт-2-ил]-4-гексеновой кислоты.
К раствору хлорангидрида кислоты, полученного согласно примеру 2, часть 1 (0,46 г, 1,3 ммоль) и неизвестному количеству соли Вильсмайера в 5 мл хлороформа при комнатной температуре в атмосфере аргона добавляют 0,25 мл (0,18 г, 1,8 ммоль) (C2H5)3N и 0,24 г (1,5 ммоль) амина.
Е. Реакционную смесь перемешивают при комнатной температуре в течение 16 ч, после чего разбавляют этилацетатом и водой. Органический слой отделяют, а водный слой экстрагируют дважды 20 мл этилацетата. Органические слои объединяют, промывают рассолом, сушат над сульфатом магния и концентрируют. После хроматографирования с испарением (градиент этилацетата в гексане, от 0 до 100% ) получают 0,20 г (выход 32%) указанного в заголовке целевого продукта в виде масла. Rf = 0,8 в 1% трифторуксусной кислоты, 1% метанола и 98% этилацетата.
Спектр ЯМР-С13 (дейтерохлороформ) δ: 172,8; 163,6; 160,2; 140,1; 135,9; 129,1; 128,3; 79,3; 79,1; 51,1; 49,4; 46,4; 43,9; 38,8; 33,5; 29,9; 29,5; 29,4; 29,1; 28,7; 27,6; 26,7; 24,1; 22,5.
П р и м е р 46. [1S-1 α,2 α (Z),3 α ,4 α]]-6-[3-[4-[[(7,7-диметилоктил)-амино] карбонил] -2-оксазолил-7- оксабицикло[2.2.1]гепт-2-ил]-4-гексеновая кислота.
Раствор сложного эфира, полученного согласно примеру 46 (0,20 г, 0,38 ммоль), в 10 мл 1 н. раствора гидроксида натрия и 10 мл тетрагидрофурана, представляющий собой реакционную смесь, перемешивают в течение 13 ч, после чего концентрируют в вакууме для удаления тетрагидрофурана и подкисляют до рН = 2 посредством 1 н. раствора соляной кислоты. К смеси добавляют этилацетат и отделяют органический слой. Водный слой дважды экстрагируют 20 мл этилацетата. Органические слои объединяют, промывают рассолом и концентрируют в вакууме, получая прозрачное масло. Это масло хроматографируют (0,1% уксусной кислоты в градиенте этилацетата в гексане, от 0 до 50%), получая в результате 0,12 г (выход 66%) указанной в заголовке кислоты в виде белого твердого вещества. Температура плавления 68-69оС. Rf = 0,18 в смеси гексан-этилацетат (1:1) с 0,05% уксусной кислоты.
[ α]Do = + 31,1 в метаноле при с = 0,46 г/100 мл.
Спектр ЯМР-С13 (дейтерохлороформ) δ: 176,8; 163,9; 160,7; 140,8; 135,7; 129,4; 128,4; 79,5; 79,3; 49,6; 46,5; 44,0; 39,1; 33,6; 30,1; 29,6; 29,4; 29,3; 28,8; 27,8; 26,8; 24,3; 22,5.
П р и м е р 47. [1S-[1 α, 2α (Z),3α , 4 α]]-6-[3-[4-[[(4-циклогексилбутил)-амино] карбонил] -2-оксазолил-7- оксабицикло[2.2.1]гепт-2-ил]-4-гексеновая кислота.
A. [1S-[1 α, 2 α (Z), 3 α, 4 α ]-7-[3-[[диметил-(1,1-диметилэтил)силил] окси] -7-оксаби- цикло[2.2.1] - гепт-2-ил]-5-гептеновая кислота, метиловый эфир.
К перемешиваемому раствору 20,57 г [1S-[1 α, 2 α (Z), 3 α, 4 α ]]-7-[3-оксиметил-7-оксабицикло[2.2.1] гепт-2-ил] -5-гептеновой кислоты в виде метилового эфира (полученного согласно описанному в патенте США N 4143054 способу) (76,8 ммоль) и 5,74 г имидазола (84,4 ммоль, 1,1 экв.) в 100 мл хлористого метилена в атмосфере аргона при 0оС добавляют 12,05 г трет-бутилдиметилсилилхлорида (79,8 ммоль, 1,04 экв.). При этом образуется осадок. Смесь нагревают до комнатной температуры и спустя 10 мин разбавляют диэтиловым эфиром, промывают водой (трижды) и рассолом (один раз), сушат над сульфатом натрия и концентрируют посредством испарения на роторном испарителе, а затем - в высоком вакууме. Остаток в количестве 30,31 г представляет собой почти чистый силиловый эфир, указанный в заголовке, и используется без дальнейшей очистки. Выход указанного в заголовке сложного эфира практически является количественным.
