Изобретение относится к химическому производству оксидов, а именно к способам производства реактивного оксида никеля, который применяется в различных отраслях народного хозяйства для получения ферритов, катализаторов, пигментов в качестве краски для стекол и др.
В настоящее время актуальной является задача получения оксида никеля определенного химического и гранулометрического состава.
Наиболее близким к предлагаемому по технической сущности является способ получения оксидов никеля путем термической обработки азотнокислого никеля в присутствии водяного пара [1] Способ получения оксидов никеля заключается в приготовлении нитрата никеля, его термической обработке в среде водяного пара, где реакция происходит по механизму гидролиза, конденсации полученных в результате реакции газов для получения раствора азотной кислоты и с наличием оксидов азота в хвостовых газах в количестве до 5-7% от теоретического баланса азотной кислоты.
Исходный нитрат никеля загружается в прокалочную печь в определенном объеме, поэтому при термическом разложении в присутствии водяного пара будет наблюдаться неравномерность взаимодействия всего объема загруженного продукта с водяным паром, что приведет к получению неоднородного по химическому и фракционному составу конечного продукта. Полученный оксид никеля представляет собой спек, который не является кондиционным продуктом. Этот спек необходимо размолоть и просеять для получения продукта определенного фракционного состава.
Получаемая в процессе реакции 6-10%-ная азотная кислота не может быть непосредственно использована на стадии приго- товления исходного раствора (растворение металлического никеля). Для использования этой кислоты необходимо подвергнуть ее дополнительной обработке с целью повышения концентрации. Для решения экологической задачи, исключения выбросов в атмосферу необходима дополнительная переработка "хвостовых" газов.
Целью изобретения является предотвращение выбросов оксидов азота в атмосферу, повышение производительности, экономичности процесса получения оксидов никеля и качества готового продукта путем безотходного получения частиц оксидов, однородных по химическому и гранулометрическому составу.
Цель достигается тем, что в известном способе получения оксидов никеля, включающем приготовление раствора нитрата никеля, его термическую обработку в присутствии водяного пара, конденсацию выделяющихся газов с образованием азотной кислоты и возвратом ее на стадию приготовления раствора, новым является то, что перед термической обработкой исходный раствор упаривают до концентрации не менее 1,1 кг/л, а водяной пар на стадии термической обработки используют в качестве ожижающего агента в аппарате кипящего слоя (КС) при соотношении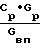 ≅ 1,68 где Ср концентрация исходного раствора, кг/кг;
≅ 1,68 где Ср концентрация исходного раствора, кг/кг;
Gр расход исходного раствора, кг/ч;
Gвп расход водяного пара, кг/ч.
Таким образом, предлагаемый способ получения оксидов никеля соответствует критерию "новизна". Благодаря наличию отличительных признаков, перечисленных выше, в предлагаемом способе проявляется новое свойство исключение выбросов оксидов азота в атмосферу.
Обычно на заводах по производству оксидов никеля (и как промежуточный этап получение нитрата никеля) для растворения металлического никеля используется азотная кислота с концентрацией не менее 25% (например, на Уральском заводе химических реактивов).
Таким образом, при массовой концентрации исходного раствора нитрата никеля менее 1,1 кг/л после конденсации образующихся газов получалась азотная кислота, концентрация которой была недостаточной (15-20%) для непосредственного использования на стадии приготовления исходного раствора, что является неэкономичным, так как для приготовления раствора нитрата никеля необходимо дополнительное проведение операций для повышения ее концентрации до ≥ 25%
Существенным является то, что при упаривании водных растворов азотной кислоты происходит насыщение паровой фазы парами азотной кислоты и тем больше, чем концентрированный раствор, т.е. концентрирование водного раствора азотной кислоты методом упарки невозможно без ее потери с паровой фазой. Причем потери растут с повышением концентрации кислоты в растворе [2]
При упаривании же водных растворов нитратов потери азотной кислоты не происходит до момента термического разложения нитрата. Максимально возможное в условиях обычной упарки содержание составляющих азотной кислоты в водном растворе нитрата никеля соответствует составу дегидрата его Ni(NO3)2.2H2O [3]
Концентрация, до которой упаривается исходный раствор, необходима для повышения производительности установки по готовому продукту (получение NiO) и обусловлена требованиями по концентрации, предъявляемыми к регенерируемой азотной кислоте.
