Изобретение относится к пиримидиновым нуклеозидам, используемым в медицине, в частности, для лечения и профилактики вирусных заболеваний герпеса.
Вирусы ДНК группы герпеса являются причиной широко распространенных вирусных заболеваний человека. Эта группа состоит из вируса герпеса простого (HSY), вируса герпеса опоясывающего генерализованного (YZY), вируса цитомегалии (CMY) и вируса Эпштейна-Барра (EBY).
Вирус герпеса опоясывающего генерализованного (YZY) является вирусом герпеса, который вызывает ветряную оспу и опоясывающий лишай. Ветряная оспа представляет собой первичное заболевание, к которому у хозяина не вырабатывается иммунитет. У детей, которые обычно легко его переносят, оно характеризуется пузырчатым высыпанием и жаром. Опоясывающий лишай является рецидивной формой этого заболевания, которым болеют взрослые, зараженные ранее вирусом герпеса опоясывающего генерализованного. Клиническим проявлением опоясывающего лишая являются невралгия и пузырчатое высыпание на коже, одностороннее по распределению. Распространение воспаления может приводить к параличу или конвульсиям. В случае поражения мягких мозговых оболочек может иметь место кома. YZY может распространяться у больных с иммунодефицитом, вызывая тяжелые, а иногда и смертельные заболевания. YZY может быть опасным для больных, получающих при трансплантации органов лекарства, подавляющие иммунную способность, или при лечении злокачественных новообразований. Он также вызывает серьезные осложнения у больных СПИДом вследствие непарности у них иммунной системы.
Как и в случае других вирусов группы герпеса, заражение CMY приводит к образованию сообщества вирус-хозяин на всю жизнь. Поэтому после первичного заражения вирус может на многие горы сохранять способность к распространению. Клиническими эффектами могут быть как смерть и тяжелые заболевания (микроцефалия, гепатоспленомегалия, желтуха, умственная отсталость), так и прекращение роста, восприимчивость к грудным и ушным заболеваниям, а также отсутствие явных признаков заболевания. Инфекция СМУ у больных СПИДом является главной причиной заболеваемости. У 80% взрослого населения он находится в латентной форме и может реактироваться у иммунно-компромиссных больных.
Вирус Эпштейна-Барра (EBY) вызывает инфекционный мононуклераг. Предполагается также, что он является причиной рака носоглотки, иммунобластной лимфомы, лимфомы Burkitt и лейкоплакии волос.
Внимание последователей было сконцентрировано на нуклеозидах-аналогах, которые можно было бы использовать для лечения инфекционных заболеваний, вызываемых вирусами группы герпеса. Одним из таких соединений, которое представляет интерес в качестве полупродукта, является 2'-деокси-5-этинилуридин. Синтез его описан Барром и др. (J. Chem. Soc. Perkin braus 1: 1978, 1268). Это соединение было испытано на антивирусную активность in vitro против коровьей оспы и герпеса простого, как это описано, например, Walker и др. (Nucleic Acid Res. Special pub. N 4, 1978) и в патенте Великобритании N 1601020. Однако об использовании его для лечения человека ничего не известно.
В европейской заявке на патент N 86305297 описано использование 2'-деокси-5-этинилуридина и его фармацевтически приемлемых производных для лечения или профилактики инфекционных заболеваний человека, вызываемых вирусом цитогалии (CMY) или вирусом герпеса опоясывающего генерализованного (YZY).
Вышеупомянутые пиримидиновые нуклеозиды могут быть описаны общей формулы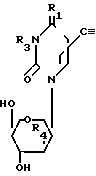
 в которой R1-оксогруппа; R2 атом водорода или метил; R3 бензоил, необязательно замещенный на атом галогена или алкокси; R4 атом водорода.
в которой R1-оксогруппа; R2 атом водорода или метил; R3 бензоил, необязательно замещенный на атом галогена или алкокси; R4 атом водорода.
Соединения предлагаемого изобретения могут быть использованы для лечения или профилактики вирусных инфекционных заболеваний, в частности заболеваний, вызываемых вирусом группы герпеса, а именно YZY, CMY и EBY.
Соединения в соответствии с настоящим изобретением могут вводиться любым способом, в зависимости от состояния больного. Подходящими способами являются оральный, ректальный, носовой, местный (включая защечный или подъязычный), вагинальный и парентеральный (включая подкожный, внутримышечный, внутривенный, внутрикожный и введение в спинно-мозговой канал). Следует отметить, что предпочтительный способ введения может изменяться, например, в зависимости от состояния больного.
