Изобретение относится к области химии N-нитрозоалифатических соединений, конкретно к усовершенствованному способу получения динатриевой соли бис(N-нитрозогидроксиламино)метана формулы
NaO-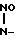 CH2-
CH2-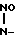 ONa (I) которая является биологически активным веществом, представляющим интерес в качестве лекарственного средства психотропного действия.
ONa (I) которая является биологически активным веществом, представляющим интерес в качестве лекарственного средства психотропного действия.
Известен способ получения динатриевой соли бис(N-нитрозогидроксиламино)метана, который осуществляется в две стадии:
1. Взаимодействием оксида азота с ацетоном в среде абсолютного спирта в присутствии этилата натря получают динатриевую соль бис(N-ниитрозогидроксиламино)ацетона:
CH3COCH3+ NO 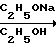
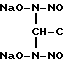 O-CH3
O-CH3
2. Гидролизом выделенной из спиртового раствора динатриевой соли бис(N-нитрозогиидроксиламино)ацетона путем нагревания в водном растворе с последующим осаждением целевого продукта спиртом: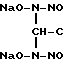 O-CH3
O-CH3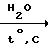
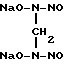 + CH3COOH
+ CH3COOH
Условия процесса: соотношение реагентов, температурный режим не указаны.
Недостатками этого способа являются сложность реакции нитролиза, которая заключается в том, что первичные продукты взаимодействия оксида азота, ацетона и алкоголята натрия подвергаются множеству вторичных реакций, что в свою очередь является причиной малого выхода целевого продукта - 25-30% , а также повышенная пожаровзрывоопасность процесса, связанная с использованием металлического натрия для получения этилата натрия и абсолютного спирта.
Целью изобретения является упрощение процесса и повышение выхода целевого продукта.
Поставленная цель достигается взаимодействием ацетона с оксидом азота в спирте в присутствии основания, отличающимся тем, что в качестве основания используют гидроксид натрия, и процесс проводят при 25-30oС и мольном соотношении оксида азота:ацетон:гидроксид натрия, равном 4-4,4:1,8-2,0:3.
Изобретение осуществляется следующим образом.
П р и м е р 1. Динатриевая соль бис(N-нитрозогидроксиламино)метана.
В реакционной колбе, снабженной мешалкой, термометром и ртутным затвором, готовят раствор 9,0 г (0,45 моль) едкого натра (ГОСТ 4328-77) в 90 мл этилового спирта, добавляют 16,5 г (0,285 моль) ацетона (ГОСТ 2603-71) и подают из газометра 18,9 г (0,63 моль) оксида азота. Реакционную смесь интенсивно перемешивают при 25-30оС на всем протяжении процесса. По окончании подачи оксида азота выпавший осадок целевого продукта отфильтровывают, промывают на фильтре 20-30 мл ацетона и сушат на воздухе. Выход продукта в виде моногидрата динатриевой соли бис(N-нитрозогидроксиламино)метана равен 12,3 г (83% в расчете на исходный едкий натр). Продукт представляет собой белое кристаллическое вещество с температурой начала разложения 216оС (со взрывом).
Найдено, %: С 6,14; Н 2,10; N 27,85.
СН2N4O4Na2 ˙ H2O (CH4N4O5Na2).
Вычислено, %: C 6,06; H 2,02; N 28,28.
ИК-спектр вещества в таблетках с КВr снимают на спектрофотометре "UR-10"; УФ спектр водного раствора продукта определяют на спектрофотометре "СФ-26"; ПМР спектры снимают в растворе дейтерированного диметилсульфоксида на спектрометре "Tesla BS-497" (100 МГц), внутренний стандарт - ТМС. ИК-спектр, ν, см-1: 1286, 1241, 966 (N2O2).
УФ спектр, λмакс = 258 нм, lg ε = 4,26.
ПМР спектр, σ , м.д.: 3,35 (C, 2H, CH2).
Приведенные экспериментальные характеристик ИК и УФ спектров полностью совпадают с литературными данными для ИК и УФ спектров динатриевой соли бис(N-нитрозогидроксиламино)метана.
