Изобретение относится к способу получения компонентов катализаторов процесса /со/сополимеризации олефинов типа СН2=СНR, где R это водород или алкильный радикал с 1-6 атомами углерода, или арильный радикал, а также к получаемым из них катализаторам.
Известны катализаторы полимеризации олефинов, полученные импрегнированием оксида металла, содержащего поверхностные группы гидроксила, например оксида кремния или алюминия, металл-органическим соединением, предпочтительно триалкилиндием или диалкилмагнием, которые вводятся в молярном избытке по отношению к поверхности гидроксильным группам, и последующим взаимодействием носителя с тетрахлоридом титана (GВ-А-1,256,851 и 1,306,044). Эти катализаторы подходят для полимеризации этилена, где, однако, они не обеспечивают достаточно высоких выходов (300-500 г полимера) г компонента катализатора в час при рабочем давлении этилена 10 атм).
При модифицировании катализаторов с помощью электронно-донорного соединения с целью придания им стереоизбирательности, что позволило бы их использовать для стереорегуляторной полимеризации пропилена или других альфа-олефинов, можно ожидать значительного снижения их активности или, как показано в случае полимеризации этилена, не слишком высокой.
Известны компоненты катализаторов процесса полимеризации пропилена и других альфа-олефинов, получаемые взаимодействием оксида металла, содержащего поверхностные гидроксильные группы (кремнезем, глинозем и др.), с металл-органическим соединением магния формулы Mg R(2-х)Хх (где R радикал гидрокарбил; Х это галоген; х число от 0,5 до 1,5), после чего оксид взаимодействует сначала с электронно-донорным соединением, а затем с тетрахлоридом титана. Металлорганическое соединение магния реагирует в молярном избытке по отношению к гидроксильным группам, тогда как электронно-донорное соединение используется в количествах до 1 моль на моль прореагировавшего соединения магния, предпочтительно в количестве 0,5-0,8 моль. Реакция с ТiСl4 проводится с избытком его.
В качестве варианта предусматривается, что оксид металла или до, или после реакции c металлорганическим соединением магния подвергается воздействию галогенирующего агента, который берется в количестве, достаточном для введения не менее одного атома галогена на каждую гидроксильную группу. Галогенирующий агент можно также вводить во время реакции с электронно-донорным соединением. Активность и стереоизбирательность таких катализаторов не настолько высока, чтобы обеспечить им привлекательность для применения в промышленном масштабе.
Катализаторы на основе галидов магния, иммобилизованных на оксидах металлов, обладающие высокой активностью в сочетании с хорошей стереоспецифичностью, не только снижают содержание нежелательных галогенированных соединений, которые остаются в полимере, но также позволяют контролировать, причем относительно простым способом, морфологию полимера. В современных промышленных способах производства полиолефинов необходимы катализаторы, обеспечивающие получение полимера с контролируемыми морфологическими характеристиками (малый разброс размеров частиц и достаточно высокая насыпная масса).
В настоящее время получены компоненты катализаторов процесса полимеризации олефинов типа СН2=СНR, где R это алкильный радикал с 1-6 атомами углерода или арил, или водород, обладающие очень высокой активностью и стереоспецифичностью. Указанные компоненты катализаторов представляют собой продукт взаимодействия галида или галогеналкоголята четырехвалентного титана и электронно-донорного соединения с твердым продуктом, полученным реакцией оксида металла, в структуре которого имеются поверхностные гидроксильные группы, предпочтительно, наряду с химически не связанной водой, с металлоорганическим соединением магния формулы:
Mg R2-хХ, где R это радикал гидрокарбил с 1-20 атомами углерода, Х галоген или группа ОR либо СОХ', где Х' это галоген, Х число в интервале от 0,5 до 1,5, которое используется в таких условиях и в таком количестве, которые не могут вызвать восстановления титана в ходе последующей реакции с соединением четырехвалентного титана.
Количество металлоорганического соединения магния, не вызывающее в условиях проведения реакции, которые будут указаны ниже, восстановления соединения четырехвалентного титана, представляет собой стехиометрическое количество (1 моль) по отношению к гидроксилу или к 1 моль воды либо выше (до примерно 2 моль соединения магния, когда в качестве соединений магния используются метилмагнийхлорид или бромид, растворенные в диэтиловом эфире или тетрагидрофуране; оно может быть значительно выше и достигать 10 моль на моль ОН-групп или воды в случае использования таких соединений, как бутил-, изоамил-, н-октилмагнийхлорид или бромид).
Выражение "не вызывает восстановления титана" означает, что не менее 80% титана в твердом продукте реакции с тетрахлоридом титана и электронно-донорным соединением находится в четырехвалентном состоянии.
Проведение процесса в условиях, позволяющих восстановление титана, то есть, например, при избытке металлоорганического соединения магния, значительно снижает активность и стереоспецифичность катализатора. Катализаторы изобретения обладают высокой активностью и стереоспецифичностью даже в тех случаях, когда оксиды металлов содержат не связанную химически воду. Действительно, все способы, применяемые до настоящего времени для получения катализаторов процесса полимеризации олефинов, где используются указанные оксиды, жестко требуют удаления из них воды. Особенностью катализаторов изобретения является также высокая их активность при полимеризации пропилена и подобных ему олефинов, но не этилена. К оксидам металлов, которые могут быть использованы при получении компонентов катализаторов, относятся кремнезем, глинозем, оксид и силикат магния, оксид титана, оксид тория, смешанные оксиды кремния-алюминия. Предпочтение отдается оксидам алюминия, кремния и смешанным оксидам кремния-алюминия. Как уже указывалось, эти оксиды содержат поверхностные гидроксильные группы, их количество может колебаться от 1 до 3 ммоль и более на 1 г оксида. Предпочтительным представляется также присутствие, кроме гидроксильных групп, химически не связанной воды в количестве до 0,015 моль на 1 г оксида. Как правило, оксиды обладают площадью поверхности (по БЕТ-методу) выше, чем 30 м2/г, в частности, в интервале от 100 до 500 м2/г, и пористостью (измеренной по азоту) от 0,5 до 3,5 см3/г. Не вступившую в реакцию воду можно удалить, выдерживая оксиды при температуре в интервале от 150 до 250оС. Тепловая выдержка оксидов при температуре от 150 до 800о позволяет регулировать содержание ОН-групп. Чем выше температура, тем ниже содержание гидроксильных групп. Химически не связанную воду можно ввести различными способами; один из предпочтительных способов заключается в том, что над оксидом, предварительно подвергнутым обезвоживанию или нет, пропускают ток влажного азота. Содержание ОН-групп предпочтительно составляет от 1 до 3 моль на 1 г оксида; в случае присутствия воды, ее количество колеблется предпочтительно от 1 до 10 моль на 1 г оксида. Определение ОН-групп осуществляется титрованием в соответствии с описанным в J. Phys, Chem. 66, 800 (1962) способом, вода определяется по Фишеру.
