Изобретение относится к новому классу простых диэфиров и способу их получения.
Простые диэфиры, являющиеся предметом настоящего изобретения, можно использовать в качестве добавок в горючее (где они приводят к увеличению октанового числа), в качестве растворителей, в качестве агентов для образования комплексов ионов металлов, и при получении катализаторов Циглера-Натта, в качестве внешних доноров. Использование предложенных диэфиров позволяет повысить выход и стереорегулярность получаемых продуктов.
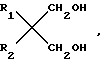
Простые диэфиры согласно изобретению имеют общую формулу 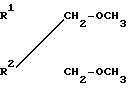 (I) в которой R1, R2 могут быть как одинаковыми, так и различными и являются разветвленными С3-С6-алкильными или С5-С6-циклоалкильными радикалами.
(I) в которой R1, R2 могут быть как одинаковыми, так и различными и являются разветвленными С3-С6-алкильными или С5-С6-циклоалкильными радикалами.
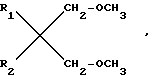
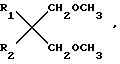
Новые простые диэфиры могут быть получены с помощью различных способов. В данном случае предлагается способ, включающий взаимодействие диолов общей формулы 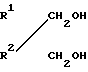 (II) где R1 и R2 имеют вышеуказанные значения, с метилгалоидом с получением целевых соединений.
(II) где R1 и R2 имеют вышеуказанные значения, с метилгалоидом с получением целевых соединений.
Далее изобретение иллюстрируется примерами.
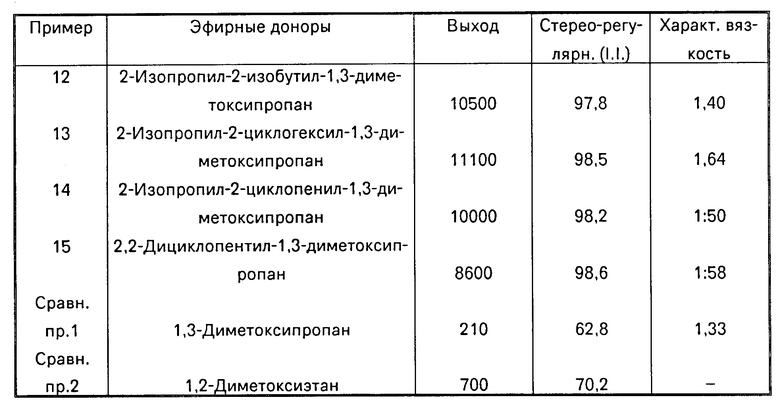
Для подтверждения полезности и преимуществ всех групп соединений формулы I, приведены примеры 12, 13, 14 и 15 и примеры сравнения 1 и 2 (табл. 1). В перечисленных примерах каталитические системы, содержащие диэфиры, подпадающие под определение настоящего изобретения, позволяют получить лучшие выход и стереорегулярность, в сравнении с известными каталитическими системами и системами из примеров для сравнения, упомянутых выше. В примерах для сравнения 1 и 2 полимеризацию проводили с диэфирами, выходящими за рамки объема настоящего изобретения (т.е. 2-диметоксиэтан и 1,3-диметоксипропан). Первый был получен от Aldrich, второй является известным соединением и получен описанным методом. Примеры 7, 8, 9 и 10 касаются синтеза диэфиров, используемых при получении каталитических систем для полимеризации.
Способ получения твердого каталитического компонента и способ полимеризации (см. пример 11) является аналогичным как в случае диэфиров согласно настоящему изобретению, так и в случае известных простых полиэфиров.
П р и м е р 1. Получение 2,2-диизобутил-1,3-диметоксипропана.
а) Получение диизобутилэтилмалоната.
В 250 мл колбу, снабженную мешалкой, холодильником, загрузочной воронкой, термометром и трубкой для подачи газов, загружают в потоке азота 100 г безводного этанола и 5 г (0,22 моль) Na. Когда растворение натрия заканчивается, подают 16 г (0,1 моль) диэтилмалоната и перемешивают при комнатной температуре в течение нескольких минут. Затем добавляют 28 г изобутилбромида (0,21 моль) и смесь подвергают дефлегмации при перемешивании в течение 6 ч. Затем добавляют 7,5 г сухого этилата натрия (0,12 моль) и 14 г изобутилбромида (0,1 моль). Перемешивание и дефлегмацию продолжают еще в течение 8 ч.