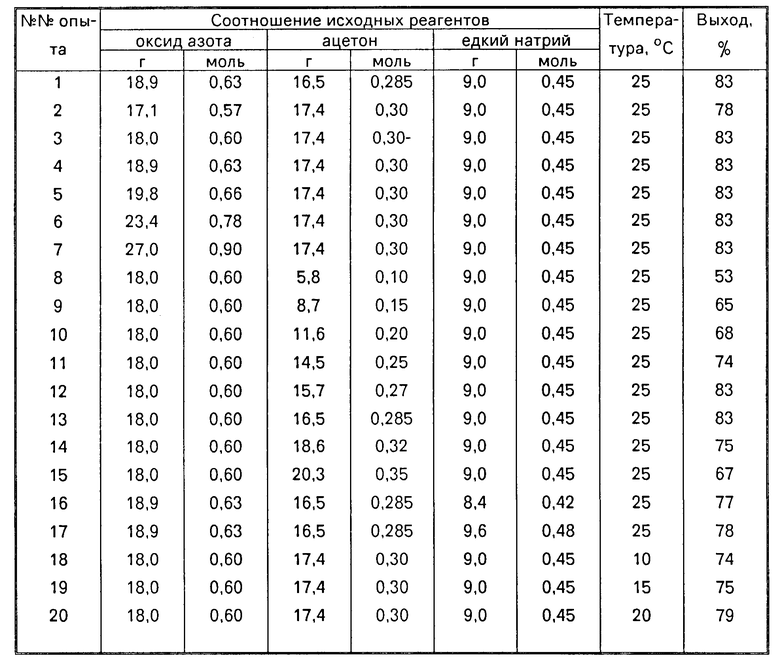
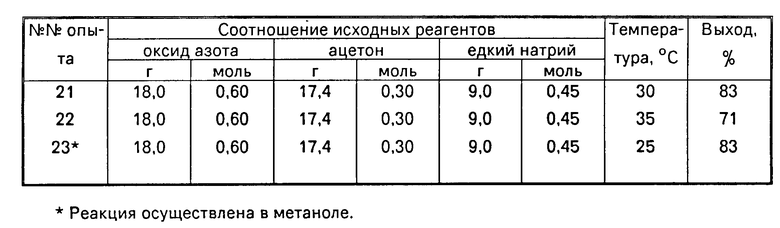
Зависимость выхода динатриевой соли бис(N-нитрозогидроксиламино)метана от соотношения исходных реагентов и температуры представлена в таблице.
П р и м е р ы 2-23. Условия проведения экспериментов аналогичны приведенным в примере 1 при вариации в каждом примере лишь одного параметра, как это показано в таблице. Данные элементного анализа на С, Н, N для полученных в примерах 2-23 продуктов соответствуют вычисленным а их ИК, УФ и ПМР спектры полностью повторяют ИК, УФ и ПМР спектры динатриевой соли бис(N-нитрозогидроксиламино)метана.
Как видно из таблицы, использование стехиометрического соотношения реагентов: оксид азота:ацетон:едкий натр, равного 4:1:3 моль (0,6:0,15:0,45 моль) обеспечивает лишь 65% выход целевого продукта (пример 9). Увеличение количества ацетона в этом соотношении до 1,8-2 моль (0,27-0,3 моль) дает максимальный выход целевого продукта 83% (примеры 3, 12 и 13). Дальнейший рост количества ацетона приводит к частичному осмолению продукта и снижению его выхода (примеры 14 и 15).
Избыток оксида азота в реакции (при исключении утечки газа) практически не сказывается на выходе целевого продукта, если используемые количества ацетона и щелочи находятся в оптимальном соотношении равном 1,8-2 моль ацетона:3 моль едкого натра (примеры 1,4-7).
При одновременном избытке оксида азота и щелочи в процессе наблюдается резкое снижение выхода продукта (пример 8).
Изменение температуры процесса с 10 до 35оС показало, что оптимальный выход продукта достигается при 25 и 30оС (примеры (1,3-5,21). При более высокой и более низких температурах выход целевого продукта значительно падает (примеры 18-22).
Замена этилового спирта в реакции на метиловый спирт не сказывается на выходе продукта, но значительно упрощает приготовление раствора едкого натра (пример 23).
Таким образом, предложенный способ является простым, безопасным, одностадийным и основан на взаимодействии отечественных сырьевых продуктов.
Использование изобретения: для синтеза биологически активного вещества. Сущность изобретения: продукт - динатриевая соль бис (N-нитрозогидроксиламино) метана NAON(NO)CH2N(NO)ONa , т. разл. 216°С (взрыв). Реагент 1 : ацетон. Реагент 2: оксид азота. Условия реакции: в спирте в присутствии NaOH при мольном соотношении реагент 1: реагент 2: NaOH, равном 4-4,4: 1,8-2,0:3. 1 табл.
СПОСОБ ПОЛУЧЕНИЯ ДИНАТРИЕВОЙ СОЛИ БИС (N-НИТРОЗОГИДРОКСИЛАМИНО)МЕТАНА формулыNaO-N(NO)CH2N(NO)-ONa взаимодействием ацетона с окисью азота в спирте в присутствии основания, отличающийся тем, что в качестве основания используют гидроксид натрия и процесс проводят при 25 - 30oС и молярном соотношении оксид азота : ацетон : гидроксид натрия 4 oC 4, 4 : 1, 8 oC 2, 0 : 3.
| Яндовский В.Н., Фролова Г.М., Целинский И.В | |||
| Азо- и азоксисоединения | |||
| YI | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| -ЖОрХ, 1982, т.18, вып.3, с.503-508. | |||