Металлоорганическое соединение магния может использоваться без комплексообразователя или в виде комплекса с электронно-донорными соединениями, такими, как простые эфиры, в частности, с диэтиловым эфиром и тетрагидрофураном. Как правило, комплексообразователь вводится в количестве от 0,5 до 3 моль, предпочтительно от 0,5 до 1 моль на 1 моль соединения магния. Примерами металлоорганических соединений магния могут служить: хлорид метилмагния, бромид метилмагния, хлорид н-бутилмагния, хлорид изобутилмагния, хлорид изоамилмагния, хлорид н-октилмагния, бромид n-пропилмагния, этоксид н-бутилмагния, метоксид этилмагния. Реакцию между оксидом металла и металлоорганическим соединением магния проводят при температуре от 0 до 100оС в инертной углеводородной атмосфере. После окончания реакции в предпочтительном исполнении изобретения твердые отделяют и промывают гексаном, гептаном или другим аналогичным углеводородом, допустимо также использовать суспензию, не отделяя твердые.
Предпочтительной методикой проведения реакции является добавление по каплям раствора соединения магния к суспензии оксида металла в гексане, гептане и аналогичных углеводородах.
После обработки металлоорганическим соединением магния металлический оксид подвергают взаимодействию с соединением четырехвалентного титана и электронно-донорным соединением. Предпочтительно, чтобы в качестве соединения титана использовался тетрахлорид титана, а при проведении реакции в качестве реакционной среды использовался также сам тетрахлорид титана. Рабочая температура реакции колеблется от 40 до 135оС, время реакции от 0,25 до 1 ч и более. После завершения реакции в горячем состоянии отделяют избыток ТiCl4 и промывают твердый продукт углеводородом (гексаном) до исчезновения ионов хлора. Рекомендуется повторить еще раз обработку ТiCl4, промывая твердый продукт описанным выше способом.
Взаимодействие с электронно-донорным соединением проводится одновременно с реакцией с соединением титана. При использовании ТiCl4 электронно-донорное соединение растворяют в его избытке, и раствор вводят в реакцию с оксидом металла. Количество электронно-донорного соединения колеблется в пределах от 0,1 до 1,5 моль на 1 г ˙ ат Мg, предпочтительно от 0,2 до 0,4 моль.
Реакцию с электронно-донорным соединением можно проводить не одновременно со взаимодействием с соединением титана, а до и после него. В случае, если она проводится позже, реакционной средой должен служить ароматический углеводород, например бензол или толуол, а электронно-донорное соединение должно использоваться в эквимолярных количествах по отношению к соединению титана, иммобилизованному на оксиде металла. Наилучшие результаты получены при проведении взаимодействия с электронно-донорным соединением одновременно с соединением титана или до него.
Для приготовления компонентов катализаторов можно использовать любое электронно-донорное соединение, обладающее способностью образовывать комплексы с галидами магния и/или галидами четырехвалентного титана. В качестве примеров соединений, применение которых возможно при осуществлении изобретения, можно привести простые и сложные эфиры, кетоны, лактоны, а также соединения, в состав которых входят атомы N, R и/или S. Предпочтение отдается эфирам дикарбоновых ароматических кислот, таких как фталовая кислота, а также эфирам малоновой, триметилуксусной, янтарной и угольной кислот. Наиболее подходящими являются сложные эфиры следующей формулы:
RO 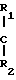 - CH2 OR где R, R1 и R2 одинаковые или разные, линейные или разветвленные алкильные группы С1-18, циклоалкильные группы C3-18, или арильные группы С6-18, или алкиларилы, либо арилалкилы С7-18, и где R1 или R2 могут также представлять собой водород. В частном случае, R может быть метилом, а R1 и R2 мoгут представлять собой одинаковые или разные радикалы из ряда этил, пропил, изопропил, бутил, изобутил, третичный бутил, неопентил, изопентил, фенил, бензил или циклогексил. Особыми сложными эфирами являются диизобутил, диоктил и дифенилфталат, бензил-бутилфталат, диизобутил- и диэтилмалонат, этиловый эфир триметилуксусной кислоты, этилфенилкарбонат, дифенилкарбонат. Представителями простых эфиров являются 2,2-диизобутил-1,3-диметоксипропан, 2-изопропил-2-изопентил-1,3-диметоксипро- пан, 2,2-bis (циклогексилметил)-1,3-диметоксипропан, 2,2-bis (циклогексил)-1,3-диметоксипропан.
- CH2 OR где R, R1 и R2 одинаковые или разные, линейные или разветвленные алкильные группы С1-18, циклоалкильные группы C3-18, или арильные группы С6-18, или алкиларилы, либо арилалкилы С7-18, и где R1 или R2 могут также представлять собой водород. В частном случае, R может быть метилом, а R1 и R2 мoгут представлять собой одинаковые или разные радикалы из ряда этил, пропил, изопропил, бутил, изобутил, третичный бутил, неопентил, изопентил, фенил, бензил или циклогексил. Особыми сложными эфирами являются диизобутил, диоктил и дифенилфталат, бензил-бутилфталат, диизобутил- и диэтилмалонат, этиловый эфир триметилуксусной кислоты, этилфенилкарбонат, дифенилкарбонат. Представителями простых эфиров являются 2,2-диизобутил-1,3-диметоксипропан, 2-изопропил-2-изопентил-1,3-диметоксипро- пан, 2,2-bis (циклогексилметил)-1,3-диметоксипропан, 2,2-bis (циклогексил)-1,3-диметоксипропан.
Содержание магния в оксидах (после обработки соединением магния) колеблется от 0,5 до 20 мас. в компонентах катализатора отношение Mg/Тi составляет от 0,5 дои 8.