Для лечения вышеперечисленных болезней необходимое количество конкретного активного компонента зависит от ряда факторов, в частности от тяжести заболевания и состояния больного, и в конечном счете определяется лечащим врачом. В общем случае однако при лечении указанных болезней эффективная ежедневная доза находится в пределах 0,1-250, предпочтительно 1-100, наиболее предпочтительно 5-30 мг на кг веса. Оптимальная ежедневная доза состаляет около 15 мг на кг веса (если это не оговорено, все количества активного компонента рассчитаны на сами соединения; в случае их солей и эфиров приведенные цифры необходимо увеличить соответствующим образом). Необходимая доза при желании может вводиться в два, три, четыре или более приемов через соответствующие промежутки времени в течение дня. Такие мелкие дозы могут вводиться в виде разовых форм, содержащих например, 10-1000, предпочтительно 20-500, наиболее предпочтительно 100-400 мг активного компонента.
Предлагаемые соединения можно вводить как сами по себе, так и (что предпочтительно) в виде фармацевтических композиций. Такие композиции в соответствии с настоящим изобретением включают по меньшей мере один вышеописанный активный компонент в сочетании с одним или несколькими приемлемыми носителями. Они, кроме того, могут содержать и другие терапевтические компоненты. Используемый носитель (или носители) должен быть "приемлемым" с точки зрения совместимости его с другими компонентами композиции и не должен оказывать вредного действия на больного.
Под композициями в соответствии с настоящим изобретением имеются в виду композиции, которые могут использоваться для орального, ректального, носового, местного (включая защечное и подъязычное), вагинального или парентерального (включая подкожное, внутримышечное, внутривенное, внутрикожное, введение в спинно-мозговой канал и внутрь оболочек спинного мозга) введения. Указанные композиции могут быть приготовлены в виде единичных доз. Они могут быть получены любым из используемых в фармацевтической промышленности способом. Эти способы включают стадию смешения активного компонента с носителем, который содержит один или несколько вспомогательных компонентов. Обычно такие композиции готовят путем тщательного смешения до получения однородной смеси активного компонента с жидким или мелко измельченным (или с тем, и с другим) носителем с последующим (при необходимости) формованием полученного продукта.
Композиции в соответствии с настоящим изобретением, пригодные для орального введения, могут выпускаться в виде единичных доз, например в виде капсул, облаток или таблеток, содержащих определенное количество активного компонента, порошков или гранул, растворов или водных или неводных суспензий, эмульсий типа вода в масле или масло в воде. Активный компонент может быть также приготовлен в виде шариков, лекарственной кашки или пасты.
Таблетки могут быть получены путем прессования или формовки, с использованием при желании одного или нескольких вспомогательных компонентов. Прессованные таблетки могут быть получены путем прессования в соответствующем устройстве активного компонента, находящегося в сыпучем состоянии, например в виде порошка или гранул, который может быть смешан со связующим (например повидоном, желатином, оксипропилметилцеллюлозой), смазывающим веществом, инертным разбавителем, консервантом, диспергатором (например, крахмалом, гликолятом натрия, сшитым повидоном, сшитой натриевой солью карбоксиметилцеллюлозы) или поверхностно-активным веществом. Таблетки, полученные путем формования, могут быть приготовлены формованием в соответствующем устройстве смеси порошкообразного соединения с жидким инертным разбавителем. Приготовленные вышеуказанными способами таблетки могут быть покрыты оболочкой или иметь надрез. Они могут быть приготовлены таким образом, чтобы при приеме их обеспечивался медленный или контролируемый переход активного компонента в организм больного, для чего они покрываются слоем оксипропилметилцеллюлозы соответствующей толщины.
Для лечения глаз или других внешних органов, например рта и кожи, предпочтительно применять композиции, приготовленные в виде мази или крема, содержащих активный компонент в количестве, например, 0,075-20, предпочтительно 0,2-15, наиболее предпочтительно 0,5-10 мас. В этом случае, если композиция представляет собой мазь, активный компонент используется в сочетании с парафиновой или смешивающейся с водой мазевой основой. На основе активных компонентов могут быть приготовлены также кремы с использованием в качестве основы эмульсии типа масло в воде.
