Изобретение относится к производным арилалкиламинов или их фармацевтически приемлемым солям новым биологически активным соединениям, которые могут найти применение в медицине, и способу их получения.
Известны аралкиловые эфиры аминокислот, например алапролат [1] с потенциальным антидепрессивным действием сочетающимся с пониженной чистотой побочных эффектов. Эти соединения оказывают свое действие путем блокирования нейронного 5-окситриптамина (5+НТ), поднимающегося в центральной нервной системе. Наряду с ингибированием 5-НТ подъема они также обладают заметным действием на холинергические ответы.
Цель изобретения способ получения новых производных арилалкиламинов или их фармацевтически приемлемых солей, малотоксичных и обладающих более высоким антидепрессивным действием.
Поставленная цель достигается способом получения производных аралкиламина общей формулы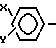 CH
CH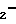
 Z
Z  CH2
CH2
 NH2 где n 0 или 1; Z -СО- или СН(ОН)-группы; Х и Y одинаковые или различные и означают галоид, трифторметил, алкил С1-С3, водород; R водород, прямая или разветвленная насыщенная алкильная группа С1-С6, или циклоалкил С3-С6, в рацемической или оптическиактивной форме, а когда Z является СН(ОН)-группой, также в форме смеси диастереомеров или чистого диастереомера, или их фармакологически приемлемых солей, заключающимся в том, что соединение формулы II:
NH2 где n 0 или 1; Z -СО- или СН(ОН)-группы; Х и Y одинаковые или различные и означают галоид, трифторметил, алкил С1-С3, водород; R водород, прямая или разветвленная насыщенная алкильная группа С1-С6, или циклоалкил С3-С6, в рацемической или оптическиактивной форме, а когда Z является СН(ОН)-группой, также в форме смеси диастереомеров или чистого диастереомера, или их фармакологически приемлемых солей, заключающимся в том, что соединение формулы II: CH
CH
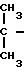 Z
Z  CH2
CH2

 (CH2)n
(CH2)n R где Z, X, Y, R и n имеют указанные значения, подвергают взаимодействию с ацетатом аммония в присутствии натрийборгидрида, натрийцианборгидрида, и при необходимости, в случае получения соединения формулы 1 со значением Z-СН(ОН)-группа, окисляют указанную группу до СО-группы, или при получении соединения формулы 1 со значением Z= СО-группа, восстанавливают указанную группу до СН(ОН)-группы, с последующим выделением целевого продукта в виде свободного основания или фармакологически приемлемой соли, в форме рацемата или оптически активного антипода.
R где Z, X, Y, R и n имеют указанные значения, подвергают взаимодействию с ацетатом аммония в присутствии натрийборгидрида, натрийцианборгидрида, и при необходимости, в случае получения соединения формулы 1 со значением Z-СН(ОН)-группа, окисляют указанную группу до СО-группы, или при получении соединения формулы 1 со значением Z= СО-группа, восстанавливают указанную группу до СН(ОН)-группы, с последующим выделением целевого продукта в виде свободного основания или фармакологически приемлемой соли, в форме рацемата или оптически активного антипода.
П р и м е р 1. 5-Амино-2,2-диметил-1-(4-фторфенил)-3-гептаноноксалат.
К раствору 3,02 г (12 ммолей) 2,2-диметил-1-(4-фторфенил)-3,5-гептандиона 7,4 г (96 ммолей) сухого ацетата аммония и 1,13 г (18 ммолей) натрийцианоборгидрида и молекулярных сит в 150 мл сухого метанола прибавляют несколько миллиметров уксусной кислоты до рН реакционной смеси 6,5. Перемешивают 30 ч при комнатной температуре, прерывают реакцию добавлением кислоты до рН 2. Реакционную смесь фильтруют, маточник упаривают, остаток растворяют в воде, раствор промывают эфиром, подщелачивают. После экстракции эфиром, сушки и упаривания получают 0,33 г сырого масла, которое осаждают в виде оксалата, перекристаллизовывают из 2-пропанола, получают 0,15 г целевого продукта. Т.пл. 125-127оС;
П р и м е р 2. Рацемат 5-амино-2,2-диметил-1-(4-фторфенил)-3-гептанона разделяют на его энантиомеры фракционной перекристаллизацией (+) и (-) тартратов из этанола. Энантиомерную чистоту определяют газовой хроматографией (GC) на амиде (S)-О-метилминдальной кислоты.
а) (+) энантиомер > 96% имеет следующие характеристики: т.пл. [(-)-тартрат] 122-122,5оС, [α]D25 +52о (основание, С 2, 7, СНСl3).
б) (-) энантиомер > 96% е.е. имеет следующие характеристики: т.пл. [(+)-тартрат] 120-121,5оС [α]D25 -42о (основание, С 3,1, CHCl3).
П р и м е р 3. 5-Амино-2,2,1-[4-фторфенил]-3-гептанол оксалат.