Электронно-донорное соединение вводят, как правило, в количестве от 5 до 20% в частности от 10 до 15% на 1 г ˙ ат магния. Общее содержание галида магния, галида или галоген-алкоголята титана и электронно-донорного соединения в компоненте катализатора колеблется от 5 до 60 мас.
Удельная поверхность (по оценке БЕТ-методом) компонента катализатора принимает, как правило, значения выше 100 м2/г; в частном случае колеблется в пределах от 100 до 300 м2/г.
Компоненты катализаторов образуют, в сочетании с соединениями алкилалюминия, предпочтительно триалкилалюминия, катализаторы, применяемые для полимеризации олефинов типа СН2=CНR, где R это водород, или алкид с 1-6 атомами углерода, или арил, а также смесей олефинов, в которых присутствуют, но не обязательно, наибольшие количества диенов. В качестве соединений триалкилалюминия можно назвать триэтил-Al, триизобутил-Al, три-n-бутил-Al, а также линейные или циклические соединения, в составе которых имеется два или более атомов Al, связанных мостиковой связью с атомами О или N либо с группами So4 и SO3. Могут быть также использованы соединения типа AlR2OR', где R' представляет собой радикал арил, замещенный в положении 2 и/или 6, а R радикал, в составе которого от 1 до 6 атомов углерода, а также соединения типа AlR2H. Соединения алкилалюминия вводятся при соотношении Al/Тi в общем случае от 1 до 1000. Во многих случаях рекомендуется, с целью повышения стереоспецифичности катализатора, использовать, одновременно с соединением алкилалюминия, электронно-донорное соединение в количестве 0,01-0,25 моль на 1 моль соединения алкил-Al.
Электронно-донорное соединение выбирают из простых или сложных эфиров или соединений кремния, в структуре которых имеется хотя бы одна связь SiOR (R радикал гидрокарбил), а также может быть использован 2,2,6,6-тетраметилпиперидин. В случаях, когда компонент катализатора содержит эфир ароматической дикарбоновой кислоты, например фталовой, или эфир малоновой, малеиновой, триметилуксусной, янтарной или угольной кислоты, то электронно-донорское соединение, которое предполагается использовать совместно с соединением алкилалюминия, выбирают предпочтительно среди соединений кремния, в структуре которых имеется хотя бы одна связь SiOR. Примерами указанных соединений могут служить фенилтриэтоксисилан, дифенилдиметоксисилан, дициклопентилдиметоксисилан, метил-трет-бутилдиметоксисилан, метилциклогексилдиметоксисилан. При наличии в составе компонента катализатора простого эфира его стереоспецифичность значительно возрастает, так что отпадает необходимость во введении совместно с соединением алкилалюминия электронно-донорного соединения.
Согласно известным способам, полимеризация олефинов осуществляется либо в жидкой фазе, а именно в среде жидкого мономера или в растворе мономера в инертном углеводородном растворителе, либо в газовой фазе, либо представляет собой комбинацию стадий, протекающих в жидкой и газовой фазах. Температура полимеризации колеблется от 0 до 150оС, предпочтительно от 60 до 100оС, давление от атмосферного до избыточного.
Предлагаемые катализаторы применяются и при гомополимеризации, и при сополимеризации олефинов. Что касается сополимеризации, то их можно использовать для получения, например, нерегулярно-кристаллических сополимеров пропилена с меньшими количествами этилена и, возможно, бутена и других более высокоатомных альфа-олефинов или эластомеров-сополимеров этилена с пропиленом, включающих, но не обязательно, малые количества диена (бутадиена, гексадиена-1,4).
Предлагаемые катализаторы могут также использоваться в процессах последовательной полимеризации пропилена и смесей пропилена и этилена и/или бутена и аналогичных более высокоатомных альфа-олефинов с целью получения полипропилена требуемой ударной вязкости. Обнаружено, что катализаторы, полученные из компонентов, в состав которых входят известные простые эфиры, особенно подходят для получения аморфных сополимеров этилена с пропиленом, содержащих, но не обязательно, небольшие количества диена, это представляет отдельный аспект предлагаемого изобретения.
Катализаторы можно предварительно, до начала полимеризации, привести в контакт с небольшим количеством олефинов (предварительная полимеризация), что осуществляется либо в суспензии в углеводородном растворителе (гексан, гептан и т. д.), проводя затем полимеризацию при температуре от комнатной до 60оС, что дает количество полимера, в 0,5-3 раза превышающее вес твердого компонента катализатора, либо в жидком мономере, что дает до 1000 г полимера на 1 г твердого компонента.
П р и м е р 1. Синтез компонента катализатора.