При желании водная фаза кремовой основы может включать, например, по меньшей мере 30 мас. многоатомного спирта, т.е. спирта, содержащего две или более гидроксильных групп, например пропиленгликоля, бутан-1,3-диола, маннитола, сорбитола, глицерола, полиэтиленгликоля или смеси указанных спиртов. Композиции для местного применения могут содержать соединение, которое способствует абсорбции или проникновению активного компонента через кожу или другие обрабатываемые поверхности. Примерами таких соединений, повышающих способность активных компонентов проникать через кожу, являются диметилсульфоксид и аналогичные ему соединения.
Масляная фаза эмульсий в соответствии с настоящим изобретением может состоять из известных компонентов и приготавливается известными способами. Такая фаза может содержать только эмульгатор. Желательно, чтобы она содержала смесь по меньшей мере одного эмульгатора с жиром или маслом, или с тем и другим. Предпочтительно, чтобы в состав ее входили гидрофильный и олефиновый эмульгатор (последний играет роль стабилизатора). Предпочтительно также, чтобы она включала и масло, и жир. Вместе взятые эмульгатор(ы) с или без стабилизатор(а)ов образуют так называемый эмульгирующий воск, а этот воск с маслом и/или жиром образуют так называемую эмульгирующую основу, являющуюся масляной фазой кремовых композиций.
Подходящими для использования в композициях в соответствии с настоящим изобретением эмульгаторами и стабили- заторами эмульсий являются Твин 60, Спан 80, цетостеариловый спирт, миристиловый спирт, глицерилмоностеарат и раурилсульфат натрия.
Выбор того или иного масла или жира для получения композиций диктуется необходимостью достижения нужных косметических свойств, поскольку растворимость активных соединений в большинстве масел, которые могут использоваться в эмульсионных композициях, очень мала. Предпочтительно, чтобы получаемый крем был нежирным, не оставлял пятен, был бы смываемым и имел бы такую консистенцию, чтобы не вытекал из тюбика(ов) или других емкостей. Можно использовать для этой цели одно- или двухатомные алкиловые эфиры с прямой или разветвленной цепью, такие как диизоадипаты, изоцетилстеарат, пропиленгликолевые эфиры жирных кислот, масла кокосового ореха, изопропилмиристат, децилолета, изопропилпальмитат, бутилстеарат, 2-этилгексилпальмитат или смесь эфиров с разветвленной цепью, известных под названием Кродамол САР. Предпочтительными являются три последние из названных эфиров. Они могут использоваться как сами по себе, так и в комбинации, причем выбор зависит от того, с какими свойствами хотят получить композицию. Можно использовать, кроме того, липиды с высокой температурой плавления, такие как белый мягкий парафин и/или жидкий парафин, или другие минеральные масла.
Композиции, пригодные для местного применения, а именно для лечения глаз, включают также глазные капли, в которых активный компонент растворен или суспендирован в подходящем носителе, в частности в водном растворителе активного компонента. Активный компонент содежится в таких композициях в количестве 0,5-20, предпочтительно 0,5-10, наиболее предпочтительно примерно 1,5 мас.
Композиции для местного применения, которые могут использоваться для лечения рта, изготавливаются в виде лепешек, содержащих активный компонент на вкусовой основе, как правило, на сахарозе, аравийской камеди или траганте, пастилок, содержащих активный компонент на инертной основе, например желатине и глицерине или сахарозе и аравийской камеди; и жидкостей для полоскания, содержащих активный компонент в среде подходящего жидкого носителя.
Композиции для ректального введения могут быть приготовлены в виде свечей на подходящей основе, например кокосовом масле или салицилате.
Композиции для введения в нос с твердым носителем содержат порошок с частицами крупного размера (например, порядка 20-500 мкм). Эти композиции вводятся таким же образом, как нюхательный табак, т.е. путем быстрой ингаляции через носоглотку порошка из емкости, которую подносят близко к носу. Подходящие композиции, в которых используется жидкий носитель и которые вводятся, например, в виде аэрозоля или капель в нос, содержат водные или масляные растворы активного компонента.
Композиции для вагинального введения могут изготавливаться в виде пессариев, тампонов, кремов, гелей, паст, пен или аэрозолей, содержащих помимо активного компонента используемые обычно для этих целей подходящие носители.