Большую часть растворителя отгоняют при пониженном давлении (50 мм рт. ст.), а остаток экстрагируют 200 мл гексана. После отгонки гексанового раствора получают 15,5 г диизобутилэтилмалоната с т. кип. 145-146оС (20 мм рт. ст. ). Этот продукт, далее, подвергали анализу на чистоту с помощью газовой хроматографии (площадь основного пика) (97,5%), что согласуется с пробой диизобутилэтилмалоната, полученного в соответствии с описанием, приведенным Бентли и Перкиным, в I. Chem. Soc., 73,61.
в) Получение 2,2-диизобутил-1,3-пропандиола.
В тот же аппарат, чтоб был описан выше в а), вводят в потоке азота 100 мл диэтилового простого эфира и 3 г LiAIH4 (0,079 моль).
Затем по каплям в течение 1 ч при одновременном энергичном перемешивании добавляют 15,5 г диизобутилэтилмалоната из а) и смесь подвергают дефлегмации в течение 30 мин.
Далее реакционную смесь выливают в сосуд, содержащий 100 г льда, подкисленного разбавленной HCl и экстрагируют 3 порциями по 100 мл этилового простого эфира.
Простой эфир выпаривают и получают 10 г сырого материала, который после кристаллизации из гексана дает 8,5 г 2,2-диизобутил-1,3-пропандиола с т. пл. 75-77оС, а элементный анализ дает С = 70,3% и Н = 12,6%.
Теоретическое значение для С11Н24О2 составляет С = 70,21% и Н = 12,7%.
с) Получение 2,2-диизобутил-1,3-диметоксипропана.
В тот же аппарат, что был описан выше в а), подают в атмосфере азота 8,5 г (0,06 моль) 2,2-диизобутил-1,3-пропандиола, 200 мл диоксана и 15,4 г (0,136 моль) трет-бутилата калия.
Смесь перемешивают при комнатной температуре в течение 30 мин, а затем по каплям добавляют 20 г СН3I (0,14 моль). Во время этой процедуры температура поднимается спонтанно до 50оС.
Спустя 2 ч добавляют дополнительное количество трет-бутилата калия (154 г, 0,136 моль) и СН3I (20 г, 0,14 моль) и смесь подвергают рефлексу в течение 1 ч. Реакционную массу фильтруют и фильтрат отгоняют при пониженном давлении. Среди других продуктов получают 7,4 г (34,3%) 2,2-диизобутил-1,3-диметоксипропана, имеющего т. кип. 100-101оС (22 мм рт.ст.), чистота которого, определенная с помощью газовой ГЖХ хроматографии, составила 99% (площадь хроматографических пиков). nD20 1,4337. Элем. анализ С 72,05%; Н 13,2%.
1Н-ЯМР (300 Мгц, CDCl3, ТМС в качестве внутреннего стандарта): сигналы в 0,89 м. д. , дублет 12Н; 1,21 м.д. дублет 4Н; 1,68 м.д., мультиплет 2Н; 3,16 м.д. синглет 4Н; 3,26 м.д., синглет 6Н.
Используя процедуры из а), в) и с) получали следующие соединения:
1) 2-Метил-2-изопропил-1,3-диметоксипропан, выход 10,1%; nD20 = 1,4209, т. кип. 160-167оС (760 мм рт.ст.), Элем. анализ: С 67,32%; Н 12,35%.
2) 2,2-Дибензил-1,3-диметоксипропан; выход 47,7% . Элем. анализ: С 79,95%; Н 8,35%. Т. точки летучести 105оС (из петролейного эфира).
3) 2,2-Диизобутил-1,3-дибутоксипропан; выход 23,5% . Элем. анализ: С 76,08%; Н 13,52; nD20 = 1,43378, т. кип. 115-117оС (1 мм рт.ст.).
4) 2,2-Диизобутил-1,3-диэтоксипропан; выход 28,3. Элем. анализ: C 73,60%; H 13,25; nD20 = 1,4302, т. кип. 118-120оС (20 мм рт.ст.).
П р и м е р 2. Получение 2,2-бис(циклогексилметил)-1,3-диметоксипропана гидрогенизацией 2,2-дибензил-1,3-диметоксипропана, полученного в примере 1.