Тонкослойная хроматография (50% этилацетата в гексане - анисовом альдегиде): Исходный продукт 0,21
Указанный в заголов- ке сложный эфир 0,79
B. [1S-[1 α, 2 α, 3 α, 4 α] ]-3-[[[(1,1-диметилэтил)диметилсилил]окси] метил]-7-окса-бицикло [2.2.1]гептан-2-ацетальдегид.
Раствор 30,31 г практически числового силильного эфира согласно разделу A (76,8 ммоль) в 200 мл хлористого метилена при -78оС обрабатывают озоном до появления устойчивой голубой окраски в течение 25 мин. После продувки избытком озона с кислородом добавляют 48,0 г (770 ммоль, 10 экв. диметилсульфида (CH3)2S и нагревают реакционную смесь до комнатной температуры. После перемешивания в течение 1 ч при комнатной температуре растворитель и избыток диметилсульфида удаляют посредством обработки в роторном испарителе. Спектр ЯМР-Н1 неочищенного продукта свидетельствует о неполном восстановлении производного озона. Поэтому после повторного растворения в хлористом метилене при комнатной температуре добавляют 20,2 г трифенилфосфина (77 ммоль, 1,0 экв.). Полученная смесь нагревается, поскольку протекающая реакция является экзотермической. После перемешивания в течение ночи большая часть растворителя отгоняется и к смеси добавляют гексан для осаждения окиси трифенилфосфина и самого трифенилфосфина. Осадок отфильтровывают, а фильтрат концентрируют, а затем хроматографируют (градиент этилацетата в гексане от 5 до 15% ), получая в результате 16,31 г чистого соединения, указанного в заголовке, в виде масла.
Тонкослойная хроматография (50% этилацетата в гексане - анисовом альдегиде):
Силильный эфир согласно разделу A 0,89
Указанный в заголовке альдегид 0,76
Метил-5-оксапен- теноат 0,51
С. [1S-( α, 2 α (E), 3 α, 4α )-[4-[3-[[[(1,1-диметилэтил)-диметилсилил)окси)метил] -7-ок- сабицикло- [2.2.1]гепт-2-ил)-2-бутеновая кислота, метиловый эфир.
Колбу, содержащую 3,3 г (37,9 ммоль) бромида лития, помещают под вакуум и нагревают посредством тепловентилятора для удаления следов влаги. После охлаждения колбу продувают аргоном и добавляют в нее 20 мл хлористого метилена. К этой смеси разбавляют при перемешивании раствор 5,44 г (29,9 ммоль) триметилфосфонацетата в 30 мл хлористого метилена, а затем 4,0 мл (28,7 ммоль) (C2H5)3N. Полученную смесь перемешивают в течение 15 мин, после чего добавляют в течение 1 мин раствор 5,4 г (19,0 ммоль максимум) неочищенного альдегида согласно разделу B в 35 мл хлористого метилена. Реакция сопровождается небольшим выделением тепла и образованием кристаллического осадка. Реакционную смесь интенсивно перемешивают при комнатной температуре в течение ночи. Затем ее перераспределяют между 200 мл гексана и 100 мл 0,3 н. соляной кислоты. Водный слой экстрагируют 100 мл диэтилового эфира. Объединенные органические экстракты сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Анализ методом тонкослойной хроматографии показывает, что реакция не прошла до конца (40-50%). Остаток растворяют в 50 мл хлористого метилена и добавляют к нему 6,3 г (18,8 ммоль) карбометоксиметилентрифенилфосфина. Этот раствор перемешивают при комнатной температуре в течение 22 ч, после чего концентрируют в вакууме. Остаток растирают с диэтиловым эфиром и разбавляют равным раствором гексана. Смесь охлаждают в холодильнике в течение 2 ч, после чего отделяют фильтрованием твердый остаток. Фильтрат концентрируют в вакууме. Неочищенный продукт подвергают очистке хроматографированием на 167 г силикагеля, используя в качестве элюента смесь гексана и диэтилового эфира 4:1, и получая в результате 5,9 г (выход 91%) указанного в заголовке сложного эфира.
Тонкослойная хроматография (силикагель, смесь гексан-эфир 2:1); Rf = 0,6 (ванилин).
Спектр ЯМР-С13 (67,8 МГц, дейтерохлороформ) δ : 148,8, 121,8, 79,9, 78,8, 61,9, 51,4, 44,8, 30,7, 29,5, 29,4, 25,9, 18,2, - 5,4.
D. [1S-(1 α, 2 α (E), 3 α, 4 α )]-4-[3-[[[(1,1-диметилэтил)диметилсилил]окси]метил]-7-оксабицикло [2.2.1](гепт-2-ил)-2-бутен-1-ол.