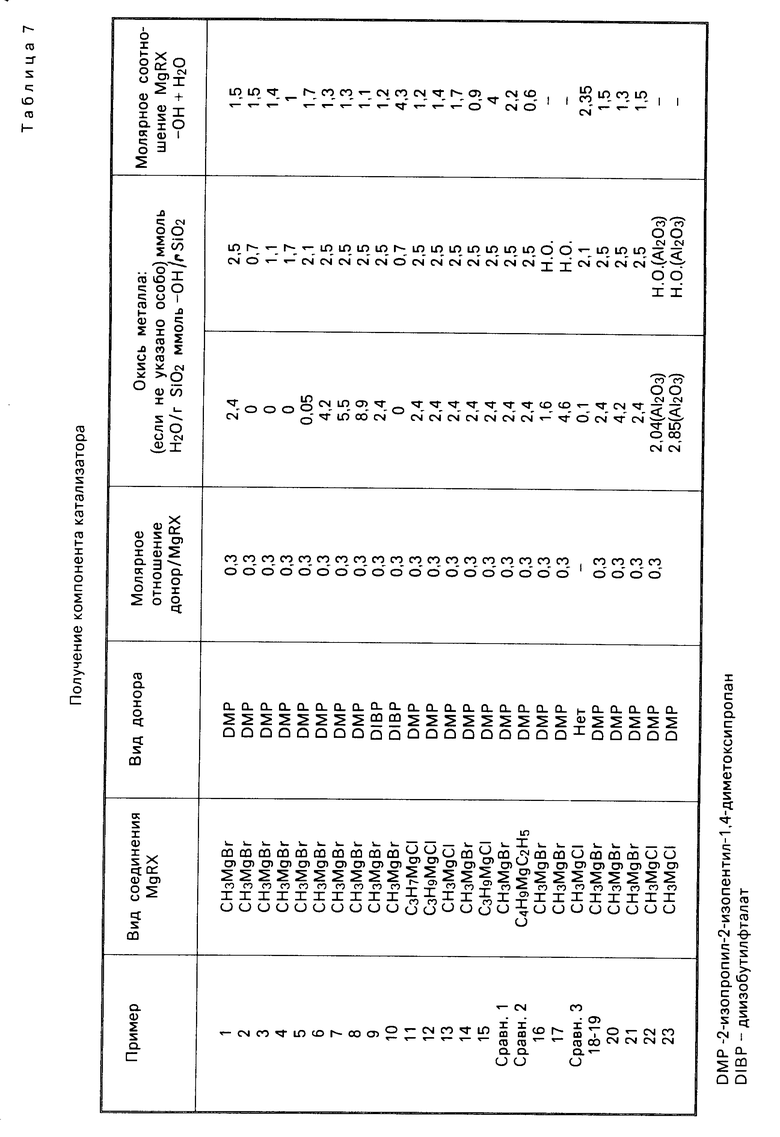
В реактор емкостью 0,350 л, снабженный фильтрующей перегородкой и приспособлением для выгрузки со дна, поместили 5 г оксида кремния марки Grace Davison 952, обладающего следующими характеристиками: удельная поверхность 290 м2/г, пористость (измеренная по азоту) 1,53 см3/г, содержание Н2О по Фишеру 4,3% а также 40 мл гексана. При непрерывном перемешивании суспензии ввели по каплям 12 мл 3 М раствора МеМg Вr в этиловом эфире (в течение около 40 мин). Затем суспензию в течение 1 ч нагревали с обратным холодильником. Далее охладили до комнатной температуры и отфильтровали, после чего твердый продукт промыли 5 раз 120 мл гексана. Опять отфильтровывали твердые и высушили в токе азота в течение 1,5 ч при 70оС. Состав полученного таким образом твердого продукта приведен в табл. 1. 5 г твердого компонента катализатора поместили в тот же самый ранее использованный реактор. Ввели 200 мл ТiСl4 при комнатной температуре и, при непрерывном перемешивании, быстро подняли температуру до 70оС, после чего ввели 2-изопропил-2-изопентил-1,3-диметоксиропан (ДМП) в молярном соотношении 1:3 по отношению к содержанию Мg в компоненте катализатора. Температуру подняли до 100оС и продолжали нагрев в течение 2 ч. Затем отфильтровывали, добавили 200 мл ТiСl4 и повторили обработку при 100оС в течение 2 ч. И последнее: после удаления ТiСl4 фильтрованием промыли твердый продукт гексаном дважды при 60оС и три раза при комнатной температуре, затем высушили, как было описано. Состав компонента катализатора приведен в табл. 1.
Полимеризация пропилена.
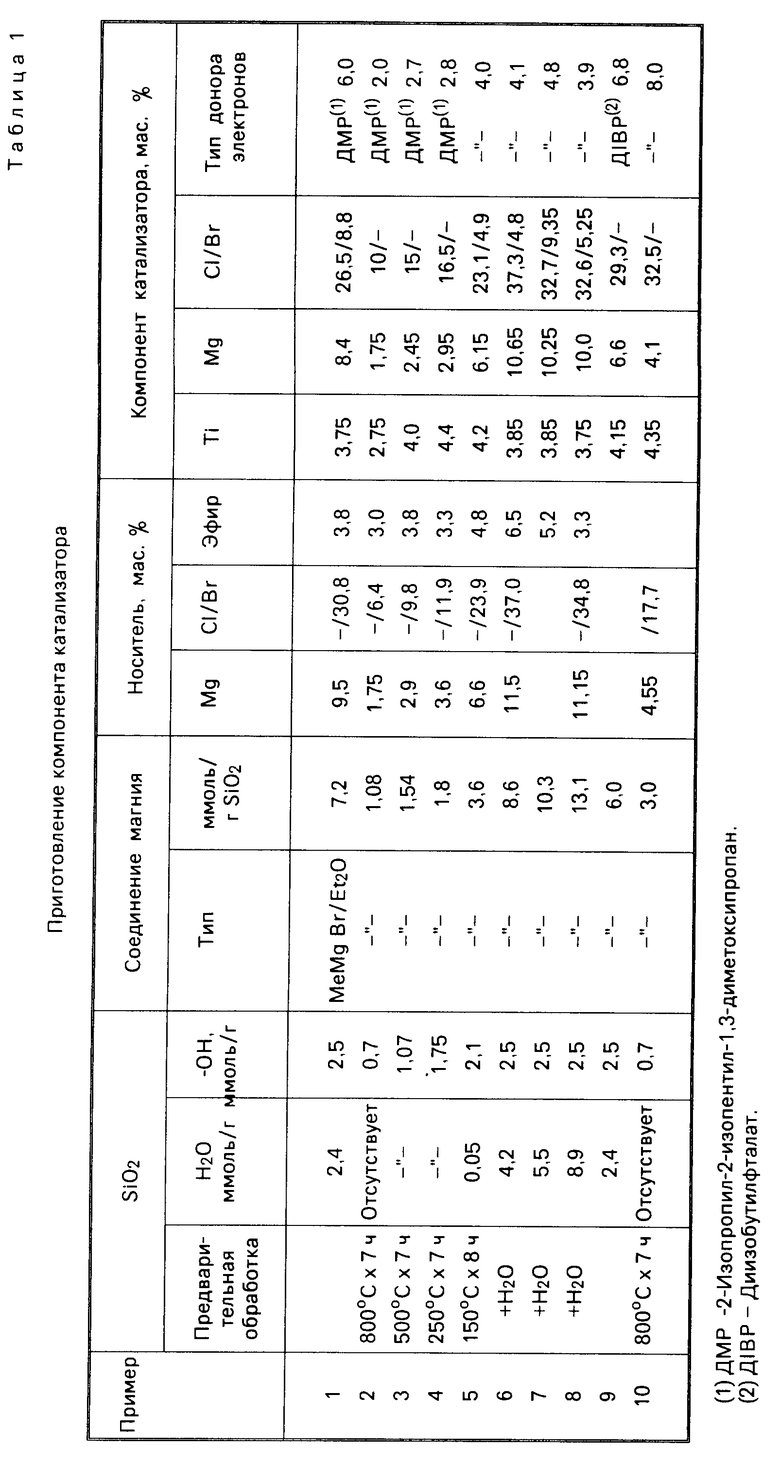
В автоклав емкостью 4 л, снабженный системами термостатирования и перемешивания, ввели при 30оС в легком токе пропилена 75 мл гексана, в котором содержалось 7 ммоль триэтил-Al, и компонент катализатора, подвергнутый в течение примерно 5 мин предварительной полимеризации в указанном в табл. 2 количестве. Автоклав закрыли и ввели 1,6 нл водорода. Включили перемешивающее устройство и ввели 1,2 кг жидкого пропилена, после чего температуру подняли до 70оС. Автоклав выдерживали в указанных условиях в течение 2 ч, затем перемешивание прекратили и быстро удаляли непрореагировавший пропилен. Далее охладили автоклав до комнатной температуры, извлекли полимер и сушили его в печи при 70оС в течение 3 ч в токе N2. Выход продукта определяли как 1 кг полимера/г компонента катализатора. Изотактический показатель определяли как процент полимера, не растворимый в кcилоле при 25оС. Показатель плавления и насыпную массу определяли соответственно по методу ASТМ Д-1238, L, и D 1895. Результаты полимеризации приводятся в табл. 2.