Композиции для парентерального введения включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиокислители, буферные добавки, средства, задерживающие рост микробов, и растворенные вещества, обеспечивающие изотоничность композиции с кровью пациента; водные и неводные стерильные суспензии, которые могут содержать диспергаторы и загустители. Такие композиции могут выпускаться в емкостях для единичных или нескольких доз, например в запаянных ампулах и пузырьках. Они могут храниться в лиофилизированном (достигнутом в результате сублимационной сушки) состоянии. Для использования их в этом случае непосредственно перед применением достаточно лишь добавить стерильный жидкий носитель. Приготовленные для непосредственного приема растворы и суспензии для инъекций могут быть получены из стерильных порошков, гранул и таблеток, описанных ранее.
Предпочтительными дозированными композициями являются композиции, содержащие, как это отмечалось, дневную или меньшую единичную дозу (или ее часть) активного компонента.
Следует отметить, что по мимо вышеперечисленных компонентов композиции в соответствии с настоящим изобретением могут содержать и другие соответствующие данному типу композиции добавки. Так, например, композиции для орального введения могут содержать вкусовые компоненты.
Соединения в соответствии с настоящим изобретением могут быть получены любым из известных для получения таких или аналогичных соединений способом (см. например, патент Великобритании N 1601020 или статью Robins M.J. и Barb. P.J.b J. Org. Chem. (1983) 48, 1854-1862), а также способом, описанным в нижеприведенных примерах.
Более конкретно в соответствии с предлагаемым способом предлагается получение соединений формулы I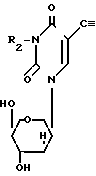
 где R1 атом водорода или метил; R2 бензоил, необязательно замещенный или монозамещенный галогеном или алкокси заместителем путем взаимодействия соединения формулы II
где R1 атом водорода или метил; R2 бензоил, необязательно замещенный или монозамещенный галогеном или алкокси заместителем путем взаимодействия соединения формулы II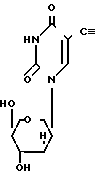
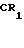 в которой R1 имеет вышеприведенные значения с хлористым бензоилом, способным давать необходимую бензоильную группу в положении 3N, при этом хлористый бензоил может быть незамещенным или монозамещенным галоидом или метоксигруппой.
в которой R1 имеет вышеприведенные значения с хлористым бензоилом, способным давать необходимую бензоильную группу в положении 3N, при этом хлористый бензоил может быть незамещенным или монозамещенным галоидом или метоксигруппой.
П р и м е р 1. 3-N-бензоил-2'-деокси-5-этинилуридин.
К перемешиваемой суспензии 0,3 г (1,19 ммоль) 2'-деокси-5-этинилуридина (J. Med. Chem. 26(5), 661-666, (1983)) в сухом ацетонитриле (8 мл) и хлортриметилсилане (0,5 мл) добавляют при охлаждении льдом 0,85 мл (6,1 ммоль) триэтиламина и перемешивают смесь в течение 2 ч при комнатной температуре. После этого к ней добавляют 0,18 мл (1,54 ммоль) хлористого бензоила, после чего продолжают перемешивание в течение еще 1,5 ч и проводят фильтрацию, фильтрат концентрируют, остаток растворяют в этаноле (10 мл) и добавляют к раствору 0,4 г ледяной уксусной кислоты. После перемешивания в течение 0,5 ч растворители отгоняют, а остаток подвергают очистке с помощью колоночной хроматограции на силикагеле с использованием в качестве элюента смесь метиленхлорида и метанола, взятых в соотношении 19:1. В результате получают 0,2 г (47% от теоретического выхода) целевого соединения с тем.пл. 156-157оС.
Результаты анализа.
Рассчитано, C 60,70, H 4,49, N 7,86.
Найдено, C 60,44, H 4,336, N 7,69.
П р и м е р 2. 3-N-бензоил-2'-деокси-5-пропинилуридин.
К перемешиваемой суспензии 0,4 г (1,5 ммоль) 2'-деокси-5-пропинилуридина и 0,54 г (4,97 ммоль) триметилсилилхлорида в 10 мл сухого ацетонитрила добавляют при 0оС 1,1 мл сухого триэтиламина. Смесь перемешивают при комнатной температуре в течение 2,5 ч, после чего добавляют к ней 0,3 мл бензоилхлорида и продолжают перемешивание в течение 5 ч. Выпадающий осадок отфильтровывают, фильтрат упаривают досуха, остаток растворяют в этаноле и добавляют к раствору 0,4 г уксусной кислоты, после чего смесь перемешивают в течение 0,5 ч при комнатной температуре. Затем ее упаривают досуха и остаток подвергают элюированию на колонке, заполненной силикагелем, смесью CH2Cl2 и MeOH (9:1). После перекристаллизации несколько раз из водного раствора этанола получают 0,14 г (25%) целевого продукта с т.пл. 137-140оС.