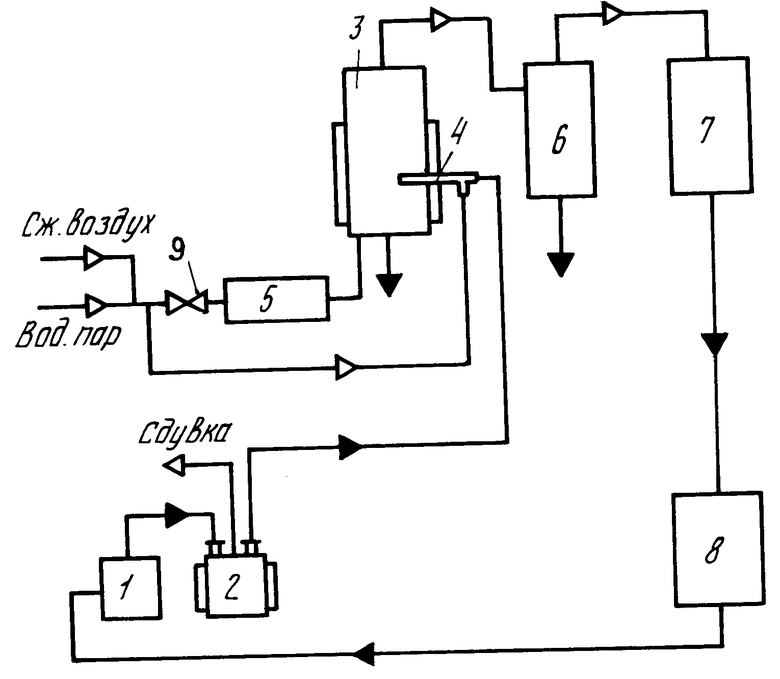
На чертеже показана установка для реализации способа получения оксида никеля. Она работает следующим образом:
нитрат никеля в виде исходного раствора нитрата никеля поступает из аппарата приготовления раствора 1 в питающий монжюс 2, снабженный паровой рубашкой для доупарки раствора до необходимой концентрации. Упаренный раствор из монжюса 2 под давлением подается в реактор кипящего слоя 3, снабженный электронагревателем, через пневматическую форсунку 4. Здесь в кипящем слое гранул готового оксида никеля при 400оС происходит термическое разложение нитрата никеля до оксида никеля по реакции
Ni(NO3)2·6H2O __→ NiO+2NO+3/2 O2+6H2O
В период разогрева установки в качестве ожидающей и распыляющей среды использовался сжатый воздух, а в период проведения рабочего процесса водяной пар.
Готовый продукт в виде гранул оксида никеля выводится из реактора 3 в процессе работы.
Образованные в процессе реакции газы из реактора 3 с частично вынесенным продуктом проходят последовательно циклон 6 и конденсатор 7.
В циклоне происходит очистка газов от оксида никеля, который по мере накопления вывозится в контейнер готового продукта. В конденсаторе 7 происходит улавливание концентрирующих газов. Они выводятся в приемную часть 8 в виде раствора азотной кислоты, которая подается в аппарат для приготовления исходного раствора нитрата никеля 1. Для псевдоожижения используется воздух и водяной пар, которые поступают из магистрали через калорифер 5 в газораспределительное устройство реактора 3.
П р и м е р. Исходный раствор нитрата никеля перед термической обработкой упаривали до массовой концентрации не менее 1,1 кг/л. После концентрации газов, полученных в результате термического разложения нитрата никеля до оксида никеля, получалась азотная кислота, концентрация которой была более 25% Кислота использовалась на начальной стадии приготовления исходного раствора при растворении металлического никеля.