П р и м е р ы 2-5. Процесс проводили в соответствии с примером 1, с той же разницей, что кремнезем предварительно прокаливали в течение 7 ч в токе безводного N2 при температуре 800, 500, 250 и 150оС соответственно. Состав компонента катализатора приведен в табл. 1, результаты полимеризации в табл. 2.
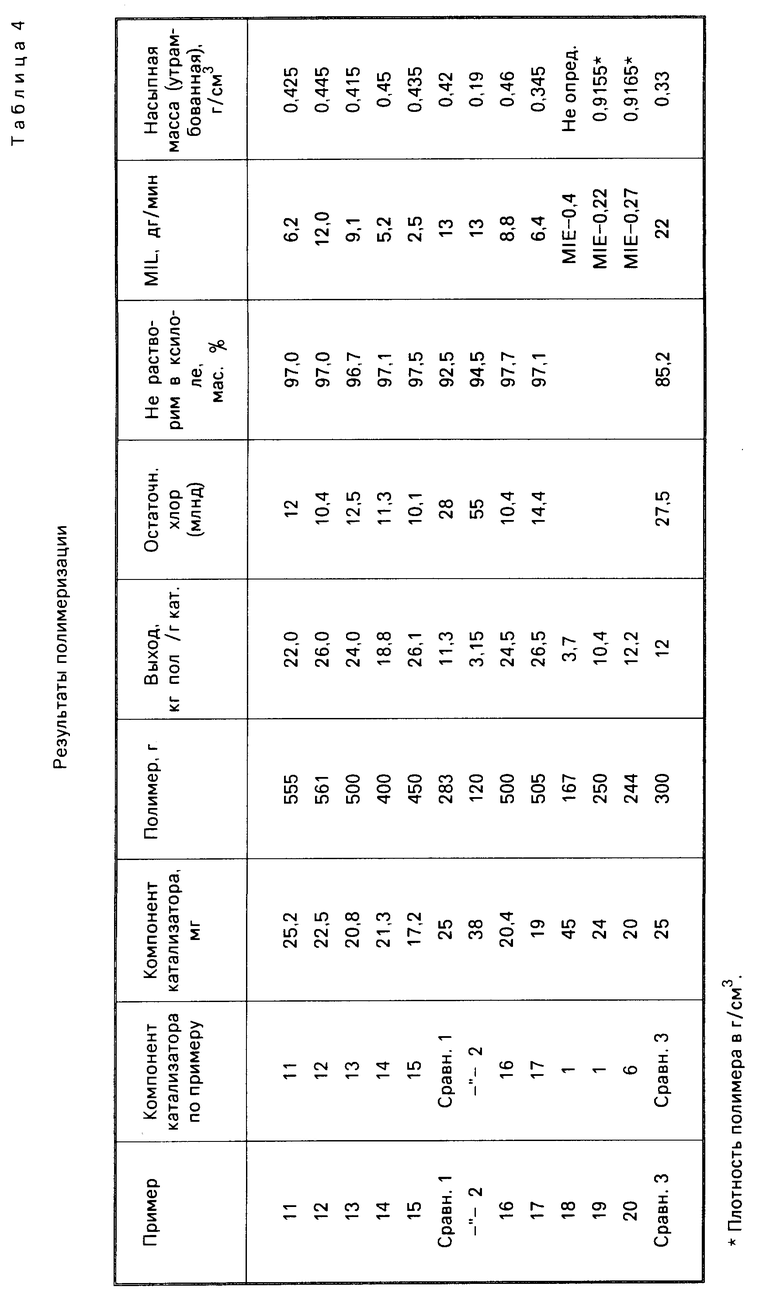
П р и м е р ы 6-8. В этих примерах использовали кремнезем, обогащенный водой с помощью обработки влажным азотом. Полученные в отношении компонента катализатора и полимеризации результаты приводятся соответственно в табл. 1 и 4.
П р и м е р ы 9-10. Импрегнирование кремнезема и синтез катализатора осуществляли по примеру 1, с той разницей, что в данном случае в качестве электронно-донорного соединения использовали диизобутилфталат и полимеризацию проводили с использованием смеси триэтил-Al/ДФМС (дифенилдиметоксисилан) в молярном отношении 20/1 вместо одного триэтилалюминия.