Результаты анализа из расчета на формулу C19H18N2O6˙ 0,2H2O
Рассчитано, C 60,96, H 4,920, N 7,489
Найдено, C 60,69, H 4,681, N 7,478.
П р и м е р 3. 2'-Деокси-5-пропинил-3N-(парафторбензоил)-уридин.
2'-Деокси-5-пропинилуридин (J. Med. Chem. 26(5) 661-666 (1983)) (0,4 г, 1,5 ммоль) в 15 мл осушенного пиридина перемешивают в ледяной бане и обрабатывают, 0,95 мл (0,81 г, 7,5 ммоль) триметилхлорсилана. После перемешивания в течение 45 мин при 0оС добавляют 0,44 мл (0,59 г, 3,75 ммоль) парафторбензоилхлорида и смесь перемешивают при комнатной температуре в течение 5 ч, после чего ее хранят в холодильнике в течение ночи. Затем смесь перемешивают в бане с теплой водой в течение 3 ч, после чего ее замораживают и реакцию прерывают, добавляя 5 мл воды. Смесь перемешивают при комнатной температуре 1 ч 30 мин, затем выпаривают до масла. Это масло распределяют между водой и эфиром. Органический слой выпаривают досуха, растворяют в этилацетате и промывают насыщенным раствором бикарбоната натрия, сушат над сульфатом натрия и выпаривают досуха. Остаток хроматографируют на силикагеле смесью 4% метанола в дихлорметане, и необходимую фракцию растирают в смеси эфира с петролейным эфиром, чтобы получить белое твердое вещество.
Т.пл. 162-165оС.
Вычислено, C 58,76, H 4,381, N 7,22, F 4,90.
Найдено, C 59,21, H 4,321, N 7,268, F 4,85.
П р и м е р 4. 2'-Деокси-5-пропинил-3N-(парахлорбензоил)-уридин.
2'-Деокси-5-пропинилуридин (J. Med. Chem. 26(5), 661-666 (1983)) (0,4 г, 1,5 ммоль) растворяют в 15 мл осушенного пиридина и перемешивают в бане со льдом. Спустя 5 мин при 0оС добавляют 0,95 мл (0,81 г, 7,5 ммоль) триметилхлорсилана и перемешивание продолжают при 0оС в течение 1 ч. Добавляют парахлорбензоилхлорид (0,48 мл, 0,66 г, 3,75 ммоль) и смесь перемешивают при комнатной температуре 6 ч, затем хранят в холодильнике в течение ночи. После перемешивания еще в течение 2 ч при комнатной температуре смесь охлаждают льдом, реакцию прерывают водой (3 мл) и затем перемешивают при комнатной температуре в течение 2 ч 30 мин. Растворители выпаривают, чтобы получить масло. Это масло распределяют между водой и эфиром. Водный раствор снова промывают эфиром. Органические промывки объединяют и выпаривают досуха, растворяют остаток в этилацетате и промывают насыщенным раствором бикарбоната натрия (2 раза по 25 мл), затем сушат над сульфатом натрия и выпаривают досуха, получая белое пенистое твердое вещество, которое подвергают кристаллизации из эфира, содержащего немного этанола.
Т.пл. 162-165оС.
Вычислено, C 56,37; H 4,203; N 6,92; Cl 8,78.
Найдено, C 56,36; H 4,066; N 6,864, Cl 3,95.
П р и м е р 5. 2'-Деокси-5-пропинил-3N-(параметоксибензоил)- уридин.
2'-Деокси-5-пропинилуридин (J. Med. Chem. 26(5), 661-666 (1983)) (0,4 г, 1,5 ммоль) растворяют в 15 мл осушенного пиридина и перемешивают при 0оС в бане со льдом, в раствор добавляют 0,95 мл (0,81 г, 7,5 ммоль) хлортриметилсилана. После перемешивания во льду в течение 1 ч добавляют 0,34 мл (3,75 ммоль) параанизолилхлорида и смесь перемешивают при комнатной температуре в течение ночи. Поскольку промежуточное вещество еще присутствует, добавляют диметиламинопиридин и продолжают перемешивание в бане с теплой водой (40оС) в течение суток. Смесь охлаждают в бане со льдом, и реакцию прерывают 3 мл воды и перемешивают при комнатной температуре 1 ч, после чего смесь выпаривают досуха, чтобы получить масло. Его растворяют в этилацетате и промывают насыщенным раствором бикарбоната натрия (3 раза по 25 мл) и водой (25 мл), сушат и выпаривают. Образовавшееся пенистое твердое вещество хроматографируют на флэшколонке, элюируемой 5% метанола в дихлорметане. Соответствующие фракции собирают, растирают в смеси эфира с гексаном, и отфильтровывают белый порошок. Выход 150 мг (25%).