К раствору 37,0 г (150 ммолей) 6,6-диметил-7-(4-фторфенил)-5- оксо-3-гептанона в 300 мл метанола, прибавляют 64 г (800 ммолей) ацетата аммония, 10,0 г (150 ммолей) натрийцианборгидрида и 1,0 г молекулярных сит (размер 3  ). Смесь перемешивают 2 ч при комнатной температуре, фильтруют и выпаривают растворитель. Остаток обрабатывают диэтиловым эфиром и 2 М HCl. Устанавливают рH 9 добавлением 2 М NaOH и разделяют фазы. Органическую фазу сушат и выпаривают. Остаток растворяют в 250 мл диизопропилового эфира и 18,9 г (150 ммолей) дигидрата щавелевой кислоты, растворенного в 60 мл этанола, прибавляют для осаждения целевого продукта.
). Смесь перемешивают 2 ч при комнатной температуре, фильтруют и выпаривают растворитель. Остаток обрабатывают диэтиловым эфиром и 2 М HCl. Устанавливают рH 9 добавлением 2 М NaOH и разделяют фазы. Органическую фазу сушат и выпаривают. Остаток растворяют в 250 мл диизопропилового эфира и 18,9 г (150 ммолей) дигидрата щавелевой кислоты, растворенного в 60 мл этанола, прибавляют для осаждения целевого продукта.
Выход 16,3 г, т.пл. 152-154оС.
П р и м е р 4. 5-Амино-1-(4-фторфенил)-2,2,6-триметил-3-гептанол оксалат, т.пл. 156-160оС.
П р и м е р 5. 5-Амино-2,2-диметил-1-(4-фторфенил)-3-деканолоксалат, т. пл. 130-132оС.
П р и м е р 6. 5-Амино-2,2-диметил-1-(4-фторфенил)-3-гептанон оксалат.
К суспензии 9,0 г (90 ммолей) оксида хрома (VI) в 150 мл пиридина прибавляют раствор 10,3 г (30 ммолей) 5-амино-2,2-диметил-1- (4-фторфенил)-3-гептанол оксалата в 100 мл пиридина при комнатной температуре в течение 15 мин. Смесь перемешивают 2,5 ч и затем выливают в 600 мл воды и 500 мл диэтилового эфира.
Устанавливают рН 9,5 и после разделения эфирную фазу обрабатывают углем, сушат и выпаривают. Остаток растворяют в 50 мл диизопропилового эфира и прибавляют раствор 2,2 г (17 ммолей) дигидрата щавелевой кислоты в 5 мл этанола. Осадок собирают, перекристаллизовывают из 45 мл смеси диизопропилового эфира/этанола 2:1, получают 2,0 г целевого соединения. Т.пл. 126-127оС.
П р и м е р 7. 5-Амино-1-(4-фторфенил)-2,2,6-триметил-3-гептанон оксалат, т.пл. 148-149оС.
П р и м е р 8. 5-Амино-2,2-диметил-1-(3-трифторметилфенил)-3-гептанон оксалат, т.пл. 98-101оС.
Соединения примеров 7 и 8 получены в условиях примера 6.
П р и м е р 9. 5-Амино-1-(4-толил)-2,2,7-триметил-3-октанол оксалат, т. пл. 173-176оС (в условиях примера 3).
П р и м е р 10. 5-Амино-1-(4-толил)-2,2,7-триметил-3-октанол оксалат, т. пл.148-150оС (получен в условиях примера 6).
П р и м е р 11. 5-Амино-1-(3,5-дихлорфенил)-2,2-диметил-3-гексанол оксалат.
К раствору 1,01 г (3,5 ммоля) 5-амино-1-(3,5-дихлорфенил)-2,2-диметил-3-гекса- нона в 20 мл этанола прибавляют 132 мг (3,5 ммоля) натрийборгидрида при 5оС. Смесь перемешивают 30 мин при 5оС, выливают в смесь разбавленной холодной соляной кислоты и эфира. Водную фазу подщелачивают и экстрагируют эфиром. Эфирную фазу сушат и выпаривают растворитель. Остаток растворяют в 15 мл диизопропилового эфира и прибавляют 353 мг (2,8 ммоля) дигидрата щавелевой кислоты, растворенных в 1 мл этанола, осаждается 720 мг целевого соединения, т.пл. 170-173оС.
Все соединения охарактеризованы с помощью стандартных спектроскопических методов (1Н-ЯМР и β13 С-ЯМР спектров).
В табл. 1. обозначены все полученные соединения и приведены их физико-химические характеристики.
Фармакологический метод.
Усиление холинергетических ответов.
Используют самцов крыс (Спраг Доулей Алаб Лабораториетянст АВ, Соллентуна, Швеция) массой 150-180 г в возрасте 35-40 дней. Животным дают вдоволь пищи (R 3, Эвос АБ, Седертайж, Швеция) и воды до начала эксперимента. Испытуемые соединения инъецируют интраперитонеально (и.п.) за 30 мин до инъекции оксотреморина агонита мускарина (ОТМN) в шею (подкожно). Интенсивность тремора была оценена визуально в течение 60 мин после инъекции ОТМN крысе, находящейся в Макролоновых клетках (25 х 25 х 30 см) (три на клетку). Пороговая доза OTMN для вызывания тремора у крысы составляет 200 мкг/кг.