В автоклав, изготовленный из нержавеющей стали, снабженный анкерной системой перемешивания, подают 5,8 г (0,02 моль) (С6Н5СH2)2(СН2ОСН3)2, полученного в соответствии с примером 1, 100 мл н-гексана и 10 г никеля Ренея, промытого декантированием 3 порциями по 50 см3 безводного этанола, а затем 3 порциями 50 см3 гексана.
В автоклаве создают давление 17 атм с помощью водорода и нагревают содержимое до 135оС (внутренняя температура) на 8 ч при перемешивании.
После охлаждения реакционную смесь фильтруют, отделяя катализатор, выпаривают в вакууме, чтобы получить в результате 5,9 г (97,5%) бесцветного масла с чистотой 99%; nD20 = 1,4790. Единственным обнаруженным соединением при помощи тонкослойной хроматографии (ТСХ) было 2,2-бис(циклогексил, метил)-1,3-диметоксипропан. Элем. анализ: С 76,51%; Н 12,40%.
1Н-ЯМР (300 МГц, CDCl3, ТМС в качестве внутреннего стандарта).
Были получены сигналы в:
0,96 м. д., мультиплет 4Н; 1,18 м.д., мультиплет 12Н; 1,63 м.д., мультиплет 10Н; 3,15 м.д., синглет 4Н; 3,27 м.д., синглет 6Н.
П р и м е р 3. Получение 2,2-дифенил-1,3-диметоксипропана.
а) Получение 2,2-дифенил-1,3-пропандиола.
В тот же аппарат, что был описан в примере 1(а), загружают 10,6 г (0,054 моль) (С6Н5)2СНСНО (Fluca), 4,03 г (0,028 моль) К2СО3, 10 см3 воды, 13,2 мл безводного формальдегида в концентрации 40% (0,176 моль) и 35 мл этанола с чистотой 99%.
Смесь перемешивают и подвергают рефлюксу в течение 6 ч, охлаждают и разбавляют 200 мл воды. Образованный таким образом осадок фильтруют, промывают водой и кристаллизуют из бензола, чтобы получить 9,6 г 2,2-дифенил-1,3-пропандиола с т. пл. 102-104оС.
в) Получение 2,2-дифенил-1,3-диметоксипропана.
В тот же аппарат, что описан в а), загружают 9,6 г 2,2-дифенил-1,3-пропандиола, растворенного в 400 мл безводного тетрагидрофурана, и перемешивают в атмосфере азота с 3,8 г NaH (55% NaH, диспергированный в вазелиновом масле) до тех пор, пока не прекратится выделение азота. В течение 20 мин добавляют 9,6 мл СН3I и перемешивание продолжают в течение 2 ч. Большую часть ТГФ отгоняют; затем продукт разбавляют водой (200 мл) и экстрагируют двумя порциями по 50 мл диэтилового простого эфира. Экстракт простого эфира дает в результате вакуумной отгонки 3,5 г (28,3%) 2,2-дифенил-1,3-диметоксипропана, имеющего температуру кипения в 188-190оС (20 мм рт.ст.), который был однороден согласно ТСХ-хроматографии и который имел nD20 1,5558. Элементный анализ: C 79,95%; Н 7,95%.
В соответствии с той же процедурой, что была описана выше в а) и в), получают следующие соединения, исходя соответственно из гексагидробензолальдегида и норборнан-2-карбоксальдегида.
А) 1,1-диметоксиметилциклогексан; выход 25,1%. Элем. анализ: C 70,75%; H 11,97%; т. кип. 97-98оС (22 мм рт.ст.); nD20 = =1,4487. 1Н-ЯМР (300 МГц, CDCl3, ТМС в качестве внутреннего стандарта): сигналы в:
1,36 м. д. , мультиплет 10Н; 3,20 м.д., синглет 4Н; 3,29 м.д., синглет 6Н.
В) (+)-) 2,2-Диметоксиметилнорборнан, выход 22,8%. Эл. анализ: С 71,85% ; Н 22,8% . Температура точки кипения 106-108оС (22 мм рт.ст.); nD20 = 1,4659 сигналы в:
0,72 м. д., дублет 1Н; 1,14 м.д.; дублет 1Н; 1,06 м.д., мультиплет 1Н; 1,34 м. д. , мультиплет 2Н; 1,51 м.д., мультиплет 3Н; 1,97 м.д., синглет (широкий) 1Н; 2,15 м. д. синглет (широкий) 1Н; 3,06 м.д., система АВ 1Н; 3,14 м. д. система АВ 1Н; 3,33 ч/млн. миллион, система АВ 1Н; 3,36 м.д. система АВ 1Н; 3,29 м.д. мультиплет 6Н.