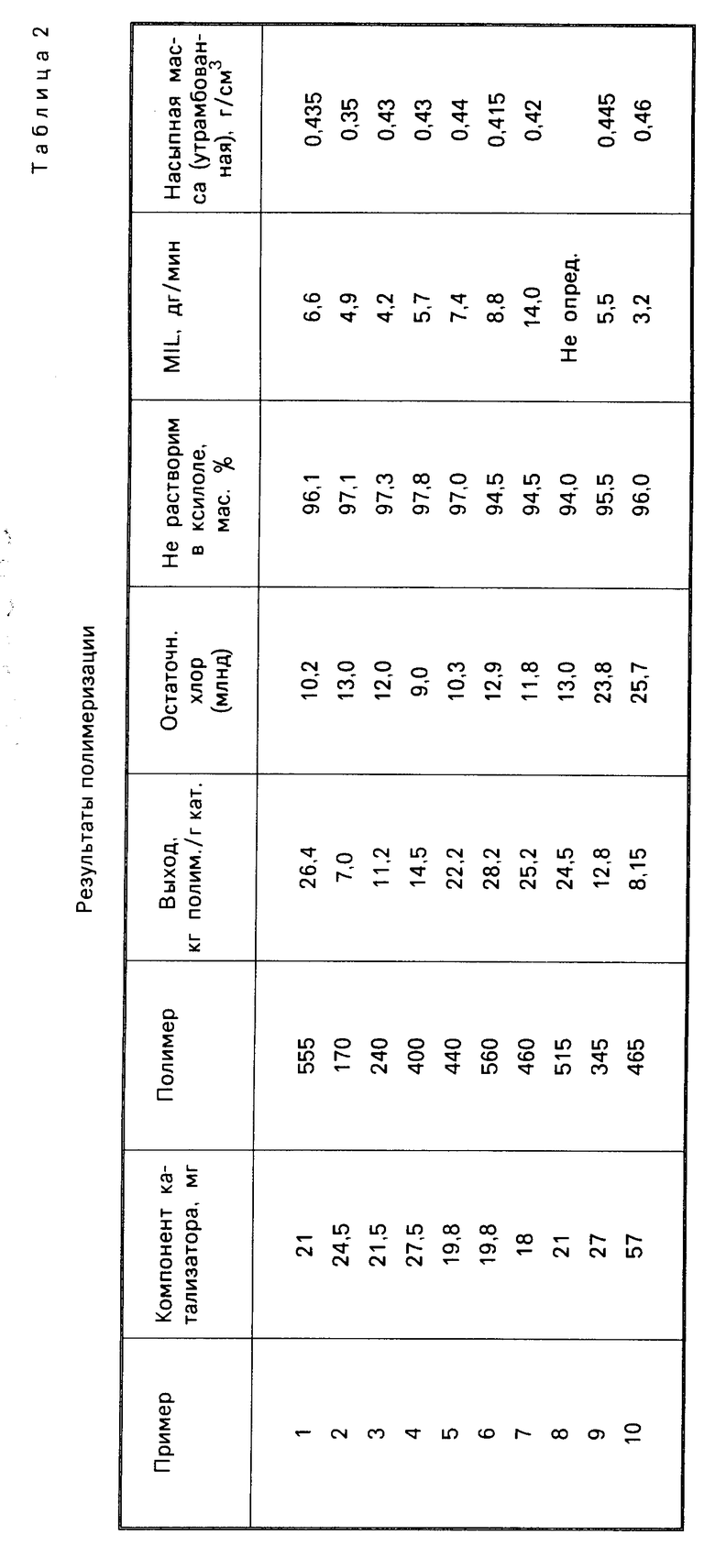
П р и м е р ы 11-13. Процесс осуществляли в описанных в примере 1 условиях, но для импрегнирования кремнезема использовали перечисленные в табл. 3 реактивы Гриньяра. Результаты полимеризации указаны в табл. 4.
П р и м е р 14 и сравнительный пример 1. Условия проведения процесса соответствуют примеру 1, но были взяты соответственно недостаток (пример 14) и избыток (сравнительный пример 1) соединения Гриньяра. Результаты полимеризации см. в табл. 4.
П р и м е р 15. Условия процесса совпадают с описанными в примере 1, использовали непрокаленный SiO2, но вводили 20 ммоль соединения Гриньяра (Вu MgCl) на грамм оксида кремния. Данные, полученные о составе катализатора и результатах полимеризации, даны в табл. 3 и 4.
П р и м е р ы 16-17. Компоненты катализатора приготовили по методике, описанной в примере 1, но использовали кремнезем высокой и низкой пористости, проводимый фирмой PQ Corporation (марки 988-1М и 850-40-5 с характеристиками, соответственно: удельная поверхность (БЕТ) 282 м2/г и пористость 2,75 см3/г, удельная поверхность 410 м2/г и пористость 1,37 см3/г). Результаты полимеризации приводятся в табл. 4.
П р и м е р 18. Для полимеризации этилена использовали 45 г компонента катализатора, приготовленного согласно примеру 1, с указанными ниже модификациями. В автоклав емкостью 2,5 л, снабженный системами термостатирования и перемешивания, который предварительно наполнили при 50оС азотом и затем водородом, поместили при температуре 45оС 850 мл 0,0025 М раствора триизобутилалюминия в безводном гексане. После чего ввели в легком токе Н2 50 мг компонента катализатора, приготовленного описанным выше способом, в виде суспензии в 150 см3 того же раствора. Автоклав закрыли, включили перемешивание и быстро подняли температуру до 75оС. Затем ввели водород до давления 4,5 бар, а затем этилен до давления 11,5 бар. Такие условия поддерживали в течение 3 ч, непрерывно восполняя затраченный этилен. По завершении процесса его выпустили и охладили до комнатной температуры. Полимер извлекали с помощью фильтрования и затем высушили при 70оС в токе азота в течение 3 ч. Результаты полимеризации приведены в табл. 4.
П р и м е р ы 19-20. В описанный в примере 1 автоклав, предварительно наполненный током пропана при 70оС в течение 40 мин и охлажденный затем до 30оС, ввели в токе пропана 10 см3 безводного гексана, в котором содержалось 0,96 г триэтил-Al и определенное количество компонента катализатора, которые указаны в табл. 4. Затем ввели 800 г пропана, одновременно включив перемешивание. Быстро подняли температуру до 75оС, после чего последовательно ввели 2 атм. Н2, 250 г бутена-1 и этилена, в результате чего установилось давление 34 бар. Эти условия поддерживали в течение 2 ч, непрерывно восполняя падение давления за счет введения смеси этилен-бутилен, состав которой по массе равен 10/1. В конце эксперимента осуществили быстрое снижение давления, после чего охладили автоклав до комнатной температуры. Полученный полимер сушили при температуре 70оС в атмосфере N2 в течение 4 ч. Результаты эксперимента по полимеризации приведены в табл. 4.