При массовой концентрации исходного раствора нитрата никеля 0,75-0,8 кг/л концентрация азотной кислоты, полученная после конденсации газов термического разложения нитрата никеля до оксида никеля составляла порядка 10-15% и не могла быть непосредственно использована на стадии приготовления исходного раствора без дополнительной обработки.
Высококонцентрированные растворы нитрата никеля, приготовленные из фильтрованных технологических растворов промышленного производства (концентрацией по нитрату никеля 0,5 кг/л) путем упарки в аппарате с паровой рубашкой, заливали в количестве 95 л в монжюс с паровой рубашкой 2. После этого в реактор кипящего слоя 3 загружали "подушку" порошка оксида никеля в количестве 35 кг со средним размером зерна 0,2 мм. Затем в форсунку 4 подавали сжатый воздух с расходом 2 м3/ч во избежание засорения ее частицами "подушки". Далее в реактор 3 через калорифер 5 подавали воздух на псевдоожижение с расходом 20 м3/ч и включали электронагрев калорифера 5 и реактора 3. После прогрева реактора до 400оС воздух, подаваемый в форсунку 4 и на псевдоожижение в реактор 3, заменяли водяным паром с расходами 2,5 кг/ч и 20 кг/ч соответственно.
После этого с помощью сжатого воздуха из монжюса 2 в форсунку 4 подавали раствор нитрата никеля с расходом от 10 до 17 л/ч, который во время разогрева реактора находился под давлением собственных паров во избежание дополнительной упарки. Температура в реакторе во время подачи раствора нитрата никеля через форсунку поддеpживалась в пределах от 350 до 450оС. Отходящие из реактора газы пропускали через циклон 6 и кожухотрубный конденсатор 7, охлаждаемый водой, которую подавали одновременно с включением электронагрева калорифера и реактора. Получаемый конденсат непрерывно сливался в емкость 8. Из реактора 3 через каждый час выводился оксид никеля в количестве 4 кг.
Проведено 65 технологических опытов. Общее время составило 286,5 ч. В ходе этих опытов переработано 3000 кг нитрата никеля в виде высококонцентрированных растворов с содержанием нитрата никеля от 0,8 до 1,4 кг/л при температуре "кипящего" слоя от 350 до 450оС.
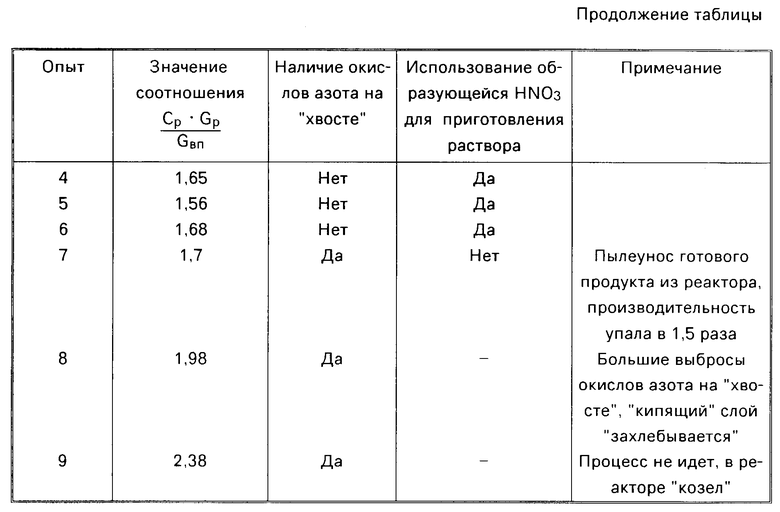
Данные определяющих опытов сведены в таблицу. Как видно из таблицы, при упаривании исходного раствора нитрата никеля от 1,1 до 1,4 кг/л (опыты 1-6) и соблюдении соотношения 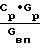 ≅ 1,68 процесс идет стабильно при отсутствии окислов азота на "хвосте" и получении кондиционной азотной кислоты на стадии конденсации. Процесс проходит устойчиво на всех ступенях передела: упарке, разложении, пылеочистке и конденсации. Из реактора кипящего слоя выгружали гранулированный порошок оксида никеля при отсутствии агломератов, спеков. Полученный сухой продукт обладал хорошей сыпучестью. Гранулометрический состав оксида никеля во всех проведенных опытах изменялся от 0,1 до 0,5 мм.