П р и м е р 4. Получение 2,2,4-Триметил-1,3-димтоксипентана.
В колбу вместимостью 2 л, снабженную мешалкой, холодильником, загрузочной воронкой, термометром и трубкой для введения газов, загружают в потоке азота:
29,2 г (0,2 моль) 2,2,4-триметил-1,3-пропандиола, 600 мл диоксана и 10 г (0,2 моль) NaH 50%-ной концентрации в вазелиновом масле. Содержимое перемешивают до тех пор, пока не прекратится выделение газа; затем нагревают до 80оС и по каплям добавляют 18 мл CH3I (0,28 моль). Через 2 ч добавляют 10 г NaH 50%-ной концентрации (0,2 моль) в вазелиновом масле и 40 см3 СН3I (0,62 моль).
После дефлегмации в течение 8 ч реакционную смесь разбавляют 1,5 л воды и экстрагируют 3 порциями гексана (по 100 мл каждая).
Экстракт промывают водой и сушат, отгоняют под вакуумом, в результате чего получают 22,5 г (64,5%), 2,2,4-триметил-1,3-диметоксипентана, имеющего т. кип. 105оС/70 мм рт.ст. чистота которого (площади пиков), определяемая с помощью ГЖХ, составляла 98,6%, а nD20 = 1,4227. Элем. анализ С 69,20%; Н 12,92%.
1Н-ЯМР (60 МГц, CDCl3, ТМС в качестве внутреннего стандарта) Сигналы в:
1,5 м. д., мультиплет 12Н; 3,7 мд, мультиплет 3Н; 4,1 м.д., мультиплет 6Н.
П р и м е р 5. Получение 2-изопентил-2-изопропил-1,3-диметоксипропана.
а) Получение изопентилиденизопентаналя.
50 г изопентанала подвергают взаимодействию в соответствии с процедурами, описанными в DRP 643341 (1993, И. Г. Фарб) и DRP 544 192 (1933, И. Г. Фарб).
Получают 27 г 2-изопентилиденизопентаналя, имеющего т. пл. 98-102оС/20 мм рт.ст.
в) Получение 2-изопентилизопентаналя.
В соответствии со способом гидрогенизации, описанными Дж. В. Брауном и Г. Манцем, Ber., 1969, 67 (1934), исходя из 27 г 2-изопетилиденизоопентаналя, получают 27 г сырого материала, который не анализировали. Измерение, поглощения водорода согласовываются с описанной реакцией.
с) Получение 2-изопропил-2-изопентил-1,3-пропандиола.
В тот же аппарат, что был описан в примере 1 а), загружают 27 г сырого альдегида из предыдущей стадии в), 16 г К2СО3, 200 мл 99% чистого этанола и 52 мл безводного СН2О 40%-ной концентрации и смесь поддерживают при температуре дефлегмации при перемешивании в течение 4 ч. Затем реакционную смесь разбавляют 1 мл воды, экстрагируют двумя порциями по 250 мл простого эфира. Простой эфир выпаривают, а экстракт простого эфира сушат и отгоняют при пониженном давлении, в результате чего получают среди других соединений 9 г 2-изопропил-2-изопентил-1,3-пропандиола, имеющего температуру кипения 165оС/20 мм рт. ст. , который согласно ТСХ, был однородным. Этот материал используют в следующей реакции без какого-либо анализа.
d) Получение 2-изопропил-2-изопентил-1,3-диметоксипропана.
В аппарат из примера 1 а) в атмосфере азота загружают 9 г сырого материала со стадии с), 200 см3 диоксана и 15 г (СН3)3 СОК (трет-бутилат калия) и перемешивают в течение примерно 30 мин. Затем в течение 1 ч подают 10 мл СН3I и содержимоe подвергают рефлюксу в течение 5 ч. Реакционную смесь разбавляют 1 л воды и экстрагируют диэтиловым простым эфиром. Простой эфир выпаривают и эфирный экстракт сушат и отгоняют при пониженном давлении. Среди других соединений получают 7,3 г (14,5%) 2-изопропил-2-изопентил-1,3-диметокси- пропана, имеющего т. кип. 130-133оС/35 мм рт.ст., чистота которого (площади пиков) составляет 98% (определяют с использованием газовой хроматографии). Элем. анализ: С 72,25%; Н 13,32%. nD20 = 1,4365.