Интенсивность тремора была оценена с использованием следующей шкалы: 0-нет тремора, 1 слабый тремор (умеренный периодический тремор): 2 сильный тремор (интенсивный, непрерывный тремор, охватывающий все тело). Животных оценивали через 5, 10, 15, 30 и 60 мин после инъекции OTMN. Тремор оценивают в течение 30 с.
Величины для каждого наблюдения были суммированы за весь период наблюдения (5-60 мин) и была вычислена средняя величина для каждой группы (общая средняя оценка). Для определения различий между контрольной OTMN группой и испытуемой группой, получавшей соединения + OTMN использовали U-тест Манн-Уитни двуследовой). Следует отметить одновременность для каждого испытуемого соединения.
Самая низкая доза, вызывающая значительное (р < 0,05) усиление OTMN ответа, была определена из кривых log дозы ответ.
В табл.2 приведены данные по испытанию усиления тремора индуцированного оксотреморином у крыс известным соединением алапроклат формулы
Cl CH
CH
 O
O 


 NH2 и соединений изобретения
NH2 и соединений изобретения
X CH
CH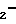
 Z
Z  CH2
CH2
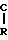 H
H  NH2
NH2
Полученные соединения в дозах, при которых наблюдался фармакологический эффект, не было обнаружено никаких признаков токсичности, т.е. они относятся к категории малотоксичных.
При этом эти соединения обладают способностью усиливать тремор, индуцируемый оксотреморином, что указывает на облегчение центральных холинергетических ответов.

Использование: в медицине, в частности в качестве веществ с антидепрессивной активностью. Сущность изобретения: продукт - производные аралкиламинов ф-лы 1, где n = 0 или 1; Z - карбонил или CH(OH); X и Y - равные или разные- галоген, трифторметил, C1-C3 -алкил, водород; R-n- или i-(C1-C6) -алкил или C3-C6 -циклоалкил, водород, в рацемической или оптически активной форме, причем когда Z = -CH(OH), то в форме смеси или чистого дистериомера кроме рацематов указанных производных, в которых X = n-Cl, Y = H, Z = -CO-, n = 0; R - метил, или их фармацевтически приемлемых солей. Способ предусматривает реакцию введения аминогруппы в соответствующее производное с помощью ацетата аммония в присутствии натрийборгидрида или натрийцианборгидрида с последующим окислением (если Z = -CH(OH), то в карбонил) или восстановлением (карбонила в группу - СН(ОН)) и выделением целевого продукта в виде свободного основания, нужной соли, в форме рацемата или оптически активного антипода. Структура ф-лы 1(см. чертеж). 2 с.п. ф-лы, 2 табл.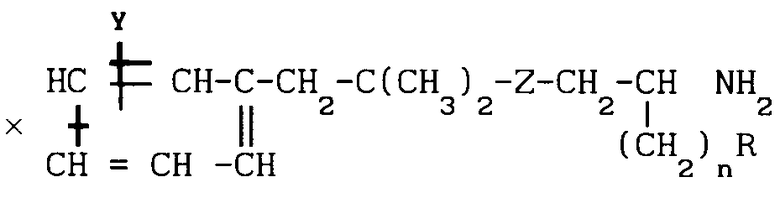

где n-0 или 1;
Z -СО или -СН(ОН);
X и Y, одинаковые или различные, галоген, трифторметил, С1 - С3-алкил, водород;
R прямая или разветвленная насыщенная С1 С6-алкильная группа или С3-С6-циклоалкильная группа, водород в рацемической или оптически активной форме, и когда Z СН(ОН)-группа, также в форме смеси диастереомеров или чистого диастереомара, кроме рацематов указанных производных, где X n-Cl, Y водород, Z-CO, n=0, R метил, или их фармацевтически приемлемые соли.
где n 0 или 1;
Z= -CO- или -CH(OH)-; X и Y, одинаковые или различные, галоген, трифторметил, С1 С3-алкил, водород;
R-прямая или разветвленная насыщенная С1-С6-алкильная группа или С3-С6-циклоалкильная группа, водород, в рацемической или оптически активной форме, и когда Z-СН(ОН)-группа, также в форме смеси диастереомеров или чистого диастеромера, кроме рецематов указанных производных, где Х- N-Cl, Y-водород, Z=-CO-, n= 0, R-метил,
или их фармацевтически приемлемых солей, отличающийся тем, что соединение общей формулы II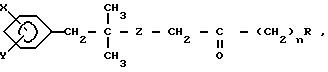
где X, Y, R и n имеют указанные значения,
подвергают взаимодействию с ацетатом аммония в присутствии натрийборгидрида или натрийцианборгидрида и при необходимости, в случае получения соединения формулы I, где Z CH(OH)-группа, окисляют указанную группу до СО-группы или при получении соединения формулы I, где Z= -CO- группа, восстанавливают указанную группу до СН(ОН)-группы с последующим выделением целевого продукта в виде свободного основания или фармакологически приемлемой соли в форме рацемата или оптически активного антипода.
| Патент США N 4237322, кл | |||
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
| Способ получения фтористых солей | 1914 |
|
SU1980A1 |