Т.пл. 179-181оС.
Вычислено, C 59,46, H 5,055, N 6,94.
Найдено, C 59,45, H 4,80, N 6,701.
П р и м е р 6. Глазные капли, г: Активный компонент 0,5 Хлорид натрия квали- фикации ч.д.а. 0,9 Тиомерсал 0,001 Очищенная вода До 100 мл рН 7,5
П р и м е р 7. Таблетки.
Нижеследующие композиции А и В готовят путем мокрой грануляции компонентов с раствором повидона, с последующим добавлением стеарата магния и прессованием.
Композиция А
мг/таб- мг/таб-
летку летку а) активный компонент 250 250 b) лактоза В.Р 210 26 с) повидон В.Р. 15 9 d) натриевый крахмал гли- колят 20 12 е) стеарат маг- ния 5 3
---- ----
500 300
Композиция В
мг/таб- мг/таб-
летку летку а) активный компонент 250 250 d) лактоза 150 с) авицел РН 101 60 26 d) повидон В.Р. 15 9 е) натриевый крахмал гли- колят 20 12 f) стеарат маг- ния 5 3
----- -----
500 300 Композиция С, мг/таблетку: Активный компонент 100 Лактоза 200 Крахмал 50 Повидон 5 Стеарат магния 4
-----
359
Нижеследующие композиции D и Е готовили путем непосредственного прессования смеси компонентов. Используемая в композиции Е лактоза предназначена для прямого прессования. Композиция D, мг/капсулу: Активный компонент 250 Предварительно же- латинизированный крахмал F15 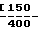 Композиция Е, мг/капсулу: Активный компонент 250 Лактоза 150 Авицел
Композиция Е, мг/капсулу: Активный компонент 250 Лактоза 150 Авицел 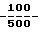
Композиция F (с контролируемым освобождением активного компонента). Эту композицию готовят путем мокрого гранулирования нижеперечисленных компонентов с раствором повидона с последующими добавлением стеарата магния и прессования. Состав композиции F, мг/таблетку: а) активный компонент 500 b) оксипропилметилцел- люлоза (Methocel K4M Premium) 112 c) лактоза В.Р. 53 d) повидон В.Р.С. 28 е) стеарат магния 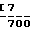
Переход активного компонента в организм происходит в течение примерно 6-8 ч и завершается через 12 ч.
П р и м е р 8. Капсулирование композиции.
Композиция А.
Капсулированную композицию получают путем смешения компонентов композиции D вышеприведенного примера 7 и заполнения этой смесью состоящей из двух частей капсулы из твердого желатина. Композицию В (инфра) готовят аналогичным образом. Композиция В, мг/капсулу: а) активный компонент 250 b) лактоза В. Р. 143 с) натриевый крахмал гликолят 25 d) стеарат магния 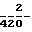 Композиция С, мг/капсулу: а) активный компонент 250 b) макрогол 4000 ВР
Композиция С, мг/капсулу: а) активный компонент 250 b) макрогол 4000 ВР 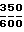
Капсулы получают путем плавления макрогола 4000 ВР, диспергирования активного компонента в расплаве и заполнения расплавом состоящей из двух частей капсулы из твердого желатина. Композиция D, мг/капсулу: Активный компонент 250 Лецитин 100 Арахисовое масло 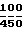
Капсулы получают путем диспергирования активного компонента в лецитине и арахисовом масле и заполнения дисперсией эластичных капсул из мягкого желатина.