1Н-ЯМР (300 МГц, СDCl3, ТМС в качестве внутреннего стандарта), сигналы в:
0,87 м. д. дублет 6Н; 8,89 м.д., дублет 6Н; 1,11 м.д., мультиплет 2Н; 1,28 м. д. мультиплет 2Н; 1,42 м.д., мультиплет 1Н; 1,76 м.д., мультиплет 1Н; 3,23 м.д., синглет 2Н; 3,24 м.д., синглет 2Н.
П р и м е р 6 иллюстрирующий использование соединения согласно изобретению.
В колбу, вместимостью 500 мл загружают при перемешивании 60 мл н-гептана и 67 мл тетра-н-бутоксититана и нагревают до 45оС. В течение 3 ч постепенно подают раствор AlEt2Cl (44,8 мл) в н-гептане (108 мл).
Температуру поднимают до 60оС за 1 ч, а затем охлаждают до комнатной температуры.
Твердое вещество отделяют и промывают четыре раза с использованием порций гептана по 100 мл, а затем сушат под вакуумом.
В колбу загружают 8,1 г этого твердого вещества вместе с 20,3 ммоль тетрахлорида титана, 20,3 мл толуола и 20,3 ммоль 2,2-диизобутил-1,3-диметоксипропана и нагревают при 60оС 1 ч, а затем при 100оС 4 ч. Реакционную смесь затем охлаждают до комнатной температуры, твердый продукт отделяют, промывают н-гептаном до тех пор, пока не исчезнут ионы хлора в фильтрате, затем сушат в печи в атмосфере азота.
В 120 мл автоклав, снабженный магнитной мешалкой, подают после сушки в атмосфере азота 250 мг AlEt2Cl, 12,4 мл твердого вещества, полученного выше, и 80 мл жидкого пропилена, и все это нагревают до 60оС, которую поддерживают в течение 1 ч при перемешивании. Избыток непрореагировавшего пропилена выгружают и получают 16,9 г полипропилена с изотактическим индексом 96,8% (экстрагирование н-гептаном, кипящим в течение 4 ч).
П р и м е р 7. Получение 2-изопропил-2-циклогексил-1,3-диметоксипропана.
а) Получение диэтилового эфира 2-изопропил-2-циклогексилмалоновой кислоты.
В колбу емкостью 250 мл, снабженную мешалкой, холодильником, загрузочной воронкой, термометром и трубкой для введения газов, загружают 100 мл п-ксилола и 5 г (0,1 моль) гидрида натрия (55%-ная дисперсия гидрида натрия в вазелиновом масле) в токе азота.
После завершения растворения гидрида натрия вводят 20,2 г (0,1 моль) диэтилового эфира изопропилмалоновой кислоты (полученного по методу Conrad, Ann. 204, 144) и перемешивают при комнатной температуре несколько минут. Затем в течение 30 мин добавляют 40 г (0,24 моль) гексилбромида. Смесь нагревают с обратным холодильником при перемешивании в течение 8 ч.
После охлаждения раствор нейтрализуют, используя 10%-ную водную серную кислоту.
Дистиллированный раствор п-ксилола дает 10,5 г диэтилового эфира 2-изопропил-2-циклогексилмалоновой кислоты, имеющей т. кип. 120-125оС/1 мм рт. ст.
в) Получение 2-изопропил-2-циклогексилпропандиола.
В тот же аппарат, как описано в пункте а) выше, вводят 10,5 г диэтилового эфира 2-изопропил-2-циклогексилмалоновой кислоты, растворенного в диэтиловом эфире, и 3 г (0,08 моль) алюмогидрата лития в потоке азота. После добавления некоторого количества воды выпаренная органическая фаза дает 7,5 г неочищенного материала.
с) Получение 2-изопропил-2-циклогексил-1,3-диметоксипропана.
В тот же аппарат, описанный в пункте а), вводят неочищенный материал, полученный в пункте в), 200 мл диоксана и затем при перемешивании 28,2 г (0,21 моль) йодистого метила. Через 2 ч добавляют дополнительное количество трет-бутилата калия (20 г, 0,17 моль) и затем йодистый метил (28,2 мг, 0,2 моль).