Раствор 5,2 г (15,3 ммоль) сложного эфира, полученного согласно разделу С, в 80 мл тетрагидрофурана охлаждают до - 78оС. К этому раствору добавляют по каплям при перемешивании 50,0 мл 1,5М раствора ДИБАЛ-Н в толуоле за промежуток времени, равный 20 мин. Реакционную смесь перемешивают в течение 5 ч при - 78оС, после чего реакцию гасят в результате введения раствора 10 мл ацетона в 10 мл толуола. Затем к раствору медленно добавляют 60 г смеси силикагеля с водой (10: 9 по массе). После введения нескольких граммов влажного силикагеля охлаждающую баню убирают. Реакционную смесь затем разбавляют 200 мл эфира. За температурой внутри реакционного сосуда внимательно наблюдают, и когда она достигнет 10оС, колбу погружают в баню со льдом. Реакционную смесь перемешивают в течение 1 ч, после чего силикагель удаляют фильтрованием. Осадок на фильтре промывают 2 порциями по 100 мл эфира. Объединенные фильтраты сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая в результате 5,05 г (более 100% от теории) неочищенного продукта, указанного в заголовке. Тонкослойная хроматография (силикагель, смесь гексан-эфир 2:1); Rf = 0,1 (ванилин).
Спектр ЯМР-С13 (67,8 МГц в дейтерохлороформе) δ: 131,6, 130,6, 79,8, 78,8, 63,3, 61,8, 49,3, 45,4, 30,5, 29,5, 29,3, 25,9, 18,2, - 5,4.
E. (1S-(1 α, 2 α(E), 3α ,4α )]-4-]3-[[[(1,1-диметилэтил)-диметилсилил(окси[метил-7-оксаби- цикло [2.2.1](гепт-2-ил)-2-бутен-1-бромид.
В колбу, содержащую 0,83 г трифенилфосфина (3,17 ммоль, 1,0 экв.), растворенного в 10 мл толуола добавляют в атмосфере аргона при 0оС 0,51 г брома (3,17 ммоль, 1,0 экв. ) в виде одной порции. При этом образуется желтый осадок и оранжевая резинообразная масса. В результате обработки шпателем резинообразная масса превращается в более легколетучий осадок. Смесь быстро нагревают до комнатной температуры, после чего вновь охлаждают до 0оС. К ней добавляют по каплям раствор 1,04 г спирта, полученного согласно разделу D (степень чистоты 95; 0,99 г, 3,17 ммоль) и 0,28 г пиридина (3,49 ммоль, 1,1 экв.) в 5 мл толуола (плюс 5 мл для промывки). Реакционную смесь перемешивают при 0оС в течение 30 мин, затем нагревают до комнатной температуры. Согласно данным тонкослойной хроматографии, реакция не доходит до конца. Спустя 4 ч (когда данные тонкослойной хроматографии не свидетельствуют более о каких-либо изменениях) реакционную смесь фильтруют и отгоняют из фильтрата растворитель. В результате хроматографирования с испарением (в градиенте этилацетата в гексане (от 3 до 50% этилацетата) получают 830 мг указанного в заголовке бромида в виде масла, а также 190 мг непрореагировавшего спирта согласно части D. Выход указанного в заголовке бромида равен 74%.
Тонкослойная хроматография (25% этилацетата в гексане - анисовом альдегиде):
Спирт согласно раз- делу D 0,21
Указанный в заго- ловке бромид 0,75
Спектр ЯМР-С13 (67,8 МГц, в дейтерохлороформе) δ : 135,6, 127,5, 79,8, 78,8, 61,8, 49,4, 45,3, 33,1, 30,5, 29,5, 29,4, 25,9, 18,2, -5,3.
F. [1S-(1 α, 2 α (E), 3 α, 4α )]-6-[3-[[[(1,1-диметилэтил)-диметилсилил] окси]метил]-7- оксабицикло[2.2.1]гепт-2-ил)-4-гексеновая кислота, 1,1-диметилэтиловый эфир.
К раствору ЛПА в тетрагидрофуране (2,70 ммоль, 1,15 экв.), полученной в результате медленного добавления 1,08 мл 2,5М раствора C4H9Li в гексане к раствору 303 мг диизопропиламина (3,0 ммоль, 1,28 экв.) в 4 мл сухого тетрагидрофурана при перемешивании в атмосфере аргона при 0оС с последующим перемешиванием смеси в течение 15 мин) добавляют по каплям при перемешивании в атмосфере аргона при - 78оС раствор 348 мг трет-бутилацетата (3,0 ммоль, 1,28 экв. ) в 3 мл сухого тетрагидрофурана в течение 15 мин. После перемешивания в течение 1 ч к смеси добавляют по каплям раствор 880 мг бромида, полученного согласно разделу Е (2,35 ммоль) в 3 мл тетрагидрофурана при двух промывках по 2 мл. Спустя 8 ч перемешивания при - 78оС тонкослойная хроматография свидетельствует лишь о частичном протекании реакции, и реакционную смесь медленно нагревают (со скоростью примерно 8оС в час) до комнатной температуры. Тонкослойная хроматография не показывает дальнейшего изменения, однако расход бромида все еще не является полным. Вслед за добавлением 1 мл насыщенного водного раствора хлорида аммония смесь сушат над сульфатом натрия и отгоняют растворитель. Остаток хроматографируют (5% этилацетата в гексане), получая в результате 550 мг практически чистого сложного эфира, указанного в заголовке (степень чистоты 97% = 534 мг). Выход указанного в заголовке сложного эфира равен 55%.