Композиции Е (капсулы с контролируемым выходом активного компонента). Нижеописанные капсулы с контролируемым выходом активного компонента получают путем экстразии компонентов, a, b и с с помощью экструдера с последующей стероидизацией и сушкой экструдата. На высушенные шарики наносят мембрану (d), контролирующую скорость проникновения через нее активного компонента, и заполняют ими состоящие из двух частей капсулы из твердого желатина. Состав композиции Е, мг/капсулу: а) активный компонент 250 b) микрокристалличес- кая целлюлоза 125 с) лактоза ВР 125 d) этилцеллюлоза 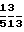
П р и м е р 9. Композиции для инъекций: Активный компонент 0,200 г стерильный, не содер- жащий пирогенных примесей фосфатный буфер (рН 7,0) До 10 мл
Активный компонент растворяют в большей части фосфатного буфера (35-40оС), раствор доводят до нужного объема и фильтруют через стерильный микропористый фильтр и стерильную ампулу объемом 10 мл из янтарного стекла (тип 1) и герметизируют ее.
П р и м е р 10. Композиция для внутримышечных инъекций, г: Активный компонент 0,20 Бензиловый спирт 0,10 Гликофурол 75 1,45 Вода для инъекций в достаточном коли- честве До 3,0 мл
Активный компонент растворяют в гликофуроле, после чего к раствору добавляют бензиловый спирт и растворяют его и добавляют воду до 3 мл. Смесь фильтруют через стерильный микропористый фильтр и заливают в стерильные стеклянные ампулы объемом 3 мл (тип 1), которые запаивают.
П р и м е р 11. Сиропообразная суспензия, г: Активный компонент 0,2500 Раствор сорбитола 1,5000 Глицерол 2,000 Диспергирующаяся целлюлоза 0,0750 Бензоат натрия 0,0050 Вкусовая добавка Peach 17.42.3169 0,0125 мл Очищенная вода в достаточном коли- честве До 5,0000 мл
Бензоат натрия растворяют в части очищенной воды и добавляют к раствору раствор сорбитола. После этого добавляют активный компонент и диспергируют его. В глицероле диспергируют загуститель (диспергирующуюся целлюлозу). Смешивают обе суспензии и доводят объем до нужной величины очищенной водой. При необходимости дальнейшее увеличение вязкости достигается путем увеличения концентрации суспензии.
П р и м е р 12. Суппозитории, мг/суппозиторий: Активный компонент (63 мкм) 250 Твердый жир, ВР (ви- тепсол Н15-Динамит (Нобель) 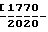
Активный компонент используется в виде порошка, в котором не менее 90% частиц имеют диаметр 63 или менее мкм.
Одну пятую витепсола Н15 плавят в поддоне с паровой рубашкой при температуре не выше 45оС. Активный компонент просеивают через сито с размером ячеек 200 мкм и добавляют при перемешивании к расплаву, используя сильверсон с режущей насадкой, до получения однородной суспензии. Поддерживая температуру смеси равной 45оС, к ней добавляют оставшийся витепсол Н15 и перемешивают суспензию для получения однородной смеси. Суспензию пропускают затем через сито из нержавеющей стали с ячейками диаметром 250 мкм и при непрерывном перемешивании дают ей остыть до 40оС. При температуре 38-40оС 2,02 г смеси заливают в пластиковые формы. Полученные суппозитории охлаждают до комнатной температуры.
П р и м е р 13. Пессарии, мг/пессарий: Активный компонент (63 мкм) 250 Ангидрат декстрозы 380 Картофельный крахмал 363 Стеарат магния 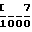
Смешивают вышеперечисленные компоненты и из полученной смеси готовят пессарии путем прямого прессования.
Испытания на токсичность и антивирусную активность.
Вирус цитомегалии человека (НСМУ) определяли в монослоях клеток МРС5 (эмбриональные клетки легкого человека) или Detroit 532 (крайняя плоть фибропласта человека) в ячеистых поддонах. Активность соединений определяли в опытах по уменьшению количества кровяных пластинок, в которых монослой клеток заражали суспензией НСМУ, после чего сверху наносили слой питательной агарозы в форме геля, для того чтобы предотвратить распространение вируса через культуру. Поверх питательной агарозы наносили растворы испытуемого соединения различной известной молярной концентрации. Для каждой концентрации определяли количество кровяных пластинок, выражая их в процентах от количества пластинок в контрольном опыте, и строили соответствующую кривую зависимости от концентрации. Из этой кривой находили концентрацию (IC50), при которой наблюдалось 50%-ное торможение.
Вирус герпеса опоясывающего генерализованного (YZY) определяли в клетках МРС5 аналогичным образом, с той разницей, что поверх монослоя клеток не наносили слой агарозы.