Реакционную массу фильтруют и фильтрат дистиллируют при пониженном давлении. Среди других продуктов получают 3 г 2-изопропил-2-циклогексил-1,3-диметокси- пропана, имеющего т. кип. 150оС/15 мм рт.ст., который согласно газовой тонкослойной хроматографии однороден. Данные элементного анализа следующие:
C 73,51% (вычислено 73,62%)
Н 12,52% (вычислено 12,35%)
П р и м е р 8. Получение 2,3-дициклопентил-1,3-диметоксипропана.
а) Получение диэтилового эфира 2-циклопентилмалоновой кислоты.
В тот же аппарат, что описан в примере 7, пункт а), вводят 240 г (1,5 моль) диэтилового эфира малоновой кислоты и 1000 мл безводного этанола и затем при перемешивании добавляют 36 г (1,44 моль) натрия. После завершения растворения натрия вводят 250 г бромистого циклопентила и смесь нагревают с обратным холодильником 4 ч.
Этанол испаряют и добавляют некоторое количество воды. Продукт реакции экстрагируют петролейным эфиром. Затем петролейный эфир испаряют и отгоняют неочищенный материал. Получают 270 г диэтилового эфира циклопентилмалоновой кислоты, имеющего точку кипения 120оС/1,0 мм рт.ст.
в) Получение диэтилового эфира 2,2-дициклопентилмалоновой кислоты.
В тот же аппарат, как описано в примере 7, пункт а), вводят 228 г диэтилового эфира циклопентилмалоновой кислоты, полученного в пункте а) выше, и 1000 мл безводного ксилола. В течение 30 мин добавляют 35 г гидрида натрия (80% -ная дисперсия гидрида натрия в вазелиновом масле) при перемешивании и поддержании температуры при 80-90оС. Затем температуру повышают до 135оС и добавляют 180 г бромистого циклопентила. Смесь непрерывно перемешивают 3 ч. Затем последовательно добавляют дополнительное количество 10 г гидрида натрия и бромистого циклопентила (60 г, 0,4 моль). После охлаждения вводят некоторое количество воды, затем органическую фазу отделяют, промывают водой, в завершение сушат и получают 300 г продукта.
с) Получение 2,2-дициклопентил-1,3-пропандиола.
В колбу, вместимостью 5 л, снабженную мешалкой, холодильником и термометром, вводят 45 г (1,2 моль) алюмогидрата лития и 2000 мл безводного этилового эфира. В течение 1 ч (по каплям вводят продукт, полученный в пункте а) выше, растворенный в 300 мл этилового эфира. После охлаждения раствор нейтрализуют, используя хлористоводородную кислоту 20%-ной концентрации в воде. Реакционную смесь отделяют от эфирной фазы, сушат, затем разбавляют 500 мл петролейного эфира. Раствор охлаждают. Образовавшийся при этом осадок отфильтровывают, промывают петролейным эфиром и сушат в вакууме. Получают 150 г 2,2-дициклопентил-1,3-пропандиола.
d) Получение 2,2-дициклопентил-1,3-димeтоксипропана.
В колбу вместимостью 2 л, оборудованную мешалкой, холодильником, термометром и трубопроводом для подведения газов, загружают 133 г 2,2-дициклопентил-1,3-пропандиола, полученного в пункте с) выше, 500 мл диметилсульфоксида и 60 г гидроксида натрия, измельченной.
Температуру повышают до 35оС и при перемешивании барботируют хлористый метил. Через 2 ч добавляют дополнительное количество 30 г (0,75 моль) гидроксида натрия. Снова в течение 3 ч барботируют хлористый метил. Затем раствор фильтруют и органическую фазу экстрагируют петролейным эфиром. Экстракт промывают водой, 2%-ным водным раствором перманганата калия, сушат, отгоняют в вакууме и получают 120 г 2,2-дициклопентил-1,3-диметоксипропана, имеющего т. кип. 150-152оС/15 мм рт.ст., чистота которого была 99% согласно тонкослойной газовой хроматографии.
С 74,8% (вычислено 74,95%)
Н 11,8% (вычислено, 11,74%)
П р и м е р 9. Получение 2-изопропил-2-циклопентил-1,3-диметоксипропана.
а) Получение диэтилового эфира 2-изопропил-2-циклопентилмалоновой кислоты.
В тот же аппарат, что описан в примере 7, пункт а), загружают 20,2 г (0,1 моль) диэтилового эфира 2-изопропилмалоновой кислоты, 100 мл п-ксилола и 5 г (0,1 моль) гидрида натрия в виде 50%-ной дисперсии в вазелиновом масле и далее добавляют 16 г (0,1 моль) бромистого циклопентила.