Тонкослойная хроматография (5% этилацетата в гексане-анисовом альдегиде):
Бромид согласно раз- делу Е 0,20
Указанный в заго- ловке сложный эфир 0,13
Спектр ЯМР-С13 (67,8 МГц, дейтерохлороформ) δ: 172,4, 130,6, 129,6, 80,0, 79,8, 78,8, 61,9, 49,4, 45,8, 35,4, 30,8, 29,6, 29,4, 28,1, 25,9, 18,2, - 5,3.
G. [1S-(1 α, 2 α (E), 3 α, 4 α )]-6-[3-(оксиметил)-7-оксабицикло[2.2.1] гепт-2-ил)-4-гексе- новая кислота, метиловый эфир.
К раствору 550 мг практически чистого сложного эфира, полученного согласно разделу F (степень чистоты 97% = 534 мг, 1,30 ммоль) в 20 мл метанола добавляют при перемешивании при комнатной температуре в атмосфере аргона 2 мл раствора сухого HCl в метаноле (полученного посредством добавления 2 капель ацетилхлорида к 2 мл метанола при комнатной температуре с последующим выдерживанием в течение 1 мин). Тонкослойная хроматография указывает на полное превращение в интермедиат в течение 1 ч. Указанный интермедиат затем очень медленно превращается в продукт. Добавление еще 10 мл HCl в метаноле ускоряет реакцию. Спустя 14 суток добавляют 2 мл (C2H5)3N и отгоняют растворитель из реакционной смеси. В результате получают 630 мг неочищенного спирта, указанного в заголовке.
Тонкослойная хроматография (50% этилацетата в гексане-анисовом альдегиде):
Сложный эфир соглас но разделу F 0,94 Интермедиат 0,33
Указанный в заголов- ке спирт 0,23
H. [1S-(1 α, 2 α (E), 3α, 4α )]-6-[3-карбокси-7-оксабицикло[2.2.1]гепт-2-ил]-4-гексено- вая кислота, метиловый эфир.
К раствору 630 мг неочищенного спирта согласно разделу G в 20 мл ацетона добавляют при 0оС в атмосфере аргона 4 мл реагента Джонса (2,6М по CrVI). К окончанию введения реагента наблюдается устойчивое красное окрашивание. Образующийся при реакции осадок нагревают до комнатной температуры в течение 20 мин, после чего вновь охлаждают и добавляют 2-пропанол для гашения избытка реагента. Все еще при температуре 0оС добавляют к смеси при перемешивании 3М раствор NaHSO3 до тех пор, пока все соли не растворятся. Затем добавляют рассол и трижды экстрагируют смесь этилацетатом. После сушки экстрактов над сульфатом натрия и отгонки растворителя остаток хроматографируют (диоксид кремния, в градиенте в гексане (от 0,25 до 50%) 5%-ной уксусной кислоты в этилацетате), получая после азеотропной отгонки уксусной кислоты с толуолом 260 мг указанной в заголовке кислоты, что соответствует выходу 75% по сложному эфиру, полученному согласно разделу F.
Тонкослойная хроматография (50% (5% уксусной кислоты в этилацетате) в гексане-анисовом альдегиде): Спирт согласно разделу G 0,32
Указанная в заголовке кислота 0,36
Спектр ЯМР-С13 (67,8 МГц, дейтерохлороформ) δ: 177,1, 173,4, 130,4, 129,2, 78,2, 78,1, 61,9, 51,3, 47,4, 33,8, 32,4, 28,9, 28,8, 27,7.
I. [1S-(1 α, 2 α (E), 3 α, 4 α ]]-6-[3-[4-[[(4-циклогексилбутил)амино] карбонил]-2-оксазо-лил]-7- оксабицикло[2.2.1]гепт-2-ил]-4-гексеновая кислота.
В соответствии с описанной в примере 1 процедурой, начиная с раздела Е, за исключением замены кислоты согласно разделу D примера 1, на кислоту согласно разделу Н данного примера, получают указанное в заголовке соединение. Температура плавления 122-125оС, тонкослойная хроматография (50%, 5% уксусной кислоты в этилацетате, в гексане; Rf для указанной в заголовке кислоты 0,33.