≅ 1,68 процесс идет стабильно при отсутствии окислов азота на "хвосте" и получении кондиционной азотной кислоты на стадии конденсации. Процесс проходит устойчиво на всех ступенях передела: упарке, разложении, пылеочистке и конденсации. Из реактора кипящего слоя выгружали гранулированный порошок оксида никеля при отсутствии агломератов, спеков. Полученный сухой продукт обладал хорошей сыпучестью. Гранулометрический состав оксида никеля во всех проведенных опытах изменялся от 0,1 до 0,5 мм.
При несоблюдении соотношения 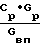 ≅ 1,68 (опыты 7-9) процесс идет неустойчиво с наличием окислов азота "на хвосте", получением азотной кислоты на стадии конденсации, непригодной непосредственно для приготовления исходного раствора и даже к образованию "козла" в реакторе кипящего слоя.
≅ 1,68 (опыты 7-9) процесс идет неустойчиво с наличием окислов азота "на хвосте", получением азотной кислоты на стадии конденсации, непригодной непосредственно для приготовления исходного раствора и даже к образованию "козла" в реакторе кипящего слоя.
Таким образом, предлагаемое изобретение имеет следующие технико-экономические преимущества по сравнению со способом-прототипом:
1. Исключает выброс окислов азота в атмосферу после конденсации отходящих газов реакции.
2. За счет использования раствора более высокой концентрации увеличивается производительность процесса по готовому продукту.
3. Повышается качество готового продукта оксида никеля за счет исключения любых примесей и необходимости дополнительной обработки.
4. Получаемая на стадии конденсации отходящих газов азотная кислота непосредственно используется для приготовления исходного раствора нитрата никеля.
5. Безотходность производства и исключение жидких и газообразных выбросов решают экологическую задачу.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ АЗОТНО-ФОСФОРНЫХ УДОБРЕНИЙ ПУТЕМ ПЕРЕРАБОТКИ РАСТВОРОВ, ПОЛУЧЕННЫХ ПРИ КИСЛОТНОЙ ЭКСТРАКЦИИ ФОСФОРСОДЕРЖАЩЕГО СЫРЬЯ | 1994 |
|
RU2078064C1 |
| СПОСОБ ПОЛУЧЕНИЯ НИТРАТА АММОНИЯ | 1990 |
|
RU2049725C1 |
| СПОСОБ КОНЦЕНТРИРОВАНИЯ РАДИОАКТИВНЫХ ОТХОДОВ | 2015 |
|
RU2596816C1 |
| СПОСОБ ПЕРЕРАБОТКИ ХЛОРИДНО-СУЛЬФАТНЫХ СТОЧНЫХ ИЛИ ПРИРОДНЫХ ВОД | 1990 |
|
RU2060973C1 |
| СПОСОБ КОНЦЕНТРИРОВАНИЯ ЭЛЕКТРОЛИТИЧЕСКОЙ КАУСТИЧЕСКОЙ ЩЕЛОЧИ | 1990 |
|
SU1835790A1 |
| Способ получения ферритообразующей шихты для магнитомягких ферритов | 1991 |
|
SU1822389A3 |
| СПОСОБ ПРОИЗВОДСТВА НИТРИТА НАТРИЯ | 1993 |