П р и м е р 21. В остальной реактор объемом 1,35 л, снабженный якорной мешалкой и наполненный предварительно газообразным пропиленом при комнатной температуре, ввели в токе пропилена 5 см3 гексана, в котором содержалось 0,6 г триэтил-Al и 104 мг компонента катализатора, полученного в примере 1. Затем добавили смесь, состоящую из 22,3 г пропилена, 4,4 г этилена и 0,44 г 1,4-гексадиена (76% транс-изомера). Соответствующее давление составило 11 бар. Температуру быстро подняли до 35оС и поддерживали на этом уровне в течение 4 ч, непрерывно компенсируя падение давления за счет введения смеси пропилен (1,4-гексадиен) этилен в массовом соотношении 67:3:30. По завершении процесса удалили непрореагировавшие газообразные мономеры, наполнили автоклав N2 и извлекли полимер, последний затем, после введения 0,1% ВНТ, высушили в токе азота при 60оС в течение 2 ч. Таким образом, было получено 135 г полимера (что соответствует выходу 1,3 кг полимера/г компонента катализатора) в виде легких сфероидных частиц следующего состава, мас. этилен-пропилен-гексадиен 35,1: 63: 1,9. Для выяснения механических характеристик полимер подвергали вулканизации (после гомогенизации в каландре при 800оС в течение 10 мин) в пластинчатом прессе при 160оС в течение 30 мин в смеси следующего состава, Полимер 51,35 ZnO 2,57 Стеариновая кислота 0,51 Сажа 28,24 100 М масла Cortis 14,50 Моносульфид тетра- метилтиоурата 0,77 Меркаптобензотиазол (МБТ) 0,39 Сера 0,77
Усадка при 100% растяжении составила 8,4; предел прочности на растяжение составил 11,3 МПа, а удлинение при разрыве 440%
Сравнительный пример 2. Методика эксперимента соответствовала описанной в примере 1, с той разницей, что в качестве соединения магния использовался диалкил (марки ВЕМ фирмы Техаs Alkyls). Результаты приведены в табл. 3 и 4.
Сравнительный пример 3. Компоненты катализаторов приготовили по примеру 1, используя кремнезем марки Davison 952, прокаленный в течение 7 ч при 150оС. Однако обработку ТiСl4 проводили в отсутствие электронно-донорного соединения. Состав компонента катализатора приводится в табл. 3. Полученный компонент катализатора испытывали в эксперименте полимеризации пропилена, используя, в соответствии с методиками примеров 9 и 10, смесь триэтил-Al/ДФМС. Результаты эксперимента см. в табл. 4.
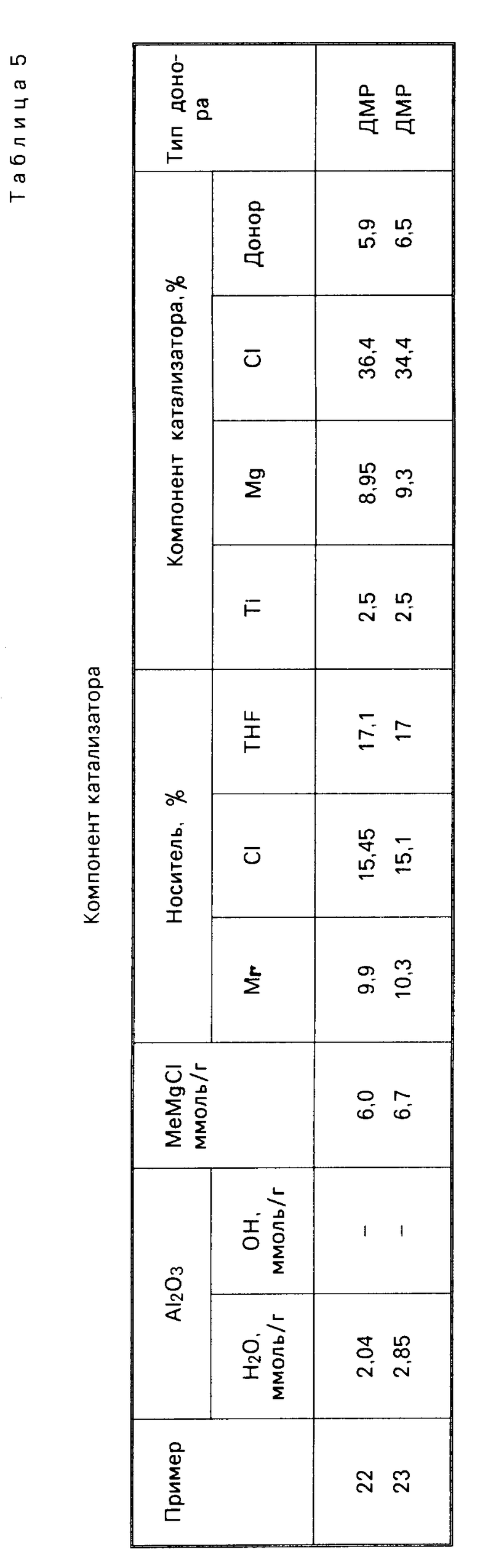
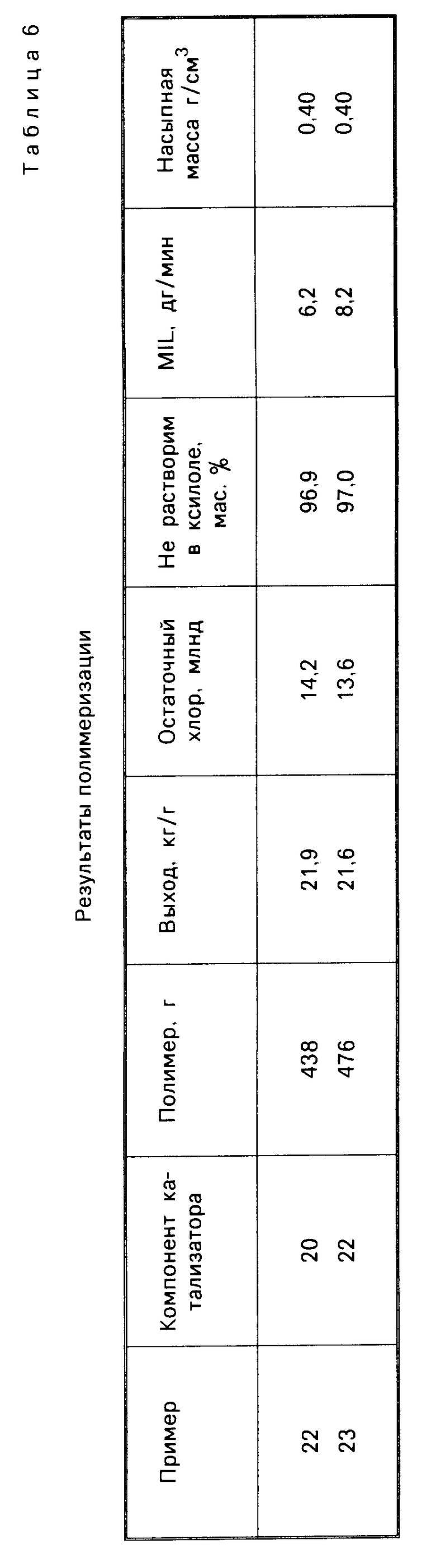
П р и м е р 22. С целью доведения содержания свободной воды до 3,55% (что соответствует 2,04 ммоль/г Al2O3), 5 г Al2O3 марки AKZO Ketjen В (в кристаллической форме псевдобемита) с характеристиками: удельная поверхность 266 м2/г, пористость 0,64 см3/г и содержание воды (по К. Фишеру) 17% подвергли предварительной обработке при 100оС под вакуумом (1 ч при давлении 100 Торр и 1 ч при 10 Торр), а затем импрегнировали 3 М раствором МеМgCl и ТНF (6 ммоль/г Al2O3) в соответствии описанной в примере 1 методикой. Далее провели обработку ТiCl4 и ДМП. Полученный таким способом катализатор используется в процессе полимеризации пропилена, как и в примере 1. Результаты проводятся в табл. 5 и 6.
П р и м е р 23. Осуществили процесс по примеру 1, за исключением того, что в данном случае Al2O3 сначала подвергли прокаливанию в течение 6 ч при 800оС (кристаллическая γ-форма), а затем воздействию воздуха в течение 5 мин, что имело целью доведение содержания свободной воды до 4,9% (это равно 2,85 ммоль/г Al2O3). Результаты проводятся в табл. 5-7.