Аналогичным образом, описанным в примере 7, пункт а), получают 15 г диэтилового эфира 2-изопропил-2-циклопентилмалоновой кислоты с т. кип. 115-120оС/1 мм рт.ст.
в) Получение диэтилового эфира 2-изопропил-2-циклопентилпропандиола.
В такой же аппарат, как описано в примере 7, пункт а), загружают диэтиловый эфир 2-изопропил-2-циклопентилмалоновой кислоты, полученного в пункте а), выше, и 8 г (0,21 моль) алюмогидрида лития.
Методом, аналогичным описанному в примере 7, пункт в), получают 2-изопропил-2-циклопентил-1,3-диметоксипропандиол.
с) Получение 2-изопропил-2-циклопентил-1,3-диметоксипропана.
В тот же аппарат, описанный в примере 7, пункт а), к реакционному продукту, полученному в пункте в), добавляют трет-бутилат калия (20 г, 0,17 моль) и затем 50 г иодистого метила. Способом, аналогичным описанному в примере 7, пункт с), получают 5,4 г 2-изопропил-2-циклопентил-1,3-диметоксипропана с т. кип. 125-130оС/15 мм рт.ст., который согласно тонкослойной газовой хроматографии был однородный.
Элементный анализ был следующий:
С 72,65% (вычислено 72,84%).
Н 12,3% (вычислено 12,2%).
П р и м е р 10. Получение 2-изопропил-2-изобутил-1,3-диметоксипропана.
а) Получение диэтилового эфира 2-изопропил-2-изобутилмалоновой кислоты.
В такой же аппарат, как описано в примере 7, пункта а), загружают 20,2 г (0,1 моль) диэтилового эфира 2-изопропилмалоновой кислоты, 100 мл толуола и 2,5 г (0,1 моль) гидрида натрия (80%-ная дисперсия гидрида натрия в вазелиновом масле) и затем добавляют 14 г (0,13 моль) бромистого изобутила.
Способом, аналогичным описанному в примере 7, пункт а), получают 15 г диэтилового эфира 2-изопропил-2-изобутил-малоновой кислоты с т. кип. 112-155оС/1 мм рт.ст.
в) Получение 2-изопропил-2-изобутилпропандиола.
В такой ж аппарат, как описано в примере 7, пункт а), загружают диэтиловый эфир 2-изопропил-2-изобутилмалоновой кислоты, полученный в пункте а), и 10 г (0,27 моль) алюмогидрата лития.
Методом, аналогичным описанному в примере 7, пункт в), получают 2-изопропил-2-изобутилпропандиол.
с) Получение 2-изопропил-2-изобутил-1,3-диметоксипропана.
В такой же аппарат, как описано в примере 7, пункт а), к продукту реакции, полученному в пункте в), добавляют трет-бутилат калия (30 г, 0,25 моль) и затем 50 г иодистого метила в диоксане. Способом, аналогичным описанному в примере 7, пункт с), получают 10 г 2-изопропил-2-изобутил-1,3-диметоксипропана с т. кип. 120-125оС/1 мм рт.ст., который согласно тонкослойной газовой хроматографии был однородным.
Элементный анализ был следующим:
С 71,1% (вычислено 71,23%)
Н 13,0% (вычислено 12,95%)
П р и м е р 11. Получение твердого компонента катализатора.
В реактор емкостью 500 мл, снабженный фильтрующий пластиной на днище, вводят 225 мл четыреххлористого титана при 0оС перемешивают в течение 15 мин и добавляют 10,1 г (54 ммоль). MgCl2 ˙2C2H5OH в форме микросфер, полученного по примеру 1.
По завершении добавления температуру повышают до 40оС и вводят 9 ммоль диизобутилфталата. Затем температуру повышают до 100оС в течение 1 ч и смесь оставляют для продолжения реакции на 2 ч. Избыток четыреххлористого титана удаляют фильтрацией. Затем добавляют 200 мл четыреххлористого титана и содержимое реактора нагревают при 120оС в течение 1 ч при перемешивании. Смесь фильтруют и твердое вещество промывают н-гептаном при 60оС до тех пор, пока ионы хлора не обнаруживаются более в фильтрате.