|
RU2069174C1 |
| СПОСОБ ПРИГОТОВЛЕНИЯ НИКЕЛЬХРОМОВОГО КАТАЛИЗАТОРА ДЛЯ ГИДРИРОВАНИЯ БЕНЗОЛА | 1992 |
|
RU2054319C1 |
| СПОСОБ ПОЛУЧЕНИЯ ОКСИДОВ МЕТАЛЛА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 1993 |
|
RU2061584C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГИДРОКСИЛАМИНСУЛЬФАТА | 2003 |
|
RU2259940C1 |
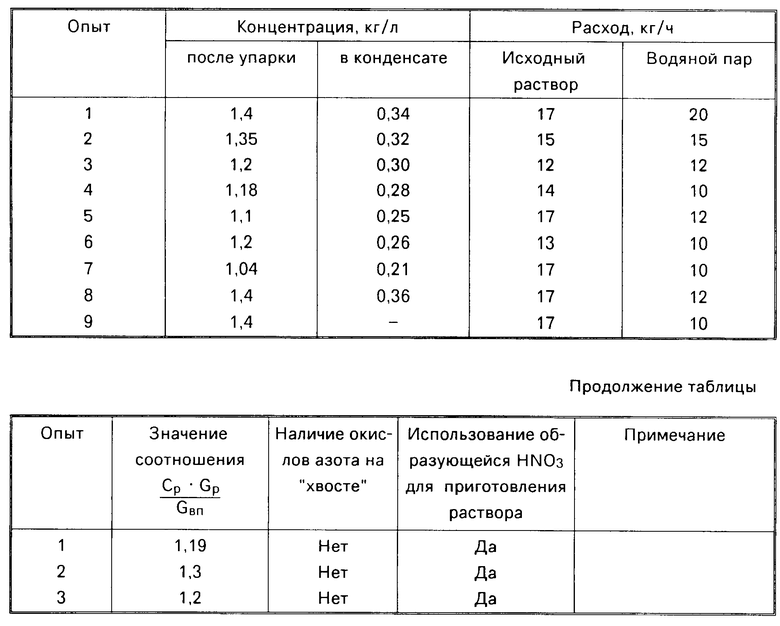
Изобретение относится к химическому производству оксидов никеля, применяемых в различных отраслях народного хозяйства для получения ферритов, катализаторов, пигментов в качестве красок для стекол и т.п. Способ включает приготовление раствора Ni(NO3)2 упаривание его до массовой концентрации не менее 1,1 кг/л, термическую обработку его в псевдоожиженном слое частиц оксида никеля. Образующуюся после конденсации газов азотную кислоту возвращают на операцию приготовления исходного раствора нитрата никеля. Водяной пар на стадии термической обработки используют в качестве ожижающего агента в аппарете кипящего слоя при соотношении 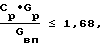 где Cр - концентрация раствора после упаривания , кг/кг; Gр - расход раствора, кг/ч; Gвп - расход водяного пара, кг/ч. Способ позволяет получать оксид определенного химического и гранулометрического состава и предотвратить выбросы оксидов азота. 1 ил., 2 табл.
где Cр - концентрация раствора после упаривания , кг/кг; Gр - расход раствора, кг/ч; Gвп - расход водяного пара, кг/ч. Способ позволяет получать оксид определенного химического и гранулометрического состава и предотвратить выбросы оксидов азота. 1 ил., 2 табл.
СПОСОБ ПОЛУЧЕНИЯ ОКСИДА НИКЕЛЯ, включающий приготовление исходного раствора нитрата никеля, его термическую обработку в присутствии водяного пара с получением целевого продукта, конденсацию выделяющихся газов с образованием азотной кислоты и возвратом ее на стадию приготовления раствора, отличающийся тем, что, с целью предотвращения выбросов оксидов азота в атмосферу, перед термической обработкой исходный раствор упаривают до концентрации не менее 1,1 кг/л, а расход водяного пара на стадии термической обработки, используемого в качестве ожижающего агента в аппарате кипящего слоя, поддерживают в соответствии с соотношением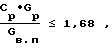
где Cр концентрация раствора после упаривания, кг/кг;
Gр расход раствора, кг/ч;
Gв.п расход водяного пара, кг/ч.
| Краткая химическая энциклопедия | |||
| М.: Советская энциклопедия, 1964, Т.3 с.466. |