| название | год | авторы | номер документа |
|---|---|---|---|
| КОМПОНЕНТ КАТАЛИЗАТОРА ПОЛИМЕРИЗАЦИИ АЛЬФА-ОЛЕФИНОВ | 1992 |
|
RU2073688C1 |
| КОМПОНЕНТ КАТАЛИЗАТОРА (СО)ПОЛИМЕРИЗАЦИИ АЛЬФА-ОЛЕФИНОВ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ | 1990 |
|
RU2045537C1 |
| КОМПОНЕНТ КАТАЛИЗАТОРА ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ | 1990 |
|
RU2032689C1 |
| ПОЛИОЛЕФИНОВАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1991 |
|
RU2036942C1 |
| ЭЛЕКТРОНОДОНОРНЫЕ КРЕМНИЙОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ И КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ α -ОЛЕФИНОВ | 1990 |
|
RU2039062C1 |
| ПОЛИОЛЕФИНОВАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1989 |
|
RU2043373C1 |
| КАТАЛИЗАТОР ПОЛИМЕРИЗАЦИИ АЛЬФА-ОЛЕФИНОВ И СПОСОБ ПОЛУЧЕНИЯ ПОЛИОЛЕФИНОВ | 1991 |
|
RU2078088C1 |
| СПОСОБ ПОЛУЧЕНИЯ СТАБИЛИЗИРОВАННОГО ПОЛИПРОПИЛЕНА | 1992 |
|
RU2070206C1 |
| СПОСОБ ОЧИСТКИ ОТ МОНООКСИДА УГЛЕРОДА α -ОЛЕФИНОВ И НАСЫЩЕННЫХ УГЛЕВОДОРОДОВ | 1994 |
|
RU2045510C1 |
| Способ получения катализатора для полимеризации олефинов | 1988 |
|
SU1811420A3 |
Использование: изобретение относится к способу получения катализатора полимеризации. Сущность изобретения: в качестве электронодонора используют 2-изопропил-2-изопентил-1,3-диметоксипропан или диизобутилфталат, осуществляют последовательно обработку твердого носителя первой порцией тетрахлорида титана, электронодонором и второй порцией тетрахлорида титана и процесс проводят при молярном отношении соединения магния к общему количеству гидроксильных групп, введенных с окислом, от 0,9 до 4,3, электронодонора к соединению магния 0,3, содержание соединения магния 1,08 20,00 ммоль на 1г окисла, молярном соотношении тетрахлорида титана и соединения магния, по крайней мере, 1 1,2. Используют двуокись кремния и окись алюминия, содержащие от 0,05 до 8,90 ммоль химически не связанной воды на 1г окисла. Взаимодействие твердого осадка с триалкилалюминием осуществляют в присутствии дифенилдиметоксисилана в количестве 0,05 моль на 1 моль триалкилалюминия. 2 з. п. ф-лы, 7 табл.
MgRX,
где R метил;
Н пропил или бутил,
Х хлор, бром,
в алкиловом эфире с последующей обработкой полученного твердого носителя тетрахлоридом титана и электронодонором, выделением, промывкой и сушкой полученного твердого осадка и взаимодействием его с триалкиалюминием, отличающийся тем, что в качестве электронодонора используют 2-изопропил-2-изопентил-1,3-диметоксипропан или диизобутилфталат, осуществляют последовательно обработку твердого носителя первой порцией тетрахлорида титана, электронодонором и второй порцией тетрахлорида титана и процесс проводят при молярном отношении соединение магния: общее количество гидроксильных групп, введенных с окислом, от 0,9 до 4,3 электронодонор: соединение магния 0,3, содержание соединения магния 1,08 20,00 ммоль на 1 г окисла и молярном отношении тетрахлорид титана: соединение магния по крайней мере 1 1,2.
| Патент США N 4263168, кл | |||
| Топка с несколькими решетками для твердого топлива | 1918 |
|
SU8A1 |
| Устройство для видения на расстоянии | 1915 |
|
SU1982A1 |