Спектр ЯМР-С13 (дейтерохлороформ, 67,8 МГц), δ: 176,9; 163,9; 160,7; 140,8; 135,6; 130,0; 128,8; 79,4; 79,0; 49,1; 46,5; 39,1; 37,4; 36,9; 33,7; 33,2; 33,0; 29,7; 29,5; 28,8; 27,5; 26,5; 26,2; 24,0.
П р и м е р 48. [1S-(1 α, 2α , 3α , 4 α)-3-[4-[[(4-циклогексилбутил)аминокарбонил] -2- оксазолил]-7- оксабицикло[2.2.1]гептан-2-гексановая кислота.
Раствор 130 мг кислоты согласно примеру 1 в 10 мл этилацетата и 1,0 мл уксусной кислоты дегазируют посредством циклической процедуры откачки - стравливания аргоном. К полученному раствору добавляют 34 мг 10%-ного палладия на активированном угле и заменяют атмосферу на водород посредством двух циклических процедур вакуумирования - заполнение системы. Некоторое избыточное давление поддерживают посредством использования баллона с водородом. Смесь перемешивают при комнатной температуре в течение 22,5 ч, разбавляют хлористым метиленом и фильтруют через поликарбонатный фильтр для удаления катализатора. Фильтрат концентрируют в вакууме. Остаток разбавляют толуолом и вновь концентрируют. После добавления к остатку этилацетата небольшое количество гелеобразного продукта не растворяется. Раствор декантируют и концентрируют в вакууме. Неочищенный продукт растворяют в минимальном количестве горячего этилацетата и разбавляют примерно 3 объемами гексана. После охлаждения появляется твердый осадок, однако, после стояния при 5оС в течение ночи образуется белый гелеобразный твердый продукт. Его отделяют фильтрованием и cушат в вакууме. Образующийcя при этом белый порошок растирают с гексаном, фильтруют и сушат в вакууме, получая в результате 61 мг чистой кислоты, указанной в заголовке. Температура плавления 97оС (размягчение).
С26H40N2O5.
Вычислено, %: C 67,79; H 8,75; N 6,08.
Найдено, %: C 67,58; H 8,79; N 5,97.
Тонкослойная хроматография (силикагель, 4% метанола в хлористом метилене): Rf = 0,35 (сульфат церия).
[ α]D = + 23 (с = 0,68, хлороформ).
Спектр ЯМР-С13 (дейтерохлороформ, 67,5 МГц) δ: 164,2, 160,8, 140,7, 135,9, 79,5, 79,4, 49,8, 47,2, 39,2, 37,5, 37,1, 33,7, 33,4, 29,9, 29,7, 29,2, 29,0, 28,1, 26,7, 26,4, 24,5, 24,2.
П р и м е р 49. [1S-[1 α, 2 α(S), 3 α , 4 α]]- 6-[3-[4-[[(-4-циклогексилбутил)амино)карбо- нил)-2-оксазолил)-7- оксабицикло(2.2.1)гепт-2-ил)-4-гексеновая кислота, метиловый эфир.
A. (1S-[1 α, 2α (Z), 3 α (R*), 4α ]]-6-[3-2-](4-циклогексилбутил)амино] -1-оксиметил-2-ок- соэтиламино] -1-оксиметил-2-оксоэтил]амино]карбонил]-7-оксабицикло[2.2.1]гепт-2- ил]-4-гексеновая кислота, метиловый эфир.
К перемешиваемой смеси кислоты согласно примеру 1, раздел D (6,70 г, 25,2 ммоль), 3,40 г (25,2 ммоль) 1-оксибензотриазола и соли трифторуксусной кислоты амина согласно примеру 1, раздел B (8,97 г, 25,2 ммоль), в 100 мл диметилформамида при 0оС в атмосфере аргона последовательно добавляют 17,6 мл (126 ммоль) (С2H5)3N и 4,82 г (25,2 ммоль) 1-(3-диметиламинопропил)-3-этилкарбодиимида в виде хлоргидрата. Реакционную смесь перемешивают при 0оС в течение 2 ч, а затем при комнатной температуре в течение 16 ч. Смесь концентрируют в вакууме и перераспределяют между 800 мл этилацетата и 1 н. раствором соляной кислоты (2 порции по 100 мл), 0,2 н. раствором гидроксида натрия (2 порции по 100 мл), насыщенным раствором NaHCO3 (1 порция 100 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Остаток хроматографируют на 180 г силикагеля-60 фирмы "Мерк", используя в качестве элюента 2% метанола в хлористом метилене и получая в результате 4,75 г (выход 39%) указанного в заголовке амида.
Тонкослойная хроматография (силикагель, 50% , 5% уксусной кислоты в этилацетате, в гексане): Rf = 0,22 (анисовый альдегид.
B. (1S-[1 α, 2 α (Z), 3 α (R*), 4α ]]-6-[3-[4-(4-циклогексилбутил)амино]карбонил-4,5- дигидро-2-оксазолил]- 7-оксабицикло[2.2.1]гепт-2-ил] -4-гексеновая кислота, метиловый эфир.