В ходе опыта продуцирующие вирус клетки (РЗНR-1) в течение 14 дней подвергали воздействию препарата, после чего определяли количество копий генома ЕВУ в клетке, для чего осуществляли специфическую на ЕВУ гибридизацию с РНК-ДНК Эпштейна-Варра определяли методами, Nomo yama и pagauo описанным в Nature New, Biology, т. 233, с. 103-4, 1971. Полученное в результате значение IC50 представляет собой концентрацию, необходимую для торможения на 50% количества геномов ЕВУ в клетке.
Клеточную токсичность определяли в опытах по торможению роста клеток. Субконфлюентные культуры клеток Yeгo, выращенные в 96-ячеистых Microtiter сосудах, подвергали воздействию препаратов с различным содержанием активного вещества и ежедневно определяли жизнеспособность клеток на репликативных культурах по поглощению тетразолиевого красителя (МТТ). Концентрация, при которой происходило 50%-ное торможение жизнеспособности клеток через 96 ч, принимали за CClD50.
Полученные результаты приведены в таблице.
| название | год | авторы | номер документа |
|---|---|---|---|
| СИНЕРГЕТИЧЕСКАЯ ФАРМАЦЕВТИЧЕСКАЯ КОМБИНАЦИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЛЕЧЕНИЯ ИЛИ ПРОФИЛАКТИКИ ВИЧ-ИНФЕКЦИЙ | 1992 |
|
RU2110993C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЗАМЕЩЕННЫХ ПУРИНОВЫХ АРАБИНОЗИДОВ ИЛИ ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ ПРОИЗВОДНЫХ | 1988 |
|
RU2039752C1 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ПРОЯВЛЯЮЩАЯ АКТИВНОСТЬ ПРОТИВ ВИРУСНОЙ ИНФЕКЦИИ И β D-АРАБИНОФУРАНОЗИЛ-2-АМИНО-6-МЕТОКСИ-9Н-ПУРИНЫ | 1993 |
|
RU2112765C1 |
| СПОСОБ ИНГИБИРОВАНИЯ ВИРУСНЫХ ИНФЕКЦИЙ ГЕПАТИТА В | 1992 |
|
RU2116789C1 |
| Способ получения пиримидиновых нуклеозидов | 1987 |
|
SU1731064A3 |
| ЭНАНТИОМЕРНЫЕ СОЕДИНЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1992 |
|
RU2091386C1 |
| СПОСОБ ПОЛУЧЕНИЯ ИМИДАЗОПИРИДАЗИНОВ | 1989 |
|
RU2017741C1 |
| СПОСОБ ЛЕЧЕНИЯ ГЕПАТИТА В | 1992 |
|
RU2104700C1 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНЫХ СОЕДИНЕНИЙ ИЛИ ИХ ПРОИЗВОДНЫХ | 1990 |
|
RU2068849C1 |
| Способ получения производных флавана или их солей | 1979 |
|
SU1072805A3 |

Сущность изобретения: продукт - пиридиновые нуклеозиды ф-лы 1, где R1 -H, CH3, R2 -бензоил, возможно монозамещен Hal или алкокси. Реагент 1 : R2 незамещенный нуклеозид. Реагент 2: хлористый бензоил, возможно замещенный. Условия реакции: при комнатной температуре. Соединения обладают противовирусной активностью (группы герпеса). 1 табл. Структура ф-лы I 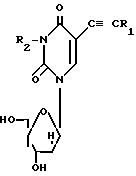
СПОСОБ ПОЛУЧЕНИЯ ПИРИДИНОВЫХ НУКЛЕОЗИДОВ общей формулы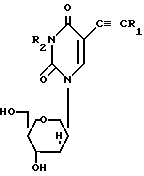
где R1 водород или метильная группа;
R2 бензоильная группа, необязательно замещенная или монозамещенная галоидом или алкокси,
отличающийся тем, что осуществляют взаимодействие соединения общей формулы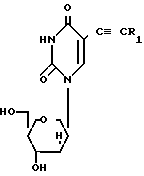
где R1 имеет указанные значения,
при комнатной температуре с хлористым бензоилом, незамещенным или монозамещенным галоидом или метокси.
| Устройство для передачи грузов между судами в море в условиях качки | 1988 |
|
SU1601020A1 |
| Способ восстановления хромовой кислоты, в частности для получения хромовых квасцов | 1921 |
|
SU7A1 |