Полимеризация
В 2000 мл автоклав из нержавеющей стали, снабженный стационарной мешалкой, вводят при 25оС в потоке пропилена 100 мл н-гептана, 5 ммоль этилата алюминия, 30 мг каталитического компонента и 1 ммоль эфирного соединения, представленного в таблице.
Автоклав закрывают. После установления давления в 1 атм вводят 0,2 атм водорода и содержимо автоклава нагревают при 70оС, подавая пропилен до создания общего давления 7 атм.
Полимеризацию проводят 2 ч. В течение этого времени поступление мономера было непрерывным. Полимер выделяют фильтрацией в конце реакционного периода и сушат в вакууме. Остающуюся часть полимера в фильтрате выделяют осаждением метанолом, сушат в вакууме и принимают в расчет при определении общего выхода с остатком, экстрагируемым н-гептаном.
Использованные эфирные доноры, результаты полимеризации (выход и общий показатель стереорегулярности II) и характеристическая вязкость полученного полимера представлены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ КАТАЛИЗАТОРА ПОЛИМЕРИЗАЦИИ | 1991 |
|
RU2050365C1 |
| КОМПОНЕНТ КАТАЛИЗАТОРА ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1990 |
|
RU2032689C1 |
| Твердый каталитический компонент для полимеризации олефинов и катализатор полимеризации олефинов | 1989 |
|
SU1836384A3 |
| КОМПОНЕНТ КАТАЛИЗАТОРА ПОЛИМЕРИЗАЦИИ АЛЬФА-ОЛЕФИНОВ | 1992 |
|
RU2073688C1 |
| КОМПОНЕНТ КАТАЛИЗАТОРА (СО)ПОЛИМЕРИЗАЦИИ АЛЬФА-ОЛЕФИНОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ | 1990 |
|
RU2045537C1 |
| СПОСОБ ПОЛУЧЕНИЯ СТАБИЛИЗИРОВАННОГО ПОЛИПРОПИЛЕНА | 1992 |
|
RU2070206C1 |
| ЭЛЕКТРОНОДОНОРНЫЕ КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ И КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ α -ОЛЕФИНОВ | 1990 |
|
RU2039062C1 |
| ТВЕРДЫЙ КАТАЛИТИЧЕСКИЙ КОМПОНЕНТ ДЛЯ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ, СОДЕРЖАЩИЙ ЕГО КАТАЛИЗАТОР И ЕГО ПРИМЕНЕНИЕ | 2003 |
|
RU2298014C2 |
| Диэтил 2,2-диалкилмалонат и 2,2-диалкил-1,3-диметоксипропан, содержащие 6,6-диметилбицикло[3.1.1]гептановый фрагмент природного происхождения, способ их получения и титан-магниевый катализатор полимеризации пропилена, содержащий эти соединения в своем составе | 2023 |
|
RU2819723C1 |
| КОМПОНЕНТ КАТАЛИЗАТОРА, ПРЕДНАЗНАЧЕННОГО ДЛЯ ПОЛИМЕРИЗАЦИИ ПРОПЕНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СОДЕРЖАЩИЙ ЕГО КАТАЛИЗАТОР | 2015 |
|
RU2690192C2 |
Описываются простые диэфиры общей формулы: 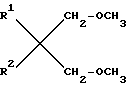 , где R1 и R2 одинаковы или различны и являются разветвленными C1-C6-алкилами или C5-C6-циклоалкилами. Эти простые диэфиры можно, в частности, использовать при получении катализаторов Циглера-Натта.Реагент 1 : диол формулы
, где R1 и R2 одинаковы или различны и являются разветвленными C1-C6-алкилами или C5-C6-циклоалкилами. Эти простые диэфиры можно, в частности, использовать при получении катализаторов Циглера-Натта.Реагент 1 : диол формулы 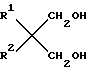 где R1 и R2 имеют выше указанные значения, подвергают метилированию галоидметилом.2 с.п. ф-лы, 1 табл.
где R1 и R2 имеют выше указанные значения, подвергают метилированию галоидметилом.2 с.п. ф-лы, 1 табл.
ПРОСТЫЕ ДИЭФИРЫ И СПОСОБ ИХ ПОЛУЧЕНИЯ.
| J | |||
| Chem | |||
| Пожарный двухцилиндровый насос | 0 |
|
SU90A1 |
| Прибор для сортирования металлических | 1925 |
|
SU1932A1 |