К перемешиваемой смеси амида согласно разделу A (4,69 г, 9,53 ммоль) в 60 мл сухого хлористого метилена добавляют при 0оС в атмосфере последовательно: 2,66 мл (19,1 ммоль) (C2H5)3N и 0,82 мл (10,5 ммоль) мезилхлорида. Реакционную смесь перемешивают при 0оС в течение 1 ч и разбавляют 100 мл хлористого метилена, после чего промывают насыщенным раствором гидрокарбоната натрия NaHCO3 (1 х 30 мл) и рассолом (1 х 30 мл). Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме.
Неочищенный мезилат растворяют в 60 мл ацетона и объединяют с 5,0 г (36,2 ммоль) карбоната калия (K2CO3). Смесь нагревают с обратным холодильником в течение 4,5 ч и охлаждают до комнатной температуры. Твердый остаток отфильтровывают и промывают ацетоном (4 порции по 40 мл). Фильтрат концентрируют в вакууме и хроматографируют на 120 г силикагеля-60 фирмы "Мерк", используя в качестве элюента 2%-ный метанол в хлористом метилене и получая в результате 3,93 г (выход 86%) указанного в заголовке оксазолина.
Тонкослойная хроматография (силикагель, 20% ацетона в толуоле): Rf = 0,29 (анисовый альдегид).
С. (1S-[1 α, 2 α (Z), 3 α, 4 α]]-6-[3-[4-[[(4-циклогексилбутил)амино] карбонил] -2-окса- золил-7- оксабицикло[2.2.1]гепт-2-ил]-4-гексеновая кислота, метиловый эфир.
К перемешиваемой смеси оксазолина согласно разделу B (3,90 г, 8,23 ммоль) в 80 мл сухого хлористого метилена добавляют в атмосфере аргона 6 г окиси никеля NiO2. Реакционную смесь перемешивают при комнатной температуре в течение 40 минут, после чего добавляют 4 г NiO2. Смесь перемешивают в течение дополнительных 70 мин и вновь добавляют 2 г NiO2. Затем смесь перемешивают при комнатной температуре в течение 2 ч и разбавляют 120 мл этилацетата, 60 мл 3 М раствора NaHSO3 и 60 мл 1 М раствора цитрата натрия. Водный слой отделяют и экстрагируют 4 порциями по 150 мл этилацетата. Объединенные экстракты в этилацетате суша над сульфатом магния, фильтруют и концентрируют в вакууме. Очистку осуществляют методом хроматографирования с испарением на 120 г силикагеля-60 фирмы "Мерк", используя в качестве элюента 2% -ный метанол в хлористом метилене и получая в результате 1,85 г (выход 48%) указанного в заголовке оксазола.
Тонкослойная хроматография (силикагель, этилацетат): Rf = 0,81 (анисовый альдегид).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПУРИНИЛ- ИЛИ ПИРИМИДИНИЛПРОИЗВОДНЫЕ МЕТИЛЕНЦИКЛОПЕНТИЛА | 1991 |
|
RU2037496C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ТРИФТОРМЕТИЛМЕРКАПТАНА | 1991 |
|
RU2024504C1 |
| СЕРУЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ МЕВИНОВОЙ КИСЛОТЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ | 1991 |
|
RU2041205C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЗОТСОДЕРЖАЩИХ ГЕТЕРОЦИКЛИЧЕСКИХ СОЕДИНЕНИЙ ИЛИ ИХ ФАРМАЦЕВТИЧЕСКИПРИЕМЛЕМЫХ СОЛЕЙ | 1990 |
|
RU2026296C1 |
| ПРОИЗВОДНЫЕ БЕНЗАЗЕПИНА ИЛИ БЕНЗОТИАЗЕПИНА | 1989 |
|
RU2090562C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ ПИРАНИЛ-ЦИАНОГУАНИДИНА | 1990 |
|
RU2057129C1 |
| СПОСОБ ПОЛУЧЕНИЯ ПРОИЗВОДНЫХ 5-АМИНО-2,4,6-ТРИИОДО- 1,3-БЕНЗОЛКАРБОНОВОЙ КИСЛОТЫ | 1990 |
|
RU2046795C1 |
| СПОСОБ ПОЛУЧЕНИЯ БИС(ГИДРОКСИМЕТИЛ)ЦИКЛОБУТИЛ ПУРИНОВ ИЛИ ПИРИМИДИНОВ | 1989 |
|
RU2041213C1 |
| ПРОИЗВОДНЫЕ ИМИДАЗОЛА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ИНГИБИРУЮЩАЯ АКТИВНОСТЬ АНГИОТЕНЗИНА II, И СПОСОБ ЛЕЧЕНИЯ ГИПЕРТОНИИ У МЛЕКОПИТАЮЩИХ | 1991 |
|
RU2111208C1 |
| СПОСОБ ПОЛУЧЕНИЯ ДИАСТЕРЕОИЗОМЕРНОЙ ПАРЫ ФОСФИНИЛУКСУСНОЙ КИСЛОТЫ | 1991 |
|
RU2044740C1 |
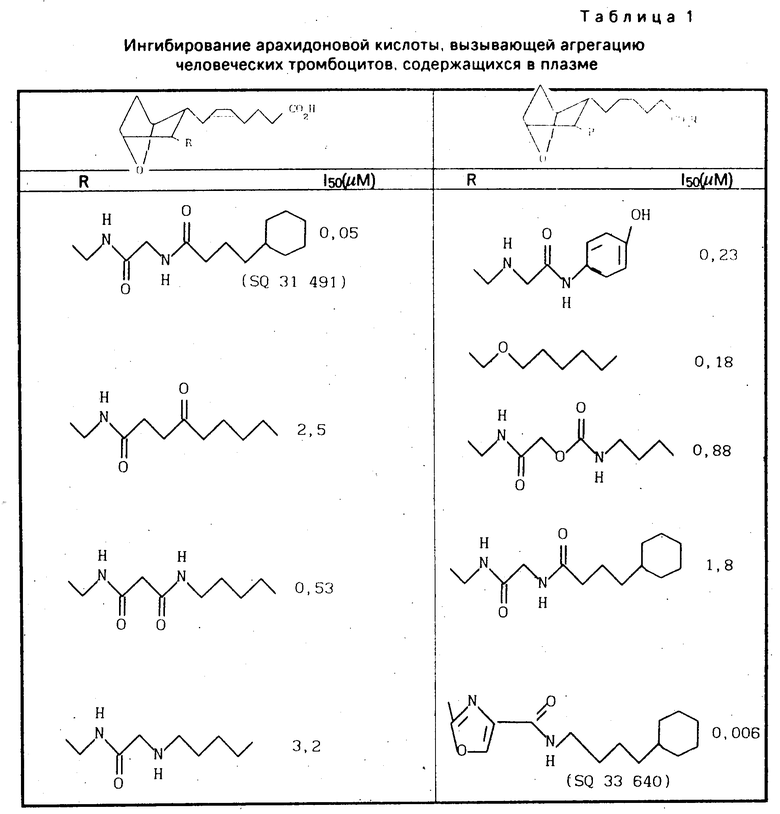
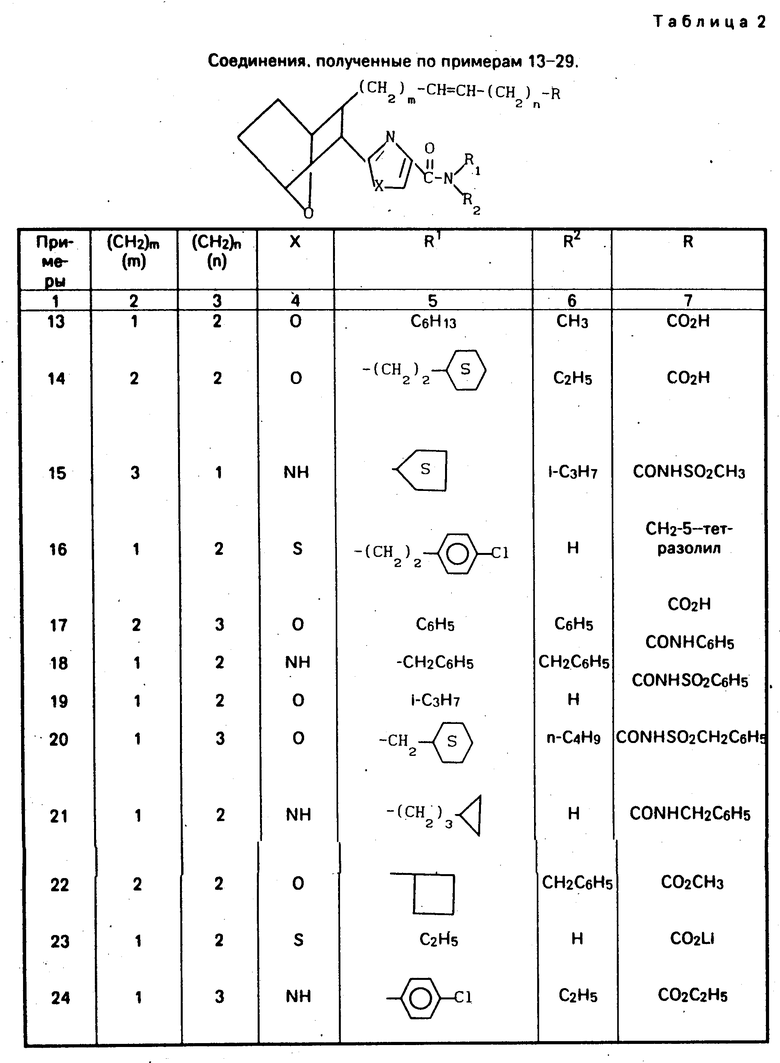
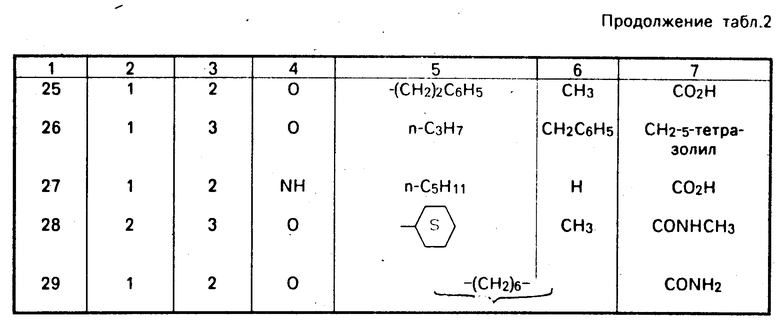
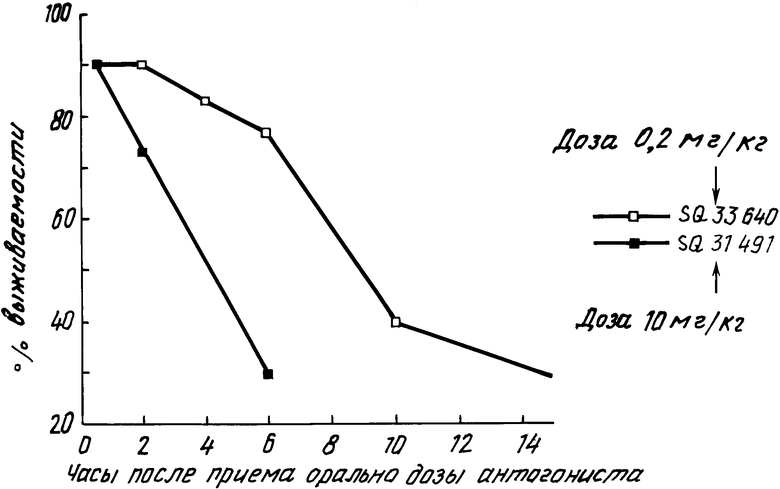
Использование: в медицине, в качестве антагонистов рецепторов тромбоксана. Сущность изобретения: продукт: 7-оксабициклогептилзамещенные гетероциклические амиды ф-лы I или их стереоизомеры, где m - 1,2 или 3; n - 0,1,2,3 или 4,Z- CH2-CH2 - или CH=CH-, при условии, когда Z- CH=CH-, n не равно 0; R - карбоксил, его соль щелочного металла, карбокси - (C1-C8) - алкил, CONHSO2(C1-C8) - алкил, CONHSO2 - фенил, CONHSO2 - бензил или CH2 -5-тетразолил; X - O, S, или NH; R1-C1-C16 - алкил, возможно замещенный C3-C8 - циклоалкилом или фенилом, который может содержать атом галогена; C3-C8 - циклоалкил или фенил, который может быть замещен галогеном; R2 - H или C1-C8 - алкил или R1 и R2, взятые вместе с соседним атомом азота, образуют 5-8-членное кольцо. 2 табл., 1 ил. Структура соединения ф-лы 1.
7-Оксабициклогептилзамещенные гетероциклические амины общей формулы I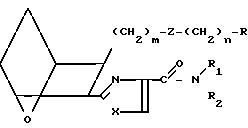
где m = 1,2 или 3;
n = 0, 1, 2, 3 или 4;
Z - группа -СН2 - СН2- или -СН=СН -, при условии, что когда Z представляет собой - СН=СН-, n не равно 0;
R - карбоксил, его соль щелочного металла, карбокси (С1 - С8)-алкил, СОNHSO2(С1 - С8)-алкил, CONHSO2-фенил; CONHSO2-бензил или -CH2 - 5-тетразолил;
Х - 0, S или NХ;
R1 - С1 - С16-алкил, который может быть замещен С3 - С8-циклоалкилом или фенилом, который может содержать атом галогена,
С3 - С8-циклоалкил, или фенил, который может быть замещен галогеном;
R2 - водород или С1 - С8-алкил, или R1 и R2, взятые вместе, с соседним атомом азота образуют 5 - 8 членное кольцо,
или их стереоизмеры в качестве антагонистов рецепторов тромбоксана.
| Приспособление к токарным резцам для отвода стружки при резании хрупких металлов | 1951 |
|
SU94792A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Гребенчатая передача | 1916 |
|
SU1983A1 |
